Embed Size (px)
Citation preview

DFCI ALL Consortium Protocol 11-001Site Initiation Visit Part I:
Protocol Overview
Lewis Silverman, MDSeptember 27, 2012

Protocol 11-001: Study Design
• Randomized study to determine Safety and Feasibility of IV Calaspargase Pegol (SC-PEG) compared with IV Oncaspar in children and adolescents with newly diagnosed ALL and lymphoblastic lymphoma

Background
• Calaspargase Pegol– EZN-2285– SC-PEG
• Similar to Oncaspar, but more stable linker (SC-linker)
• Soon to replace Oncaspar as only commercially available form of PEG-ASP in North America

COG AALL07P4
• Randomized comparison of SC-PEG vs Oncaspar• HR ALL • COG Augmented BFM backbone
– Induction: 1 dose– IM#1: 2 doses (Days 2 + 22)– DI#1: 2 doses (Days 4 + 43)– IM #2: 2 doses (Days 2 + 22)– DI #2: 2 doses (Days 4 + 43)
Only SER

COG AALL07P4
Day after dose Oncaspar SC-PEG
15 100% 100%
22 85% 100%
29 22% 100%
• Induction dose: Day 4• Initial Comparison: Each dosed at 2500 IU/m2• Toxicity data similar to IV Oncaspar• SC-PEG appears to have longer half-life
Percentage of patients with serum asparaginase activity level > 0.1 IU/mL on COG Protocol AALL07P4

SC-PEG on DFCI Backbone
• Can SC-PEG be given every 3-weeks during post-induction treatment phases?
– Goal: Achieve 30 weeks of asparagine depletion
• How will this dosing compare with every 2-week IV Oncaspar?
– NSAA– Toxicity

11-001: Primary Objective
Primary Objective• To assess the safety and feasibility
associated with the administration of IV SC-PEG given as a single dose during Remission Induction and every 3-weeks for 30 weeks during post-induction therapy
• Toxicity• NSAA

Protocol 11-001: Study design
• Randomized comparison of IV SC-PEG vs IV Oncospar– Induction: 1 dose (by randomization) at 2500 IU/m2
– Post-Induction• SC-PEG 2500 IU/m2 every 3-weeks• Oncospar 2500 IU/m2 every 2-weeks
• Total planned accrual: 240 patients

Primary Objective: Safety
• Safety: Determining that SC-PEG is not more toxic than IV Oncaspar
• Primary endpoint is not comparison of randomized arms
• Each arm will be compared to baseline IV Oncaspar toxicity rate on 05001– Study is powered to determine whether SC-PEG arm is
more toxic than baseline IV Oncaspar (05-001)– IV Oncaspar arm on 11-001 is to ensure that baseline
toxicity on 11-001 is not significantly different than 05001

Primary Objective: Feasibility
• Pharmacokinetics of every 3-week SC-PEG– “Feasible”: Proportion of pts with at least one
NSAA > 0.1 IU/mL during consolidation phase with SC-PEG is not significantly lower than with IV Oncaspar
– Of first 114 pts treated on 05-001, 100% had at least one NSAA > 0.1 IU/mL

Primary Objective: Feasibility
• Pharmacokinetics of every 3-week SC-PEG– Direct comparison of 2 arms– Assume 99% of pts on IV Oncaspar arm have
at least one NSAA > 0.1 IU/mL– With 112 evaluable pts on each arm, there is
89% power to detect a 10% reduction in proportion of pts with at least one NSAA > 0.1 IU/mL

SC-PEG: Secondary Objectives
• To determine the proportion of samples with NSAA > 0.025, ≥ 0.10, ≥ 0.20, and ≥ 0.40 IU/mL prior to each dose for both arms
• To directly compare NSAA levels between the two arms at weeks 7, 13, 19, and 25 for every 2-week IV Oncaspar and compare them to every 3-week IV SC-PEG.

Early Stopping Rules
• Safety (Toxicity)• MRD• ASP PK

Early Stopping Rules
• Safety Monitoring– Safety data reviewed every 6 months by DMC– Monitor proportion of pts with ASP-related
toxicity to ensure rate does not significantly differ from 27%
– Early stopping rules: Trial to stop if toxicity on either arm appears to exceed 27%:
• eg, 30 pts/arm: 12 or more (40%) with toxicity (80% CI: 28-53%)

Early Stopping Rules
• MRD Monitoring– Of first 432 B-ALL pts on 05001,12.5% with
high MRD ( > 0.001 by PCR)– 11-001: Early stopping if proportion of pts with
high end-induction MRD exceeds 12.5%• eg, 30 pts/arm, assuming 28 evaluable, study
stopped if > 7/28 (25%, 80% CI: 14-38%) with high MRD

Early Stopping Rules
• Induction Asp PK– Ensure that most pts treated with SC-PEG have
level > 0.1 IU/mL 18 days after induction dose– On Protocol 05-001: 88% of pts had this level
18 days after IV Oncaspar– Will monitor level, and stop if proportion of pts
with this level on either arm is lower than 88%• eg, 30 pts/arm, it would be unacceptable if < 23
(77%, 80% CI: 64-87%) have level >0.1 IU/mL

Protocol 11-001: Secondary Objectives
• To determine the feasibility of administering antibiotic prophylaxis during the remission induction phase.
• To describe the outcome of children and adolescents with lymphoblastic lymphoma treated with a DFCI ALL Consortium treatment regimen.
• To explore the impact on outcome of changing therapy for patients with B-ALL based on end-induction minimal residual disease (MRD) and/or high-risk cytogenetics (MLL-gene rearrangements or low hypodiploidy).

Protocol 11-001: Secondary Objectives
• To assess the feasibility of vitamin D screening and supplementation.
– To determine the prevalence of vitamin D deficiency at the following time-points: diagnosis, end of remission induction, start of continuation, conclusion of therapy, and one year after the conclusion of therapy.
– To assess the feasibility of correcting vitamin D deficiency with supplementation of vitamin D and calcium in patients found to have vitamin D deficiency.
– To explore the relationship between vitamin D status and skeletal toxicity (fracture and osteonecrosis) in children and adolescents undergoing therapy for ALL.
– To explore risk factors for vitamin D deficiency, including demographic variables such as age, sex, and ethnicity, as well as geographic location (geographic latitude) and season of measurement.

Protocol 11-001: Biology Objectives
• To determine the feasibility of prospective screening for absence of biallelic TCRg deletions (ABGD) via qPCR in patients with T-ALL
– To determine the frequency of ABGD in patients with T-cell ALL at diagnosis– To explore the correlation between early T-cell precursor (ETP) phenotype and
ABGD• To determine the feasibility of prospective screening for abnormalities
(eg, mutations, deletions, rearrangements) of IKZF1, CRLF2 and JAK1/2 in patients with newly diagnosed B-ALL.
– To determine the frequency of these abnormalities in standard-risk and high-risk patients
– To explore the correlation between the presence of these abnormalities and end-induction MRD levels.
• To explore the relationship between mitochondrial BCL-2 family preconditions and response to chemotherapy as measured by induction response, end-induction MRD level and event-free survival (EFS).
• To obtain patient samples for the development of primary xenograft mouse models with the goal of identifying novel therapies for high-risk (including those with high end-induction MRD, T-ALL with ABGD) and relapsed patients.

11-001: Antibiotic Prophylaxis
• Documented infections (primarily bacteremias) in > 25% of patients during Induction
• 2% of patients die during induction of infection (~4% of HR patients)
• Episodes of bacteremia during induction are significant source of morbidity– prolonged hospitalization– ICU/significant complications– dose reductions and delays in chemotherapy

11-001: Antibiotic Prophylaxis
• In 2005, two large double-blind, placebo controlled trials using levofloxacin in adult cancer patients with neutropenia were published.– Both demonstrated benefit of levofloxacin (fewer
episodes of fever, fewer infections) in cancer patients, especially those with acute leukemia
• In 2008, the National Comprehensive Cancer Network (NCCN) recommended the use of fluoroquinolone prophylaxis in high risk cancer patients, including leukemia patients receiving induction chemotherapy.

11-001: Antibiotic Prophylaxis
• Non-randomized testing of fluoroquinolone prophylaxis during induction phase.
• All afebrile patients will begin either levofloxacin or moxifloxacin during steroid prophase– Continue until count recovery of (ANC > 200/mm3 or
APC > 500/mm3) or initiation of broad-spectrum antibiotics for fever.
• Patients unable to tolerate fluoroquinolone should receive cefipime instead
• Prophylaxis is mandatory

11-001: Antibiotic Prophylaxis
Statistical Plan: • Rates of bacteremia will be compared to historic
controls (25% rate of bacteremia on 05-001)• Only infections in ALL patients will be considered • Will consider infection rate in each randomized
arm separately• With 103 evaluable ALL patients per arm, we
would have 82% power to detect a 12% reduction in the rate of bacteremia

Eligibility (Section 3)
• Confirmed Diagnosis of ALL or Lymphoblastic Lymphoma
– ALL• >30% marrow involvement• If circulating blasts: flow cytometry confirmation of ALL sufficient
for registration; marrow should be performed “as soon as feasible, preferably prior to initiation of any therapy”
• Excluded: Mature B-cell ALL, Leukemia of Ambiguous Lineage– Lymphoblastic Lymphoma: Radiographic evidence of
lymphoma with:• Biopsy of involved site• Cytology from pleural or other fluid• Marrow aspirate with < 30% lymphoblasts

Eligibility
• Prior Therapy: None allowed, except– Corticosteroids: < 7 days within 4-weeks preceding
registration (should treate without prophase)• Not Eligible if > 7 days steroid within previous 4-weeks, or
more than 28 days of steroids within previous 6 months
– IT Cytarabine (single dose)– Emergent Radiation Therapy: Mediastinum, other life-
threatening masses

Eligibility
• Age 365 days to < 18 years• Direct bilirubin < 1.4 mg/dL• Signed, informed consent

Exclusion Criteria
• Chronic steroid pre-treatment, as defined above• Any other prior cancer and/or any prior
chemotherapy or radiation (at any time in the past)– Exception: Pts treated for cancer with surgey only > 5
years preceding registration
• Currently on an investigational agent• Known HIV-positivity (HIV testing not required
for enrollment)

Exclusion Criteria
• Uncontrolled intercurrent illness– Infection with sepsis– Life-threatening tumor lysis syndrome (eg, renal
failure)– Congestive hear failure– Uncontrolled bleeding (intracranial hemorrhage)
• Psychiatric or Social situation that would limit compliance
• Pregnancy– Pregnancy test required of females of childbearing
potential prior to start of therapy

Informed Consent
• Copy of signed, dated informed consent document must be sent to DFCI for patient to be registered.
• ONLY physicians who are listed on the 1572 form may consent participants to 11-001
• ONLY attending physicians may consent participants to 11-001
• Per DFCI Consenting Policy: fellows, nurses, pharmacists may not consent to 11-001 without attending co-sign

Informed Consent
• Informed Consent Document is necessary but not sufficient to document informed consent process
• Review of Consent Discussion must be in medical record• Essential elements of note:
– Research study was fully explained to the participant and/or parents
– Risks and alternative treatment options were discussed – The participant and/or parents had all of his/her questions
regarding the study answered. – They understand they can withdraw consent at any time– The participant and/or parents signed the research consent form
and was/were given a copy of the signed consent form

Registration
• Note: Only patients who consent to Asparaginase randomization will be enrolled on study– No “direct assignments” for patients who refuse
randomization
• Weekdays: Randomization performed at time of registration
• Off-hours: Registration allows start of therapy; Randomized assignment made and communicated to site on next business day

Risk Classification: Differences from 05-01
– Essentially same criteria for SR, HR, VHR– Slight modification in definition of
“Hypodiploidy”• Now < 44 chromosomes (formerly < 45
chromosomes
– Lymphoblastic lymphoma risk-stratified like ALL, but no MRD

Risk Group Classification (Section 5)
• Initial Risk Group: Age, WBC, Immunophenotype, CNS status (if known)
• Final Risk Group: Cytogenetics, MRD (B-ALL only)

Standard Risk
All of the criteria must be met:• Age: 365 days to < 10 years.• WBC count: Highest pre-treatment WBC <50,000/mm3 (prior to
registration).• CNS leukemia: No evidence of CNS leukemia, defined by meeting
all of the following criteria:– Diagnostic lumbar puncture (Day 1) without any CSF blast cells on
cytospin (CNS-1) or fewer than 5 WBC/hpf in CSF with blast cells noted on cytospin (CNS-2). If traumatic tap (>10 RBC's on CSF cell count) with > 5 WBC cells are seen, use Steinherz/Bleyer algorithm (see 5.1.6) to determine if patients should be considered CNS-2 or CNS-3 for purposes of risk group assignment.
– CNS-1 CSF on Days 18 and 32. Absence of a cranial nerve palsy at diagnosis.

Risk Group Classification: Standard Risk
• Immunophenotype: Predominance of B-precursor cell surface antigens on lymphoblasts.
• Chromosomal abnormalities: Absence of t(9;22), MLL gene translocations and hypodiploidy < 44 chromosomes as determined by karyotype, PCR or FISH analysis. If chromosomal data unavailable, may continue to be treated as SR if all other risk group criteria are met.
• MRD (leukemia patients only): MRD level < 0.001 on a marrow sample obtained at end of remission induction therapy (Day 32). SR patients whose end-of-induction MRD status cannot be determined will remain SR.

Risk Group Classification: High Risk
Any of the following
• Age: 10 to < 18 years• WBC count: Highest pre-treatment WBC >50,000/mm3 (prior to
registration).• CNS leukemia: Evidence of CNS leukemia, defined by meeting any of
the following criteria:– Diagnostic lumbar puncture (Day 1) with 5 or greater WBC/hpf and blast
cells on cytospin (CNS-3). If traumatic tap (>10 RBC's on CSF cell count) with > 5 WBC cells are seen, use Steinherz/Bleyer algorithm (see 5.1.6) to determine if patients should be considered CNS-2 or CNS-3 for purposes of risk group assignment.
– CNS-2 on Day 18 or 32. – CNS-3 on Day 18. – Presence of a cranial nerve palsy at diagnosis.

High Risk
• Immunophenotype: Predominance of T-cell markers on lymphoblasts.
• MRD (B-ALL only): – B-precursor HR patients with MRD level < 0.001 on a
marrow sample obtained at Day 32 will continue to be treated as HR.
– B-precursor HR patients with MRD levels > 0.001 at Day 32 will be re-classified and re-consented as very high risk
– B-precusor HR patients whose end-of-induction MRD status cannot be determined will remain HR.

Very High Risk
Any of the following• Chromosomal abnormalities:
– MLL gene translocations [such as t(4;11)] – Hypodiploidy < 44 chromosomes by karyotype
or FISH analysis.
• MRD status (leukemia patients only): B-ALL patients with MRD >0.001 at Day 32.

Ph+ ALL
• Patients with t(9;22) will be removed from study prior to Day 15.
• These patients will be eligible to enroll on the open COG Protocol for Philadelphia chromosome-positive ALL.– Diagnostic sample from DFCI MRD lab to be sent to
COG lab to allow MRD determination on COG study– Site responsible for sending flow histograms from
diagnosis to MRD Flow Cytometry Lab

Lymphoblastic Lymphoma: Risk Group Classification
• Patients will be classified as Standard Risk, High Risk or Very High Risk based on
– Age, immunophenotype, CNS status and cytogenetics (same as for ALL patients)
– MRD will not be used for risk-group classification.

Steinherz/Bleyer Algorithm to Interpret Traumatic LP’s
• Traumatic LP: >10 RBC per high power field with > 5 WBC/hpf:
• Traumatic CSF specimens should be treaetd as CNS-3 if:
CSF WBC/CSF RBC > 2x (Blood WBC/Blood RBC)
• All other traumatic CSF specimens with blasts should be treated as CNS2.

Cytogenetics/FISH/PCR
• Required Studies:– Karyotype– FISH (PCR insufficient screening test):
• TEL/AML1 (also screens for iAMP21)• MLL (screens for all fusion partners)
– FISH or PCR:• BCR-ABL

Cytogenetics/FISH/PCR
• Suggested Studies (esp if normal or uninterpretable karyotype)– FISH for trisomies 4,10,17– FISH for 9p deletions– FISH or PCR for t(1;19)

Cytogenetics/FISH/PCR
• Reports must be sent to DFCI for central review prior to Day 15
• Please ensure that all tests that were performed are clearly listed on report(s)

Final Risk Group Assignment
• Final Risk Group Assignment will be made no later than 21 days after start of Consolidation I phase, and only after receipt of following source documents:
– Path reports confirming diagnosis (and scan reports of involved sites for LL)
– Path and scan reports confirming complete remission– Diagnostic flow cytometry reports (ALL only)– Diagnostic cytogenetics, FISH, PCR (ALL only)– CBC with differential from diagnosis and from end of induction– CSF cell count/cytospin (and/or cytopath) from diagnosis and end of
induction
• DFCI Study Team will call/email verification of MRD results (if applicable) and final risk group assignment to participating site

Protocol 11-001: Treatment (Section 6)
– Steroid Prophase– Induction– Consolidation I– CNS Phase– Consolidation II – Continuation

11-001: Treatment
• Section 7.2.1: Allowable variations in dose/timing– Dose: variations within 10% of calculated dose (rounding)– Timing (treatment/procedures):
• Induction, Consol I, CNS, Consol II: +/- 3 days• Continuation: +/- 7 days
– Schedule (drugs given multiple times/day)• +/- 3 hours from specified intervals
• Any variation in dosing outside of these parameters are considered protocol violations– Anything can be delayed/modified for significant illness
(must be discussed in advance)

11-001 Induction: Differences from 05-01
• Induction Phase– Essentially the same as 05-01 (including prophase)– Randomized: SC-PEG vs IV-PEG (Day 7)
• PK samples required pre-dose, weekly after dose x 4
– Antibiotic Prophylaxis (begin during prophase)– Steroid taper at end of induction to begin at Day 32
• Adrenal suppression after 1 month of steroids• ?reduce fever/illness during Consol I phase

11-001: Prophase
• Methylpred Day 1-3• IT cytarabine Day 1
– Twice-weekly until 3 consecutive clear if • CNS-2• CNS-3• Traumatic LP with blasts• Cranial nerve palsy (continue until Day 18)
– CSF sample to Peter Cole (optional)• Begin antibiotic prophylaxis (if afebrile)
– Continue until ANC > 200 or APC > 500, or fever develops

11-001 Induction
• Days 4-32: Methylpred 3x/day or pred 2-3x/day– Day 32: Begin taper (should be off steroids by Day 39)– Taper should be administered even if Consol I delayed
• Days 4,11,18,25: Vincristine• Days 4,5: Doxorubicin +/- Dexrazoxane (HR)• Day 6: methotrexate (low dose)• Day 18: IT MAH

11-001: Induction
• Day 7: Asparaginase by Randomization• SC-PEG vs Oncaspar
– Each dosed at 2500 IU/m2– Each given over 1 hour
• Labs (Required): ASP PK/Antibody– Pre-dose– End of infusion– Days 11, 18, 25 and 32

11-001: End of Induction
• ALL patients: – Marrow must be done at Day 32 (send sample for MRD)– CXR/Chest CT if AMM at diagnosis– LP with IT MTX if counts recovered
• CSF sample to Peter Cole (optional)
• Lymphoma patients:– Repeat scans of involved sites– Marrow only if involved at diagnosis– LP with IT MTX if counts recovered
• CSF sample to Peter Cole (optional)

Definition of Complete Remission
• ALL– Count recovery: APC > 1000, platelets > 100K– Interpretable marrow with < 1% blasts– No peripheral blood lymphoblasts– No blasts in CSF– > 70% reduction in size of any mass present at diagnosis (scan)
• Lymphoblastic Lymphoma– > 70% reduction in size of largest nodes/masses at diagnosis– No evidence disease on exam– Interpretable marrow with < 1% blasts (if involved at diagnosis)– No blasts in CSF

Induction Failure
• > 1% blasts in marrow (confirmed by flow, FISH, other studies)
• CNS-3• < 70% reduction in size of any mass noted at
diagnosis, or any new, biopsy-proven mass
• All patients with induction failure are removed from protocol treatment

Delayed Recovery
• If no peripheral blood criteria at Day 32– Marrow at Day 32, send for MRD (ALL only)– No LP– Give vincristine 1.5 mg/m2/dose (max 2 mg)
weekly until count recovery– Repeat marrow must be done when counts
recover– CR must be documented by Day 53 for patients
to remain on study

11-001 Consolidation I: Differences from 05-01
• Consolidation I: No change – End-induction steroid taper during 1st week if
Consol I begins at Day 32
• VHR Consol IB and IC: No change– ASP randomization begins during Consol IC

11-001: Consolidation I
• Starting Criteria– Documented CR– APC > 1000, platelets > 100K– SGOT < 8x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)– Normal creatinine for age– No mucositis– No ascites/effusions/significant edema
• May be delayed for significant illness

11-001: Consolidation I
• VCR (Day 1)• 6MP (Days 1-14)• Dox/Dexrazoxane (Day 1): HR only• HD MTX over 24 hours
– SR: Day 1– HR: 8-24 hours after doxorubicin
• IT MTX (if > 72 hours since Day 32 IT MTX)• Steroid Taper
– Should be completed by Day 39

Consolidation I
• Final Risk group classification should be made no later than 21-days after start of Consolidation I

11-001: Consolidation IB (VHR only)
• To begin approx 21 days after start of Consol IA
• Starting Criteria– APC > 750, platelets > 75K– SGOT < 8x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)– Normal creatinine for age– No or nearly fully resolved mucositis

11-001: Consolidation IB (VHR only)
• Cyclophosphamide (Day 1)• 6MP (Days 1-14)• Low dose cytarabine:Days 2-5, 9-12
– Once 4-day course commences, give all 4-doses regardless of counts
– Delay Day 9 if APC < 500 or platelets < 50K– If Day 9 cannot be given by Day 23, omit the second 4-
day course

11-001: Consolidation IC (VHR)
• To begin approx 21 days after start of Consol IB
• Starting Criteria– APC > 750, platelets > 75K– SGOT < 8x normal– No or nearly fully resolved mucositis– Suggestion: check amylase prior to ASP

11-001: Consolidation IC (VHR)
• HD araC 2 gm/m2 q12 hours x 4 doses (Days 1-2)
• Etoposide daily Days 3-5• Dexamethasone 18 mg/m2/day oral or IV
divided twice daily: Days 1-5• ASP by randomization Day 8

11-001: Consolidation IC (VHR)
• ASP by randomization (Day 8)– SC PEG 2500 IU/m2 IV over 1 hour
• Continue every 3 weeks for 10 doses (30 weeks)
– Oncaspar 2500 IU/m2 IV over 1 hour• Continue every 2 weeks for 15 doses (30 weeks)
• Required samples: ASP PK/Antibody– Prior to every dose, including 1st dose

11-001 CNS phase: Differences from 05-01
• CNS Phase– Marrow on Day 1 for VHR patients (MRD)– CSF samples to Peter Cole (4th LP)
• Optional
– New criteria for cranial radiation• Criteria for XRT
• VHR• T-ALL• CNS-3
• HR B-ALL with WBC > 100K no longer a criterion

11-001: CNS Phase
• 21 days from start of Consolidation I• Starting Criteria:
– APC > 1000, platelets > 100K– Direct bilirubin < 1.4 mg/dL (23. 9 micromoles/L)
– SGOT < 8x normal– No mucositis– Amylase prior to ASP (Suggested)

11-001: CNS Phase-SR
• VCR: Day 1• 6MP: Days 1-14• Dexamethasone 6 mg/m2/day (Days 1-5)• Twice weekly Triple IT x 4 doses
– Dose by age
• ASP by randomization (Day 1)

11-001: CNS Phase-HR
• VCR: Day 1• Doxorubicin/Dexrazoxane: Day 1• 6MP: Days 1-14• Dexamethasone 18 mg/m2/day (Days 1-5)• Twice weekly Triple IT x 4 doses
– Dose by age
• ASP by randomization (Day 1)

11-001: CNS Phase (SR, HR)
• ASP by randomization (Day 1)– SC PEG 2500 IU/m2 IV over 1 hour
• Continue every 3 weeks for 10 doses (30 weeks)
– Oncaspar 2500 IU/m2 IV over 1 hour• Continue every 2 weeks for 15 doses (30 weeks)
• Required samples: ASP PK/Antibody– Prior to every dose, including 1st dose

CNS Phase: Radiation
• No XRT– SR ALL/LL– HR B-precursor ALL/LL: CNS1, CNS2– T-cell LL: CNS1, CNS2
• 12 Gy XRT– T-cell ALL: CNS1, CNS2– VHR ALL/LL: CNS1, CNS2
• 18 Gy XRT– CNS3 at diagnosis– Cranial nerve palsy at diagnosis– CNS2 at Day 18

11-001 Consolidation II/ Continuation: Differences from 05-01
• Consolidation II/Continuation– New criteria to start cycles
• APC > 750/mm3 (instead of 1000)• Platelets > 75,000/mm3 (instead of 100K)
– IV/IM Methotrexate to be given on same day as ASP• PO MTX allowed up to 3x during protocol (for vacation, etc)
– HR/VHR: Only 10 cycles of high-dose dexamethasone

11-001: Consolidation II
• Starting Time: 21 days for start of CNS phase
• Criteria to start each 3-week cycle– APC > 750, platelets > 75K– Mucositis: None or mild– SGOT < 8 x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)

11-001: Consolidation II
• Criteria to continue each 3-week cycle (Days 2-21)– APC > 500, platelets > 50K– Mucositis: None or mild– SGOT < 8 x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)

11-001: Consolidation II
• Criteria to administer asparaginase– No clinical pancreatitis– No new, untreated DVT– Direct bili < 1.4 mg/dL (23.9 micromoles/L)
• Must be checked prior to each dose
– Triglycerides < 2000 (<22.8 mmol/L)• Does not need to be checked, but if value known, must hold
if > 2000
– May administer regardless of blood counts, mucositis or SGOT

Consolidation II
• SR: Every 3-week cycle– VCR: Day 1– Dex: 6 mg/m2/day divided twice-daily: Days 1-5
• If taper used, total dose given each cycle should not exceed 30 mg/m2
– 6MP: Days 1-14• Daily dose may be adjusted to accommodate available tablets
to achieve 14-day total
– MTX: Days 1, 8, 15– ASP by randomization to complete 30 total weeks
(including CNS Phase)

Consolidation II
• HR: Every 3-week cycle– VCR: Day 1– Doxorubicin/Dexrazoxane: Day 1
• Until total cumulative dose 300 mg/m2 +/- 15 mg/m2• After total dose achieved, begin weekly MTX
– Dex: 18 mg/m2/day divided twice-daily: Days 1-5• If taper used, total dose given each cycle should not exceed 90 mg/m2• 10 cycles only, then give 6 mg/m2/day twice-daily
– 6MP: Days 1-14• Daily dose may be adjusted to accommodate available tablets to
achieve 14-day total– ASP by randomization to complete 30 total weeks (including CNS
Phase)

Consolidation II, SR and HR
• Triple IT chemotherapy– No XRT: Every 9 weeks x 6 doses, then every 18
weeks• 1st dose 9 weeks after CNS LP #1• Always administer at start of 3-week cycle• No IV MTX on same day as Triple IT chemo
– XRT: Every 18 weeks• 1st dose 18 weeks after CNS LP#1• Always administer at start of 3-week cycle• No IV MTX on same day as Triple IT chemo
– CSF to Peter Cole (LP#1 of Consol II phase)--optional

11-001: Consolidation II (SR, HR)
• ASP by randomization (Day 1)– SC PEG 2500 IU/m2 IV over 1 hour
• Continue every 3 weeks for 10 doses (30 weeks)
– Oncaspar 2500 IU/m2 IV over 1 hour• Continue every 2 weeks for 15 doses (30 weeks)
• Required samples: ASP PK/Antibody– Prior to every dose

11-001: Continuation
• Starting Time: Completion of all components of Consolidation II– SR: All doses of ASP– HR/VHR: All doses of ASP, cumulative dox
300 +/- 15 mg/m2, 10 cycles with high-dose dex (including CNS phase)

11-001: Continuation
• Criteria to start each 3-week cycle– APC > 750, platelets > 75K– Mucositis: None or mild– SGOT < 8 x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)
• Criteria to continue each 3-week cycle (Days 2-21)– APC > 500, platelets > 50K– Mucositis: None or mild– SGOT < 8 x normal– Direct bili < 1.4 mg/dL (23.9 micromoles/L)

Continuation (All risk groups)
• Every 3-week cycle– VCR: Day 1– Dex: 6 mg/m2/day divided twice-daily: Days 1-5
• If taper used, total dose given each cycle should not exceed 30 mg/m2
– 6MP: Days 1-14• Daily dose may be adjusted to accommodate available tablets
to achieve 14-day total
– MTX: Days 1, 8, 15

Continuation (All risk groups)
• Triple IT chemotherapy– No XRT: Every 9 weeks x 6 doses, then every 18
weeks• 1st dose 9 weeks after CNS LP #1• Always administer at start of 3-week cycle• No IV MTX on same day as Triple IT chemo
– XRT: Every 18 weeks• 1st dose 18 weeks after CNS LP#1• Always administer at start of 3-week cycle• No IV MTX on same day as Triple IT chemo

Duration of Therapy (Section 6.17)
• 104 weeks (+/- 2 weeks) after CR documented– Even if significant delays or dose modifications
during therapy– Therapy should not be stopped in midst of a 3-
week continuation cycle

Duration of Therapy
• Removed from Protocol Therapy if:– Ph+ ALL– Induction Failure– Relapse– SMN– Withdrawal by participant– Investigator feels that further treatment is unacceptable
(change in condition, compliance)• Duration of follow-up (Section 6.18): Indefinite
– Including those who stop treatment early, unless withdrawal of consent

Supportive Care (Section 6.16)
• Mandatory infection prophylaxis during prophase/induction
– Levofloxacin/Moxifloxacin– Cefipime if allergic
• Bactrim prophylaxis after achieving CR• GCSF, GMCSF allowed at discretion of treating physician• Use of corticosteroids for other conditions
– Allowed: Blood pre-med, allergic rxn, RAD– Must be discussed: Prolonged use (more than a few days at a time)– Prohibited: Steroids as antiemetics

Supportive Care (Section 6.16)
• Leucovorin after IT chemotherapy: Allowed at discretion of treating clinician for: – Down Syndrome– History of excessive toxicity with prior IT
MTX

Dose Modifications: Consolidation II/Continuation (Section 7.2.8)
• APC < 750 or platelets < 75K when cycle is due:– Hold cycle (including vcr, dexamethasone, IT chemo) – Proceed with ASP if ASP-criteria met– Consider dose reduction of MTX/6MP by 20% when
counts recover– HR/VHR: Reduce 6MP first, then doxorubicin
• May only reduce Doxorubicin once (to 80% dose)
– Consider TPMT testing for persistent or excessive myelosuppression

Dose Modifications: Consolidation II/Continuation (Section 7.2.8)
• APC < 500 or platelets < 50K during cycle:– Hold MTX and 6MP for remainder of cycle (do
not restart)– Consider dose reduction of both agents at start
of cycle by 20%– HR/VHR: Reduce 6MP first, then doxorubicin
• May only reduce Doxorubicin once (to 80% dose)– Consider TPMT testing for persistent or
excessive myelosuppression

Dose Modifications: Consolidation II/Continuation (Section 7.2.8)
• SGOT > 8 x normal, direct bili > 1.4 mg/dL or Mucositis:– Hold start of cycle (Hold ASP for direct bili, not SGOT
or mucositis)– If during cycle, hold MTX and 6MP until within
acceptable range; may restart during same cycle– Consider dose reduction of both agents at start of next
cycle or when resumed by 20%– HR/VHR: Reduce 6MP first, then doxorubicin
• May only reduce Doxorubicin once (to 80% dose)

Dose Modifications: Asparaginase (Section 7.2.8.5)
• Clinical Allergy– Switch to Erwinia 25000 IU/m2 IM twice-
weekly to complete 30 weeks of asparaginase.• If Erwinia allergy, stop all ASP
• Silent Allergy– If 2 consecutive NSAA are non-detectable
(<0.025 IU/mL), switch to twice-weekly Erwinia as above

Dose Modifications: Asparaginase (Section 7.2.8.5)
• Thrombosis (Appendix II)– Hold ASP– Begin anticoagulation– Resume ASP after coagulation status stabilized
and clinical symptoms resolved• May permanently discontinue after severe CNS
event (no restart)
– If recurrence after restart, permanently stop

Dose Modifications: Asparaginase (Section 7.2.8.5)
• Lipemic Blood– Triglyceride levels not required (discouraged)– Hold if found to be high (>2000 or 22.8
mmol/L)– Resume when levels are within acceptable
range– Lipid lowering agents may be used at discretion
of treating clinician

Dose Modifications: Asparaginase (Section 7.2.8.5)
• Pancreatitis– Asymptomatic enzyme elevation: Hold ASP if > 3 x normal,
resume when below this level• If receiving cranial radiation, check lipase
– Mild/Moderate: symptoms < 72 hours duration with amylase and/or lipase elevation
• Hold ASP• Resume when signs, symptoms, enzymes return to baseline• May permanently discontinue after recurrent episodes of
mild/moderate pancreatitis– Severe: symptoms > 72 hours with amylase/lipase elevation, or
pseudocyst, or life-threatening complications• Permanetly discontinue

Dose Modifications: Asparaginase (Section 7.2.8.5)
• Asparaginase Intolerance: Pt permanently stops ASP having received 10 or fewer weeks– SR: Add 3 cycles of HR Consol II
(Doxorubicin + dexrazoxane, high-dose dex)– HR: No changes; Continue rest of doxorubicin.
Need to give 10 cycles with high-dose dexamethasone

Dose Modifications: Dexamethasone (Section 7.2.8.7)
• Taper allowed for withdrawal pain: do not exceed total intended dose for the cycle
• Permanently discontinue for symptomatic osteonecrosis (symptoms + scan)
• Hold for fracture; resume when healed (scan, symptoms, clearance by orthopedist); do not make up missed doses
• May be held for pancreatitis • Severe, intolerable behavioral changes
– HR/VHR: may be reduced to lower dose during Consol II– SR: may be stopped or given less frequently (need to discuss first)– May switch to prednisone
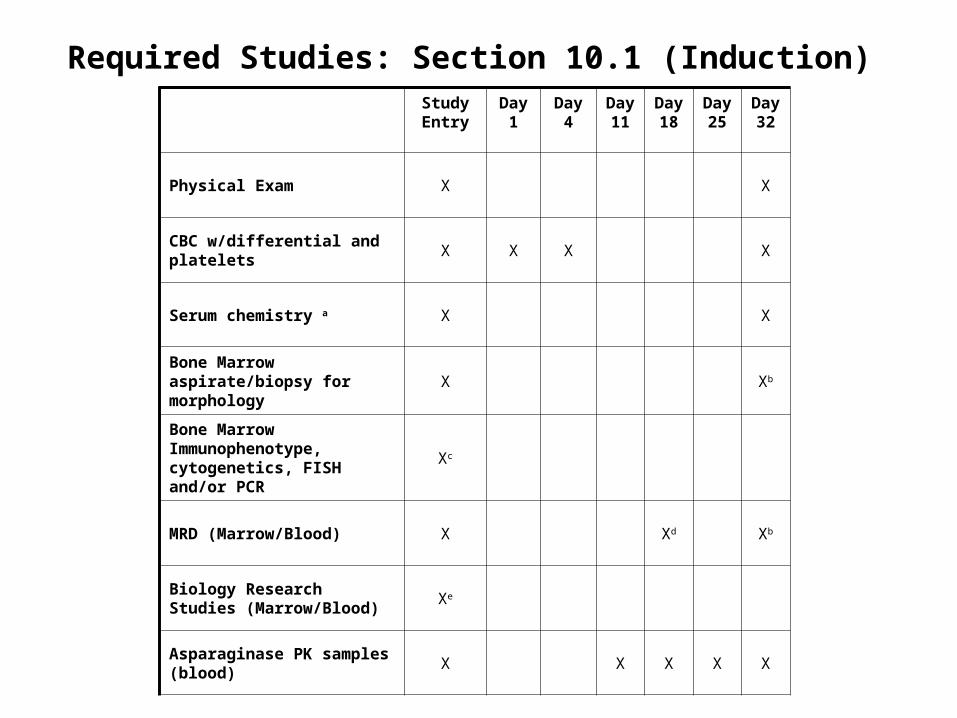
Study Entry
Day 1
Day4 Day 11
Day 18
Day 25
Day 32
Physical Exam X X
CBC w/differential and platelets X X X X
Serum chemistry a X X
Bone Marrow aspirate/biopsy for morphology
X Xb
Bone Marrow Immunophenotype, cytogenetics, FISH and/or PCR
Xc
MRD (Marrow/Blood) X Xd Xb
Biology Research Studies (Marrow/Blood)
Xe
Asparaginase PK samples (blood)
X X X X X
Required Studies: Section 10.1 (Induction)

Study Entry
Day 1
Day4
Day 11
Day 18
Day 25
Day 32
Vitamin D 25 OH level (blood) Xf Xf
CSF for cell count, cytospin Xk X Xk
Chest x-ray X Xg
CT scans (Neck, chest, abdomen, pelvis) Xh Xi
Echocardiogram (recommended, not required)
X
B-HCG (urine or serum) Xj
AE/Toxicity Assessment L X X X X X X X
Required Studies: Section 10.1 (Induction), cont’d

CNS Phase
Consolidation II
Continuation End of Therapy
Physical Exam X X
CBC w/differential and platelets X Xa Xa
Serum chemistryb X Xa Xa
Bone Marrow aspirate for morphology
Xc
MRD (Marrow/Blood) Xc
Asparaginase PK samples (blood) Xd Xd
Vitamin D level (blood) Xe Xe
Echocardiogram Xf
CSF Xg Xg
AE/Toxicity Assessmenth X X X X
Required Studies: Post-Induction

Outside Affiliate Sites (Section 15)
• Some standard-of-care lab studies and treatments may be performed at satellite sites affiliated with a Participating Site
• It is the responsibility of the Participating Institution to which the satellite is affiliated to monitor all care that is performed at the satellite in order to ensure protocol compliance and screen for AEs.
• Only those Labs and Treatments Listed in Protocol (Section 15) can be performed at Affiliate Site

Affiliate Sites: Allowed Labs and Treatment
• Laboratory Studies– Standard-of-care labs used to determine whether starting criteria
for chemotherapy are met and/or to monitor for side effects• CBC, LFTs, amylase, lipase, triglyceride levels
• Therapy– Standard-of-care chemotherapy agents (eg, vincristine,
doxorubicin/dexrazoxane) administered during Consolidation II or Continuation phases
– IM Erwinia asparaginase (for patients who develop allergy to PEG asparaginase)
– Oral agents (eg, prednisone, dexamethasone, 6-mercaptopurine) may be administered in the home or at a satellite site.
– IV/IM methotrexate (Consolidation II or Continuation phase only) – Low-dose IV cytarabine during the Consolidation IB phase

Procedures
• Diagnostic Procedures and Radiographic Studies– Diagnostic marrow, nodal and/or mass biopsies and
scans that have already been performed at an outside institution do not have to be repeated prior to study entry
– Pathologic/radiographic interpretation of these studies confirming protocol eligibility must be performed at Lead Institution or Participating Institution
• All subsequent marrows, restaging studies and LP’s with IT chemo may only be done at Participating Site (not at Affiliate)

Labs/Therapies that Cannot be Performed at Affiliate Site
The following can only be performed at Participating Site (not allowed at Affiliate Site)
• Lab Studies– All Research Labs– MTX levels (Consolidation I)
• Treatment– All doses of Oncaspar or SC-PEG– All IV chemo during Prophase, Induction,
Consolidation I and CNS phase (except low-dose araC during Consol IB)
– All IT chemotherapy (any phase)

DFCI ALL Consortium Protocol 11-001Site Initiation Visit Part II:
Regulatory Overview
Lewis Silverman, MDOctober 11, 2012

Process to Open 11-001 at Outside Sites
• DFCI IRB approved protocol is provided to outside sites and submitted to local IRB
• Consortium site IRB approval is submitted to DFCI IRB adding consortium site onto the study
• DFCI IRB approval of the addition of the consortium site is submitted to FDA with consortium site FDA Form 1572
• SIV (teleconference)• Sigma-Tau approves all required regulatory documents
and ships drug to consortium site• 11-001 may be open to enrollment

Timeline to Open at Outside Sites
US Sites• In process• Contract• LOI with Sigma-Tau• IRB Approvals at Consortium Sites
Canadian Sites• End of October 2012
– Sigma Tau to submit final Drug Master File (DMF) to Health Canada for SC-PEG
– Beginning of November: CTA to be submitted to Health Canada (immediately after DMF submission)
• 60 day review period for the CTA• 11-001 can open at Canadian sites upon CTA approval

Required Regulatory Documents
• Must be submitted to DFCI prior to activation of study at each site:– 1572 (DFCI will provide Template)– Site PI and Co-Investigators CV, FDFs, Licenses– Lab Documents
• CLIA/CAP/Lab Normal Ranges/Lab Directors CV and License
– Initial IRB Approval – IRB Membership List– Delegation of Authority Log– Protocol Signature Page– IRB Letter of Assurance (FWA)

Site Regulatory Binder
• Protocol• Investigator Brochure• Consent Form• FDA 1572• CV/Licenses/Financial
Disclosure• Laboratory Documents• Delegation of Authority
Log• Protocol Training• Monitoring Log/Reports• Screening/Enrollment Log
• Drug Accountability Records• Initial IRB Application• Initial IRB/SRC Review • Amendments• Protocol Deviations/Violations• Continuing Review• Study Close Out• SAEs• IND Safety Reports• DMC Reports• General Correspondence

Site Initiation Visit
• DFCI leads SIV via teleconference • Expected attendees (must be documented)
– Site PI– All site co-investigators (other MD’s who will consent patients)
• Only co-investigators can sign informed consent, so any MD who will consent participants must be listed as co-investigator
– Site study staff (eg, research nurse, CRA, pharmacists, etc)• Topics to be covered
– Objectives, rationale, study design, eligibility, registration, required data, treatment schedule, side effects, SAE reporting, dose modification
• SIV must be completed before site is activated

On-going Training
• On-going training (Documented in Training Log at each site)– Must be performed and documented for
• Individuals unable to join the SIV • Anyone joining the study team in the future
– Training Log documents • What study information was reviewed• Training was conducted by a person with
appropriate level of knowledge

Monthly Teleconferences• Study Updates
• Expected Attendees:– Site PI, study staff
• Agenda: Discuss study-related issues, including– Study Accrual– SAE’s/AE’s– Deviations/Violations– Protocol Amendments– Clinical issues
• Review Minor Deviation/Violation Logs
• Teleconference minutes will be posted on the ALL Consortium Website

Ordering Study Drug
• Initial order– DFCI receives initial required regulatory documents– Regulatory documents are provided to Sigma-Tau– Initial shipment of drug will be sent directly to site
from Sigma-Tau
• Future orders– Re-order directly through Sigma-Tau

Protocol Review and Amendments (Section 14.1)
• DFCI will initiate all protocol changes and distribute updated IRB approved protocols to outside sites
• Current version of the Protocol will be posted on the ALL Consortium Website
• Amendments must be submitted to local IRB within 30 days of receipt (DFCI Policy)

Consent Form Changes (Section 14.2)
• DFCI will initiate consent form changes and distribute finalized updated Model Consent Form to all sites
• Reference DFCI SOP regarding what language can/can not be changed
• All revised consent forms must be approved by DFCI prior to IRB submission
• Current DFCI Model Consent Form will be posted on DFCI ALL Consortium Website

Adverse Event Reporting Requirements (Section 12)
• All AE’s and SAE’s must be documented in medical record and in case-report form
• Grading using CTCAE version 4.0

Adverse Event
• An adverse event (AE) is any undesirable sign, symptom or medical condition or experience that develops or worsens in severity after starting the first dose of study treatment or any procedure specified in the protocol, even if the event is not considered to be related to the study.

Adverse Event: Exceptions (Section 12.1.1)
The following expected toxicities related to the diagnosis and treatment of ALL but thought to be unrelated to asparaginase do not need to be recorded as AEs, unless they meet definition of an SAE:
– Blood count abnormalities (any grade)– Electrolyte abnormalities (any grade)– Uric Acid (any grade)– Transaminitis without clinical symptoms– Mucositis (grade 2 or below)– Nausea (any grade)– Vomiting (any grade)– Fatigue (any grade)– Anorexia (any grade)– Fever (any grade)– Constipation (any grade)– Diarrhea (any grade)– Hypertension thought to be related to steroids (grade 2 or below)– Hyperglycemia related to steroids that does not require the use of insulin– Neuropathy – sensory, motor, or cranial (grade 3 or below and thought to be related
to Vincristine)

Serious Adverse Event (Section 12.1.2)
Any adverse event, regardless of causality that: • Results in death• Is life-threatening. • Requires intensive inpatient medical interventions,
including mechanical ventilation, pressor support, and/or fluid resuscitation, or emergent surgical operations.
– Unplanned hospital admissions for febrile neutropenia, bacteremia or other documented infections, seizure, pancreatitis, thrombosis, nausea/vomiting, diarrhea, and other expected toxicities from the study treatment will not be considered a SAE unless the severity of the condition is considered life-threatening and/or lead to intensive medical interventions or surgical procedures.

Serious Adverse Event
• Results in persistent or significant disability/incapacity. Disability is defined as a substantial disruption of a person’s ability to conduct normal life functions.
• Is a congenital anomaly or birth defect; or• Is an important medical event that may jeopardize the
participant and require medical or surgical intervention to prevent one of the outcomes listed above.
– An example of such a medical events is allergic bronchospasm requiring intensive treatment in an emergency room or in the hospital.

SAE: Exceptions
• Events not considered to be serious adverse events are hospitalizations for:
– Routine treatment or monitoring of the studied indication, not associated with any deterioration in condition, or for elective procedures
– Elective or pre-planned treatment for a pre-existing condition that did not worsen (including pre-planned hospital admissions and/or surgeries), provided condition did not deteriorate in an unexpected manner during trial
– Toxicities that are expected from the study treatment and are not considered life-threatening (ie, do not require intensive medical or surgical management, as defined above).
– Emergency outpatient treatment for an event not fulfilling the serious criteria outlined above and not resulting in inpatient admission
– Respite care

SAE Reporting
• Must report all SAE’s that occur after initial dose of study treatment, during treatment or within 30 days of last dose of treatment
• Report to Overall PI (Dr. Silverman) must be made within 24 business hours of learning of the event
• Events required to be reported are outlined in protocol section 12.1.2 and also include the following:
– All Grade 2 and 3 events that are unexpected and at least possibly related to study treatment
– All Grade 4 events that are unexpected or not specifically exempted in protocol
– All Grade 5 (fatal) events

SAE Reporting
• SAE report must be completed on the MedWatch 3500 or MedWatch 3500A form. • Report must be emailed or faxed to the Overall PI and DFCI Research Nurse WITHIN
24 BUSINESS HOURS OF INITIAL STUDY TEAM NOTIFICATION (this includes Investigator notification).
– Lewis Silverman, MD: 617-632-6191, [email protected]– Cailin Toomey: 617-632-3960, [email protected] – Email: [email protected]– FAX: 617-632-3977
• SAE reports MUST include the following information within the narrative (which can be attached if it does not fit on the Medwatch form):
– CTCAE V4 Event Name (for every serious event reported)– Grade of Event– Relationship (not related, unlikely, possibly, probably, definitely)– Expectedness (is the event listed in the consent or IB as risk?)– Date of study team notification of event– Participant study ID and Initials– Date of study Enrollment– Current phase of treatment
• DFCI is responsible for submitting all SAEs to FDA, Sigma Tau, and Health Canada• If Sigma-Tau requires additional information, they will follow up directly with sites

SAE Reporting to DFCI IRB
• The following SAE’s must be submitted to DFCI IRB within 10 business days: – Grade 2 and 3 events that are unexpected and possibly, probably, or definitely
related/associated with the intervention– All grade 4 events – unless expected AND specifically listed in the protocol as not
requiring reporting – All grade 5 events, regardless of relationship ** Note: Grade 2 and 3 laboratory abnormalities that are considered by the investigator to
be clinically insignificant and do not require therapy, or adjustment in prior therapy, do not need to be reported to DFCI IRB as an SAE.
• Each site is responsible for completing the DFCI IRB SAE Reporting Form and submitting to DFCI Study Team for IRB submission
• Sites must use current form available on the DFCI ALL Consortium Website
• SAE’s submitted to DFCI IRB will be posted on website
• Sites responsible for reporting any SAE’s to their own IRB as per their institutional policies

Investigator’s Brochure: Calaspargase pegol
(EZN-2285, SC-PEG E. coli L-asparaginase)
• Current version:– Edition number: 6.0– Release date: October 17, 2011
• Updated version will be released annually by Sigma-Tau• If new risks are identified, consent form changes will be
initiated by DFCI• Site PIs must review and sign/date updated versions of IB• Sites should submit updated Ibs to their IRBs as required
by their local IRB

IND Safety Reports
• All study staff will be added to Sigma Tau’s email distribution list to receive SC-PEG IND Safety Reports
• All IND Safety Reports will be posted on the DFCI ALL Consortium Website
• Each site will submit IND Safety Reports to their IRB per their local IRB policy
• DFCI will distribute information about all SAEs that occur on 11-001 to all outside sites
– Posted on DFCI ALL Consortium Website
• If new risks are identified, consent form changes will be initiated by DFCI

Safety Monitoring by DMC
• Independent committee• Meets twice-yearly• Reviews safety and efficacy data from
participating sites to assess treatment benefit or harm to study participants– Review study conduct to date (summary report)– Review all SAE reports
• Determines if study may proceed as planned (no safety concerns)
• Report posted on ALL Website

Data Submission Requirements
• Missing forms reports will be posted for each site on the DFCI ALL Consortium Website
• Timely data submission is required

Data Submission: New DMC Requirements
• Overall Missing Forms for the trial must be <20%• Overall Missing Toxicity Forms for the trial must be <20%• Each individual site must have < 35% of forms missing• Each individual site must have < 35% of toxicity forms
missing.• More stringent protocol specific reporting rules may be
added as necessary to a protocol.• If any of the above are not met, they MUST be met at the
next running of the report or they will be reported to the DMC. The DMC may suspend registration to either the protocol/site with the issue OR to all ALL Consortium Trials in which the site participates.

Violations and Deviations (Section 20.7)
• Protocol Deviation: Any departure from the defined procedures set forth in the IRB-approved protocol which is prospectively approved prior to its implementation.
• Protocol Violation: Any protocol deviation that was not prospectively approved by the IRB prior to its occurrence.

Minor Deviations/Violations
• Minor Deviations/Violations• Each site will maintain Minor Deviation/Violation Log • Minor deviation log submitted to DFCI Study Team Monthly
• Must include detail of deviation and corrective action plan• Submitted to DFCI IRB at time of Continuing Review
• Examples: • Timing of treatment exceeds allowable deviation (eg, medication
was > 3 hours late)• Cycle started although not all specified criteria were met
• Note: if you get permission in advance to start cycle, this is not a violation (allowed in protocol)

Minor Deviations/Violations
Minor Deviation/Violations submitted to DFCI IRB as a Major Deviation/violation if: 3 for more minor deviations for the same
subject (or of the same type) Impact the safety of participants Compromise the integrity of the study data
and/or affect subject’s willingness to participate in the study

Major Deviations/Violations
• Must be reported to DFCI and local IRB in a timely manner
– Site reports to DFCI study team and to their local IRB– DFCI study team submits to DFCI IRB
• Outside sites to complete DFCI Major Deviation/Violation Form and submit to DFCI study team
– Must include corrective action plan!
• Examples:– Enrollment of participant who did not meet all inclusion/exclusion
criteria– Study visit or procedure conducted outside of required timeframe
that, in the opinion of the PI, may affect participant safety or data integrity

Study Monitoring (Section 20.8)
• Virtual Monitoring of each site
• On-Site Monitoring

Virtual Monitoring
• Participating institutions will be required to submit source documents to the Lead Institution for virtual monitoring:– All eligibility source documentation – Registration Documents – Consent form, eligibility
checklist – Pathology reports confirming diagnosis– All screening laboratory reports and scans– All results of the “Required Assessments”– Any extra source documentation (i.e., admission notes,
scans, culture results) pertaining to a serious adverse event (SAE).All results of the “Required Assessments” at study entry and at the date of remission assessments

On-Site Monitoring
• Annually, after 3 participants have been enrolled• Review of all
– Regulatory documentation– Source documentation (Diagnosis, Informed consent, CR,
AE/SAE, required labs)– Drug accountability
• Note: All eligibility requirements must be found either in source documentation (eg, lab reports) or must be included in MD note (eg, “no known history of HIV”).
– Eligibility checklist is not source documentation

On-Site Monitoring
• More frequent on-site monitoring visits will be made if sub-standard performance is discovered. For example:– Data not entered on time– Unreported adverse events– Enrolling ineligible participants

Audits (Section 20.9.1)
• Auditing: Systematic and independent examination of all trial-related activities and documents
– To determine if evaluated activities were appropriately conducted
• DFCI QACT will conduct on-site auditing
• Scheduled at each site after the first 10 subjects are accrued and then annually if 10 subjects accrue since the time of the last audit.
– If a site enrolls less than 10 participants, they will undergo one audit over the course of the study.
• Approximately 3-4 subjects would be audited at the site over a 2 day
period

Audits
• QACT Audit Team submits reports to DF/HCC Audit Committee with audit rating and recommendations
• PI/Site will receive final copy of report– May require implementation of corrective
action plans, further follow-up

Sub-Standard Performance (Section 20.9.4.1)
“Participating Institutions that fail to meet the performance goals of accrual, submission of timely accurate data, adherence to protocol requirements, and compliance with state, federal, and Good Clinical Practice guidelines, will be recommended for a six- month probation period. Such institutions must respond with a corrective action plan and must demonstrate during the probation period that deficiencies have been corrected, as evidenced by the improved performance measures. Participating Institutions that fail to demonstrate significant improvement will be considered by the Protocol Chair for revocation of participation.”

Electronic Data Capture System
• Inform Version 5.5• Training:
– Must complete training for Inform version 5.5 and provide training completion certificate to DFCI study team
– Contact Annette Dalton for access to training: [email protected]
• Queries are expected to be resolved in real time

STUDY SAMPLES

141
Asparaginase PK Samples- Mandatory
• Obtain 3ml of peripheral blood in one red top tube– Samples should be processed as soon as possible
• Schedule– Induction Phase:
• Day 7 (1 sample collected just prior to first dose of Asparaginase; 2nd sample collected 5-10 min after completion of Asparaginase infusion)
• Day 11, Day 18, Day 25 and Day 32
– Post Induction: Just PRIOR to each dose of asparaginase administered during post induction treatment phases
– If patient is switched to twice-weekly Erwinia, samples should be obtained every 3-weeks (or at the start of the chemotherapy cycle if cycle is delayed) just prior to the Erwinia dose

142
Asparaginase PK Samples- MandatoryProcessing (Protocol Section 9.1): • Allow the tube to stand at room temperature for 30 min to clot (do not
place the tube in ice).• Centrifuge the clotted blood sample at 1,100-1,300 x g for 10 minutes
at 25º C. Record the time that the sample was centrifuged.• Using a disposable pipette, carefully remove the serum (uppermost
clear layer) without disturbing the pelleted blood cells and buffy coat, and transfer it into a 2 mL self-standing polypropylene cryogenic tube with external threads.
• Immediately place the labeled cryotube in a freezer maintained at -70ºC or lower until packaged for shipment. Record the time that the sample was placed in the freezer.
• Ship on dry ice to Dr. Jeff Supko at MGH Monday through Wednesday (can be batched weekly)

143
MRD Samples-Leukemia patients Only
• Marrow- One purple top (EDTA) tube, with 2-3mL of bone marrow aspirate• Blood-One purple top (EDTA) tube , with 2-3mL of peripheral blood• Schedule:
– At time of study entry (pre-treatment): Required of all leukemia patients
– Induction Phase, Day 18 (if consent obtained for Day 18 bone marrow study)
– Induction Phase, Day 32: Required of all leukemia patients– VHR patients only:
• Consolidation IB, Day 1: Peripheral blood• Consolidation IC, Day 1: Peripheral blood• CNS Phase, Day 1: Bone Marrow and Peripheral blood
– At time of suspected relapse• Shipping: Send at room temperature, same day overnight to DFCI

144
Biology Samples- optional
• Marrow: 1-2 purple top (EDTA) tubes each with 2-3 mL of bone marrow aspirate in purple top (EDTA) tube
• Peripheral Blood: 1-2 purple top (EDTA) tubes, each with 2-3 mL of blood
• Schedule:• At time of study entry (pre-treatment)• At time of suspected relapse
• Shipping: Send at room temperature, same day overnight to DFCI

145
CSF Samples-Optional• Obtain 1-3 mL of leftover CSF (collected at the time of a therapeutic
lumbar puncture, just prior to the administration of intrathecal chemotherapy)
• Place on ice immediately after sample acquisition• Within 1 hour of collection, centrifuge specimen for 5 minutes then
aliquot into cryovials and immediately freeze at -80 degrees• Schedule:
– Day 1 Lumbar Puncture (will be changed to Day 18 in future amendment)
– Day 32 (end-induction) Lumbar puncture– 4th Lumbar puncture during the CNS phase– 1st Lumbar puncture performed during the Consolidation II phase
• Shipping: – Supernatant should be shipped on dry ice– Ship to Dr. Peter Cole at Albert Einstein College of Medicine

146
Vitamin D Samples-Optional
• Schedule:– Prior to starting treatment– End of induction– At the start of the 1st or 2nd cycle of
Continuation– End of treatment– Recheck according to guidelines in protocol for
those with low Vitamin D levels– To be sent to local institutional lab

QUESTIONS?





























