Embed Size (px)
Citation preview

Eur. J. Biochem. 223, 947-956 (1994) 0 FEBS 1994
High affinity of ergopeptides for cytochromes P450 3A Importance of their peptide moiety for P450 recognition and hydroxylation of bromocriptine
Marie-Anne PEYRONNEAU', Marcel DELAFORGE', RenC RIVIERE', Jean-Paul RENAUD' and Daniel MANSUY Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques Unit6 de Recherche AssociCe au CNRS (URA 400), Universitk RenC Descartes, France
* Service Hospitalier Frederic Joliot, Departement de Biologie, CEN Saclay, Gif Sur Yvette, France
(Received March 9May 16, 1994) - EJB 94 0327/4
The interaction between rat and human liver cytochromes P450 with a series of lysergic acid derivatives and ergopeptide alkaloids was studied by difference visible spectroscopy. Ergopeptides, like bromocriptine, ergocryptine and dihydroergotamine, strongly interacted with rat liver micro- somes with the appearance of a difference spectrum which is characteristic of their binding to a protein site close to the heme. The intensity of this spectrum was clearly dependent on the amounts of P450s 3A in the microsomes and was at its maximum in dexamethasone-treated rat microsomes. All the ergopeptides studied exhibited a high affinity for rat P450s 3A (K- around 1 pM), although lysergic acid derivatives not bearing the tripeptide moiety failed to give significant interactions with these P450s. A cyclic azatripeptide exhibiting a structure very similar to that of the tripeptide moiety of ergopeptides also interacted with P450s 3A with appearance of an intense type I difference spectrum. Very similar results were observed with two allelic forms of human liver P450 3A4, P450 NF25 and P450 hPCN1, produced in yeast. In both cases all the ergopeptides studied showed high affinities for the P450s (K? 0.6-2.2 pM) and an intense shift from the low-spin to the high-spin state upon substrate binding (60- 100% spin shift). Lysergic acid derivatives not bearing the tripep- tide group of ergopeptides also completely failed to interact with P450s 3A4.
Liver microsomes from rats pretreated with dexamethasone, a specific inducer of P450 3A, were found to be particularly active for the hydroxylation of bromocriptine, which occurs at the level of its tripeptide moiety. Human liver microsomes as well as P450 NF25 and P450 hPCNl also exhibited a high activity for bromocriptine hydroxylation at this level.
These results show that ergopeptides exhibit a particularly high affinity for P450s of the 3A subfamily. The tripeptide moiety of ergopeptides is essential for their recognition by P450s 3A and binds at a site close to P450 heme, producing type-I difference spectra. Accordingly, at least one of the studied ergopeptides, bromocriptine, is hydroxylated by P450s 3A at the proline ring of the cyclopeptide moiety. As cyclosporine is known to be a good substrate of P450s 3A, these results suggest that P450s 3A may be especially prone in a general manner to recognize and oxidize peptides or pseudopeptides.
Cytochrome P450 enzymes constitute a superfamily of heme-thiolate proteins that catalyse the primary oxidation of a wide variety of natural endogenous substrates like steroids, fatty acids, prostaglandins, leukotrienes and lipid hydroper- oxides. They also play an important role in the metabolism of exogenous compounds like drugs, procarcinogens, solvents, anesthetics and environmental pollutants. Their broad sub- strate specificity is now well understood on the basis of en- zyme multiplicity (Gonzalez, 1992). More than 220 P450s have been sequenced and characterized; they have been clas- sified on the basis of primary amino acid sequence similarity (Nelson et al., 1993). Changes in the P450-dependent metab- olism of a drug very often occur when a co-administered
Correspondence to D. Mansuy, URA 400 CNRS, UniversitC RenC Descartes, 45 Rue des Saints-Pkres, F-75270 Paris Cedex 06, France
Fax: +33 142868387. Abbreviation. P450, cytochrome P450. Enzyme. Cytochrome P450 (EC 1.14.14.1).
drug acts as an inducer or inhibitor of the same P450 iso- zyme. This may lead to dramatic changes of the pharmacoki- netic parameters of the drug and, from time to time, to the appearance of severe secondary toxic effects. In order to pre- dict such drug interactions in man, it is very important to determine the substrate specificity of each human P450 iso- zyme.
CYP 3A4, a P450 subfamily highly expressed in human liver, is important from a pharmacological and toxicological point of view, not only because of its relative abundance in human liver (Guengerich and Turvy, 1991) but also because it is involved in the metabolism of many widely used drugs including nifedipine (Guengerich et al., 1986a), quinidine (Guengerich et al., 1986b), erythromycin and troleandomycin (Pessayre et a]., 1982; Watkins et al., 1985; Combalbert et al., 1989; Brian et al., 1990; Renaud et al., 1990), cyclo- sporin A (Kronbach et al., 1988; Aoyama et al., 1989; Com- balbert et al., 1989), 17 a-ethynylestradiol (Guengerich, 1988), midazolam (Kronbach et al., 1989), lidocaine (Bar-

948
Bromocriptine R 1 = H R z = H
M1 and M2 metabolites R I = O H R 2 = H
M3 and M4 metabolites R l = O H R 2 = O H
Fig. 1. Chemical structure of bromocriptine and its major oxi- dized metabolites in rats and humans. The structures are taken from Maurer et al. (1982, 1983).
getzi et al.. 1989; Imaoka et al., 1990), and diltiazem (Pichard et al., 1990).
Several cDNA clones related to CYP 3A4 have been iso- lated: NF25 (Beaune el al., 1986), lzPCNl (Gonzalez et al., 1988), and NFlO (Bork et al., 1989), showing only point differences in the translated amino acid sequences. NF25 and hPCNl were isolated from different liver cDNA libraries and are considered as allelic variants. They have been expressed in yeast (Brian et al., 1990; Renaud el al., 1990; Peyronneau et al., 1993). hPCNl has also been expressed in COS cells (Gonzalez et al., 1988) and in Hep G2 cells (Aoyama et al., 1989) and some catalytic activities of the corresponding pro- teins have been studied. P4.50~ of the 3A subfamily have been implicated in several drug interactions involving certain macrolide antibiotics like erythromycin and troleandomycin (Babany et al., 1988; Periti et al., 1992; Mansuy and Dela- forge, 1993). This is due to the strong inhibitory effect of these macrolides which are oxidized by P450s 3A to nitro- soalkane metabolites that strongly bind to P450-iron(II).
The metabolism in man of two ergot alkaloids of the ergopeptide type, dihydroergotamine and bromocriptine, has been shown to be inhibited by co-administration of erythro- mycin or troleandomycin (Nelson et al., 1990; Hayton, 1969; Martinet and Kiechel, 1983; Varoquaux et al., 1981). This suggests that these alkaloids may be oxidized by P45Os 3A in human liver.
Bromocriptine is an ergot alkaloid with dopaminergic agonist properties. It is able to inhibit prolactin secretion and is widely used in the treatment of hyperprolactinaemia. Bro- mocriptine has also been shown to be effective in the treat- ment of Parkinson's disease. In addition, this drug can correct abnormal secretions of growth hormone. Its metabolism has been studied in animals and man, in vivo and in cell cultures (Maurer et al., 1982, 1983). Primary oxidation occurs on the proline ring of the pseudopeptide moiety of the molecule and leads to 8'-hydroxy-bromocriptines. Further oxidation leads to 8',9'-dihydroxy derivatives (Fig. 1 ). A similar pathway was proposed for the biotransformation of dihydroergotam- ine in man, except that, in contrast to bromocriptine, dihy- droergotamine is also oxidized at the level of the indole part of the molecule (Maurer and Frick, 1984).
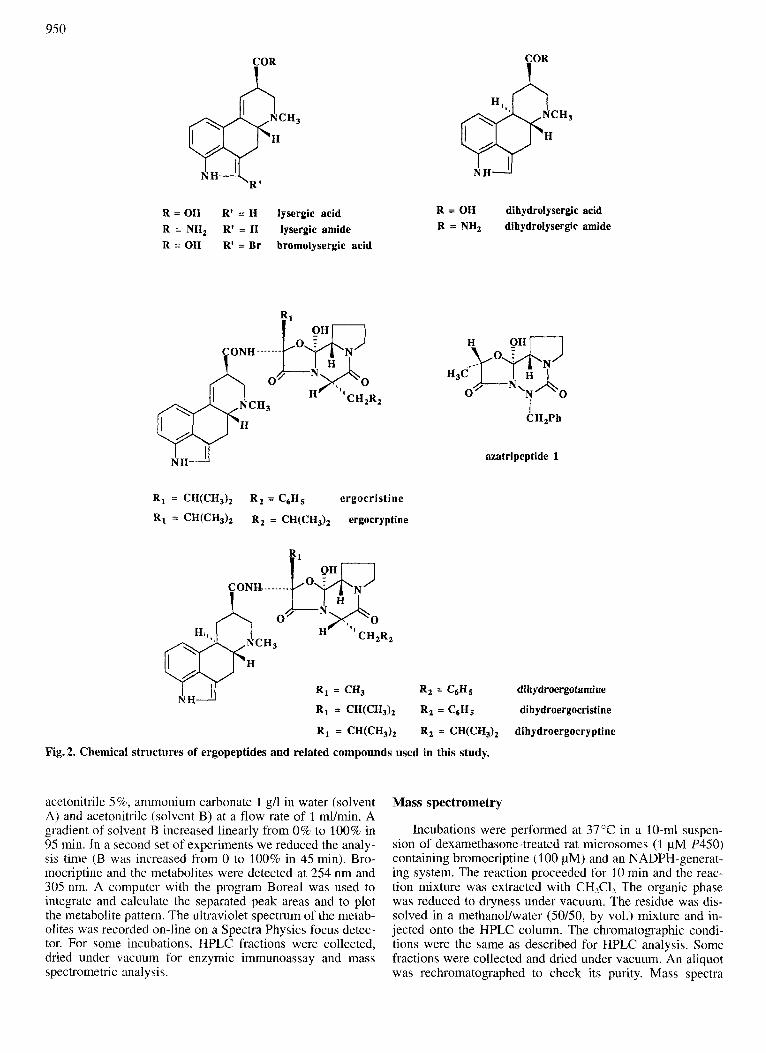
Ergopeptides are amides formed between lysergic acid and cyclic tripeptides containing a proline residue (Fig. 2). Their interactions with cytochromes P450 have not been studied so far, except for a preliminary work concerning the interaction of only one of them, dihydroergotamine, with rat liver P4SOs (Delaforge et a]., 1989). Moreover, the nature of the P450 isozymes responsible for the oxidation of these ergopeptides in humans or in rats has not been determined.
Since previously reported interactions between erythro- mycin and ergopeptides in humans suggested that ergopep- tides could be oxidized by P450s 3A, we have studied the interactions of eleven compounds related to ergot alkaloids and lysergic acid with rat liver P450s and human liver P45Os 3A4 expressed in yeast, as well as the oxidation of one of them, bromocriptine, by these cytochromes. This paper re- ports results showing that P45Os 3A exhibit a particularly high affinity for ergopeptides. It demonstrates for the first time that the tripeptide moiety of ergopeptides plays a key role in their interaction with P45Os 3A active site, as lysergic acid derivatives not bearing this moiety do not bind to P450s 3A and an azatripeptide, exhibiting a structure very similar to the tripeptide moiety of ergopeptides but not bearing the lysergic acid moiety, does bind to P450s 3A. Moreover, it provides evidence showing that an ergopeptide, bromocrip- tine, is hydroxylated at the level of its tripeptide ring by P45Os 3A.
EXPERIMENTAL PROCEDURES
Chemicals Bromocriptine, ergocryptine and 3-methylcholanthrene
were purchased from Sigma Chemical Co. ; NADPH, NADP, glucose 6-phosphate, glucose 6-phosphate dehydrogenase were obtained from Boehringer Mannheim. Dihydrolysergic acid, bromolysergic acid, dihydroergocristine, dihydroer- gocryptine and dihydroergotamine were obtained from CEN (CEA, Saclay). The amides of lysergic acid and dihydrolys- ergic acid were kindly provided by P. Dostert from Farmitalia Carlo Erba and the azatripeptide 1 by Dr G. Lucente (Univer- sity of Rome, Italy). Dexamethasone and clofibrate were ob- tained from Janssen Chemicals. Troleandomycin was a gift from Pfizer.
All molecular biological reagents were of analytical grade. The ingredients for yeast culture media were purchased from Difco. Rabbit liver cytochrome b, was puri- fied according to Strittmatter et al. (1978) and was kindly provided by Dr D. Pompon (CGM, Gif-sur-Yvette, France) as well as the yeast strains W(R) engineered in their genomic DNA (Truan et al., 1993). Other chemicals and solvents used were of the highest quality commercially available.
Enzyme preparation Preparation of rut liver microsomes
Male Sprague-Dawley rats (200 - 250 g) were provided laboratory chow and water ad libitum. After 10 days of adap- tation, animals were treated intraperitoneally either with 3- methylcholanthrene (20 mgkg, in corn oil, for 4 days), phe- nobarbital (80 mgkg in 0.9% saline, for 4 days), dexametha- sone (100 mgkg, in corn oil, for 4 days), clofibrate (500 mg/ kg, in corn oil, for 4 days), troleandomycin (500 mgkg, in corn oil, for 3 days). The control animals were treated with corn oil (0.5 ml).

949
Rat liver microsomes were prepared as reported pre- viously (Kremers et al., 1981) and stored at -80°C before use.
Human liver microsomes
Human liver samples were obtained from a kidney donor (H97 as described by Dansette et al., 1991); microsomes were prepared as previously described (Kremers et al., 1981).
Yeast strain, yeast transformation, cell culture and microsome preparation
Expression vector construction, yeast strains, yeast trans- formation and culture media were previously described (Pompon, 1988; Cullin and Pompon, 1988; Renaud et al., 1990, Peyronneau et al., 1992). Human P450 NF25 cDNA or P450 hPCNl cDNA, which were kindly provided by F. P. Guengerich (Beaune et al., 1986) and F. J. Gonzalez (Gonza- lez et al., 1988), were inserted into the YeDP1/8-2 (V8) expression vector based on the yeast 2 p origin of replication and on the URA3 selection marker. The heterologous cDNA was placed under the transcriptional control of the galactose- inducible GALlO-CYCl hybrid yeast promoter (Guarente et al., 1982). In this study, we used yeast microsomes contain- ing a human liver P450 3A4 [NF25-V8-W(R) or hPCN1-V8- W(R)] expressed in Saccharomyces cerevisiae as well as yeast control microsomes [VS-W(R)]. The W(R) strain, (MAT a, le~2~," ' , his3",", ura3', ade2-1, trpl', canR, cyr') which overexpresses yeast endogenous NADPH-P450 re- ductase (Truan et al., 1993) was used for expression.
Transformations were performed according to a modified LiCl method (Cullin and Pompon, 1988). Transformed yeast were grown at 28°C in a minimal medium containing 2% (madvol.) D-galactose, 0.7% (mass/vol.) yeast nitrogen base without amino acids, 0.1 % (mass/vol.) bactocasaminoacids, 0.002% (mass/vol.) tryptophan and 0.004% (masdvol.) ade- nine (Urban et al., 1990). Yeast microsomes were prepared as previously described (Peyronneau et al., 1992) using glass beads to mechanically disrupt the yeast cell walls. The microsomal pellet was homogenized in 50 mM Tris/HCl pH 7.4 containing 1 mM EDTA and 20% glycerol (by vol.), aliquoted, frozen in liquid N, and stored at -80°C until use.
Quantitation of the different enzymes in microsomal fractions
Protein content in microsomal suspensions was deter- mined by the method of Lowry et al. (1951) using bovine serum albumin as standard. Total P450 concentration was measured according to Omura and Sato (1964).
Study of substrate binding to microsomal P450s by difference visible spectroscopy
Yeast microsomes were suspended in 50 mM Tris/HCI, 1 mM EDTA, pH 7.4 to obtain a P450 NF25 concentration of 200 nM; the solution (1 ml) was equally divided between the two cuvettes of a Kontron 820 spectrophotometer. After recording the baseline, aliquots (0.5 -2 pl) of substrate solu- tions (0.1 - 10 mM in dimethyl sulfoxide) were added to the sample cuvette, the same volume of solvent being added to the reference cuvette. The difference spectrum was recorded
between 380-500 nm at 20°C; 0-10 mM substrate concen- tration range was thus covered. The experiments carried out on rat microsomes were similar except that a P450 concen- tration of 2 pM was used.
Bromocriptine oxidation assays
Rat and human microsome incubations
Typical incubations, containing (final volume of 1 ml) rat or human liver microsomal suspensions (equivalent to 1 pM P450), bromocriptine (200 pM added from a fresh solution in methanol) and an NADPH-generating system (NADP' 0.5 mM, glucose 6-phosphate 5 mM and 1 U glucose-6-phos- phate dehydrogenase) diluted in a 0.1 M potassium phos- phate, 0.1 mM EDTA pH 7.4 buffer, were performed at 37 "C. The oxidation reaction was initiated by addition of the NADPH-generating system after a preincubation of 2 min at 37"C, proceeded for 10 or 30 min and was stopped by addi- tion of 1 ml acetonitrile. Samples were centrifuged at 13000 rpm and 40 pl of the supernatant analysed by HPLC. The main metabolites observed in all experiments, M,, M2, M, and M4, exhibited an ultraviolet spectrum identical to that of bromocriptine ; their amounts were calculated on the basis of their absorbance at 254 nm. In some incubations, the for- mation of isobromocriptine and its hydroxylated metabolites was detected. These compounds derived from an epimeriza- tion at position 8 of the corresponding bromocriptine deriva- tives; they have been observed in the in vivo metabolism of bromocriptine (Maurer et al., 1982, 1983). The relative amounts of these isobromocriptine derivatives greatly de- pended on the time spent between the incubation and HPLC analysis. If the samples were injected into the HPLC just after the end of the incubation, they were formed in very low amounts.
Yeast microsomes incubations
Typical incubations performed at 37 "C included bro- mocriptine (200 pM, 10 pl from a fresh stock solution 10 mM in methanol), yeast microsomes containing P450 NF25 (0.2 pM), NADPH (0.5 mM) and 50 mM Tris/HCl pH 7.4, containing 1 mM EDTA in a final volume of 0.5 ml. When rabbit liver cytochrome b, was added (0.2 pM, 5 pl from a 20 pM stock solution), microsomal suspension diluted in buffer was first preincubated for 15 min on ice with cyto- chrome b, then with the substrate for 3 min at 37°C. The reaction was initiated by addition of NADPH, proceeded for 10 min at 37°C and was then quenched by the addition of 800 p1 CH,Cl,. The reaction mixture was extracted and the organic phase reduced to dryness under a N, stream at room temperature. The residue was dissolved in 200 p1 of a metha- nol/water (50/50, by vol.) mixture for HPLC analysis.
In all experiments, microsomes from the same yeast strain containing only the expression vector without the NF25 cDNA were used in parallel as a negative control by including equivalent amounts of microsomal protein in the incubation mixture.
HPLC analysis
HPLC separations were performed at room temperature on a C18 Ultrabase 5-pm column (250X4.6mm; SFCC, Neuilly Plaisance, France). The mobile phase included
950
COR 1 FOR
( $ C H 3
N H
R = OH R‘ = H lysergic acid R = NH, R‘ = H lysergic amide R = OH R’ = Br bromolysergic acid
R = OH dihydrolysergic acid R = NH, dihydrolysergic amide
azatripeptide 1
R , = CH(CH3)z R2 = C6H5 ergocristine
R i = CH(CH3)z R, = CH(CH3), ergocryptine
CH,R,
R , = CH3 R2 = C6Hs di hydroergotarnine
R, = CH(CH,), R2 = C6H5 di hydroergocristine
R, = CH(CH3), R, = CH(CH,), dihydroergocryptine
Fig. 2. Chemical structures of ergopeptides and related compounds used in this study.
acetonitrile 5 % , ammonium carbonate 1 g/l in water (solvent A) and acetonitrile (solvent B) at a flow rate of 1 ml/min. A gradient of solvent B increased linearly from 0% to 100% in 95 min. In a second set of experiments we reduced the analy- s is time (B was increased from 0 to 100% in 45 min). Bro- mocriptine and the metabolites were detected at 2.54 nm and 305 nm. A computer with the program Boreal was used to integrate and calculate the separated peak areas and to plot the metabolite pattern. The ultraviolet spectrum of the metab- olites was recorded on-line on a Spectra Physics focus detec- tor. For some incubations, HPLC fractions were collected, dried under vacuum for enzymic immunoassay and mass spectrometric analysis.
Mass spectrometry
Incubations were performed at 37°C in a 10-ml suspen- sion of dexamethasone-treated rat microsomes (1 pM P450) containing bromocriptine (100 pM) and an NADPH-generat- ing system. The reaction proceeded for 10 inin and the reac- tion mixture was extracted with CH,CI, The organic phase was reduced to dryness under vacuum. The residue was dis- solved in a methanol/water (.50/50, by vol.) mixture and in- jected onto the HPLC column. The chromatographic condi- tions were the same as described for HPLC analysis. Some fractions were collected and dried under vacuum. An aliquot was rechromatographed to check its purity. Mass spectra

951
Table 1. Spectral interactions of ergot alkaloid derivatives with liver microsomes from rats pretreated with various inducers. Difference spectra were obtained by addition of increasing amounts (0.1 - 100 pM) of substrate to microsomal suspensions containing 2 pM P450 from rats either untreated or treated with phenobarbital, dexamethasone, 3-methylcholanthrene, clofibrate or troleandomycin. These suspensions contained 1 .O, 2.3, 1.8, 1.4, 1.4 and 1.2 nmol P450 . mg protein-', respectively. +K, indicates that the microsomes from troleandomycin-treated rats were preincubated at 0°C for 5 min in the presence of 15 pM potassium ferricyanide in order to dissociate the inhibitory P450-iron-metabolite complex formed in vivo (2.4 nmol P450 . mg protein-'). The three compounds gave type-I difference spectra (A,,, = 390 nm, A,,, = 420 nm). Maximum absorbances at saturating concentrations of the compounds are given in this table. Apparent spectral dissociation constants, Ks, were determined from double-reciprocal plots of AA~390--420nm) versus [substrate]. The reported AA values were means of four measurements (SD = 15%). K, values were means +- SD from four measurements also; ns, not significant (AA < 0.005).
Treatment AA,,,lP450 (KJ for substrate
bromocriptine ergocryptine dihydroergotamine
nmol-' (pM)
None Phenobarbital Dexamethasone 3-Methylcholanthrene Clofibrate Troleandomycin Troleandomycin + K,
ns ns 0.01 (2 +- 0.3) 0.015 ns 0.016 (1.7 +- 0.4) 0.045 (0.7 +- 0.2) 0.030 (1.5 5 0.3) ns ns ns ns ns ns 0.01 0.023 (1 +- 0.3) 0.015 (2.5 +- 0.3) 0.05 (0.5 +- 0.2) 0.035 (0.5 +- 0.3)
0.045 (2 5 0.3)
0.04 (2.3 5 0.4)
were carried out on a Ribermag 10.10 mass spectrometer under chemical ionization using NH,.
Enzymic immunoassay Polyclonal antibodies against the bromolysergic acid
moiety were produced by injection of rabbits with bromolys- ergic acid linked to bovine serum albumin (Grognet et al., 1991). Enzymic immunoassay was performed as described previously (Grognet et al., 1991).
RESULTS Structure of the compounds used in this study
All the ergot alkaloid derivatives used in this study in- volve a tetracyclic structure related to lysergic acid, dihydro- lysergic acid or bromolysergic acid (Fig. 2). The ergopep- tides, like ergocristine, ergocryptine and bromocriptine, are amides formed between lysergic (or bromolysergic) acid and cyclic tripeptides always involving a proline residue (Fig. 2). For comparison, an aza analog of the cyclic tripeptide moiety of ergopeptides (Pinnen et al., 1993), called azatripeptide 1 in Fig. 2, was also studied.
Interaction of ergot derivatives with rat liver P450s Interaction of three ergopeptides, bromocriptine, er-
gocryptine and dihydroergotamine, with liver microsomes from rats pretreated with various inducers was studied by difference visible spectroscopy (Table 1). The three com- pounds produced classical type-I difference spectra with a peak around 390 nm and a trough around 420 nm, which is indicative of a low-spin+high-spin transition of cytochrome P450. The maximum absorbance observed with dihydroer- gotamine was very low for microsomes from rats treated with 3-methylcholanthrene and clofibrate but markedly increased when passing from control to rats treated with phenobarbital and dexamethasone. The apparent spectral dissociation con- stant, K,, of dihydroergotamine was similar for the various microsomes (around 2 pM).
These data, which are in agreement with previous results (Delaforge et al., 1989), suggest that dihydroergotamine mainly interacts with P450s of the 3A subfamily that are present in relatively low amounts in control rat microsomes and those treated with 3-methylcholanthrene or clofibrate, are slightly induced by phenobarbital and strongly induced by dexamethasone (Guengerich et al., 1982). This was con- firmed by experiments performed with liver microsomes from rats treated with troleandomycin, another strong inducer of P450s 3A. In the troleandomycin-treated rat microsomes, a large part of P450 3A exists as an inactive P450-Fe(II)- nitrosoalkane complex that is formed in vivo (Pessayre et al., 1981; Delaforge et al., 1983; Pershing and Franklin, 1982). P450 3A is easily regenerated as an active form by treatment of microsomes with ferricyanide which dissociates the P450- nitrosoalkane complex. As shown in Table 1, addition of di- hydroergotamine to troleandomycin-treated rat microsomes led to a type-I difference spectrum, the intensity of which was only slightly higher than that obtained with control microsomes, whereas its intensity was dramatically increased when using troleandomycin-treated rat microsomes preincu- bated with ferricyanide. This result was expected because of regeneration of the largest part of P450 3A after ferricyanide treatment.
Bromocriptine and ergocryptine exhibited a similar be- haviour as they only interacted weakly with phenobarbital- treated rat microsomes but gave intense type-I spectra with dexamethasone- and troleandomycin-treated rat microsomes. Moreover they failed to give any significant spectral interac- tions with microsomes from control rats and those treated with 3-methylcholanthrene or clofibrate.
Since these first results suggested a specific interaction of ergot alkaloids with P450s 3A, we have studied the in- teractions of a series of ergot derivatives with dexametha- sone-treated rat liver microsomes. As shown in Table 2, all the ergopeptides used in this study, including ergocristine, ergocryptine, ergotarnine and bromocriptine derivatives, gave intense type-I difference spectra (AA390-420nm > 0.03/nmol P450) and high affinities (K,=l pM) for the corresponding P450s. Quite interestingly, all the lysergic acid derivatives

952
Table 2. Spectral interaction of lysergic acid derivatives and ergopeptides with liver microsomes from dexamethasone-treated rats. Conditions as in Table 1 with ns = not significant (AA < 0.005); n.m. = not measurable. d A and K, values were means ? SD from four measurements.
Substrate Spectrum K, AA,,,lP450 tY Pe
PM nmol-' Ergocristine I 1.7 ? 0.3 0.035 -C 0.005 Ergocry ptine I 1.5 ? 0.3 0.03 ? 0.005 Bromocriptine 1 0.7 ? 0.1 0.045 -C 0.008 Dihydro-
ergocristine I 3.0 -+ 0.4 0.05 ? 0.007 Di hydro-
ergocryptine 1 0.9 -+ 0.2 0.05 ? 0.007 Dihydro-
ergotamine I 2 + 0.3 0.045 ? 0.008 Bromolysergic
aci d n.m. ns Dihydro-
n.m. ns Dihydro-
~
lysergic acid .-
lysergic amide I 55 5 10 0.005 t 0.005 Lysergic amide - n.m. ns Azatripeptide 1 I 14 -+ 3 0.03 ? 0.005
used in this study that do not contain the ergopeptide group (lysergic amide, dihydrolysergic acid and amide, and bromo- lysergic acid) either completely failed to produce a spectral interaction or exhibited a very low affinity (Table 2). These results indicate that the tripeptide moiety of ergopeptides is crucial for their strong binding to P450s 3A. Accordingly, the azatripeptide 1, which has a structure very similar to that of the cyclic tripeptide moiety of ergopeptides and is the closest stable analog of this tripeptide moiety available (Pin- nen et al., 1993), also interacted with P450s 3A with the appearance of an intense type-I difference spectrum, al- though with a lower affinity (K5 = 14 pM). These results sug- gest that the cyclic tripeptide moiety of ergopeptides binds to P450s 3A at a site sufficiently close to the heme to induce
the loss of coordinated water of originally low-spin P450 Fe(II1) and the formation of high-spin pentacoordinate P450 Fe(II1). However, the lower K, observed for ergopeptides when compared to azatripeptide 1 shows that the lysergic part of ergopeptides, although unable by itself to produce a P450 spin shift, markedly contributes to the strong binding of ergopeptides to P450s 3A.
In order to further confirm such a specific binding to P450s 3A, we have studied the interactions of lysergic acid derivatives with well-defined human P450s 3A4 expressed in yeast.
Interaction of lysergic acid derivatives with human P450s NF25 and hPCN1
Addition of dihydroergotamine to microsomes of yeast producing either one of two allelic forms of P450 3A4, P450 NF25 or P450 hPCNl, led to a type-I difference visible spectrum with a K, value around 0.7 pM (Renaud et al., 1990; Peyronneau et al., 1993). As shown in Table 3, all the ergopeptides used in this study led to classical type-I differ- ence spectra both with P450 NF25 and P450 hPCNl . These difference spectra corresponded to a shift of 60-100% of P450 originally in the low-spin state to the high-spin state. The apparent dissociation constants varied between 0.6- 2.2 pM as a function of the ergopeptide structure. Bro- mocriptine was the most potent compound toward P450 NF25 with a K, of 0.6 pM and a 100% shift of the spin state. Only small differences were observed between P450 NF25 and P450 hPCN1, as the ratios between the K, values for a given compound were always below 2.
Interestingly, all the lysergic acid derivatives not bearing the cyclic tripeptide moiety of ergopeptides completely failed to give a spectral interaction with P450 NF25 and P450 hPCNl, although the azatripeptide 1 produced a clear type-I difference spectrum (30% spin shift and K,=30 pM) (Table 3). These data are in good agreement with those ob- served with P450s from rat liver microsomes (Table 2), and strongly suggest that the cyclic tripeptide moiety of ergopep- tides is an important determinant of the recognition of ergot alkaloids by P450s of the 3A subfamily.
Table 3. Interactions of lysergic acid derivatives with microsomes of yeast producing P450 NF25 or P450 hPCN1, studied by difference visible spectroscopy. Conditions as in Table 1, except for the P450 concentration used = 0.2 pM: n.m., not measurable; corresponding to the case where no difference spectrum appears even for [substrate] 2 1 mM. The spin shift i s the maximum amount of P450 undergoing a low-spin to high-spin shift upon addition of saturating concentrations of substrate, it was calculated by using L i l ~ ~ ~ ~ ~ - ~ ~ ~ ~ , , , , , ) = 126 c n - ' mM-' (Peterson, 1971). The indicated values are means t SD from at least three independent experiments.
Substrate P450 NF25 in yeast NF25-W(R) P450 hPCNl in yeast hPCNl -W(R)
Ergocristine Ergocryptine Bromocriptine Dihydroergocristine Dihydroergocryptine Dihydroergotamine Bromolysergic acid Dihydrolysergic acid Dihydrolysergic amide Lysergic amide Azatripeptide 1
nm PM % nm PM %
420 388 1.6 t 0.2 81 420 388 2.2 t 0.3 70 422 390 0.6 +- 0.2 100 422 388 1.9 t 0.2 60 420 390 1.3 -C 0.1 82 420 388 0.7 -C 0.1 84
- n.m. n.m. n.m. n.m.
- -
- - -
- - -
- - -
420 388 30 t- 10 30
41 8 420 420 420 41 8 420
390 388 388 390 388 388
0.9 ? 0.2 1.5 ? 0.2 1.1 ? 0.2 1.6 t 0.2 0.7 t- 0.1 0.6 5 0.2 n.m. n.m. n.m. n.m.
70 84 79 61 60
100

953
t ime (min)
7 1 0 1000 Q d : I 500
'IKT - E
0 20 40 60 8 0
time (min)
Fig. 3. Typical HPLC chromatogram from incubations of bro- mocriptine with dexamethasone-treated rat liver microsomes in the presence of NADPH. (A) Spectroscopic detection at 254 nm; (B) detection by enzyme immunoassay using an antibody raised against bromolysergic acid, measuring the products recognized by the antibody in 1-min HPLC fractions. Incubations contained 1 pM P450 from rat liver microsomes and 200 pM bromocriptine (BKT) and were performed at 37°C for 30 min, as described in Experimen- tal Procedures. Peaks x, y, z and k were found in control incubations in the absence of NADPH.
Similar experiments performed with other human liver P450s expressed in yeast, P450 1A1, 1A2 (Gautier et al., 1993) and 2C9 (Brian et al., 1989; Guengerich et al., 1991) showed that bromocriptine either failed to give any differ- ence spectrum (P450 2C9) or gave weak reverse type-I spectra (with a peak at 420 nm and a trough around 390 nm, in the case of P450 1Al and P450 1A2). In any case, the reverse type-I spectra observed with P450s 1A were weak (4A(420-390 .,,/nmol P450 < 0.003) and were similar to that also obtained with lysergic amide (data not shown). Thus, the small interactions observed with P450s 1A are very dif- ferent from the specific intense interactions of ergopeptides with P450s 3A that absolutely required the cyclic tripeptide moiety.
Oxidation of bromocriptine by rat and human liver microsomes
The HPLC profile of incubates of bromocriptine with dexamethasone-treated rat liver microsomes in the presence of NADPH showed the formation of four main polar metabo- lites (Fig. 3). The presence of NADPH or an NADPH-gener- ating system was absolutely required for the formation of these metabolites. As shown in Fig. 4, bromocriptine was al- most totally consumed after a 60-min incubation under the conditions employed (1 pM P450, 100 pM bromocriptine),
50 6ol 30 40i
I \
0 20 40 60 80
time (min)
Fig. 4. Bromocriptine oxidation by dexamethasone-treated rat liver microsomes as a function of time. (0) Consumption of bro- mocriptine; (0) formation of metabolites M, and M,; (0) formation of metabolites M, and M,. Incubations (0.5 ml) contained 1 pM P450, 100 pM bromocriptine and a NADPH-generating system (see Experimental Procedures).
Table 4. Main characteristics of the mass spectra of bromocrip- tine and its microsomal metabolites. The conditions are described in Experimental Procedures.
Ion mlz for "Br (*lBr) for compound
MH+ 654 (656) 670 (672) 686 (688) MH+-H,O 636 (638) 652 (654) MH+-16 670 (672) Lysergic amide
fragment 346 (348) 346 (348) 346 (348) Main peak (100%) 211 346 (348) 654 (656)
while metabolites M, and M, rapidly appeared, reached a maximum level after 15 min and then decreased. In contrast, metabolites M, and M, slowly appeared during the first 15 min and increased in a linear manner between 20- 60 min. These data suggest that M, and M, are primary oxi- dized metabolites which are then further oxidized into metab- olites M, and M,.
The four metabolites exhibited ultraviolet spectra with A,,,- at 235 nm and 305 nm that were almost identical to that of bromocriptine (data not shown). This result showed that the indole chromophore of bromocriptine remained intact during microsomal biotransformation. The mass spectra of the four metabolites exhibited several peaks with an isotopic cluster characteristic of the presence of a Br substituent (Ta- ble 4). Metabolites MI and M, exhibited very similar mass spectra with a molecular ion at mlz 670 (for 79Br) correspond- ing to bromocriptine having incorporated one oxygen atom. Metabolites M, and M, also exhibited similar mass spectra but with a molecular ion at mlz 686 corresponding to bro- mocriptine having incorporated two oxygen atoms. The low- est mass fragment containing a Br substituent observed for
954
T
no PR DEX 3-MC CLO TAO+K3
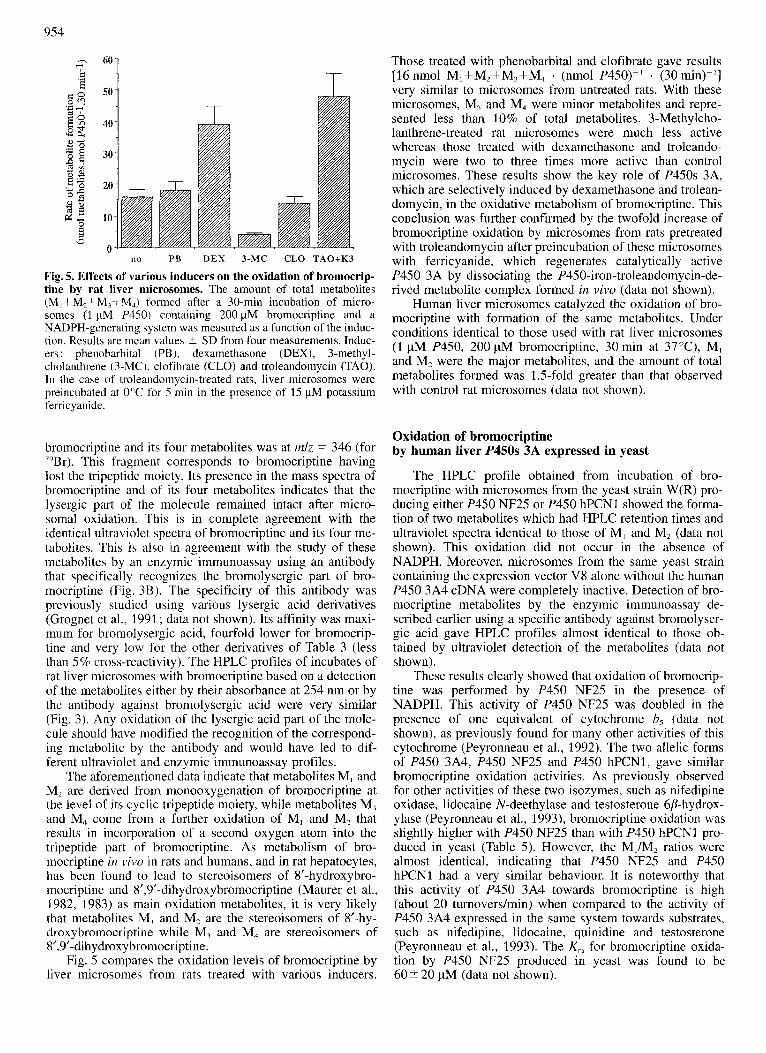
Fig. 5. Effects of various inducers on the oxidation of bromocrip- tine by rat liver microsomes. The amount of total metabolites (M,+M,+M,+M,,) formed after a 30-min incubation of micro- somes (1 pM P450) containing 200 pM bromocriptine and a NADPH-generating system was measured as a function of the induc- tion. Results are mean values 2 SD from four measurements. Induc- ers : phenobarbital (PB), dexamethasone (DEX), 3-methyl- cholanthrene (3-MC), clofibrate (CLO) and troleandomycin (TAO). In the case of troleandomycin-treated rats, liver microsomes were preincubated at 0°C for 5 min in the presence of 15 pM potassium femcyanide.
bromocriptine and its four metabolites was at mlz = 346 (for 79Br). This fragment corresponds to bromocriptine having lost the tripeptide moiety. Its presence in the mass spectra of bromocriptine and of its four metabolites indicates that the lysergic part of the molecule remained intact after micro- soma1 oxidation. This is in complete agreement with the identical ultraviolet spectra of bromocriptine and its four me-
This is also in agreement with the study of these es by an enzymic immunoassay using an antibody
that specifically recognizes the bromolysergic part of bro- mocriptine (Fig. 3B). The specificity of this antibody was previously studied using various lysergic acid derivatives (Grognet et a]., 1991; data not shown). Its affinity was maxi- mum for bromolysergic acid, fourfold lower for bromocrip- tine and very low for the other derivatives of Table 3 (less than 5% cross-reactivity). The HPLC profiles of incubates of rat liver microsomes with bromocriptine based on a detection of the metabolites either by their absorbance at 254 nm or by the antibody against bromolysergic acid were very similar (Fig. 3). Any oxidation of the lysergic acid part of the mole- cule should have modified the recognition of the correspond- ing metabolite by the antibody and would have led to dif- ferent ultraviolet and enzymic immunoassay profiles.
The aforementioned data indicate that metabolites M, and M, are derived from monooxygenation of bromocriptine at the level of its cyclic tripeptide moiety, while metabolites M, and M, come from a further oxidation of M, and M, that results in incorporation of a second oxygen atom into the tripeptide part of bromocriptine. As metabolism of bro- mocriptine in vivo in rats and humans, and in rat hepatocytes, has been found to lead to stereoisomers of 8'-hydroxybro- mocriptine and 8',9'-dihydroxybromocriptine (Maurer et al., 1982, 1983) as main oxidation metabolites, it is very likely that metabolites MI and M, are the stereoisomers of 8'-hy- droxybromocriptine while M, and M, are stereoisomers of 8',9'-dihydroxybromocriptine.
Fig. 5 compares the oxidation levels of bromocriptine by liver microsomes from rats treated with various inducers.
Those treated with phenobarbital and clofibrate gave results [16 nmol M,+M,+M,+M, . (nmol P450)-' . (30 fin)- ' ] very similar to microsomes from untreated rats. With these microsomes, M, and M, were minor metabolites and repre- sented less than 10% of total metabolites. 3-Methylcho- lanthrene-treated rat microsomes were much less active whereas those treated with dexamethasone and troleando- mycin were two to three times more active than control microsomes. These results show the key role of P450s 3A, which are selectively induced by dexamethasone and trolean- domycin, in the oxidative metabolism of bromocriptine. This conclusion was further confirmed by the twofold increase of bromocriptine oxidation by microsomes from rats pretreated with troleandomycin after preincubation of these microsomes with ferricyanide, which regenerates catalytically active P450 3A by dissociating the P45O-iron-troleandomycin-de- rived metabolite complex formed in vivo (data not shown).
Human liver microsomes catalyzed the oxidation of bro- mocriptine with formation of the same metabolites. Under conditions identical to those used with rat liver microsomes (1 pM P450, 200 pM bromocriptine, 30 min at 37"C), M, and M, were the major metabolites, and the amount of total metabolites formed was 1.5-fold greater than that observed with control rat microsomes (data not shown).
Oxidation of bromocriptine by human liver P450s 3A expressed in yeast
The HPLC profile obtained from incubation of bro- mocriptine with microsomes from the yeast strain W(R) pro- ducing either P450 NF25 or P450 hPCNl showed the forma- tion of two metabolites which had HPLC retention times and ultraviolet spectra identical to those of M, and M, (data not shown). This oxidation did not occur in the absence of NADPH. Moreover, microsomes from the same yeast strain containing the expression vector V8 alone without the human P450 3A4 cDNA were completely inactive. Detection of bro- mocriptine metabolites by the enzymic immunoassay de- scribed earlier using a specific antibody against bromolyser- gic acid gave HPLC profiles almost identical to those ob- tained by ultraviolet detection of the metabolites (data not shown).
These results clearly showed that oxidation of bromocrip- tine was performed by P450 NF25 in the presence of NADPH. This activity of P450 NF25 was doubled in the presence of one equivalent of cytochrome b, (data not shown), as previously found for many other activities of this cytochrome (Peyronneau et al., 1992). The two allelic forms of P450 3A4, P450 NF25 and P450 hPCNl, gave similar bromocriptine oxidation activities. As previously observed for other activities of these two isozymes, such as nifedipine oxidase, lidocaine N-deethylase and testosterone 6P-hydrox- ylase (Peyronneau et al., 1993), bromocriptine oxidation was slightly higher with P450 NF25 than with P450 hPCNl pro- duced in yeast (Table 5) . However, the M , N , ratios were almost identical, indicating that P4.50 NF2.5 and P4.50 hPCNl had a very similar behaviour. It is noteworthy that this activity of P450 3A4 towards bromocriptine is high (about 20 turnoverdmin) when compared to the activity of P450 3A4 expressed in the same system towards substrates, such as nifedipine, lidocaine, quinidine and testosterone (Peyronneau et al., 1993). The K,, for bromocriptine oxida- tion by P450 NF25 produced in yeast was found to be 60 2 20 pM (data not shown).

955
Table 5. Hydroxylation of bromocriptine by microsomes of yeast producing P450 NF25 or P450 hPCN1. Formation of metabolites M, and M2 was measured after a 10-min incubation of yeast micro- somes (0.2 pM P450) with 200 pM bromocriptine, 0.2 pM cyto- chrome b, purified from rabbit liver and 0.5 mM NADPH. In the case of the control yeast V8-W(R) transformed by the V8 vector not containing a P450 cDNA, equivalent amounts of microsomal protein were used. Results are mean values 2 SD from three independent experiments (in duplicate).
Yeast Activity of
nmol . (nmol P450)-‘ (10 min)-’
NF25-V8-W(R) 161 2 6 4 3 t 1 204 2 5 hPCNl -V8-W(R) 109 2 4 38 t 6 148 t 10 V8-W(R) <10 <1 <10
DISCUSSION
A family of ergot alkaloids, the ergopeptides, constitutes a new class of high-affinity substrates for rat liver micro- soma1 P450s 3A and for human liver P450 3A4. All the compounds investigated, namely ergocristine, ergocryptine and their dihydroderivatives, as well as bromocriptine and dihydroergotamine, exhibit spectral dissociation constants for rat liver P450s 3A and the human P450s 3A4 isozymes, P450 NF25 and P450 hPCN1, of around 1 pM (Tables 2 and 3). The specificity of interaction of these compounds with P450s of the 3A subfamily was shown by their very weak or weak spectral interactions with liver microsomes from un- treated rats or rat treated with classical inducers of the 1A (3- methylcholanthrene), 2B (phenobarbital) and 4 (clofibrate) families, and their strong spectral interactions with micro- somes of rats treated with classical inducers of P450s 3A (dexamethasone and troleandomycin; Tables 1 -3). The cy- clic tripeptide moiety of ergopeptides was found to be essen- tial for their recognition by P450s 3A since lysergic acid derivatives, which do not bear this moiety, failed to induce type-I spectral interactions with P450s 3A, whereas the azapeptide 1 did produce type-I interactions.
P450s 3A from rat liver microsomes and yeast-expressed human liver P450s 3A4 were found to selectively hydroxyl- ate one of these ergopeptides, bromocriptine, at the level of its peptide group, presumably at position 8’ of the proline residue (Fig. 1 ; this work). Dihydroergotamine has been found previously to be hydroxylated at position 8’ of its tripeptide moiety both in vivo in man and in human liver microsomes (Maurer and Frick, 1984) and has also been found to exhibit a high affinity for P450s 3A (Delaforge et al., 1989; this work). All these results suggest that ergot alka- loids, like bromocriptine and dihydroergotamine, could be metabolized at least in part by P450 3A4 in human liver. This would explain certain problems encountered in patients treated simultaneously with these ergopeptides and a macro- lide antibiotic such as erythromycin or troleandomycin (Hay- ton, 1969; Varoquaux et al., 1981; Ludden, 1985; Nelson et al., 1990). These macrolide antibiotics have been found to greatly inhibit P450s 3A in man by forming stable P450- iron-metabolite complexes (Pessayre et al., 1982; Larrey et al., 1983). This leads to a dramatic decrease in the oxidative metabolism of bromocriptine (or dihydroergotamine), which depends on the same P450 isozyme. This phenomenon could
be the origin of the increase of the plasma levels of these alkaloids observed in patients also treated with erythromycin or troleandomycin (Nelson et al., 1990).
Many substrates of human P450 3A4 have been de- scribed in the literature (Guengerich et al., 1991 ; Wrighton et al., 1990). Cyclosporin is the only pseudopeptidic substrate reported so far (Kronbach et al., 1988; Combalbert et al., 1989; Aoyama et al., 1989). This cyclopeptide exhibits a high affinity for P450 3A4 (Ks around 2 pM; Peyronneau et al., 1993) and is hydroxylated on several substituents of the cyclopeptide ring (Aoyama et al., 1989). The ergopeptide family is only the second example of pseudopeptide com- pounds that act as high-affinity substrates for P450s 3A and particularly P450s 3A4. Their cyclopeptide moiety should bind at a site close to P450 heme as the azatripeptide 1 and the ergopeptides induce a loss of water bound to low-spin P450 Fe(II1) and give type-I difference spectra. This is in agreement with the hydroxylation of bromocriptine by P450s 3A which seems to occur on the proline ring. The tripeptide moiety of the ergopeptide molecule is able by itself to induce a spin shift of P450s 3A whereas the second part of the ergopeptide molecule, the lysergic acid moiety, is not. How- ever, this lysergic acid moiety should contribute to the strong binding of ergopeptides to P450s 3A (K, of ergopeptides around 1 pM as compared to Ks of 1 of 14 pM for rat P450s 3A and of 30 pM for P450 3A4) and should participate in the positioning of the tripeptide moiety of ergopeptides close to the heme. Interestingly, a lysergic acid derivative bearing a NHSO,NEt, substituent at position 8 was found to be N- deethylated at the level of this substituent by P450 3A4 (Ball et al., 1992).
The results described for cyclosporin and ergopeptides suggest that P450s of the 3A subfamily may well recognize peptides or pseudopeptide compounds in a more general manner. Recent preliminary results show that many com- pounds of this type, like leucyl-phenylalanine, leucyl-proline and leucine-enkephalin, exhibit high affinities for P450 3A4 (M. Delaforge, C . Bensoussan and D. Mansuy, unpublished results). The biological consequences of these results con- cerning the possible roles of P450s 3A toward endogenous substrates remain to be determined.
We thank Prof. F. P. Guengerich (Nashville, TN) and P. H. Beaune (Paris, France) for the gift of NF25 cDNA and yeast ex- pressing P450 2C9, and Dr F. J. Gonzalez (Bethesda, MD) for the hPCNl cDNA. We also thank Dr D. Pompon (Gif sur Yvette, France) for providing us with the W(R) yeast strains, Dr P. Dostert (Farmitalia, Italy) for samples of the amides of lysergic and dihydro- lysergic acid, and Dr G. Lucente (Rome, Italy) for a sample of the azatripeptide 1.
REFERENCES Aoyama, T., Yamano, S., Waxman, D. J., Lapenson, D. P., Meyer,
U. A., Fischer, V., Tyndale, R., Inaba, T., Kalow, W., Gelboin, H. V. & Gonzalez, F. J. (1989) J. Biol. Chem. I I , 61-68.
Babany, G., Larrey, D. & Pessayre, D. (1988) Progl: Drug Metab. 11, 61-98.
Ball, S. E., Maurer, G., Zollinger, M., Ladona, M. & Vickers, A. E. M. (1992) Drug Metab. Disp. 20, 56-63.
Bargetzi, M. J., Aoyama, T., Gonzalez, F. J. & Meyer, U. A. (1989) Clin. Pharmacol. Ther. 46, 521 -521.
Beaune, P. H., Umbenhauer, D., Bork, R. W., Lloyd, R. S. & Guen- gerich, F. P. (1986) Proc. Nut1 Acad. Sci. USA 83, 8064-8068.
Bork, R. W., Muto, T., Beaune, P., Srivastata, P. K., Lloyd, R. S. & Guengerich, F. P. (1989) J. Bid. Chem. 264, 910-919.

956
Brian, W. R., Snvastava, P. K., Umbenhauer, D. R., Lloyd, R. S. & Guengerich, F. P. (1989) Biochemistry 28, 4993-4999.
Brian, W. R., Sari, M. A,, Iwasaki, M., Shimada, T., Kaminsky, L. & Guengerich, F. P. (1 990) Biochemistry 29, 11 280- 11 292.
Combalbert, J., Fabre, I., Fabre, G., Dalet, I., Derancourt, J., Cano, J. P. & Maurel, P. (1989) Drug Metab. Dispos. 17, 197-207.
Cullin, C. & Pompon, D. (1988) Gene 65, 203-217. Dansette, P., Amar, C., Valadon, P., Pons, C., Beaune, P. H. & Man-
suy, D. (1991) Biochem. Pharmacol. 41, 553-560. Delaforge, M., Jaouen, M. & Mansuy, D. (1983) Biochem. Pharma-
col. 32, 2309-2318. Dekdforge, M., Riviere, R., Sartori, E., Doignon, J. L. & Grognet, J.
M. (1989) Xenobiotica 19, 1285-1295. Gautier, J. C., Urban, P., Beaune, P. & Pompon, D. (1993) Euc J.
Biochem. 211, 63 - 72. Gonzalez, F. J., Schmidt, B. J., Umeno, M., MacBride, 0. W., Hard-
wick, J. P., Meyer. U. A,, Gelboin, H. V. & Idle, J. R. (1988) DNA 7, 79-86.
Gonzalez, F. J. (1992) Trends Pharmacol. Sci. 13, 346-352. Grognet, J. M., Istin, M., Zanetti, A,, Mailland, F. & Coppi, G.
(1991) Arzneim. Forsch. 41, 689-691. Guarente, L., Yocum, R. R. & Gifford, P. (1982) Proc. Natl Acad.
Sci. USA 79, 7410-7414. Guengerich, F. P., Dannan, G. A,, Wright, S. J., Martin, M. J. &
Kaminsky, L. S. (1982) Biochemistry 21, 6019-6030. Guengerich, F. P., Martin, M. V., Beaune, P., Kremers, P., Wolff,
T. & Waxman, D. J. (1986a) J. Biol. Chem. 261, 5051 -5060. Guengerich, F. P., Miiller-Enoch, D. &Blair, I. A. (1986) Mol. Phar-
mucol. 30, 287-295. Guengerich, F. P. (1988) Mol. Pharmacol. 33, 500-508. Guengerich, F. P., Brian, W. R., Sari, M. A. & Ross, J. T. (1991)
Guengerich, F. P. & Turvy, C. G. (1991) J. Phurmacol. Exp. Ther.
Hayton, A. (1969) N. Z. Med. J. 69, 42. Imaoka, S., Enomoto, K.. Oda, Y., Asada, A,, Fujimori, M., Shi-
mada, T., Fujita, S., Guengerich, F. P. & Funae, Y. (1990) J. Pharmacol. Exp. Ther. 255, 1385 - 1391.
Kremers, P., Beaune, P., Gresteil, T., De Graeve, J., Columelli, S., Leroux, J. P. & Gielen. J. E. (1981) Eur. J. Biochem. 118, 599- 606.
Kronbach, T., Fisher. V. & Meyer, U. A. (1988) Clin. Pharnzacol. Ther. 43, 630-635.
Kronbach, T., Mathys. D.. Umeno, M., Gonzalez, F. J. & Meyer, U. A. (1989) Mol. Pharmricol. 36, 89-96.
Larrey, D., Funck Brentano, C., Breil, P., Vitaux, J., Theodore C., Babany, G. & Pessayre, D. (1983) Biochem. Pharmacol. 32, 1063 - 1068.
Lowry, 0. H., Rosenbrough, N. J., Farr, A. L. & Randall, R. J. (1951) J. Biol. Chem. 193. 265-275.
Ludden, T. M. (1985) Clin. Pharmacokin. 10, 63-79.
Methods Enzpnol. 206, 130-145.
256, 1189-1194.
Mansuy, D. & Delaforge, M. (1993) Macrolides: chemistry, phurma- cology and clinical uses (Bryskier A,, Butzler, J. P., Neu, H. C. & Pulkens, P. M., eds) pp. 635-646, Arnette Blackwell, Paris.
Martinet, M. & Kiechel, J. R. (1983) Eur. .I. Drug Metab. Pharma- cokin. 8, 261-267.
Maurer, G., Schreier, E., Delaborde, S., Loosli, H. R., Nufer, R. & Shukla, A. P. (1982) Eur: J. Drug Metab. Pharmacokin. 7, 281 - 292.
Maurer, G., Schreier, E., Delaborde, S., Nofer, R. & Shukla, A. P. (1983) Eur. J. Drug Metab. Pharmacokin. 8, 51-62.
Maurer, G. & Frick, W. (1984) Eur. J. Clin. Pharmacol. 26, 463- 470.
Nelson, M. V., Berchou, R. C., Kareti, D. & LeWitt, P. A. (1990) Clin. Pharmacol. Ther. 47, 694-697.
Nelson, D. R., Kamataki, T., Waxman, D. J., Guenguerich, F. P., Estabrook, R. W., Feyereisen, R., Gonzalez, F. J., Coon, M. J., Gunsalus, I. C., Gotoh, O., Okuda, K. & Nebert, D. W. (1993) DNA Cell Biol. 12, 1-51,
Omura, T. & Sato, R. (1964) J. Biol. Chern. 239, 2370-2378. Periti, P., Mazzei, T., Mini, E. & Novelli, A. (1992) Clin. Pharma-
Pershing, L. K. & Franklin, M. R. (1982) Xenobiotica 12, 687-699. Pessayre, D., Descatoire, V., Konstantinova Mitcheva, M., Cobert,
B., Level, R., Benhamou, J. P., Jaouen, M. & Mansuy, D. (1981) Biochem. Pharmacol. 30, 553-558.
Pessayre, D., Larrey, D., Vitaux, J., Breil, P., Belghiti, J. & Benha- mou, J. P. (1982) Biochem. Pharmacol. 31, 1699-1704.
Peterson, J. A. (1971) Arch. Biochem. Bioplzys. 144, 678-693. Peyronneau, M. A., Renaud, J. P., Truan, G., Urban, P., Pompon,
D. & Mansuy, D. (1992) Euc J. Biochem. 207, 109-116. Peyronneau, M. A,, Renaud, J. P., Jaouen, M., Urban, P., Cullin, C.,
Pompon, D. & Mansuy, D. (1993) Eur. J . Biochenz. 218, 355- 361.
Pichard, L., Gillet, G., Fabre, I. , Dalet-Beluche, I., Bonfils, C., Thenot, J. P. & Maurel, P. (1990) Drug Metah. Dispos. 18, 711 - 719.
Pinnen, F., Luisi, G., Lucente, G., Gavuzzo, E. & Cerrini, S. (1993) J. Chem. Soc. Perkin Trans. 1, 819-824.
Pompon, D. (1988) Eur. J. Biochem. 177, 285-293. Renaud, J. P., Cullin, C., Pompon, D., Beaune, P. & Mansuy, D.
Strittmatter, P., Fleming, P., Connors, M. & Corcoran, D. (1978)
Truan, G., Cullin, C., Reisdorf, P., Urban, P. & Pompon, D. (1993)
Urban, P., Cullin, C. & Pompon, D. (1990) Biochimie 72,463-472. Varoquaux, O., Advenier, L. & Renier, E. (1981) Gaz. Med. Fr. 88,
1625-1629. Watkins, P. B., Wrighton, S. A,, Maurel, P., Schuetz, E. J., Mendez
Picon, G. & Guzelian, P. S. (1985) Proc. Nut1 Acud. Sci. USA 82, 6310-6314.
Wrighton, S . A,, Brian, W. R., Sari, M. A,, Iwasaki, M., Guengerich, F. P., Rancy, J. L., Molowa, D. J. & Vanderbranden, M. (1990) Mol. Pharmacol. 38, 207-213.
cokinet. 23, 106-131.
(1990) Eur. J. Biochem. 194, 889-896.
Methods Enzymol. 52, 97-101.
Gene 125, 49-55.





























