Embed Size (px)
Citation preview

Incontinentia Pigmenti Achromians as Part of a Neurocutaneous Syndrome:
A Case Report
Keisuke Hamada, MD, Tomoko Tanaka, MD, Shozo Ohdo, MD,
Kunio Hayakawa, MD, Ichiro Kikuchi , MD, and Hiroaki Katsuya, MD
A case of incontinentia pigmenti achromians associated with mental retardation, epilepsy , short stature and ocular anomalies was reported.
A study of this case together with a review of the 38 cases in the literature revealed that this entity has been associated with central nervous system involvem ents at a high frequency . It is necessary, therefore, to consider incontinentia pigmenti achromians as a neurocutaneous syndrome from the viewpoint of pediatric neurology.
Hamada K, Tanaka T, Ohdo S, Hayakawa K , Kikuchi I, Katsuy a H: Incantinentia pigmenti achromians as part of a n eurocutaneous syndrome: A case report. Brain Dev 4: 313-31 7, 1979.
Incontinentia pigmenti achromians (IPA) is a rare but striking disorder of pigmentation of skin, first described by Ito in 1952 [5] .
Up to now about 40 cases of this disorder have been reported. As a result , it has been clarified that in addition to specific fmdings of the skin, IPA is frequently associated with anomalies of the central nervous system in the same manner as incontinentia pigmenti (IP : BlochSulzberger syndrome).
The purposes of this paper are to report a case of IP A and to stress that it is necessary to d~al with IPA as a neurocutaneous syndrome.
Case Report
A 7-year-old Japanese girl was admitted to our
From Departments of Pediatrics (KH, TT, SO, KH), Dermatology (IK), and the Second Department of Pathology (HK), Miyazaki Medical College, Miyazaki.
Received for publication: November 2,1979. Accepted for publication. December 29 , 1979.
Key words. Incontinentia pigmenti achromians, inc0/1-tinentia pigmenti, noncutaneous associated anomalies, neurocutaneous syndrome. Correspondence address: Dr. Keisuke Hamada, Department of Pediatrics, Miyazaki Medical College, Kiyotake-cho, Miyazaki 889-16 , Japan .
hospital for evaluation of convulsions, short stature, mental retardation and hypopigmented patches on the skin.
She was the third child of three from the same parents of a non-consanguineous marriage. Neither the same skin abnormalities nor any disorder worthy of note were found in her siblings and relatives.
She was born at term weighing 2,450 g after a normal delivery. Developmental milestones in infancy were remarkably retarded. At 2 years of age she began to develop generalized tonic and clonic convulsions, which were unaffected by various antiepileptics. At 5 years of age she developed hypopigmented patches scattered over the right lateral abdominal and the gluteal regions and therfiht half of the back. These skin conditions were not preceded by prodromal skin manifestations such as vesiculation or erythema.
She had a well-proportioned short stature with a score of -3.4 SD and was free from striking external anomalies. Nothing abnormal was recognized in the hair, nails and teeth. Hypopigmen ted ' patches were scattered over the right half of the body ; the anterior pectoral and abdominal regions and the scapular, dorsal and gluteal regions. These regions had many streaks and spots on the surface with irregular demar-
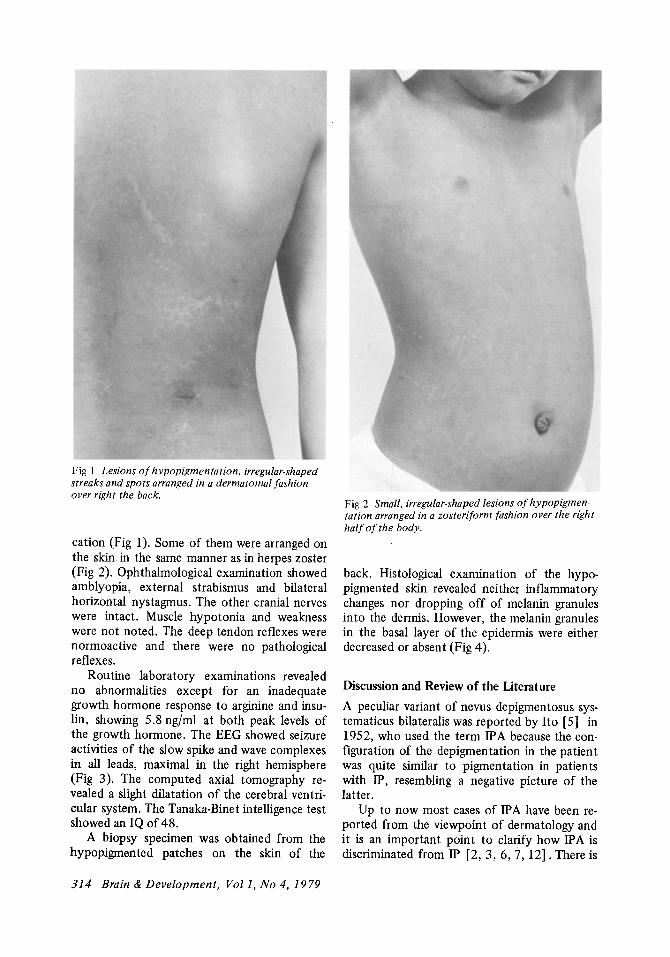
Fig 1 Lesions of hypopigmentation, irregular· shaped streaks and spots arranged in a derma to mal fashion over right the back.
cation (Fig 1). Some of them were arranged on the skin. in the same manner as in herpes zoster (Fig 2). Ophthalmological examination showed amblyopia, external strabismus and bilateral horizontal nystagmus. The other cranial nerves were intact. Muscle hypotonia and weakness were not noted. The deep tendon reflexes were normoactive and there were no pathological reflexes.
Routine laboratory examinations revealed no abnormalities except for an inadequate growth hormone response to arginine and insulin, showing 5.8 ng/ml at both peak levels of the growth hormone. The EEG showed seizure activities of the slow spike and wave complexes in all leads, maximal in the right hemisphere (Fig 3). The computed axial tomography revealed a slight dilatation of the cerebral ventricular system. The Tanaka-Binet intelligence test showed an IQ of 48.
A biopsy specimen was obtained from the hypopigmented patches on the skin of the
314 Brain & Development, Vall, No 4,1979
Fig 2 Small, irregular-shaped lesions of hypopigmen· tation arranged in a zosteriform fashion over the right half of the body.
back. Histological examination of the hypopigmented skin revealed neither inflammatory changes nor dropping off of melanin granules into the dermis. However, the melanin granules in the basal layer of the epidermis were either decreased or absent (Fig 4).
Discussion and Review of the Literature
A peculiar variant of nevus depigmentosus systematicus bilateralis was reported by Ito [5] in 1952, who used the term IPA because the configuration of the depigmentation in the patient was quite similar to pigmentation in patients with IP, resembling a negative picture of the latter.
Up to now most cases of IP A have been reported from the viewpoint of dermatology and it is an important point to clarify how IPA is discriminated from IP [2,3,6,7,12]. There is
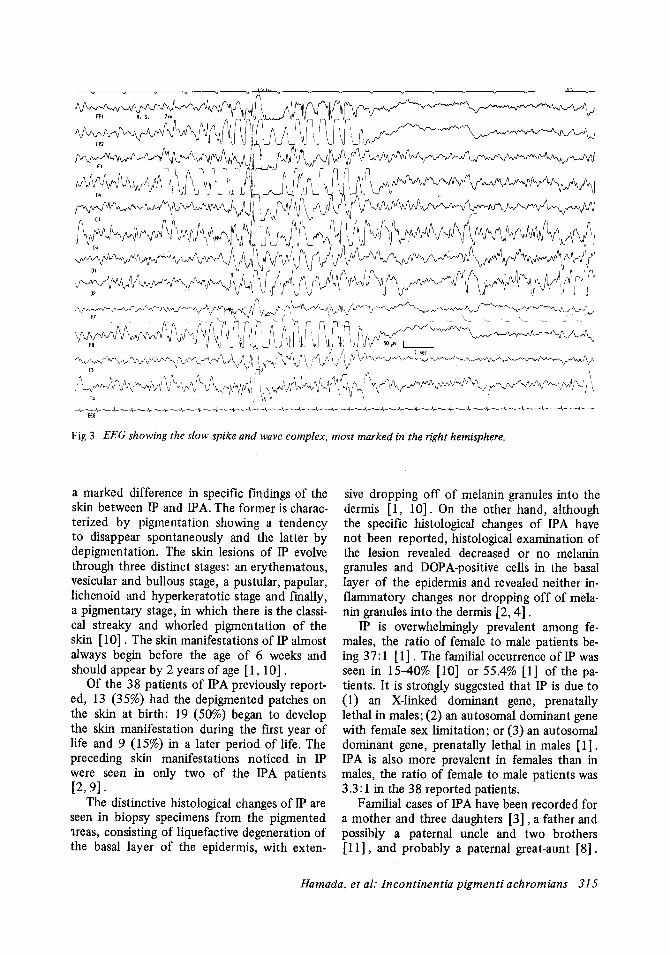
Fig 3 EEG showing the slow spike and wave complex, most marked in the right hemisphere.
a marked difference in specific fmdings of the skin between IP and IP A. The former is characterized by pigmentation showing a tendency to disappear spontaneously and the latter by depigmentation. The skin lesions of IP evolve through three distinct stages: an erythematous, vesicular and bullous stage, a pustular, papular, lichenoid and hyperkeratotic stage and fmally , a pigmentary stage, in which there is the classical streaky and whorled pigmentation of the skin [10]. The skin manifestations of IP almost always begin before the age of 6 weeks and should appear by 2 years of age [1, 10] .
Of the 38 patients of IP A previously reported, 13 (35%) had the depigmented patches on the skin at birth: 19 (50%) began to develop the skin manifestation during the first year of life and 9 (l5%) in a later period of life. The preceding skin manifestations noticed in IP were seen in only two of the IPA patients [2,9] .
The distinctive histological changes of IP are seen in biopsy specimens from the pigmented 1feas, consisting of liquefactive degeneration of the basal layer of the epidermis, with exten-
sive dropping off of melanin granules into the dermis [1, 10]. On the other hand, although the specific histological changes of IP A have not been reported, histological examination of the lesion revealed decreased or no melanin granules and DOPA-positive cells in the basal layer of the epidermis and revealed neither inflammatory changes nor dropping off of melanin granules into the dermis [2,4] .
IP is overwhelmingly prevalent among females, the ratio of female to male patients being 37: 1 [1]. The familial occurrence of IP was seen in 15-40% [H)] or 55.4% [1] of the patients. It is stroiigly suggested that IP is due to (l) an X-linked dominant gene, prenatally lethal in males; (2) an autosomal dominant gene with female sex limitation; or (3) an autosomal dominant gene, prenatally lethal in males [1]. IPA is also more prevalent in females than in males, the ratio of female to male patients was 3.3 :1 in the 38 reported patients.
Familial cases of IP A have been recorded for a mother and three daughters [3] , a father and possibly a paternal uncle and two brothers [11], and probably a paternal great-aunt [8] .
Hamada. et al: Incontinentia pigmenti achromians 315

Fig 4 Histological examination of a hypopigmented lesion in the gluteal region showing decreased or no melanin granules (arrows) in the basal layer of the epidermis. Neither inflammatory changes nor dropping off of melanin granules into the dermis were seen. Fontana Masson stain, x 100.
The familial occurrence of IPA suggests that it is not due to an X-linked dominant gene, but due to an autosomal dominant gene. However, it seems necessary to accumulate further cases before any definite conclusion can be drawn about the hereditary nature of IF A.
Carney (1] reported that associated anomalies were seen in 79.8% of the patients with IP. Of these associated anomalies, dental anomalies including partial anodontia and pegged and malformed teeth were predominant and seen in 64.7% of the patients. Diffuse alopecia was seen in 37.8% of these patients, ocular anomalies including strabismus, nystagmus, cataracts and retrolental fibroplasia in 35.2~, central nervous system involvements including spastic and paralytic disorders, mental retardation, slow motor development, convulsive disorders and abnormalities in EEG in 30.5%, and structural anomalies including skull deformities, dwarfism, clubfoot, spina bifida, cleft palate, cleft lip and ear anomalies in 13.7%.
Of the 38 patients with IP A, 29 patients
316 Brain & Development, Vol 1, No 4, 1979
(76.4%) had associated noncutaneous anomalies. Predominant types of associated anomalies in IPA were central nervous system involvements including mental retardation, convulsive disorders, and abnormalities in EEG, which were seen in 50% of the patients: Furthermore, structural anomalies including skull deformities, ear anomalies, spina bifida occuita, cleft palate, congenital dislocation of the hip, scoliosis, syndactyly and leg discrepancy were seen in 36.8%, ocular anomalies including strabismus, nystagmus, retinal pigmentary abnormality, tessellated fundus , pupillary dislocation, choroidal atrophy, opaque cornea, optic atrophy and microphthalmia in 31.5%, dental anomalies including malformation, malalignment of teeth and dental dysplasia in 13.5% and diffuse alopecia in 2.6%.
There is a considerable difference in frequency of the appearance of each associated anomaly between IP and IPA. However, there is very good agreement in the types of such anomalies between IP and IP A.

This agreement in the types of associated anomalies seems to suggest that IP and IP A may have the same pathogenesis [4] . Nevertheless, the review of 38 cases with IPA seems to indicate that IPA belongs to a syndrome entity which is clearly different clinically, histologically and genetically from that to which IP belongs. In particular, IP A has been associated with central nervous system involvements at a high frequency. Therefore, it is necessary to consider IPA as a neurocutaneous syndrome from the viewpoint of pediatric neurology.
References
1. Carney RG Jr: Incontinentia pigmenti. Arch Dermatol1l2: 535-542, 1976.
2. Griffiths A: Incontinentia pigmenti achromians. Arch Dermatollll: 751-752, 1975.
3. Grosshans EM, Stoebner P, Bergond H, et ai: Incontinentia pigmenti achromians (Ito). Dermatologica 142: 65-78, 1978.
4. Hamada T, Saito T, Sugai T, et al : Incontinentia
pigmenti achromians (Ito). Arch Dermatol 96: 673-676,1967.
5. Ito M: Studies on melanin XI. Incontinentia pigmenti achromians: A singular case of nevus depigmentosus systematicus bilateralis. Tohoku J Exp Med (Sendai) 55 (Suppl): 57-59, 1952.
6. Jelinik JE, Bart RS, Schiff GM: Hypomeianosis of Ito ("lncontinentia pigmenti achromians") . Arch Dermatol107: 596-601, 1973.
7. Maize JC , Headington JH, Lynch PJ, et al : Systematized hypochromic nevus. Arch Dermatol 106: 884-885 , 1972.
8. Masumizu T: lncontinentia pigmenti achromians (Ito). Jpn J Dermatol (Tokyo) Ser B 73 : 303, 1963 .
9. Mittal R, Handa F, Sharma SC : Incontinentia pigmenti et achromians. Dermatologica 150: 355-359, 1975.
10. Morgan JO : lncontinentia pigmenti (BlochSulzberger syndrome). Am J Dis Child 122: 294-300, 1971.
11. Rubin MB: lncontinentia pigmenti achromians. Arch Dermatol 105: 424-425, 1972.
12. Schwartz ME Jr, Esterly , NB, Fretzin OF, et al: Hypomelanosis of Ito (lncontinentia pigmenti achromians) : A neurocutaneous syndrome. J Pediatr90: 236-240,1977.
Hamada, et al: Incontinentia pigmenti achromians 317

![Tnfa Signaling Through Tnfr2 Protects Skin Against ...eprints.whiterose.ac.uk/81541/1/Tnfa signaling through tnfr2 protects... · genodermatosis incontinentia pigmenti (IP) [17]](https://img.pdfslide.net/doc/110x75/5f3bedf6651a4c137761035c/tnfa-signaling-through-tnfr2-protects-skin-against-signaling-through-tnfr2-protects.jpg)



























