Embed Size (px)
Citation preview

Page1

Page2
Table-1 Raw Herbs
Table-1 Raw Herbs
Table-2 Herbal Extracts
Table-3 Processed
Herbs/Excipeints/ Pharma Aids
1. Acacia 2. Ajwain 3. Amalaki 4. Amra 5. Anantmula 6. Arjuna 7. Artemisia 8. Ashwagandha 9. Belladonna Leaf 10. Bhibhitaki 11. Bhringraj 12. Bhuiamla 13. Brahmi 14. Coleus 15. Daruharidra
Roots 16. Daruharidra
Stems 17. Garcinia 18. Gokhru 19. Gudmar 20. Guduchi 21. Guggul 22. Haridra 23. Haritaki 24. Ispaghula Husk*
25. Kalmegh 26. Kunduru 27. Kutki 28. Lasuna 29. Lavang 30. Mandukaparni 31. Manjistha 32. Maricha 33. Methi* 34. Neem* 35. Pippali Large 36. PippaliSmall 37. Punarnava 38. Sarpagandha 39. Saunf 40. Senna Leaf 41. Senna Pods 42. Shatavari 43. Shati 44. Sunthi 45. Tulasi 46. Vasaka 47. Yasti
48. Amla Juice Powder* 49. Arjuna Dry Extract* 50. Ashwagandha Dry Extract*
51. Belladonna Dry Extract 52. Bhibhitaki_ Aqueous_Extract* 53. Brahmi Extract* 54. Coleus Dry Extract* 55. Garcinia Aqueous Extract*
56. Gugulipid 57. Haritaki Aqueous Extract* 58. Haridra Dry Extract* 59. Haritaki Extract* 60. Kalmegh Dry Extract* 61. Sarpagandha Powder* 62. Senna Dry Extract 63. Sunthi Extract* 64. Tulasi Dry Extract* 65. Vasaka Extract* 66. Yasti Dry Extract 67. Opium
68. Arachis Oil 69. Castor Oil* 70. Clove Oil* 71. Coconut Oil* 72. Coriander Oil* 73. Eucalyptus Oil* 74. Guar Gum* 75. Hydrogenated_
Castor Oil* 76. Malt Extract* 77. Mentha Oil* 78. Papain* 79. Peppermint Oil* 80. Shellac* 81. Starch* 82. Tolu Balsam* 83. Tragacanth

Challenges: New Science and Tools under Evaluation
DNA Based Identification/Evaluation. Enhanced activity in this area. Macroscopy/Microscopy usage is a question. Bar Code possibility Under Evaluation by IP along with Manipal’s School of Life Sciences. DNA bar coding using ITS2 region as barcode candidate Samples of Shatavari [ Asparagus racemosus (AGR/30),
Asparagus (AGR/31), Saariva (Hemidesmus indicus), Saariva-Substitute analyzed
Page3

Page4

Challenges: New Science and Image Analysis Tools under Evaluation
Number of parameters in Raw herbs/powders can be used in the identification process. Starch grains and their structures, lignified parenchyma, cork and their structures,
calcium oxalate crystals, stomata and differences and stomatal index, the most famous trichomes, vessels and tracheids, stone cells, type and varieties of fibers pollen grains, and others.
“Microcomputer as an aid in Drug Microscopy” for over 100 plants of western origin. (Trease and Evans, Pharmacognosy, 13th edition, ELBS, Chapter 43, p 784-797)
Today’s PC assisted techniques are available based on Image analysis- Viz. For Chromosomal aberration testing, for Mineralogical & Metallurgical studies in alloys, etc
Can they not be adopted to Plants for identification?
Page5

Page6

Page7

Page8

Page9

11
Page11
19. IP GUIDELINES FOR THE QUALITY OF HERBAL DRUGS AND ITS PRODUCTS
Introduction Around 80% of the population of developing countries uses herbal medicine for the treatment of various diseases. India is the hub for herbal drugs and also leads in the production of phytopharmaceuticals. Therefore Indian Pharmacopoeia (IP) lay down monograph for herbs and herbal products to maintain to maintain their quality. Appearance of herbal drug monograph does not mean its approval as a drug under the law; it is to provide qualitative and quantitative standards of quality for the herb its use either as a food item or food ingredient or food supplement/nutraceuticals, as a drug, and/or as an ingredient in cosmetics. Each such use would need to comply with applicable regulations. This document covers criteria for inclusion of herbal drug monograph in IP and various tests and assay methods to be applied to maintain the quality of herbal drugs and its products. Inclusion Criteria To introduce a monograph for an herb in the Indian Pharmacopoeia, the criteria that would be kept in mind, but not limited to are:
a. Herbs with specific name and a definitive botanical identity up to species b. Availability and usage in trade and commerce c. Regulatory interest d. Knowledge of and availability of specific chemical compound of well characterised
structure [either responsible for the biological activity of the herb (bio-marker) or a chemical compound known to be present in the herb even if not responsible for biological activity (chemical/analytical marker)
e. Availability of a quantitative method for estimation of such a compound f. Knowledge of safety of the herb and its sustainability.
Types of herbal Drug Monograph a. Crude herbs b. Processed herbs: semisolid & solid extracts, Tinctures and Juices c. Formulations
Crude Herbs This term means, unless specified otherwise, mainly whole, fragmented or cut, plants, parts of plants, algae, fungi, and lichen in a form which is not processed. Herbs are usually in dried form, but sometimes, when specified, may also be in a fresh form. In specific cases exudates which have not been processed further also are covered under the term herbs. Processed Herbs
Obtained by subjecting herb to treatment such as extraction, distillation, expression, fractionation, purification, concentration and partial or full fermentation
Standardized extract, a term commonly employed, would for Pharmacopoeial purposes, mean an extract adjusted with an acceptable tolerance to a given content of biomarker or chemical/analytical marker.
Standardization may be achieved by adjusting the extracts with approved inert mateiral or by blending one or more batches of extracts

12
Page12
The difference between extracts and tincture would be, in the type of solvents used for extracting a herb and tincture would normally mean an extract where aqueous ethanol is used as a solvent for extraction
Dry extracts usually have a loss on drying or water content not greater than 5 per cent w/w unless specified otherwise in any monograph.
Extracts shall be free from solvents used for extraction and shall comply with the respective limits as given in Appendix 5.4 (IP 2010, vol 2 ; p 650-651
Harmful and carcinogenic solvents shall not be used for extraction process. Solvents and solvent systems may include use of propylene glycol, glycerin, sorbitol and such other polyhydroxy alcohols, as long as the content of such polyhydroxy alcohol are within safe limit in the final product.
The extract which consists of a single chemical compound of more than at least 70 per cent purity, such extracts shall be treated as an active pharmaceutical ingredient of a food additive or a cosmetic ingredient and would be required to meet relevant laws.
Herbs may also be extracted using vegetable oils (approved by Food Law) for extraction purposes and such extracts shall specify the oil used for processing.
Approved preservatives or preservatives system may be used during preparation of extracts.
Herbal Formulations
A dosage form consisting of one or more herbs or processed herb(s) specified quantities to provide specific nutritional, cosmetic, and/or other benefits meant for use to diagnose, treat, mitigate diseases of human beings or animals and/or to alter the structure or physiology of human beings or animals.
Herbal formulation shall be labelled to comply with relevant labelling requirements under food or drug or cosmetics laws as applicable. Additionaly, adequate information shall be provided on label os such formulations to include the name of the herb, parts used, nature and type of extract or processed herb used, nature and type of extract or processed herb used, extraction ratios, quantity per unit dose or per serving, name(s) of inert excipients used and any preservatives added shall be provided on the label.
Definition In monograph of the vegetable drugs the definition indicates whether the subject of the monograph is for example, the whole drug or the drug in powdered form. Identification This section includes tests performed to identify the drug
a. Macroscopic : The important macroscopic botanical character of the drug are specified to permit a clear identification. When two species/subspecies of the same plant are included in the definition, the individual difference between them are indicated.
b. Microscopic : The microscopic examination of the drug reduced to a powder describes the dominant or the most specific characters including stomata and stomatal index.
c. TLC : It is prescribed for the identification of the herbal drugs (2.4.17).
Tests a. Foreign Organic Matter : It is the material consisting of any or all of the following :
Parts of the organ or organs from which the drug is derived other than the parts named in the definition and description or for the limit is prescribed in the individual monograph
Any organs other than those named in the definition and description

13
Page13
Matter not coming from the source plant and Moulds, insects or other animal contamination
b. Physio-Chemical Evaluation: It is an important parameter in detecting adulteration or
improper handling of drugs. It can serve as a valuable source of information and provide appropriate standard to establish the quality of herbs. Those are :
Ethanol-soluble extractive Water-soluble extractive Total ash Acid-insoluble ash
c. Heavy metals : The test is prescribed where there is the potential for contamination of heavy metals. The limit for heavy metals is indicated in the individual monograph in terms of ppm, i.e., the parts if lead, Pb, per million parts (by weight) of the substance under examination
d. Loss on drying : It is the loss of weight expressed as per centage w/w resulting form water and volatile matter of any kind that can be driven off under specified conditions. Determined on 5.0 g by drying in an oven at 105°.
e. Microbial contamination : The sample should complies with the microbial contamination tests as specified in IP 2.2.9.
Assay Carried out by using UV spectrophotometry, LC, GC, HPTLC Indian Pharmacopoeia Botanical Reference Substance These are the authentic specimens chosen and verified on the basic of their suitability for intended use as prescribed in the Pharmacopoeia and are not necessarily suitable in other circumstans. Author: Dr. V. Kalaiselvan,
Senior Scientific Officer, Indian Pharmacopeia Commission, Ghaziabad Email: [email protected]
----------------------

14
Page14
20. GUIDELINES FOR INDIAN PHARMACOPOEIA REFERENCE SUBSTANCES (IPRS)
1. INTRODUCTION
These guidelines will provide assistance and information to industry as well as all testing
laboratories on how to comply with respect to using Indian Pharmacopoeia Reference Substances
during testing. Alternative approaches may be acceptable provided they are supported by scientific
justification.
Standards and reference substances play a vital role in evaluating the quality of active
pharmaceutical ingredients and finished products. They are highly characterized specimens of drug
substance, excipients, impurities and degradation products that are used to conduct tests such as:
Identity, Purity and Potency.
Reference standards are frequently necessary to achieve adequate quality control of substances for
pharmaceutical use and pharmaceutical preparations. Indian Pharmacopoeia Reference Substances
are established using suitable procedures and protocols and their continued suitability for use is
monitored according to a predefined programme. Where an Indian Pharmacopoeia Reference
Substances is referred in a monograph or general chapter, it refers to the official substance which is
alone authentic.
2. TERMINOLOGY
2.1 Primary Reference Standard. A standard shown to have suitable properties for the intended
use, the demonstration of suitability being made without comparison to an existing standard. This
reference standard is supplied by the I.P. Commission and is referred to as Indian Pharmacopoeia
Reference Substance (IPRS). IPRSs are Primary Reference Standards.
2. Secondary Reference Standard. A standard established by comparison with a primary
Reference Standard. Stake holders may use Secondary Reference Standard established by
comparison with Indian Pharmacopoeia Reference Substances for the day to day routine
uses.
3. Working Reference Standard. For preparing adequate number of working Reference
Standards for day to day analytical uses/comparison to adherence of quality requirements of
an specified monograph. The user may make their own working Reference Standards with
their quality parameters calibrated with reference to the IPRS of the same material.
4. SOURCE MATERIAL

15
Page15
Source material of satisfactory quality is to be selected from a batch (lot) of the substance
originating from the normal production process, if the purity is acceptable. Further purification
techniques may be needed to render the material acceptable for use as a chemical reference
substance.
The purity requirements for an IP reference substance depend upon its intended use. An IP
reference substance proposed for an identification test does not require meticulous purification,
since the presence of a small percentage of impurities in the substance often has no noticeable effect
on the test
On the other hand, IP reference substances that are to be used in assays should a high degree of
purity. As a guiding principle, a purity of 99.5% or higher is desirable, calculated on the basis of the
material in its anhydrous form or free of volatile substance. In making a decision about the
suitability of an IP reference substance, the most important consideration is the influence of the
impurity on the attribute measured in the assay when used in a non-specific assay procedure.
Impurities with physicochemical characteristics similar to those of the main component will not
impair the usefulness of a IP reference substance, whereas even traces of impurities with
significantly different properties may render a substance unsuitable as IP reference substance.
When source material to be used as a IP reference substance is obtained from a supplier, the
following should be supplied with the material:
− Certificate of analysis
− Information on optimal storage conditions
− Results of any hygroscopicity study and/or statement of the hygroscopicity of the source
material.
− Results of any accelerated stability studies.
− Identification of detected impurities
− Updated Material Safety Data Sheet outlining any health hazards.
4. ESTABLISHMENT OF IP REFERENCE SUBSTANCES
4.1 Primary Standard. A substance or preparation to be established as a primary standard is
characterised by a variety of analytical techniques chosen to demonstrate its suitability for use.
Following test programmes are usually applied.
→ Characterisation of the substance (structural elucidation) by appropriate chemical attributes such
as structural formula, empirical formula and molecular weight. A number of techniques may be
used including:
− Nuclear magnetic resonance spectrometry;
− Mass spectrometry;
− Infrared spectrophotometry;

16
Page16
− Elemental analysis.
→ Determination of the purity:
− Determination of the content of organic impurities by an appropriate separation technique or
spectrometric method, where applicable;
− Quantitative determination of water;
− Determination of the content of residual solvents;
− Determination of loss on drying, which may in certain circumstances replace the
determinations of water and residual solvents;
− Determination of inorganic impurities (test for heavy metals, sulphated ash, atomic
spectrometry, inductively coupled plasma spectrometry, X-ray fluorescence); the results are
not used in determining an assigned content, except where they would have an appreciable
impact upon it;
− Determination of the purity by an absolute method.
The candidate materials are tested against a wide variety of analytical methods. The extent of
testing and the number laboratories involved depends on the use of the reference standard.
Compliance with the relevant monograph is usually required, unless otherwise justified.
Where a collaborative trial is carried out during establishment, a protocol is provided for each
participant and only valid results derived according to the protocol are used for establishing an
assigned value or otherwise confirming suitability.
For IP reference substance, relevant parts of the following programme are typically applied.
4.1.1 Identification Test. In general, a batch selected from the normal production of the substance
is satisfactory. It is shown to comply with the requirements of the monograph full structural
elucidation is carried out for the first batch.
4.1.2 Related Substance Test. A reference substance corresponding to an impurity is characterised
for identity and purity. Where a reference standard is used to determine the content of a given
impurity, the preferred minimum content is 95.0 per cent; where this is achieved no assign value is
given, the content being considered as 100.0 per cent.
If an impurity is not available in a sufficient quantity to establish a reference standard, a number of
other options exist:
− Preparation of a reference standard that contains a mixture of the compound(s) and the impurity
or impurities;
− Preparation of a reference substance containing a mixture of specified impurities.
Where such a mixture is also used to determine the content of a given impurity, the content of the
impurity in the reference substance is determined by appropriate separation methods and a value
assigned to the reference substance.
5. Assay.

17
Page17
Chemical assay. When a reference substance is used for quantitative determination of an active
substance an �hysic�tre (assay standard), the extent of testing is great. In general, several
collaborating laboratories examine the proposed substance, following a detailed protocol that
describes the procedures to be followed. The results obtained are used to assign a content. It is
particularly important to quantify the impurities if a selective assay is employed.
A protocol is prepared and must be strictly followed by the participants of the collaborative trial to
assign the content. The protocol usually requires:
− Determination of water (or loss on drying);
− Estimation of the organic impurities (including residual solvents when appropriate) using the
prescribed separation techniques;
− And possibly, determination of the content of the substance by an absolute method.
The protocol also indicates the system suitability test and acceptance criteria for each of the tests
performed.
Unless otherwise stated, an assigned value is given for the substance or preparation as presented in
the container (‘as such’), and the contents are not to be dried before use. For assay standards
prepared by lyophilisation the content of the pure substance is indicated in milligrams or
International Units per vial.
4.1.4 Establishment report. A report containing the results of the establishment study as well as
information concerning the use of the reference standard is prepared. The report for a chemical
assay standard has a value assigned to the substance with the rationale for attributing that value.
The estimated uncertainty of the assigned value is calculated, and where it is less than a predefined
value, which is considered to be negligible in relation to the acceptance criteria for the assay, then
the study is accepted. Otherwise, the trial may be repeated, in whole or in part, or the limits define
for the pharmaceutical substance may be widened. The uncertainty of the assigned value is not
given as part of the information provided with the reference substance, since the precision of the
method and the uncertainty of the value attributed to reference substance are taken into account
when setting the limit(s) in a monograph.
4.2 Secondary Standard. A secondary standard should exhibit the same property or properties as
the primary standard, relevant for the test(s) for which it is established. The extent of testing is not
so great as is required for the establishment of a primary standard. The secondary standard is
established by comparison with the primary standard to which it is traceable. An official primary
standard is used wherever possible for establishment of secondary standards.
4.2.1 Identification.
− For use in infrared spectrophotometry: the absorbance bands correspond in position and relative
size to the absorbance bands of the primary standard.

18
Page18
− For use in separation techniques: the migration distance, migration time and retention time of
the secondary standard are the same as those of the primary standard for thin-layer
chromatography or electrophoresis, capillary electrophoresis and gas or liquid chromatography
respectively.
4.2.2 Purity Test. For use in separation techniques: as for identification but when used for
quantification, a content relative to the signal from the primary standard is to be established.
4.2.3 Assay. Secondary standards are assayed against a primary standard with an assigned content
or potency. The property for which a value is to be assigned for the secondary standard is similar in
magnitude to that of the primary reference standard with which it is compared. Both the number of
independent replicate determinations to be performed and the acceptance criteria to be applied are
predefined.
6. LABELLING, STORAGE AND DISTRIBUTION
All operations are carried out according to the relevant norms of best practices to ensure the
traceability and integrity of the reference substance. Reference substances are filled and sealed
under appropriate conditions, to ensure the integrity of the reference standard. The containers
employed may be multi-use or single use, but the latter is preferred to minimize the risk of
decomposition, contamination, or water uptake.
7. LABELLING.
The label bears the name of the reference substance, the name of the supplier, the Lot number,
Catalogue No., direction for its use and any other information necessary to the proper use of the
reference standard. If used as an assay standard the following information is also given:
− The assigned percentage content;
− The content in milligrams or �hysic�tres of the chemical entity in the container;
− The assigned potency (for biological assays or microbiological assays) in units either per
milligram or per vial.
For Indian Pharmacopoeia Reference Substances, no re-test or expiry date is given since the re-test
programme monitors continued fitness for use.
8. STORAGE AND DISTRIBUTION.
Reference Substances are to be stored and distributed in conditions suitable to preserve their
properties and to ensure optimal stability. The reference materials shall be identified preserved and
segregated from all other reference materials from the time of preparation to their distribution to
users. I.P. Commission ensures proper primary packaging of all reference materials (air free,
moisture free or inert gas filled) and provides proper secondary for protection from heat, light,

19
Page19
moisture etc and stores in secure storage areas designed and equipped to prevent damages or
deteriorate and preserve the properties of the materials. The qualities of all stored reference
materials are assessed at appropriate intervals throughout their storage life for detection of any
possible deterioration/ change of properties etc.
Indian Pharmacopoeia reference substances are generally stored at temperature between 2 – 8° C.
For materials needing other storage conditions, appropriate storage conditions are provided.
9. USE OF IP REFERENCE SUBSTANCES
IP Reference Substances are employed in the identification, purity testing and assay of substances
for pharmaceutical use and pharmaceutical preparations as per Indian Pharmacopoeia. Reference
standards certified for their intended purposes may not be suitable for other purposes. Any value
assigned to a reference substance is valid for the intended use only and not for other uses.
An Indian Pharmacopoeia Reference Substance with an assigned content/potency for use in the
assay of a substance for pharmaceutical use may be suitable to determine the content of that
substance in a pharmaceutical preparation where all the following conditions are fulfilled:
− The chromatographic assay method described in the active substance monograph is
employed;
− The user verifies the application of the method to the particular pharmaceutical preparation.
− Any pre-treatment of the sample is validated for the particular pharmaceutical preparation;
− The use is approved by the competent authority.
IP Reference Substances are also established for the determination of the content of components of
herbal drug and herbal drugs preparations. These may be the active principles themselves; marker
constituents used for quantification; or extracts. Reference standards consisting of extracts are
established using well-characterised samples of active principles or marker constituents.
It is the policy of the Indian Pharmacopoeia Commission to supply IP reference substances in
adequate quantities for immediate use after opening of the container. Use in other conditions is the
responsibility of the user. If an unopened container is stored in the recommended conditions, it
remains suitable for use till the end of its shelf life. Storage of solutions of reference standards is
not recommended unless the suitability has been demonstrated by the user.
Secondary standards. A secondary standard may be used for routine quality control purposes for
any of the uses described for its primary standards, provided that it is established with reference to
the I.P. Reference Substance. A secondary standard is established and employed to reduce the use
of the primary standard, which requires more extensive characterisation and evaluation and may be
available only in a limited quantity. A secondary standard is used only for the same purpose as the
primary standard with reference to which it has been established.

20
Page20
10. REVALIDATION SCHEME
A system is established and implemented to ensure the continued fitness-for-use of the IP reference
substances. Normally, a re-test programme is applied, taking into account the known �hysic-
chemical properties and the stability data for the reference standard. Reference standards are
periodically tested for stability during storage. A monitoring programme is applied that is designed
to detect at an early stage any sign of decomposition using appropriate analytical techniques.
The periodicity and extent of re-testing of reference standards depends on a number of factors
including:
− Stability;
− Container and closure;
− Storage conditions;
− Hygroscopicity;
− Physical form;
− Intended use;
Most IP reference Substances are presented in powder form but some are prepared as solutions.
Preferably, reference standards are presented as single-use units. However, if the standard is
presented in multi-use containers then re-testing may be more frequent for hygroscopic or oxygen-
sensitive substances. The testing methods include the determination of water and decomposition
products. The re-test period may be lengthened with the support of sufficient data. The maximum
permitted variation from the assigned value should be pre-defined, and if exceeded, the lot should
be re-established or replaced.
The monitoring programme of I.P. Commission includes a selection of the following tests, chosen
for their rapidity, sensitivity and applicability to small quantities:
− Determination of water, loss on drying and/or thermogravimetric analysis;
− Estimation of impurities by stability-indicating separation techniques;
− Where appropriate, determination of the molar purity by differential scanning calorimetry;
− Application of other specific tests for detecting impurities.
Author: Dr. Anil Kumar Teotia,
Senior Scientific Officer, Indian Pharmacopeia Commission, Ghaziabad Email: [email protected]
------------------------------

21
Page21
21. GUIDANCE DOCUMENT FOR BIOTECHNOLOGY DERIVED THERAPEUTIC PRODUCTS
Biotechnology derived therapeutic products generally refer to protein products produced via rDNA
technology in microbial or mammalian cells. The major difference between a biotechnological
product and other pharmaceutical products is with respect to the complexity of the product itself
and the process utilized to manufacture it. Biotechnology therapeutic products are from genetically
modified living organisms, which therefore make it different from other protein products obtained
by direct isolation from natural sources such as plasma, serum, tissue, or chemical synthesis.
The basic requirements such as process validation, environmental control, aseptic manufacturing
and quality control/ quality assurance for biotechnology derived therapeutic products are same as
that for protein pharmaceuticals and classically isolated/ synthesized protein products. However
complexity of these biotechnology derived therapeutic products are greater due to its cell
propagation process, complicated purification methods and analytical controls that are required to
ensure homogeneity, lot-to-lot consistency, safety and efficacy.
Quality Control
Quality control systems for biotechnological products are very similar to those of other traditional
pharmaceutical products but the method used to determine identity, consistency, purity and perform
impurity profiling is complex when compared to traditional pharmaceutical products. Moreover
quality control of biotechnological products depends on both final product and validated in-process
testing and impurity removal. These products generally require a detailed characterization of the
producing organism, a complete analysis of cell growth/ propagation and final product recovery
process.
The complexity of the product depends on its size and structure and the manufacturing process i.e.
products produced from prokaryotic system require less complex analytical methods when
compared to eukaryotic system. A proper combination of methods to determine identity, purity and
potency is required for analytical characterization of rDNA products.
Analytical methods play a vital role in quality control of biotechnological products in determination
of their identity, purity and potency. Aminoacid analysis, peptide mapping, protein sequencing,
glycoprotein analysis, electrophoresis (Sodium dodecyl sulphate- polyacrylamide gel
electrophoresis, Western blot analysis and isoelectric focusing) are some of the important analytical
methods that are used to evaluate purity, identity, homogeneity and stability of biotech products.

22
Page22
Chromatographic methods like reverse phase– high performance liquid chromatography (RP-
HPLC), size exclusion chromatography are important in determination of purity and impurity
profile of the product. Other biological techniques like immunoassay, DNA hybridization and
polymerase chain reactor (PCR) are proved to be useful tools for detection and identification of host
cell impurities. BET (Bacterial Endotoxin Test) is the most sensitive, specific and better test to
detect pyrogen in a product. Capillary electrophoresis is utilized for measurement of impurities
created due to denaturation, aggregation, oxidation, deamidation etc. The potency of
biotechnological product is generally assessed by specific assay methods based on its nature and its
therapeutic application. Assay method includes animal based biological assay, cell culture based
biological assay, biochemical assay and ligand binding assay. Determination of potency strictly
requires traceable reference substances calibrated in international units.
Indian Pharmacopoeia Reference Substances (Biotechnological)
The use of suitable reference substances is extremely important for the analysis of biotechnological
products. Indian Pharmacopoeia Reference Substances (IPRS) are reference substances certified by
Indian Pharmacopoeia Commission (IPC) and distributed by the Commission or by the laboratories
authorized by the IPC. If letter RS appear after italicized name of substance (Eg. Interferon alpha -2
RS) in a test or assay in the IP monograph, the relevant IPRS should be used. IPRS is reference
substance that has appropriate quality within specified content and is accepted without any
comparison. In case of non-availability of IPRS relevant reference material from European
Pharmacopoeia, United States Pharmacopoeia or WHO can be used.
This presentation is for the guidance of users in the field of biotechnologically derived therapeutic
products.

23
Page23
Table: Summary of analytical methods used for measurement of the quality of biotechnology
products
Quality parameters Analytical methods
Identity Protein sequencing, Aminoacid analysis, Peptide mapping, SDS-
PAGE, Western blot analysis, Isoelectric focusing, Capillary zone
electrophoresis
Purity SDS-PAGE, High Performance Liquid Chromatography (HPLC),
Peptide mapping
Impurity Immunoassay, DNA Hybridisation, PCR, LAL, RP-HPLC, SDS-
PAGE
Potency Animal model assay, Cell culture based bioassay, Physico-
chemical assay Author: Dr. Anurag Rathore, Associate Professor, Indian Institute of Technology, New Delhi. Email: [email protected]
Mrs. M.Kalaivani, Pharmacopoeial associate, Indian Pharmacopeia Commission, Ghaziabad
---------------------------------------

24
Page24
22. QUALITY OF WATER SYSTEM FOR PHARMACEUTICAL PURPOSES
1. INTRODUCTION
Water is the most used and least understood raw material ingredient in the pharmaceutical industries. It may be used as excipient, used for reconstitution of products, during synthesis, during production of finished products and as a cleaning agent for rinsing vessels, equipments, primary packing materials etc. Continuous environmental monitoring of water systems is an unequivocal regulatory requirement and a major cost strain on company personnel and resources. Proper water system planning with personnel knowledge in all the physical, chemical, microbiology and engineering issues associated with water is essential. It is the user responsibility to assure that pharmaceutical water and its production meet applicable regulations and compendia specifications for the type of water used in pharmaceutical preparations. Control of the chemical and microbiological purity of water is important for safety and efficacy of the pharmaceutical products.
2. SCOPE
This document is intended to provide guidance to the pharmaceutical industries on the pharmaceutical use of different grade of water in the manufacturing of active pharmaceutical ingredients and pharmaceutical preparations for human and veterinary use.
3. TYPES OF PHARMACEUTICAL GRADE WATER
There are many different grades of water used for pharmaceutical purposes. Mainly waters can be divided into two general types: Bulk Waters, which are typically produced on site where they are used; and Packaged Waters, which are produced, packaged, and sterilized to preserve microbial quality throughout their packaged shelf life. Bulk water includes Drinking Water, Purified Water, Hot Purified Water, Water for Injection, Water for Hemodialysis, whereas Packed Water includes Sterile Purified Water, Sterile Water for Injection, Bacteriostatic Water for Injection, Sterile Water for Irrigation, Sterile Water for Inhalation. Beside these types another category of water i.e. Analytical Waters is used in specific analytical methods. Analytical grade waters includes Distilled Water, Deionized Water, Deionized Distilled Water, Filtered Distilled or Deionized Water, Filtered Water, High Purity Water, Ammonia-Free Water, Carbon Dioxide-Free Water, Ammonia- and Carbon Dioxide-Free Water, Deaerated Water, Recently Boiled Water, Oxygen-Free Water, LAL Reagent Water, Organic-Free Water, Lead-Free Water, Chloride-Free Water, Hot Water.
In general, water for pharmaceutical use is divided into following four main categories:
i. Potable Water- It is not covered by pharmacopoeia but must comply with the regulations on water laid down by the competent authority. Testing should be carry out at the manufacturing site to conform the quality of water. Potable water may be used in chemical synthesis and in the early stages of cleaning pharmaceutical manufacturing equipments unless there are special technical or quality requirements for higher grades of water. It is the prescribed source feed water for the production of pharmacopoeial grade waters.
ii. Purified Water- Purified water is used for the preparation of medicinal products other than those that require the use of water which is sterile and/or apyrogenic. It is used as an excipient in the production of nonparenteral preparations and in other pharmaceutical applications, such as cleaning of certain equipment and nonparenteral product contact components. Purified Water

25
Page25
must meet the requirements for ionic and organic chemical purity and must be protected from microbial contamination. The minimal quality of source or feed water for the production of Purified Water is Drinking Water. This source water may be purified using unit operations that include deionization, distillation, ion exchange, reverse osmosis, filtration, or other suitable purification procedures. Purified water systems must be validated to reliably and consistently produce and distribute water of acceptable chemical and microbiological quality. These systems require frequent sanitization and microbiological monitoring to ensure water of appropriate microbiological quality at the points of use.
iii. Highly Purified Water- It is intended for use in preparation of products where water of high biological quality is needed, except where water for injection is required. Highly purified water is obtained from the water that complies with the regulations on water intended for human consumption laid down by the competent authority. Current production methods include, for example, double pass reverse osmosis, coupled with other suitable techniques such as ultra filtration and deionization. Highly purified water meets the same as WFI but the production methods are considered less reliable than distillation and thus it is considered unacceptable for use as WFI.
iv. Water for Injection- It is used as an excipient in the production of parenteral and other preparations where product endotoxin content must be controlled, and in other pharmaceutical applications, such as cleaning of certain equipment and parenteral product-contact components. The minimum quality of source or feed water for the generation of Water for Injection is Drinking Water as defined by the U.S. EPA, EU, Japan, or the WHO. This source water may be pre-treated to render it suitable for subsequent distillation. The finished water must meet all of the chemical requirements for Purified Water as well as an additional bacterial endotoxin specification. Since endotoxins are produced by the kinds of Gram negative bacteria that are prone to inhabit water, the equipment and procedures used by the system to purify, store, and distribute Water for Injection must be designed to minimize or prevent microbial contamination as well as remove incoming endotoxin from the starting water.
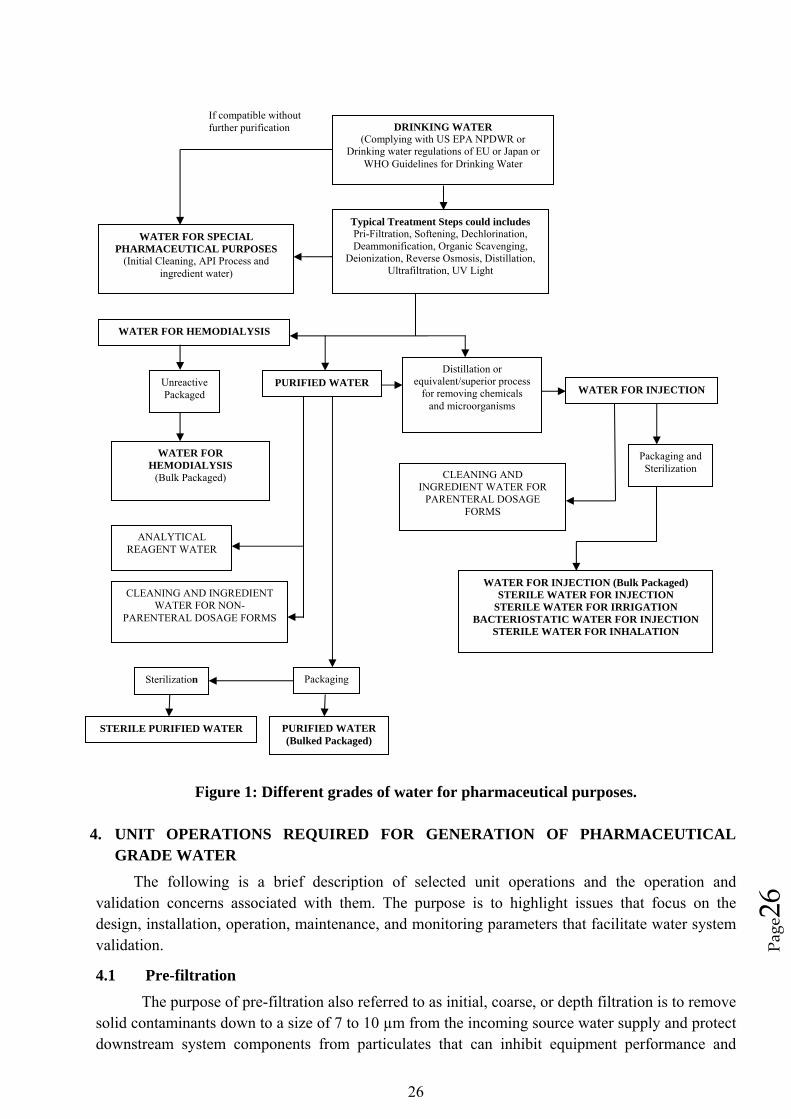
26
Page26
If compatible without further purification DRINKING WATER
(Complying with US EPA NPDWR or Drinking water regulations of EU or Japan or
WHO Guidelines for Drinking Water
Typical Treatment Steps could includes Pri-Filtration, Softening, Dechlorination, Deammonification, Organic Scavenging,
Deionization, Reverse Osmosis, Distillation, Ultrafiltration, UV Light
WATER FOR SPECIAL PHARMACEUTICAL PURPOSES
(Initial Cleaning, API Process and ingredient water)
WATER FOR HEMODIALYSIS
Distillation or equivalent/superior process
for removing chemicals and microorganisms
Unreactive Packaged
PURIFIED WATER WATER FOR INJECTION
WATER FOR HEMODIALYSIS
(Bulk Packaged)
Packaging and Sterilization
Figure 1: Different grades of water for pharmaceutical purposes.
CLEANING AND INGREDIENT WATER FOR
PARENTERAL DOSAGE FORMS
ANALYTICAL REAGENT WATER
WATER FOR INJECTION (Bulk Packaged) STERILE WATER FOR INJECTION
STERILE WATER FOR IRRIGATION BACTERIOSTATIC WATER FOR INJECTION
STERILE WATER FOR INHALATION
CLEANING AND INGREDIENT WATER FOR NON-
PARENTERAL DOSAGE FORMS
Packaging Sterilization
PURIFIED WATER (Bulked Packaged)
STERILE PURIFIED WATER
4. UNIT OPERATIONS REQUIRED FOR GENERATION OF PHARMACEUTICAL GRADE WATER
The following is a brief description of selected unit operations and the operation and validation concerns associated with them. The purpose is to highlight issues that focus on the design, installation, operation, maintenance, and monitoring parameters that facilitate water system validation.
4.1 Pre-filtration
The purpose of pre-filtration also referred to as initial, coarse, or depth filtration is to remove solid contaminants down to a size of 7 to 10 µm from the incoming source water supply and protect downstream system components from particulates that can inhibit equipment performance and

27
Page27
shorten their effective life. This coarse filtration technology utilizes primarily sieving effects for particle capture and a depth of filtration medium that has a high “dirt load” capacity. Removal efficiencies and capacities differ significantly, from granular bed filters such as multimedia or sand for larger water systems, to depth cartridges for smaller water systems.
4.2 Activated Carbon
Granular activated carbon beds adsorb low molecular weight organic material and oxidizing additives, such as chlorine and chloramine compounds, removing them from the water. They are used to achieve certain quality attributes and to protect against reaction with downstream stainless steel surfaces, resins, and membranes. The chief operating concerns regarding activated carbon beds include the propensity to support bacteria growth, the potential for hydraulic channelling, the organic adsorption capacity, appropriate water flow rates and contact time, the inability to be regenerated in situ, and the shedding of bacteria, endotoxins, organic chemicals, and fine carbon particles.
4.3 Additives
Chemical additives are used in water systems:
(a) To control microorganisms by use of sanitants such as chlorine compounds and ozone.
(b) To enhance the removal of suspended solids by use of flocculating agents.
(c) To remove chlorine compounds.
(d) To avoid scaling on reverse osmosis membranes.
(e) To adjust pH for more effective removal of carbonate and ammonia compounds by reverse osmosis.
These additives do not constitute “added substances” as long as they are either removed by subsequent processing steps or are otherwise absent from the finished water.
4.4 Organic Scavengers
Organic scavenging devices use macroreticular weakly basic anion-exchange resins capable of removing organic material and endotoxins from the water. They can be regenerated with appropriate biocidal caustic brine solutions. Operating concerns are associated with organic scavenging capacity, particulate, chemical and microbiological fouling of the reactive resin surface, flow rate, regeneration frequency, and shedding of resin fragments.
4.5 Softeners
Water softeners may be located either upstream or downstream of disinfectant removal units. They utilize sodium-based cation-exchange resins to remove water-hardness ions, such as calcium and magnesium that could foul or interfere with the performance of downstream processing equipment such as reverse osmosis membranes, deionization devices, and distillation units. Water softeners can also be used to remove other lower affinity cations, such as the ammonium ion, that may be released from chloramine disinfectants commonly used in drinking water and which might otherwise carryover through other downstream unit operations. If ammonium removal is one of its purposes, the softener must be located downstream of the disinfectant removal operation, which itself may liberate ammonium from neutralized chloramine disinfectants. Water softener resin beds are regenerated with concentrated sodium chloride solution (brine).

28
Page28
4.6 Deionization
Deionization (DI), is effective method of improving the chemical quality attributes of water by removing cations and anions. Deionization systems have charged resins that require periodic regeneration with an acid and base. Typically, cationic resins are regenerated with either hydrochloric or sulphuric acid, which replace the captured positive ions with hydrogen ions. Anionic resins are regenerated with sodium or potassium hydroxide, which replace captured negative ions with hydroxide ions. Because free endotoxin is negatively charged, there is some removal of endotoxin achieved by the anionic resin. Both regenerant chemicals are biocidal and offer a measure of microbial control. The system can be designed so that the cation and anion resins are in separate or “twin” beds or they can be mixed together to form a mixed bed. Twin beds are easily regenerated but de-ionize water less efficiently than mixed beds, which have a considerably more complex regeneration process. Rechargeable resin canisters can also be used for this purpose.
4.7 Reverse Osmosis
Reverse osmosis (RO) units employ semipermeable membranes. The “pores” of RO membranes are actually intersegmental spaces among the polymer molecules. They are big enough for permeation of water molecules, but too small to permit passage of hydrated chemical ions. However, many factors including pH, temperature, and differential pressure across the membrane affect the selectivity of this permeation. With the proper controls, RO membranes can achieve chemical, microbial, and endotoxin quality improvement. The process streams consist of supply water, product water (permeate), and wastewater (reject). Depending on source water, pre-treatment and system configuration variations and chemical additives may be necessary to achieve desired performance and reliability.
4.8 Ultrafiltration
Ultrafiltration is a technology most often employed in pharmaceutical water systems for removing endotoxins from a water stream. It can also use semipermeable membranes, but unlike RO, these typically use polysulfone membranes whose intersegmental “pores” have been purposefully exaggerated during their manufacture by preventing the polymer molecules from reaching their smaller equilibrium proximities to each other. Depending on the level of equilibrium control during their fabrication, membranes with differing molecular weight “cutoffs” can be created such that molecules with molecular weights above these cutoffs ratings are rejected and cannot penetrate the filtration matrix.
4.9 Charge-Modified Filtration
Charge-modified filters are usually microbially retentive filters that are treated during their manufacture to have a positive charge on their surfaces. Microbial retentive filtration will be described in a subsequent section, but the significant feature of these membranes is their electrostatic surface charge. Such charged filters can reduce endotoxin levels in the fluids passing through them by their adsorption (owing to endotoxin's negative charge) onto the membrane surfaces. Though ultrafilters are more often employed as a unit operation for endotoxin removal in water systems, charge-modified filters may also have a place in endotoxin removal particularly where available upstream pressures are not sufficient for ultra filtration and for a single, relatively short term use.
4.10 Microbial-Retentive Filtration

29
Page29
Microbial-retentive membrane filters have experienced an evolution of understanding in the past decade that has caused previously held theoretical retention mechanisms to be reconsidered. These filters have a larger effective “pore size” than ultrafilters and are intended to prevent the passage of microorganisms and similarly sized particles without unduly restricting flow. This type of filtration is widely employed within water systems for filtering the bacteria out of both water and compressed gases as well as for vent filters on tanks and stills and other unit operations. In water applications, microbial retentive filters may be used downstream of unit operations that tend to release microorganisms or upstream of unit operations that are sensitive to microorganisms. Microbial retentive filters may also be used to filter water feeding the distribution system.
4.11 Ultraviolet Light
The low-pressure UV lights that emit a 254-nm wavelength is use for microbial control. This 254-nm wavelength is also useful in the destruction of ozone. With intense emissions at wavelengths around 185 nm (as well as at 254 nm), medium pressure UV lights have demonstrated utility in the destruction of the chlorine containing disinfectants used in source water as well as for interim stages of water pre-treatment. High intensities of this wavelength alone or in combination with other oxidizing sanitants, such as hydrogen peroxide, have been used to lower TOC levels in recirculating distribution systems.
4.12 Distillation
Distillation units provide chemical and microbial purification via thermal vaporization, mist elimination, and water vapour condensation. A variety of designs is available including single effect, multiple effect, and vapour compression. The latter two configurations are normally used in larger systems because of their generating capacity and efficiency. Distilled water systems require different feed water controls than required by membrane systems. For distillation, due consideration must be given to prior removal of hardness and silica impurities that may foul or corrode the heat transfer surfaces as well as prior removal of those impurities that could volatize and condense along with the water vapour. In spite of general perceptions, even the best distillation process cannot afford absolute removal of contaminating ions and endotoxin.
4.13 Storage Tanks
Storage tanks are included in water distribution systems to optimize processing equipment capacity. Design and operation considerations are needed to prevent or minimize the development of biofilm, to minimize corrosion, to aid in the use of chemical sanitization of the tanks, and to safeguard mechanical integrity. These considerations may include using closed tanks with smooth interiors, the ability to spray the tank headspace using spray balls on recirculating loop returns, and the use of heated, jacketed/insulated tanks. This minimizes corrosion and biofilm development and aids in thermal and chemical sanitization. Storage tanks require venting to compensate for the dynamics of changing water levels. This can be accomplished with a properly oriented and heat-traced filter housing fitted with a hydrophobic microbial retentive membrane filter affixed to an atmospheric vent. Alternatively, an automatic membrane-filtered compressed gas blanketing system may be used. In both cases, rupture disks equipped with a rupture alarm device should be used as a further safeguard for the mechanical integrity of the tank. Areas of concern include microbial growth or corrosion due to irregular or incomplete sanitization and microbial contamination from unalarmed rupture disk failures caused by condensate-occluded vent filters.
4.14 Distribution Systems

30
Page30
Distribution system configuration should allow for the continuous flow of water in the piping by means of recirculation. Use of nonrecirculating, dead-end, or one-way systems or system segments should be avoided whenever possible. If not possible, these systems should be periodically flushed and more closely monitored. Pumps should be designed to deliver fully turbulent flow conditions to facilitate thorough heat distribution (for hot water sanitized systems) as well as thorough chemical sanitant distribution. Components and distribution lines should be sloped and fitted with drain points so that the system can be completely drained. In stainless steel distribution systems where the water is circulated at a high temperature, dead legs and low-flow conditions should be avoided, and valved tie-in points should have length-to-diameter ratios of six or less. If constructed of heat tolerant plastic, this ratio should be even less to avoid cool points where biofilm development could occur. Water exiting from the distribution system should not be returned to the system without first passing through all or a portion of the purification train. The distribution system should permit sanitization for microorganism control. The system may be continuously operated at sanitizing conditions or sanitized periodically.
5. VALIDATION AND QUALIFICATION OF WATER PURIFICATION, STORAGE, AND DISTRIBUTION SYSTEMS
An appropriate period of monitoring and observation is required for establishing the dependability of pharmaceutical water purification, storage, and distribution systems. An assessment of the consistency of the water's chemical purity over time must be part of the validation program. However, even with the most well controlled chemical quality, it is often more difficult to consistently meet established microbiological quality criteria owing to phenomena occurring during and after chemical purification. A typical program involves intensive daily sampling and testing of major process points for at least one month after operational criteria have been established for each unit operation, point of use, and sampling point. An overlooked aspect of water system validation is the delivery of the water to its actual location of use. If this transfer process from the distribution system outlets to the water use locations is defined as outside the water system, then this transfer process still needs to be validated to not adversely affect the quality of the water to the extent it becomes unfit for use. A validation program qualifies and documents the design, installation, operation, and performance of equipment. It begins when the system is defined and moves through several stages: installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ).
A validation plan for a water system typically includes the following steps: (1) Establishing standards for quality attributes of the finished water and the source water. (2) Defining suitable unit operations and their operating parameters for achieving the desired
finished water quality attributes from the available source water. (3) Selecting piping, equipment, controls, and monitoring technologies. (4) Developing an IQ stage consisting of instrument calibrations, inspections to verify that the
drawings accurately depict the final configuration of the water system and, where necessary, special tests to verify that the installation meets the design requirements.
(5) Developing an OQ stage consisting of tests and inspections to verify that the equipment, system alerts, and controls are operating reliably and that appropriate alert and action levels are established (This phase of qualification may overlap with aspects of the next step.).

31
Page31
(6) Developing a prospective PQ stage to confirm the appropriateness of critical process parameter operating ranges (During this phase of validation, alert and action levels for key quality attributes and operating parameters are verified.).
(7) Assuring the adequacy of ongoing control procedures, e.g., sanitization frequency. (8) Supplementing a validation maintenance program (also called continuous validation life cycle)
that includes a mechanism to control changes to the water system and establishes and carries out scheduled preventive maintenance including recalibration of instruments (In addition, validation maintenance includes a monitoring program for critical process parameters and a corrective action program.).
(9) Instituting a schedule for periodic review of the system performance and requalification.
(10) Completing protocols and documenting Steps 1 through 9.
DEFINE WATER QUALITY ATTRIBUTES
Define Systems and Subsystems Including Processing Technologies, Operating Parameters and Corrective Actions Features to Meet Quality Parameters Attributes
Installation Qualification (IQ)
Install Equipment Piping and Control Systems
Operational Qualification (OQ)
Performance Qualification (PQ)
Identify Critical Process Parameters and Establish Operating Ranges
Establish Alert and Action Levels for Key Quality Attributes
Establish Corrective Action Responses
Prospective Phase-Confirm Appropriateness of Critical Process Parameter Operating Ranges
Validation Maintenance Change Control Periodic Review
Concurrent/Retrospective Phase Establish Reproducibility and Reliability of
System Evaluate Effect of Seasonal Changes Confirm Appropriateness of Alert and Action
Levels and Corrective Action Program
SYSTEM/EQUEPMENT/CHGS/ADJ
CHGS
CHGS
CHGS
Figure 2: Water system validation life cycle.

32
Page32
Microbial control in water systems is achieved primarily through sanitization practices. Systems can be sanitized using either thermal or chemical means. Thermal approaches to system sanitization include periodic or continuously circulating hot water and the use of steam. Temperatures of at least 80°C are most commonly used for this purpose, but continuously recirculating water of at least 65°C has also been used effectively in insulated stainless steel distribution systems when attention is paid to uniformity and distribution of such self-sanitizing temperatures. These techniques are limited to systems that are compatible with the higher temperatures needed to achieve sanitization. Although thermal methods control biofilm development by either continuously inhibiting their growth or, in intermittent applications, by killing the microorganisms within biofilms, they are not effective in removing established biofilms. Killed but intact biofilms can become a nutrient source for rapid biofilm regrowth after the
6. OPERATION, MAINTENANCE, AND CONTROL OF WATER SYSTEM
A preventive maintenance program should be established to ensure that the water system remains in a state of control. The program should include:
(1) Procedures for operating the system.
(2) Monitoring programs for critical quality attributes and operating conditions including calibration of critical instruments.
(3) Schedule for periodic sanitization.
(4) Preventive maintenance of components.
(5) Control of changes to the mechanical system and to operating conditions.
Operating Procedures-Procedures for operating the water system and performing routine maintenance and corrective action should be written, and they should also define the point when action is required. The procedures should be well documented, detail the function of each job, assign who is responsible for performing the work, and describe how the job is to be conducted. The effectiveness of these procedures should be assessed during water system validation.
Monitoring Program-Critical quality attributes and operating parameters should be documented and monitored. The program may include a combination of in-line sensors or automated instruments (e.g., for TOC, conductivity, hardness, and chlorine), automated or manual documentation of operational parameters (such as flow rates or pressure drop across a carbon bed, filter, or RO unit), and laboratory tests (e.g., total microbial counts). The frequency of sampling, the requirement for evaluating test results, and the necessity for initiating corrective action should be included.
Sanitization-Depending on system design and the selected units of operation, routine periodic sanitization may be necessary to maintain the system in a state of microbial control. Technologies for sanitization are described above.
Preventive Maintenance-A preventive maintenance program should be in effect. The program should establish what preventive maintenance is to be performed, the frequency of maintenance work, and how the work should be documented.
Change Control-The mechanical configuration and operating conditions must be controlled. Proposed changes should be evaluated for their impact on the whole system. The need to requalify the system after changes are made should be determined. Following a decision to modify a water system, the affected drawings, manuals, and procedures should be revised.
7. WATER SYSTEM SANITIZATION

33
Page33
sanitizing conditions are removed or halted. In such cases, a combination of routine thermal and periodic supplementation with chemical sanitization might be more effective. The more frequent the thermal sanitization, the more likely biofilm development and regrowth can be eliminated. Chemical methods, where compatible, can be used on a wider variety of construction materials. These methods typically employ oxidizing agents such as halogenated compounds, hydrogen peroxide, ozone, peracetic acid, or combinations thereof. Halogenated compounds are effective sanitizers but are difficult to flush from the system and may leave biofilms intact. Compounds such as hydrogen peroxide, ozone, and peracetic acid oxidize bacteria and biofilms by forming reactive peroxides and free radicals (notably hydroxyl radicals). The short half-life of ozone in particular, and its limitation on achievable concentrations require that it be added continuously during the sanitization process. Hydrogen peroxide and ozone rapidly degrade to water and oxygen; peracetic acid degrades to acetic acid in the presence of UV light. In fact, ozone's ease of degradation to oxygen using 254-nm UV lights at use points allow it to be most effectively used on a continuous basis to provide continuously sanitizing conditions.
In-line UV light at a wavelength of 254 nm can also be used to continuously “sanitize” water circulating in the system, but these devices must be properly sized for the water flow. Such devices inactivate a high percentage (but not 100%) of microorganisms that flow through the device but cannot be used to directly control existing biofilm upstream or downstream of the device. However, when coupled with conventional thermal or chemical sanitization technologies or located immediately upstream of a microbially retentive filter, it is most effective and can prolong the interval between system sanitizations.
It is important to note that microorganisms in a well-developed biofilm can be extremely difficult to kill, even by aggressive oxidizing biocides. The less developed and therefore thinner the biofilm, the more effective the biocidal action. Therefore, optimal biocide control is achieved by frequent biocide use that does not allow significant biofilm development between treatments.
Sanitization steps require validation to demonstrate the capability of reducing and holding microbial contamination at acceptable levels. Validation of thermal methods should include a heat distribution study to demonstrate that sanitization temperatures are achieved throughout the system, including the body of use point valves. Validation of chemical methods require demonstrating adequate chemical concentrations throughout the system, exposure to all wetted surfaces, including the body of use point valves, and complete removal of the sanitant from the system at the completion of treatment. Methods validation for the detection and quantification of residues of the sanitant or its objectionable degradants is an essential part of the validation program. The frequency of sanitization should be supported by, if not triggered by, the results of system microbial monitoring. Conclusions derived from trend analysis of the microbiological data should be used as the alert mechanism for maintenance. The frequency of sanitization should be established in such a way that the system operates in a state of microbiological control and does not routinely exceed alert levels.
8. SAMPLING OF WATER
Monitoring of water systems should be done at a frequency that is sufficient to ensure that the system is in control and continues to produce water of acceptable quality. Samples should be taken from representative locations within the processing and distribution system. Established sampling frequencies should be based on system validation data and should cover critical areas including unit operation sites. The sampling plan should take into consideration the desired attributes of the water being sampled. Analyses of water samples often serve two purposes:

34
Page34
In-Process Control Assessments and final Quality Control Assessments. In-process control analyses are usually focused on the attributes of the water within the system. Quality control is primarily concerned with the attributes of the water delivered by the system to its various uses. Samples should be collected from use points using the same delivery devices, such as hoses, and procedures, such as preliminary hose or outlet flushing, as are employed by production from those use points.
Samples containing chemical sanitizing agents require neutralization prior to microbiological analysis. Samples for microbiological analysis should be tested immediately, or suitably refrigerated to preserve the original microbial attributes until analysis can begin. Samples of flowing water are only indicative of the concentration of planktonic (free floating) microorganisms present in the system. Microorganisms in biofilms represent a continuous source of contamination and are difficult to directly sample and quantify. Consequently, the planktonic population is usually used as an indicator of system contamination levels and is the basis for system Alert and Action Levels.
Sampling for chemical analyses is also done for in-process control and for quality control purposes. However, unlike microbial analyses, chemical analyses can be and often are performed using on-line instrumentation. Such on-line testing has unequivocal in-process control purposes because it is not performed on the water delivered from the system.
9. MICROBIAL CONTAMINATION AND THEIR MONITORING PROGRAM IN WATER SYSTEM
The main source of microbial contamination of pharmaceutical water is feed water. Feed water quality must meet the quality attributes of Drinking Water. Wide variety Gram-negative bacteria, chiefly coliforms may be present in the incoming water. Examples of other potential exogenous sources of microbial contamination include unprotected vents, faulty air filters, ruptured rupture disks, backflow from contaminated outlets, unsanitized distribution system, inadequate drain & air-breaks, and replacement activated carbon, deionizer resins, & regenerant chemicals. In these situations, the exogenous contaminants may not be normal aquatic bacteria but rather microorganisms of soil or even human origin. Endogenous microbial contamination can be take place during Unit operations. Microorganisms present in feed water may adsorb to carbon bed, deionizer resins, filter membranes and other unit operation surfaces and initiate the formation of a biofilm.
Distribution system itself acts as another source of endogenous microbial contamination. Microorganisms can colonize pipe surfaces, rough welds, badly aligned flanges, valves, and unidentified dead legs, where they proliferate, forming a biofilm which becomes a continuous source of microbial contamination. Gram-negative bacteria that form biofilms can become a source of endotoxins in pharmaceutical waters. Endotoxins may occur as clusters of lipopolysaccharide molecules associated with living microorganisms, fragments of dead microorganisms or the polysaccharide slime surrounding biofilm bacteria, or as free molecules. Endotoxin levels may be minimized by controlling the introduction of free endotoxins and microorganisms in the feed water and minimizing microbial proliferation in the system. Control methods for endotoxin includes water system sanitization, use of ultrafilters or charge-modified filters, either in-line or at the point of use.
The main objective of microbiological monitoring program for water system is to provide sufficient information to control and assess the microbiological quality of the pharmaceutical grade water. The monitoring program and methodology should indicate adverse trends and detect microorganisms that are potentially harmful to the finished product, process, or consumer. Several criteria should be considered when selecting a method to monitor the microbial content of a

35
Page35
pharmaceutical water system. These include method sensitivity, range of organism types or species recovered; sample processing throughput, incubation period, cost, and methodological complexity.
The number of detectable planktonic bacteria within the sample will tend to either die or to irretrievably adsorb to the container walls reducing the number of viable planktonic bacteria that can be withdrawn from the sample for testing. The opposite effect can also occur if the sample container is not clean and contains a low concentration of some microbial nutrient that could promote microbial growth within the sample container. Because the number of recoverable bacteria in a sample can change positively or negatively over time after sample collection, it is best to test the samples as soon as possible after being collected. If it is not possible to test the sample within about 2 hours of collection, the sample should be held at refrigerated temperatures (2 °C to 8 °C) for a maximum of about 12 hours to maintain the microbial attributes until analysis. In situations where even this is not possible (such as when using off-site contract laboratories), testing of these refrigerated samples should be performed within 48 hours after sample collection.
10. CHEMICAL CONTAMINANTS IN WATER SYSTEM
Pharmaceutical grade water must meet the requirements for ionic and organic chemical purity. The two contemporary analytical technologies employed were TOC and Conductivity for the detection of chemical contaminants. The TOC test replaced the test for Oxidizable substances that primarily targeted organic contaminants. A multistaged Conductivity test which detects ionic (mostly inorganic) contaminants replaced, with the exception of the test for Heavy metals, all of the inorganic chemical tests (i.e., Ammonia, Calcium, Carbon dioxide, Chloride, Sulfate). In general the TOC and Conductivity tests are used for “on-line” measurements. The TOC and Conductivity tests can also be performed “off-line” in the laboratories using collected samples, though sample collection tends to introduce opportunities for adventitious contamination that can cause false high readings
Total solids and pH are the only tests not covered by conductivity testing. The test for Total solids was considered redundant because the nonselective tests of Conductivity and TOC could detect most chemical species other than silica, which could remain undetected in its colloidal form. If silica is a significant component in the source water, and the purification unit operations could be operated or fail and selectively allow silica to be released into the finished, then either silica-specific or a total solids type testing should be utilized to monitor and control this rare problem.
Packaged waters present a particular dilemma relative to the attributes of Conductivity and TOC. The package itself is the source of chemicals (inorganics and organics) that leach over time into the water and can easily be detected. The irony of organic leaching from plastic packaging is that when the Oxidizable substances test was the only “organic contaminant” test for both bulk and packaged waters, that test's insensitivity to those organic leachable rendered their presence in packaged water at high concentrations virtually undetectable. Similarly, glass containers can also leach inorganics, such as sodium, which are easily detected by conductivity, but are undetected by the wet chemistry tests for water (other than pH or Total solids). The attributes of conductivity and TOC tend to reveal more about the packaging leachables than they do about the water's original purity.
Pharmacopoeial quality control specification (chemical and microbiology) for different pharmaceutical grade water is given in Appendix I. Water is the main source of cross contamination for all the pharmaceutical preparations. Hence the stakeholder should adhere to use water as per pharmacopoeial requirements.

36
Appendix I PHARMACOPOEIAL QUALITY CONTROL SPECIFICATION FOR PHARMACEUTICAL GRADE WATER
a) Purified Water Specification
IP 2010 USP 2009 BP 2011 Ph. Eur 2011 S. No. Test
Purified Water Purified Water
Sterile Purified water
Purified Water in
Bulk
Purified Water in Containers
Highly purified Water
Purified Water in
Bulk
Purified Water in Containers
Highly purified Water
1 Acidity or Alkalinity
Resulting sol. should not be Red and Blue in colour respectively
- - -
Resulting sol. should not be Red and Blue in colour respectively - -
Resulting sol. should not be Red and Blue in colour respectively
-
2 Ammonium Sol. should not be more than intensely coloured
- - - Max. 0.2 ppm - - Max. 0.2 ppm -
3 pH - - - - - 5-7 - - 5-7
4 Conductivity - As per requirement
Max. 25 µS/cm in 10 ml or < 10 ml Vol.at 25 oC,
Max. 5 µS/cm in >10 ml Vol. A 25 oC
5.1 µS/cm at 25 oC - 1.3 µS/cm
at 25 oC 5.1 µS/cm at 25 oC - 1.3 µS/cm at
25 oC
5 Total organic carbon - As per
requirement - 0.5 mg/ltr - 0.5 mg/ltr 0.5 mg/ltr - 0.5 mg/ltr
6 Calcium & Magnesium
Resulting sol. should produce Blue colour
- - - Resulting sol. should produce Blue colour - -
Resulting sol. should produce Blue colour -
7 Heavy Metals 0.1 ppm - - Max. 0.1 ppm - - Max. 0.1 ppm - -
8 Chlorides
Appearance of the solution does not change for at least 15 min.
- - -
Appearance of the solution does not change for at least 15 min.
- -
Appearance of the solution does not change for at least 15 min.
-
9 Nitrates Blue colour in the solution should not more intense
- - Max. 0.2 ppm - Max. 0.2 ppm Max. 0.2 ppm - Max. 0.2 ppm
10 Sulphates
Appearance of the solution does not change for at least 1 hour
- - -
Appearance of the solution does not change for at least 1 hour.
- -
Appearance of the solution does not change for at least 1 hour
-
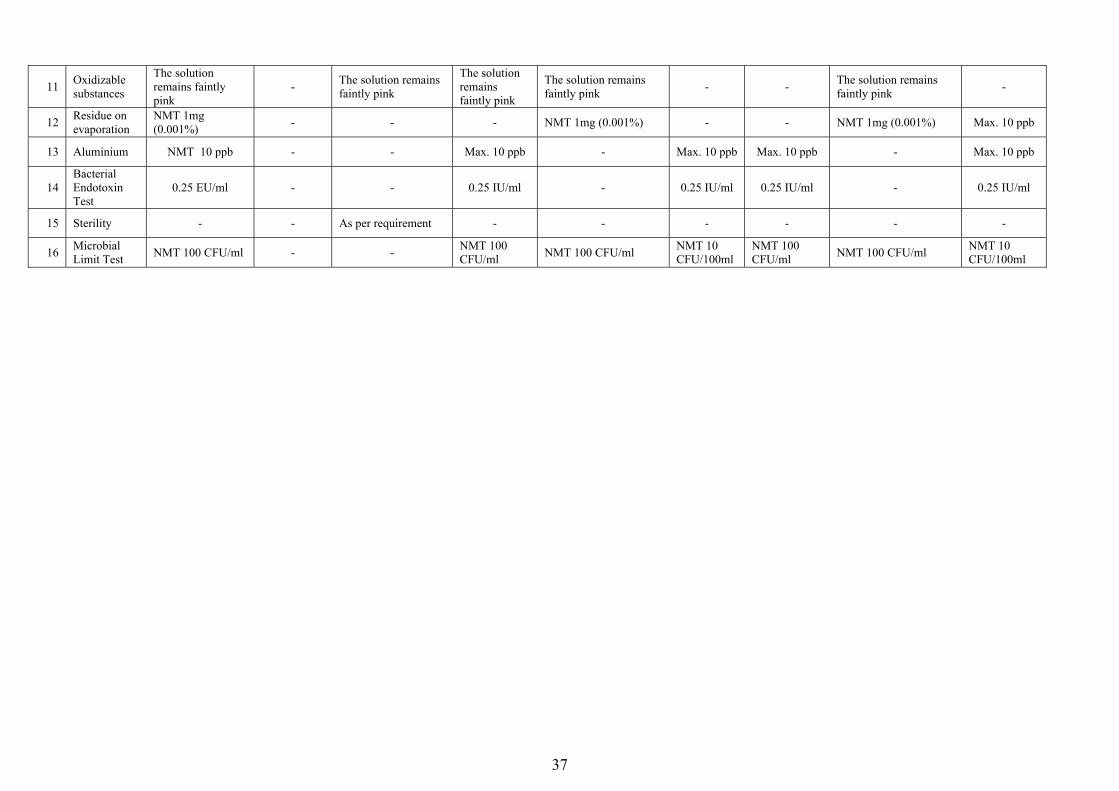
37
11 Oxidizable substances
The solution remains faintly pink
- The solution remains faintly pink
The solution remains faintly pink
The solution remains faintly pink - - The solution remains
faintly pink -
12 Residue on evaporation
NMT 1mg (0.001%) - - - NMT 1mg (0.001%) - - NMT 1mg (0.001%) Max. 10 ppb
13 Aluminium NMT 10 ppb - - Max. 10 ppb - Max. 10 ppb Max. 10 ppb - Max. 10 ppb
14 Bacterial Endotoxin Test
0.25 EU/ml - - 0.25 IU/ml - 0.25 IU/ml 0.25 IU/ml - 0.25 IU/ml
15 Sterility - - As per requirement - - - - - -
16 Microbial Limit Test NMT 100 CFU/ml - - NMT 100
CFU/ml NMT 100 CFU/ml NMT 10 CFU/100ml
NMT 100 CFU/ml NMT 100 CFU/ml NMT 10
CFU/100ml

b
38
) Water for Injection (WFI) Specification
IP 2010 USP 2009 BP 2011 Ph. Eur 2011 S. No. Test
WFI in Bulk Sterile WFI WFI Sterile WFI WFI in Bulk Sterile WFI WFI in Bulk Sterile WFI
1 Acidity or Alkalinity
Resulting sol. should not be Red and Blue in colour respectively
Resulting sol. should not be Red and Blue in colour respectively
- - - Resulting sol. should not be Red and Blue in colour respectively
- Resulting sol. should not be Red and Blue in colour respectively
2 Ammonium Sol. should not be more than intensely coloured
Sol. should not be more than intensely coloured
-
Max. 0.6 mg / ltr (50 ml or � 50 ml vol.)
Max. 0.3 mg / ltr (>50 ml vol.)
-
Max. 0.6 ppm (50 ml or � 50 ml vol.)
Max. 0.2 ppm (>50 ml vol.)
-
Max. 0.6 ppm (50 ml or � 50 ml vol.) Max. 0.2 ppm (>50 ml vol.)
3 pH - - - 5-7 5-7 - 5-7 -
4 Conductivity As per requirement - As per requirement - 1.3 µS/cm at 25 oC
Max. 25 µS/cm in 10 ml or < 10 ml Vol. at 25 oC,
Max. 5 µS/cm in >10 ml Vol. at 25 oC
1.3 µS/cm at 25 oC
Max. 25 µS/cm in 10 ml or < 10 ml Vol. at 25 oC,
Max. 5 µS/cm in >10 ml Vol. at 25 oC
5 Total organic carbon NMT 0.5 mg/ltr - As per requirement - NMT 0.5
mg/ltr - Max. 0.5 mg/ltr -
6 Calcium & Magnesium
Resulting sol. should produce Blue colour
Resulting sol. should produce Blue colour
- No turbidity produced - Resulting sol. should
produce Blue colour - Resulting sol. should produce Blue colour
7 Heavy Metals 0.1 ppm 0.1 ppm - - - - - -
8 Chlorides
Appearance of the solution does not change for at least 15 min.
Appearance of the solution does not change for at least 15 min.
- 0.5 mg /ltr - Max. 0.5 ppm - Max. 0.5 ppm
9 Nitrates Blue colour in the solution should not more intense
Blue colour in the solution should not more intense
- - Max. 0.2 ppm Max. 0.2 ppm Max. 0.2 ppm Max. 0.2 ppm
10 Sulphates
Appearance of the solution does not change for at least 1 hour
Appearance of the solution does not change for at least 1 hour
- No turbidity produced -
Appearance of the solution does not change for at least 1 hour
-
Appearance of the solution does not change for at least 1 hour
11 Oxidizable substances - The solution
remains faintly pink - The solution remains faintly pink - The solution remains
faintly pink - The solution remains faintly pink
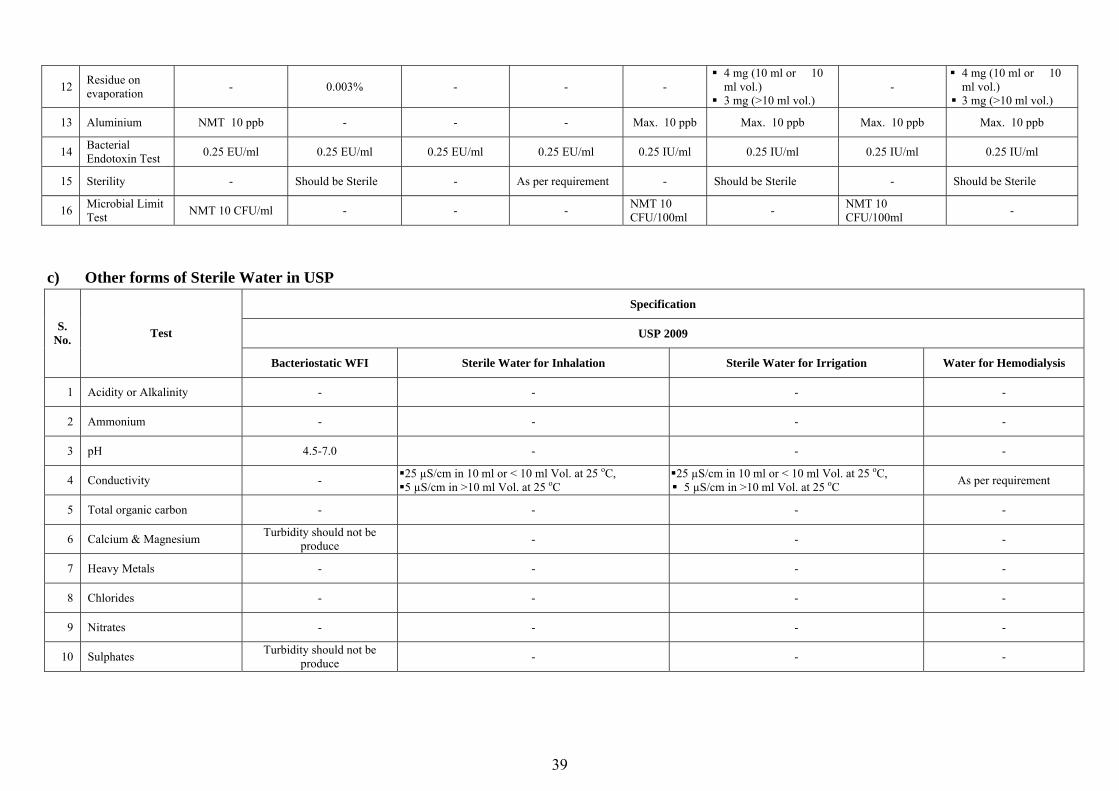
39
12 Residue on evaporation - 0.003% - - -
4 mg (10 ml or � 10 ml vol.)
3 mg (>10 ml vol.) -
4 mg (10 ml or � 10 ml vol.)
3 mg (>10 ml vol.)
13 Aluminium NMT 10 ppb - - - Max. 10 ppb Max. 10 ppb Max. 10 ppb Max. 10 ppb
14 Bacterial Endotoxin Test 0.25 EU/ml 0.25 EU/ml 0.25 EU/ml 0.25 EU/ml 0.25 IU/ml 0.25 IU/ml 0.25 IU/ml 0.25 IU/ml
15 Sterility - Should be Sterile - As per requirement - Should be Sterile - Should be Sterile
16 Microbial Limit Test NMT 10 CFU/ml - - - NMT 10
CFU/100ml - NMT 10 CFU/100ml -
c) Other forms of Sterile Water in USP Specification
USP 2009 S. No. Test
Bacteriostatic WFI Sterile Water for Inhalation Sterile Water for Irrigation Water for Hemodialysis
1 Acidity or Alkalinity - - - -
2 Ammonium - - - -
3 pH 4.5-7.0 - - -
4 Conductivity - 25 µS/cm in 10 ml or < 10 ml Vol. at 25 oC, 5 µS/cm in >10 ml Vol. at 25 oC
25 µS/cm in 10 ml or < 10 ml Vol. at 25 oC, 5 µS/cm in >10 ml Vol. at 25 oC As per requirement
5 Total organic carbon - - - -
6 Calcium & Magnesium Turbidity should not be produce - - -
7 Heavy Metals - - - -
8 Chlorides - - - -
9 Nitrates - - - -
10 Sulphates Turbidity should not be produce - - -

40
11 Oxidizable substances - The solution remains faintly pink The solution remains faintly pink The solution remains faintly pink
12 Residue on evaporation - - - -
13 Aluminium - - - -
14 Bacterial Endotoxin Test 0.5 EU/ml 0.5 EU/ml 0.25 EU/ml 2 EU/ml
15 Sterility As per requirement As per requirement As per requirement -
16 Microbial Limit Test - - - NMT 100 CFU/ml
Reference: 1. (EMEA)-The European Agency for the evalution of medicinal products. Landon (UK), 19th Oct 2010 2. Indian Phramacopoeia 2010 3. European Pharamcopoeia 2010 4. Bitish Pharmacopoeia 2011 5. United State Phrmacopoeia 2009 6. US-EPA water quality standard guideline
Author: Dr. Nishant Dafale,
Senior Scientific Officer, Indian Pharmacopeia Commission, Ghaziabad Email:

Annexure I
I P 2010 GENERAL NOTICES VOLUME I General Notices
General Statements The General Notices provide the basic guidelines for the interpretation and application of the standards, tests, assays, and other specifications of the Indian Pharmacopoeia (IP), as well as to the statements made in the monographs and other texts of the Pharmacopoeia.
A monograph is to be constructed in accordance with any general monograph or notice or any appendix, note or other explanatory material that is contained in this Pharmacopoeia and that is applicable to that monograph. All statements contained in the monograph, except where a specific general notice indicates otherwise and with the exceptions given hereafter, constitute standards for the official articles. An article is not of pharmacopoeial quality unless it complies with all of the requirements stated.
Exceptions to the General Notices do exist, and where they do, the wording in the individual monograph or an appendix takes precedence and specifically indicates directions or the intent. Thus, the specific wording of standards, tests, assays and other specifications is binding wherever deviations from the General Notices exist. Likewise, where there is no specific mention to the contrary, the General Notices apply.
Name. The full name or title of this book, including addenda thereto, is Indian Pharmacopoeia 2010, abbreviated to IP 2010. In the texts, the term “Pharmacopoeia” or “IP” without qualification means the Indian Pharmacopoeia 2010 and any addenda thereto.
Official and Official Articles. The word ‘official’ wherever used in this Pharmacopoeia or with reference thereto, is synonymous with ‘pharmacopoeial’, with ‘IP’ and with ‘compendial’. The designation IP in conjunction with the official title on the label of an article is an indication that the article purports to comply with IP standards.
The following terms are used where the articles for which monographs are provided are to be distinguished.
An official substance is a single drug or a drug entity or a pharmaceutical aid for which the monograph title includes no indication of the nature of a dosage form.
An official preparation is a drug product (dosage form) and is the finished or partially finished preparation or product of one or more official substances formulated for use on the patient.
41

An article is an item for which a monograph is provided, whether an official substance or an official preparation.
Official Standards. The requirements stated in the monographs apply to articles that are intended for medicinal use but not necessarily to articles that may be sold under the same name for other purposes.
The active pharmaceutical ingredients (drug substances), excipients (pharmaceutical aids), pharmaceutical preparations (dosage forms) and other articles described in the monographs are intended for human and veterinary use (unless explicitly restricted to one of these uses).
The requirements given in the monographs are not framed to provide against all possible impurities, contaminants or adulterants; they provide appropriate limitation of potential impurities only.
A preparation must comply throughout the shelf-life assigned to it by the manufacturer; for opened or broached containers the maximum period of validity for use may sometimes be stated in the individual monograph. Nevertheless, the responsibility for assigning the period of validity shall be with the manufacturer.
Added Substances. An official substance, as distinguished from an official preparation, contains no added substances except when specifically permitted in the individual monograph. Unless otherwise specified in the individual monograph, or elsewhere in the General Notices, suitable substances may be added to an official preparation to enhance its stability, usefulness or elegance, or to facilitate its preparation. Such auxiliary substances shall be harmless in the amounts used, shall not exceed the minimum quantity required to provide their intended effect, shall not impair the therapeutic efficacy or the bioavailability or safety of the preparation and shall not interfere with the tests and assays prescribed for determining compliance with the official standards. Particular care should be taken to ensure that such substances are free from harmful organisms. The freedom to the manufacturers to add auxiliary substances imposes on them the responsibility of satisfying the licensing authorities on the purpose of the addition and the innocuity of such substances.
Alternative Methods. The tests and assays described are the official methods upon which the standards of the Pharmacopoeia are based. Alternative methods of analysis may be used for control purposes, provided that the methods used are shown to give results of equivalent accuracy and enable an unequivocal decision to be made as to whether compliance with the standards of the monographs would be achieved if the official methods were used. Automated procedures utilising the same basic chemistry as the test procedures given in the monograph may also be used to determine compliance. Such alternative or automated procedures must be validated.
42

In the event of doubt or dispute, the methods of analysis of the Pharmacopoeia are alone authoritative and only the result obtained by the procedure given in this Pharmacopoeia is conclusive.
Meanings of Terms
Alcohol. The term “alcohol” without qualification means ethanol (95 per cent). Other dilutions of ethanol are indicated by the term “ethanol” or “alcohol” followed by a statement of the percentage by volume of ethanol (C2H6O) required.
Desiccator. A tightly-closed container of suitable size and design that maintains an atmosphere of low moisture content by means of silica gel or phosphorus pentoxide or other suitable desiccant.
Drying and ignition to constant weight. Two consecutive weighings after the drying or igniting operations do not differ by more than 0.5 mg, the second weighing following an additional period of drying or of ignition respectively appropriate to the nature and quantity of the residue.
Ethanol. The term “ethanol” without qualification means anhydrous ethanol or absolute alcohol.
Filtration. Unless otherwise stated, filtration is the passing of a liquid through a suitable filter paper or equivalent device until the filtrate is clear.
Freshly prepared. Made not more than 24 hours before it is issued for use.
Label. Any printed packing material, including package inserts that provide information on the article.
Negligible. A quantity not exceeding 0.50 mg.
Solution. Where the name of the solvent is not stated, “solution” implies a solution in water. The water used complies with the requirements of the monograph on Purified Water. The term ‘distilled water’ indicates Purified Water prepared by distillation.
Temperature. The symbol ‘º’ used without qualification indicates the use of the Celsius thermometric scale.
Water. If the term is used without qualification it means Purified Water of the Pharmacopoeia. The term ‘distilled water’ indicates Purified Water prepared by distillation.
Water-bath. A bath of boiling water unless water at another temperature is indicated. Other methods of heating may be used provided the required temperature is approximately maintained but not exceeded.
Provisions Applicable To Monographs and Test Methods
43

Expression of Contents. Where the content of a substance is defined, the expression “per cent” is used according to circumstances with one of two meanings:
— per cent w/w (percentage, weight in weight) expressing the number of grams of substance in 100 grams of final product,
— per cent v/v (percentage, volume in volume) expressing the number of millilitres of substance in 100 millilitres of final product.
The expression “parts per million” refers to the weight in weight, unless otherwise stated.
Where the content of a substance is expressed in terms of the chemical formula for that substance an upper limit exceeding 100 per cent may be stated. Such an upper limit applies to the result of the assay calculated in terms of the equivalent content of the specified chemical formula. For example, the statement ‘contains not less than 99.0 per cent and not more than 101.0 per cent of C7H6O2 implies that the result of the assay is not less than 99.0 per cent and not more than 101.0 per cent, calculated in terms of the equivalent content of C7H6O2.
Where the result of an assay or test is required to be calculated with reference to the dried, anhydrous, ignited substance, or the substance free from solvent, the determination of loss on drying, water content, loss on ignition, content of the specified solvent, respectively is carried out by the method prescribed in the relevant test in the monograph.
Expression of Concentrations. The following expressions in addition to the ones given under Expression of Content are also used:
— per cent w/v (percentage, weight in volume) expressing the number of grams of substance in 100 millilitres of product
— per cent v/w (percentage, volume in weight) expressing the number of millilitres of substance in 100 grams of product.
Usually, the strength of solutions of solids in liquids is expressed as percentage weight in volume, of liquids in liquids as percentage volume in volume, of solids in semi-solid bases (e.g. creams) and of gases in liquids as percentage weight in weight.
When the concentration of a solution is expressed as parts of dissolved substance in parts of solution, it means parts by weight (g) of a solid in parts by volume (ml) of the final solution; as parts by weight (g) of a gas in parts by weight (g) of the final solution.
When the concentration of a solution is expressed in molarity designated by the symbol M preceded by a number, it denotes the number of moles of the stated solute contained in sufficient Purified Water (unless otherwise stated) to produce 1 litre of solution.
Abbreviated Statements. Incomplete sentences are employed in parts of the monographs for directness and brevity (for example, Iodine Value. Not more than ……;
44

Relative Density. …….to……..) Where the tests are abbreviated, it is to be understood that the test method referred to in brackets provides the method to be followed and that the values specified are the applicable limits.
Weights and Measures. The metric system of weights and measures is employed in the Pharmacopoeia. All measures are required to be graduated at 25º and all measurements in tests and assays, unless otherwise stated, are to be made at that temperature. Graduated glass apparatus used in analytical operations shall comply with the requirements stated in Chapter 2.1.6
Monographs
General Monographs
General monographs on dosage forms include requirements of general application and apply to all preparations within the scope of the Introduction section of the general monograph, except where a preamble limits the application. The requirements are not necessarily comprehensive for a given specific preparation; additional requirements may sometimes be given in the individual monograph for it.
Production. Statements given under the heading Production relate to particular aspects of the manufacturing process and are not necessarily comprehensive. However, they are mandatory instructions to manufacturers. They may relate, for example, to source materials, to the manufacturing process and its validation and control, to any in-process testing that is to be carried out by the manufacturer on the final product either on selected batches or on each batch prior to release. All this cannot be verified on a sample of the final product by an independent analyst. It is for the licensing authority to verify that the instructions have been followed.
The absence of a section on Production does not imply that attention to features such as those given above is not required. An article described in a monograph of the Pharmacopoeia is to be manufactured in accordance with the principles of good manufacturing practice and in accordance with the requirements of the Drugs and Cosmetics Rules, 1945. The general principles applicable to the manufacture and quality assurance of drugs and preparations meant for human use apply equally to veterinary products as well.
Manufacture of Drug Products. The opening definitive statement in certain monographs for drug products is given in terms of the active ingredient(s) only. Any ingredient(s) other than those included in the statement, must comply with the general notice on Excipients and the product must conform to the Pharmacopoeial requirements.
Official preparations are prepared only from ingredients that comply with the requirements of the pharmacopoeial monographs for those individual ingredients for which monographs are provided.
45

Excipients. Any substance added in preparing an official preparation shall be innocuous, shall have no adverse influence in the therapeutic efficacy of the active ingredients and shall not interfere with the tests and assays of the Pharmacopoeia. Care should be taken to ensure that such substances are free from harmful organisms.
Individual Monographs
Drug products that are the subject of an individual monograph are also required to comply with the tests given in the general monographs.
Titles. The main title for a drug substance is the International Non-proprietary Name (INN) approved by the World Health Organization. Subsidiary names and synonyms have also been given in some cases; where included, they have the same significance as the main title.
The main titles of drug products are the ones commonly recognised in practice. Synonyms drawn from the full non-proprietary name of the active ingredient or ingredients have also been given. Where, however, a product contains one or the other of different salts of an active molecule, the main title is based on the full name of the active ingredient. For example, Chloroquine Phosphate Tablets and Chloroquine SulphateTablets.
Chemical Formulae. When the chemical structure of an official substance is known or generally accepted, the graphic and molecular formulae are normally given at the beginning of the monograph for information. This information refers to the chemically pure substance and is not to be regarded as an indication of the purity of the official material. Elsewhere, in statement of purity and strength and in descriptions of processes of assay, it will be evident from the context that the formulae denote the chemically pure substances.
Where the absolute stereochemical configuration is specified, the International Union of Pure and Applied Chemistry (IUPAC) R/S and E/Z systems of designation have been used. If the substance is an enantiomer of unknown absolute stereochemistry, the sign of the optical rotation, as determined in the solvent and under the conditions specified in the monograph, has been attached to the systematic name. An indication of sign of rotation has also been given where this is incorporated in a trivial name that appears on an IUPAC preferred list. Atomic and Molecular Weights. The atomic weight or molecular weight is shown , as and when appropriate at the top right hand corner of the monograph. The atomic and molecular weights and graphic formulae do not constitute analytical standards for the substances described. Definition. The opening statement of a monograph is one that constitutes an official definition of the substance, preparation or other article that is the subject of the
46

monograph. In certain monographs for pharmaceutical preparations the statement is given in terms of the principal ingredient(s). In monographs on vegetable drugs, the definition indicates whether the subject of the monograph is, for example, the whole drug or the drug in powdered form. Certain pharmaceutical substances and other articles are defined by reference to a particular method of manufacture. A statement that a substance or article is prepared or obtained by a certain method constitutes part of the official definition and implies that other methods are not permitted. A statement that a substance may be prepared or obtained by a certain method, however, indicates that this is one possible method and does not imply that other methods are not permissible. Statement of content. The limits of content stated are those determined by the method described under Assay. Category. The statement of category is provided for information and is indicative of the medical or pharmaceutical basis for recognition in the Pharmacopoeia. It generally represents an application of the best known pharmacological action of the article or of its active ingredient. In the case of pharmaceutical aids it may indicate the more common usage of the article. The statement is not intended to limit in any way the choice or use of the article nor to indicate that it has no other activity or use. Dose. Doses mentioned in the Pharmacopoeia are intended merely for general guidance and represent, unless otherwise stated, the average range of quantities which are generally regarded as suitable for adults when administered by mouth. They are not to be regarded as binding upon the prescribers. The medical practitioner will exercise his own judgment and act on his own responsibility in respect of the amount of any therapeutic agent he may prescribe or administer or the frequency of its administration. If it is usual to administer a drug by a method other than by mouth, the single dose suitable for that method of administration is mentioned. In the case of some preparations notes have been given below the statement of doses to show the approximate quantities of active ingredients contained in the maximal doses as information for the prescriber. Usual Strength. The statement on the usual strength(s) of a preparation given in the individual monograph indicates the strength(s) usually marketed for information of the pharmacist and the medical practitioner. It does not imply that a strength other than the one(s) mentioned in the individual monograph meeting all the prescribed requirements cannot be manufactured and marketed with the approval of the appropriate authority.
Description. The statements under the heading Description are not to be interpreted in a strict sense and are not to be regarded as official requirements.
Solubility. Statements on solubility are given in Chapter 2.4.26 and are intended as information on the approximate solubility at a temperature between 15º and 30º, unless otherwise stated, and are not to be considered as official requirements. However, a test
47

for solubility stated in a monograph constitutes part of the standards for the substance that is the subject of that monograph.
Test Methods
References to general methods of testing are indicated by test method numbers in brackets immediately after the heading of the test or at the end of the text.
Identification. The tests given under the heading Identification are not necessarily sufficient to establish absolute proof of identity. They provide a means of verifying that the identity of the material under examination is in accordance with the label on the container.
In certain monographs alternative series of identification tests are given; compliance with either one or the other set of tests is adequate to verify the identity of the article.
When tests for infrared absorption are applied to material extracted from formulated preparations, strict concordance with the specified reference spectrum may not always be possible, but nevertheless a close resemblance between the spectrum of the extracted material and the specified reference spectrum should be achieved.
Tests and Assays
The tests and assays are the official methods upon which the standards of the Pharmacopoeia depend. The requirements are not framed to take into account all possible impurities. It is not to be presumed, for example, that an impurity that is not detectable by means of the prescribed tests is tolerated. Material found to contain such an impurity is not of pharmacopoeial quality if the nature or amount of the impurity found is incompatible with good pharmaceutical practice.
Pharmacopoeial methods and limits should be used merely as compliance requirements and not as requirements to guarantee total quality assurance. Tests and assays are prescribed for the minimum sample available on which the attributes of the article should be measured. Assurance of quality must be ensured by the manufacturer by the use of statistically valid sampling and testing programmes.
Tests. Unless otherwise stated, the assays and tests are carried out at a temperature between 20º and 30º.
Where it is directed that an analytical operation is to be carried out ‘in subdued light’, precautions should be taken to avoid exposure to direct sunlight or other strong light. Where a procedure is directed to be performed ‘protected from light’ precautions should be taken to exclude actinic light by the use of low-actinic glassware, working in a dark room or similar procedures.
For preparations other than those of fixed strength, the quantity to be taken for a test or an assay is usually expressed in terms of the active ingredient. This means that the quantity
48

of the active ingredient expected to be present and the quantity of the preparation to be taken are calculated from the strength stated on the label.
Other Tests. In the monographs on dosage forms and certain preparations, under the sub-heading ‘Other tests’ it is stated that the article complies with the tests stated under the general monograph of the relevant dosage form or preparation. Details of such tests are provided in the general monographs.
Limits. The limits given are based on data obtained in normal analytical practice. They take into account normal analytical errors, of acceptable variations in manufacture and of deterioration to an extent that is acceptable. No further tolerances are to be applied to the limits for determining whether or not the article under examination complies with the requirements of the monograph.
Quantities. Unless otherwise stated, the quantities to be taken for assays, limit tests and other tests are of the substance under examination.
In tests with numerical limits and assays, the quantity stated to be taken for testing is approximate. The amount actually used, which may deviate by not more than 10 per cent from that stated, is accurately weighed or measured and the result of analysis is calculated from this exact quantity. In tests where the limit is not numerical but usually depends upon comparison with the behaviour of a reference in the same conditions, the stated quantity is taken for testing. Reagents are used in the prescribed amounts.
Quantities are weighed or measured with an accuracy commensurate with the indicated degree of precision. For weighings, the precision is plus or minus 5 units after the last figure stated. For example, 0.25 g is to be interpreted as 0.245 g to 0.255 g. For the measurement of volumes, if the figure after the decimal point is a zero or ends in a zero, e.g. 10.0 ml or 0.50 ml, the volume is measured using a pipette, a volumetric flask or a burette, as appropriate; in other cases, a graduated measuring cylinder or a graduated pipette may be used. Volumes stated in microlitres are measured using a micropipette or microsyringe.
The term ‘transfer’ is used generally to indicate a quantitative operation.
Apparatus. Measuring and weighing devices and other apparatus are described in the chapter entitled ‘Apparatus for Tests and Assays’. A specification for a definite size or type of container or apparatus in a test or assay is given merely as a recommendation.
Unless otherwise stated, comparative tests are carried out using identical tubes of colourless, transparent, neutral glass with a flat base, commonly known as Nessler cylinders.
Reagents and Solutions. The reagents required for the tests and assays of the Pharmacopoeia are defined in the various chapters showing their nature, degree of purity and the strengths of the solutions to be made from them. The requirements set out are not
49

intended to imply that the materials are suitable for use in medicine; reagents not covered by monographs in the pharmacopoeia shall not be claimed to be of IP quality.
The term ‘analytical reagent grade of commerce’ implies that the chemical is of a high degree of purity wherein the limits of various impurities are known. Where it is directed to use a ‘general laboratory reagent grade of commerce’ it is intended that a chemically pure grade material, not necessarily required to be tested for limiting or absence of certain impurities, is to be used.
Indicators. Where the use of an indicator solution is mentioned in an assay or test, approximately 0.1 ml of the solution shall be added, unless otherwise directed.
Reference Substances. Certain monographs require the use of a chemical reference substance or a biological reference preparation or a reference spectrum These are authentic specimens chosen and verified on the basis of their suitability for intended use as prescribed in the Pharmacopoeia and are not necessarily suitable in other circumstances.
IP Reference Substances, abbreviated to IPRS (and referred to as RS in the individual monographs) are issued by the Indian Pharmacopoeia Commission (IPC). They are the official standards to be used in cases of arbitration. Secondary Standards (Working Standards) may be used for routine analysis, provided they are standardized at regular intervals against the Reference Substances
Biological Reference Substances, also abbreviated to IPRS and Standard Preparations of antibiotics are issued by agencies authorised by the IPC. They are standardized against the International Standards and Reference Preparations established by the World Health Organization (WHO). The potency of these preparations is expressed in International Units.
Reference spectra are published by the IPC and they are accompanied by information concerning the conditions used for sample preparation and recording of the spectra.
Test Animals. Unless otherwise directed, animals used in a test or an assay shall be healthy and are drawn from a uniform stock, and have not previously been treated with any material that will interfere with the test or the assay.
Calculation of Results. In determining compliance with a numerical limit in assay or test, the result should be calculated to one decimal place more than the significant figures stated and then rounded up or down as follows: if the last figure calculated is 5 to 9, the preceding figure is increased by 1; if it is 4 or less, the preceding figure is left unchanged.
Storage. Statements under the side-heading Storage constitute non-mandatory advice. The articles of the Pharmacopoeia are to be stored under conditions that prevent contamination and, as far as possible, deterioration. Precautions that should be taken in relation to the effects of the atmosphere, moisture, heat and light are indicated, where appropriate, in the individual monograph.
50

Specific directions are given in some monographs with respect to the temperatures at which Pharmacopoeial articles should be stored, where it is considered that usage at a lower or higher temperature may produce undesirable results. The storage conditions are defined by the following terms:
— Store in a dry, well-ventilated place at a temperature not exceeding 30º
— Store in a refrigerator (2º to 8º). Do not freeze
— Store in a freezer (-2º to -18º)
— Store in a deep freezer (Below -18º)
Storage conditions not related to temperature are indicated in the following terms:
— Store protected from light
— Store protected from light and moisture
Where no specific storage directions or limitations are given in the monograph or by the manufacturer, it is to be understood that the storage conditions include protection from moisture, freezing and excessive heat (any temperature above 40º). Storage Containers. The requirements, guidance and information on containers for pharmaceutical use are given in the chapter entitled Containers (6.1) In general, an article should be packed in a well-closed container i.e. one that protects the contents from contamination by extraneous solids, liquids or vapours and from loss of the article under normal conditions of handling and storage. Where, additionally, loss or deterioration of the article from effervescence, deliquescence or evaporation under normal conditions of storage is likely, the container must be capable of being tightly closed, and re-closed after use. In certain cases, special requirements of pack have been indicated in some monographs under Storage, using expressions that have been defined in chapter 6.1. Labelling. The labelling of drugs and pharmaceuticals is governed by the Drugs and Cosmetics Rules, 1945. The statements that are given in the monographs under the side-heading ‘Labelling’ are not comprehensive. Only those that are necessary to demonstrate compliance or otherwise with the monograph have been given and they are mandatory. For example, in the monograph on Betamethasone Sodium Tablets the labelling statement is “The label states the strength in terms of the equivalent amount of betamethasone”. Any other statements are included as recommendations.
51

Annexure II A SOPS FOR LABORATORIES
The SOPs to be prepared by adopted by laboratories as specified in Schedule L1 of the Drugs & Cosmetics Rule, 1945 are listed below:
1. Standard Operating Procedure for preventive maintenance of machine or equipment or apparatus shall be prepared by the laboratory (Sch L1 – 4(g)
2. There shall be written instructions in the form of Standard Operating Procedures for the operation, maintenance and calibration of instruments (Sch L1 – 4(d)
3. Maintenance procedure in the form of Standard Operating Procedures must be prepared (Sch L1 – 4(i)
4. Standard Operating Procedure for preparation and standardisation on stock solutions, standard solutions, volumetric solutions must be prepared for the guidance of staff Sch L1 5(c).
5. Standard Operating Procedure for safety, house-keeping and loss prevention shall be prepared in accordance with the various rules, and regulations of the Government of India (Sch L1-6(b))
6. For most of the equipments and instruments, Standard Operating Procedures for calibration and calibration schedule be prepared (Sch L1- 7(c))
7. Standard Operating Procedure for maintenance of microbial culture and sub-culture must be prepared by the laboratories. Sch L1- 9(a)
8. Internal audits are done to assure the integrity of the analysis and such audits shall be conducted periodically with a predetermined schedule and procedure with appropriate checklist Sch L1- 11(a)
9. Schedule L1 -13. Standard Operating Procedures:- (a) Standard Operating Procedures are written procedures for different activities being conducted in a laboratory and shall include the following characteristics: (i) they shall be written in a chronological order listing different steps leading to an analysis of drugs or calibration of an instrument: (ii) testing laboratories shall have Standard Operative Procedure manuals and have its periodic review; (iii) it shall be user friendly documents and shall include designation of the person responsible for intended activity. (b) Standard Operating Procedures in addition to those recommended under various activities shall also be prepared to the minimum in respect of the following: (i) sample handling and accountability; (ii) receipt identification, storage, mixing and method sampling of the test and control articles;
52

(xxvi) record keeping, reporting, storage and retrieval of data; (xxvi) coding of different studies, handling of data including use of computerized
data system; (xxvi) operation of technical audit personnel in performing and reporting audits,
inspections and final report reviews; (vi) routine inspection of cleaning, maintenance, testing, calibration and standardisation of instruments; (xxvi) action to be taken in respect of equipment failure; (viii) analytical data methods; (ix) the raw data; (x) data handling and storage retrieval; (xi) health and safety protection; (xii) animal room preparations; (xiii) animal care; (xiv) storage and maintenance of microbial cultures; (xv) maintenance of sterility room (i.e. constant maintenance and monitoring of Aseptic condition of sterility room); (xvi) use and storage of reference standards (xvii) procurement of stores and equipment; (xviii) monitoring of testing of samples; (xix) method of retention of unexpended samples, their location, maintenance and disposal; (xx) document control; (xxi) redressal of technical complaints; (xxii) housing-keeping; (xxiii) corrective and preventing action; (xxiv) working procedure (test methods); (xxv) calibration Manual; and (xxvi) training manual.
The format that can be adopted for Sops and the SOP for preparing SOPs as adopted by the IP Laboratory (Specimen only for guidance – Not to be copied or reproduced) are furnished below (Annexure IIB):
53

Annexure IIB
INDIAN PHARMACOPOEIA LABORATORY Page No. 54 of 84
STANDARD OPERATING PROCEDURE SOP No. IPC/QSP/001
Section Quality Assurance Revision No. 00
Effective date Immediate after training Review date two years w.e.d.
Title: Preparation and Control of Standard Operating Procedure 1.0 OBJECTIVE To lay down the procedure for Preparation, Review, Approval, Distribution,
Withdrawal, Archiving and Disposition of Standard Operating Procedure.
2.0 SCOPE This Standard Operating procedure (SOP) shall be applicable to all SOPs of Indian
Pharmacopoeia Commission.
3.0 RESPONSIBILITY 3.1 All the Officers and section Incharge of IPC shall ensure that this parent SOP has
been reflected in the section / departmental SOPs. 3.2 Quality Manager / Quality Assurance person designated by CEO / Director shall
ensure overall compliance. 4.0 PROCEDURE 4.1 Construction of SOP (Refer Annexure I) SOP is constructed mainly in three parts, viz. header, front page and body. The
contents of each parts of SOP shall be as follows:
Name Designation Signature Date
Prepared by Dept. Personnel
Reviewed by HOD/ QM/TM
Approved by Director 4.1.1 Header
54

The header shall provide the following details: - A. Organization logo The logo of the institute shall be written. B. Name of the laboratory Name of the Indian Pharmacopoeia Laboratory. C. Section Standard Operating Procedure shall be written in this cell. D. Page No. Page No. indicates serial no. of pages in “X of Y” pattern. Formats attached as
annexure shall not be given page number in continuation of SOP pages. However, any annexure of more than one page shall be given the page number in the similar pattern of “X of Y” (Independent for each format).
E. SOP No. SOP No shall be an alphanumeric unique number, consisting of maximum 15
characters. F. Section Particular section of the department or operation (if any) where the SOP shall be
applicable. If any specific section does not exist in any department then word “General” shall be written under this column.
G. Revision No.
Revision no shall be numeric, consisting of maximum two digits.
H. Effective Date This date shall specify the date on which the SOP is effective from, the difference
between approval date and effective date shall not be more than 30 days.
I. Review date This column shall specify the month, in which the individual SOP is to be
reviewed. Review date should be two years from the month of the effective date. However any SOP can be reviewed earlier than this date as per the requirement.
J. Title:
55

Indicates the title of the SOP, mentioning heading of the document. 4.1.2 Footer on Each Page (K) Footer on each page of the SOP shall have Prepared by, Reviewed by and
Approved by following with Name, Designation, Signature and Date apart from the header details.
4.1.3 Body The SOP shall be prepared as per the below given sequence under the following
headings. Objective Shall write in a nutshell the purpose and use of the SOP, starting with “to lay down
a procedure or prescribed for ……” Scope Shall specify the boundary limits for the application of the SOP. Responsibility Shall specify the personnel responsible for the development, implementation and
monitoring of the SOPs. Procedure Procedure shall provide the step wise actions to be taken for implementation of the
SOP. It shall also provide the applicable references to other quality documents as and when required.
Safety & precautions (If any) Shall mention on the salient feature of operational and behavioral precautions and
safety measures. Revision History Shall trace the chronological changes, reasons thereof at least last three revisions
from the current version in short form or in single line. Revision history shall contain the change control No.
Reference Mention the document(s), if reference has been made into the preparation of the
particular SOP.
56

Abbreviation Full form of an abbreviation shall appear in the text at the place where it appeared
for the first time before start of use of an abbreviation. However, a list of full form of all the abbreviations used in the document shall also to be detailed under this column.
Annexure The list of annexure related to the document shall be mentioned under this heading
along with individual specific format number of listed annexure but without mention of revision number e.g. annexure - III of this document shall be written as “Record of Distribution, Withdrawal and Disposition of Document XYZ / 03” where X is the code of department and YZ is the number of this document and “03” is for annexure -III.
Distribution The list of departments to which the SOP & documents shall be distributed as a
controlled copy shall be provided in a tabular form in annexure III, XYZ / 04. 4.2 Formatting of the SOP 4.2.1 The SOP shall be prepared in accordance with the guideline given in this SOP.
4.2.2 The title and text shall be clearly understood and written in clear, plain English so that everyone who carries out a particular activity /operation /function shall perform in the same way and safely.
4.2.3 The language used shall be in directive form that gives step-wise instructions, the words like “shall”, “must” are to be used rather than “should”, “may” or “might” to the possible extent.
4.2.4 The page setting and text shall conform to the following specifications.
Paper Size A4 Size Language English Font Type/Size (Header) Times New Roman, Bold/ 12 Point Font Type/Size (Text) Times New Roman, plain / 12 Point Font size (Sections) Times New Roman, bold / 12 Point Printing One side Space Between Paragraph Single
57

4.2.5 Text shall be divided into logical sections numbered 1.0, 2.0, and 3.0 etc. whereas 1.0 relates to Purpose; 2.0 relate to Scope; 3.0 relates to Responsibility etc. Type the section head in Uppercase (Capital) letters.
4.2.6 These sections shall be sub-divided into logical sections. 1.1; 1.2; 2.1; 2.2; 2.3 etc. Where required, these sub-divisions may further be sub-divided, as 1.1.1; 1.1.2; 1.1.3 etc. Type the subsections in sentence case. In case, in any sub section does not have any guideline or in complete sentence is intended to be written, bullets can be used instead of use of numeric system.
4.2.7 Each SOP shall be allocated a unique SOP No. by the concerned department and same shall be verified by QA personnel. The number shall be alphanumeric with maximum 15 characters. The SOP Number shall be written in the following sequence:
O/XXX/YYY/ZZ
Where,
“IPC” : The First characters in alphabetic digits shall be used for mention of organization (location) code e.g. IPC/XXXX/001/00
“/” : The second character shall be used as a “Slash”
“XXXX” : The third characters in alphabetic digits shall be used for mention of SOP’s category code e.g. (QSP, INST, and GEN), (IPC/QSP/001/00, IPC/INST/001/00, IPC/GEN/001/00).
“/” : The fifth character shall be used as a “Slash”
“YYY” : The next three characters in numeric digits shall be used for allotting the serial number to SOP in ascending order starting from 001.
“/” : The next character shall be used as a “slash”
“ZZ” : The last two characters in two numeric digits shall be used for mention of version/revision number of SOP starting from 00.
E.g. the document number of First version of first SOP shall be allotted by QA manager.
4.2.8 All annexure should be numbered as Annexure I, II, and III as follows. All annexures/formats be a part of respective SOP and shall be attached to the SOP. Each annexure/ format of the SOP shall be numbered as follows:
O/XX/NNN/YY/nn
Where,
O is Organization / location code
58

XXXX is SOP category code,
YYY is serial no of the SOP,
ZZ is the version no.
Eg. IPC/QSP/001/02 is the second revision of the first format of this SOP.
4.2.9 The Name, Designation, Signature and Date shall be entered in blue pen only. Effective date and Review due on must be handwritten. Date shall be written as alphanumeric, starting with the day as two numeric characters, followed by the month in alphabetic characters and the year in two numeric characters showing the last two numbers of the year.
4.2.10 The gap between date mentioned in the control copy stamp and effective date shall be enough, to take care of time taken in distribution of SOP to other departments. SOP shall be made effective only after training
4.3 Writing of SOP
4.3.1 Person designated by the Director / section Incharge of concerned department shall write SOP.
4.3.2 After completion of draft of SOP, the concerned person shall circulate it to the persons/departments being involved in the operation of the SOP, for inviting their opinion /comments.
4.3.3 Person responsible for preparation of SOP in consultation of section Incharge shall incorporate the relevant portion of received comments / opinion in the draft SOP.
4.4 Review of the SOP
4.4.1 Section Incharge of the concerned department shall designate the person for review of the draft SOP.
4.4.2 In case, other department(s) is getting affected by the procedure of SOP under question, draft SOP shall also be sent to the related department(s) for review purpose.
4.4.3 Reviewer’s further comments, if any, shall be incorporated in the final version of SOP in consultation with section Incharge of concerned department(s).
4.4.4 The reviewer(s) shall sign the final version in the provided columns. Number of reviewers may be more than two, depending upon the Cross functional activities.
59

4.4.5 Person responsible for preparation of SOP shall arrange to send the final version of SOP duly signed by him/her and by the reviewer(s), to the QA or to the person designated by Quality Manager / Director for final approval.
4.5 Approval of the SOP 4.5.1 Director / Quality Manager or to the person designated by Director shall approve
the SOP. 4.5.2 Quality Manager or the person designated by Director shall arrange the mention
of effective date and review due on in respective columns and affixing “MASTER COPY” (Black in color) on the bottom of the right corner of each page in the original signed copy.
4.5.3 QA, as a custodian shall retain this “MASTER COPY”. 4.5.4 Functional department shall organize for training of the concerned personnel
responsible before executing the SOP.
4.5.5 Distribution of SOP 4.6.1 Copies of the SOP shall be distributed to all the concerned sectional /
departmental Incharge. QA personnel shall take photocopies from Master Copy for the desired number of copies.
4.6.2 QA personnel shall stamp the Photocopy of SOP as “CONTROLLED COPY” (Blue in colour) on the bottom near to Master copy of each page the “Copy No.” shall given by QA personnel as per codification of departments.
4.6.3 Incase, SOP is required to be submitted to an external party, QA personnel responsible of maintaining the master copy, shall take photocopy of master copy and shall stamp “UNCONTROLLED COPY” [Red in color.] on the bottom near to Master of each page.
4.6.4 The impression of all the stamps is attached as Annexure VI. 4.7 Revision of SOP 4.7.1 All SOPs shall be reviewed on or before date mentioned as review due on.
4.7.2 Any revision in the SOP or annexure(s) shall be through Change control system. Refer SOP on change control.
4.7.3 In case of any revision in the existing SOP, same procedure shall be adopted for preparation of revised SOP as mentioned in the procedure of preparation of SOP starting from clause 4.3.
4.7.4 In case ,during or before periodic review of SOP , any change / revision in the SOP or in Annexure(s) or in both is required ,then SOP or Annexure(s) or both as per the case shall be reprinted after incorporating suggested and approved changes as per change control system.
60

4.7.5 In the case of revision in the main SOP {with or without annexure(s)}, a next revision number shall be given to the SOP and the summary of revisions made shall be documented under the column of revision history.
4.7.6 In case of revision only in the annexure(s) no next revision number to the SOP shall be given. In this case next revision number shall be given to a particular revised annexure(s). The revision history and effective date of annexure(s) shall be maintained in the Master list of Records .In this case only revised annexure(s) shall be printed for master copy and distribution.
4.7.7 Concerned department shall arrange to send the print of revised annexure(s) to QM or person designated by Director for further action of attachment with master copy and for distribution.
4.7.8 QM or person designated by Director shall affix the stamp of master copy on the original copy of revised annexure(s) and shall attach with the already issued master copy of SOP.
4.7.9 QM or person designated by Director shall take photocopy (ies) of issued annexure(s) and distribute to the concerned department(s) and retrieving the old version.
4.7.10 In case, during periodic review of SOP and Any master documents, no revision is required in the existing version of SOP, annexure(s) and any master documents, the same form of SOP along with annexure(s) and any master documents shall be extended for use by any department with the approval of QM or the person designated by Director.
4.7.11 In case of 4.7.10, concerned department shall get the Review Report and Extension Authorization (Annexure-IV, ) issued from QA, QM or the person designated by Director for allowing its use before or up to next review due on.
4.7.12 QM or the person designated by Director shall stamp master Copy on the original copy of Review Report and Extension Authorization (Annexure-IV, ) and attached with the master document and shall issue the controlled copy of this form to the concerned department(s) to which earlier copy of SOP was distributed.
4.7.13 Personal responsible for maintaining the SOPs in the concerned department(s), shall ensure that received copy of Review Report and Extension Authorization (Annexure-V is attached with the controlled copy of said SOP, already available in the department.
61

4.8 Withdrawal & archiving of SOP
4.8.1 Withdrawal & archiving of SOP and or related annexure(s) shall be done by QA department. The controlled copy of the previous version of the SOP and or related annexure(s) shall be withdrawn by QA Department before effective date of issues document. All the retrieved Controlled Copies shall be destroyed by QA or the person designated by Director. The activity is recorded on annexure II.
4.8.2 The master copy of archived SOP and or related annexure(s) shall be stamped ‘‘OBSOLETE’’ (Green in color), on the middle of each page of the SOP. Obsolete copy of master SOP shall be preserved for any future reference by QM or person designated by Director. The record of Obsolete SOP shall be maintained as per Annexure - III.
5.0 SAFETY AND PRECAUTION
Do not use any SOP if it is not signed and issued by QA Personnel’s or the authorized signatories. Do not use pencil for recording on SOP. Do not use adhesive tape or whitener on SOP.
6.0 REVISION HISTORY
7.0 REFERENCES In-House 8.0 ABBREVIATIONS
SOP : Standard Operating Procedure QM : Quality Manager QA : Quality Assurance Dep. : Department
9.0 ANNEXURE(s) Annexure I : Format for SOP preparation QA/001/01 Annexure II : Record of Distribution, Withdrawal and Disposition of
Document(s) QA/001/02 Annexure III : Record of Obsolete Document(s) QA/001/03
62

Annexure IV : Review Report and Extension Authorization QA/001/04 Annexure V : Impression of Stamps QA/001/05/ Annexure VI : Organization / Departmental Code QA/001/06
Annexure III
ACCREDITED CALIBRATION LABORATORIES/AGENCIES IN INDIA.
North Zone 1) National Physical Laboratory
Dr. K.S. Krishnan Marg New Delhi - 110012 India Phone: 91-11-45609212, 91-11-25742610, 91-11-25742611, 91-11-25742612 Fax: 91-11-45609310, 91-11-25726938 Email: [email protected] or [email protected]
Website-http://www.nplindia.org/ 2) Calibration Laboratory, Glassco Laboratory Equipments, Ambala
Address: Manglai, P.O. Khudda Kalan, Ambala Cantt. Ambala, Punjab, India. Pin - 133 004 Tel No. 0171-6451250/ 98964 23304 Email: [email protected]
3) Calibration Laboratory, Jain Scientific Glass Works, Ambala Cantt Address: 14, HSIDC Industrial Estate Ambala Cantt. Ambala, Haryana, India. Pin - 133006 Tel No. 0171 - 2698184, 2698185 Fax No. 0171 - 2698390 Email: [email protected]
4) DVG Laboratories & Consultants Pvt. Ltd., Wazirabad, Gurgaon book
Address: Hospital Road, Wazirabad, Near Sector 56, Gurgaon, Haryana, India. Pin – 122011 Tel No. 0124-4112677 Fax No. 0124-4112677 Email [email protected]
5) Electrometer Corporation, Delhi
Address: 34 Main, Patel Nagar Road , New Delhi, India. Pin – 110008
63

Tel No. 011 – 25892548 Fax No. 011 – 25892548 Email [email protected]
6) Indian Calibration Services, Delhi
Address: 313, ‘Laxmi Deep’, 9, District Centre, Laxmi Nagar, Delhi, India. Pin – 110092 Tel No. 011-22453259, 22434362 Fax No. 011-22453259, 22434362
7) Jyoti Test House, Faridabad Address: B-1/111, Sector-11,Faridabad, Haryana, India. Pin – 121006 Tel No. 0129-5008795 Fax No. 0129-5008795
8) National Research & Technology Consortium (C-ACT), Parwanoo (H.P.) Address: HPCED Building, Deptt. of Industries Complex, Sector-1, Parwanoo, HP, India. Pin - 173220 Tel No. 01792-233675 Fax No. 01792-234107 Email [email protected]
9) S.V. Engineeing Centre, Faridabad Address: Plot No. E- 3, Friends Industrial complex Street Sector 23, Faridabad, Haryana, India. Pin - 121005 Tel No. 0129-4156748, 9811177048 Fax No. 0129-4156748 Email [email protected]
10) SARRC Test House, Faridabad
Address: Plot No. 100, Sector 29, Faridabad, Haryana, India. Pin - 121008
Tel No. 0129 - 2500404,2501021 Fax No. 0129 - 25278136 Email [email protected] 11) Shriram Institute for Industrial Research, Delhi
Address: 19, University Road, P. B. No. 2122, New Delhi, Delhi, India. Pin - 110007 Tel No. 011-27667267 / 27667860 Email [email protected]
12) Spectro Analytical Labs Ltd., Delhi
64

Address: E-41, Okhla Industrial Area, Phase – II, New Delhi, India. Pin - 110020 Tel No. 41611000, 40522000, 9873001515
South Zone 1) Fluid Control Research Institute, Palakkad, Kerala
Address: Kanjikode West, Palghat, Kerala, India. Pin - 678623 Tel No. 0491 - 2566206,25661202566119 Fax No. 0491 - 2566326 Email [email protected]
2) Northlab (India) Pvt. Ltd., Chennai
Address: Flat No. 25, II Floor, LUZ Ginza Complex 140 Royapettah High Road Mylapore, Chennai, Tamil Nadu, India. Pin - 600004 Tel No. 044 - 24987763/24981539,9840977514 Fax No. 044 - 27109824 Email [email protected]
3) Perfect Calibration Centre Pvt. Ltd., Chennai
Address: No. 84, Ist Floor, "Times Corner" Gengu Reddy Road, Egmore, Chennai, India. Pin - 600 008 Tel No. 044-28193130,/2530/2930, 09381010843, 09972697755 Fax No. 044-42142898 Email [email protected]
4) Shriram Institute for Industrial Research, Bangalore
Address: 14-15, Sadarmangala Industrial Area, Whitefield Road, Bangalore, Andhra Pradesh, India. Pin - 560048 Tel No. 080-28410165/28410172/ 9845875539 Fax No. 080-28410189
5) Sree Chitra Tirunal Institute for Medical Sciences & Technology, Biomedical
Technology Wing, Trivand Address: Sree Chitra Titunal Institute for Medical Sciences And Technology Bio Medical Technology Wing Poojappura, Thiruvananthapuram, Kerala, India. Pin - 695012 Tel No. 0471-2520217 / 2340801 Fax No. 0471 2341814 Email [email protected]
65

6) Transcaal Engineers India Pvt. Limited, Bangalore
Address: #18, Miltan Corner, 2nd Main, 2nd cross, SSI Area, 5th Block, Rajajinagar, Bangalore, Karnataka, India. Pin - 560 010 Tel No. 080-23350716 Fax No. 022-23355238 Email [email protected]
7) Godrej & Boyce Mfg. Co. Ltd., Lawkim Motors Group, Chennai Address: No.1, SIDCO Industrial Estate, Ambattur, Chennai, Madras, India. Pin – 600098 Tel No. 044-66544651-53 Fax No. 044-66544651 Email [email protected]
8) Macro Calibration Services, Chennai
Address: 4/7, Ist Floor, Hasthinapuram main road nehru nagar, Chromepet, Chennai, India. Pin - 600 044 Tel No. 044-65362575, 64550704
East Zone 1) Eastern Calibrators, Kolkata
Address: 1, Anil Roy Road, Kolkata, West Bengal, India. Pin – 700029 Tel No. 033 - 24647740 Fax No. 033 - 24655927 Email [email protected]
2) Electrometer Corporation, Kolkata
Address: P- 5, CIT Road , Scheme LV 9th Floor, Moulali, Kolkata, West Bengal, India. Pin - 700014 Tel No. 033 - 22469024/22485516/22164401 Fax No. 033 - 22469824 Email [email protected]
3) Electronics Regional Test Laboratory (E), Kolkata Address: STQC Directorate, DN Block Sector V, Salt Lake City, Kolkata, West Bengal, India. Pin – 700091 Tel No. 033 - 23678974/23672366/23677543 Fax No. 033 - 23679472
66

4) Measure Techno Lab, Kolkata
Address: 32A, Ganesh Chandra Avenue, 8th Floor, Kolkata, West Bengal, India. Pin - 700013 Tel No. 033-32943376, 22159687, 09831190974 Email [email protected]
5) National Test House, Salt Lake, Kolkata
Address: Block-CP, Sector-V, Salt Lake, Kolkata, West Bengal, India. Pin - 700091 Tel No. 033-23679741/3871/3308 Fax No. 033-23673871/3308 Email [email protected]
6) Young Engg. & Calibration Services Pvt. Ltd., Howrah Address: ‘Utsav Bhawan’, Kamardanga Road, Ichapur, Howrah, West Bengal, India. Pin – 7111049 Tel No. 033-26771411/26771412 Fax No. 033-26771411 Email [email protected]
West Zone 1) Calitron Calibration Laboratory, Pune
Address: 207, Kohinoor Arcade, Tilak Chowk Nigdi, Pune, Maharashtra, India. Pin – 411044 Tel No. 020-27650791, 9822455541 Fax No. 020-27650791 Email [email protected]
2) Godrej & Boyce Mfg. Co. Limited, Lawkim Motor Group, Mumbai
Address: B-302/303 Pratik Industrial Estate Mulund Goregaon Limk Rd, Next to Wockhardt Hospital, Bhandup (W), Mumbai, Maharashtra, India. Pin - 400078 Tel No. 022 -25934575 / 6/9821698504 Fax No. 022 -25664575/76/75 Email [email protected]
3) Institute for Design of Electrical Measuring Instruments, Mumbai Address: Swatantryaveer Tatya Tope Marg, Chunabhatti,Sion P.O., Mumbai, Maharashtra, India. Pin – 400022 Tel No. 022 - 25220302/25220303/25220304 Fax No. 022 - 25229016 Email [email protected]
67

4) Jain R & D, Jain Irrigation Systems Limited, Jalgaon, Maharashtra
Address: Agripark, Jain Hills, Shirsoli Road, Jalgaon,, Maharashtra, India. Pin – 425001 Tel No. 0257-2260011/22/33 Fax No. 0257-2264920
5) National Centre for Quality Calibration, Ahmedabad
Address: 903-904, Samudra Annexe, Near Hotel Klassic Gold, Sardar Patel Nagar, Off.C. G. Raod, Ahmedabad, Gujarat, India. Pin – 380006 Tel No. 079 - 2656 5405,2656 1104+,98250 31523 Fax No. 079 - 26561104 Email [email protected]
6) Reliable Analytical Laboratories Pvt. Ltd., Thane Address: Indian Corporation Complex, Building No. 125&139, opp. Gajanan Petrol Pump, Gundavi, Mankoli Naka, Bhiwandi Thane , Thane, Maharashtra, India. Pin – 421302 Tel No. 02522-398100 Fax No. 02522-398100 Email [email protected]
• Reference NABL Site. • Note – Present status of above laboratory/agencies can be verified from the
NABL office /website.
68

Annexure IV
LIST OF EQUIPMENTS REQUIRED FOR A DRUGS TESTING LABORATORY
The equipments listed here are the minimum requirements for performing the tests specified under the various monographs and for various dosage forms. The actual requirements of instruments and their specifications will vary depending upon the activities to be carried out in a laboratory. The analyst performing the tests shall suggest instrument specifications as per requirements. The design of the laboratory layout and space should be in accordance with the principles of GLP. Adequate area shall be provided for sensitive and sophisticated instruments employed for analysis. Equipments shall be installed at locations designed, constructed, adapted and maintained to suit the operation to be carried out. The instruments shall be installed in dust-free and safe environment and conditions of temperature and humidity shall be maintained and periodic check on temperature and humidity shall be made and recorded. Instruments requiring the calibration shall be calibrated at regular intervals. There shall be written instructions in the form of SOP for operation, maintenance and calibration of instruments. Most instruments need vibration proof flat level and hard surface for installation. Uninterrupted power supply is also needed to perform the operations properly. The Equipment Manual provided by the manufacturer shall be referred to for the various qualification criteria, operative and maintenance procedures to generate good quality data. Schedule L1 of the Drugs & Cosmetics Act 1940 and Rules thereunder applies to the statutory requirements in the matter of use of instruments.
List of instruments required for analytical laboratory
1. Electronic Balance (sensitivity 0.0001g)
2. Electronic Balance (sensitivity 0.1g)
3. Double beam UV Visible Spectrophotometer
4. ELISA Reader 5. IR Spectrophotometer 6. HPLC system
69

7. TLC System 36. Water Bath Serological 8. Centrifuge 9. Muffle Furnace 10. Water Bath 11. Distillation Apparatus 12. Vacuum Distillation 37. Water Bath
38. Dry bath 13. Disintegration Machine 39. Autoclave 14. Dissolution Apparatus 40. Electric Hot Plate 15. Gas Chromatograph 41. Microwave oven 16. UV Lamp 42. Vacuum Pump 17. Flame Photometer 43. Filtration Units for Sterility Test 18. Melting Range Apparatus 44. Incubator 19. Desiccators 45. Air Handling Unit/Air
conditioner 20. pH Meter 21. Fluorimeter
46. Distillation Set for Glass Distilled Water /Milli Q water system
22. Potentiometric Titrator 23. Refractrometer 24. Refrigerator
47. Vortex Mixture 25. Sonicator 48. Micro Pipettes 26. Hot air Oven 49. Media (As per pharmacopoeial
requirement) 27. Vacuum Oven 28. Karl Fisher Instrument
50. Microbial strains 29. Laminar Flow Bench 51. Dehumidifier 30. Bunsen Burner 52. Kjeldhal Apparatus 31. Gas Cylinder/Gas Supply
32. Microscope 33. Air Particle Counter
34. Zone Reader 35. Colony counter
Author: Mr. Alok Sharma,
Scientific Officer, Indian Pharmacopeia Commission, Ghaziabad
70

Annexure V
LIST OF EXPERTS AND PARTICIPANTS OF IPC WORKSHOPS Workshop: Challenges and opportunities in compliance of current standards prescribed in IP 2010 held at IPC, Ghaziabad on 8th and 9th of Dec 2010 S. No. Name of Expert Affiliation
1. Dr Surinder Singh Drugs Controller General (India) 2. Dr. Y. K Gupta Head, Department of Pharmacology,AIIMS 3. Dr. Madhur Gupta National Project Officer, WHO -Country Office, New Delhi 4. Dr. Anurag Rathore Professor, Dept of Chemical Engineering, IIT.,New Delhi, 5. Dr. Murali,
Senior Manager- Quality Control, Natural Remedies, Bangalore
5. Dr. D Roy DyDC(I), CDSCO North Zone 6. Dr. R. A Singh Director, RDTL,Sector-39 C, Chandigarh- 160036 7. Dr. Jai Prakash Principal scientific Officer, IPC, Ghaziabad 8. Dr. Mayank Rawat
Division of Biological Standardization,Indian Veterinary Research Institute (IVRI), Izatnagar (U.P)
9. Dr.Prasad V.Kanitkar Director, Plant operations, Pfizer limited, Navi Mumbai 10. Dr.Prashant Dixit Watson Pharma Pvt. Ltd. Dist. Thane, Pin-421506 11. Mr. Vinod Kalani Managing Director, CRIS Pharma (India) Ltd., Rajasthan 12. Dr. Raman M. Singh Principal Scientific Officer, IPC, Ghaziabad 13. Dr. (Mrs) Gopa Ghosh Director In-charge, CDTL, Mumbai,Mumbai 400008 14. Dr. S K Gupta
Delhi Institute of Pharmaceutical Sciences and Research (DIPSAR), New Delhi
15. Dr.S S Agarwal DIPSAR, New Delhi 16. Dr.S S Yadav Ex-General Manager, IDPL 17. Dr.Jotna Sokhey Director , National Institute of Biologicals, Noida 18. Mrs Archana Mudgal Secretary-cum-Registrar, (PCI), New Delhi 19. Dr P K Gupta Ex-Drugs Controller, India 20. Dr N Chodankare Group CEO Pharma Business Excel Industries Ltd. Mumbai. 21. Dr G N Singh Secretary-cum-Scientific Director, IPC, Ghaziabad 22. Dr Manish Dare Principal Scientific Officer,IPC, Ghaziabad
71

S. No.
Name of Participant
Affiliation
1. Mr. Arvind Kukreti Assistant Drug Controller (India), CDSCO 2. Mr.A.P. Jain
Ex-Director Grade I,(Quality Assurance), Ministry of Defence
3. Smt.K.Bhubneswari Drug Inspector,CDSCO North Zone 4. Mr. G. S. Bedi
Manager & Head- Production/ Formulation development, IDPL, Rishikesh
5. Dr.Hemal Patel
Asst.General Manager (Regulatory affairs),Torrent Pharmaceutical Ltd., Gujarat
6. Mr. S. L. Jat Federation of Pharma Entrepreneurs(FOPE) 7. Dr. Kanchan Kohli
Reader, Deptt of Pharmaceutics, Jamia Hamdard University, Hamdard Nagar, New Delhi
8. Mr. P.Manavalan Drug Inspector, CDSCO North Zone
10. Dr. Meenkshi Bajpai
Principal, Raj Kumar Goel Institute of Technology, Ghaziabad-201003.
11. Dr. RK Pawar
Research Assistant, Pharmacopoeial Laboratory for Indian Medicine, Dept. of AYUSH, Ghaziabad-201002
12. Dr. Prakash Joshi,
Deputy Director, Homeopathic Pharmacopoeia Laboratory, Ghaziabad
13. Dr. Prem Kumar Gupta Ex- Drug Controller (India), New Delhi-110025 14. Ms.Poonam Raghav Tech. Data Associate CDSCO North Zone 15. Mr.Pradeep Kumar Tech. Data Associate, CDSCO North Zone 16. Dr. Renu Jain Scientist, National Institute of Biologicals, Noida-201307 17. Dr.Ravinder Singh
Asstt. Director (Chem), Pharmacopoeial Laboratory for Indian Medicine, Dept. of AYUSH, Ghaziabad-201002
18. Mr.S.L.Nasa Registrar, Delhi Pharmacy Council, New Delhi. 19. Ms.Stuti Bansal Tech. Data Associate, CDSCO North Zone 20. Mr.S.R. Kulkarni Baxter (India) Pvt. Ltd.,IMT – Manesar 21. Mr. Satish Gupta Drug Controller, J&K 22. Mr. Surinder Mohan Asstt. Drugs Controller, J&K 23. Mr. D. K. Shringi Drugs Controller, Rajasthan 24. Mr. Subhash Gupta Cris Pharma (India) Ltd,Dehradun 25. Mr. S S Venkatakrishnan Ex- Drugs Controller, Kerala, Thiruvananthapuram-695013 26. Mr. Vinod Arora Vice President, Ranbaxy Research Laboratories, Gurgaon 27. Mr. Vijay Kr Arora Managing Director, Arbro Pharmaceuticals, New Delhi 28. Mrs. Devki Pant Homoeopathic Pharmacopoeia Lab, Dept. of AYUSH,
72

Ghaziabad-201002 29. Mr. Satish Kumar
Homoeopathic Pharmacopoeia lab Dept. of AYUSH, Ghaziabad-201002
30. Mr. Prakash Gupta
Homoeopathic Pharmacopoeia lab, Dept. of AYUSH, Ghaziabad-201002
31. Mr. Abhishank Singh Research Scholar, Ranbaxy Laboratories Ltd., Gurgaon. 32. Dr. D. Linga Rao Executive Vice President, Corporate Quality Control &
Assurance, NATCO, Hyderabad 33. Dr. Brijesh Chandra
Gautam Ex-Managing Direcor, Rajasthan Drugs and Pharmaceuticals
34. Mr. Abhishek Chawla CDSCO (NZ), Ghaziabad 35. Dr. R.A. Singh Director, Regional Drugs Testing Laboratory, Chandigarh. 36. Dr. Nand Kumar
Chodankar Group CEO, Pharma Business Excel Industries Ltd. Mumbai.
37. Mr. Saurabh Arora Director, Arbro Pharmaceuticals Ltd., New Delhi 38. Dr. V.Kalaiselvan SSO, IPC 39. Dr. Robin kumar SSO, IPC 40. Dr. Anil Kumar Teotia SSO, IPC 41. Mr. Pawan Kr. Saini SA, IPC 42. Dr. Kanchan Kohli Reader, Jamia Hamdard 43. Dr. SC Mathur SO, IPC 44. Dr. D.K.Sharma SA, IPC 45. Dr. Nishant Dafale SSO, IPC 46. Mr. Anuj Prakash Yadav SSO, IPC 47. Mr. Alok Sharma SO, IPC 48. Dr. V.K. Sharma BIT 49. Dr. Jogi 50. Mr. IJS Oberoi IPC
Coordinator of the workshop:
Mr. S S Venkatakrishnan Ex- Drugs Controller, Kerala, Thiruvananthapuram
Workshop: “Role of IPC in ensuring quality of medicines”
List of Experts 1. Dr.Surinder Singh DCG(I), FDA Bhawan, Kotla Road, New Delhi-110002 2. Dr.G.N.Singh Secretary-cum-Scientific Director,IPC, Ghaziabad 3. Dr. D Roy DyDC(I),CDSCO North Zone 4. Dr.Madhur Gupta National Project Officer, WHO -Country Office, New Delhi
73

5. Dr. Jai Prakash Principal scientific Officer,IPC, Raj Nagar, Ghaziabad 6. Dr. D.B.A. Narayana Chairman, Herbs and Herbal Products Committee of IPC 7. Mr. Kapil Bhargava Rtd.DyDC(I), Mumbai 8. Dr. Raman M. Singh Principal scientific Officer, IPC, Ghaziabad 9. Dr. Manish Dare Principal scientific Officer,IPC, Ghaziabad
10. Mr.S.S.Venkatakrishnan Rtd. Drugs Controller, Kerala 11. Dr. Rajesh Madan, Executive Director, Medicamen Biotech Ltd., Bhiwadi 12. Mr.N.R.Munjal President, IDMA, Mumbai - 400 018. 13. Mr. Lotika Khajuria Dy. Drugs Controller, J&K 14. Dr. K. K. Bhutani, Officiating Director, Professor & Head, Natural Products,
NIPER, Mohali. 15. Dr. Saranjit Singh
Professor and Head, Department of Pharmaceutical Analysis, NIPER, Mohali
16. Dr. R. A Singh Director RDTL, Sector-39 C, Chandigarh- 160036 17. Dr. Girish Sahni,
Director, Institute of Microbial Technology, Sector 39A, Chandigarh 160036
18. Dr.Pramil Tiwari
Prof. & Head, Department of Pharmacy Practice, NIPER, Mohali
19. Dr.Shyam Sunder Sharma
Associate Professor, Department of Pharmacology and Toxicology, NIPER, Mohali.
20. Mr Bhag Singh Drugs Controller, Punjab 21. Mr Ajay Singla Assistant. Drugs Controller Punjab
Participants:
22. Dr.Sanjay Kumar Scientist –II, NIPER, Mohali 23. Rajinder Singh T.A., NIPER 24. Inder Singh Rawat R.D.T.L. Chandigarh 25. Ashok Kumar Sharma J R.D.T.L. Chandigarh 26. Mr Hitesh Khare J.S.A., RDTL, Chandigarh 27. Ms. Amandeep Kaur Bench Chemist RDTL, Chandigarh. 28. Dr. Amit Kumar RDTL, Chandigarh 29. Mr Pankaj Rao Gautam RDTL, Chandigarh 30. Mr. Dinesh Kumar RDTL, Chandigarh 31. Mr Vikas Sharma RDTL, Chandigarh 32. Ms. Vrinda Goyal RDTL, Chandigarh 33. Sh. Sandeep Kumar RDTL, Chandigarh 34. Dr. Saranjit Institute of Microbial Technology, Chandigarh 35. Mr Rampal Singh Mandloi RDTL, Chandigarh. 36. Mr Ankit Mittal (TDA) CDSCO, Sub Zone, Chandigarh 37. Dr. Jagdish Chandra Prof. and Head, Deptt. of Microbiology,GMCH, Chandigarh
74

38. Mr. Suyog Jain Managing Director, Vardhman Chemtech Ltd.,Chandigarh 39. Dr.Supritika Tripathi, General Manager, Unichem laboratories, Baddi (HP) 40. Dr.Harish Tyagi, Manager-QC, Abbott Healthcare pvt.ltd.,Baddi, Solan, HP 41. Dr.Shailendra Kumar, GM-QC, Abbott Healthcare Pvt.Ltd., Baddi, Dist Solan 42. Dr. Kamal Sharma Haryana Veterinary Vaccine Institute, Barwala Road, Hisar 43. Dr. Ajit Kukreja Haryana Veterinary Vaccine Institute, Barwala Road, Hisar 44. Dr. Sumit Gupta Meridian Medicare Ltd. Solan (HP) 45. Dr. Nishant Dafale Senior Scientific Officer, IPC, Ghaziabad 46. Dr. Surinder Mohan Assistant Drugs Controller (HQ) J & K 47. Sh. B. Kumar Asst. Drugs Controller 48. Sh. CHV Subbarao Chimak Health Care, Solan (HP) 49. Mr P.K. Lama (Retd.) M/s Sauraw Chemicals Ltd., Darabassi 50. Mr Kamlesh Desai GM Manufacturing, ALKEM labs Ltd.Baddi 51. Mr Rajinder Kr Harna Assistant Drugs Controller, FDA, Haryana 52. Mr B.K. Gupta Medicamen Biotech, Bhiwadi 53. Sh. Jagdeep Singh President, Punjab Drugs Manufacturers Association 54. Mr Ashutosh Jain Executive Director, Venus Remedies Ltd. Panchkula 55. Mr Parveen Goyal Managing Director, Saurav Chem. Ltd., Panchkula 56. Mr. Udai Pal IPC, Ghaziabad 57. Mr. I.J.S.Oberai IPC, Ghaziabad 58. Mr. Chandan Kumar IPC, Ghaziabad 60. Mr. Munendra Kumar IPC, Ghaziabad 61. Mr. Munesh Bindal IPC, Ghaziabad
Programme coordinator: S.S.Venkatakrishnan
Workshop for government analysts on ip 2010 on 26th and 27th may 2011
S. No. Experts Designation and Affiliation
1. Mr Sudhir Kumar Under Secretary to the Govt. of India, Ministry of Health & Family Welfare
2. Mr D.K. Shringi Drugs Controller, Rajasthan 3. Mr P. R. Uttarwar Joint Commissioner and Drugs Controller, Maharashtra 4. Dr P.K. Guha Director, CDL, Kolkata 5. Mr Rajan Pol Asst. Director, Drug Control Lab, Maharashtra 6. Dr Madhur Gupta National Project Officer, WHO -Country Office, New Delhi 7. Dr K.V. Jogi Ex-Director, CDTL, Mumbai 8. Mr S.K. Gaind Lead Assessor, NABL, New Delhi
9. Mr Sipahimalani Chairman, Regulatory & Technical & Quality Management Subcommittees, IDMA, Mumbai
75

10. Dr Jai Prakash Principal Scientific Officer, IPC, Ghaziabad
11. Mr Giddy Asrani Chairman of PEG and Vice Chairman, Quality Management Sub-committee, IDMA
12. Dr Raman Mohan Singh Principal Scientific Officer, IPC, Ghaziabad 13. Mr Kapil Bhargava Retd DyDC(I), CDSCO 14. Dr ( Mrs ) Gopa Ghosh Director, CDTL , Mumbai
Participants
15. Mrs. Nilima Mishal Jr. Scientific Officer (Drug), Govt. Analyst, Food & Drugs Testing Laborartory, Bombolim, Goa
16. Mrs. Snehaprabha Amonkar
Chemist, Food & Drugs Testing Laboratory, Bombolim, Goa
17. Dr. M.F.A. Beg SSO, CDL Kolkata 18. Mr. C. Hariharan Bacteriologist (SST), CDL Kolkata 19. Dr. Saroj Kumar Ghosh Biochemist (SSO), CDL Kolkata 20. Dr. Subir Kumar Basu Technical Officer (Bacteriology), CDL Kolkata 21. Dr. Kajal Dumar Datta Associate Biochemist, CDL Kolkata 22. Mr. Sheju Purushothaman Analyst Grade-I (Govt.), Drugs Testing Laboratories, Kerala 23. Mr. T.S. Krishna Kumar Analyst Grade-II, Drugs Testing Laboratories, Kerala 24. Mr. Vijay Kumar Sharma Govt. Analyst, Composite Testing Laboratory, H.P 25. Mr. Arvind Kumar Sharma Sr. Scientist, Composite Testing Laboratory, H.P 26. Mr V.C. Patro Sr. Govt. Analyst, SDTRL, Odisha 27. Mr K.C. Rath Sr. Govt. Analyst, SDTRL, Odisha 28. Mr Gopal S. Magadsum Scientific Officer, Drugs Testing Laboratory, Karnataka 29. Mr Rajendra C.E. Scientific Officer, Drugs Testing Laboratory, Karnataka 30. Mrs. S.M. Saptarshi Senior Scientific Officer, Drug Control Lab, Mumabi 31. Mrs. H.G. Sopte Drug Control Lab, Mumbai 32. Mr A.M. Rawal Govt. Analyst, Drug Control Lab, Mumbai 33. Mrs. A.A. Vengurlekar Scientific Officer/Govt. Analyst, Drug Control Lab, Mumbai 34. Mrs. A.G. Rao Drug Control Lab, Mumbai 35. Mrs. S.S. Dalal Govt. Analyst, Drug Control Lab, Mumbai 36. Mrs. Kusum Nehra Govt. Analyst, Food & Drugs Laboratory, Haryana 37. Mr K.N. Patel Govt. Analyst, Food & Drugs Control Admn., Gujarat 38. Miss S.K. Bhole Govt. Analyst, food & Drugs Control Admn., Gujarat 39. Mr Nazir Ahmad Wani Dy. Controller, Drugs & Food, Kashmir 40. Mrs. Irfana Ahmad Drug Analyst, Drug Testing Laboratory, Kashmir 41. Mrs. Bina Debbarma JSO & G.A., SDTL, Agartala, Tripura 42. Mr Biplab Chakma JSO, SDTL, Agartala, Tripura 43. Mr Yatendra Raj Mehta Dy. Director, DTL, Jaipur, Rajasthan 44. Mr Girish Kumar Asstt. Drug Analyst, DTL, Jaipur, Rajasthan
76

45. Mrs. S. Uma Maheswari Bench Chemist, CDTL, Hyderabad 46. Mr Naveen Kumar Bench Chemist, CDTL, Hyderabad 47. Mrs. S. Sunita Bench Chemist, CDTL, Hyderabad 48. Mr K. Ashok Kumar Bench Chemist, CDTL, Hyderabad 49. Mr Suresh Jadhav Asstt. Director, FDA, Aurangabad, Maharashtra 50. Mr V.C. Kharat Sr. Scientific Officer, FDA, Aurangabad, Maharashtra 51. Mrs. Mule S.O./G.A., FDA, Aurangabad, Maharashtra 52. Mr D.B. Parmar SSA, GMSD, Mumbai 53. Mrs. Smita Pimpale JSA, GMSD, Mumbai 54. Mrs .M. M.Patel SSO (I) , Chemistry , CDTL Mumbai. 55. Mrs.S.U. Warde SSO ( II) , Microbiology, CDTL Mumbai 56. Mr. T.K. Bhattacharya JSO , CDTL Mumbai 57. DR. M.Viyay Kumar SSA, CDTL Mumbai 58. Mrs.S. A.Navaratne SSA , CDTL Mumbai 59. Mrs.A.S.Paranje SSA,CDTL Microbiology
77

Annexure VI
LIST OF DRUGS ADDED AND OMITTED IN IP-2010 Monographs on drug substances, dosage forms and pharmaceutical aids Acepromazine Maleate Allantoin Aluminium Magnesium Silicate S-Amlodipine Besylate S-Amlodipine Tablets Liposomal Amphotericin B Injection Anastrozole Anastrozole Tablets Anhydrous Lactose Artesunate Atazanavir Sulphate Atazanavir Capsules Benzoic Acid Solution Betamethasone Dipropionate Betamethasone Cream Betamethasone Lotion Betamethasone Ointment Bifonazole
Bifonazole Cream Bumetanide Bumetanide Injection Bumetanide Oral solution Bumetanide Tablets Butylparaben Calcium Chloride Injection Capecitabine Capecitabine Tablets Cefamandole Nafate Cefamandole Injection Cetrimide Cream Cetyl Palmitate Chlorothiazide Chlorothiazide Oral Suspension Chlorothiazide Tablets Chymotrypsin Cilastatin Sodium
Clindamycin Hydrochloride Clindamycin Capsules Codeine Phosphate Tablets Cyproterone Acetate Cyproterone Tablets Daunorubicin Hydrochloride Daunorubicin Injection Dexchlorpheniramine Maleate Dexchlorpheniramine Oral Solution Dexchlorpheniramine Tablets Dextropropoxyphene Hydrochloride Dextropropoxyphene Capsules Dextropropoxyphene Napsilate Diacerein Diacerein Capsules
Diazoxide Diazoxide Tablets Dicloxacillin Sodium Dicloxacillin Capsules Dicloxacillin Oral Suspension Diethanolamine Dihydroergocristine Mesylate Dihydroergotamine Mesylate Dimethicone Disopyramide Disopyramide Capsules Disopyramide Phosphate Capsules Disopyramide Phosphate Sustained-release Capsules Divalproex Sustained-release Tablets
78

Docetaxel Trihydrate Docetaxel Injection Domperidone Doxofylline Doxofylline Tablets Enoxaparin Sodium Enoxaparin Injection Escitalopram Oxalate Escitalopram Tablets Estradiol and Norethisterone Tablets Etodolac Etodolac Capsules Etodolac Tablets Famotidine Famotidine Tablets Felodipine Felodipine Sustained-release Tablets Fenbendazole Fenofibrate Fentanyl Fentanyl Citrate Fentanyl Injection Finasteride Finasteride Tablets Fluconazole Fluconazole Capsules Fluconazole Tablets Flucytosine Flucytosine Capsules Flucytosine Oral Suspension Flucytosine Tablets Fluorescein Injection Flutamide Flutamide Capsules Fumaric Acid Gefitinib Gefitinib Tablets Gemifloxacin Mesylate Gemifloxacin Tablets
Gliclazide Gliclazide Tablets Glimepiride Glimepiride Tablets Homatropine Methylbromide Homatropine Methylbromide Tablets Hyoscyamine Sulphate Hyoscyamine Injection Hyoscyamine Oral Solution Hyoscyamine Tablets Ibuprofen Cream Ibuprofen Gel Imatinib Mesylate Imatinib Capsules Indapamide Tablets Isobutane Isopropyl Myristate Lactulose Lamotrigine Sustained-release Tablets Lansoprazole Lansoprazole Sustained-release Capsules Lecithin Levosalbutamol Sulphate Linezolid Linezolid Tablets Losartan Potassium and Amlodipine Tablets Losartan Potassium and Hydrochlorothiazide Tablets Maleic Acid Malic Acid Maltitol Liquid Maltitol Maltodextrin Mefloquine Hydrochloride Meloxicam Oral Suspension Menthol and Benzoin Inhalation Metformin Hydrochloride Sustained-release Tablets Methadone Linctus
79

Metronidazole Sterile Suspension Miconazole Microcrystalline Cellulose and Carboxymethylcellulose Sodium Misoprostol Mometasone Furoate Mometasone Aqueous Nasal Spray Mometasone Cream Mometasone Ointment Montelukast Sodium Montelukast Tablets Mycophenolate Mofetil Mycophenolate Mofetil Capsules Myristic Acid Naloxone Hydrochloride Naloxone Injection Naltrexone Hydrochloride Naltrexone Tablets Naproxen Naproxen Oral Suspension Naproxen Suppositories Naproxen Sustained-release Tablets Naproxen Tablets Neotame Ondansetron Orally Disintegrating Tablets Ondansetron Oral Solution Pantoprazole Sodium Pantoprazole Sustained-release Tablets Perphenazine Perphenazine Tablets Phenoxyethanol Phenylpropanolamine Hydrochloride Phenytoin Phenytoin Capsules Phenytoin Oral Suspension Pimozide Pimozide Tablets Piperacillin Piperacillin Intravenous Infusion
Poloxamers Polyoxyl 35 Castor Oil Polyoxyl 40 Hydrogenated Castor Oil Potassium Sorbate Pravastatin Sodium Pravastatin Tablets Praziquantel Praziquantel Tablets Prednisolone Acetate Pregabalin Pregabalin Capsules Pregelatinised Starch Progesterone Injectable Suspension Promazine Tablets Propane Propionic Acid Propofol Propofol Injection Protriptyline Hydrochloride Protriptyline Tablets Pyrimethamine Tablets Quiniodochlor Cream Quiniodochlor Ointment Quiniodochlor and Hydrocortisone Cream Quiniodochlor and Hydrocortisone Ointment Ramipril and Hydrochlorothiazide Tablets Ribavirin Ribavirin Inhalation Solution Serratiopeptidase Serratiopeptidase Tablets Sildenafil Citrate Sildenafil Tablets Simvastatin Simvastatin Tablets Sorbitan Oleate Sucralose Sumatriptan
80

Sumatriptan Injection Vancomycin Oral Solution Telmisartan Xanthan Gum Telmisartan Tablets Zoledronic Acid Temozolomide Zoledronic Acid Injection Temozolomide Capsules Herbal Monographs Terazosin Hydrochloride
Amla Juice Powder Thiocolchicoside
Arjuna Dry Extract Thiocolchicoside Capsules
Ashwagandha Dry Extract Ticarcillin and Clavulanic Acid Injection
Belladonna Tincture Tolazamide
Bhibhitaki Aqueous Extract Tolazamide Tablets
Brahmi Extract Tolnaftate
Coconut Oil Tolnaftate Cream
Coleus Dry Extract Tolnaftate Gel
Coriander Oil Tolnaftate Topical Powder
Garcinia Aqueous Extract Tolnaftate Topical Solution
Haridra Dry Extract Tolterodine Tartrate
Haritaki Extract Tramadol Hydrochloride
Haritaki Aqueous Extract Tramadol Capsules
Ipecac Tincture Trandolapril
Lavang Trandolapril Tablets
Methi Travoprost
Neem Travoprost Eye drops
Sarpagandha Powder Tributyl Citrate
Sarpagandha Tablets Trichloromonofluoromethane
Sunthi Extract Triethyl Citrate Tulasi Dry Extract Trimetazidine Hydrochloride Vasaka Extract Valproate Injection
Veterinary Monographs Valproic Acid Valproic Acid Capsules Infectious Bursal Disease Vaccine , Live Valproic Acid Oral Solution Infectious Chicken Aneamia Vaccine,
Inactivated Valsartan Infectious Chicken Aneamia Vaccine, Live
Valsartan Tablets Valsartan and Hydrochlorothiazide Tablets Marek’s Disease Vaccine, Live
Reo Virus Vaccine, Inactivated Vancomycin Hydrochloride Reo Virus Vaccine, Live Vancomycin Capsules Salmonella Vaccine, Inactivated Vancomycin Intravenous Infusion
Sterile Diluent for Live Vaccines
81

Omissions Adenine Aluminium Sulphate Analgin Analgin Tablets Butylated Hydroxyanisole Caramel Cyclopropane Deslanoside Deslanoside Injection Dibutyl Phthalate Emetine Hydrochloride Emetine Injection Ephedrine Erythromycin Estolate Erythromycin Estolate Tablets Fusidic Acid Oral Solution 2- Deoxy- D- Glucose Protamine Zinc Insulin Injection Lanatoside C Lanatoside C Tablets Laryngotracheitis Vaccine, Live Menadione Methdilazine Hydrochloride Methdilazine Tablets Oxyphenbutazone Oxyphenbutazone Tablets
Phenindamine Tartrate Phenindamine Tablets Phenylbutazone Phenylbutazone Tablets Propantheline Bromide Propantheline Tablets Sodium Aurothiomalate Sodium Aurothiomalate Injection Sodium Cromoglycate Sodium Cromoglycate Powder for Inhalation Sodium Fusidate Capsules Prepared Storax Sulphadimethoxine Sulphadimethoxine Tablets Sulphadimidine Sulphadimidine Sodium Sulphadimidine Injection Sulphadimidine Tablets Sulphafurazole Sulphafurazole Tablets Sulphalene Sulphaphenazole Sulphaphenazole Tablets Sulphobromophthalein Sodium Sulphobromophthalein Sodium Injection
82

FEEDBACK FORM Name Designation Organization Address Telephone, FAX, E-mail and Mobile phone numbers
Contents of the Manual:
a. In your experience or views, what the problems that the user of the I P generally faces?
b. Your view on the contents of the manual as to whether they address the problems that an user generally faces while using IP?
c. If not, what other areas need attention?
d. If yes, has the Manual resolved the problems to an extent that the user of IP can perform the tests correctly?
e. If any matter(s) has/have been left out in your views, please state the same.
1.
f. Your views on the presentations in general.
2. Outline the benefits that you have derived out of this manual.
Excellent Very good Good Average
3. Your Rating of the Manual: ( ) Tick in appropriate column
Below Average
Signature
83

Date
84







