Embed Size (px)
Citation preview

Involvement of Upregulated P53-Induced Death DomainProtein (PIDD) in Neuronal Apoptosis after Rat TraumaticBrain Injury
Chunhua Wan & Junkang Jiang & Hui Mao &
Jianhua Cao & Xiaohong Wu & Gang Cui
Received: 25 April 2013 /Accepted: 11 June 2013# Springer Science+Business Media New York 2013
Abstract Traumatic brain injury (TBI) initiates a series ofcomplicated pathological events that could eventually lead toneuronal apoptosis. Recent studies indicated that p53 playeda crucial role in neuronal apoptosis and regeneration follow-ing TBI. However, the detailed mechanism of p53-inducedneuronal apoptosis in TBI remains largely elusive. In thisstudy, we identified that p53-induced death domain protein(PIDD), whose transcription could be rapidly induced by p53activation, was significantly upregulated after TBI. Westernblot and immunohistochemistrical analyses revealed that theexpression of PIDD was gradually increased, reached a peakat 3 days, and then decreased gradually to basal level afterbrain trauma. Further, double immunofluorescent analysisshowed that PIDD was distributed predominantly in neu-rons, and the number of PIDD-positive neurons was signif-icantly elevated in injured brain cortex. In addition, we foundthat PIDD was mainly distributed in active caspase-3-positive neurons, implicating a possible involvement ofPIDD in the regulation of neuronal apoptosis during TBI.Finally, we showed that the expressions of p53 and Bax werealtered correlatively with PIDD after brain trauma, implyingthat the upregulation of PIDD after TBI might be a result ofp53 activation. Taken together, these findings suggested thatPIDD might be an important regulator and potential thera-peutic target of TBI.
Keywords PIDD . Traumatic brain injury . Neuronalapoptosis . Rat
AbbreviationsPIDD p53-induced death domain proteinTBI Traumatic brain injuryCNS Central neural systemNeuN Neuronal nucleiRAIDD RIP-associated Ich-1/CED homologous protein
with death domainATM Ataxia–telangiectasia-mutated proteinBax Bcl-2-associated X proteinGFAP Glial fibrillary acidic proteinGAPDH Glyceraldehyde-3-phosphate dehydrogenase
Background
Traumatic brain injury (TBI) represents a serious public healthproblem and a major cause of death and disability in youngpeople worldwide. It is estimated that there are approximatelyten million people who suffer from TBI worldwide each year,of which some 2.2–3.6 million people encounter moderate orsevere TBI (Langlois et al. 2006). The pathophysiology oftraumatic brain injury is complicated and could be dividedmainly into two injury phases, the primary injury and second-ary injury (Werner and Engelhard 2007). The primary injuryrepresents immediate cell death and injury due to mechanicaldamage. The secondary injury occurs usually within fewhours or days following the initial insults and is mainly causedby free radical damage, ischemia, microglial activation, reac-tive astrocytes, the secretion of reactive oxygen species, andpro-inflammatory cytokines that exacerbate the initial damage(Morganti-Kossmann et al. 2002). In this phase, a massive
Chunhua Wan and Junkang Jiang contribute equally to this work.
C. Wan : J. JiangSchool of Public Health, Nantong University, Nantong, People’sRepublic of China
H. Mao : J. Cao :X. Wu :G. Cui (*)Department of Neurosurgery, the First Affiliated Hospital ofSoochow University, No. 188, Shixin Street, Suzhou 215006,People’s Republic of Chinae-mail: [email protected]
J Mol NeurosciDOI 10.1007/s12031-013-0050-4

neuronal apoptosis might occur in focal injury or diffuse brainregion, which was supposed to be a key determinant of theextent of neurological injury and recovery after TBI(Raghupathi et al. 2000; Conti et al. 1998). However, theunderlying molecular mechanism of neuronal apoptosis fol-lowing TBI remains largely elusive.
It has been widely accepted that p53 was a master regulatorof cell apoptosis and viability. p53 signaling was found to beinvolved in the pathology of kinds of central nervous system(CNS) diseases (Culmsee and Mattson 2005; Martin 2001).JA Napieralski reported that p53 was upregulated in TBI andparticipated in the repair and apoptotic process of neurons(Napieralski et al. 1999). Further studies using p53 null micerevealed that p53 activation following TBI was a major path-ological event that led to neuronal apoptosis, while p53knockout resulted in significantly reduced neuronal apoptosisand better prognosis (Martin et al. 2001). Controversially,other studies found that though p53 knockout improvedneuromotor function, it did not prevent neuronal cell deathfollowing experimental brain trauma, suggesting a complicat-ed role of p53 in neuronal apoptosis after TBI (Tomasevicet al. 2010). Though some of p53 target genes, such as Bcl-2-associated X protein (Bax) and Noxa, were identified to beimplied in the regulation of p53-dependent neuronal apoptosisafter brain injury, much of p53 downstream signaling in theprocess remains unclear (Kiryu-Seo et al. 2005). Thus, a betterunderstanding of the molecular mechanism of p53-inducedneuronal apoptosis would help clarify the neuropathologicalprocess during TBI and thus provide potential opportunitiesfor an improved therapy of TBI.
The p53-induced death domain protein (PIDD) was initial-ly identified as a p53-response gene and contributed to p53-induced apoptosis under stress conditions (Lin et al. 2000).Later, it was found that PIDD could interact with caspase-2and RIP-associated Ich-1/CED homologous protein withdeath domain (RAIDD) to form a protein complex, calledPIDDosome, to trigger apoptosis (Tinel and Tschopp 2004;Berube et al. 2005). PIDDosome promotes the activation ofcaspase-2, which then leads to BH3 interacting-domain deathagonist (Bid) cleavage, Bax translocation, and mitochondria-dependent apoptosis. Increased expression of PIDD resultedin spontaneous caspase-2 activation and sensitization to stress-induced apoptosis. Notably, p53-induced PIDD expressionand caspase-2 activation was found to be responsible for theinitial stage of p53-mediated apoptosis, highlighting PIDD asan important mediator of p53-triggered apoptotic process(Baptiste-Okoh et al. 2008). Besides, PIDD was shown toactivate NF-κb to promote cells for repair in response togenotoxic stimuli, which provided a direct link between p53and nuclear factor kappa-light-chain-enhancer of activated Bcells (NF-κb) activation (Berube et al. 2005). Besides, it wasreported that PIDD could be post-transcriptionally regulatedand cleaved to exert its function (Ando et al. 2012; Tinel et al.
2007). All of these findings indicated that PIDD could be animportant downstream target in p53-induced apoptosis andrepair. However, whether PIDD was involved in p53-mediated neuronal apoptosis after TBI remains unknown.
In the study, we found a significant elevation of PIDDexpression following TBI. Further, we showed that the ex-pression of PIDD was relevant to caspase-3 activation afterTBI. Our findings suggested that PIDD might be involved inthe regulation of neuronal apoptosis during brain trauma,shedding new light on the pathophysiology of TBI.
Materials and Methods
Establishment of Rat Traumatic Brain Injury Model
Experiments were performed in accordance with National In-stitutes of Health Guidelines for the Care andUse of LaboratoryAnimals. The experimental protocol was approved by theCommittee on Animal Care and Use of Nantong University.All efforts were made to minimize the number of animals usedand their suffering. TBI model was established as previousreports with minor modifications (Logan et al. 1992; Liu et al.2010; Chen et al. 2011). Briefly, adult male Sprague–Dawleyrats (n=48; body weight, 220–275 g) were anesthetized withketamine (90 mg/kg)/xylazine (10 mg/kg) and subject to sur-gery under aseptic conditions. An anteroposterior surgical inci-sion (5-mm long, 3-mmdeep, and 1-mmwide) was then carriedout by inserting a microknife into the right cortex 3 mm lateralfrom the midline (n=42). Sham-treated rats (n=6) were subjectto identical procedures as the experimental rats, except that theydid not receive surgical incision with a microknife. The scalpswere immediately closed in layers with 4-0 silk sutures andstaples, and the animals were recovered on a 30 °C heating pad.After the surgery, animals were housed with a 12-h light/darkcycle and allowed to get water and food freely. Animals weresacrificed at 12 h, 1, 3, 5, 7, 14, and 28 days following injury,and sham-treated rats were killed at day 3.
Western Blot Analysis
To obtain protein samples for Western blot analysis, ratswere sacrificed at indicated time points after operation (eachtime point, n=3), and the brain tissue surrounding the epi-center (extending 2 mm to the incision) as well as an equalregion of the contralateral, sham-operated cortex was dis-sected out and immediately stored at −80 °C until use. Next,frozen tissue samples were thawed and minced with eyescissors on ice. The samples were then homogenized in ahomogenization buffer containing 50 mmol/L Tris, pH 7.5,5 mmol/L EDTA, 1 % sodium deoxycholate, 0.2 % Triton X-100, 1 % NP-40, and complete protease inhibitor cocktail(Roche Diagnostics, Basel, Switzerland). The lysates were
J Mol Neurosci
centrifuged for 20 min at 13,000×g, 4 °C. After the determina-tion of protein concentration with the Bradford assay(Bio-Rad), the resulting supernatant was separated withSDS-PAGE. The separated proteins were then trans-ferred to a polyvinylidene difluoride membrane (Millipore,MA, USA) by a transfer apparatus. The membrane was thenblocked with 5 % nonfat milk and incubated with primaryantibodies against PIDD (rabbit, 1:1,000; Santa Cruz),glyceraldehyde-3-phosphate dehydrogenase (GAPDH; rabbit,1:1,000; Santa Cruz), p53 (rabbit, 1:1,000; Santa Cruz),Bax (rabbit, 1:1,000; Cell Signaling), Bcl-2 (rabbit, 1:1,000;Cell Signaling), active caspase-3 (rabbit, 1:1,000; Cell Sig-naling). After secondary antibody incubation with a horse-radish peroxidase-conjugated donkey anti-rabbit IgG (JacksonImmunoResearch, Pennsylvania, USA), protein was visual-ized using an enhanced chemiluminescence system (ECL,Pierce Company, IL, USA).
Sections and Immunohistochemistry
After a defined survival time, rats were terminally anesthe-tized with chloral hydrate and perfused through the ascend-ing aorta with saline, followed by 4 % paraformaldehyde atdifferent survival time (each time point, n=3). Thereafter, thebrains were removed and fixed in 4 % paraformaldehyde for6 h and then replaced with 20 % sucrose for 1 day, followed
by 30 % sucrose for 2–3 days. Next, the tissues were em-bedded in OCT compound, and 10-μm frozen cross sectionswere prepared and examined. All of the sections were blockedwith 10 % donkey serum with 0.3 % Triton X-100 and 1 %(w/v) bovine serum albumin (BSA) for 2 h at room tempera-ture and incubated overnight at 4 °C with anti-PIDD antibody(1:200, Santa Cruz), followed by incubation with biotinylatedsecondary antibody (Vector Laboratories, Burlingame, CA,USA). The sections were visualized with a DAB reagent(Vector Laboratories). Cells with strong or moderate brownstaining were counted as positive, cells with no staining werecounted as negative.
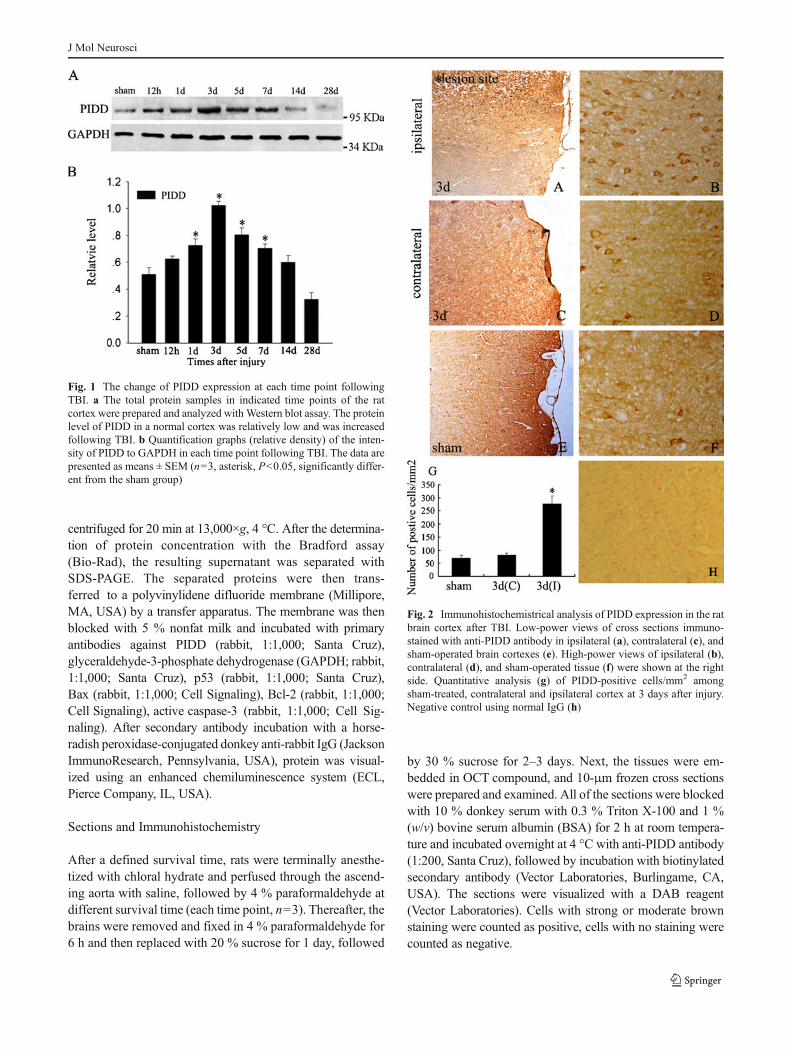
Fig. 1 The change of PIDD expression at each time point followingTBI. a The total protein samples in indicated time points of the ratcortex were prepared and analyzed with Western blot assay. The proteinlevel of PIDD in a normal cortex was relatively low and was increasedfollowing TBI. b Quantification graphs (relative density) of the inten-sity of PIDD to GAPDH in each time point following TBI. The data arepresented as means ± SEM (n=3, asterisk, P<0.05, significantly differ-ent from the sham group)
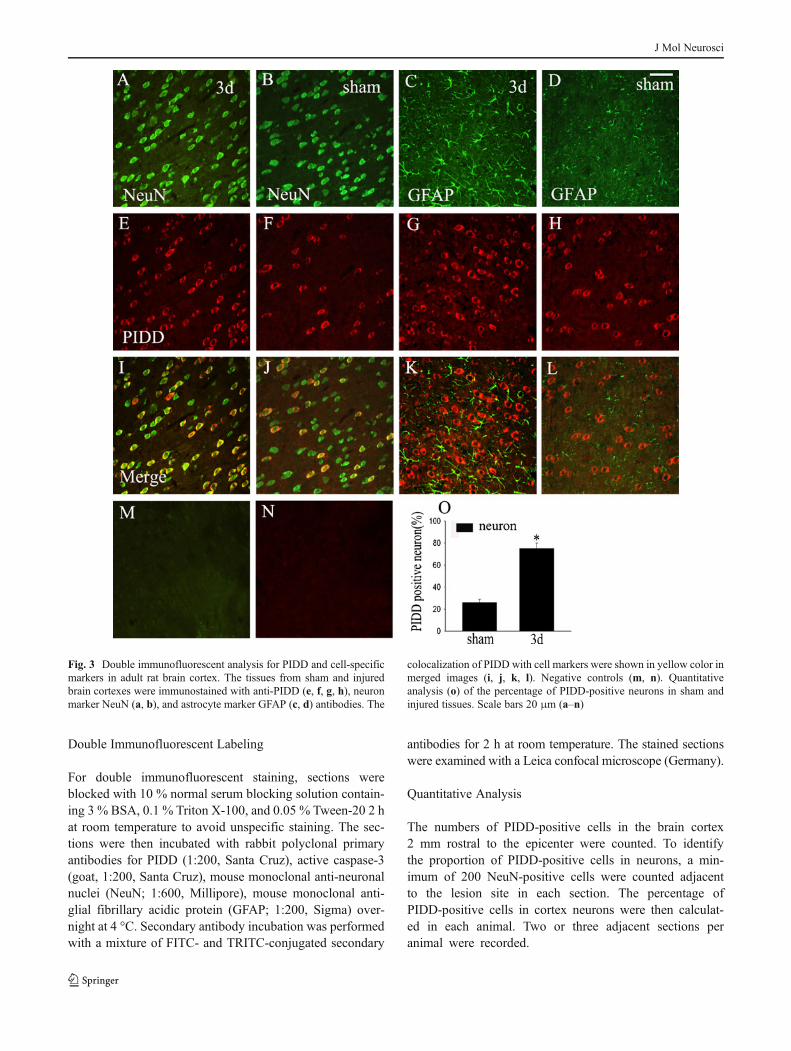
Fig. 2 Immunohistochemistrical analysis of PIDD expression in the ratbrain cortex after TBI. Low-power views of cross sections immuno-stained with anti-PIDD antibody in ipsilateral (a), contralateral (c), andsham-operated brain cortexes (e). High-power views of ipsilateral (b),contralateral (d), and sham-operated tissue (f) were shown at the rightside. Quantitative analysis (g) of PIDD-positive cells/mm2 amongsham-treated, contralateral and ipsilateral cortex at 3 days after injury.Negative control using normal IgG (h)
J Mol Neurosci
Double Immunofluorescent Labeling
For double immunofluorescent staining, sections wereblocked with 10 % normal serum blocking solution contain-ing 3 % BSA, 0.1 % Triton X-100, and 0.05 % Tween-20 2 hat room temperature to avoid unspecific staining. The sec-tions were then incubated with rabbit polyclonal primaryantibodies for PIDD (1:200, Santa Cruz), active caspase-3(goat, 1:200, Santa Cruz), mouse monoclonal anti-neuronalnuclei (NeuN; 1:600, Millipore), mouse monoclonal anti-glial fibrillary acidic protein (GFAP; 1:200, Sigma) over-night at 4 °C. Secondary antibody incubation was performedwith a mixture of FITC- and TRITC-conjugated secondary
antibodies for 2 h at room temperature. The stained sectionswere examined with a Leica confocal microscope (Germany).
Quantitative Analysis
The numbers of PIDD-positive cells in the brain cortex2 mm rostral to the epicenter were counted. To identifythe proportion of PIDD-positive cells in neurons, a min-imum of 200 NeuN-positive cells were counted adjacentto the lesion site in each section. The percentage ofPIDD-positive cells in cortex neurons were then calculat-ed in each animal. Two or three adjacent sections peranimal were recorded.
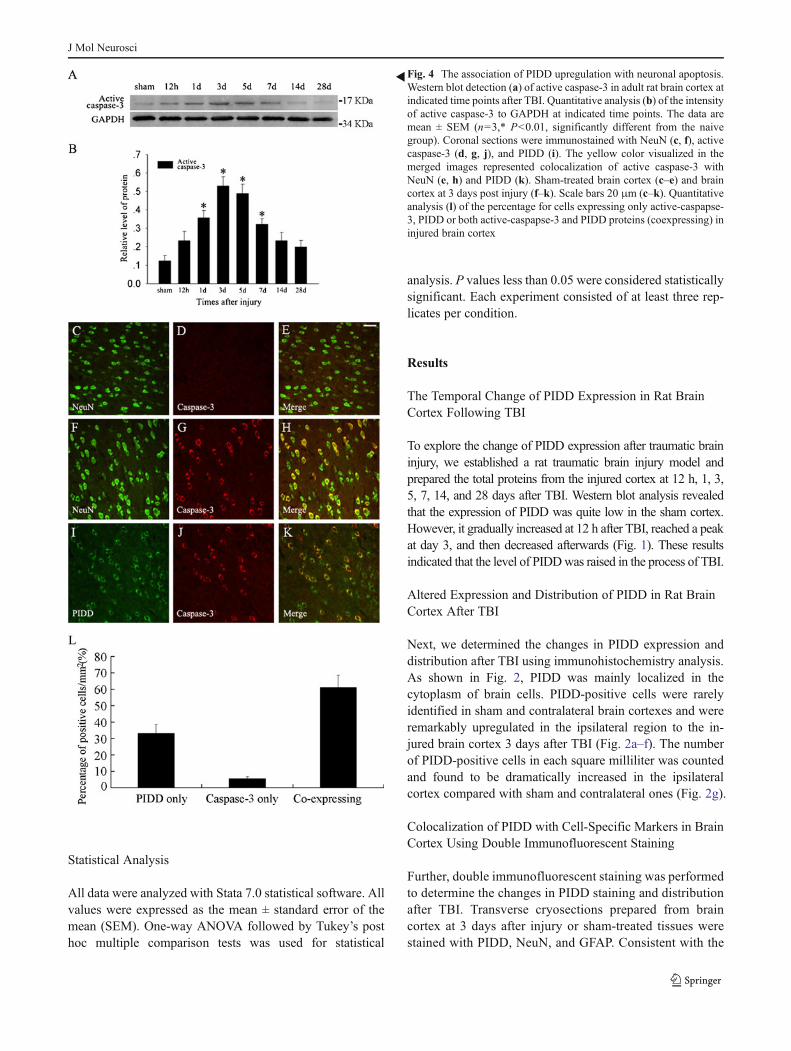
Fig. 3 Double immunofluorescent analysis for PIDD and cell-specificmarkers in adult rat brain cortex. The tissues from sham and injuredbrain cortexes were immunostained with anti-PIDD (e, f, g, h), neuronmarker NeuN (a, b), and astrocyte marker GFAP (c, d) antibodies. The
colocalization of PIDD with cell markers were shown in yellow color inmerged images (i, j, k, l). Negative controls (m, n). Quantitativeanalysis (o) of the percentage of PIDD-positive neurons in sham andinjured tissues. Scale bars 20 μm (a–n)
J Mol Neurosci
Statistical Analysis
All data were analyzed with Stata 7.0 statistical software. Allvalues were expressed as the mean ± standard error of themean (SEM). One-way ANOVA followed by Tukey’s posthoc multiple comparison tests was used for statistical
analysis. P values less than 0.05 were considered statisticallysignificant. Each experiment consisted of at least three rep-licates per condition.
Results
The Temporal Change of PIDD Expression in Rat BrainCortex Following TBI
To explore the change of PIDD expression after traumatic braininjury, we established a rat traumatic brain injury model andprepared the total proteins from the injured cortex at 12 h, 1, 3,5, 7, 14, and 28 days after TBI. Western blot analysis revealedthat the expression of PIDD was quite low in the sham cortex.However, it gradually increased at 12 h after TBI, reached a peakat day 3, and then decreased afterwards (Fig. 1). These resultsindicated that the level of PIDDwas raised in the process of TBI.
Altered Expression and Distribution of PIDD in Rat BrainCortex After TBI
Next, we determined the changes in PIDD expression anddistribution after TBI using immunohistochemistry analysis.As shown in Fig. 2, PIDD was mainly localized in thecytoplasm of brain cells. PIDD-positive cells were rarelyidentified in sham and contralateral brain cortexes and wereremarkably upregulated in the ipsilateral region to the in-jured brain cortex 3 days after TBI (Fig. 2a–f). The numberof PIDD-positive cells in each square milliliter was countedand found to be dramatically increased in the ipsilateralcortex compared with sham and contralateral ones (Fig. 2g).
Colocalization of PIDD with Cell-Specific Markers in BrainCortex Using Double Immunofluorescent Staining
Further, double immunofluorescent staining was performedto determine the changes in PIDD staining and distributionafter TBI. Transverse cryosections prepared from braincortex at 3 days after injury or sham-treated tissues werestained with PIDD, NeuN, and GFAP. Consistent with the
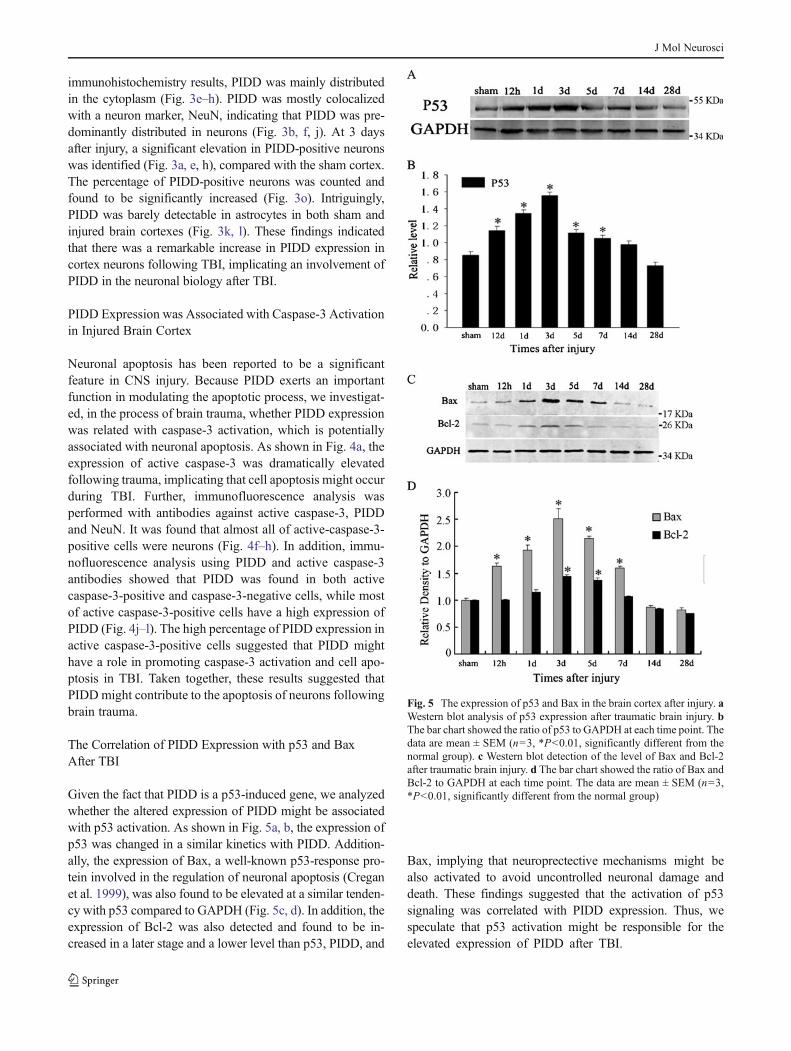
�Fig. 4 The association of PIDD upregulation with neuronal apoptosis.Western blot detection (a) of active caspase-3 in adult rat brain cortex atindicated time points after TBI. Quantitative analysis (b) of the intensityof active caspase-3 to GAPDH at indicated time points. The data aremean ± SEM (n=3,* P<0.01, significantly different from the naivegroup). Coronal sections were immunostained with NeuN (c, f), activecaspase-3 (d, g, j), and PIDD (i). The yellow color visualized in themerged images represented colocalization of active caspase-3 withNeuN (e, h) and PIDD (k). Sham-treated brain cortex (c–e) and braincortex at 3 days post injury (f–k). Scale bars 20 μm (c–k). Quantitativeanalysis (l) of the percentage for cells expressing only active-caspapse-3, PIDD or both active-caspapse-3 and PIDD proteins (coexpressing) ininjured brain cortex
J Mol Neurosci
immunohistochemistry results, PIDD was mainly distributedin the cytoplasm (Fig. 3e–h). PIDD was mostly colocalizedwith a neuron marker, NeuN, indicating that PIDD was pre-dominantly distributed in neurons (Fig. 3b, f, j). At 3 daysafter injury, a significant elevation in PIDD-positive neuronswas identified (Fig. 3a, e, h), compared with the sham cortex.The percentage of PIDD-positive neurons was counted andfound to be significantly increased (Fig. 3o). Intriguingly,PIDD was barely detectable in astrocytes in both sham andinjured brain cortexes (Fig. 3k, l). These findings indicatedthat there was a remarkable increase in PIDD expression incortex neurons following TBI, implicating an involvement ofPIDD in the neuronal biology after TBI.
PIDD Expression was Associated with Caspase-3 Activationin Injured Brain Cortex
Neuronal apoptosis has been reported to be a significantfeature in CNS injury. Because PIDD exerts an importantfunction in modulating the apoptotic process, we investigat-ed, in the process of brain trauma, whether PIDD expressionwas related with caspase-3 activation, which is potentiallyassociated with neuronal apoptosis. As shown in Fig. 4a, theexpression of active caspase-3 was dramatically elevatedfollowing trauma, implicating that cell apoptosis might occurduring TBI. Further, immunofluorescence analysis wasperformed with antibodies against active caspase-3, PIDDand NeuN. It was found that almost all of active-caspase-3-positive cells were neurons (Fig. 4f–h). In addition, immu-nofluorescence analysis using PIDD and active caspase-3antibodies showed that PIDD was found in both activecaspase-3-positive and caspase-3-negative cells, while mostof active caspase-3-positive cells have a high expression ofPIDD (Fig. 4j–l). The high percentage of PIDD expression inactive caspase-3-positive cells suggested that PIDD mighthave a role in promoting caspase-3 activation and cell apo-ptosis in TBI. Taken together, these results suggested thatPIDD might contribute to the apoptosis of neurons followingbrain trauma.
The Correlation of PIDD Expression with p53 and BaxAfter TBI
Given the fact that PIDD is a p53-induced gene, we analyzedwhether the altered expression of PIDD might be associatedwith p53 activation. As shown in Fig. 5a, b, the expression ofp53 was changed in a similar kinetics with PIDD. Addition-ally, the expression of Bax, a well-known p53-response pro-tein involved in the regulation of neuronal apoptosis (Creganet al. 1999), was also found to be elevated at a similar tenden-cy with p53 compared to GAPDH (Fig. 5c, d). In addition, theexpression of Bcl-2 was also detected and found to be in-creased in a later stage and a lower level than p53, PIDD, and
Bax, implying that neuroprectective mechanisms might bealso activated to avoid uncontrolled neuronal damage anddeath. These findings suggested that the activation of p53signaling was correlated with PIDD expression. Thus, wespeculate that p53 activation might be responsible for theelevated expression of PIDD after TBI.
Fig. 5 The expression of p53 and Bax in the brain cortex after injury. aWestern blot analysis of p53 expression after traumatic brain injury. bThe bar chart showed the ratio of p53 to GAPDH at each time point. Thedata are mean ± SEM (n=3, *P<0.01, significantly different from thenormal group). c Western blot detection of the level of Bax and Bcl-2after traumatic brain injury. d The bar chart showed the ratio of Bax andBcl-2 to GAPDH at each time point. The data are mean ± SEM (n=3,*P<0.01, significantly different from the normal group)
J Mol Neurosci

Discussion
In the present study, we found p53-response protein PIDDwas elevated in experimental traumatic brain injury. Immuno-fluorescence analysis indicated that PIDD was predominantlydistributed in neurons and colocalized with the apoptoticmarker, active caspase-3, after TBI. Finally, we showed thatthe expression of PIDD was related with p53, Bax. Takentogether, our results indicated that PIDD might be an impor-tant regulator and promising therapeutic target of TBI.
Traumatic brain injury is caused by a mechanical forcewhich traumatically injures the brain. The injury then induceda variety of biological events, such as oxidative stress,microglial activation, blood–brain barrier breakdown, andinflammation response, which might resulted in neuronalapoptosis and neurological dysfunction after traumatic braininjury (Readnower et al. 2010; Ghirnikar et al. 1998). Thus, abetter knowledge with the molecular mechanism of neuronapoptosis would benefit the development of neuroprotectivetherapies for the treatment of TBI.
Serving as a key regulator of cellular stress response, p53pathway was supposed to play a crucial role in regulatingneuronal apoptosis and viability during TBI (Liou et al.2003). Earlier studies indicated that p53 was rapidly inducedfollowing experimental traumatic brain injury, implicating aninvolvement of p53 in both injury-induced neuronal repair andapoptosis procedures (Napieralski et al. 1999). However, themechanism of how p53 determines the fate of neurons duringTBI remains elusive. Our studies identified PIDD to be a genecorrelatively expressed with p53 and Bax. Besides, both of p53and PIDDwasmainly found in neurons and coincident with theactivation of caspase-3, suggesting that PIDDmight be activat-ed directly by p53 and participate in p53-regulated neuronalstress response. Thus, our findings might help clarify the mech-anism of p53 in modulating the pathological process of TBI.
The role of upregulated PIDD in neurons of injured braincortex remains largely elusive. It has been documented thatPIDD was involved in the formation of PIDDosome to triggerp53-induced apoptosis. The PIDDosome complex then activatescaspase-2, resulting in subsequent Bid cleavage, cytochrome crelease, and cell apoptosis (Tinel and Tschopp 2004). Thus,PIDD might exert a key function in the regulation of neuronalapoptosis after brain trauma. Indeed, our data showed that PIDDwas mainly localized in active caspase-3-positive cells, provid-ing a possible link between PIDD upregulation and neuronalapoptosis during TBI. In keeping with the findings, graduallyenhanced interaction between RAIDD and caspase-2 has beenreported following brain trauma, and the elevated expression ofPIDDmight be involved in the process (Qiu et al. 2002). Furtherinvestigation on the association between PIDD expression andcaspase-2 activation might shed new light on the mechanism ofneuronal apoptosis in brain injury. Moreover, PIDD-mediatedactivation of NF-κb might be crucial for cell repair (Janssens
et al. 2005). In this regard, PIDD upregulation might be impor-tant not only for neuronal apoptosis but also for neuron repairand survival. In line with this hypothesis, we found that PIDDwas expressed not only in apoptotic neurons but also in neuronsthat did not undergo apoptosis. In fact, prolonged activation ofNF-κb signalingwas observed in cortex neurons following braintrauma, and PIDDmight play a vital role in this process (Nonakaet al. 1999). These data suggested that PIDDmight be importantfor the regulation of both neuronal apoptosis and survival afterTBI. Of note, Kiyohiro Ando reported that ataxia–telangiecta-sia-mutated protein (ATM)-mediated PIDD phosphorylationcould control the balance between pro-death and pro-survivalsignaling (Ando et al. 2012). In unphosphorylated state, PIDDonly interacts with RIP1 to promote NF-κb activation and cellsurvival. However, when phosphorylated by ATM, PIDD failsto bind RIP1 but engages RAIDD and caspase-2 to facilitate celldeath instead. Thus, we proposed that PIDD could influence thechoice between neuron survival and apoptosis and might be avital determinant for neuron fate decision following TBI.
In this study, we for the first time identified PIDD to be aprotein involved in the pathology of TBI. Based on the factthat PIDD was a p53-inducible protein involved in both cellrepair and apoptosis, we infer that PIDD could be an impor-tant regulator of neuron fate determination in TBI. However,the detailed mechanism of PIDD upregulation and functionin neurons remains largely unclear. Further investigations areneeded to elucidate the molecular mechanism of neuronapoptosis in TBI and how PIDD plays a role in this regard.
Acknowledgments This work was supported by a grant of theAdministration of Science and Technology of Nantong (BK2012090).
References
Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT,Wang J, Bouchier-Hayes L, Sidi S (2012) PIDD death-domainphosphorylation by ATM controls prodeath versus prosurvivalPIDDosome signaling. Mol Cell 47(5):681–693
Baptiste-Okoh N, Barsotti AM, Prives C (2008) A role for caspase 2and PIDD in the process of p53-mediated apoptosis. Proc NatlAcad Sci U S A 105(6):1937–1942
Berube C, Boucher LM, Ma W, Wakeham A, Salmena L, Hakem R, YehWC,Mak TW, Benchimol S (2005) Apoptosis caused by p53-inducedprotein with death domain (PIDD) depends on the death adapterprotein RAIDD. Proc Natl Acad Sci U S A 102(40):14314–14320
Chen J, Wu X, Shao B, Zhao W, Shi W, Zhang S, Ni L, Shen A (2011)Increased expression of TNF receptor-associated factor 6 after rattraumatic brain injury. Cell Mol Neurobiol 31(2):269–275
Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK (1998) Ex-perimental brain injury induces regionally distinct apoptosis dur-ing the acute and delayed post-traumatic period. J Neurosci18(15):5663–5672
Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW,Park DS, Slack RS (1999) Bax-dependent caspase-3 activation is akey determinant in p53-induced apoptosis in neurons. J Neurosci19(18):7860–7869
J Mol Neurosci

Culmsee C, Mattson MP (2005) p53 in neuronal apoptosis. BiochemBiophys Res Commun 331(3):761–777
Ghirnikar RS, Lee YL, Eng LF (1998) Inflammation in traumatic braininjury: role of cytokines and chemokines. NeurochemRes 23(3):329–340
Janssens S, Tinel A, Lippens S, Tschopp J (2005) PIDD mediates NF-kappaB activation in response to DNA damage. Cell 123(6):1079–1092
Kiryu-Seo S, Hirayama T, Kato R, Kiyama H (2005) Noxa is a criticalmediator of p53-dependent motor neuron death after nerve injuryin adult mouse. J Neurosci 25(6):1442–1447
Langlois JA, Rutland-Brown W, Wald MM (2006) The epidemiologyand impact of traumatic brain injury: a brief overview. J HeadTrauma Rehabil 21(5):375–378
Lin Y, Ma W, Benchimol S (2000) Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis.Nat Genet 26(1):122–127
Liou AK, Clark RS, Henshall DC, Yin XM, Chen J (2003) To die or notto die for neurons in ischemia, traumatic brain injury and epilepsy:a review on the stress-activated signaling pathways and apoptoticpathways. Prog Neurobiol 69(2):103–142
Liu Y, Wang Y, Cheng C, Chen Y, Shi S, Qin J, Xiao F, Zhou D, Lu M,Lu Q, Shen A (2010) A relationship between p27(kip1) and Skp2after adult brain injury: implications for glial proliferation. JNeurotrauma 27(2):361–371
Logan A, Frautschy SA, Gonzalez AM, Baird A (1992) A time coursefor the focal elevation of synthesis of basic fibroblast growth factorand one of its high-affinity receptors (flg) following a localizedcortical brain injury. J Neurosci 12(10):3828–3837
Martin LJ (2001) Neuronal cell death in nervous system development,disease, and injury (Review). Int J Mol Med 7(5):455–478
Martin LJ, Kaiser A, Yu JW, Natale JE, Al-Abdulla NA (2001) Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. JComp Neurol 433(3):299–311
Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T (2002)Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care 8(2):101–105
Napieralski JA, Raghupathi R, McIntosh TK (1999) The tumor-suppressor gene, p53, is induced in injured brain regions followingexperimental traumatic brain injury. Brain Res Mol Brain Res71(1):78–86
Nonaka M, Chen XH, Pierce JE, Leoni MJ, McIntosh TK, Wolf JA,Smith DH (1999) Prolonged activation of NF-kappaB followingtraumatic brain injury in rats. J Neurotrauma 16(11):1023–1034
Qiu J, Whalen MJ, Lowenstein P, Fiskum G, Fahy B, Darwish R,Aarabi B, Yuan J, Moskowitz MA (2002) Upregulation of theFas receptor death-inducing signaling complex after traumaticbrain injury in mice and humans. J Neurosci 22(9):3504–3511
Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after trau-matic brain injury. J Neurotrauma 17(10):927–938
Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR,McCarron RM, Sullivan PG (2010) Increase in blood–brain barrierpermeability, oxidative stress, and activated microglia in a ratmodel of blast-induced traumatic brain injury. J Neurosci Res88(16):3530–3539
Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B,Quadroni M, Tschopp J (2007) Autoproteolysis of PIDD marksthe bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J 26(1):197–208
Tinel A, Tschopp J (2004) The PIDDosome, a protein complex impli-cated in activation of caspase-2 in response to genotoxic stress.Science 304(5672):843–846
Tomasevic G, Raghupathi R, Scherbel U, Wieloch T, McIntosh TK(2010) Deletion of the p53 tumor suppressor gene improvesneuromotor function but does not attenuate regional neuronal cellloss following experimental brain trauma in mice. J Neurosci Res88(15):3414–3423
Werner C, Engelhard K (2007) Pathophysiology of traumatic braininjury. Br J Anaesth 99(1):4–9
J Mol Neurosci








![Original Article EMMPRIN, SP1 and microRNA-27a mediate ... · pathways and mechanisms, including loss of function of the tumor suppressor p53 [10], upregulated vascular endothelial](https://img.pdfslide.net/doc/110x75/5e22380d3a89c23c53196456/original-article-emmprin-sp1-and-microrna-27a-mediate-pathways-and-mechanisms.jpg)




















