Embed Size (px)
Citation preview

Hereditary Motor and Sensory Neuropathywith Agenesis of the Corpus Callosum
Nicolas Dupre, MD, MSc,1 Heidi C. Howard, PhD,1 Jean Mathieu, MD, MSc,2 George Karpati, MD,3
Michel Vanasse, MD,4 Jean-Pierre Bouchard, MD,5 Stirling Carpenter, MD,6 and Guy A. Rouleau, MD, PhD1
Hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum (OMIM 218000) is an auto-somal recessive disease of early onset characterized by a delay in developmental milestones, a severe sensory-motorpolyneuropathy with areflexia, a variable degree of agenesis of the corpus callosum, amyotrophy, hypotonia, and cogni-tive impairment. Although this disorder has rarely been reported worldwide, it has a high prevalence in the Saguenay-Lac-St-Jean region of the province of Quebec (Canada) predominantly because of a founder effect. The gene defectresponsible for this disorder recently has been identified, and it is a protein-truncating mutation in the SLC12A6 gene,which codes for a cotransporter protein known as KCC3. Herein, we provide the first extensive review of this disorder,covering epidemiological, clinical, and molecular genetic studies.
Ann Neurol 2003;54:9–18
Hereditary motor and sensory neuropathy associatedwith agenesis of the corpus callosum (HMSN/ACC)(OMIM 218000), also known as peripheral neuropa-thy associated with agenesis of the corpus callosum(ACCPN) or Andermann syndrome, is an autosomalrecessive disease of early onset. It is characterized by adelay in developmental milestones, a severe progressivesensory-motor neuropathy with areflexia, a variable de-gree of agenesis of the corpus callosum, amyotrophy,hypotonia, cognitive impairment, an atypical psychosis,and occasional dysmorphic features (high arched pal-ate, syndactyly). Howard and colleagues1 recently iden-tified the gene responsible for HMSN/ACC, solute car-rier family 12 member 6 (SLC12A6), which codes forK�-Cl� cotransporter 3 (KCC3). In this review, wepresent a complete summary on the current state ofknowledge of HMSN/ACC. Although rare, this disor-der is particularly interesting because it represents aunique example of a gene defect causing both develop-mental and neurodegenerative problems involving boththe central and the peripheral nervous systems. Thecallosal agenesis indicates a defect in axonal migrationduring embryogenesis, whereas the progressive neurop-athy along with the cognitive involvement and theatypical psychotic occurrences suggest a degenerative
process. The severe sensory-motor neuropathy is strik-ing both for its distinctive pathological features (axonalswelling) and for the distribution of its abnormalities(cranial nerves, nerve roots, peripheral nerves). HMSN/ACC is an ion transporter disorder that is quite differ-ent from other diseases caused by dysfunctional ionchannels. The fact that the K-Cl cotransporter 3 pro-tein is at least indirectly involved in early axonal mi-gration across the corpus callosum and in maintenanceof the peripheral nervous system (PNS) throughout lifecan lead the way to a generation of new hypotheses onion transporter function.
Epidemiological StudiesHMSN/ACC is found mainly in the French Canadian(FC) population of Quebec, and specifically in two re-gions of northeastern Quebec, the Saguenay-Lac-St-Jean (SLSJ) region, and the Charlevoix County.2,3
In a study based on 101 individuals distributed in 82families, De Braekeleer and colleagues2 analyzed thegeographical distribution of HMSN/ACC in northeast-ern Quebec. The overall incidence at birth was 1 in2,117 live births and the carrier rate was 1 in 23 in-habitants. Genealogical reconstruction was attemptedin 161 obligate carriers, and a set of 22 founders was
From the 1Centre for Research in Neurosciences and the Depart-ment of Neurology and Neurosurgery, McGill University, Mon-treal; 2Hotel-Dieu de Chicoution, Chicoutioni; 3Montreal Neuro-logical Hospital and Institute, Department of Neurology andNeurosurgery, McGill University, Montreal; 4Hopital Ste-Justine,Universite de Montreal, Montreal; 5Department of Neurology, Ho-pital de l’Enfant-Jesus, Laval University, Quebec City, Quebec,Canada; and 6Hospital Sao Joao, Porto, Portugal.
Received Jan 2, 2003, and in revised form Mar 20. Accepted forpublication Mar 20, 2003.
Address correspondence to Dr Rouleau, Centre for Research inNeurosciences and the Department of Neurology and Neurosurgery,McGill University, 1650 Cedar Avenue, Montreal, Quebec, H3GIA4 Canada. E-mail: [email protected]
NEUROLOGICAL PROGRESS
© 2003 American Neurological Association 9Published by Wiley-Liss, Inc., through Wiley Subscription Services

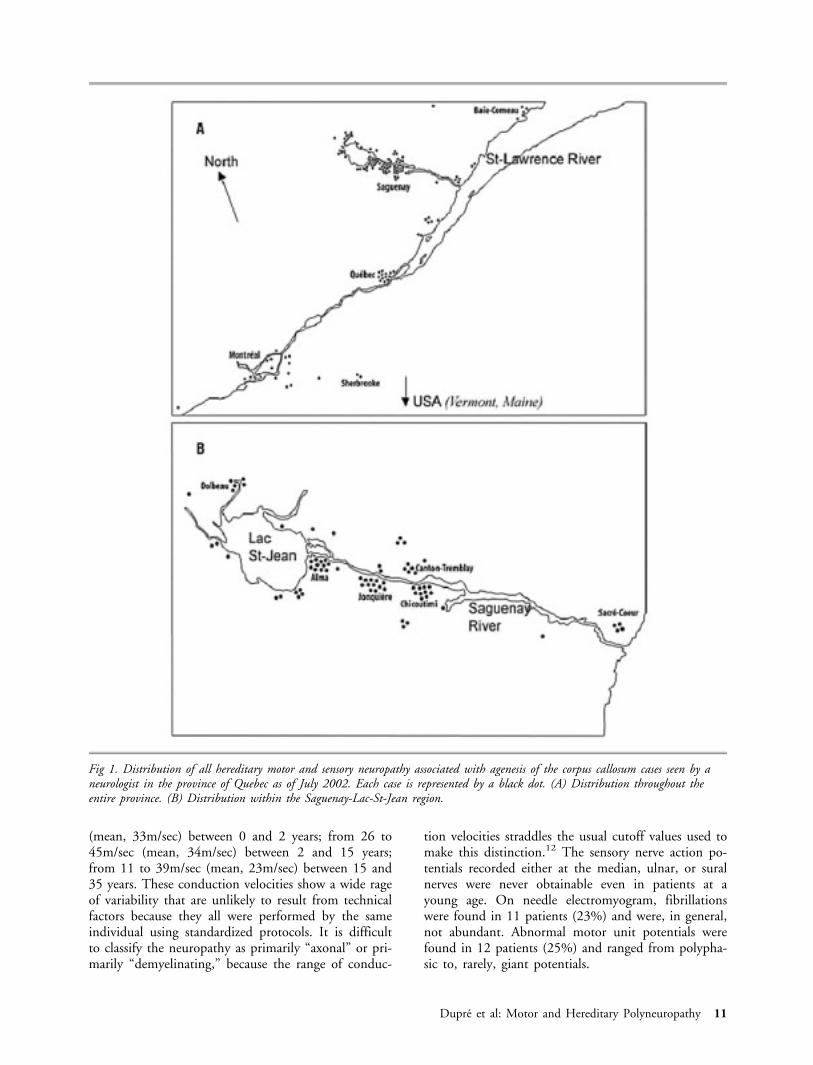
common to all of them. All these founders originatedfrom France, including 3 from Poitou, 11 from Perche,and 4 from unknown regions. The authors concludedthat based on genealogical data, the founders whocame from France in the late 17th century probablyestablished themselves in the Charlevoix County wherethey lived until their descendants moved to SLSJ in theearly 19th century. The Charlevoix County contrib-uted 70% of the immigrants to SLSJ during the first30 years of settlement, and, because migration wasmainly of familial type,4 many child carriers of theHMSN/ACC gene may have entered the SLSJ regionwith their parents, contributing to the high gene fre-quency in the region. The most represented foundercouple in SLSJ was found in only 8 of the 75 completegenealogies, indicating that quite a few carriers musthave introduced the HMSN/ACC gene in this region.We recently performed an updated survey of the geo-graphical distribution of HMSN/ACC cases acrossQuebec, by collecting data from each major pediatrichospital and rehabilitation center of the province (Fig1). These results show that despite important migra-tion within the province over the past 20 years, thedisease remains concentrated in the SLSJ region.
Clinical StudiesClinical DescriptionIn 1966, Leblanc and colleagues5 first reported the ex-istence of HMSN/ACC. He recognized that this “syn-drome of callosal agenesis” had a familial componentand was particularly frequent in the SLSJ region ofQuebec. Subsequently, Andermann and colleagues6 de-scribed HMSN/ACC as an autosomal recessive diseasecharacterized by agenesis of the corpus callosum, a mo-tor neuropathy (which they thought was compatiblewith anterior horn cell disease), mental retardation,areflexia, amyotrophy, hypotonia, and dysmorphic fea-tures. Subsequent descriptions of the disease recognizedthat it involved a sensory-motor neuropathy ratherthan an anterior horn cell disease.7 Mathieu and col-leagues8 studied 64 patients between the ages of 2 and34 years old. Forty-five (70%) of the 64 patients fromthis phenotypic study were later tested for the HMSN/ACC gene mutation. They were all found to be ho-mozygous for a single nucleotide deletion in exon 18 ofthe SLC12A6 gene (c.2436delG, Thr813fsX813),thereby providing molecular confirmation of the diag-nosis and confirming the genetic homogeneity of thispopulation (N. Dupre, unpublished data). Mathieuand colleagues8 found the following clinical character-istics in these patients: ptosis (59%), upper gaze palsy(30%), facial asymmetry (34%), areflexia (100%), hy-potonia (36%), amyotrophy (86%), tremor (25%), sei-zures (17%), early Achilles’ tendon retraction (47%),scoliosis (86%). Dysmorphism also was evaluated in a
systematic way, with the following characteristicsamong their 64 patients: brachycephaly (16%), high-arched palate (39%), overriding of the first toe (16%),and partial syndactyly of second to third toes (8%).Clinical evolution was quantified with the followingmeasures: average age of onset of walking 3.8 years (47patients), average age of end of walking capacity 13.8years (35 patients), average age of appearance of scoliosis10.4 years (22 patients), and average age of death 24.8years (6 patients). We recently reviewed 49 FC HMSN/ACC cases (subjects who are now deceased) and foundthe average age of death to be 33 years; this is a longersurvival period than previously estimated (N. Dupre,unpublished data). The main features of ACCPN in theFC population can be found in Table 1.
Cognitive FunctionThe cognitive function of individuals with HMSN/ACC has been addressed in relatively few studies. Intheir clinical study of 64 cases, Mathieu and col-leagues8 evaluated 53 subjects for cognitive dysfunc-tion. Using the clinical classification of Taft9 to stratifythe degree of mental retardation, they found that 8%of cases had normal intelligence, 49% had mild mentalretardation, 40% had moderate mental retardation,and 4% had severe mental retardation. They did not,however, indicate the age at which the patients wereevaluated, which, in light of the progressive nature ofthe disease, would be relevant information. They alsoreported that 39% of patients developed “psychotic ep-isodes” after the age of 15 years. Paranoid delusions,depressive states, visual hallucinations, auditory hallu-cinations, or “autistic-like” features characterized theseepisodes. Filteau and colleagues10 studied a cohort of62 patients to look at the relationship between imagingfeatures and the appearance of “psychotic episodes” inHMSN/ACC. They identified 20 patients (32%) whohad presented such episodes, with a mean age at thetime of evaluation of 27.6 years and a mean age ofonset of psychosis of 19.5 years. The psychiatric eval-uation was performed using a semistructured interviewaccording to Diagnostic and Statistical Manual III-Rcriteria. They found that all 20 identified patients hadparanoid delusions often accompanied by visual or au-ditory hallucinations described as “monsters” or “per-secutors.” Interestingly, it appears that psychosis wasmore common in HMSN/ACC patients (32%) than inadult populations of mentally retarded individuals(18%).11
ElectrophysiologyWe recently reviewed the electrophysiological studies of48 patients (aged 0 to 35 years) evaluated in Chi-coutimi by one of the authors (J.M) over the last 20years (Fig 2). We found that the median motor nerveconduction velocities ranged from 16 to 57m/sec
10 Annals of Neurology Vol 54 No 1 July 2003
(mean, 33m/sec) between 0 and 2 years; from 26 to45m/sec (mean, 34m/sec) between 2 and 15 years;from 11 to 39m/sec (mean, 23m/sec) between 15 and35 years. These conduction velocities show a wide rageof variability that are unlikely to result from technicalfactors because they all were performed by the sameindividual using standardized protocols. It is difficultto classify the neuropathy as primarily “axonal” or pri-marily “demyelinating,” because the range of conduc-
tion velocities straddles the usual cutoff values used tomake this distinction.12 The sensory nerve action po-tentials recorded either at the median, ulnar, or suralnerves were never obtainable even in patients at ayoung age. On needle electromyogram, fibrillationswere found in 11 patients (23%) and were, in general,not abundant. Abnormal motor unit potentials werefound in 12 patients (25%) and ranged from polypha-sic to, rarely, giant potentials.
Fig 1. Distribution of all hereditary motor and sensory neuropathy associated with agenesis of the corpus callosum cases seen by aneurologist in the province of Quebec as of July 2002. Each case is represented by a black dot. (A) Distribution throughout theentire province. (B) Distribution within the Saguenay-Lac-St-Jean region.
Dupre et al: Motor and Hereditary Polyneuropathy 11

ImagingIn their first report of the disease, Leblanc and col-leagues5 had recognized the callosal agenesis based onpneumoencephalography. This was confirmed in laterstudies with the use of computed tomography (CT)imaging, and then with magnetic resonance imaging(Fig 3). Unfortunately, because few magnetic resonanceimages have been performed on these patients, wemust rely mainly on the data provided by CT imagingto assess the extent of anatomical abnormalities in thebrain. Mathieu and colleagues8 performed CT imagingon 64 patients with HMSN/ACC and found no evi-dence of ACC in 33% of cases, partial ACC in 9%,and complete ACC in 58%. They also found 4.7% ofpatients with diffuse cerebral atrophy and 17% withcerebellar atrophy. However, CT criteria are not idealfor assessing the degree of callosal agenesis. Althoughsigns of complete or near complete ACC are unlikelyto be missed, it is much more difficult to assess partialACC. In addition, even in cases where the corpus cal-losum is anatomically well preserved, axonal loss is seenon pathology (see autopsy findings below), which sug-gests that most, if not all cases of HMSN/ACC havesome degree of callosal abnormality.
PathologyWe reviewed five autopsies of patients who died be-tween 26 and 37 years of age, as well as six sural nervesof patients biopsied between the ages of 2 and 12years. Two of the cases autopsied have had moleculartesting performed and were found to be homozygousfor the typical FC mutation, c.2436delG mutation, inexon 18 of SLC12A6.
Table 1. Features of HMSN/ACC in the French CanadianPatients
Clinical featuresDysmorphic traits
Hypertelorism (usually mild)Syndactyly of second and third toesBrachycephalyHigh-arched palateOverriding of the first toe
Cognitive FunctionMild to severe mental retardation“Psychotic episodes” appearing usually during adolescence
Cranial NervesEyelid ptosis (symmetrical or asymmetrical)Facial plegia (symmetrical or asymmetrical; sometimes
associated with hemifacial atrophy)Esotropia or exotropia (due to variable combinations of
oculomotor nerve palsies)Upper gaze palsyHorizontal nystagmus
MotorProgressive distal and proximal symmetrical limb weak-
ness with muscle atrophy (the infant is invariably hy-potonic in the first year of life, learns to walk with adevice between 2 and 5 yr old and eventually becomesconfined to a wheelchair)
Diffuse limb tremor (probably due to the polyneurop-athy)
ReflexesInvariably absent from infancy
SensoryAll modalities are moderately to severely affected from
infancyOther
ScoliosisPulmonary restrictive syndromeSeizures (infrequent)
InvestigationLumbar puncture
Usually mild elevation of proteinsNerve conductions and EMG in young children
Invariably absent sensory potentials (sural, median, cubi-tal) from infancy
Variable motor nerve conduction velocities (median, cubi-tal, tibial)
FibrillationsEEG
Usually normalImaging
May have no ACC, partial ACC, or complete ACCMay see mild cortical or cerebellar atrophy in older sub-
jectsSural nerve biopsy and muscle biopsy
Sural nerve biopsies show a lack of large myelinated fi-bers, signs of axonal loss (ovoids of Wallerian degener-ation), and some enlarged axons that on electron mi-croscopy show a decreased density of neurofilaments
Muscle biopsies show nonspecific signs of chronic dener-vation atrophy
AutopsyThe hallmark of autopsies is the swollen axons demon-
strated in cranial nerve samples (third and seventhespecially) as well as in dorsal and ventral nerve roots.Swollen axons can also be found scattered in thewhite matter
The brain shows either no ACC, partial ACC, or com-plete ACC with preservation of Probst bundle
HMSN/ACC � hereditary motor and sensory neuropathy withagenesis of the corpus callosum; EMG � electromyogram; EEG �electroencephalogram.
Fig 2. Graph representing the median motor nerve conductionvelocity (m/sec) as a function of the age in 48 French Cana-dian patients with hereditary motor and sensory neuropathyassociated with agenesis of the corpus callosum. Most patientswere evaluated at a young age when they presented with de-layed motor development and hypotonia. MNCV � motornerve conduction velocity.
12 Annals of Neurology Vol 54 No 1 July 2003
AUTOPSY FINDINGS. In two cases, the corpus callosumwas entirely absent. One of these tested positive for thec.2436delG mutation in SLC12A6; the other was nottested. In a third case, only a small rounded portion ofcorpus callosum was present anteriorly and was in con-tact with a thin lamina terminalis. The gyral pattern on
the medial side of the hemisphere corresponded to thatusually associated with callosal agenesis. Probst’s bun-dles were prominent in these brains. In the last twocases, the corpus callosum was present in its entirety,although the posterior portion was thinner than theanterior (Fig 4A). One of these cases had had molecu-lar testing and was found to have the typical FC mu-tation. The neocortex appeared grossly normal in allpatients, as did the hemispheric white matter.
Microscopic examination showed scattered smalloval vacuoles in the white matter of the brain in allcases. They could be mistaken for Buscaino bodies, ex-cept that they never contained basophilic or metachro-matic material, but were optically empty or containedan eosinophilic web. The evidence suggested that theyhad resulted from axonal swelling. The corpus callo-sum of the two patients in which it was completelypresent appeared normal in its anterior portion exceptfor occasional vacuoles. In the posterior part, there wasprogressively more evidence of axonal loss, with in-creased packing density of oligodendroglial cells andproliferation of astrocytes. Where axonal loss was great-est, vacuoles were absent. Occasional pinkish, slightlyenlarged axons could be identified on hematoxylin andeosin stains.
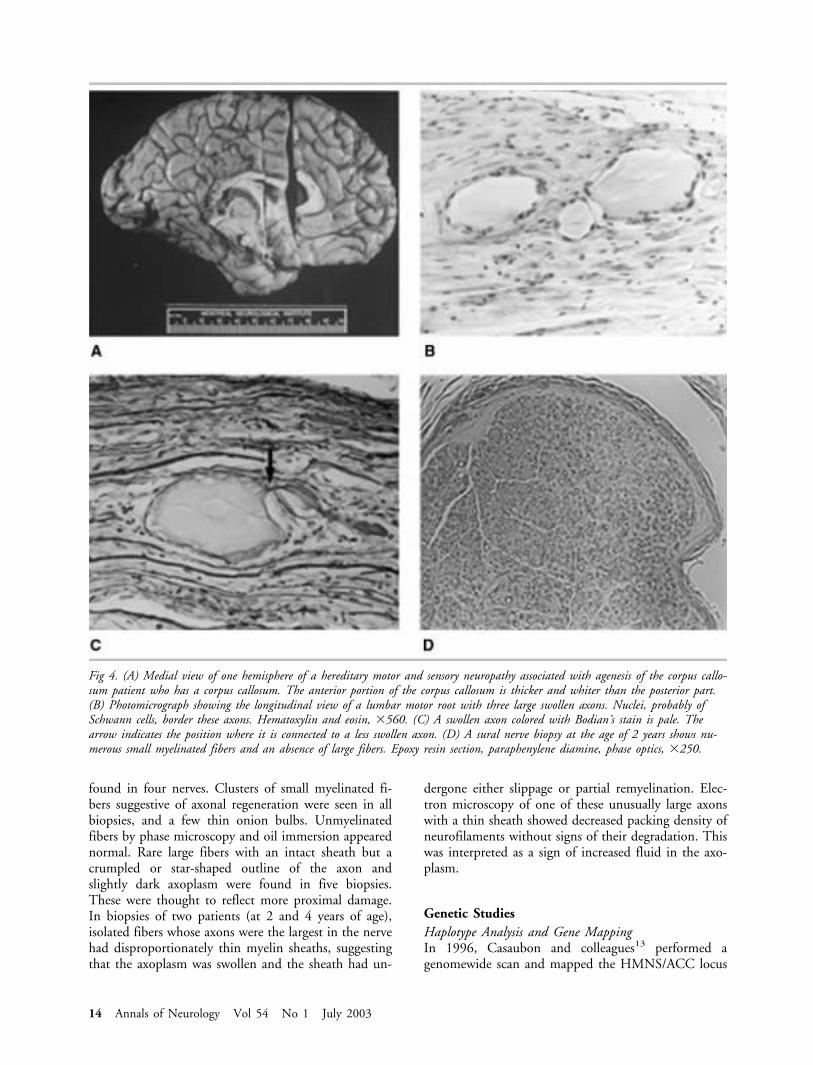
In contrast with the subtle microscopic changes inthe cerebral white matter, there were striking abnor-malities in the spinal nerve roots and cranial nerves.Massively enlarged axons up to 120�m in diametercould be identified on paraffin sections (see Fig 4B).On stains for axons, such as Bodian’s (see Fig 4C),these immense axons were a pale gray, but numerousaxons with a lesser degree of enlargement showeddarker staining that approached that of normal axons.In some sections of roots, there were numerous tinyaxons, sometimes with a random or recurrent coursethat suggested aberrant regeneration. Moderate axonalloss per unit area was noted. No myelin could bestained around the largest axons, whereas moderatelyenlarged ones had abnormally thin sheaths. Epoxy resinsections confirmed the findings of paraffin sections onthe roots and showed numerous onion bulb forma-tions. Teased fibers from motor roots showed thegreatest degree of abnormality at approximately 1cmfrom the spinal cord. Axonal enlargement tended topersist through several internodes, although focal ac-centuation of enlargement occurred. Balls of Schwanncells adhered to teased fiber bundles.
BIOPSY FINDINGS IN SURAL NERVES. All biopsiesshowed an almost total lack of large myelinated fibers.Small myelinated fibers from the three oldest patients(6, 7, and 12 years old) were somewhat reduced innumbers, whereas in two patients (2 and 3 years old)small myelinated fibers were increased compared withcontrols (see Fig 4D). A few Wallerian ovoids were
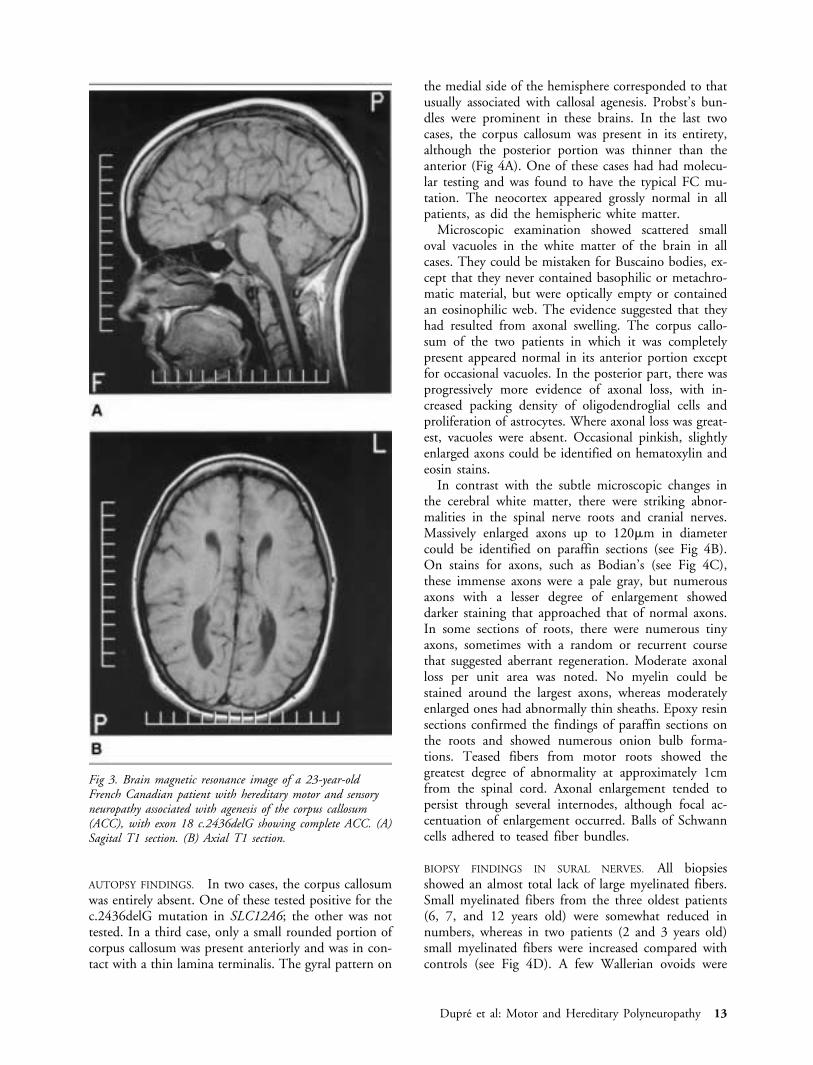
Fig 3. Brain magnetic resonance image of a 23-year-oldFrench Canadian patient with hereditary motor and sensoryneuropathy associated with agenesis of the corpus callosum(ACC), with exon 18 c.2436delG showing complete ACC. (A)Sagital T1 section. (B) Axial T1 section.
Dupre et al: Motor and Hereditary Polyneuropathy 13
found in four nerves. Clusters of small myelinated fi-bers suggestive of axonal regeneration were seen in allbiopsies, and a few thin onion bulbs. Unmyelinatedfibers by phase microscopy and oil immersion appearednormal. Rare large fibers with an intact sheath but acrumpled or star-shaped outline of the axon andslightly dark axoplasm were found in five biopsies.These were thought to reflect more proximal damage.In biopsies of two patients (at 2 and 4 years of age),isolated fibers whose axons were the largest in the nervehad disproportionately thin myelin sheaths, suggestingthat the axoplasm was swollen and the sheath had un-
dergone either slippage or partial remyelination. Elec-tron microscopy of one of these unusually large axonswith a thin sheath showed decreased packing density ofneurofilaments without signs of their degradation. Thiswas interpreted as a sign of increased fluid in the axo-plasm.
Genetic StudiesHaplotype Analysis and Gene MappingIn 1996, Casaubon and colleagues13 performed agenomewide scan and mapped the HMNS/ACC locus
Fig 4. (A) Medial view of one hemisphere of a hereditary motor and sensory neuropathy associated with agenesis of the corpus callo-sum patient who has a corpus callosum. The anterior portion of the corpus callosum is thicker and whiter than the posterior part.(B) Photomicrograph showing the longitudinal view of a lumbar motor root with three large swollen axons. Nuclei, probably ofSchwann cells, border these axons. Hematoxylin and eosin, �560. (C) A swollen axon colored with Bodian’s stain is pale. Thearrow indicates the position where it is connected to a less swollen axon. (D) A sural nerve biopsy at the age of 2 years shows nu-merous small myelinated fibers and an absence of large fibers. Epoxy resin section, paraphenylene diamine, phase optics, �250.
14 Annals of Neurology Vol 54 No 1 July 2003

to a 5cM region on chromosome 15. The FC foundereffect suggested by previous epidemiological studiessubsequently was confirmed by haplotype and linkagedisequilibrium analyses.13,14 Furthermore, Howard andcolleagues1 established that the most common disease-associated haplotype covering 4cM on chromosome 15is present on approximately 64% of disease chromo-somes of FC HMSN/ACC patients. In addition,marker D15S1232, which shared the greatest linkagedisequilibrium with the HMNS/ACC status, waspresent in more than 97% of affected chromosomes.1
Howard and colleagues1 reduced the HMNS/ACCcritical region to approximately 1,000kb and proceededto test genes within this interval. The SLC12A6 gene,which codes for a potassium chloride cotransporter(KCC3) protein, was found to have a guanine deletionin exon 18 (c.2436delG, Thr813fsX813) on both ho-mologs in all but one FC HMNS/ACC patient.1 ThisFC HMNS/ACC patient was found to be a compoundheterozygote with one chromosome 15 homolog bear-ing the c.2436delG mutation, whereas the other ho-molog has adjacent cytosine and thymine deletedwith a guanine insertion (c.1584_1585delCTinsG,Phe529fs532 mutation) in exon 11 of the SLC12A6gene.1 This patient’s phenotype does not differ signif-icantly from the classic HMNS/ACC phenotype. Fur-thermore, as expected, all patients homozygous forc.2436delG mutation in SLC12A6 were homozygousfor at least a portion of the founder haplotype, whereasthe compound heterozygote had one founder haplo-type and one nonfounder haplotype at flanking mark-ers. A total of 110 unaffected individuals from theSLSJ region were screened for the exon 18(c.2436delG) mutation. Four individuals (3.6%) werefound to be carriers of the mutation, therefore con-firming the carrier rate reported in this region by pre-vious epidemiological studies.2
Although it is clear that a geographical cluster existsin the SLSJ region of the Province of Quebec, very fewsimilar cases have been described outside Quebec (Ta-ble 2). Although the genealogical studies suggest an an-cestral couple coming from France in the late 17thcentury, no cases of HMSN/ACC have so far been re-ported in France. Various centers in France have beencontacted, and a few cases have been identified withatypical yet mildly comparable phenotypes, but no mu-tations in SLC12A6 were detected in these individualssuggesting that these are not authentic HMSN/ACCcases. As for other cases described in the literatureworldwide, we found only six15–17 that have clinicalcharacteristics completely compatible with those de-scribed in our large sample of FC HMSN/ACC pa-tients.8 These reported non-FC cases have yet to betested for the HMSN/ACC gene mutation. We have,however, formed collaborations with clinicians follow-ing up patients with typical characteristics of HMSN/ACC (unreported in the literature) and were able toidentify mutations in the HMSN/ACC gene in twonon-FC families.
A brother (4 years old) and his sister (5 years old)from the region of Verona (Italy) born from unrelated,unaffected parents presented with developmental delay,a sensory-motor axonal polyneuropathy, and callosalagenesis. Both have the same mutation in exon 15 ofSLC12A6 (c.2023C-T, Arg675X).
Two boys of Turkish origin born from unaffectedparents who are second-degree cousins presented withdevelopmental delay, areflexia, hypotonia, a sensory-motor polyneuropathy, and complete callosal agenesis.Both have the same nonsense mutation in exon 22 ofSLC12A6 (c.3031C-T, Arg1011X).
The finding of non-FC cases is significant because itconfirms the existence of HSMN/ACC outside of FCand predicts the existence of other non-FC cases.
Table 2. Published Cases with a Phenotype Similar to That of French Canadian HMSN/ACC
EthnicOrigin
No. ofSiblings Clinical Features Imaging NCS/EMG Nerve Biopsy
Inheritance(SLC12A6mutation) Reference
Italian(Veneto)
1 Developmental delay,hypotonia,areflexia, amyotro-phy, bilateral pto-sis, strabismus
Complete ACC onCT
Axonal motor poly-neuropathy
Fewer myelinated fibers,“demyelination”
AR (not tested) 15
Austrian 2 Developmental delay,areflexia, amyotro-phy
Complete ACC onMRI
Axonal sensory-motorpolyneuropathy
Fewer myelinated fibers,thin myelin sheaths,onion bulbs
AR (not tested) 16
Tanzanian(Mwanza)
3 Developmental delay,hypertelorism,areflexia, amyotro-phy
Complete or partialACC on CT
Axonal sensory-motorpolyneuropathy
Degeneration and vacu-olation of axons
AR (not tested) 17
Italian(Veneto)
2 Developmental delay ACC Axonal sensory-motorpolyneuropathy
Not done AR (exon 15,c.2023C-T)
1
Turkish 2 Developmental delay,hypotonia,areflexia
Complete ACC onMRI
Sensory-motor poly-neuropathy (axonal?)
Not done AR (exon 22,c.3031C-T)
1
HMSN/ACC � hereditary motor and sensory neuropathy with a genesis of the corpus callosum; NCS � nerve conduction studies; EMG �electroencephalomyogram; CT � computed tomography; AR � autosomal recessive; MRI � magnetic resonance imaging.
Dupre et al: Motor and Hereditary Polyneuropathy 15
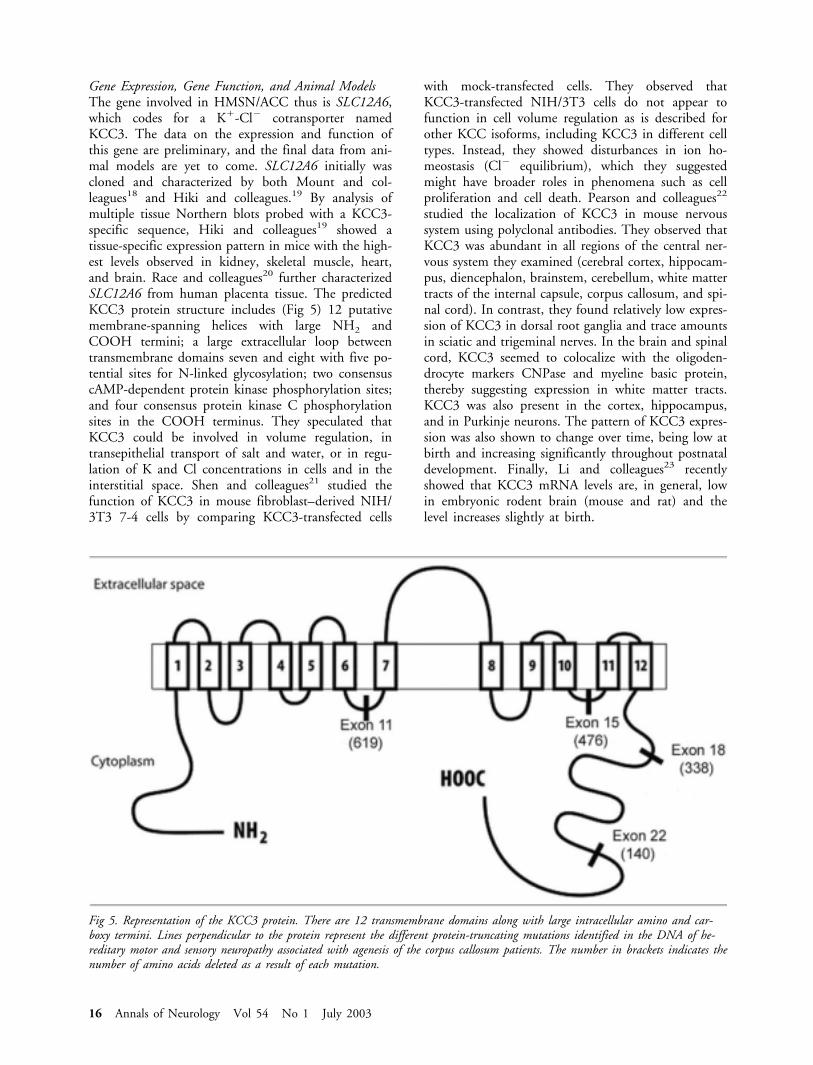
Gene Expression, Gene Function, and Animal ModelsThe gene involved in HMSN/ACC thus is SLC12A6,which codes for a K�-Cl� cotransporter namedKCC3. The data on the expression and function ofthis gene are preliminary, and the final data from ani-mal models are yet to come. SLC12A6 initially wascloned and characterized by both Mount and col-leagues18 and Hiki and colleagues.19 By analysis ofmultiple tissue Northern blots probed with a KCC3-specific sequence, Hiki and colleagues19 showed atissue-specific expression pattern in mice with the high-est levels observed in kidney, skeletal muscle, heart,and brain. Race and colleagues20 further characterizedSLC12A6 from human placenta tissue. The predictedKCC3 protein structure includes (Fig 5) 12 putativemembrane-spanning helices with large NH2 andCOOH termini; a large extracellular loop betweentransmembrane domains seven and eight with five po-tential sites for N-linked glycosylation; two consensuscAMP-dependent protein kinase phosphorylation sites;and four consensus protein kinase C phosphorylationsites in the COOH terminus. They speculated thatKCC3 could be involved in volume regulation, intransepithelial transport of salt and water, or in regu-lation of K and Cl concentrations in cells and in theinterstitial space. Shen and colleagues21 studied thefunction of KCC3 in mouse fibroblast–derived NIH/3T3 7-4 cells by comparing KCC3-transfected cells
with mock-transfected cells. They observed thatKCC3-transfected NIH/3T3 cells do not appear tofunction in cell volume regulation as is described forother KCC isoforms, including KCC3 in different celltypes. Instead, they showed disturbances in ion ho-meostasis (Cl� equilibrium), which they suggestedmight have broader roles in phenomena such as cellproliferation and cell death. Pearson and colleagues22
studied the localization of KCC3 in mouse nervoussystem using polyclonal antibodies. They observed thatKCC3 was abundant in all regions of the central ner-vous system they examined (cerebral cortex, hippocam-pus, diencephalon, brainstem, cerebellum, white mattertracts of the internal capsule, corpus callosum, and spi-nal cord). In contrast, they found relatively low expres-sion of KCC3 in dorsal root ganglia and trace amountsin sciatic and trigeminal nerves. In the brain and spinalcord, KCC3 seemed to colocalize with the oligoden-drocyte markers CNPase and myeline basic protein,thereby suggesting expression in white matter tracts.KCC3 was also present in the cortex, hippocampus,and in Purkinje neurons. The pattern of KCC3 expres-sion was also shown to change over time, being low atbirth and increasing significantly throughout postnataldevelopment. Finally, Li and colleagues23 recentlyshowed that KCC3 mRNA levels are, in general, lowin embryonic rodent brain (mouse and rat) and thelevel increases slightly at birth.
Fig 5. Representation of the KCC3 protein. There are 12 transmembrane domains along with large intracellular amino and car-boxy termini. Lines perpendicular to the protein represent the different protein-truncating mutations identified in the DNA of he-reditary motor and sensory neuropathy associated with agenesis of the corpus callosum patients. The number in brackets indicates thenumber of amino acids deleted as a result of each mutation.
16 Annals of Neurology Vol 54 No 1 July 2003

Howard and colleagues1 studied the functional con-sequence of the predominant FC mutation(c.2436delG) of SLC12A6 in Xenopus laevis oocytes.They observed that the truncated mutant was appro-priately glycosylated and expressed at the cellular mem-brane, but that it was nonfunctional. A KCC3 knock-out mouse was generated in which exon 3 of SLC12a6was removed and substituted with a �-galactosidase/neomycin cassette, therefore deleting the remainder ofthe protein. KCC3 expression was significantly reducedin heterozygous mice and was absent in homozygousmice. Starting at 2 weeks of age, KCC3-null miceshowed an abnormal locomotor function comparedwith controls and heterozygotes. They had a low pos-ture with limb weakness and disorganized limb move-ments. Behavioral tests (exploratory locomotion andprepulse inhibition) were conducted to assess centralnervous system function: both heterozygous and ho-mozygous mice exhibited a significantly lower level ofexploratory behavior; prepulse inhibition was abnormalboth in heterozygous and in homozygous mice, butmore so in the later. The researchers performed de-tailed anatomical studies on the brain and spinal cordof three homozygous mice and three control mice: ho-mozygous mice showed no specific abnormalities frommorphometric analysis (cortex, corpus callosum, cere-bellum) and immunostaining. Interestingly, however,sciatic nerves of homozygous mice showed many verylarge axons with thin myelin sheaths, reminiscent ofthose seen in HMSN/ACC patient’s biopsies, as well asextensive Wallerian degeneration.
DiscussionIn light of the HMSN/ACC gene discovery, manyquestions need to be addressed. What is the role ofKCC3 in the development and maintenance of thecentral nervous system and PNS? Is HMSN/ACC pri-marily a disease of the axon or the myelin sheath? Canall the features of HMSN/ACC be explained by a sin-gle gene defect, or are other genes involved, eitherwithin the same chromosomal region or on other chro-mosomes as modifier genes? Can environmental factorsinfluence the phenotypic expression of HMSN/ACC?
Lack of KCC3 in the developing nervous systemmay increase the susceptibility of damaging the fibersmigrating across the midline close to the subarachnoidspace to form the corpus callosum. As this structureforms during embryogenesis, absence of KCC3 musthave phenotypic effects early during neuronal develop-ment. One possible mechanism was proposed by Shenand colleagues,21 who showed that KCC3 may be in-volved in ion homeostasis (Cl� equilibrium) with apossible role in cell proliferation via ion-sensitive ki-nases. The occurrence of cases without callosal agenesisleaves open the question of interacting genetic and en-vironmental factors, such as prenatal tobacco or alcohol
exposure.24 The findings in the PNS, on the otherhand, are progressive and do not suggest any migratoryabnormality. The site of maximum damage in the PNSappears to be in the nerve roots, where nerve fibers arebathed in cerebrospinal fluid. This is where the greatmajority of swollen axons are encountered, along withaberrant regeneration and Schwann cell proliferation.
The presence of some onion bulbs in peripheralnerve biopsies25 and in nerve roots raises the questionas to whether Schwann cell or axon is the primary siteof damage. �-Iminodiproprionitrile is a toxin that leadsto the formation of neurofilamentous masses that fo-cally distend axons.26 Animals treated continuouslywith this toxin have shown axonal swellings in nerveroots along with segments of demyelination and accu-mulation of onion bulbs.26 Axonal swelling has notbeen described as a result of demyelination, and acutelydemyelinated axons tend to show reduced cross sec-tional area. The pathological evidence in HMSN/ACCpoints to the axon as the primary structure damagedand suggests that watery swelling of the axon is theearliest morphological abnormality. This would beconsistent with abnormal electrolyte gradients, whichmay occur with dysfunction of an electrolyte channelin the axonal membrane. Acute swelling of axons atnodes of Ranvier has been reported to occur with tox-ins that inactivate sodium channels.27
Selective large myelinated fiber loss as seen in thenerve biopsies could be a useful diagnostic clue in caseswhere the corpus callosum is present, although it isseen also in Friedreich’s ataxia and some children con-sidered to have the neuronal type of hereditary motorand sensory neuropathy, especially with onset in earlychildhood.28 In patients with HMSN/ACC, it almostcertainly reflects greater susceptibility of large axons inthe roots with their more extensive axolemma. Varioussecondary changes in axons, such as accumulation ofdamaged organelles, probably distal to the site of max-imal damage, could be observed, but this could be eas-ily distinguished from the accumulation of neurofila-ments in giant axonal neuropathy and of variousabnormal organelles in neuroaxonal dystrophy.
Now that we know the gene defect responsible forHMSN/ACC, it will be important to continue detailedclinical studies as well as pursue detailed molecularstudies to better understand the pathophysiology ofthis disease. In addition, it will now be possible to de-termine the prevalence of this disorder outside of Que-bec.
This work was supported by “La Fondation des Jumelles Coudees”and the Canadian Institute of Health Research (#15459, G.A.R.).
Dupre et al: Motor and Hereditary Polyneuropathy 17

References1. Howard H, Mount D, Rochefort D, et al. Mutations in the
K-Cl cotransporter KCC3 cause a severe peripheral neuropathyassociated with agenesis of the corpus callosum. Nat Genet2002;32:384–392.
2. De Braekeleer M, Dallaire A, Mathieu J. Genetic epidemiologyof sensorimotor polyneuropathy with or without agenesis of thecorpus callosum in northeastern Quebec. Hum Genet 1993;91:223–227.
3. Andermann E, Andermann F, Bergeron D, et al. Familial agen-esis of the corpus callosum with sensorimotor neuronopathy:genetic and epidemiological studies of over 170 patients. CanJ Neurol Sci 1979;6:400.
4. Gauvreau D, Guerin M, Hamel M. De Charlevoix Saguenay:mesure et characteristiques du mouvement migratoire avant1991. In: Bouchard D, De Braekeleer M, eds. Histoire d’ungenome: population, societe et genetique dans l’est du Quebec.Sillery: Presses de l’Universite du Quebec, 1991:76–106.
5. Leblanc G, Mortezai M, Popez-Pinto C. Agenesie du corps cal-leux (12 cas). Neurochirurgie 1966;7:789.
6. Andermann E, Andermann F, Joubert M, et al. Familial agen-esis of the corpus callosum with anterior horn cell disease. Asyndrome of mental retardation, areflexia and paraplegia. TransAm Neurol Assoc 1972;97:242–244.
7. Andermann E. Sensorimotor neuronopathy with agenesis of thecorpus callosum. In: Teopoulos M, ed. Handbook of clinicalneurology. Vol 42. Amsterdam: North-Holland, 1981:100–103.
8. Mathieu J, Bedard F, Prevost C, Langevin P. NeuropathieSensitivo-Motrice Hereditaire avec ou sans Agenesie du CorpsCalleux : Etude Radiologique et Cinique de 64 cas. Can J Neu-rol Sci 1990;17:103–108.
9. Taft L. Mental retardation: an overview. Pediatr Ann 1973;2:10–24.
10. Filteau M, Pourche E, Bouchard R, et al. Corpus callosumagenesis and psychosis in Andermann syndrome. Arch Neurol1991;48:1275–1280.
11. Linaker O, Nitter R. Psychopathology in institutionalized men-tally retarded adults. Br J Psychiat 1990;156.
12. Dyck P, Chance P, Leno R, Carney J. Hereditary motorand sensory neuropathies. In: Dyck P, Thomas P, Griffin J,eds. Peripheral neuropathy. Philadelphia: Saunders, 1993:1094–1136.
13. Casaubon L, Melancon M, Lopes-Cendes I, et al. The generesponsible for a severe form of peripheral neuropathy andagenesis of the corpus callosum maps to chromosome 15q.Am J Hum Genet 1996;58:28–34.
14. Howard H, Dube M-P, Prevost C, et al. Fine mapping thecandidate region for peripheral neuropathy with or withoutagenesis of the corpus callosum in the French Canadian popu-lation. Eur J Hum Genet 2002;10:406–412.
15. Battistella P, Drigo P, Laverda A, et al. The Andermann syn-drome. Progressive neuropathy, mental retardation with agene-sis of the corpus callosum. Ital J Pediatrics 1987;13:200–202.
16. Hauser E, Bittner P, Liegl C, et al. Occurrence of Andermannsyndrome out of French Canada—agenesis of the corpus callo-sum with neuronopathy. Neuropediatrics 1993;24:107–110.
17. Deleu D, Bamanikar S, Muirhead D, Louon A. Familial pro-gressive sensorimotor neuropathy with agenesis of the corpuscallosum (Andermann syndrome): a clinical, neuroradiologicaland histopathological study. Eur Neurol 1997;37:104–109.
18. Mount D, Mercado A, Song L, et al. Cloning and character-ization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem 1999;274:16355–16362.
19. Hiki K, D’Andrea R, Furze J, et al. Cloning, characterization,and chromosomal location of a novel human K-Cl cotrans-porter. J Biol Chem 1999;274:10661–10667.
20. Race J, Makhlouf F, Logue P, et al. Molecular cloning andfunctional characterization of KCC3, a new K-Cl cotransporter.Am J Physiol 1999;277:C1210–C1219.
21. Shen M, Chou C, Hsu K, et al. The KCl cotransporter isoformKCC3 can play an important role in cell growth regulation.Proc Natl Acad Sci USA 2001;98:14714–14719.
22. Pearson M, Lu J, Mount D, Delpire E. Localization of theK-Cl cotransporter, KCC3, in the central and peripheral ner-vous system: expression in the choroid plexus, large neuronsand white matter tracts. Neuroscience 2001;103:481–491.
23. Li H, Tornberg J, Kaila K, et al. Patterns of cation-chloridecotransporter expression during embryonic rodent CNS devel-opment. Eur J Neurosci 2002;16:2358–2370.
24. Sowell E, Mattson S, Thompson P, et al. Mapping callosalmorphology and cognitive correlates: effects of heavy prenatalalcohol exposure. Neurology 2001;57:235–244.
25. Carpenter S. The pathology of the Andermann syndrome. In:Lassonde M, Jeeves M, eds. Callosal agenesis: a natural splitbrain? New York: Plenum, 1994:27–30.
26. Griffin J, Price D. beta, beta�-iminodiproprionitrile andhexacarbon neuropathies. Evidence for an axonal influence. LabInvest 1981;45:130–141.
27. Love S, Cruz-Hofling M. Acute swelling of nodes of Ranviercaused by venoms which slow inactivation of sodium channels.Acta Neuropathol 1986;70:1–9.
28. Ouvrier R, McLeod J, Pollard J. Peripheral neuropathy inchildhood. New York: Raven Press, 1990:91–97.
18 Annals of Neurology Vol 54 No 1 July 2003





























