Embed Size (px)
Citation preview

EVALUATIONS ON EFFICACY AND IMMUNO-
STIMULATORY PROPERTIES OF TROPOMYOSIN
RECEPTOR KINASE C TARGETED PEPTIDOMIMETIC
LIGAND-DIIODO-BORON DIPYRROMETHENE HYBRIDS IN
PHOTODYNAMIC ANTICANCER THERAPY
KUE CHIN SIANG
FACULTY OF MEDICINE
UNIVERSITY OF MALAYA
KUALA LUMPUR
2016

EVALUATIONS ON EFFICACY AND IMMUNO-
STIMULATORY PROPERTIES OF TROPOMYOSIN
RECEPTOR KINASE C TARGETED PEPTIDOMIMETIC
LIGAND-DIIODO-BORON DIPYRROMETHENE
HYBRIDS IN PHOTODYNAMIC ANTICANCER
THERAPY
KUE CHIN SIANG
THESIS SUBMITTED IN FULFILMENT OF THE
REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY
FACULTY OF MEDICINE
UNIVERSITY OF MALAYA
KUALA LUMPUR
2016

UNIVERSITY OF MALAYA
ORIGINAL LITERARY WORK DECLARATION
Name of Candidate: KUE CHIN SIANG (I.C/Passport No: 850717-10-5661)
Registration/Matric No: MHA 130018
Name of Degree: DOCTOR OF PHILOSOPHY
Title of Project Paper/Research Report/Dissertation/Thesis (“this Work”):
EVALUATIONS ON EFFICACY AND IMMUNO-STIMULATORY PROPERTIES OF
TROPOMYOSIN RECEPTOR KINASE C TARGETED PEPTIDOMIMETIC LIGAND-
DIIODO-BORON DIPYRROMETHENE HYBRIDS IN PHOTODYNAMIC
ANTICANCER THERAPY
Field of Study: PHARMACOLOGY
I do solemnly and sincerely declare that:
(1) I am the sole author/writer of this Work;
(2) This Work is original;
(3) Any use of any work in which copyright exists was done by way of fair
dealing and for permitted purposes and any excerpt or extract from, or
reference to or reproduction of any copyright work has been disclosed
expressly and sufficiently and the title of the Work and its authorship have
been acknowledged in this Work;
(4) I do not have any actual knowledge nor do I ought reasonably to know that
the making of this work constitutes an infringement of any copyright work;
(5) I hereby assign all and every rights in the copyright to this Work to the
University of Malaya (“UM”), who henceforth shall be owner of the
copyright in this Work and that any reproduction or use in any form or by any
means whatsoever is prohibited without the written consent of UM having
been first had and obtained;
(6) I am fully aware that if in the course of making this Work I have infringed
any copyright whether intentionally or otherwise, I may be subject to legal
action or any other action as may be determined by UM.
Candidate’s Signature Date:
Subscribed and solemnly declared before,
Witness’s Signature Date:
Name:
Designation:

iii
ABSTRACT
Photodynamic therapy (PDT) utilises the administration of photosensitiser (PS) along
with focal light activation at specific wavelengths to generate singlet oxygen species to
kill tumour cells. Currently, most of the available photosensitisers have poor tumour
selectivity. This has led to poor therapeutic outcome. In order to improve the tumour
selectivity of the PS, a Tropomyosin receptor kinase C (TrkC) receptor based active
targeting ligand-PS complex was synthesised. TrkC is being targeted due to its
overexpression in cancer including breast, melanoma, pancreatic, neuroblastoma etc. In
this study, a synthetic isoleucine-tyrosine-isoleucine-tyrosine based TrkC receptor
ligand (IY-IY) was linked to a model photosensitiser diiodo-boron dipyrromethene I2-
BODIPY to form a TrkC targeting PS derivative IYIY-I2-BODIPY. Thereafter, tumour
targeting properties, in vivo antitumour efficacy and immune stimulatory properties of
conjugates in TrkC positive (4T1) and TrkC negative (67NR) breast cancer models in
pre and post-PDT scenarios were evaluated. Data showed that IYIY-I2-BODIPY, but
not a scrambled control (YIYI-I2-BODIPY) and free drug (I2-BODIPY) selectively
induced photocytotoxicity in a dose-dependent manner in 4T1 cells upon irradiation.
Bio-distribution studies in 4T1 mouse model showed that IYIY-I2-BODIPY
accumulated high in tumours at 1 h post intravenous administration and maintained high
up to 6 h, at a level which was approximately 2-fold higher compared to YIYI-I2-
BODIPY. Antitumour activity of IYIY-I2-BODIPY in 4T1 showed 96% reduction in
tumour volume at day-6 post PDT at 10 mg/kg. Moreover, 71% of IYIY-I2-BODIPY
treated mice were “healed” from aggressive breast cancer for up to 90 days post-PDT
with no evidence of metastasis, indicating complete remission. This observation was
neither found in YIYI-I2-BODIPY nor I2-BODIPY treated mice. The selectivity of
IYIY-I2-BODIPY was further confirmed when 67NR breast tumours in mice showed
only slight tumour reduction followed by tumour re-growth upon PDT using all three

iv
compounds. At a similar therapeutic dosage, IYIY-I2-BODIPY (strong response) and
YIYI-I2-BODIPY (weak response), but not I2-BODIPY, in non-irradiation condition
selectively increases the pro-inflammatory cytokines IL-2, IL-6, IL-17 and suppresses
immunosuppressive cytokines TGF-β at 2 h post administration. Moreover, the
conjugates increase both CD4 and CD8 T-lymphocyte populations with phenotype of
IFN-γ (Th1, CTL) and IL-17 (Th17, Tc17), and decreases immunosuppressive
granulocytic myeloid-derived suppressor cells (G-MDSC) and regulatory T cells, which
were known to be highly increased in cancer. Only IYIY-I2-BODIPY induced tumor
growth delay (~20% smaller size) in mice when administrated daily for 5 days. When
illuminated with light to produce effects associated with PDT, IYIY-I2-BODIPY
induced even stronger immune responses. In addition, IYIY-I2-BODIPY and light
treated mice had higher levels of immune effector T-cells compared to photoirradiated
YIYI-I2-BODIPY and I2-BODIPY controls. Adoptive transfer of splenocytes and
lymphocytes from IYIY-I2-BODIPY treated survivor mice that were photoirradiated
gave significantly delayed tumour growth in recipient mice. Our data provide evidences
that TrkC ligand conjugate, alone and in combination with PDT modulates immune
responses that are conducive to suppressing tumour growth. Thus this conjugate can act
as an immune-stimulatory PDT agent with potential applications in cancer treatment.

v
ABSTRAK
Terapi fotodinamik (photodynamic therapy, PDT) menggunakan molekul fotosensitif
dengan sinaran cahaya yang mempunyai gelombang spesifik untuk menjana oksigen
molekul singlet reaktif yang berupaya memusnahkan sel-sel tumor dan meningkatkan
sistem imunisasi pada masa yang sama. Kebanyakan molekul fotosensitif yang
digunakan di tahap klinikal mempunyai kelemahan dalam selektiviti tumor,
mengakibatkan hasil terapi yang tidak memuaskan. Oleh yang demikian, penyasaran
reseptor TrkC dengan menggunakan ligan bermolekular kecil (IYIY) yang berkonjugasi
dengan diiodo-boron dipyrromethene (IYIY-I2-BODIPY) telah dihasilkan. Kelampauan
expresasi TrkC dalam kebanyakan kanser, termasuk payudara, melanoma, pankreas,
neuroblastoma dan lain-lain jenis kanser menjadikannya sebagai sasaran dalam
pengajian ini. Konjugasi IYIY-I2-BODIPY dalam pemilihan tambatan dan
fotositotoksisiti terhadap TrkC in vitro telah pun dijalankan. Dalam pengajian ini,
penilaian IYIY-I2-BODIPY dalam keberkesanan antitumor dan kebolehannya sebagai
pendorong imunisasi di kanser payudara tikus TrkC positif (4T1) dan TrkC negatif
(67NR) sebelum dan selepas PDT. IYIY-I2-BODIPY secara terpilih telah meningkatkan
fotositotoksisiti di sel 4T1 semasa proses penyinaran. Pengajian bio-distribusi pada
tikus membuktikan IYIY-I2-BODIPY banyak terkumpul di 4T1-tumor secepat 1 jam
dan kuantitinya tetap sehingga 6 jam selepas suntikan intravena, lebih kurang 2 kali
ganda dibandingkan dengan YIYI-I2-BODIPY. Aktiviti antitumor IYIY-I2-BODIPY
dalam 4T1 menunjukkan pengurangan saiz tumor sebanyak 96% pada hari ke-6 selepas
PDT (10 mg/kg). Tambahan lagi, 71% daripada tikus yang dirawati dengan IYIY-I2-
BODIPY “sembuh” daripada kanser selepas 90 hari proses PDT tanpa sebarang tanda-
tanda metastasis, menujukkan penyembuhan yang sempurna. Pemantauan ini tidak
didapati samada di tikus yang dirawati dengan YIYI-I2-BODIPY atau I2-BODIPY.
Kemampuan pemilihan IYIY-I2-BODIPY terhadap TrkC dibuktikan dengan selanjutnya
apabila pengurangan saiz yang sikit didapati di TrkC negatif tumor payudara 67NR

vi
selepas PDT, diikuti dengan penumbuhan semula tumor, bersamaan dengan YIYI-I2-
BODIPY. Pada dos terapi yang sama, IYIY-I2-BODIPY (kuat) dan YIYI-I2-BODIPY
(lemah) tapi bukan I2-BODIPY, meningkatkan sitokin pro-inflamatori IL-2, IL-6, IL-17
dan mengehadkan sitokin penekan-imunisasi TGF-β selepas 2 jam kompaun inokulasi
dalam keadaan tanpa penyinaran. Kedua-dua konjugasi ini juga meningkatkan
kumpulan CD4+ dan CD8+ T-sel dengan fenotip IFN-γ (Th1, CTL) and IL-17 (Th17,
Tc17), dan mengurangkan populasi immunosuppressive granulocytic myeloid-derived
suppressor cells (G-MDSC) dan regulatory T-cells yang biasanya akan meningkat
dalam kanser. Tambahan, tikus yang dirawat dengan IYIY-I2-BODIPY menyumbang
kepada penglewatan penumbuhan tumor apabila inokulasi setiap hari sebanyak 5 hari.
Rawatan IYIY-I2-BODIPY bersama dengan PDT meningkatkan reaksi imunisasi yang
lebih kuat berbanding dengan rawatan konjugasi bersendirian. Tambahan lagi, tikus
yang dirawat dengan IYIY-I2-BODIPY dan PDT mempunyai memori T-sel yang tinggi
berbanding dengan kontrol yang difotoradiasi. Pemindahan adoptif sel imunisasi dari
tikus yang sembuh daripada rawatan photoradiasi IYIY-I2-BODIPY kepada tikus
penerima menunjukkan kesan yang ketara dalam pengurangan saiz tumor. Data
penyelidikan kami membuktikan bahawa IYIY-I2-BODIPY bersendiriannya atau
bersamaan dengan PDT memyumbangkan kepada peningkatan imunisasi yang dapat
mengehadkan penumbuhan tumor. Oleh yang demikian, konjugasi ini dapat bertindak
sebagai agen PDT perangsang-imunisasi yang berpotensi dalam aplikasi rawatan kanser.

vii
ACKNOWLEDGEMENTS
I would like to take this opportunity to extend my sincere gratitude to individuals that
have made this thesis possible. Without them, the completion of my studies is
impossible. My foremost appreciation is to my PhD supervisors Prof. Dr. Chung Lip
Yong and Dr. Kiew Lik Voon for being supportive of my PhD research. Their patience,
guidance, motivation, insightful ideas and knowledge have led me to become a
successful PhD candidate. Beside my advisor, I would like to thank to Prof. Dr. Nor
Azizan Abdullah, Heads of Departments of Pharmacology, and Cancer Research
Malaysia (formerly known as Cancer Research Initiative Foundation, CARIF) for
providing the well-equipped laboratory spaces and facilities for me to conduct the
research. I also appreciate our collaborator Prof. Dr. Kevin Burgess and their team,
especially Anyanee Kamkaew at Department of Chemistry, Texas A&M University, US,
for their providing us the compound and synthetic conjugates.
I am eternally grateful to Dr. Lee Hong Boon, who is the consultant of Photodynamic
therapy (PDT) aspect of my PhD study. Dr. Lee was the Leader of Drug Discovery
Group in CARIF then. She introduced and guided me on cancer photosensitisers and
PDT, and gradually raised my interest on PDT as an alternative cancer therapy approach.
Thank for her insightful comments, encouragement, and strong guidance force which
had trained me to think from different perspectives for my research direction. My
appreciation is also extended to Dr. Lim Siang Hui, who guided me on PDT
experimental protocols. Special thanks to Ms. Voon Siew Hui for sharing knowledge,
stimulating discussion and as a good companion in the laboratory. Also, thanks to
colleagues Ms. Kiew Siaw Fui, Ms. Ng Shie Yin, Ms. Cheah Hoey Yan, Mr. Saw Wen
Shang and Ms. Chong Yee Ying for their continuous support, for being helpful and for
good companionship, as well as make my laboratory life became more colorful.

viii
I would like to give special thanks to my wife Ms. Peh Bee Ching for always being
there for me. Through her love, faith and companionship, I have been able to finish my
PhD on time. Thanks to my family members for their love and support on everything
that I do.
Last but not least, I would like to express my deepest appreciation to the Bright
Spark Unit for scholarship support throughout my study. My extend appreciation to the
Tien Te Lee Biomedical Foundation for awarding me the Excellent Scientific Paper
Award 2015. This is one of the motivations for me to do a good science in future.

ix
TABLE OF CONTENTS
Abstract ............................................................................................................................ iii
Abstrak .............................................................................................................................. v
Acknowledgements ......................................................................................................... vii
Table of Contents ............................................................................................................. ix
List of Figures ................................................................................................................ xiv
List of Tables.................................................................................................................. xvi
List of Symbols and Abbreviations ............................................................................... xvii
List of Appendices ......................................................................................................... xix
CHAPTER 1: INTRODUCTION .................................................................................. 1
1.1 Overview.................................................................................................................. 1
1.2 Aim and Objectives ........................................................................................ 4
CHAPTER 2: LITERATURE REVIEWS ................................................................... 5
2.1 Active Targeting in Cancer ...................................................................................... 5
2.1.1 Rationale of Active Targeting .................................................................... 7
2.1.2 Advantages of Small Molecule in Active Targeting..................................9
2.1.3 Parameters Determining the Efficacy of Active Targeting Ligand-Drug
Conjugate ............................................................................................... .. 10
2.1.4 Small Molecule Conjugates for Active Targeting in Cancer ...................12
2.1.5 Other Impacts of Active Targeting ........................................................... 25
2.1.5.1 Immune-modulation .................................................................. 25
2.1.5.2 Non-targeted Toxicity ............................................................... 26
2.2 Tropomyosin Receptor Kinase (Trk) ..................................................................... 27
2.2.1 Receptor Biochemistry ............................................................................. 27

x
2.2.2 Trk Receptors and Tumourigenesis..........................................................28
2.2.3 Trk Receptors, Neurotrophins and Immune System ................................ 29
2.3 Photodynamic Therapy (PDT) ............................................................................... 30
2.3.1 Background .............................................................................................. 30
2.3.2 Components of PDT.................................................................................31
2.3.3 Mechanisms of PDT ................................................................................. 33
2.3.4 Biological Target of PDT ......................................................................... 34
2.3.4.1 Direct Cytotoxicity .................................................................... 35
2.3.4.2 Vascular Effect .......................................................................... 37
2.3.4.3 Immune Responses .................................................................... 37
2.4 Cancer Immunology .............................................................................................. 39
2.4.1 Tumour Antigen in Trigerring Immune System ....................................... 39
2.4.2 Cancer Immunoediting ............................................................................. 40
2.4.2.1 Tumour Elimination .................................................................. 41
2.4.2.2 Tumour Equilibrium .................................................................. 43
2.4.2.3 Tumour Escape .......................................................................... 44
CHAPTER 3: TROPOMYOSIN RECEPTOR KINASE-C (TRKC) LIGAND
CONJUGATED DIIODO-BORON DIPYRROMETHENE ERADICATES TRKC
EXPRESSING TUMOUR IN PHOTODYNAMIC THERAPY (PDT) .................. 47
3.1 Introduction............................................................................................................ 47
3.2 Materials and Methods .......................................................................................... 51
3.2.1 Compounds and Cell Lines ...................................................................... 51
3.2.2 In vitro Photocytotoxicity Assay .............................................................. 51
3.2.3 Animal Model. .......................................................................................... 52
3.2.4 In vivo Toxicity Study .............................................................................. 52

xi
3.2.5 Biodistribution Study.. ............................................................................ 52
3.2.6 Tumour Cells Inoculation and PDT in Mouse ......................................... 53
3.2.7 Histology Sample Preparation.... ............................................................. 54
3.2.8 Statistical Analysis.............. .................................................................... 54
3.3 Results........ ........................................................................................................... 54
3.3.1 In vitro photocytotoxicity of ligated conjugates and unconjugated I2-
BODIPY ................................................................................................... 54
3.3.2 In vitro photocytotoxicity of ligated conjugates at different incubation
time in TrkC positive and TrkC negative cancer cells ............................. 58
3.3.3 Toxicity profile of I2-BODIPY and ligated conjugates in murine model.. 60
3.3.4 In vivo biodistribution study in 4T1 tumour bearing mouse .................... 61
3.3.5 In vivo PDT antitumour efficacy of IYIY-I2-BODIPY in TrkC positive
4T1 tumour bearing mouse ....................................................................... 64
3.3.6 In vivo PDT antitumour efficacy of IYIY-I2-BODIPY in TrkC negative
67NR tumour bearing mouse.................................................................... 67
3.3.7 Histopathological analysis of IYIY-I2-BODIPY treated survivor mice at
90 days post-PDT ..................................................................................... 68
3.4 Discussion........ ...................................................................................................... 69
3.5 Conclusion........ ..................................................................................................... 71
CHAPTER 4: IMMUNE RESPONSES INDUCTION AND ANTITUMOUR
ACTIVITY OF TROPOMYOSIN RECEPTOR KINASE-C (TRKC) RECEPTOR
TARGETED PHOTODYNAMIC THERAPY(PDT) CONJUGATES ................... 73
4.1 Introduction............................................................................................................ 73
4.2 Materials and Methods .......................................................................................... 76
4.2.1 Compounds...............................................................................................76

xii
4.2.2 Animal model ........................................................................................... 76
4.2.3 Tumour model development.. ................................................................... 76
4.2.4 Compounds administration ....................................................................... 77
4.2.5 Blood sampling.... ..................................................................................... 77
4.2.6 Flow cytometry quantification of plasma cytokines .............................. 77
4.2.7 Cell isolation from lymphoid organs and tumour tissues........................ .78
4.2.8 Staining and flow cytometry quantification of immune cells..................78
4.2.9 In vivo TrkC blocking studies . .............................................................. 79
4.2.10 Photodynamic therapy (PDT) in mice ..................................................... 80
4.2.11 Adoptive transfer for antitumour immunity... ......................................... 80
4.2.12 Statistical analysis....................................................................................81
4.3 Results........ ........................................................................................................... 81
4.3.1 Th1/Th2/Th17/Treg cytokines level in blood plasma of TrkC positive 4T1
tumour bearing mice post compounds administration. ............................ 81
4.3.2 Myeloid cell subtypes quantification in compounds treated mice ........... 85
4.3.3 Inflammatory cell (neutrophils) population quantification in compounds
treated mice.. ........................................................................................... 87
4.3.4 CD4+ T-helper cell subtypes (Th1/Th2/Th17/Treg) quantification in
compounds treated mice .......................................................................... .89
4.3.5 CD8+ T-cells quantification in compounds treated mice ......................... 92
4.3.6 Confirmation of immunomodulatory effects by using IYIY-TEG (ligand
without photosensitiser) .......................................................................... 93
4.3.7 IYIY-I2-BODIPY mediated immune modulations upon TrkC receptor
blocking. .................................................................................................. 94
4.3.8 Effect of conjugates administration in the dark on TrkC positive 4T1
tumour growth .......................................................................................... 96

xiii
4.3.9 Combination effect of conjugates and PDT in cytokine productions ...... 97
4.3.10 Effect of IYIY-I2-BODIPY and irradiation on neutrophil populations .. 101
4.3.11 Combination effect of conjugates and PDT in adaptive immune
responses…………………………………. ........................................... 102
4.3.12 Effector T cells quantification in compounds treated mice at 20 days post-
PDT ........................................................................................................ 105
4.3.13 Antitumour immunity in IYIY-I2-BODIPY treated survivor mice post-
PDT ........................................................................................................ 106
4.4 Discussion........ .................................................................................................... 108
4.5 Conclusion........ ................................................................................................... 113
CHAPTER 5: CONCLUSION AND FUTURE PERSPECTIVES ....................... 114
5.1 Overall Conclusion .............................................................................................. 114
5.2 Future Perspectives .............................................................................................. 116
References.....................................................................................................................119
List of Publications and Papers Presented.....................................................................149
Appendices....................................................................................................................155

xiv
LIST OF FIGURES
Figure 2.1: Passive and active targeting for cancer therapeutic ................................... 6
Figure 2.2: Common classes of ligands and cargoes used for active targeting in
cancer…………………………………………………………………...7
Figure 2.3: Conjugate uptake and receptor trafficking pathway…………………….10
Figure 2.4: Other targeting of ligand conjugates ....................................................... .26
Figure 2.5: Schematic picture of tropomyosin receptor kinase ................................. .28
Figure 2.6: Treatment procedures for cancer PDT .................................................... .32
Figure 2.7: Schematic illustration of mechanism involved in PS activation and PDT
................................................................................................................ .34
Figure 2.8: Three mechanism of PDT mediated tumour destruction. ........................ 35
Figure 2.9: Three processes (Elimination, Equilibrium and Escape) of immuno-
editing in cancer ...................................................................................... 40
Figure 3.1: Structure of synthetic peptide Isoleucine-tYrosine (IY) to bind TrkC
receptor .................................................................................................... 48
Figure 3.2: Structure of photosensitiser diiodinated BODIPY used in this study ...... 49
Figure 3.3: Structure of synthetic TrkC targeted conjugate (IYIY-I2-BODIPY) and
non-TrkC targeted conjugate as scrambled control (YIYI-I2-BODIPY). 50
Figure 3.4: Tested compounds were not toxic to cells in non-irradiated condition ... 55
Figure 3.5: IYIY-I2-BODIPY was photocytotoxic only to TrkC positive cell lines .. 57
Figure 3.6: Compounds induced non-selective photocytotoxicity in TrkC positive
and TrkC negative cells when treated for prolonged periods .................. 59
Figure 3.7: IYIY-I2-BODIPY was not toxic to mice at 20 mg/kg ............................. 60
Figure 3.8: IYIY-I2-BODIPY has high accumulation in tumour tissue at 1 hour and
cleared from the body at 72 hours post-administration ........................... 63
Figure 3.9: IYIY-I2-BODIPY effectively suppressed the growth of TrkC positive 4T1
tumour...................................................................................................... 65
Figure 3.10: IYIY-I2-BODIPY and YIYI-I2-BODIPY has equal antitumour efficacy
on TrkC negative 67NR tumour ............................................................ 67

xv
Figure 3.11: IYIY-I2-BODIPY treated survivor mice have no metastasis sign……..68
Figure 4.1: IYIY-I2-BODIPY conjugate increases IL-2, IL-6, IL-17 and decreases
TGF-β cytokine levels ............................................................................. 84
Figure 4.2: Lymphoid organs and tumour microenvironment has lowered populations
of G-MDSC in IYIY-I2-BODIPY treated mice. ...................................... 86
Figure 4.3: IYIY-I2-BODIPY treated mice have lowered neutrophils population .... 88
Figure 4.4: IYIY-I2-BODIPY increases CD4+ T-lymphocytes expressing IFN-γ and
IL-17, and decreases regulatory T-cells .................................................. 91
Figure 4.5: IYIY-I2-BODIPY increases CD8+ T-lymphocytes expressing IFN-γ and
IL-17 ........................................................................................................ 92
Figure 4.6: Control TrkC ligand (IYIY-TEG) has similar immunomodulatory
activities as IYIY-I2-BODIPY ................................................................. 94
Figure 4.7: IYIY-I2-BODIPY mediated myeloid cells reduction is TrkC dependent.95
Figure 4.8: Multiple boli i.v. administration of IYIY-I2-BODIPY transiently delays
tumour growth ......................................................................................... 97
Figure 4.9: IYIY-I2-BODIPY conjugate increases IL-6, IL-17 and decreases TGF-β
cytokine levels post-PDT ...................................................................... 100
Figure 4.10: IYIY-I2-BODIPY enhances neutrophils population post-PDT ........... 102
Figure 4.11: IYIY-I2-BODIPY combined with PDT induces stronger adaptive
immunity ............................................................................................. 104
Figure 4.12: CD4+ and CD8
+ effector T-cells in TDLN and spleen at 20 days post-
PDT…………………………………………………………………..106
Figure 4.13: Adoptive transfer of immune cells in IYIY-I2-BODIPY treated survivor
mice delays tumour growth ................................................................ .108
Figure 4.14: Summary findings of TrkC targeted PDT agent on immune responses
............................................................................................................. 113
Figure 5.1: Summary findings of TrkC targeted peptidomimetic ligand-diiodo-boron
dipyrromethene hybrid in TrkC expressing breast cancer...................116

xvi
LIST OF TABLES
Table 2.1 Small Molecule Conjugates for Cancer Therapeutics in the Clinic and in
Preclinical Animal Models......................................................................13
Table 2.2 Small Molecule Conjugates for Cancer Imaging in the Clinic and in
Preclinical Animal Models......................................................................20

xvii
LIST OF SYMBOLS AND ABBREVIATIONS
α
: alpha
β
: beta
γ
µM
J
: gamma
: microMolar
: Joule
AR
: androgen receptor
APC
BDNF
: antigen presenting cell
: brain-derived neurotrophin factor
BODIPY
: boron dipyrromethene
CaIX
: carbonic anhydrase 9
CCK2R
: cholecystokinin 2 receptor
CD
: cluster of differentiation
CTL
: cytotoxic T lymphocyte
DLI
: drug-light interval
ER
: estrogen receptor
FR
: folate receptor
GLUT
: glucose transport system
IC50
IDO
: half maximum inhibitory concentration
: Indolamine-2,3-dioxygenase
IFN
: interferon
IL
: interleukin
i.v : intravenous

xviii
IYIY
: Isoleucin-tYrosine-Isoleucin-tYrosine
MDSC
: myeloid derived suppressor cell
MHC
MTT
NGF
nm
: major histocompatibility complex
: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
: neurotrophin growth factor
: nanometer
NT-3
PBS
: neurotrophin-3
: phosphate buffered saline
PDT
: photodynamic therapy
PgR
: progesterone receptor
PMA
PS
: Phorbol 12-Myristate 13-Acetate
: photosensitizer
PSMA
ROS
: prostate-specific membrane antigen
: reactive oxygen species
SOG
: singlet oxygen generation
TGF
: transforming growth factor
Th
: T helper
TM
: tumour microenvironment
TNF
: tumour necrosis factor
Treg
: regulatory T
Trk
: tropomyosin receptor kinase
YIYI : tYrosine-Isoleucin-tYrosine-Isoleucin

xix
LIST OF APPENDICES
Appendix A: Synthesis of I2-BODIPY Derivatives……………………………….…..155
Appendix B: Characterisation of IYIY-I2-BODIPY and YIYI-I2-BODIPY………….161
Appendix C: Animal Ethic Approval Letter (Study-1)……………………………....165
Appendix D: Animal Ethic Approval Letter (Study-2) ………………………….…...166
Appendix E: Excellent Scientific Paper Awards 2015 by Tien Te Lee Biomedical
Foundation………………………………………………………….….167

1
CHAPTER 1: INTRODUCTION
1.1 Overview
Conventional cancer therapies such as radiation, chemotherapy and most of the
clinical approved anticancer drugs have shown promise in treating certain cancers.
However, these anticancer therapies often lack killing selectivity on tumour cells, and
have long been associated with immune response silencing in cancer patients (Zitvogel
et al., 2008). An ideal goal of anticancer therapy is to selectively destroyed the tumour
cells while allowing normal cells to remain unharmed, and at the same time to stimulate
anti-tumour immune response in the host immune.
Photodynamic therapy (PDT) is considered as an effective alternative treatment
option for superficial cancer. The drug used in PDT is called photosensitiser (PS),
which is non-toxic to cells unless activated by the light at specific wavelengths. Upon
activation, PS will generate the cytotoxic singlet oxygen species in presence of oxygen
molecules, and the generated singlet oxygen will have direct killing activity on tumour
cells, disrupt tumour vasculature and elevates systemic immune response. It is less toxic
to normal tissues upon activation because only the local tumour lesion is irradiated to
activate the administered photosensitiser, and stimulates tumour antigen specific
immune responses (Brackett & Gollnick, 2011; Castano et al., 2006). PDT has become
one of the clinical treatment options for early stages of certain cancer types such as
nasopharyngeal, gastroenterological, brain and gynecological cancer (Dolmans et al.,
2003; Huang, 2005). Other than PDT, targeted delivery of therapeutic agent (active or
passive targeted cancer therapy) is another anticancer therapy approach to increase drug
accumulation in tumour sites (Srinivasarao et al., 2015; Torchilin, 2010). In active

2
targeted therapy, the designed conjugate (composed of targeting ligand linked to
cytotoxic agent) targets the cell surface molecules that are overexpressed in cancers
(generally survival or metastasis biomarkers including folate receptor, estrogen receptor,
prostate specific membrane antigen, sigma-2 receptor, tropomyosin receptor kinase,
etc.), with minimum binding to normal cells. The conjugate will then be internalised by
receptor mediated internalisation, which in turn causes the drug to be released
intracellularly for its cytotoxic action once it is degraded by lysosomes.
Among the cell surface molecules overexpressed in cancer cells, Tropomyosin
receptor kinase (Trk) is selected in this study due to its limited treatment option
(chemotherapy using Trk inhibitors). Trk consists of three common receptor tyrosine
kinases, TrkA-C (Segal, 2003). Trk receptor and their natural ligands (neurotrophins)
interaction has also been reported not only to be associated with cancer progression
(Nakagawara, 2001), but also can modulate immune responses. Among the Trk
receptors, TrkC is highly correlated with cancer growth of different types.
Immunohistochemistry studies showed that TrkC is a useful biomarkers for prognosis of
tumour progression and invasion (Vaishnavi et al., 2015) in neuroblastoma (Brodeur et
al., 1997; Yamashiro et al., 1997), glioblastoma (Kumar & de Vellis, 1996; Wang et al.,
1998), thyroid cancer (McGregor et al., 1999), melanoma (Xu et al., 2003) and breast
cancer (Blasco-Gutierrez et al., 2007; Jin et al., 2010). In immunology, neurotrophins
were reported to modulate cytokines secretion such as increasing interleukin (IL)-6 and
IL-4, and impairing transforming growth factor-β signaling (Jin et al., 2007). In immune
cells, neurotrophins and Trk were known to regulate the balancing of CD4+ T-
lymphocytes subtypes by promoting growth of Th2 but not Th1, due to the relatively
high TrkC expression in Th2 compared to Th1 (Rezaee et al., 2010; Sekimoto et al.,
2003; Vega et al., 2003).

3
A synthetic peptidomimetic ligand which is selective to TrkC receptor has been
designed (Kamkaew & Burgess, 2013). The preliminary study has shown that the
synthetic TrkC receptor targeted peptidomimetic ligand (Isoleucine - tYrosine -
Isoleucine - tYrosine, IYIY) selectively binds to TrkC receptor in TrkC transfected
NIH-3T3 cells. Moreover, when comparing the effect of IYIY and neurotrophin-3
(TrkC natural ligand), both were able to induce signaling cascades to promote neuronal
cell growth and differentiation (Brahimi et al., 2014; Chen et al., 2009). Based on the
interesting preliminary findings, a TrkC targeted peptidomimetic ligand-photosensitiser
conjugate, which is IYIY-diiodo-boron dipyrromethene (IYIY-I2-BODIPY) has been
constructed (Kamkaew & Burgess, 2013). The ligated conjugate was hypothesised to
have a better targeting in TrkC+ cancer than the unconjugated free photosensitiser
diiodo-boron dipyrromethene (I2-BODIPY). On top of that, systemic immune responses
might also be affected upon the administration of the IYIY-I2-BODIPY conjugate. In
this study, the efficacies of IYIY-I2-BODIPY conjugate including in vitro binding
selectivity, biodistribution, antitumour activity as well as immunological impacts in
TrkC expressing (4T1) and non-TrkC expressing (67NR) murine breast cancer models,
under non-irradiated (without PDT) and irradiated (PDT) conditions will be carried out.
The breast cancer model is used in this study due to its distinct TrkC expression among
metastatic and non-metastatic cell lines. Moreover, tumour can be induced
orthotopically at the mammary gland, and hence it can be monitored easily, especially
during photodynamic therapy.
The above study is important because this is the first compound designed to target
TrkC tumour in active targeting, and may expand the understanding on the systemic
impact of active targeted ligand-drug conjugate in high TrkC expressing cancer.

4
1.2 Aim and Objectives
The aim of this study is to investigate the binding selectivity, biodistribution,
antitumour efficacy and systemic immune responses of TrkC receptor targeted
conjugate (Isoleucine - tYrosine - Isoleucine - tYrosine, IYIY-I2-BODIPY) in
comparison to the non-TrkC receptor targeted scrambled control tYrosine – Isoleucine –
tYrosine - Isoleucine, YIYI-I2-BODIPY (reversion of amino acid arrangement) and the
free drug I2-BODIPY in TrkC expressing (4T1) and non-TrkC expressing (67NR)
murine breast tumour model under non-irradiated and irradiated condition.
The specific objectives of this study are as follows:
i. To compare the in vitro photocytotoxicity and targeting selectivity of IYIY-I2-
BODIPY, YIYI-I2-BODIPY and I2-BODIPY in 4T1 and 67NR cell lines at
different treatment time points.
ii. To determine the in vivo toxicity profile (maximal tolerated dose, MTD) of
IYIY-I2-BODIPY, YIYI-I2-BODIPY and I2-BODIPY in healthy BALB/c mice.
iii. To compare the biodistribution pattern and antitumour efficacy of IYIY-I2-
BODIPY and YIYI-I2-BODIPY in 4T1 tumour bearing BALB/c mice.
iv. To quantify the cytokines secretion from T helper cells in IYIY-I2-BODIPY,
YIYI-I2-BODIPY and I2-BODIPY in 4T1 tumour bearing BALB/c mice under
non-irradiated and irradiated condition.
v. To characterise the immune cell populations, including myeloid cells (innate
immune responses) and T-lymphocytes (adaptive immune responses) in 4T1
tumour bearing BALB/c mice under non-irradiated and irradiated condition.
vi. To study the populations of effector T-cells and long-term immunity in IYIY-I2-
BODIPY, YIYI-I2-BODIPY and I2-BODIPY treated 4T1 tumour bearing
BALB/c mice post photodynamic therapy.

5
CHAPTER 2: LITERATURE REVIEWS
2.1 Active Targeting in Cancer
Chemotherapy of cancer destroys tumour tissues, or at least restricts its growth and
metastatic spread. In general, chemotherapy drugs are either small molecule inhibitors
with low molecular weight that interfere the up-regulated biochemical pathway in
cancer cells or monoclonal antibodies that bind to cellular surface proteins (mechanistic
or direct targeted therapy). These therapeutic agents tend to diffuse into all tumour and
healthy tissues with low selectivity. In fact in drug discovery, many potent cytotoxic
compounds fail as medicines due to their poor tissues selectivity and induced intolerable
side-effects. Targeted delivery of therapeutic agent can overcomes the limitations of
conventional cancer chemotherapy due to their (i) ability to identify the location of the
primary and metastasis tumour, and (ii) selectively accumulate and kill the cancer cells
than normal cells.
There are generally two types of drug targeting to deliver therapeutic cargoes to
tumour sites (Torchilin, 2010). Passive targeting uses nano-carriers to ferry the drugs to
tumour tissues via selective extravasation from the leaky tumour vasculature, an effect
called enhanced permeability and retention. Active targeting links targeted moieties
such as ligands and monoclonal antibodies (targeting agent) with cargoes (cytotoxic
agent or imaging probes) to bind molecules selectively overexpressed on tumour cell-
surfaces. The cargoes will then be internalised into tumour cells. Figure 2.1 outlines the
passive targeting drug delivery and active targeting ligand-drug conjugate.

6
Figure 2.1: Passive and active targeting for cancer therapeutic.
Passive targeting relies on enhanced permeability and retention effect to deliver the
drugs-encapsulated nano-structures or vesicles to tumour tissues via leaky tumour
vasculatures. The acidic tumour microenvironment causes the drugs to be released in
tumour cells. Active targeting generally using monoclonal antibody or receptor ligand
as the targeting agent, conjugated with cargoes (e.g. cytotoxic agent, imaging agent,
drug-encapsulated nanocarriers) to bind receptors that are overexpressed on tumour
cells. Diagram modified from Salim et al., 2014.
Conjugation of receptor ligands either to the anticancer therapeutic agents for
therapeutic purpose, or radiolabeled imaging probes for diagnostic purpose may
increase the binding affinity of conjugates to the cancer cells’ surface receptor. Binding
of the ligand to respective receptor enhances the endocytic internalisation by the cancer
cells (Alexis et al., 2008; Byrne et al., 2008) and increases the tumour accumulation and
residence time of the drug (Cheng et al., 2012; des Rieux et al., 2013; Koshkaryev et al.,
2013).

7
2.1.1 Rationale of Active Targeting
There are two important aspects to consider when designing an active targeting
ligand-drug conjugate, which are binding selectivity and sizes. Both aspects are
reflected in the biodistribution of ligated conjugate (Bertrand et al., 2014). In order to
have a high binding selectivity towards the targeted lesion, recognition of ligands by the
targeted cellular biomolecules is important. Targeting ligands employed so far in the
development of targetable ligand-drug conjugates include natural ligands such as
hormones, vitamins, sugar derivatives, peptides, monoclonal antibodies as well as
synthetic small molecules that possess high binding affinity to the targeted cellular
substrate (Figure 2.2). The targeted substrates are proteins, sugars or lipids on
overexpressed on cancer cells, which can reduce the binding on non-targeted tissues.

8
Figure 2.2: Common classes of ligands and cargoes used for active targeting in
cancer.
The common targeting agents (red) used are vitamins (biotin, folate), hormones
(estradiol, DHT, progestin), peptide ligands, glucose derivatives, and synthetic small
molecule ligands. These targeting agents are known to have high binding affinity to
their respective receptors. The common cargoes (purple) used for anti-cancer therapy
are alkylating agents (chlorambucil, isosfamide), antibiotic (mitomycin-C,
geldanamycin), antimetabolites (5-fluorouracil), apoptotic agents (Bim, Smac), DNA
intercalating/damaging agents (histone deacetylase, cisplatin), mitotic inhibitors
(paclitaxel, maytansinoids, desacetylvinblastine monohydrazine, tubulysin b hydrazide),
photosensitisers (Boron-dipyrromethene, pheophorbide-a), protein kinase inhibitor
(mitogen activated protein kinase kinase inhibitor) and topoisomerase inhibitor
(indenoisoquinolines, camptothecin). For imaging of cancer using
PET/SPECT/CT/Optical or fluorescence imaging system, the common agents (yellow)
used are bromine-77, carbon-11, copper-64, fluorine-18, gallium-68, indium-111,
iodine-123, technetium 99m, and near Infrared dye. Diagram obtained from Kue et al.
2016.
Another aspect to consider is the size of the ligand-drug conjugate. The pivotal issue
is that the construct should preferentially accumulate in the tumour tissue. Size matters
for the ease of penetration into tumours, and the ideal size is usually small (molecular
weight < 5000) (Srinivasarao et al., 2015; Vlashi et al., 2013). For these reasons, small
molecules can have significant advantages over antibodies due to the reason of
pharmacokinetics (Vlashi et al., 2013). In general, high molecular weight active
targeting agent has low tumour penetration, tumour retention and slow clearance
compared to smaller targeting agent. The details comparison between high molecular
weight protein (antibodies) and small peptides used in active targeting as delivering
agent will be discussed in following section 2.1.2.

9
2.1.2 Advantages of Small Molecule in Active Targeting
Active targeting in cancer has been increasingly recognised as an effective strategy
to elevate the therapeutic efficacy of anticancer drugs. Monoclonal antibodies (mAb)
have been extensively studied as a potential targeting agent due to their specificity in
antigen binding. To date, many mAbs have been used clinically to target therapeutic
agents to tumour tissue (Scott et al., 2012) for treatment purposes, but they have serious
limitations. Paramount amongst these is that mAb has low penetration into tumours
(Cabral et al., 2011; Dreher et al., 2006; Jain & Stylianopoulos, 2010) due to its relative
big size (> 4 nm) to leave the blood vessels and efficiently diffuse into tumour tissue.
Furthermore, mAbs that diffuse into tumour tissue tend to interact with the antigens that
are located on the surface of the perivascular tumour cells. This prevents their
permeation into the deeper tumour mass (Dennis et al., 2007) - a phenomenon known as
“antigen barrier” (Adams et al., 2001; Rudnick et al., 2011; Saga et al., 1995).
In addition, the clearance of mAbs from the body is relatively slow, resulting in
undesired exposure to normal tissues such as excretory organs (Baluk et al., 2003; di
Tomaso et al., 2005; O'Connor, 2007). Moreover, the antibody tends to be taken up by
macrophages via surface Fc receptor (Kamps & Scherphof, 1998), which leads to the
low deposition of mAbs in the target lesion. mAbs can also trigger antibody-dependent
complement-mediated cytotoxicity (Sapra & Allen, 2003), as well as immunogenicity
(van Schouwenburg et al., 2010) to cause hypersensitivity (Carrasco-Triguero et al.,
2013).
In comparison, small molecule targeting entities are not or less constrained by the
factors outlined above for mAbs. For instance, fluorescence studies have shown that
small molecules folate-rhodamine conjugate can saturate folate receptors on tumours

10
within five minutes of intravenous injection (Vlashi et al., 2009). The antigen-barrier
can still perceptibly impact folate-small molecule conjugates, but it has a negligible
effect at saturating doses (Vlashi et al., 2009). This implies that small molecule
conjugates can be ideal for rapid accumulation in solid tumours, and for brisk clearance
afterwards. They can be synthesised in large scale and tend to be cheaper than mAbs.
2.1.3 Parameters Determining the Efficacy of Active Targeting Ligand-Drug
Conjugate
Practically, after ligand binds to its target receptor to form a ligand-receptor complex,
the complex will be internalised via receptor mediated endocytosis, then passes through
an acidified endosome for sorting. The receptor in endosome is either recycled back to
surface or underwent enzymatically degradation upon lysosome fusion, depending on
the cellular requirement. In fact, this ligand-targeted internalisation happens more
rapidly compared to untargeted complexes (Bareford & Swaan, 2007) (Figure 2.3).

11
Figure 2.3: Conjugate uptake and receptor trafficking pathway.
Once the conjugate binds to receptor, the receptor–ligand complexes are internalised
and fused with early endosome. An early endosome is formed, followed by budding off
separation to form late endosomes with receptors and targeted ligands in separate units.
The vesicle with receptors will return to the plasma membrane (recycling). The late
endosome with targeting ligands will fuse with lysosome for subsequent degradation.
Diagram obtained from Kue et al., 2016.
There are four factors known to regulate receptor internalisation and trafficking,
which then determine the efficacy of active targeting agents for cancer therapy: (i) type
and binding rate of ligand-drug conjugate to receptor, (ii) rate of receptor recycling, (iii)
receptor occupancy and saturation, and (iv) rate of drug release from ligand-drug
conjugate upon internalisation (Bandara et al., 2014; Paulos et al., 2004a).
The rate of receptor internalisation, trafficking and recycling to cell surface is
important for the selection of optimal durations of ligand-drug conjugate infusion. The
correlation between the rate of receptor internalisation and the receptor occupancy was
investigated by Roettger and colleagues (Roettger et al., 1995). They found that high
level of receptor occupancy may induce receptor internalisation and ended in lysosome
for degradation. In contrast, low level of receptor occupancy may stimulate the
trafficking of the internalised receptor to recycling endosomes and subsequently back to
cell surface.
The type of linker used in ligand-drug conjugates for successful release of cargo in
endosomes is another key factor determining the efficacy of ligand-drug conjugate.
Studies have revealed that linkers with shorter cleavage half-lives or pH sensitive N-
ethoxybenzylimidazole (NEBI) linker can influence the rate and efficiency of cargo

12
release in acidic endosomes (Cao & Yang, 2014; Yang et al., 2006). For instance,
endosome cleavable pH sensitive acyl hydrazone bond (Leamon et al., 2006) and
reducible disulfide bond (Vlahov et al., 2006; Yang et al., 2006) were used to design
different folate-drug conjugates for comparison. Results revealed that reduction-
mediated release of the cargo from the disulfide bond was highly efficient within
endosomes of cancer cells, with cleavage half-life of 1 hour and complete cleavage
within 6 hours (Vlahov et al., 2006; Yang et al., 2006). Conversely, pH sensitive
acylhydrazone bond has a cleavage half-life of 5.5 hours under acidic endosome
environment (pH 5.5) (Leamon et al., 2006).
2.1.4 Small Molecule Conjugates for Active Targeting in Cancer
Research on small molecules (synthetic or natural ligands) as active-drug targeting in
cancer was started as early as 1990. These ligands have been designed to target either
cytotoxic or fluorescence agents to up-regulated molecules on plasma membrane of
cancer cells for therapeutic and diagnostic (imaging) purposes, respectively. The
receptors reported so far include folate, biotin, estrogen, progesterone, glucose transport
system, androgen and integrin receptors. Recently, number of new synthetic ligands has
been developed to target drugs to new receptors, such as sigma-2, prostate-specific
membrane antigen (PSMA), carbonic anhydrase IX, cholecystokin and TrkC receptor.
Table-2.1 summarises all the small molecule ligand-drug conjugates for cancer
therapeutic that target different surface receptors arranged according to cancer type.
Table 2.2 summarises all the small molecule ligand-imaging probe conjugates for
diagnostic imaging based on cancer type.
13
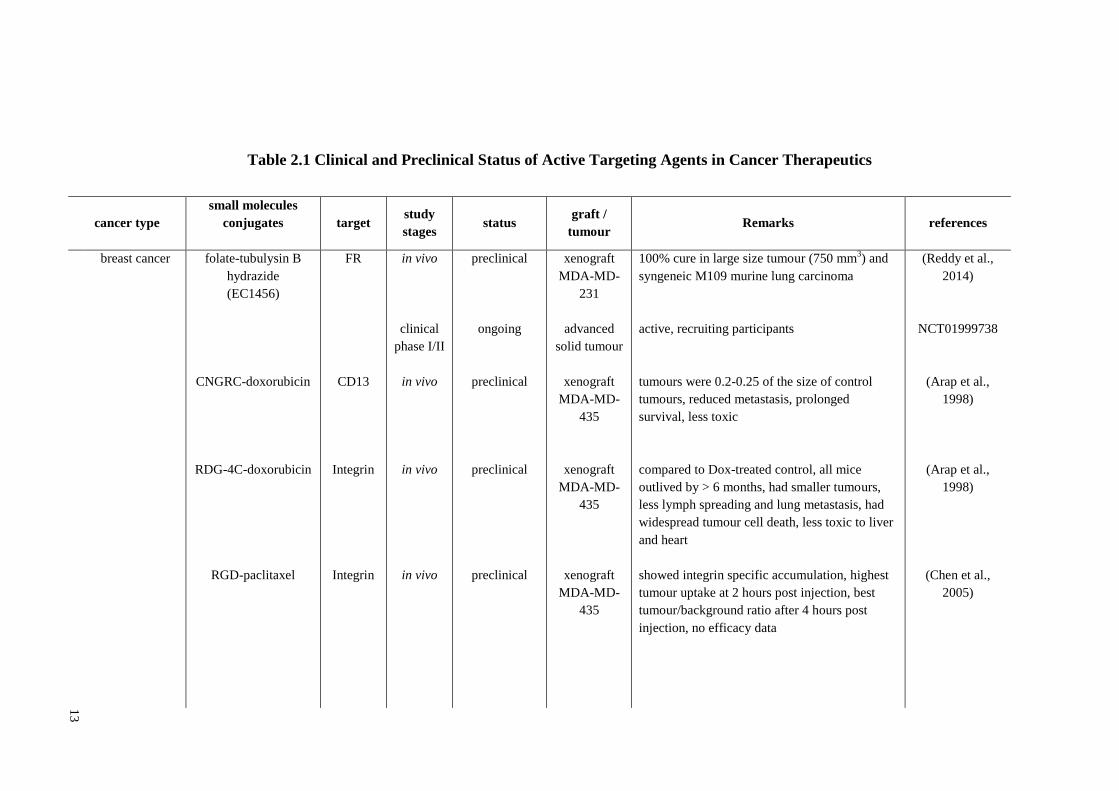
Table 2.1 Clinical and Preclinical Status of Active Targeting Agents in Cancer Therapeutics
cancer type
small molecules
conjugates
target study
stages status
graft /
tumour Remarks references
breast cancer
folate-tubulysin B
hydrazide
(EC1456)
CNGRC-doxorubicin
RDG-4C-doxorubicin
RGD-paclitaxel
FR
CD13
Integrin
Integrin
in vivo
clinical
phase I/II
in vivo
in vivo
in vivo
preclinical
ongoing
preclinical
preclinical
preclinical
xenograft
MDA-MD-
231
advanced
solid tumour
xenograft
MDA-MD-
435
xenograft
MDA-MD-
435
xenograft
MDA-MD-
435
100% cure in large size tumour (750 mm3) and
syngeneic M109 murine lung carcinoma
active, recruiting participants
tumours were 0.2-0.25 of the size of control
tumours, reduced metastasis, prolonged
survival, less toxic
compared to Dox-treated control, all mice
outlived by > 6 months, had smaller tumours,
less lymph spreading and lung metastasis, had
widespread tumour cell death, less toxic to liver
and heart
showed integrin specific accumulation, highest
tumour uptake at 2 hours post injection, best
tumour/background ratio after 4 hours post
injection, no efficacy data
(Reddy et al.,
2014)
NCT01999738
(Arap et al.,
1998)
(Arap et al.,
1998)
(Chen et al.,
2005)
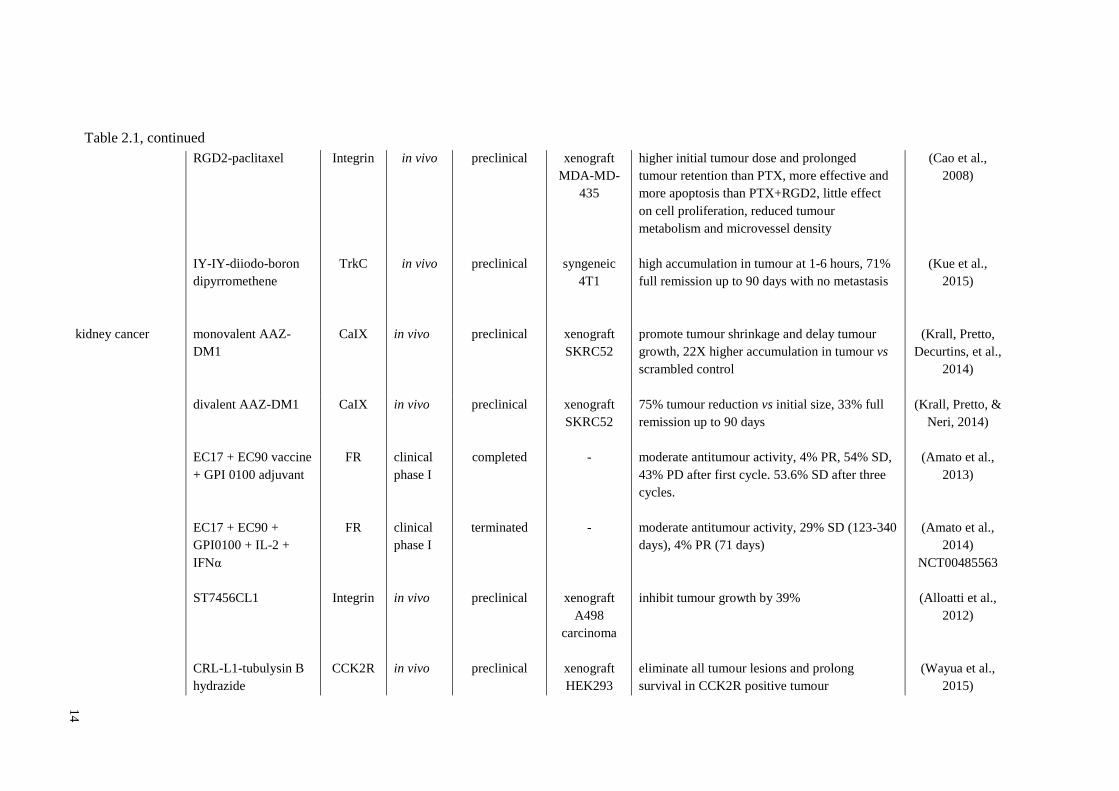
14
RGD2-paclitaxel
IY-IY-diiodo-boron
dipyrromethene
Integrin
TrkC
in vivo
in vivo
preclinical
preclinical
xenograft
MDA-MD-
435
syngeneic
4T1
higher initial tumour dose and prolonged
tumour retention than PTX, more effective and
more apoptosis than PTX+RGD2, little effect
on cell proliferation, reduced tumour
metabolism and microvessel density
high accumulation in tumour at 1-6 hours, 71%
full remission up to 90 days with no metastasis
(Cao et al.,
2008)
(Kue et al.,
2015)
kidney cancer monovalent AAZ-
DM1
CaIX in vivo preclinical xenograft
SKRC52
promote tumour shrinkage and delay tumour
growth, 22X higher accumulation in tumour vs
scrambled control
(Krall, Pretto,
Decurtins, et al.,
2014)
divalent AAZ-DM1
EC17 + EC90 vaccine
+ GPI 0100 adjuvant
EC17 + EC90 +
GPI0100 + IL-2 +
IFNα
ST7456CL1
CRL-L1-tubulysin B
hydrazide
CaIX
FR
FR
Integrin
CCK2R
in vivo
clinical
phase I
clinical
phase I
in vivo
in vivo
preclinical
completed
terminated
preclinical
preclinical
xenograft
SKRC52
-
-
xenograft
A498
carcinoma
xenograft
HEK293
75% tumour reduction vs initial size, 33% full
remission up to 90 days
moderate antitumour activity, 4% PR, 54% SD,
43% PD after first cycle. 53.6% SD after three
cycles.
moderate antitumour activity, 29% SD (123-340
days), 4% PR (71 days)
inhibit tumour growth by 39%
eliminate all tumour lesions and prolong
survival in CCK2R positive tumour
(Krall, Pretto, &
Neri, 2014)
(Amato et al.,
2013)
(Amato et al.,
2014)
NCT00485563
(Alloatti et al.,
2012)
(Wayua et al.,
2015)
Table 2.1, continued
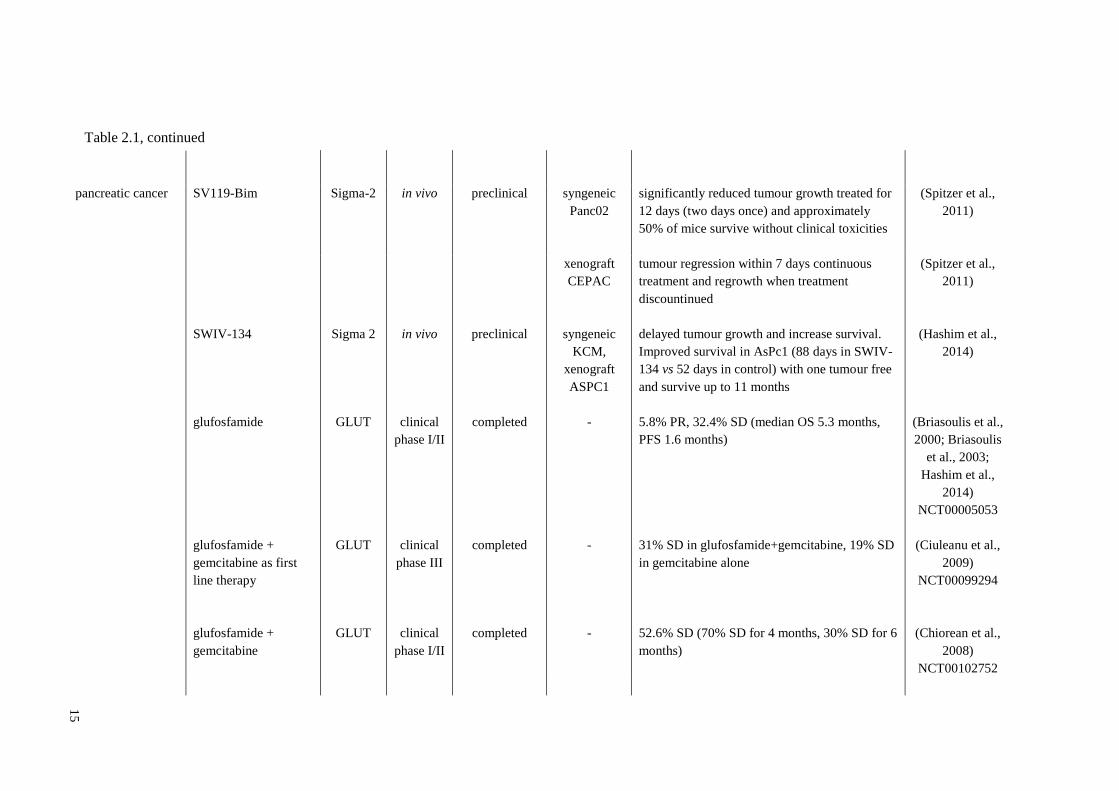
15
pancreatic cancer SV119-Bim Sigma-2 in vivo preclinical syngeneic
Panc02
significantly reduced tumour growth treated for
12 days (two days once) and approximately
50% of mice survive without clinical toxicities
(Spitzer et al.,
2011)
xenograft
CEPAC
tumour regression within 7 days continuous
treatment and regrowth when treatment
discountinued
(Spitzer et al.,
2011)
SWIV-134 Sigma 2 in vivo preclinical syngeneic
KCM,
xenograft
ASPC1
delayed tumour growth and increase survival.
Improved survival in AsPc1 (88 days in SWIV-
134 vs 52 days in control) with one tumour free
and survive up to 11 months
(Hashim et al.,
2014)
glufosfamide GLUT clinical
phase I/II
completed - 5.8% PR, 32.4% SD (median OS 5.3 months,
PFS 1.6 months)
(Briasoulis et al.,
2000; Briasoulis
et al., 2003;
Hashim et al.,
2014)
NCT00005053
glufosfamide +
gemcitabine as first
line therapy
GLUT clinical
phase III
completed - 31% SD in glufosfamide+gemcitabine, 19% SD
in gemcitabine alone
(Ciuleanu et al.,
2009)
NCT00099294
glufosfamide +
gemcitabine
GLUT clinical
phase I/II
completed - 52.6% SD (70% SD for 4 months, 30% SD for 6
months)
(Chiorean et al.,
2008)
NCT00102752
Table 2.1, continued
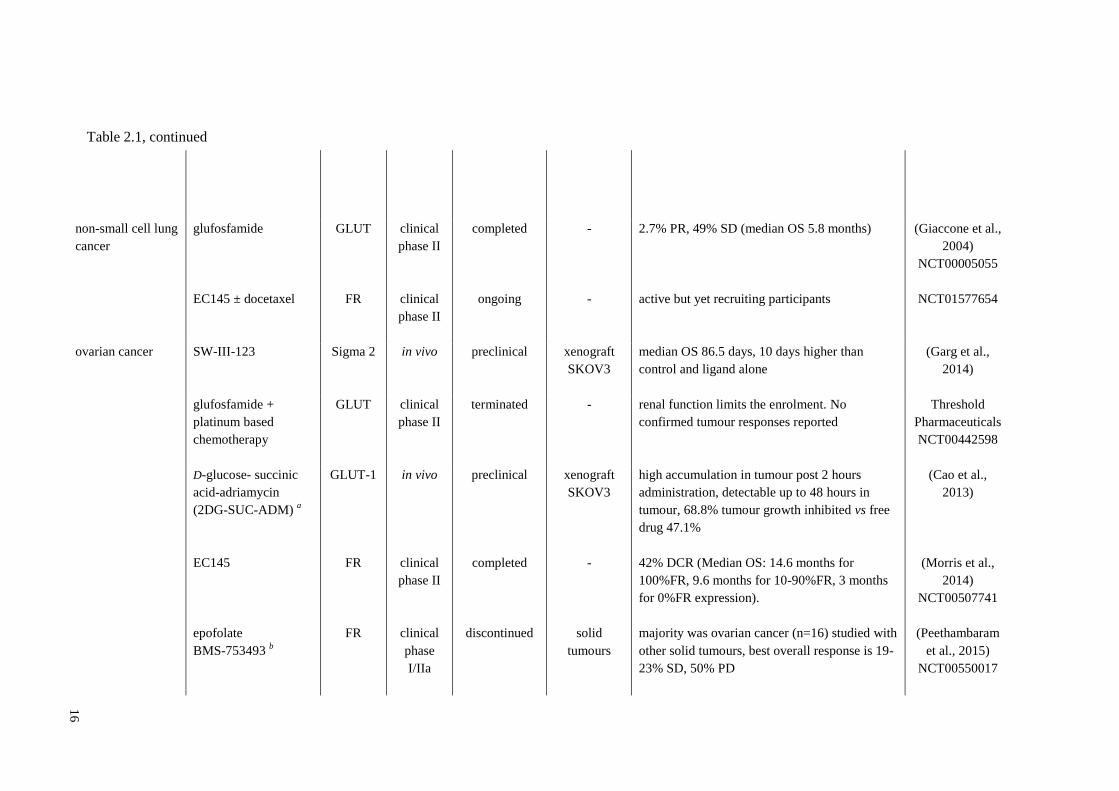
16
non-small cell lung
cancer
glufosfamide
EC145 ± docetaxel
GLUT
FR
clinical
phase II
clinical
phase II
completed
ongoing
-
-
2.7% PR, 49% SD (median OS 5.8 months)
active but yet recruiting participants
(Giaccone et al.,
2004)
NCT00005055
NCT01577654
ovarian cancer SW-III-123 Sigma 2 in vivo preclinical xenograft
SKOV3
median OS 86.5 days, 10 days higher than
control and ligand alone
(Garg et al.,
2014)
glufosfamide +
platinum based
chemotherapy
GLUT clinical
phase II
terminated - renal function limits the enrolment. No
confirmed tumour responses reported
Threshold
Pharmaceuticals
NCT00442598
D-glucose- succinic
acid-adriamycin
(2DG-SUC-ADM) a
EC145
epofolate
BMS-753493 b
GLUT-1
FR
FR
in vivo
clinical
phase II
clinical
phase
I/IIa
preclinical
completed
discontinued
xenograft
SKOV3
-
solid
tumours
high accumulation in tumour post 2 hours
administration, detectable up to 48 hours in
tumour, 68.8% tumour growth inhibited vs free
drug 47.1%
42% DCR (Median OS: 14.6 months for
100%FR, 9.6 months for 10-90%FR, 3 months
for 0%FR expression).
majority was ovarian cancer (n=16) studied with
other solid tumours, best overall response is 19-
23% SD, 50% PD
(Cao et al.,
2013)
(Morris et al.,
2014)
NCT00507741
(Peethambaram
et al., 2015)
NCT00550017
Table 2.1, continued
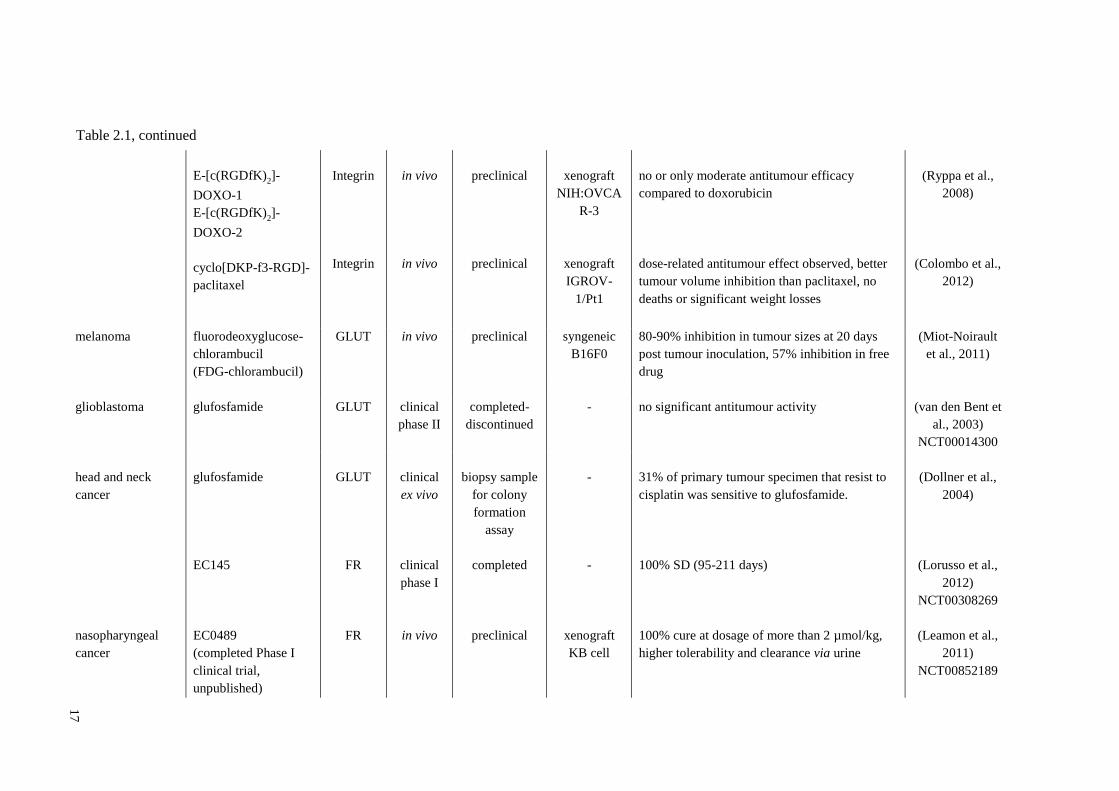
17
E-[c(RGDfK)2]-
DOXO-1
E-[c(RGDfK)2]-
DOXO-2
cyclo[DKP-f3-RGD]-
paclitaxel
Integrin
Integrin
in vivo
in vivo
preclinical
preclinical
xenograft
NIH:OVCA
R-3
xenograft
IGROV-
1/Pt1
no or only moderate antitumour efficacy
compared to doxorubicin
dose-related antitumour effect observed, better
tumour volume inhibition than paclitaxel, no
deaths or significant weight losses
(Ryppa et al.,
2008)
(Colombo et al.,
2012)
melanoma fluorodeoxyglucose-
chlorambucil
(FDG-chlorambucil)
GLUT in vivo preclinical syngeneic
B16F0
80-90% inhibition in tumour sizes at 20 days
post tumour inoculation, 57% inhibition in free
drug
(Miot-Noirault
et al., 2011)
glioblastoma glufosfamide GLUT clinical
phase II
completed-
discontinued
- no significant antitumour activity (van den Bent et
al., 2003)
NCT00014300
head and neck
cancer
nasopharyngeal
cancer
glufosfamide
EC145
EC0489
(completed Phase I
clinical trial,
unpublished)
GLUT
FR
FR
clinical
ex vivo
clinical
phase I
in vivo
biopsy sample
for colony
formation
assay
completed
preclinical
-
-
xenograft
KB cell
31% of primary tumour specimen that resist to
cisplatin was sensitive to glufosfamide.
100% SD (95-211 days)
100% cure at dosage of more than 2 µmol/kg,
higher tolerability and clearance via urine
(Dollner et al.,
2004)
(Lorusso et al.,
2012)
NCT00308269
(Leamon et al.,
2011)
NCT00852189
Table 2.1, continued
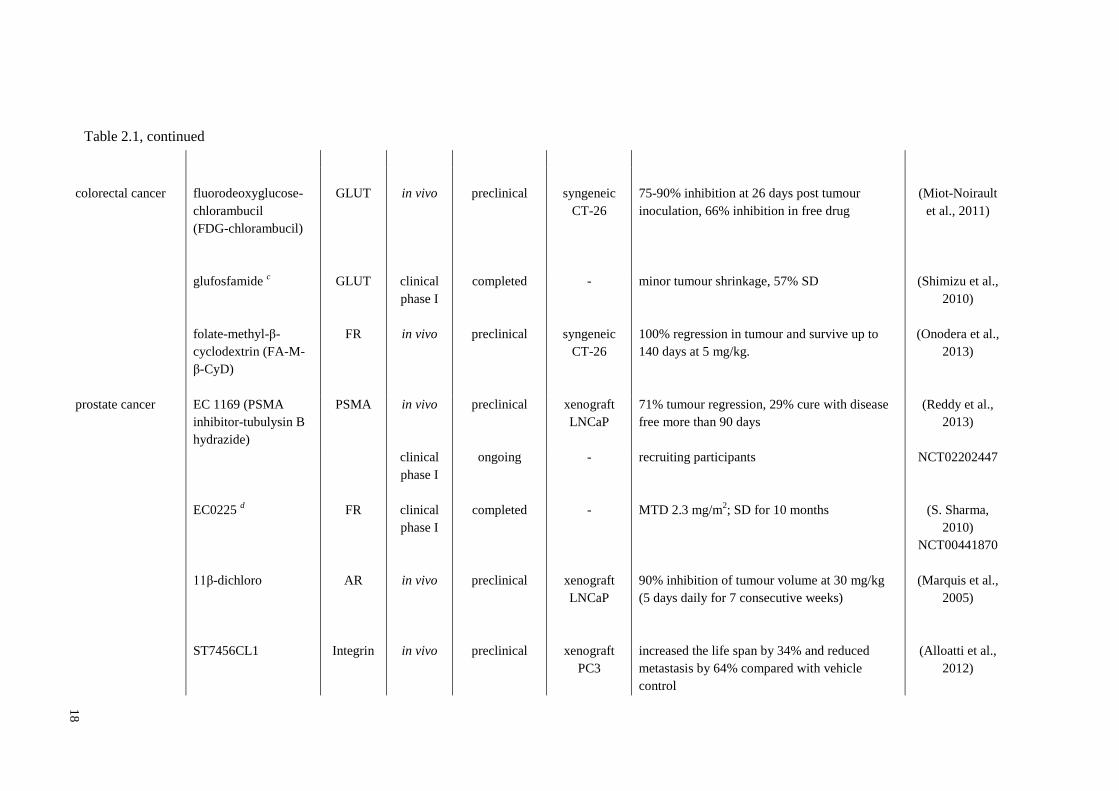
18
colorectal cancer
fluorodeoxyglucose-
chlorambucil
(FDG-chlorambucil)
GLUT
in vivo
preclinical
syngeneic
CT-26
75-90% inhibition at 26 days post tumour
inoculation, 66% inhibition in free drug
(Miot-Noirault
et al., 2011)
glufosfamide c
folate-methyl-β-
cyclodextrin (FA-M-
β-CyD)
GLUT
FR
clinical
phase I
in vivo
completed
preclinical
-
syngeneic
CT-26
minor tumour shrinkage, 57% SD
100% regression in tumour and survive up to
140 days at 5 mg/kg.
(Shimizu et al.,
2010)
(Onodera et al.,
2013)
prostate cancer EC 1169 (PSMA
inhibitor-tubulysin B
hydrazide)
PSMA in vivo preclinical xenograft
LNCaP
71% tumour regression, 29% cure with disease
free more than 90 days
(Reddy et al.,
2013)
EC0225 d
11β-dichloro
ST7456CL1
FR
AR
Integrin
clinical
phase I
clinical
phase I
in vivo
in vivo
ongoing
completed
preclinical
preclinical
-
-
xenograft
LNCaP
xenograft
PC3
recruiting participants
MTD 2.3 mg/m2; SD for 10 months
90% inhibition of tumour volume at 30 mg/kg
(5 days daily for 7 consecutive weeks)
increased the life span by 34% and reduced
metastasis by 64% compared with vehicle
control
NCT02202447
(S. Sharma,
2010)
NCT00441870
(Marquis et al.,
2005)
(Alloatti et al.,
2012)
Table 2.1, continued
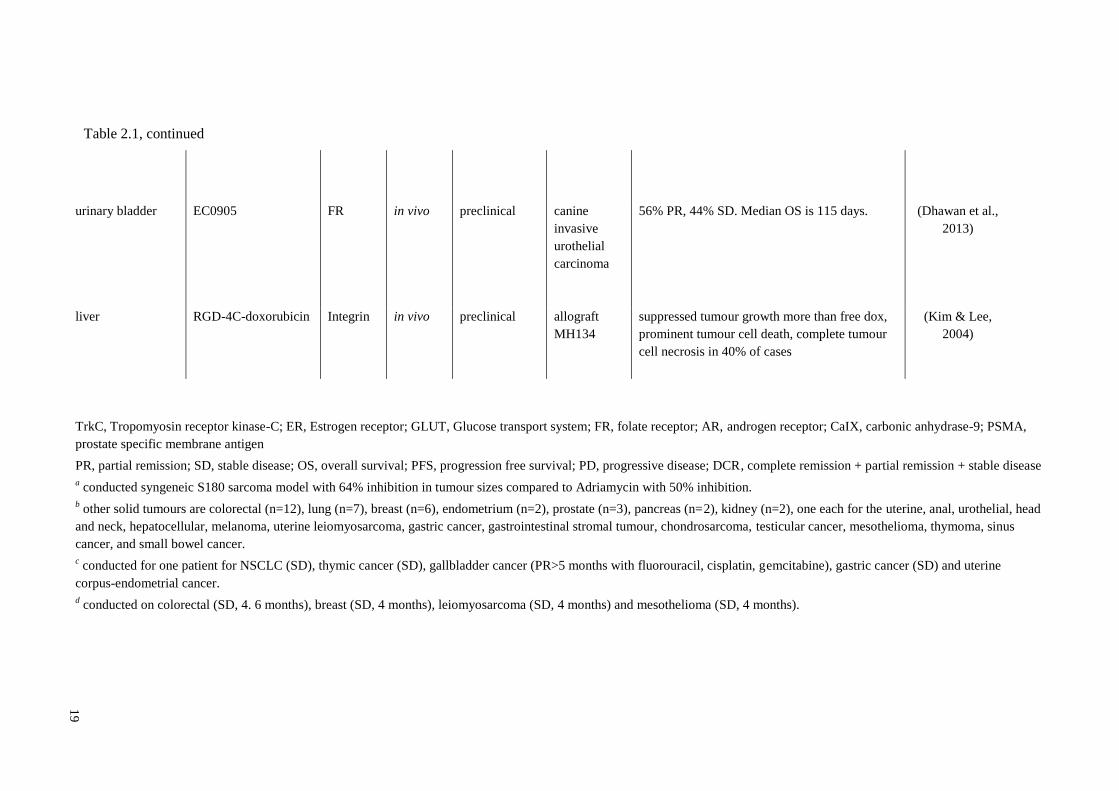
19
urinary bladder EC0905 FR in vivo preclinical canine
invasive
urothelial
carcinoma
56% PR, 44% SD. Median OS is 115 days.
(Dhawan et al.,
2013)
liver RGD-4C-doxorubicin Integrin in vivo preclinical allograft
MH134
suppressed tumour growth more than free dox,
prominent tumour cell death, complete tumour
cell necrosis in 40% of cases
(Kim & Lee,
2004)
TrkC, Tropomyosin receptor kinase-C; ER, Estrogen receptor; GLUT, Glucose transport system; FR, folate receptor; AR, androgen receptor; CaIX, carbonic anhydrase-9; PSMA,
prostate specific membrane antigen
PR, partial remission; SD, stable disease; OS, overall survival; PFS, progression free survival; PD, progressive disease; DCR, complete remission + partial remission + stable disease
a conducted syngeneic S180 sarcoma model with 64% inhibition in tumour sizes compared to Adriamycin with 50% inhibition.
b other solid tumours are colorectal (n=12), lung (n=7), breast (n=6), endometrium (n=2), prostate (n=3), pancreas (n=2), kidney (n=2), one each for the uterine, anal, urothelial, head
and neck, hepatocellular, melanoma, uterine leiomyosarcoma, gastric cancer, gastrointestinal stromal tumour, chondrosarcoma, testicular cancer, mesothelioma, thymoma, sinus
cancer, and small bowel cancer.
c conducted for one patient for NSCLC (SD), thymic cancer (SD), gallbladder cancer (PR>5 months with fluorouracil, cisplatin, gemcitabine), gastric cancer (SD) and uterine
corpus-endometrial cancer.
d conducted on colorectal (SD, 4. 6 months), breast (SD, 4 months), leiomyosarcoma (SD, 4 months) and mesothelioma (SD, 4 months).
Table 2.1, continued
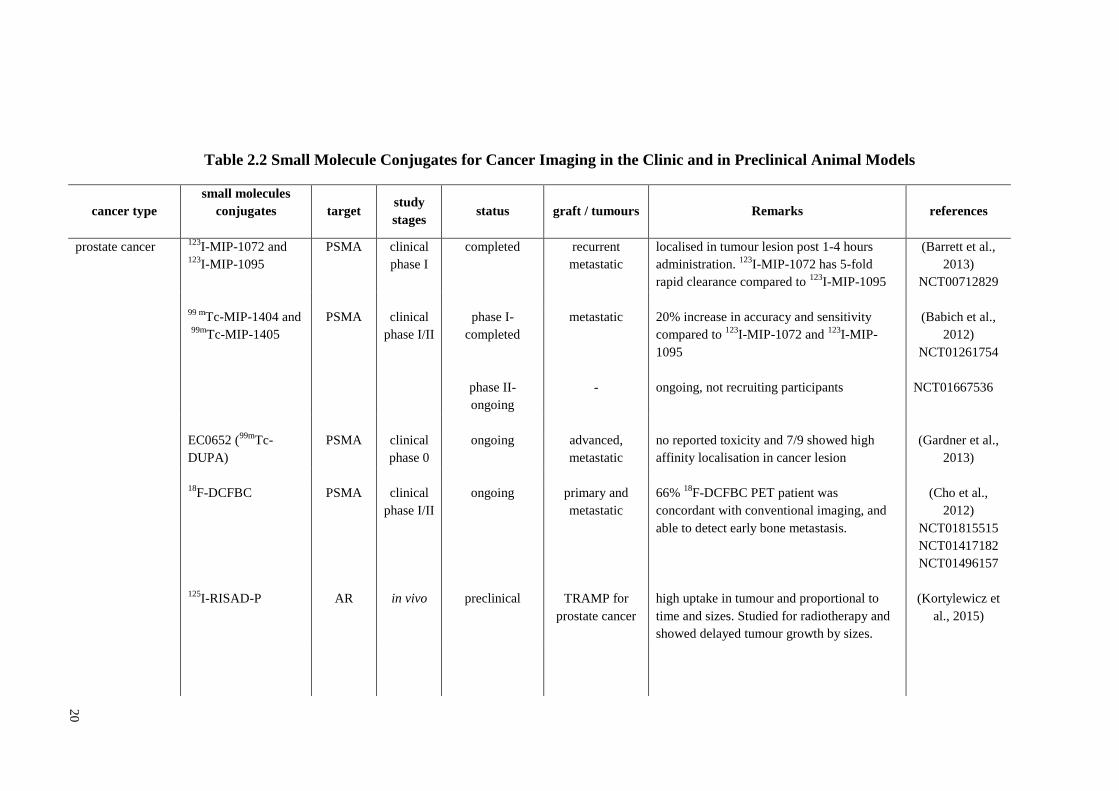
20
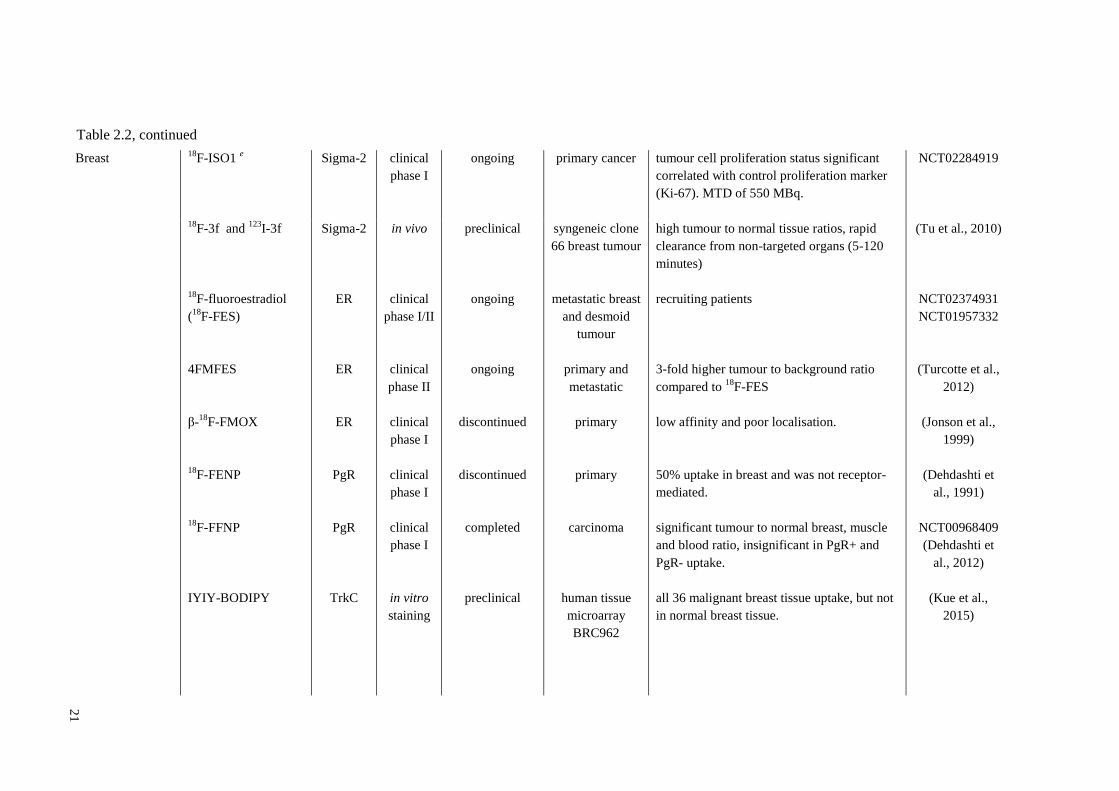
Table 2.2 Small Molecule Conjugates for Cancer Imaging in the Clinic and in Preclinical Animal Models
cancer type
small molecules
conjugates
target study
stages status graft / tumours Remarks references
prostate cancer 123
I-MIP-1072 and 123
I-MIP-1095
PSMA clinical
phase I
completed recurrent
metastatic
localised in tumour lesion post 1-4 hours
administration. 123
I-MIP-1072 has 5-fold
rapid clearance compared to 123
I-MIP-1095
(Barrett et al.,
2013)
NCT00712829
99 m
Tc-MIP-1404 and
99m
Tc-MIP-1405
PSMA clinical
phase I/II
phase I-
completed
phase II-
ongoing
metastatic
-
20% increase in accuracy and sensitivity
compared to 123
I-MIP-1072 and 123
I-MIP-
1095
ongoing, not recruiting participants
(Babich et al.,
2012)
NCT01261754
NCT01667536
EC0652 (99m
Tc-
DUPA)
PSMA clinical
phase 0
ongoing advanced,
metastatic
no reported toxicity and 7/9 showed high
affinity localisation in cancer lesion
(Gardner et al.,
2013)
18F-DCFBC
125
I-RISAD-P
PSMA
AR
clinical
phase I/II
in vivo
ongoing
preclinical
primary and
metastatic
TRAMP for
prostate cancer
66% 18
F-DCFBC PET patient was
concordant with conventional imaging, and
able to detect early bone metastasis.
high uptake in tumour and proportional to
time and sizes. Studied for radiotherapy and
showed delayed tumour growth by sizes.
(Cho et al.,
2012)
NCT01815515
NCT01417182
NCT01496157
(Kortylewicz et
al., 2015)
21
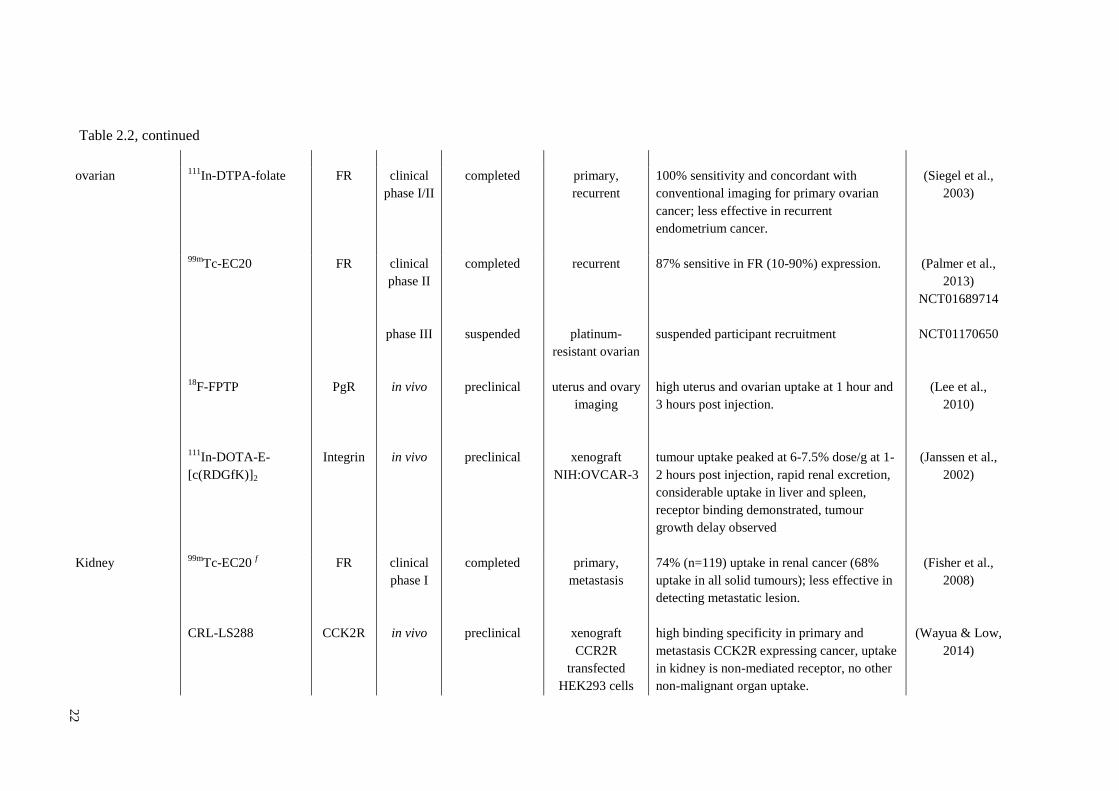
Breast 18
F-ISO1 e Sigma-2 clinical
phase I
ongoing primary cancer tumour cell proliferation status significant
correlated with control proliferation marker
(Ki-67). MTD of 550 MBq.
NCT02284919
18F-3f and
123I-3f
18
F-fluoroestradiol
(18
F-FES)
4FMFES
β-18
F-FMOX
18
F-FENP
18
F-FFNP
IYIY-BODIPY
Sigma-2
ER
ER
ER
PgR
PgR
TrkC
in vivo
clinical
phase I/II
clinical
phase II
clinical
phase I
clinical
phase I
clinical
phase I
in vitro
staining
preclinical
ongoing
ongoing
discontinued
discontinued
completed
preclinical
syngeneic clone
66 breast tumour
metastatic breast
and desmoid
tumour
primary and
metastatic
primary
primary
carcinoma
human tissue
microarray
BRC962
high tumour to normal tissue ratios, rapid
clearance from non-targeted organs (5-120
minutes)
recruiting patients
3-fold higher tumour to background ratio
compared to 18
F-FES
low affinity and poor localisation.
50% uptake in breast and was not receptor-
mediated.
significant tumour to normal breast, muscle
and blood ratio, insignificant in PgR+ and
PgR- uptake.
all 36 malignant breast tissue uptake, but not
in normal breast tissue.
(Tu et al., 2010)
NCT02374931
NCT01957332
(Turcotte et al.,
2012)
(Jonson et al.,
1999)
(Dehdashti et
al., 1991)
NCT00968409
(Dehdashti et
al., 2012)
(Kue et al.,
2015)
Table 2.2, continued
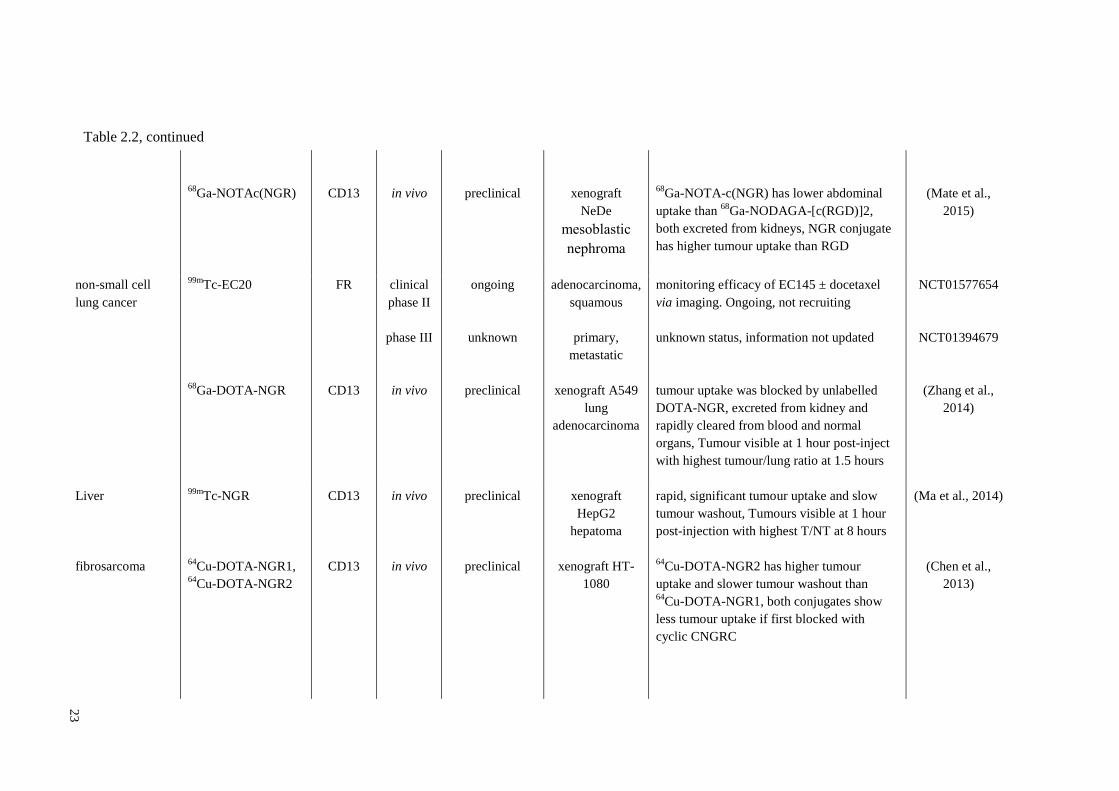
22
ovarian 111
In-DTPA-folate FR clinical
phase I/II
completed primary,
recurrent
100% sensitivity and concordant with
conventional imaging for primary ovarian
cancer; less effective in recurrent
endometrium cancer.
(Siegel et al.,
2003)
99m
Tc-EC20
18
F-FPTP
111
In-DOTA-E-
[c(RDGfK)]2
FR
PgR
Integrin
clinical
phase II
phase III
in vivo
in vivo
completed
suspended
preclinical
preclinical
recurrent
platinum-
resistant ovarian
uterus and ovary
imaging
xenograft
NIH:OVCAR-3
87% sensitive in FR (10-90%) expression.
suspended participant recruitment
high uterus and ovarian uptake at 1 hour and
3 hours post injection.
tumour uptake peaked at 6-7.5% dose/g at 1-
2 hours post injection, rapid renal excretion,
considerable uptake in liver and spleen,
receptor binding demonstrated, tumour
growth delay observed
(Palmer et al.,
2013)
NCT01689714
NCT01170650
(Lee et al.,
2010)
(Janssen et al.,
2002)
Kidney 99m
Tc-EC20 f
CRL-LS288
FR
CCK2R
clinical
phase I
in vivo
completed
preclinical
primary,
metastasis
xenograft
CCR2R
transfected
HEK293 cells
74% (n=119) uptake in renal cancer (68%
uptake in all solid tumours); less effective in
detecting metastatic lesion.
high binding specificity in primary and
metastasis CCK2R expressing cancer, uptake
in kidney is non-mediated receptor, no other
non-malignant organ uptake.
(Fisher et al.,
2008)
(Wayua & Low,
2014)
Table 2.2, continued
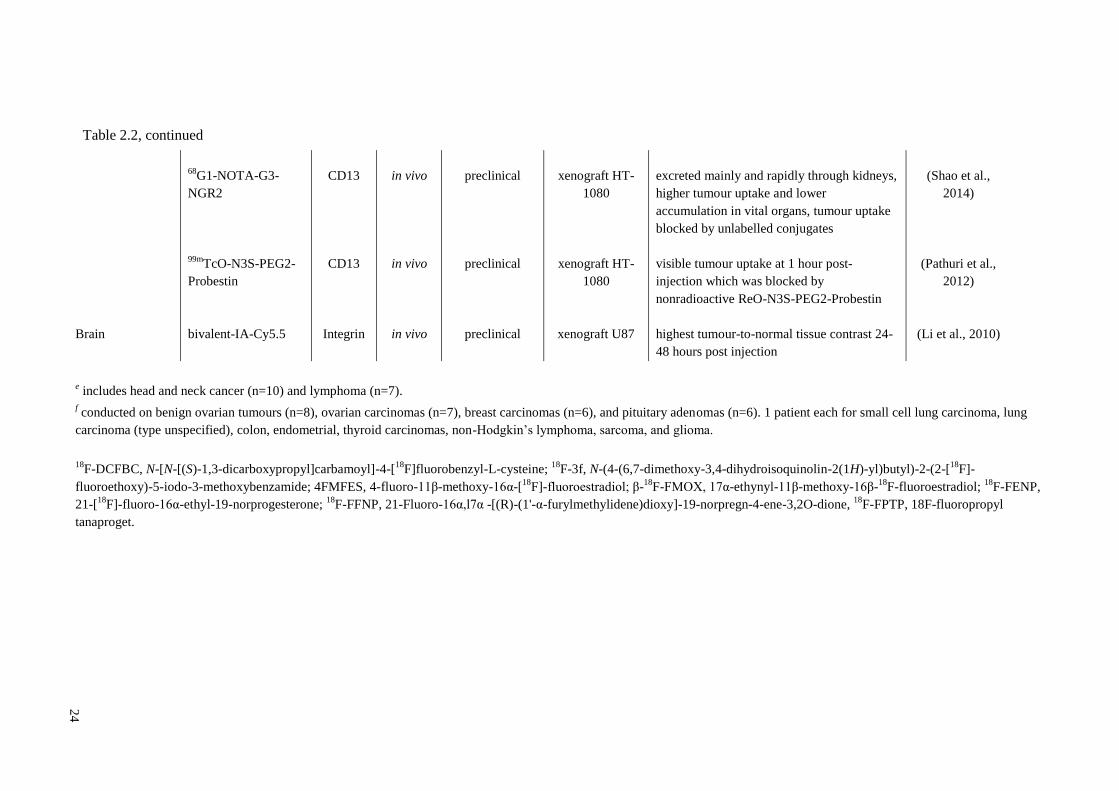
23
68
Ga-NOTAc(NGR)
CD13
in vivo
preclinical
xenograft
NeDe
mesoblastic
nephroma
68
Ga-NOTA-c(NGR) has lower abdominal
uptake than 68
Ga-NODAGA-[c(RGD)]2,
both excreted from kidneys, NGR conjugate
has higher tumour uptake than RGD
(Mate et al.,
2015)
non-small cell
lung cancer
99mTc-EC20
68
Ga-DOTA-NGR
FR
CD13
clinical
phase II
phase III
in vivo
ongoing
unknown
preclinical
adenocarcinoma,
squamous
primary,
metastatic
xenograft A549
lung
adenocarcinoma
monitoring efficacy of EC145 ± docetaxel
via imaging. Ongoing, not recruiting
unknown status, information not updated
tumour uptake was blocked by unlabelled
DOTA-NGR, excreted from kidney and
rapidly cleared from blood and normal
organs, Tumour visible at 1 hour post-inject
with highest tumour/lung ratio at 1.5 hours
NCT01577654
NCT01394679
(Zhang et al.,
2014)
Liver
fibrosarcoma
99mTc-NGR
64
Cu-DOTA-NGR1, 64
Cu-DOTA-NGR2
CD13
CD13
in vivo
in vivo
preclinical
preclinical
xenograft
HepG2
hepatoma
xenograft HT-
1080
rapid, significant tumour uptake and slow
tumour washout, Tumours visible at 1 hour
post-injection with highest T/NT at 8 hours
64
Cu-DOTA-NGR2 has higher tumour
uptake and slower tumour washout than 64
Cu-DOTA-NGR1, both conjugates show
less tumour uptake if first blocked with
cyclic CNGRC
(Ma et al., 2014)
(Chen et al.,
2013)
Table 2.2, continued
24
Brain
68
G1-NOTA-G3-
NGR2
99m
TcO-N3S-PEG2-
Probestin
bivalent-IA-Cy5.5
CD13
CD13
Integrin
in vivo
in vivo
in vivo
preclinical
preclinical
preclinical
xenograft HT-
1080
xenograft HT-
1080
xenograft U87
excreted mainly and rapidly through kidneys,
higher tumour uptake and lower
accumulation in vital organs, tumour uptake
blocked by unlabelled conjugates
visible tumour uptake at 1 hour post-
injection which was blocked by
nonradioactive ReO-N3S-PEG2-Probestin
highest tumour-to-normal tissue contrast 24-
48 hours post injection
(Shao et al.,
2014)
(Pathuri et al.,
2012)
(Li et al., 2010)
e includes head and neck cancer (n=10) and lymphoma (n=7).
f conducted on benign ovarian tumours (n=8), ovarian carcinomas (n=7), breast carcinomas (n=6), and pituitary adenomas (n=6). 1 patient each for small cell lung carcinoma, lung
carcinoma (type unspecified), colon, endometrial, thyroid carcinomas, non-Hodgkin’s lymphoma, sarcoma, and glioma.
18F-DCFBC, N-[N-[(S)-1,3-dicarboxypropyl]carbamoyl]-4-[
18F]fluorobenzyl-L-cysteine;
18F-3f, N-(4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2-(2-[
18F]-
fluoroethoxy)-5-iodo-3-methoxybenzamide; 4FMFES, 4-fluoro-11β-methoxy-16α-[18
F]-fluoroestradiol; β-18
F-FMOX, 17α-ethynyl-11β-methoxy-16β-18
F-fluoroestradiol; 18
F-FENP,
21-[18
F]-fluoro-16α-ethyl-19-norprogesterone; 18
F-FFNP, 21-Fluoro-16α,l7α -[(R)-(1'-α-furylmethylidene)dioxy]-19-norpregn-4-ene-3,2O-dione, 18
F-FPTP, 18F-fluoropropyl
tanaproget.
Table 2.2, continued

25
2.1.5 Other Impacts of Active Targeting
Active targeting tends to have some limitations because most of the targeting agents
used to date are natural ligands and some of the targeted molecules are expressed on
normal healthy cells, albeit at lower levels than in cancer cells. Other impacts of active
targeting may include immune-modulation and non-targeted toxicity, which are
mediated by the ligands.
2.1.5.1 Immune-modulation
Some ligands used for active tumour targeting were found to possess some
immunomodulation properties. For instance, folate, which was classically used as a
targeting ligand against folate receptor expressing tumours, was found capable of
promoting the survival of the activated monocytes and lymphocytes that expressed
folate receptors in rheumatoid arthritis (Nakashima-Matsushita et al., 1999; Paulos et al.,
2004b). Estradiol on the other hand suppresses inflammatory cytokines such as tumour
necrosis factor-α and interferon-γ (Matejuk et al., 2001) while promoting anti-
inflammatory mediators such as interleukin-10 cytokine secretion and enhancing
regulatory T cells function via up-regulation of programmed death-1 and FoxP3
expressions (Wang et al., 2009). Such immunomodulation properties may either assist
the development or exacerbate the antitumour immunity in the host when these ligands
are used as the targeting counterpart in an active-targeting ligand-drug conjugate
(Amato et al., 2014; Siebels et al., 2011). Figure 2.4A shows some possible impacts
induced by active targeting ligand on immune cells.

26
2.1.5.2 Non-targeted Toxicity
A certain receptors or cell surface proteins that are overexpressed on the tumour cells
may be found expressed at a high level in some of the normal body tissue. For example,
folate receptors that are often overexpressed in breast cancers may be abundantly found
in kidney tissue. Hence, administration of ligand-drug conjugates that targets cell
surface proteins may led to collateral accumulation of the conjugates in the normal
tissue, and thus induces toxicity (Fisher et al., 2008). Figure 2.4B shows the possible
toxicity induced by active targeting agent on non-targeted tissues.
Figure 2.4: Other targeting of ligand conjugates.
A. Immunomodulation is one of the possible effects of active targeting. Targeted
ligands generally possess similar abilities as the natural ligands and can transduce
signals upon binding to receptors that are expressed on normal cells, for e.g. immune
cells. The ligands are known to be able to modulate immune responses in two different
ways, (I) direct binding on monocytes to promote or inhibit the release of cytokines,
which can then alter the cytokine milieu systemically, thereby affecting the adaptive
immune cell (T lymphocytes) differentiation, and (II) binding of ligand conjugates to

27
different subtypes of T-lymphocytes to regulate their survival and functions. In both
ways, the effect may be either pro-tumour or anti-tumour depending on the ligand and
receptor involved. In addition, the therapeutic cargo carried by the ligand conjugate may
also modulate the immune response, for e.g. cyclophosphamide (an anticancer drug
with immune modulation properties such as reduces T-suppressor cells). B. Non-target
tissue toxicity is another possible effect of active targeting. Some receptors that are
overexpressed in cancer cells are also highly expressed in the normal cells in healthy
organs. Therefore, while killing cancer cells with overexpressed targeted molecules,
mild or acute toxicity to non-targeted tissues might also happen in active targeted
therapy.
2.2 Tropomyosin Receptor Kinase (Trk)
2.2.1 Receptor Biochemistry
Tropomyosin kinase (Trk) or neurotrophin receptors consist of three subtypes,
namely TrkA, TrkB and TrkC. These receptors bind to their respective ligand
neurotrophins, i.e. TrkA binds to neurotrophin growth factor (NGF), TrkB binds to
brain-derived neurotrophin factor (BDNF) and neurotrophin-4 (NT-4), and TrkC binds
to neurotrophin-3 (NT-3). Trk is transmembrane receptors containing an intracellular
domain and an extracellular domain. Extracellular domain of Trk receptors include a
combination of two cysteine clusters, a tandem array of leucine-rich motifs, and two
immunoglobulin-like domain in the membrane proximal region (Schneider &
Schweiger, 1991) (Figure 2.5). Specificity and affinity to neurotrophin ligands is
dictated by the second immunoglobulin like domain (residues 266-381) of the receptor
(Urfer et al., 1995), whereas the leucine rich motif serves as a binding site for
neurotrophins (Windisch et al., 1995).

28
Figure 2.5: Schematic picture of tropomyosin receptor kinase.
Diagram obtained and modified from Marchetti et al., 2015.
Trk receptors are mainly found in neuronal populations, where TrkA is found in
basal forebrain cholinergic, dorsal root ganglion and sympathetic neurons, whereas
TrkB and TrkC are found predominantly in the central and peripheral nervous system
(Longo & Massa, 2013). These receptors are known to promote neuron cell survival and
differentiation upon neurotrophins binding (Huang & Reichardt, 2003). Other roles of
Trk receptors and neurotrophins are in tumourigenesis (section 2.2.2) and immune
response (section 2.2.3).
2.2.2 Trk Receptors and Tumourigenesis
Trk receptor was first identified as a product of colon derived oncogene. This
oncogene (genomic DNA rearrangement or mutation) was generated from the fusion of

29
a tropomyosin gene with a tyrosine kinase-related locus (Trk proto-oncogene) (Martin-
Zanca et al., 1986). Proto-oncogene of Trk was also reported to play a role in regulating
invasiveness of cancer (Vaishnavi et al., 2015) in neuronal cancer such as
neuroblastoma (Brodeur et al., 1997; Yamashiro et al., 1997) and medulloblastoma as
well as non-neuronal melanoma (Xu et al., 2003), breast (Blasco-Gutierrez et al., 2007;
Jin et al., 2010) and pancreatic (Sakamoto et al., 2001) cancer. TrkA-C receptors exist
with their respective ligands to regulate the differentiation and survival of tumour cells,
and conferring potency to invasion and metastasis. Among the Trk receptors, TrkC is
the one that showed a high correlation with various cancer growth and became a
biomarker for prognosis of tumour progression and invasion (Vaishnavi et al., 2015) in
neuroblastoma (Brodeur et al., 1997; Yamashiro et al., 1997), glioblastoma (Kumar &
de Vellis, 1996; Wang et al., 1998), thyroid cancer (McGregor et al., 1999), melanoma
(Xu et al., 2003) and breast cancer (Blasco-Gutierrez et al., 2007; Jin et al., 2010).
Trk receptors mediated tumourigenesis depends on tumour types. For instance, in
neuroblastoma, TrkA and NGR promote apoptosis of neuroblastoma cells, and hence
aggressive behavior of neuroblastoma were found to have low TrkA expression, thus
reduced TrkA expression was said to be a good prognosis for neuroblastoma.
Conversely, neuroblastoma expresses high TrkB and BDNF to increase their survival
(Chou et al., 2000). Thus, suggested that Trk receptors expression is depends on tumour
types and metastasis level.
2.2.3 Trk Receptors, Neurotrophins and Immune System
Trk receptor expression is not only restricted to neuronal cell, but also reported to be
expressed in immune cells such as monocytes, mast cells and lymphocytes, but at lower

30
levels compared to neurons (Vega et al., 2003). In the light of this, neurotrophins and
Trk receptor may play a role in the regulation of inflammation and the development of
antitumour immunity. Previously, neurotrophins have been reported to regulate
hypersensitivity reaction by up-regulating the release of hypersensitive-associated
mediators such as histamine and IL-5 from immune cells upon their binding to the Trk
receptors (Bischoff & Dahinden, 1992). Neurotrophins also studied to stimulate T
helper 2 (Th2) cell differentiations, and release the pro-inflammatory cytokine IL-6
from stromal cells (Rezaee et al., 2010; Sekimoto et al., 2003; Vega et al., 2003) which
affects the systemic T-lymphocyte subtypes populations. In detail, a study reported that
Th2 subtype expressed higher level of TrkC than other T-lymphocyte subtypes such as
Th1. This suggests the possible role of neurotrophins in regulating the survival and
function of Th2 cells, especially during the allergic and hypersensitivity responses.
Moreover, transforming growth factor (TGF)-β cytokine is also regulated by TrkC;
activated TrkC receptor will block the TGF-β receptor-mediated Smad2/3
phosphorylation and thus cease all the TGF-β production and its downstream signaling
(Jin et al., 2007).
2.3 Photodynamic Therapy (PDT)
2.3.1 Background
Photodynamic therapy (PDT), a therapy using filtered light from a carbon-arc lamp
was first reported by Danish physician, Niels Finsen in 1901 for the treatment of
tubercular condition of skin (Lupus Vulgaris). In 1978, Dougherty and co-worker at
Roswell Park cancer institute, Buffalo, New York, clinically tested photosensitiser
hematoporphyrin derivative in treating cutaneous or subcutaneous malignant tumour,

31
and found 98% of tumours (21 of 24 patients) had complete or partial response
(Dougherty et al., 1978).
Photofrin® (porfimer sodium), which is a hematoporphyrin derivative is the first
FDA approved photosensitiser. To date, there are only a few other FDA approved
anticancer photosensitising drugs including Foscan®
(temoporfin, meta-
tetrahydroxyphenylchlorin; Biolitec AG), Visudyne®
(verteporfin, benzoporphyrin
derivative monoacid ring A; Novartis Pharmaceuticals), Levulan® (5-aminolevulinic
acid; DUSA Pharmaceuticals, Inc.), and Metvix® (methyl aminolevulinate; PhotoCure
ASA) (Voon et al., 2014). PDT has become one of the clinical treatment options for
early stages of cancer such as nasopharyngeal, gastroenterological, brain and
gynecological cancer (Dolmans et al., 2003; Huang, 2005).
2.3.2 Components of PDT
PDT is non-invasive. It consists of components which are non-toxic when exist
individually. The components of PDT include (i) photosensitiser (PS), a light sensitive
drug that is administrated systemically or locally. After systemic distribution and
accumulation in the tumour, the tumour lesion is then focally irradiated with (ii) light at
a specific wavelengths to activate the PS, and in the presence of (iii) oxygen molecules
to generate singlet oxygen species through the photochemical reaction. (Dolmans et al.,
2003; Dougherty et al., 1998). Ideal photosensitisers are generally activated by red light
(630 – 700 nm), corresponding to light penetration depth of 0.5 – 1.5 cm (Salva, 2002).
Figure 2.6 illustrates the procedures of cancer PDT.
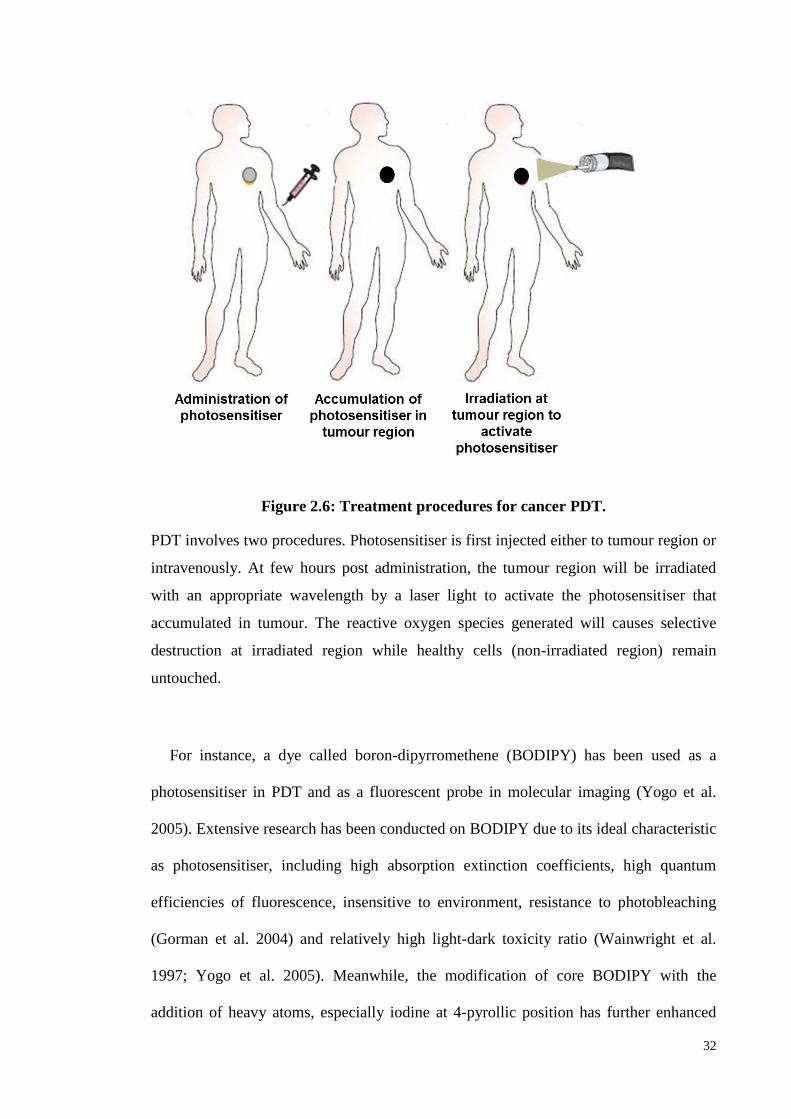
32
Figure 2.6: Treatment procedures for cancer PDT.
PDT involves two procedures. Photosensitiser is first injected either to tumour region or
intravenously. At few hours post administration, the tumour region will be irradiated
with an appropriate wavelength by a laser light to activate the photosensitiser that
accumulated in tumour. The reactive oxygen species generated will causes selective
destruction at irradiated region while healthy cells (non-irradiated region) remain
untouched.
For instance, a dye called boron-dipyrromethene (BODIPY) has been used as a
photosensitiser in PDT and as a fluorescent probe in molecular imaging (Yogo et al.
2005). Extensive research has been conducted on BODIPY due to its ideal characteristic
as photosensitiser, including high absorption extinction coefficients, high quantum
efficiencies of fluorescence, insensitive to environment, resistance to photobleaching
(Gorman et al. 2004) and relatively high light-dark toxicity ratio (Wainwright et al.
1997; Yogo et al. 2005). Meanwhile, the modification of core BODIPY with the
addition of heavy atoms, especially iodine at 4-pyrollic position has further enhanced

33
the singlet oxygen production, and exhibited potent photo-induced cytotoxicity with low
IC50 concentration compared to core BODIPY (Lim et al. 2010). The absorption
maximum and excitation wavelength of I2-BODIPY are 534 nm, and emission at 552
nm in methanol (Voon et al. 2016; Kamkaew et al. 2012), which is not suitable to treat
the deeper tissue. In addition, the free photosensitiser has poor selectivity at the targeted
sites, which leads to low therapeutic efficacy. Recently, a study on the designs of the
nanoparticles-coated I2-BODIPY for passive delivery to the tumour region was
conducted, which is an approachable strategy to improve the limitations of the BODIPY.
This strategy was significantly improved the accumulation of I2-BODIPY to the
targeted regions, and thus enhanced antitumour efficacy compared to free I2-BODIPY
(Voon et al. 2016). Another approach to target the photosensitiser to the tumour sites by
conjugating with an antibody or receptor-ligand by active targeting.
2.3.3 Mechanisms of PDT
Upon irradiation, the photosensitiser at ground singlet state is activated to highly
energised and stable triplet state, which enables it to interact with surrounding
molecules for generation of various cytotoxic species (1O2). There are two types of
photochemical reactions to generate cytotoxic species, as illustrated in Figure 2.7. Type
I reaction involves electron transfer reactions between the excited PS with organic
substrate molecules in the cellular microenvironment to form highly reactive free
radicals, which subsequently interact with oxygen molecules to produce singlet oxygen
such as superoxide, hydroxyl radicals and hydrogen peroxide that are cytotoxic (Foote,
1991).
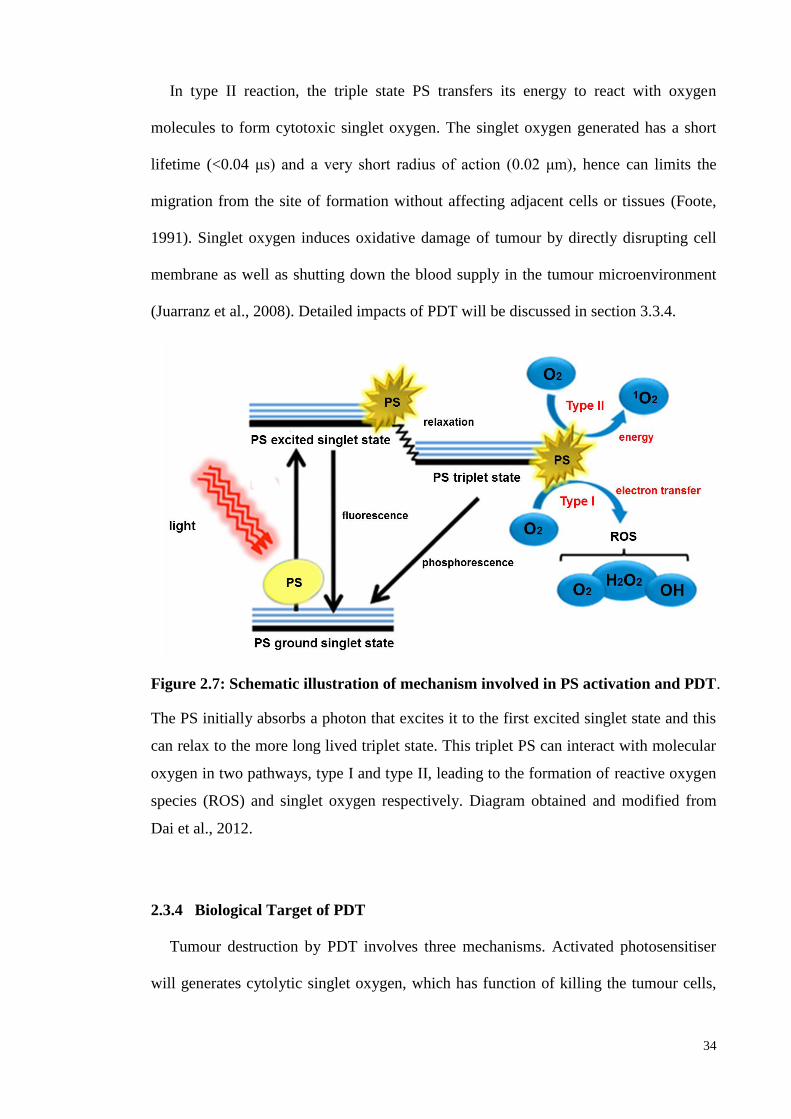
34
In type II reaction, the triple state PS transfers its energy to react with oxygen
molecules to form cytotoxic singlet oxygen. The singlet oxygen generated has a short
lifetime (<0.04 μs) and a very short radius of action (0.02 μm), hence can limits the
migration from the site of formation without affecting adjacent cells or tissues (Foote,
1991). Singlet oxygen induces oxidative damage of tumour by directly disrupting cell
membrane as well as shutting down the blood supply in the tumour microenvironment
(Juarranz et al., 2008). Detailed impacts of PDT will be discussed in section 3.3.4.
Figure 2.7: Schematic illustration of mechanism involved in PS activation and PDT.
The PS initially absorbs a photon that excites it to the first excited singlet state and this
can relax to the more long lived triplet state. This triplet PS can interact with molecular
oxygen in two pathways, type I and type II, leading to the formation of reactive oxygen
species (ROS) and singlet oxygen respectively. Diagram obtained and modified from
Dai et al., 2012.
2.3.4 Biological Target of PDT
Tumour destruction by PDT involves three mechanisms. Activated photosensitiser
will generates cytolytic singlet oxygen, which has function of killing the tumour cells,
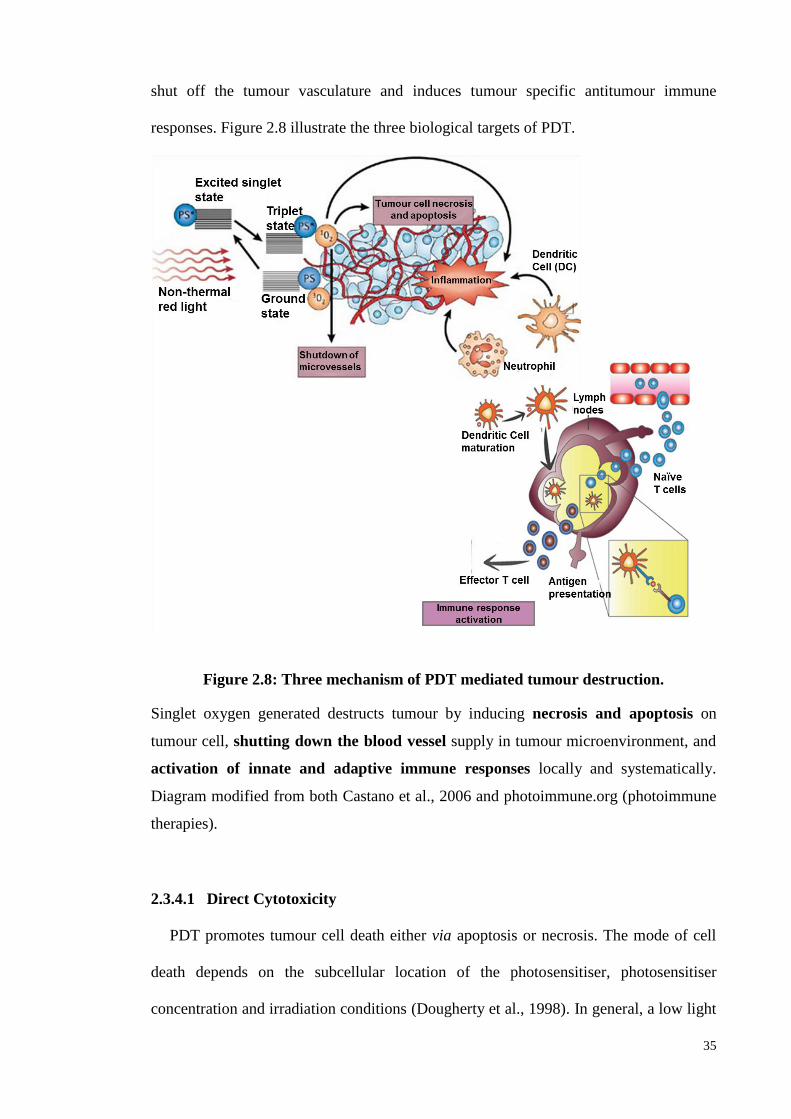
35
shut off the tumour vasculature and induces tumour specific antitumour immune
responses. Figure 2.8 illustrate the three biological targets of PDT.
Figure 2.8: Three mechanism of PDT mediated tumour destruction.
Singlet oxygen generated destructs tumour by inducing necrosis and apoptosis on
tumour cell, shutting down the blood vessel supply in tumour microenvironment, and
activation of innate and adaptive immune responses locally and systematically.
Diagram modified from both Castano et al., 2006 and photoimmune.org (photoimmune
therapies).
2.3.4.1 Direct Cytotoxicity
PDT promotes tumour cell death either via apoptosis or necrosis. The mode of cell
death depends on the subcellular location of the photosensitiser, photosensitiser
concentration and irradiation conditions (Dougherty et al., 1998). In general, a low light

36
dose is associated with apoptosis, while a higher light dose results in necrosis. Another
crucial factor in determining PDT-mediated cytotoxicity is the subcellular localisation
of photosensitiser, which is determined by the chemical properties, formulation, route of
delivery, concentration of the photosensitiser, and microenvironment of the lesion as
well as phenotype of target cells (Huang et al., 2008). Organelles such as endoplasmic
reticulum, Golgi apparatus, lysosomes, mitochondria, nucleus, and cell membrane have
been identified as the subcellular targets for photosensitisers. Among the organelles,
mitochondria are the primary subcellular target for most of the photosensitisers (Morgan
& Oseroff, 2001). Mitochondria cytochrome-c release, photodamage to Bcl-2 (pro-
survival proteins), and phospholipase activation (Agarwal et al., 1993) are example of
the apoptosis pathways that have been reported for PDT.
The localisation of photosensitiser in plasma membrane has been studied leads to cell
necrosis upon the photosensitisation process (Du et al., 2003; Mroz et al., 2011). Upon
irradiation, singlet oxygen species damage the plasma membrane proteins, lipids and
even cytoplasmic proteins. This leads to loss of membrane integrity and intracellular
ATP supplies. In addition, studies showed that activation of photosensitiser stimulates
autophagic cell death (Kessel & Reiners, 2007). The autophagy process in PDT has dual
functions, either in promoting survival by degrading or recycling the damaged
organelles, or by inducing cell death (Scherz-Shouval & Elazar, 2007), depending on
the severity of the damages mediated by generated singlet oxygen. In general,
autophagy cell death happens in tumour cells that lack apoptotic proteins (caspases-3,
Bax, Bac) (Buytaert et al., 2006). Therefore, autophagy can be an alternative cell death
pathway in cells deficient in apoptotic proteins.

37
2.3.4.2 Vascular Effect
Tumour microenvironment obtains their nutrients and oxygen primarily via the blood
capillaries, which are generated by the tumour itself through angiogenesis. Abnormal
neovascularisation in tumour lesions not only leads to enhanced supply of nutrients for
tumour growth, but also constitute an important point in cancer progression (Folkman,
2002). Microvascular destruction is an important mechanism for cancer PDT (Siemann
et al., 2005). Microvascular shutdown in tumour microenvironment upon PDT leads to
formation of hypoxic environment and nutrient deprivation, and ultimately tumour
regression (Henderson & Fingar, 1987).
Factors that affect the microvascular damage in PDT are the type of photosensitiser
used and the duration of drug-light interval (DLI), which refers to the time between
drug administration and irradiation. Photosensitisers that are suitable for use in vascular
targeting are photofrin (Allison et al., 2006), talaporfin sodium (Juzeniene, 2009),
verteporfin (Houle & Strong, 2002). They have been studied to be rapidly circulated and
accumulated in the vascular system after injection. PDT-mediated microvascular
damage regulated by DLI is depends on the pharmacokinetic and biodistribution of
photosensitisers in different tissues at different time points. In general, short DLI (0.5 h)
has better vascular targeting compared to long DLI (> 1 h) (Chen et al., 2002).
2.3.4.3 Immune Responses
Unlike conventional chemotherapy and radiotherapy, which are known to be
immune-suppressive (Penn & Starzl, 1973), PDT has been studied to stimulate antigen
specific immune responses, which can work synergistically with photosensitisers in

38
mediating short-term tumour killing, as well as long-term tumour immunity (Pizova et
al., 2012; Korbelik, 1996; Korbelik and Dougherty, 1999).
Preclinical studies have shown that PDT damages the tumour cells and causes the
release of damage-associated molecular pattern (DAMP) molecules which mediate an
inflammation (Korbelik, 2006). The release of DAMPs attracts the recruitment of
inflammatory cells such as neutrophils, mast cells and monocytes to the site of
irradiation after PDT. Inflammatory cytokines such as IL-1β, IL-2, IL-6, tumour
necrosis factor (TNF)-α, IL-10 and granulocytic-colony stimulatory factor (G-CSF) are
also reported to be actively secreted by immune cells and tumour stroma post-PDT,
which may regulate the systemic and tumour microenvironment immune responses
(Gollnick et al., 2003). In addition, PDT was reported to increase the release of tumour
antigens, which are then taken up by antigen presenting cells (APC), leading to the
presentation of tumour antigens via MHC class I protein complex to lymphocytes. This
initiates the onset of adaptive immune responses such as stimulation of tumour antigen
specific CD8+ T-cells with the help of helper CD4
+ T-cells. The activated T-cells will
undergo proliferation and differentiation into different T-cell subtypes based on the
surrounding cytokine milieu (Abdel-Hady et al., 2001). The detail of cancer
immunology will be discussed in section 2.4.
Recent research has led to the introduction of PDT-generated tumour lysate as
vaccines, which is a highly favourable method to stimulate and maximise the host
antitumour immune responses (Korbelik, 2011).

39
2.4 Cancer Immunology
The immune system plays a critical role in protecting the host from various diseases,
including cancer, through immune surveillance. Section 2.4.1 to 2.4.2 outlines the
activation mechanisms of the tumour antigen specific immune responses, and explains
the role played by an immune system to restrict tumour growth.
2.4.1 Tumour Antigen in Triggering Immune System
The benign tumour may turn to malignancy when they developed mechanisms to
escape the host’s immune surveillance. Despite the immunosuppressive chemotherapy,
there are some cases where the anti-tumour immune response can be elicited via the
drug-mediated cell killing that releases the tumour antigen (Casares et al., 2005).
Antigen presenting cells (APCs) including dendritic cells, macrophages and B cells are
responsible for processing the engulfed tumour antigens into short peptides and
presenting them to CD8 T-cells via major-histocompatibility complex (MHC) class-I
molecules. Self-antigen (tumour antigen) present their peptides in the form of MHC
class I molecules, whereas non-self-antigen present peptides in the form of MHC class-
II molecules to CD4 T helper cells.
Another factor that can activate the immune responses is tumour immunogenicity,
defined as the ability of tumour to elicit adaptive immune responses in vivo. Tumour
immunogenicity varies among cancer types and different individuals (Blankenstein et
al., 2012). There are three possible ways for self-antigens to become tumour antigens
(Finn, 2008): First is mutation that causes failure of cells to repair DNA damage,
resulting in abnormal peptide generation and presentation on the surface of tumour cells.
Secondly, mutated tumour cells overproduce proteins and presented them on cell
surface at highly abnormal levels. Thirdly, abnormal protein post-translation processes
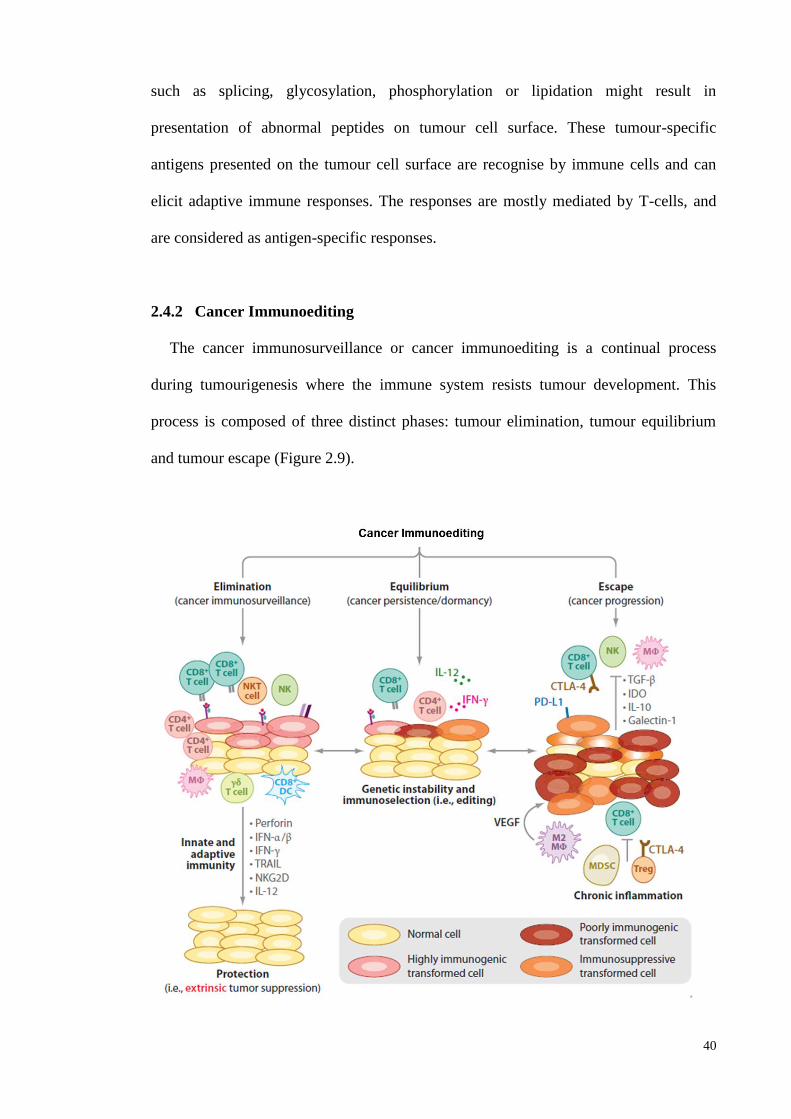
40
such as splicing, glycosylation, phosphorylation or lipidation might result in
presentation of abnormal peptides on tumour cell surface. These tumour-specific
antigens presented on the tumour cell surface are recognise by immune cells and can
elicit adaptive immune responses. The responses are mostly mediated by T-cells, and
are considered as antigen-specific responses.
2.4.2 Cancer Immunoediting
The cancer immunosurveillance or cancer immunoediting is a continual process
during tumourigenesis where the immune system resists tumour development. This
process is composed of three distinct phases: tumour elimination, tumour equilibrium
and tumour escape (Figure 2.9).

41
Figure 2.9: Three processes (Elimination, Equilibrium and Escape) of immuno-
editing in cancer.
Elimination involves tumour suppression by both innate and adaptive immune cells
such as NK cells, macrophage, dendritic cells, CD4+ and CD8+ T helper and NK T-
cells, with high levels of inflammatory and cytolytic molecules including IFN-g, IL-12,
TRAIL, NKG2D, and perforin. Equilibrium proceeded when tumour cells started to
undergo genetic instability and mutation to become new variants that are poor
immunogenics and resist to immune attack, at the same time immune cells still act on
highly immunogenic tumour cells. Escape occurs when poor immunogenic and
immunosuppressive transformed new variant tumour cells become dominant. Tumour
evades from immunosurveillance by expressing PD-L1, secretes immunesuppressive
mediators including IDO, TGF-b and IL-10 to encourage recruitment and differentiation
of immunosuppressive Treg cells to attenuate the effector T-cells function. Diagram
modified from Vesely et al., 2011.
2.4.2.1 Tumour Elimination
Tumour elimination phase is an example of immunosurveillance, which involves the
cytokines and cells of both innate and adaptive immunity to destroy the tumour cells
before it become clinically apparent (Vesely et al., 2011). When the tumour cells grow
invasively, they start to produce stromagenic and angiogenic proteins such as fibroblast
growth factor (FGF) and vascular endothelial growth factor (VEGF) to enhance the
blood nutrient supply to their microenvironment (Hanahan & Folkman, 1996). The
growth of the tumour cells cause the disruption of the tumour surrounding tissues and
induce inflammatory signaling to recruit the infiltration of immune cells into tumour
microenvironment. The major effector immune cells that infiltrate into tumour
microenvironment are natural killer cells (NK), dendritic cells (DC), macrophages,
neutrophils, eosinophils, basophils, mast cells, and cytotoxic T-lymphocytes. These
immune cells will be stimulated to produce inflammatory cytokines including
interferon-γ (IFN-γ), interleukin (IL)-12 and tumour necrosis factor (TNF) (Dranoff,

42
2004). The IFN-γ was said to be the pivotal cytokine in tumour elimination, as it can
induce CXCL-9, CXCL-10 and CXCL-11 chemokines production to attract more T-
lymphocytes infiltrate into the tumour site and generate antiangiogenic activity
(Angiolillo et al., 1995; Coughlin et al., 1998). All these factors will kill the tumour
cells by mechanisms including tumour necrosis factor-related apoptosis inducing
ligands (TRAIL), perforin, reactive oxygen and nitrogen intermediates (Ikeda et al.,
2002; Takeda et al., 2002; Trinchieri, 1995).
The process continues when dead tumour cells were engulfed by APCs, processed
and presented to naive T-lymphocytes in lymph nodes to prime more tumour-specific
CD4+ and CD8+ T-lymphocytes. Dendritic cell is APC that bridge between innate and
adaptive immune response, and may stimulate different CD4+ T-lymphocytes mediated
inflammatory responses such as T helper (Th) 1, Th2, Th17 and regulatory T-cells
(Treg). In general, the tumour antigen primed CD4+ T-lymphocytes will differentiate to
IFN-γ expressing Th1 to help the development and enhance the function of tumour-
specific CD8+ cytotoxic T lymphocytes (CTL). CTLs kill the remaining tumour cells by
secreting the cytolytic mediator perforin (Russell & Ley, 2002), and the surrounding
IFN-γ increases the sensitivity of tumour cells to CTLs-secreted perforin. Studies have
proved that IFN-γ and perforin are the two essential mediators for tumour elimination.
IFN-γ and perforin knockout mice respectively have significant incidence of lung
adenocarcinoma and lymphoma development compared to wild-type mice (Street et al.,
2002), and less effective in regulating the initiation, growth and metastasis of solid
tumour (Street et al., 2001; van den Broek et al., 1996). Other immune regulatory
cytokines including IL-12, IL-18 and TNF are the other important cytokines in
eliminating tumour (Street et al., 2002).

43
2.4.2.2 Tumour equilibrium
Those tumour cells that have survived the elimination process will enter into an
equilibrium process. The equilibrium state is originally from study of transplantation of
certain tumour cell lines into pre-immunised mice, where the immunity restrained the
outgrowth of inoculated tumour cells (Weinhold et al., 1977). In this state, the tumour
cells are considered “dormant”, as the elimination process continues and some of the
tumour cells undergo mutation and transformation to form a new variant with resistance
to immune attack (immune-escape). Some tumour cells in equilibrium are immunogenic
and susceptible to be cleared by immune system, whereas those that have undergone
transformation have attenuated immunogenicity and can escape immune surveillance.
Converse to the elimination process that requires both innate and adaptive immunity,
the equilibrium process depends fully on adaptive immunity, which is the T-
lymphocytes (Koebel et al., 2007). Koebel and colleagues reported that administration
of anti-CD4, anti-CD8, anti-IFN-γ and anti-IL-12 (cytokine critical for IFN-γ
production) promoted sarcoma outgrowth in mice which were in the equilibrium state.
The equilibrium state was proposed because the mice injected with a single low dose of
chemical carcinogen 3’-methylcholantrene (MCA) displayed small stable masses at the
site of MCA injection even up to 200-230 days post MCA inoculation. Under similar
experiment condition, tumour outgrowth was not observed in mice that deplete innate
immunity including NK cell recognition (NKG2D) or tumour necrosis factor-related
apoptosis inducing ligands (TRAIL). In contrast, the mice with Rag2 knockout gene
(gene that regulates adaptive T- and B-lymphocytes development, but possess innate
immunity) had progressive tumour outgrowth compared to wild-type in equilibrium
state.

44
Equilibrium state has the longest time periods among the three states where
immunity controls the cancer growth. The cancer in equilibrium state can become
functionally dormant and clinically unapparent to host (Vesely et al., 2011). Hence,
cancer immunotherapy can be applied in this state to enhance the adaptive immunity to
further improve tumour control.
2.4.2.3 Tumour escape
The new tumour variants developed during equilibrium state are less immunogenic,
and thus can survive and enter the escape state. Due to their insensitivity to
immunesurveillance, the new variant tumour cells begin to expand and growth in an
uncontrollable manner and become malignant (Dunn et al., 2002). In addition, tumour
evades from immunesurveillance by suppressing both the local and systemic immunities
via the active production of immunosuppressive molecules such as transforming growth
factor (TGF)-β, IL-10, soluble Fas ligand to induce CTL apoptosis, immunosuppressive
enzyme indolamine-2,3-dioxygenase and programmed death-ligand 1 (PD-L1).
Studies have reported that tumour cells produce high level of TGF-β and tumour
microenvironment has high population of immunosuppressive regulatory T-cells (Treg)
compared to effector T-cells. The Treg cells actively produce immunosuppressive
cytokines TGF-β and IL-10 to suppress the effector immune cells in both systemic and
tumour microenvironment. Moreover, induced-Tregs (iTregs) are produced alongside
increase in TGF-β, which is the main cytokine in priming naive or effector CD4+ T to
Treg cells (Liu et al., 2007). Thus, tumour evades from immune-mediated destruction
and become malignant. This was reported in many cancer types including lung, ovarian
(Woo et al., 2001), head and neck (Chikamatsu et al., 2007), melanoma (Cesana et al.,

45
2006), breast (Okita et al., 2005), pancreatic (Yamamoto et al., 2012) and
gastrointestinal cancer (Sasada et al., 2003).
Indolamine-2,3-dioxygenase (IDO) is an enzyme responsible for tryptophan
catabolism amino acid through the kynurenine pathway. It is an immunosuppressive
enzyme as it induces tryptophan catabolism. This suppresses T-lymphocytes, NK cells
and B-lymphocytes proliferation by reducing the supply of essential tryptophan
(Frumento et al., 2002; Moffett & Namboodiri, 2003). Also, the tryptophan metabolite
kynurenine and 3-hydroxyanthranilic acid (3HAA) are toxic to activated immune cells
(Fallarino et al., 2003). In addition, IDO can trigger the generation of iTreg from naive
T-cells via plasmacytoid dendritic cells (PDCs) which express high levels of IDO (Chen
et al., 2008). Clinical studies reported that IDO was highly expressed in 56.7% of
surgically resected ovarian cancer tissues and its expression was correlated with a
reduced number of CD8+ tumour infiltrated lymphocytes (Inaba et al., 2009). Moreover,
ovarian cancer that were positive for IDO staining was found to be correspondence with
the primary lesion and metastatic site, suggested the increase of IDO had impaired
survival in ovarian cancer patient (Takao et al., 2007). Another study involving
transfections of IDO vector into non-IDO expressing human ovarian cancer cell line
OMC-1 revealed that it could promote tumour growth, inhibits NK cells in tumour
microenvironment and induce angiogenesis (Nonaka et al., 2011).
Another molecule overexpressed in cancer cells is programmed death ligand-1 (PD-
L1) (Maine et al., 2014). PD-L1 binds to PD1 receptors that are expressed on activated
T-lymphocytes, B-lymphocytes and myeloid cells (Agata et al., 1996). PD1 down-
regulates activated immune cells to reduce autoimmunity and promotes self-tolerance.
Many cancer types including ovarian (Maine et al., 2014), kidney (Thompson et al.,

46
2004), breast (Ghebeh et al., 2006), melanoma (Hino et al., 2010), esophageal (Ohigashi
et al., 2005) and pancreatic (Nomi et al., 2007) express high PD-L1 and this is
associated with poor prognosis in patient. The high PD-L1 is associated with low
tumour infiltrated lymphocytes into tumour microenvironment (Hamanishi et al., 2007).
Other than Treg, myeloid derived suppressor cell (MDSC) is another
immunosuppressive cell that negatively regulates immune responses in cancer. MDSC
is a heterogeneous population of activated myeloid cells characterised by a mixture of
granulocytic and monocytic cells lacking the surface markers associated with fully
differentiated monocytes, macrophage or dendritic cells (Youn et al., 2008). MDSC is
highly expanded in cancer due to the active secretion of expansion factors such as IL-6
(Bunt et al., 2007), prostaglandin (Sinha et al., 2007), TGF-β (Terabe et al., 2003; Yang
et al., 2008), macrophage-colony stimulating factor (Menetrier-Caux et al., 1998) and
VEGF (Gabrilovich et al., 1998) by tumour cells. MDSC functions by disrupting the
binding of specific MHC peptide in APC to CD8+ T-lymphocytes (Nagaraj et al., 2007).
Preclinical (Donkor et al., 2009; Younos et al., 2012) and clinical (Diaz-Montero et al.,
2009; Porembka et al., 2012; Wang et al., 2013) studies have revealed that the number
of MDSC in systemic circulation is correlated with poor prognosis, tumour
angiogenesis and tumour escape from immunity.

47
CHAPTER 3: TROPOMYOSIN RECEPTOR KINASE-C (TRKC) LIGAND
CONJUGATED DIIODO-BORON DIPYRROMETHENE ERADICATES TRKC
EXPRESSING TUMOUR IN PHOTODYNAMIC THERAPY (PDT)
3.1 Introduction
Active targeting ligand-drug conjugate in cancer refers to the specific delivery of
therapeutic drugs or imaging probes that are conjugated with the targeting ligand to the
tumour cells. This therapeutic approach is different from mechanistic or direct targeting,
that uses cytotoxic agent intended to target surface molecules or intracellular pathways
that are up-regulated in cancer cells (Agarwal et al., 2008; Minko et al., 2004). To date,
targeting ligand such as folic acid and prostate specific membrane antigen (PSMA)
inhibitor to target folate receptor and PSMA, respectively (Hilgenbrink & Low, 2005;
Low & Kularatne, 2009; Lu & Low Philip, 2012; Xia & Low, 2010) are probably the
most widely appreciated examples. These PSMA and folate receptor conjugates have
been brought to clinic for therapeutic and imaging of solid tumours (Srinivasarao et al.,
2015). However, there are no clinically approved small molecule active targeting agents
for delivering therapeutics to breast cancer (Meng & Li, 2013).
Other than targeted cancer therapy using active targeting ligand-drug conjugate,
photodynamic therapy (PDT) is considered as another selective anticancer therapy. PDT
requires administration of photosensitiser (PS), follow by the illumination of tumour at
specific wavelengths to activate the PS that had been accumulated in the tumour to
generate cytolytic singlet oxygen species to mediate tumour cell killing (Dolmans et al.,
2003; Dougherty et al., 1998). PDT has minimal invasiveness and consider specific due
to the only targeted region being illuminated, which can reduce the non-selective
toxicity to normal cells that uptake the PS. However, the low to moderate accumulation

48
and localised of PS in tumours to fully eradicate irradiated tumour region is the
limitation of PDT.
IY-monomer
Figure 3.1: Structure of synthetic peptide Isoleucine-tYrosine (IY) to bind TrkC
receptor.
Tropomyosin receptor kinase C (TrkC) is a cell surface receptor that is
overexpressed in breast cancer and plays an essential role in breast cancer growth and
metastasis (Ivanov et al., 2013; Stephens et al., 2005). Studies have shown that
suppression of TrkC expression in highly metastatic mammary carcinoma cells
inhibited their growth in vitro and their ability to metastasis from the mammary gland to
the lung in vivo (Jin et al., 2010). The high expression of TrkC has made it a good
biomarker for therapeutic purposes in cancer. A novel molecular fragment IY (Figure
3.1), of which consist of amino acid Isoleucine (I) and tYrosine (Y) has been designed
as a TrkC targeting ligand (Brahimi et al., 2009; Chen et al., 2009; Liu et al., 2010). A
preliminary study had shown that two fragments of IY (IYIY)-ligand gave good
adherent to TrkC receptor, whereas one fragment IY-ligand induces weak functional
effects (Chen et al., 2009).

49
I2-BODIPY
Figure 3.2: Structure of photosensitiser diiodinated BODIPY used in this study.
Following the successful targeting ability of the IYIY-ligand to TrkC expressing
cancer (Chen et al., 2009), the ligand had then conjugated with photosensitiser diiodo-
boron dipyrromethene (I2-BODIPY) as a cargo (Figure 3.2). Boron dipyrromethene
(BODIPY) and derivatives have excellent attributes for PDT with high extinction
coefficients, favorable light-to-dark toxicity ratios, high antitumour efficacies in vivo,
and good body clearance (Awuah & You, 2012; Byrne et al., 2009; Kamkaew et al.,
2012; Lim et al., 2010; Yogo et al., 2005). The conjugate, named IYIY-I2-BODIPY
(Figure 3.3, upper panel) was used to investigate the binding selectivity, toxicity profile,
biodistribution and in vivo efficacy on TrkC expressing murine 4T1 breast cancer cells
in PDT. In this study, non-TrkC targeted scrambled control, named YIYI-I2-BODIPY
(Figure 3.3, lower panel) and free drug I2-BODIPY were used as negative controls.
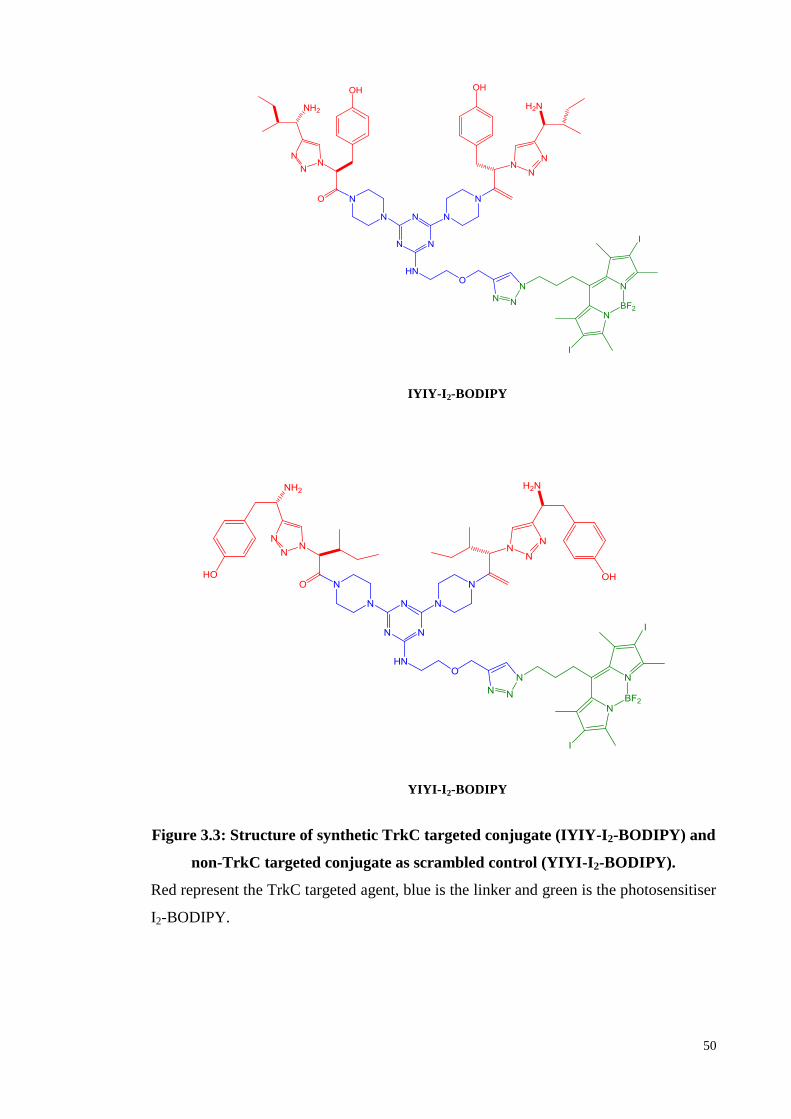
50
IYIY-I2-BODIPY
YIYI-I2-BODIPY
Figure 3.3: Structure of synthetic TrkC targeted conjugate (IYIY-I2-BODIPY) and
non-TrkC targeted conjugate as scrambled control (YIYI-I2-BODIPY).
Red represent the TrkC targeted agent, blue is the linker and green is the photosensitiser
I2-BODIPY.

51
3.2 Materials and Methods
3.2.1 Compounds and Cell Lines
Compounds IYIY-I2-BODIPY, YIYI-I2-BODIPY and I2-BODIPY were a gift from
Prof. Dr. Kevin Burgess from Texas A&M University, USA. 4T1 murine mammary
carcinoma cell line was purchased from American Type Culture Collection, ATCC
(Manassas, VA, USA) and cultured in RPMI media with 10% FBS. 67NR murine
mammary carcinoma cell line was purchased from Barbara Ann Karmanos Cancer
Institute, Detroit, MI, USA; and cultured in DMEM high glucose medium with 10%
FBS. All cells were maintained at 37oC in a 5% CO2 incubator.
3.2.2 In vitro Photocytotoxic Assay
4T1 and 67NR cell lines were respectively cultured in 96 wells plate at density of
4000 cells per well for 24 hours. Compounds IYIY-I2-BODIPY, YIYI-I2-BODIPY and
I2-BODIPY were dissolved in DMSO and diluted to the range of 0.1-10 μM. The cell
lines were incubated respectively with compounds at prepared concentrations for 2, 4
and 6 hours. At the end of incubation, all cells were washed twice with PBS. Fresh
media was added to the culture wells and irradiated with a light dose of 7.3 J/cm2 from a
halogen light source at the fluence rate of 12.2 mW/cm2 (Kamkaew & Burgess, 2013),
and further incubated for 24 hours. Briefly, 20 uL of MTT solutions (5 mg/mL) were
added to each well and incubated for 4 hours in 37oC. The solutions were removed and
100 uL of DMSO was added to dissolve the formazan crystal formed. Plate was read at
absorbance of 570 nm using SpectraMax® M4 Multi-Mode Microplate Reader
(Molecular Devices, Sunnyvale, CA, USA). The viability of each group of treatment in
different cell lines was calculated as percentage of dead cells = 100 − (OD treated / OD
untreated control) × 100.

52
3.2.3 Animal Model
Female 8- to 10-week old wildtype BALB/c mice were purchased from Monash
University, Malaysia campus (Sunway, Selangor, Malaysia) for in vivo studies. The
mice were maintained under AAALAC accredited satellite animal facility in
Department of Pharmacology, Faculty of Medicine, University of Malaya, Kuala
Lumpur, Malaysia. All animal experiments were performed according to protocol
approved by the Faculty of Medicine Institutional Animal Care and Use Committee,
University of Malaya (FOM IACUC). (Ethics Approval No. 2013-05-07/PHAR/KLV,
appendix A).
3.2.4 In vivo Toxicity Study
The toxicity profiles of all compounds were determined. Mice (n=2) were injected
intravenously with IYIY-I2-BODIPY, YIYI-I2-BODIPY and I2-BODIPY at 20-100
mg/kg via tail vein. Toxicity was observed based on the Berlin test of typical symptoms
such as apathy, horrent fur, behavior changes and a loss of body weight for two weeks.
3.2.5 Biodistribution Study
4T1 tumour bearing female BALB/c mice at the tumour volume of 80 mm3 were
divided into 2 groups, and intravenously injected with 10 mg/kg of IYIY-I2-BODIPY
and YYI-I2-BODIPY respectively. Mice (n=3) were then sacrificed at different time
points (0, 0.25, 1, 3, 6, 24, 48 and 72 hours) and major organs such as liver, spleen, lung,
kidney, draining lymph nodes, skin, eye and tumour tissues were isolated. Organs and
tissues were imaged using an In Vivo MS FX PRO imager (Carestream Molecular
Imaging, Woodbridge, CT, USA) with an excitation filter at 530 nm and an emission
filter at 550 nm. Mice with saline treatment were used as control. Fluorescent intensity
of each organs and tissues were quantified using Carestream Molecular Imaging

53
software 5.0 (CT, USA). The time point that gives a high accumulation in tumour
tissues and large differences with the scrambled control was chosen for PDT.
3.2.6 Tumour cells inoculation and PDT in mouse
The fur of the mouse was first shaved. Murine 4T1 and 67NR at density of 5 x 105
tumour cells in 0.1 mL of medium was then orthotopically injected into the mammary
fat pad of the mice (18-20 g, 8-10 weeks old) respectively. The mice were then
randomly divided to groups for PDT at 8 day post tumour inoculation when the tumour
size reaches approximately 60-80 mm3. Compounds (IYIY-I2-BODIPY, YIYI-I2-
BODIPY and I2-BODIPY) at 2-10 mg/kg body weight were dissolved in cocktail of 2.5%
ethanol and 2.5% Cremophore EL. The mixture was then further dissolved using saline
to volume of 0.2 mL and administered by intravenous tail vein injection to the mice
(n=7). Mice were then kept in dark for 1 hour. Anesthetic with 90 mg/kg of ketamine
and 10 mg/kg of xylazine cocktail was then given to mice. Thereafter, PDT was
performed using Lumacare LC-122A fibre optic light delivery system (Lumacare
Medical Group, Newport Beach, CA, USA) emitting light at 530 nm. Glass slide with
thickness of 4mm was used as a barrier to avoid direct photo-thermal effect on tumour.
Illuminating spot was positioned at the tumour region and the surrounding was covered
using black cloth to avoid PDT effect on other part of body. PDT was conducted at 100
J/cm2 with the fluence rate of 160 mW/cm
2. Mice were kept in dark and tumour sizes
were monitored 3 times per week. Tumour volume changes were determined by caliper
measurements with tumour volume, mm3 = (L x W
2 / 2), where L is the longest
dimension and W is the shortest dimension. The experimental mice were euthanised
when either tumour diameter reached the 20 mm ethical limit approved by IACUC
Guidelines for Maximum Tumour Bearing in Laboratory Rodents, 2012, or at the end of
the 90 day of monitoring period for survivor mice.

54
3.2.7 Histology Sample Preparation
Survivor mice were sacrificed at 90 days post therapy, and major organs such as liver,
kidney, spleen, draining lymph node, lung, and heart were isolated for histological
analysis. The isolated specimens were fixed with 10% formalin solution for a minimum
of 48 hours at room temperature, following which, tissues were trimmed into
representative segments and then dehydrated using an ascending series of alcohol,
cleared in xylene, and embedded in paraffin. Microtomy was performed using a Leica
RM2255 microtome (Leica Microsystems, Germany). Sections were cut at 5 μm
thickness and placed onto appropriately labeled microscope slides. The slides were then
stained with haematoxylin and eosin (H&E) and coverslipped, and then evaluated by a
board certified veterinary pathologist at the Institute of Molecular and Cell Biology,
Agency for Science, Technology and Research, in Singapore.
3.2.8 Statistical Analysis
In vitro and in vivo experiments were performed to compare the efficacy of three
compounds. Results were analysed using one-way ANOVA with Dunnett’s Multiple
Comparisons when comparing between three groups of compounds using SPSS with p
< 0.05 is considered as significant. Student t-test was used to analyse between two
groups, with p < 0.05 considered as significant.
3.3 Results
3.3.1 In vitro photocytotoxicity of ligated conjugates and unconjugated I2-
BODIPY
TrkC targeted dipeptidomimetic ligand (IYIY) has been studied to transduce signals
in promoting neuronal cell survival and differentiation in a similar pattern as TrkC
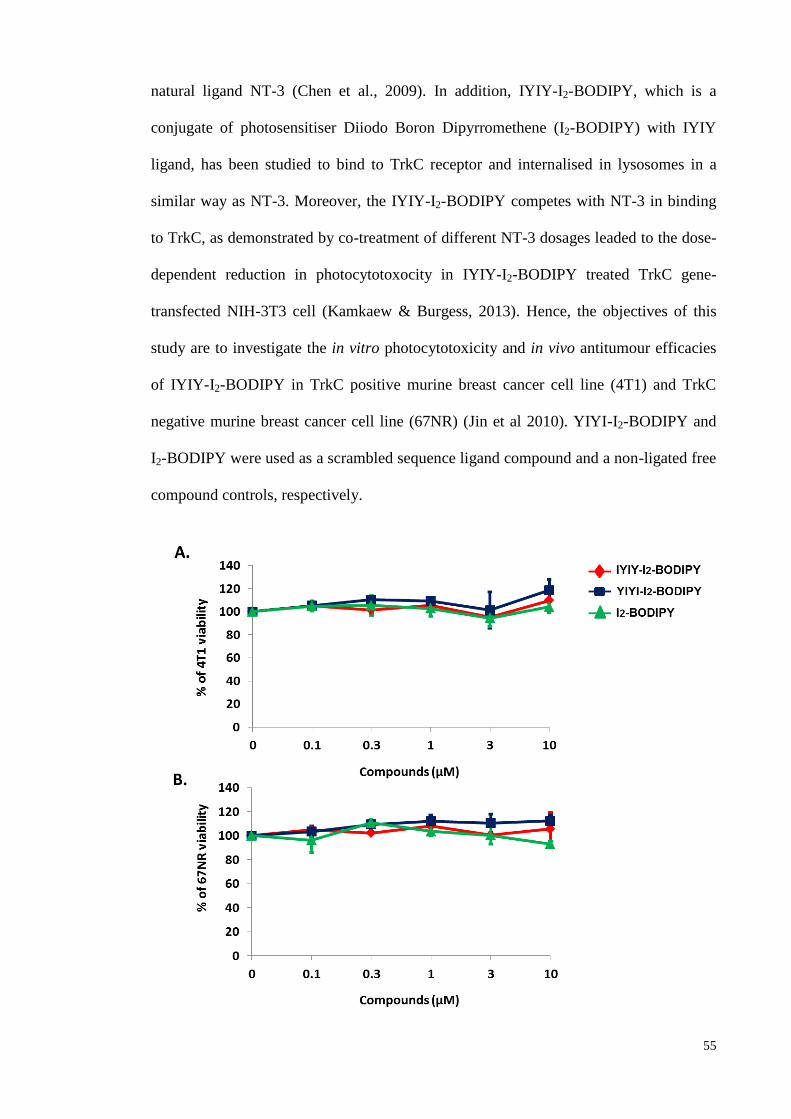
55
natural ligand NT-3 (Chen et al., 2009). In addition, IYIY-I2-BODIPY, which is a
conjugate of photosensitiser Diiodo Boron Dipyrromethene (I2-BODIPY) with IYIY
ligand, has been studied to bind to TrkC receptor and internalised in lysosomes in a
similar way as NT-3. Moreover, the IYIY-I2-BODIPY competes with NT-3 in binding
to TrkC, as demonstrated by co-treatment of different NT-3 dosages leaded to the dose-
dependent reduction in photocytotoxocity in IYIY-I2-BODIPY treated TrkC gene-
transfected NIH-3T3 cell (Kamkaew & Burgess, 2013). Hence, the objectives of this
study are to investigate the in vitro photocytotoxicity and in vivo antitumour efficacies
of IYIY-I2-BODIPY in TrkC positive murine breast cancer cell line (4T1) and TrkC
negative murine breast cancer cell line (67NR) (Jin et al 2010). YIYI-I2-BODIPY and
I2-BODIPY were used as a scrambled sequence ligand compound and a non-ligated free
compound controls, respectively.

56
Figure 3.4: Tested compounds were not toxic to cells in non-irradiated condition.
(A) 4T1 and (B) 67NR cells were cultured in 96-well plates. After 24 hours, IYIY-I2-
BODIPY, YIYI-I2-BODIPY and I2-BODIPY with indicated concentrations were added
to cells and incubated for 2 hours. The media was removed and the cells were then
washed with PBS before new media was added. MTT was carried out 24 hours after
further incubation. Data represent mean ± SEM of three independent experiments in
PDT.
The conjugates and free drug photosensitiser were first tested for dark cytotoxicity.
As shown in Figure 3.4, all compounds tested were not toxic to 4T1 and 67NR cells in
non-irradiated condition. When exposed to light, IYIY-I2-BODIPY induced significant
photocytotoxicity in 4T1 cell line (IC50 = 0.325 M) in a dose-dependent manner
compared to control YIYI-I2-BODIPY and I2-BODIPY (indeterminable IC50 values)
(Figure 3.5A). Importantly, IYIY-I2-BODIPY, YIYI-I2-BODIPY as well as I2-BODIPY
showed no photocytotoxic effect in TrkC negative cell line 67NR (Figure 3.5B). These
results suggest that IYIY-I2-BODIPY induces selective photocytotoxicity in TrkC
expressing cells via TrkC targeting.
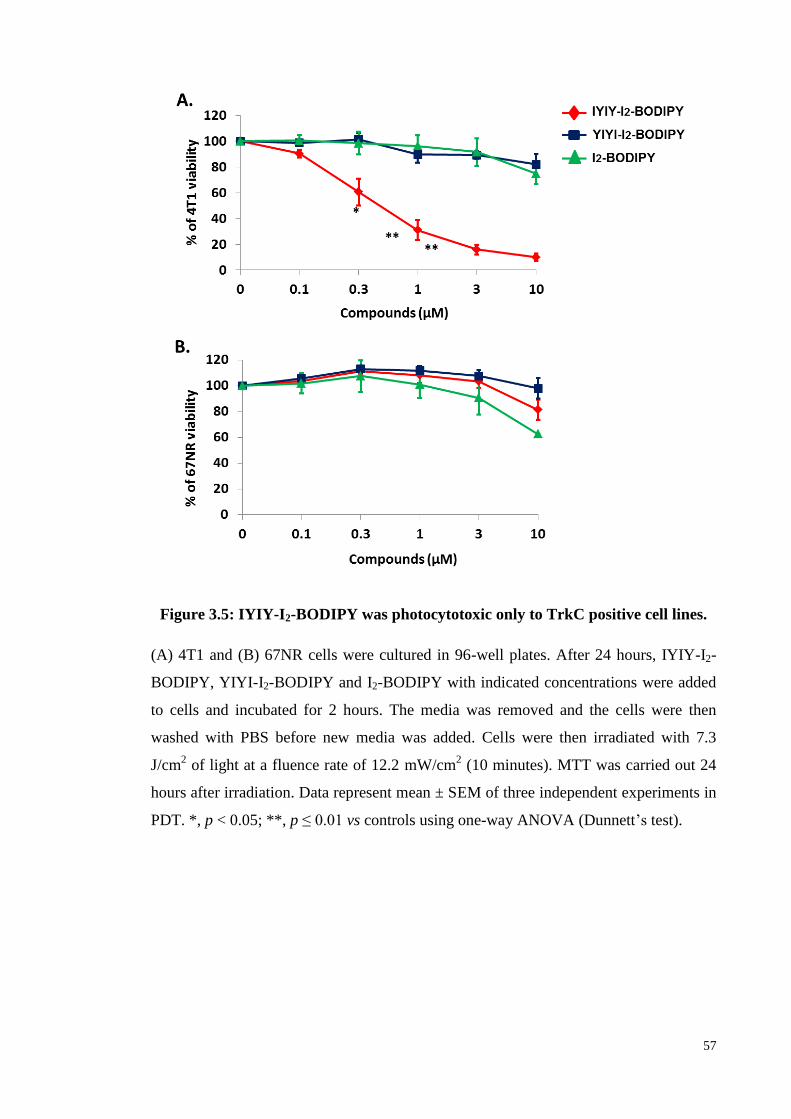
57
Figure 3.5: IYIY-I2-BODIPY was photocytotoxic only to TrkC positive cell lines.
(A) 4T1 and (B) 67NR cells were cultured in 96-well plates. After 24 hours, IYIY-I2-
BODIPY, YIYI-I2-BODIPY and I2-BODIPY with indicated concentrations were added
to cells and incubated for 2 hours. The media was removed and the cells were then
washed with PBS before new media was added. Cells were then irradiated with 7.3
J/cm2 of light at a fluence rate of 12.2 mW/cm
2 (10 minutes). MTT was carried out 24
hours after irradiation. Data represent mean ± SEM of three independent experiments in
PDT. *, p < 0.05; **, p ≤ 0.01 vs controls using one-way ANOVA (Dunnett’s test).

58
3.3.2 In vitro photocytotoxicity of ligated conjugates at different incubation time
in TrkC positive and TrkC negative cancer cells.
All the photocytotoxicity experiments described above involved adding the test
compounds for 2 hours incubation, followed by illumination. The effects of prolonging
the incubation with the cells on the cell viability post-illumination were also studied.
Interestingly, the data revealed that IYIY-I2-BODIPY mediated selective cell death was
achieved for Trk positive 4T1 compared to TrkC negative 67NR at 2 hours incubation
(Figure 3.6A), and the difference becomes less noticeable when 4- and 6-hours
incubation was used. This observation leads to the conclusion that a shorter incubation
time is optimal for selective photo-killing by the TrkC seeking conjugate. For a full
comparison, the same time course experiments for the untargeted YIYI-I2-BODIPY
(Figure 3.6B) and the parent I2-BODIPY was conducted (Figure 3.6C). As expected,
both compounds increased photo-killing with increasing incubation time. However, the
extent of cell death observed between 4T1 and 67NR cells were similar across the
different time points for incubation, implying no selective binding to cell surface
receptors.
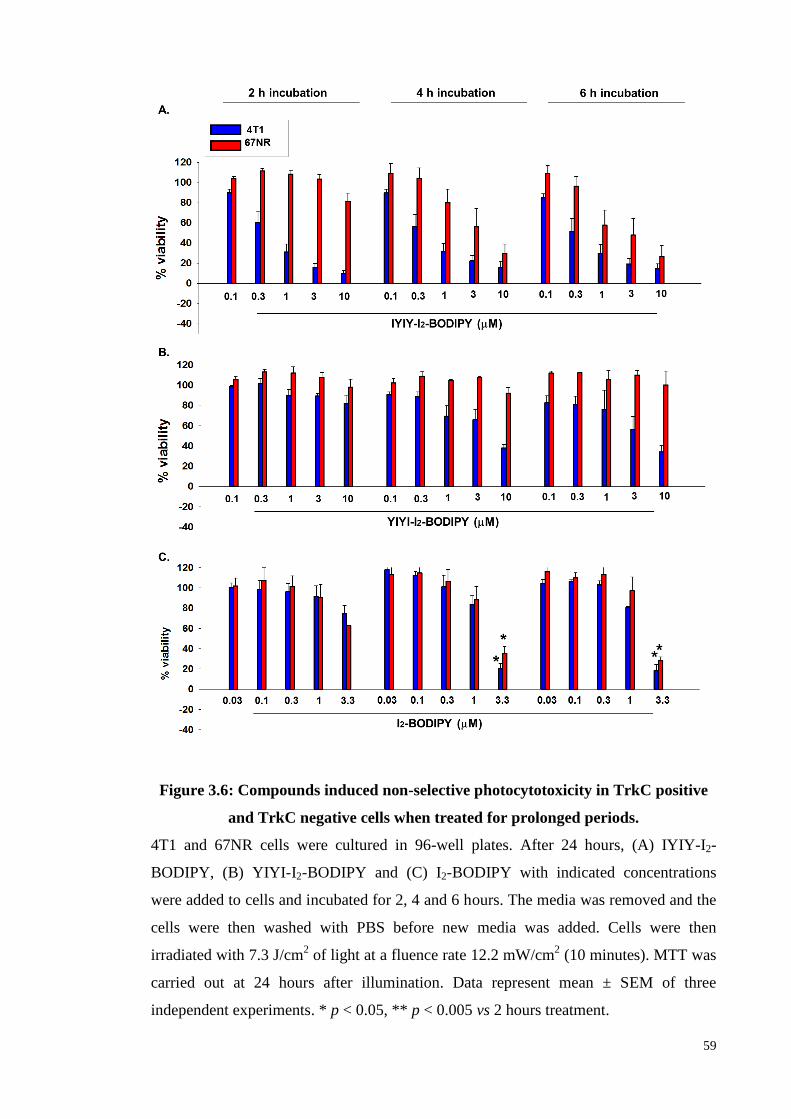
59
Figure 3.6: Compounds induced non-selective photocytotoxicity in TrkC positive
and TrkC negative cells when treated for prolonged periods.
4T1 and 67NR cells were cultured in 96-well plates. After 24 hours, (A) IYIY-I2-
BODIPY, (B) YIYI-I2-BODIPY and (C) I2-BODIPY with indicated concentrations
were added to cells and incubated for 2, 4 and 6 hours. The media was removed and the
cells were then washed with PBS before new media was added. Cells were then
irradiated with 7.3 J/cm2 of light at a fluence rate 12.2 mW/cm
2 (10 minutes). MTT was
carried out at 24 hours after illumination. Data represent mean ± SEM of three
independent experiments. * p < 0.05, ** p < 0.005 vs 2 hours treatment.
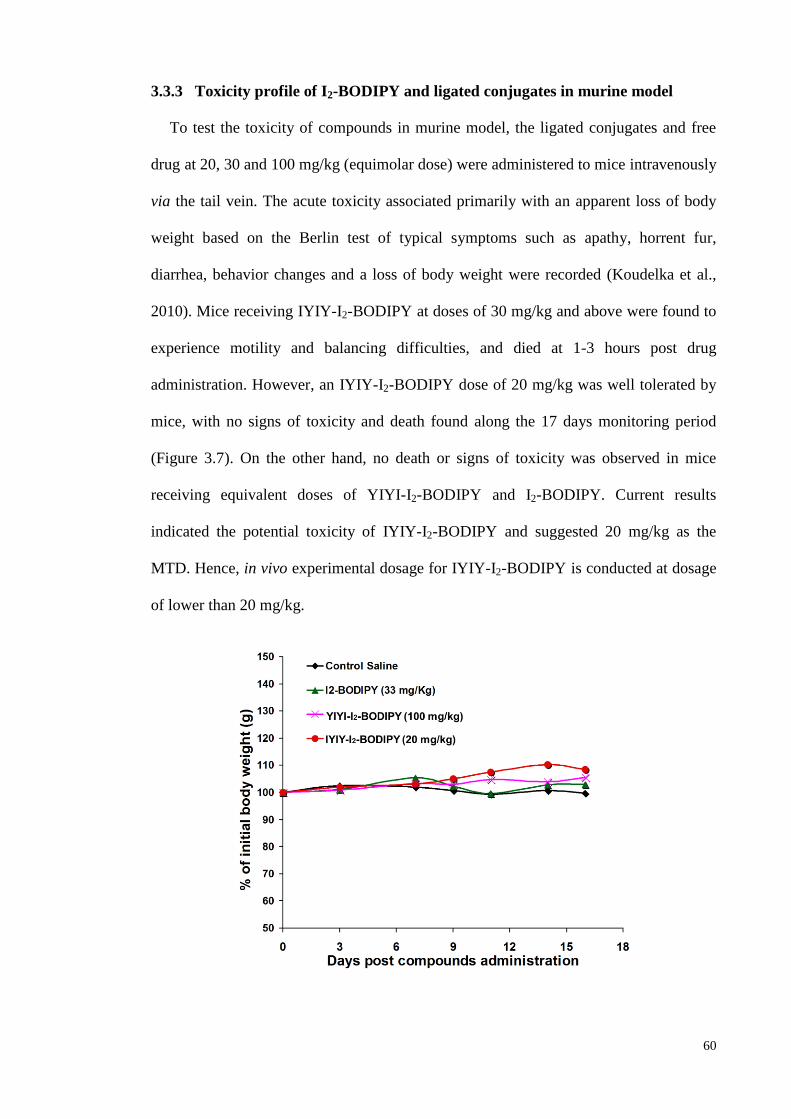
60
3.3.3 Toxicity profile of I2-BODIPY and ligated conjugates in murine model
To test the toxicity of compounds in murine model, the ligated conjugates and free
drug at 20, 30 and 100 mg/kg (equimolar dose) were administered to mice intravenously
via the tail vein. The acute toxicity associated primarily with an apparent loss of body
weight based on the Berlin test of typical symptoms such as apathy, horrent fur,
diarrhea, behavior changes and a loss of body weight were recorded (Koudelka et al.,
2010). Mice receiving IYIY-I2-BODIPY at doses of 30 mg/kg and above were found to
experience motility and balancing difficulties, and died at 1-3 hours post drug
administration. However, an IYIY-I2-BODIPY dose of 20 mg/kg was well tolerated by
mice, with no signs of toxicity and death found along the 17 days monitoring period
(Figure 3.7). On the other hand, no death or signs of toxicity was observed in mice
receiving equivalent doses of YIYI-I2-BODIPY and I2-BODIPY. Current results
indicated the potential toxicity of IYIY-I2-BODIPY and suggested 20 mg/kg as the
MTD. Hence, in vivo experimental dosage for IYIY-I2-BODIPY is conducted at dosage
of lower than 20 mg/kg.

61
Figure 3.7: IYIY-I2-BODIPY was not toxic to mice at 20 mg/kg.
Healthy 7−8 weeks old BALB/c female mice (n=2/group) were administered
intravenously via tail vein respectively with IYIY-I2-BODIPY and YIYI-I2-BODIPY at
20, 30, and 100 mg/kg (I2-BODIPY content equivalent to 6.25, 10, and 33 mg/kg,
respectively, i.e., corrected for MW), and the parent I2-BODIPY (33 mg/kg). The mice
were then kept in the dark and observed for 16 days. Data represent the average body
weight (grams) of two mice/treatment group.
3.3.4 In vivo biodistribution study in 4T1 tumour bearing mouse
The biodistribution of IYIY-I2-BODIPY and the isomeric YIYI-I2-BODIPY were
monitored in 4T1 tumour bearing mice (n = 3/ each time point, 0, 0.25, 1, 3, 6, 12, 24,
48, 72 hours). At 1 hour post administration, the fluorescence intensity of tumours in the
mice treated with IYIY-I2-BODIPY was the highest among the time points investigated,
and significant higher (2.1-fold) than the corresponding intensities for tumours treated
with the YIYI-I2-BODIPY (416000 ± 43000). Among the organs and tissues
investigated, tumour tissue was the third highest in IYIY-I2-BODIPY accumulation
after excretory organs liver and kidney. Interestingly, the accumulation of IYIY-I2-
BODIPY dye intensity in tumour tissue remained significant high compared with YIYI-
I2-BODIPY for up to 6 hours (p = 0.038). The dye started to diminish from tumour
tissue at 24 hours onwards with no significant differences between IYIY-I2-BODIPY
and YIYI-I2-BODIPY (Figure 3.8).
The accumulation of both ligated conjugates in non-targeted organs such as liver,
kidney and lung within the first 3 hours post administration was observed in high
intensity (Figure 3.8), but these accumulations dissipated swiftly in the subsequent
monitoring period. Similar accumulation and clearance pattern of both conjugates in

62
these organs is typical of small molecular weight compounds and indicates that the
accumulation in these organs was random and not due to TrkC receptor binding.
In addition, non-selective accumulation of ligated conjugates was also observed in
lymphoid organs such as spleen and lymph node, but at a much lower level. This
observation supported by other study on low to negligible TrkC expression for these
organs (Levanti et al., 2001). Other organs including eye and skin have relatively low
TrkC receptor expression (Cui et al., 2002), and TrkC for skin mainly concentrated at
nerve bundle portions (Botchkarev et al., 1999) (Figure 3.8). The maximum
accumulation of IYIY-I2-BODIPY at 1 hour post administration led us to adopt a drug-
to-light interval of 1 hour in determining in vivo antitumour efficacies in the subsequent
studies.
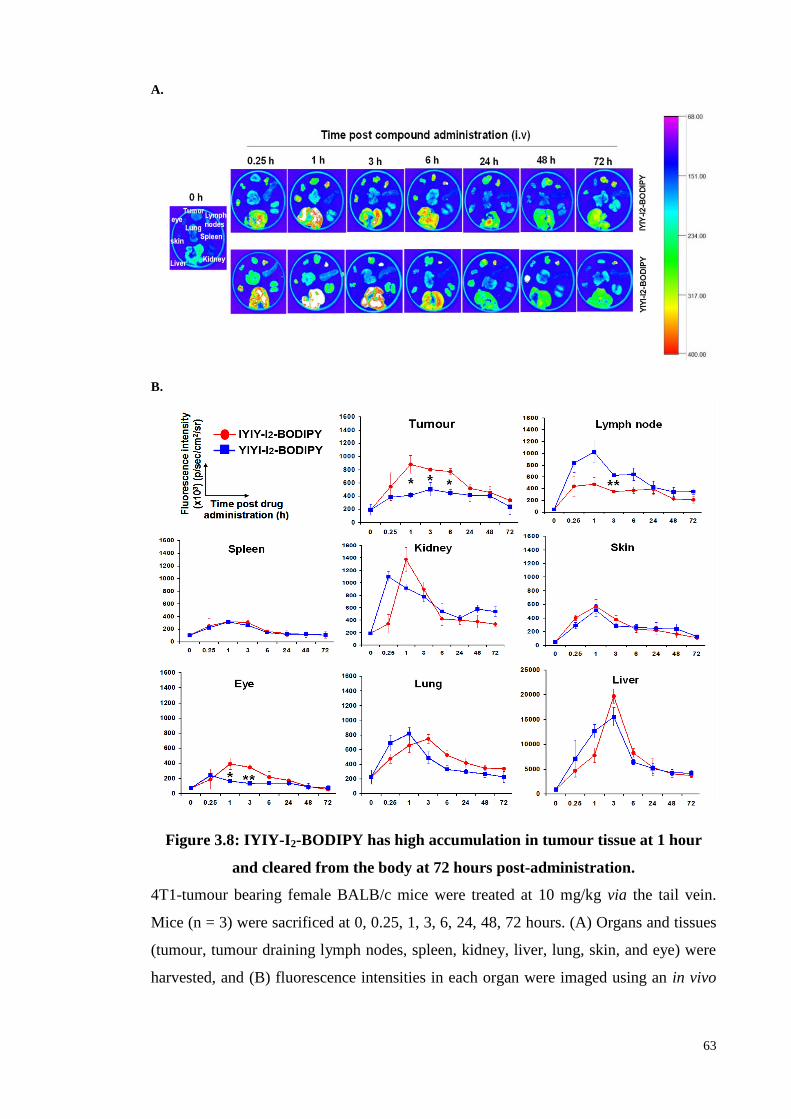
63
A.
B.
Figure 3.8: IYIY-I2-BODIPY has high accumulation in tumour tissue at 1 hour
and cleared from the body at 72 hours post-administration.
4T1-tumour bearing female BALB/c mice were treated at 10 mg/kg via the tail vein.
Mice (n = 3) were sacrificed at 0, 0.25, 1, 3, 6, 24, 48, 72 hours. (A) Organs and tissues
(tumour, tumour draining lymph nodes, spleen, kidney, liver, lung, skin, and eye) were
harvested, and (B) fluorescence intensities in each organ were imaged using an in vivo

64
imager (data represent mean ± SEM of three mice at each time point). * p < 0.05; ** p <
0.01 vs YIYI-I2-BODIPY (student’s t-test).
3.3.5 In vivo PDT antitumour efficacy of IYIY-I2-BODIPY in TrkC positive 4T1
tumour bearing mouse
The efficacies of compounds in eradicating the TrkC positive tumour in BALB/c
mouse model were examined. Aggressive TrkC positive murine breast carcinoma 4T1
cells were subcutaneously injected to the mammary fat pad, followed by intravenous
injection of compounds (2 mg/kg and 10 mg/kg) when the sizes reached 60-80 mm3. At
1 hour post compound administration, the mice will be anaesthetised and irradiated at
100 J/cm2 (Figure 3.9A).
The data revealed that there was significant reduction of tumour volume in IYIY-I2-
BODIPY treated group at 4−6 days post illumination, and healed from eschar by day
13−15. In contrast, YIYI-I2-BODIPY and I2-BODIPY treated mice showed a reduction
in tumour size post PDT, followed by tumour regrowth at day 13-15 at the peripheral of
necrotic tumour tissues (Figure 3.9B). In details, there were 61% and 96% maximum
tumour reduction in mice treated with 2 and 10 mg/kg of IYIY-I2-BODIPY compared to
pre-treatment tumour size at 4-6 days post PDT. Conversely, YIYI-I2-BODIPY and I2-
BODIPY treatment induced only moderate tumour size reduction within the first 6 days
after illumination (20% reduction in mice treated with 10 mg/kg of YIYI-I2-BODIPY,
and 11% reduction in mice treated with 3.3 mg/kg of I2-BODIPY, equimolar dose).
Most importantly, 1 out of 7 mice (14%), treated with 2 mg/kg, and 5 out of 7 mice
(71%), treated with 10 mg/kg of IYIY-I2-BODIPY showed no palpable tumour for up to
90 days post-treatment. Conversely, no tumour remission was found in both the YIYI-
I2-BODIPY and I2-BODIPY treated groups (Figure 3.9C).
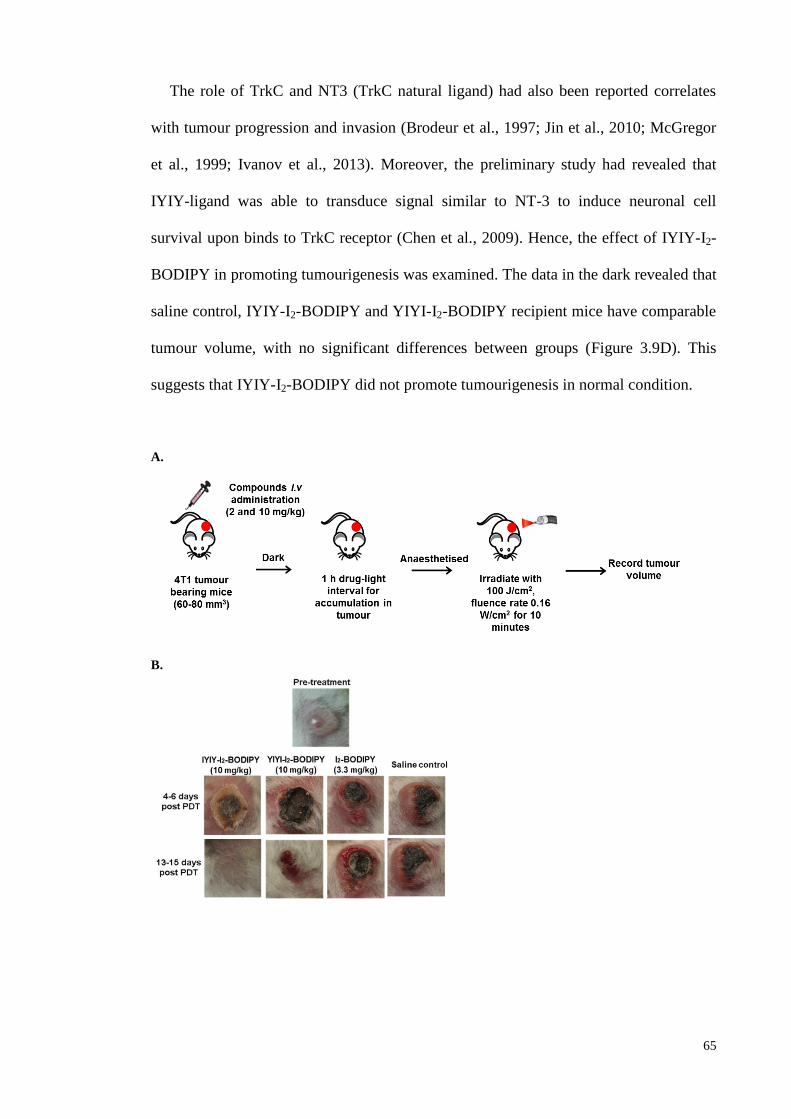
65
The role of TrkC and NT3 (TrkC natural ligand) had also been reported correlates
with tumour progression and invasion (Brodeur et al., 1997; Jin et al., 2010; McGregor
et al., 1999; Ivanov et al., 2013). Moreover, the preliminary study had revealed that
IYIY-ligand was able to transduce signal similar to NT-3 to induce neuronal cell
survival upon binds to TrkC receptor (Chen et al., 2009). Hence, the effect of IYIY-I2-
BODIPY in promoting tumourigenesis was examined. The data in the dark revealed that
saline control, IYIY-I2-BODIPY and YIYI-I2-BODIPY recipient mice have comparable
tumour volume, with no significant differences between groups (Figure 3.9D). This
suggests that IYIY-I2-BODIPY did not promote tumourigenesis in normal condition.
A.
B.
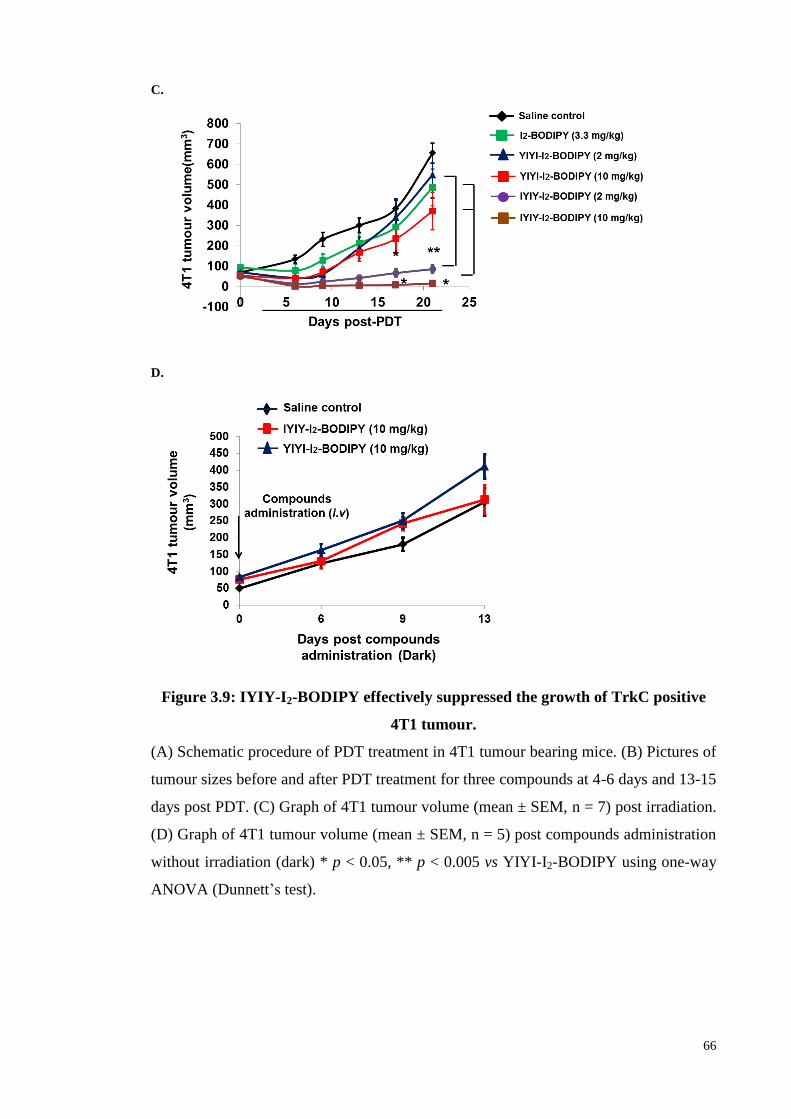
66
C.
D.
Figure 3.9: IYIY-I2-BODIPY effectively suppressed the growth of TrkC positive
4T1 tumour.
(A) Schematic procedure of PDT treatment in 4T1 tumour bearing mice. (B) Pictures of
tumour sizes before and after PDT treatment for three compounds at 4-6 days and 13-15
days post PDT. (C) Graph of 4T1 tumour volume (mean ± SEM, n = 7) post irradiation.
(D) Graph of 4T1 tumour volume (mean ± SEM, n = 5) post compounds administration
without irradiation (dark) * p < 0.05, ** p < 0.005 vs YIYI-I2-BODIPY using one-way
ANOVA (Dunnett’s test).
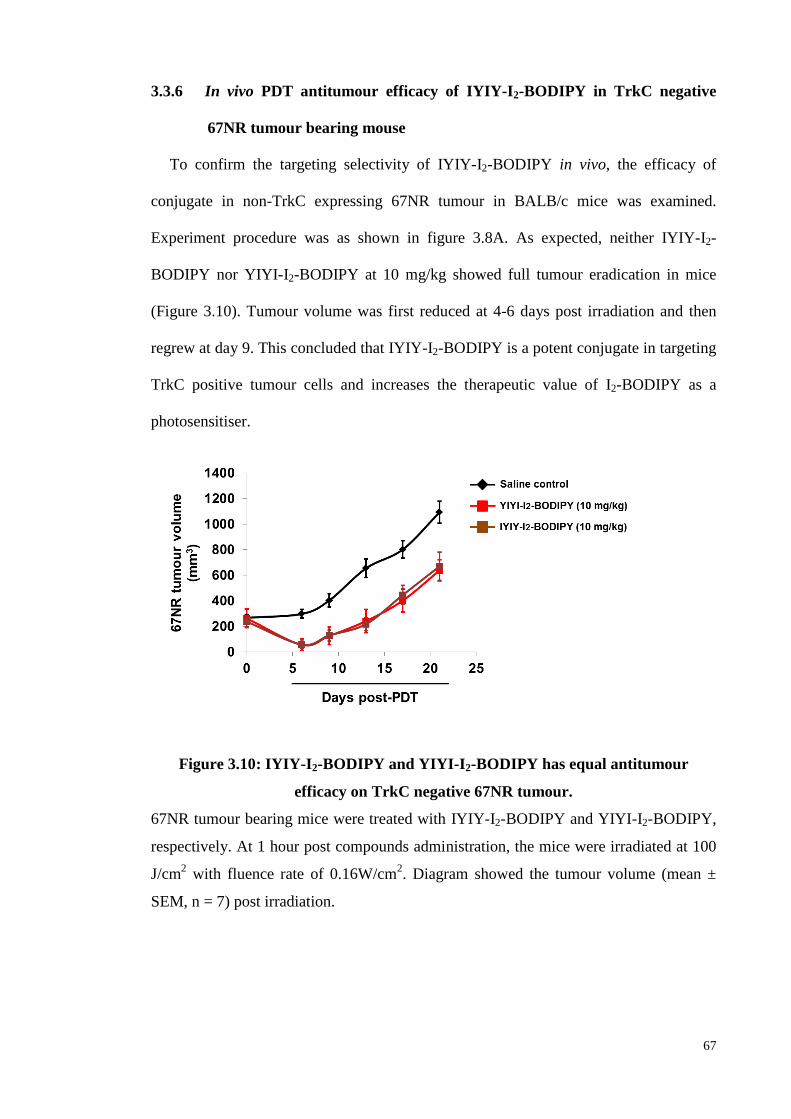
67
3.3.6 In vivo PDT antitumour efficacy of IYIY-I2-BODIPY in TrkC negative
67NR tumour bearing mouse
To confirm the targeting selectivity of IYIY-I2-BODIPY in vivo, the efficacy of
conjugate in non-TrkC expressing 67NR tumour in BALB/c mice was examined.
Experiment procedure was as shown in figure 3.8A. As expected, neither IYIY-I2-
BODIPY nor YIYI-I2-BODIPY at 10 mg/kg showed full tumour eradication in mice
(Figure 3.10). Tumour volume was first reduced at 4-6 days post irradiation and then
regrew at day 9. This concluded that IYIY-I2-BODIPY is a potent conjugate in targeting
TrkC positive tumour cells and increases the therapeutic value of I2-BODIPY as a
photosensitiser.
Figure 3.10: IYIY-I2-BODIPY and YIYI-I2-BODIPY has equal antitumour
efficacy on TrkC negative 67NR tumour.
67NR tumour bearing mice were treated with IYIY-I2-BODIPY and YIYI-I2-BODIPY,
respectively. At 1 hour post compounds administration, the mice were irradiated at 100
J/cm2 with fluence rate of 0.16W/cm
2. Diagram showed the tumour volume (mean ±
SEM, n = 7) post irradiation.
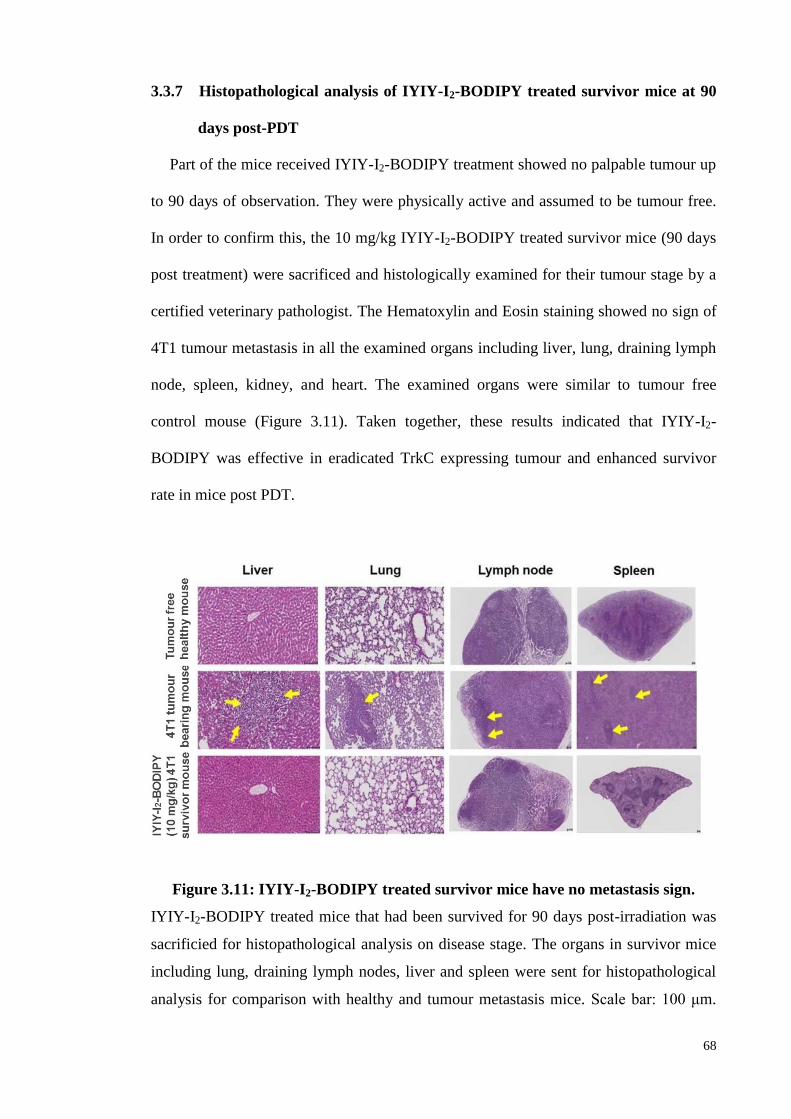
68
3.3.7 Histopathological analysis of IYIY-I2-BODIPY treated survivor mice at 90
days post-PDT
Part of the mice received IYIY-I2-BODIPY treatment showed no palpable tumour up
to 90 days of observation. They were physically active and assumed to be tumour free.
In order to confirm this, the 10 mg/kg IYIY-I2-BODIPY treated survivor mice (90 days
post treatment) were sacrificed and histologically examined for their tumour stage by a
certified veterinary pathologist. The Hematoxylin and Eosin staining showed no sign of
4T1 tumour metastasis in all the examined organs including liver, lung, draining lymph
node, spleen, kidney, and heart. The examined organs were similar to tumour free
control mouse (Figure 3.11). Taken together, these results indicated that IYIY-I2-
BODIPY was effective in eradicated TrkC expressing tumour and enhanced survivor
rate in mice post PDT.
Figure 3.11: IYIY-I2-BODIPY treated survivor mice have no metastasis sign.
IYIY-I2-BODIPY treated mice that had been survived for 90 days post-irradiation was
sacrificied for histopathological analysis on disease stage. The organs in survivor mice
including lung, draining lymph nodes, liver and spleen were sent for histopathological
analysis for comparison with healthy and tumour metastasis mice. Scale bar: 100 μm.

69
The current results had been verified by certified veterinary pathologist. Yellow arrows
indicate the metastatic tumour cells.
3.4 Discussion
BODIPY and its derivatives have been studied to be effective in inducing
photocytotoxicity in cancer cells and promoting vascular damage in the Chick
Chorioallantoic Membrane (CAM) model upon irradiation (Lim et al., 2010). BODIPY
derivative ADMP06 had been reported effective in eradicating breast tumour in
xenograft mice following PDT (Byrne et al., 2009). These studies were using free
photosensitizer BODIPY for therapeutic. In order to increase the therapeutic efficacy of
BODIPY, a targeting model for photosensitiser I2-BODIPY was constructed by
conjugating it with TrkC ligand. The resultant conjugate was reported specific and
effective in targeting TrkC+ cells with no effect on TrkC- cells (Kamkaew & Burgess,
2013). This study was concordant with the in vitro preliminary study, which shown a
significant selection of TrkC positive cells. Moreover, the in vivo studies revealed that
the conjugate has a good biodistribution profile (cleared from major organs at 24 hours
post administration), high accumulation in tumour site with outstanding antitumour
activity on TrkC positive cells. Altogether, suggested that selective binding of IYIY-I2-
BODIPY was correlated with natural levels of TrkC expression in breast cell lines.
The in vitro cell study was extended for a longer interval between compounds
incubation and illumination (4 – 6 hours, relative to 2 hours). Data showed that longer
incubation of cells with conjugates tend to decrease binding specificity, perhaps due to
relatively slow and non-selective interaction with cells (Peer et al., 2007; Veenhuizen et
al., 1997). In addition, I2-BODIPY was observed to reduce the cell viability at longer

70
incubation periods, and at concentrations higher than 1 µM at 2 h. This was due to the
uptake of the I2-BODIPY by the cells in a non-specific manner that has been reported
by Lim et al. 2010, as observed using confocal microscope; photocytotoxicity of I2-
BODIPY at high concentrations and long incubation periods is possible. Hence, a
shorter incubation period is optimum for ligand-mediated targeting agent in order to
reduce undesired photocytotoxicity, due to non-specific binding. The in vitro tumour
cell viability disaggregated from the tumour region post-PDT was not conducted in this
study. The low cell viability is predicted based on higher survival rate and delayed
tumour growth post-PDT in IYIY-I2-BODIPY treated mice.
The in vitro observation was concordant with the in vivo biodistribution study, in
which non-targeted YIYI-I2-BODIPY slowly accumulated in tumour tissues at longer
time points (3 and 6 hours post administration). However, the low specificity for
prolonged incubation cannot be compared with the in vivo antitumour efficacy. It is
remarkable that IYIY-I2-BODIPY at 10 mg/kg caused, on average, 96% tumour volume
reduction in the mice bearing TrkC positive tumour at 6 days post-PDT. The fact that
IYIY-I2-BODIPY was ineffective for suppressing TrkC negative 67NR tumours in mice
supports the assertion that this conjugate targets TrkC expressing tissue in vivo.
Toxicity issue relates to PDT is the photosensitivity. An intravenous administration
of photosensitisers can result in accumulation in the different tissues, especially skin
and eye, hence induces undesirable photosensitivities (Solban et al., 2006). This can be
overcome by targeting PDT agents to tumour that express targeted molecules.
Nevertheless, the TrkC targeted IYIY-I2-BODIPY conjugate is toxic to mouse at dosage
more than 20 mg/kg, causes motility and balancing difficulties in mouse at 15 minutes
post IYIY-I2-BODIPY injection. In this case, TrkC could be anticipated since NT-3 was

71
known to promote neuronal cell survival, differentiation and synapse transmission. A
claim that high doses of neurotrophins (including NT3) promote relatively rapid
excitotoxic necrosis of neurons (Koh et al., 1995) might be pertinent. However, IYIY-
I2-BODIPY requires further structural modifications because the light wavelength to
excite this conjugate is optimally around 530 nm, which has low tissue penetration
compared to longer wavelength (700 nm) which has deeper tissue penetration (Ballou et
al., 2005; Frangioni, 2003; Rao et al., 2007; Sevick-Muraca et al., 2002).
Throughout this study, an assumption of similar function between targeting ligand
(IYIY) itself and conjugate IYIY-I2-BODIPY in the dark was proposed. This is because
the photosensitiser is not active in the dark and thus has been excluded for the use of
core IYIY- and YIYI- ligand as another control. Given the apparent role of TrkC in
tumourigenesis of breast, neuroblastoma and pancreatic cancer, targeting the TrkC in
PDT maybe benefit for cancer patient that overexpressed TrkC in tumour cells. This
study features dosing with a targeted-PDT agent alone, but there is also the intriguing
possibility of combination therapies featuring Trk inhibitors currently in trials as
chemotherapeutic agents {e.g. Lestaurtinib (Chan et al., 2008; Iyer et al. 2010) and
PLX7486.
3.5 Conclusion
Current chemotherapy drugs used in the clinic today are toxic in nature, which lead
to the non-specific killing, undesirable side effects, morbidity and even death in cancer
patients. In this study, the synthesised TrkC targeted agent is the first to develop to
target TrkC expressing cancer using active targeting approach. The conjugated
photosensitiser will become active once activated by the light and subsequently kills the
cells via generation of singlet oxygen. This active targeted PDT agent (IYIY-I2-

72
BODIPY) can enhance the therapeutic indices by selectively accumulates in the
tumours but not in healthy tissues, suggesting that IYIY-I2-BODIPY can become
clinically important for the treatment of TrkC expressing cancer.

73
CHAPTER 4: IMMUNE RESPONSES INDUCTION AND ANTITUMOUR
ACTIVITY OF TROPOMYOSIN RECEPTOR KINASE-C (TRKC) RECEPTOR
TARGETED PHOTODYNAMIC THERAPY (PDT) CONJUGATE
4.1 Introduction
Conventional chemotherapy of cancer is nearly always associated with limitations
that include non-selective toxicity, treatment resistance and immune response silencing
(Hirsch, 2006; Weir et al., 2011). These restrictions generally lessen the effectiveness of
chemotherapy. Actively targeted cancer therapies guide the agents to cell surface
molecules (proteins, sugar or lipids), and thus increasing their cellular uptake through
the endocytic internalisation to improve selectivity and counter resistance (Byrne et al.,
2008). Many ligand-drug conjugates had been designed to selectively bind
overexpressed cell surface molecules (generally survival or metastasis biomarker in
cancer) such as biotin, folate, sigma-2, carbonic anhydrase IX, glucose receptors and
others (Srinivasarao et al., 2015).
This paper is focus on the TrkC receptor due to its tumourigenesis characteristic and
limited drugs to treat TrkC positive cancer. These receptors are found in neurons where
they regulate the neuronal cell survival and growth, proliferation, differentiation and
synaptic strength and plasticity (Huang & Reichardt, 2003), but also in neuroblastoma
(Brodeur et al., 1997; Yamashiro et al., 1997), glioblastoma (Kumar & de Vellis, 1996;
Wang et al., 1998), thyroid cancer (McGregor et al., 1999), melanoma (Xu et al., 2003)
and breast cancer (Blasco-Gutierrez et al., 2007; Jin et al., 2010) where they impact
malignancy. The expression and function of Trk subtypes are dependent on the tumour
type. For instance, in neuroblastoma, TrkC expression correlates with good prognosis,
but in breast, prostate and pancreatic cancers, the expression of the same Trk subtype is

74
associated with cancer progression and metastasis (Jin et al., 2010; Miknyoczki et al.,
2002). Furthermore, ligands binding Trk receptors activate downstream intracellular
signalling pathways that enhance tumour cell mitogenicity and survival (Brodeur et al.,
1997; Yamashiro et al., 1997). Inhibition of Trk signaling significantly reduced
tumourigenicity and invasive capability of tumour cells in vitro and in vivo xenograft
models (Iyer et al., 2010; Jin et al., 2010) (Miknyoczki et al., 2002). Several Trk
receptors targeted chemotherapeutic drugs which are inhibitors of all TrkA/B/C
receptors, are currently in clinical trials for treatment of solid tumours (Vaishnavi et al.,
2015).
Trk receptors and their ligands have been reported to modulate the immune system.
Trk receptors are expressed in small quantities in monocytes and lymphocytes. The
natural ligands of Trk receptors, neurotrophins, which include neurotrophin-3/-4 (NT-
3/NT-4), brain-derived neurotrophin factor (BDNF) and neurotrophin growth factors
(NGF), can function as non-cytokine mediators to modulate both innate and adaptive
immune responses. Such modulations include increasing the pluripotent cytokine
interleukin (IL) -6 secretion in bone marrow stromal cells (Marshall et al., 1999; Rezaee
et al., 2010). In addition, neurotrophins have been reported to enhance differentiation of
granulocytes (e.g. eosinophils, mast cells and basophils) during haematopoiesis
(Matsuda et al., 1988). In T-lymphocytes, neurotrophins regulate T-cell subtypes
balancing upon binding to TrkC expressing T helper (Th) 2 cell by promoting IL-4
release, which in turn blocks Th1 subtype and IFN-γ production (Sekimoto et al., 2003).
Other than neurotrophins, TrkC was also reported to suppress TGF-β signaling by
directly binding to type II TGF-β receptor and to reduce TGF-β mediated downstream
Smad2/3 phosphorylation in TrkC expressing cells (Jin et al., 2007). Despite evidence
that links Trk receptors to modulation of the immune system, there are currently no

75
reports that explore Trk receptors in the context of possible strategies for immune
therapy.
TrkC receptor targeted ligand conjugated with photosensitiser diiodo-boron
dipyrromethene (I2-BODIPY), termed as IYIY-I2-BODIPY (Kamkaew & Burgess, 2013)
is designed for use in PDT of cancer. The IYIY-ligand induces some biological
properties that are similar to neurotrophin-3 including internalisation into lysosomes,
and transduction of signals that regulate neuronal cell survival and differentiation (Chen
et al., 2009). Based on this and the previously reported immunological effects of TrkC
and NT-3, this study investigated the systemic immunological impacts seen on the
administration of the IYIY-I2-BODIPY conjugate in a preclinical model. Specifically,
the systemic modulation of cytokine levels, characterisation of myeloid innate immune
responses and adaptive immune T-cell subtypes populations upon compounds
administration under the non-irradiated conditions (dark) will be examined. As PDT
agents are not active in the dark, studying the conjugate without photoirradiation can
help to elucidate the immune-regulatory function of this TrkC ligand itself. This may be
important, especially for PDT agents that have long drug-light interval (DLI) since the
ligand in the conjugate may already begin to exert its therapeutic effect even before
photoirradiation. This is the first study on the immunomodulation properties and anti-
tumour activity of a TrkC ligand. In addition, the combination effect of conjugates and
PDT will be examined as PDT has been known to contribute to the development of
systemic antitumour immune responses (Nowis et al., 2005; Reginato et al., 2014).

76
4.2 Materials and Methods
4.2.1. Compounds
TrkC targeted IYIY-I2-BODIPY (Isoleucin-Tyrosine-Isoleucin-Tyrosine, IYIY
conjugated I2-BODIPY), non-TrkC targeted scrambled control YIYI-I2-BODIPY
(Tyrosine-Isoleucin-Tyrosine-Isoleucin, YIYI conjugated I2-BODIPY), free
photosensitiser I2-BODIPY and TrkC targeted ligand IYIY-TEG (Isoleucin-Tyrosine-
Isoleucin-Tyrosine conjugated to triethylene glycol (TEG) without I2-BODIPY) were a
gift from Prof. Kevin Burgess (Kamkaew & Burgess, 2013).
4.2.2 Animal Model
Female 8-10 week old, wild type BALB/c mice were purchased from Taconic and
InVivos Pte Ltd, Singapore and maintained in the AAALAC accredited satellite animal
facility at the Department of Pharmacology, Faculty OF Medicine, University of
Malaya. All animal experiments were performed according to protocols approved by the
Faculty of Medicine Institutional Animal Care and Use Committee, University of
Malaya (FOM IACUC). Ethics approval numbers: 20150303/PHAR/R/KCS (Appendix
B).
4.2.3 Tumour Model Development
The fur of the BALB/C mouse was shaved and murine breast carcinoma 4T1 cell line
(ATCC) at a density of 5 x 105 cells in 0.1 mL of RPMI medium was orthotopically
injected into the mammary fat pad of the mice. The mice were monitored for tumour
development every day and were then randomly divided into groups for compounds
administration when the size reached around 60-80 mm3.

77
4.2.4 Compounds Administration
IYIY-I2-BODIPY, YIYI-I2-BODIPY at 10 mg/kg of body weight (consist 3.3 mg
equivalent/kg of I2-BODIPY and 6.7 mg equivalent/kg of IYIY-TEG) and 3.3 mg/kg of
I2-BODIPY were dissolved respectively in a cocktail of 2.5% ethanol and 2.5%
CremophoreEL in saline. The mixture was then further dissolved using saline to a
volume of 0.2 mL and intravenously administrated to tumour bearing mice via tail vein.
The mice were then kept in an environment away from bright light for 2 hours and 24
hours (n = 5 per treatment group for every time point).
4.2.5 Blood sampling
At 2 hours (represented early innate immune response) and 24 hours (represented
late or onset of adaptive immune response) time points post compounds administration,
mice from each group (n = 5 per time point) were anaesthetised with anaesthesia
cocktail (90 mg/kg of ketamine and 10 mg/kg of xylazine). 0.5 mL of blood was
withdrawn via cardiac puncture after the onset of anaesthesia.
4.2.6 Flow Cytometry Quantification of Plasma Cytokines
The withdrawn blood was centrifuged at 5000 rpm for 10 minutes to separate the
plasma and peripheral blood mononuclear cells. Cytokine levels in blood plasma were
then determined using a BD Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17
Cytokine Kit (BD Biosciences, San Jose, CA, USA) according to manufacturer’s
instructions. Cytokines IL-2, IL-4, IL-6, IFN-γ, TNF-α, IL-17A and IL-10 were
quantified using FACSCanto II (BD Biosciences) and analysed using FCAP array
software (BD Biosciences). TGF-β cytokine in blood plasma was quantified using

78
Human/Mouse TGF-β1 sandwich ELISA (eBiosciences, San Diego, CA, USA) based
on manufacturer’s instructions.
4.2.7 Cell Isolation from Lymphoid Organs and Tumour Tissues
After the blood sampling at each indicated time points, mice (n = 5 per time point)
were sacrificed and tumour draining lymph nodes (TDLN), spleen and tumour tissues
were harvested. Single cell suspensions from TDLN and spleen were obtained by
mincing the organs using frosted glass slides, followed by red blood cell lysis using red
blood cell lysis buffer. Single cell suspensions from tumour tissues were obtained by
mincing tumour tissue with frosted glass slides, followed by digestion with 0.5 mg/mL
of collagenase type XI (Sigma Aldrich, St Louis, MO) for 30 minutes in a 37 oC shaker
at 200 rpm. The cell suspensions were then filtered to eliminate the debris. All the
single cell suspensions from TDLN and tumour tissues were stimulated for 4 hours with
50 ng/mL of Phorbol 12-Myristate 13-Acetate (PMA), 1 g/mL of Ionomycin (Sigma
Aldrich, St Louis, MO, USA) and 3 g/mL of brefeldin A (BD Bioscience) (Kue et al.,
2012).
4.2.8 Staining and Flow Cytometry Quantification of Immune Cells
The stimulated single cell suspensions from tumour tissues, TDLN and spleen were
washed with PBS, and resuspended (1 x 106 cells) in FACS washing buffer (0.5% FBS
and 0.05% sodium azide in PBS). The cells were then stained with a panel of
fluorochromes conjugated monoclonal antibodies purchased from BD Pharmingen to
detect specific surface antigen that are FITC-anti mouse CD11b (553310), PE-anti
mouse Ly6G (551461), APC-anti mouse F4/80 (17-4801), FITC-anti mouse CD4
(553729), PE-anti mouse CD25 (553075), FITC anti mouse CD8 (553031). The cells
were fixed and permeabilised using Cytofix/Cytoperm kit (BD Biosciences) according

79
to the manufacturer’s instructions. The surface antigens stained cells were then labelled
for intracellular cytokine using PE-anti-mouse IFN-γ (554412), PerCP-Cy5.5-anti-
mouse IL-4 (560700), APC-anti-mouse IL-17 (17-7177), and APC-anti-mouse FoxP3
(17-5773) with different fluorochrome combination to avoid overlapping. We quantified
the cells of our interest based on surface markers, Th1 (CD4+IFN-γ
+), Th2 (CD4
+IL-4
+),
Th17 (CD4+IL-17
+), Treg (CD4
+CD25
+FoxP3
+), CTL (CD8
+IFN-γ
+), Tc-17 (CD8
+IL-
17+), granulocytic-MDSC (CD11b
+Ly6G
+), neutrophils (CD11b
+Ly6G
+F4/80
-).
Effector T-cells were stained using PE-Cy5 rat anti-mouse CD44 (553135) for
CD4+CD44
high and CD8
+CD44
high populations. A total of 10,000 cells were analysed for
every experiment. The stained cells was analysed in a FACS Canto-II Flow Cytometry
(BD Biosciences). The data were analysed using Cell Quest software (BD Biosciences).
4.2.9 In vivo TrkC blocking studies
For TrkC blocking study, the tumour bearing mice (n = 4 per group) were treated
with IYIY-I2-BODIPY (10 mg/kg) with or without mouse TrkC antibody (R&D
systems, Minneapolis, MN, USA) at a dosage of 10 µg/mouse via tail vein and kept
away from bright light as a non-irradiated study. Tumour bearing mice receiving isotype
control Immunoglobulin G (R&D systems) at dosage of 10 µg/mouse (n = 4) was used
as a antibody control in this study. At 2 hours post IYIY-I2-BODIPY and antibodies
administration, the mice were sacrificed and tumour tissues, spleen and TDLN were
harvested, stained with respective fluorescence conjugates antibodies and phenotyped
for granulocytic-MDSC, neutrophils, CD4+ and CD8
+ T-cell subtypes using flow
cytometry.

80
4.2.10 Photodynamic therapy (PDT) in Mice
For irradiation, IYIY-I2-BODIPY (10 mg/kg), YIYI-I2-BODIPY (10 mg/kg), I2-
BODIPY (3.3 mg/kg) and saline were given respectively to the mice (n = 5 per group).
The mice were kept away from bright light post compound administration for 1 hour.
Thereafter, anaesthesia cocktail of 90 mg/kg of ketamine and 10 mg/kg of xylazine was
given to the mice. Upon the onset of anaesthesia, PDT was performed using a Lumacare
LC-122A fiber optic light delivery system (standard fiber optic probe model LUM V,
400−700 nm, Lumacare Medical Group, Newport Beach, CA, USA, with a 500/585 nm
bandpass filter from Omega Optical) to emit light at 530 nm. A 4 mm thick glass slide
was used as a barrier to avoid direct photothermal effect on tumour. The illuminating
spot was positioned at the tumour and the surrounding was covered using a black cloth
to avoid PDT effect on non-tumour region of body. PDT was conducted at 100 J/cm2
with a fluence rate of 160 mW/cm2, for 10 minutes. The irradiated mice were then
sacrificed for blood and lymphoid organ isolation at 2 hours and 24 hours post-PDT.
The methods of samples processing in irradiated mice were same as methods described
above (blood sampling, flow cytometry quantification of plasma cytokines, cell
isolation from lymphoid organs and tumour tissues, staining and flow cytometry
quantification of immune cells).
4.2.11 Adoptive Transfer for Antitumour Immunity
For adoptive transfer studies, spleen and TDLN from healthy mice (n = 5) and the
survivor mice (tumour free 60 days post PDT) in IYIY-I2-BODIPY treated group of
mice (n = 5) were harvested. Splenocytes at a density of 2 × 107 cells/0.2 mL/mouse and
lymphocytes from TDLN at a density of 1.5 × 107 cells/0.2 mL/mouse from each mouse
were injected into different recipients of healthy syngeneic mice (n = 5) via the tail vein.
After 2 days of adoptive transfer, the recipient mice were inoculated subcutaneously

81
with 4T1 tumour cells at a density of 5 × 105 cells/0.1 mL/mouse. The tumour growth in
the mice that received survivor splenocytes and lymphocytes was monitored and
compared with the mice that received splenocytes and lymphocytes from the healthy
mice.
4.2.12 Statistical Analysis
The Student’s t-test and one-way ANOVA (Dunnett’s test) were used to determine
the statistical differences between various experimental and control groups. A p value of
< 0.05 was considered significant.
4.3 Results
4.3.1 Th1/Th2/Th17/Treg cytokines level in blood plasma of TrkC positive 4T1
tumour bearing mice post compounds administration
In the initiation phase of the adaptive immune response, antigen-specific
lymphocytes (T and B cells) are activated when they are exposed to antigens
presentation. Such activation of T-helper (Th) CD4+ lymphocytes causes them to
differentiate into four different subtypes (Th1, Th2, Th17, regulatory T-cells) depending
on the availability of the different types of differentiation factors (cytokines) in the
environment. The differentiated subtypes of T-cells then secrete various specific
cytokines. For instance, Th1 secretes interleukin (IL)-2, interferon (IFN)-γ, tumour
necrosis factor (TNF)-α, Th2 secretes IL-4, IL-6, IL-10, Th17 secretes IL-17 and
regulatory T-cells (Treg) secretes transforming growth factor (TGF)-β. The released
cytokines by differentiated T-cell subtypes will further stimulate CD8+ T-cells such as
cytotoxic T lymphocytes (CTL), Tc17 cells function. The following experiments were
conducted to examine if IYIY-I2-BODIPY induces the same effects.

82
First, the cytokine levels in TrkC positive murine 4T1 breast cancer model (Jin et al.,
2010) that has been treated with compound IYIY-I2-BODIPY, YIYI-I2-BODIPY or I2-
BODIPY, the latter two as controls were examined. Blood plasma samples were
collected from 4T1 tumour bearing mice via cardiac puncture at 2 hours and 24 hours
post compound administration with no exposure to light (hereafter referred to as dark
treatment) to examine early and late responses respectively. All treatment groups had
slightly increased levels of Th1 cytokine IL-2 at 2 hours and 24 hours (0.7 – 1.9 pg/mL)
compared to saline (0 – 0.3 pg/mL; p = 0.05 for IYIY-I2-BODIPY and YIYI-I2-
BODIPY; Figure 4.1A). Since IL-2 is usually produced to promote activation and
proliferation of T-cells, elevation of that interleukin suggests that IYIY-I2-BODIPY in
the dark induced T-cell responses. Increases in levels of other Th1 cytokines such as
IFN-γ (Figure 4.1B) and TNF-α (Figure 4.1C) were hardly significant in all treatment
groups compared to the saline control, suggesting that Th1-mediated inflammation was
mild.
Th2 cytokines such as IL-4, was increased (1 to 2-fold) in all mice treated with the
IYIY-I2-BODIPY, YIYI-I2-BODIPY and I2-BODIPY at 2 hours only compared to mice
treated with saline control (Figure 4.1D), in a non-compound specific manner. IL-6 is a
set of pluripotent pro-inflammatory cytokines that regulate differentiation and functions
of T lymphocytes as well as expansion of myeloid cells (Meyer et al., 2011; Mundy-
Bosse et al., 2011). In this work, levels of IL-6 were significantly and selectively
elevated in IYIY-I2-BODIPY treated mice (approximately 7.5-, 2.6- and 4.6-fold
compared to mice treated with saline control, I2-BODIPY and YIYI-I2-BODIPY
respectively at 2 hours; p = 0.000). The increase of IL-6 level did not persist, as the
level at 24 hours was almost as low as the other groups at 24 hours (Figure 4.1E). The
increase of IL-6 was the same as other studies where neurotrophins binding to Trk

83
receptor increased IL-6 secretion in bone marrow stromal cells (Marshall et al., 1999;
Rezaee et al., 2010). Levels of the Th2 immunosuppressive cytokine IL-10 induced by
the compounds was comparable in these experiments (Figure 4.1G). Together, the data
indicate that the Th2 cytokines (IL-4, IL-6) were transiently elevated, whereas the Th1
responses (very weak IFN-γ and TNF-α) were very mild.
Another proinflammatory cytokine IL-17A which is specifically secreted by IL-17+
T-cells was mild elevated (1.3-fold) only in IYIY-I2-BODIPY treated group at 24 hours
compared to other treatment and control groups. There was no significant change in the
IL-17A levels at 2 hours post compound administration when compared to I2-BODIPY
(Figure 4.1F). Increased IL-17 in the IYIY-I2-BODIPY treated group is the same as the
selective increase in the IL-6 level in the same group, probably because IL-6 is one of
the essential cytokines for differentiating naive T-cells to IL-17+ T-cells. Based on the
non-persistent increases of IL-2 and IL-6, suggest that ligated conjugate treated mice
selectively provoke transient inflammation. Among the compounds investigated, IYIY-
I2-BODIPY had the highest immune modulation activity.
In addition to the above findings, levels of immunosuppressive cytokine TGF-β was
significantly suppressed in both IYIY-I2-BODIPY and YIYI-I2-BODIPY treated groups
at 2 hours (4.0-fold and 3.0-fold respectively, Figure 4.1H) compared to I2-BODIPY.
Previous study had shown that YIYI-I2-BODIPY possessed positive but lower targeting
selectivity on TrkC positive cells compared to IYIY-I2-BODIPY when cells exposed to
compound in prolong period (Figure 3.6), so it is conceivable that the YIYI-ligand
could elicit similar but weaker immune responses compared to IYIY as reflected by the
levels of IL-2, IL-4 and TGF-β. I2-BODIPY alone in the dark only caused mild immune
responses (as reflected by the lack of significant changes in cytokine levels post
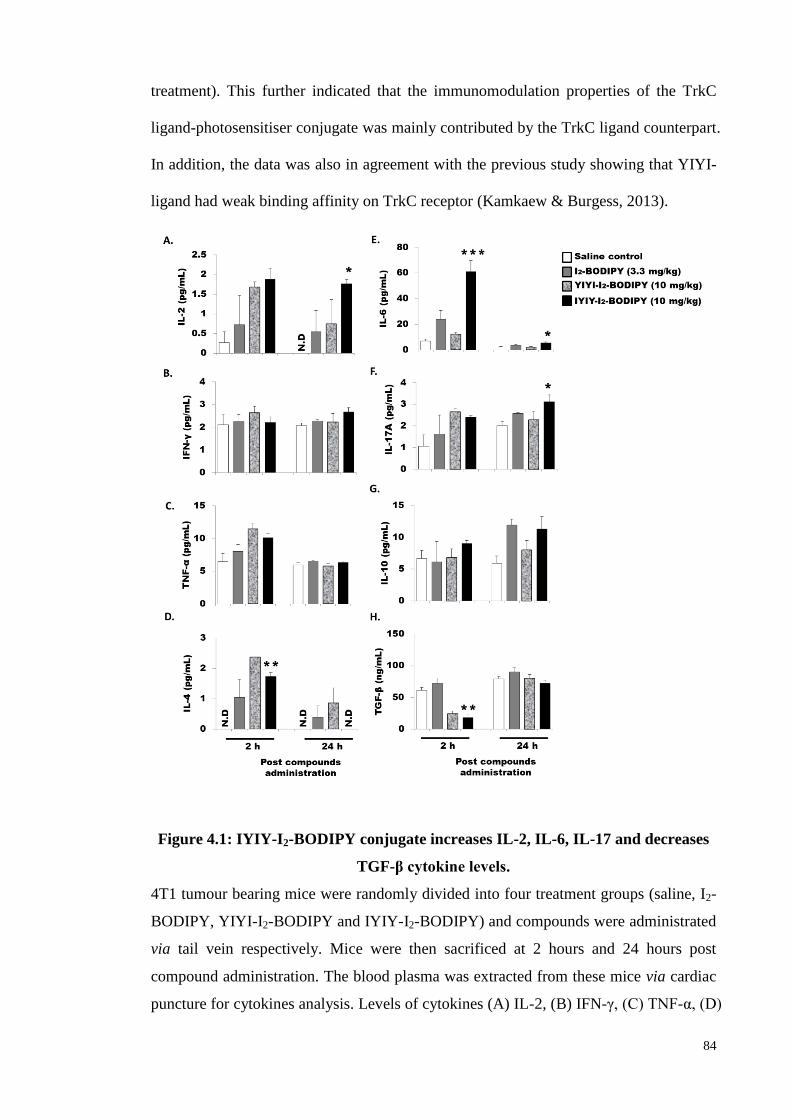
84
treatment). This further indicated that the immunomodulation properties of the TrkC
ligand-photosensitiser conjugate was mainly contributed by the TrkC ligand counterpart.
In addition, the data was also in agreement with the previous study showing that YIYI-
ligand had weak binding affinity on TrkC receptor (Kamkaew & Burgess, 2013).
Figure 4.1: IYIY-I2-BODIPY conjugate increases IL-2, IL-6, IL-17 and decreases
TGF-β cytokine levels.
4T1 tumour bearing mice were randomly divided into four treatment groups (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) and compounds were administrated
via tail vein respectively. Mice were then sacrificed at 2 hours and 24 hours post
compound administration. The blood plasma was extracted from these mice via cardiac
puncture for cytokines analysis. Levels of cytokines (A) IL-2, (B) IFN-γ, (C) TNF-α, (D)

85
IL-4, (E) IL-6, (F) IL-17A, (G) IL-10 and (H) TGF-β are shown. Data represent mean ±
SEM with minimum of four mice per group. * p < 0.05, ** p < 0.005 vs I2-BODIPY
using one-way ANOVA (Dunnett’s test).
4.3.2 Myeloid cell subtypes quantification in compounds treated mice
Cancer growth is normally accompanied by expansion of a heterogeneous group of
immunosuppressive cells collectively known as myeloid derived suppressor cells
(MDSCs). In particular, the granulocytic-MDSC (G-MDSC) subtype promotes tumour
relapse by impairing the effectiveness of host immunity through inhibition of T-cell
activation when primed by the tumour antigen (Khaled et al., 2013; Nagaraj et al., 2007).
The expansion and differentiation of MDSCs is known to be mainly regulated by
cytokines IL-6 (Meyer et al., 2011; Mundy-Bosse et al., 2011). Following the increase
of the levels of these cytokines in IYIY-I2-BODIPY treated mice, the changes in G-
MDSCs cell populations in these animals was then examined. The data revealed that
IYIY-I2-BODIPY treated mice had significant lower level of G-MDSCs (CD11b+Ly6G
+)
in spleen (4.9% ± 0.5%; p = 0.032) (Figure 4.2A) and tumour microenvironment (TM)
(1.1% ± 0.3%; p = 0.035) at 2 hours (Figure 4.2B), at approximately 2-fold and 4-fold
lower respectively compared to the other three control groups. Comparing the two
ligated conjugates, YIYI-I2-BODIPY treated mice showed low G-MDSC population
only at 24 hours, whereas IYIY-I2-BODIPY treated group had low G-MDSC population
at 2 hours and remained low up to 24 hours (Figure 4.2). This data suggests that
conjugates selectively reduce the populations of immunosuppressive G-MDSC that is
usually highly up-regulated in cancer, and could serve as a cancer immunotherapeutic
agent.
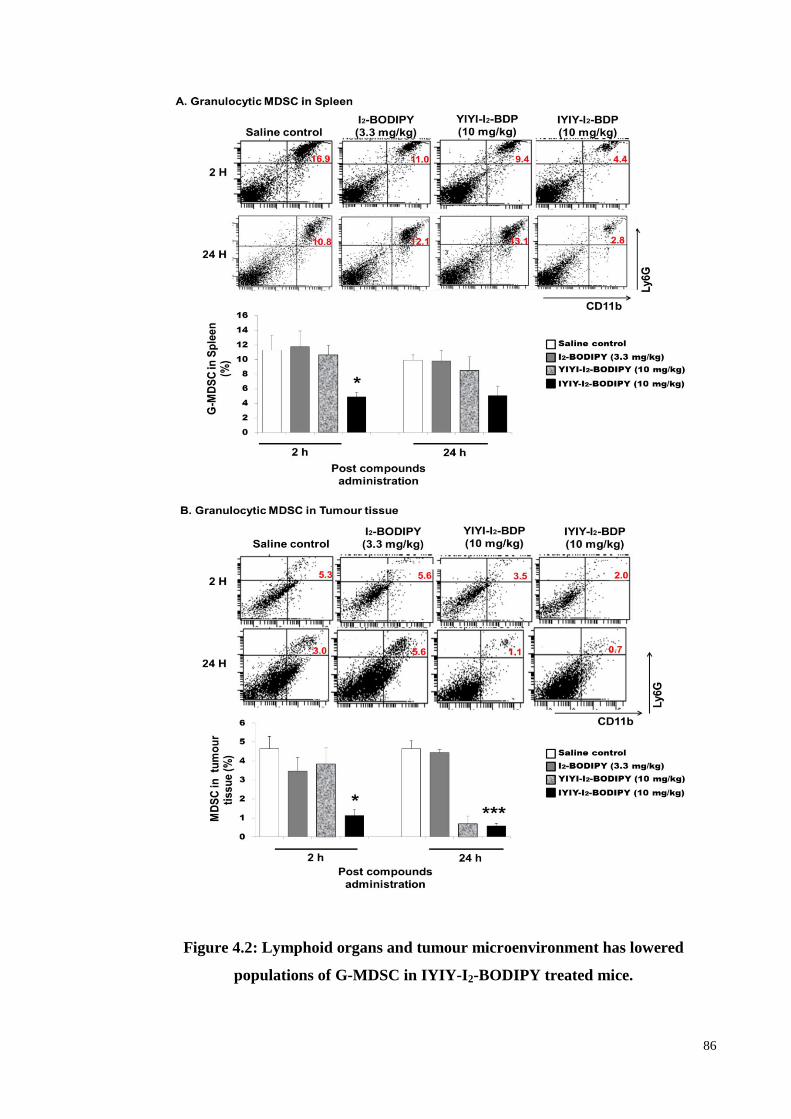
86
Figure 4.2: Lymphoid organs and tumour microenvironment has lowered
populations of G-MDSC in IYIY-I2-BODIPY treated mice.

87
4T1 tumour bearing mice were randomly divided into four treatment groups (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) and compounds were administrated
via tail vein respectively. Mice were then sacrificed at 2 hours and 24 hours post
compound administration. At scheduled time points post compound administration,
mice were sacrificed, followed by isolation of TDLN, spleen and tumour tissues.
Immuno-phenotyping of G-MDSC was conducted using surface staining of
fluorescence conjugated antibodies and quantified using flow cytometry. (A) G-MDSC
in spleen and (B) tumour tissues. Data represent mean ± SEM with minimum of four
mice per group. * p < 0.05, *** p <0.001 vs I2-BODIPY using one-way ANOVA
(Dunnett’s test).
4.3.3 Inflammatory cell (neutrophils) population quantification in compounds
treated mice
Neutrophil, which is another subtype of myeloid cell, was examined to confirm the
suppressive activity of IYIY-I2-BODIPY on myeloid cell subsets. Neutrophils are
defined by the expression of surface antigens CD11b and Ly6G, with the absence of the
macrophage marker F4/80 (Rose et al., 2012). Similar to G-MDSC, the neutrophil
population at 2 hours was reduced. The reduction was 50% in spleen (p = 0.02) and
24% in TM (statistically insignificant) of IYIY-I2-BODIPY treated mice compared to
I2-BODIPY treated mice (Figure 4.3). In YIYI-I2-BODIPY treated mice, the neutrophil
population was reduced by 13% at 2 hours compared to I2-BODIPY and at 24 hours, the
reduction was approximately 44% in spleen. This suggests that the ligand-conjugates
had direct impact on myeloid cells, rather than through IL-6 regulation.
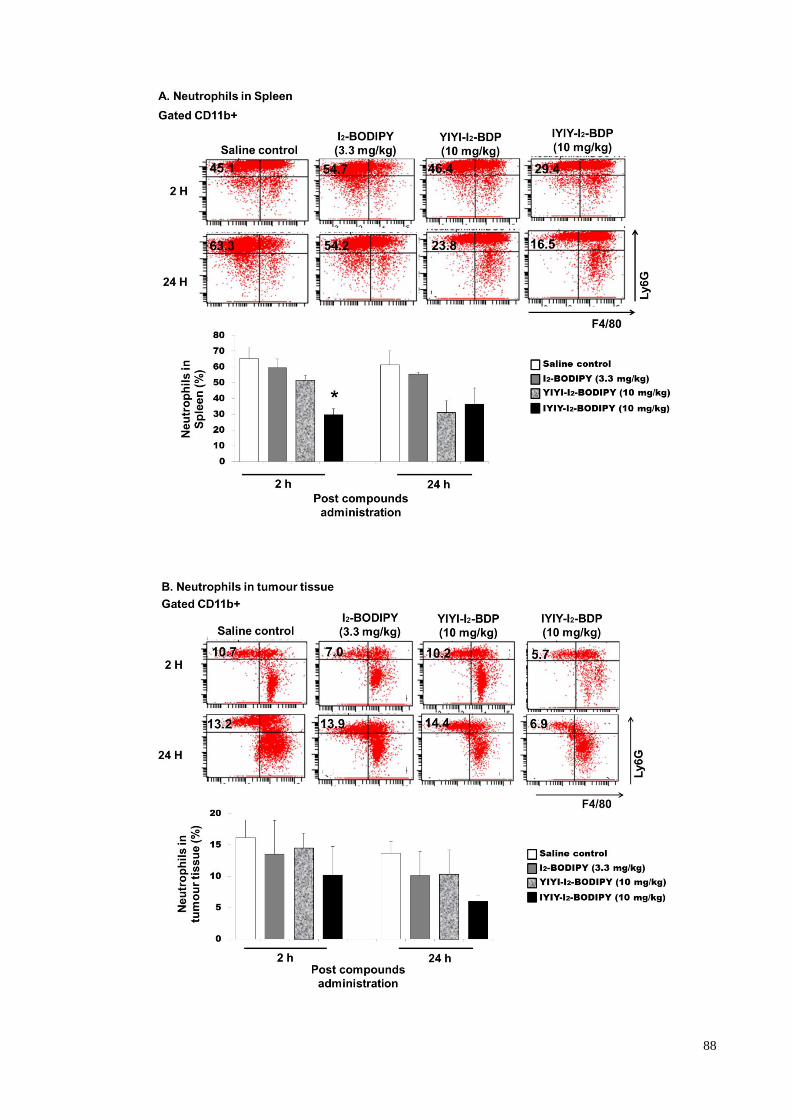
88

89
Figure 4.3: IYIY-I2-BODIPY treated mice have lowered neutrophils population.
4T1 tumour bearing mice were randomly divided into four treatment groups (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) and compounds were administrated
via tail vein respectively. Mice were then sacrificed at 2 hours and 24 hours post
compound administration. At desired time points post compound administration, mice
were sacrificed, followed by isolation of TDLN, spleen and tumour tissues. Immuno-
phenotyping of neutrophil was conducted using surface staining of fluorescence
conjugated antibodies and quantified using flow cytometry. (A) Neutrophil in spleen
and (B) tumour tissues were shown. Data represent mean ± SEM with minimum of four
mice per group. * p < 0.05 vs I2-BODIPY using one-way ANOVA (Dunnett’s test).
4.3.4 CD4+ T-helper cell subtypes (Th1/Th2/Th17/Treg) quantifications in
compounds treated mice
Different T-lymphocyte subtypes secrete cytokines into systemic circulation, and
these cytokines especially IL-2 can affect the differentiation, expansion and survival of
T-lymphocytes via autocrine or paracrine systems (Zhu et al., 2010). In addition, T
helper lymphocytes are able to differentiate into four different subtypes, which are Th1,
Th2, Th17 and regulatory T-cells (Treg) based on type of diseases and cytokines in the
environment. Consequently, the impact of ligand-drug conjugates on T-lymphocyte
subtypes populations was examined. Populations of CD4+ T helper (Th) cell subsets and
antigen specific CD8+ T-cell subsets were determined in the tumour draining lymph
node (TDLN) and tumour microenvironment (TM). Th1 cell populations (CD4+IFN-γ
+)
in IYIY-I2-BODIPY treated mice were 2-fold higher in TDLN at 24 hours and in TM at
2 hours (p = 0.045), compared to I2-BODIPY treated mice (Figure 4.4A). In the YIYI-
I2-BODIPY treated group, the Th1 subset was moderately increased only at 24 hours in
both TDLN (1.7-fold) and TM (1.3-fold) compared to I2-BODIPY (Figure 4.4A). Th2
(CD4+IL-4
+) populations in TDLN and TM were comparable among the treatment and
control groups and saline control at 2 hours. There was a moderate increase in TDLN at

90
24 hours in IYIY-I2-BODIPY and YIYI-I2-BODIPY-treated mice compared to mice
treated with I2-BODIPY and saline. Unlike TDLN, TM had lower Th2 populations in
both IYIY-I2-BODIPY and YIYI-I2-BODIPY-treated mice at 24 h compared to mice
treated with I2-BODIPY and saline (Figure 4.4B).
The Th17 cell population was increased compared to I2-BODIPY by 2.4- and 2.0-
fold at 24 hours in TDLN (p = 0.024) and TM respectively (Figure 4.4C). This
observation coincided with the increase of IL-17 cytokine. Unlike Th1, the increase in
Th17 cell was selective only in IYIY-I2-BODIPY treated group. This is similar for IL-6
level which was also only elevated in IYIY-I2-BODIPY treated group, supporting that
IL-6 mainly skewed the differentiation of naive T-cells to the Th17 subset. Taken
together, the T-helper population was concordant with the cytokines profiles in Figure
4.1, and in agreement with the conclusion that ligated conjugates increased Th1 and
Th17 subsets of T-cells population.
Immunosuppressive regulatory T cell (Treg) was known to suppress antitumour T-
cell responses as a method of escaping immune-surveillance during cancer development.
In the case of Treg cells, the population in IYIY-I2-BODIPY mice was found to
decrease by approximately 1.4-fold in TDLN at 24 hours (p = 0.027) and 2-fold in TM
at 24 hours (p = 0.045) (Figure 4.4D). Similar observations were made in experiments
featuring YIYI-I2-BODIPY treated mice only at 24 hours. This decrease in Treg
concurred with the depressed levels of the immunosuppressive cytokine TGF-β in both
groups. Together, the data suggest that IYIY-I2-BODIPY was more rapid in reducing
the populations of immunosuppressive Treg compared to YIYI-I2-BODIPY, which was
low only at the later time point (24 hours).
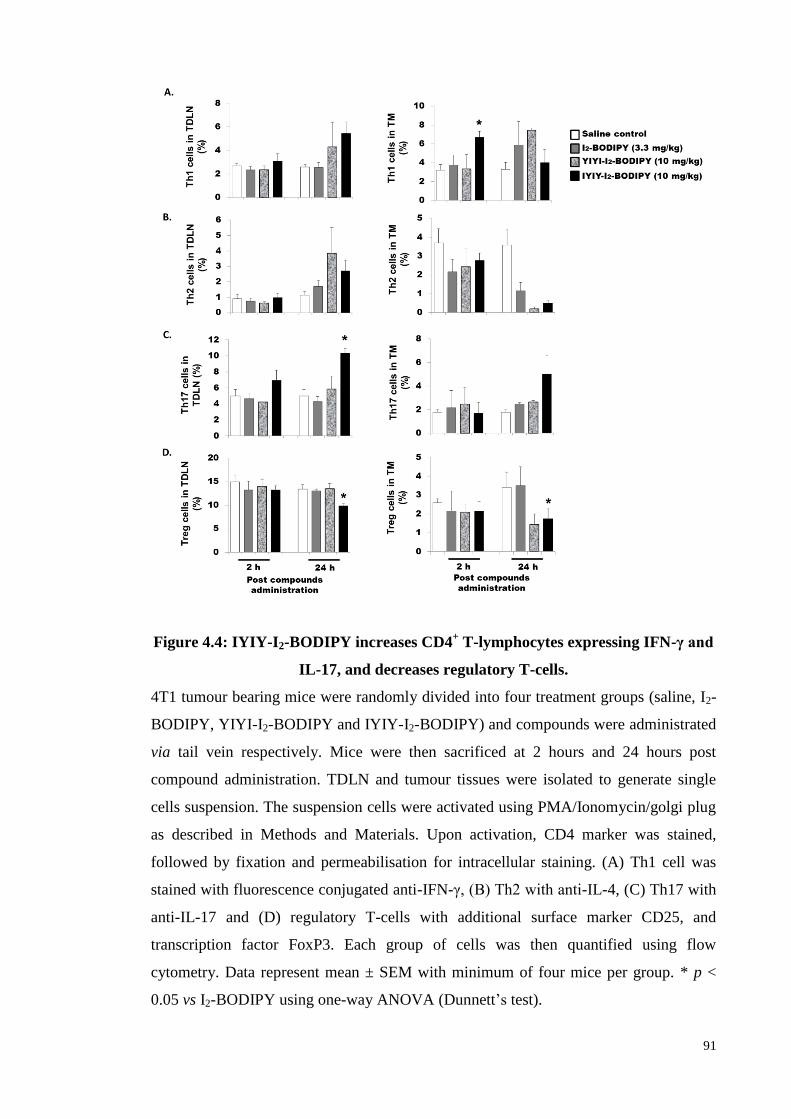
91
Figure 4.4: IYIY-I2-BODIPY increases CD4+ T-lymphocytes expressing IFN-γ and
IL-17, and decreases regulatory T-cells.
4T1 tumour bearing mice were randomly divided into four treatment groups (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) and compounds were administrated
via tail vein respectively. Mice were then sacrificed at 2 hours and 24 hours post
compound administration. TDLN and tumour tissues were isolated to generate single
cells suspension. The suspension cells were activated using PMA/Ionomycin/golgi plug
as described in Methods and Materials. Upon activation, CD4 marker was stained,
followed by fixation and permeabilisation for intracellular staining. (A) Th1 cell was
stained with fluorescence conjugated anti-IFN-γ, (B) Th2 with anti-IL-4, (C) Th17 with
anti-IL-17 and (D) regulatory T-cells with additional surface marker CD25, and
transcription factor FoxP3. Each group of cells was then quantified using flow
cytometry. Data represent mean ± SEM with minimum of four mice per group. * p <
0.05 vs I2-BODIPY using one-way ANOVA (Dunnett’s test).
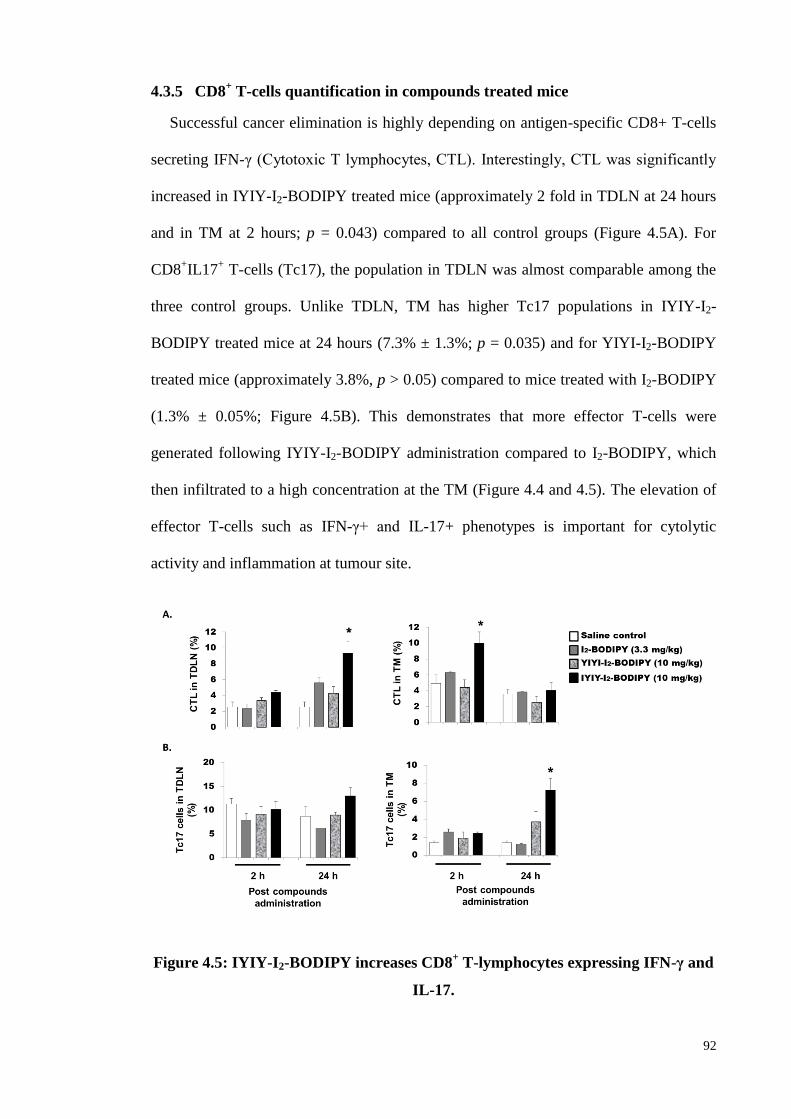
92
4.3.5 CD8+ T-cells quantification in compounds treated mice
Successful cancer elimination is highly depending on antigen-specific CD8+ T-cells
secreting IFN-γ (Cytotoxic T lymphocytes, CTL). Interestingly, CTL was significantly
increased in IYIY-I2-BODIPY treated mice (approximately 2 fold in TDLN at 24 hours
and in TM at 2 hours; p = 0.043) compared to all control groups (Figure 4.5A). For
CD8+IL17
+ T-cells (Tc17), the population in TDLN was almost comparable among the
three control groups. Unlike TDLN, TM has higher Tc17 populations in IYIY-I2-
BODIPY treated mice at 24 hours (7.3% ± 1.3%; p = 0.035) and for YIYI-I2-BODIPY
treated mice (approximately 3.8%, p > 0.05) compared to mice treated with I2-BODIPY
(1.3% ± 0.05%; Figure 4.5B). This demonstrates that more effector T-cells were
generated following IYIY-I2-BODIPY administration compared to I2-BODIPY, which
then infiltrated to a high concentration at the TM (Figure 4.4 and 4.5). The elevation of
effector T-cells such as IFN-γ+ and IL-17+ phenotypes is important for cytolytic
activity and inflammation at tumour site.
Figure 4.5: IYIY-I2-BODIPY increases CD8+ T-lymphocytes expressing IFN-γ and
IL-17.

93
4T1 tumour bearing mice were randomly divided into four treatment groups (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) and compounds were administrated
via tail vein respectively. Mice were then sacrificed at 2 hours and 24 hours post
compound administration. TDLN and tumour tissues were isolated to generate single
cells suspension. The suspension cells were activated using PMA/Ionomycin/golgi plug
as described in Methods and Materials. Upon activation, CD8 marker was stained,
followed by fixation and permeabilisation for intracellular staining. (A) CTL was
stained with fluorescence conjugated anti-IFN-γ, (B) Tc17 with anti-IL-17. Each group
of cells was then quantified using flow cytometry. Data represent mean ± SEM with
minimum of four mice per group. * p < 0.05 vs I2-BODIPY using one-way ANOVA
(Dunnett’s test).
4.3.6 Confirmation of immunomodulatory effects by using IYIY-TEG (ligand
without photosensitiser)
In order to confirm the immunomodulatory activity of conjugate is derived from the
TrkC ligand but not from the photosensitiser I2-BODIPY, a study of using another
control, IYIY-TEG (ligand without photosensitiser) was performed. Data showed that
IYIY-TEG reduced immunosuppressive G-MDSC and Treg cells (Figure 4.6A), and
increased CD4+ (Figure 4.6B) and CD8
+ (Figure 4.6C) expressing IFN-γ and IL-17 in
lymphoid organs at 2 hours and 24 hours dark. These effects mirror those produced by
both ligated conjugates, especially for IYIY-I2-BODIPY. Based on this and the lack of
immune activity from free I2-BODIPY as observed in Figure 4.1-4.5, suggests that IYIY
ligand itself (i) reduces immunosuppressive mediators TGF-β, G-MDSC and Treg; and,
(ii) elevates an adaptive immune response with IFN-γ and IL-17 phenotypes.
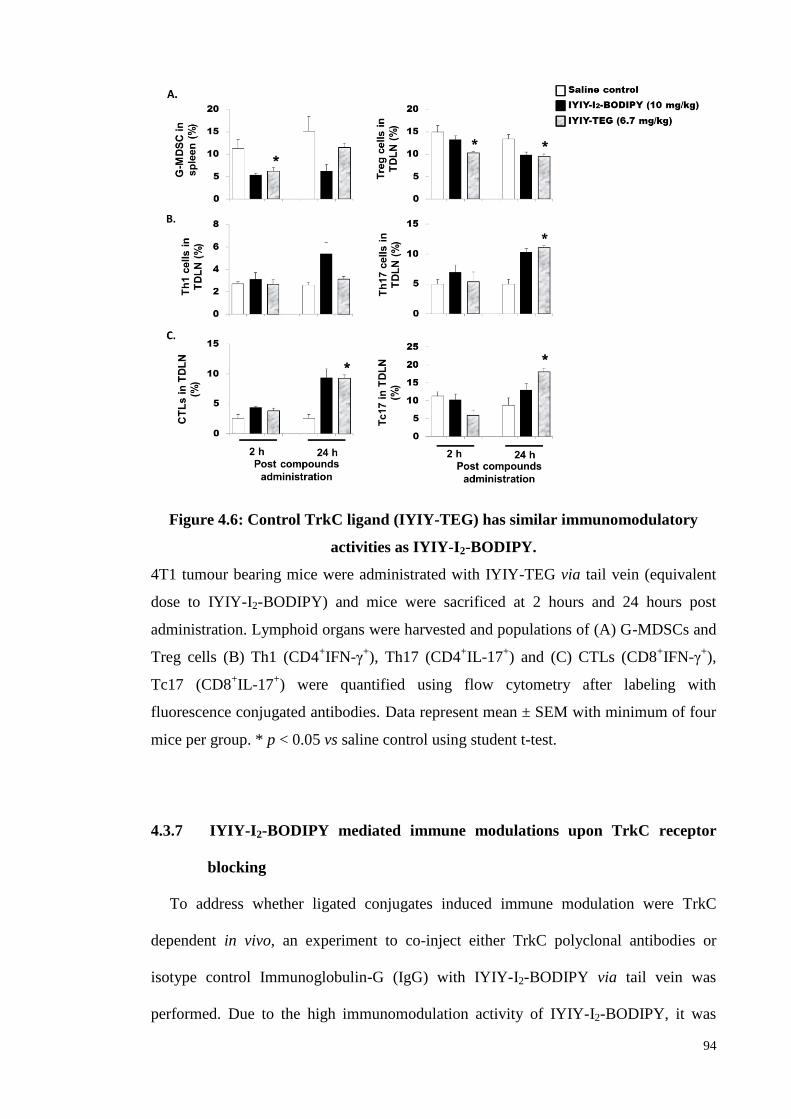
94
Figure 4.6: Control TrkC ligand (IYIY-TEG) has similar immunomodulatory
activities as IYIY-I2-BODIPY.
4T1 tumour bearing mice were administrated with IYIY-TEG via tail vein (equivalent
dose to IYIY-I2-BODIPY) and mice were sacrificed at 2 hours and 24 hours post
administration. Lymphoid organs were harvested and populations of (A) G-MDSCs and
Treg cells (B) Th1 (CD4+IFN-γ
+), Th17 (CD4
+IL-17
+) and (C) CTLs (CD8
+IFN-γ
+),
Tc17 (CD8+IL-17
+) were quantified using flow cytometry after labeling with
fluorescence conjugated antibodies. Data represent mean ± SEM with minimum of four
mice per group. * p < 0.05 vs saline control using student t-test.
4.3.7 IYIY-I2-BODIPY mediated immune modulations upon TrkC receptor
blocking
To address whether ligated conjugates induced immune modulation were TrkC
dependent in vivo, an experiment to co-inject either TrkC polyclonal antibodies or
isotype control Immunoglobulin-G (IgG) with IYIY-I2-BODIPY via tail vein was
performed. Due to the high immunomodulation activity of IYIY-I2-BODIPY, it was
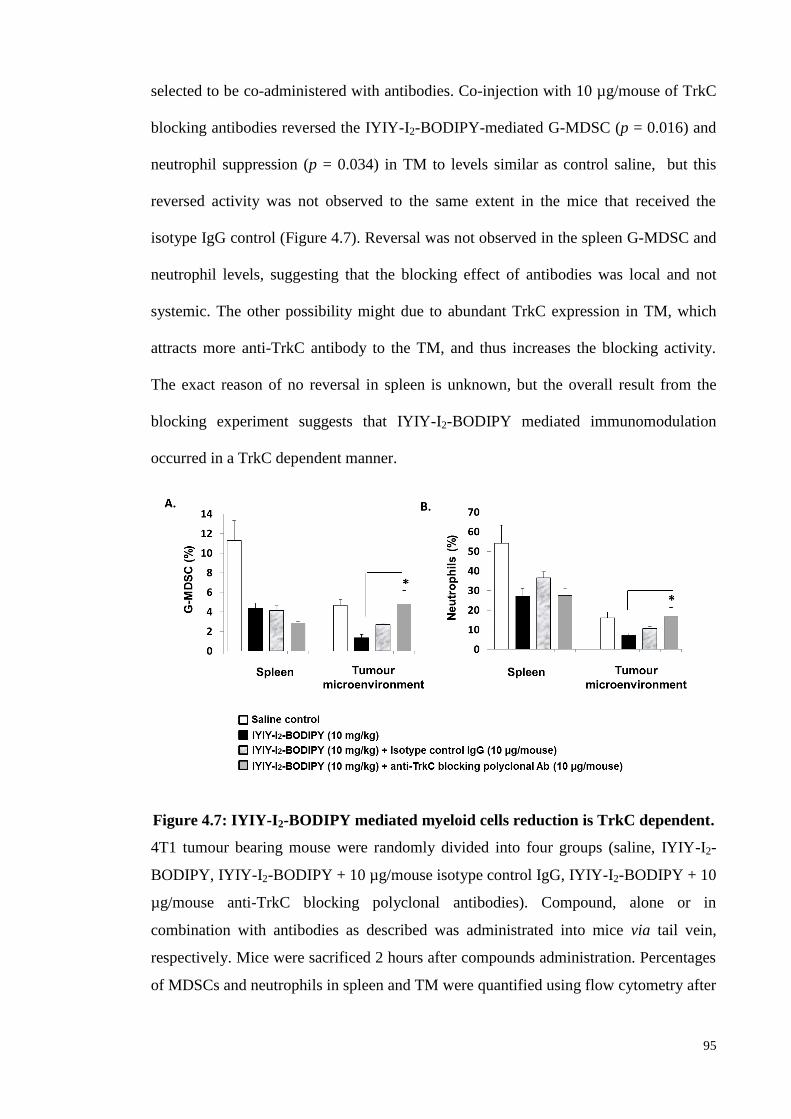
95
selected to be co-administered with antibodies. Co-injection with 10 µg/mouse of TrkC
blocking antibodies reversed the IYIY-I2-BODIPY-mediated G-MDSC (p = 0.016) and
neutrophil suppression (p = 0.034) in TM to levels similar as control saline, but this
reversed activity was not observed to the same extent in the mice that received the
isotype IgG control (Figure 4.7). Reversal was not observed in the spleen G-MDSC and
neutrophil levels, suggesting that the blocking effect of antibodies was local and not
systemic. The other possibility might due to abundant TrkC expression in TM, which
attracts more anti-TrkC antibody to the TM, and thus increases the blocking activity.
The exact reason of no reversal in spleen is unknown, but the overall result from the
blocking experiment suggests that IYIY-I2-BODIPY mediated immunomodulation
occurred in a TrkC dependent manner.
Figure 4.7: IYIY-I2-BODIPY mediated myeloid cells reduction is TrkC dependent.
4T1 tumour bearing mouse were randomly divided into four groups (saline, IYIY-I2-
BODIPY, IYIY-I2-BODIPY + 10 µg/mouse isotype control IgG, IYIY-I2-BODIPY + 10
µg/mouse anti-TrkC blocking polyclonal antibodies). Compound, alone or in
combination with antibodies as described was administrated into mice via tail vein,
respectively. Mice were sacrificed 2 hours after compounds administration. Percentages
of MDSCs and neutrophils in spleen and TM were quantified using flow cytometry after

96
cell surface staining. Data represent mean ± SEM of four mice. *p < 0.05 vs IYIY-I2-
BODIPY was analysed using student t-test.
4.3.8 Effect of conjugates administration in the dark on TrkC positive 4T1
tumour growth
Up to now, the data have shown that the ligated conjugates inhibited immuno-
suppressive mediators and increased adaptive immune responses in breast tumour
bearing mice. Next, the effect of conjugates to cause the delay in tumour growth in the
dark was carried out. The experiment involved single bolus injections of 10 mg/kg of all
three compounds via tail vein, and recordings of tumour volume three times per week.
The data shown in Figure 4.8 was quantified in comparison to the initial volume of the
respective tumours to eliminate tumour-to-tumour variation. As illustrated in Figure
4.8A, single bolus injection had no effect on delaying tumour growth, as the tumour
sizes were comparable with saline control in all treated groups. However, multiple
injections of three compounds with a regime of 10 mg/kg every day for five consecutive
days resulted in marginal delay in tumour growth only after IYIY-I2-BODIPY
administration at the first three days following treatment (days 7-9 after tumour cells
inoculation), and significant delay at days 10 (~ 16%) and 11 (~ 20%) post tumour cells
injection compared to all controls (Figure 4.8B). Unfortunately, the growth delay did
not persist, as the tumour sizes became comparable when the administration of IYIY-I2-
BODIPY was stopped. Even though the delay in tumour growth was statistically
significant, the effect may not be strong or prolonged enough for clinical relevance. In
addition, only IYIY-I2-BODIPY, but not YIYI-I2-BODIPY transiently delayed tumour
growth, as well as elicited strong immune responses, thus demonstrated that tumour
control was contributed by immune modulation. Previous study by Jin et al observed
that suppression of TGF-β by ligand-activated TrkC promoted tumourigenesis (Jin et al.,
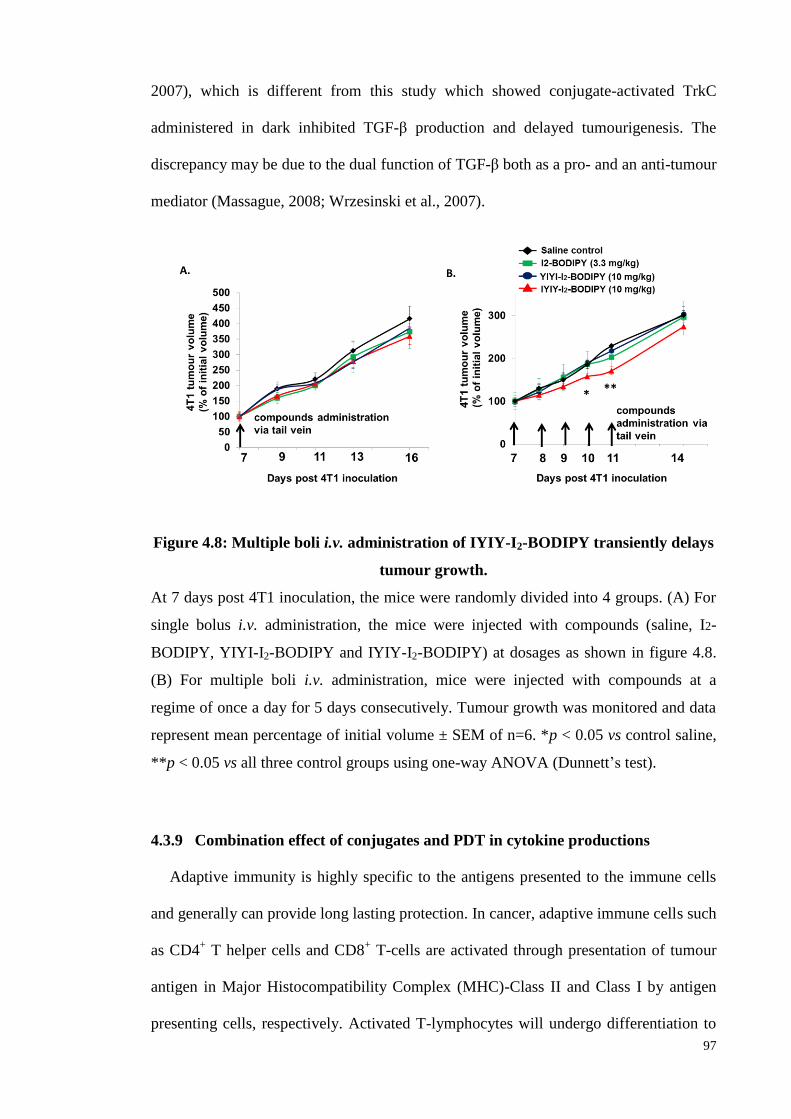
97
2007), which is different from this study which showed conjugate-activated TrkC
administered in dark inhibited TGF-β production and delayed tumourigenesis. The
discrepancy may be due to the dual function of TGF-β both as a pro- and an anti-tumour
mediator (Massague, 2008; Wrzesinski et al., 2007).
Figure 4.8: Multiple boli i.v. administration of IYIY-I2-BODIPY transiently delays
tumour growth.
At 7 days post 4T1 inoculation, the mice were randomly divided into 4 groups. (A) For
single bolus i.v. administration, the mice were injected with compounds (saline, I2-
BODIPY, YIYI-I2-BODIPY and IYIY-I2-BODIPY) at dosages as shown in figure 4.8.
(B) For multiple boli i.v. administration, mice were injected with compounds at a
regime of once a day for 5 days consecutively. Tumour growth was monitored and data
represent mean percentage of initial volume ± SEM of n=6. *p < 0.05 vs control saline,
**p < 0.05 vs all three control groups using one-way ANOVA (Dunnett’s test).
4.3.9 Combination effect of conjugates and PDT in cytokine productions
Adaptive immunity is highly specific to the antigens presented to the immune cells
and generally can provide long lasting protection. In cancer, adaptive immune cells such
as CD4+ T helper cells and CD8
+ T-cells are activated through presentation of tumour
antigen in Major Histocompatibility Complex (MHC)-Class II and Class I by antigen
presenting cells, respectively. Activated T-lymphocytes will undergo differentiation to

98
various subtypes depending on the cytokines milieu present. In PDT, antigen specific
adaptive immune responses are activated by release of high quantities of tumour
antigens by the damaged tumour tissues (Nowis et al., 2005; Reginato et al., 2014).
Data obtained until this point suggest that the IYIY-ligand had immuno-stimulatory
properties, hence we hypothesized that photo-activation of the I2-BODIPY
photosensitizer counterpart of IYIY-I2-BODIPY and YIYI-I2-BODIPY (hereafter
referred to as light treatment) following the administration of the conjugates into the
mice may induce stronger adaptive immune responses. To explore this idea, tumour
bearing mice were administrated with the IYIY-I2-BODIPY, YIYI-I2-BODIPY and I2-
BODIPY (10mg/kg equivalent dose) and illuminated with 100 J/cm2 of light after a
drug-light interval of 1 hour. Mice were then sacrificed 2 hours and 24 hours after PDT.
The levels of cytokines IL-2 (Figure 4.9A), IFN-γ (Figure 4.9B), TNF-α (Figure
4.9C), IL-4 (Figure 4.9D) and IL-10 (Figure 4.9G) post-PDT were found to be higher
compared to in dark, but similar among the groups including saline control, indicating
the presence of some light-induced immune effects. The level of IL-6 in plasma of
IYIY-I2-BODIPY treated mice was significantly high, at 2.4-fold higher at 2 hours (p =
0.039) and the level was elevated further at 24 hours to 5.2-fold higher compared to I2-
BODIPY treated mice. Conversely, the YIYI-I2-BODIPY and I2-BODIPY treated
groups showed increased IL-6 level (3.6- and 2-fold respectively) compared to saline
control only at 2 hours but not at 24 hours (Figure 4.9E). The increase of IL-6 in all
treatment groups following PDT is concordant with literatures (Du et al., 2006;
Gollnick et al., 2003). As before, the moderately high IL-6 level in YIYI-I2-BODIPY
treated group compared to I2-BODIPY treated group is probably due to the weak
binding of the conjugate to TrkC receptor.

99
Similar to trends in the dark experiments (Figure 4.1F), the level of IL-17A level
rose from low (4.8% ± 0.4%) at 2 hours to 2-fold higher at 24 hours (Figure 4.9F) solely
in IYIY-I2-BODIPY treated group. Similar levels of TGF-β inhibition by both IYIY-I2-
BODIPY and YIYI-I2-BODIPY were observed in the light (Figure 4.9H) compared to
in the dark at 2 hours (Figure 4.1H), suggesting that the ligand-mediated inhibition
effect was not intensified by light irradiation.
For ease of comparison, the data for IYIY-I2-BODIPY (highest immunomodulatory
activity) treated mice has been re-tabulated in terms of fold changes in the blood
cytokines in light treatment compared to those in dark, at 2 and 24 hours post-PDT.
Among the cytokines investigated, the biggest fold changes were: (i) 5.5-fold down-
regulation of TGF-β and (ii) 13-fold up-regulation of IL-6, both at 24 hours. High TGF-
β suppression is similar to the previously reported effect of ligand-activated TrkC
receptor in inhibiting TGF-β signaling (Jin et al. 2007), whereas the elevation of IL-6
might be due to the effect of ligand in promoting TrkC-induced IL-6 secretion (Rezaee
et al. 2010; Marshall et al. 1999). IL-17A was 2.8-fold increased in the light compared
to in the dark at 24 hours post-PDT, further suggesting that the high IL-6 level induced
by IYIY-I2-BODIPY had facilitated IL-17+ T-cell differentiation and IL-17 secretion.
The rest of the cytokines investigated had fold changes ranging from 1.25 to 2.4 (Figure
4.9I).
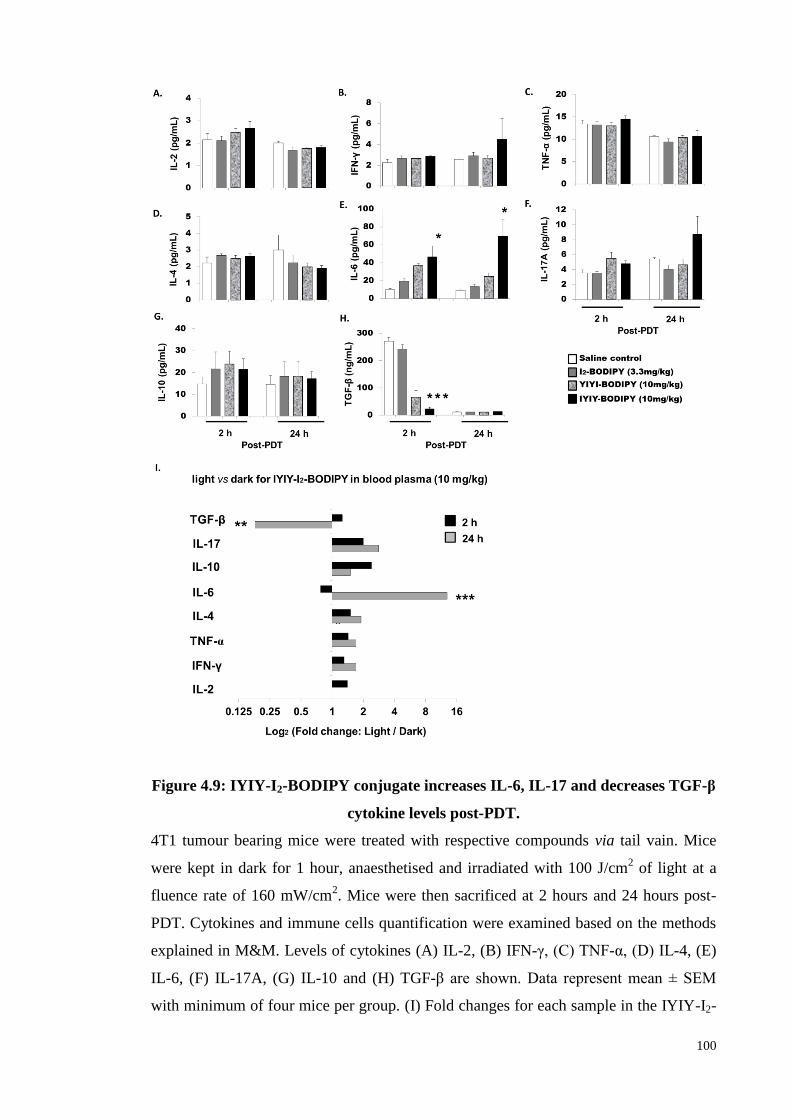
100
Figure 4.9: IYIY-I2-BODIPY conjugate increases IL-6, IL-17 and decreases TGF-β
cytokine levels post-PDT.
4T1 tumour bearing mice were treated with respective compounds via tail vain. Mice
were kept in dark for 1 hour, anaesthetised and irradiated with 100 J/cm2 of light at a
fluence rate of 160 mW/cm2. Mice were then sacrificed at 2 hours and 24 hours post-
PDT. Cytokines and immune cells quantification were examined based on the methods
explained in M&M. Levels of cytokines (A) IL-2, (B) IFN-γ, (C) TNF-α, (D) IL-4, (E)
IL-6, (F) IL-17A, (G) IL-10 and (H) TGF-β are shown. Data represent mean ± SEM
with minimum of four mice per group. (I) Fold changes for each sample in the IYIY-I2-

101
BODIPY treated group were obtained by dividing the percentage in the light over the
percentage in the dark. * p < 0.05, *** p < 0.001 vs I2-BODIPY using one-way
ANOVA (Dunnett’s test).
4.3.10 Effect of IYIY-I2-BODIPY and irradiation on neutrophil populations
In the TM sample from the light experiment, the population of myeloid precursor
cells including G-MDSC and neutrophils were examined. Data revealed that G-MDSC
populations decreased as much as those in the dark experiment, which demonstrated
that irradiation did not further affect ligand-mediated suppression (Figure 4.10A). In
PDT, acute inflammation at the treatment site occurs as a result of production of pro-
inflammatory cytokines and the presence of neutrophils (Cecic et al., 2001; Cecic et al.,
2006; de Vree et al., 1996). Hence the neutrophil population in TM post-PDT was
examined. Interestingly, IYIY-I2-BODIPY, but not other control compounds,
significantly increased the neutrophils in TM (p = 0.035) compared to saline, YIYI-I2-
BODIPY and I2-BODIPY (Figure 4.10B). This suggests that IYIY-I2-BODIPY recruits
more neutrophils to TM post-PDT. This can be explained by the good antitumour
efficacy of IYIY-I2-BODIPY in causing tumour damage as observed in figure 3.8 and
3.10, leading to the local accumulation of inflammatory neutrophils. This is concordant
with de Vree et al. 1996 who proposed that high PDT efficacy is associated with
increased population of neutrophils in systemic circulation.
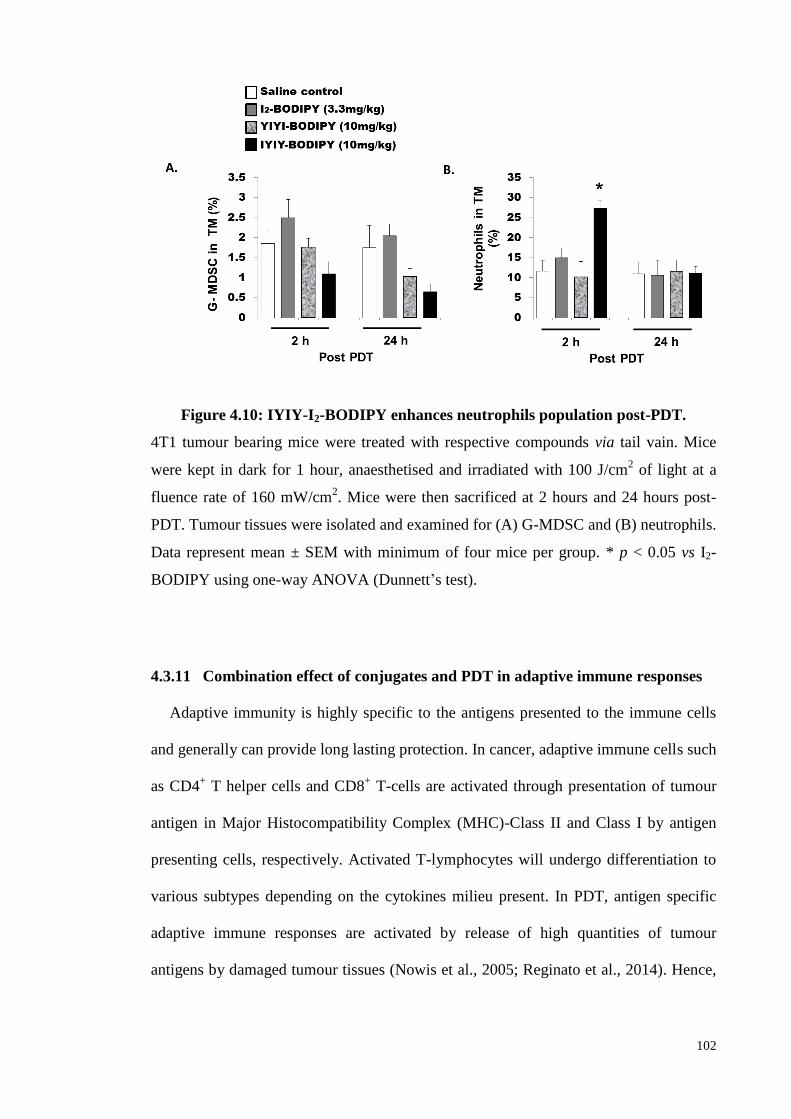
102
Figure 4.10: IYIY-I2-BODIPY enhances neutrophils population post-PDT.
4T1 tumour bearing mice were treated with respective compounds via tail vain. Mice
were kept in dark for 1 hour, anaesthetised and irradiated with 100 J/cm2 of light at a
fluence rate of 160 mW/cm2. Mice were then sacrificed at 2 hours and 24 hours post-
PDT. Tumour tissues were isolated and examined for (A) G-MDSC and (B) neutrophils.
Data represent mean ± SEM with minimum of four mice per group. * p < 0.05 vs I2-
BODIPY using one-way ANOVA (Dunnett’s test).
4.3.11 Combination effect of conjugates and PDT in adaptive immune responses
Adaptive immunity is highly specific to the antigens presented to the immune cells
and generally can provide long lasting protection. In cancer, adaptive immune cells such
as CD4+ T helper cells and CD8
+ T-cells are activated through presentation of tumour
antigen in Major Histocompatibility Complex (MHC)-Class II and Class I by antigen
presenting cells, respectively. Activated T-lymphocytes will undergo differentiation to
various subtypes depending on the cytokines milieu present. In PDT, antigen specific
adaptive immune responses are activated by release of high quantities of tumour
antigens by damaged tumour tissues (Nowis et al., 2005; Reginato et al., 2014). Hence,

103
the populations of CD4+ and CD8
+ T lymphocytes and subtypes in conjugates treated
mice post-irradiation were investigated.
In tumour microenvironment, the number of Th1 was significantly more in IYIY-I2-
BODIPY treated mice at 2 hours post-PDT in TM compared to I2-BODIPY treated mice
(3.4-fold; p = 0.024). At 24 hours, the Th1 populations dropped to become comparable
among the groups. Similar but weaker increase in Th1 population was observed in
YIYI-I2-BODIPY treated mice. For CD8+ T-cells, IYIY-I2-BODIPY treated mice had
higher populations of CTL, which was 2.1-fold (p = 0.033) higher than I2-BODIPY.
(Figure 4.11A). YIYI-I2-BODIPY treated mice have 1.6-fold higher CTL population
compared to I2-BODIPY treated mice. The increase of CTL might be due to the PDT
effect, as CTL was not increased in YIYI-I2-BODIPY treated mice in the dark. Similar
in the dark, IYIY-I2-BODIPY treated mice had higher Th17 and Tc17 cells, which were
3.4-fold and 2.9-fold higher compared to I2-BODIPY treated mice at 24 hours,
respectively (Figure 4.11B). Regulatory T-cell that was reported decreases post IYIY-I2-
BODIPY treatment was found to be decreased as well post-irradiation, suggest that PDT
did not affect the ligand-mediated Treg inhibition (Figure 4.11C). Th2 cell population
was reduced in the light at 2 hours post-PDT compared to in the dark (Figure 4.11D).
The decrease of the Th2 cells post-PDT and increase of the Th1 cell population are in
line with the known subtype balancing of Th1/Th2 cells, where activation of either cell
type can down-regulate the other (Kidd P, 2003). The fold changes in immune cell
populations in tumour microenvironment in the light treatment compared to those in the
dark was tabulated as shown in Figure 4.11E. The Th1, Th17, CTL and Tc17
populations investigated in IYIY-I2-BODIPY treated group were in increasing trend at 2
h and 24 h post-PDT compared to in the dark, ranging from 1.4- to 1.9-fold in Th1 and
Th17 and 1.2- to 1.8-fold in CTL and Tc17. Altogether suggests the combine between
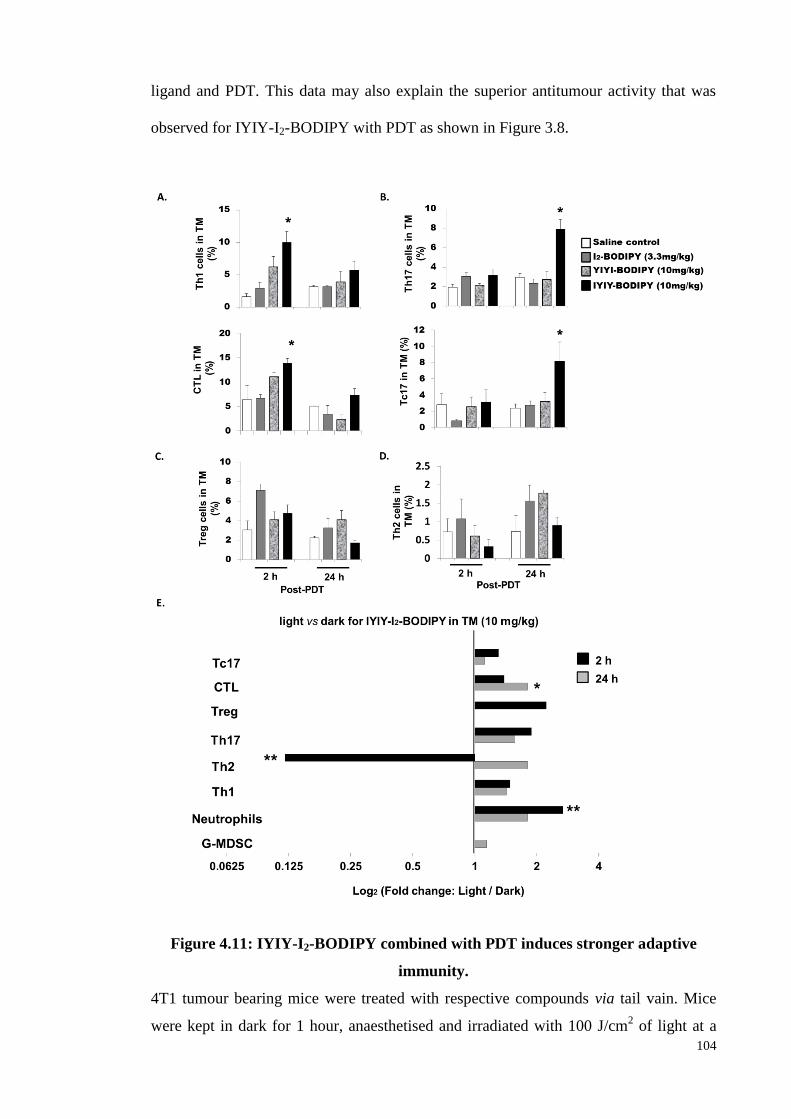
104
ligand and PDT. This data may also explain the superior antitumour activity that was
observed for IYIY-I2-BODIPY with PDT as shown in Figure 3.8.
Figure 4.11: IYIY-I2-BODIPY combined with PDT induces stronger adaptive
immunity.
4T1 tumour bearing mice were treated with respective compounds via tail vain. Mice
were kept in dark for 1 hour, anaesthetised and irradiated with 100 J/cm2 of light at a

105
fluence rate of 160 mW/cm2. Mice were then sacrificed at 2 hours and 24 hours post-
PDT. Immune cells in tumour microenvironment were quantification based on the
methods explained in M&M. Data of (A) Th1, CTL; (B) Th17, Tc17; (C) Treg cell and
(D) Th2 cell was quantified using flow cytometry. Data represent mean ± SEM of
minimum four mice in each group. (E) Fold changes for each cell type in the IYIY-I2-
BODIPY treated group were obtained by dividing the percentage of population in the
light over the percentage of population in the dark. * p < 0.05 vs I2-BODIPY using one-
way ANOVA.
4.3.12 Effector T cells quantification in compounds treated mice at 20 days post-
PDT
Effective PDT of cancer would induce antitumour immunity through inflammation
and adaptive immune responses (Cecic et al., 2001; de Vree et al., 1996), which could
further lead to the development of immune effector against the tumour (Reginato et al.,
2014). As in figure 3.8, 71% of IYIY-I2-BODIPY treated mice were cured from tumour
post PDT. Thus, an experiment was performed to study whether IYIY-I2-BODIPY
treated mice have increased CD4+ and CD8
+ effector T-cell populations post PDT.
Compounds treated mice underwent PDT and at 20 days post-PDT, TDLN and spleen
was isolated and quantified for effector T-cells using CD44 as a surface marker. As
expected, IYIY-I2-BODIPY treated mice had significant high CD4+ (Figure 4.12A) and
CD8+ (Figure 4.12B) effector T-cells in TDLN and spleen compared to the other three
controls. However, no increase in effector T cells was observed in YIYI-I2-BODIPY
treated mice. This implies that IYIY-I2-BODIPY was a more effective PDT agent in
inducing antitumour effector T-cells than the YIYI conjugate.
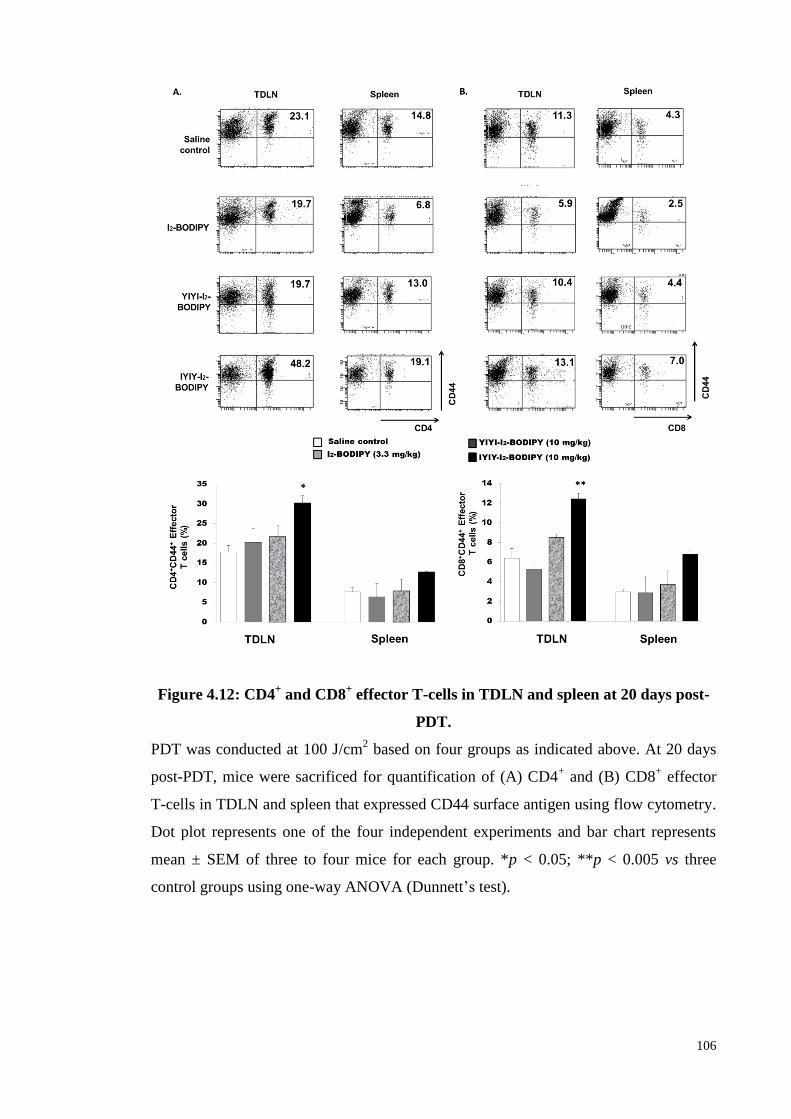
106
Figure 4.12: CD4+ and CD8
+ effector T-cells in TDLN and spleen at 20 days post-
PDT.
PDT was conducted at 100 J/cm2 based on four groups as indicated above. At 20 days
post-PDT, mice were sacrificed for quantification of (A) CD4+ and (B) CD8
+ effector
T-cells in TDLN and spleen that expressed CD44 surface antigen using flow cytometry.
Dot plot represents one of the four independent experiments and bar chart represents
mean ± SEM of three to four mice for each group. *p < 0.05; **p < 0.005 vs three
control groups using one-way ANOVA (Dunnett’s test).

107
4.3.13 Antitumour immunity in IYIY-I2-BODIPY treated survivor mice post-PDT
Lastly, the presence of long term immunity against aggressive 4T1 tumour cells in
IYIY-I2-BODIPY treated survivor mice post-PDT was examined. The splenocytes and
lymphocytes (suspension cells from TDLN) of healthy and survivor mice were isolated
and adoptively transferred into syngeneic healthy recipient mice via tail vein. Recipient
mice were challenged by subcutaneous injection of 4T1 tumour cells at 2 days post
adoptive transfer as described in the schematic plan in figure 4.13A. Interestingly,
growth delay was observed in the mice receiving splenocytes and lymphocytes from the
survivor mice, compared to controls (splenocytes and lymphocytes from healthy mice).
In brief, the delay in tumour growth was transient at days one to four post tumour
inoculation and became significant (~ 50%) in both splenocytes and lymphocytes
recipient mice at 7 days post inoculation (Figure 4.13B). The delay in tumour growth
continued, with approximately 40% smaller tumours sizes in mice receiving immune
cells from survivor mice at both 9 and 11 days post inoculation compared to mice with
cells from healthy donors. Unfortunately, there was no full immunity in the mice after
adoptive transfer, probably due to the highly aggressive and metastatic nature of 4T1
cells. However, the delayed tumour growth was significant as compared to controls,
suggesting the presence of anti-tumour immune effector T cells. Taken together, the
higher survivor rate previously observed in IYIY-I2-BODIPY PDT treated mice
compared to the other controls (Figure 3.8) might have been due to the enhanced
immune effector T cells caused by the combined TrkC and PDT therapy.
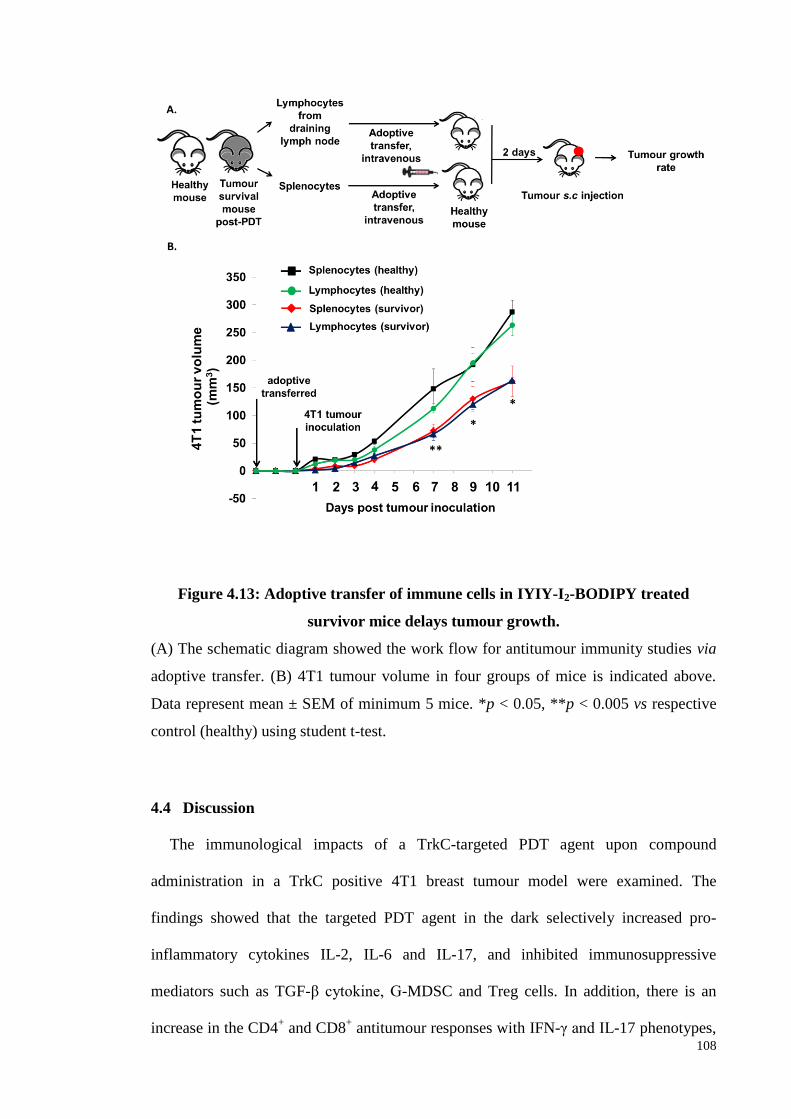
108
Figure 4.13: Adoptive transfer of immune cells in IYIY-I2-BODIPY treated
survivor mice delays tumour growth.
(A) The schematic diagram showed the work flow for antitumour immunity studies via
adoptive transfer. (B) 4T1 tumour volume in four groups of mice is indicated above.
Data represent mean ± SEM of minimum 5 mice. *p < 0.05, **p < 0.005 vs respective
control (healthy) using student t-test.
4.4 Discussion
The immunological impacts of a TrkC-targeted PDT agent upon compound
administration in a TrkC positive 4T1 breast tumour model were examined. The
findings showed that the targeted PDT agent in the dark selectively increased pro-
inflammatory cytokines IL-2, IL-6 and IL-17, and inhibited immunosuppressive
mediators such as TGF-β cytokine, G-MDSC and Treg cells. In addition, there is an
increase in the CD4+ and CD8
+ antitumour responses with IFN-γ and IL-17 phenotypes,

109
and growth delay of aggressive 4T1 tumours in mice. While IYIY-BODIPY in the dark
activated the immune system and decreased tumour growth to some extent, the assault
by reactive oxygen species upon PDT accentuated these effects, probably because the
sensitivity of the immune system was increased due to formation of damaged tissue.
Upon PDT, this conjugate also enhanced the population of effector T-cells and delayed
the tumour growth through adoptive transfer of immune cells. This is the first report of
antitumour immune responses elicited by a targeted ligand designed primarily to
actively direct a therapeutic agent to cancer.
Throughout the study, the scrambled control YIYI-I2-BODIPY showed similar, but
weaker immune-modulation properties than IYIY-I2-BODIPY. This might be due to the
isomeric characteristics of scrambled ligands (reversed order of amino acids), leading to
similar, albeit weaker binding affinity to the targeted receptor compared to the
unscrambled ligands (Koolpe et al., 2002). Another possibility is the presence of
multiple bioactive conformations in peptide ligands that can be recognised by more than
one receptor types (Pedragosa-Badia et al., 2013). It is therefore possible that the YIYI-
I2-BODIPY scrambled control adopts a bioactive conformation that binds to other
unknown receptors to modulate immune response.
IYIY-I2-BODIPY induced IL-6 secretion and reduced G-MDSCs and neutrophils
both in dark and light treatments. The concurrent reduction in G-MDSCs and
neutrophils however is in contrast as in other studies that reported correlative increases
in IL-6 with MDSC expansion and inflammation (Bunt et al., 2007; Meyer et al., 2011;
Mundy-Bosse et al., 2011). It is possible that the TrkC ligand mediated MDSC
suppression was independent of IL-6 cytokine, but through direct binding to myeloid
cells, since blocking with TrkC antibodies could reverse the IYIY-I2-BODIPY-mediated

110
MDSC suppression. Another explanation for the mechanism of MDSC suppression may
involve transcription factor STAT-3 which is an essential mediator for regulating
myeloid progenitor proliferations (Zhang et al., 2010), as well as a downstream
mediator of Trk signaling (Ng et al., 2006). In tumour bearing hosts with inflammation,
MDSC is considered a subpopulation of neutrophils that have acquired suppressive
phenotype (Pillay et al. 2013). Thus, suggests that the conjugates mediated MDSC and
neutrophils suppression is concurrent.
Studies have revealed that the balance of Treg and Th17 differentiation is regulated
by the relative abundance of differentiation factors TGF-β and IL-6. A high level of
TGF-β generally promotes Treg differentiation and inhibits Th17 (Zhou et al., 2008),
whereas a high IL-6 level stimulates Th17 differentiation and antagonises Treg
formation (Zhou et al., 2007). The increased of Th17 upon IYIY-I2-BODIPY
administration both in dark and light treatments were in concordance with the latter
scenario. Th17 cells secrete IL-17 cytokine that is known to possess both protumour and
antitumour functions (Murugaiyan & Saha, 2009). The IYIY-BODIPY mediated IL-17+
cells in this study probably functioned in an antitumour way based on the observed
delay in tumour growth post IYIY-I2-BODIPY administration, the reduction of TGF-β
which is known to block the production of vascular endothelial growth factor (VEGF), a
potent angiogenic factor (Huang & Lee, 2003; Jeon et al., 2007) and, increased in Th1
and CTL cell populations which have been reported in the presence of IL-17
(Benchetrit et al., 2002; Muranski et al., 2008).
TGF-β signaling has dual functions in cancer: (i) as a tumour suppressor to mediate
growth arrest and apoptosis in cancer cells (antitumour) (Derynck et al., 2001; Pardali &
Moustakas, 2007), and (ii) as a potent immunosuppressive agent to suppress both innate

111
and adaptive immune responses comprising of CD4+ effector T-cells (Th1 and Th2),
CD8+ cytotoxic T cells (CTLs), NK cells and antigen presenting cells, as well as to
stimulate the generation of regulatory T-cells which inhibit effector T-cell functions
(protumour) (Massague, 2008). Jin and co-workers reported TrkC and its tyrosine
kinase activity mediate tumourigenesis via suppression of the TGF-β signaling (Jin et al.,
2007), suggesting that TrkC acts as a pro-tumour agent by inhibiting tumour suppressor
TGF-β. Contrary to this, our data suggests that TrkC and TGF-β played an anti-tumour
role. To explain, the binding of TrkC by the IYIY conjugate might have activated the
tyrosine kinase activity of TrkC and subsequently blocked the TGF-β signalling and
inhibited TGF-β production at an early time point, which in turn reduced the Treg cells
in a late time point to overall delay the tumour growth.
TrkC has also been reported to play a role in regulating the systemic balance of T
helper subtypes. A study from Sekimoto and colleagues revealed that neurotrophin-3
(TrkC natural ligand) increased the secretion of IL-4 by Th2 subtype, but did not affect
the Th1 population IFN-γ, as Th1 did not express TrkC but Th2 did expressed
(Sekimoto et al., 2003). Other studies reported that neurotrophins increase IL-6
secretion via Trk receptors (Marshall et al., 1999; Rezaee et al., 2010). Hence, the
increase in IL-4 and IL-6 levels observed upon IYIY-I2-BODIPY administration is in
agreement with what has been reported in literature. However, this increase in IL-4 and
Th2 cells was also observed in I2-BODIPY and YIYI-I2-BODIPY control, suggesting
that the increased in control groups might due to other unknown factors mediated by
ligands or I2-BODIPY itself.
PDT can lead to systemic induction of IL-6 (Du et al., 2006; Gollnick et al., 2003),
and this cytokine has been studied to possess dual functions, either in enhancing PDT

112
efficacy (Barathan et al., 2013; Usuda et al., 2001; Wei et al., 2007) or inhibiting PDT
mediated antitumour immunity (Brackett et al., 2011) via regulation of apoptotic
proteins. The results in this study suggest that IL-6 was an antitumour factor, likely by
enhancing PDT efficacy via upregulation of Th17 and Tc17 cells. This is contrary to the
findings by Brackett et al. 2011, where they reported IL-6 attenuated PDT mediated
antitumour immune memory in an IL-6 knockout mouse model implanted with 4T1
breast mammary tumour. The differences in findings might due to the different drugs
administered (the ligand in IYIY-I2-BODIPY has additional immunomodulation
properties) or the extreme difference in total systemic IL-6 level in different genetic
background of mice (IL-6 knockout vs normal). The role of IL-6 in PDT mediated
immune responses may depend on the severity of inflammation and its action in
mediating the balance between differentiation of Th17 and Treg cells.
As shown in figure 4.8, single bolus administration has no antitumour effect, whereas
multiple bolus administration only induced transient effect, and diminished when IYIY-
I2-BODIPY was stopped giving to the mice. Thus, predicted that adoptively transfer of
the immune cells from IYIY-I2-BODIPY treated donor mice without irradiation has no
antitumour effect in recipient mice. Although the adoptive transferred of immune cells
from survivor donor mice only induced delay in tumour growth, but not full immunity
in recipient mice. However, this is sufficient to demonstrate the presence of effector T
cells. Perhaps the antitumour effect would be further enhanced via in vitro expansion of
the isolated T cells, which is a recognised adoptive immune cell therapy for cancer
treatment.

113
4.5 Conclusion
Stimulation of inflammatory IL-6, IL-17 cytokines and inhibition of
immunosuppressive mediators are potential contributors of immune stimulatory effects
in IYIY-ligand, as summarised in figure 4.14. Hence, the conjugate has promise as a
therapeutic agent for cancer treatment to selectively destroy the tumour cells in PDT
and simultaneously stimulates antitumour immunity in the host.
Figure 4.14: Summary findings of TrkC targeted PDT agent on immune responses.

114
CHAPTER 5: OVERALL CONCLUSION AND FUTURE PERSPECTIVES
5.1 Overall Conclusion
Enhanced delivery of anticancer drugs to the tumour tissue through the conjugation
of the drugs to small molecule ligands that target the molecules overexpressed on cancer
cell surface is one among the increasingly favorable approaches in anticancer therapy
research. Active targeting of ligand-drug conjugates can be used to target cancer cells
that are resistant to the free drug so as to enhance the anticancer effects (Yang et al.,
2009; Zhang et al., 2008).
Previously, IYIY, a synthetic TrkC ligand has been shown to possess good
selectivity in binding to cancer cells over-expressing TrkC receptor in vitro (Kamkaew
and Burgess, 2013). These studies further proved that IYIY retained its selectivity in
tumour cell binding upon conjugation to a model photodynamic therapy (PDT) agent I2-
BODIPY. The formed ligand-drug conjugate, i.e. IYIY-I2-BODIPY has shown selective
cell binding and cytotoxicity to TrkC expressing cancer cells, and have selectively
accumulated in TrkC expressing tumour in vivo, with maximum accumulation at 1 hour
post compound administration and at a significantly higher level (2.1-fold) compared to
a scrambled control. In addition, among the major organs investigated, tumour tissue is
the third highest in IYIY-I2-BODIPY accumulation after excretory organs liver and
kidney. In contrast, accumulation of YIYI-I2-BODIPY in tumour was lower than
excretory organs, lymph nodes and lung. This suggests the selectivity of IYIY-I2-
BODIPY in TrkC positive cells. In antitumour evaluation using TrkC expressing
tumour model, the IYIY-I2-BODIPY possesses superior antitumour activity with 71%
mice showing no sign of primary tumour regrowth and metastasis for up to 90 days of

115
observation, and was confirmed by histopathological evaluation. The selectivity was
further confirmed when moderate antitumor activity was observed in non-TrkC
expressing tumour model.
As the IYIY was able to mimic the TrkC natural ligand NT-3 in activating signal
transduction that regulate neuronal cell survival and differentiation (Chen et al., 2009);
NT-3 can function as non-cytokine mediators to modulate both innate and adaptive
immune responses (Matsuda et al., 1988; Marshall et al., 1999; Sekimoto et al., 2003;
Rezaee et al., 2010). Hence, further investigation was conducted to access the
immunomodulation capability of the IYIY-I2-BODIPY. This study shown that TrkC
targeted ligand-drug conjugates (IYIY-I2-BODIPY and YIYI-I2-BODIPY) modulates
immune responses through (i) the inhibition of immunosuppressive mediators (TGF-β,
granulocytic-MDSC and Treg cells) and (ii) the stimulation of antitumour immune
responses (Th1, CTL, Th17, Tc17). Moreover, IYIY-I2-BODIPY treated mice have
higher effector T-cells post-light irradiation compared to all control groups. Adoptive
transfer of immune cells from the IYIY-I2-BODIPY treated tumour survivor mice to
other tumour bearing mice significantly delayed the tumour growth in these mice
compared to mice receiving immune cells from healthy donors (Figure 5.1).
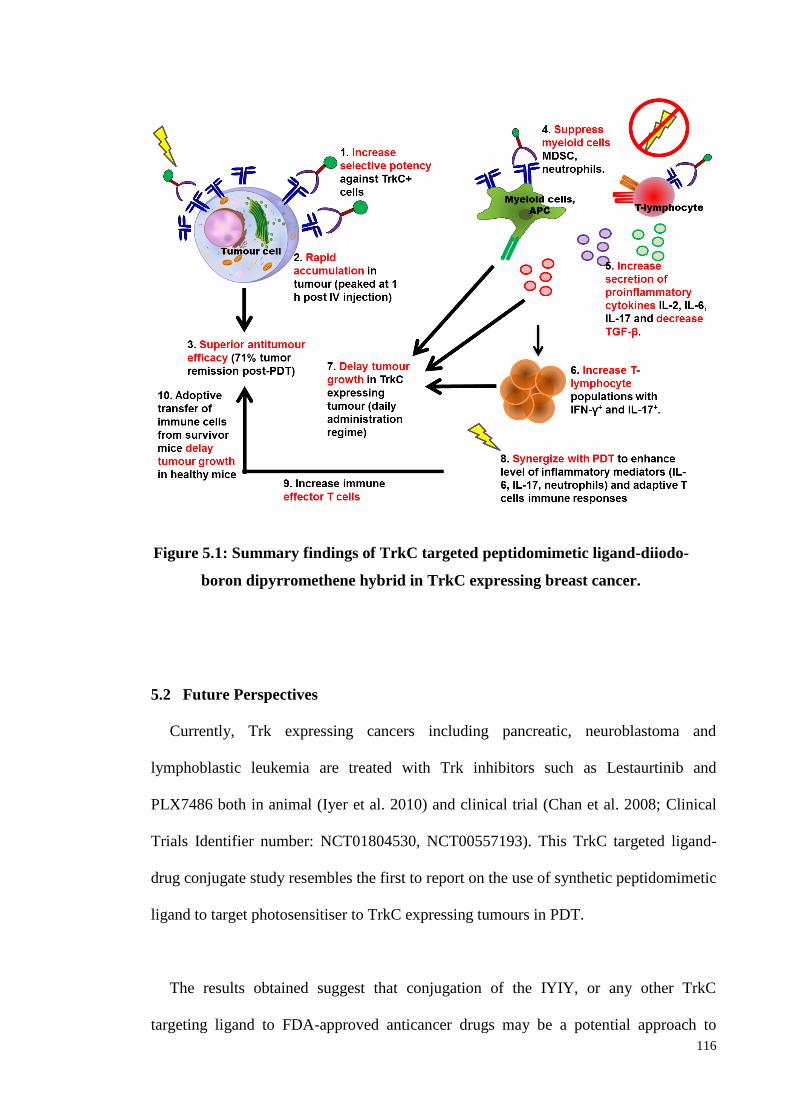
116
Figure 5.1: Summary findings of TrkC targeted peptidomimetic ligand-diiodo-
boron dipyrromethene hybrid in TrkC expressing breast cancer.
5.2 Future Perspectives
Currently, Trk expressing cancers including pancreatic, neuroblastoma and
lymphoblastic leukemia are treated with Trk inhibitors such as Lestaurtinib and
PLX7486 both in animal (Iyer et al. 2010) and clinical trial (Chan et al. 2008; Clinical
Trials Identifier number: NCT01804530, NCT00557193). This TrkC targeted ligand-
drug conjugate study resembles the first to report on the use of synthetic peptidomimetic
ligand to target photosensitiser to TrkC expressing tumours in PDT.
The results obtained suggest that conjugation of the IYIY, or any other TrkC
targeting ligand to FDA-approved anticancer drugs may be a potential approach to

117
improve the selectivity and efficacy profiles of the anticancer therapeutics in the
treatment of TrkC expressing cancer. In addition, studies reported that small molecule
Trk inhibitors were able to suppress the activities of TrkA/B/C kinases mediated
survival pathway in cancer and subsequently inhibited tumour growth (Iyer et al., 2010;
Minturn et al., 2011). Thus, combination therapy by using IYIY-I2-BODIPY with Trk
inhibitors (Lestaurtinib, PLX7486) is possible in order to increase the therapeutic
efficacy on TrkC expressing cancer. Besides, mice treated with the synthetic TrkC
targeted IYIY-I2-BODIPY were found to develop significant antitumor immune
responses post-PDT. This further suggests the possible use of the IYIY-ligand as a
systemic photoimmunotherapeutic agent when combined with therapies such as
photothermal therapy and sonodynamic therapy that can provoke the abundant release
of tumour antigens. The shortcoming of this study is lacking of biodistribution data on
brain organ. This is important to verify, as TrkC receptor is found predominantly in
brain and neuronal cells. The ability of conjugates across the blood brain barrier to the
brain is interesting to explore, perhaps the IYIY-ligand can be used to treat glioblastoma,
in which the treatment remains one of the most challenging tasks in clinical oncology.
TrkC ligand can also be conjugated with imaging probes for theranostic purposes
such as tracing biodistribution of drugs and detection of local and metastatic TrkC
expressing tumours in a more precise way. This could be very useful in clinical for
monitoring the status of TrkC in cancer patient. Despite of neurotoxicity characteristics
of the IYIY-I2-BODIPY prototype (probably via binding to Trk receptors of neuronal
cells), the antitumor efficacy on TrkC+ tumour was superior at half-maximum tolerated
dose. The toxicity problem can be reduced by conjugating IYIY ligand (active targeting)
on nano-carrier (passive targeting). Through a combination of active and passive
targeting approach, the ligand-nano-carrier complexes tend to accumulate more in the

118
tumour region via enhanced permeation and retention (EPR) effect compared to non-
targeted neuron cells. Moreover, the amount of IYIY-ligand used as active targeting in
this approach can be reduced, and hence reduced the undesired toxicity. This study
showed that IYIY-ligand is very potent against TrkC+ tumour cells, targeting TrkC
using the IYIY-ligand maybe benefit for TrkC expressing human cancer. Perhaps
combination effect of IYIY-ligand-drug conjugate with Trk inhibitors such as
lestaurtinib and PLX7486 (Chan et al., 2008; Iyer et al. 2010) can enhance the
antitumour effect. In term of therapeutic purposes in PDT, further modification of IYIY-
conjugate such as using a longer wavelength (600 nm and above) photosensitiser for
deeper tissue penetration (Salva, 2002; Ballou et al., 2005; Frangioni, 2003; Rao et al.,
2007; Sevick-Muraca et al., 2002) would be ideal.
Among the available anticancer therapies, immunotherapy has been recognised as an
effective approach for anticancer therapy (American Association of Cancer Research).
Recently, a study was conducted by Spiegel’s group on the use of antibody-recruiting
small molecule (ARM-U2) as an immunotherapeutic agent for treatment of urokinase-
type plasminogen activator receptor (uPAR) expressing cancer. The ARM-U2 act as a
targeting agent to bind uPAR expressed on cancer cells, whereas the antibody will
attract immune cells to ARM-U2/uPAR complex in cancer cells and mediates antibody-
dependent cellular phagocytosis (Rullo et al. 2016). Perhaps this approach can be
applied in the treatment of TrkC expressing cancer by conjugating similar antibody to
TrkC targeting-ligand for effective therapeutic outcomes, particularly when newer TrkC
ligand that are more selective in receptor targeting is developed.

119
REFERENCES
Abdel-Hady, E. S., Martin-Hirsch, P., Duggan-Keen, M., Stern, P. L., Moore, J. V.,
Corbitt, G., et al. (2001). Immunological and viral factors associated with the
response of vulval intraepithelial neoplasia to photodynamic therapy. Cancer
Research, 61(1), 192-196.
Adams, G. P., Schier, R., McCall, A. M., Simmons, H. H., Horak, E. M., Alpaugh, R.
K., et al. (2001). High affinity restricts the localization and tumor penetration of
single-chain fv antibody molecules. Cancer Research, 61(12), 4750-4755.
Agarwal, A., Saraf, S., Asthana, A., Gupta, U., Gajbhiye, V., & Jain, N. K. (2008).
Ligand based dendritic systems for tumor targeting. International Journal of
Pharmaceutics, 350(1-2), 3-13.
Agarwal, M. L., Larkin, H. E., Zaidi, S. I., Mukhtar, H., & Oleinick, N. L. (1993).
Phospholipase activation triggers apoptosis in photosensitized mouse lymphoma
cells. Cancer Research, 53(24), 5897-5902.
Agata, Y., Kawasaki, A., Nishimura, H., Ishida, Y., Tsubat, T., Yagita, H., et al. (1996).
Expression of the PD-1 antigen on the surface of stimulated mouse T and B
lymphocytes. International Immunology, 8(5), 765-772.
Alexis, F., Pridgen, E., Molnar, L. K., & Farokhzad, O. C. (2008). Factors affecting the
clearance and biodistribution of polymeric nanoparticles. Molecular
Pharmaceutics, 5(4), 505-515.
Allison, R. R., Bagnato, V. S., Cuenca, R., Downie, G. H., & Sibata, C. H. (2006). The
future of photodynamic therapy in oncology. Future Oncology, 2(1), 53-71.
Alloatti, D., Giannini, G., Vesci, L., Castorina, M., Pisano, C., Badaloni, E., et al.
(2012). Camptothecins in tumor homing via an RGD sequence mimetic.
Bioorganic and Medicinal Chemistry Letters, 22(20), 6509-6512.
Amato, R. J., Shetty, A., Lu, Y., Ellis, P. R., Mohlere, V., Carnahan, N., et al. (2014). A
Phase I/Ib study of folate immune (EC90 vaccine administered with GPI-0100
adjuvant followed by EC17) with interferon-alpha and interleukin-2 in patients
with renal cell carcinoma. Journal of Immunotherapy, 37(4), 237-244.
Amato, R. J., Shetty, A., Lu, Y., Ellis, R., & Low, P. S. (2013). A phase I study of folate
immune therapy (EC90 vaccine administered with GPI-0100 adjuvant followed

120
by EC17) in patients with renal cell carcinoma. Journal of Immunotherapy,
36(4), 268-275.
Angiolillo, A. L., Sgadari, C., Taub, D. D., Liao, F., Farber, J. M., Maheshwari, S., et al.
(1995). Human interferon-inducible protein 10 is a potent inhibitor of
angiogenesis in vivo. The Journal of Experimental Medicine, 182(1), 155-162.
Arap, W., Pasqualini, R., & Ruoslahti, E. (1998). Cancer treatment by targeted drug
delivery to tumor vasculature in a mouse model. Science, 279(5349), 377-380.
Awuah, S. G., & You, Y. (2012). Boron dipyrromethene (BODIPY)-based
photosensitizers for photodynamic therapy. RSC Advances, 2(30), 11169-11183.
Babich, J., Coleman, R. E., Van Heertum, R., Vallabhajosula, S., Goldsmith, S.,
Osborne, J., et al. (2012). Small molecule inhibitors of prostate specific
membrane antigen (PSMA) for SPECT: Summary of phase I studies in patients
with prostate cancer (PCa). Journal of Nuclear Medicine,
53(1_MeetingAbstracts), 179.
Ballou, B., Ernst, L. A., & Waggoner, A. S. (2005). Fluorescence imaging of tumors in
vivo. Current Medicinal Chemistry, 12(7), 795-805.
Baluk, P., Morikawa, S., Haskell, A., Mancuso, M., & McDonald, D. M. (2003).
Abnormalities of basement membrane on blood vessels and endothelial sprouts
in tumors. The American Journal of Pathology, 163(5), 1801-1815.
Bandara, N. A., Hansen, M. J., & Low, P. S. (2014). Effect of receptor occupancy on
folate receptor internalization. Molecular Pharmaceutics, 11(3), 1007-1013.
Barathan, M., Mariappan, V., Shankar, E. M., Abdullah, B. J., Goh, K. L., & Vadivelu,
J. (2013). Hypericin-photodynamic therapy leads to interleukin-6 secretion by
HepG2 cells and their apoptosis via recruitment of BH3 interacting-domain
death agonist and caspases. Cell Death & Disease, 4, e697.
Bareford, L. M., & Swaan, P. W. (2007). Endocytic Mechanisms for Targeted Drug
Delivery. Advanced Drug Delivery Reviews, 59(8), 748-758.
Barrett, J. A., Coleman, R. E., Goldsmith, S. J., Vallabhajosula, S., Petry, N. A., Cho, S.,
et al. (2013). First-in-man evaluation of 2 high-affinity PSMA-avid small
molecules for imaging prostate cancer. Journal of Nuclear Medicine, 54(3), 380-
387.

121
Benchetrit, F., Ciree, A., Vives, V., Warnier, G., Gey, A., Sautes-Fridman, C., et al.
(2002). Interleukin-17 inhibits tumor cell growth by means of a T-cell-
dependent mechanism. Blood, 99(6), 2114-2121.
Bertrand, N., Wu, J., Xu, X., Kamaly, N., & Farokhzad, O. C. (2014). Cancer
nanotechnology: the impact of passive and active targeting in the era of modern
cancer biology. Advanced Drug Delivery Reviews, 66, 2-25.
Bischoff, S. C., & Dahinden, C. A. (1992). Effect of nerve growth factor on the release
of inflammatory mediators by mature human basophils. Blood, 79(10), 2662-
2669.
Blankenstein, T., Coulie, P. G., Gilboa, E., & Jaffee, E. M. (2012). The determinants of
tumour immunogenicity. Nature Review. Cancer, 12(4), 307-313.
Blasco-Gutierrez, M. J., Jose-Crespo, I. J., Zozaya-Alvarez, E., Ramos-Sanchez, R., &
Garcia-Atares, N. (2007). TrkC: a new predictive marker in breast cancer?
Cancer Investigation, 25(6), 405-410.
Botchkarev, V. A., Metz, M., Botchkareva, N. V., Welker, P., Lommatzsch, M., Renz,
H., et al. (1999). Brain-derived neurotrophic factor, neurotrophin-3, and
neurotrophin-4 act as "epitheliotrophins" in murine skin. Laboratory
Investigation, 79(5), 557-572.
Brackett, C. M., & Gollnick, S. O. (2011). Photodynamic Therapy Enhancement of
Anti-Tumor Immunity. Photochemical and Photobiological Sciences, 10(5),
649-652.
Brackett, C. M., Owczarczak, B., Ramsey, K., Maier, P. G., & Gollnick, S. O. (2011).
IL-6 potentiates tumor resistance to photodynamic therapy (PDT). Lasers in
Surgery and Medicine, 43(7), 676-685.
Brahimi, F., Ko, E., Malakhov, A., Burgess, K., & Saragovi, H. U. (2014).
Combinatorial assembly of small molecules into bivalent antagonists of TrkC or
TrkA receptors. PloS One, 9(3), e89617.
Brahimi, F., Malakhov, A., Lee, H. B., Pattarawarapan, M., Ivanisevic, L., Burgess, K.,
et al. (2009). A peptidomimetic of NT-3 acts as a TrkC antagonist. Peptides, 30,
1833-1839.
Briasoulis, E., Judson, I., Pavlidis, N., Beale, P., Wanders, J., Groot, Y., et al. (2000).
Phase I trial of 6-hour infusion of glufosfamide, a new alkylating agent with

122
potentially enhanced selectivity for tumors that overexpress transmembrane
glucose transporters: a study of the European Organization for Research and
Treatment of Cancer Early Clinical Studies Group. Journal of Clinical Oncology,
18(20), 3535-3544.
Briasoulis, E., Pavlidis, N., Terret, C., Bauer, J., Fiedler, W., Schoffski, P., et al. (2003).
Glufosfamide administered using a 1-hour infusion given as first-line treatment
for advanced pancreatic cancer. A phase II trial of the EORTC-new drug
development group. European Journal of Cancer, 39(16), 2334-2340.
Brodeur, G. M., Nakagawara, A., Yamashiro, D. J., Ikegaki, N., Liu, X. G., Azar, C. G.,
et al. (1997). Expression of TrkA, TrkB and TrkC in human neuroblastomas.
Journal of Neuro-Oncology, 31(1-2), 49-55.
Bunt, S. K., Yang, L., Sinha, P., Clements, V. K., Leips, J., & Ostrand-Rosenberg, S.
(2007). Reduced inflammation in the tumor microenvironment delays the
accumulation of myeloid-derived suppressor cells and limits tumor progression.
Cancer Research, 67(20), 10019-10026.
Buytaert, E., Callewaert, G., Hendrickx, N., Scorrano, L., Hartmann, D., Missiaen, L.,
et al. (2006). Role of endoplasmic reticulum depletion and multidomain
proapoptotic BAX and BAK proteins in shaping cell death after hypericin-
mediated photodynamic therapy. FASEB Journal, 20(6), 756-758.
Byrne, A. T., O'Connor, A. E., Hall, M., Murtagh, J., O'Neill, K., Curran, K. M., et al.
(2009). Vascular-targeted photodynamic therapy with BF2-chelated Tetraaryl-
Azadipyrromethene agents: a multi-modality molecular imaging approach to
therapeutic assessment. British Journal of Cancer, 101(9), 1565-1573.
Byrne, J. D., Betancourt, T., & Brannon-Peppas, L. (2008). Active targeting schemes
for nanoparticle systems in cancer therapeutics. Advanced Drug Delivery
Reviews, 60(15), 1615-1626.
Cabral, H., Matsumoto, Y., Mizuno, K., Chen, Q., Murakami, M., Kimura, M., et al.
(2011). Accumulation of sub-100 nm polymeric micelles in poorly permeable
tumours depends on size. Nature Nanotechnology, 6(12), 815-823.
Cao, J., Cui, S., Li, S., Du, C., Tian, J., Wan, S., et al. (2013). Targeted cancer therapy
with a 2-deoxyglucose-based adriamycin complex. Cancer Research, 73(4),
1362-1373.
Cao, Q., Li, Z. B., Chen, K., Wu, Z., He, L., Neamati, N., et al. (2008). Evaluation of
biodistribution and anti-tumor effect of a dimeric RGD peptide-paclitaxel

123
conjugate in mice with breast cancer. European Journal of Nuclear Medicine
and Molecular Imaging, 35(8), 1489-1498.
Cao, Y., & Yang, J. (2014). Development of a folate receptor (FR)-targeted
indenoisoquinoline using a pH-sensitive N-ethoxybenzylimidazole (NEBI)
bifunctional cross-linker. Bioconjugate Chemistry, 25(5), 873-878.
Carrasco-Triguero, M., Yi, J.-H., Dere, R., Qiu, Z. J., Lei, C., Li, Y., et al. (2013).
Immunogenicity assays for antibody-drug conjugates: case study with ado-
trastuzumab emtansine. Bioanalysis, 5(9), 1007-1023.
Casares, N., Pequignot, M. O., Tesniere, A., Ghiringhelli, F., Roux, S., Chaput, N., et al.
(2005). Caspase-dependent immunogenicity of doxorubicin-induced tumor cell
death. The Journal of Experimental Medicine, 202(12), 1691-1701.
Castano, A. P., Mroz, P., & Hamblin, M. R. (2006). Photodynamic therapy and anti-
tumour immunity. Nature Review Cancer, 6(7), 535-545.
Cecic, I., Parkins, C. S., & Korbelik, M. (2001). Induction of systemic neutrophil
response in mice by photodynamic therapy of solid tumors. Photochemistry and
Photobiology, 74(5), 712-720.
Cecic, I., Stott, B., & Korbelik, M. (2006). Acute phase response-associated systemic
neutrophil mobilization in mice bearing tumors treated by photodynamic therapy.
International Immunopharmacology, 6(8), 1259-1266.
Cesana, G. C., DeRaffele, G., Cohen, S., Moroziewicz, D., Mitcham, J., Stoutenburg, J.,
et al. (2006). Characterization of CD4+CD25+ regulatory T cells in patients
treated with high-dose interleukin-2 for metastatic melanoma or renal cell
carcinoma. Journal of Clinical Oncology, 24(7), 1169-1177.
Chan, E., Mulkerin, D., Rothenberg, M., Holen, K. D., Lockhart, A. C., Thomas, J., et al.
(2008). A phase I trial of CEP-701 + gemcitabine in patients with advanced
adenocarcinoma of the pancreas. Investigational New Drugs, 26(3), 241-247.
Chen, B., Roskams, T., & de Witte, P. A. (2002). Antivascular tumor eradication by
hypericin-mediated photodynamic therapy. Photochemistry and Photobiology,
76(5), 509-513.
Chen, D., Brahimi, F., Angell, Y., Li, Y. C., Moscowicz, J., Saragovi, H. U., et al.
(2009). Bivalent peptidomimetic ligands of TrkC are biased agonists and

124
selectively induce neuritogenesis or potentiate neurotrophin-3 trophic signals.
ACS Chemical Biology, 4(9), 769-781.
Chen, K., Ma, W., Li, G., Wang, J., Yang, W., Yap, L. P., et al. (2013). Synthesis and
evaluation of 64Cu-labeled monomeric and dimeric NGR peptides for
MicroPET imaging of CD13 receptor expression. Molecular Pharmaceutics,
10(1), 417-427.
Chen, W., Liang, X., Peterson, A. J., Munn, D. H., & Blazar, B. R. (2008). The
Indoleamine 2,3-Dioxygenase Pathway Is Essential for Human Plasmacytoid
Dendritic Cell-Induced Adaptive T Regulatory Cell Generation. Journal of
Immunology, 181(8), 5396-5404.
Chen, X., Plasencia, C., Hou, Y., & Neamati, N. (2005). Synthesis and biological
evaluation of dimeric RGD peptide-paclitaxel conjugate as a model for integrin-
targeted drug delivery. Journal of Medicinal Chemistry, 48(4), 1098-1106.
Cheng, Z., Al Zaki, A., Hui, J. Z., Muzykantov, V. R., & Tsourkas, A. (2012).
Multifunctional Nanoparticles: Cost Versus Benefit of Adding Targeting and
Imaging Capabilities. Science, 338(6109), 903-910.
Chikamatsu, K., Sakakura, K., Whiteside, T. L., & Furuya, N. (2007). Relationships
between regulatory T cells and CD8+ effector populations in patients with
squamous cell carcinoma of the head and neck. Head and Neck, 29(2), 120-127.
Chiorean, E. G., Dragovich, T., Hamm, J., Langmuir, V. K., Kroll, S., Jung, D. T., et al.
(2008). A Phase 1 dose-escalation trial of glufosfamide in combination with
gemcitabine in solid tumors including pancreatic adenocarcinoma. Cancer
Chemotherapy and Pharmacology, 61(6), 1019-1026.
Cho, S. Y., Gage, K. L., Mease, R. C., Senthamizhchelvan, S., Holt, D. P., Jeffrey-
Kwanisai, A., et al. (2012). Biodistribution, tumor detection, and radiation
dosimetry of 18F-DCFBC, a low-molecular-weight inhibitor of prostate-specific
membrane antigen, in patients with metastatic prostate cancer. Journal of
Nuclear Medicine, 53(12), 1883-1891.
Chou, T. T., Trojanowski, J. Q., & Lee, V. M. (2000). A novel apoptotic pathway
induced by nerve growth factor-mediated TrkA activation in medulloblastoma.
The Journal of Biological Chemistry, 275(1), 565-570.
Ciuleanu, T. E., Pavlovsky, A. V., Bodoky, G., Garin, A. M., Langmuir, V. K., Kroll, S.,
et al. (2009). A randomised Phase III trial of glufosfamide compared with best

125
supportive care in metastatic pancreatic adenocarcinoma previously treated with
gemcitabine. European Journal of Cancer, 45(9), 1589-1596.
Colombo, R., Mingozzi, M., Belvisi, L., Arosio, D., Piarulli, U., Carenini, N., et al.
(2012). Synthesis and biological evaluation (in vitro and in vivo) of cyclic
arginine-glycine-aspartate (RGD) peptidomimetic-paclitaxel conjugates
targeting integrin alphaVbeta3. Journal of Medicinal Chemistry, 55(23), 10460-
10474.
Coughlin, C. M., Salhany, K. E., Gee, M. S., LaTemple, D. C., Kotenko, S., Ma, X., et
al. (1998). Tumor cell responses to IFNgamma affect tumorigenicity and
response to IL-12 therapy and antiangiogenesis. Immunity, 9(1), 25-34.
Cui, Q., Tang, L. S., Hu, B., So, K. F., & Yip, H. K. (2002). Expression of trkA, trkB,
and trkC in injured and regenerating retinal ganglion cells of adult rats.
Investigative Ophthalmology and Visual Science, 43(6), 1954-1964.
Dai, T., Fuchs, B. B., Coleman, J. J., Prates, R. A., Astrakas, C., St. Denis, T. G., et al.
(2012). Concepts and Principles of Photodynamic Therapy as an Alternative
Antifungal Discovery Platform. Frontiers in Microbiology, 3, 120.
de Vree, W. J., Essers, M. C., de Bruijn, H. S., Star, W. M., Koster, J. F., & Sluiter, W.
(1996). Evidence for an important role of neutrophils in the efficacy of
photodynamic therapy in vivo. Cancer Research, 56(13), 2908-2911.
Dehdashti, F., Laforest, R., Gao, F., Aft, R. L., Dence, C. S., Zhou, D., et al. (2012).
Assessment of progesterone receptors in breast carcinoma by PET with 21-18F-
fluoro-16alpha,17alpha-[(R)-(1'-alpha-furylmethylidene)dioxy]-19-norpregn- 4-
ene-3,20-dione. Journal of Nuclear Medicine, 53(3), 363-370.
Dehdashti, F., McGuire, A. H., Van Brocklin, H. F., Siegel, B. A., Andriole, D. P.,
Griffeth, L. K., et al. (1991). Assessment of 21-[18F]fluoro-16 alpha-ethyl-19-
norprogesterone as a positron-emitting radiopharmaceutical for the detection of
progestin receptors in human breast carcinomas. Journal of Nuclear Medicine,
32(8), 1532-1537.
Dennis, M. S., Jin, H., Dugger, D., Yang, R., McFarland, L., Ogasawara, A., et al.
(2007). Imaging Tumors with an Albumin-Binding Fab, a Novel Tumor-
Targeting Agent. Cancer Research, 67(1), 254-261.
Derynck, R., Akhurst, R. J., & Balmain, A. (2001). TGF-[beta] signaling in tumor
suppression and cancer progression. Nature Genetics, 29(2), 117-129.

126
des Rieux, A., Pourcelle, V., Cani, P. D., Marchand-Brynaert, J., & Preat, V. (2013).
Targeted nanoparticles with novel non-peptidic ligands for oral delivery.
Advanced Drug Delivery Reviews, 65(6), 833-844.
Dhawan, D., Ramos-Vara, J. A., Naughton, J. F., Cheng, L., Low, P. S., Rothenbuhler,
R., et al. (2013). Targeting folate receptors to treat invasive urinary bladder
cancer. Cancer Research, 73(2), 875-884.
di Tomaso, E., Capen, D., Haskell, A., Hart, J., Logie, J. J., Jain, R. K., et al. (2005).
Mosaic Tumor Vessels: Cellular Basis and Ultrastructure of Focal Regions
Lacking Endothelial Cell Markers. Cancer Research, 65(13), 5740-5749.
Diaz-Montero, C. M., Salem, M. L., Nishimura, M. I., Garrett-Mayer, E., Cole, D. J., &
Montero, A. J. (2009). Increased circulating myeloid-derived suppressor cells
correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-
cyclophosphamide chemotherapy. Cancer Immunology, Immunotherapy : CII,
58(1), 49-59.
Dollner, R., Dietz, A., Kopun, M., Helbig, M., Wallner, F., & Granzow, C. (2004). Ex
vivo responsiveness of head and neck squamous cell carcinoma to glufosfamide,
a novel alkylating agent. Anticancer Research, 24(5a), 2947-2951.
Dolmans, D. E., Fukumura, D., & Jain, R. K. (2003). Photodynamic therapy for cancer.
Nature Reviews Cancer, 3(5), 380-387.
Donkor, M. K., Lahue, E., Hoke, T. A., Shafer, L. R., Coskun, U., Solheim, J. C., et al.
(2009). Mammary tumor heterogeneity in the expansion of myeloid-derived
suppressor cells. International Immunopharmacology, 9(7-8), 937-948.
Dougherty, T. J., Gomer, C. J., Henderson, B. W., Jori, G., Kessel, D., Korbelik, M., et
al. (1998). Photodynamic therapy. Journal of the National Cancer Institute,
90(12), 889-905.
Dougherty, T. J., Kaufman, J. E., Goldfarb, A., Weishaupt, K. R., Boyle, D., &
Mittleman, A. (1978). Photoradiation therapy for the treatment of malignant
tumors. Cancer Research, 38(8), 2628-2635.
Dranoff, G. (2004). Cytokines in cancer pathogenesis and cancer therapy. Nature
Reviews Cancer, 4(1), 11-22.

127
Dreher, M. R., Liu, W., Michelich, C. R., Dewhirst, M. W., Yuan, F., & Chilkoti, A.
(2006). Tumor Vascular Permeability, Accumulation, and Penetration of
Macromolecular Drug Carriers. Journal of the National Cancer Institute, 98(5),
335-344.
Du, H., Bay, B. H., Mahendran, R., & Olivo, M. (2006). Hypericin-mediated
photodynamic therapy elicits differential interleukin-6 response in
nasopharyngeal cancer. Cancer Letters, 235(2), 202-208.
Du, H. Y., Olivo, M., Tan, B. K., & Bay, B. H. (2003). Hypericin-mediated
photodynamic therapy induces lipid peroxidation and necrosis in
nasopharyngeal cancer. International Journal of Oncology, 23(5), 1401-1405.
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., & Schreiber, R. D. (2002). Cancer
immunoediting: from immunosurveillance to tumor escape. Nature Immunology,
3(11), 991-998.
Fallarino, F., Grohmann, U., Vacca, C., Orabona, C., Spreca, A., Fioretti, M. C., et al.
(2003). T cell apoptosis by kynurenines. Advances in Experimental Medicine
and Biology, 527, 183-190.
Finn, O. J. (2008). Cancer Immunology. The New England Journal of Medicine,
358(25), 2704-2715.
Fisher, R. E., Siegel, B. A., Edell, S. L., Oyesiku, N. M., Morgenstern, D. E., Messmann,
R. A., et al. (2008). Exploratory study of 99mTc-EC20 imaging for identifying
patients with folate receptor-positive solid tumors. Journal of Nuclear Medicine,
49(6), 899-906.
Folkman, J. (2002). Role of angiogenesis in tumor growth and metastasis. Seminars in
Oncology, 29(6 Suppl 16), 15-18.
Foote, C. S. (1991). Definition of type I and type II photosensitized oxidation.
Photochemistry and Photobiology, 54(5), 659.
Frangioni, J. V. (2003). In vivo near-infrared fluorescence imaging. Current Opinion in
Chemical Biology, 7, 626-634.
Frumento, G., Rotondo, R., Tonetti, M., Damonte, G., Benatti, U., & Ferrara, G. B.
(2002). Tryptophan-derived catabolites are responsible for inhibition of T and
natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. Journal
of Experimental Medicine, 196(4), 459-468.

128
Gabrilovich, D., Ishida, T., Oyama, T., Ran, S., Kravtsov, V., Nadaf, S., et al. (1998).
Vascular endothelial growth factor inhibits the development of dendritic cells
and dramatically affects the differentiation of multiple hematopoietic lineages in
vivo. Blood, 92(11), 4150-4166.
Gardner, T. A., Fletcher, J. W., Ko, S.-C., Low, P. S., & Ratliff, T. L. (2013). Abstract
LB-90: DUPA-99mTc localizes to prostate cancer in men with locally advanced
and metastatic disease. Cancer Research, 73(8 Supplement), LB-90.
Garg, G., Vangveravong, S., Zeng, C., Collins, L., Hornick, M., Hashim, Y., et al.
(2014). Conjugation to a SMAC mimetic potentiates sigma-2 ligand induced
tumor cell death in ovarian cancer. Molecular Cancer, 13, 50.
Ghebeh, H., Mohammed, S., Al-Omair, A., Qattan, A., Lehe, C., Al-Qudaihi, G., et al.
(2006). The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in
breast cancer patients with infiltrating ductal carcinoma: correlation with
important high-risk prognostic factors. Neoplasia, 8(3), 190-198.
Giaccone, G., Smit, E. F., de Jonge, M., Dansin, E., Briasoulis, E., Ardizzoni, A., et al.
(2004). Glufosfamide administered by 1-hour infusion as a second-line
treatment for advanced non-small cell lung cancer; a phase II trial of the
EORTC-New Drug Development Group. European Journal of Cancer, 40(5),
667-672.
Gollnick, S. O., Evans, S. S., Baumann, H., Owczarczak, B., Maier, P., Vaughan, L., et
al. (2003). Role of cytokines in photodynamic therapy-induced local and
systemic inflammation. British Journal of Cancer, 88(11), 1772-1779.
Gorman, A., Killoran, J., O’Shea, C., Kenna, T., Gallagher, W. M. & O’Shea, D. F.
(2004). In vitro demonstration of the heavy-atom effect for photodynamic
therapy. Journal of the American Chemical Society, 126(34): 10619-10631.
Hamanishi, J., Mandai, M., Iwasaki, M., Okazaki, T., Tanaka, Y., Yamaguchi, K., et al.
(2007). Programmed cell death 1 ligand 1 and tumor-infiltrating CD8(+) T
lymphocytes are prognostic factors of human ovarian cancer. Proceedings of the
National Academy of Sciences of the United State of America, 104(9), 3360-
3365.
Hanahan, D., & Folkman, J. (1996). Patterns and emerging mechanisms of the
angiogenic switch during tumorigenesis. Cell, 86(3), 353-364.

129
Hashim, Y. M., Spitzer, D., Vangveravong, S., Hornick, M. C., Garg, G., Hornick, J. R.,
et al. (2014). Targeted pancreatic cancer therapy with the small molecule drug
conjugate SW IV-134. Molecular Oncology, 8(5), 956-967.
Henderson, B. W., & Fingar, V. H. (1987). Relationship of tumor hypoxia and response
to photodynamic treatment in an experimental mouse tumor. Cancer Research,
47(12), 3110-3114.
Hilgenbrink, A. R., & Low, P. S. (2005). Folate receptor-mediated drug targeting: From
therapeutics to diagnostics. Journal of Pharmaceutical Sciences, 94(10), 2135-
2146.
Hino, R., Kabashima, K., Kato, Y., Yagi, H., Nakamura, M., Honjo, T., et al. (2010).
Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic
factor for malignant melanoma. Cancer, 116(7), 1757-1766.
Hirsch, J. (2006). An anniversary for cancer chemotherapy. JAMA, 296(12), 1518-1520.
Houle, J. M., & Strong, A. (2002). Clinical pharmacokinetics of verteporfin. Journal of
Clinical Pharmacology, 42(5), 547-557.
Huang, E. J., & Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal
transduction. Annual Review of Biochemistry, 72, 609-642.
Huang, X., & Lee, C. (2003). Regulation of stromal proliferation, growth arrest,
differentiation and apoptosis in benign prostatic hyperplasia by TGF-beta.
Frontiers in Bioscience, 8, s740-749.
Huang, Z. (2005). A Review of Progress in Clinical Photodynamic Therapy.
Technology in Cancer Research and Treatment, 4(3), 283-293.
Huang, Z., Xu, H., Meyers, A. D., Musani, A. I., Wang, L., Tagg, R., et al. (2008).
Photodynamic therapy for treatment of solid tumors – potential and technical
challenges. Technology in Cancer Research and Treatment, 7(4), 309-320.
Ikeda, H., Old, L. J., & Schreiber, R. D. (2002). The roles of IFN gamma in protection
against tumor development and cancer immunoediting. Cytokine and Growth
Factor Reviews, 13(2), 95-109.

130
Inaba, T., Ino, K., Kajiyama, H., Yamamoto, E., Shibata, K., Nawa, A., et al. (2009).
Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the
progression of ovarian carcinoma. Gynecologic Oncology, 115(2), 185-192.
Ivanov, S. V., Panaccione, A., Brown, B., Guo, Y., Moskaluk, C. A., Wick, M. J., et al.
(2013). TrkC signaling is activated in adenoid cystic carcinoma and requires
NT-3 to stimulate invasive behavior. Oncogene, 32(32), 3698-3710.
Iyer, R., Evans, A. E., Qi, X., Ho, R., Minturn, J. E., Zhao, H., et al. (2010). Lestaurtinib
Enhances the Anti-tumor Efficacy of Chemotherapy in Murine Xenograft
Models of Neuroblastoma. Clinical Cancer Research, 16(5), 1478-1485.
Jain, R. K., & Stylianopoulos, T. (2010). Delivering nanomedicine to solid tumors.
Nature Review Clinical Oncology, 7(11), 653-664.
Janssen, M. L., Oyen, W. J., Dijkgraaf, I., Massuger, L. F., Frielink, C., Edwards, D. S.,
et al. (2002). Tumor targeting with radiolabeled alpha(v)beta(3) integrin binding
peptides in a nude mouse model. Cancer Research, 62(21), 6146-6151.
Jeon, S. H., Chae, B. C., Kim, H. A., Seo, G. Y., Seo, D. W., Chun, G. T., et al. (2007).
Mechanisms underlying TGF-beta1-induced expression of VEGF and Flk-1 in
mouse macrophages and their implications for angiogenesis. Journal of
Leukocyte Biology, 81(2), 557-566.
Jin, W., Kim, G. M., Kim, M. S., Lim, M. H., Yun, C., Jeong, J., et al. (2010). TrkC
plays an essential role in breast tumor growth and metastasis. Carcinogenesis,
31(11), 1939-1947.
Jin, W., Yun, C., Kwak, M. K., Kim, T. A., & Kim, S. J. (2007). TrkC binds to the type
II TGF-[beta] receptor to suppress TGF-[beta] signaling. Oncogene, 26(55),
7684-7691.
Jonson, S. D., Bonasera, T. A., Dehdashti, F., Cristel, M. E., Katzenellenbogen, J. A., &
Welch, M. J. (1999). Comparative breast tumor imaging and comparative in
vitro metabolism of 16alpha-[18F]fluoroestradiol-17beta and 16beta-
[18F]fluoromoxestrol in isolated hepatocytes. Nuclear Medicine and Biology,
26(1), 123-130.
Juarranz, A., Jaen, P., Sanz-Rodriguez, F., Cuevas, J., & Gonzalez, S. (2008).
Photodynamic therapy of cancer. Basic principles and applications. Clinical and
Translational Oncology, 10(3), 148-154.

131
Juzeniene, A. (2009). Chlorin e6-based photosensitizers for photodynamic therapy and
photodiagnosis. Photodiagnosis and Photodynamic Therapy, 6(2), 94-96.
Kamkaew, A., & Burgess, K. (2013). Double-targeting using a TrkC ligand conjugated
to dipyrrometheneboron difluoride (BODIPY) based photodynamic therapy
(PDT) agent. Journal of Medicinal Chemistry, 56(19), 7608-7614.
Kamkaew, A., Lim Siang, H., Lee Hong, B., Kiew Lik, V., Chung Lip, Y., & Burgess,
K. (2012). BODIPY dyes in photodynamic therapy. Chemical Society Reviews,
42, 77-88.
Kamps, J. A., & Scherphof, G. L. (1998). Receptor versus non-receptor mediated
clearance of liposomes. Advanced Drug Delivery Reviews, 32(1-2), 81-97.
Kessel, D., & Reiners, J. J., Jr. (2007). Apoptosis and autophagy after mitochondrial or
endoplasmic reticulum photodamage. Photochemistry and Photobiology, 83(5),
1024-1028.
Khaled, Y. S., Ammori, B. J., & Elkord, E. (2013). Myeloid-derived suppressor cells in
cancer: recent progress and prospects. Immunology and Cell Biology, 91(8),
493-502.
Kidd, P. (2003) Th1/Th2 balance: the hypothesis, its limitations, and implications for
health and disease. Alternative Medicine Review, 8(3), 223-246.
Kim, J. W., & Lee, H. S. (2004). Tumor targeting by doxorubicin-RGD-4C peptide
conjugate in an orthotopic mouse hepatoma model. International Journal of
Molecular Medicine, 14(4), 529-535.
Koebel, C. M., Vermi, W., Swann, J. B., Zerafa, N., Rodig, S. J., Old, L. J., et al. (2007).
Adaptive immunity maintains occult cancer in an equilibrium state. Nature,
450(7171), 903-907.
Koh, J.-Y., Gwag, B. J., Lobner, D., & Choi, D. W. (1995). Potentiated necrosis of
cultured cortical neurons by neurotrophins. Science, 268(5210), 573-575.
Koolpe, M., Dail, M., & Pasquale, E. B. (2002). An ephrin mimetic peptide that
selectively targets the EphA2 receptor. The Journal of Biological Chemistry,
277(49), 46974-46979.

132
Korbelik, M. (1996). Induction of tumor immunity by photodynamic therapy. Journal of
clinical laser medicine and surgery, 14(5), 329-334.
Korbelik M. & Dougherty, G. J. (1999). Photodynamic therapy-mediated immune
response against subcutaneous mouse tumors. Cancer research, 59(8), 1941-
1946.
Korbelik, M. (2006). PDT-associated host response and its role in the therapy outcome.
Lasers in Surgery and Medicine, 38(5), 500-508.
Korbelik, M. (2011). Cancer vaccines generated by photodynamic therapy.
Photochemical and Photobiological Sciences, 10(5), 664-669.
Kortylewicz, Z. P., Mack, E., Enke, C. A., Estes, K. A., Mosley, R. L., & Baranowska-
Kortylewicz, J. (2015). Preclinical evaluation of investigational
radiopharmaceutical RISAD-P intended for use as a diagnostic and molecular
radiotherapy agent for prostate cancer. Prostate, 75(1), 8-22.
Koshkaryev, A., Sawant, R., Deshpande, M., & Torchilin, V. (2013).
Immunoconjugates and long circulating systems: origins, current state of the art
and future directions. Advanced Drug Delivery Reviews, 65(1), 24-35.
Koudelka, S., Turanek-Knotigova, P., Masek, J., Korvasova, Z., Skrabalova, M.,
Plockova, J., et al. (2010). Liposomes with high encapsulation capacity for
paclitaxel: Preparation, characterisation and in vivo anticancer effect. Journal of
Pharmaceutical Sciences, 99(5), 2309-2319.
Krall, N., Pretto, F., Decurtins, W., Bernardes, G. J., Supuran, C. T., & Neri, D. (2014).
A small-molecule drug conjugate for the treatment of carbonic anhydrase IX
expressing tumors. Angewandte Chemie. International ed. in English, 53(16),
4231-4235.
Krall, N., Pretto, F., & Neri, D. (2014). A bivalent small molecule-drug conjugate
directed against carbonic anhydrase IX can elicit complete tumour regression in
mice. Chemical Science, 5(9), 3640-3644.
Kue, C. S., Jung, M. Y., Cho, D., & Kim, T. S. (2012). C6-ceramide enhances
Interleukin-12-mediated T helper type 1 cell responses through a
cyclooxygenase-2-dependent pathway. Immunobiology, 217(6), 601-609.

133
Kue, C. S., Kamkaew, A., Burgess, K., Kiew, L. V., Chung, L. Y., & Lee, H. B. (2016).
Small molecules for active targeting in cancer. Medicinal research reviews,
36(3), 494-575.
Kue, C. S., Kamkaew, A., Lee, H. B., Chung, L. Y., Kiew, L. V., & Burgess, K. (2015).
Targeted PDT agent eradicates TrkC expressing tumors via photodynamic
therapy (PDT). Molecular Pharmaceutics, 12(1), 212-222.
Kumar, S., & de Vellis, J. (1996). Neurotrophin activates signal transduction in
oligodendroglial cells: expression of functional TrkC receptor isoforms. Journal
of Neuroscience Research, 44(5), 490-498.
Leamon, C. P., Reddy, J. A., Klein, P. J., Vlahov, I. R., Dorton, R., Bloomfield, A., et al.
(2011). Reducing undesirable hepatic clearance of a tumor-targeted vinca
alkaloid via novel saccharopeptidic modifications. The Journal of Pharmacology
and Experimental Therapeutics, 336(2), 336-343.
Leamon, C. P., Reddy, J. A., Vlahov, I. R., Kleindl, P. J., Vetzel, M., & Westrick, E.
(2006). Synthesis and biological evaluation of EC140: a novel folate-targeted
vinca alkaloid conjugate. Bioconjugate Chemistry, 17(5), 1226-1232.
Lee, J. H., Zhou, H. B., Dence, C. S., Carlson, K. E., Welch, M. J., & Katzenellenbogen,
J. A. (2010). Development of [F-18]fluorine-substituted Tanaproget as a
progesterone receptor imaging agent for positron emission tomography.
Bioconjugate Chemistry, 21(6), 1096-1104.
Levanti, M. B., Germana, A., Catania, S., Germana, G. P., Gauna-Anasco, L., Vega, J.
A., et al. (2001). Neurotrophin receptor-like proteins in the bovine (Bos taurus)
lymphoid organs, with special reference to thymus and spleen. Anatomia,
Histologia, Embryologia, 30(4), 193-198.
Li, F., Liu, J., Jas, G. S., Zhang, J., Qin, G., Xing, J., et al. (2010). Synthesis and
evaluation of a near-infrared fluorescent non-peptidic bivalent integrin
alpha(v)beta(3) antagonist for cancer imaging. Bioconjugate Chemistry, 21(2),
270-278.
Lim, S. H., Thivierge, C., Nowak-Sliwinska, P., Han, J., van den Bergh, H., Wagnières,
G., et al. (2010). In Vitro and In Vivo Photocytotoxicity of Boron
Dipyrromethene Derivatives for Photodynamic Therapy. Journal of Medicinal
Chemistry, 53(7), 2865-2874.
Liu, J., Brahimi, F., Saragovi, H. U., & Burgess, K. (2010). Bivalent Diketopiperazine-
based TrkC Antagonists. Journal of Medicinal Chemistry, 53, 5044-5048.

134
Liu, V. C., Wong, L. Y., Jang, T., Shah, A. H., Park, I., Yang, X., et al. (2007). Tumor
evasion of the immune system by converting CD4+CD25- T cells into
CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. Journal of
Immunology, 178(5), 2883-2892.
Longo, F. M., & Massa, S. M. (2013). Small-molecule modulation of neurotrophin
receptors: a strategy for the treatment of neurological disease. Nature Reviews.
Drug Discovery, 12(7), 507-525.
Lorusso, P. M., Edelman, M. J., Bever, S. L., Forman, K. M., Pilat, M., Quinn, M. F., et
al. (2012). Phase I study of folate conjugate EC145 (Vintafolide) in patients with
refractory solid tumors. Journal of Clinical Oncology, 30(32), 4011-4016.
Low, P. S., & Kularatne, S. A. (2009). Folate-targeted therapeutic and imaging agents
for cancer. Current Opinion in Chemical Biology, 13(3), 256-262.
Lu, Y., & Low Philip, S. (2012). Folate-mediated delivery of macromolecular
anticancer therapeutic agents. Advanced Drug Delivery Reviews, 64, 342-352.
Ma, W., Wang, Z., Yang, W., Ma, X., Kang, F., & Wang, J. (2014). Biodistribution and
SPECT Imaging Study of 99mTc Labeling NGR Peptide in Nude Mice Bearing
Human HepG2 Hepatoma. BioMed Research International, 2014, 618096.
Maine, C. J., Aziz, N. H., Chatterjee, J., Hayford, C., Brewig, N., Whilding, L., et al.
(2014). Programmed death ligand-1 over-expression correlates with malignancy
and contributes to immune regulation in ovarian cancer. Cancer Immunology,
Immunotherapy : CII, 63(3), 215-224.
Marchetti, L., Luin, S., Bonsignore, F., de Nadai, T., Beltram, F., & Cattaneo, A. (2015).
Ligand-Induced Dynamics of Neurotrophin Receptors Investigated by Single-
Molecule Imaging Approaches. International Journal of Molecular Sciences,
16(1), 1949-1979.
Marquis, J. C., Hillier, S. M., Dinaut, A. N., Rodrigues, D., Mitra, K., Essigmann, J. M.,
et al. (2005). Disruption of gene expression and induction of apoptosis in
prostate cancer cells by a DNA-damaging agent tethered to an androgen receptor
ligand. Chemical Biology, 12(7), 779-787.
Marshall, J. S., Gomi, K., Blennerhassett, M. G., & Bienenstock, J. (1999). Nerve
growth factor modifies the expression of inflammatory cytokines by mast cells

135
via a prostanoid-dependent mechanism. Journal of Immunology, 162(7), 4271-
4276.
Martin-Zanca, D., Hughes, S. H., & Barbacid, M. (1986). A human oncogene formed by
the fusion of truncated tropomyosin and protein tyrosine kinase sequences.
Nature, 319(6056), 743-748.
Massague, J. (2008). TGFbeta in Cancer. Cell, 134(2), 215-230.
Mate, G., Kertesz, I., Enyedi, K. N., Mezo, G., Angyal, J., Vasas, N., et al. (2015). In
vivo imaging of Aminopeptidase N (CD13) receptors in experimental renal
tumors using the novel radiotracer (68)Ga-NOTA-c(NGR). European Journal of
Pharmaceutical Sciences, 69, 61-71.
Matejuk, A., Adlard, K., Zamora, A., Silverman, M., Vandenbark, A. A., & Offner, H.
(2001). 17 beta-estradiol inhibits cytokine, chemokine, and chemokine receptor
mRNA expression in the central nervous system of female mice with
experimental autoimmune encephalomyelitis. Journal of Neuroscience Research,
65(6), 529-542.
Matsuda, H., Coughlin, M. D., Bienenstock, J., & Denburg, J. A. (1988). Nerve growth
factor promotes human hemopoietic colony growth and differentiation.
Proceedings of the National Academy of Sciences of the United States of
America, 85(17), 6508-6512.
McGregor, L. M., McCune, B. K., Graff, J. R., McDowell, P. R., Romans, K. E.,
Yancopoulos, G. D., et al. (1999). Roles of trk family neurotrophin receptors in
medullary thyroid carcinoma development and progression. Proceedings of the
National Academy of Sciences of the United States of America, 96(8), 4540-4545.
Menetrier-Caux, C., Montmain, G., Dieu, M. C., Bain, C., Favrot, M. C., Caux, C., et al.
(1998). Inhibition of the differentiation of dendritic cells from CD34(+)
progenitors by tumor cells: role of interleukin-6 and macrophage colony-
stimulating factor. Blood, 92(12), 4778-4791.
Meng, Q., & Li, Z. (2013). Molecular imaging probes for diagnosis and therapy
evaluation of breast cancer. International Journal of Biomedical Imaging, 2013,
230487.
Meyer, C., Sevko, A., Ramacher, M., Bazhin, A. V., Falk, C. S., Osen, W., et al. (2011).
Chronic inflammation promotes myeloid-derived suppressor cell activation
blocking antitumor immunity in transgenic mouse melanoma model.

136
Proceedings of the National Academy of Sciences of the United States of
America, 108(41), 17111-17116.
Miknyoczki, S. J., Wan, W., Chang, H., Dobrzanski, P., Ruggeri, B. A., Dionne, C. A.,
et al. (2002). The neurotrophin-trk receptor axes are critical for the growth and
progression of human prostatic carcinoma and pancreatic ductal adenocarcinoma
xenografts in nude mice. Clinical Cancer Research, 8(6), 1924-1931.
Minko, T., Dharap, S. S., Pakunlu, R. I., & Wang, Y. (2004). Molecular targeting of
drug delivery systems to cancer. Current Drug Targets, 5(4), 389-406.
Minturn, J. E., Evans, A. E., Villablanca, J. G., Yanik, G. A., Park, J. R., Shusterman, S.,
et al. (2011). Phase I trial of lestaurtinib for children with refractory
neuroblastoma: a new approaches to neuroblastoma therapy consortium study.
Cancer Chemotherapy and Pharmacology, 68(4), 1057-1065.
Miot-Noirault, E., Reux, B., Debiton, E., Madelmont, J. C., Chezal, J. M., Coudert, P.,
et al. (2011). Preclinical investigation of tolerance and antitumour activity of
new fluorodeoxyglucose-coupled chlorambucil alkylating agents.
Investigational New Drugs, 29(3), 424-433.
Moffett, J. R., & Namboodiri, M. A. A. (2003). Tryptophan and the immune response.
Immunology and Cell Biology, 81(4), 247-265.
Morgan, J., & Oseroff, A. R. (2001). Mitochondria-based photodynamic anti-cancer
therapy. Advanced Drug Delivery Reviews, 49(1-2), 71-86.
Morris, R. T., Joyrich, R. N., Naumann, R. W., Shah, N. P., Maurer, A. H., Strauss, H.
W., et al. (2014). Phase II study of treatment of advanced ovarian cancer with
folate-receptor-targeted therapeutic (vintafolide) and companion SPECT-based
imaging agent (99mTc-etarfolatide). Annals of Oncology, 25(4), 852-858.
Mroz, P., Yaroslavsky, A., Kharkwal, G. B., & Hamblin, M. R. (2011). Cell death
pathways in photodynamic therapy of cancer. Cancers, 3(2), 2516-2539.
Mundy-Bosse, B. L., Young, G. S., Bauer, T., Binkley, E., Bloomston, M., Bill, M. A.,
et al. (2011). Distinct myeloid suppressor cell subsets correlate with plasma IL-6
and IL-10 and reduced interferon-alpha signaling in CD4(+) T cells from
patients with GI malignancy. Cancer Immunology, Immunotherapy : CII, 60(9),
1269-1279.

137
Muranski, P., Boni, A., Antony, P. A., Cassard, L., Irvine, K. R., Kaiser, A., et al.
(2008). Tumor-specific Th17-polarized cells eradicate large established
melanoma. Blood, 112(2), 362-373.
Murugaiyan, G., & Saha, B. (2009). Protumor vs antitumor functions of IL-17. Journal
of Immunology, 183(7), 4169-4175.
Nagaraj, S., Gupta, K., Pisarev, V., Kinarsky, L., Sherman, S., Kang, L., et al. (2007).
Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in
cancer. Nature Medicine, 13(7), 828-835.
Nakagawara, A. (2001). Trk receptor tyrosine kinases: a bridge between cancer and
neural development. Cancer Letters, 169(2), 107-114.
Nakashima-Matsushita, N., Homma, T., Yu, S., Matsuda, T., Sunahara, N., Nakamura,
T., et al. (1999). Selective expression of folate receptor beta and its possible role
in methotrexate transport in synovial macrophages from patients with
rheumatoid arthritis. Arthritis and Rheumatism, 42(8), 1609-1616.
Ng, Y. P., Cheung, Z. H., & Ip, N. Y. (2006). STAT3 as a downstream mediator of Trk
signaling and functions. The Journal of Biological Chemistry, 281(23), 15636-
15644.
Nomi, T., Sho, M., Akahori, T., Hamada, K., Kubo, A., Kanehiro, H., et al. (2007).
Clinical significance and therapeutic potential of the programmed death-1
ligand/programmed death-1 pathway in human pancreatic cancer. Clinical
Cancer Research, 13(7), 2151-2157.
Nonaka, H., Saga, Y., Fujiwara, H., Akimoto, H., Yamada, A., Kagawa, S., et al. (2011).
Indoleamine 2,3-dioxygenase promotes peritoneal dissemination of ovarian
cancer through inhibition of natural killercell function and angiogenesis
promotion. International Journal of Oncology, 38(1), 113-120.
Nowis, D., Stokłosa, T., Legat, M., Issat, T., Jakóbisiak, M., & Gołąb, J. (2005). The
influence of photodynamic therapy on the immune response. Photodiagnosis
and Photodynamic Therapy, 2(4), 283-298.
O'Connor, R. (2007). The pharmacology of cancer resistance. Anticancer Research,
27(3A), 1267-1272.
Ohigashi, Y., Sho, M., Yamada, Y., Tsurui, Y., Hamada, K., Ikeda, N., et al. (2005).
Clinical significance of programmed death-1 ligand-1 and programmed death-1

138
ligand-2 expression in human esophageal cancer. Clinical Cancer Research,
11(8), 2947-2953.
Okita, R., Saeki, T., Takashima, S., Yamaguchi, Y., & Toge, T. (2005). CD4+CD25+
regulatory T cells in the peripheral blood of patients with breast cancer and non-
small cell lung cancer. Oncology Reports, 14(5), 1269-1273.
Onodera, R., Motoyama, K., Okamatsu, A., Higashi, T., & Arima, H. (2013). Potential
use of folate-appended methyl-beta-cyclodextrin as an anticancer agent.
Scientific Reports, 3, 1104.
Palmer, E., Scott, J., & Symanowski, J. (2013). Reliability and reproducibility of
etarfolatide as a folate receptor (FR)-targeted diagnostic imaging agent. Journal
of Nuclear Medicine, 54(2_MeetingAbstracts), 400.
Pardali, K., & Moustakas, A. (2007). Actions of TGF-beta as tumor suppressor and pro-
metastatic factor in human cancer. Biochimica et Biophysica Acta, 1775(1), 21-
62.
Pathuri, G., Hedrick, A. F., Disch, B. C., Doan, J. T., Ihnat, M. A., Awasthi, V., et al.
(2012). Synthesis and evaluation of novel Tc-99m labeled probestin conjugates
for imaging APN/CD13 expression in vivo. Bioconjugate Chemistry, 23(1), 115-
124.
Paulos, C. M., Reddy, J. A., Leamon, C. P., Turk, M. J., & Low, P. S. (2004a). Ligand
binding and kinetics of folate receptor recycling in vivo: impact on receptor-
mediated drug delivery. Molecular Pharmacology, 66(6), 1406-1414.
Paulos, C. M., Turk, M. J., Breur, G. J., & Low, P. S. (2004b). Folate receptor-mediated
targeting of therapeutic and imaging agents to activated macrophages in
rheumatoid arthritis. Advanced Drug Delivery Reviews, 56(8), 1205-1217.
Pedragosa-Badia, X., Stichel, J., & Beck-Sickinger, A. G. (2013). Neuropeptide Y
receptors: how to get subtype selectivity. Frontiers in Endocrinology, 4, 5.
Peer, D., Karp, J. M., Hong, S., Farokhzad, O. C., Margalit, R., & Langer, R. (2007).
Nanocarriers as an emerging platform for cancer therapy. Nature
Nanotechnology, 2(12), 751-760.
Peethambaram, P. P., Hartmann, L. C., Jonker, D. J., de Jonge, M., Plummer, E. R.,
Martin, L., et al. (2015). A phase I pharmacokinetic and safety analysis of
epothilone folate (BMS-753493), a folate receptor targeted chemotherapeutic

139
agent in humans with advanced solid tumors. Investigational New Drugs, 33(2),
321-331.
Penn, I., & Starzl, T. E. (1973). Immunosuppression and Cancer. Transplantation
Proceedings, 5(1), 943-947.
Pillay, J., Tak, T., Kamp, V. M., & Koenderman, L. (2013). Immune suppression by
neutrophils and granulocytic myeloid-derived suppressor cells: similarities and
differences. Cellular and Molecular Life Sciences, 70(20), 3813-3827.
Pizova, K., Tomankova, K., Daskova, A., Binder, S., Bajgar, R., & Kolarova, H. (2012).
Photodynamic therapy for enhancing antitumour immunity. Biomedical Papers
of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia,
156(2), 93-102.
Porembka, M. R., Mitchem, J. B., Belt, B. A., Hsieh, C. S., Lee, H. M., Herndon, J., et
al. (2012). Pancreatic adenocarcinoma induces bone marrow mobilization of
myeloid-derived suppressor cells which promote primary tumor growth. Cancer
Immunology, Immunotherapy : CII, 61(9), 1373-1385.
Rao, J., Dragulescu-Andrasi, A., & Yao, H. (2007). Fluorescence imaging in vivo:
recent advances. Current Opinion in Biotechnology, 18(1), 17-25.
Reddy, J. A., Bloomfield, A., Dorton, R., Nelson, M., Vetzel, M., Hahn, S., et al. (2013).
Abstract 2145: PSMA-specific anti-tumor activity of the targeted-tubulysin
conjugate, EC1169. Cancer Research, 73(8 Supplement), 2145.
Reddy, J. A., Bloomfield, A., Nelson, M., Dorton, R., Vetzel, M., & Leamon, C. P.
(2014). Abstract 832: Pre-clinical development of EC1456: A potent Folate
targeted Tubulysin SMDC. Cancer Research, 74(19 Supplement), 832.
Reginato, E., Wolf, P., & Hamblin, M. R. (2014). Immune response after photodynamic
therapy increases anti-cancer and anti-bacterial effects. World Journal of
Immunology, 4(1), 1-11.
Rezaee, F., Rellick, S. L., Piedimonte, G., Akers, S. M., O'Leary, H. A., Martin, K., et al.
(2010). Neurotrophins regulate bone marrow stromal cell IL-6 expression
through the MAPK pathway. PloS One, 5(3), e9690.
Roettger, B. F., Rentsch, R. U., Pinon, D., Holicky, E., Hadac, E., Larkin, J. M., et al.
(1995). Dual pathways of internalization of the cholecystokinin receptor. The
Journal of Cell Biology, 128(6), 1029-1041.

140
Rose, S., Misharin, A., & Perlman, H. (2012). A novel Ly6C/Ly6G-based strategy to
analyze the mouse splenic myeloid compartment. Cytometry A, 81(4), 343-350.
Rudnick, S. I., Lou, J., Shaller, C. C., Tang, Y., Klein-Szanto, A. J. P., Weiner, L. M., et
al. (2011). Influence of Affinity and Antigen Internalization on the Uptake and
Penetration of Anti-HER2 Antibodies in Solid Tumors. Cancer Research, 71(6),
2250-2259.
Rullo, A.F., Fitzgerald, K.J., Muthusamy, V., Liu, M., Yuan, C., Huang, M., et al.
(2016). Re-engineering the immune response to metastatic cancer: antibody-
recruiting small molecules targeting the urokinase receptor. Angewandte Chemie
(International ed. In English), 55(11), 3642-3646.
Russell, J. H., & Ley, T. J. (2002). Lymphocyte-mediated cytotoxicity. Annual Review
of Immunology, 20, 323-370.
Ryppa, C., Mann-Steinberg, H., Fichtner, I., Weber, H., Satchi-Fainaro, R., Biniossek,
M. L., et al. (2008). In vitro and in vivo evaluation of doxorubicin conjugates
with the divalent peptide E-[c(RGDfK)2] that targets integrin alphavbeta3.
Bioconjugate Chemistry, 19(7), 1414-1422.
Saga, T., Neumann, R. D., Heya, T., Sato, J., Kinuya, S., Le, N., et al. (1995). Targeting
cancer micrometastases with monoclonal antibodies: a binding-site barrier.
Proceedings of the National Academy of Sciences of the United States of
America, 92(19), 8999-9003.
Sakamoto, Y., Kitajima, Y., Edakuni, G., Sasatomi, E., Mori, M., Kitahara, K., et al.
(2001). Expression of Trk tyrosine kinase receptor is a biologic marker for cell
proliferation and perineural invasion of human pancreatic ductal
adenocarcinoma. Oncology Reports, 8(3), 477-484.
Salim, M., Minamikawa, H., Sugimura, A., & Hashim, R. (2014). Amphiphilic designer
nano-carriers for controlled release: from drug delivery to diagnostics. Medicinal
Chemical Communications, 5(11), 1602-1618.
Salva, K. A. (2002). Photodynamic therapy: unapproved uses, dosages, or indications.
Clinics in Dermatology, 20(5), 571-581.
Sapra, P., & Allen, T. M. (2003). Ligand-targeted liposomal anticancer drugs. Progress
in Lipid Research, 42(5), 439-462.

141
Sasada, T., Kimura, M., Yoshida, Y., Kanai, M., & Takabayashi, A. (2003).
CD4+CD25+ regulatory T cells in patients with gastrointestinal malignancies:
possible involvement of regulatory T cells in disease progression. Cancer, 98(5),
1089-1099.
Scherz-Shouval, R., & Elazar, Z. (2007). ROS, mitochondria and the regulation of
autophagy. Trends in Cell Biology, 17(9), 422-427.
Schneider, R., & Schweiger, M. (1991). A novel modular mosaic of cell adhesion
motifs in the extracellular domains of the neurogenic trk and trkB tyrosine
kinase receptors. Oncogene, 6(10), 1807-1811.
Scott, A. M., Wolchok, J. D., & Old, L. J. (2012). Antibody therapy of cancer. Nature
Reviews. Cancer, 12(4), 278-287.
Segal, R. A. (2003). Selectivity in neurotrophin signaling: theme and variations. Annual
Review of Neurosciences, 26, 299-330.
Sekimoto, M., Tsuji, T., Matsuzaki, J., Chamoto, K., Koda, T., Nemoto, K., et al. (2003).
Functional expression of the TrkC gene, encoding a high affinity receptor for
NT-3, in antigen-specific T helper type 2 (Th2) cells. Immunology Letters, 88(3),
221-226.
Sevick-Muraca, E. M., Houston, J. P., & Gurfinkel, M. (2002). Fluorescence-enhanced,
near infrared diagnostic imaging with contrast agents. Current Opinion in
Chemical Biology, 6(5), 642-650.
Shao, Y., Liang, W., Kang, F., Yang, W., Ma, X., Li, G., et al. (2014). A direct
comparison of tumor angiogenesis with (68)Ga-labeled NGR and RGD peptides
in HT-1080 tumor xenografts using microPET imaging. Amino Acids, 46(10),
2355-2364.
Sharma, S., LoRusso, E. A. S., P., Vogelzang, N. J., Samlowski, W. E. Carter, J.,
Forman, K., Bever, K., Messmann, R. A. (2010). A phase I study of EC0225
administered weeks 1 and 2 of a 4-week cycle. Journal of Clinical Oncology,
28:15s, (supplement abstract 3082).
Shimizu, T., Okamoto, I., Tamura, K., Satoh, T., Miyazaki, M., Akashi, Y., et al. (2010).
Phase I clinical and pharmacokinetic study of the glucose-conjugated cytotoxic
agent D-19575 (glufosfamide) in patients with solid tumors. Cancer
Chemotherapy and Pharmacology, 65(2), 243-250.

142
Siebels, M., Rohrmann, K., Oberneder, R., Stahler, M., Haseke, N., Beck, J., et al.
(2011). A clinical phase I/II trial with the monoclonal antibody cG250
(RENCAREX(R)) and interferon-alpha-2a in metastatic renal cell carcinoma
patients. World Journal of Urology, 29(1), 121-126.
Siegel, B. A., Dehdashti, F., Mutch, D. G., Podoloff, D. A., Wendt, R., Sutton, G. P., et
al. (2003). Evaluation of 111In-DTPA-folate as a receptor-targeted diagnostic
agent for ovarian cancer: initial clinical results. Journal of Nuclear Medicine,
44(5), 700-707.
Siemann, D. W., Bibby, M. C., Dark, G. G., Dicker, A. P., Eskens, F. A., Horsman, M.
R., et al. (2005). Differentiation and definition of vascular-targeted therapies.
Clinical Cancer Research, 11(2 Pt 1), 416-420.
Sinha, P., Clements, V. K., Fulton, A. M., & Ostrand-Rosenberg, S. (2007).
Prostaglandin E2 promotes tumor progression by inducing myeloid-derived
suppressor cells. Cancer Research, 67(9), 4507-4513.
Solban, N., Rizvi, I., & Hasan, T. (2006). Targeted photodynamic therapy. Lasers in
Surgery and Medicine, 38(5), 522-531.
Spitzer, D., Simon, P. O., Kashiwagi, H., Xu, J., Zeng, C., Vangveravong, S., et al.
(2011). Use of Multifunctional Sigma-2 Receptor Ligand Conjugates to Trigger
Cancer-Selective Cell Death Signaling. Cancer Research, 72(1), 201-209.
Srinivasarao, M., Galliford, C. V., & Low, P. S. (2015). Principles in the design of
ligand-targeted cancer therapeutics and imaging agents. Nature Reviews. Drug
Discovery, 14(3), 203-219.
Stephens, P., Edkins, S., Davies, H., Greenman, C., Cox, C., Hunter, C., et al. (2005). A
screen of the complete protein kinase gene family identifies diverse patterns of
somatic mutations in human breast cancer. Nature Genetics, 37(6), 590-592.
Street, S. E., Cretney, E., & Smyth, M. J. (2001). Perforin and interferon-gamma
activities independently control tumor initiation, growth, and metastasis. Blood,
97(1), 192-197.
Street, S. E., Trapani, J. A., MacGregor, D., & Smyth, M. J. (2002). Suppression of
lymphoma and epithelial malignancies effected by interferon gamma. The
Journal of Experimental Medicine, 196(1), 129-134.

143
Takao, M., Okamoto, A., Nikaido, T., Urashima, M., Takakura, S., Saito, M., et al.
(2007). Increased synthesis of indoleamine-2,3-dioxygenase protein is positively
associated with impaired survival in patients with serous-type, but not with other
types of, ovarian cancer. Oncology Reports, 17(6), 1333-1339.
Takeda, K., Smyth, M. J., Cretney, E., Hayakawa, Y., Kayagaki, N., Yagita, H., et al.
(2002). Critical role for tumor necrosis factor-related apoptosis-inducing ligand
in immune surveillance against tumor development. The Journal of
Experimental Medicine, 195(2), 161-169.
Terabe, M., Matsui, S., Park, J. M., Mamura, M., Noben-Trauth, N., Donaldson, D. D.,
et al. (2003). Transforming growth factor-beta production and myeloid cells are
an effector mechanism through which CD1d-restricted T cells block cytotoxic T
lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor
recurrence. The Journal of Experimental Medicine, 198(11), 1741-1752.
Thompson, R. H., Gillett, M. D., Cheville, J. C., Lohse, C. M., Dong, H., Webster, W.
S., et al. (2004). Costimulatory B7-H1 in renal cell carcinoma patients: Indicator
of tumor aggressiveness and potential therapeutic target. Proceedings of the
National Academy of Sciences of the United States of America, 101(49), 17174-
17179.
Torchilin, V. P. (2010). Passive and active drug targeting: drug delivery to tumors as an
example. Handbook of Experimental Pharmacology(197), 3-53.
Trinchieri, G. (1995). Interleukin-12: a proinflammatory cytokine with
immunoregulatory functions that bridge innate resistance and antigen-specific
adaptive immunity. Annual Review of Immunology, 13, 251-276.
Tu, Z., Xu, J., Jones, L. A., Li, S., Zeng, D., Kung, M. P., et al. (2010). Radiosynthesis
and biological evaluation of a promising sigma(2)-receptor ligand radiolabeled
with fluorine-18 or iodine-125 as a PET/SPECT probe for imaging breast cancer.
Applied Radiation and Isotopes, 68(12), 2268-2273.
Turcotte, E., Paquette, M., Lavallee, E., Langlois, R., Dubreuil, S., Senta, H., et al.
(2012). Comparison of 4FMFES-PET with FES-PET in a phase II trial to detect
estrogen receptor-positive breast cancer: Preliminary results. Journal of Nuclear
Medicine, 53(1_MeetingAbstracts), 281.
Urfer, R., Tsoulfas, P., O'Connell, L., Shelton, D. L., Parada, L. F., & Presta, L. G.
(1995). An immunoglobulin-like domain determines the specificity of
neurotrophin receptors. The EMBO Journal, 14(12), 2795-2805.

144
Usuda, J., Okunaka, T., Furukawa, K., Tsuchida, T., Kuroiwa, Y., Ohe, Y., et al. (2001).
Increased cytotoxic effects of photodynamic therapy in IL-6 gene transfected
cells via enhanced apoptosis. International Journal of Cancer, 93(4), 475-480.
Vaishnavi, A., Le, A. T., & Doebele, R. C. (2015). TRKing down an old oncogene in a
new era of targeted therapy. Cancer Discovery, 5(1), 25-34.
van den Bent, M. J., Grisold, W., Frappaz, D., Stupp, R., Desir, J. P., Lesimple, T., et al.
(2003). European Organization for Research and Treatment of Cancer (EORTC)
open label phase II study on glufosfamide administered as a 60-minute infusion
every 3 weeks in recurrent glioblastoma multiforme. Annals of Oncology, 14(12),
1732-1734.
van den Broek, M. E., Kagi, D., Ossendorp, F., Toes, R., Vamvakas, S., Lutz, W. K., et
al. (1996). Decreased tumor surveillance in perforin-deficient mice. The Journal
of Experimental Medicine, 184(5), 1781-1790.
van Schouwenburg, P. A., Bartelds, G. M., Hart, M. H., Aarden, L., Wolbink, G. J., &
Wouters, D. (2010). A novel method for the detection of antibodies to
adalimumab in the presence of drug reveals "hidden" immunogenicity in
rheumatoid arthritis patients. Journal of Immunological Methods, 362(1-2), 82-
88.
Veenhuizen, R., Oppelaar, H., Ruevekamp, M., Schellens, J., Dalesio, O., & Stewart, F.
(1997). Does Tumour Uptake of Foscan Determine PDT Efficacy? International
Journal of Cancer, 73, 236-239.
Vega, J. A., García-Suárez, O., Hannestad, J., Pérez-Pérez, M., & Germanà, A. (2003).
Neurotrophins and the immune system. Journal of Anatomy, 203(1), 1-19.
Vesely, M. D., Kershaw, M. H., Schreiber, R. D., & Smyth, M. J. (2011). Natural innate
and adaptive immunity to cancer. Annual Review of Immunology, 29, 235-271.
Vlahov, I. R., Santhapuram, H. K., Kleindl, P. J., Howard, S. J., Stanford, K. M., &
Leamon, C. P. (2006). Design and regioselective synthesis of a new generation
of targeted chemotherapeutics. Part 1: EC145, a folic acid conjugate of
desacetylvinblastine monohydrazide. Bioorganic and Medicinal Chemistry
Letters, 16(19), 5093-5096.
Vlashi, E., Kelderhouse, L. E., Sturgis, J. E., & Low, P. S. (2013). Effect of Folate-
Targeted Nanoparticle Size on Their Rates of Penetration into Solid Tumors.
ACS Nano, 7(10), 8573-8582.

145
Vlashi, E., Sturgis, J. E., Thomas, M., & Low, P. S. (2009). Real Time, Noninvasive
Imaging and Quantitation of the Accumulation of Ligand-Targeted Drugs into
Receptor-Expressing Solid Tumors. Molecular Pharmaceutics, 6(6), 1868-1875.
Voon, S. H., Kiew, L. V., Lee, H. B., Lim, S. H., Noordin, M. I., Kamkaew, A., et al.
(2014). In vivo studies of nanostructure-based photosensitizers for
photodynamic cancer therapy. Small, 10(24), 4993-5013.
Voon, S. H. Tiew, S. X., Kue, C. S., Lee, H. B., Kiew, L. V., Misran, M., et al. (2016).
Chitosan-Coated Poly(lactic-co-glycolic acid)-Diiodinated Boron-
Dipyrromethene Nanoparticles Improve Tumor Selectivity and Stealth
Properties in Photodynamic Cancer Therapy. Journal of Biomedical
Nanotechnology 12, 1431-1452.
Wainwright, M., Phoenix, D. A., Rice, L., Burrow, S. M. & Waring, J. (1997).
Increased cytotoxicity and phototoxicity in the methylene blue series via
chromophore methylation. J Photochem Photobiol B. 40(3): 233-239.
Wang, C., Dehghani, B., Li, Y., Kaler, L. J., Proctor, T., Vandenbark, A. A., et al.
(2009). Membrane estrogen receptor regulates experimental autoimmune
encephalomyelitis through up-regulation of programmed death 1. Journal of
Immunology, 182(5), 3294-3303.
Wang, L., Chang, E. W., Wong, S. C., Ong, S. M., Chong, D. Q., & Ling, K. L. (2013).
Increased myeloid-derived suppressor cells in gastric cancer correlate with
cancer stage and plasma S100A8/A9 proinflammatory proteins. Journal of
Immunology, 190(2), 794-804.
Wang, Y., Hagel, C., Hamel, W., Muller, S., Kluwe, L., & Westphal, M. (1998). Trk A,
B, and C are commonly expressed in human astrocytes and astrocytic gliomas
but not by human oligodendrocytes and oligodendroglioma. Acta
Neuropathology, 96(4), 357-364.
Wayua, C., & Low, P. S. (2014). Evaluation of a cholecystokinin 2 receptor-targeted
near-infrared dye for fluorescence-guided surgery of cancer. Molecular
Pharmaceutics, 11(2), 468-476.
Wayua, C., Roy, J., Putt, K. S., & Low, P. S. (2015). Selective Tumor Targeting of
Desacetyl Vinblastine Hydrazide and Tubulysin B via Conjugation to a
Cholecystokinin 2 Receptor (CCK2R) Ligand. Molecular Pharmaceutics, 12(7),
2477-2483.

146
Wei, L. H., Baumann, H., Tracy, E., Wang, Y., Hutson, A., Rose-John, S., et al. (2007).
Interleukin-6 trans signalling enhances photodynamic therapy by modulating
cell cycling. British Journal of Cancer, 97(11), 1513-1522.
Weinhold, K. J., Goldstein, L. T., & Wheelock, E. F. (1977). Tumour-dormant states
established with L5178Y lymphoma cells in immunised syngeneic murine hosts.
Nature, 270(5632), 59-61.
Weir, G. M., Liwski, R. S., & Mansour, M. (2011). Immune Modulation by
Chemotherapy or Immunotherapy to Enhance Cancer Vaccines. Cancers, 3(3),
3114-3142.
Windisch, J. M., Marksteiner, R., & Schneider, R. (1995). Nerve growth factor binding
site on TrkA mapped to a single 24-amino acid leucine-rich motif. The Journal
of Biological Chemistry, 270(47), 28133-28138.
Woo, E. Y., Chu, C. S., Goletz, T. J., Schlienger, K., Yeh, H., Coukos, G., et al. (2001).
Regulatory CD4+CD25+ T Cells in Tumors from Patients with Early-Stage
Non-Small Cell Lung Cancer and Late-Stage Ovarian Cancer. Cancer Research,
61(12), 4766-4772.
Wrzesinski, S. H., Wan, Y. Y., & Flavell, R. A. (2007). Transforming growth factor-
beta and the immune response: implications for anticancer therapy. Clinical
Cancer Research, 13(18 Pt 1), 5262-5270.
Xia, W., & Low, P. S. (2010). Folate-Targeted Therapies for Cancer. Journal of
Medicinal Chemistry, 53(19), 6811-6824.
Xu, X., Tahan, S. R., Pasha, T. L., & Zhang, P. J. (2003). Expression of neurotrophin
receptor Trk-C in nevi and melanomas. Journal of Cutaneous Pathology, 30(5),
318-322.
Yamamoto, T., Yanagimoto, H., Satoi, S., Toyokawa, H., Hirooka, S., Yamaki, S., et al.
(2012). Circulating CD4+CD25+ regulatory T cells in patients with pancreatic
cancer. Pancreas, 41(3), 409-415.
Yamashiro, D. J., Liu, X. G., Lee, C. P., Nakagawara, A., Ikegaki, N., McGregor, L. M.,
et al. (1997). Expression and function of Trk-C in favourable human
neuroblastomas. European Journal of Cancer, 33(12), 2054-2057.
Yang, J., Chen, H., Vlahov, I. R., Cheng, J. X., & Low, P. S. (2006). Evaluation of
disulfide reduction during receptor-mediated endocytosis by using FRET

147
imaging. Proceedings of the National Academy of Sciences of the United States
of America, 103(37), 13872-13877.
Yang, L., Huang, J., Ren, X., Gorska, A. E., Chytil, A., Aakre, M., et al. (2008).
Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-
1+CD11b+ myeloid cells that promote metastasis. Cancer Cell, 13(1), 23-35.
Yang, W., Cheng, Y., Xu, T., & Wen, L.P. (2009). Targeting cancer cells with biotin-
dendrimer conjugates. European Journal of Medicinal Chemistry, 44(2), 862-
868.
Yogo, T., Urano, Y., Ishitsuka, Y., Maniwa, F., & Nagano, T. (2005). Highly efficient
and photostable photosensitizer based on BODIPY chromophore. Journal of the
American Chemical Society, 127, 12162-12163.
Youn, J. I., Nagaraj, S., Collazo, M., & Gabrilovich, D. I. (2008). Subsets of myeloid-
derived suppressor cells in tumor-bearing mice. Journal of Immunology, 181(8),
5791-5802.
Younos, I. H., Dafferner, A. J., Gulen, D., Britton, H. C., & Talmadge, J. E. (2012).
Tumor regulation of myeloid-derived suppressor cell proliferation and
trafficking. International Immunopharmacology, 13(3), 245-256.
Zhang, H., Nguyen-Jackson, H., Panopoulos, A. D., Li, H. S., Murray, P. J., &
Watowich, S. S. (2010). STAT3 controls myeloid progenitor growth during
emergency granulopoiesis. Blood, 116(14), 2462-2471.
Zhang, J., Lu, X., Wan, N., Hua, Z., Wang, Z., Huang, H., et al. (2014). 68Ga-DOTA-
NGR as a novel molecular probe for APN-positive tumor imaging using
MicroPET. Nuclear Medicine and Biology, 41(3), 268-275.
Zhang, X. G., Miao, J., Dai, Y.Q., Du, Y.Z., Yuan, H., & Hu, F.Q. (2008). Reversal
activity of nanostructured lipid carriers loading cytotoxic drug in multi-drug
resistant cancer cells. International Journal of Pharmaceutics, 361(1-2), 239-
244.
Zhou, L., Ivanov, II, Spolski, R., Min, R., Shenderov, K., Egawa, T., et al. (2007). IL-6
programs T(H)-17 cell differentiation by promoting sequential engagement of
the IL-21 and IL-23 pathways. Nature Immunology, 8(9), 967-974.

148
Zhou, L., Lopes, J. E., Chong, M. M., Ivanov, II, Min, R., Victora, G. D., et al. (2008).
TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing
RORgammat function. Nature, 453(7192), 236-240.
Zhu, J., Yamane, H., & Paul, W. E. (2010). Differentiation of Effector CD4 T Cell
Populations. Annual Review of Immunology, 28, 445-489.
Zitvogel, L., Apetoh, L., Ghiringhelli, F., & Kroemer, G. (2008). Immunological
aspects of cancer chemotherapy. Nature Reviews. Immunology, 8(1), 59-73.

149
LIST OF PUBLICATIONS AND PAPERS PRESENTED
A) Research Presentations
i. Poster presentation at The International Congress on Photodynamic
Applications (ICPA), The Caird Hall, Dundee, Scotland, 25th
-28th
May 2014.
TrkC Ligand Conjugated Boron-Dipyrromethene (BODIPY) Improves
Efficacy and Eradicates TrkC Expressing Tumour in Photodynamic
Therapy
Kue, C. S.1, Kamkaew, A.
2, Burgess, K.
2, Lee, H. B.
3, Chung, L. Y.
3, and Kiew,
L. V1.
1 Department of Pharmacology, Faculty of Medicine, University Malaya,
Lembah Pantai, Kuala Lumpur, 50603, Malaysia.
2 Department of Chemistry, Texas A&M University, College Station Texas
77842, United States.
3 Department of Pharmacy, Faculty of Medicine, University of Malaya, Lembah
Pantai, Kuala Lumpur, 50603, Malaysia.

150
4T
1 T
um
or
Vo
l.(%
In
itia
l V
ol.)
0
200
400
600
800
1000
1200
6 9 13 17 21
Days post-PDT
Control saline YIYI-BODIPY(2 mg/kg)
YIYI-BODIPY (10 mg/kg)
IYIY-BODIPY (2 mg/kg) IYIY-BODIPY (10 mg/kg)
I2-BODIPY (3.3 mg/kg
0
* **
* *
TRKC LIGAND CONJUGATED BORON DIPYRROMETHENE
(BODIPY) IMPROVES EFFICACY AND ERADICATES TRKC
EXPRESSING TUMOR IN PHOTODYNAMIC THERAPY
Chin Siang KUE1, Anyanee KAMKAEW2, Hong Boon LEE3, Lip Yong CHUNG3, Kevin BURGESS2, Lik Voon KIEW1
1. Department of Pharmacology, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia
2. Department of Chemistry, Texas A&M University, Box 30012, College Station, TX77842, USA
3. Department of Pharmacy, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia
Intro and aims: Tumor targeting of photosensitizers can be improved through the use of ligands that targets receptors that are overexpressed on the cancer cells. Therefore, in this study, we explored the
use of peptidomimetics ligand to enhance the efficacy of anticancer therapeutics. We linked a synthetic peptidomimetic ligand (IYIY) that binds to TrkC receptors that are overexpressed in metastatic breast
cancer to the photosensitizer I2BODIPY to construct a proof of concept peptidomimetic ligand-drug conjugate (IYIY-I2BODIPY).
Results : IYIY-I2BODIPY was found to cause selective in vitro phototoxicity to TrkC+4T1 breast cancer cells (IC50 = 0.33M) but not TrkC- 67NR breast cancer cells. Biodistribution study in TrkC+ 4T1 tumor
bearing Balb/C mice indicated that IYIY-I2BODIPY showed selective and prolonged (up to 24hr) accumulation in TrkC+4T1 tumor compared to scrambled YIYI-I2BODIPY control. Significant tumor
regression was also found in TrkC+4T1 tumor bearing Balb/C mice at 6 days post intravenous administration of 10mg/kg IYIY-I2BODIPY followed by 10 mins irradiation of tumor with green light (530nm, 100
J/cm2 at fluence rate of 0.16 W/cm2). Tumor remission was found in 71% (5 out of 7) of the treated animal at day 13, with no sign of tumor regrowth or metastasis to key organs observed during the 90 days
study period. Such observation was neither found in YIYI-I2BODIPY and I2BODIPY recipient mice nor 67NR tumor bearing mice treated with both IYIY-I2BODIPY and YIYI-I2BODIPY respectively.
Conclusion: The current findings demonstrate the significant enhancement of the antitumor efficacy of I2BODIPY photosensitizer model drug by TrkC receptor targeting IYIY, and indicate the potential in
adopting IYIY or similar peptidomimetics as tumor targeting ligands for anticancer therapeutics.
Abstract
1. IYIY-BODIPY binds to TrkC receptors expressed on 4T1 cells and is taken up into the lysosomes. It competes with NT3 (natural TrkC receptor ligand) and IYIY-TEG (without BODIPY) for the same
receptor.
2. The optimum drug-light interval for IYIY-BODIPY to show selective targeting of TrkC+ tumor cells in mice was 2 h, and prolonged drug-light interval (4 – 6 h) gave reduced selectivity.
3. IYIY-BODIPY was not toxic to experimental mice at therapeutic doses. The maximum tolerated dose (MTD) of IYIY-BODIPY was 20 mg/kg whereas YIYI-BODIPY (scrambled control) was up to 100
mg/kg.
4. Five out of seven (71%) IYIY-BODIPY (10 mg/kg) treated 4T1 tumor bearing mice post PDT showed full remission with no metastasis post 90 days of PDT. This indicates that IYIY-BODIPY
successfully targets and eradicates TrkC receptor expressing 4T1 tumor cells upon PDT. In dark, IYIY-BODIPY did not promote 4T1 tumor growth, whereas literature states binding of ligands to TrkC
promotes tumor survival and growth.
5. The study shows the potential of peptidomimetic TrkC ligand IYIY or similar peptidomimetics as tumor targeting molecules.
Acknowledgement: Ministry of Higher Education of Malaysia High Impact Research Grants (MoHE-HIR, UM.C/625/1/HIR/MOHE/MED/17 & UM.C/625/1/HIR/MOHE/MED/33).
IYIY-BODIPY dose dependently induces photocytotoxicity in TrkC+ (4T1)
but not in TrkC- (67NR) cells
Figure 2
Tumor stained with IYIY-BODIPY peaked at 1 hour post intravenous
administration, but low in YIYI-BODIPY
Figure 3
Figure 4
Results
C.
IYIY-BODIPY induced selective
photocytotoxicity in 4T1 cells.
Photocytotoxicity was reduced in
the presence of NT-3 and IYIY-TEG
(without BODIPY).
5 out of 7 IYIY-BODIPY (10mg/kg) treated mice showed full remission with no
metastasis development post 90 days of PDT.
Survivor mice
gave no
evidence of
metastasis
compared to
control (yellow
arrowhead)
C. Histopathology of IYIY-BODIPY (10 mg/kg) treated survivor mice
A. B.
Discussion & Conclusion
IY-IY-Bodipy LysoTracker overlay
4T1 D4T1
age 30, malignant,
phyllodes sarcoma
age 50,
normal pathology
Membrane
staining
age 48, malignant, invasive
ductal carcinoma
cytoplasm
staining
IYIY-BODIPY binds to TrkC receptor of tumor cells of mouse and human
Figure 1
IYIY-BODIPY internalized into lysosomes
same as natural ligand NT3
IYIY-BODIPY stained in human
malignant tumor, but not in
normal tissue.
A. B.
% o
f d
ea
d c
ell
-40
-20
0
20
40
60
80
100
120
0.1 0.3 1.0 3.0 10
compounds (M)
*
** ***
B. A.
% o
f d
ead
cell
-40
-20
0
20
40
60
80
100
120
0.1 0.3 1.0 3.0 10
compounds (M)
4T1 IY-IY-BODIPY LIGHT
4T1 YI-YI-BODIPY LIGHT
4T1 I2-BODIPY LIGHT
67NR IY-IY-BODIPY LIGHT
67NR YI-YI-BODIPY LIGHT 67NR I2-BODIPY LIGHT
0.25 h 1 h 3 h 6 h 24 h 48 h 72 h
0 h
Time post compound administration (i.v)
1-P
DT
2-P
DT
Liver Kidney
Spleen
Lymph
nodes
Tumor eye
skin
Lung IYIY
-BO
DIP
Y
(10
mg/
kg)
YIY
I-BO
DIP
Y
(10
mg/
kg)
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
Skin Kidney Spleen
Lymph node
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
Eye
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
Lung
0
5000
10000
15000
20000
25000
0 0.25 1 3 6 24 48 72
Liver
0
200
400
600
800
1000
1200
1400
1600
0 0.25 1 3 6 24 48 72
Tumor
* * * **
* **
Time post drug
administration (h)
Fluo
resc
ence
inte
nsity
(x10
3 ) (p/
sec/
cm2 /s
r)
IYIY-BODIPY
10 mg/kg
YIYI-BODIPY
10 mg/kg
67
NR
Tu
mo
r V
ol.
(% I
nit
ial
Vo
l.)
0
100
200
300
400
500
600 Control saline
IYIY-BODIPY (10 mg/kg)
YIYI-BODIPY (10 mg/kg)
6 9 13 17 21
Days post-PDT
0
IYIY
-BO
DIP
Y
(10m
g/k
g)
4T1
surv
ivo
r m
ou
se
Tu
mo
r fr
ee
hea
lth
y m
ou
se
4T1
tum
or
bea
rin
g
mo
use
Liver Lung Lymph nodes Spleen 100
90
80
70
60
50
40
30
20
10
0
-10
-20
-30
1 0.8 0.6 0.4 0.2 0.1 0.08 0.06 0.04 0.02
4T1
4T1 + NT3 (200ng/ml)
4T1 + IYIY-TEG(20M)
% o
f d
ea
d c
ell
s
Concentration of IYIY-PDT (M) Concentrations of IYIY-BODIPY (M)

151
ii. Oral presentation at Controlled Release & Drug Delivery
Symposium (CRDDS) 2015 in conjunction with The 1st MyCRS
Scientific Conference: Targeted Delivery – Translating Ideas into
New Technology, Kuala Lumpur, Malaysia, 15th
– 16th
August 2015.
TrkC Receptor Targeted Delivery of PDT Agent Inhibits Immune-
suppressive Leukocytes and Enhances Adaptive Immune Response Before
and After Photodynamic Therapy (PDT)
Kue, C. S.1, Kamkaew, A.
2, Voon, S. H.
1, Kiew, L. V.
1, Chung, L. Y.
3, Burgess,
K.2, Lee, H. B.
3
1 Department of Pharmacology, Faculty of Medicine, University Malaya,
Lembah Pantai, Kuala Lumpur, 50603, Malaysia.
2 Department of Chemistry, Texas A&M University, College Station Texas
77842, United States.
3 Department of Pharmacy, Faculty of Medicine, University of Malaya, Lembah
Pantai, Kuala Lumpur, 50603, Malaysia.

152
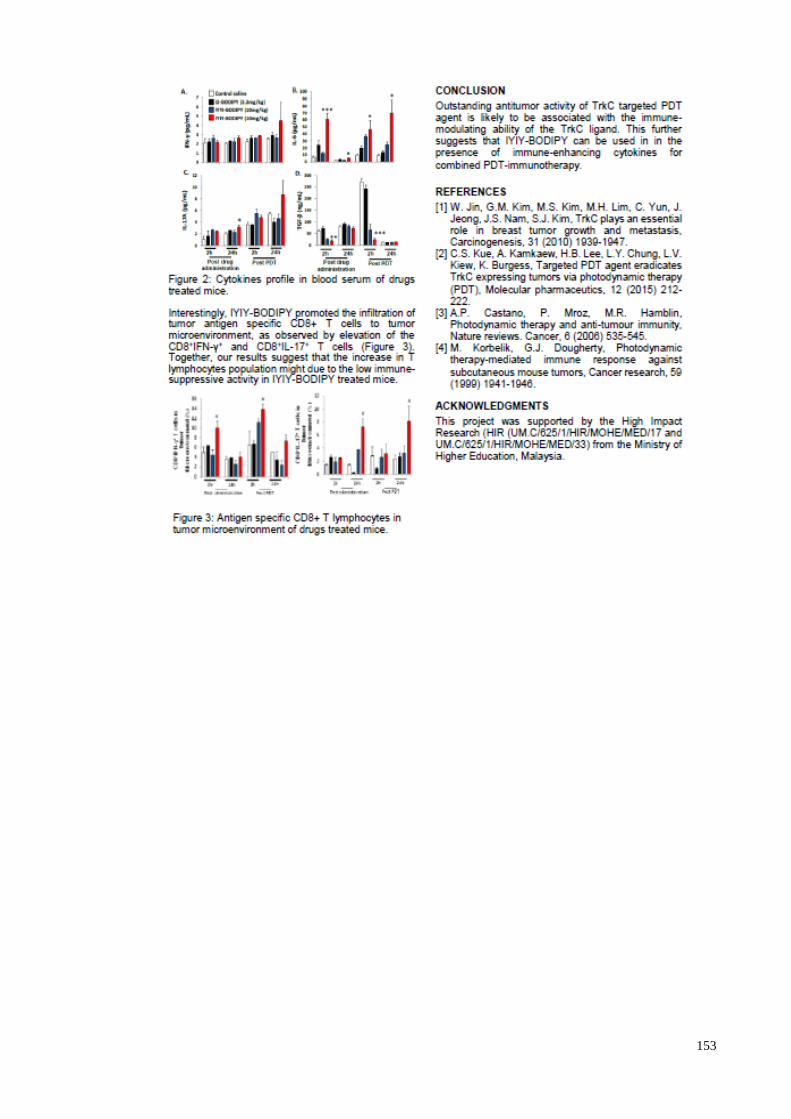
153

154
B) Publications
i. Kue, C.S., Kamkaew, A., Lee, H.B., Chung, L.Y, Kiew, L.V., and Burgess,
K. (2015). Targeted PDT Agent Eradicates TrkC Expressing Tumors via
Photodynamic Therapy (PDT). Molecular Pharmaceutics 12, 212-222. (IF
2015: 4.342)
ii. Kue, C.S., Kamkaew, A., Burgess, K., Kiew, L.V., Chung, L.Y., Lee, H.B.
(2016) Small Molecules for Active Targeting in Cancer. Medicinal
Research Reviews 36(3), 494-575. (IF 2015: 9.135)
iii. Kue, C.S., Kamkaew, A., Voon, S.H., Kiew, L.V., Chung, L.Y., Burgess, K.,
and Lee, H.B.. (2016). Tropomyosin Receptor Kinase C Targeted Delivery
of a Peptidomimetic Ligand-Photosensitizer Conjugate Induces Antitumor
Immune Responses Following Photodynamic Therapy. Scientific Reports 6,
37209. (IF 2015: 5.228)

155
APPENDICES
Appendix A: Synthesis of I2-BODIPY Derivatives
I2-BODIPY derivatives were synthesised by collaborator based on the protocols
published by Anyanee and Burgess, 2013.
10-(3-Chloropropyl)-5,5-difluoro-1,3,7,9-tetramethyl-5H-4λ4,5λ
4-dipyrrolo[1,2-
c:2',1'-f][1,3,2]diazaborinine (3). 2,4-Dimethylpyrrole (2.0 mL, 19 mmol) was added
to a solution of 4-chlorobutanoyl chloride (0.99 mL, 8.8 mmol) in CH2Cl2 (20 mL) over
10 min at 0 ºC. The reaction was stirred at 0 ºC for 30 min, then warmed up to 25 ºC
and stirred for an additional 30 min. Triethylamine (3.7 mL, 26 mmol) was then added
in small portions at 0 ºC and the mixture was stirred at 25 ºC for 10 min. BF3•OEt2 (5.5
mL, 44 mmol) was then added in portions and the mixture was stirred at 25 ºC for 14 h.
The reaction was quenched with careful addition of H2O (20 mL) and the system was
stirred vigorously for 15 min. The aqueous layer was extracted with CH2Cl2 (3 × 10
mL). The organic layers were combined and washed with H2O (2 × 10 mL) and brine
(10 mL). The organic layer was dried over MgSO4, filtered, and the solvent was
156
removed under reduced pressure. The brown powder was dissolved in small amount of
CH2Cl2 and filtered through a short plug of silica. The solvent was removed under
reduced pressure and the remaining powder was recrystallised from ethyl acetate to
yield 0.90 g (32 %) of 4 as a red needles. 1H NMR (300 MHz, CDCl3) δ 6.06 (s, 2H),
3.70 (t, J = 6.0 Hz, 2H), 3.13 (t, J = 8.4 Hz, 2H), 2.52 (s, 6H), 2.43 (s, 6H), 2.08-2.16 (m,
2H). 13
C (100 MHz, CDCl3) δ 154.4, 144.4, 140.3, 131.4, 121.8, 44.7, 34.0, 25.9, 16.5,
14.4. HRMS (ESI) calcd for C16H19BClF2N2 {M-H}- 323.1298, found 323.1287.
1H-NMR of compound 3
157
13C-NMR of compound 3
10-(3-Azidopropyl)-5,5-difluoro-1,3,7,9-tetramethyl-5H-4λ4,5λ
4-dipyrrolo[1,2-
c:2',1'-f][1,3,2]diazaborinine (4). Sodium azide (240 mg, 3.6 mmol) was added to a
solution of 4 (600 mg, 1.9 mmol) in DMF (36 mL). The mixture was stirred at 40 ºC for
36 h. Water (20 mL) was then added and the suspension was extracted with ethyl
acetate (3 × 20 mL). The combined organic layers were washed with H2O (2 × 20 mL)
and brine (20 mL). The organic layer was dried over MgSO4, filtered and the solvent
was removed under reduced pressure to afford 600 mg (99 %) of 5 as an orange powder.
1H NMR (500 MHz, CDCl3) δ 6.06 (s, 2H), 3.49 (t, J = 6.0 Hz, 2H), 3.03 (t, J = 8.4 Hz,
2H), 2.51 (s, 6H), 2.46 (s, 6H), 1.86-1.93 (m, 2H). 13
C (100 MHz, CDCl3) δ 154.3,
144.6, 140.3, 131.4, 121.9, 51.5, 31.4, 25.6, 16.5, 14.4. HRMS (ESI) calcd for
C16H19BF2N5 {M-H}- 330.1702, found 330.1712. FT-IR (NaCl): N3 band at 2098 cm
-1.
158
1H-NMR of compound 4
13C-NMR of compound 4
10-(3-Azidopropyl)-5,5-difluoro-2,8-diiodo-1,3,7,9-tetramethyl-5H-4λ4,5λ
4-
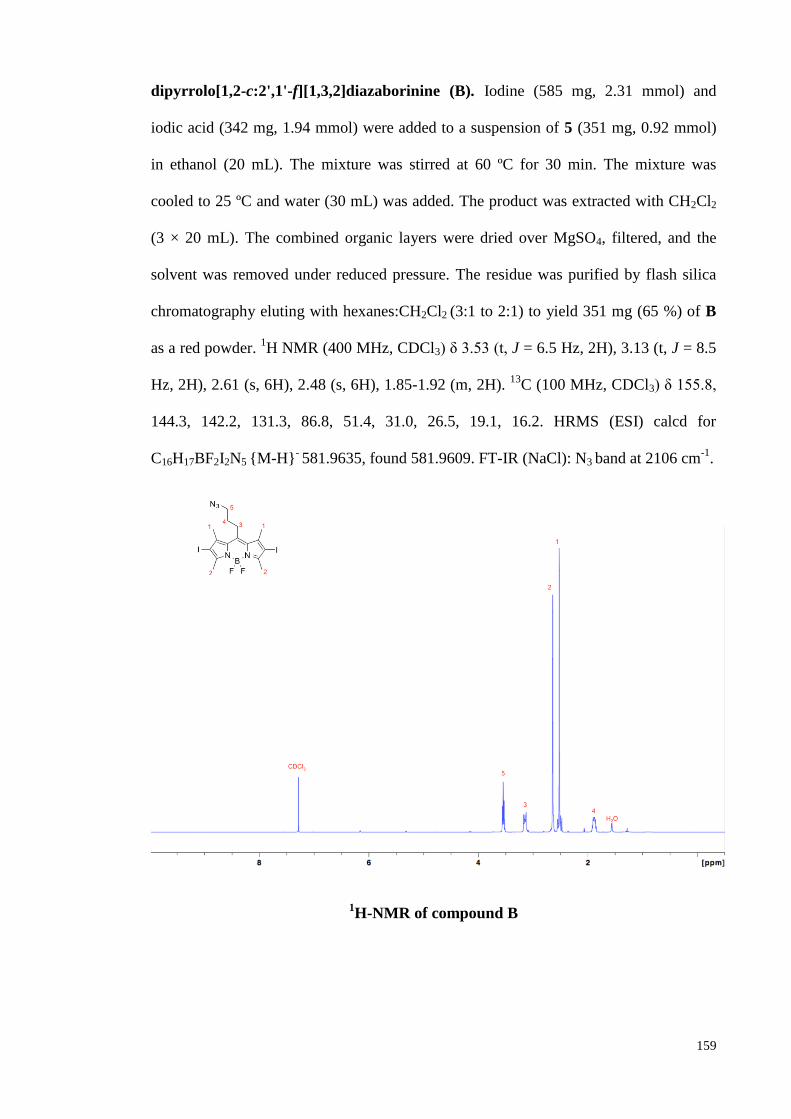
159
dipyrrolo[1,2-c:2',1'-f][1,3,2]diazaborinine (B). Iodine (585 mg, 2.31 mmol) and
iodic acid (342 mg, 1.94 mmol) were added to a suspension of 5 (351 mg, 0.92 mmol)
in ethanol (20 mL). The mixture was stirred at 60 ºC for 30 min. The mixture was
cooled to 25 ºC and water (30 mL) was added. The product was extracted with CH2Cl2
(3 × 20 mL). The combined organic layers were dried over MgSO4, filtered, and the
solvent was removed under reduced pressure. The residue was purified by flash silica
chromatography eluting with hexanes:CH2Cl2 (3:1 to 2:1) to yield 351 mg (65 %) of B
as a red powder. 1H NMR (400 MHz, CDCl3) δ 3.53 (t, J = 6.5 Hz, 2H), 3.13 (t, J = 8.5
Hz, 2H), 2.61 (s, 6H), 2.48 (s, 6H), 1.85-1.92 (m, 2H). 13
C (100 MHz, CDCl3) δ 155.8,
144.3, 142.2, 131.3, 86.8, 51.4, 31.0, 26.5, 19.1, 16.2. HRMS (ESI) calcd for
C16H17BF2I2N5 {M-H}- 581.9635, found 581.9609. FT-IR (NaCl): N3 band at 2106 cm
-1.
1H-NMR of compound B
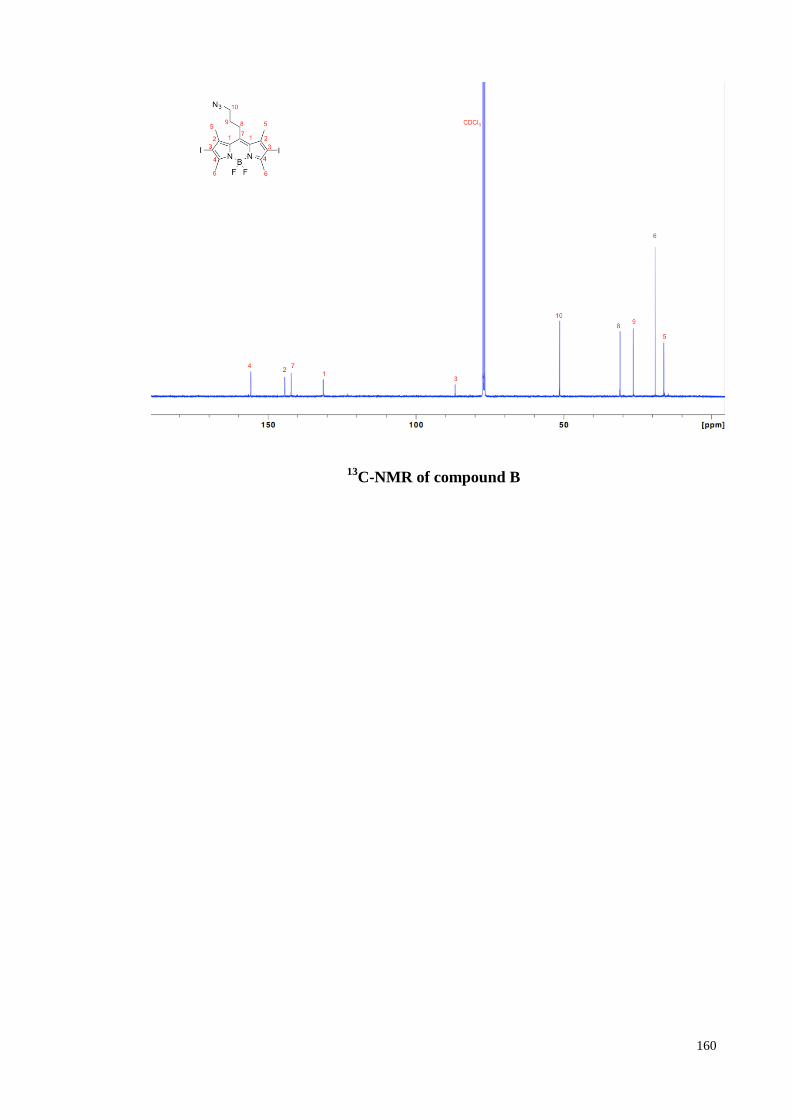
160
13C-NMR of compound B

161
Appendix B: Characterisation of IYIY-I2-BODIPY and YIYI-I2-BODIPY
Solution of diiodo-BODIPY (1.5 equiv) and IY-IY-TEG or YIYI-TEG (1.0 equiv) in
DMSO:H2O:CH2Cl2 (2:1:2, 0.01 M) were added CuSO4 (0.1 equiv, from 0.05 M
aqueous solution) and fresh Na ascorbate (0.4 equiv, from 0.05 M aqueous solution) at
25 °C. The mixture was stirred at 25 °C for 24 h. The reactions were monitored by
analytical HPLC. The crude compounds were lyophilized to remove DMSO, and then
purified by RP-preparative HPLC to obtain the bivalent mimics IYIY-I2-BODIPY or
YIYI-I2-BODIPY. The final products were lyophilised three times in 1.0 % acetic acid
to remove TFA.
IYIY-I2-BODIPY
After purification and lyophilisation, the product was obtained as a red powder (27-35 %
yield). The purity was found to be ca. >99% by HPLC analysis, retention time 16.083.
1H NMR (400 MHz, DMSO-d6) δ 8.38 (br, 4H), 8.29 (s, 2H), 8.23 (s, 1H), 7.00 (d, 4H,
J= 8.5 Hz), 6.78 (br, 2H), 6.61 (d, 4H, J = 8.4 Hz), 6.17 (t, 2H, J = 7.3 Hz), 4.63-4.54 (m,
2H), 4.38 (t, 2H, J = 5.6 Hz), 3.64-3.42 (m, 30H), 3.34-3.19 (m, 4H), 3.06-2.98 (m,
2H),2.53 (s, 6H), 2.25 (s, 6H), 2.14-2.07 (m, 2H), 1.98-1.87 (m, 2H), 1.40-1.30 (m, 2H),
1.08-0.94 (m, 2H), 0.93-0.86 (m, 6H), 0.79-0.70 (m, 6H)
13C NMR (100 MHz, DMSO-d6) δ 166.4, 158.6, 158.3, 156.8, 155.2, 145.7, 144.6,
143.3, 142.0, 131.3, 130.7, 125.9, 125.0, 115.5, 115.5, 88.1, 70.3, 70.3, 70.2, 70.1, 70.0,
69.5, 69.4, 63.9, 51.2, 49.6, 45.4, 42.7, 42.2, 37.7, 31.8, 25.6, 19.0, 16.3, 15.2, 14.1,
11.3
Hi-Res MALDI-MS: m/z calcd for C68H93BF2I2N21O7+
{M+H}+ 1618.5723 found
1618.5836.
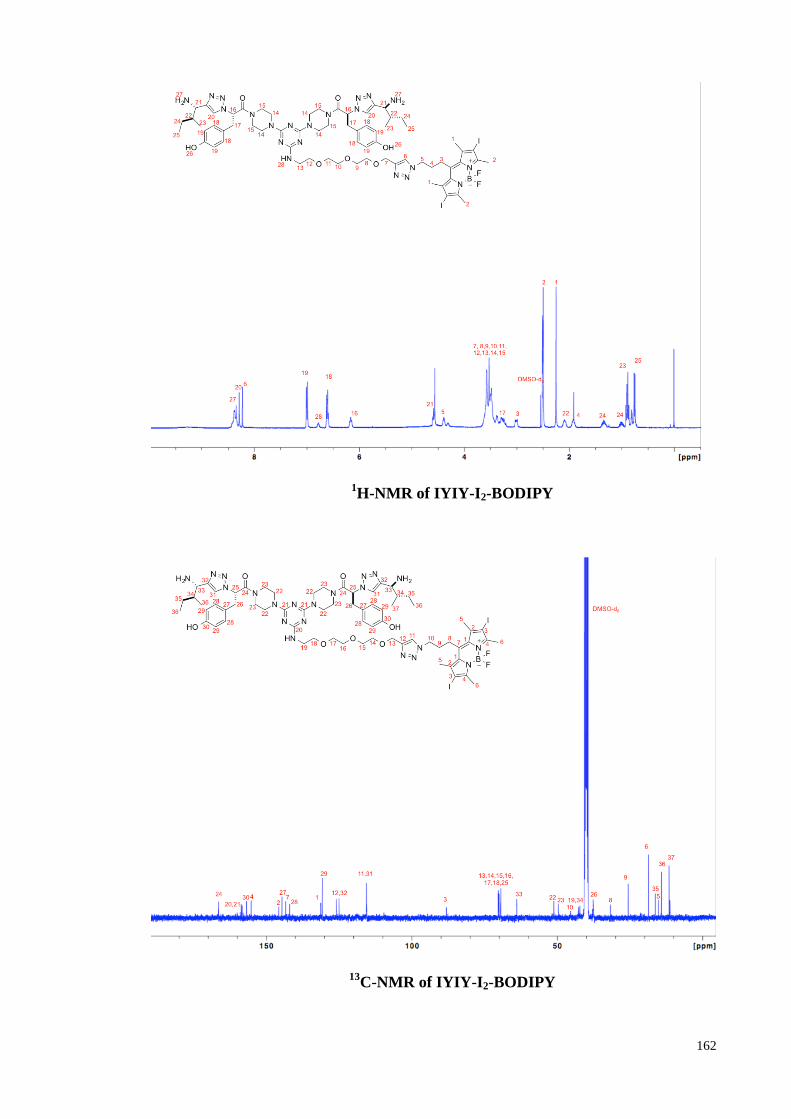
162
1H-NMR of IYIY-I2-BODIPY
13C-NMR of IYIY-I2-BODIPY
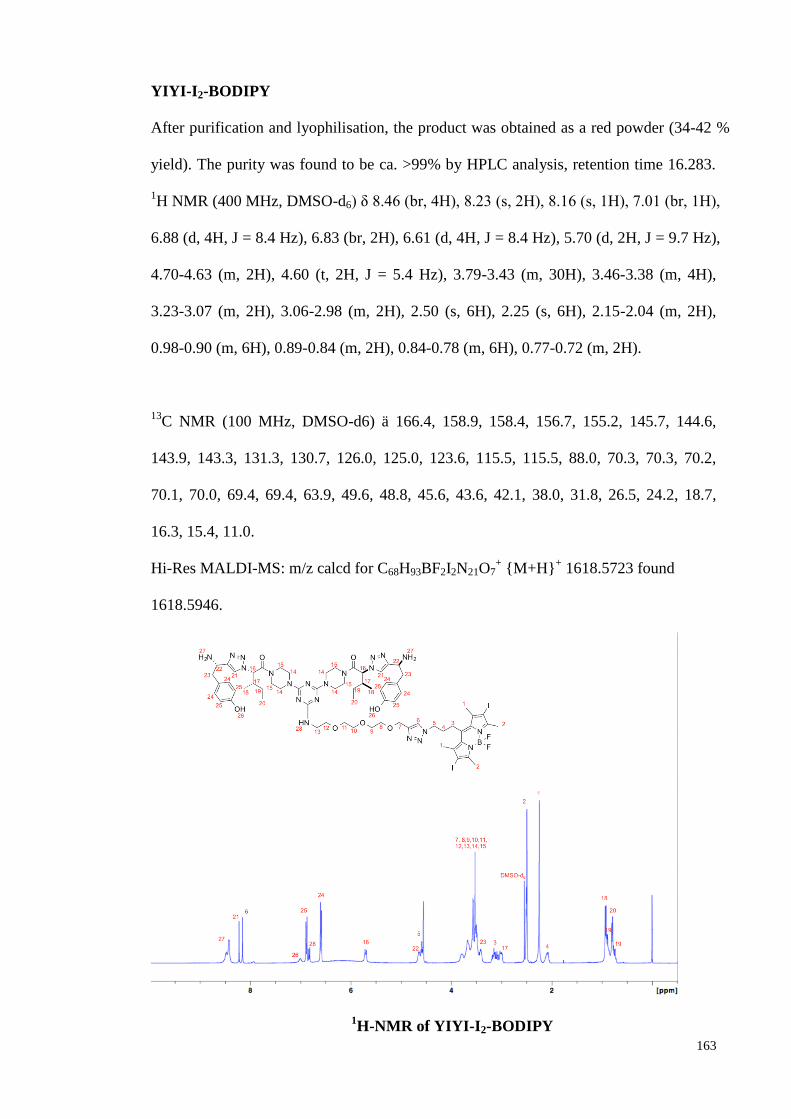
163
YIYI-I2-BODIPY
After purification and lyophilisation, the product was obtained as a red powder (34-42 %
yield). The purity was found to be ca. >99% by HPLC analysis, retention time 16.283.
1H NMR (400 MHz, DMSO-d6) δ 8.46 (br, 4H), 8.23 (s, 2H), 8.16 (s, 1H), 7.01 (br, 1H),
6.88 (d, 4H, J = 8.4 Hz), 6.83 (br, 2H), 6.61 (d, 4H, J = 8.4 Hz), 5.70 (d, 2H, J = 9.7 Hz),
4.70-4.63 (m, 2H), 4.60 (t, 2H, J = 5.4 Hz), 3.79-3.43 (m, 30H), 3.46-3.38 (m, 4H),
3.23-3.07 (m, 2H), 3.06-2.98 (m, 2H), 2.50 (s, 6H), 2.25 (s, 6H), 2.15-2.04 (m, 2H),
0.98-0.90 (m, 6H), 0.89-0.84 (m, 2H), 0.84-0.78 (m, 6H), 0.77-0.72 (m, 2H).
13C NMR (100 MHz, DMSO-d6) ä 166.4, 158.9, 158.4, 156.7, 155.2, 145.7, 144.6,
143.9, 143.3, 131.3, 130.7, 126.0, 125.0, 123.6, 115.5, 115.5, 88.0, 70.3, 70.3, 70.2,
70.1, 70.0, 69.4, 69.4, 63.9, 49.6, 48.8, 45.6, 43.6, 42.1, 38.0, 31.8, 26.5, 24.2, 18.7,
16.3, 15.4, 11.0.
Hi-Res MALDI-MS: m/z calcd for C68H93BF2I2N21O7+ {M+H}
+ 1618.5723 found
1618.5946.
1H-NMR of YIYI-I2-BODIPY
164
13C-NMR of YIYI-I2-BODIPY

165
Appendix C: Animal Ethics Approval Letter (Study-1)

166
Appendix D: Animal Ethics Approval Letter (Study-2)

167
Appendix E: Excellent Scientific Paper Awards 2015 by Tien Te Lee Biomedical
Foundation





























