Embed Size (px)
Citation preview

Dr. Nabila Hamdi
MD, PhD
Lecture 8 Neoplasia II

ILOs • Understand the definition of neoplasia.
• List the classification of neoplasia.
• Describe the general characters of benign tumors.
• Understand the nomenclature of benign and malignant tumors.
• Recall the most important epidemiological aspects of cancer.
• Discuss the etiology of malignant tumors.
• Recognize the definition, microscopic changes and types of dysplasia.
• Understand the pathogenesis of tumor formation.
• Describe general characters of malignant tumors.
• Understand the methods of grading and staging of malignant neoplasms.
• Understand the definition of Carcinoma in-situ
• Explain the molecular basis of cancer
• Discuss the methods of spread of malignant tumors.
• Discuss the laboratory diagnosis of malignant tumors.
• Be aware of the effects of tumors on the host.

Outline
1. Overview 2. Nomenclature 3. Characteristics of Benign and Malignant Tumors 4. Epidemiology 5. Carcinogenesis: The Molecular Basis of Cancer • Self-Sufficiency in Growth Signals • Insensitivity to Growth-Inhibitory Signals • Evasion of Apoptosis • Limitless Replicative Potential • Sustained Angiogenesis • Invasion and Metastasis • Role of DNA Repair Genes
6. Etiology of Cancer: Carcinogenic Agents 7. Host Defense Against Tumors: Tumor Immunity 8. Clinical Aspects of Neoplasia 3
Neoplasia 2
Neoplasia 3
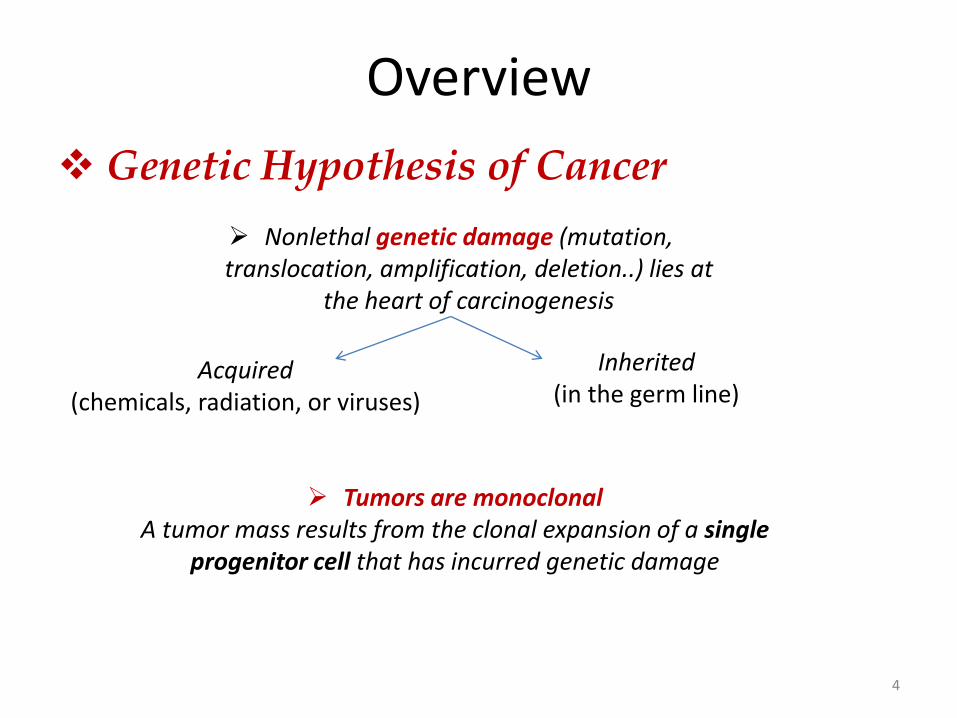
Overview
Nonlethal genetic damage (mutation, translocation, amplification, deletion..) lies at
the heart of carcinogenesis
4
Genetic Hypothesis of Cancer
Acquired (chemicals, radiation, or viruses)
Inherited (in the germ line)
Tumors are monoclonal A tumor mass results from the clonal expansion of a single
progenitor cell that has incurred genetic damage

Overview
Protooncogenes
(Growth-promoting)
Tumor suppressor genes
(Growth-inhibiting)
Genes that regulate apoptosis
Genes that regulate repair of
damaged DNA
Oncogenes dominant
Cancer autonomous cell growth
(transformed phenotype)
mutation
recessive Cells refractory to cell inhibition
dominant
recessive
Down regulation of pro-apoptotic genes
Up regulation of anti-apoptotic genes
Widespread mutation in the genome
Four classes of normal regulatory genes
Mostly recessive
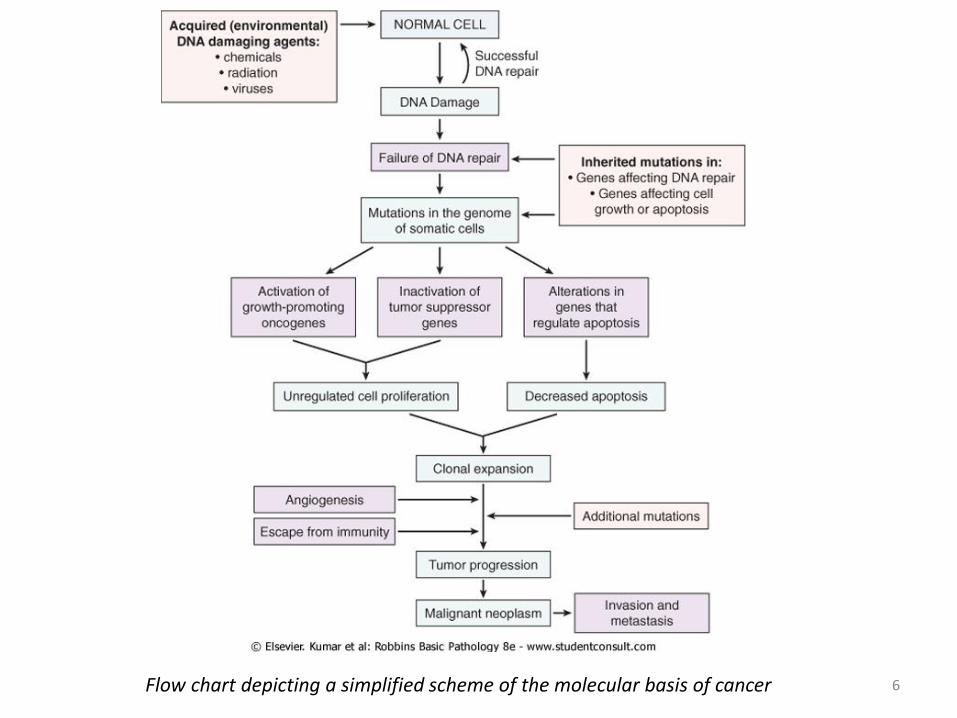
6 Flow chart depicting a simplified scheme of the molecular basis of cancer
7
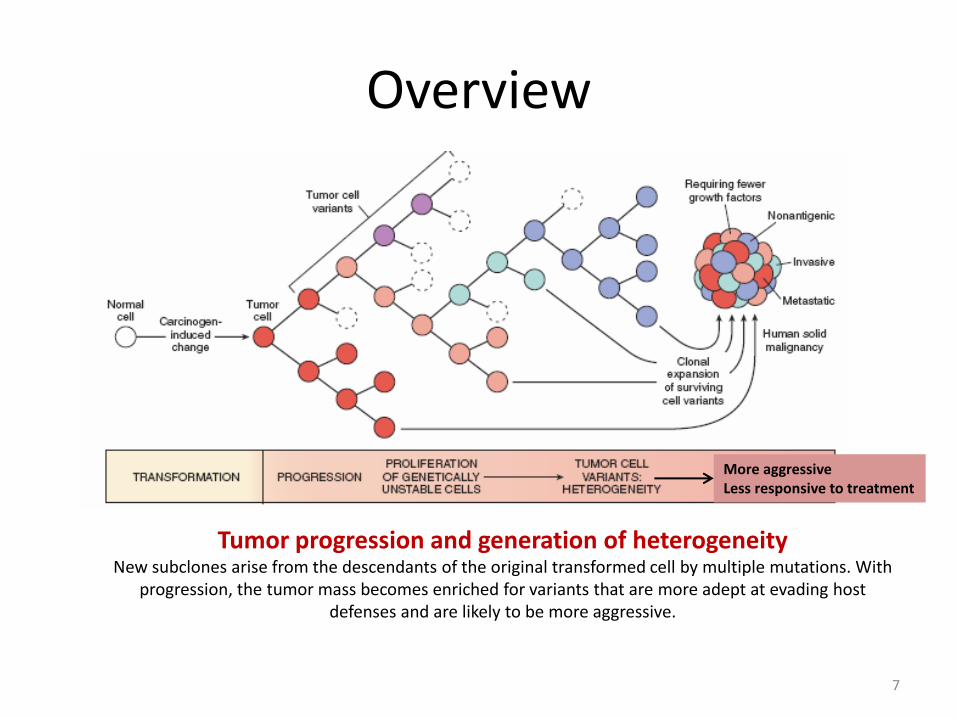
Overview
Tumor progression and generation of heterogeneity New subclones arise from the descendants of the original transformed cell by multiple mutations. With
progression, the tumor mass becomes enriched for variants that are more adept at evading host defenses and are likely to be more aggressive.
More aggressive Less responsive to treatment
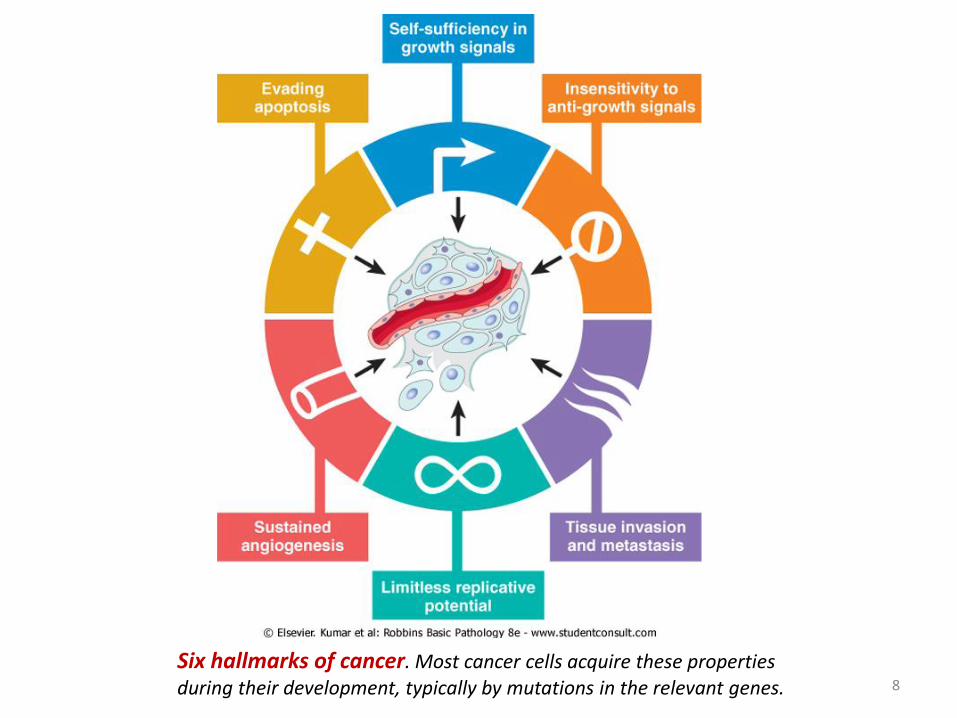
8
Six hallmarks of cancer. Most cancer cells acquire these properties during their development, typically by mutations in the relevant genes.

Self-Sufficiency in Growth Signals
9

Self-Sufficiency in Growth Signals
Growth Factors
10
Cancer cells acquire growth self-sufficiency by: Synthesizing the same growth factors to which they are responsive (autocrine loop): • Glioblastomas secrete platelet-derived growth factor (PDGF) and express the PDGF receptor. • Sarcomas make both transforming growth factor-α (TGF-α) and its receptor.
Interacting with stroma: in some cases, tumor cells send signals to activate normal cells in
the supporting stroma, which in turn produce growth factors that promote tumor growth.
In normal cells, most soluble growth factors are made by one cell type and act on a neighboring cell to stimulate proliferation (paracrine action).

Self-Sufficiency in Growth Signals
Growth Factor Receptors
11
Mutant receptor proteins deliver continuous mitogenic signals to cells, even in the absence of the growth factor in the environment.
Overexpression of growth factor receptors is more common than mutations: cancer cells are hyper-responsive to levels of the growth factor that would not normally trigger proliferation. Example: epidermal growth factor (EGF) receptors ERBB1 is overexpressed in 80% of squamous cell carcinomas of the lung, 50% of glioblastoma and 80 to 100% of epithelial tumors of head and neck HER2/NEU (ERBB2), is amplified in 25% to 30% of breast cancers and adenocarcinomas of the lung, ovary, and salivary glands.
High level of HER2 protein in breast cancer cells is a harbinger of poor prognosis!!
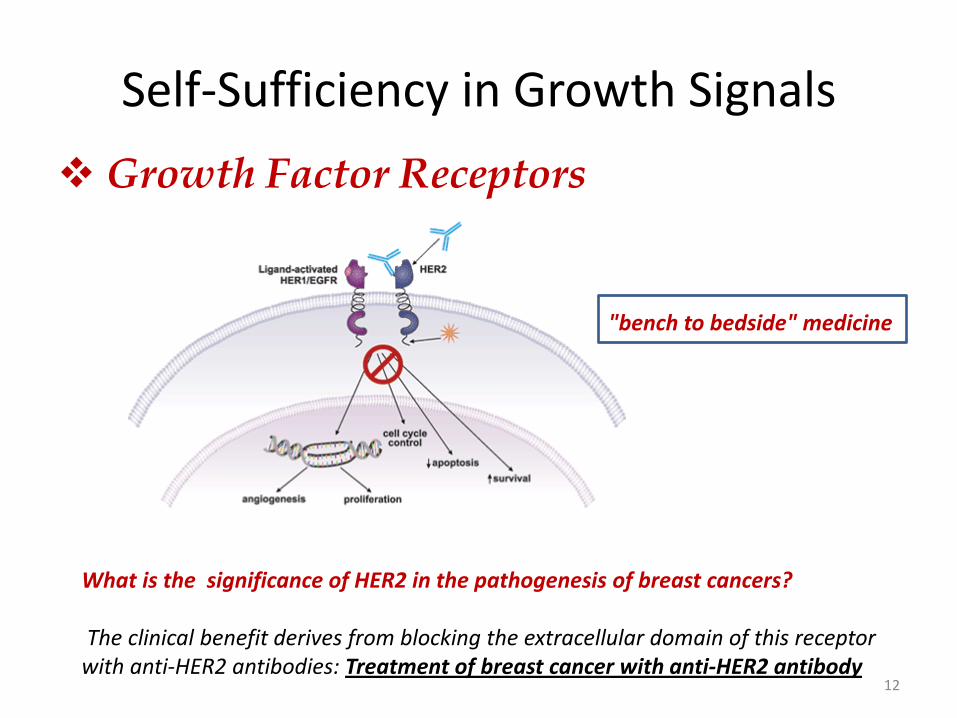
Self-Sufficiency in Growth Signals
Growth Factor Receptors
12
What is the significance of HER2 in the pathogenesis of breast cancers? The clinical benefit derives from blocking the extracellular domain of this receptor with anti-HER2 antibodies: Treatment of breast cancer with anti-HER2 antibody
"bench to bedside" medicine

Self-Sufficiency in Growth Signals
Signal-Transducing Proteins: RAS
13
Approximately 30% of all human tumors contain mutated versions of the RAS gene
GTPase-activating proteins (molecular brakes)
Intrinsic GTPase activity

Self-Sufficiency in Growth Signals
Signal-Transducing Proteins: RAS
14
Normal RAS proteins flip back and forth between an excited signal-transmitting state and a quiescent state
RAS proteins are inactive when bound to GDP; stimulation of cells by growth
factors leads to exchange of GDP for GTP and subsequent conformational changes that generates active RAS
The activated RAS in turn stimulates down-stream regulators of proliferation,
such as the RAF-mitogen-activated protein (MAP) kinase mitogenic cascade, which floods the nucleus with signals for cell proliferation.

Self-Sufficiency in Growth Signals
Signal-Transducing Proteins: RAS
15
The excited signal-emitting stage of the normal RAS protein is short-lived, because its intrinsic guanosine triphosphatase (GTPase) activity hydrolyzes GTP to GDP.
The GTPase activity of activated RAS protein is magnified dramatically by a family of
GTPase-activating proteins (GAPs), which act as molecular brakes that prevent uncontrolled RAS activation by favoring hydrolysis .
The mutant RAS protein (point mutations) is permanently activated because of inability
to hydrolyze GTP, leading to continuous stimulation of cells without any external trigger. RAS is thus trapped in its activated GTP-bound form, and the cell is forced into a
continuously proliferating state.
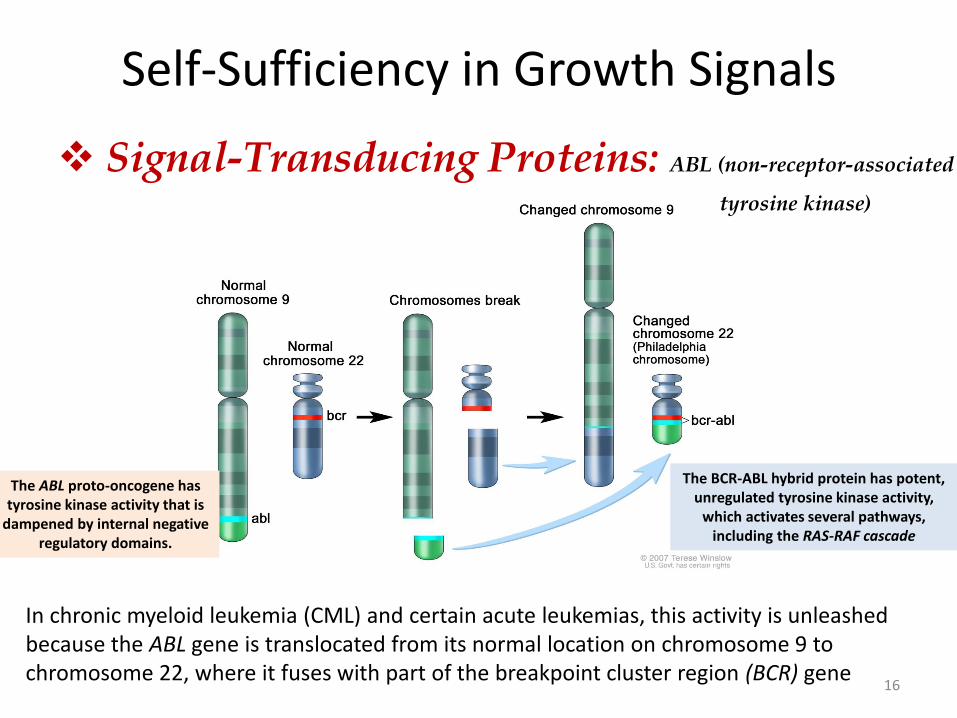
Self-Sufficiency in Growth Signals
Signal-Transducing Proteins: ABL (non-receptor-associated
16
In chronic myeloid leukemia (CML) and certain acute leukemias, this activity is unleashed because the ABL gene is translocated from its normal location on chromosome 9 to chromosome 22, where it fuses with part of the breakpoint cluster region (BCR) gene
The ABL proto-oncogene has tyrosine kinase activity that is
dampened by internal negative regulatory domains.
The BCR-ABL hybrid protein has potent, unregulated tyrosine kinase activity,
which activates several pathways, including the RAS-RAF cascade
tyrosine kinase)

Self-Sufficiency in Growth Signals
Signal-Transducing Proteins: ABL
17
A cell with BCR-ABL fusion gene is dysregulated by inappropriate tyrosine kinase activity that leads to growth autonomy.
The crucial role of BCR-ABL in transformation has been confirmed by the dramatic clinical response of patients with chronic myeloid leukemia after therapy with an inhibitor of the BCR-ABL fusion kinase called imatinib mesylate (Gleevec, STI571)
Mechanism of action of STI571: it Inhibits growth by inhibiting the kinase activity.

Self-Sufficiency in Growth Signals
Nuclear Transcription Factors
18
Growth autonomy may thus occur as a consequence of mutations affecting genes that regulate transcription of DNA: MYC, MYB, JUN, FOS, and REL oncogenes
regulate the expression of growth-promoting genes, such as cyclins. The MYC gene is involved most commonly in human tumors.
The MYC proto-oncogene is expressed in virtually all cells, and the MYC protein is induced rapidly when quiescent cells receive a signal to divide.

Self-Sufficiency in Growth Signals
Nuclear Transcription Factors
19
Betrayed by Nature: The War on Cancer, Dr. Robin Hesketh
Cyclins, CDKs
In normal cells, MYC levels decline to near basal level when the cell cycle begins. Oncogenic versions of the MYC gene are associated with persistent expression or overexpression, contributing to sustained proliferation.
Cell cycle activation
+ CDKIs _
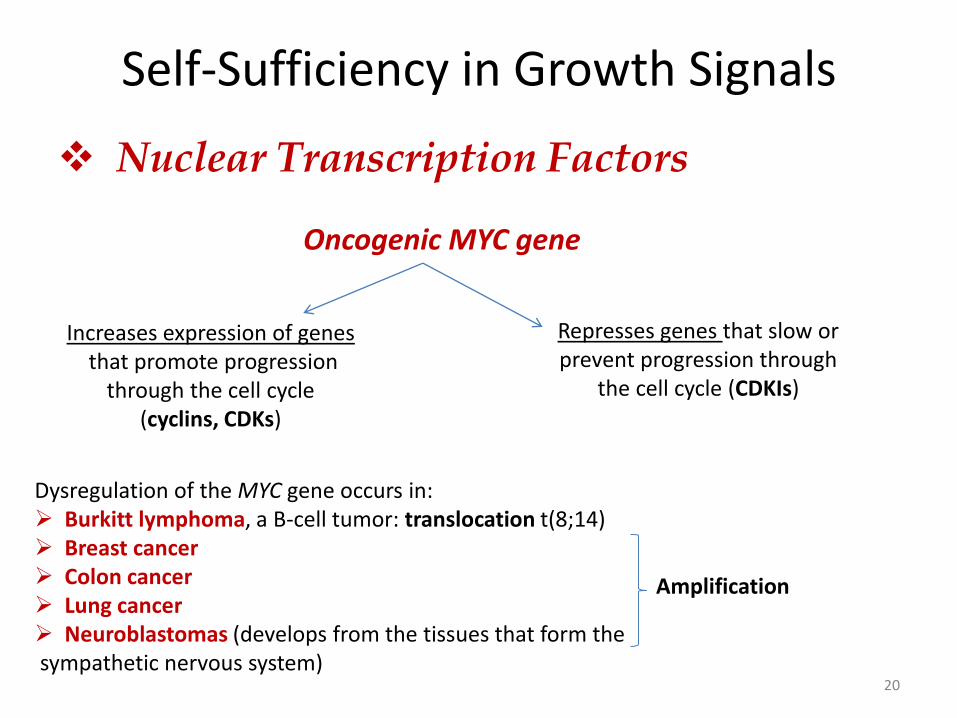
Self-Sufficiency in Growth Signals
Nuclear Transcription Factors
20
Dysregulation of the MYC gene occurs in: Burkitt lymphoma, a B-cell tumor: translocation t(8;14) Breast cancer Colon cancer Lung cancer Neuroblastomas (develops from the tissues that form the sympathetic nervous system)
Oncogenic MYC gene
Increases expression of genes that promote progression
through the cell cycle (cyclins, CDKs)
Represses genes that slow or prevent progression through
the cell cycle (CDKIs)
Amplification
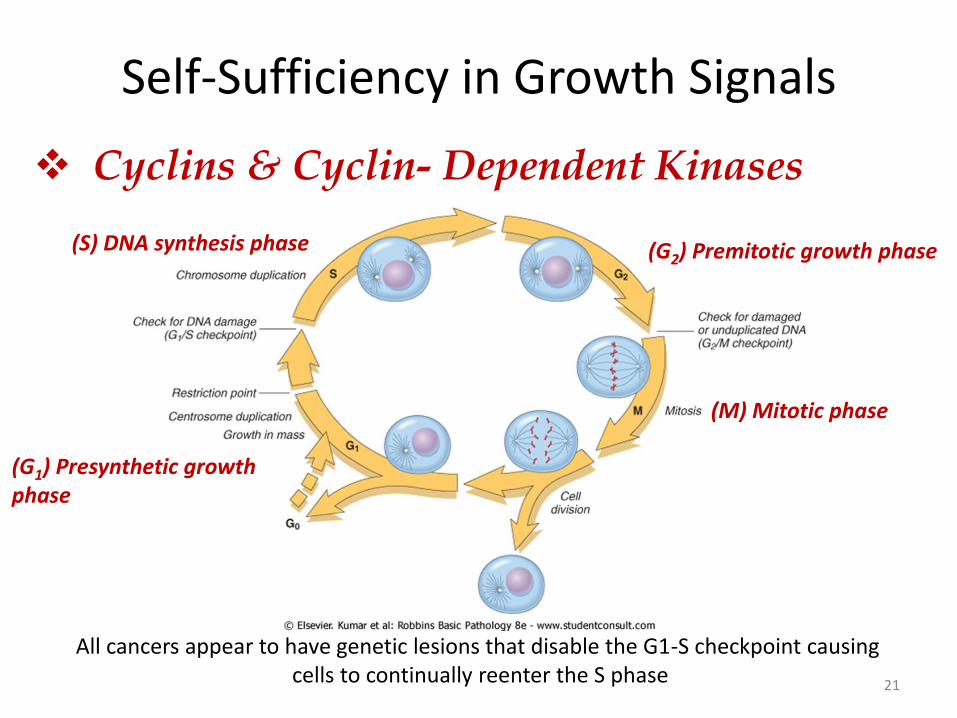
Self-Sufficiency in Growth Signals
21
(S) DNA synthesis phase (G2) Premitotic growth phase
(G1) Presynthetic growth phase
(M) Mitotic phase
Cyclins & Cyclin- Dependent Kinases
All cancers appear to have genetic lesions that disable the G1-S checkpoint causing cells to continually reenter the S phase

Self-Sufficiency in Growth Signals
Cyclins & Cyclin- Dependent Kinases
22
Cyclins D, E, A, and B appear sequentially during the cell cycle and bind to one or more CDK
Cyclin D genes: overexpressed in many cancers, including those affecting the breast, esophagus, liver, and a subset of lymphomas. CDK4 gene is amplified in melanomas, sarcomas, and glioblastomas. Cyclin B and cyclin E are mutated in some malignant neoplasms, but they are much less frequent than those affecting cyclin D/CDK4. CDKIs are disabled by mutation, gene silencing, deletions …in many cancers.
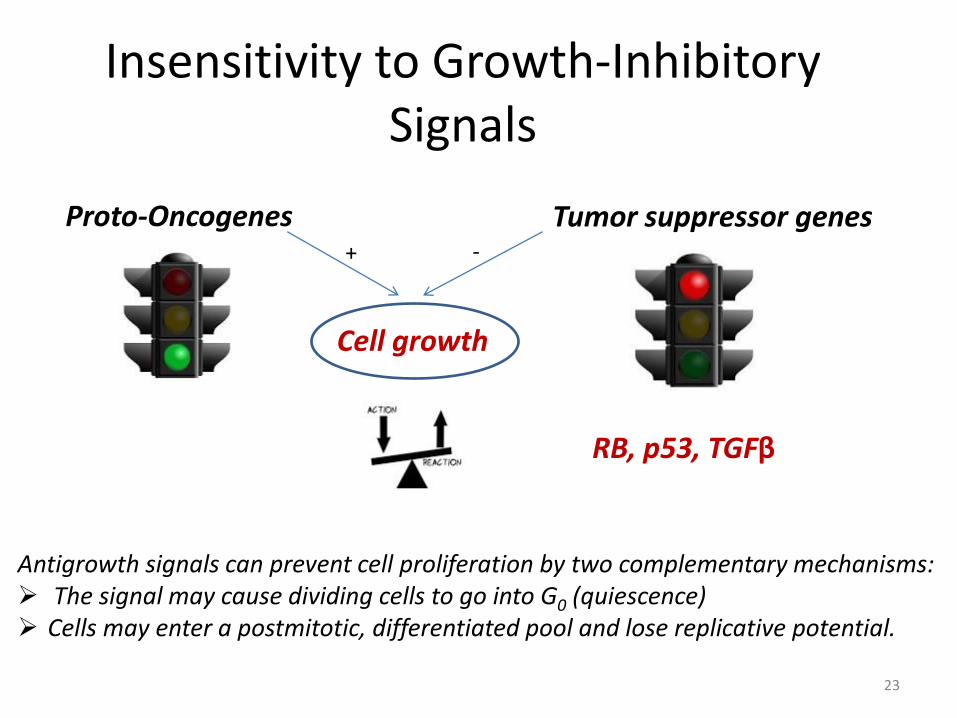
Insensitivity to Growth-Inhibitory Signals
23
Cell growth
Proto-Oncogenes Tumor suppressor genes
RB, p53, TGFβ
Antigrowth signals can prevent cell proliferation by two complementary mechanisms: The signal may cause dividing cells to go into G0 (quiescence) Cells may enter a postmitotic, differentiated pool and lose replicative potential.
+ -

Insensitivity to Growth-Inhibitory Signals
Retinoblastoma Gene (RB)
Two-hit hypothesis (Knudson, 1974) Two mutations (hits) of RB gene (chromosome 13q14) are
required to produce retinoblastoma
In familial cases (40%), children inherit one defective copy in the germ line; the other copy is normal. Retinoblastoma develops when the normal RB gene is lost in retinoblasts as a result of somatic mutation.
In sporadic cases (60%), both normal RB alleles are lost by somatic mutation in one of the retinoblasts.
24
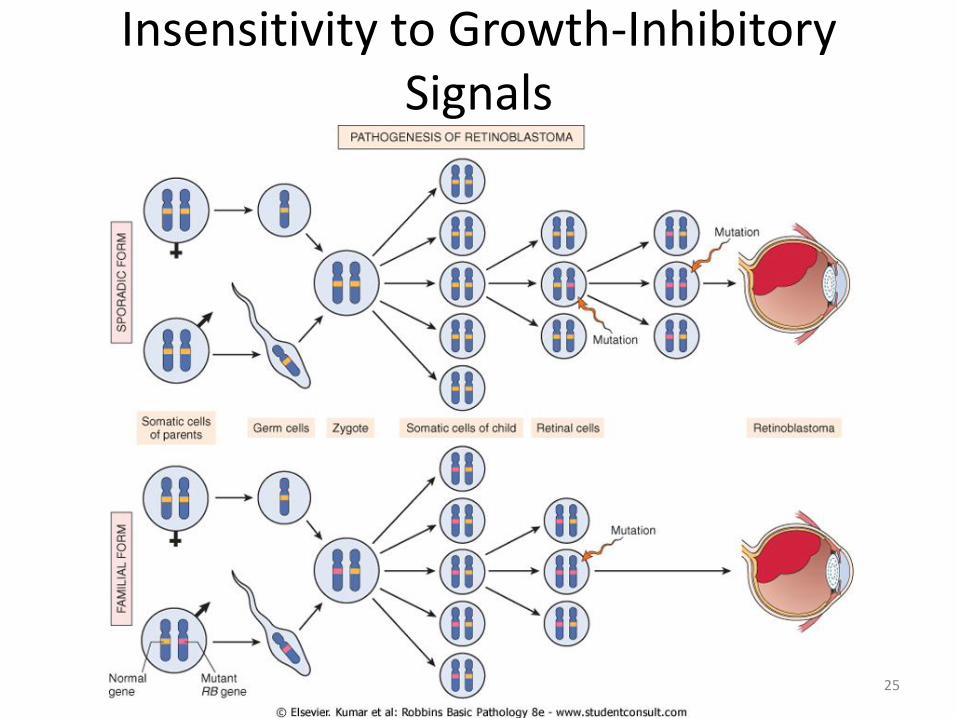
Insensitivity to Growth-Inhibitory Signals
25

Insensitivity to Growth-Inhibitory Signals
Retinoblastoma Gene (RB)
A cell heterozygous at the RB locus is not neoplastic. Tumors develop when the cell becomes homozygous for the mutant allele or, in other words, loses heterozygosity of the normal RB gene.
it is now evident that homozygous loss of this gene is a fairly
common event in several tumors:
• Breast cancer
• Small-cell cancer of the lung
• Bladder cancer 26

Insensitivity to Growth-Inhibitory Signals
Retinoblastoma Gene (RB)
27
• When RB is phosphorylated by the cyclin D-CDK4/6 and cyclin E-CDK2 complexes, it releases E2F.
• E2F then activates transcription of S-phase genes. • The phosphorylation of RB is inhibited by CDKIs, because they inactivate cyclin-CDK
complexes
Hypophosphorylated RB in complex with the E2F transcription factors binds to DNA, recruits chromatin remodeling factors (histone deacetylases and histone methyltransferases), and inhibits transcription of genes whose products are required for the S phase of the cell cycle.
Mutations in other genes that control RB phosphorylation can mimic the effect of RB loss (CDK4, cyclin D, CDKI)
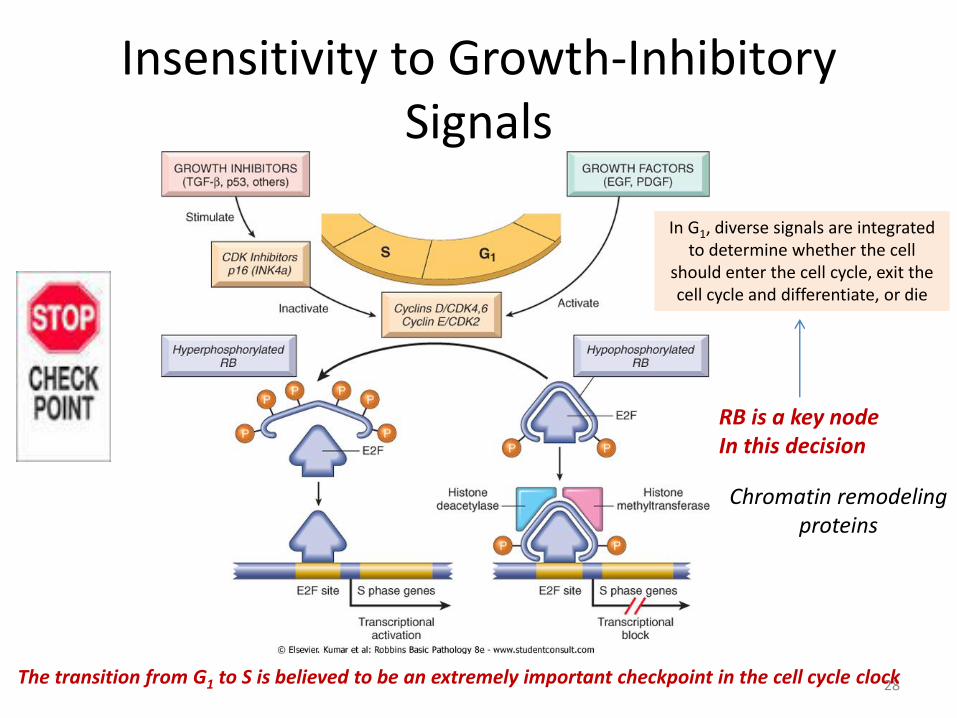
Insensitivity to Growth-Inhibitory Signals
28 The transition from G1 to S is believed to be an extremely important checkpoint in the cell cycle clock
In G1, diverse signals are integrated to determine whether the cell
should enter the cell cycle, exit the cell cycle and differentiate, or die
RB is a key node In this decision
Chromatin remodeling proteins
Insensitivity to Growth-Inhibitory Signals
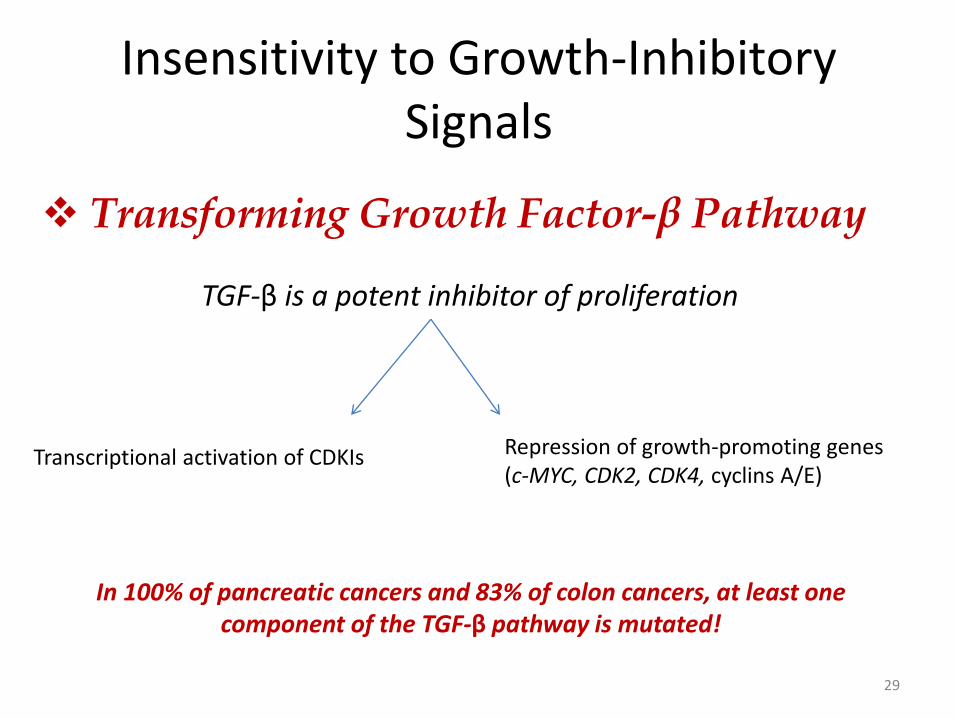
29
Transforming Growth Factor-β Pathway
TGF-β is a potent inhibitor of proliferation
Transcriptional activation of CDKIs Repression of growth-promoting genes (c-MYC, CDK2, CDK4, cyclins A/E)
In 100% of pancreatic cancers and 83% of colon cancers, at least one component of the TGF-β pathway is mutated!

Insensitivity to Growth-Inhibitory Signals
30
p53: Guardian of the Genome
p53 senses DNA damage and assists in DNA repair by causing G1 arrest and inducing DNA repair genes.
A cell with damaged DNA that cannot be repaired is directed by p53 to either enter senescence or undergo apoptosis.
With homozygous loss of p53, DNA damage goes unrepaired, mutations become fixed in dividing cells, and the cell turns onto a one-way street leading to malignant transformation.

31
p53: Guardian of the Genome

References
• Cover image http://guardianlv.com/2013/11/prostate-cancer-and-evolution-how-breakthrough-can-lead-to-new-treatments/
• Basic Pathology 8th Edition, by Kumar, Cotran and Robbins
• Basic Pathology 9th Edition, by Kumar, Cotran and Robbins
32