Embed Size (px)
Citation preview

Functional Genomics of Plant Phosphorylation

Bioinformatics
Michael Gribskov
Douglas W. Smith

Bioinformatics
• Develop a plant phosphorylation database
• Completely classify and annotate plant protein kinases and phosphatases
• Develop data models and implement data handling procedures for experimental subprojects

Bioinformatics
• Timeline– Year 1: Implement Plant PP Functional Genomics Database
Data design and implementation for knockouts projectClassify Kinase and Phosphatases
– Year 2: Data design and implementation for proteomicsContinue knockout acquisitionScreen completed genome for additional targetsClassify and annotate functional domains
– Year 3: Data design and implementation for interactionsContinue knockout and add proteomics data acquisitionClassify and annotate functional domains
– Year 4: Complete knockoutsMap A. thaliana functional data to other plant genomesIntegrate expression data from external sourcesContinue proteomics and interaction data acquisition
– Year 5: Continue proteomics and interaction data acquisitionUpdate and extend classifications

Experimental Subprojects
• Gene Knockouts
– Isolate knockout mutations for all the protein kinases and phosphatases encoded by the arabidopsis genome
• Proteomics
– Create a two-dimensional gel phosphoprotein database
– Integrate the phosphoprotein database with gene sequences
• Interactions of Signaling Components
– Develop three-hybrid and split-hybrid screens for the analysis of plant protein kinases
– Begin genome-wide screening with individual arabidopsis protein kinases using the three-hybrid and split-hybrid approaches

Communication
• Electronic communication will be used to facilitate interactions between PPP functional genomics personnel
• Features of COW
– Responses organized by Topics
– Topics permit detailed conversations focused on specific PPP issues or projects
– Provides archive of conversations
– Coordination & management of issues
– Text and HTML possible: Web links to data

COW Interface and PPP Conference:

Tools and Approaches
• Bioinformatics group will draw on a broad range of community experience and resources.
• Resources developed at UCSD include
– Protein Kinase Resource
– Molecular Information Agent
– Family Pairwise Search
– Molecular Pattern Recognition
• Profile/MEME/MAST
– DictyDB
– INFO

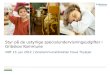
Plant PP Functional Genomics Database
• http://www.sdsc.edu/mpr/plant_p
• Data definitions in STAR for sequences and features derived from Protein Kinase Resource
• Relational database (MySQL)
• Currently populated with 850 plant kinases and kinase substrates
• Preliminary classification into Hanks and Quinn groups

Plant PP Functional Genomics Database

Plant PP Functional Genomics Database
Plant PP Functional Genomics Database

Plant PP Functional Genomics Database

Sequence
UIDName, alternatesProperties
Sequence
SourceURLDates
Features List
Feature 1Feature 2
Feature 3Feature 4PositionsRanges
Feature
UID Name
Feature Members List
Sequence 1PositionsRanges
Sequence2PositionsRanges
Sequence 3PositionsRanges
Sequence 4PositionsRanges
AlignmentMethod
PKR/PPFGDB Data Model

PKR/PPFGDB Data Model
• STAR data definition language
– Used for CIF and mmCIF
– Data model for PDB at SDSC/Rutgers/NIST
• Sequence and features dictionaries include methods (PERL) to derive data from online sources (SwissProt, PIR, NCBI)

Plant Phosphoprotein Functional Genomics Database
• 850 sequences - all matches to keyword “protein kinase” in Entrez
– 770 kinases
– 80 other (mostly kinase substrates)
• Classification (Hanks and Quinn, 1994)– AGC (9) 72
I VIII Other1 46 25
– CAMK (3) 204I II Other177 16 11
– CMGC (6) 157I II III IV V54 45 35 9 14
– PTK (23) 23
– OPK (14) 297II IV VIII X XII Other7 10 5 251 5 19
– Unassigned 17

Plant Phosphoprotein Functional Genomics Database
• Classification
– 1449 kinases from PKR
– Remove very similar sequences using WU Purge program
• 141 probe sequences
– Use FPS to calculated P-value of match to Kinase, class, and subclass
• 141 x 1850 SW comparisons on Biocellerator

Family Pairwise Search (FPS)
• Combines information from multiple queries
– Identifies family membership based on a known panel
• Does not require multiple alignment or “training”
• Identifies motifs and folds using known panel
– SCOP families
– PROSITE motifs
– identifies homologs based on similarity to the entire group of sequences
• Not sensitive to spurious matches
– Sensitive “product of P-values” statistic and effective family size
• Server fps.sdsc.edu

Plant Phosphoprotein Functional Genomics Database
• Fibronectin type-iii domain• Sh2 domain• Sh3 domain• Phorbol-ester and DAG binding domain• Ig-like domain• C2 domain• Pleckstrin homology domain• Lim (lin-11 isl-1 mec-3) domain• Dhr domain• Guanylate kinase domain• Phorbol-ester and dag binding domain• F5/8 type c (phospholipid-binding) domain• Dimerization and phosphorylation domain• Transmitter domain • Calmodulin-binding domain• Phospholipid binding domain• P21-binding domain• Nuclear localization signal • Egf-like domain• Polo-homology domain• Collagen-like• Death domain
• Gap domain• Myosin domain• Cbs domain• Sam domain• Fha domain• Cub domain• Actin-binding domain• Rii-beta subunit binding domain• P10 binding site domain• Drbm domain• Receiver domain • Pac motif• Zinc-fingers• Mads domain• Metallopeptidase domain• Heat repeats domain
• Leucine-rich repeats• Coiled coil • Many low entropy regions
Domains and Features found with Kinase catalytic domains

Protein Kinase Resource
• Http://www.sdsc.edu/Kinases

Protein Kinase Resource
• PKR Goal
– Integrate sequence, structure, genetics, function and disease information
• How many kinases are there?
– Currently about 4500 kinase sequences (approximately 2-fold redundancy)
– 50+ three-dimensional structures

Protein Kinase Resource

Protein Kinase Resource

Protein Kinase Resource
• CAMK II

Protein Kinase Resource

Protein Kinase Resource
• The SH3 (Src Homology 3) domain is a small conserved sequence of about 60 amino acid residues that interacts with proline-rich peptides to form protein aggregates.
• Structurally, the SH3 domain folds as a compact beta-barrel of five to six anti-parallel beta-strands. The hydrophobic beta-strands are connected by hydrophilic loops to form two orthogonal beta-sheets bringing the amino and the carboxyl termini of the domain close to each other. The ligands of the SH3 domains are peptides containing a ten residue consensus sequence, XPXXPPPFXP (where X is any amino acid residue, F is phenylalanine and P is peoline). This peptide forms a left-handed polyproline (PPII) helix that lies along the binding site of the SH3 domain, with its prolines interacting with the aromatic residues on the hyrophobic face of the SH3 domain.
• Functionally, the SH3 domain is involved in cell-cell communication and signal transduction from the cell surface to the nucleus. It acts as part of an adapter molecule and recruits downstream proteins in a signalling pathway. For example, in the eye development pathway in the Drosophila (Sevenless Pathway), a ligand from the R8 cell, Boss (Bride of Sevenless), binds two molecules of Sev (Sevenless) receptors on the surface of the R7 cell. This binding dimerizes the receptors which are Protein Tyrosine Kinases, so now they are close to one another and can transphosphorylate each other.

STAR Formatted Data
A Sample Data Block Using the Sequence Dictionary

STAR Data Block (Sequence)
data_ABL1_HUMANloop__sequence.id_sequence.type_sequence.name_sequence.date_create_sequence.update_sequence_sequence.update_annotation_sequence.synonym_sequence.citation_sequence.length_sequence.mol_weight_sequence.sequencePKRPSEQ000008PROTEINABL1_HUMAN1986-07-211990-04-011997-02-01'ABL1, ABL'; RN [1] RP SEQUENCE FROM N.A. RC TISSUE=FIBROBLAST; RX MEDLINE; 90082420. [, Geneva] RA FAINSTEIN E., EINAT M., GOKKEL E., MARCELLE C., CROCE C.M., RA GALE R.P., CANAANI E.; RL ONCOGENE 4:1477-1481(1989).;1130122944; MLEICLKLVGCKSKKGLSSSSSCYLEEALQRPVASDFEPQGLSEAARWNSKENLLAGPSE NDPNLFVALYDFVASGDNTLSITKGEKLRVLGYNHNGEWCEAQTKNGQGWVPSNYITPVN SLEKHSWYHGPVSRNAAEYLLSSGINGSFLVRESESSPGQRSISLRYEGRVYHYRINTAS DGKLYVSSESRFNTLAELVHHHSTVADGLITTLHYPAPKRNKPTVYGVSPNYDKWEMERT DITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQ LLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFI;

STAR Data Block (uid)
loop__uid.id_uid.status_uid.date_create_uid.update_uid.keywords_uid.description_short_uid.description_longPKRPSEQ000008ACTIVE1997-08-41997-08-4;TRANSFERASE, TYROSINE-PROTEIN KINASE, PROTO-ONCOGENE, ATP-BINDING,PHOSPHORYLATION, SH2 DOMAIN, SH3 DOMAIN, CHROMOSOMAL TRANSLOCATION,3D-STRUCTURE, ALTERNATIVE SPLICING;;PROTO-ONCOGENE TYROSINE-PROTEIN KINASE ABL (EC ) (P150)(C-ABL);;CC -!- CATALYTIC ACTIVITY: ATP + A PROTEIN TYROSINE = ADP +CC PROTEIN TYROSINE PHOSPHATE.CC -!- SUBCELLULAR LOCATION: CYTOPLASMIC.CC -!- TISSUE SPECIFICITY: WIDELY EXPRESSED.CC -!- DISEASE: PARTICIPATES IN A T(9;22)(Q34;Q11) CHROMOSOMALCC TRANSLOCATION THAT PRODUCES A BCR-ABL ONCOGENE RESPONSIBLE FORCC CHRONIC MYELOID LEUKEMIA (CML), ACUTE MYELOID LEUKEMIA (AML), ANDCC ACUTE LYMPHOBLASTIC LEUKEMIA (ALL).CC -!- ALTERNATIVE PRODUCTS: TWO FORMS, IA AND IB HAVE ALTERNATIVE AMINOCC TERMINI.CC -!- SIMILARITY: TO OTHER PROTEIN-TYROSINE KINASES IN THE CATALYTICCC DOMAIN. BELONGS TO THE ABL SUBFAMILY.CC -!- SIMILARITY: CONTAINS A COPY EACH OF THE SH2 AND SH3 DOMAINS.;

STAR Data Block (xref)
_xref.id PKRPSEQ000008loop_ _xref.dbname _xref.primary _xref.acc _xref.name _xref.date _xref.type _xref.source_db _xref.source_accession _xref.comment SWISS yes P00519 ABL1_HUMAN 1997-02-01 PROTEIN . . 'blah blah' EMBL no X16416 . 1997-02-01 NUCLEIC SWISS P00519 . EMBL no M14752 . 1997-02-01 NUCLEIC SWISS P00519 . EMBL no S69223 . 1997-02-01 NUCLEIC SWISS P00519 . PIR no A25582 TVHUA 1997-02-01 PROTEIN SWISS P00519 . PDB no 1AB2 1AB2 1997-02-01 STRUCTURE SWISS P00519 . PDB no 1ABL 1ABL 1997-02-01 STRUCTURE SWISS P00519 . OMIM no 189980 . 1997-02-01 NUCLEIC SWISS P00519 .

STAR Data Block (feature table)
_feature_table.id PKRPSEQ000008loop_ _feature_table.feature_id _feature_table.feature_name _feature_table.feature_location _feature_table.feature_type _feature_table.feature_source PKRFEAT85 SP-SH3 61-121 DOMAIN PRIMARY PKRFEAT86 SP-SH2 127-217 DOMAIN PRIMARY PKRFEAT87 SP-PROTEIN_KINASE 242-493 DOMAIN PRIMARY PKRFEAT88 SP-NUCLEAR_LOCALIZATION 605-609 DOMAIN PRIMARY PKRFEAT89 SP-PRO-RICH 782-1019 DOMAIN PRIMARY PKRFEAT90 SP-ATP 248-256 SITE PRIMARY PKRFEAT91 SP-ATP 271-271 SITE PRIMARY

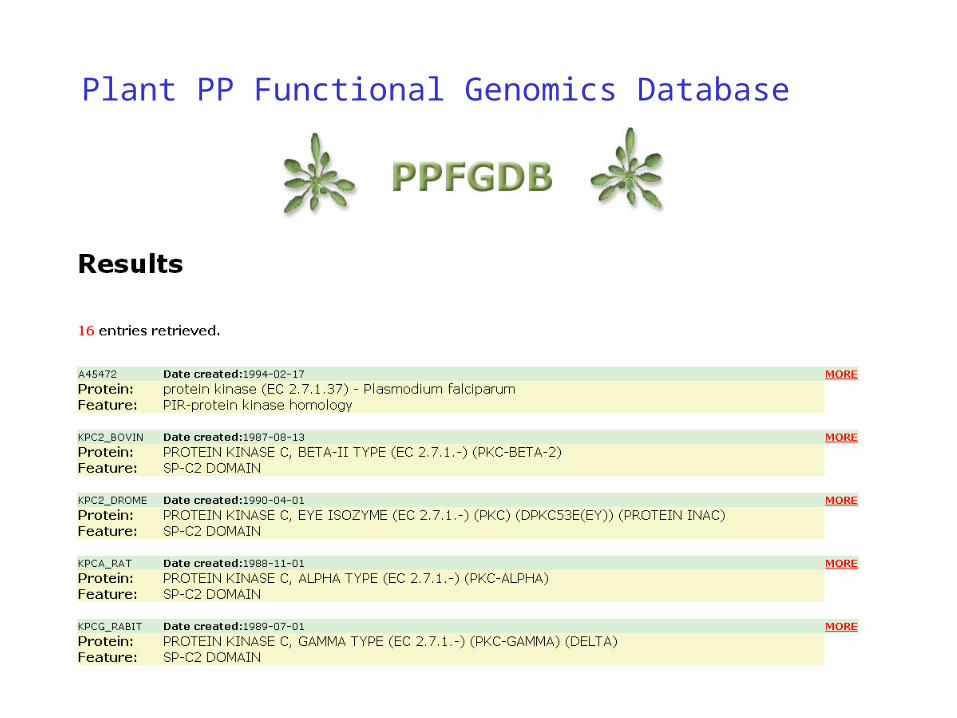
Molecular Information Agent
Transparently Linking Electronic Data Resources

Molecular Information Agent
• MIA - Molecular Information Agent
– Web resources often change URLs
– Web resources often change available services
– Laboratory scientists may only use services occasionally
• May not know what services are available
• May not know how to efficiently use them
• Want quick answers (where possible)

Molecular Information Agent
• MIA - Molecular Information Agent
– Search for linked information starting from a single item
– Automatic location and validation of web links
• All links are visited and the presence of a usable page confirmed
• Useful information extracted and used as a basis of further searches
– Basis of sequence/structure/information crosslinking in MSD

TemplateResource
QueryParser
Query Manager Resource
ParserKeywords
Synopsis &Reference
Molecular Information Agent

PATH_MAP_NUM
DSSP
IMB JenaImage
PDBSum
PDB
BiochemicalPathways
PDB Report 3Dee ProMotif
Protein Motions
ProCheckMacromolecular
File
STING GRASSColumbia
Pic
CATH FSSP SCOP
TOPS
MMDBPDB_CODE
EC_UNPRECISE
EC_PRECISE
ENZYME
WIT Swiss2D PAGE
SwissModel
SEView
PeptideMass
ProtScale
Swiss 3dImage
Compute pI/MW
ProfileScan
DOMO
ProtPattern ProDom
ProtoMap
Swiss-Prot
Swiss-Prot
Scan PROSITE
SP_Name
SP_ID
PROSITE
GI_Nuc
GI_PROT
NCBI
PRODOC
NCBI_UID
MGI
EMBL_UID
Prosite
MouseGenome DB
EMBL
Medline
ProDocOMIM
GDB
GeneCards
OMIM
OMIM
GENE_SYM
BLAST
PIR_IDPIR_ENTRY
PIRPIRProClass
NCBITaxonomy
NCBI_TAXONOMY

Motifs/Domains
Other
Sequence
Structure
NCBI
EMBL
SWISS_PROT
PIR
PDB
SCOP
CATH
Medline
OMIM
FSSP
HSSP
PROSITE PRODOM
PROTOMAP
DOMO
EC Enzyme
TGD
DSSP
MMDB
WIT
MIA - Selected Data Resources

Molecular Information Agent
• Limit Searches
• Do not requery resources
• Time-outs and availability issues

MIA - Molecular Information Agent
– Problems
• Web resources often change URLs
• Web resources often change available services
– Laboratory scientists may only use services occasionally
• May not know what services are available
• May not know how to efficiently use them
• Want quick answers (where possible)

MIA - Molecular Information Agent
• Simplifies finding and using information
– Search for all linked information beginning from a single item
– Automatic location and validation of web links
• All links are visited and the presence of a usable page confirmed
• Useful information extracted and used as a basis of further searches
– Basis of sequence/structure/information crosslinking in PDB

Query: Gene Name
ProteinSequences
DNASequences
Motifs andDomains

Motifs andDomains
PhysicalParameters
LiteratureReferences
Images, Genes,Taxonomy ...

Macromolecular Pattern Recognition
Profile Analysis, MEME & MAST

Molecular Pattern Recognition
Genes
Sequences
AlignedSequences
Motifs
FKEAFSLFD
KDGDGTITTK
ELGTVMRSL
FKEAFSLFDKDGDGCITTKELGTVMRSLIREAFRVFDKDGNGYISAAELRHVMTNLIKAIIQKADANKDGKIDREEFMKLIKS.IDAIIKKADGNNDGKIRVQEFVKMIESSFNKAFELYDQDGDGYIDENELDALLKDL
FKEAFSLFDKDGDGCITTKELGTVMRSL
IREAFR
VFDKDG
NGYISA
AELRHV
MTNLI
KAII
QK ADANKDG KI
DREE F
MKLI
KS.
FNKAFELYDQDGDGYIDENELDALLKDL

Molecular Pattern Recognition
MCEG FKEAFSLFDKDGDGTITTKELGTVMRSLMCDO FKEAFSLFDKDGDGSITTKELGTVMRSLMCSP FKEAFSLFDKDGDGCITTKELGTVMRSLMCCHM IREAFRVFDKDGNGYISAAELRHVMTNLMCUR1C IKAIIQKADANKDGKIDREEFMKLIKS.MCUR2C IDAIIKKADGNNDGKIRVQEFVKMIESSKLBOB FNKAFELYDQDGDGYIDENELDALLKDLKLCHI FNKAFEMYDQDGNGYIDENELDALLKDLKLBOI LDELFEELDKNGDGEVSFEEFQVLVKKIKLPGI LDDLFQELDKNGNGEVSFEEFQVLVKKI
Ligands to Calcium
Hydrophobic patches that stabilize structure
Calcium Binding EF-Hand Structure

Molecular Pattern Recognition
• Motif Learning/Description
– Profile
• Analytical calculation of motif description using finite mixture model
• Learning is fast - seconds on workstation
• Database search is slow - hours on workstation, about 90 sec on Compugen Bio-XLP
– MEME
• Unsupervised learning using expectation maximization
• Learning is quadratic - typically minutes on T3E
• Searching database is linear - seconds on workstation

Profile Analysis
• Describes protein structural and sequence motifs using a position specific scoring matrix and position specific gap penalties calculated from a sequence or multiple sequence alignment
• Evolutionary profile - sequence information
• Structural Profile - structural information
• Evolutionary/Structural Profile sequence and structure

Evolutionary Profile
• Mixture distribution using a biologically relevant model
• Explicit evolutionary model for each aligned column
– Sequences weighted for similarity
• Find the group of preferred residues at each position
• Weight mixture components by probability of observed data given the model distribution
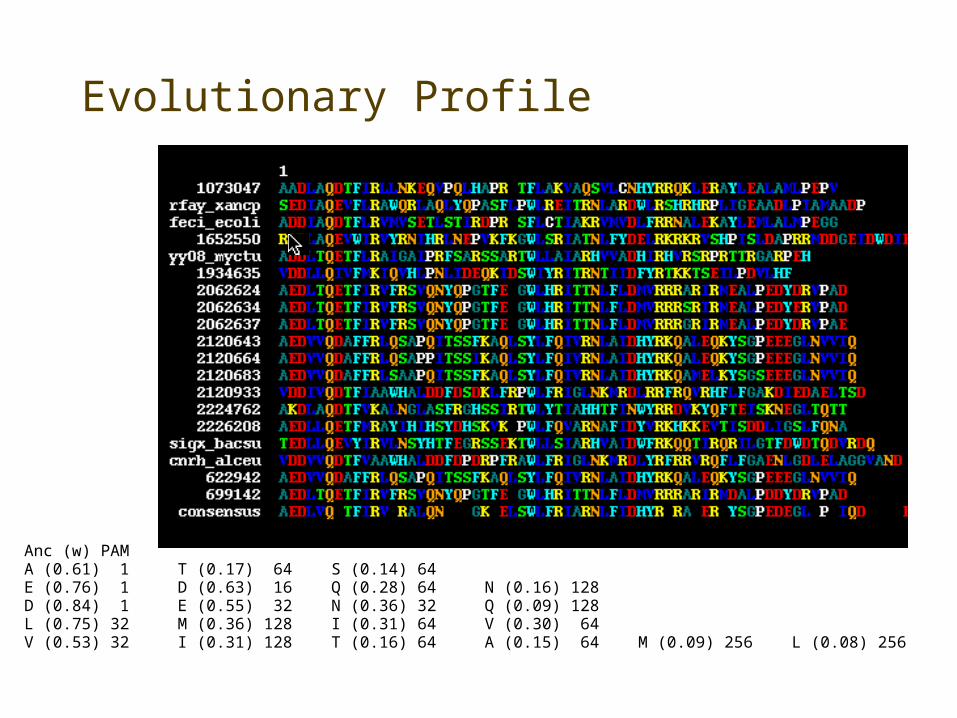
Anc (w) PAMA (0.61) 1 T (0.17) 64 S (0.14) 64E (0.76) 1 D (0.63) 16 Q (0.28) 64 N (0.16) 128D (0.84) 1 E (0.55) 32 N (0.36) 32 Q (0.09) 128L (0.75) 32 M (0.36) 128 I (0.31) 64 V (0.30) 64V (0.53) 32 I (0.31) 128 T (0.16) 64 A (0.15) 64 M (0.09) 256 L (0.08) 256
Evolutionary Profile

MAST/MEME
• Describes protein sequence motifs using a position specific scoring matrix derived from an unsupervised EM algorithm applied to unaligned sequences
• MEME - motif learning
• MAST - database searching
• Servers
– MEME/MAST
– ParaMEME - parallel MEME
– MetaMEME - multiple motif models)

MEME/MAST
• Expectation maximization
– Unsupervised learning
– Unaligned sequences
– Automatic pattern width determination
– Automatic extraction of multiple motifs
• Learning is quadratic in the size of training data - typically minutes on CRAY T3E
• Searching database is linear in length of database - seconds on workstation

A C D E F G H I K L M N P Q R S T V W Y 0.40 -6.97 2.23 3.16 -6.50 -4.35 -3.58 -5.13 -0.13 -5.04 -4.24 -3.35 -4.07 -1.69 -3.90 -3.70 -3.74 -4.35 -6.43 -5.43 -4.59 -4.83 4.20 -2.45 -5.37 -4.85 -3.79 -5.41 -5.13 -5.65 -5.06 -2.28 -5.62 -4.70 -5.05 -4.61 -5.05 -5.20 -5.05 -4.95 -4.28 -3.74 -7.04 -6.66 -3.22 -6.76 -6.22 1.56 -6.54 2.57 -2.01 -6.41 -6.43 -5.98 -6.49 -6.24 -4.25 2.02 -5.13 -4.96 1.71 -2.64 -5.41 -4.90 -2.97 -4.21 -4.04 -1.21 -4.64 0.75 -1.90 -4.40 -4.72 -4.27 -4.51 -3.43 1.97 2.23 -3.89 -3.56 -5.22 -5.32 -5.80 -4.06 -6.24 -5.91 -2.89 -6.23 -5.34 -5.44 -4.37 -5.05 -5.55 4.57 -4.72 -5.54 -5.56 -6.11 -5.29 -6.00 -2.99 -6.93 3.01 2.91 -6.50 -4.34 -3.59 -0.31 -2.63 -5.06 -4.24 -3.31 -4.08 -1.72 -3.93 -3.70 -3.75 -4.36 -6.44 -5.43 1.49 -2.76 -5.58 -5.67 -4.92 -3.44 -5.03 -3.16 -5.48 -4.51 -3.80 -4.34 -4.62 -4.79 -5.16 -2.35 3.20 1.66 -5.36 -5.41 -3.72 -2.67 -4.68 -4.69 4.40 -4.93 -3.37 -2.70 -4.71 -1.98 -2.24 -4.41 -4.39 -4.60 -4.72 -3.71 -4.35 -2.98 0.74 0.33 -4.25 -3.86 -6.79 -6.24 2.25 -6.20 -5.12 2.82 -5.96 0.95 2.14 -5.90 -5.93 -5.09 -5.65 -5.54 -4.21 0.76 -4.02 -4.10 -0.94 -3.01 -4.44 -4.17 -4.79 -4.21 -1.99 -4.15 0.00 -4.10 -3.65 -3.24 -3.84 -2.22 4.01 -3.77 -3.76 -4.63 -3.16 -4.12
bits 6.6 6.0 5.3 4.7 Information 4.0 content 3.3 * * * (28.2 bits) 2.7 * * * 2.0 *** ****** 1.3 ********** 0.7 ********** 0.0 ----------
Multilevel EDLVQETFIRconsensus D VA DA sequence T
MEME/MAST
• Log-Odds form of a MEME motif

MEME/MAST
• Aligned training data for one motif

Motif Width Consensus Sites
1 10 EDLVQDTFIR 19 Core Binding 2 16 WLYRITRNLFIDHYRR 19 Strand Opening 3 36 REAFVLCDIHGLSYKEIAKTLGVSLGTVRSRIHRAR 19 -35 region 4 24 PSPEQQYHNYETLECIQAALDELP 19 ? Region 3.2 5 11 WLAYRLTGCQH 18 ? Region 1 6 10 QNYQPGTFEG 18 -10 region 7 14 RCDMPSWDELVRQH 17 8 15 MQAMPQDYDSFPADE 16 9 20 HWREPSDEPQGTAVFDATGD 8 10 8 LHALPDHS 12 11 8 MAEQLSTS 12.8
MEME/MAST
• RNA polymerase sigma factor motifs
• Top motifs are all known biologically important regions!

DictyDB
ACeDB Based Interface to Genomic Data
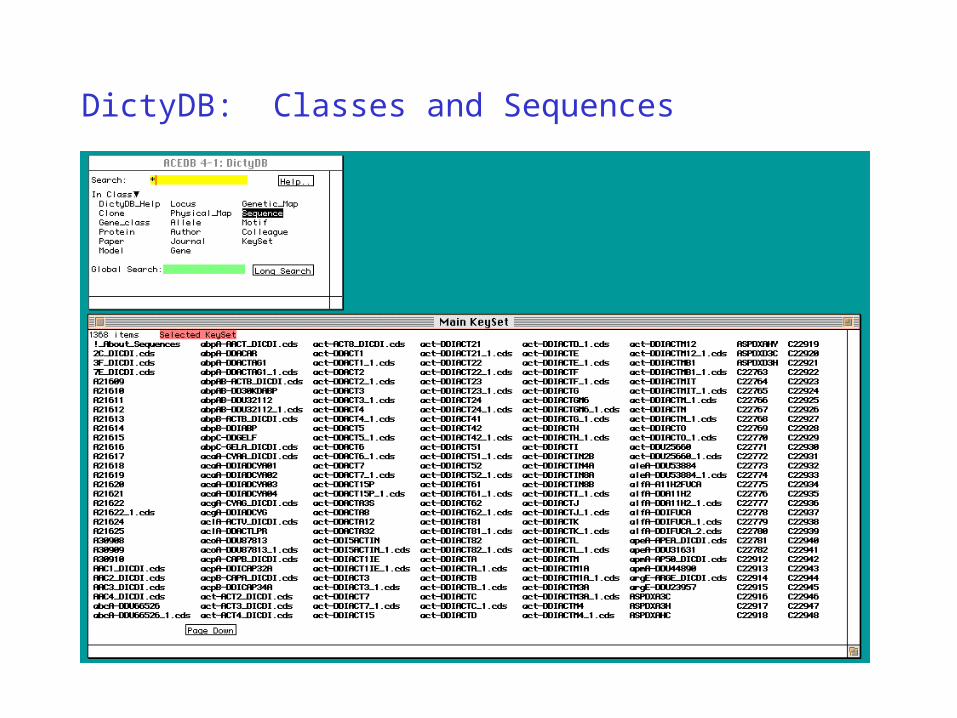
DictyDB: Classes and Sequences
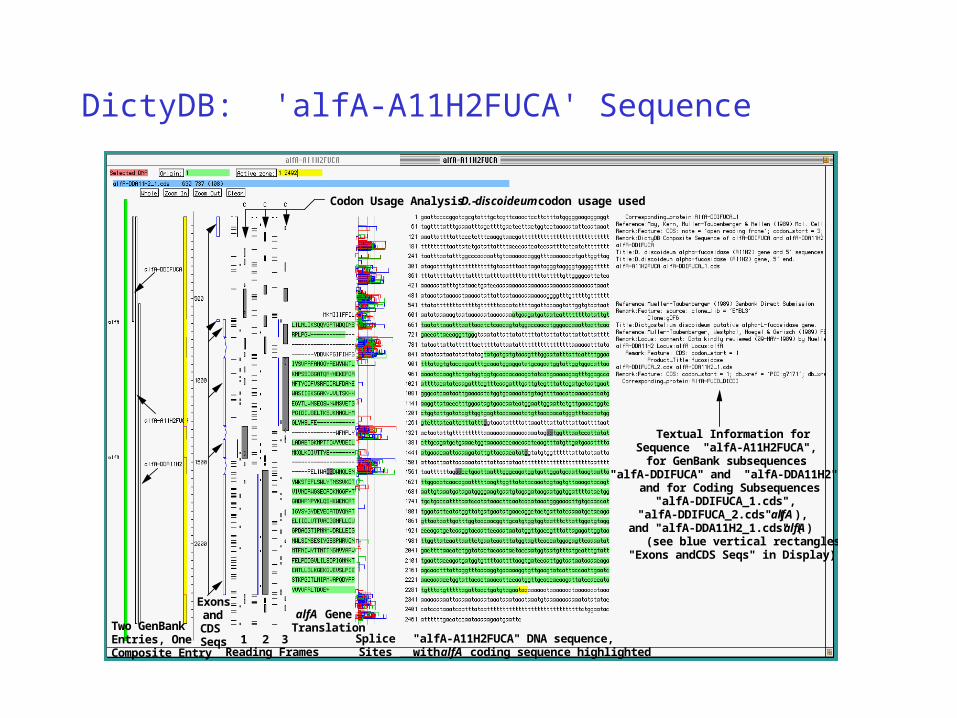
DictyDB: 'alfA-A11H2FUCA' Sequence
Reading Frames1 2 3 Splice
Sites
Codon Usage Analysis - D. discoideum codon usage used
Exons and CDS Seqs
alfA Gene Translation
"alfA-A11H2FUCA" DNA sequence, with alfA coding sequence highlighted
Two GenBank Entries, One Composite Entry
Textual Information for Sequence "alfA-A11H2FUCA",
for GenBank subsequences "alfA-DDIFUCA" and "alfA-DDA11H2",
and for Coding Subsequences "alfA-DDIFUCA_1.cds",
"alfA-DDIFUCA_2.cds" (alfA), and "alfA-DDA11H2_1.cds" (alfA)
(see blue vertical rectangles "Exons andCDS Seqs" in Display)
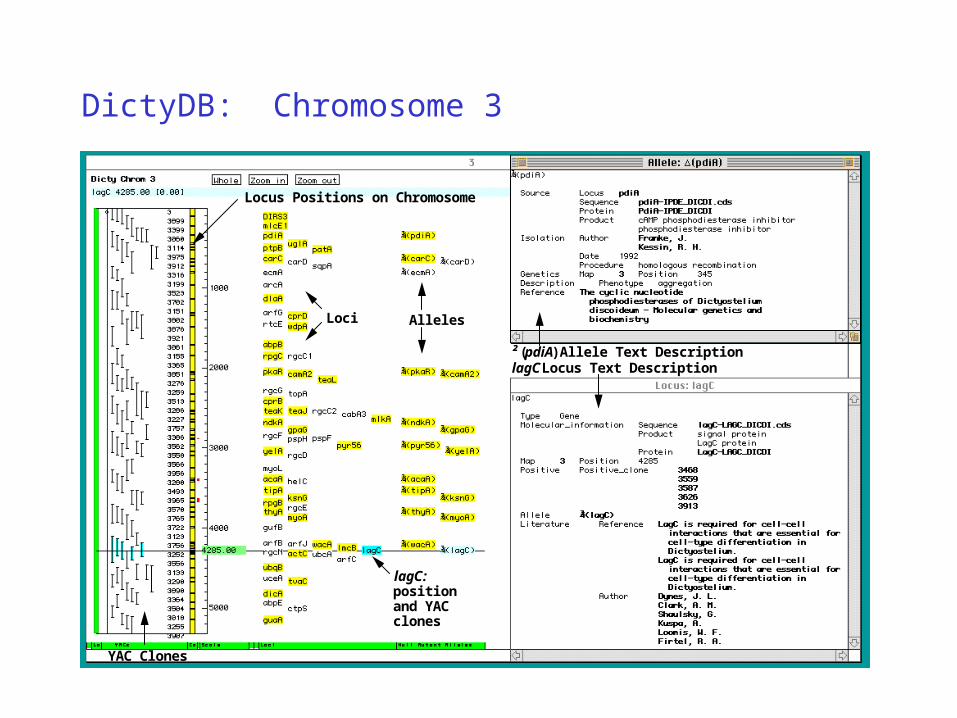
DictyDB: Chromosome 3
lagC: position and YAC clones
Locus Positions on Chromosome
AllelesLoci
lagC Locus Text Description² (pdiA) Allele Text Description
YAC Clones

DictyDB: Sequence and Protein Text

DictyDB Home Page:

DictyDB: Size Increases

DictyDB Web Objects:

DictyDB via Webace: Main Pages

DictyDB via Webace: Graphics

Genomic View: DictyDB and SGD

Genomic View Map Displays:

Java Genomic View Interface:

INFO
Interruption Finder and Organizer

INFO: INterruption Finder and Organizer
• Identifies interruptions in one sequence relative to DB sequences
• Task 1: DNA coding sequence vs Protein Databases
• Task 2: Alternative Splice Sites
• Task 3: Protein Secondary Structure vs alpha helix/beta strand Databases

INFO: Coding Sequence Algorithm
• Input DNA translated into 6 frames
• Each 25 aa Window of each translate compared to each 25 aa Window of each DB entry via ktup and Distance Matrix
• Hits above Min Score kept; overlapping Hits to given DB entry comprise a Cluster
• Overlapping Clusters against many DB entries comprise a MegaCluster
• End points of MegaCluster define Exon / Gene

INFO: Three Runs refine Results
• Run 1: High Min Score - no False Positives
• MiniLibrary built from DB entry Hits of Run 1
• Run 2: Low Min Score -run against MiniLibrary - high Specificity - refine MegaCluster ends
• Run 3: Reverse Sequence - Sequences of input translates and MiniLibrary entries are reversed - refines Leading Edge end points
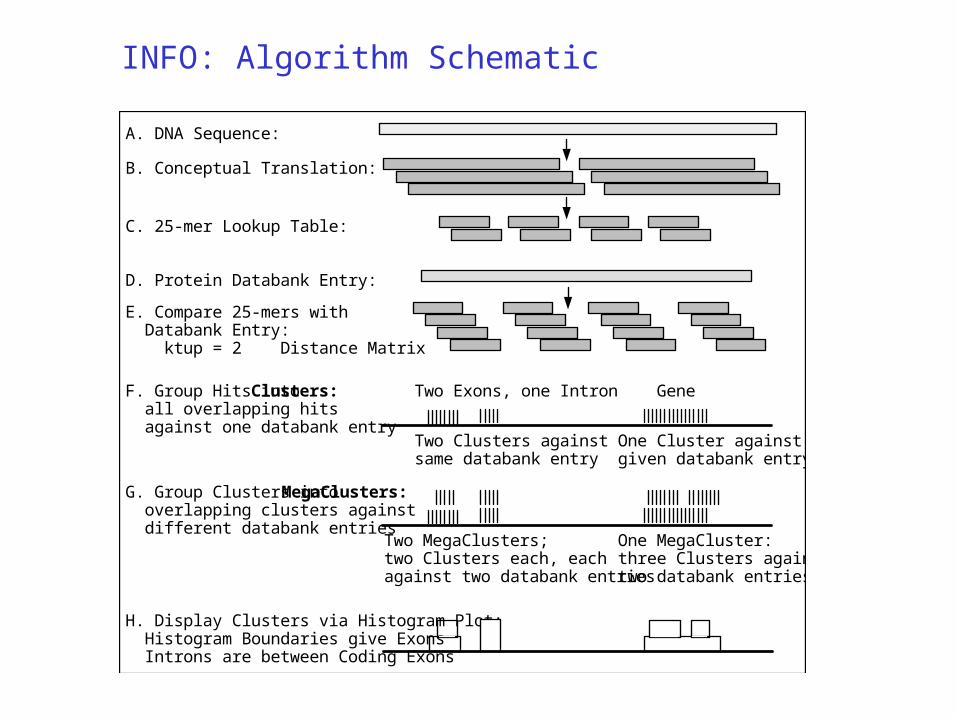
INFO: Algorithm Schematic
A. DNA Sequence:
B. Conceptual Translation:
C. 25-mer Lookup Table:
D. Protein Databank Entry:
E. Compare 25-mers with Databank Entry: ktup = 2 Distance Matrix
F. Group Hits into Clusters: all overlapping hits against one databank entry
Two Clusters against same databank entry
One Cluster against given databank entry
GeneTwo Exons, one Intron
G. Group Clusters into MegaClusters: overlapping clusters against different databank entries
Two MegaClusters; two Clusters each, each against two databank entries
One MegaCluster: three Clusters against two databank entries
H. Display Clusters via Histogram Plot: Histogram Boundaries give Exons Introns are between Coding Exons
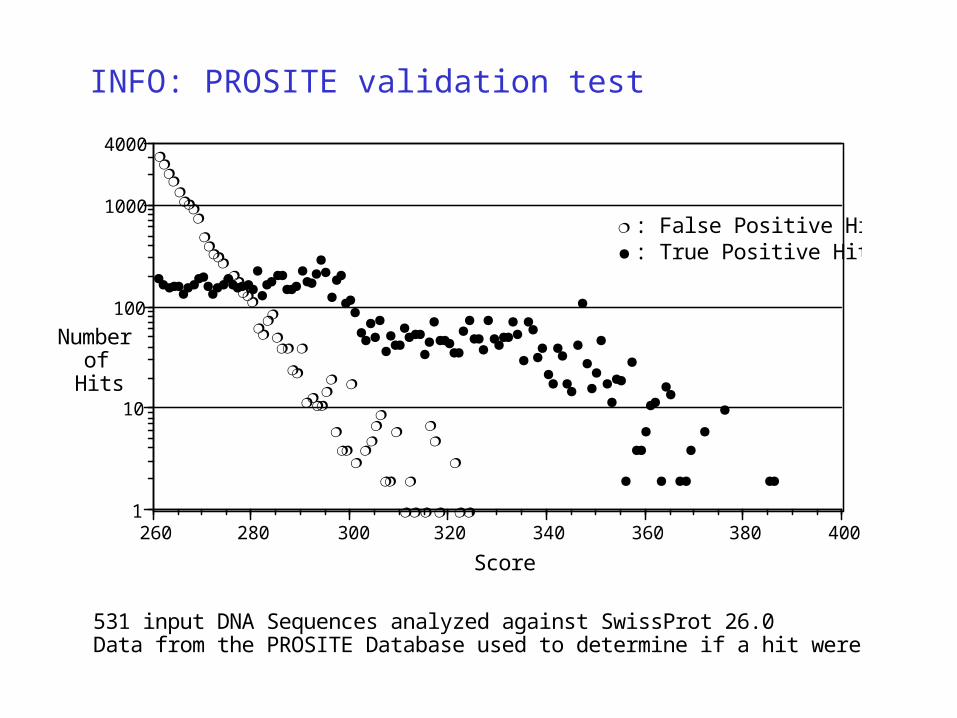
INFO: PROSITE validation test
1
10
100
1000
4000
260 280 300 320 340 360 380 400
Score
Number of
Hits
: True Positive Hits
: False Positive Hits
531 input DNA Sequences analyzed against SwissProt 26.0 Data from the PROSITE Database used to determine if a hit were Positive or Negative

INFO: Exon Sizes reported
0
20
40
60
80
0 200 400 600 800 1000 0 50 100 2001500
0.2
0.4
0.6
0.8
Exon Size (bases)
1.0
Exon Count Fraction Found per 10 Bases33
33 75 bases

INFO: Three Human ExamplesC
lust
er
Count
0
4
8
0 4 8 12 16
HUMPAIA(J03764)
0
4
8
0 4 8 12
HSFESFPS(X06292)
0
4
8
0 10 20 30
HUMTHB(M17262)
Sequence Position (kb)

INFO: Splice Junction Precision

INFO: Three Alternative Splicings
0 1 2 3 4 5
Peripherin-2:
Peripherin-1:
0 1 2 3 4 5
Sequence Position (kb)
Peripherin-3:
0 1 2 3 4 5
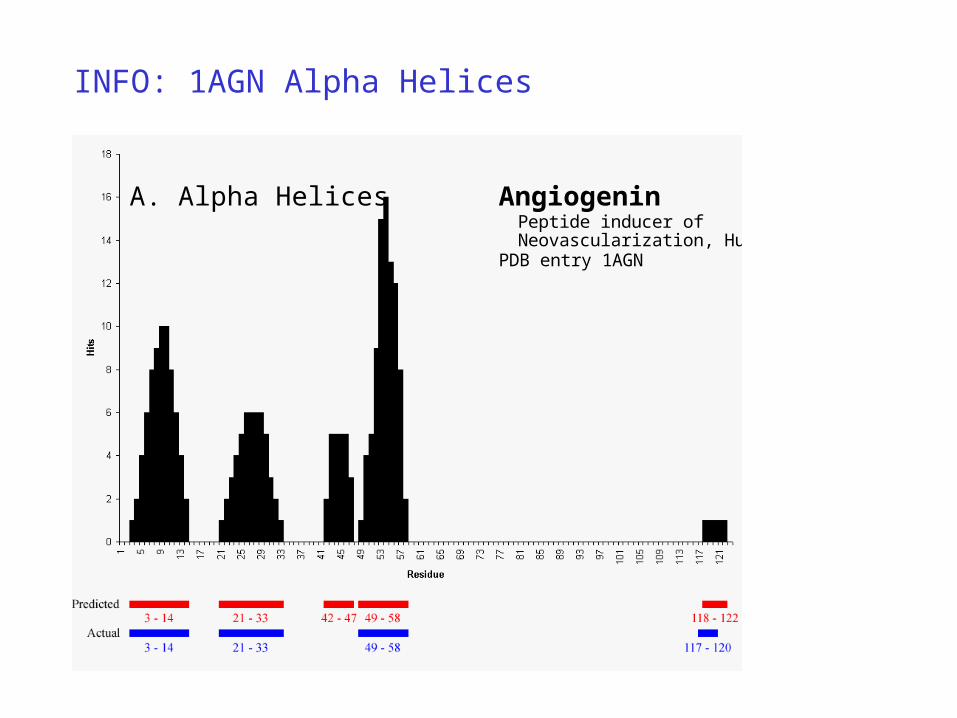
INFO: 1AGN Alpha Helices
A. Alpha Helices AngiogeninPeptide inducer of Neovascularization, Human
PDB entry 1AGN

INFO: 1AGN Beta Strands
B. Beta Strands AngiogeninPeptide inducer of Neovascularization, Human
PDB entry 1AGN

Extra UCSD icons