Embed Size (px)
Citation preview

ORIGINAL PAPER
Genetics of canine anal furunculosis in the German shepherd dog
Jonathan Massey & Andrea D. Short & Brian Catchpole &
Arthur House & Michael J. Day & Hannes Lohi &William E. R. Ollier & Lorna J. Kennedy
Received: 5 August 2013 /Accepted: 25 February 2014 /Published online: 14 March 2014# Springer-Verlag Berlin Heidelberg 2014
Abstract Canine anal furunculosis (AF) is characterised byulceration and fistulation of perianal tissue and is a disease thatparticularly affects German shepherd dogs (GSDs). There aresome similarities between AF and perianal Crohn’s disease(CD) in man. An immune-mediated aetiopathogenesis for AFhas been suggested due to tissue pathology, a major histocom-patibility complex (MHC) association and clinical response tociclosporin. Genome-wide association studies (GWAS) can beconducted in dogs with fewer markers and individuals than
would be required in a human study. A discovery GWAS wasperformed on 21 affected and 25 control GSDs from the UK.No SNPs reached genome-wide significance levels at thisstage. However, 127 nominally associated SNPs were geno-typed in further 76 cases and 191 controls from the UK andFinland. Sequencing of these regions was undertaken to dis-cover novel genetic variation. Association testing of thesevariants in the UK and Finnish cohorts revealed nine signifi-cantly associated SNPs, six of which cause non-synonymouschanges in protein sequence. The ADAMTS16 and CTNND2gene regions were most significantly associated with disease.Members of the butyrophilin protein family, important inintestinal inflammatory regulation, were also associated withdisease, but their independence from theMHC region remainsto be established. TheCTNND2 gene region is also interestingas this locus was implicated in human ulcerative colitis andCD, albeit at a different candidate gene:DAP. We suggest thatthis represents a common association between inflammatorybowel disease-related conditions in both species and believethat future studies will strengthen this link.
Keywords Canine anal furunculosis . German shepherd dog .
Crohn’s disease . Perianal . Inflammatory bowel disease
Introduction
Canine anal furunculosis (AF) is characterised by inflamma-tion and ulceration of perianal tissue. Similarities have beenrecognised between AF and perianal Crohn’s disease (CD) inman. Perianal lesions are reported in 13–43 % of patients withCD (Galandiuk et al. 2005) and include perianal fissures,fistulas and abscesses (Sandborn et al. 2003). Over 80 % ofcases of AF occur in German shepherd dogs (GSDs) (Day andWeaver 1992). A complex immune-mediated pathology issuspected due to the nature of the tissue pathology (Day
Electronic supplementary material The online version of this article(doi:10.1007/s00251-014-0766-5) contains supplementary material,which is available to authorized users.
J. Massey (*) :A. D. Short :W. E. R. Ollier : L. J. KennedyCentre for Integrated GenomicMedical Research (CIGMR), Instituteof Population Health, Faculty of Medical and Human Sciences,University of Manchester, 2.722 Stopford Building, Oxford Road,Manchester M13 9PT, UKe-mail: [email protected]
B. CatchpoleDepartment of Pathology and Infectious Diseases, Royal VeterinaryCollege, University of London, Hatfield, UK
A. HouseDepartment of Veterinary Clinical Sciences, Royal VeterinaryCollege, University of London, Hatfield, UK
M. J. DaySchool of Veterinary Sciences, University of Bristol, Bristol, UK
H. LohiDepartment of Veterinary Biosciences, Research Programs Unit,Molecular Neurology, University of Helsinki, Helsinki, Finland
H. LohiThe Folkhälsan Institute of Genetics, Helsinki, Finland
Present Address:A. HouseCentre for Animal Referral and Emergency, Collingwood, Australia
Immunogenetics (2014) 66:311–324DOI 10.1007/s00251-014-0766-5

1993; Day and Weaver 1992), the clinical response to immu-nosuppressive drugs (e.g. ciclosporin and prednisone) (Harkinet al. 1996; Mathews et al. 1997; Mathews and Sukhiani1997), a cytokine mRNA expression profile typical of a T-cell mediated inflammatory process (House et al. 2003; Tiverset al. 2008) and an association between the disease and class IImolecules of the major histocompatibility complex (MHC)(the dog leukocyte antigen [DLA] system) (Kennedy et al.2008).
Genetic investigations of AF outside the MHC region havehad limited success and so far have focused on a candidategene approach. House et al. (2009) used a microsatellite andsingle nucleotide polymorphism (SNP) genotyping approachto test for the association of NOD1, NOD2 and several TLRgenes with AF in the GSD. They showed no SNP associationsbut did identify restricted allelic variation thought to haveresulted from selective breeding. Barnes et al. (2009) typedseveral SNPs within the TNF-α gene and found one haplotypeto be associated with AF. However, this association was foundto be in an extended haplotype with DLA-DRB1*001:01 andtherefore secondary to the DLA association identified previ-ously. The authors conclude that there are likely to be manynon-MHC genes that will be discovered with genome-wideassociation studies (GWAS).
Several GWAS in human CD have identified novel geneticrisk factors including IL23R (Duerr et al. 2006), ATG16L1(Rioux et al. 2007) and several genes involved in the Th17pathway (STAT3, JAK2 and ICOSLG) (Barrett et al. 2008).The latest meta-analysis of CD and ulcerative colitis (UC)brings the total number of susceptibility loci to 163 (Jostinset al. 2012). No studies have been conducted solely on theperianal phenotype in CD. The environmental factors in-volved in human CD have been more extensively studied thanthose involved in canine AF. There are many conflictingstudies in this arena, but it is generally accepted that smoking,gut biota and certain medications all play a role, reviewed byVatn (2012).
The dog is an excellent genetic model for human diseasesand is particularly suited to genome-wide SNP genotyping.This is largely due to ‘founder effects’ introduced by breedcreation (~200 years ago) and the maintenance of thesebreeds. This means that causal variants act on a geneticbackground that is less heterogeneous (Boyko 2011), i.e. thereare likely to be relatively few variants each with large effect.Additionally, there is extensive linkage disequilibrium withina breed, and this enables trait mapping with fewer markersthan would be required in a human study (Lindblad-Toh et al.2005). Proof-of-principle studies showed that using 27,000SNPs, a Mendelian trait could be mapped using around tenaffected and ten control dogs (Karlsson et al. 2007).
For a multigenic trait, such as AF, 100 cases and 100controls would have 97 % power to detect an allele increasingrisk by a multiplicative factor (λ) of fivefold, and the power at
λ=2 is 50 % (Lindblad-Toh et al. 2005). Researchers have,however, been successful with fewer samples than this. Using81 affected and 57 control dogs, several genes involved in theNF-AT transcription factor pathway were identified in a ca-nine systemic lupus erythematous (SLE)-related-complexGWAS, which may have implications for human disease(Wilbe et al. 2010).
We present the first GWAS of AF in the GSD. Using asmall number of samples (21 affected and 25 controls),GWAS was used as a discovery tool to identify regionsnominally associated with disease. We then followed upSNPs within these regions in two independent cohorts ofaffected dogs from the UK and Finland. A joint analysis ofthe GWAS and independent cohort studies was undertaken.Targeted next-generation sequencing was utilised to discovernovel genetic variations in these regions. Additional genotyp-ing was performed to assess the association of these discov-ered variants with disease.
Results
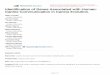
An overview of the different stages of the study is shown inFig. 1.
GWAS
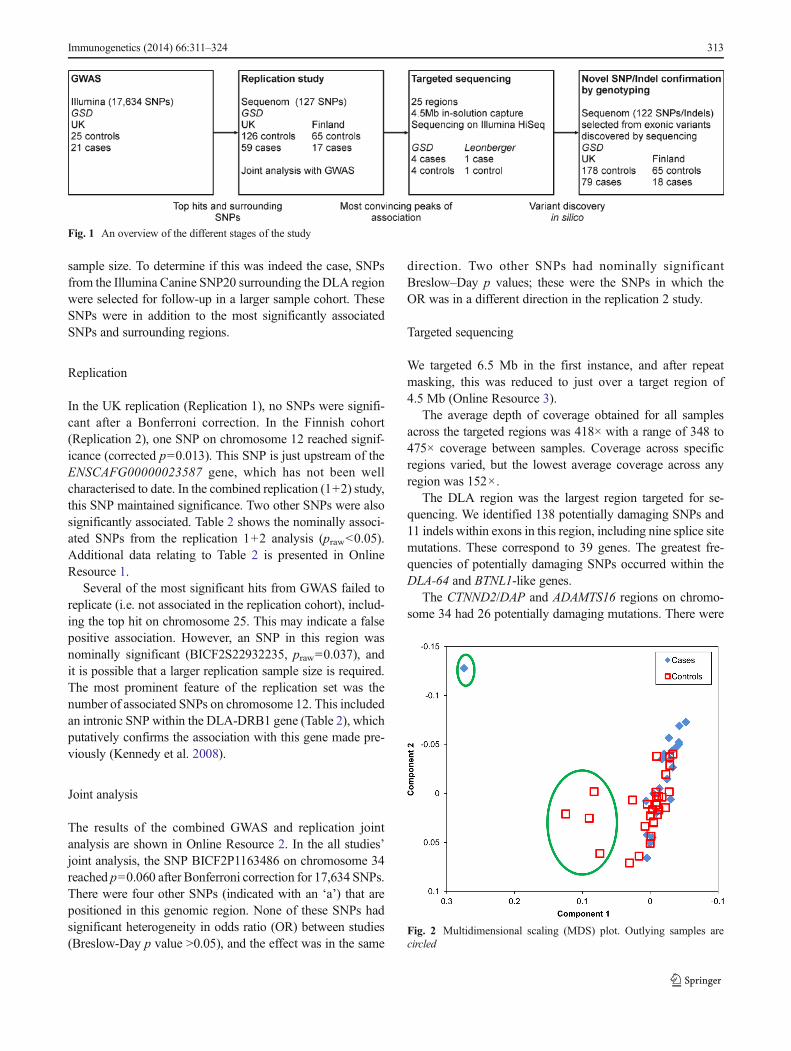
The multidimensional scaling plot (MDS) (Fig. 2) showedfive outlier samples (one affected and four controls), whichwere removed from further analysis. Two further controlsamples were removed for low genotyping (<95 %), leaving21 affected and 25 controls for analysis with 17,634 SNPs.Fisher’s exact test for association was performed.
A quantile–quantile (QQ) plot of the data is shown inFig. 3. The genomic inflation factor (λ) was 1.00, whichindicates that population stratification has been adequatelyaccounted for. The QQ plot did not indicate any major tech-nical concerns with the study.
The results with a p value <1×10−3 are shown in Table 1(additional data relating to Table 1 is presented in OnlineResource 1). There were four SNPs at the 10−5 level and afurther 23 at the 10−4 level. There were no genome-widesignificant SNPs after 10,000 maxT permutations to correctfor multiple testing. The most statistically significant SNP(lowest p value) was BICF2P1012648 on chromosome 25which is upstream of a gene cluster that includes AADATand MFAP3L. AADAT codes for aminoadipate aminotransfer-ase, which is involved in lysine catabolism. MFAP3L codesfor microfibrillar-associated protein 3-like.
It is interesting that the MHC class II region, previouslyidentified to be associated with AF (Kennedy et al. 2008), wasnot detected as significant in the GWAS. This is due to a lackof coverage of the region on the array and possibly small
312 Immunogenetics (2014) 66:311–324
sample size. To determine if this was indeed the case, SNPsfrom the Illumina Canine SNP20 surrounding the DLA regionwere selected for follow-up in a larger sample cohort. TheseSNPs were in addition to the most significantly associatedSNPs and surrounding regions.
Replication
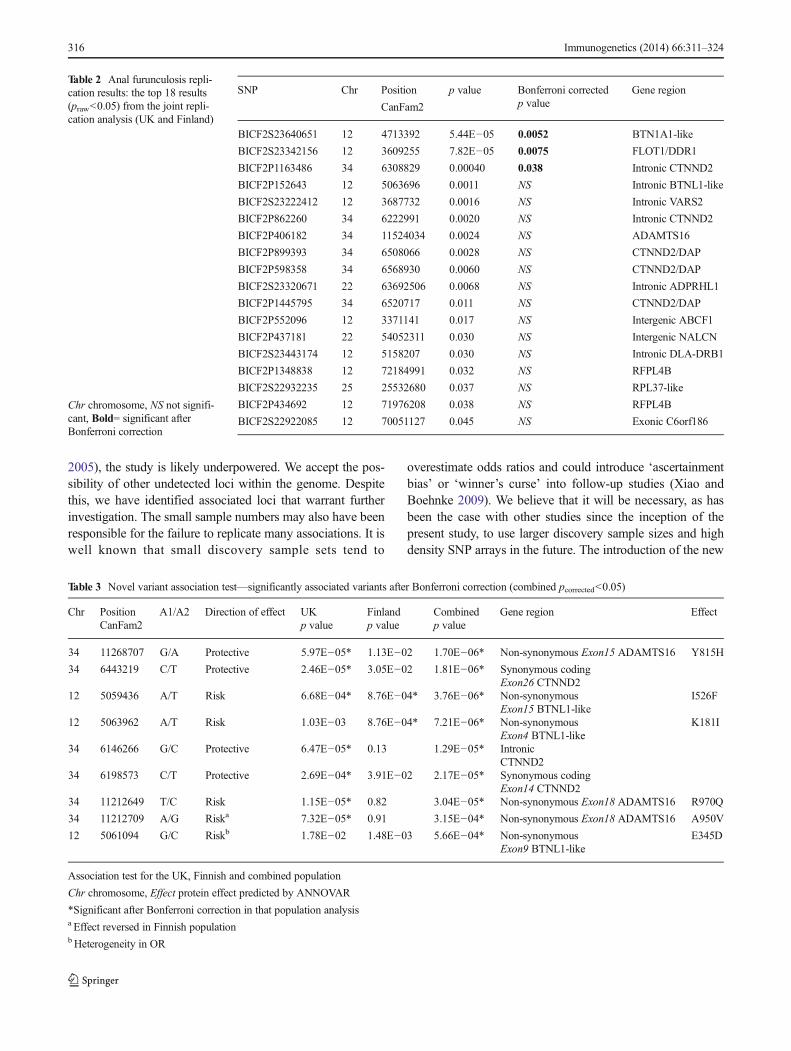
In the UK replication (Replication 1), no SNPs were signifi-cant after a Bonferroni correction. In the Finnish cohort(Replication 2), one SNP on chromosome 12 reached signif-icance (corrected p=0.013). This SNP is just upstream of theENSCAFG00000023587 gene, which has not been wellcharacterised to date. In the combined replication (1+2) study,this SNP maintained significance. Two other SNPs were alsosignificantly associated. Table 2 shows the nominally associ-ated SNPs from the replication 1+2 analysis (praw<0.05).Additional data relating to Table 2 is presented in OnlineResource 1.
Several of the most significant hits from GWAS failed toreplicate (i.e. not associated in the replication cohort), includ-ing the top hit on chromosome 25. This may indicate a falsepositive association. However, an SNP in this region wasnominally significant (BICF2S22932235, praw=0.037), andit is possible that a larger replication sample size is required.The most prominent feature of the replication set was thenumber of associated SNPs on chromosome 12. This includedan intronic SNP within the DLA-DRB1 gene (Table 2), whichputatively confirms the association with this gene made pre-viously (Kennedy et al. 2008).
Joint analysis
The results of the combined GWAS and replication jointanalysis are shown in Online Resource 2. In the all studies’joint analysis, the SNP BICF2P1163486 on chromosome 34reached p=0.060 after Bonferroni correction for 17,634 SNPs.There were four other SNPs (indicated with an ‘a’) that arepositioned in this genomic region. None of these SNPs hadsignificant heterogeneity in odds ratio (OR) between studies(Breslow-Day p value >0.05), and the effect was in the same
direction. Two other SNPs had nominally significantBreslow–Day p values; these were the SNPs in which theOR was in a different direction in the replication 2 study.
Targeted sequencing
We targeted 6.5 Mb in the first instance, and after repeatmasking, this was reduced to just over a target region of4.5 Mb (Online Resource 3).
The average depth of coverage obtained for all samplesacross the targeted regions was 418× with a range of 348 to475× coverage between samples. Coverage across specificregions varied, but the lowest average coverage across anyregion was 152×.
The DLA region was the largest region targeted for se-quencing. We identified 138 potentially damaging SNPs and11 indels within exons in this region, including nine splice sitemutations. These correspond to 39 genes. The greatest fre-quencies of potentially damaging SNPs occurred within theDLA-64 and BTNL1-like genes.
The CTNND2/DAP and ADAMTS16 regions on chromo-some 34 had 26 potentially damaging mutations. There were
Fig. 1 An overview of the different stages of the study
Fig. 2 Multidimensional scaling (MDS) plot. Outlying samples arecircled
Immunogenetics (2014) 66:311–324 313

no non-synonymous coding SNPmutations discovered withinthe CTNND2 gene region. However, there were three frame-shift indels. According to the Ensembl gene annotation, theseindels are within the ENSCAFG00000025498 gene, whichappears to be a CTNND2 pseudo-gene. This may be a mistakein the Ensembl annotation as if one takes the RefSeq annota-tions (http://www.ncbi.nlm.nih.gov/RefSeq), all of theseindels reside in exon 11 of the CTNND2 gene. In theADAMTS16 region, there were nine non-synonymous muta-tions within the KIAA0947 gene and seven within theADAMTS16 gene itself. This was in addition to frameshiftindels within exon 1 of the KIAA0947 gene.
Targeted sequencing—novel variant validation by genotyping
The GWAS samples and replication samples from the UK andFinland (plus extra controls) were genotyped for 122 variantsdiscovered from sequencing. These were mainly plausiblyfunctional variants within exons. The raw cluster data fromSequenom suggested that 61 gave satisfactory qualitygenotyping data, 14 were monomorphic, 13 failed outrightand the remainder gave lower quality genotyping (it wasconsidered a best practice to remove these lower qualityvariants).
A combined analysis of the UK and Finnish datasets re-vealed that nine SNPs remained significant after a Bonferronicorrection (Table 3 and Online Resource 1). These correspondto two regions on chromosome 34 within the ADAMTS16 andCTNND2 gene, as well as one gene within the DLA region:BTNL1-like.
The most significant SNP in the combined analysis was anon-synonymous SNP in exon 15 of the ADAMTS16 gene,causing a Y815H (tyrosine>histidine) mutation in the proteinsequence. This was in addition to two other associated non-synonymous SNPs in exon 18 of this gene: Chr34:11212649(R970Q) and Chr34:11212709 (A950V). The Y815 and A950residues are not conserved across Eutherian mammals, but theR970 residue is generally conserved (Online Resource 4). Theassociation of protective and risk SNPs within ADAMTS16suggests a haplotype effect. Using Haploview 4.2 (Barrettet al. 2005) and a solid spine of linkage disequilibrium (LD)to define haplotypes, a 112-kb haplotype consisting of sixSNPs can be identified in this region. In the UK population,the CTAACG haplotype was the most associated (p=2.98×
Fig. 4 Manhattan plot of thep values for the GWAS analysis.Twenty-one cases; 25 controls.Upper line genome-wide signifi-cance; lower line suggestivesignificance
Fig. 3 Quantile–quantile (QQ) plot for the GWAS analysis
314 Immunogenetics (2014) 66:311–324

10−6), incorporating the risk alleles Chr34:11212649 (T),Chr34:11212709 (A) and Chr34:11268707 (A) (OnlineResource 5).
Non-synonymous SNPs were also associated in BTNL1-like and are generally well conserved across Eutherianmammals (Online Resource 4). There were no non-synonymous SNPs discovered in the CTNND2 gene fromsequencing. However, there were two synonymous SNPsin exon 14 and 26 of this gene, associated in the com-bined analysis; these two SNPs are in high LD (r2=0.95).A 433-kb haplotype can be identified in this region fromChr34:6146266 to Chr34:6579573, but the frequencies didnot differ significantly between cases and controls (datanot shown).
Online Resource 6 shows the remainder of the variants, fromthe analysis above, that had a p value<0.05 but did not maintainsignificance after a Bonferroni correction. This shows onefurther non-synonymous SNP within the ADAMTS16 gene.This is in addition to further four synonymous SNPs withintheCTNND2 gene, as well as nine SNPs in the widerCTNND2/
DAP region, including an SNP within a non-coding RNA(ncRNA). Two regions on chromosome 22 were also nominally associated: there were five SNPs within theNALCN/ITGBL1region and two in the ‘coagulation factor VII’ gene region. The‘Furin-like’ gene on chromosome 25 appears to be importantwith three non-synonymous SNP mutations and one frameshiftindel mutation nominally associated with disease risk. Two ofthese SNP mutations affect conserved amino acid sequencesacross Eutherian mammals, namely Chr25:24519288(P165L—P conserved) andChr25:24519548 (A189V—A con-served) (Online Resource 4). There was one SNP that wasnominally associated within the ABCF1 gene that is locatedwithin the DLA class I region on chromosome 12.
Discussion
Considering the calculations that for a multigenic trait, 100cases and 100 controls would be optimal (Lindblad-Toh et al.
Table 1 Anal furunculosisGWAS result: the top 27 results(p<1×10−3) from the GWASanalysis
The gene/region was determinedfrom Ensembl release 64
OR odds ratio, Chr chromosome
SNP Chr Position
CanFam2
p value OR Gene region
BICF2P1012648 25 24184481 2.08E−05 7.25 Intergenic AADAT
BICF2G630214311 17 29466390 5.39E−05 0.15 Intronic LTBP1 (Q2PPL3)
BICF2S23714577 25 24882846 6.32E−05 0.16 Intergenic XM847016.1
BICF2P1093883 22 57921758 7.62E−05 24.50 Gene desert
BICF2P347366 14 42053749 1.10E−04 0.16 Intergenic NPVF/RPL39L
BICF2P434692 12 71976208 1.98E−04 5.88 Intergenic RFPL4B
BICF2P1232275 22 57130167 2.37E−04 12.00 Gene desert
BICF2P1348838 12 72184991 3.01E−04 5.78 Intergenic RFPL4B
BICF2G63095769 25 29967508 3.01E−04 5.78 Intergenic XKR6
BICF2P983044 17 28851946 3.15E−04 5.14 Intronic BIRC6
BICF2S23060299 25 29438154 3.30E−04 5.57 Intergenic BLK/FAM167A
BICF2P406182 34 11524034 3.63E−04 0.20 Intergenic ADAMTS16
BICF2G630565169 7 65960916 3.66E−04 5.52 Intergenic SS18/ZNF521
BICF2P381161 7 41294985 4.06E−04 0.21 Intronic ITPKB
BICF2P1445795 34 6520717 5.05E−04 0.16 Intergenic CTNND2/DAP
BICF2G630673540 2 17620028 5.23E−04 0.13 Intergenic EPC1
BICF2P1111230 X 104310053 6.89E−04 0.15 Intergenic ELF4/AIFM1
BICF2P874837 7 25634323 6.98E−04 0.21 Intronic ASTN1
BICF2G63096050 25 30313696 7.05E−04 4.63 Non-synonymous coding RP1L1
BICF2P794055 25 8778796 7.29E−04 0.18 Intergenic WTAP
BICF2P675990 20 58237305 7.62E−04 0.22 Intronic SH3GL1
BICF2S22952229 34 10542936 7.62E−04 0.22 Intergenic MED10
BICF2P890748 29 5576204 7.72E−04 0.15 Intergenic SNTG1
BICF2S23148152 17 36651445 7.75E−04 N/A Intergenic PKDCC
BICF2S23148152 25 29257802 7.87E−04 5.01 Intergenic GATA4/BLK
BICF2G630214253 17 29513227 8.48E−04 0.23 Intronic LTBP1 (Q2PPL3)
BICF2P1163486 34 6308829 8.48E−04 0.23 Intronic CTNND2
Immunogenetics (2014) 66:311–324 315
2005), the study is likely underpowered. We accept the pos-sibility of other undetected loci within the genome. Despitethis, we have identified associated loci that warrant furtherinvestigation. The small sample numbers may also have beenresponsible for the failure to replicate many associations. It iswell known that small discovery sample sets tend to
overestimate odds ratios and could introduce ‘ascertainmentbias’ or ‘winner’s curse’ into follow-up studies (Xiao andBoehnke 2009). We believe that it will be necessary, as hasbeen the case with other studies since the inception of thepresent study, to use larger discovery sample sizes and highdensity SNP arrays in the future. The introduction of the new
Table 2 Anal furunculosis repli-cation results: the top 18 results(praw<0.05) from the joint repli-cation analysis (UK and Finland)
Chr chromosome, NS not signifi-cant, Bold= significant afterBonferroni correction
SNP Chr Position
CanFam2
p value Bonferroni correctedp value
Gene region
BICF2S23640651 12 4713392 5.44E−05 0.0052 BTN1A1-like
BICF2S23342156 12 3609255 7.82E−05 0.0075 FLOT1/DDR1
BICF2P1163486 34 6308829 0.00040 0.038 Intronic CTNND2
BICF2P152643 12 5063696 0.0011 NS Intronic BTNL1-like
BICF2S23222412 12 3687732 0.0016 NS Intronic VARS2
BICF2P862260 34 6222991 0.0020 NS Intronic CTNND2
BICF2P406182 34 11524034 0.0024 NS ADAMTS16
BICF2P899393 34 6508066 0.0028 NS CTNND2/DAP
BICF2P598358 34 6568930 0.0060 NS CTNND2/DAP
BICF2S23320671 22 63692506 0.0068 NS Intronic ADPRHL1
BICF2P1445795 34 6520717 0.011 NS CTNND2/DAP
BICF2P552096 12 3371141 0.017 NS Intergenic ABCF1
BICF2P437181 22 54052311 0.030 NS Intergenic NALCN
BICF2S23443174 12 5158207 0.030 NS Intronic DLA-DRB1
BICF2P1348838 12 72184991 0.032 NS RFPL4B
BICF2S22932235 25 25532680 0.037 NS RPL37-like
BICF2P434692 12 71976208 0.038 NS RFPL4B
BICF2S22922085 12 70051127 0.045 NS Exonic C6orf186
Table 3 Novel variant association test—significantly associated variants after Bonferroni correction (combined pcorrected<0.05)
Chr PositionCanFam2
A1/A2 Direction of effect UKp value
Finlandp value
Combinedp value
Gene region Effect
34 11268707 G/A Protective 5.97E−05* 1.13E−02 1.70E−06* Non-synonymous Exon15 ADAMTS16 Y815H
34 6443219 C/T Protective 2.46E−05* 3.05E−02 1.81E−06* Synonymous codingExon26 CTNND2
12 5059436 A/T Risk 6.68E−04* 8.76E−04* 3.76E−06* Non-synonymousExon15 BTNL1-like
I526F
12 5063962 A/T Risk 1.03E−03 8.76E−04* 7.21E−06* Non-synonymousExon4 BTNL1-like
K181I
34 6146266 G/C Protective 6.47E−05* 0.13 1.29E−05* IntronicCTNND2
34 6198573 C/T Protective 2.69E−04* 3.91E−02 2.17E−05* Synonymous codingExon14 CTNND2
34 11212649 T/C Risk 1.15E−05* 0.82 3.04E−05* Non-synonymous Exon18 ADAMTS16 R970Q
34 11212709 A/G Riska 7.32E−05* 0.91 3.15E−04* Non-synonymous Exon18 ADAMTS16 A950V
12 5061094 G/C Riskb 1.78E−02 1.48E−03 5.66E−04* Non-synonymousExon9 BTNL1-like
E345D
Association test for the UK, Finnish and combined population
Chr chromosome, Effect protein effect predicted by ANNOVAR
*Significant after Bonferroni correction in that population analysisa Effect reversed in Finnish populationbHeterogeneity in OR
316 Immunogenetics (2014) 66:311–324

CanFam3 genome assembly might also offer further insights,but this was released after study completion.
In both the replication analysis and in the novel variantassociation analysis, SNPs within or close to the ADAMTS16gene were associated with AF. The three SNPs associated inthe latter analysis were significant after Bonferroni correctionand would cause non-synonymous SNP mutations withinexons 15 and 18 of the gene. A 112-kb haplotype incorporat-ing the risk alleles of these three SNPs can be identified in theUK population, with a p value lower than the single-pointanalyses. The Y815 and A950 residues are not conservedacross Eutherian mammals, but the R970 residue is generallyconserved (except in guinea pig, Cavia porcellus—Q978).However, as the R970Q mutation is not significantly associ-ated in the Finnish population alone, it might indicate anothercausal mutation in the region, a non-common genetic associ-ation between populations, or simply that the statistical powerin the Finnish sample was not great enough to detect thiseffect. ADAMTSs (a disintegrin and metalloproteinase withthrombospondin motifs) are a family of proteases, which areencoded by 19 genes in the human genome (Porter et al.2005). Little is known about the function of ADAMTS16,but it is known to be highly expressed in the kidneys, brainand fetal lung (Surridge et al. 2009). Functional roles havebeen attributed using chondrosarcoma cells transfected withan ADAMTS16 construct. Surridge et al. (2009) were able toshow that the overexpression of ADAMTS16 led to reducedcell proliferation and migration. Additionally, they found thatthere was an inverse relationship between the expression ofADAMTS16 and matrix metalloproteinase (MMP) 13 andthat transforming growth factor (TGF)-β could induceADAMTS16 expression. The involvement of MMP13 is par-ticularly interesting as this was one of the metalloproteinaseswith significantly increased expression in canine AF lesions(House et al. 2007). There is also a role for MMP13 in thepathogenesis of human CD and UC (Rath et al. 2006; Vizosoet al. 2006). ADAMTS16 has also been identified as a posi-tional candidate for human hypertension (Joe et al. 2009). Thishas been extended to a rat model of hypertension, andADAMTS16 is thought to act in this capacity by changes invasculature (Gopalakrishnan et al. 2012).
The top hit in the GWAS was on chromosome 25 at posi-tion 24,184,481 close to the MFAP3L/AADAT gene cluster.There was a suggestive association in a wider regionencompassing a gene desert, so the entire region was se-quenced. This narrowed the region of association to twoframeshift deletions and one insertion in exon 8, and a furtherinsertion and splicing deletion in exon 3 of a proproteinconvertase family gene with a start position at 24,518,772.There was a nominal association in this region, in the com-bined analysis of sequencing discovered variants, with threenon-synonymous SNP mutations and one frameshift deletion.Two of these SNP mutations affect conserved amino acid
sequences ac ross Euthe r i an mammals , name lyCh r 2 5 : 2 4 5 1 9288 ( P 1 6 5L—P con s e r v e d ) a n dChr25:24519548 (A189V—A conserved). Although moreinvestigation will be required in larger cohorts, these associa-tions are interesting. The gene is incompletely characterised,but it appears in both the Ensembl and RefSeq annotations.There is no syntenic alignment to the human genome, butorthologue mapping and syntenic alignment to the mouse(Mus musculus) genome reveal that this gene is most similarto Furin, which was found to be essential in the maintenanceof peripheral immunological tolerance; conditional deletion ofFurin in T cells impaired the function of both regulatory andeffector T cells (Pesu et al. 2008). Additionally, these authorsshowed that furin-deficient regulatory T cells were less pro-tective in a colitis model. Interestingly, furin activatesADAMTS16 and other members of this protein family byproteolytic cleavage at specific sites (Joe et al. 2009). Beforefirm conclusions can be drawn, further mapping and investi-gation of the gene in this region would have to be performed.
Another associated region on chromosome 34 wasCTNND2/DAP. In the joint replication analysis, there werefive protective associations within this region, of which threeare intronic within the CTNND2 gene (encodes for δ-catenin),while the remaining SNPs are intergenic downstream of thisgene. In the analysis of the sequencing discovered variants,two highly correlated (r2=0.95) synonymous coding muta-tions within CTNND2 were significantly associated with dis-ease, as well as one intronic SNP (r2=0.72 with exon 26SNP). This was in addition to nominally associated SNPswithin the wider region, including two SNPs in the 3′ untrans-lated (UTR) region of DAP. The identified haplotypeencompassing CTNND2 did not extend as far as DAP; thehighest LD r2 value between the exon 26 CTNND2 SNP andthe 3′ UTR DAP SNPs is 0.56.
DAP encodes for death-associated protein. An intronic SNP(rs267939) within the DAP gene was associated in a GWASmeta-analysis in human UC (Anderson et al. 2011), pointing toa possible shared susceptibility locus between human UC andcanine AF. The candidate gene identified by Anderson andcolleagues within the 5p15 locus was the DAP gene itself,due to the intronic SNP’s correlation with expression of DAPin a control population (cis-eQTL) (Anderson et al. 2011).DAPalso has a purported role in negatively regulating autophagy(Koren et al. 2010); other genes involved in autophagic pro-cesses have been associated with CD andUC, reviewed in Kabiet al. (2012). In a follow-up study, a proxy SNP (rs2930047, r2
=0.871) of the previous SNP association, also intronic withintheDAP gene, reached p=1.03×10−8 in a combined analysis ofCD and UC studies conducted on GWA arrays and theImmunochip (Jostins et al. 2012). In the analysis of CD alone,the best p value was 1.45×10−7.
Despite the associations with both human CD and UC andreported candidate function of DAP, variants in the DAP gene
Immunogenetics (2014) 66:311–324 317

were only nominally associated with AF in the dog. Theresults presented here perhaps suggest a role for theCTNND2 gene in the canine disease pathogenesis. This be-comes ever more pertinent considering the extension of δ-catenin’s role from seemingly neurone specific (De Strooperet al. 1998) to epithelial cell expression (Lu et al. 1999) andcancer (Burger et al. 2002; Lu et al. 2005; Zeng et al. 2009).More recently, DeBusk et al. (2010), working from an onco-logical perspective, reported that mice deficient for δ-catenin(either homozygous or heterozygous deficiency) had inhibitedtumour growth. In addition, they showed that δ-catenin defi-ciency led to a reduced rate of wound healing in a skin biopsypuncture model. This was shown to be due to the impairmentof angiogenesis. Furthermore, they revealed that δ-cateninexpression is regulated by the inflammatory cytokines, inter-leukin (IL)-1 and tumour necrosis factor (TNF)-α, throughnuclear factor (NF)-κB signalling.
The angiogenic-related findings have been supported byHe et al. (2013), who showed that prostate cancer cells over-expressing δ-catenin had increased angiogenic potential, pos-sibly through actions on vascular endothelial growth factor(VEGF). Taken together, these findings build a convincingpicture of a role for δ-catenin in the pathogenesis of AFthrough regulation of angiogenesis. In support of a generalangiogenesis dysregulation model, there is evidence thatmany GSDs affected by anal furunculosis also suffer frompannus (chronic superficial keratitis), a disease characterisedby angiogenesis/vascularisation of the cornea. The evidence ismainly anecdotal, but in one study, pannus was an extra-intestinal manifestation in almost a quarter of dogs with AF(Stanley and Hauptman 2009). It is interesting that both lociassociated on chromosome 34, CTNND2 and ADAMTS16,possibly point to a change in vasculature as part of the pathol-ogy of AF.
The CTNND2/DAP region is undoubtedly important withregards to AF, as well as human CD and UC. The results ofthis study imply causality to CTNND2, while the humanstudies have identified DAP as the causative gene. This couldreflect differences in the specific pathology being studied inAF; an analysis of the human data that included only thoseindividuals with perianal involvement might give differentresults. It would be interesting to study eQTL effects in thisregion in the dog.
Chromosome 12, perhaps unsurprisingly given that thischromosome includes the MHC, is also important in thep a t h o g e n e s i s o f AF. T h e B ICF 2S 2 3 6 4 0 6 5 1(Chr12:4713392) SNP, which was significant in both thesecond replication set and the joint replication study(Table 3), is upstream of a gene that is not well annotated.Orthologue mapping suggests that this gene is most similar tothe BTN1A1 (butyrophilin subfamily 1 member A1) gene. Thegene is part of the butyrophilin superfamily and contains animmunoglobulin V-set domain, giving them structural
homologywith the B7 family of immunological costimulatorymolecules (Arnett et al. 2009), which includes ICOSLG.Another SNP, BICF2P152643 (Chr12: 5063696), which wasnominally significant in the joint replication study (praw=0.0011, Table 3), is intronic within another BTN family mem-ber gene, most similar to BTNL1. The association analysis ofsequencing-discovered SNPs narrowed the focus to three non-synonymous SNPs within exons 4, 9 and 15 of the BTNL1-like gene. The mutations in exon 15 (Chr12:5059436—I526F) and exon 4 (Chr12:5063962—K181I) are generallywell conserved across Eutherian mammals.
Both of these chromosome 12 loci are upstream of the mainDLA class II genes and reside in a gene cluster with othermembers of the butyrophilin family, which makes definingcausative genes difficult. However, expression of BTN familymembers has been demonstrated in immune cells and thehuman intestine (Arnett et al. 2009). However, genes withinthis region, including BTNL1 and BTNL2, are thought to be instrong linkage disequilibrium with the MHC class II region.Some studies have found non-independent effects in thisregion (Orozco et al. 2005), while others have demonstratedindependence (Rybicki et al. 2005), which could reflect ge-netic variances between the different populations tested. Asthe SNPs on chromosome 12 were not associated in theGWAS and only selected for follow-up based on MHC prox-imity, more work will be required to determine if these vari-ants are independent of the DLA-DRB1*001:01 association.These findings also indicate the low power of the GWAS.
The region on chromosome 22 identified by theChr22:63692506 SNP (joint p=9.31×10−5) had 13 non-synonymous SNPs from sequencing. These were found acrossfive genes within the region: MCF2L, GRTP1, TMCO3, co-agulation factor VII and coagulation factor X fragment.Frameshift deletions were also discovered in the latter twogenes. Two non-synonymous SNP mutations and one frame-shift indel were nominally associated in the analysis of vari-ants discovered from sequencing. This is particularly interest-ing given the hypothesis of small blood vessel thrombosis inthe aetiopathogenesis of IBD (Alkim et al. 2011). Plasmaconcentration of coagulation factor VII was significantlyhigher in patients with CD and UC compared with controls(Hudson et al. 1996), but this was not supported by a laterstudy (Alkim et al. 2011). This nominal association within thecoagulation factor VII gene would support the thrombosishypothesis, but further fine-mapping of the association willbe required.
A limitation of this work is that the complete samplephenotype information was lacking for many samples. Theinclusion of parameters such as severity and age of onsetcould help to identify subsets of disease and new geneticassociations. One caveat of this is that given the small samplenumbers to start with, this would reduce power further. Aproblem related to the lack of phenotype information was that
318 Immunogenetics (2014) 66:311–324

the relatedness of dogs was not always known. Despite at-tempts in the GWAS to identify closely related individuals andoutliers/stratification viaMDS, this same analysis could not beperformed on Sequenom genotype data. Where informationwas available, unrelated dogs were selected. Considering thatthe samples were drawn from across the UK and Finland, webelieve the cohorts are representative of the general popula-tions. This is supported by very few significant differences inheterogeneity in OR between the UK GWAS population andthe replication cohort, and between the UK population as awhole and the Finnish dogs.
Another limitation of this work regards the confirmation ofsequencing-discovered SNPs. Although many variants wereconfirmed with Sequenom genotyping, conventional sequenc-ing of mutations should be conducted. This is particularlynecessary where SNP assays may have failed due toSequenom assay design problems or the limitations ofSequenom technology. Another consideration that must betaken into account is that the sequencing confirmation focusedonly on plausibly functional variants within exons, largelyignoring intergenic and intronic variation. Such non-exonicvariation could possibly be functional with regard to generegulation, but until a comprehensive map of such sites hasbeen established in the dog, it will be difficult to study.Equally, the identification of structural variation (SV), suchas copy number variants (CNVs), could be important in dis-ease pathology. However, given the small sample numbersand the difficulty of identifying and confirming novel SVs,this was considered beyond the scope of the study. It isunlikely that we have identified the causal variants in thisstudy due to the limited number of markers tested in eachregion, but we hope that this research provides a platform forfuture canine and human studies.
Future studies could also investigate the effect of serumIgA levels on AF. It is known that serum IgA levels are lowerin GSD than other breeds, which could explain the highprevalence of immune-mediated and inflammatory disorders,such as atopic dermatitis (Tengvall et al. 2013). The measure-ment of serum IgA in a cohort of GSDs with AF could proveuseful in identifying subsets of disease. This could reveal acommon genetic basis for the inflammatory conditions seen inthe breed.
In conclusion, several novel loci associated with AF in theGSD have been identified by a GWAS, follow-up genotypingand sequencing approach. Presented is further evidence of arole in AF pathogenesis for variants beyond DLA-DRB1within the MHC region and for the emerging role played bybutyrophilins in intestinal inflammatory disease. One putativeassociated locus, the most significant association in theGWAS, possibly points to the gene Furin, the product ofwhich is involved in the maintenance of peripheral immuno-logical tolerance. This gene links to the discovery ofADAMTS16 on chromosome 34, which is a gene that possibly
ties this research with the expression profiling already con-ducted in AF relating toMMP13. δ-catenin was identified as aconvincing candidate gene that probably would not have beenconsidered before this study, being seemingly neurone specif-ic. This research makes it imperative to further study the linkbetween AF and human CD/UC at this genomic locus.
Methods
An overview of the different stages of the study is shown inFig. 1.
Animals
Affected samples from the United Kingdomwere collected byveterinary surgeons at clinics across the country, using thesame diagnostic criteria as used previously (Kennedy et al.2008). Parameters such as age of onset and severity wereunknown. Samples were residual EDTA blood from diagnos-tic tests. Control samples were obtained from the UK DNAArchive for Companion Animals at the Centre for IntegratedGenomic Medical Research (CIGMR) at the University ofManchester. Samples from Finland were collected byHannes Lohi’s research group from the University of Helsinki.
DNA extraction
DNA was extracted using Qiagen QIAmp DNA Blood MidiKits (Crawley, UK), following the manufacturer’s standardprotocol. DNA concentration was measured using a NanodropND-1000 spectrophotometer.
Sample management
Samples were logged into a Nautilus Laboratory InformationManagement System (LIMS) (Thermo Scientific) andbarcoded with a unique identifier. DNA samples were storedin 96-well 2D barcoded Matrix plates (Thermo Scientific) at−80 °C until required.
GWAS
The Illumina Canine SNP20 was used for the GWAS. This isbased on BeadArray technology and includes 22,362 SNPsfrom the CanFam2.0 genome assembly. The SNP array wasanalysed by The Genome Centre at Barts and the LondonSchool of Medicine and Dentistry according to the manufac-turer’s instructions.
BC|Gene (Biocomputing Platforms Ltd, Espoo, Finland)was used to store genotype and phenotype data. BC|Gene wasalso used for data quality control (QC) and analysis using aPLINK extension (v1.07) (Purcell et al. 2007). An Identity-
Immunogenetics (2014) 66:311–324 319

by-state (IBS) was calculated to show any relatedness betweensamples. All the GWAS data were subject to the following QCprocedures: only SNPs which conformed to Hardy-Weinbergexpectations (HWE) (p≤0.0001) in control samples, exhibitedcall rates of ≥95% and had a minor allele frequency (MAF)≥0.01 were included in the analysis. Additionally, only sam-ples with a ≥95% genotyping success rate were included. Thepresence of population stratification was assessed by multidi-mensional scaling (MDS) analysis with four dimensions ex-tracted. Outlying individuals were removed from subsequentanalysis. Thirty-one control and 23 affected GSDs from theUK were genotyped on the array. After QC procedures, therewere 25 control and 21 affected dogs and 17,634 SNPs foranalysis.
Fisher’s exact test for association was performed. To cor-rect for multiple testing, 10,000 permutations (maxT) wereperformed. AManhattan plot which plots the−log10 unadjust-ed p values against chromosome position was produced usingHaploview 4.2 (Barrett et al. 2005). A quantile–quantile (Q-Q) plot to show the difference between observed and expectedresults was also generated using GenABEL (Aulchenko et al.2007).
The 50 most significantly associated SNPs from the anal-ysis were selected for follow-up. SNPs surrounding theseregions (‘shoulder’ SNPs of the top associated ‘peak’ SNP)as well as 23 additional SNPs from chromosome 12 (sur-rounding the DLA region) were selected, giving 127 SNPsin total.
Sequenom genotyping
Sequenom Assay Design software was used to design the PCRprimers and extension primers (probes). These were thensynthesised by Metabion (Germany) or Sigma-Aldrich(UK). Sequenom PCR, cleanup and extension reactions werethen performed according to the manufacturer’s standard in-structions. A Sequenom nanodispenser (Samsung) was usedto spot each sample onto a SpectroCHIP Bioarray. ASequenom Matrix-assisted-laser-desorption/ionization time-of-flight (MALDI-TOF) mass spectrophotometer was usedto read the array. MassARRAY Workstation version 3.3 soft-ware was used to interpret this data. Sequenom Typer analyserversion 3.4 was used to manually review and analyse all rawcluster data.
GWAS replication study
Two replication studies were conducted using SequenomiPLEX technology. The data were analysed using PLINK(v1.07), with SNP QC procedure: Hardy-Weinberg (HWE)expectations (p≤0.001) in control samples, call rate of ≥80 %and MAF ≥0.01. Additionally, the sample genotyping successrate of ≥80 % was used. Post-QC, replication 1 consisted of
126 control and 59 affected GSDs from the United Kingdom.Replication 2 consisted of 65 control and 17 affected GSDsfrom Finland.
The 46 samples successfully genotyped on the Illuminaarray were also genotyped by Sequenom for these SNPs. Thelevel of genotype concordance between the two platforms wasassessed on a per sample and per SNP basis. Average sampleconcordance was 99 %, but ten SNPs showing <90 % con-cordance were excluded from subsequent analysis.
Fisher’s exact test for association was performed on eachreplication dataset. The Bonferroni correction method wasapplied for the number of SNPs successfully tested in eachdataset.
Joint analysis
ACochran–Mantel–Haenszel (2×2×K,K=2) test was used tocombine the data from each replication dataset.
Finally, the GWAS, replication 1 and replication 2 sampleswere combined in a Cochran–Mantel–Haentzel test (K=3). ABreslow-Day test p value for each SNP set at >0.05 was usedas an assurance of no significant heterogeneity in odds ratiosbetween strata in the joint analysis.
Targeted sequencing
Twenty-five regions totalling 6.5 Mb were selected for se-quencing. These regions included those surrounding SNPsthat were nominally replicated in the Sequenom follow-upstudy and interesting candidate genes, viz. CTNND1,ARVCF (both members of the p120 family), and MMP9,MMP13 (matrix metalloproteinases found to be up-regulatedin AF lesions (House et al. 2007)) and PTPN22.
All sequencing preparations and reactions were carried outas a commercial service at ARK-Genomics, Roslin Institute,The University of Edinburgh. Target enrichment was carriedout using custom-designed SureSelect in-solution capture(Agilent, Wokingham, UK). The libraries were hybridised tothe custom-designed biotinylated RNA baits. Streptavidin-coated magnetic beads were then used to capture thehybridised libraries. Unique index barcode tags were addedto enable multiple individuals to be sequenced in a single laneon the sequencer. Cluster generation was carried out using thecBot robot before 2×100 bp paired-end DNA sequencing onthe Illumina HiSeq 2000.
Dogs were selected for sequencing based on DNA qualityand quantity available. Four affected and four control GSDswere selected. This was in addition to one affected and onecontrol Leonberger, a dog breed that also suffers from anincreased prevalence of AF. Genomic DNA samples weresupplied at a concentration of 100 ng/μl and volume of 100 μl.
320 Immunogenetics (2014) 66:311–324

Targeted sequencing data analysis
The raw reads for each sample from each of the two Flow Cellsequencing lanes were separately aligned to the CanFam2.0reference genome with Burrows-Wheeler Aligner (BWA) ver-sion 0.6.1 (Li and Durbin 2009). The files were output in theSAM format. Samtools version 0.1.12 (Li et al. 2009) wasused to convert this file to the BAM format and local realign-ment to better distinguish between SNPs, and indels wasperformed using the ‘GATK indel-realigner package version1.3’ (DePristo et al. 2011). Duplicates were removed using theSamtools rmdup function before GATK was used to recali-brate the base quality scores. Samtools was then used tocombine the BAM files generated on the per lane basis so thatthere was now only one BAM file per sample.
‘GATK Unified Genotyper version 1.3’ was used to callSNPs and indels from the BAM input files. The CanFam2.0reference genome was used. SNPs and indels were calledseparately in the targeted regions. The standard GATK param-eters were otherwise used: stand_call_conf = 50,stand_emit_conf=10, dcov=100. Additionally, the BaseAlignment Quality (BAQ) was recalculated, which greatlyimproves SNP discovery by measuring the probability of abase being wrongly aligned (Li 2011). The results were outputin the VCF format.
During the variant calling procedure, various quality scoreswere tagged to each call. The next stage used the ‘GATKVariant Filtration’ function to refine the data by filtering outlow quality calls. The parameters used here follow the ‘BestPractice Variant Detection with the GATK v3’ issued by theBroad Institute:
http://gatkforums.broadinstitute.org/discussion/15/retired-best-practice-variant-detection-with-the-gatk-v3`
The called variants that passed the above QC filters werethen functionally annotated using ANNOVAR version2011Nov20 ‘annotate_variation.pl’ function (Wang et al.2010).
Targeted sequencing—novel variant association testing
The samples used for the replication study (plus extra con-trols) were assayed for 16 indel and 106 SNP variants discov-ered from sequencing. Sequenom iPLEX technology wasused to genotype these variants. The data were analysed usingPLINK (v1.07), with SNP QC procedure: Hardy-Weinberg(HWE) expectations (p≤0.001) in control samples, call rate of≥80 % and MAF ≥0.01. Additionally, the sample genotypingsuccess rate of ≥80 % was used. Post-QC, there were 178control and 79 affected GSDs from the United Kingdom.Additionally, there were 65 control and 18 affected GSDsfrom Finland.
Fisher’s exact test for association was performed on eachreplication dataset. The Bonferroni correction method was
applied for the number of SNPs successfully tested in eachdataset.
A Cochran–Mantel–Haenszel (2×2×K, K=2) test wasused to combine the data from each geographic dataset.
Protein conservation across Eutherian mammals
Jalview 2.8 was used to assess the conservation of proteinsequences across Eutherian mammals, where sequences wereavailable. Protein sequences were taken from Ensembl release67.
Ethics statement
Collection of blood samples from animals solely for researchpurposes, without a Home Office project licence, is prohibitedin the United Kingdom. However, residual blood remainingafter a diagnostic test may be used for research, and this doesnot require a licence. Residual blood was submitted by theattending veterinary surgeon with owner permission. Samplesfrom Finlandwere collected with owner consent and under thepermission of the animal ethical committee of the CountyAdministrative Board of Southern Finland (ESLH-2009-07827/Ym-23).
Acknowledgments We would like to acknowledge the Kennel Club(UK) for funding the GWAS, the American Kennel Club for funding theGWAS replication and sequencing and the University of Manchester forfunding the sequencing confirmation study (through an ‘Investing inSuccess’ award made to JM). We thank Turlough O’Neill for diagnosingmany of the UK samples and Hazel Platt for assistance with genotyping.We would also like to extend thanks to the technical staff supporting theUK DNA Archive for Companion Animals.
Conflict of interest The authors declare that they have no conflict ofinterest.
References
Alkim H, Ayaz S, Alkim C, Ulker A, Sahin B (2011) Continuous activestate of coagulation system in patients with nonthrombotic inflam-matory bowel disease. Clin Appl Thromb Hemost 17(6):600–604.doi:10.1177/1076029611405034
Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD,Lee JC, Goyette P, Imielinski M, Latiano A, Lagace C, Scott R,Amininejad L, Bumpstead S, Baidoo L, Baldassano RN, BarclayM,Bayless TM, Brand S, Buning C, Colombel JF, Denson LA, De VosM, Dubinsky M, Edwards C, Ellinghaus D, Fehrmann RS, FloydJA, Florin T, Franchimont D, Franke L, Georges M, Glas J, GlazerNL, Guthery SL, Haritunians T, Hayward NK, Hugot JP, Jobin G,Laukens D, Lawrance I, LemannM, Levine A, Libioulle C, Louis E,McGovern DP, Milla M, Montgomery GW, Morley KI, Mowat C,Ng A, NewmanW, Ophoff RA, Papi L, Palmieri O, Peyrin-BirouletL, Panes J, Phillips A, Prescott NJ, Proctor DD, Roberts R, RussellR, Rutgeerts P, Sanderson J, Sans M, Schumm P, Seibold F, SharmaY, Simms LA, Seielstad M, Steinhart AH, Targan SR, van den BergLH, Vatn M, Verspaget H, Walters T, Wijmenga C, Wilson DC,
Immunogenetics (2014) 66:311–324 321

Westra HJ, Xavier RJ, Zhao ZZ, Ponsioen CY, Andersen V, TorkvistL, Gazouli M, Anagnou NP, Karlsen TH, Kupcinskas L,Sventoraityte J, Mansfield JC, Kugathasan S, Silverberg MS,Halfvarson J, Rotter JI, Mathew CG, Griffiths AM, Gearry R,Ahmad T, Brant SR, Chamaillard M, Satsangi J, Cho JH,Schreiber S, Daly MJ, Barrett JC, Parkes M, Annese V,Hakonarson H, Radford-Smith G, Duerr RH, Vermeire S,Weersma RK, Rioux JD (2011) Meta-analysis identifies 29 addi-tional ulcerative colitis risk loci, increasing the number of confirmedassociations to 47. Nat Genet 43(3):246–252. doi:10.1038/ng.764
Arnett HA, Escobar SS, Viney JL (2009) Regulation of costimulation inthe era of butyrophilins. Cytokine 46(3):370–375. doi:10.1016/j.cyto.2009.03.009
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: anR library for genome-wide association analysis. Bioinformatics23(10):1294–1296. doi:10.1093/bioinformatics/btm108
Barnes A, O'Neill T, Kennedy LJ, Short AD, Catchpole B, House A,Binns M, Fretwell N, Day MJ, Ollier WE (2009) Association ofcanine anal furunculosis with TNFA is secondary to linkage dis-equilibrium with DLA-DRB1*. Tissue Antigens 73(3):218–224
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis andvisualization of LD and haplotype maps. Bioinformatics 21(2):263–265. doi:10.1093/bioinformatics/bth457
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, BrantSR, Silverberg MS, Taylor KD, Barmada MM, Bitton A,Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO,Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH,Targan SR, Xavier RJ, Libioulle C, Sandor C, Lathrop M, BelaicheJ, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, VanGossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M,Vermeire S, Louis E, Cardon LR, Anderson CA, Drummond H,NimmoE,AhmadT, Prescott NJ, Onnie CM, Fisher SA,Marchini J,Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P,Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M,GeorgesM, DalyMJ (2008) Genome-wide association definesmorethan 30 distinct susceptibility loci for Crohn's disease. Nat Genet40(8):955–962. doi:10.1038/ng.175
Boyko AR (2011) The domestic dog: man's best friend in the genomicera. Genome Biol 12(2):216. doi:10.1186/gb-2011-12-2-216
Burger MJ, Tebay MA, Keith PA, Samaratunga HM, Clements J, LavinMF, Gardiner RA (2002) Expression analysis of delta-catenin andprostate-specific membrane antigen: their potential as diagnosticmarkers for prostate cancer. Int J Cancer 100(2):228–237. doi:10.1002/ijc.10468
Day MJ (1993) Immunopathology of Anal Furunculosis in the Dog. JSmall Anim Pract 34(8):381–389. doi:10.1111/j.1748-5827.1993.tb02726.x
Day MJ, Weaver BMQ (1992) Pathology of surgically resected tissuefrom 305 cases of anal furunculosis in the dog. J Small Anim Pract33(12):583–589
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G,Annaert W, Von Figura K, Van Leuven F (1998) Deficiency ofpresenilin-1 inhibits the normal cleavage of amyloid precursor pro-tein. Nature 391(6665):387–390. doi:10.1038/34910
DeBusk LM, Boelte K, Min Y, Lin PC (2010) Heterozygous deficiencyof delta-catenin impairs pathological angiogenesis. J Exp Med207(1):77–84. doi:10.1084/jem.20091097
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C,Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A,Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, GabrielSB, Altshuler D, Daly MJ (2011) A framework for variation dis-covery and genotyping using next-generation DNA sequencingdata. Nat Genet 43(5):491–498. doi:10.1038/ng.806
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ,Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T,Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP,
Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, ChoJH (2006) A genome-wide association study identifies IL23R as aninflammatory bowel disease gene. Science 314(5804):1461–1463
Galandiuk S, Kimberling J, Al-Mishlab TG, Stromberg AJ (2005)Perianal Crohn disease: predictors of need for permanent diversion.Ann Surg 241(5):796–801, discussion 801-792
Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, Kalinoski AL,Morgan EE, Gohara AF, Nauli SM, Filipiak WE, Saunders TL,Joe B (2012) Targeted disruption of Adamts16 gene in a rat geneticmodel of hypertension. Proc Natl Acad Sci U S A 109(50):20555–20559. doi:10.1073/pnas.1211290109
Harkin KR, Walshaw R, Mullaney TP (1996) Association of perianalfistula and colitis in the German shepherd dog: response to high-dose prednisone and dietary therapy. J Am Anim Hosp Assoc 32(6):515–520
He Y, Kim H, Ryu T, Kang Y, Kim JA, Kim BH, Lee JH, Kang K, Lu Q,Kim K (2013) delta-catenin overexpression promotes angiogenicpotential of CWR22Rv-1 prostate cancer cells via HIF-1alpha andVEGF. FEBS Lett 587(2):193–199. doi:10.1016/j.febslet.2012.11.024
House A, Gregory SP, Catchpole B (2003) Expression of cytokinemRNA in canine anal furunculosis lesions. Vet Rec 153(12):354–358
House AK, Catchpole B, Gregory SP (2007) Matrix metalloproteinasemRNA expression in canine anal furunculosis lesions. Vet ImmunolImmunopathol 115(1–2):68–75. doi:10.1016/j.vetimm.2006.10.018
House AK, Binns MM, Gregory SP, Catchpole B (2009) Analysis ofNOD1, NOD2, TLR1, TLR2, TLR4, TLR5, TLR6 and TLR9 genesin anal furunculosis of German shepherd dogs. Tissue Antigens73(3):250–254. doi:10.1111/j.1399-0039.2008.01190.x
Hudson M, Chitolie A, Hutton RA, Smith MS, Pounder RE, WakefieldAJ (1996) Thrombotic vascular risk factors in inflammatory boweldisease. Gut 38(5):733–737
Joe B, Saad Y, Dhindaw S, Lee NH, Frank BC, Achinike OH, Luu TV,Gopalakrishnan K, Toland EJ, Farms P, Yerga-Woolwine S,Manickavasagam E, Rapp JP, Garrett MR, Coe D, Apte SS,Rankinen T, Perusse L, Ehret GB, Ganesh SK, Cooper RS,O'Connor A, Rice T, Weder AB, Chakravarti A, Rao DC,Bouchard C (2009) Positional identification of variants ofAdamts16 linked to inherited hypertension. Hum Mol Genet18(15):2825–2838. doi:10.1093/hmg/ddp218
Jostins L, Ripke S,Weersma RK, Duerr RH, McGovern DP, Hui KY, LeeJC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M,Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P,Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L,Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L,Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, BuningC, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL,Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, FranchimontD, Fransen K, Gearry R, Georges M, Gieger C, Glas J, HarituniansT, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L,Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW,Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, NewmanW, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M,Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, SchreiberS, Simms LA, Sventoraityte J, Targan SR, Taylor KD, TremellingM, Verspaget HW, De Vos M, Wijmenga C, Wilson DC,Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, ZhaoH, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A,Parkes M, Vermeire S, Barrett JC, Cho JH (2012) Host-microbeinteractions have shaped the genetic architecture of inflammatorybowel disease. Nature 491(7422):119–124. doi:10.1038/nature11582
Kabi A, Nickerson KP, Homer CR, McDonald C (2012) Digesting thegenetics of inflammatory bowel disease: insights from studies of
322 Immunogenetics (2014) 66:311–324

autophagy risk genes. Inflamm Bowel Dis 18(4):782–792. doi:10.1002/ibd.21868
Karlsson EK, Baranowska I,Wade CM, SalmonHillbertz NH, ZodyMC,Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ 3rd,Comstock KE, Keller ET, Mesirov JP, von Euler H, Kampe O,Hedhammar A, Lander ES, Andersson G, Andersson L, Lindblad-Toh K (2007) Efficient mapping of mendelian traits in dogs throughgenome-wide association. Nat Genet 39(11):1321–1328
Kennedy LJ, O'Neill T, House A, Barnes A, Kyostila K, Innes J, FretwellN, Day MJ, Catchpole B, Lohi H, Ollier WE (2008) Risk of analfurunculosis in German shepherd dogs is associated with the majorhistocompatibility complex. Tissue Antigens 71(1):51–56
Koren I, Reem E, Kimchi A (2010) DAP1, a novel substrate of mTOR,negatively regulates autophagy. Curr Biol 20(12):1093–1098. doi:10.1016/j.cub.2010.04.041
Li H (2011) Improving SNP discovery by base alignment quality.Bioinformatics 27(8):1157–1158. doi:10.1093/bioinformatics/btr076
Li H, Durbin R (2009) Fast and accurate short read alignment withBurrows-Wheeler transform. Bioinformatics 25(14):1754–1760.doi:10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G,Abecasis G, Durbin R, Proc GPD (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. doi:10.1093/bioinformatics/btp352
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB,Kamal M, Clamp M, Chang JL, Kulbokas EJ 3rd, Zody MC,Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, PontingCP, Galibert F, Smith DR, DeJong PJ, Kirkness E, Alvarez P, BiagiT, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ,DeCaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M,Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP,Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, WebberC, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M,Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H,Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M,Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, BoguslavskiyL, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N,Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A,Cooke P, Dawoe T, Daza R, Decktor K, DeGray S, Dhargay N,Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, DupesA, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D,Ferreira P, Fisher S, FitzGerald M, Foley K, Foley C, Franke A,Friedrich D, Gage D, Garber M, Gearin G, Giannoukos G, Goode T,Goyette A, Graham J, Grandbois E, Gyaltsen K, Hafez N, HagopianD, Hagos B, Hall J, Healy C, Hegarty R, Honan T, Horn A, HoudeN, Hughes L, Hunnicutt L, Husby M, Jester B, Jones C, Kamat A,Kanga B, Kells C, Khazanovich D, Kieu AC, Kisner P, Kumar M,Lance K, Landers T, Lara M, LeeW, Leger JP, Lennon N, Leuper L,LeVine S, Liu J, Liu X, Lokyitsang Y, Lokyitsang T, Lui A,Macdonald J, Major J, Marabella R, Maru K, Matthews C,McDonough S, Mehta T, Meldrim J, Melnikov A, Meneus L,Mihalev A, Mihova T, Miller K, Mittelman R, Mlenga V, MulrainL, Munson G, Navidi A, Naylor J, Nguyen T, Nguyen N, Nguyen C,Nguyen T, Nicol R, Norbu N, Norbu C, Novod N, Nyima T, OlandtP, O'Neill B, O'Neill K, Osman S, Oyono L, Patti C, Perrin D,Phunkhang P, Pierre F, Priest M, Rachupka A, Raghuraman S,Rameau R, Ray V, Raymond C, Rege F, Rise C, Rogers J, RogovP, Sahalie J, Settipalli S, Sharpe T, Shea T, Sheehan M, Sherpa N,Shi J, Shih D, Sloan J, Smith C, Sparrow T, Stalker J, Stange-Thomann N, Stavropoulos S, Stone C, Stone S, Sykes S, TchuingaP, Tenzing P, Tesfaye S, Thoulutsang D, Thoulutsang Y, Topham K,Topping I, Tsamla T, Vassiliev H, Venkataraman V, Vo A,Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A,Yadav S, Yang S, Yang X, Young G, Yu Q, Zainoun J, Zembek L,Zimmer A, Lander ES (2005) Genome sequence, comparative
analysis and haplotype structure of the domestic dog. Nature438(7069):803–819
Lu Q, Paredes M, Medina M, Zhou J, Cavallo R, Peifer M,Orecchio L, Kosik KS (1999) delta-catenin, an adhesivejunction-associated protein which promotes cell scattering. JCell Biol 144(3):519–532
Lu Q, Dobbs LJ, Gregory CW, Lanford GW, Revelo MP, Shappell S,Chen YH (2005) Increased expression of delta-catenin/neuralplakophilin-related armadillo protein is associated with the down-regulation and redistribution of E-cadherin and p120ctn in humanprostate cancer. Hum Pathol 36(10):1037–1048. doi:10.1016/j.humpath.2005.07.012
Mathews KA, Sukhiani HR (1997) Randomized controlled trial of cy-closporine for treatment of perianal fistulas in dogs. J Am Vet MedAssoc 211(10):1249–1253
Mathews KA, Ayres SA, Tano CA, Riley SM, Sukhiani HR, Adams C(1997) Cyclosporin treatment of perianal fistulas in dogs. Can Vet J38(1):39–41
Orozco G, Eerligh P, Sanchez E, Zhernakova S, Roep BO, Gonzalez-Gay MA, Lopez-Nevot MA, Callejas JL, Hidalgo C, Pascual-Salcedo D, Balsa A, Gonzalez-Escribano MF, Koeleman BP,Martin J (2005) Analysis of a functional BTNL2 polymorphismin type 1 diabetes, rheumatoid arthritis, and systemic lupuserythematosus. Hum Immunol 66(12):1235–1241. doi:10.1016/j.humimm.2006.02.003
Pesu M, Watford WT, Wei L, Xu LL, Fuss I, Strober W, Andersson J,Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y,Creemers J, O'Shea JJ (2008) T-cell-expressed proproteinconvertase furin is essential for maintenance of peripheral immunetolerance. Nature 455(7210):246. doi:10.1038/Nature07210
Porter S, Clark IM, Kevorkian L, Edwards DR (2005) The ADAMTSmetalloproteinases. Biochem J 386(Pt 1):15–27. doi:10.1042/BJ20040424
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D,Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC (2007)PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575. doi:10.1086/519795
Rath T, Roderfeld M, Graf J, Wagner S, Vehr AK, Dietrich C, Geier A,Roeb E (2006) Enhanced expression of MMP-7 and MMP-13 ininflammatory bowel disease: a precancerous potential? InflammBowel Dis 12(11):1025–1035. doi:10.1097/01.mib.0000234133.97594.04
Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A,Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, GriffithsAM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL,Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, ChoJH, Daly MJ, Brant SR (2007) Genome-wide association studyidentifies new susceptibility loci for Crohn disease and implicatesautophagy in disease pathogenesis. Nat Genet 39(5):596–604. doi:10.1038/ng2032
Rybicki BA, Walewski JL, Maliarik MJ, Kian H, Iannuzzi MC (2005)The BTNL2 gene and sarcoidosis susceptibility in AfricanAmericans and Whites. Am J Hum Genet 77(3):491–499. doi:10.1086/444435
SandbornWJ, Fazio VW, Feagan BG,Hanauer SB (2003)AGA technicalreview on perianal Crohn's disease. Gastroenterology 125(5):1508–1530
Stanley BJ, Hauptman JG (2009) Long-term prospective evaluation oftopically applied 0.1 % tacrolimus ointment for treatment of perianalsinuses in dogs. J AmVetMedAssoc 235(4):397–404. doi:10.2460/javma.235.4.397
Surridge AK, Rodgers UR, Swingler TE, Davidson RK, Kevorkian L,Norton R, Waters JG, Goldring MB, Parker AE, Clark IM (2009)Characterization and regulation of ADAMTS-16.Matrix Biol 28(7):416–424. doi:10.1016/j.matbio.2009.07.001
Immunogenetics (2014) 66:311–324 323

Tengvall K, Kierczak M, Bergvall K, Olsson M, Frankowiack M, FariasFH, Pielberg G, Carlborg O, Leeb T, AnderssonG, Hammarstrom L,Hedhammar A, Lindblad-Toh K (2013) Genome-wide analysis inGerman shepherd dogs reveals association of a locus on CFA 27with atopic dermatitis. PLoS Genet 9(5):e1003475. doi:10.1371/journal.pgen.1003475
TiversMS, Catchpole B, Gregory SP, House AK (2008) Interleukin-2 andinterferon-gamma mRNA expression in canine anal furunculosislesions and the effect of ciclosporin therapy. Vet ImmunolImmunopathol 125(1–2):31–36. doi:10.1016/j.vetimm.2008.04.018
Vatn M (2012) Environmental factors in the epidemiology of inflamma-tory bowel disease. In: Baumgart DC (ed) Crohn’s Disease andulcerative colitis. Springer, US, pp 17–38. doi:10.1007/978-1-4614-0998-4_2
Vizoso FJ, Gonzalez LO, Corte MD, Corte MG, Bongera M, Martinez A,Martin A, Andicoechea A, Gava RR (2006) Collagenase-3 (MMP-13) expression by inflamed mucosa in inflammatory bowel disease.
Scand J Gastroenterol 41(9):1050–1055. doi:10.1080/00365520600554667
Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotationof genetic variants from high-throughput sequencing data. NucleicAcids Res 38(16):e164. doi:10.1093/nar/gkq603
WilbeM, Jokinen P, Truve K, Seppala EH, Karlsson EK, Biagi T, HughesA, Bannasch D, Andersson G, Hansson-Hamlin H, Lohi H,Lindblad-Toh K (2010) Genome-wide association mapping iden-tifies multiple loci for a canine SLE-related disease complex. NatGenet 42(3):250–254. doi:10.1038/ng.525
Xiao R, Boehnke M (2009) Quantifying and correcting for the winner'scurse in genetic association studies. Genet Epidemiol 33(5):453–462. doi:10.1002/gepi.20398
ZengY, Abdallah A, Lu JP,Wang T, ChenYH, Terrian DM, KimK, LuQ(2009) delta-Catenin promotes prostate cancer cell growth andprogression by altering cell cycle and survival gene profiles. MolCancer 8:19. doi:10.1186/1476-4598-8-19
324 Immunogenetics (2014) 66:311–324