Embed Size (px)
Citation preview

DaltonTransactions
Dynamic Article Links
Cite this: Dalton Trans., 2012, 41, 2659
www.rsc.org/dalton PAPER
Exchange coupling and magnetic anisotropy of exchanged-biased quantumtunnelling single-molecule magnet Ni3Mn2 complexes using theoreticalmethods based on Density Functional Theory†
Silvia Gómez-Coca and Eliseo Ruiz*
Received 27th July 2011, Accepted 21st November 2011DOI: 10.1039/c2dt11426g
The magnetic properties of a new family of single-molecule magnet Ni3Mn2 complexes were studiedusing theoretical methods based on Density Functional Theory (DFT). The first part of this study isdevoted to analysing the exchange coupling constants, focusing on the intramolecular as well as theintermolecular interactions. The calculated intramolecular J values were in excellent agreement with theexperimental data, which show that all the couplings are ferromagnetic, leading to an S = 7 ground state.The intermolecular interactions were investigated because the two complexes studied do not showtunnelling at zero magnetic field. Usually, this exchange-biased quantum tunnelling is attributed to thepresence of intermolecular interactions calculated with the help of theoretical methods. The resultsindicate the presence of weak intermolecular antiferromagnetic couplings that cannot explain theferromagnetic value found experimentally for one of the systems. In the second part, the goal is to analysemagnetic anisotropy through the calculation of the zero-field splitting parameters (D and E), using DFTmethods including the spin–orbit effect.
Introduction
Since the discovery in 1993 of single-molecule magnet (SMM)behaviour by Gatteschi and co-workers1 in a Mn12 compound, inwhich one single molecule behaves like a magnet, many researchgroups have intensively searched for new molecules showingsuch an appealing property.2 SMM complexes show a splittingof Ms states due to the presence of the zero-field splittingphenomenon (ZFS) caused by spin–orbit contributions. Theenergy difference between the highest and the lowest Ms statescaused by the loss of degeneracy could be understood as anenergy barrier (Ueff ), whose height is directly related to thesquare of the total spin (S) of the molecule and its magneticaxial anisotropy (D), which must be overcome in order tochange the spin direction (from +Ms to −Ms states). This Dvalue must be negative to have a barrier instead of a single well:the higher the barrier is, the more difficult it is to change thespin direction. Slow relaxation of magnetization at low tempera-ture is responsible for the presence of a hysteresis loop in magne-tization curves, which also display some irregular shapes due tothe presence of thermally-assisted quantum tunnelling to crossthe energy barrier.3,4 These tunnelling effects are also directlyrelated to the magnitude of the magnetic rhombic anisotropy (E).
Such transition metal complexes showing SMM behaviour aremuch sought-after synthetic targets, due to their potential assystems that could eventually lead to future applications in high-density information storage systems and quantum computing.5,6
Logically, one of the goals in this research field was to lookfor molecules with a high spin-flipping barrier. Since the mag-netic anisotropy barrier depends on the square of the total spinof the ground state, the presence of ferromagnetic couplings is ofcrucial importance to reaching a large S value. However, ferro-magnetic couplings in the exchange interaction between para-magnetic transition metal centres are scarce, withantiferromagnetism as the common case. Among the few SMMsthat show only ferromagnetic couplings, it is worth noting thosebased in Mn10 (4MnII–6MnIII) supertetrahedrons,7 and thefamily of Mn6 complexes8 containing exclusively MnIII cations.One of these Mn6 complexes shows the highest magnetic aniso-tropy barrier for a SMM.9 Recently, Das et al.10 synthesized twoNi3Mn2 complexes with NiII and MnIII cations, [Mn2Ni3X2L4
(LH)2(H2O)2] (H2L = 2-{3-(2-hydroxyphenyl)-1H-pyrazol-1-yl}ethanol, X = Cl: 1; X = Br: 2), showing an S = 7 ground statewith only ferromagnetic coupling between the paramagneticcentres (see Fig. 1). The two complexes are relatively similar,with just the change in one of the axial ligands of the MnIII
cations replacing chloride by bromide anions.In addition, the lack of tunnelling effects at zero-field is very
appealing in these two Ni3Mn2 complexes. This exchange-biased quantum tunnelling behaviour was noticed for the firsttime in a dimer of Mn4 complexes11,12 showing relatively strongintermolecular interactions, which are the origin of such
†Electronic supplementary information (ESI) available. See DOI:10.1039/c2dt11426g
Departament de Química Inorgànica and Institut de Recerca deQuímica Teòrica i Computacional, Universitat de Barcelona, Diagonal645, E-08028, Spain. E-mail: [email protected]
This journal is © The Royal Society of Chemistry 2012 Dalton Trans., 2012, 41, 2659–2666 | 2659
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online / Journal Homepage / Table of Contents for this issue

conduct. Such behaviour is particularly important for potentialtechnological applications of SMM because the presence of tun-nelling effects at zero-field leads to possible loss of information.It is also worth noting that the steps in magnetization due to theavalanche caused by the tunnelling effects, in the case ofcomplex 1, is shifted to positive values of the magnetic field,whereas in 2 a negative value appears, probably reflecting theopposite nature of the intermolecular exchange interactions inthe two complexes.
Theoretically, it is possible to determine the two main contri-butions to the magnetic anisotropy barrier. The exchange coup-ling constants that control the total spin of the ground state canbe determined by calculating the energy for different spin distri-butions with methods based on Density Functional Theory(DFT) without including spin–orbit contributions. Meanwhile, itis possible to calculate the zero-field splitting parameters (D andE) by adding the spin–orbit term.13,14 Theoretical methods canhelp to properly assign the intramolecular J values to the differ-ent interactions present in this kind of system, because in somecases it is not possible to perform the assignment: for instance, ifthere are J values similar to the interaction present in the Ni3Mn2complexes studied. Previously, we also analysed the role of theintermolecular interactions in systems that have no magnetizationstep at zero field, such as dimers of Fe9 complexes.15 Thus, thegoals of this paper are, first, to analyse the intramolecular andintermolecular exchange coupling constants of the two Ni3Mn2complexes by using theoretical calculations and, in the secondpart, to study the zero-field splitting parameters that quantify themagnetic anisotropy of the molecule.
Computational details
To study the exchange interactions, a phenomenological Heisen-berg Hamiltonian was used, excluding the terms relating to mag-netic anisotropy, to describe the exchange coupling in thepolynuclear complex:
bH ¼ �Xa,b
Jab bSa bSb ð1Þ
where Ŝa and Ŝb are the spin operators of the different paramag-netic centres. The Jab parameters are the pairwise coupling con-stants between the paramagnetic centres of the molecule.Basically, we need to calculate the energy of n + 1 spin distri-butions for a system with n different exchange coupling con-stants. These energy values allow us to build up a system of nequations in which the J values are the unknowns. In the presentstudy, four calculations were performed in order to obtain thetwo exchange coupling constants of the Ni3Mn2 complexes (seeFig. 1). They correspond to the high-spin Sz = 7 solution, oneS = 5 wave function flipping the spin of the central nickel atom,one S = 3 with the spin inversion of the two external nickelatoms, and finally an S = 1 corresponding to the spin inversionof the three nickel atoms.
Gaussian0316 calculations were performed with the hybridB3LYP functional17 using a guess function generated with theJaguar 7.0 code,18 which employs a procedure that allows us todetermine individually the local charges and multiplicities of theatoms, including the ligand field effects. A triple-ζ all-electronGaussian basis set19 was used for all the atoms.
The calculation of the zero-field splitting parameters using DFTmethods was introduced by Pederson and Khanna.14 We only showin this section the most important details of such an approach.This method introduces the spin–orbit effect through second-orderperturbation theory with the following spin–orbit operator:
Uðr;L; SÞ ¼ 1
2c2S� L
1
r
dΦðrÞdr
ð2Þ
where S is the spin moment operator, L is the angular momentoperator, r is the distance and Φ(r) the Coulomb potential oper-ator. Thus, we can express the matrix elements, considering thespatial functions ϕj(r) and spin functions χ as:
Uj;σ;k;σ0 ¼ hφjχjjUðr;L;SÞjφkχσ0 i ð3Þ
Uj;σ;k;σ0 ¼ �ihφjjVxjφkihχσ jSxjχσ0 i ð4Þwhere Vx is defined as:
ϕjjVxjϕkD E
¼ 1
2c2dϕjdz
jΦj dϕkdy
� �� dϕj
dyjΦj dϕk
dz
� �� �ð5Þ
second-order perturbative energy can be expressed as:
Eð2Þ ¼ Δ2 ¼Xσσ0
Xij
M σσ0ij Sσσ
0i Sσ
0σj ð6Þ
where σ considers all the spins, while i and j correspond to thedifferent directions of space. The matrix elements are defined as:
Sσσ0
ij ¼ χσ jSi jχσ0D E
;
M σσ0ij ¼ �
Xkl
φlσ jVijφkσ0h i φkσ0 jVjjφlσ� �
εlσ � εkσ0
ð7Þ
for the full and empty orbitals, respectively, with εσ and εkσ thecorresponding energies. Due to the similarity of eqn (5) with theHamiltonian anisotropy term, depending on the zero-field split-ting parameter (eqn (8)), it is possible to compute such a D value:
Ĥ ¼ Ŝ D Ŝ ð8Þ
Fig. 1 Molecular structure of complex 1 [Mn2Ni3Cl2L4(LH)2(H2O)2](H2L = 2-{3-(2-hydroxyphenyl)-1H-pyrazol-1-yl}ethanol).10 Violet,grey, green, light blue, red and brown spheres correspond to manganese,nickel, chlorine, nitrogen, oxygen and carbon atoms, respectively, whilehydrogen atoms are omitted for clarity.
2660 | Dalton Trans., 2012, 41, 2659–2666 This journal is © The Royal Society of Chemistry 2012
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online

Thus, for a diagonal form of the tensor, the followingexpression can be obtained:X
σσ0
Xij
M σσ0ij Sσσ
0i Sσ
0σj ¼ DxxS
2x þ DyyS
2y þ DzzS
2z ð9Þ
which will let each component be obtained independently.
DiiS2i ¼
Xσσ0
M σσ0ii Sσσ
0i Sσ
0σj ð10Þ
Using the most common form of the Hamiltonian,
bH ¼ DðbSz2 � 1
3bS2 Þ þ EðbSx2 � bSy2Þ ð11Þ
we can obtain the final expression for the D and E zero-fieldsplitting parameters from the diagonal terms of the D tensor:
D ¼ Dzz � 1
2ðDxx þ Dyy Þ ð12Þ
E ¼ 1
2ðDxx � Dyy Þ ð13Þ
This method was implemented in the NRLMOL code,20 usingGGA functionals21 and its own basis set. The procedure wasrevisited by Neese13 and van Wüllen,22 starting with a differentapproach. Neese, employing the spin–orbit mean-field (SOMF)method originally proposed by Hess et al.,23 reached the follow-ing expression for the spin–orbit contribution to the DKL
elements:
DðSOC;NÞKL ¼ � 1
S2X
bðSb¼SÞ
Ψ 0S jhK;SOCjΨ b
Sb
D EΨ 0
S jhL;SOC jΨ bSb
D EEb � E0
� 1
ðS þ 1Þð2S þ 1ÞX
bðSb¼Sþ1Þ
Ψ 0S jhK;SOCjΨ b
Sb
D EΨ 0
S jhL;SOC jΨ bSb
D EEb � E0
� 1
Sð2S � 1ÞX
bðSb¼S�1Þ
Ψ 0S jhK;SOC jΨ b
Sb
D EΨ 0
S jhL;SOC jΨ bSb
D EEb � E0
ð14Þwhere Ψ0
S corresponds to the ground state, while there are threeterms depending on the difference of spin Sb = S and the excitedstates Ψb
Sb with different prefactors. The operator hK;SOC
represents the Kth component (K = x,y,z) of the spatial part ofan effective one-electron SOC operator. Using a similar
notation, the Pederson–Khanna approach14 is a sum-over-statesexpression:
DðSOC;PKÞKL ¼ � 1
4S2X
σ;σ0¼α;β
ð�1Þσþσ0Xiσ ;aσ0
ψσi jhK;SOCjψσ0
a
� �ψσ0a jhL;SOCjψσ
i
� �εaσ0 � εiσ
ð15Þ
where ψσi and ψσ′
a are occupied (i) and virtual (a) canonicalKohn–Sham orbitals of spin σ,σ′ = αβ and orbital energies εaσ′and εiσ, respectively. Note that this equation is only valid in theabsence of Hartree–Fock exchange or other non-local potentials.
Recently, van Wüllen et al.24 analysed both approaches andfound some deficiencies: the underestimation of some contri-butions in the Pederson–Khanna approach14 and the spuriouscontribution of diamagnetic species in the Neese procedure.13
The new set of equations is similar to those proposed by Neese,but with the same prefactor for all the terms, resulting in a sum-over-states formula:
DðSOC;νW ÞKL ¼ � 1
Sð2S � 1ÞXb
XSb�Sb
Ψ 0S jhK;SOC jΨ b
Sb
D EΨ 0
S jhL;SOC jΨ bSb
D EEb � E0
ð16Þ
The results using the new expression are very similar to thoseobtained with eqn (14), but correct the spurious contribution ofdiamagnetic species in the Neese procedure with the new prefac-tor. Using the Orca code25 and on the basis of the ground stateSlater determinant, the spin–spin contribution of the D tensorcan be calculated according to the McWeeny and Mizunoformula:26
DKL ¼ � g2e16
α2
Sð2S � 1ÞXμνκτ
PμνPκτ � PμκPντ
� μνjr�5
12 3r12;Kr12;L � δKLr212
� jκτ� �ð17Þ
where Pα−β = Pα − Pβ is the spin density matrix with Pσμν =
Σpσcσμpc
σνp; c
σ is the MO coefficient matrix for spin σ; and α is thefine structure constant (∼1/137 in atomic units). The spin–orbitcalculations with ORCA 2.8.0 were performed using theSOCtype option 1,2,2,1 in order to avoid spurious results for thebromine system with the hybrid functional with the suggested1,3,3,1 option.
Results and discussion
Exchange interaction in Ni3Mn2 complexes
The intramolecular magnetic interactions in the Ni3Mn2 com-plexes were studied with a Heisenberg Hamiltonian (see
Table 1 Structural and magnetic data of the Mn2Ni3X2 complexes (X = Cl: 1; X = Br: 2). Distances, angles and J values are in Å, degrees andcm−1, respectively
Complex Bridging ligands M⋯M M–O–M Mn–O Jexp Jcalc
1Ni⋯Ni 2(μ2-OCH2R) 3.109 96.1, 100.5 — +15.3 +12.3Mn⋯Ni 2(μ2-OPhR) 3.045 100.7, 101.4 1.888, 1.889 +32.0 +36.02Ni⋯Ni 2(μ2-OCH2R) 3.111 96.4, 100.8 — +12.5 +11.2Mn⋯Ni 2(μ2-OPhR) 3.046 100.6, 101.4 1.886, 1.891 +29.2 +31.6
This journal is © The Royal Society of Chemistry 2012 Dalton Trans., 2012, 41, 2659–2666 | 2661
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online
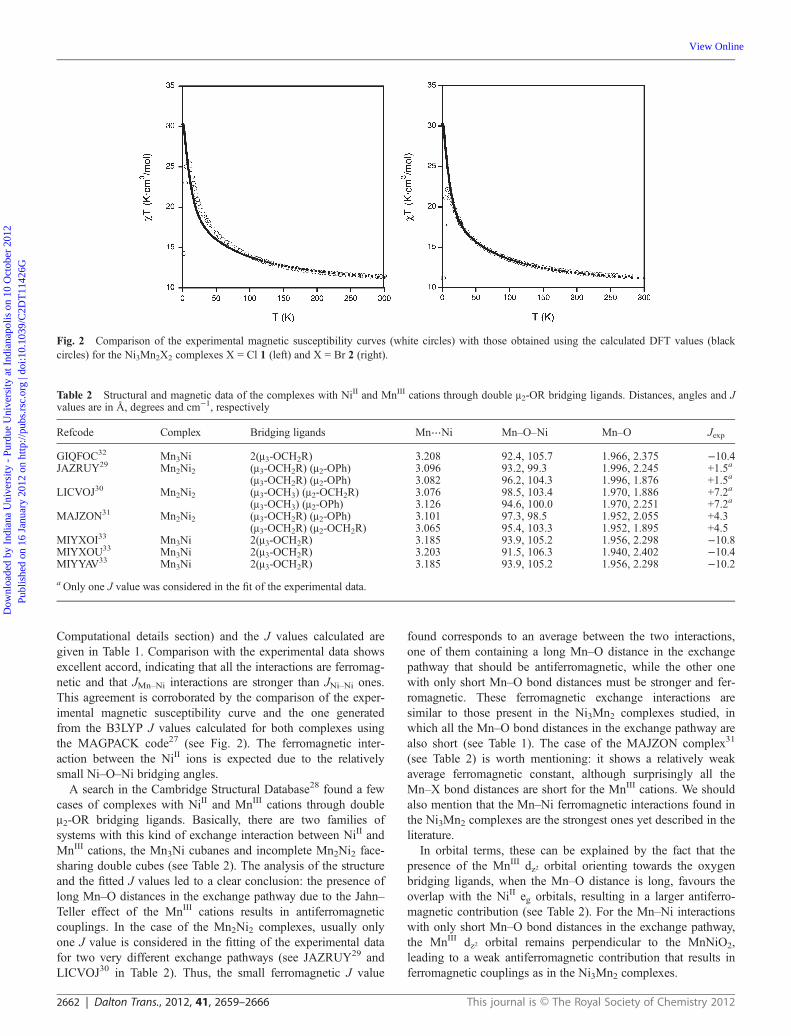
Computational details section) and the J values calculated aregiven in Table 1. Comparison with the experimental data showsexcellent accord, indicating that all the interactions are ferromag-netic and that JMn–Ni interactions are stronger than JNi–Ni ones.This agreement is corroborated by the comparison of the exper-imental magnetic susceptibility curve and the one generatedfrom the B3LYP J values calculated for both complexes usingthe MAGPACK code27 (see Fig. 2). The ferromagnetic inter-action between the NiII ions is expected due to the relativelysmall Ni–O–Ni bridging angles.
A search in the Cambridge Structural Database28 found a fewcases of complexes with NiII and MnIII cations through doubleμ2-OR bridging ligands. Basically, there are two families ofsystems with this kind of exchange interaction between NiII andMnIII cations, the Mn3Ni cubanes and incomplete Mn2Ni2 face-sharing double cubes (see Table 2). The analysis of the structureand the fitted J values led to a clear conclusion: the presence oflong Mn–O distances in the exchange pathway due to the Jahn–Teller effect of the MnIII cations results in antiferromagneticcouplings. In the case of the Mn2Ni2 complexes, usually onlyone J value is considered in the fitting of the experimental datafor two very different exchange pathways (see JAZRUY29 andLICVOJ30 in Table 2). Thus, the small ferromagnetic J value
found corresponds to an average between the two interactions,one of them containing a long Mn–O distance in the exchangepathway that should be antiferromagnetic, while the other onewith only short Mn–O bond distances must be stronger and fer-romagnetic. These ferromagnetic exchange interactions aresimilar to those present in the Ni3Mn2 complexes studied, inwhich all the Mn–O bond distances in the exchange pathway arealso short (see Table 1). The case of the MAJZON complex31
(see Table 2) is worth mentioning: it shows a relatively weakaverage ferromagnetic constant, although surprisingly all theMn–X bond distances are short for the MnIII cations. We shouldalso mention that the Mn–Ni ferromagnetic interactions found inthe Ni3Mn2 complexes are the strongest ones yet described in theliterature.
In orbital terms, these can be explained by the fact that thepresence of the MnIII dz2 orbital orienting towards the oxygenbridging ligands, when the Mn–O distance is long, favours theoverlap with the NiII eg orbitals, resulting in a larger antiferro-magnetic contribution (see Table 2). For the Mn–Ni interactionswith only short Mn–O bond distances in the exchange pathway,the MnIII dz2 orbital remains perpendicular to the MnNiO2,leading to a weak antiferromagnetic contribution that results inferromagnetic couplings as in the Ni3Mn2 complexes.
Fig. 2 Comparison of the experimental magnetic susceptibility curves (white circles) with those obtained using the calculated DFT values (blackcircles) for the Ni3Mn2X2 complexes X = Cl 1 (left) and X = Br 2 (right).
Table 2 Structural and magnetic data of the complexes with NiII and MnIII cations through double μ2-OR bridging ligands. Distances, angles and Jvalues are in Å, degrees and cm−1, respectively
Refcode Complex Bridging ligands Mn⋯Ni Mn–O–Ni Mn–O Jexp
GIQFOC32 Mn3Ni 2(μ3-OCH2R) 3.208 92.4, 105.7 1.966, 2.375 −10.4JAZRUY29 Mn2Ni2 (μ3-OCH2R) (μ2-OPh) 3.096 93.2, 99.3 1.996, 2.245 +1.5a
(μ3-OCH2R) (μ2-OPh) 3.082 96.2, 104.3 1.996, 1.876 +1.5a
LICVOJ30 Mn2Ni2 (μ3-OCH3) (μ2-OCH2R) 3.076 98.5, 103.4 1.970, 1.886 +7.2a
(μ3-OCH3) (μ2-OPh) 3.126 94.6, 100.0 1.970, 2.251 +7.2a
MAJZON31 Mn2Ni2 (μ3-OCH2R) (μ2-OPh) 3.101 97.3, 98.5 1.952, 2.055 +4.3(μ3-OCH2R) (μ2-OCH2R) 3.065 95.4, 103.3 1.952, 1.895 +4.5
MIYXOI33 Mn3Ni 2(μ3-OCH2R) 3.185 93.9, 105.2 1.956, 2.298 −10.8MIYXOU33 Mn3Ni 2(μ3-OCH2R) 3.203 91.5, 106.3 1.940, 2.402 −10.4MIYYAV33 Mn3Ni 2(μ3-OCH2R) 3.185 93.9, 105.2 1.956, 2.298 −10.2aOnly one J value was considered in the fit of the experimental data.
2662 | Dalton Trans., 2012, 41, 2659–2666 This journal is © The Royal Society of Chemistry 2012
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online
The spin distribution for complex 1 is seen in Fig. 3. For theNiII cations, the predominant mechanism is spin delocalisationdue to the large mixing of the antibonding eg orbitals of the Ni
II
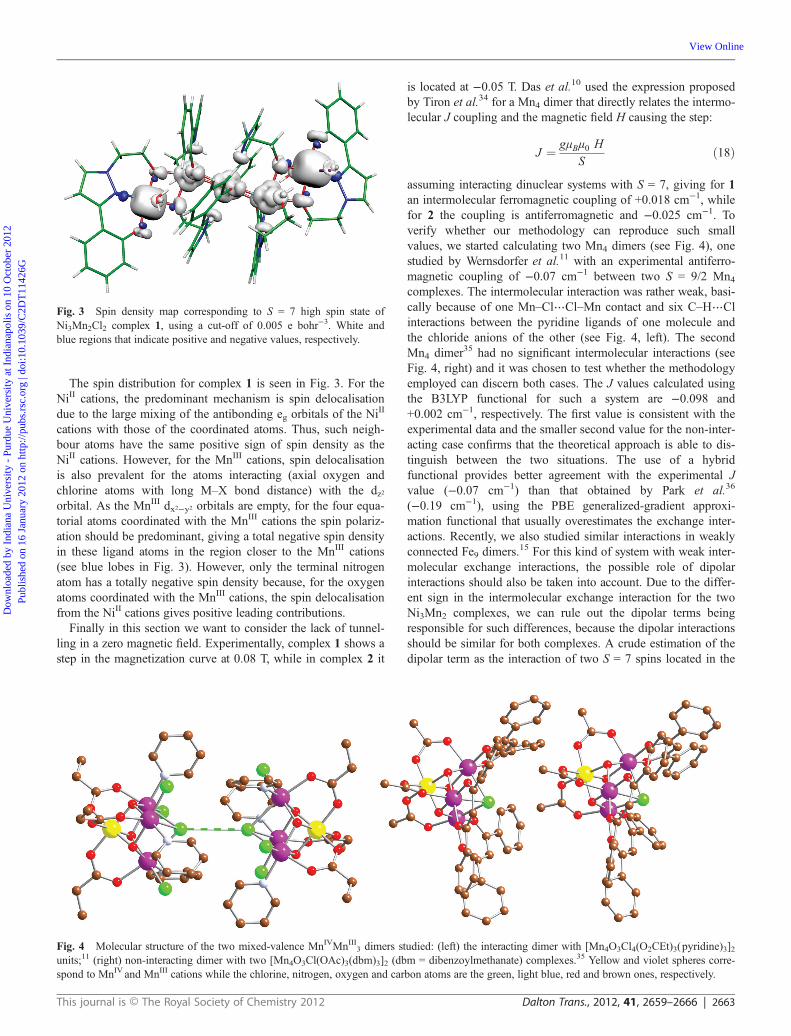
cations with those of the coordinated atoms. Thus, such neigh-bour atoms have the same positive sign of spin density as theNiII cations. However, for the MnIII cations, spin delocalisationis also prevalent for the atoms interacting (axial oxygen andchlorine atoms with long M–X bond distance) with the dz2orbital. As the MnIII dx2−y2 orbitals are empty, for the four equa-torial atoms coordinated with the MnIII cations the spin polariz-ation should be predominant, giving a total negative spin densityin these ligand atoms in the region closer to the MnIII cations(see blue lobes in Fig. 3). However, only the terminal nitrogenatom has a totally negative spin density because, for the oxygenatoms coordinated with the MnIII cations, the spin delocalisationfrom the NiII cations gives positive leading contributions.
Finally in this section we want to consider the lack of tunnel-ling in a zero magnetic field. Experimentally, complex 1 shows astep in the magnetization curve at 0.08 T, while in complex 2 it
is located at −0.05 T. Das et al.10 used the expression proposedby Tiron et al.34 for a Mn4 dimer that directly relates the intermo-lecular J coupling and the magnetic field H causing the step:
J ¼ gμBμ0 H
Sð18Þ
assuming interacting dinuclear systems with S = 7, giving for 1an intermolecular ferromagnetic coupling of +0.018 cm−1, whilefor 2 the coupling is antiferromagnetic and −0.025 cm−1. Toverify whether our methodology can reproduce such smallvalues, we started calculating two Mn4 dimers (see Fig. 4), onestudied by Wernsdorfer et al.11 with an experimental antiferro-magnetic coupling of −0.07 cm−1 between two S = 9/2 Mn4complexes. The intermolecular interaction was rather weak, basi-cally because of one Mn–Cl⋯Cl–Mn contact and six C–H⋯Clinteractions between the pyridine ligands of one molecule andthe chloride anions of the other (see Fig. 4, left). The secondMn4 dimer35 had no significant intermolecular interactions (seeFig. 4, right) and it was chosen to test whether the methodologyemployed can discern both cases. The J values calculated usingthe B3LYP functional for such a system are −0.098 and+0.002 cm−1, respectively. The first value is consistent with theexperimental data and the smaller second value for the non-inter-acting case confirms that the theoretical approach is able to dis-tinguish between the two situations. The use of a hybridfunctional provides better agreement with the experimental Jvalue (−0.07 cm−1) than that obtained by Park et al.36
(−0.19 cm−1), using the PBE generalized-gradient approxi-mation functional that usually overestimates the exchange inter-actions. Recently, we also studied similar interactions in weaklyconnected Fe9 dimers.15 For this kind of system with weak inter-molecular exchange interactions, the possible role of dipolarinteractions should also be taken into account. Due to the differ-ent sign in the intermolecular exchange interaction for the twoNi3Mn2 complexes, we can rule out the dipolar terms beingresponsible for such differences, because the dipolar interactionsshould be similar for both complexes. A crude estimation of thedipolar term as the interaction of two S = 7 spins located in the
Fig. 3 Spin density map corresponding to S = 7 high spin state ofNi3Mn2Cl2 complex 1, using a cut-off of 0.005 e bohr−3. White andblue regions that indicate positive and negative values, respectively.
Fig. 4 Molecular structure of the two mixed-valence MnIVMnIII3 dimers studied: (left) the interacting dimer with [Mn4O3Cl4(O2CEt)3(pyridine)3]2units;11 (right) non-interacting dimer with two [Mn4O3Cl(OAc)3(dbm)3]2 (dbm = dibenzoylmethanate) complexes.35 Yellow and violet spheres corre-spond to MnIV and MnIII cations while the chlorine, nitrogen, oxygen and carbon atoms are the green, light blue, red and brown ones, respectively.
This journal is © The Royal Society of Chemistry 2012 Dalton Trans., 2012, 41, 2659–2666 | 2663
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online

centre of the Ni3Mn2 molecules leads to an energy value ofaround 0.1 cm−1, while the energy associated with the intermole-cular interactions is one order of magnitude larger.
In order to analyse the sign and strength of the intermolecularinteraction in the two Ni3Mn2 complexes, we have performedthe calculations of the exchange constants using two Ni3Mn2molecules through two different contacts (see Fig. 5, contacts Aand B in dashed green and blue lines, respectively). The Acontact corresponds to a MnIII⋯MnIII exchange through a π–πstacking intermolecular interaction and the calculated J value is−0.003 and −0.004 cm−1 for the two complexes, 1 and 2,respectively. We have also considered a second intermolecularexchange pathway B for the two complexes that corresponds totwo NiII⋯MnIII interactions through a hydrogen bond C–H⋯Cl(or Br) intermolecular interaction (see Fig. 5). The J values forsuch interactions for 1 and 2 are still antiferromagnetic, −0.003and −0.008 cm−1, respectively. Antiferromagnetic values areexpected for intermolecular exchange pathways, and it would bein qualitative agreement with the antiferromagnetic value pro-posed experimentally for 2 despite our calculated value beingsmaller. Thus, the calculated J values for the closest intermolecu-lar contacts using the mentioned methodology always results inweak antiferromagnetic couplings for both complexes.
Magnetic anisotropy in Ni3Mn2 complexes
Magnetic anisotropy is usually quantified through the zero-fieldsplitting parameters (D and E). These two parameters are crucialfor the single-molecule magnet properties of the complexes.Most attention is paid to the D value because the spin-flippingenergy barrier is |D|S2. However, although the E parameters arevery small, the tunnelling effects are proportional to their value.The values calculated using NRLMOL20 and Orca25 codes aregiven in Table 3. Despite the basis set and grid differences, in
qualitative terms the results with the two programs using thesame PBE21 functional were relatively close. It is worth keepingin mind that the sign of the zero-field splitting parameters (D andE) depends on a very subtle change in the components of the Dtensor. Thus, the PBE results for complex 2 are very similar butwith different sign. The D values were reasonably consistentwith the experimental data obtained from fitted magnetizationvalues. It is worth noting that accurate experimental values canonly be obtained by using high-field electronic paramagnetic res-onance experiments. The values of E calculated are very small incomparison with the “experimental” value, which was not deter-mined due to technical problems, and a maximum value of 10|E|≤ D was proposed. The use of a hybrid functional gave valuescloser to the experimental data than the PBE functional for thetwo complexes. The spin–spin contributions were relativelysmall in comparison with the spin–orbit terms, around 20–30%of the total D value. For complex 1, the main spin–orbit contri-butions corresponded to the α–α channel, involving excitedstates corresponding to dz2 to dx2−y2 transitions of the MnIII
cations. The largest contribution was the α–β channel, whichprobably corresponds to quintet-triplet local excitation of theMnIII centres. These two contributions are also the predominantones in mononuclear MnIII complexes.13 The low contributioncorresponding to the β–β channel usually indicated that the exci-tations of the β electrons from the t2g to eg NiII orbitals play aminor role for complex 1. Surprisingly, such contributions wereconsiderably more important for complex 2, even though thesubstitution of chloride for bromide is in the MnIII coordinationsphere. The D values using the Orca code reproduced the exper-imental trend that the magnetic anisotropy is smaller forcomplex 2.
To check the origin of the differences between the two com-plexes we repeated the calculations, replacing the halides, e.g. incomplex 1, replacing the chloride by bromide anions while
Fig. 5 Molecular structure of intermolecular contacts (A and B, dashed green and blue lines respectively) for complex 1 [Mn2Ni3Cl2L4(LH)2(H2O)2](H2L = 2-{3-(2-hydroxyphenyl)-1H-pyrazol-1-yl}ethanol).10 Violet, grey, green, light blue, red, brown and white spheres correspond to manganese,nickel, chlorine, nitrogen, oxygen, carbon and hydrogen atoms, respectively. Only hydrogen atoms involved in the contacts are represented.
2664 | Dalton Trans., 2012, 41, 2659–2666 This journal is © The Royal Society of Chemistry 2012
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online

retaining the structure of the rest of the complex; and then theopposite change for complex 2. The results for these two newmodel systems (1·Br and 2·Cl) were almost identical to the orig-inal complex with the other anion. Thus, complex 1·Br had Dand E contributions practically (not shown) equal to complex 2(and the same for the 2·Cl and 1 systems). These results indicatethat the changes in the contributions of D and E in complex 2are related to the presence of a relatively heavy atom likebromine, and not to changes in the levels of the NiII and MnIII
paramagnetic centres for structural rearrangements. The analysisof the orbital energies corresponding to the MnIII cations indi-cated that the values remain almost constant when chlorideanions are replaced by bromide anions, confirming the assump-tion that the differences in D and E contributions between thetwo complexes are not related to the levels of the metals.
Conclusions
The intramolecular exchange couplings of the Ni3Mn2 com-plexes studied using the B3LYP functional correlated closelywith the experimental data, confirming the ferromagnetic natureof all the couplings and the S = 7 ground state, even thoughJMn–Ni interactions are stronger than JNi–Ni ones. It is worthnoting that the nature of the exchange interactions between NiII
and MnIII cations is directly related to the presence of longMn–O distances in the exchange pathway. Such long bond dis-tances due to the Jahn–Teller effect of the MnIII cations result inantiferromagnetic couplings. The Ni3Mn2 complexes show thestrongest ferromagnetic coupling for exchange interactionsbetween NiII and MnIII cations reported up to now.
One of the most appealing features of these two Ni3Mn2 com-plexes is the lack of tunnelling effects at zero-field. Thisexchange-biased quantum tunnelling behaviour was attributed tothe presence of intermolecular exchange interactions.
Experimentally, the magnetization step shifts a positive magneticfield for the chloride Ni3Mn2 complex 1, while the analogouscomplex with bromide anions 2 shows shift to negative magneticfield values. Despite the scarcity of complexes showing this be-haviour, the second case is the most common one, because suchbehaviour is attributed to the presence of antiferromagnetic inter-molecular interactions, while for complex 1, ferromagnetic inter-molecular coupling is required to explain such behaviour. Tocheck whether the approach employed reproduces the tinyexchange couplings due to intermolecular interactions, westudied two Mn4 dimers. The first was the original system whereexchange-biased quantum tunnelling behaviour was detected andthe second system had no important intermolecular interactions.The intermolecular J values calculated using the B3LYP func-tional were −0.098 and +0.002 cm−1, respectively, with the firstvalue close to the figure proposed in the experimental data,−0.07 cm−1. These results clearly indicated that the method-ology employed is able to handle such weak exchange inter-actions. However, for the Ni3Mn2 complexes the calculatedintermolecular J values corresponding to two different pathwaysalways give weak antiferromagnetic couplings, one order ofmagnitude smaller than the experimental ones. These antiferro-magnetic coupling are at least qualitatively in agreement forcomplex 2, however we can not obtain ferromagnetic intermole-cular interactions expected for complex 1. Also, the calculationof the dipolar interaction indicates that the magnitude is oneorder smaller than that involved in the intermolecular couplingsand that the dipolar terms cannot explain the differences betweenthe two complexes studied.
The last part of our study is devoted to the magnetic aniso-tropy of the two Ni3Mn2 complexes. The results for both com-plexes are quite consistent, independently of the approach,reproducing the experimental negative D value and the largervalues found the complex 1. The results using the hybrid B3LYP
Table 3 Zero-field splitting parameters (D and E, in cm−1) for the Mn2Ni3X2 complexes (X = Cl: 1; X = Br: 2). The fitted experimental data (D and|E|) is provided for comparison. In the calculations of the NRLMOL,20 only the spin–orbit term (eqn (10)–(12)) was considered, whereas with theOrca25 code, the different contributions to the D value are indicated (eqn (14) and (17))
Complex
1 2
D E D E
PBE-NRLMOL −0.19 0.057 0.11 0.026
PBE-Orca −0.235 −0.001 −0.153 −0.024
Spin–spin −0.042 0.007 −0.041 0.003Spin–orbit −0.193 −0.008 −0.112 −0.027α–α −0.066 0.000 0.002 0.013β–β 0.056 −0.028 0.322 −0.047α–β −0.182 0.019 −0.447 0.019β–α −0.000 0.001 0.012 −0.013B3LYP-Orca −0.394 −0.089 −0.251 0.081
Spin–spin −0.045 −0.001 −0.046 −0.003Spin–orbit −0.349 −0.088 −0.205 −0.017α–α −0.073 0.009 −0.013 0.027β–β 0.013 −0.030 0.273 −0.001α–β −0.283 −0.062 −0.467 −0.015β–α −0.007 −0.005 0.002 −0.028
Experimental −0.35 0.035 −0.28 0.028
This journal is © The Royal Society of Chemistry 2012 Dalton Trans., 2012, 41, 2659–2666 | 2665
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online

functional are quite close to the available experimental data. Theanalysis of the contributions indicated that the α–α and α–β con-tributions of the MnIII cations are the main features responsiblefor anisotropy in such systems. However, for complex 2 surpris-ingly due to the presence of bromine atoms, there are larger β–βchannel contributions that are not present in complex 1, and thiscould be attributed to the excitations from the t2g to eg NiII
orbitals.
Acknowledgements
We thank Prof. Franc Meyer for providing us with the crystalstructures prior to their publication. The research reported herewas supported by Spain’s Ministerio de Ciencia e Innovaciónand the Generalitat de Catalunya through grants CTQ2008-06670-C02-01 and 2009SGR-1459, respectively. S. G. C. thanksthe Ministerio de Educación y Ciencia for a postgraduate fellow-ship. The authors gratefully acknowledge the computerresources, technical expertise and assistance provided by theCentre de Supercomputació de Catalunya, for the calculations ofthe exchange interactions, and the Barcelona SupercomputerCentre, for the study of magnetic anisotropy.
References
1 R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993,365, 141.
2 G. Aromi and E. K. Brechin, Struct. Bonding, 2006, 122, 1.3 D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268.4 D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, OxfordUniversity Press, Oxford, 2006.
5 G. Rogez, B. Donnio, E. Terazzi, J.-L. Gallani, J.-P. Kappler, J.-P. Bucherand M. Drillon, Adv. Mater., 2009, 21, 4323.
6 E. R. Winpenny and E. J. L. McInnes, in Molecular Materials, ed.D. W. Bruce, D. O’Hare and R. I. Walton, Wiley, Chichester, 2010.
7 T. C. Stamatatos, K. A. Abboud, W. Wernsdorfer and G. Christou, Angew.Chem., Int. Ed., 2006, 45, 4134.
8 R. Inglis, L. F. Jones, C. J. Milios, S. Datta, A. Collins, S. Parsons,W. Wernsdorfer, S. Hill, S. P. Perlepes, S. Piligkos and E. K. Brechin,Dalton Trans., 2009, 3403.
9 C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S.P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007,129, 2754.
10 A. Das, K. Gieb, Y. Krupskaya, S. Demeshko, S. Dechert, R. Klingeler,V. Kataev, B. Buechner, P. Mueller and F. Meyer, J. Am. Chem. Soc.,2011, 133, 3433.
11 W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou,Nature, 2002, 416, 406.
12 S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science,2003, 302, 1015.
13 F. Neese, J. Chem. Phys., 2007, 127, 164112.14 M. R. Pederson and S. N. Khanna, Phys. Rev. B, 1999, 60, 9566.15 S. Gomez-Coca, E. Ruiz and J. Kortus, Chem. Commun., 2009, 4363.16 M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb,
J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin,J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone,B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson,H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa,M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li,J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo,J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin,R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma,G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich,A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck,K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul,S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko,P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith,M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe,P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez andJ. A. Pople, GAUSSIAN 03 (Revision D.02), Gaussian, Inc., Wallingford,CT, 2004
17 A. D. Becke, J. Chem. Phys., 1993, 98, 5648.18 Jaguar 7.0, Schrödinger, Inc, Portland, 200919 A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829.20 M. R. Pederson, D. V. Porezag, J. Kortus and D. C. Patton, Phys. Status
Solidi B, 2000, 217, 197.21 J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77,
3865.22 C. van Wüllen, J. Chem. Phys., 2009, 130, 194109.23 B. A. Hess, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys.
Lett., 1996, 251, 365.24 S. Schmitt, P. Jost and C. van Wuellen, J. Chem. Phys., 2011, 134,
194113.25 Orca 2.8.0, F. Neese, University of Bonn, Germany, 2010.26 R. McWeeny and Y. Mizuno, Proc. R. Soc. London, Ser. A, 1961, 259,
554.27 J. J. Borras-Almenar, J. M. Clemente-Juan, E. Coronado and
B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985.28 F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380.29 M. Koikawa, M. Ohba and T. Tokii, Polyhedron, 2005, 24, 2257.30 M. Nihei, A. Yoshida, S. Koizumi and H. Oshio, Polyhedron, 2007, 26,
1997.31 H. Oshio, M. Nihei, S. Koizumi, T. Shiga, H. Nojiri, M. Nakano,
N. Shirakawa and M. Akatsu, J. Am. Chem. Soc., 2005, 127, 4568.32 P. L. Feng, C. C. Beedle, W. Wernsdorfer, C. Koo, M. Nakano, S. Hill
and D. N. Hendrickson, Inorg. Chem., 2007, 46, 8126.33 P. L. Feng, C. C. Beedle, C. Koo, W. Wernsdorfer, M. Nakano, S. Hill
and D. N. Hendrickson, Inorg. Chem., 2008, 47, 3188.34 R. Tiron, W. Wernsdorfer, D. Foguet-Albiol, N. Aliaga-Alcalde and
G. Christou, Phys. Rev. Lett., 2003, 91, 227203.35 S. Y. Wang, H. L. Tsai, E. Libby, K. Folting, W. E. Streib, D.
N. Hendrickson and G. Christou, Inorg. Chem., 1996, 35, 7578.36 K. Park, M. R. Pederson and N. Bernstein, J. Phys. Chem. Solids, 2004,
65, 805.
2666 | Dalton Trans., 2012, 41, 2659–2666 This journal is © The Royal Society of Chemistry 2012
Dow
nloa
ded
by I
ndia
na U
nive
rsity
- P
urdu
e U
nive
rsity
at I
ndia
napo
lis o
n 10
Oct
ober
201
2Pu
blis
hed
on 1
6 Ja
nuar
y 20
12 o
n ht
tp://
pubs
.rsc
.org
| do
i:10.
1039
/C2D
T11
426G
View Online





























