Embed Size (px)
Citation preview

JMB—MS 565 Cust. Ref. No. RH 007/95 [SGML]
J. Mol. Biol. (1995) 249, 785–799
Crystal Structure of Escherichia coli QOR QuinoneOxidoreductase Complexed with NADPH
Jennifer M. Thorn, John D. Barton, Nicholas E. Dixon, David L. Ollisand Karen J. Edwards*
The crystal structure of the homodimer of quinone oxidoreductase fromCentre for MolecularEscherichia coli has been determined using the multiple isomorphousStructure and Functionreplacement method at 2.2 Å resolution and refined to an R-factor of 14.1%Research School of Chemistry
Australian National The crystallographic asymmetric unit contains one functional dimer with theUniversity, Canberra two subunits being related by a non-crystallographic 2-fold symmetry axis.ACT. 0200, Australia The model consists of two polypeptide chains (residues 2 through 327), one
NADPH molecule and one sulphate anion per subunit, and 432 watermolecules. Each subunit consists of two domains: a catalytic domain and anucleotide-binding domain with the NADPH co-factor bound in the cleftbetween domains. Quinone oxidoreductase has an unusual nucleotide-bind-ing fingerprint motif consisting of the sequence AXXGXXG. The overallstructure of quinone oxidoreductase shows strong structural homology tothat of horse liver alcohol dehydrogenase.
Keywords: quinone oxidoreductase; alcohol dehydrogenase; z-crystallin;*Corresponding author crystal structure; NADPH
Introduction
In a recent report we identified a soluble enzymewith NAD(P)H-dependent quinone oxidoreductase(QOR) activity in Escherichia coli (Lilley et al., 1993).The enzyme reduces a variety of synthetic quinonesubstrates, is inactive towards menadione, anaturally occurring quinone, and is inhibited bydicumerol which is a common inhibitor of quinoneoxidoreductases (Lilley et al., unpublished results).Although membrane-bound quinone oxidoreduc-tases are present in bacteria, few examples of solublequinone oxidoreductases are to be found. In higherorganisms there appear to be two distinct classes ofsoluble quinone oxidoreductase. QOR has significantsequence identity to one of these, z-crystallin, andshows similar reactivity to this enzyme. Both QORand z-crystallin (Rodokanaki et al., 1989) showsequence similarity to horse liver alcohol dehydro-genase (LADH), suggesting that they may be part ofa super-family of structurally related enzymes.
Membrane-bound quinone oxidoreductases foundin bacteria are involved in the respiratory chain andresemble the mitochondrial NADH-quinone oxido-
reductase enzymes (Yagi, 1991). These enzymes areNADH-dependent, require either FMN or FAD andmay also possess iron-sulphur clusters as prostheticgroups. By contrast, the soluble quinone reductasesare simpler enzymes and have quite differentproperties and functions. The properties of tworepresentative enzymes, DT-diaphorase (EC 1.6.99.2)and z-crystallin are summarized in Table 1. A majordifference between these enzymes is their respectivereductive mechanism. DT-diaphorase is a widelydistributed enzyme which catalyses the NAD(P)H-dependent two-electron reduction of quinonesubstrates (Ernster et al., 1987) and requires FAD asan additional prosthetic group. This reductionresults in the formation of hydroquinones which canthen be inactivated by glucuronidation (Presteraet al., 1993). Available evidence suggests that thisenzyme is involved in protecting cells from thecytotoxic effects of quinones which occur widely innature (Ernster et al., 1987; Bellomo et al., 1987). Bycontrast, Rao et al. (1992) have shown that z-crystallinacts through a one-electron transfer process toproduce a semiquinone radical. This radical maythen be further non-enzymatically reduced to thehydroquinone or oxidized back to quinone in thepresence of O2, resulting in the production of H2O2.The cellular distribution of z-crystallin suggeststhat it may serve a dual role in mammalian cells.z-Crystallin is a major taxon-specific structural eyelens protein of some hystricomorphic rodents and
Abbreviations used: QOR, quinone oxidoreductase;LADH, liver alcohol dehydrogenase; GDH, glucosedehydrogenase; MIR, multiple isomorphousreplacement; SA, simulated annealing; NCS,non-crystallographic symmetry.
0022–2836/95/240785–15 $08.00/0 7 1995 Academic Press Limited

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase786
Table 1. Properties of soluble classes of quinoneoxidoreductaseExample DT-diaphorase z-Crystallin
Mechanism 2-Electron 1-ElectronCo-factor NAD(P)H NADPHrequirements FAD
absent in QOR and z-crystallin, and the threeresidues which bind the catalytic Zn atom in LADHhave been replaced by both enzymes. The likelyabsence of the catalytic Zn atom thus explains thelack of alcohol dehydrogenase activity in both QORand z-crystallin.
We report here the refined structure of the binarycomplex of QOR and NADPH and give details of thecofactor–enzyme interaction. We also identify theprobable site of substrate binding.
Results and Discussion
Quality of the structure
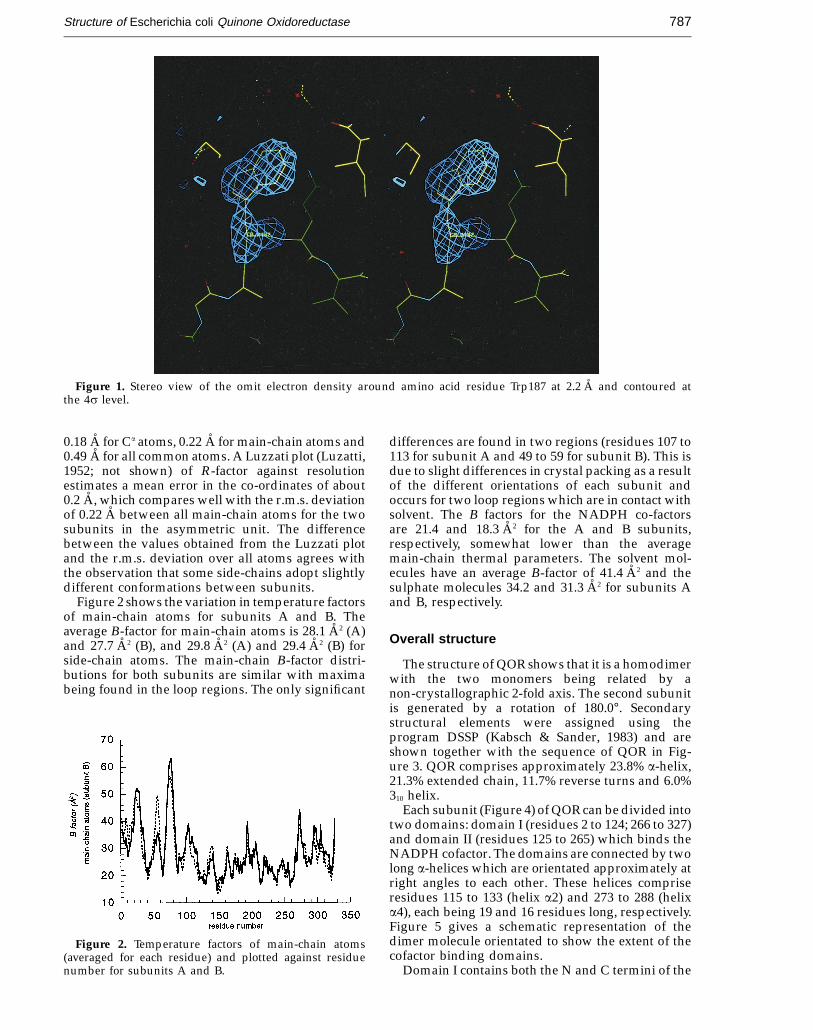
Although the sequence of QOR was predicted tostart with Met1, there was no suggestion of thisresidue in any of our electron density maps.Subsequent N terminus sequencing (Lilley et al.,unpublished results) showed that this residue wasabsent from the polypeptide chain. All residues weretherefore located in our model. Figure 1 shows asection of electron density from an omit map aroundTyr187. The main-chain density in the final 2Fo − Fc,ac map is contiguous at the 1s level for all main-chainatoms of the model except for a few minor breaks insurface loop regions. A number of polar side-chains,28 out of a total of 652 residues, have weak orunobserved density and may thus exist in multipleconformations.
The geometries of the main-chain and side-chainswere analysed using the program PROCHECK(Laskowski et al., 1993). Ramachandran plots(Ramachandran & Sasikharan, 1968; not shown) ofthe f–c conformational torsion angles of subunits Aand B showed that the overall distribution ofconformations in both subunits was very similar. Forboth subunits all non-glycine residues are found infavourable regions, with the exception of tworesidues in subunit A (Leu209 and Asp141), and oneresidue in subunit B (Leu209), being found inpartially allowed regions. The final 2Fo − Fc mapsshow good electron density for these residues. Thereare no residues in disallowed regions. One of theproline residues, Pro58, in both A and B chains formsa cis-peptide bond.
Superposition of the A and B subunits byleast-squares methods gave an r.m.s. deviation of
camels (Rao et al., 1992). In addition, it has beenfound at enzymatic levels in the liver and kidneytissues of both humans and guinea pig (Rao &Zigler, 1992; Gonzalez et al., 1993), suggesting ametabolic role or possibly a role in the detoxificationof quinones (Tumminia et al., 1993). AlthoughQOR has significant sequence homology and showssimilar reactivity to z-crystallin, its mechanistic simi-larity to this enzyme has yet to be confirmed and itsphysiological role elucidated.
QOR belongs to a structural class of enzymes thatinclude LADH (Eklund et al., 1976) and glucosedehydrogenase (GDH: John et al., 1994) as shown inTable 2. QOR, in common with z-crystallin, exhibitshigh sequence similarity to LADH (Borras et al.,1989). This is significant in the NAD-binding domainwhere major residues are conserved, but moresurprising is the high degree of identity shown in thecatalytic domain, particularly the N-terminal region.Absent from both QOR and z-crystallin is the largeloop found at the surface of the catalytic domain inLADH which binds the structural zinc atom (Eklund& Branden, 1987). It is interesting to note thatQOR (Lilley et al., unpublished results) and LADH(Eklund et al., 1976) consist of dimers of identicalsubunits, whereas z-crystallin and GDH have atetrameric quaternary structure (Huang et al., 1987;John et al., 1994). Previous observations (Borras et al.,1989; Eklund et al., 1985; Jornvall et al., 1978) havecorrelated the absence of the structural Zn-bindingloop with tetrameric enzyme structure as found inz-crystallin and yeast alcohol and mammaliansorbitol dehydrogenases. GDH, by contrast, doespossess a structural Zn atom in addition to thecatalytic Zn but the binding loop for the structural Znatom is in a different orientation from that for LADH.This Zn-containing structural lobe is indeed thesecond site for subunit–subunit interactions in thetetrameric GDH structure. The four cysteine residueswhich bind the structural Zn atom in LADH are
Table 2. Properties of four enzymes: LADH (liver alcohol dehydrogenase), GDH (glucosedehydrogenase), QOR (quinone oxidoreductase) and z-crystallin, belonging to the same structuralclass
LADH GDH QOR z-Crystallin
No. amino acid residues 374 352 327 328Subunit weight (kDa) 40 40 35 35Quaternary structure Dimer Tetramer Dimer TetramerMetal requirements 2 Zn 2 Zn — —Cofactor NAD + NAD(P) + NAD(P)H NADPHActivity Alcohol Glucose Quinone Quinone
dehydrogenase dehydrogenase reductase reductaseX-ray structure known Yes Yes Yes NoSequence identity to QOR (%) 21 17 — 31
JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 787
Figure 1. Stereo view of the omit electron density around amino acid residue Trp187 at 2.2 A and contoured atthe 4s level.
0.18 A for Ca atoms, 0.22 A for main-chain atoms and0.49 A for all common atoms. A Luzzati plot (Luzatti,1952; not shown) of R-factor against resolutionestimates a mean error in the co-ordinates of about0.2 A, which compares well with the r.m.s. deviationof 0.22 A between all main-chain atoms for the twosubunits in the asymmetric unit. The differencebetween the values obtained from the Luzzati plotand the r.m.s. deviation over all atoms agrees withthe observation that some side-chains adopt slightlydifferent conformations between subunits.
Figure 2 shows the variation in temperature factorsof main-chain atoms for subunits A and B. Theaverage B-factor for main-chain atoms is 28.1 A2 (A)and 27.7 A2 (B), and 29.8 A2 (A) and 29.4 A2 (B) forside-chain atoms. The main-chain B-factor distri-butions for both subunits are similar with maximabeing found in the loop regions. The only significant
differences are found in two regions (residues 107 to113 for subunit A and 49 to 59 for subunit B). This isdue to slight differences in crystal packing as a resultof the different orientations of each subunit andoccurs for two loop regions which are in contact withsolvent. The B factors for the NADPH co-factorsare 21.4 and 18.3 A2 for the A and B subunits,respectively, somewhat lower than the averagemain-chain thermal parameters. The solvent mol-ecules have an average B-factor of 41.4 A2 and thesulphate molecules 34.2 and 31.3 A2 for subunits Aand B, respectively.
Overall structure
The structure of QOR shows that it is a homodimerwith the two monomers being related by anon-crystallographic 2-fold axis. The second subunitis generated by a rotation of 180.0°. Secondarystructural elements were assigned using theprogram DSSP (Kabsch & Sander, 1983) and areshown together with the sequence of QOR in Fig-ure 3. QOR comprises approximately 23.8% a-helix,21.3% extended chain, 11.7% reverse turns and 6.0%310 helix.
Each subunit (Figure 4) of QOR can be divided intotwo domains: domain I (residues 2 to 124; 266 to 327)and domain II (residues 125 to 265) which binds theNADPH cofactor. The domains are connected by twolong a-helices which are orientated approximately atright angles to each other. These helices compriseresidues 115 to 133 (helix a2) and 273 to 288 (helixa4), each being 19 and 16 residues long, respectively.Figure 5 gives a schematic representation of thedimer molecule orientated to show the extent of thecofactor binding domains.
Domain I contains both the N and C termini of the
Figure 2. Temperature factors of main-chain atoms(averaged for each residue) and plotted against residuenumber for subunits A and B.

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase788
Figure 3. Sequence of E. coli QOR determined from the cloned gene (Lilley et al., unpublished results) together withthe secondary structure assignments for subunits A and B as assigned by the program DSSP (Kabsch & Sander, 1983).Abbreviations: H, a-helix; E, b-strand; G, 310-helix; T, turn; B, bridge; S, bend. Helices comprise four or more residues andstrands three or more residues. Secondary structure elements have been labelled so as to be consistent with that of LADH(Eklund et al., 1976).
polypeptide chain. Based largely on analogies toLADH and also from the position of the catalytichydride on the nicotinamide ring of NADPH, weassume that the catalytic site is encompassed bythis domain and it will be referred to as thecatalytic domain. Inspection of this domain alsoshows the presence of a large cleft near thenicotinamide group of the cofactor. This domainhas a complex secondary structural arrangementand is formed by three b-sheets, bI, bII, bIII, andtwo a-helices, a1 and a5. The simplest and leastsignificant sheet, bIII, consists of two antiparallelstrands, b1 and b2. Sheet bI is comprised of sixstrands; two parallel strands, b11 and b12, at oneend and four strands (b4, b5, b7 and b10) whichrun antiparallel to each other and b12. b11 and b12form a typical bab-type fold and are connected viahelix a5. The only other helix within this domainis found as part of the large loop region connectingb4 and b5. The third sheet (bII) is formed by fourantiparallel strands, b3, b6, b8, b9, and lies acrossand approximately at right angles to sheet bI. Thereis a region of poorly defined extended chain which
runs parallel to b6 but is not in hydrogen-bondedcontact.
Sheets bI and bII together form a barrel-like motifmade up of four strands each, in which the sheets areorientated almost at right angles to each other. Theconnections between antiparallel strands in thisdomain are characterized by a number of tight turnscreating a very compact region of predominantlyb-character. Figure 6 shows a schematic diagram ofthe two stretches of b-sheet (bI and bII) forming thebarrel-like motif in the catalytic domain. This barrelappears to consist of two Greek-key motifs. One ofthese strands (b8) does not consist entirely ofextended coil character but in an ideal situationwould form the fourth strand in this sheet andenables us to describe the structure in simple terms.
Domain II exhibits the classic ‘‘Rossman’’ fold(Rao & Rossman, 1973) and consists of two bababfolds which is common among nucleotide bindingdomains. There is thus a central b sheet (bIV) whichconsists of six parallel b-strands (bA to bF) connectedby a-helices (aA to aF). Helix a2 links the catalyticand NAD domains and crosses over to strand bA at

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 789
Figure 4. (a) Stereo view of the Ca
trace of the QOR monomer colour-coded to show the extent of domainI (cyan) and domain II (magenta).The NADPH cofactor is colouredgreen. Every 20th amino acid isnumbered. (b) Schematic diagramin the same orientation showing thenomenclature of secondary structureelements. Circles represent a-helicesand triangles represent b-sheets.
(a)
(b)

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase790
Figure 5. Schematic representation of the QOR dimer drawn using MOLSCRIPT (Kraulis, 1991) with labelling forsecondary structure elements.
the start of sheet bIV. bA, bB and bC are linked byhelices aB and aC to form the first babab unit. Thereis then a long loop region which contains anadditional helix (aD) before beginning the secondbabab unit consisting of strands bD, bE and bF linkedby helices aE and aF. From bF, the polypeptide chainthen crosses back over to the catalytic domain via ashort helix (a3) prior to the second, long linker helixa4. Domain II forms the interface with the secondsubunit and together the coenzyme-binding regionsof the two subunits form a 12-stranded b-sheetextending across the dimeric molecule. The bFstrands of each subunit run in opposite directionsperpendicular to the non-crystallographic 2-fold axis
and are joined together by hydrogen bonds to formtwo strands of antiparallel structure.
The overall structure of QOR is remarkably similarto that of LADH. There are, however, a number ofdifferences mainly in the catalytic domain due to theabsence of the large structural zinc-binding loop.This loop contributes secondary structure elementsto the structure of LADH which are missing in QOR.A description of the sheet structure for QOR istherefore somewhat different from that of LADH.Differences in the nucleotide-binding domain aredue to slightly different orientations of helices, loopsand off-setting of strands bD, bE and bF with respectto LADH.

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 791
Dimer interface
There are two main regions of contact between thetwo subunits in the QOR dimer, both of which occurin domain II. There are numerous potentialnon-bonded contacts between subunits in addition topredominantly main-chain hydrogen-bonded con-tacts. The first of these regions contains residues 258to 265 and is located between the bF strands of thetwo subunits. These strands lie antiparallel to eachother, are in hydrogen-bonded contact and form theextended 12-stranded b-sheet across the dimerinterface. There are additional contacts betweenthe flanking helices aF (residues 251 to 255) anda3 (residues 268 to 271) of strand bF and thecorresponding bF strand in the second subunitincluding contacts to strand bE (residues 232 to 237)and the turn (residues 230 to 231) between bEand aE.
The second region of contact is the loop region,found directly behind the two bF strands andconnecting strands bE and bF in each subunit. Thisloop forms a short region of extended chain (residues245 to 250) in antiparallel hydrogen-bonded contactto residues from the second subunit.
Solvent-accessible areas were calculated by themethod of Lee & Richards (1971) as implementedby X-PLOR using a probe of radius 1.4 A. Thesolvent-accessible area for the dimer molecule is22,788 A2 and for each subunit is 12,723 A2 and12,654 A2, for A and B, respectively, when treated inisolation. Dimer formation thus buries 2589 A2 ofaccessible protein surface area.
Water structure
In addition to the large number of water moleculesfound on the surface of the enzyme there aresignificant numbers of water molecules buriedwithin the protein. There are a total of 43 buriedwater molecules with an accessible surface area of0.0 A2 for the dimer molecule. Internal watermolecules are thought to play a structural role andprovide hydrogen bond interactions betweenpeptide units (Teeter, 1991). A number of these water
molecules appear to stabilize bent regions ofpolypeptide chain which often occur before or afterb-strands in this structure. They appear to aid in thepositioning of terminal regions of strands so as to becorrectly orientated for b-sheet formation. Examplesof buried water molecules are Wat192, whichprovides the necessary contacts to maintain strandsbA and bD in hydrogen bond contact for sheetformation, and Wat79 and Wat80, which mediatecontact between residues at the terminal ends ofstrands bD and bE and helix aE. In addition, there arenetworks of buried water molecules, particularlyaround the NADPH molecules, which mediateinteractions between the cofactor and protein.
NADPH binding domain
Figure 7 shows the density from an omit map forone of the NADPH molecules and Figure 8 a stereoview of NADPH bound to QOR in the NADPH-bind-ing site. There is strong continuous electron densityover the entire molecule and all side-chains aroundthe cofactor are in well-defined electron density. TheNADPH molecule refines to a similar position ineach subunit with an r.m.s. deviation of 0.14 Abetween the molecules in subunits A and B. Thebinding of NADPH to QOR is similar in bothsubunits. NADPH is bound in the open, extendedconformation in QOR. The distance between the C-6atom of the adenine ring and the C-2 atom of thenicotinamide ring has often been used as a measureof the extension of the coenzyme molecule (Rossmanet al., 1975) and is 13.8 A (subunit A) and 13.6 A(subunit B) for NADPH bound to QOR. Thiscompares to a distance of 15 A for NADH in LADH(Eklund et al., 1984) and 17.1 A for NADPH indihydrofolate reductase (Matthews et al., 1979). Thedistance from PN to N1N is 5.66 A and 5.46 A forsubunits A and B, respectively, and that from PA toN9A is 6.82 A (subunit A) and 6.85 A (subunit B),implying that the nicotinamide half of the moleculeis more bent.
The adenine ribose sugars adopt a C-1'-endoconformation with the adenine bases being syn(x = 52.9° and 54.5° for subunits A and B,
Figure 6. Schematic representationof the b-‘‘barrel-like’’ motif foundin the catalytic domain of QOR.b-Strands are depicted as arrows(bI shaded and bII unshaded) andhelices as filled spheres.

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase792
Figure 7. Omit electron density map for NADPH bound to the B subunit of QOR and contoured at 4s.
respectively) with respect to the glycosidic linkage ineach subunit. By contrast, the ribose sugars for thenicotinamide portion of the molecule adopt a C-4'-exo conformation in subunit A and an O-4'-endoconformation in subunit B. Both of the bases are anti(x = −117.8° and − 115.9° for each subunit A and B,respectively) with respect to the glycosidic linkage.
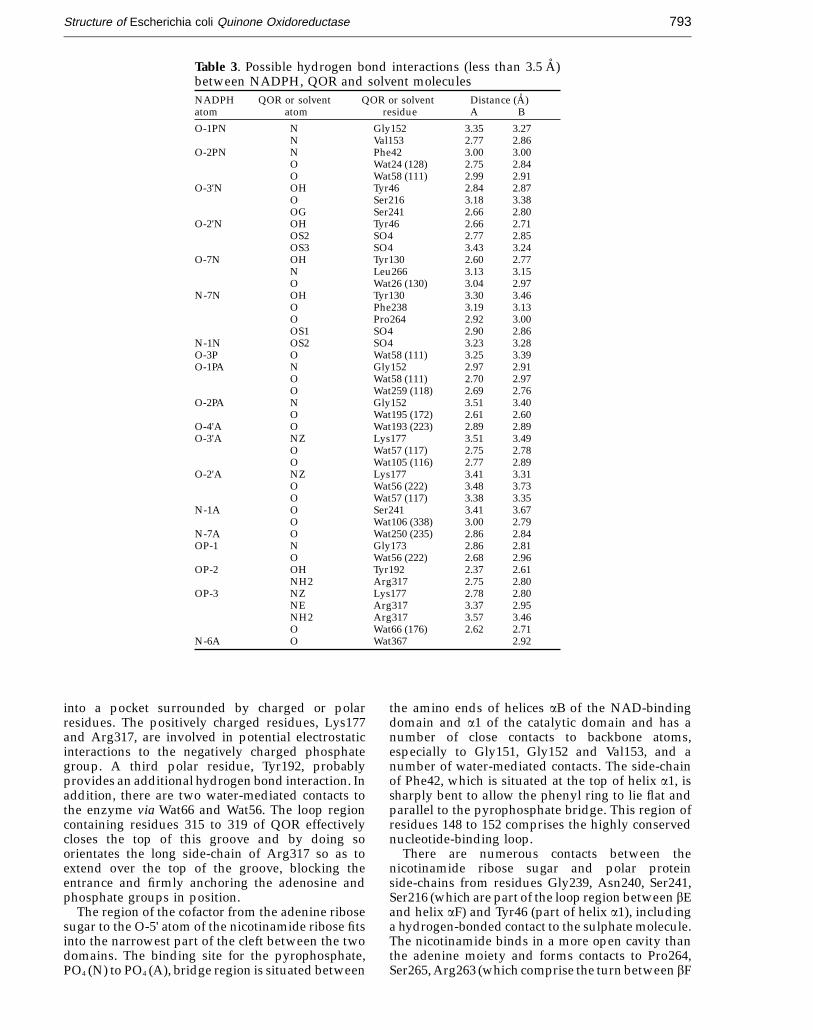
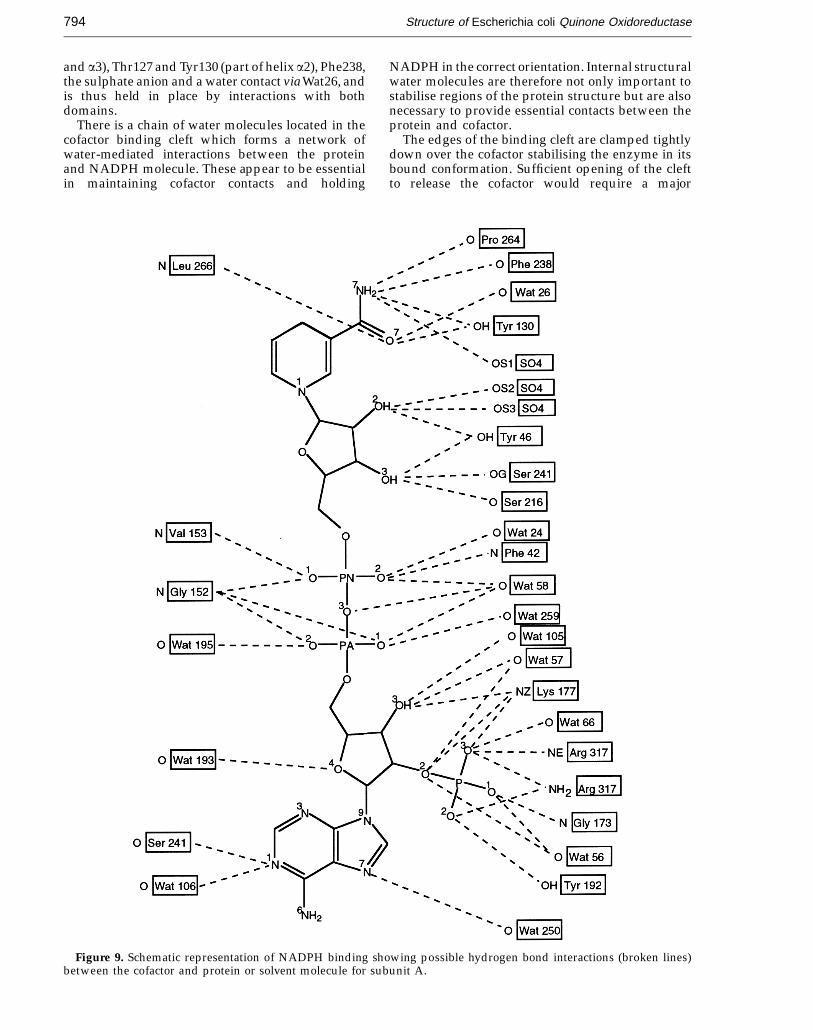
The NADPH molecule binds in the large cleftbetween the two domains of each subunit andinteracts with residues from both domains. NADPHbinds to the protein via hydrogen bond interactionsand non-bonded contacts and also indirectly via anumber of water-mediated contacts. The NADPHmolecule fits tightly into the binding cleft and isalmost completely encompassed by the enzyme.Table 3 gives a list of possible hydrogen-bondingdistances between NADPH, QOR and solvent andFigure 9 shows a schematic representation ofNADPH binding to the enzyme.
The adenine ring of the coenzyme is located in acleft which is part of the coenzyme-binding domainand is of a predominantly polar nature. The NH2
group is orientated to point away from the proteinmolecule and into solution. The adenine fits in aparallel orientation and between a tight turn formedby two serine residues, 241 and 242, which lie at theend of strand bE, and the long, extended side-chainof Arg317, which is part of a tight turn immediatelyadjacent to helix a5. There are a number ofwater-mediated contacts (via Wat106 and Wat250)between the adenine ring and the protein in additionto van der Waal’s contacts to main-chain atoms ofSer242 and side-chain atoms of Arg317. The onlydirect contact to the adenine ribose sugar is apotential hydrogen bond to Lys177. There are,however, three contacts to the protein via watermolecules (Wat193, Wat105 and Wat57).
The adenine ribose phosphate group fits neatly
Figure 8. Stereo view of NADPH bound to QOR on subunit B. Amino acid residues which interact with the adenineribose phosphate are labelled.
JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 793
Table 3. Possible hydrogen bond interactions (less than 3.5 A)between NADPH, QOR and solvent moleculesNADPH QOR or solvent QOR or solvent Distance (A)atom atom residue A B
O-1PN N Gly152 3.35 3.27N Val153 2.77 2.86
O-2PN N Phe42 3.00 3.00O Wat24 (128) 2.75 2.84O Wat58 (111) 2.99 2.91
O-3'N OH Tyr46 2.84 2.87O Ser216 3.18 3.38OG Ser241 2.66 2.80
O-2'N OH Tyr46 2.66 2.71OS2 SO4 2.77 2.85OS3 SO4 3.43 3.24
O-7N OH Tyr130 2.60 2.77N Leu266 3.13 3.15O Wat26 (130) 3.04 2.97
N-7N OH Tyr130 3.30 3.46O Phe238 3.19 3.13O Pro264 2.92 3.00OS1 SO4 2.90 2.86
N-1N OS2 SO4 3.23 3.28O-3P O Wat58 (111) 3.25 3.39O-1PA N Gly152 2.97 2.91
O Wat58 (111) 2.70 2.97O Wat259 (118) 2.69 2.76
O-2PA N Gly152 3.51 3.40O Wat195 (172) 2.61 2.60
O-4'A O Wat193 (223) 2.89 2.89O-3'A NZ Lys177 3.51 3.49
O Wat57 (117) 2.75 2.78O Wat105 (116) 2.77 2.89
O-2'A NZ Lys177 3.41 3.31O Wat56 (222) 3.48 3.73O Wat57 (117) 3.38 3.35
N-1A O Ser241 3.41 3.67O Wat106 (338) 3.00 2.79
N-7A O Wat250 (235) 2.86 2.84OP-1 N Gly173 2.86 2.81
O Wat56 (222) 2.68 2.96OP-2 OH Tyr192 2.37 2.61
NH2 Arg317 2.75 2.80OP-3 NZ Lys177 2.78 2.80
NE Arg317 3.37 2.95NH2 Arg317 3.57 3.46O Wat66 (176) 2.62 2.71
N-6A O Wat367 2.92
into a pocket surrounded by charged or polarresidues. The positively charged residues, Lys177and Arg317, are involved in potential electrostaticinteractions to the negatively charged phosphategroup. A third polar residue, Tyr192, probablyprovides an additional hydrogen bond interaction. Inaddition, there are two water-mediated contacts tothe enzyme via Wat66 and Wat56. The loop regioncontaining residues 315 to 319 of QOR effectivelycloses the top of this groove and by doing soorientates the long side-chain of Arg317 so as toextend over the top of the groove, blocking theentrance and firmly anchoring the adenosine andphosphate groups in position.
The region of the cofactor from the adenine ribosesugar to the O-5' atom of the nicotinamide ribose fitsinto the narrowest part of the cleft between the twodomains. The binding site for the pyrophosphate,PO4 (N) to PO4 (A), bridge region is situated between
the amino ends of helices aB of the NAD-bindingdomain and a1 of the catalytic domain and has anumber of close contacts to backbone atoms,especially to Gly151, Gly152 and Val153, and anumber of water-mediated contacts. The side-chainof Phe42, which is situated at the top of helix a1, issharply bent to allow the phenyl ring to lie flat andparallel to the pyrophosphate bridge. This region ofresidues 148 to 152 comprises the highly conservednucleotide-binding loop.
There are numerous contacts between thenicotinamide ribose sugar and polar proteinside-chains from residues Gly239, Asn240, Ser241,Ser216 (which are part of the loop region between bEand helix aF) and Tyr46 (part of helix a1), includinga hydrogen-bonded contact to the sulphate molecule.The nicotinamide binds in a more open cavity thanthe adenine moiety and forms contacts to Pro264,Ser265, Arg263 (which comprise the turn between bF
JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase794
and a3), Thr127 and Tyr130 (part of helix a2), Phe238,the sulphate anion and a water contact via Wat26, andis thus held in place by interactions with bothdomains.
There is a chain of water molecules located in thecofactor binding cleft which forms a network ofwater-mediated interactions between the proteinand NADPH molecule. These appear to be essentialin maintaining cofactor contacts and holding
NADPH in the correct orientation. Internal structuralwater molecules are therefore not only important tostabilise regions of the protein structure but are alsonecessary to provide essential contacts between theprotein and cofactor.
The edges of the binding cleft are clamped tightlydown over the cofactor stabilising the enzyme in itsbound conformation. Sufficient opening of the cleftto release the cofactor would require a major
Figure 9. Schematic representation of NADPH binding showing possible hydrogen bond interactions (broken lines)between the cofactor and protein or solvent molecule for subunit A.

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 795
Table 4. Possible hydrogen bond interactions for thesulphate anionsSulphate Contact Contact Distance (A)atom atom residue A B
OS-1 O Phe238 3.20 3.89NH1 Arg263 2.87 3.14NH2 Arg263 3.50 4.01N7N NADPH 2.90 2.86O Wat315 (94) 3.98 3.39O Wat26 (130) 3.59 3.16
OS-2 O2'N NADPH 2.77 2.85N1N NADPH 3.23 3.28O Wat25 (129) 3.20 3.35O Wat260 (131) 2.56 2.52
OS-3 OH Tyr52 2.97 3.46NH1 Arg263 3.95 3.38NH2 Arg263 3.21 2.90O2'N NADPH 3.43 3.24
OS-4 OH Tyr52 3.81 2.71O Wat26 (130) 3.08 4.71O Wat260 (131) 3.20 4.33O Wat315 (94) 2.92 3.10
four contacts to the NADPH molecule, although anumber of these are different between subunits. Inaddition, there are five hydrogen bonds to watermolecules which mediate in further interactions toeither the protein or NADPH. The positions of watermolecules in each subunit are slightly different.
Binding of the quinone substrate would requiredisplacement of the structured water moleculesbound in the active site. Docking of a naphtho-quinone substrate into the active site region showsthat the aromatic rings of the quinone molecule couldlie parallel to the nicotinamide ring so as to becorrectly orientated for electron transfer from thecatalytic hydride of NADPH. Two orientations for thequinone molecule are possible. The active site cleft inQOR is fairly open and appears to allow easy accessto the site by the substrate.
Evolution of enzymes
The structure of tetrameric glucose dehydrogen-ase (GDH) from Thermoplasma acidophilum has beenreported (John et al., 1994). Although GDH doesnot show significant sequence similarity to otherNAD(P)H-binding proteins it contains the GXGXXAfingerprint motif for NADP binding. A surprisingresult from the structure is that it not only exhibitedthe classical Rossman fold, but revealed that therewas extensive homology between GDH and LADHin regions of the catalytic domain. QOR also showssignificant structural homology to both LADH andGDH, the main difference being the absence of thelarge zinc-binding loop and lack of zinc atoms.Figure 10 shows a superposition of the Ca traces forthe monomers of LADH, GDH and QOR.
The structures of three enzymes, LADH, GDHand QOR, which catalyse different reactions yetshow substantial overall structural homology arenow known. The results obtained from these struc-tures should allow us to study the evolutionaryrelationships between these enzymes and theconservation of stable folds. It appears for theseenzymes at least that their homology is not limitedto similarity in the NAD-binding domains but tostrong resemblances in the catalytic domain as well.
The structure of QOR has also shown that it hasan unusual nucleotide binding motif, consisting ofthe sequence AXXGXXG, as a result of the insertionof an alanine residue in the fingerprint region of thenucleotide-binding domain. The implications of thisanomaly, as well as a comparison of nucleotidebinding between QOR and LADH, will be publishedelsewhere, as will a detailed description of thestructural comparisons between QOR, LADH andGDH.
Materials and Methods
Crystallization and data collection
Crystals of QOR were obtained as described (Edwardset al., 1994) by vapour diffusion from ammonium sulphatein the presence of NADPH. The crystals were stored in a
conformational change. This suggests that theenzyme must have a different conformation in thebound and unbound states. We were unable tocrystallise QOR in the absence of NADPH andattempts to co-crystallise QOR with NADH werealso unsuccessful. We cannot, therefore, make anydirect comparisons between the bound andunbound states of the molecule. We assume that, asfor LADH, the molecule undergoes a conformationalchange upon cofactor binding which results in muchtighter binding of cofactor to the enzyme. It appearsthat the reduced form of the cofactor is capable ofstabilising the closed conformation of the enzyme.
Catalytic site
The substrate binding site is thought to be locatedin a cleft situated between the coenzyme andcatalytic domains, adjacent to the NADPH molecule.Residues from both domains which line the activesite pocket are of a predominantly polar natureespecially in the region closer to the nicotinamidemoiety. These residues are Asn41, Tyr46, Tyr52,Asn240, Arg263, Ser265, and AsnB254 from thesecond subunit. The lower region of the active siteconsists mostly of hydrophobic residues, Ile43,Leu123, Leu226 and Thr63, mainly from the catalyticdomain. In this structure the active site cleft is filledwith structured water molecules. The presence of asulphate anion has been detected in the structure andis located in the active site but away from the catalytichydride of NADPH. The sulphate anion is held inplace by a number of hydrogen-bonded interactions(Table 4) to side-chain atoms, the nicotinamide andnicotinamide ribose sugar and also a number ofwater-mediated contacts. It therefore appears to befairly tightly held in position in the active site region.The orientation of the sulphate anion in each subunitis slightly different. Each sulphate anion has fourpotential hydrogen bond contacts to the protein and

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase796
Figure 10. Stereo view of the superimposed Ca traces for the monomers of LADH (green), GDH (red) and QOR (black).
2.5 M ammonium sulphate stabilizing buffer at pH 6.5.QOR crystals belong to the orthorhombic space groupP212121 with a = 107.44 A, b = 104.06 A and c = 77.45 A.There are two independent QOR subunits in theasymmetric unit.
Data were collected on a Rigaku RAXIS-IIC imagingplate detector mounted on a Rigaku RU 200 rotating anodeX-ray generator operating at 50 kV and 100 mA. A 2.5 Anative data set was collected and used for structuresolution and initial model building. A second crystal wasthen used to collect two further native data sets (at x = 0°and 45°) to a resolution of 2.0 A. The three data sets were
merged to give a final data set containing 40,569 uniquereflections to a resolution of 2.2 A with an overall mergingR factor of 7.3%. The data are 90.6% complete to 2.2 Aresolution with 75.6% in the 2.2 to 2.5 A shell. Datacollection statistics are given in Table 5.
Multiple isomorphous structure determination
Heavy-atom derivatives were prepared by soakingpregrown crystals in the stabilizing solution containingheavy-atom compounds. An extensive search for heavy-atom derivatives to solve the structure by multiple
Table 5. Native and derivative data collection statisticsCompleteness
Soaking Resolution Rmergea of data Temperature
Crystal conditions (A) (%) (%) (°C)
Native 1 2.5 6.3 90.0b 18Native 2a 2.0 5.8 78.6c (90.3)b 18Native 2b 2.0 8.2 74.2c (84.8)b 18Overall native 2.2 7.3 90.6c (97.2)b 18HgCl2 (10 mM, 3.5 h at 4°C) 3.0 7.1 78.6d 4KAu(CN)2 (10 mM, 3.5 h at 4°C) 3.0 6.3 93.1d 4Na2AuCl4 (1:2 Sate, 4 h at 4°C) 3.0 5.0 91.5d 4
a Rmerge = S=I − �I�=/SI.b To 2.5 A.c To 2.2 A.d To 3.0 A.e Saturated.

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 797
Table 6. Summary of heavy-atom phasing analysisPhasing Mean figure
Derivative No. of sites powera of merit
HgCl2 2 1.82 (1.26)b
KAu(CN)2 6 1.92 (1.73)b
NaAuCl4 2 2.44 (1.89)b
0.73a Phasing power = r.m.s. (=FH=/E); FH, heavy-atom structure
factor amplitude; E, residual lack of closure.b The number in parentheses represents the phasing power in
the highest resolution shell 3.36 to 3 A.
obtained from the Brookhaven Protein Data Bank(Bernstein et al., 1977; accession number PDB8ADH.ENT),to aid in the construction of a Ca model for one subunit (A)of QOR. The density for secondary structure elementsof QOR was identified and the corresponding Ca atomsfrom LADH fitted into the density as a rigid body. Asthe secondary structural elements of LADH and QORgenerally had different lengths, the structure from theLADH Ca atoms was then modified according to theobserved electron density. The model of the loop regionsof QOR was built independently of the structure ofLADH.
A partial polyalanine model for one QOR subunit wasconstructed using the Ca atoms as a guide. Although thebackbone density was fairly well defined, the density formany side-chains was weak, and in some regions it wasdifficult to account for the known amino acid sequence. Wetherefore decided to improve the map by averaging thedensity for the two independent subunits. The QOR modelwas fitted to the electron density of the second subunit (B).Rigid-body refinement of the model for subunit (B) usingX-PLOR v3.1 (Brunger, 1992), followed by least-squaresfitting to subunit (A) enabled us to calculate an accuratetransform relating the two subunits. Twofold averaging(using the program PROTEIN) of the MIR electron densitymap resulted in a much improved map into which a morecomplete model could be built. The density averaged mapwas consistent with the known amino acid sequence andenabled us to build a model containing residues 18 to 323with side-chains for most residues. We were careful at thisstage not to include any side-chains for which the densitywas not well defined.
Refinement
The program X-PLOR was used for refinement andmanual adjustments to the model made using TOM. At theend of each refinement step a number of maps werecalculated to check the course of the refinement andmanual rebuilding carried out. Early in the refinement,averaged phase combined maps (calculated using modeland MIR phases with PROTEIN) were used for modelbuilding. In the later stages of refinement 2Fo − Fc, ac andFo − Fc, ac maps were used to make small adjustments to themodel and for fitting water molecules.
The initial model (consisting of residues 18 to 323) wasrefined against 3 A native X-ray data with strictnon-crystallographic symmetry restraints being applied.The resolution limits of the data were then extended to2.5 A to give an R-factor of 36.8%. A more complete modelconsisting of residues 3 to 327 was then refined bysimulated annealing (SA). The model was slow-cooledfrom 4000 K to 300 K reducing the temperature by 25 Kwith a time-step of 0.5 fs, followed by positionalrefinement to give an R-factor of 27.6%. A model forNADPH was unambiguously fitted into clearly definedelectron density in the nucleotide binding domain of the2.5 A Fo − Fc, ac map. Grouped thermal parameters wererefined initially followed by individual B-factor refinementto give an R-factor of 21.8%. The resolution of the nativedata was increased to 2.2 A. Side-chains were carefullyplaced as their density improved and in the later stages ofrefinement partial side-chains consisting only of atomswhich were clearly visible in the electron density map wereincluded in the model. The NCS restraints were thenremoved permitting refinement of all atoms in the dimer.A further round of SA with slow cooling from 2000 K to300 K, followed by further positional and thermalparameter refinement reduced the R-factor to 20.2%.
isomorphous replacement (MIR) eventually yielded threesuitable derivatives. Anomalous data for all derivativeswere collected to 3.0 A resolution at 4°C. It was importantto maintain the crystals at 4°C to prevent non-isomorphouscell changes. Data collection statistics for the heavy-atomscan be found in Table 5.
The positions of heavy-atom binding sites were locatedfrom inspection of Patterson and difference Fourier maps.The positions of the heavy-atom peaks was confirmed byapplication of the vector verification method. Heavy-atomparameters were refined and phases calculated for the datafrom 20 to 3 A resolution using the program PROTEIN(Steigemann, 1992) to give an overall mean figure of meritof 0.73 for all derivatives. Table 6 gives a summary of theheavy-atom phasing analysis. An electron density map wascalculated at 3 A resolution using the MIR phases. Thismap was very clean and of a sufficiently good quality totrace the polypeptide chain. It was clear from an inspectionof this map that the QOR structure was similar to that ofLADH as suggested from sequence comparisons.
Model building
The program TOM v3.0 (Israel & Chirino, 1994), runningon an IRIS 4D/310GVX workstation, was used for allmodel building. Since the QOR structure appeared to besimilar to LADH, we made use of a model of LADH
Table 7. Refinement statistics for QORResolution range (A) 5.0 − 2.2No. of reflections F > 2s (F) 36270Completeness (%) 89.7R-factor (%) 14.1No. of atoms
Total 5394Protein 4856NADPH 96Sulphate 10Solvent 432
Average B-factors (A2)Main-chain
(A) 28.1(B) 27.6
Side-chain(A) 29.8(B) 29.4
Solvent 41.4r.m.s. deviations of geometry from ideal values
Bonds (A) 0.015Angles (°) 2.024Dihedrals (°) 25.187Impropers (°) 1.797

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase798
Solvent molecules were included in the model once theR-factor had dropped to 20.0%.
A large spherical region of electron density was locatednear the nicotinamide end of each NADPH molecule. Thissphere of electron density was larger than that expected fora single water molecule. From a consideration of thecrystallization conditions we concluded that this wasprobably a sulphate anion. Subsequent difference mapsconfirmed our assignment. Several cycles of conventionalpositional refinement with the addition of further watermolecules was then carried out. Thermal parameters andoccupancies of solvent molecules were refined and smallmanual adjustments made to the structure. Simulatedannealing (with slow cooling from 2000 K to 300 K) of twoloop regions, which were less well defined (residues 20 to30 and 71 to 81 for subunit A; residues 17 to 29 and 71 to80 for subunit B), was undertaken and omit maps of theregions calculated to check positioning of the polypeptidechain. Omit maps of the NADPH molecules were alsocalculated in order to check the accuracy of the model. Thequality of our omit maps showed the structure to bewell-defined, indicating satisfactory refinement to yield anaccurate structure.
The current model consists of a dimer, each subunit ofwhich contains residues 2 to 327, one NADPH and onesulphate anion, and a total of 432 water molecules. The finalR-factor is 14.1% at 2.2 A for 36,270 reflections between 5and 2.2 A with F > 2sF. Table 7 gives the final refinementstatistics and stereochemical parameters for the model.Co-ordinates have been deposited with the BrookhavenProtein Data Bank (accession number PDB1QOR.ENT),and are available from the authors upon request.
AcknowledgementsWe thank Mike Carson for supplying the NADPH
parameters for X-PLOR refinement and the A.N.U.Supercomputer Centre for providing us with time on theFujitsu VP2200.
ReferencesBellomo, G., Thor, H., Eklow-Lastbom, L., Nicotera, P. &
Orrenius, S. (1987). Oxidative stress—mechanisms ofcytotoxicity. Chem. Scripta, 27A, 117–120.
Bernstein, C. F., Koetzle, T. F., Williams, G. J. B., Meyer,E. F., Jr, Brice, M. D., Rodgers, J. R., Kennard, O.,Shimanouchi, T. & Tasumi, M. (1977). The Protein DataBank: a computer-based archival file for macromol-ecular structures. J. Mol. Biol. 112, 535–542.
Borras, T., Persson, B. & Jornvall, H. (1989). Eye lenszeta-crystallin dehydrogenases. Protein trimming andconservation of stable parts. Biochemistry, 28, 6133–6139.
Brunger, A. T. (1992). X-PLOR v3.1 Manual. A system forX-ray crystallography and NMR, Yale UniversityPress, New Haven.
Edwards, K. J., Thorn, J. M., Daniher, J. A., Dixon, N. E. &Ollis, D. L. (1994). Crystallization and preliminaryX-ray diffraction studies on a soluble Escherichia coliquinone oxidoreductase. J. Mol. Biol. 240, 501–503.
Eklund, H. & Branden, C.-I. (1987). Alcohol dehydrogen-ase. Biological Macromolecules and Assemblies, vol. 3,John Wiley & Sons, New York.
Eklund, H., Nordstrom, B., Zeppezauer, E., Soderlund, G.,Ohlsson, I., Boiwe, T., Soderbury, B.-O., Tapla, O.,Branden, C.-I. & Akeson, A. (1976). Three-dimen-
sional structure of horse liver alcohol dehydrogenaseat 2.4 A resolution. J. Mol. Biol. 102, 27–59.
Eklund, H., Samama, J.-P. & Jones, T. A. (1984).Crystallographic investigations of nicotinamide dinu-cleotide binding to horse liver alcohol dehydrogenase.Biochemistry, 23, 5982–5996.
Eklund, H., Horjales, E., Jornvall, H., Branden, C.-I. &Jeffrey, J. (1985). Molecular aspects of functionaldifferences between alcohol and sorbitol dehydrogen-ases. Biochemistry, 24, 8005–8012.
Ernster, L., Estabrook, R. W., Hochstein, P. & Orrenius, S.(1987). Editors of DT Diaphorase; a quinone reductasewith special functions in cell metabolism anddetoxification. Chem. Scripta, 27A, 1–207.
Gonzalez, P., Rao, P. V. & Zigler, J. S. (1993). Molecularcloning and sequencing of zeta-crystallin/quinonereductase from cDNA from human liver. Biochem.Biophys. Res. Commun. 191, 902–907.
Huang, Q.-L., Russell, P., Stone, S. H. & Zigler, J. S., Jr(1987). Zeta-crystallin, a novel lens protein fromguinea pig. Curr. Eye Res. 6, 725–732.
Israel, M. & Chirino, A. J. (1994). TOM/FRODO—amolecular modelling program for the IRIS, Alberta/Caltech version 3.0.
John, J., Crennel, S. J., Hough, D. W., Danson, M. J. &Taylor, G. L. (1994). The crystal structure of glucosedehydrogenase from Thermoplasma acidophilum.Structure, 2, 385–393.
Jornvall, H., Eklund, H. & Branden, C.-I. (1978). Subunitconformation of yeast alcohol dehydrogenase. J. Biol.Chem. 253, 8414–8419.
Kabsch, W. & Sander, C. (1983). Dictionary of proteinsecondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22,2577–2637.
Kraulis, P. J. (1991). MOLSCRIPT: a program to produceboth detailed and schematic plots of proteins. J. Appl.Crystallogr. 24, 946–950.
Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallogr. 6, 283–291.
Lee, B. & Richards, F. M. (1971). The interpretation ofprotein structure: estimation of static accessibility.J. Mol. Biol. 55, 379–400.
Lilley, P. E., Stamford, N. P. J., Vasudevan, S. G. & Dixon,N. E. (1993). The 92-min region of the Escherichia colichromosome: location of the ubiA and alr genes. Gene,129, 9–16.
Luzzati, P. V. (1952). Traitement statistique des erreurs dansla determination des structures cristallines. ActaCrystallogr. 5, 802–810.
Matthews, D. A., Alden, R. A., Freer, S. T., Xuong, N. H.& Kraut, J. (1979). Dihydrofolate reductase fromLactobacillus casei: stereochemistry of NADPH bind-ing. J. Biol. Chem. 254, 4144–4151.
Prestera, T., Holtzclaw, W. D., Zhang, Y. & Talalay, P. (1993).Chemical and molecular recognition of enzymes thatdetoxify carcinogens. Proc. Nat. Acad. Sci. USA, 90,2965–2969.
Ramachandran, G. N. & Sasisekharan, V. (1968).Conformation of polypeptides and proteins. Advan.Protein Chem. 23, 283–437.
Rao, P. V. & Zigler, J. S. (1992). Purification andcharacterization of zeta-crystallin/quinone reductasefrom guinea pig liver. Biochim. Biophys. Acta, 1117,315–320.
Rao, P. V., Krishna, C. M. & Zigler, J. S. (1992). Identificationand characterization of the enzymatic activity of

JMB—MS 565
Structure of Escherichia coli Quinone Oxidoreductase 799
zeta-crystallin from guinea pig lens. J. Biol. Chem. 267,96–102.
Rao, S. T. & Rossman, M. G. (1973). Comparison ofsuper-secondary structures in proteins. J. Mol. Biol. 76,241–256.
Rodokanaki, A., Holmes, R. K. & Borras, T. (1989).Zeta-crystallin, a novel protein from guinea pig lensis related to alcohol dehydrogenases. Gene, 78,215–224.
Rossman, M. G., Liljas, A., Branden, C.-I. & Banaszak, L. J.(1975). Evolutionary and structural relationshipsamong dehydrogenases. Enzymes, 11, 61–102.
Steigemann, W. (1992). PROTEIN v3.1, Max-PlanckInstitut fuer Biochemie, Martinsried bei Muenchen,FRG.
Teeter, M. M. (1991). Water-protein interactions: theory andexperiment. Annu. Rev. Biophys. Biophys. Chem. 20,577–600.
Tumminia, S. J., Rao, P. V., Zigler, J. S. & Russell, P. (1993).Xenobiotic induction of quinone oxidoreductaseactivity in lens epithelial cells. Biochim. Biophys. Acta,1203, 251–259.
Yagi, T. (1991). Bacterial NADH-quinone oxidoreductases.J. Bioenerg. Biomembr. 23, 211–225.
Edited by R. Huber
(Received 3 January 1995; accepted 27 March 1995)



![Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase ...Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase CYP82Q3 Converts Jasmolone To Pyrethrolone1[OPEN] Wei Li,a](https://img.pdfslide.net/doc/110x75/5e2d08c0200c602a86070292/pyrethrin-biosynthesis-the-cytochrome-p450-oxidoreductase-pyrethrin-biosynthesis.jpg)
























