Embed Size (px)
Citation preview

LOCALIZZAZIONE E RUOLO DI FATTORI CHE INTERVENGONO NELLA
PROLIFERAZIONE DELL’EPITELIO BILIARE E RELAZIONI CON LE
CARATTERISTICHE ULTRASTRUTTURALI NEL FEGATO NORMALE E
POLICISTICO
DOTT.SSA ROMINA MANCINELLI, DIP ANATOMIA UMANA, “SAPIENZA”
UNIVERSITÀ DI ROMA
DOTTORATO DI RICERCA IN EPATOLOGIA SPERIMENTALE E CLINICA
XXI CICLO
DIPARTIMENTO DI ANATOMIA UMANA “SAPIENZA” UNIVERSITÀ DI
ROMA
DIRETTORE: PROF. EUGENIO GAUDIO
TUTORE: DOTT. ANTONIO FRANCHITTO, DIP. ANATOMIA UMANA,
“SAPIENZA” UNIVERSITÀ DI ROMA
DOCENTI ESAMINATORI: PROF. ADOLFO FRANCESCO ATTILI, DIP. DI
MEDICINA CLINICA, “SAPIENZA” UNIVERSITÀ DI ROMA;
PROF. ADRIANO DE SANTIS, DIP. DI MEDICINA CLINICA, “SAPIENZA”
UNIVERSITÀ DI ROMA;
PROF. MAURIZIO RIPANI, DIP. DI SCIENZE DEL MOVIMENTO UMANO E
DELLO SPORT, ISTITUTO UNIVERSITARIO DI SCIENZE MOTORIE, ROMA

2
ABSTRACT
I colangiociti sono cellule normalmente dormienti, ma proliferano in alcuni modelli sperimentali (BDL) e alcune colangiopatie umane. Molti fattori, come gli ormoni sessuali regolano la crescita biliare. Scopo del presente studio è stato quello di valutare il ruolo dell’ormone follicolo stimolante (FSH) nella regolazione della proliferazione dell’epitelio biliare. A tal fine abbiamo trattato ratti femmine e maschi normali e BDL con (i) soluzione salina, (ii) FSH, (iii) Antide (un antagonista dell’ormone rilasciante le gonadotropine), (iv) un anticorpo anti-FSH, or (v) siero non immune per una settimana. Successivamente abbiamo valutato: (i) l’espressione dell’FSH recettore (FSHR), (ii) proliferazione e apoptosi colangiocitaria, (iii) livelli del cAMP basali e dopo secretina e fosforilazione delle proteine ERK1/2 e Elk-1. In aggiunta, abbiamo studiato le caratteristiche ultrastrutturali dell’epitelio biliare umano normale e in corso di malattia policistica del fegato (PLD). In vitro, linee cellulari di colangiociti normali di ratto (NRICC), di colangiociti normali umani (H69) e di colangiociti provenienti da cisti epatiche (LCDE) sono state stimolate con FSH in presenza o assenza di preincubazione con un anticorpo prodotto contro il recettore dell’FSH (anti-FSHR) o con PD98056 (un inibitore del pathway ERK/MAPK), per valutarne i cambi nell’espressione di FSH e FSHR, insieme agli effetti sulla proliferazione. Di seguito, abbiamo valutato le conseguenze del silenziamento di FSH in NRICC sulla proliferazione, apoptosi e sulla fosfotilazione di ERK1/2. I risultati ottenuti hanno dimostrato che il trattamento cronico con FSH aumenta l’espressione del FSHR, la massa biliare e il pathway del cAMP. Questo aumento viene bloccato con Antide o con un anticorpo anti-FSH. Questi dati si associano ad alterazione della morfologia del ciglio primario nell’epitelio biliare umano in corso di PLD. Anche in vitro, l’FSH presenta un’azione di stimolo della proliferazione nelle linee cellulari considerate. Il silenziamento dell’FSH in NRICC diminuisce la crescita dei colangiociti e la fosforilazione di ERK1/2. L’azione svolta dall’ormone follicolo stimolante sulle capacità proliferative dei colangiociti in corso di colestasi puó porre, quindi, le basi morfologiche per suggerire potenziali strategie terapeutiche basate sulla regolazione dell’ormone follicolo stimolante.
ABSTRACT (english)
Cholangiocytes are normally mitotically quiescient but proliferate in animal model of cholestasis (BDL) and in human cholangiopathies. A number of factors including sex hormones regulate biliary growth. No data exists on the role of follicle-stimulating hormone (FSH) on biliary epithelium. Normal and BDL female and male rats were treated with: (i) saline; (ii) FSH; (iii) Antide (a gonadotropin releasing hormone antagonist); (iv) a neutralizing FSH antibody; or (v) non-immune serum for 1 week. We evaluated: (i) expression FSH receptor (FSHR); (ii) cholangocyte proliferation and apoptosis; (iii) basal and secretin cAMP levels and ERK1/2 and Elk-1 phosphorylation. In addition, we studied the ultrastructural characteristics of normal human biliary epithelium and in course of polycystic liver disease (PLD) by scanning electron microscope. In vitro, cell lines of normal rat cholangiocytes (NRICC), normal human cholangiocytes (H69) and cholangiocytes from hepatic cysts (LCDE) were stimulated with FSH before to evaluate the effects on proliferation in absence/presence of pre-incubation with an antibody anti-FSHR or PD98056 (an ERK/MAPK pathway inhbitor). Then, we evaluated the effects of FSH silencing in NRICC proliferation, apoptosis and ERK1/2 phosphorylation. Chronic administration of FSH increased expression of FSHR, ductal mass and cAMP pathway. It was blocked by administration of Antide or anti-FSH antibody. These findings are associated with changes of primary cilium morphology in the biliary epithelium during PLD. In vitro, FSH increased the proliferation of the cell lines considered. Silencing of FSH gene decreases cholangiocyte proliferation and ERK1/2 phosphorylation. FSH may
3
be a key regulator of cholangiocyte growth and its regulation may suggest some therapeutic strategies in the management of cholestatis.
INTRODUZIONE
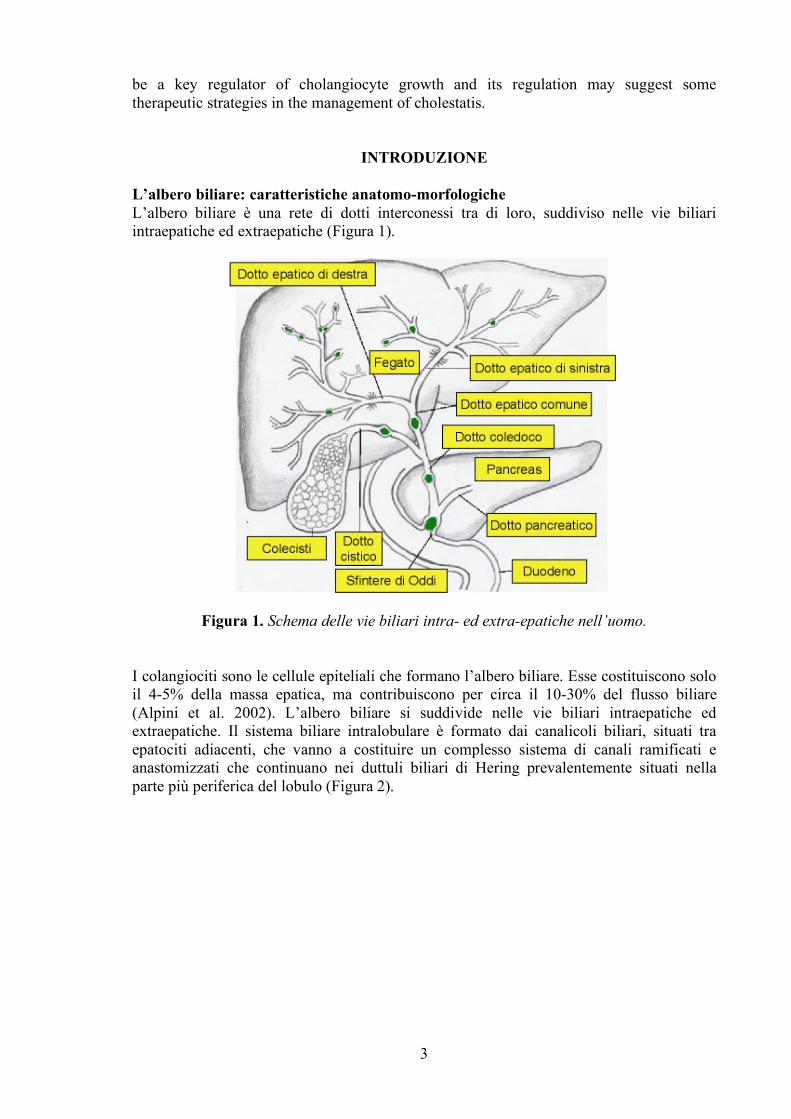
L’albero biliare: caratteristiche anatomo-morfologiche L’albero biliare è una rete di dotti interconessi tra di loro, suddiviso nelle vie biliari intraepatiche ed extraepatiche (Figura 1).
Figura 1. Schema delle vie biliari intra- ed extra-epatiche nell’uomo. I colangiociti sono le cellule epiteliali che formano l’albero biliare. Esse costituiscono solo il 4-5% della massa epatica, ma contribuiscono per circa il 10-30% del flusso biliare (Alpini et al. 2002). L’albero biliare si suddivide nelle vie biliari intraepatiche ed extraepatiche. Il sistema biliare intralobulare è formato dai canalicoli biliari, situati tra epatociti adiacenti, che vanno a costituire un complesso sistema di canali ramificati e anastomizzati che continuano nei duttuli biliari di Hering prevalentemente situati nella parte più periferica del lobulo (Figura 2).

4
Figura 2. (A) rappresentazione schematica dell’origine a fondo cieco delle vie biliari intraepatiche. I canalicolli biliari sono formati dalla giustapposizione di due docce presenti sulle pareti cellulari dei poli biliari di due epatociti adiacenti. (B) Ai canalicoli seguono i canali di Hering (non indicati), composti in parte da epatociti e in parte da colangiociti, che terminano nei dotti biliari interlobulari dello spazio portale.
Tali duttuli contengono, come dimostrato recentemente, le cellule epatiche progenitrici bipotenziali (HPC) (Theise et al. 1999) (Alvaro et al. 2007). Essi sono formati da poche cellule epiteliali cubiche con citoplasma basofilo poggianti su una delicata membrana basale. I duttuli biliari iniziano unendosi, da un lato, agli epatociti che delimitano i capillari, realizzando, così, la giunzione duttulo-canalicolare. I duttuli di Hering, quindi, vanno a costituire veri e propri canali di raccordo tra epatociti e colangiociti e, convogliano la bile verso i dotti biliari dello spazio portale, dove si aprono nei dotti biliari interlobulari. In questi i colangiociti, che ne costituiscono la parete, formano uno strato continuo, appaiono di forma più cilindrica e presentano un nucleo sferico in posizione basale. I dotti interlobulari si affiancano, all’interno del connettivo contenuto negli spazi portobiliari, alle ramificazioni dell’arteria epatica, della vena porta, ai nervi e ai vasi linfatici, quindi confluiscono in condotti di calibro progressivamente maggiore, fino a formare due voluminosi condotti intraepatici che drenano i lobi destro e sinistro del fegato e che, a livello dell’ilo, danno origine alle vie biliari extraepatiche. Queste ultime si suddividono in via biliare principale e via biliare accessoria. La prima è costituita dai dotti epatici di destra e di sinistra, che si uniscono dando origine al dotto epatico comune. Il quale, dopo aver ricevuto il dotto cistico, continua con il dotto coledoco. La via biliare accessoria e’ rappresentata dalla colecisti e dal dotto cistico (Trattato di Anatomia Umana; Edi Ermes). Sulla base di differenze morfologiche, fenotipiche e funzionali tra i dotti di differente diametro, il sistema biliare intraepatico è stato suddiviso in: piccoli dotti (< 15 µm in diametro) e grandi dotti (> 15 µm in diametro) (Alpini et al. 1996). I piccoli dotti sono composti da 4-5 colangiociti e sono caratterizzati dalla presenza di un grande nucleo, scarso citoplasma, di una membrana basale, tight junctions e microvilli che si proiettano nel lume del dotto biliare. I colangiociti poi diventano progressivamente più grandi con un
5
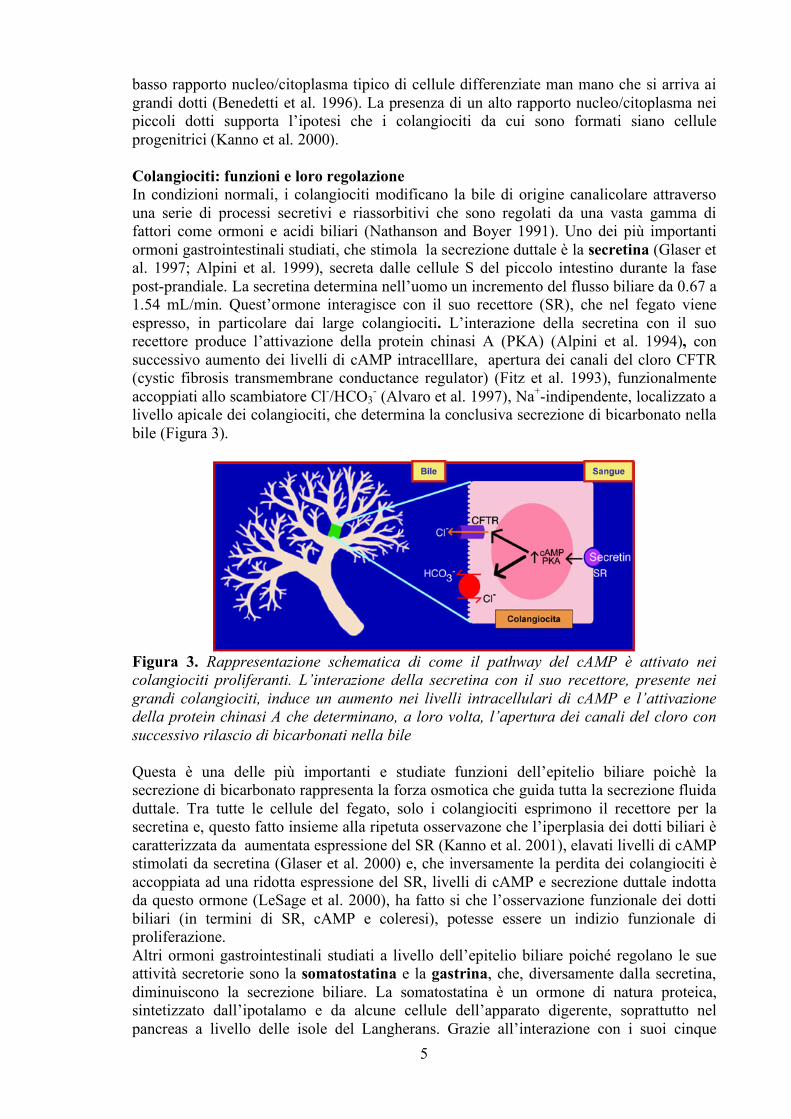
basso rapporto nucleo/citoplasma tipico di cellule differenziate man mano che si arriva ai grandi dotti (Benedetti et al. 1996). La presenza di un alto rapporto nucleo/citoplasma nei piccoli dotti supporta l’ipotesi che i colangiociti da cui sono formati siano cellule progenitrici (Kanno et al. 2000). Colangiociti: funzioni e loro regolazione In condizioni normali, i colangiociti modificano la bile di origine canalicolare attraverso una serie di processi secretivi e riassorbitivi che sono regolati da una vasta gamma di fattori come ormoni e acidi biliari (Nathanson and Boyer 1991). Uno dei più importanti ormoni gastrointestinali studiati, che stimola la secrezione duttale è la secretina (Glaser et al. 1997; Alpini et al. 1999), secreta dalle cellule S del piccolo intestino durante la fase post-prandiale. La secretina determina nell’uomo un incremento del flusso biliare da 0.67 a 1.54 mL/min. Quest’ormone interagisce con il suo recettore (SR), che nel fegato viene espresso, in particolare dai large colangiociti. L’interazione della secretina con il suo recettore produce l’attivazione della protein chinasi A (PKA) (Alpini et al. 1994), con successivo aumento dei livelli di cAMP intracelllare, apertura dei canali del cloro CFTR (cystic fibrosis transmembrane conductance regulator) (Fitz et al. 1993), funzionalmente accoppiati allo scambiatore Cl-/HCO3
- (Alvaro et al. 1997), Na+-indipendente, localizzato a livello apicale dei colangiociti, che determina la conclusiva secrezione di bicarbonato nella bile (Figura 3).
Figura 3. Rappresentazione schematica di come il pathway del cAMP è attivato nei colangiociti proliferanti. L’interazione della secretina con il suo recettore, presente nei grandi colangiociti, induce un aumento nei livelli intracellulari di cAMP e l’attivazione della protein chinasi A che determinano, a loro volta, l’apertura dei canali del cloro con successivo rilascio di bicarbonati nella bile
Questa è una delle più importanti e studiate funzioni dell’epitelio biliare poichè la secrezione di bicarbonato rappresenta la forza osmotica che guida tutta la secrezione fluida duttale. Tra tutte le cellule del fegato, solo i colangiociti esprimono il recettore per la secretina e, questo fatto insieme alla ripetuta osservazone che l’iperplasia dei dotti biliari è caratterizzata da aumentata espressione del SR (Kanno et al. 2001), elavati livelli di cAMP stimolati da secretina (Glaser et al. 2000) e, che inversamente la perdita dei colangiociti è accoppiata ad una ridotta espressione del SR, livelli di cAMP e secrezione duttale indotta da questo ormone (LeSage et al. 2000), ha fatto si che l’osservazione funzionale dei dotti biliari (in termini di SR, cAMP e coleresi), potesse essere un indizio funzionale di proliferazione. Altri ormoni gastrointestinali studiati a livello dell’epitelio biliare poiché regolano le sue attività secretorie sono la somatostatina e la gastrina, che, diversamente dalla secretina, diminuiscono la secrezione biliare. La somatostatina è un ormone di natura proteica, sintetizzato dall’ipotalamo e da alcune cellule dell’apparato digerente, soprattutto nel pancreas a livello delle isole del Langherans. Grazie all’interazione con i suoi cinque

6
recettori, questo ormone regola numerose funzioni corporee, come esercitare un potente effetto inibente sulla secrezione dell’ormone della crescita (GH) e della prolattina (PRL). La somatostatina legandosi ai suoi recettori (SSTR2) espressi dai grandi colangiociti, sia di ratto che di uomo, esercita il suo effetto inibitorio sulla proliferazione e sulla secrezione basale e stimolata dalla secretina, diminuendo i livelli intracellulari di cAMP (Tietz et al. 1995). La gastrina è un ormone polipeptidico prodotto dalle cellule G dello stomaco con funzione principale di regolare la secrezione gastrica. Tale ormone diminuisce la secrezione duttale di bile e i livelli di cAMP indotti da secretina mediante l’interazione con i suoi recettori CCK-A e CCK-B presenti sulla membrana basolaterale dei colangiociti (Glaser, Benedetti et al. 2000). Tra gli altri fattori che agiscono sull’epitelio biliare studiati, non possono non essere citati gli estrogeni, i principali ormoni sessuali femminili prodotti maggiormente dai follicoli ovarici, dal corpo luteo e dalla placenta. La sintesi di tali ormoni inizia dal colesterolo, passa per l’androstenedione fino all’estradiolo tramite l’enzima aromatasi. Negli ultimi anni è stato molto studiato il ruolo svolto dagli estrogeni sulle capacità proliferative e secretorie dell’albero biliare. Infatti i colangiociti presentano entrambi i recettori: ERα e ERβ. La loro espressione è correlata alla proliferazione biliare con una overespressione in condizioni patologiche (Alvaro et al. 2000; Alvaro et al. 2002; Alvaro et al. 2004). Inoltre, la riduzione della massa biliare indotta da ovariectomia in ratti BDL è associata ad una diminuita espressione degli stessi, insieme ad un aumento nei livelli di apoptosi (Alvaro et al. 2002). In definitiva, nei tessuti che esprimono i recettori degli estrogeni, la proliferazione può avvenire tramite: (i) un pathway genomico diretto, in cui l’attivazione del recettore induce i meccanismi trascrizionali nel nucleo, (ii) un pathway indiretto non genomico, in cui viene attivata una cascata di interazioni tra diverse proteine con differenti fattori trascrizionali (Filardo et al. 2000; Alvaro et al. 2002). In vari organi, come per esempio nell’utero, gli estrogeni sono in grado di modulare la proliferazione attivando una serie di eventi fosforilativi legati al pathway Ras/Raf/MAPK (Ruzycky 1996). Durante la proliferazione colangiocitaria, Alvaro et al. hanno dimostrato che una Src/Shc/ERK pathway viene attivata dagli estrogeni. Una cascata di eventi tipicamente coinvolta anche nell’azione di molti fattori di crescita, come l’IGF1, attraverso recettori tirosin-chinasici, che suggerisce un possibile sinergismo tra estrogeni e tali fattori nella modulazione della crescita dell’epitelio biliare (Adesanya et al. 1999; Alvaro et al. 2005). In aggiunta agli studi su modelli sperimentali, gli estrogeni sono stati anche indicati come induttori di crescita in diversi tipi di cancro. Il colangiocarcinoma di origine intraepatico esprime i recettori per gli estrogeni e cooperano nel regolare la crescita cellulare e l’apoptosi (Alvaro et al. 2006). Recentemente, molte osservazioni inducono a pensare che questi ormoni sono fondamentali nella neo-angiogenesi di tumori estrogeno-sensibili. Infatti è stato mostrato il ruolo del VEGF come mediatore degli effetti proliferativi causati dagli estrogeni nel colangiocarcinoma intraepatico (Hyder 2002; Mancino et al. 2009). Le colangiopatie I colangiociti hanno fisiologicamente una bassa attività replicativa ma, in alcune condizioni patologiche, come nelle cosidette colangiopatie diventano il target in alcune condizioni patologiche, le cosiddette colangiopatie o anche conosciute come “Vanishing Bile Duct Syndromes”, caratterizzate dalla progressiva scomparsa dei dotti biliari che conduce ad un grave grado di duttopenia con gravissime conseguenze per la funzionalità epatica. Le colangiopatie sono, infatti, responsabili di più del 20% dei trapianti di fegato negli adulti e dell’80% delle indicazioni di trapianto di fegato in età pediatrica. Esempi di queste patologie sono la Cirrosi Biliare Primitiva (PBC) (Alvaro, Invernizzi et al. 2004) e la Colangite Sclerosante Primitiva (PSC) (Chapman et al. 1980). La PBC è una malattia autoimmune, anche se la precisa eziopatogenesi è ancora sconosciuta, che colpisce maggiormente le donne da 35 a 60 anni (Talwalkar and Lindor
7
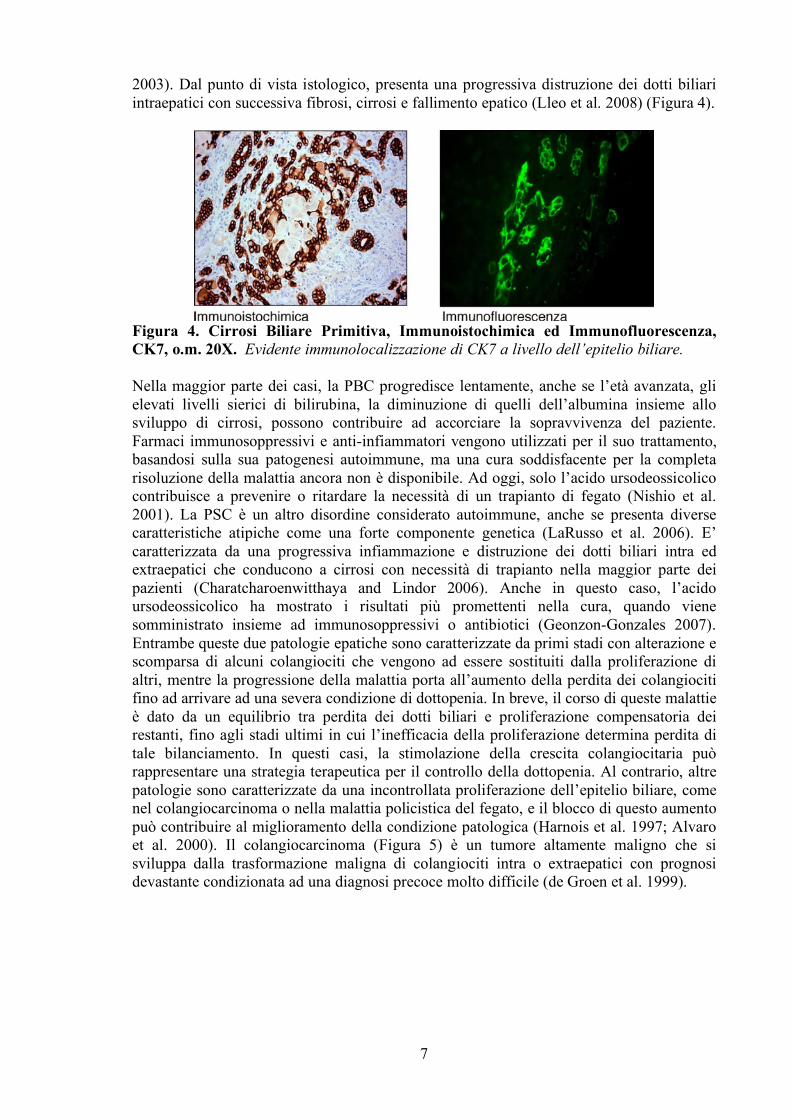
2003). Dal punto di vista istologico, presenta una progressiva distruzione dei dotti biliari intraepatici con successiva fibrosi, cirrosi e fallimento epatico (Lleo et al. 2008) (Figura 4).
Figura 4. Cirrosi Biliare Primitiva, Immunoistochimica ed Immunofluorescenza, CK7, o.m. 20X. Evidente immunolocalizzazione di CK7 a livello dell’epitelio biliare.
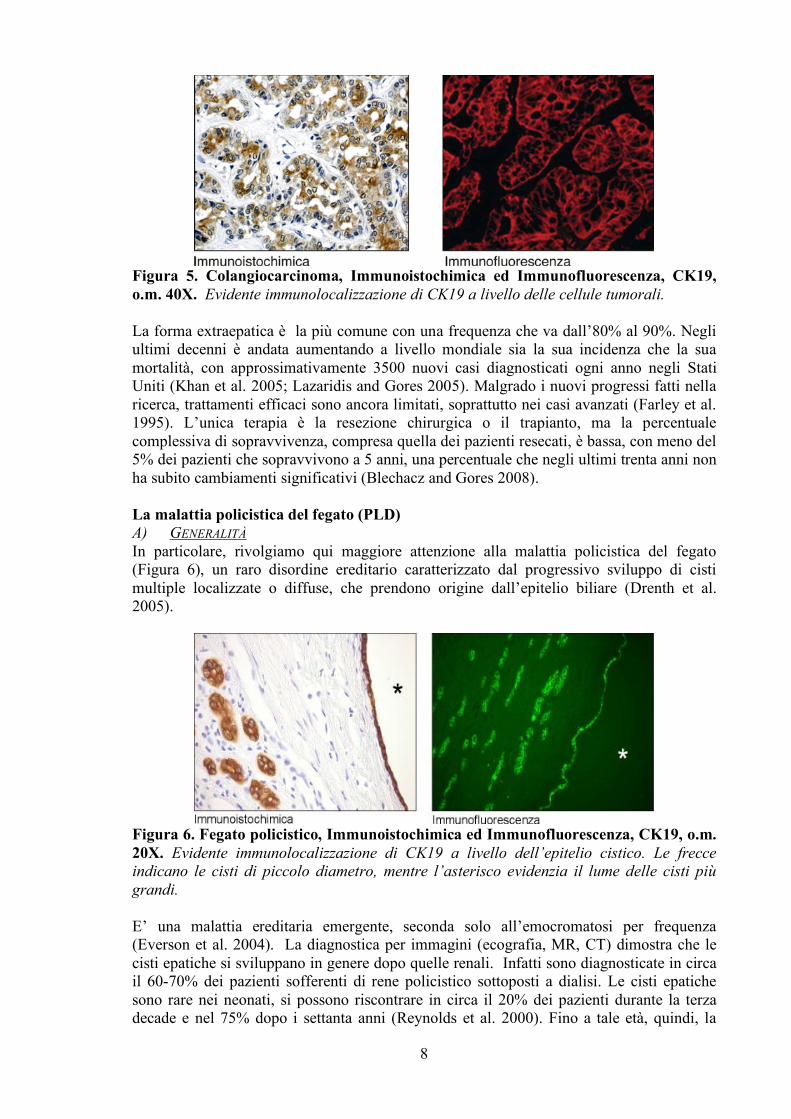
Nella maggior parte dei casi, la PBC progredisce lentamente, anche se l’età avanzata, gli elevati livelli sierici di bilirubina, la diminuzione di quelli dell’albumina insieme allo sviluppo di cirrosi, possono contribuire ad accorciare la sopravvivenza del paziente. Farmaci immunosoppressivi e anti-infiammatori vengono utilizzati per il suo trattamento, basandosi sulla sua patogenesi autoimmune, ma una cura soddisfacente per la completa risoluzione della malattia ancora non è disponibile. Ad oggi, solo l’acido ursodeossicolico contribuisce a prevenire o ritardare la necessità di un trapianto di fegato (Nishio et al. 2001). La PSC è un altro disordine considerato autoimmune, anche se presenta diverse caratteristiche atipiche come una forte componente genetica (LaRusso et al. 2006). E’ caratterizzata da una progressiva infiammazione e distruzione dei dotti biliari intra ed extraepatici che conducono a cirrosi con necessità di trapianto nella maggior parte dei pazienti (Charatcharoenwitthaya and Lindor 2006). Anche in questo caso, l’acido ursodeossicolico ha mostrato i risultati più promettenti nella cura, quando viene somministrato insieme ad immunosoppressivi o antibiotici (Geonzon-Gonzales 2007). Entrambe queste due patologie epatiche sono caratterizzate da primi stadi con alterazione e scomparsa di alcuni colangiociti che vengono ad essere sostituiti dalla proliferazione di altri, mentre la progressione della malattia porta all’aumento della perdita dei colangiociti fino ad arrivare ad una severa condizione di dottopenia. In breve, il corso di queste malattie è dato da un equilibrio tra perdita dei dotti biliari e proliferazione compensatoria dei restanti, fino agli stadi ultimi in cui l’inefficacia della proliferazione determina perdita di tale bilanciamento. In questi casi, la stimolazione della crescita colangiocitaria può rappresentare una strategia terapeutica per il controllo della dottopenia. Al contrario, altre patologie sono caratterizzate da una incontrollata proliferazione dell’epitelio biliare, come nel colangiocarcinoma o nella malattia policistica del fegato, e il blocco di questo aumento può contribuire al miglioramento della condizione patologica (Harnois et al. 1997; Alvaro et al. 2000). Il colangiocarcinoma (Figura 5) è un tumore altamente maligno che si sviluppa dalla trasformazione maligna di colangiociti intra o extraepatici con prognosi devastante condizionata ad una diagnosi precoce molto difficile (de Groen et al. 1999).
8
Figura 5. Colangiocarcinoma, Immunoistochimica ed Immunofluorescenza, CK19, o.m. 40X. Evidente immunolocalizzazione di CK19 a livello delle cellule tumorali. La forma extraepatica è la più comune con una frequenza che va dall’80% al 90%. Negli ultimi decenni è andata aumentando a livello mondiale sia la sua incidenza che la sua mortalità, con approssimativamente 3500 nuovi casi diagnosticati ogni anno negli Stati Uniti (Khan et al. 2005; Lazaridis and Gores 2005). Malgrado i nuovi progressi fatti nella ricerca, trattamenti efficaci sono ancora limitati, soprattutto nei casi avanzati (Farley et al. 1995). L’unica terapia è la resezione chirurgica o il trapianto, ma la percentuale complessiva di sopravvivenza, compresa quella dei pazienti resecati, è bassa, con meno del 5% dei pazienti che sopravvivono a 5 anni, una percentuale che negli ultimi trenta anni non ha subito cambiamenti significativi (Blechacz and Gores 2008). La malattia policistica del fegato (PLD) A) GENERALITÀ In particolare, rivolgiamo qui maggiore attenzione alla malattia policistica del fegato (Figura 6), un raro disordine ereditario caratterizzato dal progressivo sviluppo di cisti multiple localizzate o diffuse, che prendono origine dall’epitelio biliare (Drenth et al. 2005).
Figura 6. Fegato policistico, Immunoistochimica ed Immunofluorescenza, CK19, o.m. 20X. Evidente immunolocalizzazione di CK19 a livello dell’epitelio cistico. Le frecce indicano le cisti di piccolo diametro, mentre l’asterisco evidenzia il lume delle cisti più grandi. E’ una malattia ereditaria emergente, seconda solo all’emocromatosi per frequenza (Everson et al. 2004). La diagnostica per immagini (ecografia, MR, CT) dimostra che le cisti epatiche si sviluppano in genere dopo quelle renali. Infatti sono diagnosticate in circa il 60-70% dei pazienti sofferenti di rene policistico sottoposti a dialisi. Le cisti epatiche sono rare nei neonati, si possono riscontrare in circa il 20% dei pazienti durante la terza decade e nel 75% dopo i settanta anni (Reynolds et al. 2000). Fino a tale età, quindi, la

9
malattia rimane di solito asintomatica, poi l’aumento del numero di cisti e la crescita delle loro dimensioni nel fegato provocano comparsa macroscopica di una protuberanza addominale con sintomatologia associata a nausea, vomito e senso di sazietà derivante dalla pressione che la ghiandola epatica determina su stomaco e pancreas (Everson, Taylor et al. 2004; Drenth, Martina et al. 2005). Lo sviluppo delle cisti epatiche è più precoce e più severo nel sesso femminile rispetto a quello maschile: inoltre, le cisti nel sesso femminile appaiono di maggiori dimensioni; le nullipare che non hanno fatto uso di estrogeni sono meno predisposte a formare cisti rispetto alle multipare o alle donne che hanno fatto uso di ormoni; infine, l’uso di estrogeni nel post-menopausa sembra favorire l’espansione delle cisti (Everson, Taylor et al. 2004; Alvaro et al. 2008). B) GENETICA L’osservazione della casistica familiare e gli studi genetici più recenti hanno dimostrato che la malattia policistica del fegato si può presentare sotto forma autosomica dominante e autosomica recessiva (Figura 7).
Figura 7. Schema riassuntivo della genetica della malattia policistica del fegato nelle sue due forme: autosomica dominante, che coinvolge mutazioni a carico dei geni PKD1, PKD2, PRKCSH e SCE63, e autosomica recessiva che interessa modificazioni nel gene PKHD1 codificante per la fibrocistina.
La più comune forma di malattia policistica epatica autosomica dominante è associata con la malattia policistica renale, comprendente quattro varianti, tre ad ereditarietà autosomica dominante (AD-PKD), che si riscontra con una frequenza 1:500-1:1000, per un numero d’individui affetti pari a circa 500.000 negli USA e 4-6 milioni nel mondo (Wilson 2001) e una variante di tipo recessivo (AR-PKD), rara anomalia di sviluppo che interessa maggiormente l’età infantile. I tipi più frequenti di ADPKD sono legati rispettivamente alla mutazione dei geni PKD1 e PKD2. PKD1 codifica per la Policistina1 (PC-1) (Figura 8), una glicoproteina integrale di membrana (Ong and Harris 2005) in grado di ricevere segnali dal compartimento extracellulare e attivare vie fosforilative nell’intracellulare che innescano, a livello nucleare, la trascrizione di specifici geni (Delmas 2004). Mutazioni in tale gene interessano l’85-90% dei casi di malattia (Anyatonwu and Ehrlich 2004). PKD2 codifica per la Policistina2 (PC-2) (Figura 8), una proteina di membrana con la porzione C terminale del dominio intracellulare tipica delle proteine che legano il calcio (Ca++). In rarissimi casi di malati di policistosi epatica che non presentavano alterazioni dei geni PDK-1 e 2, è stata più recentemente dimostrata l’alterazione di due diversi e distinti geni, il gene protein-kinase-C-substrate-80KH/ PRKCSH, che codifica per la proteina epatocistina (Drenth et al. 2004), ed il gene SEC63 (Figura 8), codificante per la proteina omonima che

10
provocano la malattia policistica epatica isolata (iPLD), non associata, cioè, alla malattia renale (Drenth, Martina et al. 2005). Infine, è stata descritta la presenza della malattia policistica autosomica recessiva, più rara e grave, legata alla mutazione di un altro gene PKHD1 codificante per la fibrocistina (Ward et al. 2003) (Figura 8). Un recettore integrale di membrana con siti extracellulari altamente glicosilati, un singolo dominio transmembrana e uno corto citoplasmatico (Ward, Yuan et al. 2003). In aggiunta, è stato anche dimostrato che co-localizza con la PC-2 sul ciglio primario, suggerendo la possibilità che queste due proteine possano intervenire su comuni pathways molecolari (Zhang et al. 2004). Il quadro clinico è dominato da una severa malattia cistica dei reni che rappresenta una delle più importanti nefropatie infantili con frequenza 1:20.000 e una mortalità del 30%, legata ad una fibrosi epatica congenita molto frequente in questi malati, oltre che alla possibile ma più rara dilatazione dei dotti biliari e formazione di cisti (Ward et al. 2002).
Figura 8. Rappresentazione schematica delle quattro proteine coinvolte nella genetica della malattia policistica del fegato. C) IL CIGLIO PRIMARIO: UN SISTEMA “GPS” DELLA CELLULA? L’espressione fenotipica dei primi due geni considerati: PKD1-PKD2 e le loro alterazioni possono essere riscontrate nel ciglio, una struttura sensoriale con una lunghezza compresa tra 5 e 7µm, diametro di circa 0,25 µm e una proiezione sulla faccia apicale (Nauli et al. 2003; Pazour and Witman 2003); ad una sezione traversa mostra struttura microtubulare interna 9 + 0 caratteristica delle ciglia primarie prive di motilità per la mancanza del paio di microtubuli centrali (Figura 9). Tali strutture sono circondate da una membrana che si continua con il plasma lemma della stessa cellula.

11
Figura 9. Struttura microtubulare delle due tipologie di ciglio esistenti. Il ciglio primario presenta una composizione 9+0, che si associa alla incapacità di movimento autonomo (es. epitelio tubulare renale). Mentre il ciglio dotato di mobilità presenta una composizione 9+2 (es. epitelio della mucosa respiratoria).
Nel fegato normale sono presenti queste ciglia a livello della faccia apicale dei colangiociti dei piccoli e grandi dotti biliari. Ogni colangiocita possiede un singolo ciglio, che funge da antenna sensoriale ed esprime molti recettori associati al controllo della proliferazione e/o apoptosi. Tanto che nell’ultimo meeting della Società Americana di Biologia Cellulare (ASCB, 48° congresso annuale, San Francisco, CA, 2008), Schneider L. et al. hanno presentato uno studio dal titolo “Primary cilium coordinates directional cell migration”, spiegando che il comportamento del ciglio primario è simile a quello del Global Positioning System che aiuta un qualsiasi veicolo a trovare la sua giusta destinazione. Questo gruppo di ricerca ha investigato il ruolo del ciglio primario nel riparo delle ferite, dimostrando che esso è orientato in modo da captare gli stimoli necessari al al processo di riparazione, se il ciglio è geneticamente danneggiato il riparo sarà molto più lento e incompleto. In generale, queste scoperte confermano che il ciglio è in grado di percepire i segnali nel compartimento extracellulare e guidare la cellula nelle opportune risposte, che coinvolgono processi fondamentali come il riparo al danno, la crescita e la morte cellulare. Per poter svolgere queste importanti attività, il ciglio possiede molti complessi recettoriali, tra cui quello formato dall’eterodimero Policistina 1/Policistina 2 (Forman et al. 2005) che recenti studi localizzano alla base delle ciglia primarie dell’epitelio renale e che è coinvolto nella trasduzione del Ca++, a seguito della mobilizzazione del ciglio stesso provocato dal flusso (Sloboda and Rosenbaum 2007) (Figura 10).

12
Figura 10. Raffigurazione schematica di ciglia primarie con l’organizzazione molecolare del complesso Policistina1-Policistina 2. Poiché in risposta a segnali esterni provenienti dal liquido tubulare, la Policistina1 attiva un meccanismo di trasmissione attraverso proteina G, che ha compito di accoppiare funzionalmente il recettore chimico di membrana, dopo sua attivazione, con un enzima – effettore capace di produrre e liberare nel citoplasma cellulare un secondo messaggero, nel caso specifico l’inositolo trifosfato (IP3). L’azione di quest’ultimo è selettivamente indirizzata alle membrane degli organuli intracellulari che fungono da serbatoi di Ca++, in particolare nel reticolo endoplasmatico (RE) dove attiva la Policistina 2 portando allo stato di apertura i suoi canali con rapido rilascio in forma libera nel citosol (Principi di Fisiologia e Biofisica della Celllula; Taglietti V, Casella C. La Gogliardica Pavese). Molte delle proteine coinvolte nella patogenesi della malattia policistica sono localizzate sulle ciglia primarie. Cellule isolate da topi transgenici che hanno perso la funzione della PC-1 formano ciglia ma non aumentano l’influsso di Ca++ in risposta al fisiologico flusso. Similarmente, bloccando la PC-2 tramite anticorpi, non si ha risposta al flusso. Questi dati suggeriscono che PC-1 e PC-2 sono molto importanti nel percepire e trasmettere i segnali che vengono dal movimento del flusso, agendo sullo stesso pathway meccanotrasduttore (Nauli, Alenghat et al. 2003). L’alterazione dell’azione di meccanotrasduttore del complesso Policistina 1/2 potrebbe essere alla base della cistogenesi, perché il Ca++ media differenti funzioni importanti della cellula, tra cui l’espressione genica, crescita, differenziamento e apoptosi (Alenghat et al. 2004; Masyuk et al. 2006). D) COME LE CISTI AUMENTANO DI VOLUME Tipicamente, per quanto riguarda la forma dominante, pochi sono i casi che presentano formazioni cistiche in età giovanile. Infatti queste iniziano a comparire con maggior frequenza dopo i 60 anni, insieme ad una significativa variabilità familiare. A suggerire che devono esistere fattori in aggiunta alla semplice mutazione germinale dei geni interessati. Infatti attualmente la teoria più accreditata sull’origine delle cisti è basata sul “two hit” model (Figura 11) in cui l’inattivazione di entrambi gli alleli del gene avviene per una mutazione germinale seguita da una somatica in una cellula epiteliale che si accresce in modo clonale nella cisti (Pei 2001). Così abbiamo una iniziale mutazione della linea germinale in una copia di PKD-1 o PKD-2 (first hit) cui segue una seconda mutazione che porta a perdita di attività nella copia funzionale del gene (second hit) (Pei et al. 1999). Quest’ultima da inizio alla proliferazione cellulare e alla formazione della cisti isolata dalla sede di origine.

13
Figura 11. Illustrazione di un ipotetico modello di cistogenesi. Le cellule dell’epitelio biliare di pazienti affetti da ADPKD sono eterozigoti per una mutazione germinale e rispondono normalmente al flusso luminale. Le cellule biliari che delimitano le cisti epatiche perdono l’abilità di percepire questi stimoli meccanici che vengono dal flusso biliare, probabilmente a causa della perdita dell’allele normale o per il danno della variazione genetica. Questa situazione conduce ad un dedifferenziamento e iperproliferazione delle cellule mutate con restringimento del lume. Tanto che la formazione e la crescita delle cisti può ulteriormente essere facilitata da un andamento anomalo del flusso biliare.
Un recente studio ha mostrato che circa l’8% delle cisti è formata da una inattivazione somatica del PKD1 e una germinale in PKD2; mentre l’inattivazione somatica di PKD2 crea il 13% delle cisti (Pei 2001). Tutte queste mutazioni somatiche sono dovute a frame-shift, delezione o inserzione di alcune coppie nucleotidiche che provocano spostamento del codice di lettura genetico e successivamente espressione alterata della proteina. Questa affascinante ipotesi permette di spiegare sia la natura dello sviluppo delle cisti che la variabilità fenotipica presente nelle diverse famiglie. Una volta che le cisti si sono formate, si è ipotizzato che esse possano aumentare di volume grazie all’accresciuto livello di fattori di crescita e di citochine sia a livello plasmatico che all’interno del fluido cistico. Infatti negli ultimi anni, gli studi riguardanti questo importante aspetto si sono moltiplicati, per scoprire che nel fluido cistico sono presenti citochine come IL-6 e IL-8 (Nichols et al. 2004), ma anche molti fattori come VEGF, Angiopoietin-1, Angiopoietin-2 (Fabris et al. 2006; Amura et al. 2007) estrogeni, IGF1 (Alvaro, Onori et al. 2008) in grado di promuovere la crescita delle cisti epatiche tramite un meccanismo sia autocrino che paracrino, inducendo anche cambiamenti e regolazione della morfogenesi e della crescita del sistema vascolare che sostiene lo sviluppo delle cisti (Ross et al. 2001). L’espressione di questi fattori angiogenici nell’epitelio biliare cistico insieme a NCAM, una molecola di adesione tipicamente presente nei colangiociti immaturi (Fabris et al. 2000), hanno concentrato l’attenzione sull’aspetto immaturo dell’epitelio biliare cistico. Collegando questa ipotesi al fatto che le due proteine PC-1 e PC-2, oltre ad essere coinvolte nella regolazione della secrezione biliare come meccanorecettori, influenzano anche la trascrizione di molte proteine regolatrici. Si può allora speculare che l’alterata funzionalità di queste proteine potrebbe causare la perdita di segnali di differenziamento favorendo il mantenimento di un fenotipo immaturo delle cellule biliari che circondano le cisti (Wilson 2001). Altri potenti agenti proliferativi, come l’ENA-70 (epithelial netrophil-activating peptide) o il GRO-alpha (growth-related oncogene-alpha), che come l’IL-8 agiscono su un recettore 2 delle chemochine (CXCR2), sono stati trovati in alte concentrazioni nelle cisti epatiche ma non in quelle renali. Supportando l’ipotesi che gli agonisti leganti il recettore CXCR2 promuovono la crescita delle cisti epatiche (Amura et al. 2008). In generale, l’accrescimento colangiocitario in corso di malattia policistica del fegato è regolata dall’ cAMP. Infatti, usando un modello in vivo ed uno in vitro di malattia policistica, sono stati misurati i livelli di cAMP in assenza o presenza di octreotide, un analogo della somatostatina che inibisce il cAMP, scoprendo che questo composto abbassa tali livelli e potrebbe essere utilizzato per bloccare la crescita cistica (Masyuk et al. 2007).

14
Modelli sperimentali in vivo Numerosi studi hanno dimostrato che l’albero biliare intraepatico gioca un ruolo critico in molte funzioni epatiche, tra cui la formazione della bile, la sua rigenerazione, il riparo al danno e l’angiogenesi. Per poter meglio studiare i meccanismi cellulari alla base di questi complessi processi, molti modelli animali sono stati prodotti. Quello più utilizzato è il modello di ratto con legatura del dotto biliare principale (BDL) per mimare ciò che succede nelle malattie colestatiche croniche. Tale legatura induce una notevole espansione della massa colangiocitaria (30% rispetto al 2% in condizioni normali) che facilita lo studio della patofisiologia biliare. Grazie a recenti studi, si è dimostrato che l’accrescimento dei colangiociti è regolato da diversi fattori di crescita (VEGF, IGF1, NGF) (Gigliozzi et al. 2004; Alvaro, Metalli et al. 2005; Gaudio et al. 2006; Gaudio et al. 2006) ormoni come estrogeni, prolattina, progesterone (Alvaro et al. 2006; Taffetani et al. 2007; Glaser et al. 2008), neuropeptidi come la serotonina (Marzioni et al. 2005) e sali biliari (Alvaro 1999; Alvaro, Gigliozzi et al. 2000; Marzioni et al. 2003). In particolare gli estrogeni sono importanti induttori di crescita e differenziamento nelle cellule che esprimono il loro recettore (ER) (Eagon et al. 1985). Per molti anni si è pensato che potessero svolgere un ruolo importante nello sviluppo e nella progressione delle patologie biliari (Ahlqvist 1980). Questa ipotesi era basata su numerose osservazioni cliniche: (i) la più comune colangiopatia, la PBC, che prevale nel sesso femminile nel periodo peri- e post- menopausa (Floreani et al. 1991), (ii) disfunzioni endocrine sono frequenti nella PBC (Stellon and Williams 1986), insieme ad un’alta incidenza di osteoporosi nel post-menopausa, segno di una deficienza estrogenica (Hodgson et al. 1985) e, (iii) la progressione della malattia policistica del fegato è significativamente influenzata dal sesso, dal numero di gravidanze e da trattamenti estrogenici esogeni (Alvaro, Onori et al. 2008). Come precedentemente accennato, diversi studi sul ratto hanno mostrato che in condizioni colestatiche sono presenti alterazioni nel metabolismo epatico degli estrogeni, inducendo aumento dei livelli serici di estradiolo, con influenza nella progressione della malattia (Chen et al. 1998). In aggiunta, questi ormoni aumentano la proliferazione e la secrezione nei colangiociti (Alvaro, Alpini et al. 2002) attivando il pathway Ras/Raf/Src/Shc/ERK1/2 sul quale agiscono in maniera sinergica differenti fattori di crescita come l’NGF e l’IGF1 (Gigliozzi, Alpini et al. 2004; Alvaro, Metalli et al. 2005). L’utilizzo di antagonisti dei recettori degli estrogeni, come il Tamoxifene, blocca l’aumento della crescita colangiocitaria dovuta a BDL inducendo apoptosi e diminuzione della fosforilazione di ERK1/2 (Alvaro, Alpini et al. 2000). (Alvaro, Alpini et al. 2000). Nelle linee cellulari di colangiocarcinoma, il Tamoxifene induce apoptosi tramite attivazione del pathway Fas ligando/recettore (Sampson et al. 1997). Presi insieme questi risultati indicano che l’antagonismo degli estrogeni può inibire la proliferazione dei colangiociti e aumentare la loro apoptosi attivando un meccanismo Fas dipendente. Contestualmente, si è visto che l’ovariectomia nei ratti BDL di sesso femminile provoca una diminuzione nel numero di dotti biliari e il trattamento con 17-β estradiolo di questi ratti previene il calo nella massa biliare intraepatica e l’espressione dei recettori degli estrogeni torna a livelli normali (Alvaro, Alpini et al. 2002). Il ruolo di altri ormoni sessuali è stato valutato, per esempio la prolattina viene secreta dai colangiociti e regola la loro crescita tramite l’attivazione del Ca++/PKC pathway. L’ultimo ormone studiato dal nostro gruppo di ricerca è stato il progesterone, il quale stimola la proliferazione e l’attività colangiocitaria attraverso un meccanismo autocrino, dato che l’epitelio biliare presenta l’intero pathway enzimatico per la steroidogenesi (Taffetani, Glaser et al. 2007; Glaser, DeMorrow et al. 2008). Altri modelli sperimentali che inducono, invece, un danno all’albero biliare sono ad esempio quelli con somministrazione di tetracloruro di carbonio (CCl4) (LeSage et al. 1999). Questo composto è in grado di distruggere i colangiociti più grandi, con conseguente attivazione e differenziazione dei piccoli colangiociti, i quali iniziano ad accrescersi assumendo un

15
fenotipo tipico dei grandi per poter compensare alla loro perdita. L’attività e la crescita dell’albero biliare è anche regolata dal sistema nervoso vegetativo parasimpatico e ortosimpatico. I colangiociti, infatti, esprimono il recettore per l’acetilcolina, responsabile del controllo dei processi di secrezione (LeSage et al. 2004). Al fine di capire meglio il ruolo del sistema nervoso parasimpatico nel regolare la crescita dell’epitelio biliare, è stato utilizzato un modello sperimentale di ratto BDL sottoposto a vagotonia. In tale modello c’è una riduzione nella proliferazione e un aumento dell’apoptosi a carico dei colangiociti, effetti che possono essere prevenuti tramite somministrazione di un acido biliare, il taurocolato (TC) (Marzioni, LeSage et al. 2003). Quindi preservare l’innervazione simpatica dei colangiociti nel ratto sottoposto ad epatoectomia parziale è fondamentale per supportare la rigenerazione epatica. Più in dettaglio, per quanto riguarda la malattia policistiica, il gruppo di LaRusso ha studiato l’espressione della fibrocistina, proteina prodotta dall’alterazione del gene PKHD1, in ratti PCK con mutazione spontanea che sviluppano PKD simile a quella umana. Studi su questi animali hanno dimostrato che: (i) l’albero biliare nel ratto PCK è marcatamente distorto, mostra dilatazioni sacculari, cisti multiple di differenti misure, ciglia più corte con strutture abberanti per presenza di estensioni bulbose; (ii) la fibrocistina è espressa nelle ciglia di colangiociti normali; (iii) i colangiociti di ratto PKC presentano ciglia con ridotte dimensioni e malformazioni e con assenza di espressione di fibrocistina; e (iv) in colangiociti isolati in cui è stata effettuata una inattivazione del gene PKHD1 mediante silenziamento dell’RNA, si è osservato un accorciamento del ciglio e la mancata espressione della fibrocistina (Masyuk et al. 2003). Questi dati permettono quindi di affermare che la fibrocistina è presente a livello del ciglio dei colangiociti normali e che una sua alterazione o down-regolazione possa essere collegata, almeno in questo modello sperimentale, ad una modificazione del ciglio stesso e alla formazione delle cisti biliari. Sembra perciò ovvio che la fibrocistina sia richiesta per mantenere la funzionalità ciliare. L’ormone follicolo stimolante (FSH) L’ormone follicolo stimolante (Follicle Stimulating Hormone - FSH) è una glicoproteina secreta dalle cellule basofile dell’adenoipofisi, sotto il controllo dell’ormone rilasciante le gonadotropine (GnRH) prodotto dall’ipotalamo. L’FSH viene anche detto gonadotropina poichè esercita i suoi principali effetti sulle gonadi. Infatti, nella donna, induce la maturazione dei follicoli e la produzione di estrogeni; che aumentando inducono inibizione del rilascio di GnRH da parte dell’ipotalamo tramite un meccanismo di feedback negativo; mentre nell’uomo, stimola la produzione di spermatozooi e di ormoni maschili da parte dei testicoli. L’FSH è un eterodimero composto da una subunita’ α, simile a quella di altri ormoni ipofisari come l’ormone luteinizzante (LH) e l’ormone tiroide stimolante (TSH) ed una subunita’ β che lo differenzia da essi. L’FSH esercita le sue funzioni legandosi al suo recettore (FSHR) (Figura 12), codificato da un esteso gene, contenente 11 esoni che favorisce estesi slicing alternativi (Tena-Sempere et al. 1999). Sono stati distinte, infatti, tre isoforme con distinti motivi strutturali (Babu et al. 2001). La forma canonica di questo recettore (FSH-R1) appartiene alla famiglia dei recettori G protein-coupled (7MSR) e sono associati all’attivazione dell’adenil ciclasi (Babu et al. 2000).

16
Figura 12. Schema della struttura esonica delle tre varianti di FSHR caratterizzate dal laboratorio di Sairam M. L’FSHR è un recettore transmembrana che interagisce con l’FSH, un eterodimero composto da due subunità: una alpha e una beta. La prima è uguale per tutti gli ormoni LH, TSH e hCG e contiene 92 aminoacidi. La subunità β, invece, è specifica per ciascuno e nel caso dell’FSH contiene 118 aminoacidi e viene detto FSHβ. Tale subunità si lega con il suo specifico recettore. Questo recettore appartiene alla famiglia dei recettori accoppiati a proteina G (GPCR) con 7 domini transmembrana (TMD) codificato da 10 esoni. FSH-R2 differisce da FSH-R1 poichè contiene 25 aminoacidi in più al C-terminale. FSH-R3 contiene gli stessi primi 8 esoni seguiti da un segmento spanning.
Dopo che il recettore si è legato al suo agonista, viene fosforilato da una chinasi accoppiata ad una proteina G, che permette il reclutamento della β-arrestina e la sua internalizzazione (Troispoux et al. 1999). Di seguito, viene stimolata l’adenilato ciclasi che determina produzione del secondo messaggero cAMP e attivazione della protein chinasi A (PKA), che fosforilando differenti attivatori trascrizionali (Zhang et al. 1991; Gromoll et al. 1996; Marion et al. 2006), conduce ai cambiamenti nell’espressione genica indotta dall’FSH (Marion, Kara et al. 2006). Una delle tre varianti derivate dallo splicing (FSH-R3) ha la tipologia del recettore per fattori di crescita e può promuovere la crescita cellulare in una via cAMP-indipendente, coinvolgente i canali per il calcio voltaggio dipendenti (Touyz et al. 2000). Recenti studi hanno dimostrato che il trattamento con FSH attiva meccanismi associati alla proliferazione in diverse tipologie di colture cellulari, come, per esempio, in quelle ovariche normali e maligne (Ji et al. 2004; Choi et al. 2005). Poco si conosce sul ruolo dell’FSH nella patofisiologia epatica, in special modo sui colangiociti. Alcuni studi hanno mostrato che la cirrosi epatica è associata a disfunzioni endocrine, particolarmente a carico dell’asse delle gonadi (Gonzales et al. 2007). Pazienti con danno epatico hanno bassi livelli di LH e FSH (Bell et al. 1995), infatti una deficienza gonadotropinica esiste anche nei casi di emocromatosi (Diamond et al. 1989). Suggerendo che sconvolgimenti nelle funzioni dell’asse ipotalamo-gonadi giocano un ruolo fondamentale nei pazienti con problemi epatici. Per tale motivo, il nostro obiettivo è stato quello di andare a valutare se l’FSH possa essere in grado di esercitare qualche azione sull’epitelio biliare di ratto normale e BDL e su quello umano normale e in corso di malattia policistica del fegato.
METODOLOGIA SPERIMENTALE Studio in vivo TRATTAMENTO DEGLI ANIMALI Ratti normali di ceppo Fischer 344 (peso compreso tra150 e 175 gm) maschi e femmine acquistati dalla Charles River (Wilmington, MA) sono stati mantenuti in un ambiente a temperatura costante di 22°C con un ciclo luce-buio di 12 ore ed alimentati ad libitum con cibo standard per ratti e con libero accesso all’acqua. Essi sono stati trattati per una

17
settimana con NaCl 0,9% o con FSH (6 µg/kg body weight) (Ritter et al. 2008) tramite minipump osmotica (IP implanted Alzet®). Allo stesso tempo, ratti BDL maschi e femmine, immediatamente dopo la legatura (Alpini et al. 1988), sono stati trattati con NaCl 0,9%, FSH, Antide (un antagonista del GnRH, che blocca la secrezione dell’FSH) (Ferris et al. 2007) o con un anticorpo specifico anti-FSH (Meachem et al. 1998). Nella tabella 1 sono riportati tutti i gruppi sperimentali utilizzati. Tabella 1:
ANIMALI
TRATTAMENTO
Normali femmine e maschi + 0.9% NaCl Ratti normali sono stati trattati tramite minipump osmotica intraperitoneale Alzet che rilasciava 0.9% NaCl per 1 settimana
Normali femmine e maschi + FSH Ratti normali sono stati trattati tramite minipump osmotica intraperitoneale Alzet che rilasciava FSH per 1 settimana
BDL femmine e maschi + siero non-immune
Appena dopo la legatura del dotto biliare ai ratti è stata impiantata una minipump osmotica intraperitoneale Alzet che rilasciava siero non-immune per 1 settimana
BDL femmine e maschi + Antide
Appena dopo la legatura del dotto biliare ai ratti è stata impiantata una minipump osmotica intraperitoneale Alzet che rilasciava Antide per 1 settimana
BDL femmine e maschi + anticorpo anti-FSH
Appena dopo la legatura del dotto biliare ai ratti è stata impiantata una minipump osmotica intraperitoneale Alzet che rilasciava anticorpo anti-FSH per 1 settimana
BDL femmine e maschi + anticorpo anti-FSH
Appena dopo la legatura del dotto biliare, i ratti sono stati trattati tramite minipump osmotica intraperitoneale Alzet che rilasciava anticorpo anti-FSH per 1 settimana
Prima di ogni procedura sperimentale, gli animali sono stati anestetizzati con sodio pentobarbital (50 mg/kg weight, IP) secondo le locali regolamentazioni. Da tutti gli animali trattati sono stati collezionati frammenti epatici e i colangiociti purificati attraverso la tecnica di separazione per immunoaffinità usando uno anticorpo monoclonale di topo gentilmente concesso dal Dr. R. Faris della Brown University, Providence, RI che riconosce un antigene di membrana non identificato, espresso da tutti i colangiociti intraepatici (Ishii et al. 1989). La sopravvivenza cellulare e la conta è stata eseguita usando Trippan blue. PRESENZA DELL’FSHR La presenza del recettore dell’FSH (FSHR) è stata valutata tramite: (i) immunoistochimica su sezioni di fegato proveniente dai gruppi di animali trattati in vivo e, (ii) real-time PCR sull’RNA totale proveniente dai colangiociti purificati da tutti i gruppi sperimentali. I frammenti di parenchima epatico, immediatamente dopo il prelievo, sono stati fissati per immersione in formalina tamponata al 10% per 24 ore a temperatura ambiente. Successivamente sono stati sottoposti alle procedure di inclusione in paraffina che prevedono disidratazione del frammento in etanolo a concentrazioni crescenti, diafanizzazione in xilolo e conseguente inclusione in paraffina a basso punto di fusione

18
(56°C). Sono, quindi, state effettuate sezioni dello spessore di 3-4 µm, poste su vetrini precedentemente trattati con L-polilisina allo 0,1% e, dopo sparaffinatura in xilolo e idratazione in alcool a concentrazioni decrescenti sono state trattate con perossido d’idrogeno al 2,5% in metanolo per 30 minuti al fine di bloccare l’attività della perossidasi endogena. Dopo lavaggio in tampone fosfato salino (PBS) a pH 7.4, i preparati sono stati incubati overnight a 4°C con anticorpo specifico anti-FSHR (santa cruz biotechnology, Inc sc-7798; 1:50) in camera umida. Il giorno successivo sono state lavate in PBS e incubate per 20 minuti con anticorpo secondario biotinilato (Dako Cytomation LSAB Plus System-HRP, code K0690, Glostrup), dopo passaggio in PBS sono state incubate per altri 20 minuti con il complesso Avidina-Biotina (Dako Cytomation Liquid DAB Plus Substrate Chromogen System, Glostrup) e lavate nuovamente con PBS. L’avvenuta immunoreazione è stata evidenziata incubando le sezioni con 3,3 diaminobenzidina (DAB) (Dako Cytomation Liquid DAB Plus Substrate Chromogen System, Glostrup). I controlli negativi sono stati ottenuti omettendo l’Ab primario. Le osservazioni sono state effettuate con un microscopio ottico BX-51 (Olympus, Tokyo, Japan) dotato di videocamera (Spot Insight; Diagnostic Instrument, Inc., Sterling Heights, MI) e processate con un programma di analisi delle immagini (IAS. Delta Sistemi, Rome, Italy). In aggiunta, abbiamo valutato l’espressione del messaggio dell’FSHR nei colangiociti purificati usando RT2 Real-Time assay da SuperArray (Frederick, MD) (Francis et al. 2008). L’RNA totale e’ stato estratto dai colangiociti (1x105) con RNeasy Mini kit (Qiagen Inc., Valencia, CA) e reverso con Reaction ReadyTM First Strand cDNA synthesis kit (SuperArray, Frederick, MD). Ad 1 µl del template sono stati aggiunti 12,5 µl di SYBR Green PCR master mix, 10,5 µl di acqua distillata e 1 µl di RT2 PCR primer (Superarray, Frederick, MD) specificamente designato per l’mRNA dell’FSHR e del gene di controllo (GAPDH). Il plate è stato posto nel real-time thermal cycler (MX30005P, Stratagene) e fatto correre a 95°C per 10 minuti e poi per 40 cicli a 95°C per 15 secondi e a 60°C per 1 minuto. I dati sono stati espressi come livelli di mRNA relativo ± SEM di FSHR su GAPDH.
EFFETTI DELL’FSH SULLA PROLIFERAZIONE COLANGIOCITARIA Per valutare la massa biliare è stato effettuato uno studio immunoistochimico in tutti i gruppi sperimentali per la citocheratina 19 (CK-19), un marker specifico dell’epitelio biliare. La colorazione è stata eseguita come precedentemente indicato, utilizzando come anticorpo primario l’anti-CK-19 (santa cruz biotechnologies, Inc; sc-33120; 1:50). Parallelamente alla crescita colangiocitaria abbiamo, anche valutato l’apoptosi delle stesse cellule dopo i diversi trattamenti in vivo. Per far ciò è stato utilizzato il metodo Tunel (terminal deoxynucleotidyl transferase-mediated triphosphate end-labeling) (ApoTag kit), secondo le indicazioni suggerite dal produttore. I controlli negativi sono stati eseguiti tramite incubazione con un adeguato siero non immune al posto dell’anticorpo primario ed hanno mostrato un’assenza dell’immunoreazione. Le immagini sono state acquisite con microscopio ottico (precedentemente descritto), per poi essere analizzate con il sistema di elaborazione d’immagine IAS 2000 (Delta Sistemi, Roma) al fine di quantificare la massa biliare e l’apoptosi.
PATHWAY INTRACELLULARE SU CUI AGISCE L’FSH In accordo con altri studi (Francis et al. 2004; LeSage, Alvaro et al. 2004) abbiamo misurato i livelli intracellulari di cAMP basali e indotti dalla stimolazione per 5 minuti con secretina. Poichè l’aumento dei livelli di cAMP dopo secretina sono stati dimostrati essere associati a cambiamenti nella proliferazione e secrezione di un gran numero di epiteli, incluso quello biliare (Alpini et al. 1997; Glaser, Benedetti et al. 2000). Così, dopo l’isolamento, i colangiociti sono stati incubati per 1 ora a 37°C per permettere

19
la rigenerazione delle proteine di membrana danneggiate dagli enzimi proteolitici delle tecniche di isolamento dellel cellule (Francis et al. 2007). Di seguito, le cellule (1x105) sono state stimolate a temperatura ambiente per 5 minuti con 0,2% di BSA (basale) o con secretina (100 nmol/L). In ciascun trattamento è stato aggiunto 3-isobutilmetilxantine, un inibitore della fosfodiesterasi per prevenire la degradazione del cAMP (Lesage et al. 1996). Infine, i livelli di cAMP (espressi in fmol per 105 cellule) sono stati misurati attraverso un kit commercialmente disponibile (cAMP [125I] Biotrak Assay System, RPA509). In aggiunta a questi dati, sono stati eseguiti anche immunoblots sugli stessi colangiociti per verificare la fosforilazione di alcune proteine che appartengono al pathway del cAMP, come ERK1/2 ed Elk-1. Le cellule sono state solubilizzate in lysis buffer contenente: 15 mM Tris HCl (pH 7.4), 5 mM EDTA, 100 mM NaCl, Igepal 1%, 2 mM PMSF (phenyl methyl sulfonyl fluorite), 2 mM benzamidina e 1% aprotina in ghiaccio per 30 minuti. Dopo una breve sonicazione i campioni sono stati centrifugati a 10.000 g per 20 secondi a 4ºC; del supernatante è stata determinata la concentrazione delle proteine mediante BIO-RAD Protein Assay-Dye Reagent (BIO-RAD Laboratories GmbH). Così 10 µg di lisato totale di colangiociti puri sono stati diluiti in 6x LSB (Laemly sample buffer) contenente 0.3 M 2-mercaptoethanolo e blu di bromofenolo e sottoposti a SDS-PAGE (SDS-polyacrylamide gel electrophoresis) in gel al 4-12%. Successivamente è stato effettuato il trasferimento su nitrocellulosa e le proteine di interesse visualizzate utilizzando anticorpi primari specifici. Infine gli immunoblots sono stati normalizzati confrontando gli immunoblots della β-actina (housekeeping protein) (Alpini et al. 2001). L’intensità delle bande è stata determinata con un densitometro a scansione video (Storm 860, GE Healthcare, Piscataway, NJ) ed analizzata con un software ImageQuant TL versione 2003.02 (GE, Healthcare, Little Chalfont, Buckinghamshire, England).
STUDIO ULTRASTRUTTURALE In questo caso, tessuto epatico umano normale e cisti di differente diametro provenienti da pazienti affetti da malattia policistica del fegato, sono stati ridotti, mediante una lama molto affilata, in piccoli frammenti non superiori a 1 mm di spessore per consentire una rapida fissazione ed una buona penetrazione delle resine da inclusione. Si è proceduto a lavaggio in soluzione tampone per rimuovere contaminanti dalla superficie del tessuto. Poi, i frammenti sono stati fissati con glutaraldeide al 2,5% in tampone fosfato per 2-6 ore a 4°C; seguita dalla post-fissazione con osmio all’1-2% per 2 ore a 4°C; e infine lavati in tampone per 20 minuti con due cambi per togliere eccesso di osmio. Dopo fissazione un lavaggio accurato in acqua evita precipitazione dei sali sulla superficie del tessuto che potrebbe essere causata dalla disidratazione. Questa viene effettuata con soluzioni scalari in alcool, successivamente i frammenti sono trasferiti all’interno di un apparecchio chiamato Critical point dryer, dove l’alcool che permea il tessuto è gradualmente sostituito da anidride carbonica liquida che viene poi portata a temperatura e pressione del punto critico, per cui i frammenti vengono a trovarsi da una fase liquida a una gassosa senza variazioni della tensione superficiale. Ora i fegati sono pronti per essere metallizzati, vengono, perciò, montati su portacampioni metallici con una colla d’argento, buona conduttrice. Per produrre una sottile pellicola d’oro, sulla superficie del preparato si usa la metodica della Stupper Coating, l’apparecchio è formato da una campana, in cui è effettuato un vuoto poco spinto (10-³ torr). All’interno rimangono molecole d’aria e di gas inerte come argon, con un catodo rivestito d’oro e un anodo su cui c’è il campione da metallizzare, generando una differenza di potenziale tramite corrente ad alto voltaggio applicata al catodo (20-40 KV). In questo modo le molecole di gas nella camera ionizzano producendo ioni positivi ed elettroni liberi, gli ioni bombardano le particelle d’oro che si trovano sul catodo facendole staccare da esso. Continuando a collidere si muovono casualmente e molte vengono in contatto con la

20
superficie del campione biologico producendo il sottile ed uniforme film (Tecniche in Anatomia Patologica; Melis M, Carpino F, Di Tondo U. Edi Ermes) (Onori et al. 2000; Gaudio et al. 2006).
Studio in vitro: LINEE CELLULARI STUDIATE La prima linea cellulare che abbiamo preso in considerazione è stata quella dei colangiociti intraepatici di ratto maschio normale (NRICC). Tali cellule sono state sviluppate e caratterizzate come precedentemente descritto (Alpini et al. 2003) e mantenute in coltura a 37°C e CO2 al 5% in DMEM con aggiunta di forskolina (4 µg/ml), triiodiotironina (3,4 µg/ml), dexamtasone (0,4 µg/ml), gentamicina (5 µg/ml), inibitore della tripsina (50 µg/ml), siero fetale bovino al 5%, fattore di crescita epidermico umano (EGF) (25 ng/ml), L-glutammina (20 nM) e 0,1 mM di soluzione MEM di aminoacidi non essenziali. Le altre due linee cellulari che abbiamo usato sono di origine umana: la H69 di colangiociti non maligni gentilmente concesseci dal Dr. GJ Gores della Mayo Clinic, Rochester MN (Grubman et al. 1994). Queste cellule sono state mantenute a 37°C con CO2 al 5% nel Dulbecco’s modified Eagle’s medium-Ham’s F-12 nutrient mixture (Cambrex Bio Science, Walkersville, MD), con aggiunta di adenina (1,8x10-4 mol/L), insulina (5 µg/ml), transferrina (5 µg/ml), triiodiotironina (2x10-9 mol/L), idrocortisone (1,1x10-6 mol/L), EGF (1,64x10-6 mol/L), epinefrina (5,5x10-6 mol/L), siero fetale bovino al 10%, 100 U/ml di penicillina e 100 µg/ml di streptomicina. L’altra linea immortalizzata umana che abbiamo usato è stata la LCDE di colangiociti isolati da cisti di pazienti affetti da malattia policistica del fegato (Perrone et al. 1997), anche questa gentile donazione del Dr. DM Jefferson della Tufts University School of Medicine, Boston, MA. La linea cellulare LCDE è stata sviluppata e mantenuta nelle stesse condizioni della H69. EFFETTI DELL’FSH SULLA PROLIFERAZIONE DI QUESTE LINEE Innanzitutto abbiamo testato in tutte e tre le linee cellulari la presenza dell’FSHR attraverso real-time PCR e immunofluorescenza. In aggiunta a questo, abbiamo anche valutato la diversa presenza del messaggio dell’FSH tramite real-time PCR come precedentemente visto. Le diverse cellule sono state seminate su un vetrino coprioggetto in un 6-well plate (500.000 cellule per well) e fatte aderire per tutta la notte. Successivamente, i vetrini coprioggetti sono stati trasferiti in un nuovo 6-well plate contenente PBS freddo. Trascorsi 5 minuti sono stati lavati per 3 volte con PBST (PBS con 0,2% di Triton X) e incubati per 1 ora in BSA al 4% in PBS. Quando la soluzione bloccante è stata rimossa, i vetrini coprioggetto sono stati incubati a 4°C con l’anticorpoo primario anti-FSHR (lo stesso usato per l’immunoistochimica) diluito in BSA 1%. Il giorno successivo le cellule sono state lavate 3 volte con PBST prima di essere incubate con l’appropriato anticorpo secondario legato a Cy3 (Jackson Immunochemicals, West Grove, PA; 1:50) per 2 ore in camera oscura. Dopo di che, i vetrini sono stati lavati con PBST, per poi essere montati su regolari vetrini con Antifade gold contenente 4,6-diamino-2-phenilindole (DAPI) come contrastante nucleare (Molecular Probes, Eugene, OR). Le immagini sono state acquisite usando un microscopio confocale Olympus IX-71. La proliferazione dopo stimolazione con concentrazioni crescenti di FSH è stata valutata tramite un kit colorimetrico che evidenzia il numero di cellule biologicamente attive (MTS assay, CellTiter 96AQueous; Promega Corp., Madison, WI) e il numero di cellule è stato valutato mediante assorbanza a 490 nm. Di seguito alla tripsinizzazione, le cellule sono state seminate in un 96-well plate (10.000 per well) per un volume finale di 200 µl di medium e fatte aderire overnight. Successivamente all’adesione, è stato cambiato il medium con uno privo di siero al fine di “affamare” le cellule, dopo 24 ore i plates sono stati stimolati con siero bovino (BSA) al 0,2% o FSH con concentrazioni comprese tra 0,1

21
ng/ml e 100 ng/ml a 37°C per 24 ore, prima di valutare la crescita cellulare tramite 3 - (4,5 -dimethylthiazol-2-yl) - 5 - (3-carboxymethoxyphenyl) - 2 - (4-sulfophenyl) - 2H -tetrazolium, inner salt (MTS) saggio di proliferazione. In esperimenti separati, le stesse cellule sono state incubate con BSA 0,2% o FSH (100 ng/ml) per 24 ore in assenza o presenza di una pre incubazione di 1 ora con PD98056 (un ERK/MAK inibitore) (Francis, Glaser et al. 2004) o con un anticorpo specifico anti-FSH, prima di valutare la crescita cellulare con MTS assay. I dati sono stati espressi come cambiamenti delle cellule trattate con FSH rispetto a quelle stimolate con BSA. La significatività statistica è stata determinate tramite t test. In aggiunta a questi esperimenti, le stesse tipologie di cellule sono state incubate a 37°C con BSA 0,2% o con FSH (100 nM) per 30 minuti, 60 minuti, 3 ore e 6 ore in assenza o in presenza degli stessi inibitori usati precedentemente, per poi andare a verificare la proliferazione tramite immunoblots per valutare l’espressione del PCNA, uno dei marker di proliferazione cellulare piu’ utilizzzati. SILENZIAMENTO DELL’FSH NEI COLANGIOCITI NORMALI DI RATTO Il ruolo dell’FSH nella proliferazione di NRICC è stato dimostrato tramite subcloni in cui abbiamo ridotto l’espressione di questo ormone. Tali nuove linee cellulari sono state stabilite usando i plasmidi SureSilencing shRNA (SABiosciences, Frederick MD) per l’FSH di ratto che conferiscono anche la resistenza alla puromicina, per la selezione di cellule stabilmente trasfettate. Dopo averle piastrate in un 6 well plate (approssimativamente al 50% di confluenza) le cellule sono state transfettate con i 5 diversi plasmidi shRNA (tra cui un controllo negativo) usando il reagente di transfezione TransIT secondo le indicazioni della compagnia (MirusBio Corporation, Madison MI). Dopo 3 ore sono stati aggiunti 5 mg/mL di puromicina per well. Ogni giorno è stato cambiato il medium (in cui è stata aggiunta puromicina) e controllata la vitalità cellulare, fino alla completa morte delle NRICC del primo well, quello in cui non è stata compiuta alcuna transfezione. Dopo di che l’RNA è stato estratto dai 5 diversi cloni per verificare, tramite real-time PCR, in quale si è realizzata maggiormente la transfezione. Questo miglior clone (chiamato NRICC-FSH) è stato fatto proliferare insieme al controllo non trasfettato con plasmidi (chiamato NRICC-puro) per poter eseguire immunoblots per PCNA, Bax, ERK1/2 e Elk-1 come precedentemente descritto.
RISULTATI
Studio in vivo L’EPITELIO BILIARE ESPRIME L’FSHR Lo studio immunoistochimico sulle sezioni di fegato (Figura 13) e la real-time PCR (Figura 14) con l’RNA estratto dai colangiociti isolati dai nostri gruppi sperimentali, ha dimostrato che l’epitelio biliare intraepatico esprime la proteina e l’mRNA del recettore per l’ ormone follicolo stimolante (FSHR). Tale espressione aumenta nei colangiociti dopo BDL e dopo trattamento con FSH per una settimana sia nei ratti maschi che femmine comparati con i normali non trattati. Mentre, dopo somministrazione dell’antagonista Antide o dell’anticorpo anti-FSH, l’espressione della proteina e del suo messaggio diminuisce.

22
Figura 13. Fegato di ratto, Immunoistochimica, FSHR, o.m. 40X. Espressione di FSHR in sezioni di fegato normale e BDL, sia di ratto maschio che femmina. E’ evidente un aumento dell’espressione del recettore dopo legatura del dotto biliare principale (BDL).
Figura 14. Real-time PCR per l’FSHR nei colangiociti isolati dai gruppi sperimentali trattati in vivo. Nelle cellule provenienti da ratti BDL il messaggio del recettore dell’FSH aumenta e viene parzialmente ridotto dopo somministrazione di Antide o di un anticorpo anti-FSH. I dati sono espressi come la media di 3 esperimenti ± ES.
23
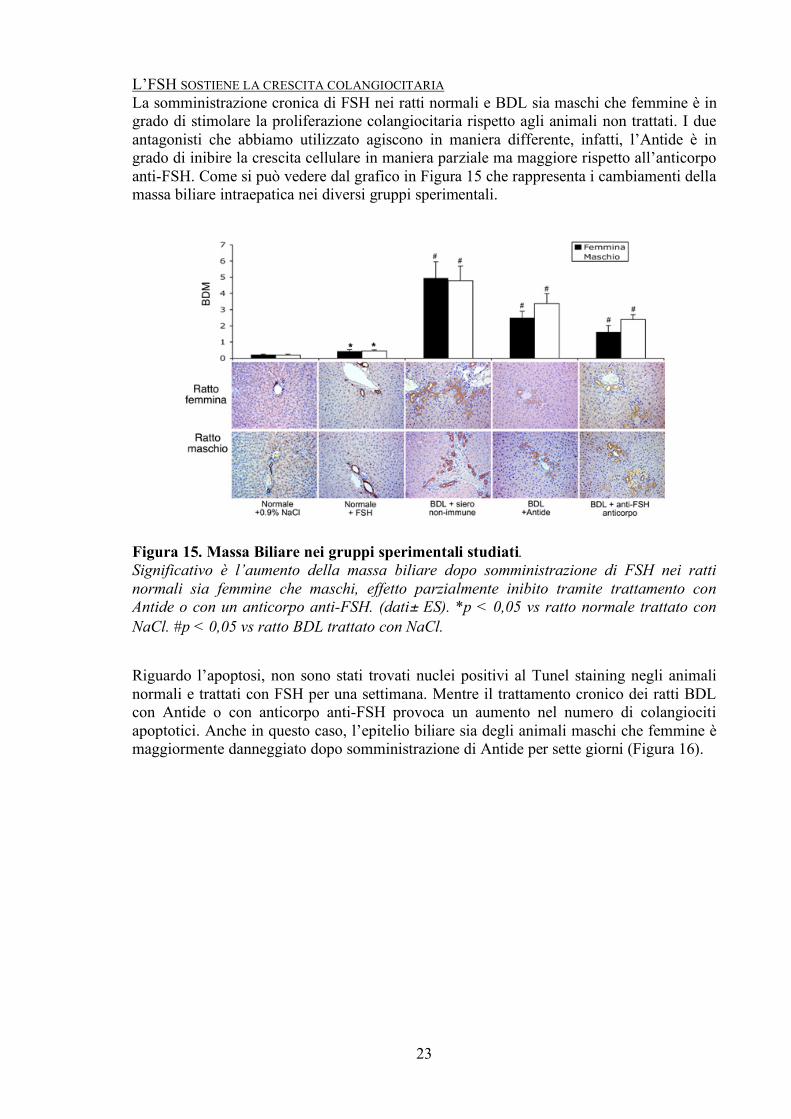
L’FSH SOSTIENE LA CRESCITA COLANGIOCITARIA La somministrazione cronica di FSH nei ratti normali e BDL sia maschi che femmine è in grado di stimolare la proliferazione colangiocitaria rispetto agli animali non trattati. I due antagonisti che abbiamo utilizzato agiscono in maniera differente, infatti, l’Antide è in grado di inibire la crescita cellulare in maniera parziale ma maggiore rispetto all’anticorpo anti-FSH. Come si può vedere dal grafico in Figura 15 che rappresenta i cambiamenti della massa biliare intraepatica nei diversi gruppi sperimentali.
Figura 15. Massa Biliare nei gruppi sperimentali studiati. Significativo è l’aumento della massa biliare dopo somministrazione di FSH nei ratti normali sia femmine che maschi, effetto parzialmente inibito tramite trattamento con Antide o con un anticorpo anti-FSH. (dati± ES). *p < 0,05 vs ratto normale trattato con NaCl. #p < 0,05 vs ratto BDL trattato con NaCl.
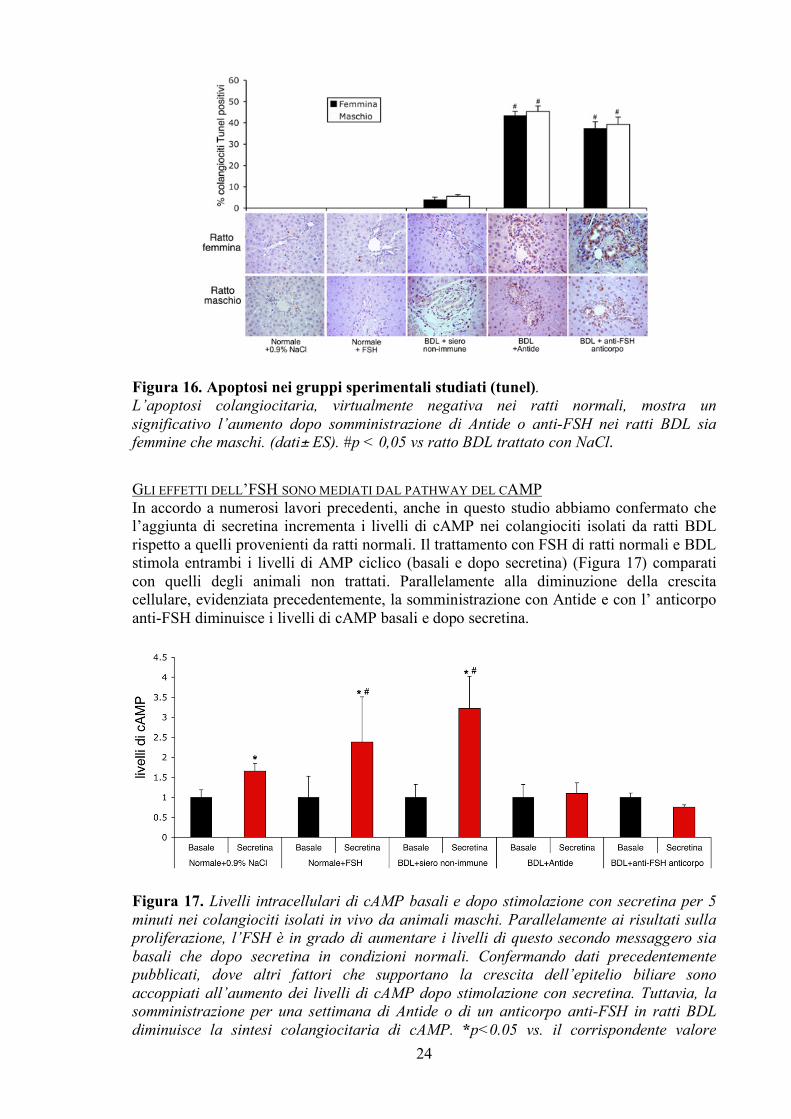
Riguardo l’apoptosi, non sono stati trovati nuclei positivi al Tunel staining negli animali normali e trattati con FSH per una settimana. Mentre il trattamento cronico dei ratti BDL con Antide o con anticorpo anti-FSH provoca un aumento nel numero di colangiociti apoptotici. Anche in questo caso, l’epitelio biliare sia degli animali maschi che femmine è maggiormente danneggiato dopo somministrazione di Antide per sette giorni (Figura 16).
24
Figura 16. Apoptosi nei gruppi sperimentali studiati (tunel). L’apoptosi colangiocitaria, virtualmente negativa nei ratti normali, mostra un significativo l’aumento dopo somministrazione di Antide o anti-FSH nei ratti BDL sia femmine che maschi. (dati± ES). #p < 0,05 vs ratto BDL trattato con NaCl.
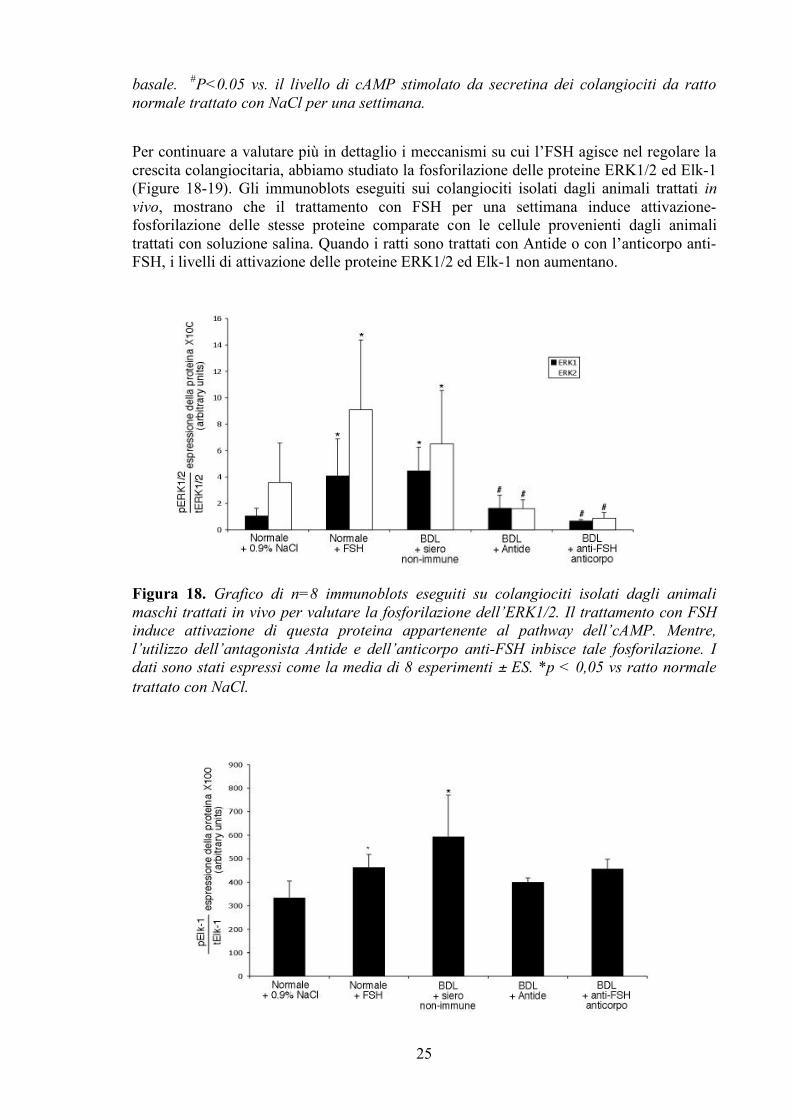
GLI EFFETTI DELL’FSH SONO MEDIATI DAL PATHWAY DEL CAMP In accordo a numerosi lavori precedenti, anche in questo studio abbiamo confermato che l’aggiunta di secretina incrementa i livelli di cAMP nei colangiociti isolati da ratti BDL rispetto a quelli provenienti da ratti normali. Il trattamento con FSH di ratti normali e BDL stimola entrambi i livelli di AMP ciclico (basali e dopo secretina) (Figura 17) comparati con quelli degli animali non trattati. Parallelamente alla diminuzione della crescita cellulare, evidenziata precedentemente, la somministrazione con Antide e con l’ anticorpo anti-FSH diminuisce i livelli di cAMP basali e dopo secretina.
Figura 17. Livelli intracellulari di cAMP basali e dopo stimolazione con secretina per 5 minuti nei colangiociti isolati in vivo da animali maschi. Parallelamente ai risultati sulla proliferazione, l’FSH è in grado di aumentare i livelli di questo secondo messaggero sia basali che dopo secretina in condizioni normali. Confermando dati precedentemente pubblicati, dove altri fattori che supportano la crescita dell’epitelio biliare sono accoppiati all’aumento dei livelli di cAMP dopo stimolazione con secretina. Tuttavia, la somministrazione per una settimana di Antide o di un anticorpo anti-FSH in ratti BDL diminuisce la sintesi colangiocitaria di cAMP. *p<0.05 vs. il corrispondente valore
25
basale. #P<0.05 vs. il livello di cAMP stimolato da secretina dei colangiociti da ratto normale trattato con NaCl per una settimana.
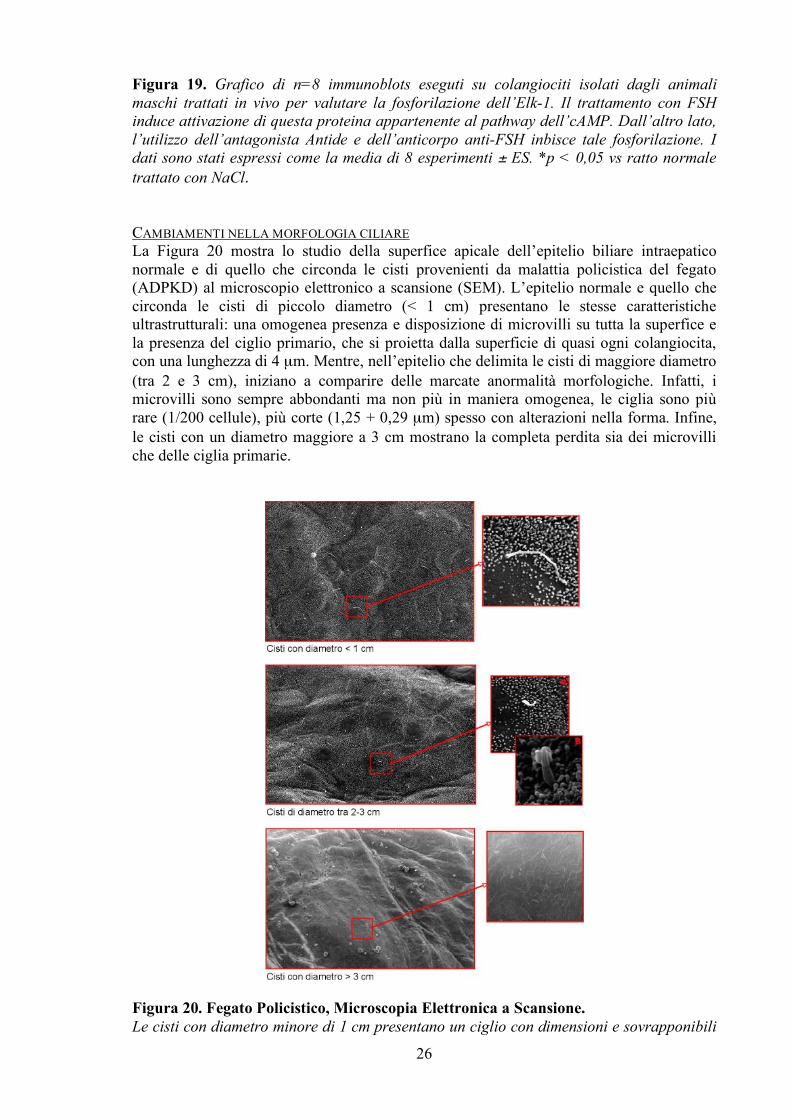
Per continuare a valutare più in dettaglio i meccanismi su cui l’FSH agisce nel regolare la crescita colangiocitaria, abbiamo studiato la fosforilazione delle proteine ERK1/2 ed Elk-1 (Figure 18-19). Gli immunoblots eseguiti sui colangiociti isolati dagli animali trattati in vivo, mostrano che il trattamento con FSH per una settimana induce attivazione-fosforilazione delle stesse proteine comparate con le cellule provenienti dagli animali trattati con soluzione salina. Quando i ratti sono trattati con Antide o con l’anticorpo anti-FSH, i livelli di attivazione delle proteine ERK1/2 ed Elk-1 non aumentano.
Figura 18. Grafico di n=8 immunoblots eseguiti su colangiociti isolati dagli animali maschi trattati in vivo per valutare la fosforilazione dell’ERK1/2. Il trattamento con FSH induce attivazione di questa proteina appartenente al pathway dell’cAMP. Mentre, l’utilizzo dell’antagonista Antide e dell’anticorpo anti-FSH inbisce tale fosforilazione. I dati sono stati espressi come la media di 8 esperimenti ± ES. *p < 0,05 vs ratto normale trattato con NaCl.
26
Figura 19. Grafico di n=8 immunoblots eseguti su colangiociti isolati dagli animali maschi trattati in vivo per valutare la fosforilazione dell’Elk-1. Il trattamento con FSH induce attivazione di questa proteina appartenente al pathway dell’cAMP. Dall’altro lato, l’utilizzo dell’antagonista Antide e dell’anticorpo anti-FSH inbisce tale fosforilazione. I dati sono stati espressi come la media di 8 esperimenti ± ES. *p < 0,05 vs ratto normale trattato con NaCl. CAMBIAMENTI NELLA MORFOLOGIA CILIARE La Figura 20 mostra lo studio della superfice apicale dell’epitelio biliare intraepatico normale e di quello che circonda le cisti provenienti da malattia policistica del fegato (ADPKD) al microscopio elettronico a scansione (SEM). L’epitelio normale e quello che circonda le cisti di piccolo diametro (< 1 cm) presentano le stesse caratteristiche ultrastrutturali: una omogenea presenza e disposizione di microvilli su tutta la superfice e la presenza del ciglio primario, che si proietta dalla superficie di quasi ogni colangiocita, con una lunghezza di 4 µm. Mentre, nell’epitelio che delimita le cisti di maggiore diametro (tra 2 e 3 cm), iniziano a comparire delle marcate anormalità morfologiche. Infatti, i microvilli sono sempre abbondanti ma non più in maniera omogenea, le ciglia sono più rare (1/200 cellule), più corte (1,25 + 0,29 µm) spesso con alterazioni nella forma. Infine, le cisti con un diametro maggiore a 3 cm mostrano la completa perdita sia dei microvilli che delle ciglia primarie.
Figura 20. Fegato Policistico, Microscopia Elettronica a Scansione. Le cisti con diametro minore di 1 cm presentano un ciglio con dimensioni e sovrapponibili
27
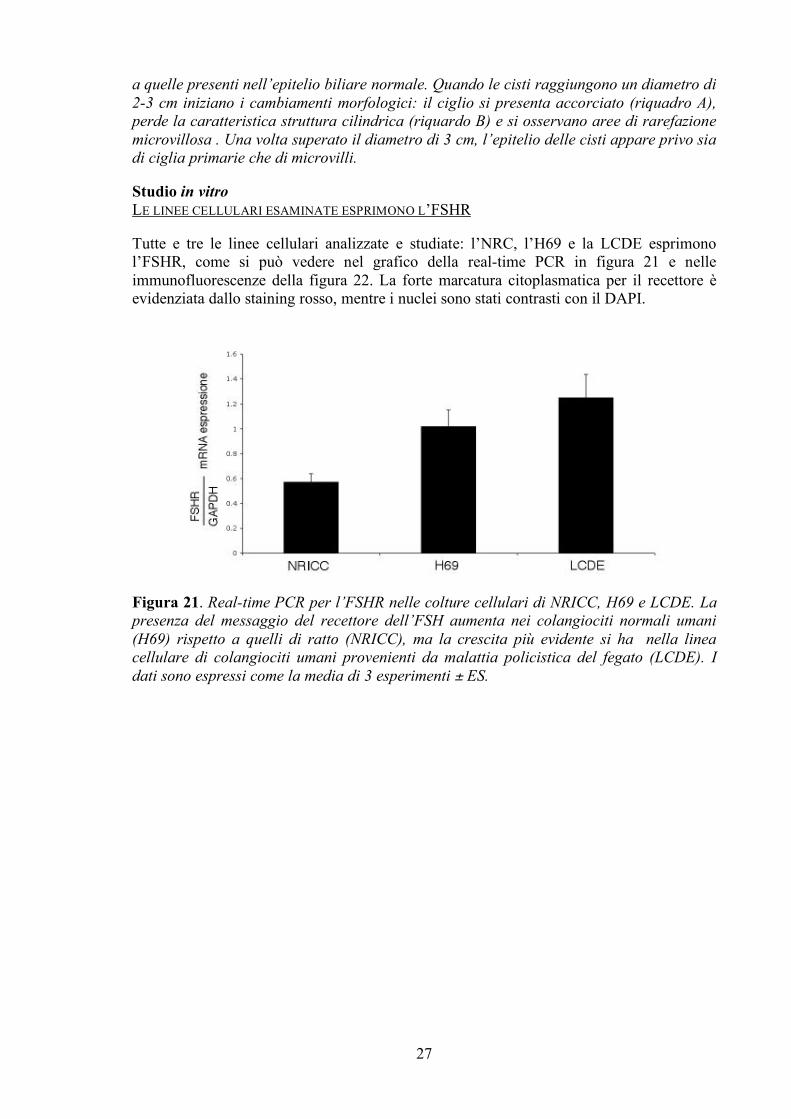
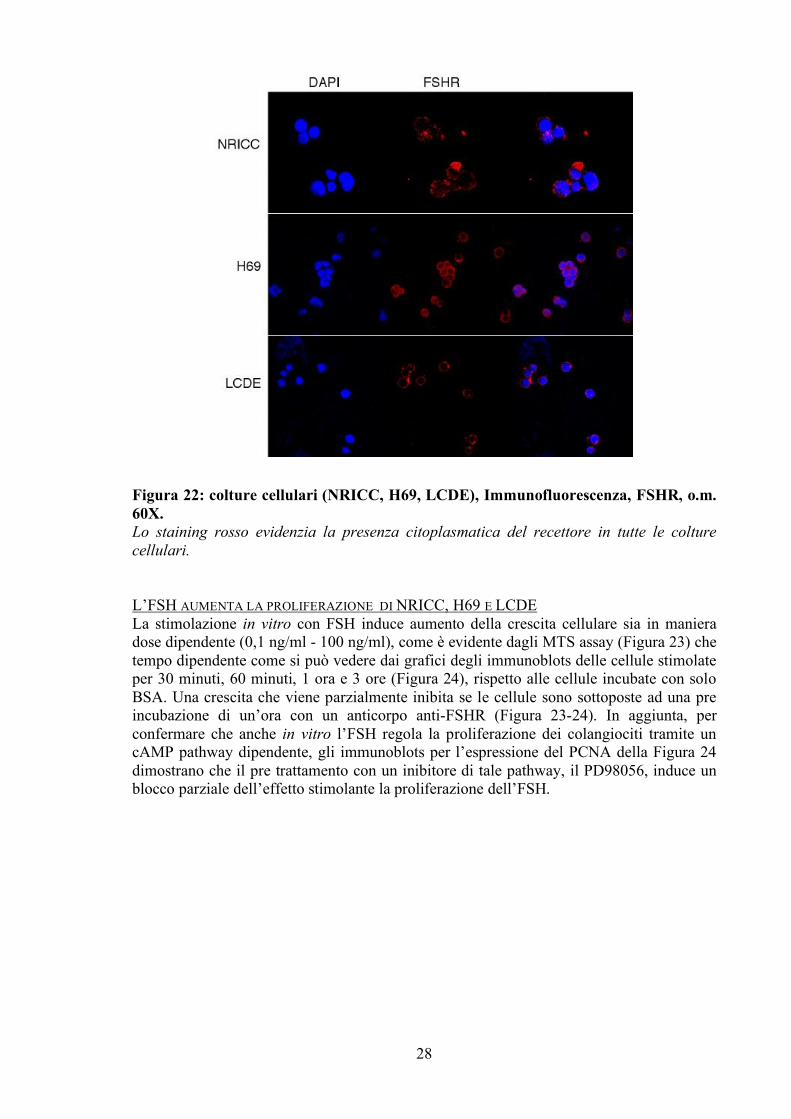
a quelle presenti nell’epitelio biliare normale. Quando le cisti raggiungono un diametro di 2-3 cm iniziano i cambiamenti morfologici: il ciglio si presenta accorciato (riquadro A), perde la caratteristica struttura cilindrica (riquardo B) e si osservano aree di rarefazione microvillosa . Una volta superato il diametro di 3 cm, l’epitelio delle cisti appare privo sia di ciglia primarie che di microvilli. Studio in vitro LE LINEE CELLULARI ESAMINATE ESPRIMONO L’FSHR Tutte e tre le linee cellulari analizzate e studiate: l’NRC, l’H69 e la LCDE esprimono l’FSHR, come si può vedere nel grafico della real-time PCR in figura 21 e nelle immunofluorescenze della figura 22. La forte marcatura citoplasmatica per il recettore è evidenziata dallo staining rosso, mentre i nuclei sono stati contrasti con il DAPI.
Figura 21. Real-time PCR per l’FSHR nelle colture cellulari di NRICC, H69 e LCDE. La presenza del messaggio del recettore dell’FSH aumenta nei colangiociti normali umani (H69) rispetto a quelli di ratto (NRICC), ma la crescita più evidente si ha nella linea cellulare di colangiociti umani provenienti da malattia policistica del fegato (LCDE). I dati sono espressi come la media di 3 esperimenti ± ES.
28
Figura 22: colture cellulari (NRICC, H69, LCDE), Immunofluorescenza, FSHR, o.m. 60X. Lo staining rosso evidenzia la presenza citoplasmatica del recettore in tutte le colture cellulari.
L’FSH AUMENTA LA PROLIFERAZIONE DI NRICC, H69 E LCDE La stimolazione in vitro con FSH induce aumento della crescita cellulare sia in maniera dose dipendente (0,1 ng/ml - 100 ng/ml), come è evidente dagli MTS assay (Figura 23) che tempo dipendente come si può vedere dai grafici degli immunoblots delle cellule stimolate per 30 minuti, 60 minuti, 1 ora e 3 ore (Figura 24), rispetto alle cellule incubate con solo BSA. Una crescita che viene parzialmente inibita se le cellule sono sottoposte ad una pre incubazione di un’ora con un anticorpo anti-FSHR (Figura 23-24). In aggiunta, per confermare che anche in vitro l’FSH regola la proliferazione dei colangiociti tramite un cAMP pathway dipendente, gli immunoblots per l’espressione del PCNA della Figura 24 dimostrano che il pre trattamento con un inibitore di tale pathway, il PD98056, induce un blocco parziale dell’effetto stimolante la proliferazione dell’FSH.

29
Figura 23. Saggio MTS nelle tre diverse linee cellulari studiate (NRICC, H69, LCDE) che dimostra un aumento della proliferazione colangiocitaria dopo trattamento con FSH a dosi crescenti, da 0,1 ng/ml fino a 100 ng/ml. Effetto che viene bloccato se le cellule vengono pre trattate con un anticorpo specifico anti-FSHR o con un inibitore del pathway MAPK/ERK (PD98056). I dati sono stati espressi come la media di 6 esperimenti ± ES. *p < 0,05 vs il valore basale.
30
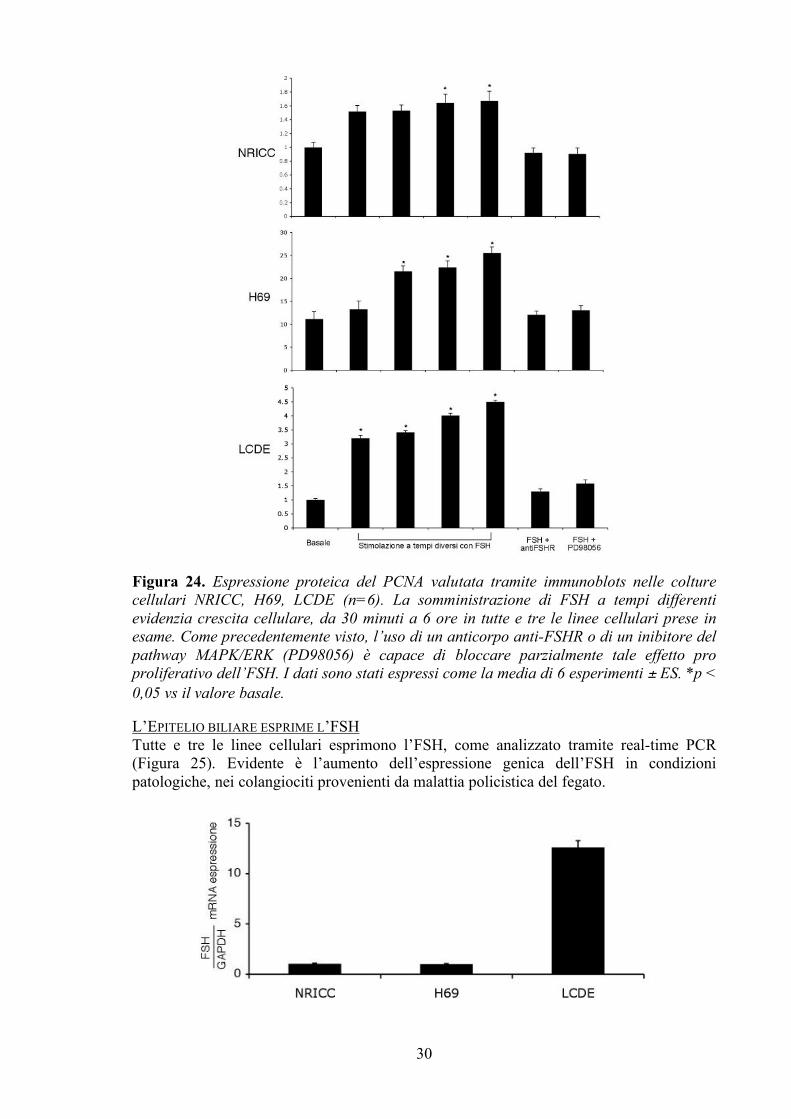
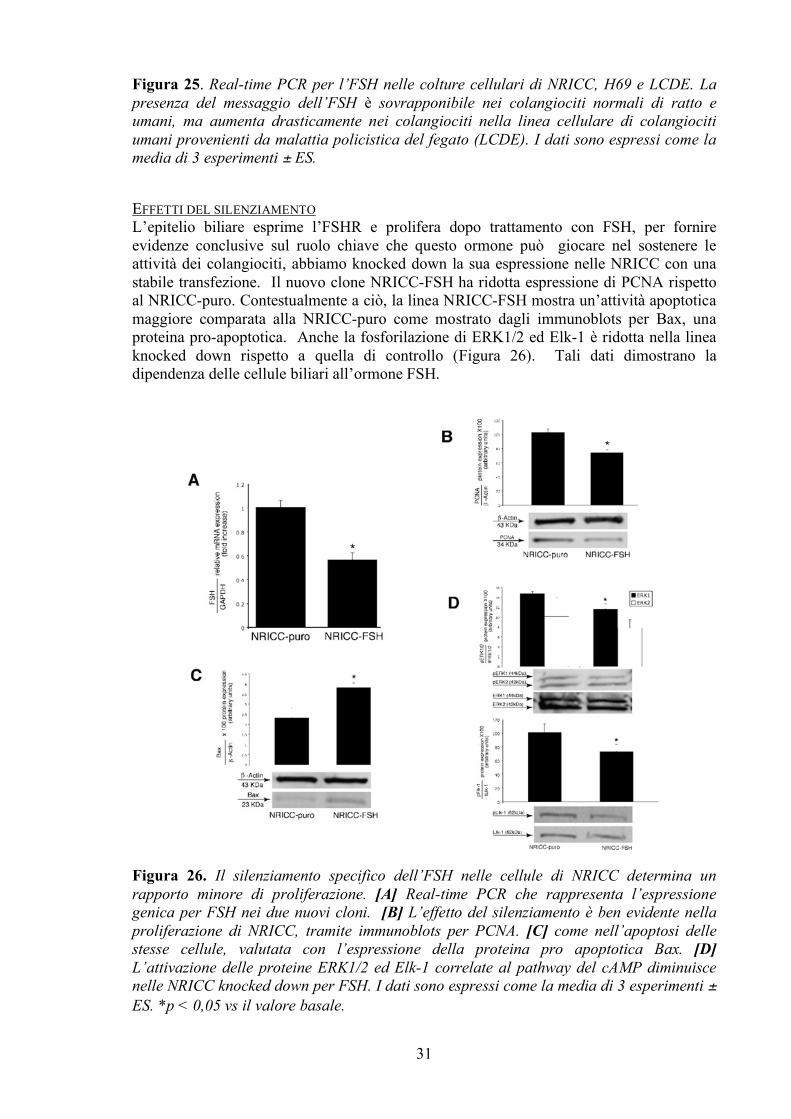
Figura 24. Espressione proteica del PCNA valutata tramite immunoblots nelle colture cellulari NRICC, H69, LCDE (n=6). La somministrazione di FSH a tempi differenti evidenzia crescita cellulare, da 30 minuti a 6 ore in tutte e tre le linee cellulari prese in esame. Come precedentemente visto, l’uso di un anticorpo anti-FSHR o di un inibitore del pathway MAPK/ERK (PD98056) è capace di bloccare parzialmente tale effetto pro proliferativo dell’FSH. I dati sono stati espressi come la media di 6 esperimenti ± ES. *p < 0,05 vs il valore basale. L’EPITELIO BILIARE ESPRIME L’FSH Tutte e tre le linee cellulari esprimono l’FSH, come analizzato tramite real-time PCR (Figura 25). Evidente è l’aumento dell’espressione genica dell’FSH in condizioni patologiche, nei colangiociti provenienti da malattia policistica del fegato.
31
Figura 25. Real-time PCR per l’FSH nelle colture cellulari di NRICC, H69 e LCDE. La presenza del messaggio dell’FSH è sovrapponibile nei colangiociti normali di ratto e umani, ma aumenta drasticamente nei colangiociti nella linea cellulare di colangiociti umani provenienti da malattia policistica del fegato (LCDE). I dati sono espressi come la media di 3 esperimenti ± ES.
EFFETTI DEL SILENZIAMENTO L’epitelio biliare esprime l’FSHR e prolifera dopo trattamento con FSH, per fornire evidenze conclusive sul ruolo chiave che questo ormone può giocare nel sostenere le attività dei colangiociti, abbiamo knocked down la sua espressione nelle NRICC con una stabile transfezione. Il nuovo clone NRICC-FSH ha ridotta espressione di PCNA rispetto al NRICC-puro. Contestualmente a ciò, la linea NRICC-FSH mostra un’attività apoptotica maggiore comparata alla NRICC-puro come mostrato dagli immunoblots per Bax, una proteina pro-apoptotica. Anche la fosforilazione di ERK1/2 ed Elk-1 è ridotta nella linea knocked down rispetto a quella di controllo (Figura 26). Tali dati dimostrano la dipendenza delle cellule biliari all’ormone FSH.
Figura 26. Il silenziamento specifico dell’FSH nelle cellule di NRICC determina un rapporto minore di proliferazione. [A] Real-time PCR che rappresenta l’espressione genica per FSH nei due nuovi cloni. [B] L’effetto del silenziamento è ben evidente nella proliferazione di NRICC, tramite immunoblots per PCNA. [C] come nell’apoptosi delle stesse cellule, valutata con l’espressione della proteina pro apoptotica Bax. [D] L’attivazione delle proteine ERK1/2 ed Elk-1 correlate al pathway del cAMP diminuisce nelle NRICC knocked down per FSH. I dati sono espressi come la media di 3 esperimenti ± ES. *p < 0,05 vs il valore basale.

32
DISCUSSIONE
Questo studio valuta il ruolo dell’FSH e del suo recettore (FSHR) sulla modulazione della proliferazione dell’epitelio biliare intraepatico utilizzando sia modelli in vivo, (ratti maschi e femmine normali e BDL), che in vitro, (linee cellulari di colangiociti normali di ratto (NRICC), di colangiociti normali umani (H69) e di colangiociti che provengono dall’epitelio che tappezza le cisti epatiche (LCDE) in corso di ADPKD (Perrone et al. 1995). Tale modulazione è associata ad un aumento dei livelli di cAMP intracellulare e della fosforilazione di proteine come ERK1/2 ed Elk-1. Questi effetti vengono parzialmente bloccati a seguito di i) somministrazione in vivo, di un antagonista del GnRH (Antide); ii) mediante uno specifico anticorpo anti-FSH; iii) mediante pre incubazione in vitro con un anticorpo anti-FSHR o con un inibitore del pathway ERK/MAPK, il PD98056. Nel modello sperimentale di BDL, molto utilizzato per valutare i meccanismi di iperplasia colangiocitaria (Alpini, Lenzi et al. 1988; LeSage, Glaser et al. 2000), vi è un aumento della massa biliare e dei livelli intracellulari di cAMP. Al contrario, nei ratti trattati con tetracloruro di carbonio (CCl4) c’è un danno a carico, soprattutto dei grandi colangiociti con diminuzione dei livelli di cAMP (LeSag et al. 1999). Negli ultimi decenni si sono intensificati gli studi sui meccanismi che regolano l’iperplasia colangiocitaria e molti di questi hanno mostrato che fattori come acetilcolina, forskolina (Francis, Glaser et al. 2004) e il fattore di crescita neuronale (NGF) (Gigliozzi, Alpini et al. 2004) sostengono la proliferazione dell’epitelio biliare accrescendo i livelli di cAMP. Dall’altro lato, fattori come gastrina (Glaser, Benedetti et al. 2000), RAMH (un agonista del recettore H3 dell’istamina) (Francis, Franchitto et al. 2007), serotonina (Marzioni, Glaser et al. 2005) e molti altri diminuiscono la crescita cellulare abbassando i livelli dello stesso secondo messaggero. Per confermare che alti livelli di cAMP nei colangiociti sono sufficienti da soli ad aumentare la crescita e l’attività secretoria cellulare, sono stati trattati ratti normali con forskolina, stimolatore dell’adenilato ciclasi, che ha indotto proliferazione associata ad aumento nei livelli di proteine quali PKA ed ERK1/2 (Francis, Glaser et al. 2004). In altri tipi cellulari, come le cellule del Sertoli e le cellule della granulosa dei follicoli ovarici, è stato mostrato che l’azione dell’FSH è legata alla fosforilazione dell'ERK1/2 (Simoni et al. 1997). Ma le conoscenze sul ruolo dell’FSH nella patofisiologia epatica è limitato (Yarney et al. 1997) e nessun dato esiste sugli effetti che l’FSH svolge nell’epitelio biliare. In questo studio, abbiamo mostrato per la prima volta che i colangiociti esprimono l’FSHR e che il trattamento con l’ormone FSH aumenta la massa biliare intraepatica in condizioni normali attivando il pathway cAMP/ERK1/2. Per meglio spiegare il ruolo del FSH nella colestasi, in accordo con altri studi in cui si è testato l’effetto dell’Antide (antagonista dell’ormone che permette il rilascio delle gonadotropine a livello ipofisario) (Ljungqvist et al. 1988; Kim et al. 2006) abbiamo trattato gli animali con questo composto. Il trattamento con Antide determina il blocco della secrezione dell’FSH determinando un’inibizione della proliferazione colangiocitaria nei ratti BDL. Inoltre, somministrando uno specifico anticorpo anti-FSH a ratti BDL per una settimana, abbiamo verificato una riduzione dell’attività mitotica dell’epitelio biliare (Gaudio, Barbaro et al. 2006; Taffetani, Glaser et al. 2007; Glaser, DeMorrow et al. 2008) L’effetto inibitorio dell’anti-FSH è minore rispetto a quello ottenuto con la somministrazione di Antide. Nell’uomo, la proliferazione colangiocitaria si ha in seguito a ostruzione biliare, nelle malattie colestatiche croniche e in molte altre forme di malattie epatiche come la malattia policistica del fegato (Alpini, McGill et al. 2002; Lazaridis et al. 2004), Precedenti studi sia sperimentali che clinici hanno dimostrato che i colangiociti proliferanti rispondono maggiormente ad ormoni, neuropeptidi, fattori di crescita, citochine e sono in grado si secernerne una gran quantità di questi attraverso un meccanismo autocrino che sostiene la crescita cellulare ed esercita un effetto anti-apoptotico (Fabris, Cadamuro et al. 2006; Alvaro, Mancino et al. 2007). In particolare gli estrogeni svolgono un ruolo chiave nel

33
supportare tale crescita (Alvaro, Alpini et al. 2000; Alvaro, Onori et al. 2002). Infatti, il blocco delle funzioni estrogeniche endogene tramite ovariectomia e somministrazione di antagonisti specifici inducono un peggioramento nella risposta proliferativa dopo BDL, situazione che si rovescia completamente ristabilendo un trattamento estrogenico. Anche la formazione delle cisti e la progressione della ADPKD si è dimostrata essere strettamente legata ai livelli di estrogeni (Chapman 2003). Infatti l’epitelio biliare che tappezza le cisti esprime i recettori per gli estrogeni (Alvaro, Metalli et al. 2005) e la stimolazione in vitro con 17β-estradiolo della linea cellulare LCDE induce un incremento nella proliferazione, non causata soltanto dall’effetto diretto degli estrogeni ma anche dalla loro potenzialità a promuovere la sintesi e il rilascio di fattori di crescita dall’epitelio cistico (Kahlert et al. 2000). In questo studio abbiamo, allo stesso modo, voluto valutare gli effetti dell’FSH sulla linea cellulare LCDE, mostrando che anche questo ormone è in grado di aumentare la crescita di questi colangiociti rispetto a quelli normali (H69). Probabilmente agendo sullo stesso pathway, in quanto la proliferazione è parzialmente bloccata con l’utilizzo di un ERK/MAPK inibitore (PD98056). Ma, molto più importante, abbiamo anche trovato che i colangiociti esprimono l’FSH con un significativo aumento in condizioni patologiche, come testato nelle LCDE. Il successivo silenziamento del gene nelle NRICC conduce ad una parziale perdita delle capacità proliferative e aumento dell’indice apoptotico, insieme ad una minore fosforilazione delle proteine ERK1/2. Insieme allo studio dei fattori che intervengono sulla progressione della crescita cistica, il nostro scopo è stato anche quello di valutare i cambiamenti nell’aspetto morfologico di queste cellule durante l’evoluzione della patologia. In particolare, abbiamo studiato il ciglio primario, la struttura solitaria non mobile (Davenport and Yoder 2005), localizzata sul versante apicale della membrana plasmatica di ogni colangiocita. L’ interesse biomedico per le caratteristiche strutturali del ciglio è recentemente aumentato, non solo perchè è essenziale nel determinare l’asse destro-sinistro del corpo durante la gastrulazione (McGrath et al. 2003), ma anche perché i due disordini genetici letali più frequenti (ADPKD e ARPKD) sono legati a modificazioni nella sua struttura (Everson, Taylor et al. 2004; Tahvanainen et al. 2005). Il ciglio primario ha la caratteristica di funzionare come un organello sensoriale che monitora i cambiamenti nel flusso biliare all’interno dei dotti biliari intraepatici e regola le risposte funzionali della cellula a tali modificazioni (Pan et al. 2005). Per questo, esso è in grado di trasmettere gli stimoli provenienti dal flusso luminale all’interno della cellula aumentando la concentrazione di Ca++ intracellulare e diminuendo quella del cAMP (Masyuk, Masyuk et al. 2006). Tale funzione meccanosensoriale dipende dalle policistine e dalle riserve di calcio extra ed intracellulari, infatti, l’iniziale aumento di calcio all’interno della cellula dipende dall’entrata di quello extracellulare attraverso la PC-2 che a sua volta stimola il rilascio da parte delle scorte interne, molto probabilmente tramite un meccanismo che coinvolge l’attivazione dei recettori del’IP3 (Hirata et al. 2002). Mentre la rimozione farmacologica del ciglio (tramite chloral hydrate o down regolando PC-1 e PC-2 con siRNA) blocca l’aumento del Ca++ indotto dal flusso biliare (Praetorius and Spring 2003). Simili risultati si ottengono nei topi con mutazioni a carico dei geni PKD1 o PKD2 (Nauli and Zhou 2004) e nella patologia umana che presenta le modificazioni a carico di uno di questi due geni. Una possibile spiegazione potrebbe essere che il fisiologico movimento del ciglio determina un livello stabile di Ca++ intracellulare, che non induce stimoli proliferativi, mentre l’assenza di questo organello nella ADPKD può danneggiare tale equilibrio, determinando un’induzione della crescita cellulare (Alvaro, Gigliozzi et al. 2000), una caratteristica patologica evidenziata sia a livello renale che epatico (Perrone, Grubman et al. 1997; Arnold and Harrison 2005). Al fine di valutare le caratteristiche morfologiche del ciglio nel paziente affetto da PLD abbiamo studiato sia l’epitelio biliare normale che in cisti di diametro crescente. Le piccole cisti, con un diametro minore di 1 cm, presentano un epitelio biliare pressoché identico a quello normale, con microvilli diffusi su tutta la superficie apicale e un ciglio ancora ben strutturato, con una lunghezza

34
compresa tra 5 - 7 µm e un diametro di circa 0,25 µm. L’aumento delle dimensioni cistiche provoca una perdita del ciglio da parte di alcuni colangiociti, modificazioni nella sua normale struttura tubulare, ma soprattutto un suo accorciamento (si passa a 1-2 µm). Infine, si osserva una sua completa scomparsa nelle cisti con diametro superiore a 3 cm. Pertanto si può concludere che le mutazioni dei geni codificanti le policistine nel modello sperimentale e le alterazioni strutturali del ciglio nella PLD, possano indurre una perdita delle caratteristiche verosimilmente meccanosensoriali del ciglio, il quale -a seguito della destrutturazione- va incontro a perdita della sua funzionalità e con essa alla perdita del normale controllo sulla proliferazione cellulare che conduce ad espansione delle cisti (Delmas 2005). La crescita delle cisti epatiche viene supportata da molti fattori. In particolare, l’FSH, insieme ad altri ormoni sessuali (estrogeni e progesterone) e ormoni ipofisari (GH e prolattina) promuovono e supportano la crescita dell’epitelio biliare. Come mostrato nel presente studio, l’FSH svolge un ruolo chiave nel modulare la risposta proliferativa dei colangiociti alla colestasi. In altre parole, i nostri risultati indicano che questo ormone può agire in un meccanismo di protezione durante il danno epatico sperimentale e clinico, suggerendo che la regolazione della sua produzione e secrezione può rappresentare la base morfologica di una potenziale strategia terapeutica di controllo del bilancio proliferazione/perdita delle cellule biliari in corso di colangiopatie. Gli effetti benefici dell’FSH nell’indurre crescita cellulare possono, quindi, essere presi in considerazione nel management di condizioni patologiche dove deve essere promossa e mantenuta la proliferazione colangiocitaria, supportando la produzione di questo ormone. Mentre, nelle colangiopatie, caratterizzate da iperplasia cellulare, la down-regolazione dell’FSH potrebbe essere di notevole importanza nel regolare il loro sviluppo e progressione. In particolare, l’utilizzo di modulatori del pathway intracitoplasmatico cAMP/ERK1/2Elk-1, su cui l’FSH agisce, insieme ad uno studio più approfondito sull’antagonismo del recettore con anticorpi anti-FSHR o recettori solubili che impediscono il normale legame della proteina al suo recettore, potrebbero permettere di sviluppare nuovi modelli terapeutici utili a contrastare l’evoluzione clinica di questi disordini displastici.
BIBLIOGRAFIA
Adesanya, O. O., J. Zhou, et al. (1999). "Insulin-like growth factor 1 is required for G2
progression in the estradiol-induced mitotic cycle." Proc Natl Acad Sci U S A 96(6): 3287-91.
Ahlqvist, J. (1980). "Can endocrine factors influence pathogenetic mechanisms in chronic active hepatitis (CAH) and primary biliary cirrhosis (PBC)? A hypothesis." Hepatogastroenterology 27(1): 64-7.
Alenghat, F. J., S. M. Nauli, et al. (2004). "Global cytoskeletal control of mechanotransduction in kidney epithelial cells." Exp Cell Res 301(1): 23-30.
Alpini, G., S. S. Glaser, et al. (1999). "Bile acid feeding induces cholangiocyte proliferation and secretion: evidence for bile acid-regulated ductal secretion." Gastroenterology 116(1): 179-86.
Alpini, G., R. Lenzi, et al. (1988). "Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules." J Clin Invest 81(2): 569-78.
Alpini, G., J. M. McGill, et al. (2002). "The pathobiology of biliary epithelia." Hepatology 35(5): 1256-68.

35
Alpini, G., J. L. Phinizy, et al. (2003). "Development and characterization of secretin-stimulated secretion of cultured rat cholangiocytes." Am J Physiol Gastrointest Liver Physiol 284(6): G1066-73.
Alpini, G., S. Roberts, et al. (1996). "Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver." Gastroenterology 110(5): 1636-43.
Alpini, G., Y. Ueno, et al. (2001). "Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes." Hepatology 34(5): 868-76.
Alpini, G., C. Ulrich, et al. (1997). "Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation." Am J Physiol 272(2 Pt 1): G289-97.
Alpini, G., C. D. Ulrich, 2nd, et al. (1994). "Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation." Am J Physiol 266(5 Pt 1): G922-8.
Alvaro, D. (1999). "Biliary epithelium: a new chapter in cell biology." Ital J Gastroenterol Hepatol 31(1): 78-83.
Alvaro, D., G. Alpini, et al. (2002). "Effect of ovariectomy on the proliferative capacity of intrahepatic rat cholangiocytes." Gastroenterology 123(1): 336-44.
Alvaro, D., G. Alpini, et al. (2002). "Alfa and beta estrogen receptors and the biliary tree." Mol Cell Endocrinol 193(1-2): 105-8.
Alvaro, D., G. Alpini, et al. (2000). "Estrogens stimulate proliferation of intrahepatic biliary epithelium in rats." Gastroenterology 119(6): 1681-91.
Alvaro, D., B. Barbaro, et al. (2006). "Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma." Am J Pathol 169(3): 877-88.
Alvaro, D., A. Gigliozzi, et al. (2000). "Regulation and deregulation of cholangiocyte proliferation." J Hepatol 33(2): 333-40.
Alvaro, D., P. Invernizzi, et al. (2004). "Estrogen receptors in cholangiocytes and the progression of primary biliary cirrhosis." J Hepatol 41(6): 905-12.
Alvaro, D., M. G. Mancino, et al. (2007). "Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver." Gastroenterology 132(1): 415-31.
Alvaro, D., M. G. Mancino, et al. (2006). "Estrogens and the pathophysiology of the biliary tree." World J Gastroenterol 12(22): 3537-45.
Alvaro, D., A. Mennone, et al. (1997). "Role of kinases and phosphatases in the regulation of fluid secretion and Cl-/HCO3- exchange in cholangiocytes." Am J Physiol 273(2 Pt 1): G303-13.
Alvaro, D., V. D. Metalli, et al. (2005). "The intrahepatic biliary epithelium is a target of the growth hormone/insulin-like growth factor 1 axis." J Hepatol 43(5): 875-83.
Alvaro, D., P. Onori, et al. (2008). "Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease." Am J Pathol 172(2): 321-32.
Alvaro, D., P. Onori, et al. (2002). "Intracellular pathways mediating estrogen-induced cholangiocyte proliferation in the rat." Hepatology 36(2): 297-304.
Amura, C. R., K. S. Brodsky, et al. (2008). "CXCR2 agonists in ADPKD liver cyst fluids promote cell proliferation." Am J Physiol Cell Physiol 294(3): C786-96.
Amura, C. R., K. S. Brodsky, et al. (2007). "VEGF receptor inhibition blocks liver cyst growth in pkd2(WS25/-) mice." Am J Physiol Cell Physiol 293(1): C419-28.
Anyatonwu, G. I. and B. E. Ehrlich (2004). "Calcium signaling and polycystin-2." Biochem Biophys Res Commun 322(4): 1364-73.
Arnold, H. L. and S. A. Harrison (2005). "New advances in evaluation and management of patients with polycystic liver disease." Am J Gastroenterol 100(11): 2569-82.

36
Babu, P. S., N. Danilovich, et al. (2001). "Hormone-induced receptor gene splicing: enhanced expression of the growth factor type I follicle-stimulating hormone receptor motif in the developing mouse ovary as a new paradigm in growth regulation." Endocrinology 142(1): 381-9.
Babu, P. S., H. Krishnamurthy, et al. (2000). "Activation of extracellular-regulated kinase pathways in ovarian granulosa cells by the novel growth factor type 1 follicle-stimulating hormone receptor. Role in hormone signaling and cell proliferation." J Biol Chem 275(36): 27615-26.
Baker, H. W., H. G. Burger, et al. (1976). "A study of the endocrine manifestations of hepatic cirrhosis." Q J Med 45(177): 145-78.
Bell, H., N. Raknerud, et al. (1995). "Inappropriately low levels of gonadotrophins in amenorrhoeic women with alcoholic and non-alcoholic cirrhosis." Eur J Endocrinol 132(4): 444-9.
Benedetti, A., C. Bassotti, et al. (1996). "A morphometric study of the epithelium lining the rat intrahepatic biliary tree." J Hepatol 24(3): 335-42.
Blechacz, B. and G. J. Gores (2008). "Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment." Hepatology 48(1): 308-21.
Chapman, A. B. (2003). "Cystic disease in women: clinical characteristics and medical management." Adv Ren Replace Ther 10(1): 24-30.
Chapman, R. W., B. A. Arborgh, et al. (1980). "Primary sclerosing cholangitis: a review of its clinical features, cholangiography, and hepatic histology." Gut 21(10): 870-7.
Charatcharoenwitthaya, P. and K. D. Lindor (2006). "Primary sclerosing cholangitis: diagnosis and management." Curr Gastroenterol Rep 8(1): 75-82.
Chen, J., G. Robertson, et al. (1998). "Effects of bile duct ligation on hepatic expression of female-specific CYP2C12 in male and female rats." Hepatology 28(3): 624-30.
Choi, J. H., K. C. Choi, et al. (2005). "Gonadotropins upregulate the epidermal growth factor receptor through activation of mitogen-activated protein kinases and phosphatidyl-inositol-3-kinase in human ovarian surface epithelial cells." Endocr Relat Cancer 12(2): 407-21.
Davenport, J. R. and B. K. Yoder (2005). "An incredible decade for the primary cilium: a look at a once-forgotten organelle." Am J Physiol Renal Physiol 289(6): F1159-69.
de Groen, P. C., G. J. Gores, et al. (1999). "Biliary tract cancers." N Engl J Med 341(18): 1368-78.
Delmas, P. (2004). "Polycystins: from mechanosensation to gene regulation." Cell 118(2): 145-8.
Delmas, P. (2005). "Polycystins: polymodal receptor/ion-channel cellular sensors." Pflugers Arch 451(1): 264-76.
Diamond, T., D. Stiel, et al. (1989). "Osteoporosis in hemochromatosis: iron excess, gonadal deficiency, or other factors?" Ann Intern Med 110(6): 430-6.
Drenth, J. P., J. A. Martina, et al. (2004). "Molecular characterization of hepatocystin, the protein that is defective in autosomal dominant polycystic liver disease." Gastroenterology 126(7): 1819-27.
Drenth, J. P., J. A. Martina, et al. (2005). "Polycystic liver disease is a disorder of cotranslational protein processing." Trends Mol Med 11(1): 37-42.
Eagon, P. K., L. E. Porter, et al. (1985). "Estrogen and androgen receptors in liver: their role in liver disease and regeneration." Semin Liver Dis 5(1): 59-69.
Everson, G. T., M. R. Taylor, et al. (2004). "Polycystic disease of the liver." Hepatology 40(4): 774-82.
Fabris, L., M. Cadamuro, et al. (2006). "Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases." Hepatology 43(5): 1001-12.

37
Fabris, L., M. Strazzabosco, et al. (2000). "Characterization and isolation of ductular cells coexpressing neural cell adhesion molecule and Bcl-2 from primary cholangiopathies and ductal plate malformations." Am J Pathol 156(5): 1599-612.
Farley, D. R., A. L. Weaver, et al. (1995). ""Natural history" of unresected cholangiocarcinoma: patient outcome after noncurative intervention." Mayo Clin Proc 70(5): 425-9.
Ferris, H. A., H. E. Walsh, et al. (2007). "Luteinizing hormone beta promoter stimulation by adenylyl cyclase and cooperation with gonadotropin-releasing hormone 1 in transgenic mice and LBetaT2 Cells." Biol Reprod 77(6): 1073-80.
Filardo, E. J., J. A. Quinn, et al. (2000). "Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF." Mol Endocrinol 14(10): 1649-60.
Fitz, J. G., S. D. Lidofsky, et al. (1993). "Regulation of hepatic Na(+)-HCO3- cotransport and pH by membrane potential difference." Am J Physiol 265(1 Pt 1): G1-8.
Floreani, A., M. Titta, et al. (1991). "Sex hormone changes in post-menopausal women with primary biliary cirrhosis (PBC) and with cryptogenic chronic liver disease." Clin Exp Obstet Gynecol 18(4): 229-34.
Forman, J. R., S. Qamar, et al. (2005). "The remarkable mechanical strength of polycystin-1 supports a direct role in mechanotransduction." J Mol Biol 349(4): 861-71.
Francis, H., A. Franchitto, et al. (2007). "H3 histamine receptor agonist inhibits biliary growth of BDL rats by downregulation of the cAMP-dependent PKA/ERK1/2/ELK-1 pathway." Lab Invest 87(5): 473-87.
Francis, H., S. Glaser, et al. (2008). "Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway." Am J Physiol Cell Physiol 295(2): C499-513.
Francis, H., S. Glaser, et al. (2004). "cAMP stimulates the secretory and proliferative capacity of the rat intrahepatic biliary epithelium through changes in the PKA/Src/MEK/ERK1/2 pathway." J Hepatol 41(4): 528-37.
Gaudio, E., B. Barbaro, et al. (2006). "Administration of r-VEGF-A prevents hepatic artery ligation-induced bile duct damage in bile duct ligated rats." Am J Physiol Gastrointest Liver Physiol 291(2): G307-17.
Gaudio, E., B. Barbaro, et al. (2006). "Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism." Gastroenterology 130(4): 1270-82.
Gaudio, E., A. Franchitto, et al. (2006). "Cholangiocytes and blood supply." World J Gastroenterol 12(22): 3546-52.
Geonzon-Gonzales, M. R. (2007). "Primary sclerosing cholangitis." Gastroenterol Nurs 30(2): 102-5; quiz 105-7.
Gigliozzi, A., G. Alpini, et al. (2004). "Nerve growth factor modulates the proliferative capacity of the intrahepatic biliary epithelium in experimental cholestasis." Gastroenterology 127(4): 1198-209.
Glaser, S., A. Benedetti, et al. (2000). "Gastrin inhibits cholangiocyte growth in bile duct-ligated rats by interaction with cholecystokinin-B/Gastrin receptors via D-myo-inositol 1,4,5-triphosphate-, Ca(2+)-, and protein kinase C alpha-dependent mechanisms." Hepatology 32(1): 17-25.
Glaser, S., S. DeMorrow, et al. (2008). "Progesterone stimulates the proliferation of female and male cholangiocytes via autocrine/paracrine mechanisms." Am J Physiol Gastrointest Liver Physiol 295(1): G124-G136.
Glaser, S. S., R. E. Rodgers, et al. (1997). "Gastrin inhibits secretin-induced ductal secretion by interaction with specific receptors on rat cholangiocytes." Am J Physiol 273(5 Pt 1): G1061-70.

38
Gonzales, P. H., C. R. Rhoden, et al. (2007). "Male gonadal function, prolactin secretion and lactotroph population in an experimental model of cirrhosis." Braz J Med Biol Res 40(10): 1383-8.
Gromoll, J., M. Simoni, et al. (1996). "An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man." J Clin Endocrinol Metab 81(4): 1367-70.
Grubman, S. A., R. D. Perrone, et al. (1994). "Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines." Am J Physiol 266(6 Pt 1): G1060-70.
Harnois, D. M., F. G. Que, et al. (1997). "Bcl-2 is overexpressed and alters the threshold for apoptosis in a cholangiocarcinoma cell line." Hepatology 26(4): 884-90.
Hirata, K., J. F. Dufour, et al. (2002). "Regulation of Ca(2+) signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms." Hepatology 36(2): 284-96.
Hodgson, S. F., E. R. Dickson, et al. (1985). "Bone loss and reduced osteoblast function in primary biliary cirrhosis." Ann Intern Med 103(6 ( Pt 1)): 855-60.
Hyder, S. M. (2002). "The role of steroid hormones on the regulation of vascular endothelial growth factor." Am J Pathol 161(1): 345-6.
Ishii, M., B. Vroman, et al. (1989). "Isolation and morphologic characterization of bile duct epithelial cells from normal rat liver." Gastroenterology 97(5): 1236-47.
Ji, Q., P. I. Liu, et al. (2004). "Follicle stimulating hormone-induced growth promotion and gene expression profiles on ovarian surface epithelial cells." Int J Cancer 112(5): 803-14.
Kahlert, S., S. Nuedling, et al. (2000). "Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway." J Biol Chem 275(24): 18447-53.
Kanno, N., G. LeSage, et al. (2001). "Regulation of cholangiocyte bicarbonate secretion." Am J Physiol Gastrointest Liver Physiol 281(3): G612-25.
Kanno, N., G. LeSage, et al. (2000). "Functional heterogeneity of the intrahepatic biliary epithelium." Hepatology 31(3): 555-61.
Khan, S. A., H. C. Thomas, et al. (2005). "Cholangiocarcinoma." Lancet 366(9493): 1303-14.
Kim, K. Y., K. C. Choi, et al. (2006). "Mechanism of gonadotropin-releasing hormone (GnRH)-I and -II-induced cell growth inhibition in ovarian cancer cells: role of the GnRH-I receptor and protein kinase C pathway." Endocr Relat Cancer 13(1): 211-20.
LaRusso, N. F., B. L. Shneider, et al. (2006). "Primary sclerosing cholangitis: summary of a workshop." Hepatology 44(3): 746-64.
Lazaridis, K. N. and G. J. Gores (2005). "Cholangiocarcinoma." Gastroenterology 128(6): 1655-67.
Lazaridis, K. N., M. Strazzabosco, et al. (2004). "The cholangiopathies: disorders of biliary epithelia." Gastroenterology 127(5): 1565-77.
LeSag, E. G., D. Alvaro, et al. (1999). "Cholinergic system modulates growth, apoptosis, and secretion of cholangiocytes from bile duct-ligated rats." Gastroenterology 117(1): 191-9.
LeSage, G., S. Glaser, et al. (2000). "Regulatory mechanisms of ductal bile secretion." Dig Liver Dis 32(7): 563-6.
LeSage, G., S. Glaser, et al. (2001). "Regulation of cholangiocyte proliferation." Liver 21(2): 73-80.
Lesage, G., S. S. Glaser, et al. (1996). "Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion." Gastroenterology 111(6): 1633-44.

39
LeSage, G. D., D. Alvaro, et al. (2004). "Alpha-1 adrenergic receptor agonists modulate ductal secretion of BDL rats via Ca(2+)- and PKC-dependent stimulation of cAMP." Hepatology 40(5): 1116-27.
LeSage, G. D., S. S. Glaser, et al. (1999). "Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from BDL rat liver." Am J Physiol 276(5 Pt 1): G1289-301.
Ljungqvist, A., D. M. Feng, et al. (1988). "Antide and related antagonists of luteinizing hormone release with long action and oral activity." Proc Natl Acad Sci U S A 85(21): 8236-40.
Lleo, A., P. Invernizzi, et al. (2008). "Etiopathogenesis of primary biliary cirrhosis." World J Gastroenterol 14(21): 3328-37.
Mancino, A., M. G. Mancino, et al. (2009). "Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor." Dig Liver Dis 41(2): 156-63.
Marion, S., E. Kara, et al. (2006). "G protein-coupled receptor kinase 2 and beta-arrestins are recruited to FSH receptor in stimulated rat primary Sertoli cells." J Endocrinol 190(2): 341-50.
Marzioni, M., S. Glaser, et al. (2005). "Autocrine/paracrine regulation of the growth of the biliary tree by the neuroendocrine hormone serotonin." Gastroenterology 128(1): 121-37.
Marzioni, M., G. D. LeSage, et al. (2003). "Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct-ligated rats." Am J Physiol Gastrointest Liver Physiol 284(5): G837-52.
Masyuk, A. I., T. V. Masyuk, et al. (2006). "Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling." Gastroenterology 131(3): 911-20.
Masyuk, T. V., B. Q. Huang, et al. (2003). "Defects in cholangiocyte fibrocystin expression and ciliary structure in the PCK rat." Gastroenterology 125(5): 1303-10.
Masyuk, T. V., A. I. Masyuk, et al. (2007). "Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate." Gastroenterology 132(3): 1104-16.
McGrath, J., S. Somlo, et al. (2003). "Two populations of node monocilia initiate left-right asymmetry in the mouse." Cell 114(1): 61-73.
Meachem, S. J., N. G. Wreford, et al. (1998). "Follicle-stimulating hormone is required for the initial phase of spermatogenic restoration in adult rats following gonadotropin suppression." J Androl 19(6): 725-35.
Nathanson, M. H. and J. L. Boyer (1991). "Mechanisms and regulation of bile secretion." Hepatology 14(3): 551-66.
Nauli, S. M., F. J. Alenghat, et al. (2003). "Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells." Nat Genet 33(2): 129-37.
Nauli, S. M. and J. Zhou (2004). "Polycystins and mechanosensation in renal and nodal cilia." Bioessays 26(8): 844-56.
Nichols, M. T., E. Gidey, et al. (2004). "Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid." Hepatology 40(4): 836-46.
Nishio, A., N. M. Bass, et al. (2001). "Primary biliary cirrhosis: from induction to destruction." Semin Gastrointest Dis 12(2): 89-102.
Ong, A. C. and P. C. Harris (2005). "Molecular pathogenesis of ADPKD: the polycystin complex gets complex." Kidney Int 67(4): 1234-47.
Onori, P., S. Morini, et al. (2000). "Hepatic microvascular features in experimental cirrhosis: a structural and morphometrical study in CCl4-treated rats." J Hepatol 33(4): 555-63.

40
Pan, J., Q. Wang, et al. (2005). "Cilium-generated signaling and cilia-related disorders." Lab Invest 85(4): 452-63.
Pazour, G. J. and G. B. Witman (2003). "The vertebrate primary cilium is a sensory organelle." Curr Opin Cell Biol 15(1): 105-10.
Pei, Y. (2001). "A "two-hit" model of cystogenesis in autosomal dominant polycystic kidney disease?" Trends Mol Med 7(4): 151-6.
Pei, Y., T. Watnick, et al. (1999). "Somatic PKD2 mutations in individual kidney and liver cysts support a "two-hit" model of cystogenesis in type 2 autosomal dominant polycystic kidney disease." J Am Soc Nephrol 10(7): 1524-9.
Perrone, R. D., S. A. Grubman, et al. (1997). "Autosomal dominant polycystic kidney disease decreases anion exchanger activity." Am J Physiol 272(5 Pt 1): C1748-56.
Perrone, R. D., S. A. Grubman, et al. (1995). "Continuous epithelial cell lines from ADPKD liver cysts exhibit characteristics of intrahepatic biliary epithelium." Am J Physiol 269(3 Pt 1): G335-45.
Praetorius, H. A. and K. R. Spring (2003). "Removal of the MDCK cell primary cilium abolishes flow sensing." J Membr Biol 191(1): 69-76.
Reynolds, D. M., C. T. Falk, et al. (2000). "Identification of a locus for autosomal dominant polycystic liver disease, on chromosome 19p13.2-13.1." Am J Hum Genet 67(6): 1598-604.
Ritter, V., B. Thuering, et al. (2008). "Follicle-stimulating hormone does not impact male bone mass in vivo or human male osteoclasts in vitro." Calcif Tissue Int 82(5): 383-91.
Ross, M. A., C. M. Sander, et al. (2001). "Spatiotemporal expression of angiogenesis growth factor receptors during the revascularization of regenerating rat liver." Hepatology 34(6): 1135-48.
Ruzycky, A. L. (1996). "Effects of 17 beta-estradiol and progesterone on mitogen-activated protein kinase expression and activity in rat uterine smooth muscle." Eur J Pharmacol 300(3): 247-54.
Sampson, L. K., S. M. Vickers, et al. (1997). "Tamoxifen-mediated growth inhibition of human cholangiocarcinoma." Cancer Res 57(9): 1743-9.
Simoni, M., J. Gromoll, et al. (1997). "The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology." Endocr Rev 18(6): 739-73.
Sloboda, R. D. and J. L. Rosenbaum (2007). "Making sense of cilia and flagella." J Cell Biol 179(4): 575-82.
Stellon, A. J. and R. Williams (1986). "Increased incidence of menstrual abnormalities and hysterectomy preceding primary biliary cirrhosis." Br Med J (Clin Res Ed) 293(6542): 297-8.
Taffetani, S., S. Glaser, et al. (2007). "Prolactin stimulates the proliferation of normal female cholangiocytes by differential regulation of Ca2+-dependent PKC isoforms." BMC Physiol 7: 6.
Tahvanainen, E., P. Tahvanainen, et al. (2005). "Polycystic liver and kidney diseases." Ann Med 37(8): 546-55.
Talwalkar, J. A. and K. D. Lindor (2003). "Primary biliary cirrhosis." Lancet 362(9377): 53-61.
Tena-Sempere, M., P. R. Manna, et al. (1999). "Molecular cloning of the mouse follicle-stimulating hormone receptor complementary deoxyribonucleic acid: functional expression of alternatively spliced variants and receptor inactivation by a C566T transition in exon 7 of the coding sequence." Biol Reprod 60(6): 1515-27.
Theise, N. D., R. Saxena, et al. (1999). "The canals of Hering and hepatic stem cells in humans." Hepatology 30(6): 1425-33.

41
Tietz, P. S., G. Alpini, et al. (1995). "Somatostatin inhibits secretin-induced ductal hypercholeresis and exocytosis by cholangiocytes." Am J Physiol 269(1 Pt 1): G110-8.
Touyz, R. M., L. Jiang, et al. (2000). "Follicle-stimulating hormone mediated calcium signaling by the alternatively spliced growth factor type I receptor." Biol Reprod 62(4): 1067-74.
Troispoux, C., F. Guillou, et al. (1999). "Involvement of G protein-coupled receptor kinases and arrestins in desensitization to follicle-stimulating hormone action." Mol Endocrinol 13(9): 1599-614.
Ward, C. J., M. C. Hogan, et al. (2002). "The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein." Nat Genet 30(3): 259-69.
Ward, C. J., D. Yuan, et al. (2003). "Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia." Hum Mol Genet 12(20): 2703-10.
Wilson, P. D. (2001). "Polycystin: new aspects of structure, function, and regulation." J Am Soc Nephrol 12(4): 834-45.
Yarney, T. A., M. H. Fahmy, et al. (1997). "Ontogeny of FSH receptor messenger ribonucleic acid transcripts in relation to FSH secretion and testicular function in sheep." J Mol Endocrinol 18(2): 113-25.
Zhang, M. Z., W. Mai, et al. (2004). "PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells." Proc Natl Acad Sci U S A 101(8): 2311-6.
Zhang, S. B., B. Dattatreyamurty, et al. (1991). "Differential roles of high and low affinity guanosine 5'-triphosphate binding sites in the regulation of follicle-stimulating hormone binding to receptor and signal transduction in bovine calf testis membranes." Endocrinology 128(1): 295-302.





























