Embed Size (px)
Citation preview

Dra Virtudes Soriano
Instituto Valenciano de Oncologia
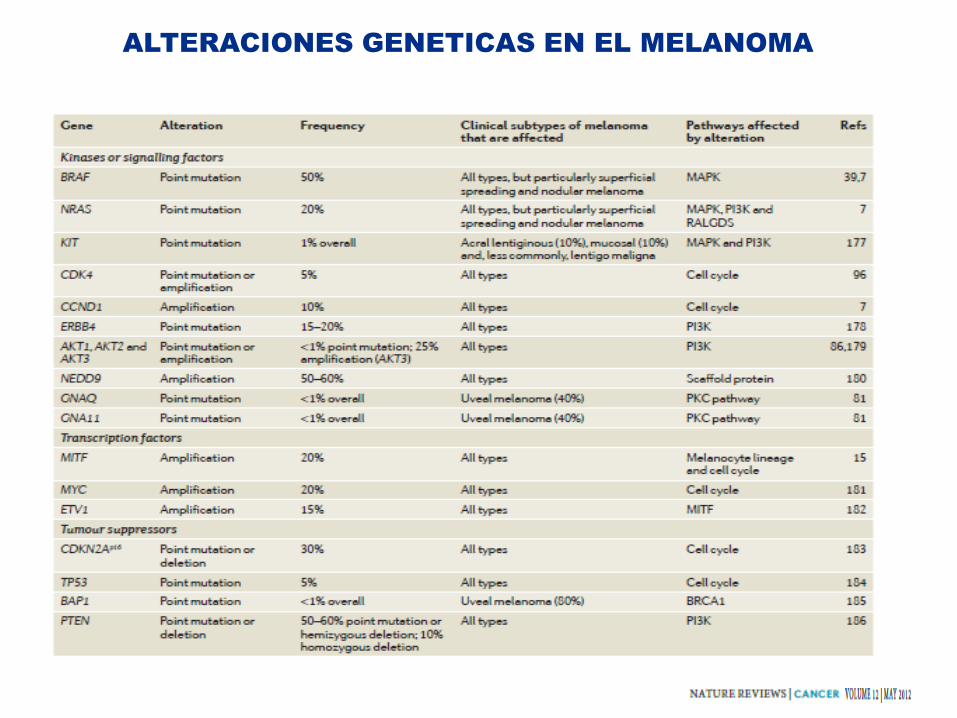
ALTERACIONES GENETICAS EN EL MELANOMA
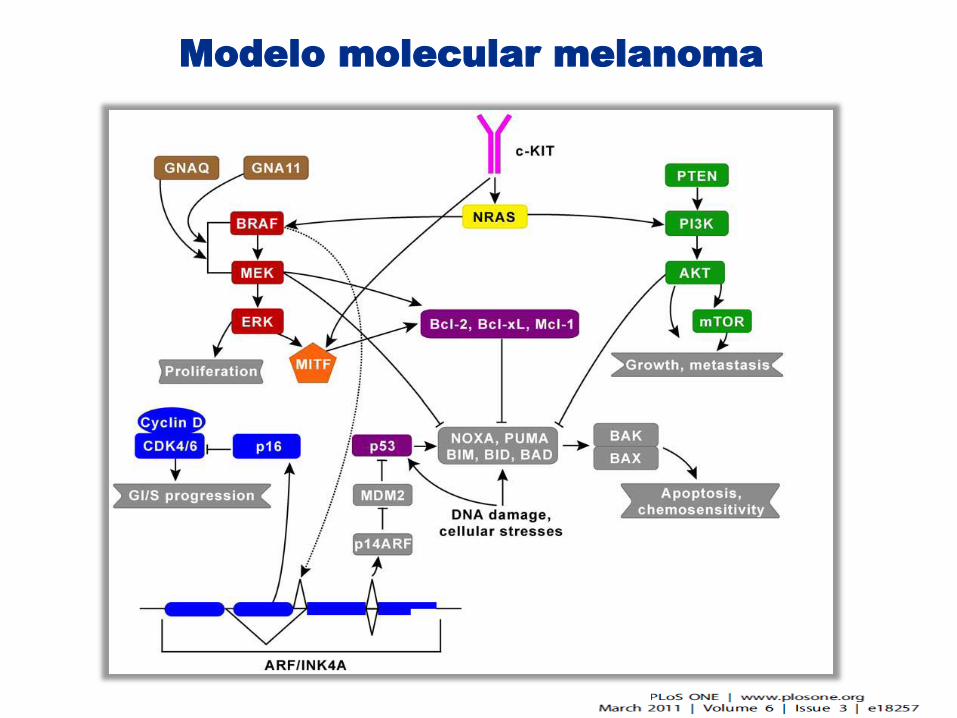
Modelo molecular melanoma
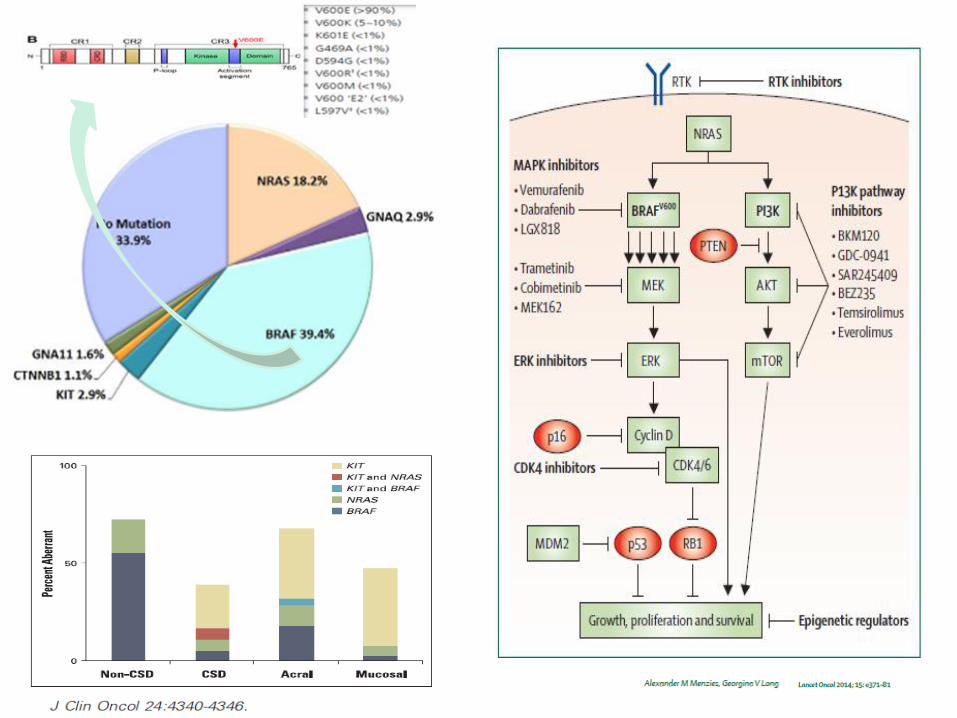
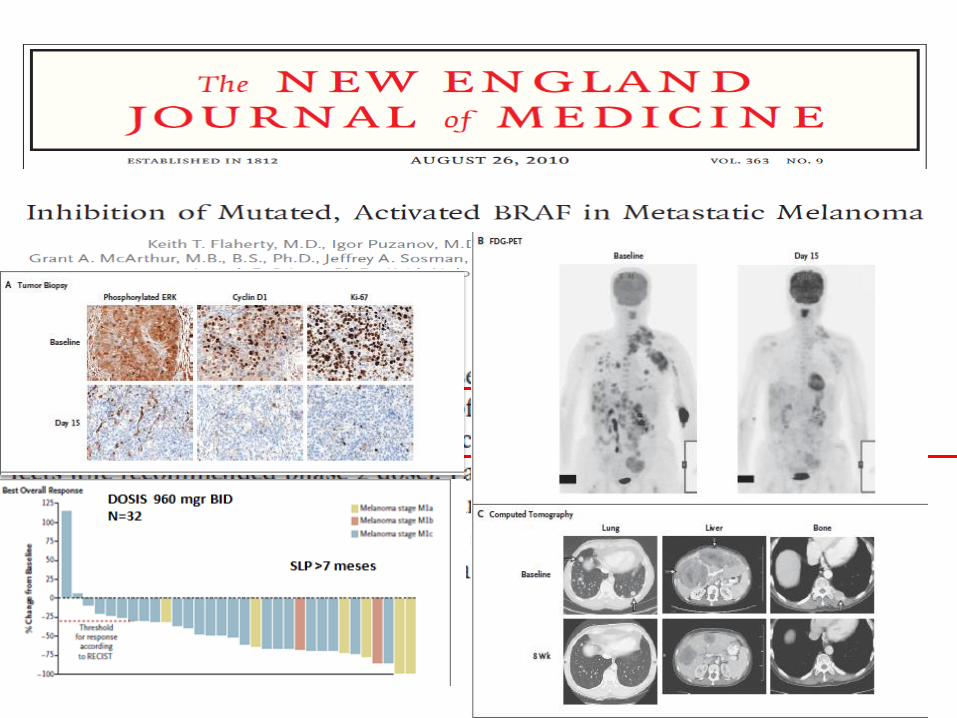
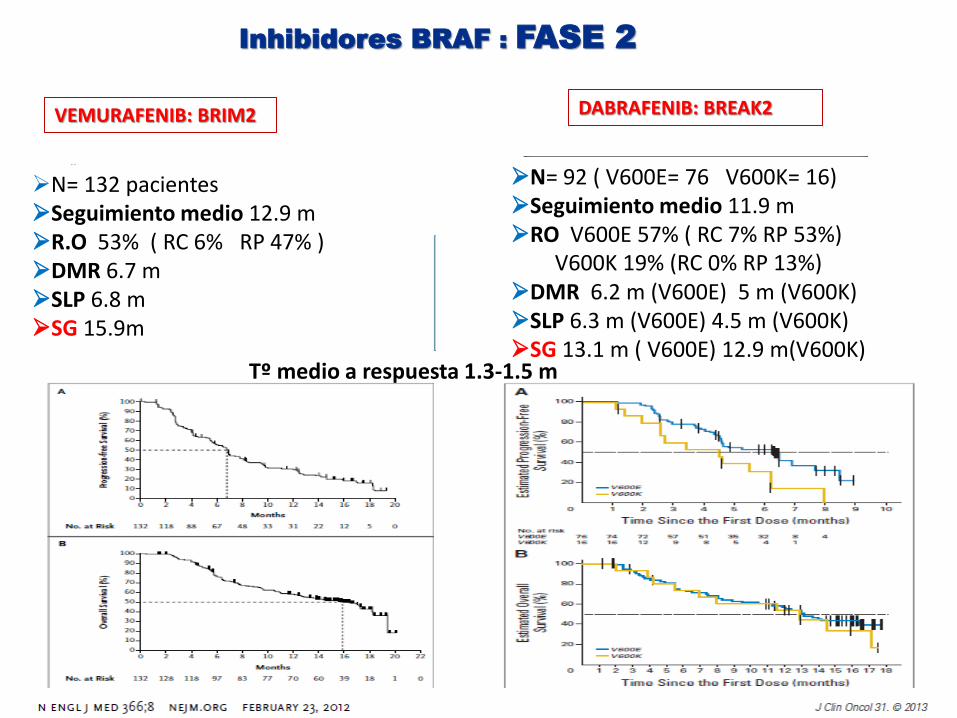
Inhibidores BRAF : FASE 2
VEMURAFENIB: BRIM2
BRAF V600E
BRAF V600K
DABRAFENIB: BREAK2
N= 132 pacientes Seguimiento medio 12.9 m R.O 53% ( RC 6% RP 47% ) DMR 6.7 m SLP 6.8 m SG 15.9m
N= 92 ( V600E= 76 V600K= 16) Seguimiento medio 11.9 m RO V600E 57% ( RC 7% RP 53%) V600K 19% (RC 0% RP 13%) DMR 6.2 m (V600E) 5 m (V600K) SLP 6.3 m (V600E) 4.5 m (V600K) SG 13.1 m ( V600E) 12.9 m(V600K)
Tº medio a respuesta 1.3-1.5 m
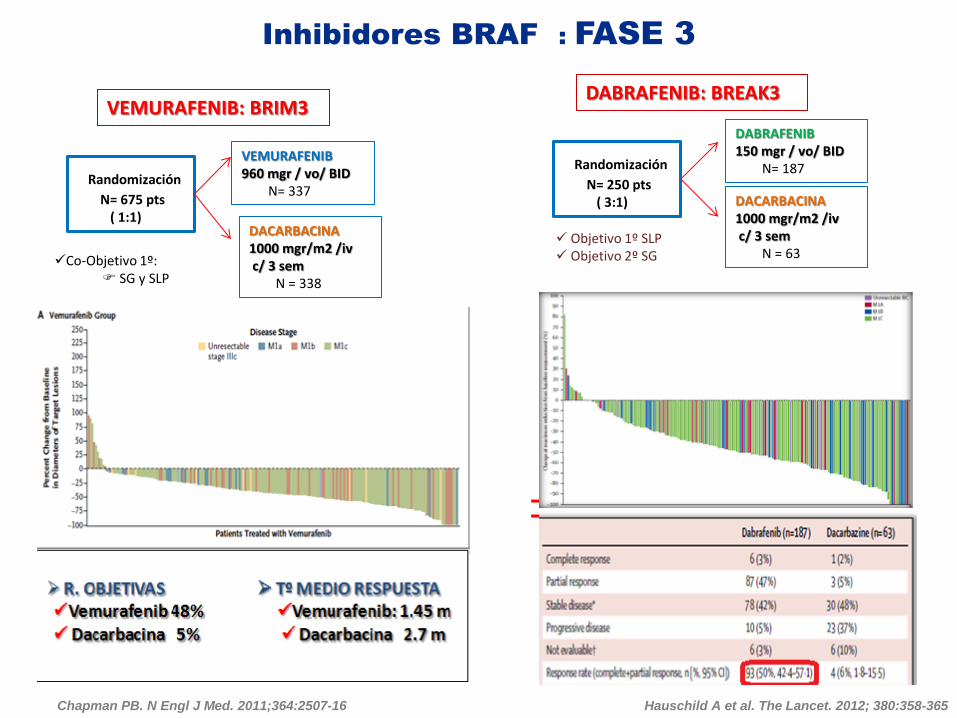
Inhibidores BRAF : FASE 3
VEMURAFENIB: BRIM3
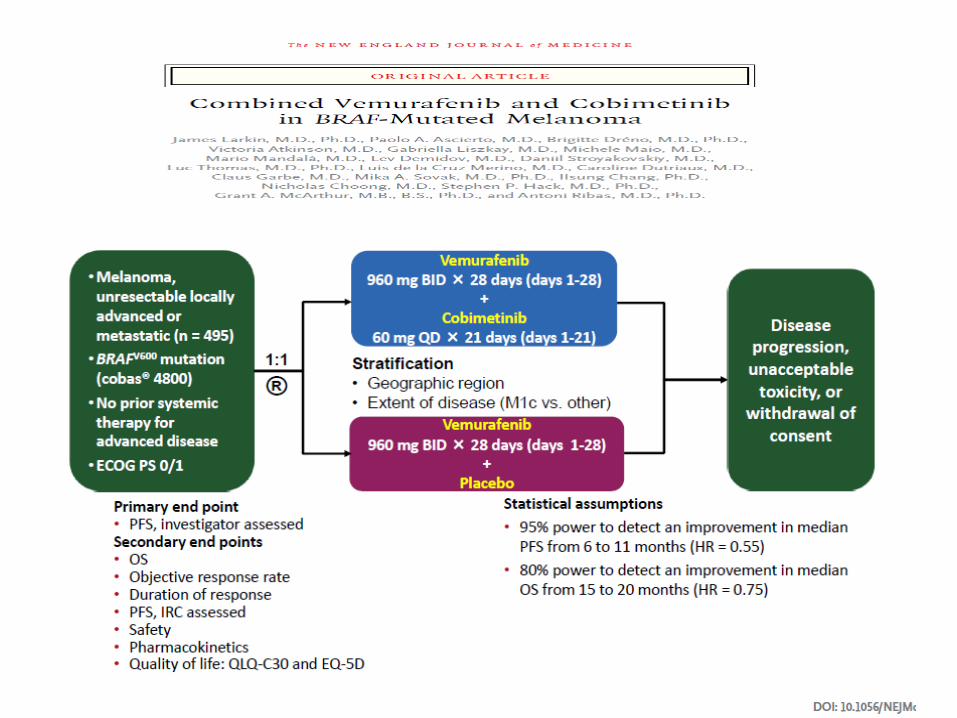
Randomización
N= 250 pts ( 3:1)
Co-Objetivo 1º: SG y SLP
VEMURAFENIB 960 mgr / vo/ BID N= 337
DACARBACINA 1000 mgr/m2 /iv c/ 3 sem N = 338
DABRAFENIB: BREAK3
Chapman PB. N Engl J Med. 2011;364:2507-16
Randomización
N= 675 pts ( 1:1)
DABRAFENIB 150 mgr / vo/ BID N= 187
DACARBACINA 1000 mgr/m2 /iv c/ 3 sem N = 63
Objetivo 1º SLP Objetivo 2º SG
Hauschild A et al. The Lancet. 2012; 380:358-365
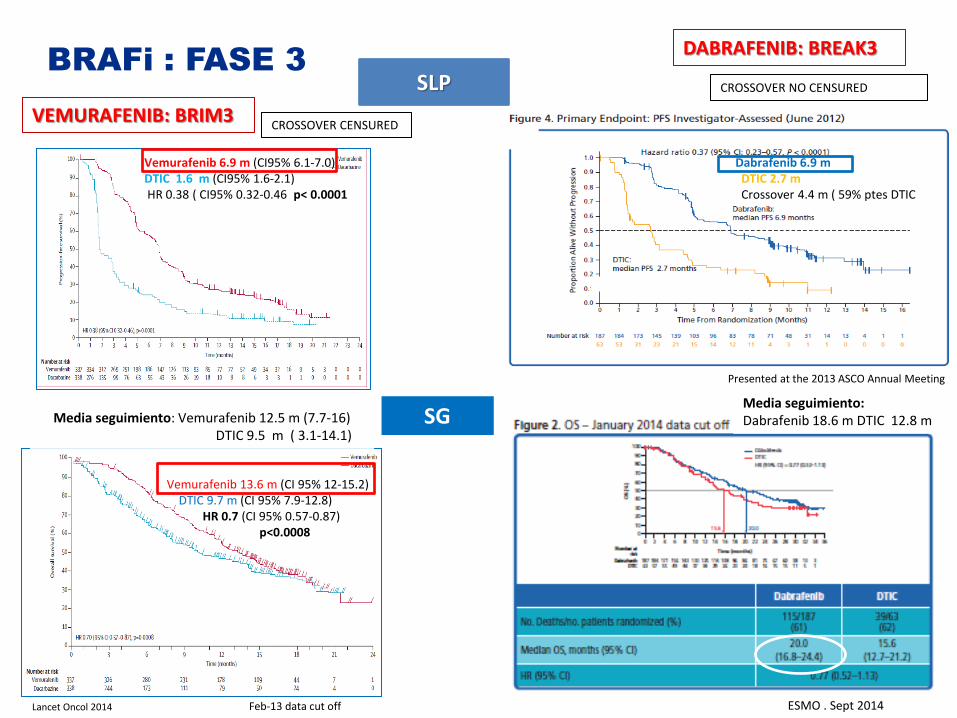
BRAFi : FASE 3
VEMURAFENIB: BRIM3
Vemurafenib 6.9 m (CI95% 6.1-7.0) DTIC 1.6 m (CI95% 1.6-2.1) HR 0.38 ( CI95% 0.32-0.46 p< 0.0001
DABRAFENIB: BREAK3
SLP
CROSSOVER CENSURED
Dabrafenib 6.9 m DTIC 2.7 m Crossover 4.4 m ( 59% ptes DTIC
SG Media seguimiento: Vemurafenib 12.5 m (7.7-16) DTIC 9.5 m ( 3.1-14.1)
Vemurafenib 13.6 m (CI 95% 12-15.2) DTIC 9.7 m (CI 95% 7.9-12.8) HR 0.7 (CI 95% 0.57-0.87) p<0.0008
CROSSOVER NO CENSURED
Lancet Oncol 2014
Presented at the 2013 ASCO Annual Meeting
ESMO . Sept 2014
Media seguimiento: Dabrafenib 18.6 m DTIC 12.8 m
Feb-13 data cut off
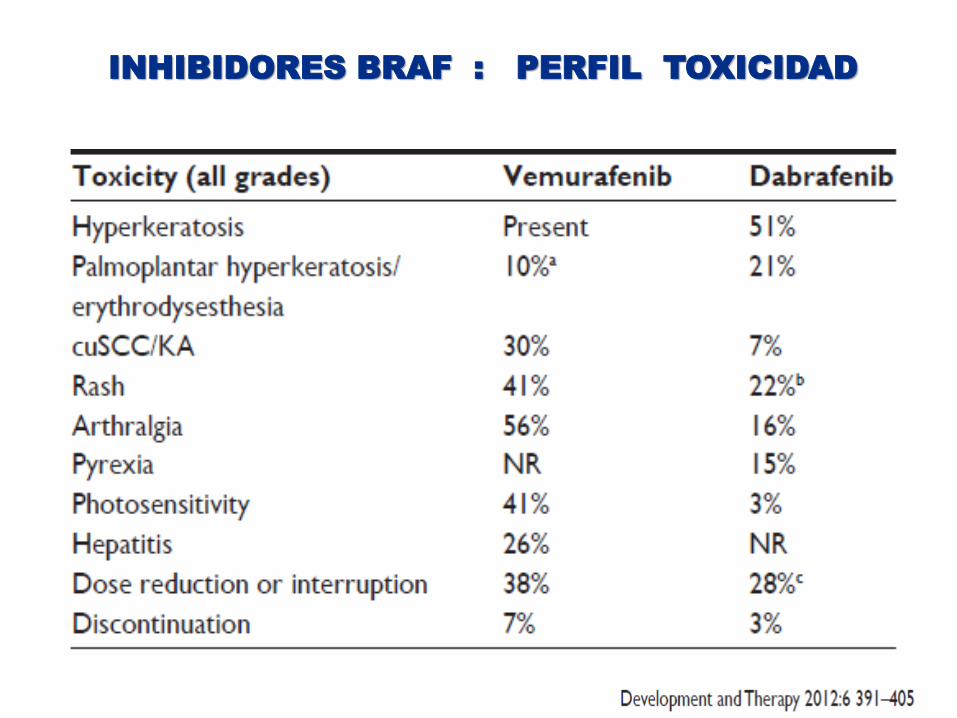
INHIBIDORES BRAF : PERFIL TOXICIDAD
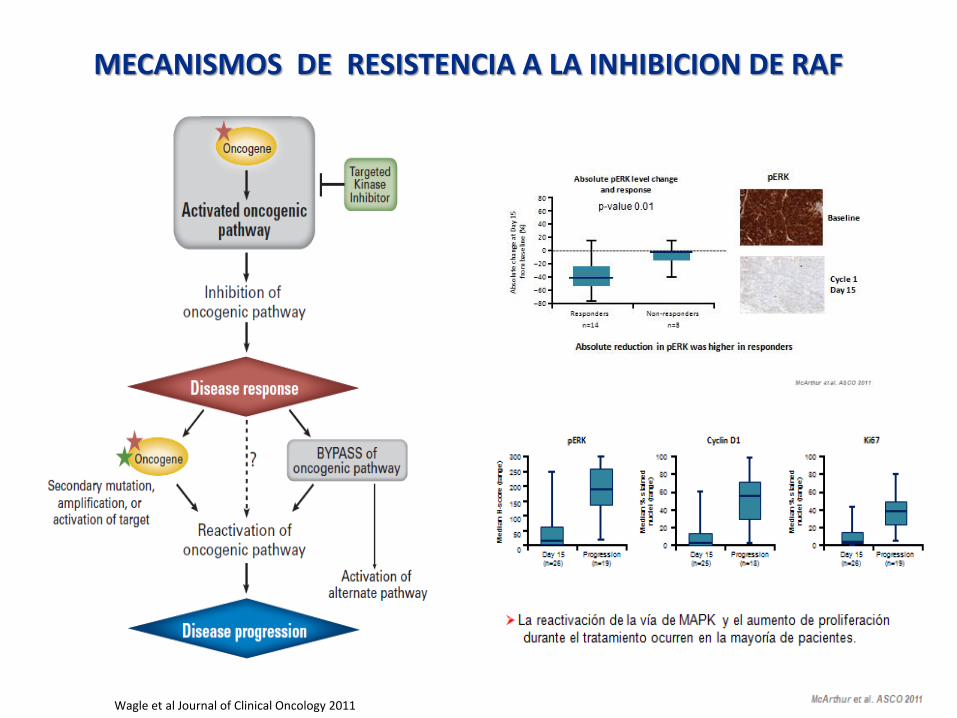
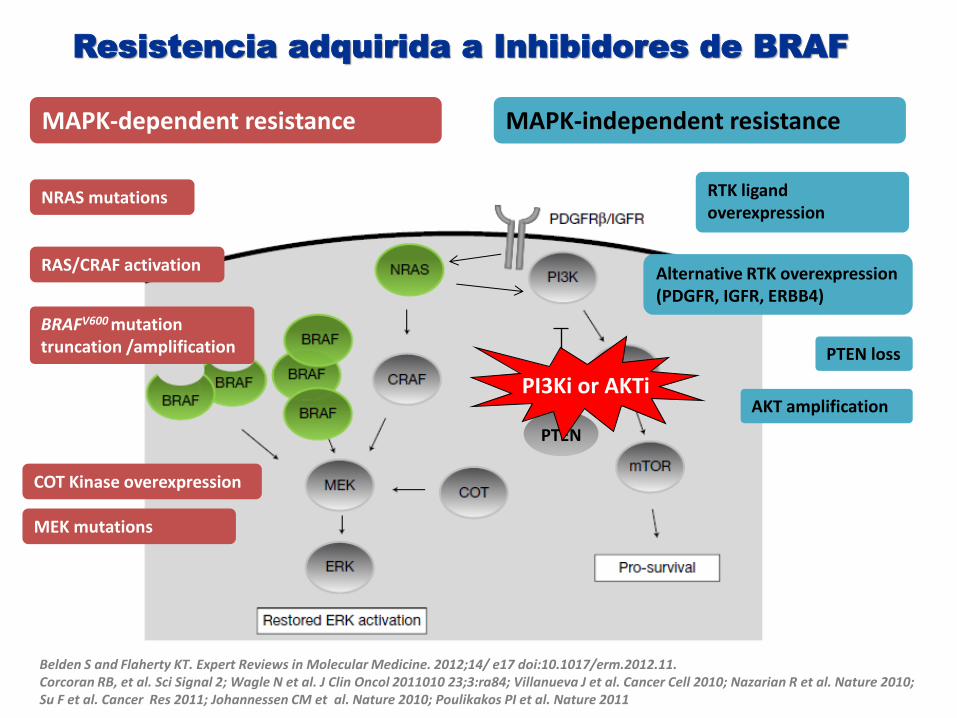
MECANISMOS DE RESISTENCIA A LA INHIBICION DE RAF
Wagle et al Journal of Clinical Oncology 2011
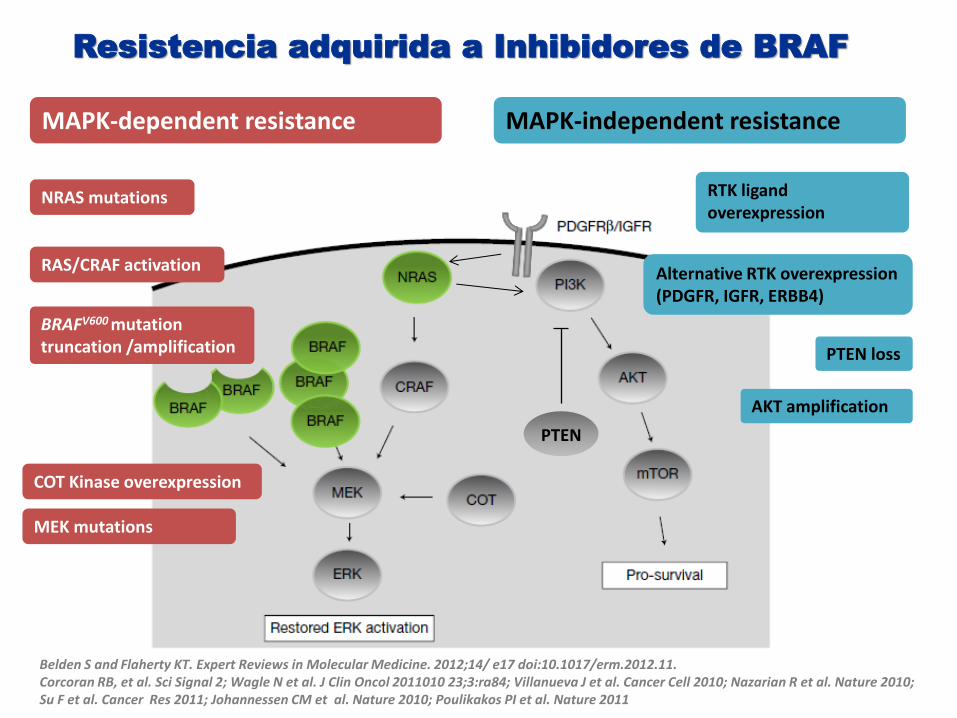
Belden S and Flaherty KT. Expert Reviews in Molecular Medicine. 2012;14/ e17 doi:10.1017/erm.2012.11. Corcoran RB, et al. Sci Signal 2; Wagle N et al. J Clin Oncol 2011010 23;3:ra84; Villanueva J et al. Cancer Cell 2010; Nazarian R et al. Nature 2010; Su F et al. Cancer Res 2011; Johannessen CM et al. Nature 2010; Poulikakos PI et al. Nature 2011
MAPK-dependent resistance MAPK-independent resistance
NRAS mutations
BRAFV600 mutation truncation /amplification
COT Kinase overexpression
MEK mutations
RTK ligand overexpression
Alternative RTK overexpression (PDGFR, IGFR, ERBB4)
RAS/CRAF activation
PTEN
PTEN loss
AKT amplification
Resistencia adquirida a Inhibidores de BRAF
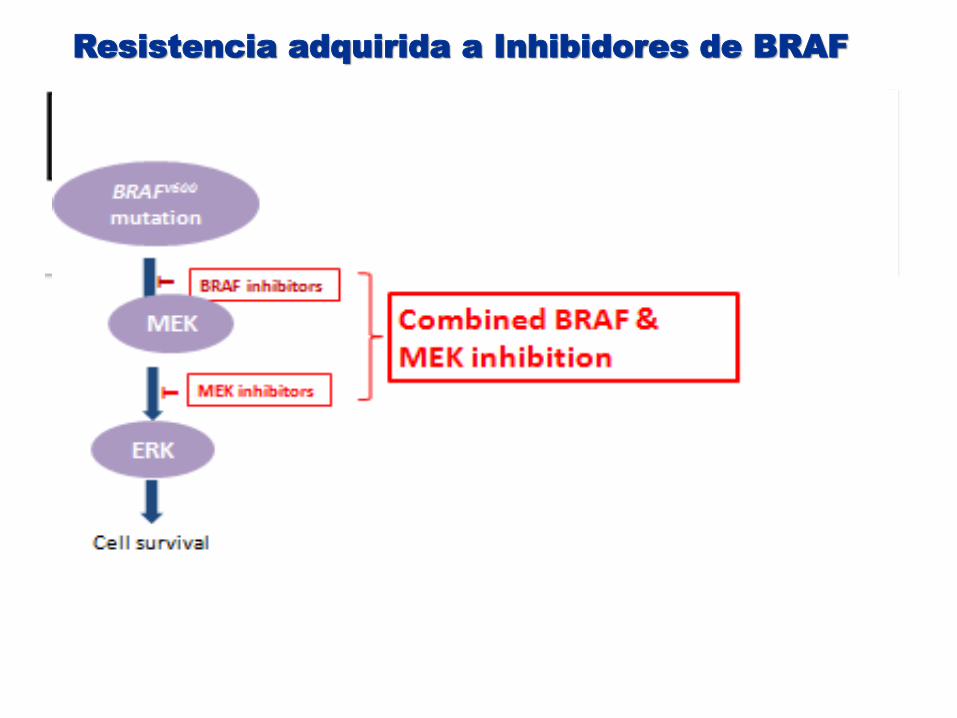
Cell survival
Trametinib Selumetinib GDC-0973 MEK 162
T
BRAFV600
mutation
ERK
MEK
MEK Inhibitors
NRAS mutations
COT overexpression
BRAFV600 mutation truncation / amplification
MEK mutations
Resistencia adquirida a Inhibidores de BRAF
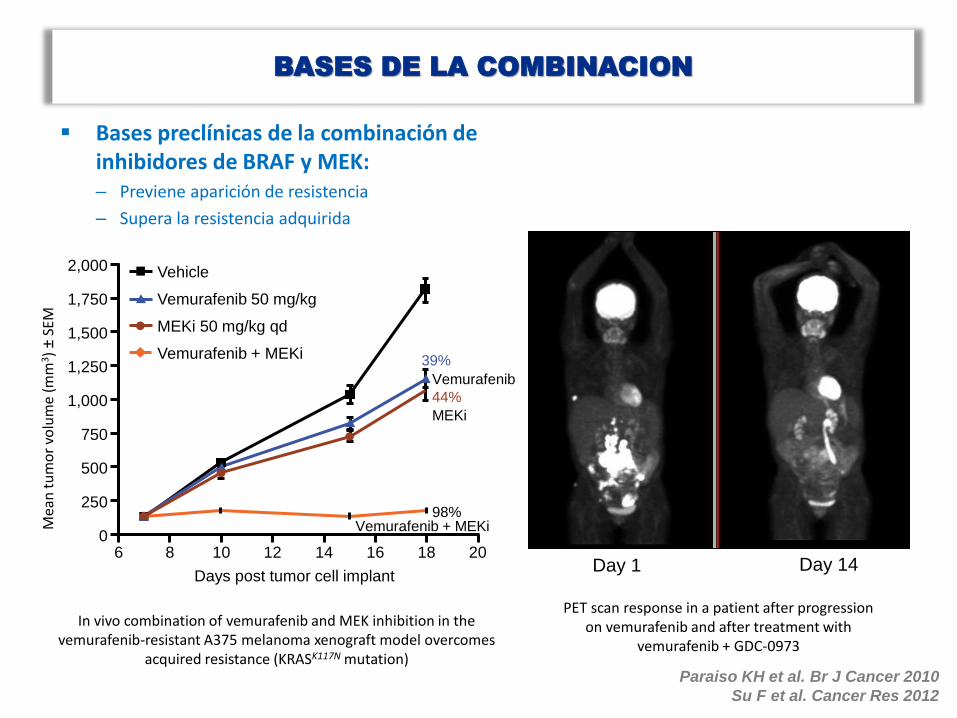
BASES DE LA COMBINACION
Bases preclínicas de la combinación de inhibidores de BRAF y MEK: – Previene aparición de resistencia
– Supera la resistencia adquirida
Paraiso KH et al. Br J Cancer 2010
Su F et al. Cancer Res 2012
In vivo combination of vemurafenib and MEK inhibition in the vemurafenib-resistant A375 melanoma xenograft model overcomes
acquired resistance (KRASK117N mutation)
PET scan response in a patient after progression on vemurafenib and after treatment with
vemurafenib + GDC-0973
2,000
1,750
1,500
1,250
1,000
750
500
250
0
Mea
n t
um
or
volu
me
(mm
3) ±
SEM
Days post tumor cell implant
6 8 10 12 14 16 18 20
39%
Vemurafenib
44%
MEKi
Vemurafenib + MEKi 98%
Vehicle
Vemurafenib 50 mg/kg
MEKi 50 mg/kg qd
Vemurafenib + MEKi
Day 1 Day 14
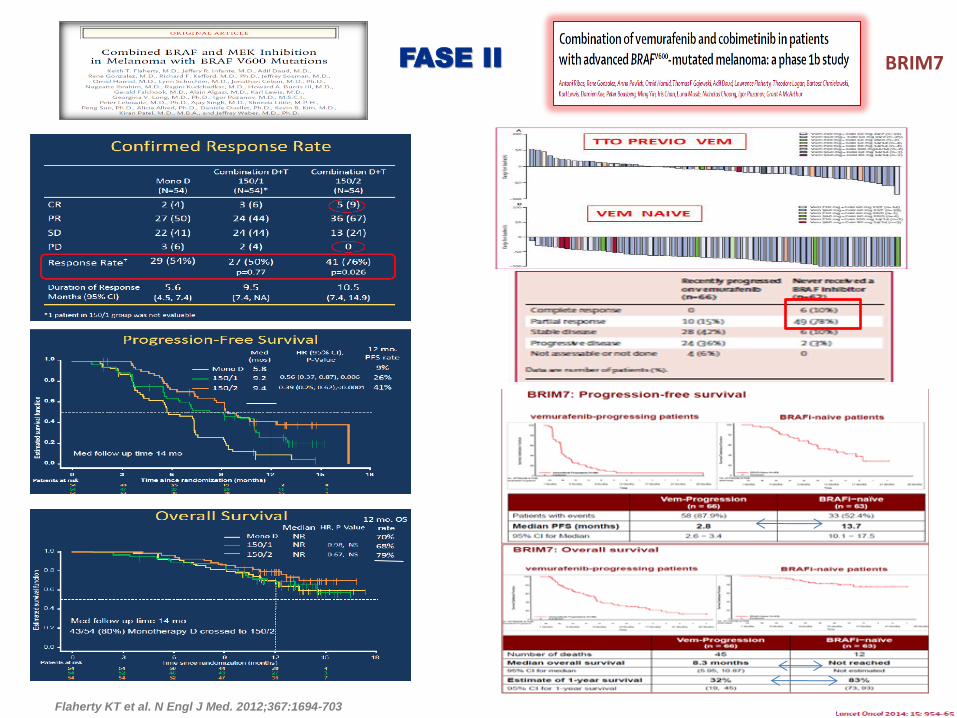
Flaherty KT et al. N Engl J Med. 2012;367:1694-703
FASE II BRIM7
Flaherty KT et al. N Engl J Med. 2012;367:1694-703
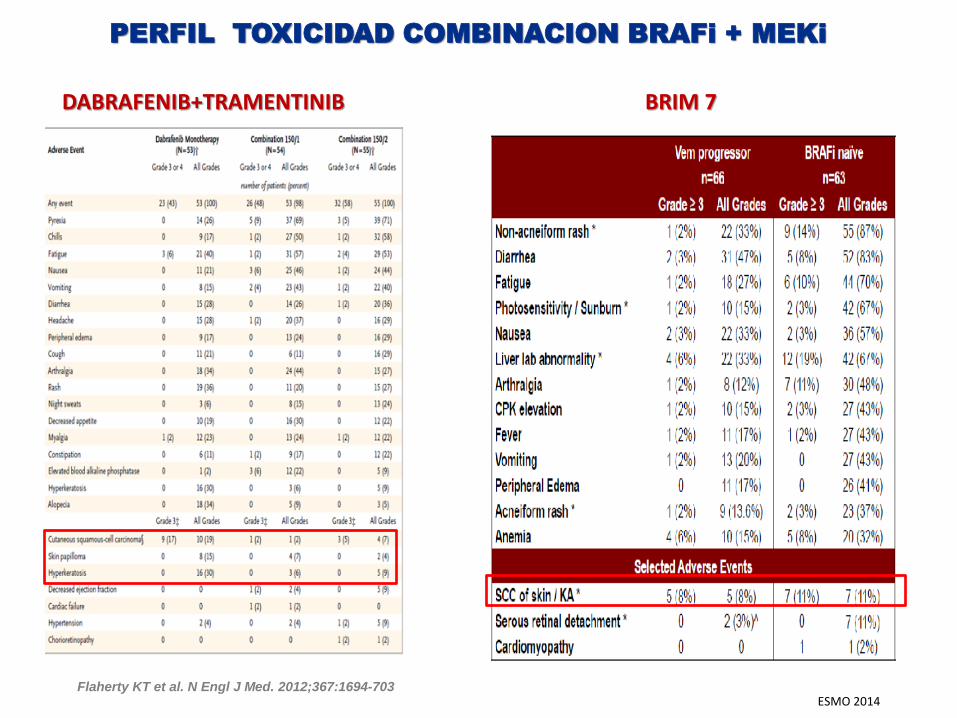
PERFIL TOXICIDAD COMBINACION BRAFi + MEKi
DABRAFENIB+TRAMENTINIB BRIM 7
ESMO 2014
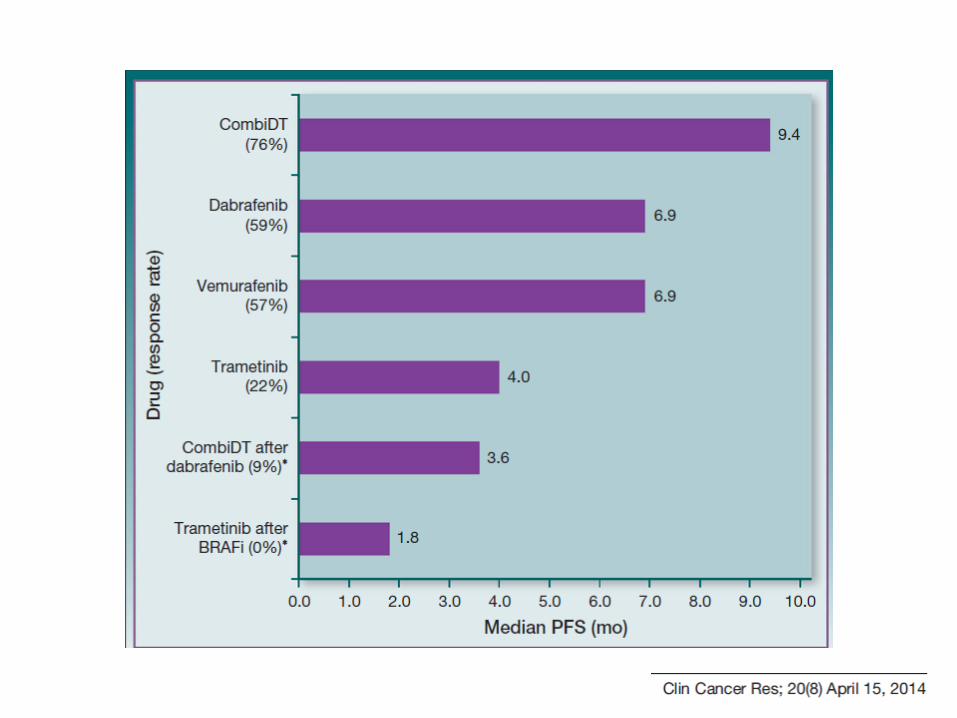

Randomization
(1:1)
N=423
DABRAFENIB ( 150 mgrBID) + TRAMETINIB (2 mg QD) n=211
DABRAFENIB (150 mgr BID) + PLACEBO n=212
BRAF V600E-K mutación Estadio III/IV no resecable O. Primario: SLP O. Secundario: ORR Duración RO Seguridad Análisis interno SG
COMBI-d
Randomization
(1:1)
N=704
DABRAFENIB ( 150 mgrBID) + TRAMETINIB (2 mg QD) n=352
VEMURAFENIB ( 960 gr BID ) n=352
Análisis final SG N=288
Estadio III/IV no resecable BRAF V600E-K mutacdo No tratamiento previo enfermedad metastasica ECOG 0-1 No M+ SNC, salvo tratadas Estables >12 sem Estratificaciión: Mutación V600E o K
LDH ( > ULN vs ≤ ULN
O. PRIMARIO: SG O. Secundario: PFS, ORR Duración RO Seguridad Prohibido crossover
COMBI-v
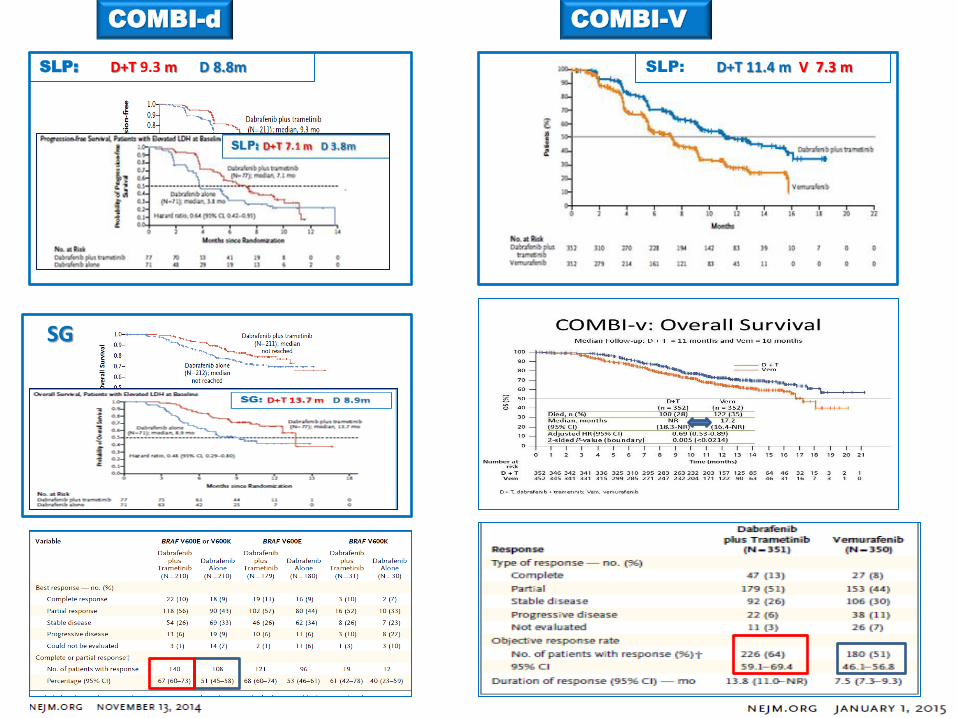
COMBI-d COMBI-V
SLP: D+T 9.3 m D 8.8m
SG
SLP: D+T 11.4 m V 7.3 m
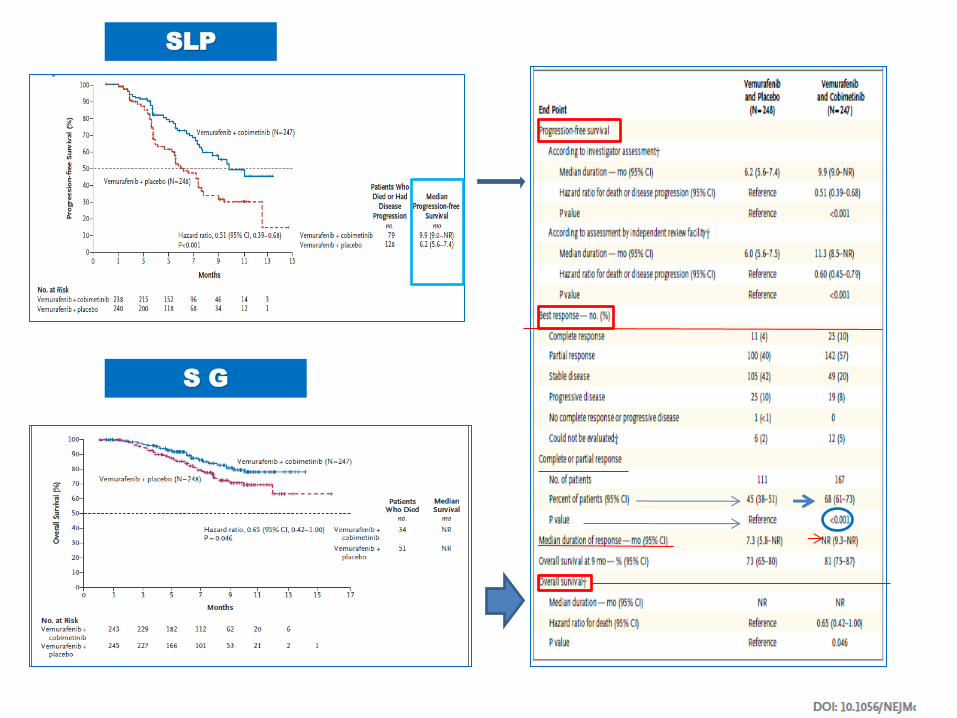
SLP
S G
Belden S and Flaherty KT. Expert Reviews in Molecular Medicine. 2012;14/ e17 doi:10.1017/erm.2012.11. Corcoran RB, et al. Sci Signal 2; Wagle N et al. J Clin Oncol 2011010 23;3:ra84; Villanueva J et al. Cancer Cell 2010; Nazarian R et al. Nature 2010; Su F et al. Cancer Res 2011; Johannessen CM et al. Nature 2010; Poulikakos PI et al. Nature 2011
MAPK-dependent resistance MAPK-independent resistance
NRAS mutations
BRAFV600 mutation truncation /amplification
COT Kinase overexpression
MEK mutations
RTK ligand overexpression
Alternative RTK overexpression (PDGFR, IGFR, ERBB4)
RAS/CRAF activation
PTEN
PTEN loss
AKT amplification PI3Ki or AKTi
Resistencia adquirida a Inhibidores de BRAF

Combined RAS/RAF/MEK pathway Inhibitors
PI3K/AKT/mTOR pathway Inhibitors
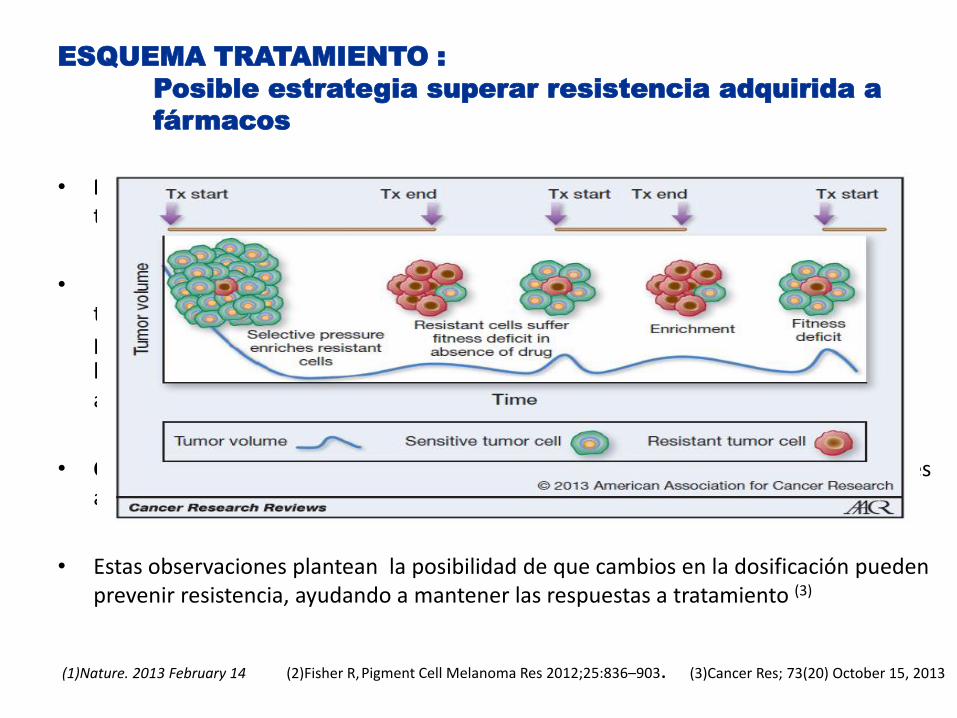
• Modelos preclinicos (1) y clinicos (2) ponen de manifiesto las células resistentes a tratamientos pueden presentar dependencia del mismo.
• Preclinica: utilizando dos modelos independientes de células resistentes al tratamiento con BRAFi (Vemurafenib) con xeno-injertos derivados de melanomas primarios resistentes a BRAFi tras administración continua de Vemurafenib , uno de los cultivos mostró dependencia al fármaco ( efecto fitness) y el cese en la administración del mismo condujo a una regresión de las células resistentes(1)
• Clinica: Fisher y col, comunicaron disminución de las lesiones en pacientes resistentes al tratamiento con Vemurafenib tras la retirada del fármaco(2)
• Estas observaciones plantean la posibilidad de que cambios en la dosificación pueden prevenir resistencia, ayudando a mantener las respuestas a tratamiento (3)
(1)Nature. 2013 February 14 (2)Fisher R, Pigment Cell Melanoma Res 2012;25:836–903. (3)Cancer Res; 73(20) October 15, 2013
ESQUEMA TRATAMIENTO :
Posible estrategia superar resistencia adquirida a
fármacos
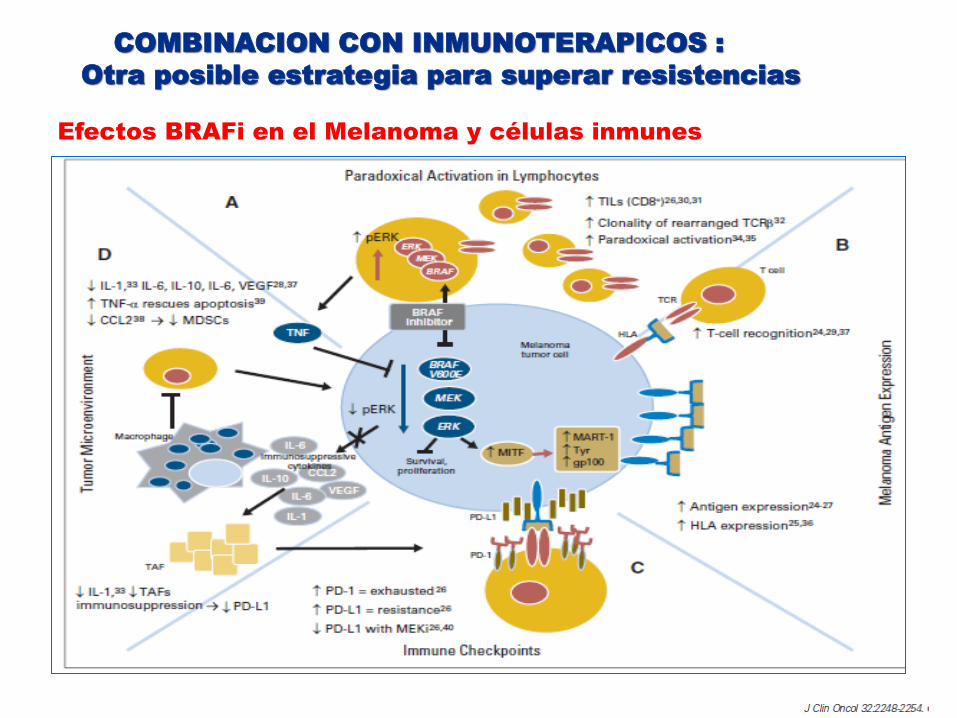
Efectos BRAFi en el Melanoma y células inmunes
COMBINACION CON INMUNOTERAPICOS :
Otra posible estrategia para superar resistencias

IMPORTANCIA MICROAMBIENTE TUMORAL
El microambiente tumoral juega un rol fundamental en la modulación de la capacidad metastásica de la mayoría de los tumores (1)
Inhibición de BRAF está asociada con un aumento en la expresión de Ag de melanoma aumento infiltrado cels T CD8+ y disminución citokinas inmunosupresoras ( IL-6 , IL-8), aumento de los marcadores de cititoxicidad de cels T(2)
(1)Nat Rev Cancer. 2009 April (2)Clin Cancer Res. 2013
Microambiente podría explicar respuestas diferentes en distintos órgano
La expresión de marcadores de agotamiento de las cels T: TIM-3 , PD1 y la PDL1 se incrementaron en el tratamiento con BRAFi (2)
El microambiente inmune puede actuar como una fuente de resistencia a los inhibidores MAPK a través TNFα derivado macrófagos a traves de MIFT
(3)Cancer Discov. 2014 October
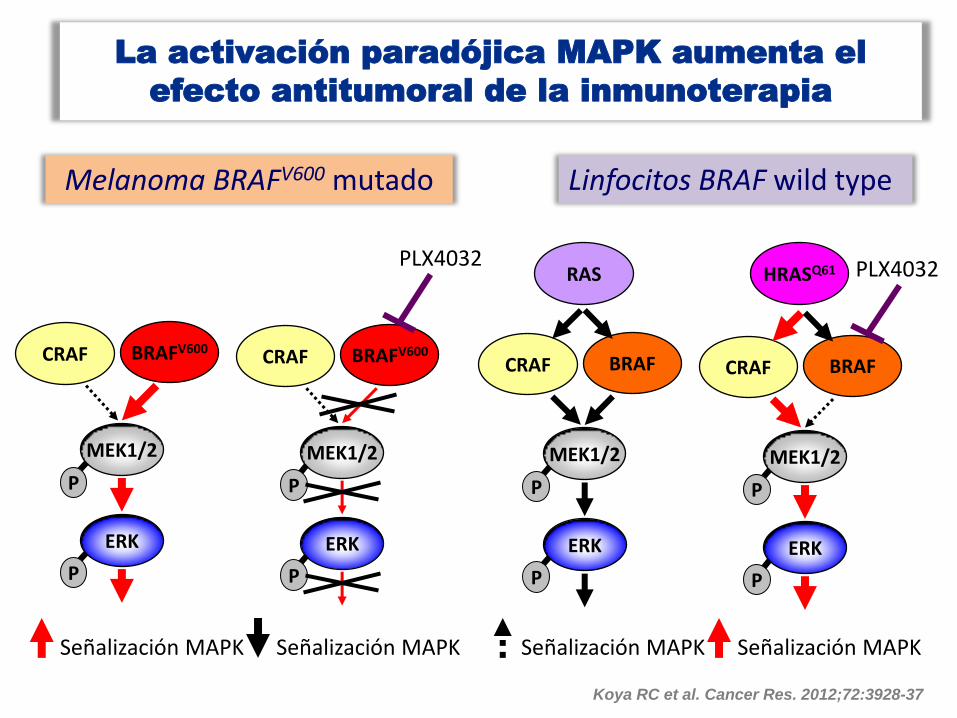
La activación paradójica MAPK aumenta el
efecto antitumoral de la inmunoterapia
CRAF
MEK1/2
ERK
P
P
BRAFV600
Melanoma BRAFV600 mutado Linfocitos BRAF wild type
Señalización MAPK
CRAF
MEK1/2
ERK
P
P
BRAFV600
PLX4032
Señalización MAPK
CRAF
MEK1/2
ERK
P
P
BRAF
Señalización MAPK
RAS
CRAF
MEK1/2
ERK
P
P
BRAF
PLX4032
Señalización MAPK
HRASQ61
Koya RC et al. Cancer Res. 2012;72:3928-37
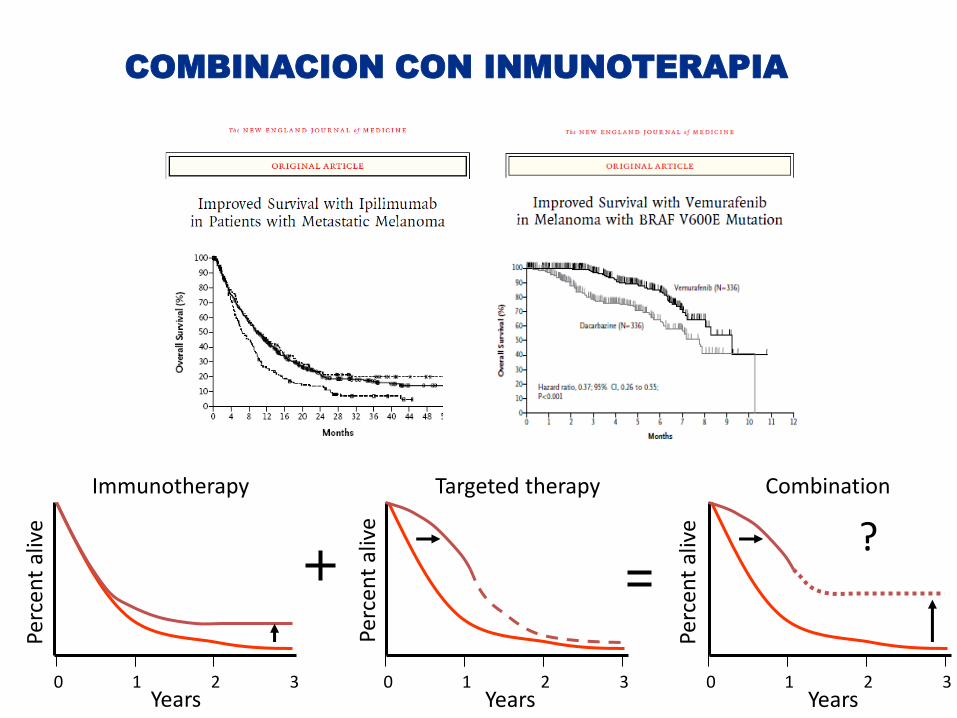
Years
Immunotherapy Targeted therapy
Perc
ent
aliv
e
Perc
ent
aliv
e
1 2 3 0 1 2 3 0 Years
Combination
Perc
ent
aliv
e
1 2 3 0 Years
?
COMBINACION CON INMUNOTERAPIA

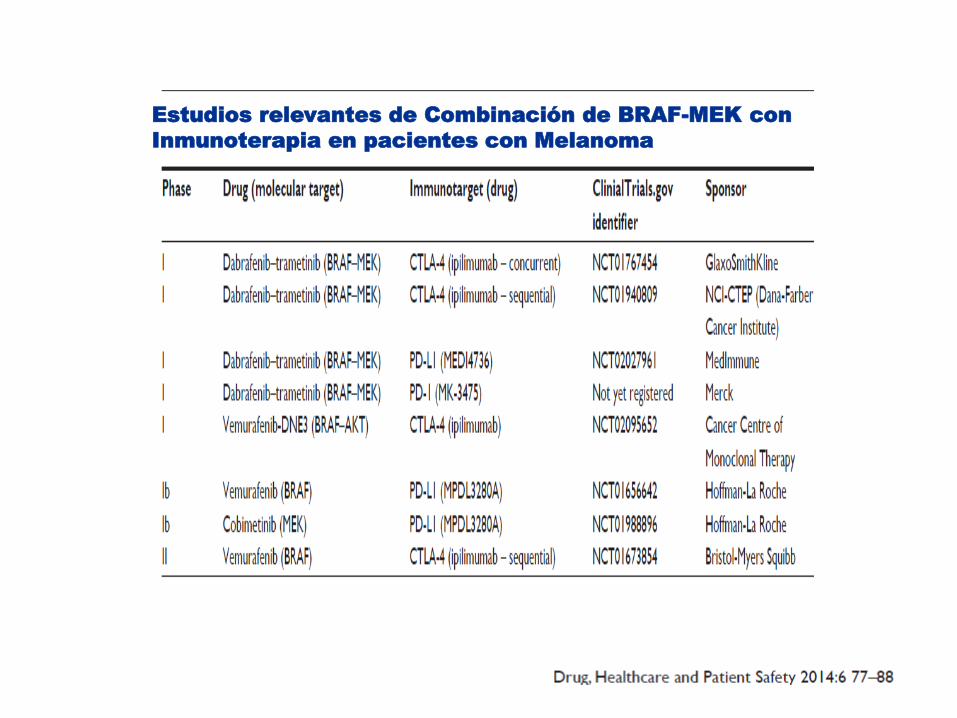
Estudios relevantes de Combinación de BRAF-MEK con
Inmunoterapia en pacientes con Melanoma

INHIBIDORES BRAF
METASTASIS CEREBRALES
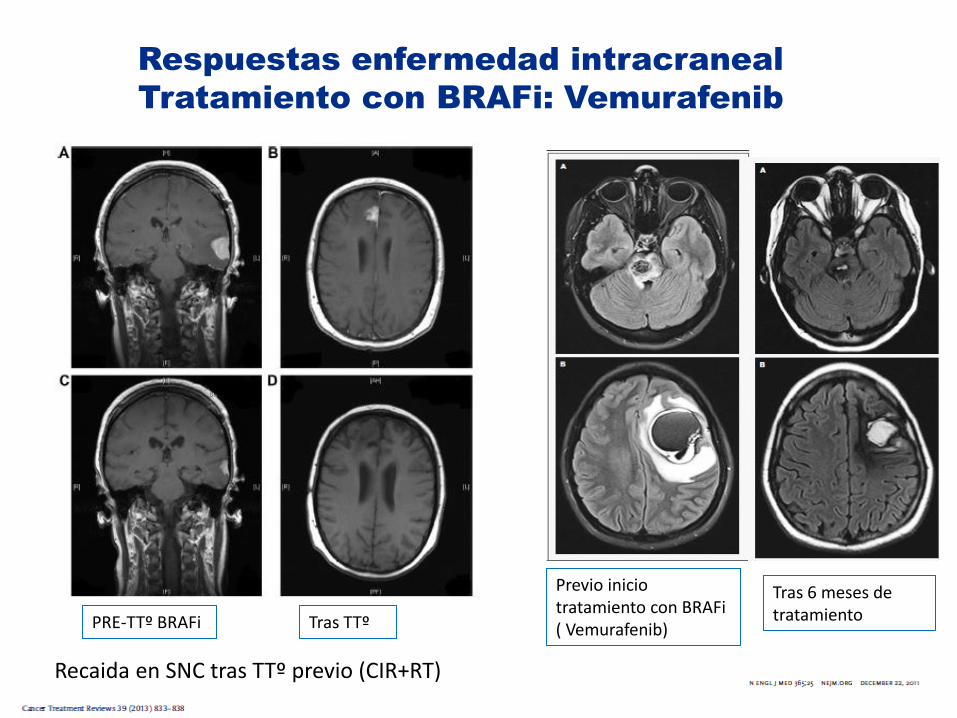
Respuestas enfermedad intracraneal
Tratamiento con BRAFi: Vemurafenib
Previo inicio tratamiento con BRAFi ( Vemurafenib)
Tras 6 meses de tratamiento
Recaida en SNC tras TTº previo (CIR+RT)
PRE-TTº BRAFi Tras TTº
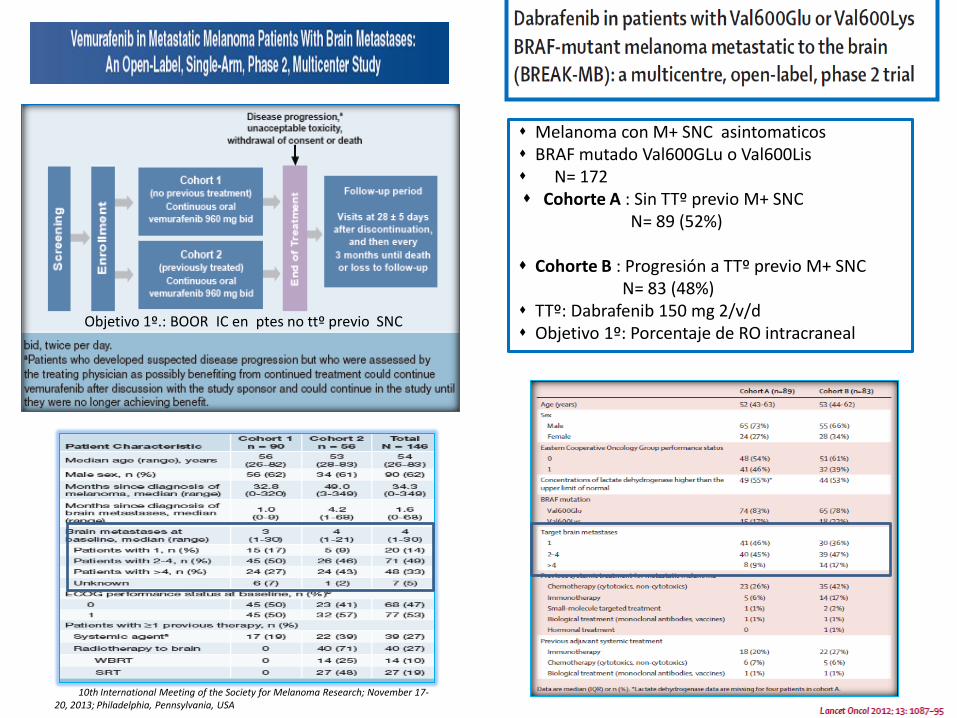
10th International Meeting of the Society for Melanoma Research; November 17-20, 2013; Philadelphia, Pennsylvania, USA
Melanoma con M+ SNC asintomaticos BRAF mutado Val600GLu o Val600Lis N= 172 Cohorte A : Sin TTº previo M+ SNC N= 89 (52%) Cohorte B : Progresión a TTº previo M+ SNC N= 83 (48%) TTº: Dabrafenib 150 mg 2/v/d Objetivo 1º: Porcentaje de RO intracraneal
Objetivo 1º.: BOOR IC en ptes no ttº previo SNC
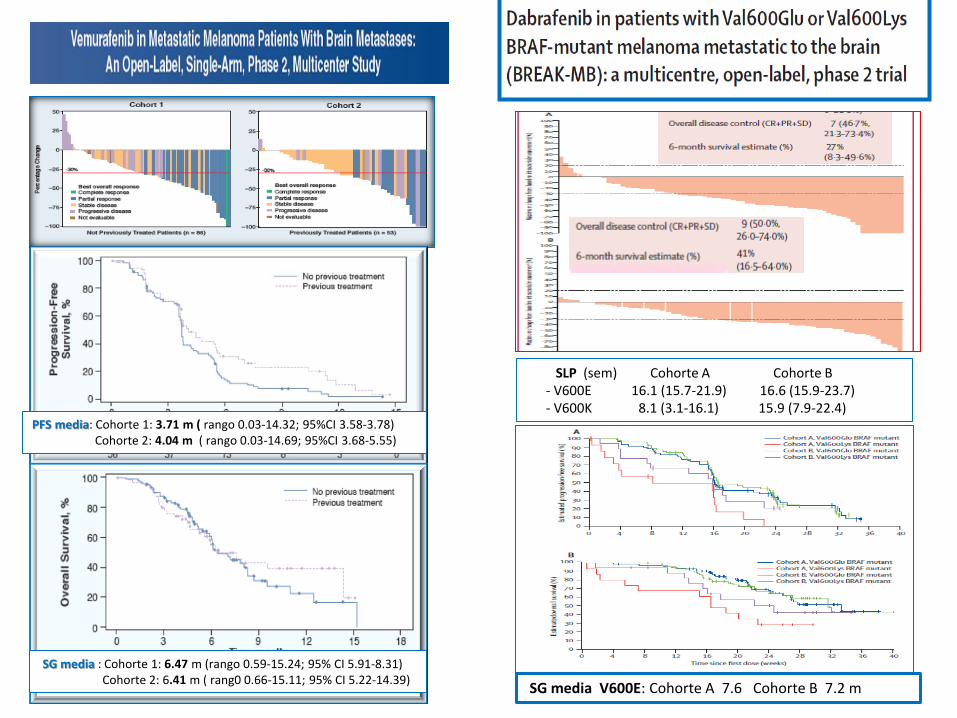
PFS media: Cohorte 1: 3.71 m ( rango 0.03-14.32; 95%CI 3.58-3.78) Cohorte 2: 4.04 m ( rango 0.03-14.69; 95%CI 3.68-5.55)
SG media : Cohorte 1: 6.47 m (rango 0.59-15.24; 95% CI 5.91-8.31) Cohorte 2: 6.41 m ( rang0 0.66-15.11; 95% CI 5.22-14.39)
SG media V600E: Cohorte A 7.6 Cohorte B 7.2 m
SLP (sem) Cohorte A Cohorte B - V600E 16.1 (15.7-21.9) 16.6 (15.9-23.7) - V600K 8.1 (3.1-16.1) 15.9 (7.9-22.4)

CONCLUSIONES
El bloqueo selectivo de la vía oncogénica BRAF-kinasa ha supuesto un gran
beneficio en el tratamiento de los melanomas con mutación con un elevado
porcentaje de respuestas y aumento en la supervivencia ( Fase III)
Perfil de toxicidad tolerable y fácilmente manejable
Actividad en la población de melanoma con metástasis cerebrales
Mecanismos de resistencia a los inhibidores, por las células tumorales incluyen
reactivación vía MAPK, y también por activación de la vía AKT/PI3K a través de
múltiples mutaciones secundarias
El doble bloqueo de BRAF y MEK previene y retrasa la resistencia al tratamiento con
BRAFi ,con menor toxicidad cutánea
Otras vÍas para revertir la resistencia adquirida:
- Asociación a fármacos inhibidores PI3K, mTOR.
- Asociación con inmunoterápicos
- Esquema de tratamiento
Son necesarios estudios para la búsqueda de biomarcadores que ayuden a una
mejor selección del tratamiento y su secuencia para obtener un mayor beneficio y
mas duradero











![Precision Medicine - 43. Deutscher Krankenhaustag€¦ · Progressionsfrei und Vemurafenib [Zelboraf ®] European Journal of Cancer 2017 Überleben in der metastasierten Situation](https://img.pdfslide.net/doc/110x75/606235c6e19a5303fe2895ff/precision-medicine-43-deutscher-progressionsfrei-und-vemurafenib-zelboraf-.jpg)


















