Embed Size (px)
Citation preview

[CANCER RESEARCH 52. 3145-3156, June I. 1992]
Development of an in Vitro Model to Study Carcinogen-induced Neoplastic
Progression of Initiated Mouse Epidermal CellsDavid Morgan, David Welty, Adam Click, David Greenhalgh,1 Henry Hennings, and Stuart H. Yuspa2
Laboratory of Cellular Carcinogenesis and Tumor Promotion, Division of Cancer Etiology, National Cancer Institute, Bethesda, Maryland 20892
ABSTRACT
Initiation and promotion in mouse skin carcinogenesis produce multiplebenign tumors, squamous papillomas, but only a few squamous cellcarcinomas. The spontaneous conversion from the benign to the malignantphenotype occurs over many months and in stages, but induced malignantconversion can be accomplished more rapidly by exposure of papilloma-
bearing mice to mutagens or by transfection of papilloma cell lines withspecific oncogenes. The analysis of genetic targets responsible for carcinogen-induced neoplastic progression would be facilitated by the development of in vitro models where the process is rapid, focal, and quantitative.To this end, primary newborn mouse keratinocytes were initiated in vitroby the introduction of the v-ror"' oncogene via a defective retrovirus.
Recipient cells produce squamous papillomas and have a high proliferation rate in culture medium with 0.05 HIM(a'*, but fail to grow in medium
with 0.5 HIMCa2* which is permissive for growth of malignant keratinocytes. When v-rfli"*-keratinocytes were exposed to mutagens in vitro,proliferative foci emerged after culture in 0.5 HIM ( :i'' for 4 weeks.
These foci stained intensely red with rhodamine stain, could be easilyquantitated, and readily incorporated bromodeoxyuridine. Dose-response
studies with several mutagens indicated that the number of foci increasedwith concentration to the point where excessive cytotoxicity developed.Mutagens varied in potency for producing foci in the following order: <•/>-diamminedichloroplatinum > benzo(a)pyrene diolexpoxide I > A'-methyl-jV'-nitro-yV-nitrosoguanidine > 4-nitroquinoline-/V-oxide > /V-acetoxy-
acetyl-aminofluorene. The tumor promoter 12-0-tetradecanoylphorbol-13-acetate was inactive in the assay. A subset of cell lines derived fromfoci produced malignant tumors in vivo, while others were not tumori-
genie. Analysis of DNA from cell lines and tumors revealed that mosttumorigenic cell lines maintained the v-ras"* genome, whereas the viral
sequences were deleted in nontumorigenic cell lines. Immunohistochem-ical analysis indicated that proliferative foci and quiescent \-ras"' keratinocytes expressed keratin 8, a marker of \-ras"* expression in culturedkeratinocytes. Cells in foci, but not \-ras"* control cells, expressed keratin
13, a marker which is strongly associated with the malignant progressionof skin tumors in vivo. This in vitro assay provides a quantitative modelto study chemically induced focal neoplastic progression at the cellularlevel and to identify agents which may be selective for enhancing malignant conversion.
INTRODUCTION
The induction of tumors on the skin of mice has been animportant model to define the stages of chemical carcinogenesis. In that model, a single exposure to specific carcinogensfollowed by repeated applications of tumor promoters resultsin multiple benign neoplasms and squamous papillomas. Anoccasional papilloma will progress to squamous cell carcinoma,generally after a long latency period. The malignant conversionof benign neoplasms is associated with the accumulation of
Received 8/23/91; accepted 3/24/92.The costs of publication of this article were defrayed in part by the payment
of page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
' Present address: Baylor College of Medicine. Houston. TX 77030.2To whom reprints should be addressed, at Laboratory of Cellular Carcino
genesis and Tumor Promotion. Division of Cancer Etiology. Building 37, Room3B25, National Cancer Institute. Bethesda. MD 20892.
3The abbreviations used are: BUdR, bromodeoxyuridine; cii-DDP. ru-diam-minedichloroplatin; MNNG, iV-methyl-iY'-nitro-iV-nitrosoguanidinc: PCR. po-lymerase chain reaction; TPA. 12-O-tetradecanoylphorbol-13-acetate; TGF.transforming growth factor.
additional genetic changes in the papilloma cell. For example,the frequency of malignant conversion is enhanced, and thelatent period reduced, by exposing papilloma-bearing mice tomutagens (1, 2). Furthermore, the introduction of specific oncogenes into cultured papilloma cells can cause malignant conversion (3, 4). When the \-ras"" oncogene alone is introduced
into normal skin or cultured keratinocytes by a retroviralvector, recipient cells produce papillomas after tumor promotion (in vivo) or when grafted to the backs of nude mice (invitro) (5, 6). Thus the v-rasHaoncogene can initiate mouse skin
tumorigenesis.In cultured mouse keratinocytes, the v-ras"a and \-fos onco
genes introduced together via retroviruses yield cells which formcarcinomas at a graft site on nude mice (7). Cytogenetic studieshave identified specific chromosomal changes which are associated with the malignant conversion of chemically inducedpapillomas (8, 9), and the relative gene dosage of mutant andnormal ras11" alÃelescommonly changes to favor the mutant
alÃelein mouse skin carcinomas (10-13). Furthermore, exam
ples of gene amplification have been associated with malignantprogression of mouse skin tumors (14, 15). Similar results havebeen reported in cultures of human keratinocytes where combinations of specific viral oncogenes or viral oncogenes andcytogenetic changes are associated with malignant progression(16, 17). Together these findings support the hypothesis that avery specific set of genetic changes is involved in malignantprogression in skin carcinogenesis.
The use of defective retroviruses as high-efficiency vectors tointroduce the ras oncogene into keratinocytes (v-ras keratino
cytes) and produce papilloma cells permits the analysis of thebenign phenotype in vitro. Expression of the ras p21 oncopro-
tein increases keratinocyte proliferation (18), alters the response pattern to phorbol ester tumor promoters (18), andinhibits keratinocyte terminal differentiation in response toincreasing concentrations of medium Ca2+ (19). In mediumwith >0.1 mM Ca2+, v-ras keratinocytes have a low proliferation
rate but do not express differentiation markers, form maturesquames, or slough from the culture dish ( 19), all characteristicsof normal keratinocytes in that medium. The v-ras"a transduced
cells have the capacity to revert to a proliferative phenotypewhen reestablished in medium with 0.05 mivi Ca2* (18),
while normal keratinocytes are irreversibly committed todifferentiate.
Malignant keratinocytes do not differentiate in medium withCa2+ > 0.1 mM (20) but are distinguished from v-ras keratino
cytes in that they continue to proliferate in this medium. Thisdifference implied that v-ras keratinocytes that progress to a
more advanced stage of neoplasia could have a growth advantage in medium with Ca2+ > 0.1 mM. Such a model might be
useful for detecting mutagens effective in causing malignantprogression and ultimately for identifying genetic changes thatspecifically contribute to progression. This report describes thedevelopment of a model designed for these purposes.
3145
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
MATERIALS AND METHODS
Chemicals. Carcinogens were obtained from the National CancerInstitute Chemical Repository, TPA was from Consolidated Midland(Eden Prairie, MM), and benzoyl peroxide was from Aldrich ChemicalCompany (Milwaukee, WI) and was used as previously described (21).Tissue culture medium was purchased from Whittaker Bioproducts(Walkersville, MD). Fetal bovine serum was from Intergen/Armour(Kankakee, IL) and was treated with Chelex Resin (BioRad, Richmond,CA) to remove Ca2* as described (22). Antibiotic-antimycotic solution
was from Gibco (Grand Island, NY).Cell Culture Methods and Establishment of the Assay. Primary new
born epidermal cells were isolated from BALB/c mice as describedpreviously (22). The cell preparation was allowed to preattach to tissueculture dishes in medium with 0.05 mM Ca2+ for l h at 37°C.A small
number of contaminating fibroblasts exist in the epidermal preparation(<0.1%), and these attach to plastic more rapidly than keratinocytesattach to plastic. After 1 h, unattached keratinocytes were replated into60-mm dishes at 4 x IO6 cells/dish in Eagle's minimum essential
medium containing 8% fetal bovine serum, I % antibiotic-antimycoticsolution, and a calcium concentration of 0.05 mM (22). For other Ca2+
concentrations, adjustments were made by adding aliquots of a 0.3 MCaClz solution. Medium Ca2* was monitored by a Perkin-Elmer atomic
absorption spectrophotometer. To prevent spontaneous activation ofthe c-ras"* gene, medium conditioned by dermal fibroblasts was added
at 10% (v/v) to all dishes (13). After 3 days of culture, the cells wereinfected with a replication-defective \-ras"* retrovirus for 72 h as
previously outlined (5). During the initial studies, mock infected cellsor cells infected with a retroviral vector carrying the neomycin resistance gene neoR were used as controls. Viral titers were adjusted to give
a multiplicity of infection of approximately I. After 2 additional daysin 0.05 mM Ca2+ medium, cultures were exposed to carcinogens orvehicle solutions for 1-24 h. Five days following the removal of thecarcinogen, triplicate dishes were trypsinized, and cells were countedin a Coulter Counter ZBI to determine the cell loss due to carcinogentreatment (23). The remaining dishes (usually 8-10/treatment group)were maintained in 0.05 mM Ca2* medium prior to a challenge with
higher-calcium medium. A variety of selection protocols were evaluated,and these will be summarized in the "Results." At the end of the high
calcium selection (usually 4 weeks), dishes were fixed in 10% formalinand stained with rhodamine to visualize dense keratinocyte colonies(23). Each densely staining colony was examined under a dissectingmicroscope to ensure that it represented epithelial cells. The numbersof foci in a test group (usually consisting of 8-10 replicate dishes/carcinogen concentration) were quantitated, corrected for cell loss dueto cytotoxic effects, and presented as corrected foci per dish for aparticular treatment (23).
Isolation of Carcinogen-induced Foci and Testing in Vivo. A numberof methods were used to evaluate the tumorigenic potential of selectedfoci. Ring cloning of individual foci resulted only rarely in survivingand expanding clones when the cells were plated on plastic or collagen-coated culture dishes. Cell lines derived by this cloning method weretested by s.c. injection or cutaneous grafting (in combination withdermal fibroblasts) of 5 x IO6 cells onto nude mice of BALB/c background (5, 13). Subcultivation of ring-cloned colonies was frequentlysuccessful when isolated cells were plated onto dishes of control keratinocytes previously transduced with the \-rasH' gene and maintainedin 0.5 m.MCa2* medium for 4 weeks prior to overplating. This method
closely approximated the culture conditions of primary foci. In severalexperiments, the entire contents of dishes containing foci were removedby exposure to 0.1% trypsin, 0.05% EDTA, and the yield from eachdish (approximately 1 x IO6 cells) was either injected s.c. or grafted
onto a nude mouse. In a third method, cells were grown and treated indishes containing a tissue-culture grade plastic liner (Falcon Plastic3006). At the end of the selection, foci were identified and circled, andthe area containing a focus attached to plastic was excised. Theseplastic disks with cells (cell number is relatively small but not determined) were then inserted s.c. into the flanks of nude mice with thecells facing inward (24).
Characterization of Carcinogen-induced Foci in Vitro. Foci were also
characterized in situ. To determine the number of cells capable of DNAsynthesis, cultures were exposed to medium containing 10^6 M BUdR
(Calbiochem, San Diego, CA) for either 1 or 24 h and fixed in methanolat 4°C.Areas of foci or background cells in treated and control dishes
were circled and analyzed by immunofluorescence microscopy using aBUdR antibody (Becton Dickinson, San Jose, CA) and a fluorescein-tagged second antibody as described (25). To detect the presence of theviral genome in foci and in control cells, cloning rings were placedaround individual foci or equivalent areas from control dishes, and cellswere released by 0.25% trypsin treatment and washed twice in phosphate-buffered saline. The cell suspension was pelleted at 12,000 x gfor 5 min and suspended in 50 n\ of lysis buffer consisting of 50 mMKC1, 10 mM Tris-HCI (pH 8.3), 10 mM Mg C12,0.002% NP40, and 60/¿g/mlProteinase K. The suspension was incubated at 50°Cfor 1 h andthen 95°Cfor 12 min to inactivate proteinase K. After a further
centrifugation, approximately 25 i¿\of clear supernatant were used asa template for PCR using 2.5 units of Amplitaq DNA polymerase/sample (Perkins Elmer Cetus, Norwalk, CT). The amplimers used were5'-CCTAAGAAGATAGTACCTGGCAACAGAAG and 3'-GAT-
GATTGTAGAGGCCGTCTCTCGTTTGG to amplify the specificviral v/30 sequence unique to the rat origin of the retroviral construct(26). A similar analysis was performed on lysates from cell lines andtumor material derived from transplanted or ring-cloned foci. NormalBALB/c cultured keratinocyte extracts were used as a negative PCRcontrol, and a region of the first intron of the mouse keratin 1 genewith the sequences 5'-CGGCTTAGCTTATGTGTGTGCATTT-GATG and 3'-AGCTGATCAAGGTGGAATCCATCTCAGAGG was
used as a positive control for the PCR reaction when sufficient samplewas available.
Control cells and foci were also examined in situ by histochemicaland immunohistochemical analysis. Dishes were fixed in 1:1 metha-nol:acetone, and individual foci were divided into with sections a waxedpencil. Double immunofluorescence analysis was performed with combinations of specific rabbit or rat antibodies and guinea pig anti-mousekeratin 14 antibody, the latter detected with Texas red-conjugated anti-guinea pig IgG in all three sections (27). Affinity-purified rabbit antibodies were used on individual sections to detect mouse keratin 1 orkeratin 13 as described (27, 28). TROMA I rat monoclonal antibodyto mouse keratin 8 was also used as described (19). Second antibodieswere fluorescein-conjugated goat anti-rabbit or anti-rat IgG. Fluorescence was monitored with a Nikon Labophot microscope as describedpreviously (27). A total of six foci and six control areas were examinedby these methods. An additional six foci, as well as control dishes, werestained for -y-glutamyltranspeptidase activity after fixation in ice-cold2-methylbutane for 20 s. The procedure of Perantoni and Berman (29)was followed for staining cultured cells, and frozen rat kidney sectionswere used as a positive control.
RESULTS
Establishment of Assay Conditions. In medium with >0.1 mMCa2+, \-ras"* keratinocytes form a monolayer of large, quiescent
cells which persist for extended culture periods (Fig. 1, A andE). In contrast, normal keratinocytes or keratinocytes infectedwith a retrovirus lacking the v-ra$Hagene but expressing a neoR
gene undergo terminal differentiation and detach from theculture dish within 3-5 days in >0.1 mM Ca2+ medium (highCa2+ medium) (18, 19). Preliminary reconstitution experiments
in which a small number of malignant keratinocytes from theSLC line (13) were mixed with v-ras"a keratinocytes in 0.05mM Ca2+ medium and then switched to 1.2 mM Ca2+ medium
indicated that malignant cell foci would expand into visiblecolonies in higher Ca2+ medium (not shown). Initial experi
ments also showed that highly cellular, dense epithelial fociwould evolve in high-Ca2+ medium if v-ras"" keratinocyte cultures were treated with carcinogens in 0.05 mw Ca2+ medium(Fig. \, B, C, and E-G). Microscopically these foci containeddensely packed epithelial cells in variable patterns of organiza-
3146
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
Fig. 1. Phase-contrast morphology of control cells and hyperproliferative foci. Primarycultures of mouse epidermal cells were infectedwith a defective retrovirus containing the v-ras"' gene in 0.05 m M Ça2*medium. After 1
week in culture, the cells were exposed to solvent (A and E) or MNNG (20.4 pM) for l hand were switched 1 week later to 0.5 mM Ca2+
medium for 4 weeks. Photographs representliving cultures examined under phase-contrastmicroscopy (x 100). /, third passage of a cellline obtained by ring-cloning a hyperproliferative focus and continuously subculturing thecells when they reached confluence.
tion and cell morphology. Some foci contained small, round,uniform cells (Fig. 1, B-D and G), others contained larger ormore angular cells (Fig. 1, F and //); some foci were homogeneous monolayers, while others were stratified or heterogeneous. The emergence of a specific phenotype did not obviouslycorrelate to a particular type or concentration of carcinogen.Furthermore, similar foci occasionally developed spontaneouslyin control \-ras"' keratinocyte cultures. All foci stained dark
red with rhodamine stain, while background cells were pink(Fig. 2). This dense rhodamine staining was also characteristicof premixed malignant cell foci (not shown). In both treatedand control \-ras"" keratinocyte cultures, some rhodamine-
stained colonies (<10%) were fibroblastic when examined microscopically. These may represent transformation of contaminating fibroblasts by the \-ras"' oncogene, and the number of
these foci was reduced by the preattachment methods. BUdRpulse-labeling experiments (Fig. 3) revealed that many cells incarcinogen-induced foci were undergoing DNA synthesis, whilenuclear BUdR labeling was virtually absent in control v-rai"a
keratinocyte cultures or in background (nonfocal) regions ofcarcinogen-treated cultures.
After initial studies indicated that foci could be induced in v-rasH" cultures by carcinogen treatment, a number of experiments were performed to validate the assay, test the reproduc-
ibility, and optimize conditions for colony development. Forstandardization studies, short exposures to the carcinogenMNNG were chosen. The importance of the v-ras"" gene in
focus formation was first examined. Conditions were established to ensure that carcinogens would not produce foci inuninfected keratinocytes (Table 1). We have previously described an assay to induce Ca2+-resistant foci in normal cells by
carcinogen treatment (23), but these foci required stepwiseincreases in Ca2+ for selection. Carcinogen exposure in 0.05mivi Ca2+, followed 1 week later by selection in 0.5 or 1.4 mMCa2+, yields numerous foci in v-ras"' keratinocytes but few in
uninfected cells (Table 1, Experiments 1 and 2, and Fig. 2).Uninfected cells slough from culture when switched directlyinto these higher-Ca2+ media whether exposed to MNNG oruntreated, while \-ras"' cells maintain a monolayer in higherCa2+without carcinogen treatment and yield foci overlaying themonolayer in the carcinogen-exposed group (Fig. 2). The number of foci produced after MNNG exposure was dose-dependent(Table 1, Experiments 3 and 4; Fig. 4), although at dosesproducing high toxicity, foci were reduced or absent. At equivalent doses of MNNG, more foci were produced by extendingcarcinogen treatment time from 1 to 3 h (not shown).
The mutagenic action of MNNG on \-rasH" keratinocytes
was presumed to be the explanation for the production of foci,3147
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

ETOH
l\ VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
MNNG ETOH MNNG
*,
* * 11
i
control rasFig. 2. Rhodamine-stained dishes after 4 weeks in 0.5 mM Ca2* medium. After 3 days in vitro, primary mouse epidermal cells were either infected with a \-ras"'
defective retrovirus (right two columns) or mock infected (left two columns) in 0.05 mM Ça2*.On the 8th day of culture, cells were treated with vehicle (first and thirdcolumns) or MNNG (20.4 JIM)for I h (second and fourth columns) and after I additional week were switched to 0.5 mM Ca2* medium. Dishes were fixed and stainedwith rhodamine after 4 weeks in 0.5 mM Ca2* medium.
Fig. 3. Incorporation of BUDR into S-phase nuclei. At the end of an assay period (seeFigs. I and 2). control dishes (A) or carcinogen-treated dishes with foci {//) were labeledwith 10~*M BUdR for 24 h. Dishes were then
fixed and stained using an anti-BUdR antibodyand visualized by immunofluorcscence as described in "Materials and Methods." Note la
beled nuclei in B but not in A. Backgroundcells in Group B dishes also did not incorporate BUdR into nuclei (not shown).
3148
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

l\ IITRO MODEL OF NEOP1.ASTIC PROGRESSION OF EPIDERMAL CELLS
Table 1 Establishment of conditions for in ritro progressionassayExperiment
Treatment12345Without\-ras"'Control
(solvent)MNNG(13.6 „¿�MxWith
\-ras"'Control
(solvent)MNNG(13.6 pMxControl
(solvent)MNNG(13.6 UMxWithout
\-ras""Control
(solvent)MNNG(13.6 „¿�MxWith
\-ras"'Control(solvent)MNNG
(13.6 MMxWith
v-raj"'Control
(solvent)MNNG(6.0 iiM x1MNNG(10.2(iM
xMNNG(13.6 ,iMxWith
\-ras"'Control
(solvent)MNNG(20.4 UMxMNNG(34.0„¿�MXWith
v-raj"'Control
(solvent)MNNG(20.4 /IMXControl
(solvent)MNNG(20.4 UMxControl
(solvent)MNNG(20.4 MMxControl
(solvent)MNNG(20.4 „¿�MxTreatment
condition(HIMCa!+)0.053h)0.053h)1.43h)0.051
h)0.051
h)0.05h)Ih)In)0.051
h)Ih)0.051
h)Ih)Ih)1
h)Corrected
Survivalcolonies/dish1.00.71.00.81.00.61.00.91.00.91.00.90.90.71.00.90.61.00.81.00.81.00.81.00.70.30.70.37.000.20003.00.10.92.26.801.1000.800.900.800.7
and this would be most effective under conditions favoringreplication at the time of exposure. This was tested by switching\-ras"* cells to 1.4 HIM Ca2* prior to MNNG exposure, sincethese cells cannot proliferate in medium with this Ca2+ level.
This protocol failed to yield foci (Table 1, Experiment 1), while\-ras"a cultures treated in 0.05 rriM Ca2+ medium prior toselection in higher-Ca2* medium responded appropriately.
Because of the nature of the assay, a number of variableswere recognized which influenced the results. Each experimentused fresh primary isolates of mouse keratinocytes, independentcollections of virus supernatants known to vary somewhat intiter, freshly prepared carcinogen solutions with limitedaqueous stability, and the usual culture variables of mediumand serum. These undoubtedly contributed to variability amongabsolute values for focus number between experiments. However, within experiments results were very reproducible. Forexample, when the same treatment protocols were repeatedthree times in one experiment using the same cell isolates andreagents (each group consisting of 8-10 dishes), the results werevirtually identical (Table 1, Experiment 5). In an attempt tocontrol for the interexperimental variation in assays, an MNNGgroup (20.4 ¿JM)was used as a positive control for each test.This was useful in estimating the sensitivity to progression ineach new experiment. Nevertheless, this control still must beconsidered within the context of the variability in handling theMNNG solutions at the time of treatment.
Many preliminary studies were performed to establish the
optimal conditions for selecting foci after carcinogen treatmentof \-ras keratinocytes. These can be summarized as follows: (a)cells maintained in 0.05 mM Ca2+ for 2 weeks after carcinogentreatment yielded more foci after high Ca2+ selection in bothtreated and untreated groups; (b) cells selected in 0.15 mM Ca2+
also yielded more spontaneous solvent control foci; (c) selectionin 1.4 mM Ca~+ medium yielded fewer foci after carcinogentreatment than selection in 0.5 mM Ca2+ medium, but sponta
neous focus formation in solvent-treated groups was low inboth. Based on these studies, a standard protocol for comparative analysis of carcinogens was established with a 1-weekpostcarcinogen expression period in 0.05 mM Ca2+ mediumand 4 weeks of selection in 0.5 mM Ca2+ medium.
Survey of Carcinogens Producing Proliferative Foci. Using astandard protocol, a number of carcinogens were evaluated fortheir potency to produce foci in this assay (Table 2). These wereselected because of their known activity to enhance malignantconversion in vivo or because of their mutagenic potency.Benzo(a)pyrene diolepoxide I had potent cytotoxic effects for\-ras"' keratinocytes but produced foci within a narrow dose
range considerably below the dose required for MNNG-inducedfoci (Table 2, Experiments 1-3). This agent has been reportedto be potent for inducing malignant conversion in vivo* Incontrast /V-acetoxyacetylaminofluorene was less cytotoxic thanbenzo(fl)pyrene diolepoxide I but did not produce a significantnumber of foci at any dose tested (Table 2, Experiments 4 and5). The potency of this compound for conversion in vivo hasnot been tested. cis-DDP is a very potent converting agent invivo (30) and was effective in vitro (Table 2, Experiments 6-9).cis-DDP was 10 times more potent at producing foci thanMNNG, although a very narrow dose range was required. Thisis of interest since in vivo m-DDP is potent as a convertingagent at 100 and 200 Mg given once but less effective whengiven repeatedly to papilloma-bearing mice (30). 4-Nitroqui-noline-/V-oxide was more cytotoxic than MNNG but effectivein producing foci in a range of doses similar to that for MNNGin vitro (Table 2, Experiments 10-11). These compounds alsohave similar potencies for malignant conversion in v/vo(l). Thetumor promoter TPA does not enhance malignant conversionin vivo (1, 2). In this in vitro assay, repeated exposure to TPAin the selection period did not induce foci in control cells (notshown) and reduced the number of foci produced by MNNGexposure (Table 2, Experiments 12 and 13). Benzoylperoxide,another skin tumor promoter, was tested for converting capacityin vitro. A single exposure to 10 mM benzoylperoxide wasineffective in enhancing the emergence of foci, and multipleexposures were toxic (not shown). This agent is reported to bean effective enhancer of malignant conversion in skin in vivo(31). Its lack of effectiveness in vitro could be related to theexcessive cytotoxicity produced by chronic exposure at biologically active concentrations in vitro, difficulties with chemicalstability in an aqueous environment, or some deficiency in theassay for detecting agents which may work via a peroxideradical.
Tumorigenic Potential of Foci. The production of proliferativefoci by carcinogens was consistent with the phenotypic changesexpected for malignant conversion based on reconstitution withcarcinoma cell lines. However, benign keratinocyte cell linesthat proliferate in media with Ca2+ > 0.1 mM have also been
described (32). Therefore several methods were used to determine whether cells in carcinogen-induced foci have the capacityto produce malignant tumors in vivo (Table 3). Cells were
4T. Slaga, personal communication.
3149
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
CONVERSION ASSAY
CONTROL 6.8uM
MNNG
13.6uM 20.4uM
Fig. 4. Rhodamine-stained dishes from an MNNG dose-response study. Protocol was as described in Fig. 2. After 4 weeks in 0.5 mM Ca2* medium, dishes werefixed and rhodamine stained as described in "Materials and Methods."
released from dishes containing foci and injected s.c. into nudemice or grafted as a reconstituted skin. These cell preparationscontained both background quiescent cells and proliferativecells from foci, but the total cell numbers were small, probably<1 x IO6. No tumors were obtained by the s.c. route. Two of
27 animals with grafts of cell preparation from dishes containing foci developed squamous cell carcinomas at the graft site.Cells from control dishes never produced tumors, but a normalskin formed which could have originated from either graftedcells or from host wound healing. The origin could not bedetermined with certainty. In other experiments, cells from fociwere subcultured by ring cloning and plated onto plastic orcollagen-coated dishes. Few subcultured foci survived as secondary cultures by these methods. From multiple attempts inseparate experiments, only five cell lines were developed. Ofthese, three were tumorigenic and produced squamous carcinomas when tested in vivo by grafting 5 x IO6 cells/mouse
(Table 3, cell lines 41 B, 27 AD, and 36 B; Fig. 5, c and h).However, greater success in subculturing was obtained whencells from ring-cloned foci were plated onto dishes containinga monolayer of keratinocytes transduced with the v-rasHa geneand grown to confluence in 0.5 mM Ca2+ medium. In these
studies, five of six cloned foci produced secondary cultures, and
four of these five became confluent in secondary culture anddeveloped into cell lines which did not require the feeder cellsby the second subculture. Of the four cell lines established bythis method, three produced squamous cell carcinomas in animals when 5 x IO6 cells were grafted to nude mice (Table 3,
cell lines B17a, 45 B3C, and 45 B6C; Fig. 5, a and b). Of theselines, 45 B3C and 45 B6C produced tumors in all animalstested, whereas B17a produced tumors in two of nine animalstested.
We considered the possibility that the removal of primarycells from the culture plate, after several months as primarycultures in 0.5 mM Ca2+ medium, might jeopardize the viability
of the cells being tested in vivo. This might also explain whypapillomas did not form from foci or background cells. Toavoid this possibility, cells were grown and treated on dishescontaining a thin plastic liner until foci were noted. On thissubstrate, few foci are formed, and foci of sufficient size did notdevelop until 11 weeks in 0.5 mM Ca2+ medium. Foci were
excised while still attached to the plastic liner, and the entiredisk with cells was placed s.c. in nude mice (Table 3). For thistechnique, liners containing background cells, homogeneousorganized foci, or heterogeneous disorganized foci were inserted. Disks with background cells or orderly foci did not give
3150
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
Table 2 Comparison of agents used to induce proliferai/ve foci
ExperimentAgent1
ControlMNNG2
ControlMNNGBPDEI3
ControlMNNGBPDEI4
ControlMNNGNA-AAF5
ControlMNNGNA-AAF6
ControlMNNGc/J-DDP7
ControlMNNGc/s-DDP8
ControlMNNGCM-DDP9
ControlMNNGa's-DDP10
ControlMNNGNQO1
1ControlMNNGNQO12
ControlMNNGTPAMNNG/TPA13
ControlMNNGTPAMNNG/TPADose(>lM)13.60.10.520.40.10.20.320.40.10.20.320.41.03.010.030.020.41.02.03.020.41.05.010.020.42.03.04.020.42.03.04.020.42.03.04.020.42.65.37.920.42.65.37.913.60.16*13.6/0.1620.40.820.4/0.8Survival1.01.01.00.51.00.81.00.90.71.00.91.01.00.81.00.81.00.90.90.61.00.41.00.90.91.00.70.90.60.61.00.80.90.70.71.00.70.70.40.31.00.50.60.50.41.00.50.90.80.41.00.60.80.70.61.00.81.00.81.00.81.00.8Corrected
foci/dish0.23.51.500000.41.60.10.50.30.40.100.60.10.10002.00.10000.800000.90.30003.00.76.11.8O.I9.93.41.000.15.70.50.61.30.13.01.62.02.30.57.00.21.00.53.00.40.4
* BPDEI, benzo(u)pyrene diolepoxide; NA-AAF, A'-acetoxyacetylaminoflu-orene; NQO, 4-nitroquinoline-A'-oxide.
* TPA given continuously during selection.
tumors. However, disorganized, heterogeneous foci producedundifferentiated carcinomas in all cases (Fig. 5d). Thus, whilecarcinogen treatment produced many foci in a dose-dependentmanner, only a few foci appeared to be converted to the fullymalignant phenotype.
Persistence of the Viral Genome. The persistent integrationof the v-ras"a gene in foci, cell lines, and tumors was evaluated
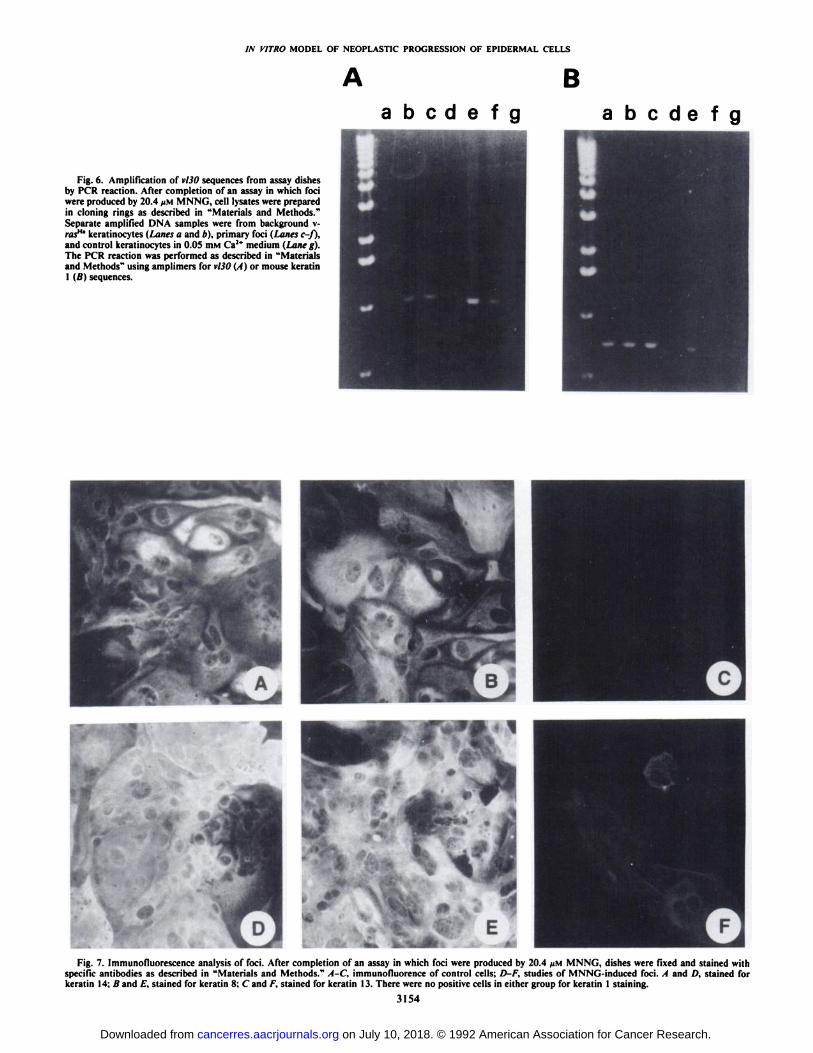
by PCR analysis using amplimers for the unique rat vl30sequences in the v-raslla genome (Table 4; Fig. 6). Amplifiedvl30 sequences were not obtained from lysates of control unin-
fected keratinocytes but were readily produced in lysates fromv-rasHa keratinocytes maintained in 0.5 niM Ca2+ medium as
untreated controls in conversion assays. Lysates from as few as5000 cells were sufficient to detect vl30 sequences in infectedcultures. Thirteen individual foci were outlined with a cloningring, and cells were released with trypsin. The pelleted cellsuspensions were lysed and processed for polymerase chainreaction as described in "Materials and Methods." Lysates of
eight foci yielded amplified vl30 sequences. Lysates from twoof five vl30 negative foci also failed to yield amplified mousekeratin 1 sequences, suggesting insufficient template in thestarting material. Because of insufficient remaining lysate, threeof the five v/50-negative foci could not be tested with the keratin1 amplimers. These results suggest that the majority, if not all,of the primary foci maintain integrated \-ras"a sequences. How
ever, lysates of cell lines derived from foci frequently lackeddetectable vl30 template, while keratin 1 sequences were amplified from the same material (Table 4). All four cell lines withthe viral genome were tumorigenic, whereas only two of fivecell lines without detectable vl30 sequences produced tumors.Tumorigenic v/JO-negative cell lines had relatively low tumorincidences (45 B17a 2 tumor in 9 animals tested; 27 AD 6tumors in 14 animals tested). The viral gene was also detectedin tumors derived by implanting hyperproliferative foci on aplastic insert. When the v/30-negative cell lines were infectedwith the replication-defective v-rasHa retrovirus and retested by
grafting in vivo, all recipient lines were tumorigenic and produced squamous cell carcinomas (Fig. 5, e-g). Furthermore,the v/JO-negative low tumorigenic cell lines 27AD and 45 B17a
produced tumors in 100% of animals tested after introductionof the \-ras genome. All tumors were positive for vl30 sequencesby PCR detection (not shown). These results suggest that theacquisition of the malignant phenotype was associated with achange which complemented the function of the v-ras"" gene.
Markers of Neoplastic Progression. Because in vivo transplantation did not support the concept that hyperproliferativefoci had invariably undergone malignant progression, cellularmarkers of neoplastic progression were evaluated in six randomly selected foci in vitro. -y-Glutamyl transpeptidase activity
increases during malignant progression of some skin tumors invivo (33, 34). There was faint staining for -y-glutamyl transpep
tidase activity in both foci and control regions, but quantitativedifferences could not be detected among six of six foci relativeto control staining. All six foci examined, as well as equivalentregions in control dishes, expressed keratin 14, confirming theirepithelial origin. Keratin 1 was not detected in either controlareas or foci, and this was an expected consequence of v-ras"*transformation (19). Background cells, control v-ras"a keratinocytes, and all foci were strongly positive for keratin 8 im-munostaining. Previously we have reported that the combinedeffects of 0.5 mM Ca2* medium and the v-ras"" oncogene
induced expression of keratins 8 and 18 in cultured keratinocytes (19). Together these results were consistent with a docu-
3151
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VÃŒTROMODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
Table 3 Tumorigenicity of cells with proliferative phenotype
Method oftestingSQ°inoculation 1 dish/animalNo.
ofexperiments4CarcinogensControl
MNNGc/i-DDPRange
offoci/dish0-5.4Total
No.animalstested6
145ResultNotumorsIn
vivoduration of
study(weeks)8-18
Grafting I dish/animal
Cell line from ring-cloned focus or subcultured dish-grafted or SQ inoculation
Insertion of focus SQ on plastic culture substrate
MNNG
MNNG
MNNG
3-10
NAC
NA
27
73
1 SCC* from each of 2
independent experiments representing 18dishes
3 lines,'' no tumors6 lines' SCC
Background monolayerNo tumors (0/3)
Homogeneous fociNo tumors (0/3)
Heterogeneous fociUndifferentiatedCarcinoma (3/3)
9-20
16-40
12
* Subcutaneous inoculation.* Squamous cell carcinoma.' NA, not available.d Designated 45B46, 41B1, and 32 A-B.' Designated 27 AD, 41B, 36B, 45 B6C, 45 B3C, and B17a.
mented v-ra5Ha phenotype in both control cells and all foci
examined. However, immunostaining of foci for keratin 13(K13) revealed that this marker, which is highly associated withthe progression of benign tumors to malignancy in vivo (28, 33,34), was positive only in foci (Fig. 7). The percentage of cellsexpressing K13 varied among foci, but all foci examined hadsome keratin 13-positive cells, and in several foci K13-positivecells constituted a substantial subpopulation (Fig. 7). This resultsuggested that the induction of hyperproliferative foci wasconsistent with neoplastic progression in vitro, although thebiological changes were not always associated with the acquisition of the malignant phenotype.
DISCUSSION
The carcinogen-induced phenotypic progression to focal hy-perproliferation of v-ras"* keratinocytes is likely to be due to
changes in specific genetic targets, since agents which are nearlyequipotent as mutagens and equally cytotoxic to keratinocyteswere variable in producing proliferative foci at comparableconcentrations. That genetic changes underly the phenotypicprogression has not been proved. However, the carcinogen-induced changes required exposure in 0.05 mM Ca2+ medium
when cell proliferation was high and did not occur with carcinogen exposure in 1.4 mM Ca2+ when cells were quiescent.
Nevertheless, it is possible that other changes induced byhigher-Ca2+ medium could be responsible for the suppression
of mutagen-induced phenotypic progression. Whatever molecular changes are involved, multiple pathways appear to bepermissive for producing hyperproliferative, Ca^-resistant foci,
since they were morphologically variable and biologically heterogeneous. Furthermore, only a subset converted to malignantgrowth or survived subcultivation. Some of this heterogeneitycould result from variable viral integration sites or loss of theviral gene. The ability of certain mutagens to induce progressionin vitro and the ineffectiveness of others which are equallycytotoxic or mutagenic are reminiscent of the variable potencyof certain initiating carcinogens which we now recognize to bebased on affinity for specific genetic targets relevant to initiation(35, 36). The current results suggest that the genetic targets for
initiation are different from the sites most relevant for progression, since agents which are potent in the conversion assays invivo and in vitro, such as as-DDP and benzo(a)pyrene diole-poxide I, are relatively weak initiating agents (37, 38).
Cells in many proliferative foci were difficult to subcultureand were not malignant when tested in vivo. Under some invivo test conditions, the number of cells applied may have beentoo low to detect the tumorigenic phenotype except for highlyaggressive malignant cells. Nevertheless, papillomas induced bychemicals applied to mouse skin are known to be heterogeneousin their risk for spontaneous and induced malignant progression(39, 40), although the biochemical basis for the variability hasnot been determined. High-risk benign tumors arise earlier andpersist after tumor promotion is discontinued, suggesting thatthey have a constitutive growth advantage (39). Enhancedgrowth alone is not sufficient to cause progression or conversion, since overexpression of TGF«in benign skin tumor cellsincreased tumor growth but did not cause phenotypic progression (41). Risk for progression in skin papillomas is not determined exclusively by the initiating mutation, since both highland low-risk tumors induced by 7,12-dimethylbenz[a]anthra-cene have the identical mutation in the c-rasHa alÃele.5However,
certain unidentified mutations associated with initiation byspecific carcinogens do appear to influence the risk for carcinoma formation in mouse skin (36). Papilloma cells initiatedby 7,12-dimethylbenz[a]anthracene and promoted by TPA ormezerein can undergo phenotypic progression involving genetically determined premalignant changes. For example, the introduction of a mutant p53 gene or the neu oncogene intopapilloma cells can cause phenotypic but not malignant progression (4), whereas introduction of the v-rasHa or \-fos onco
gene causes both (3, 4, 13). Chromosomal aberrations or genecopy number changes also commonly occur in persistent benignskin tumors but are often associated with phenotypic progression (e.g., dysplasia, changes in tumor markers) rather thanmalignant conversion (8, 9). Thus the heterogeneous responseto carcinogen exposure observed in this assay may be useful forisolating individual phenotypes representing varying stages of
' Unpublished observations.
3152
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

M VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
malignant progression in the skin model.The difficulty in subculturing cells released from foci and
plated into fresh culture dishes was obviated when we foundthat a feeder layer of \-ras"' keratinocytes maintained in 0.5mM Ca2* medium supported clonal expansion of secondary
cultures. This suggests that the neighboring cells providedconditions essential for the continued growth and survival ofcells undergoing malignant progession, at least until theyachieved substantial numbers of their own. This could be relevant to the evolution of skin carcinomas from benign lesions invivo. Cooperative effects among cells within benign tumorscould be an important underlying process which distinguisheshigh-risk from low-risk premalignant lesions. The analysis ofgrowth requirements for cell lines and foci should facilitate thecharacterization of these putative intermediate stages of progression and factors required for their propagation.
A puzzling aspect of the results obtained in this study was
Table 4 Detection ofvl30 sequences in cells andtumorsSource
ofsampleControl
keratinocytesv-raj"" keratinocytesMNNG-induced fociTumorigenic cell linesNontumorigenic cell linesTumorsPCR
detectionv/.il)positiu'/nn.tested0/4
8/88/13°
4/60/35/7*
" The keratin I sequences could not be amplified in two negative samples. The
remaining three negative samples were not tested for keratin 1 sequences becauseof insufficient volume.
* Two v/.?0negative tumors were derived from (wo vl30 negative cell lines. The
tumors derived from implanting cells on a plastic insert (see Table 3) containedthe viral sequence and account for the extra tumor type in this group.
Fig. 5. Tumors produced from cell lines, a-d and A, sections from tumors produced bygrafting 5 x IO6cells from cell lines 45 B3C(a). 45 B6C (b), 36B (c), LD3 (d). e-g, sectionsfrom tumors produced by introducing the \-ras"' retroviral construct into nontumorigeniccell lines 45B4b (e), 41 B' (/), and 32 A-B (g).
h, a tumor from cell line 27 AD which lacksVÌÌOsequences. H & E, x 160.
•¿�*<'.. <
fc^TrsswE&Ã-J. *'¿„¿�<attest»
d
3153
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
B
Fig. 6. Amplification of vl30 sequences from assay dishesby PCR reaction. After completion of an assay in which fociwere produced by 20.4 /JM MNNG, cell lysates were preparedin cloning rings as described in "Materials and Methods."Separate amplified DNA samples were from background v-ras"' keratinocytes (Lanes a and 6), primary foci (Lanes c-f),and control keratinocytes in 0.05 HIMCa2* medium (Lane g).The PCR reaction was performed as described in "Materialsand Methods" using amplimers for vl30 (A) or mouse keratin1 i />'s sequences.
Fig. 7. Immunofluorescence analysis of foci. After completion of an assay in which foci were produced by 20.4 ¿IMMNNG, dishes were fixed and stained withspecific antibodies as described in "Materials and Methods." A-C, immunofiuorence of control cells; D-F, studies of MNNG-induced foci. A and £),stained for
keratin 14; A and E, stained for keratin 8; C and F. stained for keratin 13. There were no positive cells in either group for keratin 1 staining.
3154
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
the absence of tumor formation in some of the cell linesobtained from ring-cloning carcinogen-induced foci. It wasexpected that all cell lines would yield papillomas even if notconverted to malignancy. Instead, normal skin formed at thegraft site. One explanation could be the loss of the v-ras"' gene
from the selected populations over time. In fact, PCR detectionof vl30 sequences in most foci prior to subculture, the absenceof keratin 1 expression, and the detection of keratin 8 in allfoci examined suggested that v-rasHa sequences are expressedin all primary foci. Nevertheless, during the course of subcul-turing, the viral genome was commonly lost either throughselection of foci lacking the viral gene or by some active processof exclusion and selection. In either case loss of the viral genomewas associated with the loss or reduction of tumorigenic potential, while a persistent viral genome was associated with malignant conversion. We have previously isolated carcinogen-treated or spontaneously developing cell lines without ras"*gene mutations that were Ca2+ resistant and proliferated in vitro
but produced normal skin when grafted to nude mice (42).While these lines were not normal, they have undergone agroup of changes which can complement but not produce theneoplastic phenotype. Furthermore, loss of the viral genomecould explain the frequent regression of papillomas producedby grafts of v-ras"' keratinocytes to nude mice (43). The introduction of the v-ras"" genome into y/50-negative cells produced
the carcinoma phenotype, further supporting the importance ofcooperation among an oncogenic ras gene and specific complementing changes in malignant conversion.
v-ras keratinocytes in vitro differ from other neoplastic epidermal cells that we have studied, since the v-ras"* cells areCa2+ resistant for terminal differentiation but Ca2+ sensitive forloss of proliferative capability (18). Most neoplastic keratino-cyte cell lines derived by treating normal skin or skin cells withchemical carcinogens and selecting for Ca2+ resistance in vitrocontinue to grow in the higher-Ca2* medium (23, 24). We nowknow that v-rasH" keratinocytes express high levels of TGF0,in 0.5 HIM Ca2+ medium and are sensitive to the growth-
inhibitory effects of this cytokine (44). The TGFßeffectorpathway therefore could be one target for carcinogens thatcould cause the phenotypic progression we have identified. Insupport of this possibility is the recent description of a continuous cell line of murine keratinocytes transformed by v-raswhich is resistant to TGF0 growth suppression (45).
The assay described here can distinguish among a group ofcarcinogens and detect those that will efficiently cause focalphenotypic changes in benign skin cells. The new phenotype isconsistently hyperproliferative but not consistently malignant.Nevertheless, the potency ranking of the limited list of agentsknown to enhance malignant conversion in mouse skin papillomas in vivo correlates well with the ability of these sameagents to induce focus formation in this assay. Since only asubpopulation of skin papillomas expressing a mutated c-rasHagene is susceptible to mutagen-induced malignant conversionin vivo, the in vitro assay parallels the biology in vivo (40). Theexpression of keratin 13 in hyperproliferative foci and its absence in control cells are also consistent with the process ofneoplastic progression in vivo(28,33,34). Since K13 expressionis not associated with hyperproliferation in cultured keratinocytes or skin in vivo, we must conclude that it is a marker ofneoplastic progression in this assay. These results suggest thatthis assay could be useful for identifying and studying agentsthat cause neoplastic progression. Since it is quantitative andrelatively rapid, it would have substantial advantages over the
current in vivo model for progression in mouse skin. The assayshould also be valuable for evaluating biochemical and geneticchanges in the converted cells responsible for the new phenotype. Clonal evolution of the neoplastic phenotype is extremelydifficult to detect in a tumor. Therefore, this model could be asignificant advance in evaluating that process.
ACKNOWLEDGMENTS
The authors wish to thank Dr. Wendy Weinberg for performing the•¿�v-glutamyltranspeptidase assays and Christina Cheng for assisting
with the immunofluorescence studies. The excellent editorial assistanceof Margaret Taylor is also acknowledged.
REFERENCES
1. Hennings, H., Shores, R., Wenk, M. L., Spangler, E. F., Tarone, R., andYuspa, S. H. Malignant conversion of mouse skin tumors is increased bytumor initiators and unaffected by tumor promoters. Nature (Lond.), 304:67-69, 1983.
2. O'Connell. J. F., Klein-Szanto, A. J. P., Digiovanni, D. M., Fries, J. W., and
Slaga, T. J. Malignant progression of mouse skin papillomas treated withethylnitrosourea, /V-methyl-W-nitro-iV-nitrosoguanidine, or 12-0-telradeca-noylphorbol-13-acetate. Cancer Lett., 30: 269-274, 1986.
3. Greenhalgh, D. A., and Yuspa, S. H. Malignant conversion of murinesquamous papilloma cell lines by transfection with the/05 oncogene. Mol.Carcinog., /: 134-143. 1988.
4. Dotto, G. P., O'Connell, J.. Patskan. G.. Conti, C.. Ariza, A., and Slaga, T.J. Malignant progression of papilloma-derived keratinocytes: differentialeffects of the ras, neu, and p53 oncogenes. Mol. Carcinog., /: 171-179, 1988.
5. Roop, D. R., Lowy, D. R., Tambourin, P. E., Strickland, J., Harper, J. R.,Balaschak, M., Spangler. E. F.. and Yuspa. S. H. An activated Harvey rasoncogene produces benign tumours on mouse epidermal tissue. Nature(Lond.), 323: 822-824, 1986.
6. Brown, K., Quintanilla, M., Ramsden, M., Kerr, I. B., Young, S., andBalmain. A. v-ras genes from Harvey and BALB murine sarcoma viruses canact as initiators of two stage mouse skin carcinogenesis. Cell, 46: 447-456,1986.
7. Greenhalgh, D. A., Welly, D. J., Player, A., and Yuspa, S. H. Two oncogenes,V-/0ÃŒand v-ras, cooperate to convert normal keratinocytes to squamous cellcarcinomas. Proc. Nati. Acad. Sci. USA, 87: 643-647. 1990.
8. Aldaz, C. M.. Conti, C. J., Klein-Szanto. A. J. P., and Slaga, T. J. Progressivedysplasia and aneuploidy are hallmarks of mouse skin papillomas: relevanceto malignancy. Proc. Nati. Acad. Sci. USA. 84: 2029-2032, 1987.
9. Aldaz, C. M., Trono, D., Larcher, F., Slaga, T. J., and Conti, C. J. Sequentiallrisoini/illuni of chromosomes 6 and 7 in mouse skin premalignant lesions.Mol. Carcinog., 2: 22-26, 1989.
10. Quintanilla, M.. Brown, K., Ramsden, M., and Balmain, A. Carcinogen-specific mutation and amplification of I la rut during mouse skin carcinogenesis. Nature (Lond.). 322: 78-80. 1986.
11. Bremner, R., and Balmain. A. Genetic changes in skin tumor progression:correlation between presence of a mutant ras gene and loss of heterozygosityon mouse chromosome 7. Cell, 61:407-417, 1990.
12. Bianchi, A. B., Aldaz. C. M., and Conti, C. J. Non-random duplication ofthe chromosome bearing a mutated Ha-ras-1 alÃelein mouse skin tumors.Proc. Nati. Acad. Sci. USA. 87: 6902-6906. 1990.
13. Greenhalgh. D. A., Welty, D. J., Strickland, J. E.. and Yuspa, S. H. Spontaneous Ha-ras gene activation in cultured primary murine keratinocytes:consequences of Ha-ras gene activation in malignant conversion and malignant progression. Mol. Carcinog., 2: 199-207, 1989.
14. Aldaz, C. M., Conti, C. J., O'Connell, J., Yuspa, S. H., Klein-Szanlo. A. J.
P., and Slaga. T. J. Cytogenetic evidence for gene amplification in mouseskin carcinogenesis. Cancer Res.. 46: 3565-3568, 1986.
15. Garte, S. J., Burns, F. J., Ashkenazi-Kimmel, T., Felber, M., and Sawey, M.J. Amplification of the c-myc oncogene during progression of radiation-induced rat skin tumors. Cancer Res., SO:3073-3077, 1990.
16. Rhim, J. S. Neoplastic transformation of human epithelial cells in vitro.Anticancer Res., 9: 1345-1365, 1989.
17. Stacey, M., Gallimore, P. H., McConville, C., and Taylor, A. M. Rearrangement of the same chromosome regions in different SV40 transformedhuman skin keratinocyte lines is associated with tumourigenicity. Oncogene,5:727-739, 1990.
18. Yuspa, S. H., Kilkenny, A. E., Stanley, J., and Lichti, U. Keratinocytesblocked in phorbol ester-responsive early stage of terminal differentiation bysarcoma viruses. Nature (Lond.), 314: 459-462, 1985.
19. Cheng, C.. Kilkenny, A. E., Roop, D., and Yuspa, S. H. The v-ras oncogeneinhibits the expression of differentiation markers and facilitates expressionof cytokeratins 8 and 18 in mouse keratinocytes. Mol. Carcinog., 3: 363-373, 1990.
20. Yuspa, S. H., Hawley-Nelson, P., Koehler, B.. and Stanley, J. R. A survey oftransformation markers in differentiating epidermal cell lines in culture.Cancer Res., 40: 4694-4703, 1980.
3155
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

IN VITRO MODEL OF NEOPLASTIC PROGRESSION OF EPIDERMAL CELLS
21. Hartley, J. A., Gibson, N. W., Kilkenny, A., and Yuspa, S. H. Mousekeratinocytes derived from initiated skin or papillomas are resistant to DNAstrand breakage by benzoyl peroxide: a possible mechanism for tumor promotion mediated by benzoyl peroxide. Carcinogenesis (Lond.). 8: 1827-1830, 1987.
22. Hennings, H., Michael, D.. Cheng, C., Steinen, P., Holbrook, K., and Yuspa,S. H. Calcium regulation of growth and differentiation of mouse epidermalcells in culture. Cell, 19: 245-254, 1980.
23. Kulesz-Martin, M. F., Koehler, B., Hennings. H., and Yuspa, S. H. Quantitative assay for carcinogen altered differentiation in mouse epidermal cells.Carcinogenesis (Lond.), /: 995-1006, 1980.
24. Kulesz-Martin. M., Kilkenny, A. E., Holbrook. K. A., Digernes, V., andYuspa, S. H. Properties of carcinogen altered mouse epidermal cells resistantto calcium-induced terminal differentiation. Carcinogenesis (Lond.), 4:1367-1377, 1983.
25. Gratzner, H. G. Monoclonal antibody to 5-bromo-and 5-iododeoxyuridine:a new reagent for detection of DNA replication. Science (Washington DC),218: 474-475, 1982.
26. Ellis. R. W., DeFeo, D., Maryak, J. M., Young, H. A., Shih, T. Y., Chang,E. H., Lowy, D. R., and Scolnick, E. M. Dual evolutionary origin for the ratgenetic sequences of Harvey murine sarcoma virus. J. Virol., 36: 408-420,1980.
27. Yuspa, S. H., Kilkenny, A. E., Steinert, P. M., and Roop, D. R. Expressionof murine epidermal differentiation markers is tightly regulated by restrictedextracellular calcium concentrations in vitro. J. Cell Biol., 709: 1207-1217,1989.
28. Nischt. R., Roop, D. R., Mehrel, T.. Yuspa, S. H., Rentrop. M., Winter. H.,and Schweizer, J. Aberrant expression during two-stage mouse skin carci-nogenesis of type I 47-kDa keratin. K13. normally associated with terminaldifferentiation of internal stratified epithelia. Mol. Carcinog.. /: 96-108.1988.
29. Perantoni. A., and Herman, J. J. Properties of Wilms' tumor line (TuWi)and pig kidney line (LLC-PK) typical of normal kidney tubular epithelium.In Vitro (Rockville). 15: 446-454. 1979.
30. Hennings, H., Shores, R. A., Poirier, M. C., Reed, E., Tarane, R. E., andYuspa, S. H. Enhanced malignant conversion of benign mouse skin tumorsby cisplatin. J. Nati. Cancer Inst., 82: 836-840, 1990.
31. O'Connell, J. F., Klein-Szanto, A. J. P.. DiGiovanni, D. M., Fries, J. W.,
and Slaga. T. J. Enhanced malignant progression of mouse skin tumors bythe free-radical generator benzoyl peroxide. Cancer Res., 46: 2863-2865.1986.
12. Strickland, J. E., Greenhalgh, D. A., Koceva-Chyla. A., Hennings. H..Restrepo, C., Balaschak. M., and Yuspa, S. H. Development of murineepidermal cell lines which contain an activated ras11' oncogene and form
papillomas in skin grafts on athymic nude mouse hosts. Cancer Res., 48:165-169, 1988.
33. Aldaz, C. M.. Conti, C. J., Larcher. F., Trono, D., Roop, D. R., Chesner, J.,Whitehead. T., and Slaga, T. J. Sequential development of aneuploidy,keratin modifications, and 7-glutamyltransferase expression in mouse skinpapillomas. Cancer Res., 48: 3253-3257, 1988.
34. Gimenez-Conti, !.. Aldaz, C. M., Bianchi, A. B., Roop, D. R., Slaga, T. J.,and Conti. C. J. Early expression of type I K13 keratin in the progression ofmouse skin papillomas. Carcinogenesis (Lond.). //: 1995-1999, 1990.
35. Balmain, A., and Brown. K. Oncogene activation in chemical Carcinogenesis.Adv. Cancer. Res., 5/: 147-182, 1988.
36. Brown, K., Buchmann. A., and Balmain, A. Carcinogen-induced mutationsin the mouse c-Ha-ras gene provide evidence of multiple pathways for tumorprogression. Proc. Nati. Acad. Sci. USA, 87: 538-542, 1990.
37. Slaga, T. J., Bracken, W. M.. Viaje, A.. Levin, W., Yagi, H., Jerina, D. M.,and Conney. A. H. Comparison of the tumor-initiating activities ofbenzo(o)pyrene arene oxides and diol epoxides. Cancer Res., 3 7:4130-4133,1977.
38. Barnhart, K. M., and Bowden, G. T. Cisplatin as an initiating agent in two-stage mouse skin Carcinogenesis. Cancer Lett., 29: 101-105, 1985.
39. Hennings. H., Shores. R., Mitchell. P., Spangler, E. F., and Yuspa, S. H.Induction of papillomas with a high probability of conversion to malignancy.Carcinogenesis (Lond.), 6: 1607-1610. 1985.
40. Hennings, H., Shores, R., Balaschak, M.. and Yuspa, S. H. Sensitivity ofsubpopulations of mouse skin papillomas to malignant conversion by ure-thane or 4-nitroquinoline A'-oxide. Cancer Res., 50: 653-657, 1990.
41. Finzi. E., Kilkenny. A.. Strickland. J. E., Balaschak. M.. Bringman, T.,Derynck, R., Aaronson. S., and Y'uspa, S. H. TGF «stimulates growth of
skin papillomas by autocrine and paracrine mechanisms but docs not causeneoplastic progression. Mol. Carcinog., /: 7-12, 1988.
42. Strickland, J. E., Ueda, M., Abhyankar, S., Greenhalgh. D. A., Hennings.H., and Yuspa. S. H. Cell lines from SENCAR mouse skin papillomasproduced by treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA)without chemical initiation. Proc. Am. Assoc. Cancer Res., 30: 191, 1989.
43. Weinberg, W. C.. Morgan. D.. George, C., and Yuspa, S. H. A comparisonof interfollicular and hair follicle derived cells as targets for the \-ras"'oncogene in mouse skin Carcinogenesis. Carcinogenesis (Lond.), 12: 1119-1124, 1991.
44. Click, A. B., Sporn, M. B., and Yuspa, S. H. Altered regulation of TGF-fi 1and TGF-rt in primary keratinocytes and papillomas expressing v-Ha-rm.Mol. Carcinog.. 4: 210-219. 1991.
45. Sipes, N. J., Lyons. R. M.. and Moses, H. L. Isolation and characterizationof Kirsten murine sarcoma virus-transformed mouse keratinocytes resistantto transforming growth factor ti. Mol. Carcinog., 3: 12-19. 1990.
3156
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1992;52:3145-3156. Cancer Res David Morgan, David Welty, Adam Glick, et al. Neoplastic Progression of Initiated Mouse Epidermal Cells
Model to Study Carcinogen-inducedin VitroDevelopment of an
Updated version
http://cancerres.aacrjournals.org/content/52/11/3145
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/52/11/3145To request permission to re-use all or part of this article, use this link
on July 10, 2018. © 1992 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from


![ClinicalValueofSerumGlycoproteinGalactosyltransferaseLevel ...cancerres.aacrjournals.org/content/43/9/4491.full.pdf · [CANCERRESEARCH43,4491-4496,September1983] ClinicalValueofSerumGlycoproteinGalactosyltransferaseLevelsin](https://img.pdfslide.net/doc/110x75/5ac139b17f8b9ac6688d1490/clinicalvalueofserumglycoproteingalactosyltransferaselevel-cancerresearch434491-4496september1983.jpg)



![TumorBehaviorinTransitionalCellCarcinomaoftheBladderinRela ...cancerres.aacrjournals.org/content/47/24_Part_1/6800.full.pdf · [CANCERRESEARCH47,6800-6805,December!5,1987] TumorBehaviorinTransitionalCellCarcinomaoftheBladderinRelationto](https://img.pdfslide.net/doc/110x75/5c085d3a09d3f23a458c00a0/tumorbehaviorintransitionalcellcarcinomaofthebladderinrela-cancerresearch476800-6805december51987.jpg)


![VirologieStudiesinHumanLeukemiaandLymphoma:The …cancerres.aacrjournals.org/content/28/7/1311.full.pdf · [CANCERRESEARCH28,1311-1318,July1968] VirologieStudiesinHumanLeukemiaandLymphoma:The](https://img.pdfslide.net/doc/110x75/5b98f37b09d3f219118d06a7/virologiestudiesinhumanleukemiaandlymphomathe-cancerresearch281311-1318july1968.jpg)
![HeterotransplantationofHumanLymphoidNeoplasmsUsingaNudeMou ...cancerres.aacrjournals.org/content/50/10/3078.full.pdf · (CANCERRESEARCH50,3078-3086.May15,1990] HeterotransplantationofHumanLymphoidNeoplasmsUsingaNudeMouse](https://img.pdfslide.net/doc/110x75/5e83f48eaae3144d7c04ca6b/heterotransplantationofhumanlymphoidneoplasmsusinganudemou-cancerresearch503078-3086may151990.jpg)










![TransientandStableComplementationofUltravioletRepairinXero ...cancerres.aacrjournals.org/content/47/11/2967.full.pdf · [CANCERRESEARCH47,2967-2971.June1,1987] TransientandStableComplementationofUltravioletRepairinXeroderma](https://img.pdfslide.net/doc/110x75/5a77350f7f8b9a4b538dd0bd/transientandstablecomplementationofultravioletrepairinxero-a-cancerresearch472967-2971june11987.jpg)



![ANewGlucocorticoidReceptorDetectedinHostRatLiverbutnotin ...cancerres.aacrjournals.org/content/47/14/3742.full.pdf · (CANCERRESEARCH47,3742-3746,July15,1987] ANewGlucocorticoidReceptorDetectedinHostRatLiverbutnotin](https://img.pdfslide.net/doc/110x75/5be5179609d3f20a668dcb73/anewglucocorticoidreceptordetectedinhostratliverbutnotin-cancerresearch473742-3746july151987.jpg)



![DNATopoisomeraseIIImmunostaininginHumanLeukemiaand ...cancerres.aacrjournals.org/content/52/15/4248.full.pdf · (CANCERRESEARCH52,4248-425.1,August1.1992] DNATopoisomeraseIIImmunostaininginHumanLeukemiaand](https://img.pdfslide.net/doc/110x75/5b6d7db17f8b9a962a8cc15a/dnatopoisomeraseiiimmunostaininginhumanleukemiaand-cancerresearch524248-4251august11992.jpg)