Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Versuche zur Synthese aus Harn isolierter Jonon-Derivate undihrer Umwandlungsprodukte
Author(s): Frick, Heinrich
Publication Date: 1949
Permanent Link: https://doi.org/10.3929/ethz-a-000116012
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Versuche zur
Synthese aus Harn isolierter Jonon-Derivate
und ihrer Umwandlungsprodukte
VON DER
EIDGENÖSSISCHEN TECHNISCHEN
HOCHSCHULE IN ZÜRICH
ZUR ERLANGUNG
DER WÜRDE EINES DOKTORS DER
TECHNISCHEN WISSENSCHAFTEN
GENEHMIGTE
PROMOTIONSARBEIT
VORGELEGT VON
HEINRICH FRICK
dipl. Ingenieur-Chemiker
von Zürich
Referent: Herr Prof. Dr. V. Prelog
Korreferent: Herr Prof. Dr. L. Rnzicka
BASEL
Buchdruckerei E. Birkhäuser & Cie., A. G.
1949

Leer - Vide - Empty

Meinen hochverehrten Lehrern
Herrn Prof. Dr. L. Buzicka
Herrn Prof. Dr. V. Prelog
möchte ich meinen herzlichsten Dank aussprechen
für das Wohlwollen und die Unterstützung, die sie
mir während dieser Arbeit stets zuteil werden liessen.

Leer - Vide - Empty

MEINEN LIEBEN ELTERN

Leer-Vide - Empty

Inhaltsverzeichnis
Seite
Theoretischer Teil 9
Über die Konstitution des Alkohols G13H20O und des Ketons Ci3HuO aus dem Harn
trächtiger Stuten 9
Über die beiden diastereomeren Tetrahydro-jonane 12
Über einige Oxo-tetrahydro-jonone 15
U.V.-Absorptionsspektra einiger synthetisch hergestellten Verbindungen und ihrer
Phenyl-semicarbazone 21
Experimenteller Teil 24
(2, 3, 6-Trimethyl-benzal)-aceton 24
l-(2, 3, 6-Trimethyl-phenyl)-butanon-(3) 25
Dihydro-cyelocitral 26
trans-l-[2,2, 6-Trimethyl-cyclohexyl-(l)]-buten-(l)-on-(3) 26
trans-Tetrahydro-jonon 27
trans-Tetrahydro-jonan,
27
cis-Tetrahydro-jonon 28
cis-Tetrahydro-jonan 28
a'-Oxo-tetrahydro-jonon 28
Methyl-(trans-2, 2, 6-trimethyl-cyclohexyl)-keton 29
/?'-Oxo-trans-tetrahydro-jonon 30
Jonon-epoxyde 30
4-Oxo-tetrahydro-jonon 33
Oxydation von Dihydro-a-jonol mit Selendioxyd 35
Zusammenfassung 38

Leer - Vide - Empty

Theoretischer Teil.
Aus dem Harn trächtiger Stuten sind in unserem Laboratorium
folgende Verbindungen mit 13 Kohlenstoff-Atomen isoliert worden,deren Konstitution unbekannt war:
1. ein Alkohol C13H20O2. ein Keton C13H16Ö3. und 4. zwei isomere Diole C13H2602 A und B
5. ein Keton C13H2202 C
6. und 7. zwei isomere Oxy-ketone C13H2402 E und G
8. und 9. zwei isomere Diketone C13H2202 F und D
Da es sich um kleine Mengen handelte, war es notwendig, zur
Konstitutionsermittlung die auf analytischem Wege erhaltenen Er¬
gebnisse durch synthetische Versuche zu vervollständigen. Diese Ver¬
suche bilden den Inhalt der vorliegenden Promotionsarbeit.
Über die Konstitution des Alkohols C13H20O und des
Ketons C13H160 aus dem Harn trächtiger Stuten. Die Syn¬these von l-(2, 3, 6-Trimethyl-phenyl)-butanon-(3).
Der aus dem Harn trächtiger Stuten isolierte linksdrehende
Alkohol C13H20O vom Smp. 94°liess sich durch Oxydation mit Chrom¬
säure in ein optisch inaktives, flüssiges Keton 013H18O überführen.
Dieses gab ein Semicarbazon vom Smp. 178—180° und ein
Phenyl-semicarbazon vom Smp. 170°. Die positive Jodoform-Reaktion
zeigte, daß in dem Keton eine CH3CO-Gruppe anwesend ist. Aus dem
Absorptionsspektrum des Semicarbazons und des Phenyl-semicar-bazons konnte man schliessen, dass ein Benzol-Ring vorhanden ist;die Carbonyl-Gruppe liegt nicht in einer Konjugation mit dem aroma¬
tischen Kern vor.
Da nur eine verhältnismässig kleine Menge des Alkohols C13H20Ozur Verfügung stand und das daraus erhaltene Keton mit keiner be¬
kannten Verbindung identisch war, hat man versucht, einige bisher
unbekannte oder nicht genügend charakterisierte Ketone C13H180synthetisch herzustellen. Von diesen erwartete man, dass sie auf
Grund der bisherigen Ergebnisse und gewisser arbeitshypothetischenAnnahmen (Isopren-Eegel) mit dem Oxydationsprodukt des Alkohols
aus dem Harn trächtiger Stuten identisch sein könnten.

— 10 —
Die zu diesem Zweck von R. Eagenbach1) zuerst hergestelltenisomeren Ketone C13H180: das (5-Methyl-2-isopropyl-phenyl)-aceton(I), das (2-Methyl-5-isopropyl-phenyl)-aceton (II) und das l-(4-iso-propyl-phenyl)-butanon-(3) (III) hatten sich von dem auf analyti¬schem Wege erhaltenen Keton als verschieden erwiesen. Der Ver¬
gleich der Absorptionsspektra der Semicarbazone lieferte aber einen
weiteren Anhaltspunkt über die gegenseitige Lage des Benzolkerns
und der Carbonyl-Gruppe des aus dem natürlichen Alkohol gewon¬
nenen Ketons. Während die Absorptionsspektra der Semicarbazone
der Verbindungen I und II mit der ß- Stellung der Carbonyl-Gruppegegenüber dem Benzolkern eine praktisch identische Absorptionzeigen, besitzen die Semicarbazone der Verbindung III und des
analytisch erhaltenen Ketons ein davon verschiedenes Absorptions¬spektrum. Deshalb wurde angenommen, dass die Carbonyl-Gruppenicht in /3-Stellung gegenüber dem Benzolkern sitzen kann2). Es wurde
deshalb das l-(2,3,6-Trimethyl-phenyl)-butanon-(3) (VI) hergestellt.Als Ausgangsprodukt diente der bekannte 2,3,6-Trimethyl-benzalde-hyd (IV). Durch Chlormethylierung des p-Xylols und Eeduktion des
auf diese Weise erhaltenen 2,5-Dimethyl-benzylchlorids wurde zuerst
reines Pseudocumol bereitet, welches nach der Vorschrift von L. J.
Smith und M. A. Kiess das 3-Brom-pseudocumol gab3). Aus diesem
konnte der 2,3,6-Trimethyl-benzaldehyd über die Grignard-Verbin¬dung nach der Ortho-ameisensäureester-Methode gewonnen werden4).
Der Aldehyd wurde mit Aceton und Natrium-äthylat als Kon¬
densationsmittel zum (2,3,6-Trimethyl-benzal)-aceton (V) konden¬
siert. Aus diesem oc, /5-ungesättigten Keton liess sich durch Hydrierungin Feinsprit mit Palladium-Bariumcarbonat als Katalysator das
gesuchte l-(2,3,6-Trimethyl-phenyl)-butanon-(3) (VI) erhalten. Das
Semicarbazon desselben zeigte einen Smp. von 178—180°, das Phenyl-semicarbazon einen Smp. von 167—171°. Die synthetischen Derivate
gaben mit den entsprechenden, aus dem analytischen Keton erhal¬
tenen Verbindungen keine Schmelzpunktserniedrigung. Die in Alkohol
aufgenommenen Absorptionsspektra (Fig. 1, Kurven 2 und 3), der
synthetisch hergestellten Derivate waren identisch mit den aus dem
Harn trächtiger Stuten gewonnenen. Es folgt daraus, dass dem
Keton C13H180, welches durch Oxydation des Alkohols C13H20O aus
dem Harn trächtiger Stuten gewonnen wurde, die Konstitution
eines l-(2,3,6-Trimethyl-phenyl)-butanon-(3) zukommt, woraus sich
!) Vgl. Helv. 30,113 (1947).2) Die Verschiedenheit der Absorptionsspektra der aromatischen Ketone mit
/^-ständigem Carbonyl von den Absorptionsspektra der Ketone mit einer grösseren Ent¬
fernung der Carbonyl-Gruppe wurde von P. Samart-Lucas und L. Labaune, Ann. chim.
(10) 16, 276 (1931) hervorgehoben.3) Am. Soc. 61,286(1939).4) L. J. Smith und J. Nichols, J. Org. Chem. 6, 501 (1939).
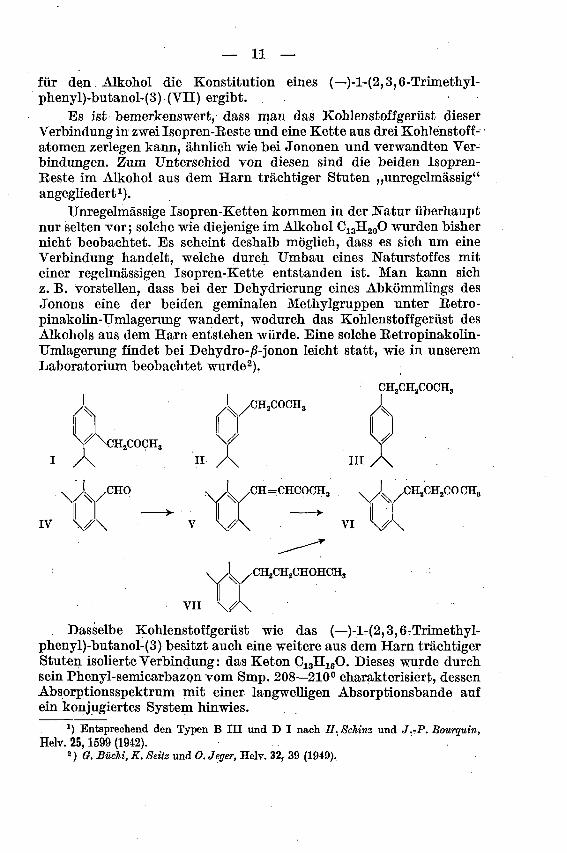
— 11 —
für den Alkohol die Konstitution eines (—)-l-(2,3,6-Trimethyl-phenyl)-butanol-(3) (VII) ergibt.
Es ist bemerkenswert, dass man das Kohlenstoffgerüst dieser
Verbindung in zwei Isopren-Beste und eine Kette aus drei Kohlenstoff¬
atomen zerlegen kann, ähnlich wie bei Jononen und verwandten Ver¬
bindungen. Zum Unterschied von diesen sind die beiden Isopren-Beste im Alkohol aus dem Harn trächtiger Stuten „unregelmässig"angegliedert1).
Unregelmässige Isopren-Ketten kommen in der Natur überhauptnur selten vor; solche wie diejenige im Alkohol C13H20O wurden bisher
nicht beobachtet. Es scheint deshalb möglich, dass es sich um eine
Verbindung handelt, welche durch Umbau eines Naturstoffes mit
einer regelmässigen Isopren-Kette entstanden ist. Man kann sich
z. B. vorstellen, dass bei der Dehydrierung eines Abkömmlings des
Jonons eine der beiden geminalen Methylgruppen unter Eetro-
pinakolin-Umlagerung wandert, wodurch das Kohlenstoffgerüst des
Alkohols aus dem Harn entstehen würde. Eine solche Betropinakolin-
Umlagerung findet bei Dehydro-/?-jonon leicht statt, wie in unserem
Laboratorium beobachtet wurde2).
v ,-CH2COCH3
^N3H2COCH3
.1 ,CHO , X /CH=CHCOCH3 .
J XiHjCHsCO CH3
IV v\
yCH2CH2CHOHCH3
VII \.
Dasselbe Kohlenstoffgerüst wie das (—)-l-(2,3,6-Trimethyl-phenyl)-butanol-(3) besitzt auch eine weitere aus dem Harn trächtigerStuten isolierte Verbindung: das Keton C13H160. Dieses wurde durch
sein Phenyl-semicarbazon vom Smp. 208—210° charakterisiert, dessen
Absorptionsspektrum mit einer langwelligen Absorptionsbande auf
ein konjugiertes System hinwies.
1) Entsprechend den Typen B HE und D I nach H. Sehinz und J.-P. Bourquin,Helv. 25,1599 (1942).
2) O. Büchi, K. Seitz und O. Jeger, Helv. 32, 39 (1949).
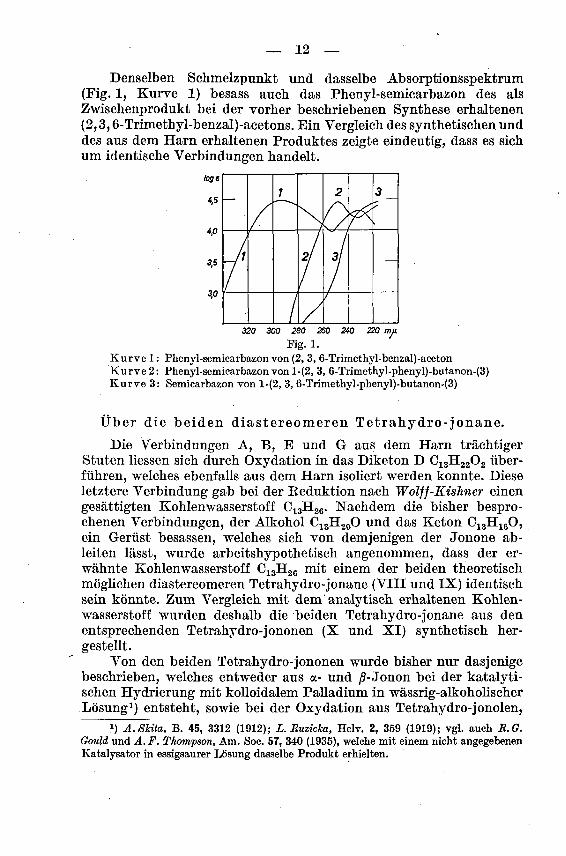
— 12 —
Denselben Schmelzpunkt und dasselbe Absorptionsspektrum(Fig. 1, Kurve 1) besass auch das Phenyl-semicarbazon des als
Zwischenprodukt bei der vorher beschriebenen Synthese erhaltenen
(2,3,6-Trimethyl-benzal)-acetons. Ein Vergleich des synthetischen unddes aus dem Harn erhaltenen Produktes zeigte eindeutig, dass es sich
um identische Verbindungen handelt.
loge
4,5
4,0
3,5
3,0
320 300 280 260 240 220 mu
Kg. 1.
Kurve 1 : Phenyl-semicarbazon von (2, 3, 6-Trimethyl-benzal)-acetonKurve 2: Phenyl-semicarbazonvon l-(2, 3, 6-Trimethyl-phenyl)-butanon-(3)Kurve 3: Semicarbazon von l-(2, 3, 6-Trimethyl-phenyl)-butanon-(3)
Über die beiden diastereomeren Tetrahydro-jonane.
Die Verbindungen A, B, E und G aus dem Harn trächtigerStuten Hessen sich durch Oxydation in das Diketon D C13H2202 über¬
führen, welches ebenfalls aus dem Harn isohert werden konnte. Diese
letztere Verbindung gab bei der Eeduktion nach Wolff-Kishner einen
gesättigten Kohlenwasserstoff C13H26. Nachdem die bisher bespro¬chenen Verbindungen, der Alkohol C13H20O und das Keton C13H160,ein Gerüst besassen, welches sich von demjenigen der Jonone ab¬
leiten lässt, wurde arbeitshypothetisch angenommen, dass der er¬
wähnte Kohlenwasserstoff C13H26 mit einem der beiden theoretisch
möglichen diastereomeren Tetrahydro-jonane (VIII und IX) identisch
sein könnte. Zum Vergleich mit dem analytisch erhaltenen Kohlen¬
wasserstoff wurden deshalb die beiden Tetrahydro-jonane aus den
entsprechenden Tetrahydro-jononen (X und XI) synthetisch her¬
gestellt.Von den beiden Tetrahydro-jononen wurde bisher nur dasjenige
beschrieben, welches entweder aus a- und /?-Jonon bei der katalyti-schen Hydrierung mit kolloidalem Palladium in wässrig-alkoholischerLösung1) entsteht, sowie bei der Oxydation aus Tetrahydro-jonolen,
*) A.Skita, B. 45, 3312 (1912); L. Ruzicka, Helv. 2, 359 (1919); vgl. auch R.O.
Gould und A. F. Thompson, Am. Soc. 57, 340 (1935), welche mit einem nicht angegebenenKatalysator in essigsaurer Lösung dasselbe Produkt erhielten.
—
; 3_
/ 2
/ //

— 13 —
welche entweder mit iücmey-Nickel bei 150—24001) oder mit Platin¬
oxyd-Katalysatoren in Eisessig bei Zimmertemperatur2) aus a-. und
/?-Jonon erhalten, wurden..
Das zweite Diastereomere liess sich auf folgendem Wege her¬
stellen. Als Ausgangsprodukt verwendete man Citral, das man nach
einer von. H. Schinz verbesserten Vorschrift von Haarmann und
Reimer3) in ein Gemisch von a- und /3-Cyclocitral überführte. Um
daraus das Dihydro-cyclocitral zu erhalten, wurde das Gemisch mit
Palladium-Bariumcarbonat hydriert, wobei jedoch nur das in dem
Gemisch vorhandene yS-Cyclocitral (XII) reagierte. Das in dem Ge¬
misch vorhandene oc-Cyclocitrâl wurde deshalb vor der Hydrierungin das /5-Cyclocitral übergeführt. Dies gelang nach einer Vorschrift
von H. Köster*), indem das Gemisch der isomeren Cyclocitrale mit
einer 8-proz. Lösung von Natriumhydroxyd in Alkohol bei einer
Temperatur von —5° behandelt wurde. Das reine yS-Cyclocitral liess
sich nun gut mit Palladium-Bariumcarbonat als Katalysator hydrie¬ren. Das erhaltene Dihydro-Cyclocitral (XIII) gab ein Semicarbazon
vom Smp. 206°. Es war also verschieden von dem Dihydro-cyclo¬
citral, welches von H. Barbier aus Dihydro-cyclogeraniol durch
Oxydation mit Chromsäure gewonnen .wurde, da dieses ein Semicar¬
bazon vom Sjnp. 185° lieferte5). Es ist möglich, dass das Produkt von
Barbier das eis- und die durch Hydrierung mit Palladium-Barium¬
carbonat erhaltene Verbindung das trans-Dihydro-cyclocitral darstellt.
Das letztere wurde mit Aceton und einer Lösung von Natrium-
äthylat kondensiert, wobei ein bisher nicht beschriebenes, krystallines
Dihydro-jonon (XIV) vom Smp. 50° mit der Doppelbindung in der
a, /S-Stellung zur Carbonyl-Gruppe entstand. Im Einklang damit
stehen die Absorptionsspektra sowohl des freien Ketons (Fig. 5,'Kurve 1) als auch seines Phenyl-semicarbazons. Bei der Hydrierungmit Palladium-Bariumcarbonat als Katalysator ging dieses krystallineDihydro-jonon unter Aufnahme von einem Mol Wasserstoff in ein
Tetrahydro-jonon (X) über.
Dieses unterscheidet sich schon durch einen etwas angenehmerenGeruch von dem sonst auf üblichem Wege aus a- oder ß-Jonon er¬
haltenen Tetrahydro-jonon. Es gibt ein Semicarbazon, welches bei
163° schmilzt, während das beschriebene Tetrahydro-jonon-semicar-bazon den Smp. 183° zeigt6). Das Phenyl-semicarbazon des neuen
!) J. Kandel, Ann. chim. (11) 11, 105 (1939). •
'
2) A. Bosshardt, Diss. ETH. Zürich 1946, S. 64.
3) D.R.P. 123747; vgl. Friedländer, Fortschritte der Teerfarbenfabrikation, Bd. 6,S. 1238; E. Kuhn und G. Wendt, Bd. 69, 1555 (1936).
4) B. 77, 558 (1944).5) Helv. 23, 529 (1940).
•'
.
t
•.; ') Nur in'einer Arbeit vonJ. Salkind, S. Sortis? und N. Blochin -wurde ein Tetra-
hydro-jonon-semicarbazon mit einem Smp. 165.-^166,5°.erwähnt; C. 1936 I 4169. : ; ,,

— .14 —
Tetrahydro-jonons schmilzt bei 133°, das bekannte Phenyl-semicar-bazon des isomeren Produktes besitzt dagegen einen Smp. 109°.
Das zu Vergleichszwecken und für die weitere Arbeit ver¬
wendete isomere Tetrahydro-jonon (XI) ist. durch Hydrierung von
a-Jonon (XV) mit Platinoxyd-Katalysator, in Eisessig von A. Boss-
hardt gewonnen und über das krystalline Semicarbazon gereinigtworden.
In der folgenden Tabelle 1 sind die Eigenschaften der beiden
isomeren Tetrahydro-jonone einander gegenübergestellt:
Tabelle 1.
df4
n22 MDSemie.
Smp.
Ph.-sem.
Smp.'
XI cis- 0,9138 1,4660 59,51 183—184° 109—110°
X trans- 0,9064 1,4634 59,69 163° 133°
Auf Grund der Bildungsweisen und der Eigenschaften (Dichteund Molekularrefraktion) kann man unter Anwendung der bekannten
Eegeln von Auwers und 8Mta der schon längerer Zeit bekannten
Verbindung die cis-Konfiguration, dem neuen Isomeren die trans-
Konfiguration zuteilen.
Durch Eeduktion der beiden isomeren, über krystalline Zwischen¬
produkte gereinigten Tetrahydro-jonone mit Hydrazinhydrat und
Natrium-äthylat bei 200° erhielt man die beiden diastereomeren
Tetrahydro-jonane (VIII und IX). Diese unterscheiden sich schon
durch ihren verschieden intensiven Geruch. Die Eigenschaften der
beiden isomeren Tetrahydrojonane und des Kohlenwasserstoffs aus
Harn sind in der Tabelle 2 angeführt:
Tabelle 2
df n20 MD
IX eis-. . . 0,8280 1,4552 59,77
VIII trans- . 0,8214 1,4531 60,02
KW aus Harn 0,8289 1,4553 59,72
Aus dieser Tabelle ist zu ersehen, dass auch hier die Zuteilung der
Konfiguration mit der J.ww?ers'schen Eegel in Einklang steht, indem
das cis-Isomere eine grössere Dichte und eine kleinere Molekular¬
refraktion aufweist als das trans-Isomere. Das cis-Tetrahydro-jonanwurde schon früher aus dem cis-Tetrahydro-jonon-semicarbazon von

0
— 15 —
L. Ruzicka und C. F. Seidel1) hergestellt. Die Eigenschaften des von
ihnen beschriebenen Kohlenwasserstoffes stimmen mit den hier
beobachteten gut überein. Die physikalischen Konstanten eines auch
von J. KandeP) durch Reduktion von a- und /?-Jonon mit Baney-Nickel bei 270° erhaltenen Tetrahydro-jonans liegen zwischen den
Werten für die beiden Diastereomeren, was auf ein Gemisch hinweist.
Eine eindeutige Unterscheidung der beiden isomeren Tetra-
hydro-jonane war auf Grund ihrer Infrarot-Absorptionsspektrenmöglich. Die Infrarot-Spektren der auf beiden Wegen erhaltenen
Präparate (Fig. 2) unterscheiden sich deutlich voneinander und
liefern die vollständige Gewissheit, dass es sich um zwei voneinander
verschiedene Isomeren handelt.
•so
40
20
cm"' 1700 1600 1500, 1400 1300 1200 H00 1000 900 600
tramrfETRAHYDRO-JONAN
iLK-nJl
-eocm-' 1700 1600 150» 1400 1300 1200 1100 1000 900 600
60
40KW C^Hx AUS HARN / \h
«20 J u
VaJU .A-A.A«..
eocm-i i7oo' lèoo' îèoa 1400 1300 1200 1100 1000 900 B00
60
40cis-TETRAHYDRCMONAN \
20^^L^^wv/S^-/ \jlA-A. jl^^
Fig. 2.
Die Eigenschaften des aus dem Diketon D erhaltenen Kohlen¬
wasserstoffes C13H26, welche neben den Eigenschaften der beiden
isomeren Tetrahydro-jonane in der Tabelle 2 angegeben sind, zeigendeutlich, dass der Kohlenwasserstoff aus Harn die Konstitution
eines cis-Tetrahydro-jonans besitzt. Noch eindeutiger ist das aus den
Infrarot-Absorptionsspektren (Fig. 2) ersichtlich. Es folgt daraus,dass das Diketon D sowie die mit diesem verknüpften VerbindungenA, B, C, E und G aus dem Harn ein cis-Tetrahydro-jonan-Gerüstbesitzen.
Über einige Oxo-tetrahydro-jonone.
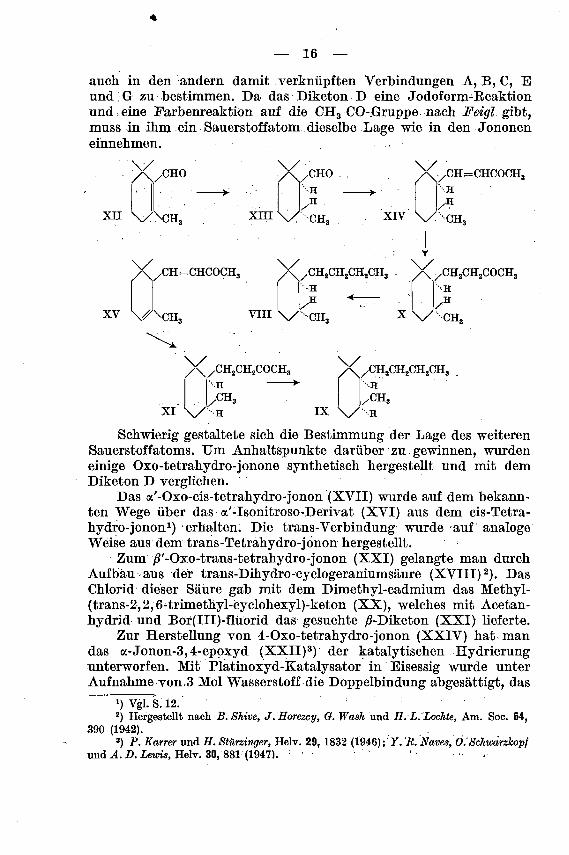
Nachdem festgestellt werden konnte, dass das Diketon D ein
cis-Tetrahydro-jonan-Derivat ist, blieb noch die Aufgabe übrig, die
Lage der beiden Sauerstoffatome, in dieser Verbindung und somit
*) Helv. 19, 432 (1936): àf = 0,8292 n^ = 1,4563.
») Ann. chim. (11) 11, 103 (1939) df = 0,8249 n^ = 1,4538.
«
— 16 —
auch in den andern damit verknüpften Verbindungen A, B, C, E
und G zu bestimmen. Da das Diketon D eine Jodoform-Eeaktion
und eine Farbenreaktion auf die CH3 CO-.Gruppe nach Feigl gibt,muss in ihm ein Sauerstoffatom dieselbe Lage wie in den Jononen
einnehmen.
XII
X/CHO
\/\CH,
X.,CHO
H
H
\y
xni\y CH< XIV
CH=CHCOCH,
X/CH=
xv \Aca
=CHCOCH, /\/CH2CH2CII2CH3H
H <
vin \y ch,
X/CH2CH2COCH3I l H
I l/HX\/ CH,
/\/CH2CH2COCH3\
H
.CH3XI v H
H
•CH.
ix \y h
Schwierig gestaltete sich die Bestimmung der Lage des weiteren
Sauerstoffatoms. Um Anhaltspunkte darüber zu gewinnen, wurden
einige Oxo-tetrahydro-jonone synthetisch hergestellt und mit dem
Diketon D verglichen.Das a'-Oxo-cis-tetrahydro-jonon (XVII) wurde auf dem bekann¬
ten Wege über das a'-Isonitroso-Derivat (XVI) aus dem cis-Tetra-
hydro-jonon1) erhalten. Die trans-Verbindung wurde auf analogeWeise aus dem trans-Tetrahydro-jonon hergestellt.
Zum /8'-Oxo-trans-tetrahydro-jonon (XXI) gelangte man durch
Aufbau aus der trans-Dihydro-cyclogeraniumsäure (XVIII)2). Das
Chlorid dieser Säure gab mit dem Dimethyl-cadmium das Methyl-(trans-2,2,6-trimethyl-cyclohexyl)-keton (XX), welches mit Acetan-
hydrid und Bor(III)-fluorid das gesuchte /?-Diketon (XXI) lieferte.
Zur Herstellung von 4-Oxo-tetrahydro-jonon (XXIV) hat man
das a-Jonon-3,4-epoxyd (XXII)3) der katalytischen Hydrierungunterworfen. Mit Platinoxyd-Katalysator in Eisessig wurde unter
Aufnahme von 3 Mol Wasserstoff die Doppelbindung abgesättigt, das
M Vgl. S. 12.
2) Hergestellt nach B. Shive, J. Horezcy, O. Wash und H. L. Lochte, Am. Soc. 64,390 (1942).
3) P. Karrer und H. Stürzinger, Helv. 29, 1832 (1946); Y. R. Naves, 0. Schwarzkopfund A. D. Lewis, Helv. 30, 881 (1947).

— 17 —
Carbonyl in die Hydroxyl-Gruppe übergeführt und der Epoxyd-Ringhydrogenolytisch gespalten. Es entstand dabei ein Gemisch von
Diolen, aus welchem eines der Isomeren krystallin erhalten werden
konnte. Dieses gab mit Ohromsäure in Eisessig ein 4-Oxo-tetrahydro-jonon (XXIV). Das krystalline Zwischenprodukt stellt demnach ein
4-Oxo-tetrahydro-jonol (XXIII) dar. Das 4-Oxo-tetrahydro-jononbildete auch das Hauptprodukt bei der Oxydation der nichtkrystal-linen Diole, woraus folgt, dass bei der Hydrogenolyse des Epoxyd-Einges in 3,4^Stellung der Sauerstoff an Kohlenstoff 4 gebundenbleibt.
X CH,CH2C0CH3
H
,CH3
\X H
^Xf/COOHH
xvm \y ch3
"X'/ch3ccoch3
XVI
H
CH.NOH
/\/COClM
H
XVII
^/\XcH2COCOCH3
\x h
x
xix \y/.CH„ XX
/C0CH3H
H
\//CH3
/\/COCH2COCH3H
H
CH,/
xxi \y
N/.CH=CHCOCH3 XXCH2CH2CHOHCH3 N/.CHjCHjCOCH,
XXII y\cH3 XXIII
r*XXIV V\CH„
y\ /CH2CH2CH2CH3H
XXV V CH3
Das 4-Oxo-tetrahydro-jonon gab bei der Reduktion nach
Wolff-Kishner das trans-Tetrahydro-jonan (XXV). Da die Kon¬
figuration am Kohlenstoffatom 3 im 4-Oxo-tetrahydro-jonon wegender benachbarten Carbonyl-Gruppe unstabil ist, kann man darausnicht mit Sicherheit schliessen, dass das Diketon ein trans-Derivat ist.
Bei einem Versuch, das a-Jonon-3,4-epoxyd mit Palladium-
Bariumcarbonat in Feinsprit zu hydrieren, wurde unter Absättigung2 Frick

— 18 —
der Doppelbindung nur 1 Mol Wasserstoff aufgenommen. Das ge¬bildete Dihydro-a-jonon-3,4-epoxyd (XXVI) geht mit alkoholischer
Schwefelsäure in ein Isomeres über, welches nicht mehr mit Car-
bonyl-Eeagentien reagiert und demnach wahrscheinlich das Di-
hydro-pyran-Derivat (XXVII) darstellt. Durch Nachhydrierung mit
einem Platinoxyd-Katalysator in Eisessig erhält man aus Dihydro-<x-jonon-3,4-epoxyd dasselbe Produkt wie aus dem a-Jonon-3,4-epo-xyd direkt.
Anders als das a-Jonon-3,4-epoxyd verhält sich bei der Hydrie-'rung mit Palladium-Bariumcarbpnat das isomere a-Jonon-a', /5'-epo-xyd (XXVIII)1). Unter Aufnahme von 1 Mol Wasserstoff wird nicht
die Doppelbindung abgesättigt, sondern der Epoxyd-Eing hydro-genolytisch gespalten. Das Hydrierungsprodukt besteht aus einem
Gemisch von Oxy-ketonen, aus dem ein Isomeres auskrystaUisierte.Dieses gibt mit alkoholischer Schwefelsäure leicht das a-Jonon
und stellt demnach sehr wahrscheinlich ein /S'-Oxy-dihydro-a-jonon(XXIX) dar. Der flüssige Eest des Hydrierungsproduktes, aus wel¬
chem sich in Form seines krystallinen Phenyl-semicarbazones ein
davon verschiedenes isomeres Oxy-Keton isolieren Hess, gibt eine
starke Eisen(III)-chlorid-Eeaktion und enthält demnach ein a'-Oxy-dihydro-a-jonon (XXX).
Im Zusammenhang mit den Versuchen zur katalytischen Hydrie¬rung der a-Jonon-epoxyde wurde auch das ß-Jonon-2,3-epoxyd(XXXI) mit Palladium-Bariumcarbonat in Feinsprit hydriert. Unter
Aufnahme von 1 Mol Wasserstoff wurde in diesem Falle die Doppel¬bindung abgesättigt ; der Epoxyd-Eing blieb dagegen intakt. Das bei
diesen Versuchen gebildete Dihydro-/S-jonon-2,3-epoxyd (XXXII)gab bei der Behandlung mit alkoholischer Schwefelsäure zwei Pro¬
dukte, von welchen das erste, durch Anlagerung von Wasser ent¬
standen, krystallisierte. Da es mit Carbonyl-Eeagentien nicht rea¬
gierte und mit Tetranitromethan keine Farbenreaktion zeigte, kann
man ihm die Konstitution eines Dioxy-tetrahydro-pyran-Derivates(XXXIII) zuschreiben. Das flüssige Produkt, welches nicht ganz
analysenrein erhalten werden konnte, war isomer mit dem Dihydro-/?-jonon-2,3-epoxyd. Es gab ebenfalls keine Derivate mit Carbonyl-Eeagentien ; dagegen war es gegenüber Tetranitromethan ungesättigt.Es handelt sich wahrscheinlich um das Oxy-dihydro-pyran-Derivat(XXXIV).
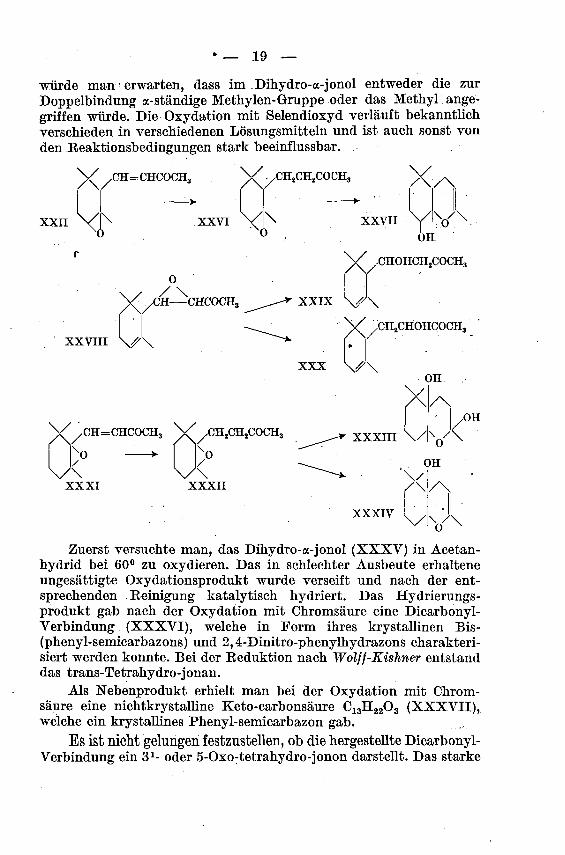
Um ein 5-Oxo-tetrahydro-jonon herzustellen, oxydierte man das
Dihydro-a-jonol mit Selendioxyd. Das Ausgangsmaterial wurde auf
übliche Weise aus dem Dihydro-oc-jonon durch Eeduktion nach
Meerwein-Pondorf mit Aluminiümisopropylat in Isopropylalkoholhergestellt. Nach den bisherigen Erfahrungen mit Selendioxyd
!) P. Karrer und H. Stürzinger, Helv. 29, 1833 (1946).
•— 19 —
würde man erwarten, dass im Dihydro-a-jonol entweder die zur
Doppelbindung a-ständige Methylen-Gruppe oder das Methyl ange¬
griffen würde. Die Oxydation mit Selendioxyd verläuft bekanntlich
verschieden in verschiedenen Lösungsmitteln und ist auch sonst von
den Beaktionsbedingungen stark beeinflussbar.
\/ ,CH=CHCOCH3
/\/3 "V'/CH2CH,!COCH3
XXII XXVI
0
V'xH CHCOCH3
XXVII
\/ yCHOHCH,COCH3
XXIX \/\
XXVIII \/\
XXX \f\
N//CHjCHOHCOCHa
OH
N//CH=CHCOCH3N0/
V\XXXI
V/CHjCRjCOCH,
XXXII
XXXIII \/o
x
OH
OH
XXXIV
/\
ox
Zuerst versuchte man, das Dihydro-a-jonol (XXXV) in Acetan-
hydrid bei 60° zu oxydieren. Das in schlechter Ausbeute erhaltene
ungesättigte Oxydationsprodukt wurde verseift und nach der ent¬
sprechenden Eeinigung katalytisch hydriert. Das Hydrierungs¬produkt gab nach der Oxydation mit Chromsäure eine Dicarbonyl-Verbindung (XXXVI), welche in Form ihres krystallinen Bis-
(phenyl-semicarbazons) und 2,4-Dinitro-phenylhydrazons charakteri¬
siert werden konnte. Bei der Beduktion nach Wolff-Kishner entstand
das trans-Tetrahydro-jonan.Als Nebenprodukt erhielt man bei der Oxydation mit Chrom¬
säure eine nichtkrystalline Keto-carbonsäure C13H2203 (XXXVII),welche ein krystallines Phenyl-semicarbazon gab.
Es ist nicht gelungen festzustellen, ob die hergestellte Dicarbonyl-Verbindung ein 31- oder 5-Oxo-tetrahydro-jonon darstellt. Das starke

— 20 —
*
Beduktionsvermögen weist auf die Anwesenheit eines Aldehydes hin,doch ist es möglich, dass ein Gemisch der beiden Isomeren vorliegt.
E. Schwenk und E. Borgwart1) oxydierten Pinen mit Selendioxydund behaupteten, daraus Yerbenon zu erhalten, welches auch bei
der Autoxydation von Pinen entsteht. G. Dupont und Mitarbeiter2)konnten jedoch zeigen, dass bei dieser Oxydation nicht das Verbenon,sondern Myrtenal und Myrtenol erhalten wird.
Gegen die Erwartung erfolgte in diesem Falle nach der Ansicht
der französischen Autoren nicht ein Angriff der zur Doppelbindung in
a-Stellung sich befindbchen Methylen-Gruppe, sondern der Methyl-Gruppe, welche sich ebenfalls in a-Stellung zu der Doppelbindungbefindet.
Die Oxydation von Dihydro-a-jonol in Dioxan führte zu einem
autoxydablen a, /5-ungesättigten Aldehyd (XXXVIII), der als Phenyl-semicarbazon erfasst werden konnte. Da die Versuche scheiterten,durch katalytische Hydrierung und Oxydation mit Chromsäure zu
einer gesättigten Dicarbonyl-Verbindung zu gelangen, wurde die
Untersuchung in dieser Eichtung abgebrochen.
N/xH2CH2CHOHCH3 S^ /CH2CH2CHOHCH3 ^ /CHjCHjCOCH,,±2^"2v.
,
- - X;\CH0 XXXVIII \^XCH3 XXXV 0^\/\
N^CHjCHüCHOHCIt,
^\CH.0H
CH2CH2CHOHCH3 V' /CH2CH2COCH3 J
\3H.OH
XXXVI
ch;ch2coch3
COOH XXXVII
Ein weiterer Versuch zur Herstellung von 5-Oxo-tetrahydro-jonon ging von Dehydro-/3-jonon aus. Dieses wurde mit 1 Mol Phtal-
monopersäure oxydiert und das Oxydationsprodukt, in welchem man
das Dehydro-/?-jonon-4,5-epoxyd vermutete, katalytisch mit Platin¬
oxyd in Eisessig hydriert. Nach der Oxydation des Hydrierungs¬produktes mit Chromsäure liess sich das 4-Oxo-tetrahydro-jonon in
Form seines Bis-(phenyl-semicarbazons) in schlechter Ausbeute iso¬
lieren. Es scheint demnach, dass der 4,5-Epoxyd-Eing in diesem
Falle hydrogenolytisch so gespalten wurde, dass der Sauerstoff am
Kohlenstoffatom 4 gebunden bleibt.
x) B. 32, 1601 (1932).
2) Bl. (5) 2, 533 (1935).

— 21 —
Zusammenstellung der TJ.V.-Absorptionsspektren einiger
synthetisch hergestellten Verbindungen und ihrer
Phenyl-semicarbazone.
Da die Ketone aus dem Harn trächtiger Stuten als Phenyl-semicarbazone isoliert worden waren, wurden von allen in diesem
Teil der Arbeit beschriebenen Phenyl-semicarbazone zum Vergleich
Absorptionsspektren aufgenommen (vgl. Fig. 3, 4, 6, 7, 8 und 10).
Diese stellten ebenso wie die Absorptionsspektren der freien Ver-
logt
<t.O
3,5
3,0
320 300 280 260 240 220 iyi
Fig. 3.
Kurve 1 : Bis-(phenyl-semicarbazon) von a'-Oxo-cis-tetrahydro-jononKurve 2: Phenyl-semicarbazon von |3'-Oxo-tetrahydro-jononKurve 3: Bis-(phenyl-semicarbazon von 4-Oxo-tetrahydro-jonon
r\^à^
—
/,A?\/j
—
/ /
loge
U,0
3,5
3,0
^f2 3
?—
/ j'
/ —
/ 1 /I
320 300 280 260 2lt0 220 ir^u,
Fig. 4.
Kurve 1: Phenyl-semicarbazon von /S-JononKurve 2: Phenyl-semicarbazon von a-Jonon
Kurve 3: Phenyl-semicarbazon von Dihydro-oc-jonon
bindungen (vgl. Fig. 5) oft eine wertvolle Hilfe bei der Zuteilung der
Konstitutionsformehl für die entsprechenden Eeaktionsprodukte dar.
Obwohl keines der hergestellten Tetrahydro-jonon-Derivate mit
einer der aus dem Harn trächtiger Stuten bisher isolierten Verbin-

— 22 —
düngen identisch war, da' diese, wie sich später herausstellte, in
Stellung 5 substituiert sind, waren die Untersuchungen der hier be¬
schriebenen Verbindungen doch für die Konstitutionsermittlung der
05E
2 5 f\ 4/
\ Vs/
/
\\l /3.5
1
1
11
hlii y
"
3.0 /
\* s/ir \
13
?511
////
1 /2.0
1 1 / /l / /
^r1
*
'^-/ 3
//
320 300 280 260 240 220 mY
Fig. 6.
Kurve 1 : Dihydro-jononKurve 2: /S'-Oxo-tetrahydro-jononKurve 3: a-Jonon-a'/3'-epoxydKurve 4: a-Jonon-3, 4-epoxydKurve 5: /?-Jonon-2, 3-epoxyd
logs
4,5 —1 3 1
—
40
r~2<
/ \
3,5
3,0
—
// 2 Il3 —
1320 300 P80 260 ZW 220 mp
Jig. 6.
Kurve 1: Phenyl-semicarbazon von a-Jonon-3,4-peoxydKurve 2: Phenyl-semicarbazon von a-Jonon-a', /S'-peoxydKurve 3: Phenyl-semicarbazon von Dihydro-a-jonon-3, 4-epoxyd
Produkte aus Harn wertvoll. Nachdem die Zahl der in Frage kom¬
menden Stellungen für die zweite Sauerstoffunktion eingeschränktwurde, Hess sich ihre Lage durch sinngemässe, von B. Schneider1)durchgeführten Abbauversuche feststellen.
>) Diss. ETH. Zürich, 1949.

— 23 —
—t 2
\s>
—
7 ,
A
/ —
/A
3,0
320 300 280 260 240 220 mj*
Fig. 7.
Kurve 1 : Phenyl-semicarbazon von /?'-Oxy-dihydro-<x-jononKurve 2: Phenyl-semicarbazon von a'-Oxy-dihydro-a-jonon
loge
4,0
3.5 —
320 300 280 260 240 220 mjt
Fig. 8.
Kurve 1 : Phenyl-semicarbazon von ß-Jonon-2, 3-epoxydKurve 2: Phenyl-semicarbazon von /3-Jonon-a', /S'-epoxydKurve 3: Phenyl-semicarbazon von Dihydro-/?-jonon-2, 3-epoxyd
—
/2
^ 1
3
_ À/
/7 1
500 hop wo t2po
eo
so2 A H\j\K
40 J V 1 ^-^VAa20
rttx npo \itLÀspo Kpo m> BpO W) «W> \§82^_A~/.
80
603
40
20XX) 1703 nM'TSOO K00 1300 12Ç0 llpO KJOO 9O0V/W^-
Fig. 9.

— 24 —
logt
4,0
3,5
0
320 300 2B0 260 XO 220 m/i
Fig. 10.
Kurve 1: Phenyl-semicarbazon von S'-Oxo-dihydro-a-jonolKurve 2: Phenyl-semicarbazon von S'-Oxo-tetrahydro-jononKurve 3: Phenyl-semicarbazon von Ketosäure
Experimenteller Teil.
(2, 3, 6-Trimethyl-benzal)-aceton (V).
Aus 300 g p-Xylol Hessen sich 160 g reines Pseudocumol herstellen, indem nach der
Vorschrift von J. v. Braun und J.Nelles1) das 2,5-Dimethyl-benzylchlorid bereitet und
dieses zu dem Kohlenwasserstoff hydriert wurde2).Aus 73 g Pseudocumol wurde die Pseudocumol-5-sulfosäure3) hergestellt und diese
bromiert4). Als Nebenprodukt wurden dabei 34 g 5-Brom-pseudocumol erhalten. Nach der
Abtrennung desselben konnte aus der zurückbleibenden 3-Brom-pseudocumol-5-sulfosäuredurch Abspaltung der Sulfo-Gruppe 17,1 g reines 3-Brom-pseudocumol erhalten werden.
Ausbeute an 3-Brom-pseudocumol: 14,3% d. Th. (auf Pseudocumol berechnet).Aus 2,9 g 3-Brom-pseudocumol wurde nach der Vorschrift von L. J. Smith und
J. Nichols1) über die Grignard-Verbindung mit Orthoameisensäure-äthylester das Acetal
hergestellt, welches nach der Verseifung den gesuchten 2,3,6-Trimethyl-benzaldehyd (P7)lieferte. Nach der Reinigung über das Semicarbazon wurden 0,5 g reiner Aldehyd erhalten
(23% d. Th.).Dieser Aldehyd gab mit Aceton und Natrium-äthylat als Kondensationsmittel das
(2,3,6-Trimethyl-benzal)-aceton. Dazu wurden 0,5 g des Aldehyds mit 3 cm3 absolutem
Aceton und einer Lösung von 35 mg Natrium in 3 cm3 absolutem Alkohol unter Eis¬
kühlung vermischt.
Das Gemisch wurde 2 Stunden bei 0° und über Nacht bei Zimmertemperatur stehen¬
gelassen. Zum Schluss wurde noch 30 Minuten auf 40° erwärmt, darauf mit verdünnter
Schwefelsäure angesäuert und das ausgeschiedene rotbraune Öl in Äther aufgenommen.Die ätherische Lösung wurde mit Natriumhydrogencarbonat-Lösung und Wasser ge¬waschen. Nach dem Trocknen und Abdampfen des Äthers destillierte man das zurück¬
bleibende Öl im Kragenkolben; Sdp.02mm 115—130° (Badtemperatur).
J) B. 67,1096 (1934).2) Vgl. Friedländer, Fortschritte der Teerfarbenfabrikation Bd. 15, S. 193.
3) L. J. Smith und 0. W. Cass, Am. Soc. 54,^1606 (1932).4) L. J. Smith und M. A. Kiess, Am. Soc. 61, 286 (1939).6) J. Org. Chem. 6, 501 (1939).
—
1 j
/2f r
.—
/ 1

— 25 —
Es wurden 0,3 g reines (2,3,6-Trimethy]-benzal)-aeeton (V) erhalten.
Zur Charakterisierung wurde aus 50 mg des ungesättigten Ketons das Semicarbazon
hergestellt. Nach dreimaligem Umkrystallisieren aus Alkohol schmolzen die farblosen,Krystalle bei 216—217°.
3,674 mg Subst. gaben 9,157 mg C02 und 2,559 mg H20C14H19ON3 Ber. 0 68,45 H 7,81%
Gef.„ 68,15 „ 7,79%
Eine Probe des ungesättigten Ketons wurde mit einer methanolischen Lösung von
Phenyl-semicarbazid versetzt. Das daraus als Nadeln erhaltene Phenyl-semicarbazonschmolz nach dem Umkrystallisieren aus Alkohol bei 208—210°.
3,714 mg Subst. gaben 10,169 mg C02 und 2,378 mg H20C20H23ON3 Ber. C 74,73 H 7,21%
Gef,„ 74,72 „ 7,16%
Dieses Phenyl-semicarbazon gab mit dem entsprechenden Derivat des aus dem Harn
erhaltenen Ketons ClsH160 keine Schmelzpunktserniedrigung.Auch das in alkoholischer Losung aufgenommene Absorptionsspektrum des synthe¬
tisch hergestellten Derivates war identisch mit dem aus dem Harn erhaltenen (Fig. 1,Kurve 1).
l-(2, 3, 6-Trimethyl-phenyl)-butanon-(3) VI.
0,3 g des (2,3,6-Trimethyl-benzal)-acetons wurden in 4 cm3 Feinsprit mit 50 mgPalladium-Bariumcarbonat hydriert. Nach der Aufnahme von 1 Mol Wasserstoff wurde
der Katalysator abfiltriert und der Alkohol im Vakuum abgedampft. Aus dem zurück¬
bleibenden ÖPtrarden die Derivate hergestellt.Das Semicarbazon wurde in Form von Blättchen erhalten, welche nach dem
Umkrystallisieren aus Alkohol bei 178—180° schmolzen.
3,790 mg Subst. gaben 9,456 mg C02 und 2,926 mg H20C14H21ON3 Ber. C 67,98 H 8,56%
Gef.„ 68,09 „ 8,64%
Dieses Derivat gab mit dem Semicarbazon des durch Oxydation des Alkohol C13H20Oaus dem Harn erhaltenen Produktes keine Schmelzpunktserniedrigung.
Das in alkoholischer Losung aufgenommene Absorptionsspektrum ist in Fig. 1
(Kurve 3) dargestellt.Das Phenyl-semicarbazon dieses gesättigten Ketons bildete aus Alkohol farblose
Nadeln vom Smp. 167—171° und gab ebenfalls mit dem Phenyl-semicarbazon des Ketons
C13H180, welches durch die Oxydation des Alkohols C13H20O gewonnen worden war, keine
Schmelzpunktserniedrigung.3,728 mg Subst. gaben 10,118 mg C02 und 2,561 mg H20
C20H25ON3 Ber. C 74,27 H 7,79%Gef.
„ 74,07 „ 7,70%Das in Alkohol aufgenommene Absorptionsspektrum, welches identisch ist mit dem
aus dem Harn erhaltenen, ist in Fig. 1 (Kurve 2) dargestellt.
Die Herstellung von trans-Tetrahydro-jonan.<x- und /S-Cyclocitral.
Nach einer von H. Schinz verbesserten Vorschrift von Haarmann und Reimer1)wurde aus Citral ein Gemisch von a- und /?-Cyclocitral hergestellt.
l) D.R.P. 123747; vgl. Friedländer, Fortschritte der Teerfarbenfabrikation. Bd. 6,S. 1238.

— 26 —
50 g frisch destilliertes Anilin wurden in 50 cm3 Äther gelöst und unter Rühren und
Eiskühlung 77 g frisch destilliertes Citral zugetropft. Das grünliche und durch ausge¬
schiedenes Wasser getrübte Reaktionsgemisch Hess man noch 2 Stunden bei 0° stehen.
Dieses wurde dann unter heftigem Rühren langsam in 500 cm3 konz. Schwefelsäure,
welche mit 20 g Eis versetzt und auf -10° gekühlt war, eingetragen. Nach zweistündigemStehen bei dieser Temperatur wurde auf 500 g Eis gegossen und anschliessend mit Wasser
auf 2 Liter verdünnt. Bei der sofort durchgeführten Destillation mit Wasserdampf de¬
stillierte das Cyclocitral-Gemisch als gelbliches öl über. Das Destillat wurde mit Äther
ausgeschüttelt, die ätherische Lösung mit Natriumcarbonat-Lösung und Wasser gewaschenund der Äther nach dem Trocknen abgedampft. Das zurückbleibende öl destillierte bei
75—95» (12 mm) Ausbeute: 55 g (65% d. Th.).
Dihydro-cyclocitral (XIII).
Es wurde zuerst versucht, das erhaltene Gemisch direkt mit Palladium-Barium-
carbonat als Katalysator zu hydrieren. Es zeigte sich aber, dass nur soviel Wasserstoff
aufgenommen wurde, wie dem Gehalt an /?-Cycloeitral entsprach.Das Cyclocitral-Gemisch wurde deshalb in /5-Cyclocitral umgelagert. Nach der Vor¬
schrift von H. Köster1) liess man 55 g Cyclocitral-Gemisch in eine 8-proz. Lösung von
Kaliumhydroxyd in 80-proz. Alkohol während 2 Stunden bei — 5° eintropfen und danach
noch 15 Stunden bei dieser Temperatur stehen.
Das rotbraune Reaktionsgemisch wurde in 1 Liter Wasser gegossen und mit Äther
ausgeschüttelt. Nach dem Eindampfen der getrockneten ätherischen Lösung blieb ein öl
zurück, welches zweimal fraktioniert wurde. Dabei konnten 39 g/3-Cyclocitral, Sdp>1293°, erhalten werden (XII).
.
•»'
Dieses wurde in 50 cm3 Feinsprit gelöst und mit einem in 30 cm3 Feinsprit vor¬
hydrierten Katalysator aus 15 g Palladium-Bariumcarbonat hydriert. Nach der Aufnahme
von 0,92 Mol erfolgte keine Aufnahme von Wasserstoff mehr. Der Katalysator wurde ab¬
filtriert und der Alkohol im Vakuum abgedampft. Bei der Destillation des zurückbleibenden
Öles konnten 36 g des farblosen Dihydro-cyclocitrals vom Sdp.12 81° erhalten werden.
Davon wurde auf die übliche Weise das Semicarbazon hergestellt. Nach dem
Umkrystallisieren aus Methanol schmolz es bei 206°.
3,670 mg Subst. gaben 8,395 mg C02 und 3,296 mg H20
CnH^ONa Ber. C 62,52 H 10,02%Gef.
„ 62,42 „ 10,05%
trans-l-[2,2, 6-Trimethyl-cyclohexyl-(l)]-buten-(l)-on-(3) (XP7)
Eine Lösung von 34 g Dihydro-cyclocitral in 80 g Aceton liess man bei — 5° zu einer-
Lösung von 5,5 g Natrium in 80 cm3 absolutem Alkohol, welche mit 80 g Aceton vermischt
worden war, unter Rühren zutropfen. Während der Reaktion wurde ein durch konz.
Schwefelsäure getrockneter Stickstoff-Strom durchgeleitet. Das Reaktionsgemisch,welches allmählich bräunlich wurde, liess man 22 Stunden bei 0° stehen. Dieses wurde dann
mit Eis zersetzt, mit verdünnter Schwefelsäure angesäuert und das ausgeschiedene Öl mit
Äther ausgeschüttelt. Der Rückstand nach dem Eindampfen der getrockneten ätherischen
Lösung wurde im Vakuum fraktioniert. Nach einem grossen Vorlauf wurde die zwischen
120—130° (12 mm) übergehende Fraktion aufgefangen und bei nochmaliger Rektifizierungein farbloses Öl vom Sdp.,„ 125—129° erhalten.
r 12mm
Nach einigem Stehen war der grösste Tfiü des Destillates krystallin geworden. Die
Krystalle konnten aus gekühltem Petroläther umkrystallisiert werden. Die Verbindungbildet farblose Blättchen von angenehmem Geruch und schmilzt bei 50°.
*) B. 77, 558 (1944).

— 27 —
,Ausbeute: 18 g (42% d. Th.).
3,720 mg Subst. gaben 10,950 mg C02 und 3,789 mg H20
C13H220 Ber. C 80,35 H 11,41%Gef.
„ 80,33 „ 11,40%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum ist in Fig. 5
(Kurve 1) dargestellt.Von einer kleinen Probe der krystallinen Substanz wurde in methanolischer Lösung
mit Phenyl-semicarbazid das Phenyl-semicarbazon hergestellt. Die aus Methanol um-
krystallisierten Nadeln schmolzen bei 195°.
3,717 mg Subst. gaben 9,988 mg C02 und 2,981 mg H20
C20H29ON3 Ber. C 73,35 H 8,93%Gef.
„ 73,33 „ 8,98%
Das Absorptionsspektrum des Phenyl-semicarbazons wurde in alkoholischer Lösung
aufgenommen und besitzt das für a, ^-ungesättigte Ketone charakteristische Maximum.
trans-Tetrahydro-jonon (trans-l-[2, 2, 6-Trimethyl-cyclohexyl-(l)]-butanon-(3)) (X).
5,5 g des a,/?-ungesättigten Ketons wurden in 30 cm3 Feinsprit gelöst und mit einem
vorhydrierten Katalysator aus 2,5 g Palladium-Bariumcarbonat in 20 cm3 Feinsprit
hydriert. Nach kurzer Zeit war genau 1 Mol Wasserstoff aufgenommen. Der Katalysator
wurde abfiltriert, der Alkohol abgedampft und der Rückstand im Vakuum destilliert. Das
Produkt war ein farbloses öl vom Sdp.,2 121° mit angenehmem, jononähnlichem
Geruch.
df= 0,9064 n^2 = 1,4634
3,672 mg Subst. gaben 10,697 mg C02 und 4,049 mg H20
C13H210 Ber. C 79,53 H 12,32%Gef.
„ 79,50 „ 12,34% ' >
Mol. Refr. MDC13H240 Ber. 60,04 Gef. 59,69
Von diesem trans-Tetrahydro-jonon wurde das Phenyl-semicarbazon herge¬stellt. Dieses krystallisierte nach Zugabe von wenig Wasser aus der methanolischen
Lösung in filzigen Nadeln vom Smp. 133°. '
3,715 mg Subst. gaben 9,913 mg C02 und 3,168 mg H20
C20H31ON3 Ber. C 72,90 H 9,48%Gef.
„ 72,82 „ 9,54%
Das Absorptionsspektrum wurde in alkoholischer Lösung aufgenommen und zeigtedas für eine Keto-Gruppe charakteristische Maximum.
Dieses Derivat gab mit dem Phenyl-semicarbazon des stereoispmeren cis-Tetrahydro-
jonon vom Smp. 109° eine Schmelzpunktserniedrigung von 5°.
Das Semicarbazon des trans-Tetrahydro-jonon schmolz, aus Methanol umkry-stallisiert, bei 163°.
3,726 mg Subst. gaben 9,065 mg C02 und 3,552 mg H20C14H2,ON3 Ber. C 66,35 H 10,74%
Gef.„ 66,39 „ 10,67%
Mit dem stereoisomeren cis-Tetrahydro-jonon-semicarbazon gibt die Verbindungkeine Schmelzpunktserniedrigung.
trans-Tetrahydro-jonan (träns-1,1, 3-Trimethyl-2-n-butyl-cyclohexan)'
(VIII).
1,0 g trans-Tetrahydro-jonon wurde mit 1 g Hydrazinhydrat und einer Lösung von
0,7 g Natrium in 15 cm3 absolutem Alkohol in einem Einschlussrohr während 8 Stunden
auf 200° erhitzt.

— 28 —
Das Reaktionsprodukt wurde mit Wasser verdünnt und mit Äther ausgeschüttelt,die ätherische Lösung mit verdünnter Salzsäure, Natriumcarbonat-Lösung und Wasser
gewaschen, mit Natriumsulfat getrocknet und eingedampft. Der Rückstand, 0,72 g eines
schwach terpenartig riechenden Öles, wurde zuerst in einer Craig-Mikrokolonne und dann
nochmals in einem Kragenkölbchen über Natrium im Vakuum destilliert (Badtemperatur98°).
df = 0,8214 n|° = 1,4531
3,698 mg Subst. gaben 11,617 mg C02 und 4,738 mg H20
C13H26 Ber. C 85,63 H 14,37%Gef.
„ 85,74 „ 14,34%Mol. Refr. MDC13H26 Ber. 60,03 Gef. 60,02
cis-Tetrahydro-jonon (cis-l-[2, 2, 6-Trimethyl-cyclohexyl-(l)]-butanon-(3)) (XI).
Zum Vergleich mit dem trans-Tetrahydro-jonon stand ein von A. Bosshardt aus
a-Jonon durch Hydrierung mit Platinoxyd-Katalysator in Eisessig und nachfolgendeOxydation mit Chromsäure hergestelltes und über das Semicarbazon gereinigtes cis-Tetra¬
hydro-jonon zur Verfügung1).
df = 0,9183 n^ = 1,4660
3,609 mg Subst. gaben 10,512 mg C02 und 3,965 mg H20
C13H240 Ber. C 79,53 H 12,32%Gef.
„ 79,49 „ 12,29%Mol. Refr. MDC13H240 Ber. 60,04 Gef. 59,51
Sein Phenyl-semicarbazon krystallisierte aus der mit wenig Wasser versetzten
methanolischen Lösung in schönen Nadeln, welche nach dem Umkrystallisieren aus
Methanol bei 109—110° schmolzen.
3,828 mg Subst. gaben 10,235 mg C02 und 3,261 mg H20
C20H31ON3 Ber. C 72,90 H 9,48%Gef.
„ 72,97 „ 9,53%
cis-Tetrahydro-jonan (cis-l,l,3-Trimethyl-2-n-butyl-cyclohexan) (IX).
Dieser Kohlenwasserstoff wurde auf analoge Weise wie die entsprechende diastere-
omere Verbindung aus 1,0 g cis-Tetrahydro-jonon hergestellt. Das über Natrium im
Vakuum destillierte öl zeigte einen starken terpenartigen Geruch.
df = 0,8280 n*° = 1,4552
3,518 mg Subst. gaben 11,026 mg C02 und 4,507 mg H20
C13H26 Ber. C 85,63 H 14,37%Gef.
„ 85,53 „ 14,34%Mol. Refr. MDC13H26 Ber. 60,03 Gef. 59,77
Die Infrarot-Absorptionsspektren von eis- und trans-,Tetrahydro-jonan sowie von
dem aus dem Harn erhaltenen Kohlenwasserstoff C13H2e sind in Fig. 2 zusammengestellt.
a'-Oxo-cis-tetrahydro-jonon (l-[2, 2, 6-Trimethyl-cyclohexyl]-butandion-(2, 3)) (XVII).
1,0 g cis-Tetrahydro-jonon in 4 cm3 absolutem Alkohol wurde nach Zugabe von
4 Tropfen Salzsäure auf 45° erwärmt2), und sofort mit 1,1 g frisch bereitetem Butyl-nitrit
x) Vgl. A. Bosshardt, Diss. ETH. Zürich, 1946, S. 67.
2) Org. Synth. 10,22(1930).

— 29 —
versetzt. Nach einem kurzen Aufsieden färbte sich das Reaktionsgemisch gelb. Es wurde
noch 2 Stunden auf 45° erwärmt, dann in Äther aufgenommen und solange mit 10-proz.
Natronlauge ausgezogen, bis sich diese alkalischen Auszüge nicht mehr gelb färbten. Die
vereinigten Natronlauge-Auszüge wurden mit Eis versetzt, mit konz. Salzsäure ange¬
säuert und das ausgeschiedene Öl in Äther aufgenommen. Die ätherische Lösung wurde
mit Wasser gewaschen, mit Natriumsulfat getrocknet und der Äther verdampft. Nach
dem Eindampfen des Äthers blieben 0,45 g der Isonitroso-Verbindung zurück (XVI).Diese wurden zur Hydrolyse mit 4 g Oxalsäure versetzt und mit Wasserdampf de¬
stilliert. Das Destillatwurde mitÄther ausgezogen und die ätherische Lösung mit Natrium-
carbonat-Lösung und Wasser gewaschen. Nach dem Abdampfen des getrockneten Äthers
erhielt man 0,25 g eines gelben Öles vom Sdp._1
98—120° (Badtemperatur).
Das öl gab mit alkoholischer Eisen(III)-chlorid-Lösung eine orangerote Färbung.60 mg des öligen Produktes wurden mit einer Lösung von 180 mg Phenyl-semi-
carbazid in 5 cm3 Methanol erwärmt. Es bildeten sich sofort gelbe, in den meisten Lösungs¬mitteln sehr schwer löslicheKrystalle des Bis-(phenyl-semicarbazons), welches .aus
Dioxan umkrystallisiert wurde; Smp. 229°.
3,756 mg Subst. gaben 9,367 mg C02 und 2,550 mg H20
C27H3602N6 Ber. C 68,04 H 7,61%Gef.
„ 67,98 „ 7,60%
Das Absorptionsspektrum des Bis-(phenyl-semiearbazon) wurde in Dioxan aufge¬nommen (Fig. 3, Kurve 1).
Auf analoge Weise konnte aus dem über das krystalline Dihydro-jonon erhaltene
trans-Tetrahydro-jonon das entsprechende a-Diketon erhalten werden. Aus diesem wurde
ein Bis-(phenyl-semicarbazon) erhalten, welches bei 233° schmolz.
3,651 mg Subst. gaben 8,957 mg C02 und 2,356 mg H20
C27H3602N6 Ber. C 68,04 H 7,61%Gef.
„ 67,62 „ 7,29%
Die beiden Bis-(phenyl-semicarbazone) gaben miteinander keine Schmelzpunkts¬erniedrigung. Die beiden Absorptionsspektren waren identisch.
Methyl-(trans-2, 2, 6-trimethyl-cyclohexyl)-keton (XX).
Als Ausgangsprodukt wurde die nach der Vorschrift von B. Shive, J. HorezcyO. Wash und H. L. Lochte1) hergestellte trans-Dihydro-cyclogeraniumsäure verwendet.
9 g der trans-Dihydro-cyclogeraniumsäure wurden mit 10 g Thionylchlorid versetzt
und über Nacht stehengelassen. Das überschüssige Thionylchlorid wurde im Vakuum ent¬
fernt und das entstandene Säurechlorid destilliert, Sdp..„ 88—90°. Das'Methyl-Ketonstellte man daraus mit Dimethyl-cadmium nach der Methode von H. Oilman und J. F.
Nelson1) her. Zu einer aus 1,2 g Magnesium und 5 g Methylbromid in absolutem Ätherbereiteten Grignard-Lösung wurde unter Kühlung mit Eis-Kochsalz 5 g Cadmiumchlorid,welches 4 Stunden bei 100° im Hochvakuum getrocknet worden war, zugefügt und während3 Stunden geschüttelt.
Unter Eiskühlung tropfte man zu dieser Lösung 3,0 g trans-Dihydro-cyclogeranium-säurechlorid in 40 cm3 absolutem Äther. Nach Entfernen des Eisbades verlief die Reaktion
unter gelindem Sieden. Das Reaktionsgemisch wurde 40 Minuten sich selbst überlassen
und dann noch 11/2 Stunden am Rückfluss erhitzt. Das Reaktionsgemisch versetzte man
vorsichtig mit Eis und säuerte mit verdünnter Salzsäure an. Die ätherische Lösung wurde
mit Wasser, Natriumhydrogencarbonat-Lösung und nochmals Wasser gewaschen, mit
Natriumsulfat getrocknet und eingedampft. Aus dem Rückstand Hessen sieh 1,9 g (71%d. Th.) Methyl-Keton als farbloses öl vom Sdp. „
86° gewinnen.
!) Am. Soc. 64,390 (1942).2) Rec. 55, 518 (1936).

— 30 —
Die erhaltene Verbindung besitzt einen intensiven, campherähnlichen Geruch und
reagiert nicht mit Semicarbazid und Phenyl-semicarbazid.
3,640 mg Subst. gaben 10,432 mg C02 und 3,843 mg H20
CnH20O Ber. C 78,51 H 11,98%Gef.
„ 78,20 „ 11,81%
/S'-Oxo-trans-tetrahydro-jonon (l-[2, 2, 6-Trimethyl-cyclohexvl]-butan^dion-fl, 3)) (XXI).
Durch eine eisgekühlte Lösung von 1,9 g Methyl-(2,2,6-trimethyl-eyclohexyl)-keton in 1,5 g Acetanhydrid wurde während einer Stunde Bortrifluorid geleitet1). Das
rötliche Reaktionsgemisch versetzte man mit Wasser und schüttelte mit Äther aus. Die
mit Natriumhydrogencarbonat-Lösung und Wasser gewaschenen ätherischen Auszügegaben bei der Destillation 1,3 g (54% d. Th.) eines farblosen Öles vom Sdp. 88°.
Das daraus hergestellte Phenyl-semicarbazon krystallisierte aus Methanol in
feinen weissen Kadern vom Smp. 187—189°.
3,708 mg Subst. gaben 9,503 mg C02 und 2,800 mg H20
C20H29O2N3 Ber. C 69,94 H 8,51%Gef.
„ 69,94 „ 8,45%
Das Absorptionsspektrum des Phenyl-semicarbazons wurde in alkoholischer Lösung
aufgenommen (Fig. 3, Kurve 2).Aus 1 g Phenyl-semicarbazon wurde durch Spaltung mit Phtalsäureanhydrid im
Wasserdampfstrom 0,45 g reines Diketon erhalten, welches zur Analyse nochmals rekti¬
fiziert wurde.
3,634 mg Subst. gaben 9,907 mg C02 und 3,372 mg H20
C13H2202 Ber. C 74,24 H 10,55%Gef.
„ 74,39 „ 10,38%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum des freien Diketons
zeigte ein Absorptionsmaximum bei 277 m/i (log e = 4,2) (Fig. 5, Kurve 2).Die Verbindung gab mit Eisen(III)-chlorid-Lösung eine weinrote Färbung.
a-Jonon-a',j?'-epoxyd (XXVIII).
Das a',/3'-Epoxyd wurde aus a-Jonon nach der Vorschrift von P. Karrer und
H. Stürzinger*) hergestellt. Es besass einen Smp. 38° und zeigte in alkoholischer Lösungein Absorptionsmaximum bei 277 m/t (log e = 1,5) (Fig. 5, Kurve 3).
3,789 mg Subst. gaben 10,394 mg C02 und 3,218 mg H20
C13H20O2 Ber. C 74,96 H 9,68%Gef.
„ 74,69 „ 9,48%
Das Phenyl-semicarbazon des Epoxydes krystallisierte aus Chloroform-Methanol
in Nadeln vom Smp. 176°.
3,800 mg Subst. gaben 9,799 mg C02 und 2,666 mg H20
C20H27O2N3 Ber. C 70,35 H 7,97%'
Gef.„ 70,37 „ 7,85%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum des Phenyl-semicarbazons war dem Absorptionsspektrum der Phenyl-semicarbazone von gesättigtenKetonen ähnlich (Fig. 6, Kurve 2).
*) Analog H. Meerwein und D. Vossen, J. pr. 141, 157 (1934).
2) Helv. 29, 1833 (1946).

— 31 —
1-(JM.2, 2, 6-Trimethyl-cyclohexenyl)-butanol-(l)-on-(3) (XXIX).und
l.(zJ5,e.2, 2, 6-Trimethyl-cyclohexenyl)-butanol-(2)-on-(3) (XXX).
8,5 g a-Jonon-a',/î'-epoxyd wurden in 30 cm3 Feinsprit mit 4,5 g in Feinsprit vor-
hydriertem Palladium-Bariumcarbonat-Katalysator hydriert. Nach der Aufnahme von
1 Mol Wasserstoff wurde der Katalysator abfiltriert und der Alkohol im Vakuum abge¬dampft. Aus dem zurückbleibenden farblosen Öl Hessen sich durch Auflösen in wenigPetroläther und Abkühlen mit Trockeneis 3,1 g in farblosen Nadeln krystallisierendesl-(J5'8-2,2,6-Trimethyl-cyclohexenyl)-butanol-(l)-on-(3) erhalten. Nach dem Abfiltrieren
der krystallinen Verbindung wurde aus der Mutterlauge noch ein öliges Produkt (4,9 g)gewonnen.
Die krystalline Verbindung, welche mit Tetranitromethan eine Gelbfärbung zeigt,wurde zur Analyse noch zweimal aus gekühltem Petroläther umkrystallisiert; Smp. 63°.
3,762 mg Subst. gaben 10,228 mg C02 und 3,562 mg H20
C13H2202 Ber. C 74,24 H 10,55%Gef.
„ .74,19 „ 10,60%
Das Absorptionsspektrum in alkoholischer Lösung besass ein für die isolierte Car-
bonyl-Gruppe charakteristisches Maximum bei 280 mfi (log e = 1,4).Mit Phenyl-semicarbazid wurde auf die übliche Weise das Phenyl-semicarbazon
hergestellt. Aus Methanol umkrystallisiert schmolzen die farblosen Nadeln bei 161°.
3,518 mg Subst. gaben 9,021 mg C02 und 2,677 mg H20
C20H29O2N3 Ber. C 69,94 H 8,51%Gef.
„ 69,99 „ 8,51%
Das, in alkoholischer Lösung aufgenommene Absorptionsspektrum ist in Fig. 7
(Kurve 1) dargestellt.'
Zur Herstellung des 3, 5-Dinitrobenzoates wurden 0,2 g des krystallinen Pro¬
duktes in 5 cm3 absolutem Äther gelöst und 0,5 g 3,5-Dinitrobenzoyl-chlorid in 5 cm3
absolutem Äther zugefügt. Nach Zugabe von 1 cm3 absolutem Pyridin" wurde 12 Stunden
stehengelassen. Dann filtrierte man den weissen Niederschlag ab. Das Filtrat wurde mit
verdünnter Salzsäure, Natronlauge und Wasser gewaschen und mit Natriumsulfat ge¬trocknet. Nach dem Abdampfen des Äthers konnte der erhaltene Rückstand aus Chloro¬
form-Methanol umkrystallisiert werden. Die gelblichen Krystalle schmolzen bei 138°.
3,653 mg Subst. gaben 7,915 mg C02 und 1,993 mg H20
C20H21O,N2 Ber. C 59,40 H 5,98%Gef.
„ 59,15 „ 6,11%
Hydrolyse1): 0,5g l-(zfM-2,2,6-Trimethyl-cyclohexenyl)-butanol-(l)-on-(3) Hess
man mit 5 cm3 20-proz. Schwefelsäure in 15 cm3 Alkohol 4 Tage stehen. Nach dem üblichen
Aufarbeiten wurde ein gelbliches Öl erhalten, welches mit Phenyl-semicarbazid das a-
Jonon-phenyl-semicarbazon gab. Dieses schmolz bei 187° und zeigte mit einem authen¬
tischen Vergleichspräparat keine Schmelzpunktserniedrigung.Das ölige Hydrierungsprodukt des a-Jonon-a',ß'-epoxyd wurde im Hochvakuum
destilliert; Sdp.Q1 77—80°. Das farblose Öl zeigte sowohl eine positive Eisen(LTI)-chlorid- wie auch eine positive Tetranitromethan-Reaktion.
Aus einer kleinen Probe konnte das Phenyl-semicarbazon, welches aus Methanol
umkrystallisiert war und bei 171° schmolz, erhalten werden.
3,778 mg Subst. gaben 9,672 mg C02 und 2,872 mg H20
C20H29O2N3 Ber. C 69,94 H 8,51%Gef.
„ 69,86 „ 8,51%Das in alkoholischer Lösung aufgenommene Absorptionsspektrum zeigte, dass die
Verbindung eine nichtkonjugierte Carbonyl-Gruppe besitzt. Das aus dem flüssigen Hydrie-
*) Vgl. P. Karrer und H. Stürzinger, Helv. 29, 1833 (1946).

— 32 —
rungsprodukt erhaltene Phenyl-semicarbazon gab mit dem aus der krystallinen Verbindungbereiteten eine Schmelzpunktserniedrigung von 25° (Absorptionsspektrum: Fig. 7,Kurve 2).
Dihydro-a-jonon-3,4-epoxyd (XXVI).
Nach der Methode von Y. B. Naves, O. Schwarzkopf und A. D. Lewis wurde das
a-Jonon-3,4-epoxyd hergestellt1). Zu 12 g a-Jonon wurde in Chloroform eine Lösung von
8,57 g Benzopersäure in Chloroform bei — 5° zugefügt. Nach dem Aufarbeiten konnten
11,1 g reines, öliges a-Jonon-3,4-epoxyd vom Sdp.o,2mm 80° erhalten werden; n^)=l,4901.3,790 mg Subst. gaben 10,434 mg C02 und 3,207 mg H20
Ci3H20O2 Ber. C 74,96 H 9,47%Gef.
„ 75,13 „ 9,68%Das Absorptionsspektrum ist in Fig! 5 (Kurve 4) dargestellt.Das Phenyl-semicarbazon fiel aus der methanolischen Lösung nach Zugabe von
wenig Wasser in Form von Prismen aus, die nach dem Umkrystallisieren aus Chloroform-
Methanol bei 196° schmolzen.
3,921 mg Subst. gaben 10,120 mg C02 und 2,824 mg H20C20H27O2N3 Ber. C 70,35 H 7,97%
Gef.„ 70,43 „ 8,06%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum zeigte Ab-
sorptionsmaxima bei 233 m/i (log e = 4,4) und 277 mß (log e = 4,5) (Fig. 6, Kurve 1).Zur Hydrierung wurden 5,0 g a-Jonon-3,4-epoxyd in Feinsprit mit 2,5 g Palladium-
Bariumcarbonat-Katalysator in einer Wasserstoff-Atmosphäre geschüttelt, wobei 1 Mol
Wasserstoff aufgenommen wurde. Nachdem der Katalysator abfiltriert und das Lösungs¬mittel im Vakuum abgedampft war, destillierte man das erhaltene Öl im Hochvakuum;
Sdp. o.imm 79—81»; n^2 = 1,4712.
Das Phenyl-semicarbazon des Hydrierungsproduktes, welches aus Methanol
umkrystallisiert wurde, schmolz bei 167°.
3,936 mg Subst. gaben 10,117 mg C02 und 2,994 mg H20Ber. C 69,94 H 8,51%Gef.
„ 70,15 „ 8,51%Das in alkoholischer Lösung aufgenommene Absorptionsspektrum besass ein Ab¬
sorptionsmaximum bei 250 m/i (log e = 4,4) (Fig. 6, Kurve 3).Hydrolyse mit alkoholischer Schwefelsäure : 0,5 g Dihydro-a-jonon-3,4-epoxyd
wurden in 20 cm3 alkoholischer Schwefelsäure (5 cm3 20-proz. Schwefelsäure in 15 cm3
Alkohol) gelöst und während 3 Tagen stehengelassen. Dann wurde die gelbliche Lösung in
Wasser gegossen und wie üblich aufgearbeitet. Der Rückstand (0,4 g) destillierte bei
135° (12 mm) und besass einen cedernartigen Geruch. Das ölige Destillat zeigte eine gelb¬braune Tetranitromethan-Reaktion und lieferte kein Phenyl-semicarbazon, nj2 = 1,4930.
3,230 mg Subst. gaben 8,766 mg C02 und 3,019 mg H20C13H2202 Ber. C 74,24 H 10,55%
Gef.„ 74,06 „ 10,46%
Es handelt sich wahrscheinlich um das Dihydro-pyran-Derivat (XXVII).
Dihydro-/S-jonon-2, 3-epoxyd (XXXII).
Nach der Methode von Y. R. Naves, 0. Schwarzkopf und A. D. Lewis wurde das
j?-Jonon-2,3-epoxyd hergestellt1) (XXXI). Zu 12 g /S-Jonon wurde bei — 5° eine Lösungvon 8,57 g Benzopersäure in Chloroform zugefügt. Nach dem Aufarbeiten konnten 6 gder krystallinen Verbindung, die bei 47° schmolz, erhalten werden.
x) Helv. 30, 881 (1947).

— 33 —
3,959 mg Subst. gaben 10,866 mg C02 und 3,484 mg H20
CiAoOj Ber. C 74,96 H 9,68%Gef.
„ 74,90 „ 9,85%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum zeigte ein Ab¬
sorptionsmaximum bei 235 m/x (log e = 4,2) (Fig. 5, Kurve 5).Das Phenyl-semicarbazon fiel aus der mit wenig Wasser versetzten methano¬
lischen Lösung in Form von Prismen aus, die nach dem Umlösen aus Chloroform-Methanol
bei 189° schmolzen.
3,996 mg Subst. gaben 10,296 mg C02 und 2,803 mg H20
C20H2,O2N3 Ber. C 70,35 H 7,97%• Gef.
„ 70,31 „ 7,85%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum besass Absorp-tionsmaxima bei 280 mfi (log e = 4,5) und 233 m/i (log e = 4,3) (Fig. 8, Kurve 1).
Zur Hydrierung wurden 3,0 g /?-Jonon-2,3-epoxyd in Feinsprit mit 2,5 g Palladium-
Bariumcarbonat-Kataly8ator hydriert, wobei 1 Mol Wasserstoff aufgenommen wurde.
Nachdem der Katalysator abfiltriert und das Lösungsmittel im Vakuum abgedampft war,
destillierte man das erhaltene öl im Hochvakuum; Sdp.p lmm 78—81°; nD = 1,4730.
Das auf die übliche Weise hergestellte Phenyl-semicarbazon, aus Methanol
umkrystallisiert, schmolz bei 158°.
3,821 mg Subst. gaben 9,812 mg C02 und 2,934 mg H20
C20H29O2N3 Ber. C 69,94 H 8,51%Gef.
„ 70,08 „ 8,59%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum zeigte das für
eine isolierte Carbonyl-Gruppe charakteristische Absorptionsmaximum (250 m/i, log e =
4,4) (Fig. 8, Kurve 3).
Hydrolyse mit alkoholischer Schwefelsäure: 0,5 g Dihydro-/?-jonon-2,3-epoxydwurden in 20 cm3 alkoholischer Schwefelsäure (5 cm3 20-proz. Schwefelsäure in 15 cm3
Alkohol) gelöst und während 3 Tagen stehengelassen. Dann wurde die Lösung in Wasser
gegossen und wie üblich aufgearbeitet. Das dabei erhaltene öl löste man in Petroläther und
kühlte mit Trockeneis, wobei sich ein krystallines Produkt (0,2 g) abschied. Nach zwei¬
maligem Umkrystallisieren aus Petroläther schmolz die in Form von Nadeln erhaltene
Verbindung (XXXIII) bei 101°.
3,637 mg Subst. gaben 9,116 mg C02 und 3,444 mg H20
ChHjÄ Ber. C 68,38 H 10,60%Gef.
„ 68,40 „ 10,60%
Aus dieser Verbindung konnte. kein Phenyl-semicarbazon erhalten werden. Das
Absorptionsspektrum in alkoholischer Lösung zeigte kein Absorptionsmaximum.Aus der Mutterlauge der krystallinen Verbindung wurde ein öliges Produkt (0,25 mg)
gewonnen, welches im Hochvakuum destilliert wurde; Sdp.01mm 85°.
3,772 mg Subst. gaben 10,335 mg C02 und 3,642 mg H20
C,3H2202 Ber. C 74,24 H 10,55%Gef.
„ 74,77 „ 10,80%
Auch aus dieser Verbindung, welche mit Tetranitromethan eine gelbbraune Färbunggab und in alkoholischer Lösung kein Absorptionsmaximum zeigte, konnte kein
Phenyl-semicarbazon erhalten werden. Es handelt sich wahrscheinlich um ein Dihydro-pyran-Derivat (XXXIV).
4-Oxy-tetrahydro-jonol (XXIII).
5,0 g <x-Jonon-3,4-epoxyd nahmen bei der Hydrierung in 20 cm3 Eisessig mit einem
Platinoxyd-Katalysator (400 mg) 3 Mol Wasserstoff auf. Nach dem Abfiltrieren des
Katalysators wurde die Eisessig-Lösung mit Wasser versetzt und mit Äther ausgeschüttelt.
3 Frlck

— 34 —
Die ätherische Lösung wurde mit Natriumcarbonat-Lösung und Wasser gewaschen und der
Äther nach dem Trocknen abgedampft. Ea blieben 4,9 g einer glasigen, zähen Masse
zurück. Diese wurde in wenig Chloroform gelöst und mit Petroläther bis zur beginnendenTrübung versetzt, wobei 0,40 g eines in Nadeln krystallisierenden Produktes ausfielen.
Nach zweimaligem Umlösen aus Chloroform-Petroläther besass die krystalline Verbindungeinen Smp. 129°. Die Analyse zeigte, dass es sich um ein von den mehreren theoretisch
möglichen diastereomeren 4-Oxy-tetrahydro-jonolen handelt.
3,986 mg Subst. gaben 10,636 mg C02 und 4,378 mg H20
C13H2602 Ber. C 72,84 H 12,33%'
Gef.„ 72,82 „ 12,29%
4-Oxo-tetrahydro-jonon (XXIV).
100 mg des krystallinen Diols wurden in 10 cms Eisessig gelöst, bei Zimmertempe¬ratur mit einer Losung von 100 mg Chromsäure in 10 cm3 Eisessig versetzt und über
Nacht stehengelassen. Nach der Aufarbeitung des Oxydationsgemisches konnte ein Öl
vom Sdp.005mm105° erhalten werden. Zur Analyse wurde nochmals im Kragenkolbendestilliert.
df = 0,9983 n^3 = 1,4779,
3,572 mg Subst. gaben 9,718 mg C02 und 3,345 mg H20C13H2202 Ber. C 74,24 H 10,55%
Gef.„ 74,25 „ 10,47%
Mol. Refr. MDC13H2202 Ber. 60,054 Gef. 59,619
Das Absorptionsspektrum im Infrarot ist in Fig. 9 (Kurve 2) dargestellt. Das in
alkoholischer Lösung aufgenommene Absorptionsspektrum im U.V. besitzt ein schwaches
Absorptionsmaximum bei 280 mfi (log e = 2,2).Das Bis-(phenyl-semicarbazon) des Diketons schied aus der methanolischen
Lösung sofort als feines, krystallines Pulver aus. Dieses konnte aus Chloroform-Methanol
umkrystallisiert werden und schmolz dann bei 207—208°.
3,604 mg Subst. gaben 8,970 mg'C02 und 2,397 mg H20
C27H3602N6 Ber. C 68,04 H 7,61%Gef.
„ 67,92 „ 7,44%
Das in Dioxan aufgenommene Absorptionsspektrum zeigte ein Absorptionsmaximumbei 248 m/t (log e = 4,8) (Fig. 3, Kurve 3). Mit dem Bis-(phenyl-semicarbazon) des KetonsD aus dem Harn trächtiger Stuten gab die Verbindung eine Schmelzpunktserniedrigungvon 10?.
Das in alkoholischer Lösung hergestellte Dioxim schmolz, aus Alkohol-Wasser
umkrystallisiert, bei 149°.
3,784 mg Subst. gaben 8,997 mg C02 und 3,388 mg H20
C13H2402N2 Ber. C 64,96 H 10,07%Gef.
„ 64,88 „ 10,02%
Da die Ausbeute an krystallinem Diol C13H2602 gering war, wurden die nicht-
krystallinen Anteile des Hydrierungsproduktes ebenfalls mit Chromsäure oxydiert. 2,1 g
des Öles wurden in Eisessig bei Zimmertemperatur mit 1,5 g Chromsäure in 10 cm3 Eis¬
essig versetzt und über Nacht stehengelassen. Nach der üblichen Aufarbeitung wurden
1,75 g eines Öles vom Sdp.0 lmm 105" erhalten (n" = 1,478).Das Bis-(phenyl-semicarbazon) dieses Oxydationsproduktes zeigte nach dem Um-
krystallisieren aus Chloroform-Methanol einen Smp. 198—200°. Es gab mit dem etwas
höher schmelzenden Produkt aus dem krystallinen Diol keine Schmelzpunktserniedrigung.
3,920 mg Subst. gaben 9,812 mg C02und 2,700 mg H20
C27H3602N„ Ber. C 68,04 H 7,61%Gef.
„ 67,93 „ 7,60%

— 35 —
0,45 g des Diketons wurden mit 1,0 g Hydrazinhydrat und einer Lösung von 0,7 gNatrium in 15 cm3 absolutem Alkohol während 8 Stunden in einem Einschlussrohr auf
200° erhitzt. Das mit Wasser versetzte Reaktionsprodukt schüttelte man mit Äther aus.
Die ätherische Lösung gab nach Waschen mit Salzsäure, Natriumcarbonat-Lösung und
Wasser, Trocknen mit Natriumsulfat, 0,31 g eines öligen Rückstandes, welcher zur Rei¬
nigung im Vakuum (12 mm) über Natrium destilliert (Badtemperatur 98°) wurde.
3,484 mg Subst. gaben 10,934 mg C02 und 4,508 mg H20
C13H26 Ber. C 85,63 H 14,37%Gef. „' 85,62 „ 14,48%
Das Absorptionsspektrum im Infrarot zeigte eindeutig, dass es sich um das trans-
Tetrahydro-jonan handelt (XXV).
Herstellung von 4-Oxo-tetrahydro-jonon aus Dehydro-/?-jonon.
2,0 g Dehydro-/}-jonon wurden bei — 5° mit einer ätherischen Lösung von Phtal-
mononpersäure, welche 1 Mol Persäure enthielt (1,92 g) versetzt und 5 Tage bei 0° stehen¬
gelassen. Nach 24-stündigenv Stehen bei Zimmertemperatur ist dann die Persäure voll¬
ständig verbraucht worden. Die entstandene Phtalsäure wurde mit Natriumcarbonat-
Lösung entfernt, die ätherische Lösung mit Wasser gewaschen, getrocknet und der Äther
abgedampft. Es blieben 1,5 g eines gelblichen Öles vom Sdp.0 lmm 78—88° zurück.
Es Hess sich daraus kein krystallines Phenyl-semicarbazon herstellen.
Bei der Hydrierung in 5 cm3 Eisessig mit 500 mg Platin-oxyd-Katalysator wurden
3,9 Mol Wasserstoff aufgenommen. Das nach dem Aufarbeiten erhaltene Öl, das sich nur
teilweise in Petroläther löste, wurde zur Reinigung über 45 g Aluminiumoxyd der Akti¬
vität III chromatographiert. Nach einem leichtflüssigen Benzol-Eluat (0,9 g) wurde mit
einem Äther-Methanol-Gemisch 100:1 0,6 g einer in Petroläther schwer löslichen, zähen
Masse eluiert, welche nicht krystallin erhalten werden konnte. 0,2 g davon wurden mit
0,2 g Chromsäure in Eisessig oxydiert. Das erhaltene farblose öl wurde fraktioniert. Die
höher siedenden Anteile (0,1 g) vom Sdp.0 lmm 102° gaben ein Bis-(phenyl-semicarbazon),welches bei 202° schmolz und mit dem aus 4-Oxo-tetrahydro-jonon bereiteten keine
Schmelzpunktserniedrigung zeigte.
3,731 mg Subst. gaben 9,246 mg C02 und 2,572 mg H20
C27H3802N« Ber. C 68,04 H 7,71%Gef.
„ 67,63 „ 7,61%
Oxydation von Dihydro-a-jonol mit Selendioxyd.Dihydro-a-jonol (XXXV).
Zur Herstellung von Dihydro-a-jonol wurden 50 g Dihydro-a-jonon, welche aus
a-Jonon durch Hydrieren in Feinsprit mit einem Palladium-Bariumcarbonat-Katalysatorerhalten wurden, in 250 cm3 absolutem Isopropylalkohol gelöst und 54 g Aluminium-
isöpropylat zugefügt. Das gebildete Aceton destillierte man bei einer Badtemperatur von
108° über eine Kolonne langsam ab. Nach 6 Stunden gab das Destillat mit einer verdünnten
Lösung von 2,4-Dinitro-phenylhydrazin keine Fällung, worauf das Reaktionsgemischim Vakuum weitgehend eingeengt wurde. Der Rückstand wurde in Wasser gegossen, mit
Salzsäure versetzt und mit Äther ausgezogen. Nach dem Abdampfen der mit verdünnter
Salzsäure und Wasser gewaschenen und mit Natriumsulfat getrockneten ätherischen
Lösung konnten 46,2 g Dihydro-a-jonol vom Sdp.I2mm 126° erhalten werden.
3,636 mg Subst. gaben 10,575 mg C02 und 3,968 mg H20
C13H240 Ber. C 79,51 H 12,34%Gef.
„ 79,37 „ 12,21%

— 36 —
5- oder 31-Oxo-tetrahydro-jonon (XXXVI).
20 g Dihydro-a-jonol in 50 g Acetanhydrid wurden mit 16 g Selendioxyd während6 Stunden bei 60° gerührt. Das vom ausgeschiedenen Selen filtrierte Reaktionsproduktgoss man in Wasser und erwärmte die wässerige Lösung zur Zerstörung des Acetanhydrid.Nach dem Erkalten wurde mit Äther ausgeschüttelt, die ätherische Lösung mit ver¬
dünnter Natronlauge gewaschen und mit Natriumsulfat getrocknet.Der nach dem Abdampfen des Äthers erhaltene zähe Rückstand gab bei der Destilla¬
tion 10,1 g einer öligen Fraktion. Diese wurde zur Verseifung in 200 cm3 15-proz. metha¬
nolischer Kalilauge gelöst und 48 Stunden stehengelassen. Nach der üblichen Aufarbeitungerhielt man 9,4 g einer glasigen Masse, welche teilweise in Petroläther löslich war. Zur
Reinigung wurde dieses Produkt in wenig Benzol gelöst und über 200 g Aluminiumoxyd(Aktivität Dil) chromatographiert. Mit Benzol und Äther konnten 6 g Dihydro-a-jonolzurückgewonnen werden. Mit einem Äther-Methanol-Gemisch 100:1 konnten 3,3 g einer
zähen, glasigen Masse eluiert werden, die sich in Petroläther nicht löste. Diese destillierte
man im Hochvakuum. Die bei 115—125° übergehende fast glasharte Masse wurde in
10 cm3 Eisessig gelöst und mit 400 mg vorhydriertem Platinoxyd-Katalysator hydriert.Bei 20° und 715 mm sollten theoretisch 354 cm3 Wasserstoff aufgenommen werden. Die
Hydrierung ging jedoch weiter, indem 510 cm3 Wasserstoff aufgenommen worden waren.
Nach dem üblichen Aufarbeiten konnten 2,9 g eines farblosen Öles erhalten werden. Das
Hydrierungsprodukt wurde in Benzol gelöst und über 90 g Aluminiumoxyd der Aktivität
ILT chromatographiert. Mit Benzol Hessen sich 1,6 g eines in Petroläther löslichen Öles
eluieren, während mit einem Äther-Methanol-Gemisch (100:1) 1,25 g glasige, zähe Masse
erhalten wurden.
1,1 g des zähen Äther-EIuates löste man in 5 cm3 Eisessig und Hess eine Lösung von
1,4 g Chromsäure in 5 cm3 Eisessig zutropfen. Nach 6 Stunden wurde auf die übliche Weise
aufgearbeitet und das Produkt in neutrale und saure Anteile getrennt.Die Neutralprodukte, 0,675 g, bildeten ein farbloses öl vom Sdp.0 lmm 100° (Kragen-
kolben.Badtemperatur), weichesausammoniakalischerSilbernitrat-Lösung Silberausschied.
df>5 = 1,0006 n|3-5= 1,4800
3,650 mg Subst. gaben 9,931 mg C02 und 3,460 mg HaO
C13H2202 Ber. C 74,24 H 10,55%Gef.
„ 74,25 „ 10,44%Mol. Refr. MDC13H2202 Ber. 60,054 Gef. 59,713
Das Absorptionsspektrum im Infrarot ist in Fig. 9 (Kurve 3) dargestellt.100 mg dieses Öles wurden mit 200 mg Phenyl-semicarbazid in 5 cm3 Methanol
versetzt. Es wurde daraus nach Zugabe von wenig Wasser in fast quantitativer Ausbeuteein weisses, krystallines Pulver ausgeschieden, welches sich aus Chloroform-Methanol
umkiystallisieren Hess. Es schmolz bei 202°.
3,718 mg Subst. gaben 9,250 mg C02 und 2,500 mg H20
C27H3602N6 Ber. C 68,04 H 7,61%Gef.
„ 67,89 „ 7,52%In alkoholischer Lösung zeigte die Verbindung ein Absorptionsmaximum bei 250 mp
(log e = 4,8) (Fig. 10, Kurve 2). Mit dem isomeren Bis-(phenyl-semicarbazon) des Di-
ketons D aus dem Harn trächtiger Stuten wurde eine Schmelzpunktserniedrigung von
8° erhalten.
Aus 50 mg des Oxydationsproduktes wurde in methanolischer Lösung mit 150 mg
2,4-Dinitro-phenylhydrazin das Bis-(2, 4-dinitrophenylhydrazon) hergestellt. Die
orangen Krystalle schmolzen nach dem Umkiystallisieren aus Chloroform-Methanol bei
227".
3,820 mg Subst. gaben 10,151 mg C02 und 1,790 mg H20
C25H30O8N8 Ber. C 52,62 H 5,30%Gef.
„ 52,46 „ 5,24%

— 37 —
Reduktion nach Wolff-Kishner: 0,45g erhitzte man während 8 Stunden mit lg
Hydrazinhydrat und einer Lösung von 0,7 g Natrium in 15 cm3 absolutem Alkohol im
Einschlussrohr auf 200°. Nach der üblichen Aufarbeitung erhielt man 0,30 g eines terpen-artig riechenden, wasserhellen Öles, welches zur Analyse zweimal über Natrium rektifiziert
wurde.
3,234 mg Subst. gaben 10,151 mg C02 und 4,126 mg H20
Ci3H28 Ber. C 85,63 H 14,37%Gef.
„ 85,66 „ 14,28% •
Das Absorptionsspektrum im Infrarot war identisch mit demjenigen des trans-
Tetrahydro-jonan.
1,1 -Dimethyl-2-(3-oxo-butyl)-cyclohexan-carbonsäure-(3) (XXXVII).
0,15 mg der zähen, in Petroläther unlöslichen Säure, die sich bei der Oxydation von
Dihydro-a-jonol in Acetanhydrid mit Selendioxyd als Nebenprodukt bildete, wurden mit
0,15 g Phenyl-semicarbazid in 5 cm1 Methanol versetzt. Nach Zugabe von wenigen TropfenWasser krystallisierte das Phenyl-semicarbazon aus. Es konnte aus Methanol um-
krystallisiert werden und schmolz bei 182°.
3,636 mg Subst. gaben 8,887 mg C02 und 2,587 mg H20
C20H29O3Ns Ber. C 66,82 H 8,13%Gef.
„ 66,70 „ 7,96%
Das in alkoholischer Lösung aufgenommene Absorptionsspektrum zeigt ein Maximumbei 250 m/t (log e = 4,3) (Fig. 10, Kurve 3).
S'-Oxo-dihydro-a-jonol (XXXVIII).
20 g Dihydro-a-jonol wurden in 50 cm3 Dioxan mit 18 g Selendioxyd während 3
Stunden am RückfIuss erhitzt. Die Lösung färbte sich dabei dunkelrot und schied schwarzes
Selen ab. Nach dem Abfiltrieren von ausgeschiedenem Selen wurde das Dioxan im Vakuum
weitgehend destilliert, der Rückstand in Äther aufgenommen und die ätherische Lösungdreimal mit verdünnter Natronlauge und Wasser gewaschen. Nach dem Abdampfen der
getrockneten ätherischen Lösung destillierte man den dunkelroten Rückstand im Hoch¬
vakuum (11,2 g).Das Destillat, Sdp.0 j mm 70—78°, wurde in 10 cm3 absolutem Methanol gelöst, mit
12 g Girard-Reagens T versetzt und nach Zugabe von 6,6 g Eisessig 1 Stunde am Rück-
flnss zum Sieden erhitzt. Zu der gekühlten Lösung fügte man 100 g Eis und eine Lösungvon 5 g Natriumcarbonat in 100 cm3 Wasser zu, worauf das Gemisch mit eiskaltem Äther
ausgezogen wurde. Die abgetrennte wässerige Lösung wurde mit 4-n Schwefelsäure ange¬säuert und nach 24 Stunden mit Äther ausgeschüttelt. Nach dem Eindampfen der ätheri¬
schen Auszüge konnten 4,1 g eines Öles vom Sdp.01 Mm 77° erhalten werden. Mit einer
ammoniakalischen Lösung von Silbernitrat gab die Verbindung einen Silberspiegel.50 mg des gelblichen Öles wurden mit einer Lösung von 100 mg Phenyl-semicarbazid
in 5 cm3 Methanol versetzt. Beim Erwärmen krystallisierte das Phenyl-semicarbazonsofort quantitativ aus. Es waren farblose Nadeln, die sich aus Chloroform-Methanol
umkrystallisieren Hessen. Sie schmolzen bei 212°.
3,566 mg Subst. gaben 9,134 mg C02 und 2,687 mg H20
C20H2902N3 Ber. C 69,94 H 8,51%Gef.
„ 69,91 „ 8,43%Das Absorptionsspektrum des Phenyl-semicarbazons zeigte die für a,ß-ungesättigte
Carbonyl-Verbindungen charakteristischen Absorptionsmaxima bei 280 m/t (log e = 4,2)und 235 m/t (log e = 4,2) (Fig. 10, Kurve 1).

— 38 —
Aus dem flüssigen Oxydationsprodukt schied sich beim Stehen an der Luft eine
krystalline Verbindung aus. Sie wurde in Chloroform aufgenommen und mit Petroläther
bis zur beginnenden Trübung versetzt. Die Verbindung krystallisierte in schönen Nadeln.
Nach zweimaligem Umlösen aus den gleichen Lösungsmitteln und Sublimieren bei 100°
(0,1 mm) schmolzen diese Nadeln bei 178°.
3,702 mg Subst. gaben 9,423 mg C02 und 2,988 mg H200,730 mg Subst. verbr. 0,03109 cm3 0,1-n Lauge (el. Titr.1)
CisHjjA, Ber. C 68,99 H 9,80% COOH 19,9%C13H20O3 Ber.
„ 69,64 „ 9,03% „ 20,08%Gef.
„ 69,46 „ 8,93% „ 19,2%
Das Absorptionsspektrum in alkoholischer Lösung zeigte kein Absorptionsmaximum.Aus der krystallinen Verbindung liess sich kein krystallisiertes Phenyl-semicarbazon ge¬
winnen.
Bei einem zweiten Ansatz wurde das ölige Oxydationsprodukt nach der Behandlungmit Girard-Reagens T zur Vermeidung der Autoxydation mit Hydrochinon versetzt.
In diesem Falle wurde die Bildung einer krystallinen Substanz nicht beobachtet.
Zusammenfassung.
1. Das (2,3,6-Trimethyl-phenyl)-butanon-(3) wurde synthetischhergestellt und erwies sich als identisch mit dem Keton C13H180,welches durch Oxydation des Alkohols Ci3H20O aus dem Harn träch¬
tiger Stuten gewonnen worden war.
2. Das bei der obigen Synthese als Zwischenprodukt erhaltene
(2,3,6-Trimethyl-benzal)-aceton war identisch mit dem Keton
C13H160 aus dem Harn trächtiger Stuten.
3. Aus den diastereomeren Tetrahydro-jononen, von welchen
bisher eines unbekannt war, wurden die beiden theoretisch möglichen
Tetrahydro-jonane hergestellt. Durch denVergleich von physikalischenEigenschaften konnte den Diastereomeren die eis- bzw. trans-Kon-
figuration zugeschrieben werden.
4. Durch Vergleich der physikalischen Eigenschaften (Dichte,Molekularrefraktion und Infrarot-Absorptionsspektren) der diastereo¬
meren Tetrahydro-jonane mit denjenigen des Kohlenwasserstoffes
C13H26, welcher nach Wolff-Kishner aus dem Diketon D gewonnen
war, Hess sich zeigen, dass dieses letztere ein cis-Tetrahydro-jonan-Gerüst besitzt. Damit wurde auch das Kohlenstoff-Gerüst anderer
mit dem Diketon D verknüpften Verbindungen aus dem Harn
trächtiger Stuten festgelegt.
5. Es wurden einige Oxo-tetrahydro-jonone, das a'-Oxo-, /S'-Oxo-,4-Oxo- und 31-(oder 5-)Oxo-tetrahydro-jonon, synthetisch herge¬stellt und mit dem Diketon D aus dem Harn verglichen, wobei keines
der synthetischen Produkte damit identisch war.
!) W. Ingold, Helv. 29, 1929 (1946).

Lebenslauf.
Am 6. August 1921 wurde ich in Zürich geboren. Wäh¬rend 6 Jahren besuchte ich die städtische Primarschule und
trat dann in das Eealgymnasium der Kantonsschule
Zürich ein, wo ich im Sommer 1940 mit der Maturitat
(Typus B) abschloss. Im Herbst 1941, nach einem durch
den Militärdienst bedingten Unterbruch, immatrikulierte
ich mich an der Abteilung für Chemie der EidgenössischenTechnischen Hochschule in Zürich. Das durch den Aktiv¬
dienst häufig unterbrochene Studium schloss ich imFrühling1946 mit dem Diplom als Ingenieur-Chemiker ab. Während
weiterer 5 Semester führte ich im Laboratorium von Herrn
Prof. Euzicka unter der Leitung von Herrn Prof. Prelog die
Promotionsarbeit durch.