Embed Size (px)
Citation preview

Gedruckt mit Unterstiitzung aus dem Jerome und Margaret Stonborough-Fonds
Die Einwirkung von Organomagnesiumverbindungen auf Phthalonitril
(I. Mitteilung) Yon
Richard WeiB und Ernst Freund Aus dem I. chemischen Laboratorium der Universit/it Wien
(Vorgelegt in der Skzung am 6./r 1924)
Von symm.etrisch orthosubstituierten Benzolderivaten sind bisher nur Abk6mmmlinge der Phthals/iure, wie z. B. das Anhydrid, der Di/ithylester und der Phthalatdehyd der Einwirkung von Organo- magnesiumverbindungen unterworfen worden. Wiihrend die ersten zwei Verbindungen als Reaktionsprodukte stets Dialkylphthalide vom Typus I liefern, gibt Phthalaldehyd bei der gleichen Reaktion symmetrisch gebaute Orthophenylenderivate der allgemeinen Formel II.
R R \ /
/ \ / c o \ /\/cHOH--R
4 o i \ / \ c o /
NH ir
/k/C-cH c0H
II NH
I II III
Es war nun von Interesse, zu untersuchen, ob das Phthalo- nitril, der Einwirkung Gringardscher Verbindungen unterworfen, symmetrische oder asymmetrische KtSrper liefern wtirde.
Zu diesem Zwecke wurde die Einwirkung des Benzyl- magnesiumchlorids auf Phthalonitril untersucht. Wtirde diese Reaktion normal verlaufen, so mtiflte das bisher unbekannte o-Phenylendiphenacetyldiketimin der Formel III entstehen.
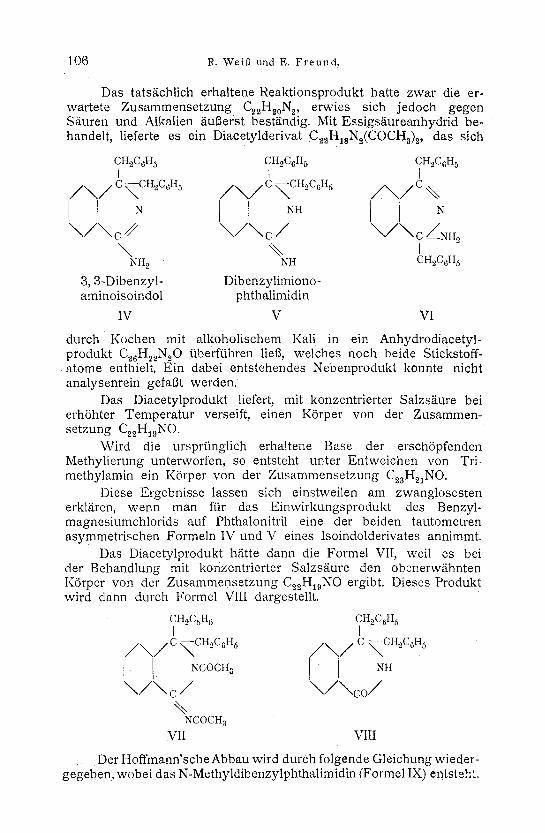
Es war abet auch vorauszusehen, daft, analog dem Dibenzyl- phthalid, alas bisher unbekannte Isoindolderivat der Formel IV oder V htitte entstehen k~Snnen.
Ferner w/ire noch ein K6rper der Formel VI denkbar, bei dem die Alkylgruppen symmetrisch angeordnet sind.
Vom Ketimin (Formel III) war zu erwarten, daft es bei der Einwirkung yon S/iuren leicht seinen Stickstoff, unter Bildung eines Diketons, vollst/indig abgeben wfirde. Das Isoindolderivat dagegen sollte bei der gleichen Behandlung oder bei der Einwirkung yon Alkalien Moil ein Stickstoffatom abspalten und das entsprechende Phthalimidinderivat bilden.
106 R. Weil3 und E: Freund,
Das tats/ichlich erhaltene Reaktionsprodukt hatte zwar die er- wartete Zusammensetzung C~H~oN2, erwies sich jedoch gegen Stiuren und AIkalien 5.ufierst bestg.ndig. Mit Essigs/iureanhydrid be- handelt, lieferte es ein Diacetylderivat C2~H,sN~(COCHs)~., das sich
CH2C6H 5 t
\ / \ c / / \
NH~
3, 3-Dibenzyl- aminoisoindol
IV
durch Kochen mit
CH2C6H5 CH2C6H 5
/ \ / c ~CH~C0H~ / \ / c %
NH I N
\ / \ c / ~ / \ c/NH,, NH CH2C6H5
Dibenzylimiono- phthalimidin
V VI
alkoholischem Kali in ein Anhydrodiacetyl- pr0dukt C26H~,2N~O tiberffihren lieB, welches noch beide Stickstoff- atome enthielt. Ein dabei entstehendes Nebenprodukt konnte nicht analysenrein getai3t werden.
Das Diacetylprodukt liefert, mit konzentrierter Salzs~iure bei erhShter Temperatur verseift, einen KSrper yon der Zusammen- setzung Ce~HjgNO.
Wird die ursprfinglich erhaltene Base der erschbpfenden Methylierung unterworfen, so entsteht unter Entweichen von Tri- methylamin ein KSrper yon der Zusammensetzung C23H21NO.
Diese Ergebnisse lassen sich einstweilen am zwanglosesten erkl/iren, wenn man ffir das Einwirkungsprodukt des Benzyl- magnesiumchlorids auf Phthalonitril eine der beiden tautometren asymmetrischen Formeln IV und V eines Isoindolderivates annimmt.
Das Diacetylprodukt h/itte dann die Formel VII, weil es bei der Behandlung mit konzentrierter Salzs/iure den obenerw/ihnten KSrper v o n d e r Zusammensetzung C2uH19NO ergibt. Dieses Produkt wird dann durch Formel VIII dargestellt.
CH~CsH 5 CH~C6H 5 I I
/ ~ / C ~CH~C~H~ / ~ , / / C ~CH2C6H 5
i I NCOCHa NH
\ / \ c / \ / \ c o / %
NCOCH a VII VIII
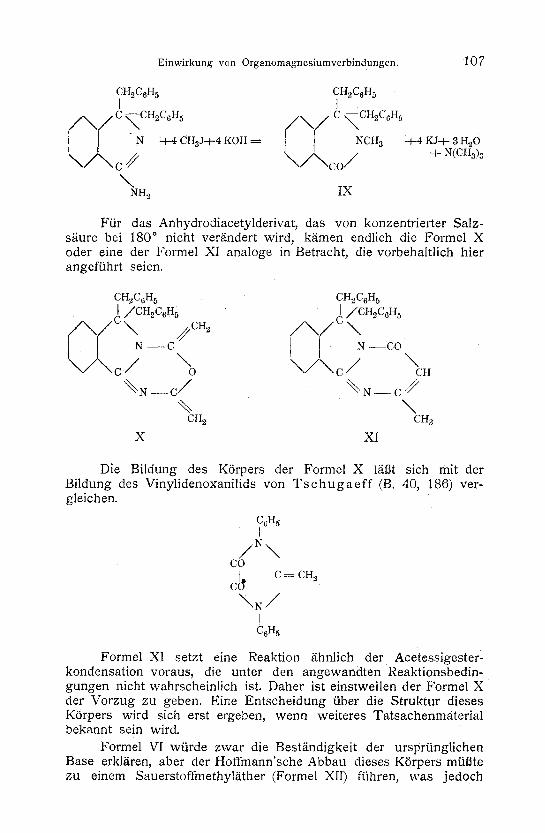
Der Hoffmann'sche Abbau wird durch folgende Gleichung wieder- gegeben, wobei das N-Methyldibenzylphthalimidin (Formel IX) entsteht.
Einwirkung von Organomagnesiumverbindungen. 107
CH2C6H 5 CH2C6H5 f
/ \ / c ~cH~c& /x, /c <cH~c0H~
' \ / \ c// \ / \ c o / + N<cH~)~ \
NH 2 I X
Ftir das Anhydrodiacetylderivat, das von konzentrierter Salz- s/iure bei 180 ~ nicht ver/indert wird, k/imen endlich die Formel X oder eine der Formel XI analoge in Betracht, die vorbehaltlich hier angeffihrt seien.
CH.~C6H 5 CH2C6H 5 I /CHoC6H 5 ] /CH~C6H 5
( \ ) ~ C . ~ " //~CH2 / x . \ / C \
c \ / \ c / "c. %N--c/ %N--c//
% \ CH 2 CH3
X XI
Die Bildung des KSrpers der Formel X 1/il3t sich mit der Bildung des Vinylidenoxanilids yon T s c h u g a e f f (B. 40, 186) ver- gleichen.
c6H5 1 /N \
co C lRv C = CH 2
\N/ I C6H5
Formel XI setzt eine Reaktion /ihnlich der Acetessigester- kondensation voraus, die unter den angewandten Reaktionsbedin- gungen nicht wahrscheinlich ist. Daher ist einstweilen der Formel X der Vorzug zu geben. Eine Entscheidung fiber die Struktur dieses K6rpers wird sich erst ergeben, wenn weiteres Tatsachenmaterial bekannt sein wird.
Formel VI wtirde zwar die Best/indigkeit der ursprtinglichen Base erkl~iren, abet der Hoffmann'sche Abbau dieses KSrpers mtiflte zu einem Sauerstoffmethyl/ither (Formel XII) ffihren, was jedoch

108 R. Weifi und E. F r e u n d ,
durch eine Methoxylbestimmung, die ein negatives Resultat gab, widerlegt worden ist.
Das -3, 3-Dibenzyl-l-aminoisoindol (Formel IV und V) biidet ein Chlorhydrat yon der Zusammensetzung Ce2H20N2HC1 , hat also
CH2C6H 5 I
/N/c% N
I CH=C6H5
XII
ausgesprochen basische Eigenschaffen. Das durch Verseifung des Diacetylproduktes entstehende Dibenzylphthalimidin (Formel VIII) bildet unter normalen Bedingungen kein Chlorhydrat, was durch die Annahme der laktamartigen Struktur dieser Verbindung gut erkl/J.rt wird.
E x p e r i m e n t e l l e s .
Darstellung des Phthalonitrils.
Zum erstenmal wurde das Phthalonitril von Joh. P i n n o w und C. S~[mann (B. 29, 630) dutch Behandlung yon o-Diazobenzo- nitril mit KupfercyanfircyankaliumlSsung in zirka 4 bis 5o/o Aus- beute erhalten.
Theodor P o s n e r (B. 30, 1698) liet~ auf o-Cyanbenzalchlorid alkoholische HydroxylaminlSsung bei erhShter Temperatur einwirken und benannte das Reaktionsprodukt f~ls~hlich o-Cyanbenzaldoxim. Aus diesem Produkte gewann er durch einst(indige Behandlung mit der ffinffachen Menge Essigsiiureanhydrid Phthalonitril. Die Ausbeute gibt P o s n e r nicht an.
A. B r a u n und $. T s c h e r n i a c (B. 40, 2710) gewannen Phthalonitril dutch einstfindige Einwirkung der vierfachen Menge Essigs~ureanhydrids auf Phthalamid in zirka 2~ Ausbeute und er- brachten in der gleichen Arbeit den Beweis, daft das in zirka 50~ Ausbeute entstehende Hauptprodukt dieser Reaktion, o-Cyan- benzamid, mit P o s n e r ' s o-Cyanbenzaldoxim identisch ist.
Ffir die vorliegende Arbeit wurde das erforderliche Phthalo- nitril auf folgende Weise erhalten.
Nach A. Braun 's und J. T s c h e r n i a c ' s Angabenwurde o-Cyan- benzamid, dargestellt und nach P o s n e r durch neuerliche Ein- wirkung der vierfachen Menge Essigsaureanhydrids in Phthalonitril (iberffihrt. Die Ausbeute betr~igt 20o/0 berechnet vom angewandten Phthalamid.

Einwirkung yon Organomagnesiumverbindungen. I09
DarsteUung des 3, 3-Dibenzyl-l-aminoisoindols.
Zu einer L/Ssung yon Phthalonitril in absolutem Benzol wurde eine ~ttherische LtSsung yon Benzylmagnesiumchlorid zutropfen gelassen.
Dabei fielen zuerst rotviolette Floeken aus, die sieh beim weiteren Zusatz tier /i.therischen Benzylmagnesiumchloridl6sung wieder aufl6sten.
Nachdem die ganze Benzylmagnesiumchloridl/Ssung zugesetzt war, zeigte die Flfissigkeit dunkelviolettrote Farbe. Da die Reaktion unter Wtirmeentbindung verlief, war W'asserkt'lhlung des Reaktions- gefg.fies notwendig.
Stets wurde ffir ein Mol Phthalonitril das Benzylmagnesium- chlorid aus 2"5 Mol Benzylehlorid dargestellt. Die grtSflte Menge Phthalonitril, die auf einmal verarbeitet wurde, betrug 20 g.
Nach dem Stehen fiber Nacht wurde das Reaktionsgemenge mit wenig Eis zersetzt, wobei die violette Farbe der /itherisch- benzolischen L/Ssung vorfibergehend in Gffin umschlug, um sehliel3- lich wieder einem roten Farbton zu weichen. Unterdessen schied sieh stets ein Teil der Substanz als rotschwarzes Harz ab.
A. S a u r e Z e r s e t z u n g .
Anfangs wurde das so erhaltene Gemenge mit verdfinnter Salzsiiure his zur sauren Reaktion versetzt. Hierauf wurde auf dem Wasserbad Ather und Benzol abdestilliert und der Rtickstand zur Entfernung des bei der Reaktion gebildeten Diphenyl~.thans einer \Vasserdampfdestillation unterworfen. Der im Kolben verbleibende Rfickstand enthielt die organischen Reaktionsprodukte neben der Salzsauren Magnesiumchloridl6sung. Die salzsaure L6sung wurde nach dem Erkalten abgegossen und alas verbleibende Harz zur Enffernung des anhaffenden Magnesiumchlorids mit kaltem W asse r ausgewasehen.
SchlieNieh wurde das Harz solange mit verdfinnter Salzsiiure gekocht und die heil]e Salzs~iurel6sung vom UngeI6sten abfiltriert, bis eine kleine Probe des Filtrats, mit Kalilauge versetzt, keine Ftillung gab.
Bei Anwendung von 10g Phthalonitril waren ungef~.hr 10 l verdfinnter Salzs/iure zur Extraktion notwendig, dabei blieb noch immer ein Tell der organischen Substanz als Unl6sliches Harz zurtick. Dieses Harz war in Eisessig und Alkohol mit dunkelroter Farbe l~Sslich und wurde nicht welter untersucht.
Nach dem Erkalten wurde die salzsaure gelbe LSsung zur Fiillung der darin enthaltenen Base mit Kalilauge bis zur alkalischen Reaktion versetzt, dabei sehied sich die freie Base in Form gelber Flocken aus, die nach dem Absetzen abgesaugt wurden.

1 1 0 R. Weil3 und E. Freund~
Zur w e i t e r e n Re in igung w u r d e die S u b s t a n z mit k o n z e n t r i e r t e r Salzsgture gekoch t , w o b e i s ich das in k o n z e n t r i e r t e r Salzsg.ure un- liSsliche C h l o r h y d r a t im g e s c h m o l z e n e n Z u s t a n d bildete. Nach d e m AbgiefSen der Sa l z s i iu re w u r d e de r R t i c k s t a n d mit k o c h e n d e m W a s s e r w i e d e r h o l t a u s g e z o g e n , w o b e i w i e d e r ein k le iner Tel l d e r S u b s t a n z h a r z i g zu r i i ckb l i eb . Die heilge, w i i s se r ige LiSsung w u r d e n u n mit k o n z e n t r i e r t e r sa lzsS.ure bis zu r b e g i n n e n d e n T r t i b u n g ve r se t z t . Beim Erka l t en der L i i sung sch ied s ich dann ein g e l b e s Kr ys t a l tmeh l ab, w e l c h e s in der g l e i chen W e i s e w e i t e r ge re in ig t w u r d e , bis e ine k le ine Probe , auf e inem Pla t inb lech ve rbrann t , ke inen R ~ i c k s t a n d hinterliel3.
Das auf d iese W e i s e da rges t e l l t e C h l o r h y d r a t ve rha rz t e bei z i rka 100 ~ Da ke in k la re r S c h m e l z p u n k t z u e rz ie len w a r und a u c h d ie freie Base n icht z u r Krys t a l l i s a t i on g e b r a c h t w e r d e n konn t e , w u r d e das so e r h a l t e n e n e C h l o r h y d r a t ana lys i e r t .
I. 0'2154g r Substanz gaben 0' 1171 g H20 und 0"5943 gr CO2. II. 0"1880g ,, ,, 0"1033,q" H~O und 0"5195g CO2.
I. Gef.: 6"080/0 H und 75"25o,/o G. II. Gef.: 6'15O/o H und 75"360/0 C.
Ber. fiir C22H2oN2HCI: 6"070/0 H und 75"720/0 C.
0"2091 g Substanz gaben bei 747 mm Druck und 17" 14'6 c m a N .
Gel.i: 8"07c o N. Ber. ftir C22H2oNzHCI: 8-040/o N.
I. 0'2236 g" Substanz gaben 0"0941 gr AgCI. 1I. 0"17233 ~ ~ 0"0723 g AgC1.
L GeE: 10"41o o C1. [L Gel.: 10"38o o C1.
Bet. for C22H2oN2Cl: 10' 17o o CI.
W u r d e das oben b e s c h r i e b e n e ge lbe C h l o r h y d r a t in w~isser iger L S s u n g e m e r s e i t s mit K a l i u m p e r m a n g a n a t oxyd ie r t , and re r se i t s mi t Zink und Sa l z s~u re ode r mi t N a t r i u m a m a t g a m in a l k o h o l i s c h e r L S s u n g reduz ie r t , so w u r d e s te t s der g le iche KSrpe r isoliert . Die A u s b e u t e n d i e s e s KSrpers , s o w o h l bei de r O x y d a t i o n als auch be i der Reduk t ion , w a r e n sehr ger ing. Z u r w e i t e r e n Re in igung w u r d e er im V a k u u m sub l imie r t und h i e r au f aus Benzo l umkrys ta l l i s i e r t . Der KSrpe r s c h m o l z be i 180 ~ und e r g a b durch A n a l y s e d i e Zu- s a m m e n s e t z u n g :
I. 0.2162 ff Substanz gaben 0" 1213 g H20 und 0'6742 gr CO2. 11 0"20203" ~ ~ 0'I1663" H20 und 0"62833 CO 2.
I. Gef.: 6" 280/0 H und 85"05o/o C, II. GeE: 6'46O/o H und 84"83(~o C,
Ber. fiir C22H2oN2: 6"46O/o H und 84'57o 0 C.
0' 1436 gr Substanz gaben bei 743 mm Druck und 21 ~ 11 "25 c m ~ N .
Gef.: 8"800/o N. Bet. fiir C22H2oNs: 8"97O/o N.

Einwirkung von Organomagnesiumverbindungen. 1 ] I
M o l e k u l a r g e w i c h t s b e s t i m m u n g nach RasL
0"0128 g Substanz gaben in 0"1286 g Kampfer gel(~st eine Depression yon 12"25~
Molekulargewicht gef. : 312" 3. ber.: 312 "i8.
Dieses Ergebnis lieB erkennen, dal3 das gelbe Krystallpulver keine einheitliche Substanz darstellte, sondern ein Gemenge yon mindestens 2 Isomeren vorliegen muI3te. Erst als nach dem voran geschilderten Verfahren Impfmaterial erhalten wurde und die Eigen- schaften des reinen -3, 3-Dibenzyl-l-aminoisoindols bekannt waren, wurde eine bequemere Darstellungsweise gefunden, bei der an Stelle der Salzs~ure AmmonchloridlSsung verwendet wurde.
B. Z e r s e t z u n g mit A m m o n c h l o r i d .
Die Einwirkung des Benzylmagnesiumchlorids auf Phthalo- nitril wurde genau wie oben beschrieben vorgenommen. Das Re- aktionsprodukt wurde, nach dem Hinzuffigen von Eiswasser, mit einer konzentrierten AmmonchloridlSsung versetzt, die organischen LSsungsmittel auf dem Wasserbad abdestilliert und die w/isserige LSsung, die neben Ammonchlorid noch das ganze Magnesium ent- hielt, abgegossen und das Zurfickbleibende rStl ichschwarze Harz mehrmals mit kaltem Wasser nachgewaschen.
Hierauf wurde die organische Substanz solange mit sehr ver- dfinnter kochender AmmonchloridlSsung ausgezogen, bis eine kleine Probe des w/isserigen Extraktes, mit Kalilauge versetzt, keine F/illung mehr gab. Ffir 10g Phthalonitril waren wieder zirka 10 I verdfinnte AmmonchloridlSsung notwendig.
Die fiber Nacht erkaltete LSsung wurde von einer geringen Menge eines gelben ausgefallenen KSrpers filtriert und das Filtrat solange mit Kalilauge versetzt, his keine weitere F/illung mehr eintrat.
Der in der Regel krystallinisch ausgefallene Niederschlag wurde abgesaugt, im Vakuum fiber Schwefels/iure getrocknet und aus wenig Benzol dreimal umkrystallisiert. Beim letzten Umkrystal- lisieren ist es vorteithaft, dem Benzol etwas hochsiedenden Petrol- /i.th er zuzusetzen.
Die auf diese Weise erhaltene Base stellte einen rein weif3en schSn krystallisierten KSrper dar, der bei 180"5 ~ scharf schmolz.
Der Mischschmelzpunkt mit dem bei tier sauren Zersetzung und darauffolgender Oxydation erhaltenen KSrper war 179 bis 180 ~
Bei der unter A angegebenen Aufarbeitung betrug die Aus- beute an Dibenzylaminoisoindol, wenn von 10 g Phthalonitril aus- gegangen wurde, 1 g'. Bei tier unter B angegebenen Aufarbeitung betrug die Ausbeute an Dibenzylaminoisoindol ftir 10g" Phthalo- nitril 6' 5 g.

112 R. WeiB und E. F r e u n d ,
0"2 g Dibenzylamin0isoindol wurden im Einschmelzrohr mit konzentr ier ter Salzs/iure auf 180 ~ erhitzt. Wie aus dem Schmelz- punkt und Mischschmelzpunkt mit dem Ausgangsmater ia l hervor- ging, blieb der KSrper unver&ndert.
Dos Dibenzylaminoisoindol ist in den meisten organischen LSsungsmit teln, mit Ausnahme yon Ligroin, leicht 15slich.
Darstellung des Dibenzylaminoisoindolehlorhydrats. 1 g Dibenzylaminoisoindol wurde in wenig Alkohol gelSst und
mit einigen Kubikzent imetern konzentrierter Salz&ure versetzt. Hierauf wurde soviel W a s s e r zugesetzt , bis eine Tr t ibung entstand. Nach einigen Stunden war das Chlorhydrat in Form von weifien Nadeln auskrystall isiert und wurde nach dem Stehen t'tber Nacht abgesaugt und mit konzentr ier ter SalzsS.ure nachgewaschen und im Vakuum tiber Schwefels/iure getrocknet.
Das Chlorhydrat ist hygroskopisch und sehmilzt unscharf yon 85 bis 9 t ~ . Die Chlorbes t immung nach C a r i u s ergab:
O' 1369 g" Substanz gaben 0 '0546 2" AgC1.
Gel.: 9" 870 0 CI.
Ber. fiir C,,oH2oN~HCI: 10' 17o o CI.
Darstellung des Diaeetyldibenzyliminophthalimidins Formel VII). 5 g Dibenzylaminoisoindol wurden mit 30 c~n ~ Essigs~ture-
anhydrid am Rtickflui3ktihler llJ~ Stunden zum gelinden Sieden er- hitzt. Die braun gefS.rbte LSsung wurde in kaltes Wasser gegossen. wobei sich ein Ol abschied, das nach der ZerstSrung des Essig- s/i.ureanhydrids nach einigen Stunden krystallinisch erstarrte.
Das nach dem Absaugen getrocknete Produkt wurde in alko- holischer LSsung mit Knochenkohle gekocht. Nach dem Filtrieren wurde dos Filtrat mit siedend heifiem Wasse r bis zur Trt ibung versetzt. Beim Auskiihlen fielen gl/i.nzend weii3e Nadeln aus, die getrocknet bet 122 bis 123 ~ schmolzen. Aus wenig Alkohol um- krystallisiert, bieb der Schmelzpunkt unver/indert. Die Ausbeute bet rug 3"6 g.
O" 2048 g Substanz gaben 0" 1140 g H,,O und 0 ' 5904 gr COx.
Gef.: 6"230 o H und 78"62o o C.
Bet. fiir C2oH24NoO2: ~-11o/0 H und 78"750 o C.
O' 1691 g Substanz gaben bet 745 mm Druek und 18~ c~n ~ N.
Gef.: 7"14o o N
Ber. fiir C2GH2tN202:7"07o/0 N.
Darstellung des Anhydrodiacetyldibenzyliminophthalimidins. (Formel X, XI.)
3 g Diacetyldibenzyliminophthalimidin wurden mit '35 cm ~ 10prozent iger alkoholischer Kalilauge eine Stunde am Riickfluflkiihler

Einwirkung yon Organomagnesiumverbindungen. 1 13
g e k o c h t und hieraut mit W a s s e r bis zu r beg innenden Tr f ibung ver- setzt. Es fielen weil3e, schuppenfSrmige Krystalle aus , die bei 162 ~ schmolzen. N a c h oftmaligem Umkrysta l l i s ieren aus w e n i g Alkohol s t ieg der Schme lz punk t au f 167 bis t68 ~ an. Beim wei teren Ver- d/.innen der ersten Mutter lauge mit W a s s e r fielen weige Krystall- d rusen aus, die unscha r f bei z i rka 135 ~ schmolzen . Es war j edoch nicht m6glich, aus dieser Frakt ion ein einheit l iches P roduk t zu gewinnen.
0 "2 g reine S u b s t a n z wurden im E inschme lz roh r 6 S tunden au f 180 ~ erhitzt. Die zurt ickerhal tene S u b s t a n z w u r d e aus Alkohol unter Zusa t z yon Kal i lauge umkrystal l is iert und schmolz wieder bei 168 ~ Ein Mischschme lzpunk t mit dem Ausgangsmate r i a l zeigte keine Depression.
I. o. 1939 g Substanz gaben 0" 1004 g H20 und 0' 5892 g CO 2. II. 0"1978g ~ ,, 0 '1036g H20 und 0"6016g CO 2.
I. Gel.: 5"79O/o H und 82'870/0 C. II. GeL: 5"86O/o H und 82 "95O/o C.
Ber. f/Jr C26H22N20: 5"86o/o H und 82'500/0 C. 0"1775g Substanz gaben bei 747 mm Druck und 17'6 ~ 11"5 c~n .~ N.
Gef.: 7" 47% N.
Bet. ffir C2oHsoN20: 7"41O/o N.
D a r s t e l l u n g des D i b e n z y l p h t h a l i m i d i n s (Formel VIII).
3 g Diacety ld ibenzyl iminophthal imidin w u r d e n mit 20prozen t ige r Salzs/ iure 6 S tunden im E inschmelz roh r au f 180 ~ erhitzt. N a c h dem Erkal ten war die in der Salzs~ture ungelSste Subs t anz te i lweise krystallisiert. Beim Hinzuf~gen yon W'asse r zerfielen die Krystal le und der Rbhreninhal t win'de pulverf6rmig. Die S u b s t a n z wurde ab- g e s a u g t und aus Alkohol , dem wen ig w/isserige Kal i lauge zugeftJgt war, umklystal l is ier t . Das Rohproduk t s chmolz bei 195 ~ Die Aus- beute be t rug 2"3 g.
Durch Verdt innen tier Mut ter lauge mit W a s s e r wurden in ger ingen Mengen Krystal le g e w o n n e n , die bei 160 ~ u n s c h a r f schmolzen . Zur Darste l lung der ana lysen re inen Subs tanz wurde das bei 195 ~ schmelzende Rohproduk t aus wen ig Alkohol umkrys ta l - lisiert. Der S c h m e l z p u n k t der reinen Subs t anz be t rug 196 bis 197 ~
Das Dibenzylphthal imidin ist in den meis ten o rgan i schen L6sungsmi t te ln , mit A u s n a h m e yon Ligroin, leicht 15slich.
o. 1949 g Substanz gaben 0" 1051 g H20 und 0"6042 g CO 2.
GeL: 6"030/0 H und 84"420/0 C. Ber. fiir C~2H:9NO: 6"12O/o H und 84"300/0 C.
0"1764g Substanz gaben bei 756 mm Druck und 18 ~ 6 '7 cm 3 N. GeL: 4"430/0 N. Ber. ftir C2.~H19NO: 4"470]0 N.

1 14 R. Weil] und E. Freund, Einwirkung ,:on Organomagnesiumverb.
Erscht ipfendr Me thy l i e rung des 3, 3 -D i benzy l - l - amino i so indo l s
und Dars te l lung des N - M e t h y l d i b e n z y l p h t h a l i m i d i n s (Formel IX).
5 g Dibenzylaminoisoindol wurden mehrere Stunden mit fiber- schtissigem Jodmethyl und Kali gekocht, wobei Tr imethylamin ent- wickelt wurde. Der nach dem Abdestiilieren des LSsungsmit tels verbleibende krystaUinische Rfickstand wurde mit Wasse r ausge- waschen und geringe Mengen anhaftenden 01s mit wenig kaltem ,~ther enffernt.
Die zurfickbleibenden Krystalle wurden aus wenig Alkohol umkrystallisiert. Die reine, in weiflen Bl~tttchen krystaliisierte Sub- stanz schmolz bei 150 bis 152 ~
0" 1972 g Substanz gaben 0" 1124 3" H20 und 0"6097 g CO, z.
Gef.: 6"38o/o H und 84"32O/o C. Ber. fiir C23H2:NO: 6"470/0 H und 84"36O/o C.
O" 1959 g Substanz gaben bei 746mm Druck und 12 ~ 7"4 ctna N.
Gel.: 4"44O/o N. Ber. ftir C2aH21NO: 4-280/0 N.



















![Einfluss von Wasser auf die Kohlenstofffaser/Epoxid ...elib.suub.uni-bremen.de/edocs/00103654-1.pdf · auf der Oberfläche der C-Faser hervorgerufen werden [10–13]. Die Einwirkung](https://img.pdfslide.net/doc/110x75/5e15453b4e4920504e4f7328/einiuss-von-wasser-auf-die-kohlenstoifaserepoxid-elibsuubuni-auf-der.jpg)









