Embed Size (px)
Citation preview



Genellikle LN’larından ve bazende herhangi bir
organdan köken alan (ekstra nodal), heterojen bir
grup B veya T hücre malignitesidir.
Sıklık:
› Tüm malign neoplastik hastalıkların yaklaşık %3-4'ünü
oluşturur.



1994’de REAL sınıflamasında lenfoid maligniteler
morfolojik, immünolojik ve genetik tekniklerle B-hücreli,
T hücreli ve Hodgkin hastalığı olarak 3 kategoride
tanımlandı.
2001 yılında WHO sınıflaması Lenfomaları gelişmiş
morfolojik, immünolojik ve genetik tekniklerle B-hücreli,
T hücreli olarak yeniden tanımladı.
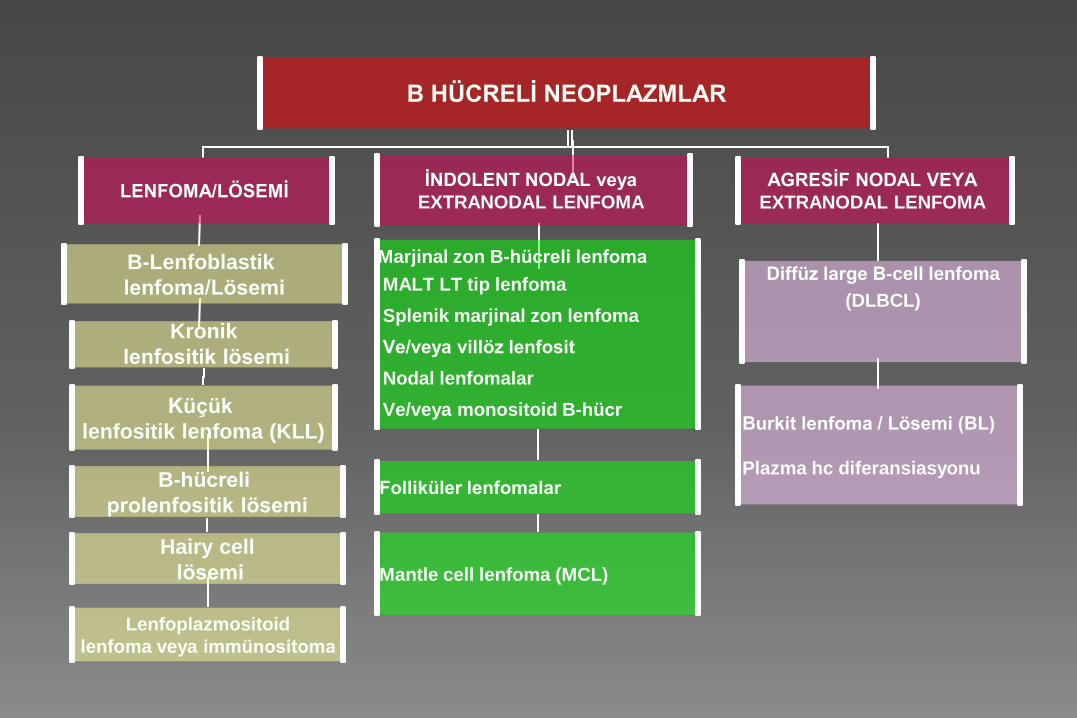
B HÜCRELİ NEOPLAZMLAR
LENFOMA/LÖSEMİ İNDOLENT NODAL veya
EXTRANODAL LENFOMA
AGRESİF NODAL VEYA
EXTRANODAL LENFOMA
B-Lenfoblastik
lenfoma/Lösemi
Marjinal zon B-hücreli lenfoma
MALT LT tip lenfoma
Splenik marjinal zon lenfoma
Ve/veya villöz lenfosit
Nodal lenfomalar
Ve/veya monositoid B-hücr
Diffüz large B-cell lenfoma
(DLBCL)
Kronik
lenfositik lösemi
Küçük
lenfositik lenfoma (KLL)
Folliküler lenfomalar
Mantle cell lenfoma (MCL)
B-hücreli
prolenfositik lösemi
Hairy cell
lösemi
Lenfoplazmositoid
lenfoma veya immünositoma
Burkit lenfoma / Lösemi (BL)
Plazma hc diferansiasyonu

T HÜCRELİ NEOPLAZMLAR
LENFOMA/LÖSEMİ NODAL veya
EXTRANODAL LENFOMA
T-Lenfoblastik
lenfoma/Lösemi
Prolenfositik lösemi
Large granüler
lenfosit lösemi
T-hücreli, NK-hücre
veya
gamma/delta T-hücreli lenfoma
CD30+ T cell cutanöz
lenfoproliferatif sendrom
NK hücreli lösemi
Mikosis fungoides
&
Sezary sendromu
Periferal T-hücreli lenfoma
Anaplastik large cell lenfoma
Lenfoma/Lösemi-
HTLV-1infeksiyona sekonder
HODGKİN LENFOMA
Lenfositden zengin
nodüler HL
Klasik varyant
Lenfosit zengin
Nodüler sklerozis
Miks –hücreli
Lenfosit fakir



Klinik inceleme
› PS (ECOG veya karnowski)
› Periferik LN
› Dalak ve kc büyümeleri
› B semptomları
Laboratuvar inceleme
› Radyoloji
Toraks, abdomen ve pelvik BT
Tümör kütlelerinin ölçülmesi
› Kİ aspirasyon ve biopsi
› Lomber ponksiyon
› Tedavi öncesi rutin laboratuvar incelemesi
CBC, PY
Kc, renal fonksiyonlar
Serum LDH
Serum B2 Mikroglobulin
› Serum protein elektroforezi
› HBV, HCV, HIV taraması

Lenfoma tanısı koymak için LN
veya extranodal tutulan yerden
biopsi şarttır.
Histopatolojik tanı biyopsi ile
konur.
› Eksizyonel biopsi tercih edilir.
› İnce iğne aspirasyon biopsisi bazı
durumlar dışında tanı için
yetersizdir
Kİ tutulumu olan intermediate
veya high grade lenfomalarda
SSS tutulumu yüksektir ve
bunlarda LP yapılmalıdır.




Tüm lenfomaların %20-25’ni oluşturur.
Hastalık genellikle yüzeyel ve derin LN’da büyüme, splenomegali ve kemik iliği
infiltrasyonu ile birliktedir.
Hastaların %80-90’ında tanı anında yaygın hastalık bulunur.
› Yaygın hastalık tablosu içinde en sık tutulan ekstranodal organ kemik iliğidir.
› Kemik iliği tutulumu olguların %75’inden daha fazlasında bulunur.
Kemik iliği tutulumu genellikle paratrabeküler bir patern gösterir.
› Tanı anında çevresel kan tutulumu %30’undan azında bulunur.
Nadiren tanı anında ekstranodal tutulum görülebilir.
Ender görülen bölgesel LAP (epitroklear, popliteal) rastlanabilir.
Folliküler lenfomalarda beklenen yaşam süresi 6-10 yıldır.


Tüm lenfomaların %5-8’ini oluşturur.
Erkeklerde ve 60 yaş civarında sıktır.
Genellikle tanı anında yaygın hastalık gözlenir; hastaların çoğunda
LN’ları ve dalakta aşırı büyüme, waldeyer halkası, kemik iliği ve kanda
infiltrasyon ve özellikle GIS (lenfomatöz polipozis) olmak üzere
ekstranodal tutulumla birliktedir.
Hastalık sıklıkla tedavi direnci ve nüksleri takiben giderek agresif bir hal
alır.


WHO sınıflamasında MZL’ler üç klinik tablo şeklinde yer alır: › Ekstranodal = MALT lenfomalar,
› Nodal lenfomalar (±monositoid B hücre)
› Splenik lenfoma (±villöz lenfositler)
› Bununla birlikte, yaygın hastalık tabloları da oluşabilir. Klinik olarak ilerlemiş hastalığa işaret ederler.
Splenik alt grup: › Tutulum alanı dalak ve buna sıklıkla eşlik eden kemik iliği ve periferik kandır.
Lösemik alt grup: › Sadece kemik iliği ve periferik kan tutulumu gösterir.
Bu hastalarda splenik lenfomaya dönüşüm sıktır.
Nodal alt grup: › Hastalık LN tutulumu ile karakterizedir (çok sınırlı veya yaygın). Kan tutulumu nadirdir.
Yaygın hastalık: › Bu olgular MALT, nodal ya da splenik alt grupların herhangi birinin geç evresinde görülebilir.


En sık görülen NHL tipidir.
Olguların %60-65 nodal, %35-40 ekstranodal (en sık mide, kemik, SSS,
böbrek ve testis) başlangıçlıdır.
Hastaların yaklaşık %30’unda hastalık lokalizedir.
Mediastinal ve abdominal tutulum sıklıkla büyük tümör kitleleri şeklinde kendini gösterir.
Bu lenfomanın invazyon yapma özelliği belirgindir, damar ve hava
yollarına bası yapabilir, periferik sinirleri tutabilirler.
Olguların %10-20’sinde kemik iliği tutulumu vardır ve bu olgularda SSS'ne
yayılma sık görülür.
Büyük hücreli lenfomaların çoğu denova gelişir
Bazıları yavaş seyirli lenfoma, folliküler lenfoma, mycosis fungoides, marjinal
zon-B-hücreli lenfoma, hatta Hodgkin hastalığına sekonder gelişmiş olabilir


Burkitt lenfoma çocuklarda sık görülmesine karşın (%40) erişkinlerde nadirdir (%5).
Erişkin olgular daha çok AIDS’li olgularda görülür.
Erkek/Kadın oranı= 2/1’dir
Endemik (Afrika tipi): yüz kemikleri, çok sıklıkla çene tutulumu görülür.
Non-endemik (avrupa/amerika): Karın bölgesi, çok sık olarakta distal ileum, çekum
ve/veya mezenter tutulum görülür.
Bazılarında ise over, meme, testis ya da periferik lenf bezi tutulumu vardır.
Kemik iliği ya da meningeal tutulumda prognoz çok kötüdür.
Kemik iliği tutulumu durumunda FAB sınıflamasına göre ALL-L3 olarak adlandırılır.
Afrikanın endemik bölgelerinde görülen olguların %100’ünde EBV infeksiyonu
bulunurken Avrupa ya da Amerikada görülen sporadik olgularda bu oran %20’dir.
Ayrıca AIDS ile ilişkili olgularda bilinmektedir, bu olgular genellikle plazmastoid
formdadır.

Ortalama başlangıç yaşı > 60, KLL’de medyan teşhis yaşı 72.
En sık görülen lösemi tipidir. İnsidans: 4.2/100.000/yıl
>80yaş ise insidans >30/1000000
%10 hastanın <55 yaş
Tüm lösemilerin %25-30’unu oluşturur.
Japonya, Çin ve diğer Asya ülkelerinde nadiren görülür.
.

KLL, matür ancak immünolojik olarak yetersiz B lenfositlerin sayısının artmasıdır. ( periferik kanda ≥5x109 /l)
Sıklıkla rutin kan testleri sırasında teşhis edilir.
Semptomlar: › Tekrarlayan enfeksiyonlar
› Anemi
› Kanama veya morluklar
› Otoimmün bozukluklar (en sık olarak OIHA ve daha az sıklıkta trombositopeni)
› Lenfadenopati
› Splenomegali
› Hepatomegali
Muller-Hermelink HK, et al. WHO Classification of Tumours 2001; 127-130.
..

Rutin tam kan sayımında lenfositoz >5x109 /L
Anemi: Başlangıçta olguların %15-20’sinde Hb<11g/dl
Trombositopeni: Başlangıçta olguların %10’unda <100.000/mm3
FM bulguları %20-30 normaldir
LAP veya HSM %40-50
Hastalığın ilerlemesi ile yaygın LAP ve HSM
LAP: lenfadenopati HSM: Hepatosplenomegali –karaciğer ve dalak büyümesi
.

Hastalığın seyrinin belirlenmesinde önemli
FISH (fluorescence in situ hybridization)
› KLL hücrelerinin kromozomal analizi
Kromozomal anormallik %82 Ortalama yaşam süresi
del13q %55 11.0 yıl
del11q %18 6.6 yıl
Trizomi12 %16 9.0 yıl
del17p % 7 2.5 yıl
..


IgVH gen mutasyonu
› KLL hücrelerinin Ig geninde mutasyon gelişmesi
CD38
› IgVH gen mutasyonu yok ise CD38
› CD38, IgVH gen mutasyonu göstergesi değil
ZAP70
› ZAP-70 tirozin kinazla ilişkili bir T-hücre reseptörü ve labil bir
protein yapısındadır.
Beta2 mikroglobulin: tümör yükünü yansıtır
› hastalığın hızlı ilerlediğini ve kısalmış yaşam süresini gösterir
BCL-2 aşırı ekspresyonu
› Antiapopitoz etkisi nedeniyle malign B hücre birikimi olur
..


**Cildin en sık görülen primer lenfomalarıdır.
Mikozis fungoides, Sezary sendromu ve diğer daha nadir görülen T-hücreli lenfomalardan oluşmaktadır.
Erkeklerde ve 50 yaş üzerinde daha sık görülür.
Tümör hücreleri genellikle serebriform bir nukleusu olan küçük hücrelerdir, az sayıda olguda büyük hücrelerde görülmektedir.
Ciltte lokalize plaklar ya da multiple nodüllerle veya yaygın eritrodermi tablosu içinde obilirler.
› Plak evresi > tümöral evre > eritrodermi evre
› Lokalize plaklar orta prognostik grup iken tümör veya eritrodermi en kötü prognozlu grubu oluşturur.
Bu hücreler epidermisi ve sıklıkla kanı infiltre eder.
› İnfiltran tümörde Langerhans hücrelerine ve ‘interdigitating’ hücrelere sıklıkla rastlanır.
› Kan infiltrasyonu vardır ve çoğunlukla minimaldir, ancak Sezary sendromunda olduğu gibi belirgin hale gelebilir.
Lenf bezlerinin veya iç organların infiltrasyonu hastalığın ileri evrelerinde ortaya çıkar ve genellikle büyük hücreli lenfomaya transformasyonu işaret eder.

Mikozis Fungoides:***Genellikle Periferik CD4(+) T-lenfositlerden ve daha
nadiren de CD8 (+) T lenfositlerinden köken alan, yaygın eritrodermi
ve/veya çok sayıda kutanöz plak ve nodüller ile kendini gösteren bir cilt
lenfomasıdır.
› Deri biyopsilerinde epidermis içinde karakteristik hücre gruplarının oluşturduğu
**Pautrier abseleri vardır.
› Lenfadenopati ve organ tutulumu hastalığın ileri evrelerinde görülür.
Sezary Sendromu; mikozis fungoides’in lösemik formu olup eritrodermi ve
periferik kanda %10'dan fazla görülen serebri form çekirdekli malign küçük
lenfosit hücreleri ile karakterizedir. Lenfodenopati ve splenomegali sıklıkta
vardır. Son dönemde büyük hücreli lenfomalara dönüşebilirler.


Çocukluk yaş grubu ile adölesan döneminde en sık görülür. › Çocukluk çağı lenfomalarının %40’ını, erişkinlerinkinin ise %5’ten azını oluşturur.
Lösemik prezentasyon çocuklarda daha sıktır.
Nodal prezentasyon adolesan çağda ve genç erkeklerde görülür.
Erişkinlerde 20-30 yaşlarında ve genellikle erkeklerde görülür.
Hastalar genellikle hızla büyüyen bir mediastinal tümörle baş vurur.
Servikal, supraklavikular ve aksiller lenfadenopati %50, mediastinal kitle %50 olguda vardır.
Mediastinal kitleye bağlı plevral effüzyon ve vena kava superior sendromu görülebilir.
%60 olguda kemik iliği infiltrasyonu ve lösemik transformasyon gelişir.
Kemik iliği ve sinir sistemi infiltrasyonu kötü prognoza işaret eder.

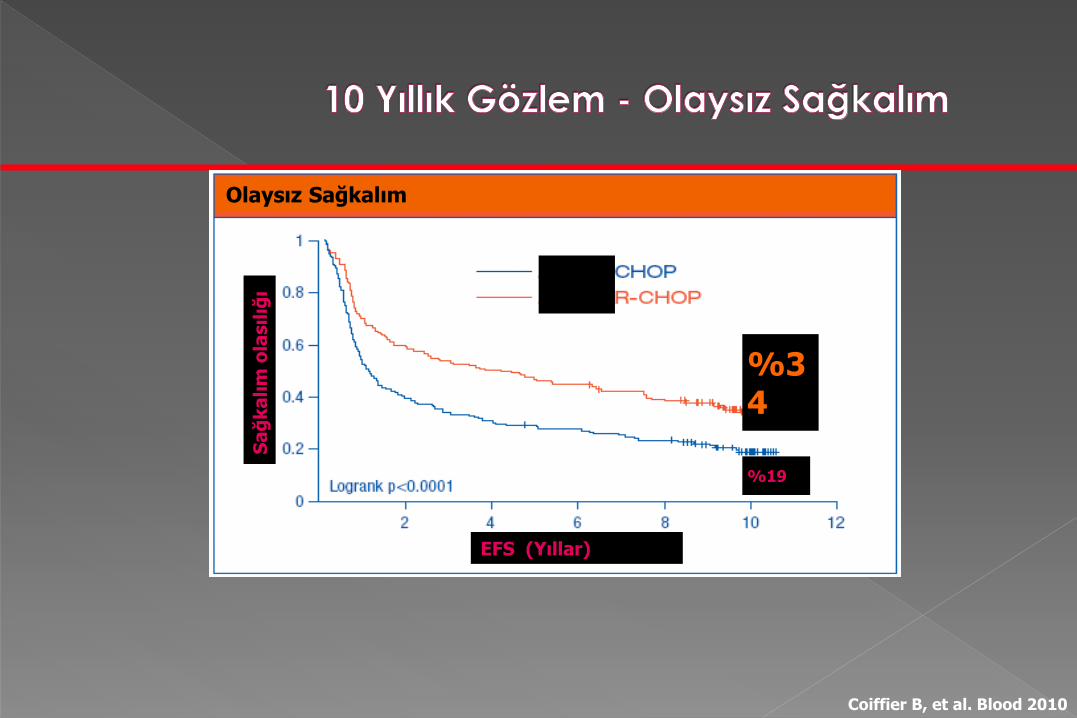
GELA LNH-98.5 çalışması, difüz büyük B-hücreli
lenfomada, R-CHOP rejiminin standart 1. basamak
tedavi olarak yerleşmesini sağlayan klinik çalışmadır
Yaşlı hastalarda yapılmış olan bu çalışma, DBBHL’li
hastalarda genel sağkalımda iyileşme sağlanan ilk
çalışmadır.
10 yıl sonra yapılan bu analizde, R-CHOP’un
CHOP’a göre avantajının, tüm alt gruplarda yıllardır
devam ettiği gösterilmektedir
Coiffier B, et al. Blood 2010

BLOOD 2010 :116(12);2040-2045

Tedavi edilmemiş
DBBHL 60 yaş n=399
8 x R-CHOP
8 x CHOP
R A N D O M I Z A S Y O N
CHOP: Siklofosfamid 750 mg/m² Doksorubisin 50 mg/m² Vinkristin 1.4 mg/m² Prednizon 40 mg/m²/gün x 5 gün R-CHOP: Rituksimab 375 mg/m2 her kürun 1.gününde
Coiffier B, et al. Blood 2010
21 günde bir
Primer sonlanım noktası olaysız sağkalım

Coiffier B, et al N Engl J Med 2002; 346:235.

Hasta özellikleri R-CHOP
n=202
CHOP
n=197
Hasta yaşı
60-70
70-75
101 (%50)
101 (%50)
110 (%55)
87 (%45)
Erkek 92 (%46) 107 (%54)
Performans skoru
0
1
>1
67 (%33)
90 (%45)
45 (%22)
70 (%36)
94 (%48)
33 (%17)
B semptomları 78 (%39) 70 (%36)
Evre 1
Evre 2
Evre 3
Evre 4
0 (%0)
41 (%20)
33 (%16)
128 (%63)
1 (%1)
39 (%20)
29 (%15)
128 (%65)
Kİ tutulumu 56 (%28) 55 (%27)
IPI skoru 0-1
IPI skoru 2
IPI skoru 3
IPI skoru 4-5
29 (%14)
64 (%32)
78 (%39)
31 (%15)
23 (%12)
69 (%35)
82 (%42)
23 (%12)
Coiffier B, et al N Engl J Med 2002; 346:235.
Coiffier B, et al. Blood 2010
Olaysız Sağkalım
Sa
ğk
alı
m o
lasıl
ığı
EFS (Yıllar)
%34
%19

Progresyonsuz Sağkalım
Sa
ğk
alı
m o
lasıl
ığı
PFS (Yıllar)
%36.5
%20
Coiffier B, et al. Blood 2010

0 2 4 6 8 10
0.2
0.4
0.6
0.8
1.0
0.0
aa IPI 0 veya 1, n = 158
CHOP: medyan 2.6 yıl
R-CHOP: medyan 9.5 yıl
p = 0.0039
Zaman (yıllar)
Pro
gre
syo
nsu
z s
ağ
ka
lım
ola
sıl
ığı
Coiffier B, et al. Blood 2010
0 2 4 6 8 10
0.2
0.4
0.6
0.8
1.0
0.0
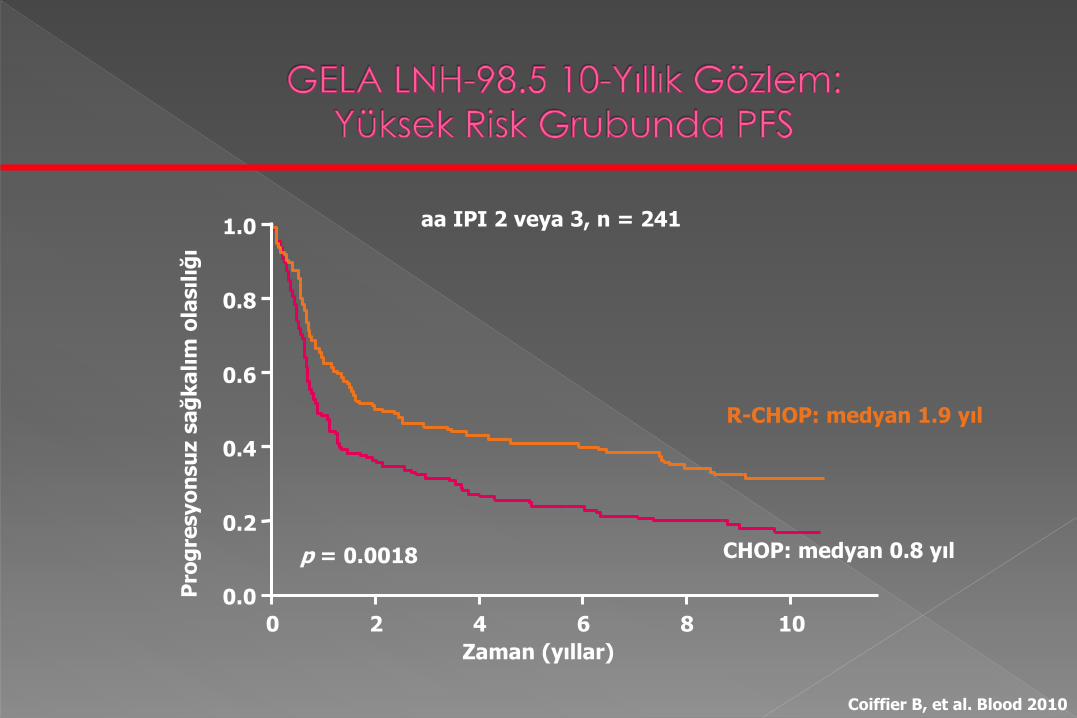
CHOP: medyan 0.8 yıl
R-CHOP: medyan 1.9 yıl
p = 0.0018
aa IPI 2 veya 3, n = 241
Zaman (yıllar)
Pro
gre
syo
nsu
z s
ağ
ka
lım
ola
sıl
ığı
Coiffier B, et al. Blood 2010
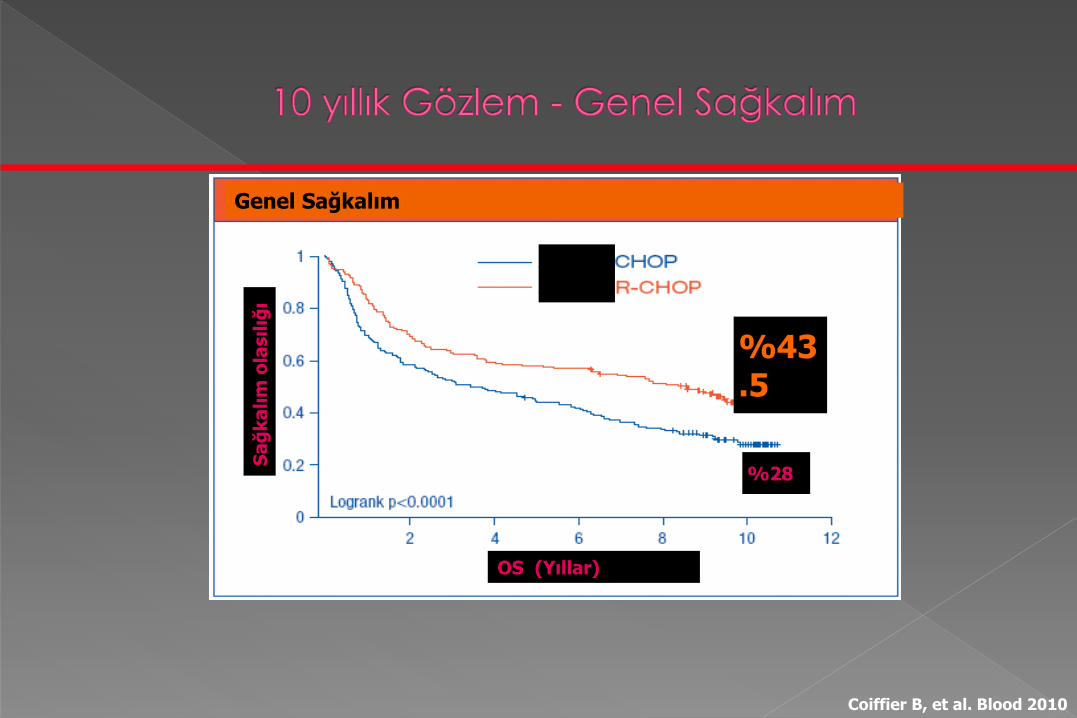
Genel Sağkalım
Sa
ğk
alı
m o
lasıl
ığı
OS (Yıllar)
%43.5
%28
Coiffier B, et al. Blood 2010

0 2 4 6 8 10
0.2
0.4
0.6
0.8
1.0
0.0
CHOP: medyan 7.3 yıl
R-CHOP: medyan ulaşılmadı
p = 0.0059
Zaman (yıllar)
Sa
ğk
alı
m o
laslı
ğı
aa IPI 0 veya 1, n = 158
Coiffier B, et al. Blood 2010

0 2 4 6 8 10
0.2
0.4
0.6
0.8
1.0
0.0
CHOP: medyan 1.8 yıl
R-CHOP: medyan 2.9 yıl
p = 0.0172
Zaman (yıllar)
aaIPI 2 veya 3, n = 241
Sa
ğk
alı
m o
laslı
ğı
Coiffier B, et al. Blood 2010


CHOP kolunda 3,5 yıl
R-CHOP kolunda 8.4 yıl
Coiffier B, et al. Blood 2010
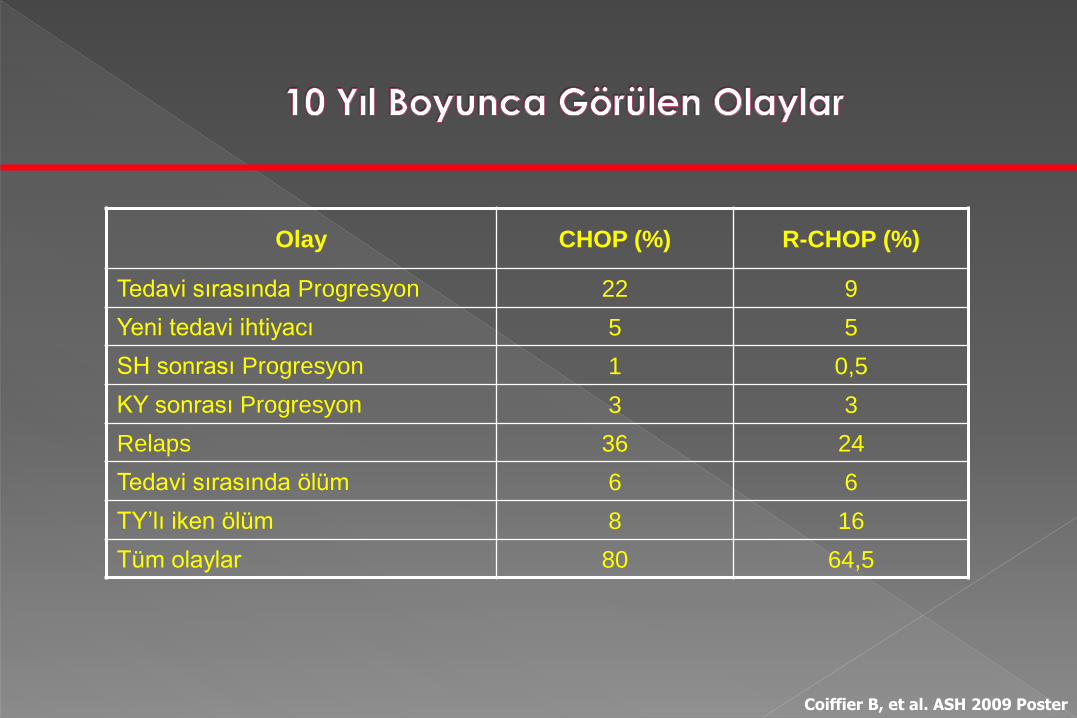
Olay CHOP (%) R-CHOP (%)
Tedavi sırasında Progresyon 22 9
Yeni tedavi ihtiyacı 5 5
SH sonrası Progresyon 1 0,5
KY sonrası Progresyon 3 3
Relaps 36 24
Tedavi sırasında ölüm 6 6
TY’lı iken ölüm 8 16
Tüm olaylar 80 64,5
Coiffier B, et al. ASH 2009 Poster

Yaşlı DBBHL’li hastalarda 8 x R-CHOP’un sağladığı progresyonsuz sağkalım ve genel sağkalım avantajının, 10 yıllık izlem süresi sonunda sürdürülebildiği görülmektedir.
Medyan PFS’de 3.6 yıl ve medyan genel sağkalımda 4.9 yıllık bir avantaj sağlanmıştır.
Hem düşük hem de yüksek risk gruplarında R-CHOP rejimi avantaj sağlamıştır.
10.yılda hastaların %40’ından fazlası hala ilk yanıtlarını koruyarak hayatta kalmışlardır.
Coiffier B, et al. Blood 2010

Lancet Oncol 2008; 9: 105–16
Michael Pfreundschuh, Joerg Schubert, Marita Ziepert, Rudolf Schmits, Martin Mohren, Eva Lengfelder, Marcel Reiser, Christina Nickenig, Michael Clemens, Norma Peter, Carsten Bokemeyer, Hartmut Eimermacher, Anthony Ho, Martin Hoff mann, Roland Mertelsmann, Lorenz Trümper, Leopold Balleisen, Ruediger Liersch, Bernd Metzner, Frank Hartmann, Bertram Glass, Viola Poeschel, Norbert Schmitz, Christian Ruebe, Alfred C Feller, Markus Loeffl er, for the German High-Grade Non-Hodgkin Lymphoma Study Group (DSHNHL).


Hastaların hepsine 7 gün boyunca “prephase
tedavi” uygulanıyor. Amaç: › İlk kemoterapi kürunun yan etkilerini azaltmak
› Hastaların performans durumlarını iyileştirmek
1 mg iv vinkristin
100 mg oral prednizon 7 gün
Pfreundschuh et al. Lancet Oncol. 2008;9:105-116

%72.5 p=0.2602
%78.1 p=0.0181
%66 p=0.8358
3 yıllık genel sağkalım
%68.8 p=0.0012
%73.4 p<0.0001
%56.9 p=0.6155
3 yıllık progresyonsuz sağkalım
%63.1 p<0.0001
%66.5 p<0.0001
%53 p=0.0365
3 yıllık olaysız sağkalım
8xCHOP14 +
8xR n=304
6xCHOP14 +
8xR n=306
8xCHOP14 n=307
Pfreundschuh et al. Lancet Oncol. 2008;9:105-116

3 yıllık olaysız sağkalım
• 8xCHOP14 kolunda %5.8
• 6xCHOP14 + 8xR kolunda %19.3
• 8xCHOP14 + 8xR kolunda %15.9 artmıştır
Pfreundschuh et al. Lancet Oncol. 2008;9:105-116

Hem olaysız sağkalım
Hem progresyonsuz sağkalım
Hem de genel sağkalım açısından 6x CHOP-14’e göre istatistiksel olarak anlamlı iyileşme sağlayan tek rejim
“8 Rituksimab + 6x CHOP-14”
Yan etkiler açısından kollar arasında anlamlı fark yaşanmamıştır.
Pfreundschuh et al. Lancet Oncol. 2008;9:105-116

Non-Hodgkin Lenfoma (NHL)
Nükseden veya kemorezistan CD20 pozitif folliküler lenfoma, diffüz
büyük B hücreli lenfoma, mantle hücreli lenfoma tanılı hastaların
tedavisinde
Daha önce tedavi edilmemiş evre III-IV folliküler lenfomalı hastalarda
kemoterapi ile kombinasyon halinde
İndüksiyon tedavisine yanıt veren folliküler lenfomalı hastalarda idame
tedavisi olarak (en fazla 2 yıl süreyle ve en fazla 8 siklus olarak)
CD20 pozitif, diffüz büyük B hücreli lenfomada CHOP kemoterapi
şemasına ek olarak kullanımı endikedir.

1. STREOİD, IVIG VE/VEYA İMMUNOSUPRESİF TEDAVİLERE DİRENÇLİ OTOİMMUN HEMOLİTİK ANEMİ
TEDAVİSİNDE, RİTUKSİMAB HAFTADA BİR 375 MG/M2, 4 HAFTA SÜREYLE
2. EN AZ BİR SERİ TEDAVİDEN EN AZ 2 KÜR ALMIŞ DİRENÇLİ VE/VEYA NÜKS MARJİNAL ZON
LENFOMA ENDİKASYONUNDA KOMBİNE KEMOTERAPİYE EK OLARAK 375 MG/M2 DOZUNDA EN
FAZLA 8 KÜR (4 KÜR SONUNDA TAM YANIT VARSA 2 KÜR DAHA DEVAM EDİLEREK TOPLAM 6 KÜR,
4 KÜR SONUNDA KISMİ YANIT VARSA 4 KÜR DAHA TEDAVİYE DEVAM EDİLEREK TOPLAM 8 KÜR)
3. EN AZ BİR SERİ TEDAVİDEN EN AZ 2 KÜR ALMIŞ DİRENÇLİ VE/VEYA NÜKS MALT LENFOMA
ENDİKASYONUNDA KOMBİNE KEMOTERAPİYE EK OLARAK 375 MG/M2 DOZUNDA EN FAZLA 8
KÜR (4 KÜR SONUNDA TAM YANIT VARSA 2 KÜR DAHA DEVAM EDİLEREK TOPLAM 6 KÜR, 4 KÜR
SONUNDA KISMİ YANIT VARSA 4 KÜR DAHA TEDAVİYE DEVAM EDİLEREK TOPLAM 8 KÜR)
4. EN AZ BİR SERİ TEDAVİDEN EN AZ 2 KÜR ALMIŞ DİRENÇLİ VE/VEYA NÜKS WALDENSTRÖM
MAKROGLOBULİNEMİSİ ENDİKASYONUNDA KOMBİNE KEMOTERAPİYE EK OLARAK 375 MG/M2
DOZUNDA EN FAZLA 8 KÜR (4 KÜR SONUNDA TAM YANIT VARSA 2 KÜR DAHA DEVAM EDİLEREK
TOPLAM 6 KÜR, 4 KÜR SONUNDA KISMİ YANIT VARSA 4 KÜR DAHA TEDAVİYE DEVAM EDİLEREK
TOPLAM 8 KÜR)
5. BURKİTT LENFOMA TANISINDA BİRİNCİ BASAMAK TEDAVİDE 375 MG/M2 DOZUNDA KEMOTERAPİ
İLAÇLARI İLE KOMBİNE OLARAK EN FAZLA 6 KÜR (4 KÜR SONUNDA TAM VEYA KISMİ YANIT
VARSA 2 KÜR DAHA TEDAVİYE DEVAM EDİLEREK TOPLAM 6 KÜR)
http://www.iegm.gov.tr/DisplayDynamicModule.aspx?mId=/PMlK+MvmJ4=




















![Sadržaj - deltaterm.net katalog [2017].pdf · Tehnički podaci Električni zidini uređaj za centralno grejanje eloBLOCK/2 Jedinica VE 6 VE 9 VE 12 VE 14 VE 18 VE 21 VE 24 VE 28](https://img.pdfslide.net/doc/110x75/5dd0f54ed6be591ccb63873f/sadraj-katalog-2017pdf-tehniki-podaci-elektrini-zidini-ureaj-za-centralno.jpg)

















