Embed Size (px)
DESCRIPTION
Low-molecular-mass Gels Responding to Ultrasounds
Citation preview

5346 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
Cite this: Chem. Soc. Rev., 2014,
43, 5346
Low-molecular-mass gels responding toultrasound and mechanical stress: towardsself-healing materials
Xudong Yu,ab Liming Chen,a Mingming Zhanga and Tao Yi*a
In this review, we focus on the types of smart supramolecular gels whose self-assembly processes are
affected or even triggered by physical forces including sonication and mechanical stress (mechanical
force). The types of gels that are responsive to sonication and mechanical stress are examined and
summarised. The gels exhibit non-covalent interactions among the gelator molecules and show
dynamic and reversible properties controlled by the stimuli. Upon stimulation, the gelators cause
instant and in situ gelation of organic solvents or water with different modes and outcomes of self-
assembly. On the other hand, sonication and mechanical stress, as external factors, can give rise to
dynamic changes in microscopic morphology, optical properties, etc. Certain thixotropic supra-
molecular gels exhibit perfect self-healing characteristics. The driving forces and the mechanism of
the self-assembly process and the responsive outcome of morphological and spectroscopic changes
are discussed. Those supramolecular gels responding to sonication and mechanical stress offer a wide
range of applications in fields such as smart and adaptive materials, switches, drug control and release,
and tissue engineering.
1. Introduction
There has been an enormous increase in interest over the pastdecades in the structures and properties of low-molecular-massorganic gelators (LMOGs, molecules whose molar mass is o3000)
and their organo-/hydrogels.1–3 These gelator molecules self-assemble in organic solvents or water through highly specific‘‘non-covalent’’ interactions, such as hydrogen bonding, hydro-phobic interactions, polar–polar interactions, electrostatic inter-actions and p–p stacking, that drive the one-dimensional growthof the gelator molecules to produce nanoscale or microscalestructures in the form of fibres, strands and tapes. These extendedobjects join together to form three-dimensional networks thatencapsulate the solvent and prevent flow.4–7 The use of theseinteractions for the directed self-assembly of a given structure
a Department of Chemistry, and Concerted Innovation Center of Chemistry for
Energy Materials, Fudan University, 220 Handan Road, Shanghai 200433, China.
E-mail: [email protected] College of Science, Hebei University of Science and Technology, Yuhua Road 70,
Shijiazhuang 050080, China
Xudong Yu
Xudong Yu completed his MSdegree at the Department ofChemistry, Nankai University, in2008 and subsequently obtainedhis PhD degree in 2011 from theDepartment of Chemistry, FudanUniversity. Currently, he isworking in the College of Science,Hebei University of Science andTechnology. His research interestsinclude supramolecular assembly ofcholesterol-based organogels andmolecular recognition. Liming Chen
Liming Chen received his BScdegree in applied chemistry fromFudan University, Shanghai,China, in 2012. In the sameyear he joined Professor Tao Yi’sgroup as a PhD candidate.Currently, he is working on theearly detection of cancer andmedical application of hydrogels.
Received 7th February 2014
DOI: 10.1039/c4cs00066h
www.rsc.org/csr
Chem Soc Rev
REVIEW ARTICLE
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article OnlineView Journal | View Issue

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5347
requires knowledge of their strength and of their dependence ondistance and directionality. In the order of decreasing strength,these interactions are as follows: (a) electrostatic interactions (ion–ion, ion–dipole and dipole–dipole interactions) and coordinativebonding (metal–ligand), (b) hydrogen bonding, (c) p–p stackingand (d) van der Waals forces. In addition, hydrophobic orsolvophobic effects, as well as the fine balance of the forces amonggelator molecules and solvent molecules, often play an importantrole. Although a single interaction is generally much weaker than acovalent bond, the cooperative action of many such interactionsmay lead to a supramolecular framework that is thermodynami-cally and kinetically stable under a variety of conditions.8 LMOGsconstructed by a self-assembled approach are much more versatile,with tuneable functionality, than those of polymer materialsbecause of their dynamic nature; therefore, it is envisioned thatmolecularly defined, intelligent materials may be constructed byusing the supramolecular concept.9–14 In particular, such materialssusceptible to the effects of external stimuli are of interest and playa crucial role as functional soft materials.15–19
When an external stimulus acts on an LMOG, a change in thechemical structure, molecular configuration or aggregation modeof the gelator may occur depending on the type and strength ofthe stimulus. Most of the common physical stimuli will notchange the chemical structure of the gelator but can modulatethe molecular configuration or the self-assembly process of thegelator except light stimulus (Fig. 1). For example, moderate heatis a typical physical stimulus extensively used to control gelformation or construct gel-related switches. Sonication andmechanical stress have recently been observed to be importantstimuli for gel-like soft materials. This type of stimulation maytrigger an imbalance in the self-assembly system toward anintermediate state, which could be partly or wholly reverted tothe initial state by external or internal stimuli. It is reasonable toconclude that gelation could occur with the aid of a physical force,such as mechanical force or sonication.20,21 Even though a largeamount of studies have been dedicated to the molecular design,synthesis and properties of LMOGs, few of them have focussed on
the physical force driving gelation. In this account, we mainlyhighlight so-called physical-stimulus-driven/-responsive LMOGs,whose self-assembly process and functionality can be controlledby physical stimuli without the change of chemical identity ofgelator molecules, which are used extensively to construct self-healing materials with reversibility.
Ultrasound waves are a kind of high frequency mechanicalwaves that require a physical medium in order to support theirpropagation.22 When ultrasound waves propagate in mediumssuch as liquid or liquid–powder suspension, cavitation whichcould create extreme physical and chemical conditions usuallyoccurs.23 This cavitation effect may provide high energy toinduce certain chemical and physical changes, which have longbeen studied in, for example, chemical reactions, such asthe degradation of halohydrocarbons,24 polymerisation reac-tions,25 oxidation–reduction reactions,26 and with respect toreaction selectivity.27 Very recently, Christopher W. Bielawskiet al. demonstrated that ultrasound could affect the previously
Fig. 1 The types of physical stimuli and their effect on gel formation.
Mingming Zhang
Mingming Zhang obtained his PhDfrom the Laboratory of AdvancedMaterials of Fudan University. HisPhD study was carried out underthe guidance of Prof. Tao Yi andhis research mainly focused onthe development of stimulusresponsive supramolecular softmaterials. He is currently a post-doctoral fellow at the RacahInstitute of Physics, HebrewUniversity of Jerusalem.
Tao Yi
Tao Yi received her BSc (1987),MS (1990) and PhD degrees(1998) from Peking University.She was a JSPS postdoctoralfellow at Kyoto University from1999 to 2001. She worked inParis University (XI) as a redposition researcher of CNRSfrom 2003 to 2004. She is nowa full professor in the Depart-ment of Chemistry, FudanUniversity. Her research interestsfocus on stimulus responsivesupramolecular soft matter and
its application in bio-imaging and drug delivery systems. She hasauthored about 120 peer reviewed publications.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5348 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
elusive retroclick reaction, transforming triazoles into their azideand alkyne precursors.28 Ultrasound has also been widely used inthe field of medical care, in vivo imaging and food and cosmeticindustries. In materials chemistry, ultrasound is used to breakand disperse particles. However, it is amazing and intriguing thatultrasound, as an external stimulus, has been observed to pro-mote gel formation.20,29 Ultrasound may be suitable for stimulat-ing gelation in a dynamic manner with a high energy barrier.Sonication-induced gelation is a rapid crystallisation process thatyields fibre-like aggregates in a metastable system. Traditional gelformation often requires a heating–cooling process. By contrast,sonication-activated gel formation can occur instantly at roomtemperature, which makes these gels highly attractive in real-world applications. In this context, the history and evolution ofultrasound-assisted gels, as well as the gelation mechanismthereof, have been explored and discussed.
In addition to ultrasound, physical shearing stress, as a weakerand purely mechanical force, can also affect gel properties at micro-or macro-levels. Most LMOGs are converted to a sol or sol-like statewhen agitated by mechanical stress, and the gel state can berecovered after a heating–cooling treatment. Notably, some LMOGsreferred to as thixotropic gels, as third-generation self-healingmaterials, have the ability to regain their consistency in situ byinternal stimuli after the removal of the external stress.30 Due to thedynamic bonding in the corresponding supramolecular systems,these thixotropic gels have become increasingly appealing as theycould be developed into autonomously, intrinsically healing materi-als that would theoretically be capable of repeatedly healing thesame damage site.9,11,31 The self-healing process in supramolecularsystems is a form of biomimicry and of great significance forindustry, fracture and fatigue, biomaterials and smart materials.32,33
Herein, we examine several types of thixotropic gel systems, theirself-healing mechanisms, as well as their extended applications.
Apart from the above-mentioned stimuli, there are also othertypes of physical stimuli, such as electrostatic interactions, mag-netism and pressure. However, these stimuli have received littleattention from researchers in the field of LMOGs. Therefore, inthis account, we mainly focus on physical organogels controlled bysonication and mechanical stress. These physical stimuli do notintroduce any new species into an organogel system for tuning orswitching gel properties, including morphology and surfacewettability, colour, emission and chirality. Moreover, the changesor switches triggered by these physical stimuli are always reversible.
The following text is divided into four parts: types of gelsthat respond to ultrasound and mechanical stress, gelationmechanisms controlled by ultrasound and mechanical stress,the application of smart organogels sensitive to ultrasound andmechanical stress, and summary and prospects.
2. Types of LMOGs responsive toultrasound and mechanical stress
The presently reported physical-stimulus-responsive organogelsgenerally exhibit multiple non-covalent intermolecular interactions,including metal–ligand interactions, dipolar forces, hydrogen
bonding, p–p interactions and hydrophobic interactions. Thedelicate balance between these interactions plays an importantrole not only in the formation of nano/micro architectures butalso in the resulting stimulus-responsive character. Therefore,small structural variations in a gelator will result in quitedifferent gelation properties. In fact, it is still a great challengeto design a gelator that can precisely respond to a physicalstimulus, particularly a mechanical force. Many physical-stimulus-responsive organogelators have been discovered acci-dentally and have been further modified and developed byorganic synthesis, resulting in systematic synthesis proceduressuch as those detailed in the following.
2.1 Metallogels
Metallogelators are generally composed of a coordination poly-hedron of a metal ion that can provide a rigid framework withfunctionality and other non-covalent interactions such as hydro-gen bonding and p–p interactions to form a flexible bond to metalions. Physical forces are not sufficiently strong to break the metal–ligand interaction but can tune the flexible linker to affect theaggregation mode of the gelators. The first example of gel for-mation induced by ultrasound was reported by Naota et al. for adinuclear Pd complex.34 The sol state of anti-1a (n = 5) in varioussolutions such as CCl4, acetone, 1,4-dioxane, ethyl acetate andtoluene could be converted to an opaque gel state when treatedwith sonication for 3–10 s due to the transformation of intra-molecular p–p stacking into intermolecular p–p stacking (Fig. 2).The resulting sonogel was reverted to a sol upon heating at aboveTg (sol-to-gel transition temperature) and subsequently cooling toroom temperature. However, the syn-1 and optically pure anti-1could not gel any solvent under sonication. This behaviour indi-cated that heterochiral aggregation (RSRS. . .) may have occurred inthe gel state. In another study, palladium complex 2a–2c contain-ing a peptide as a linker was designed.35 This complex could gelateesters or chlorobenzene with the aid of sonication by a switchablesol–gel transition. Kinetic studies suggested that the gelationprocess was composed of two steps: a sonication-triggered initialstep and a spontaneous propagation step. Without sonication,intramolecular H-bonding involving the chloro ligand 2a preventsthe formation of a dipeptide by intermolecular H-bonding. Ultra-sound irradiation releases this self-locked structure and promotesthe formation of semi-stable initial aggregates, and these initialdomains undergo spontaneous b-sheet aggregation with excessunaggregated complexes. The sonication time is an importantfactor affecting the fibre order of such gels. Increasing the durationof sonication increases the concentration of the active domain,leading to accelerated gelation rates and the formation of higher-order nanostructures with heat-resistant properties.
By replacing the palladium ion of 1 with platinum, theprecise control of phosphorescent emission was achieved byultrasound-induced gelation of organic liquids (anti-3a, n = 5,Fig. 2B).36 Nonemissive solutions of racemic, shortlinked anti-3a and optically pure, long-linked anti-3c (n = 7) in organicliquids are transformed immediately into stable phosphores-cent gels upon brief irradiation with ultrasound. Emission fromthe gels can be controlled by adjusting the sonication time,
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5349
linker length and optical activity of the complexes. Spectroscopicand structural analyses indicate that structure-dependent homo-and heterochiral aggregations and ultrasound control of theaggregate morphology are key factors for emission enhancement.
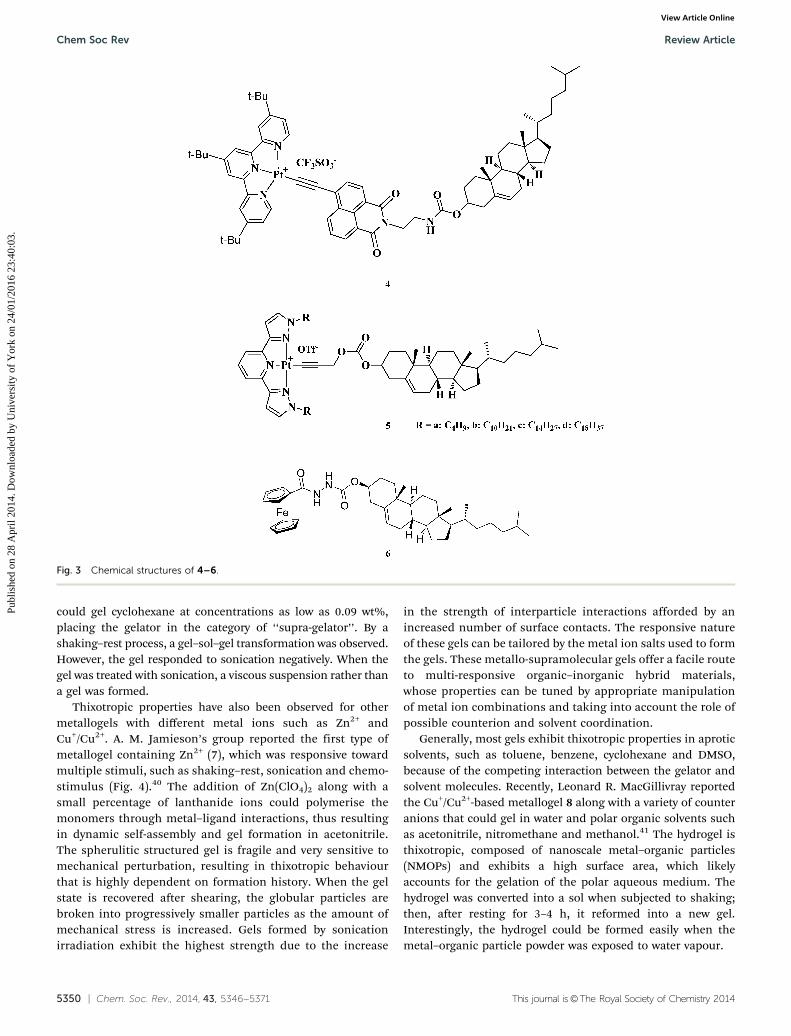
Two organometallic terpyridyl platinum gelators containinghydrophobic cholesterol with or without large steric hindrancewere synthesised and characterised by our group (Fig. 3).37 Thecomplex with three bulky tert-butyl groups (4) can form a stablegel in several types of solvents such as ethyl acetate, tolueneand n-propanol when triggered by sonication at 40 1C with aslow sol-to-gel transition lasting over 40 min. The sonogel inn-propanol of 4 displayed different colours (from yellow to red)and luminescence enhancement for the precipitate obtained bya heating–cooling process. The morphology, as well as thesurface wettability, of 4 can also be reversibly adjusted bysonication and heating. Mechanistic studies indicated thatsonication may transform the molecular conformation of 4,
via a thermally dynamic process, from a conformation with the1,8-naphthalimide perpendicular to tBu3tpy to a conformationof the complex with the 1,8-naphthalimide ring coplanar totBu3tpy, which induces the ionic dipolar and hydrophobicinteractions required for gelation and thus can switch theoptical properties of the complex in the aggregated state. Bothanti-3 and 4 showed suitable configurations for adjusting thephotophysical properties of the gel system by physical stimulisuch as sonication. With a similar structure, the complex 5a–5dshowed thixotropic properties.38 For example, shaking the gelof 5d (10 mg mL�1 in cyclohexane obtained by a heating–cooling process) with a vibrator for 5 min would disturb thegel completely, a state that would persist for half an hour, andthe gel would then reform.
Another multi-responsive metallogelator, 6, containingferrocene, carbonyl amide and a cholesterol group as a neutralcomplex was reported by Fang’s group.39 The metallogelator
Fig. 2 (A) Chemical structures of 1–3; (B) emission colour change between solution and sonogel of (�)-anti-3a (n = 4) in cyclohexane (1.5 � 10�3 M)with different sonication time. (Reprinted with permission from ref. 36. Copyright 2011, American Chemical Society.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online
5350 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
could gel cyclohexane at concentrations as low as 0.09 wt%,placing the gelator in the category of ‘‘supra-gelator’’. By ashaking–rest process, a gel–sol–gel transformation was observed.However, the gel responded to sonication negatively. When thegel was treated with sonication, a viscous suspension rather thana gel was formed.
Thixotropic properties have also been observed for othermetallogels with different metal ions such as Zn2+ andCu+/Cu2+. A. M. Jamieson’s group reported the first type ofmetallogel containing Zn2+ (7), which was responsive towardmultiple stimuli, such as shaking–rest, sonication and chemo-stimulus (Fig. 4).40 The addition of Zn(ClO4)2 along with asmall percentage of lanthanide ions could polymerise themonomers through metal–ligand interactions, thus resultingin dynamic self-assembly and gel formation in acetonitrile.The spherulitic structured gel is fragile and very sensitive tomechanical perturbation, resulting in thixotropic behaviourthat is highly dependent on formation history. When the gelstate is recovered after shearing, the globular particles arebroken into progressively smaller particles as the amount ofmechanical stress is increased. Gels formed by sonicationirradiation exhibit the highest strength due to the increase
in the strength of interparticle interactions afforded by anincreased number of surface contacts. The responsive natureof these gels can be tailored by the metal ion salts used to formthe gels. These metallo-supramolecular gels offer a facile routeto multi-responsive organic–inorganic hybrid materials,whose properties can be tuned by appropriate manipulationof metal ion combinations and taking into account the role ofpossible counterion and solvent coordination.
Generally, most gels exhibit thixotropic properties in aproticsolvents, such as toluene, benzene, cyclohexane and DMSO,because of the competing interaction between the gelator andsolvent molecules. Recently, Leonard R. MacGillivray reportedthe Cu+/Cu2+-based metallogel 8 along with a variety of counteranions that could gel in water and polar organic solvents suchas acetonitrile, nitromethane and methanol.41 The hydrogel isthixotropic, composed of nanoscale metal–organic particles(NMOPs) and exhibits a high surface area, which likelyaccounts for the gelation of the polar aqueous medium. Thehydrogel was converted into a sol when subjected to shaking;then, after resting for 3–4 h, it reformed into a new gel.Interestingly, the hydrogel could be formed easily when themetal–organic particle powder was exposed to water vapour.
Fig. 3 Chemical structures of 4–6.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5351
2.2 Cholesterol-based organogels
Cholesterol derivatives are very important gelators in supra-molecular chemistry.42 The first rational synthesis of cholesterol-based LMOGs was proposed by RG Weiss and coworkers.43,44
They showed that molecular systems comprising an aromatic (A)moiety connected to a steroidal (S) group through a function-alised link (L) could display effective gelation ability. Some ofthese gelators are sensitive to sonication or mechanical force.The first sonication-triggered sol–gel conversion in ALS systemswas reported by our group.45 Compounds 9–10 with differentacyl amino linkages and different hydrogen bonding sites havebeen designed and synthesized (Fig. 5). Compound 9a failed to
gelate any tested solvent due to the lack of intermolecularhydrogen bonding, whereas 9b formed gels in alcohols exclu-sively upon ultrasound irradiation; other external stimuli such asquick heating or cooling did not initiate aggregation. However,10 could form stable gels in a variety of solvents via a slowheating–cooling process or upon ultrasound irradiation for onlya few seconds (0.16 W cm�2, 40 kHz). The different stimuliyielded different self-assembled morphologies, from the largemultiporous vesicle morphology of gels formed via traditionalheating–cooling treatment (T-gels) to the smooth surface ofsonogels (S-gels). Repaired by heat as an external stimulus, thesonogel could be reverted to the initial state. The effects of
Fig. 5 Chemical structures of 9–11.
Fig. 4 (A) Chemical structure of 7; (B) the typical multi-stimuli responsive behavior of gel 7 + Zn2+ (11 wt% in acetonitrile); (C) Cu+/Cu2+ based metallogel 8.(Reprinted with permission from ref. 40. Copyright 2006, American Chemical Society.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5352 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
sonocrystallisation and sonogelation were further discussed withrespect to the compounds 10a–10c, which showed structuralchanges in the alkyl diamine group. It was clear that thelocations of hydrogen bonds (by different alkyl chain lengths)showed a strong effect on the solubility and gelling properties ofthese compounds. A careful thermodynamic balance among themain driving forces for self-assembly, hydrogen bonding andhydrophobic and p–p interactions, was rationalised in terms ofthe solubility and cooperative self-assembly of molecular-scalebuilding blocks induced by sonication and thermal processes.46
In another study, a chiral amino acid was introduced into thelinker in 11a and 11b.47 The result showed that the position ofL-alanine in the linker affected intermolecular hydrogen bondingand molecular polarity, thus resulting in different gelationproperties. The precipitate-to-gel transition of the gelator 11aand the gel-to-gel transition of the gelator 11b, with the aid ofultrasound, were monitored and studied. Both processes werehighly dependent on temperature. Compared with the sol–geltransition, the direct and instant gel-to-gel transitions are moreconvenient for controlled release, functional expression andengineering applications.
The first thixotropic cholesterol-based organogel CNC (12a)was reported by Weiss et al. (Fig. 6).48,49 The thixotropy of thegel is highly dependent on morphology. The gelation abilitiesof 12a and 12b are quite different, although they are structu-rally very similar. The spherulitic aggregates of 12a, composedof highly branched, small fibres in n-alkane at an incubation
temperature o28 1C or concentrations above 1.5 wt%, arethixotropic and are transformed into more stable fibrils afterseveral cycles of repeated self-healing. In contrast, the fibre gelnetwork of 12b in ethyl acetate undergoes irreversible phaseseparation upon mechanical agitation. The double bond at C5forces the B-ring of the steroid part of 12b to adopt a boatconformation, which affects the overall shape of the moleculeand thus its aggregation behaviour.
ALS2–LS2 systems have attracted great attention in recentyears due to their thixotropic behaviour. Thixotropic propertieshave been observed in most cholesterol-based derivatives withan ALS2 structure. The gelator 13 prepared by our group couldform a transparent gel in toluene and an opaque gel in isopropa-nol, both of which exhibited thixotropic properties.50 Whentreated with external mechanical stress, gel 13 (0.86–1.70 wt%)in toluene lost most of its viscosity and transformed into a sol;after resting for at least 20 minutes at room temperature, the gelcould be recovered. The self-healing process that occurred afterthe gel-to-sol transition, which was induced by mechanical stress,could be repeated many times. The thixotropy of the gel in toluenestrongly depended on concentration: below 0.86 wt%, a viscousliquid was observed; in contrast, above 1.70 wt%, the gel could notbe destroyed simply by shaking by hand. It is interesting to notethat the gelation also occurred by a simple shaking–restingprocess in a solvent–solid system at room temperature withouta heating–cooling process, which was referred to as agitation-induced gelation. The self-healing process of compound 13 in
Fig. 6 (A) Chemical structures of 12–13; (B) AFM images of gel 13 in its original, broken and repaired state (scale bar, 2.5 mm). (Reprinted with permissionfrom ref. 50. Copyright 2012, Royal Society of Chemistry.)
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5353
toluene was manifested as the destruction and regeneration ofnanoscale structures. Shaking by hand destroyed the cross-linkednanoring structures with diameters ranging from 150 to 300 nminto biased arcs, thus resulting in a gel–sol transition. After20 min, the repaired gel displayed more distinct and larger ringstructures with diameters ranging from 400 to 600 nm (Fig. 6B).3D and phase AFM images also provided information regardingthis breaking–repair behaviour, which revealed that the morpho-logy was repaired in diverging directions and that mass flowtransport also occurred from thicker walls to damaged areas. Thisgel network was also sensitive to sonication. The ultrasound-induced gel-to-gel transition process was monitored by AFM andSEM images and it was shown that the original ring structure wasdestroyed as expected, and smaller nanorings with pores measur-ing approximately 9–30 nm emerged from the original shellssimultaneously.
Fang’s group intensively studied the effect of chiral residueson the gel formation and thixotropic properties of LS2 (14–17)and ALS2 systems (18–19) (Fig. 7). In those gelators, D/L-alanineand D/L-cholesterol residues as well as the structure of thespacer were used to tune the gelation behaviour.51 The resultsshow that gelators containing D-cholesterol residues (14b, 15b)are more efficient gelators than their analogues with oppositechirality (14a, 15a). For compounds with longer spacers (16, 17),
however, those containing L-alanine residues (16a, 17a) aresuperior to the corresponding ones with D-alanine residues(16b, 17b). As expected, these gels are thixotropic, as revealedby rheological studies, and the mechanical properties of thegels can be adjusted either by altering the spacer length or bychanging the gelator concentration. Surprisingly, 17a couldform a gel in triethylamine, butyl acetate, n-alkane (7–10 carbons)or commercial oils spontaneously at room temperature within10 min, indicating thixotropic properties. 18b and 18c can gelseveral solvents spontaneously at room temperature. These gelspossess thixotropic properties and exhibit the selective gelation ofsolvents from their mixtures with water.52 The gelation behaviourcan also be adjusted by modifying the spatial structure of thelinker with a benzene ring. Compounds 19a–c can gel in a widevariety of organic solvents. Specifically, 19a is a supergelator forDMSO (CGC = 0.04% w/v). The 19a–DMSO gel possesses excellentmechanical strength and very smart thixotropic properties.53
On replacing the aromatic group with a Calix[4]arene groupin an ALS2 system, gelator 20 did not exhibit gelation in any puresolvent (Fig. 8).54 However, it could gel (2.5% w/v) in solvents ofn-decane and acetonitrile mixed in a ratio of 1 : 1. The mechan-ical strength of the gel could be adjusted by varying the concen-tration of the gel and composition of the mixed solvents. Thesegels possess smart and fully reversible thixotropic properties.
Fig. 7 Chemical structures of 14–19.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online
5354 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
In another work, a chiral phenylalanine residue was incorpo-rated into the linker instead of the ethyl diamine. The authorsobserved that mechanical agitation or sonication could reducethe duration of the gelation process of 21b from 30 h by atypical heating–cooling process to 12 min. Moreover, thestrength of gel was also enhanced.55
2.3 Organogels containing p units and phenyl alkyl ethers
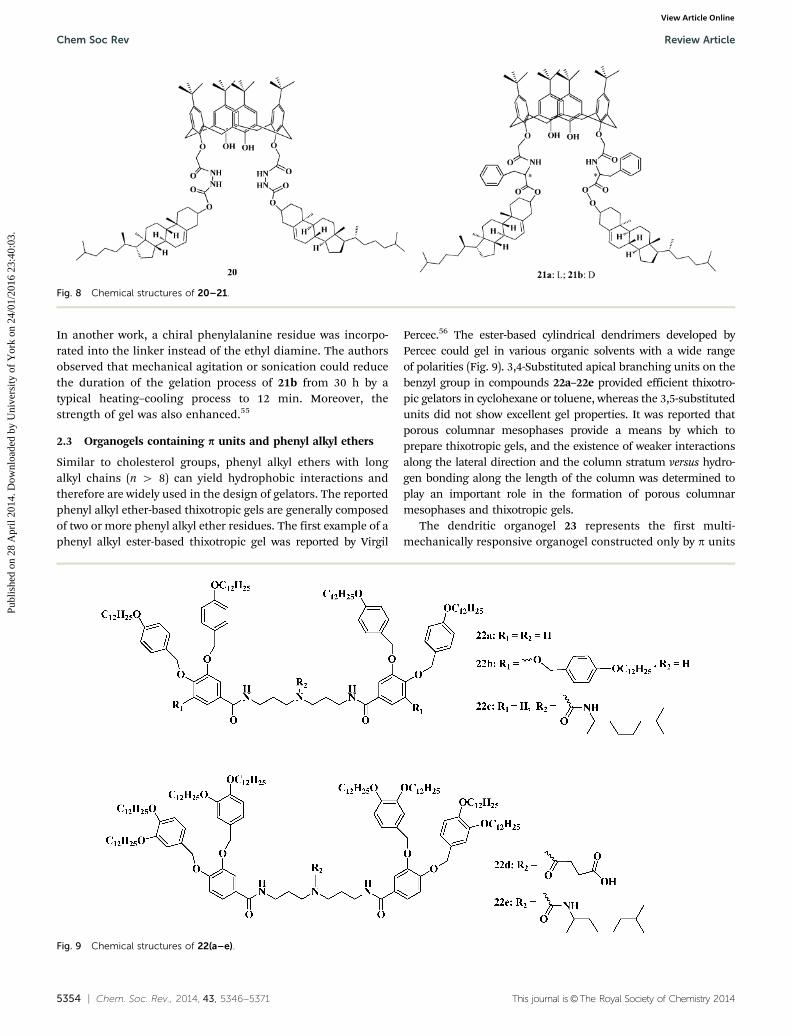
Similar to cholesterol groups, phenyl alkyl ethers with longalkyl chains (n 4 8) can yield hydrophobic interactions andtherefore are widely used in the design of gelators. The reportedphenyl alkyl ether-based thixotropic gels are generally composedof two or more phenyl alkyl ether residues. The first example of aphenyl alkyl ester-based thixotropic gel was reported by Virgil
Percec.56 The ester-based cylindrical dendrimers developed byPercec could gel in various organic solvents with a wide rangeof polarities (Fig. 9). 3,4-Substituted apical branching units on thebenzyl group in compounds 22a–22e provided efficient thixotro-pic gelators in cyclohexane or toluene, whereas the 3,5-substitutedunits did not show excellent gel properties. It was reported thatporous columnar mesophases provide a means by which toprepare thixotropic gels, and the existence of weaker interactionsalong the lateral direction and the column stratum versus hydro-gen bonding along the length of the column was determined toplay an important role in the formation of porous columnarmesophases and thixotropic gels.
The dendritic organogel 23 represents the first multi-mechanically responsive organogel constructed only by p units
Fig. 8 Chemical structures of 20–21.
Fig. 9 Chemical structures of 22(a–e).
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5355
without hydrogen bonding (Fig. 10).57 The compound wasdemonstrated to be a very efficient organogelator that couldgel more than 15 types of organic solvents after sonication for1–5 min. Notably, the gelator could gel in tetrachloromethanewith a CGC of 0.05 wt%, placing the gelator in the category ofsupergelators. In most of the solvents, the gel displayed thixo-tropic properties. For example, nanofibres with the bundlestructure of gel 23 were separated when subjected to vortexing.The regenerated fibres were thicker than those of the fresh gelafter resting for approximately 24 hours. With the addition ofan azobenzene group, the gel could also undergo a reversiblegel-to-sol transition triggered by UV- and visible-light irradia-tion, which made the gel multi-stimuli responsive.
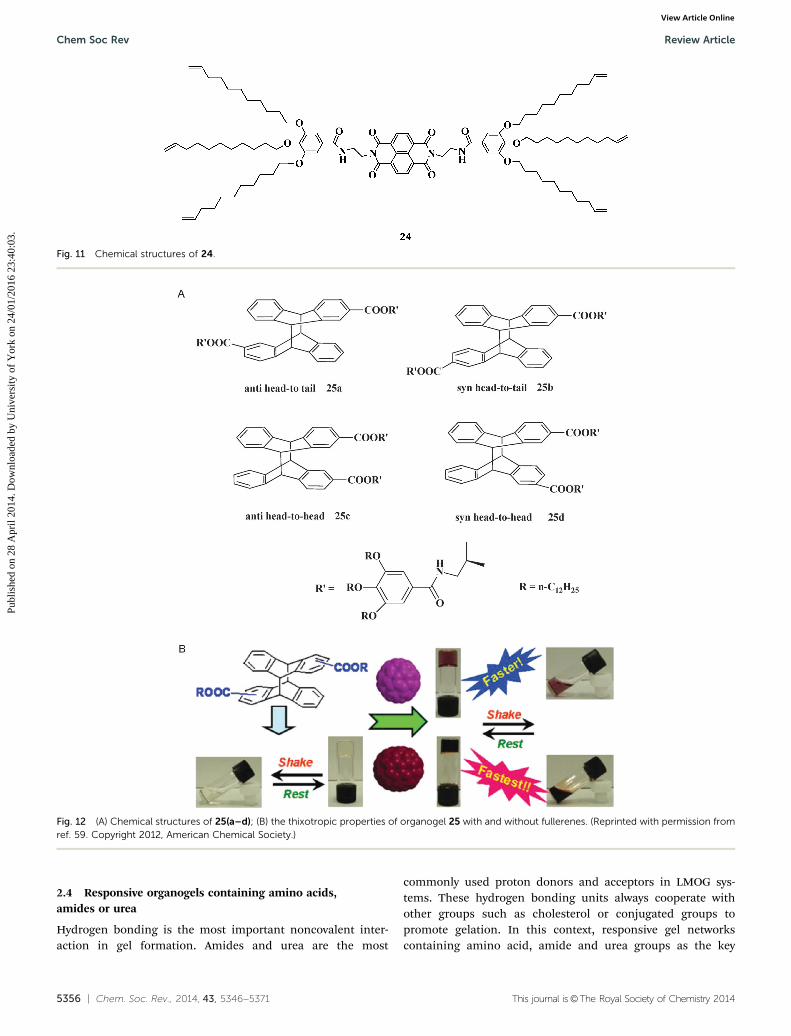
The self-healing properties of thixotropic gels (with an organo-gelator as the host) also rely heavily on concentration, and theguest species is regarded as a junction for the rapid repair of thedestroyed assembly. For example, a naphthalenediimide-basedgelator tailed with phenyl alkyl ether (24) was reported byP. Mukhopadhyay (Fig. 11). This gelator could form a stable,thixotropic and transparent gel in cyclohexane.58 By changingthe concentration, the recovery time of the sol–gel processcould be tuned over a wide range, from a few seconds todays. Rheological experiments suggested that the recoveredgel was weaker than the fresh gel (representing 72% of theoriginal amount). For the regeneration of injured nerve fibrestriggered by cAMP and neurotrophins, the authors employedelectron-rich 1,3- or 1,6-dihydroxynaphthalenes as adhesivesto regenerate the gel fibres positively via donor–acceptor inter-actions, thus achieving the stimulus-induced regeneration
and self-healing of artificial tissues. This work intensivelystudied the self-healing process under the effects of internaland external factors and also provided a new model formimicking nerve fibre regeneration.
Another exciting example was reported by Shinkai andcoworkers.59 Gels 25a–25d, composed of dianthracene units,were prepared from a parent 2-substituted anthracenecarb-oxylic acid moiety by light irradiation (Fig. 12). The gelationproperties of the irradiated products are strongly dependent onthe conformation of the molecules. The photodimers domi-nated by the head-to-tail orientation form a thixotropic gel(25a, b), whereas the system composed of mostly head-to-head photodimers (25c, d) appears as a fluid. The thixotropicproperties of compounds 25a–b were exhibited in solventssuch as decalin, n-hexane and methylcyclohexane. The gelformation is highly dependent on temperature. From �20 to�5 1C, the compounds were not observed to gel in any solventtested, even after a week; in contrast, at 20 1C, a transparentgel could be formed within an hour, which indicated thattemperature played an important role in the formation of one-dimensional aggregates. Notably, the gelation occurs even bymixing the solvent and gelator at room temperature. Further-more, fullerenes such as C60 or C70, as molecular adhesives,were used to enhance the thixotropy of the gels; the processwas accompanied by the reversible integration and disintegra-tion of the host–guest interaction upon mechanical agitation.C70 appears to be a superior guest in terms of propertyenhancement because its size better fits the concave-shapedhost (Fig. 12B).
Fig. 10 The chemical structure of 23 and its multi-responsive behavior.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online
5356 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
2.4 Responsive organogels containing amino acids,amides or urea
Hydrogen bonding is the most important noncovalent inter-action in gel formation. Amides and urea are the most
commonly used proton donors and acceptors in LMOG sys-tems. These hydrogen bonding units always cooperate withother groups such as cholesterol or conjugated groups topromote gelation. In this context, responsive gel networkscontaining amino acid, amide and urea groups as the key
Fig. 11 Chemical structures of 24.
Fig. 12 (A) Chemical structures of 25(a–d); (B) the thixotropic properties of organogel 25 with and without fullerenes. (Reprinted with permission fromref. 59. Copyright 2012, American Chemical Society.)
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5357
components will be discussed as examples to demonstrate theimportance of complementary hydrogen bonding for the for-mation of self-assembly networks and the resulting stimuli-responsive behaviour.
2.4.1 Amino acid based gelators. A number of aminoacid based building blocks have been used to fabricate func-tional supramolecular architectures because of the chirality-magnifying effect that occurs between the single-molecule andmacroscopic levels.60–66 Moreover, amino acids, which aregenerally used as linkers or as functional groups, representone of the best candidates for biological applications and cansupply facile hydrogen bonding modes in supramolecular gels.The simple N-acylated amino acid 26 can form a turbid gel atCGCs of approximately 6 wt% in octanol and 2 wt% in pro-pylene glycol (Fig. 13).67 Ultrasound stimulation is used duringthe early stages of gelation to promote not only the gelationprocess but also the formation of homogeneous 3D inter-connected fibre networks. This effect is particularly significantfor gelling systems with gelator concentrations near or belowthe CGC. For example, the cooling of a hot solution of 3 wt%26–octanol under quiescent conditions was observed to resultin a nongelled liquid, which was stored for two more daysunder ambient conditions. However, after sonication for approxi-mately 1 min, gelation occurred rapidly to form a stable gelwithin a few hours, and the CGC was reduced from B6 toB2 wt% for octanol. Furthermore, the elasticity of the sonogelof 26–octanol was proved to be approximately two orders ofmagnitude higher than that of the sample not subjected tosonication. Moreover, when a heating–cooling treatment wasapplied to the ultrasound-induced gel sample, the nongelled
liquid was obtained again. This result implies that the gellingstate and the corresponding fibre network structure benefitfrom the effect of ultrasound irradiation.
In addition to 26, most amino acid based sonogels arecomposed of p systems such as phenyl, naphthyl and dipyridylgroups. For example, the dispersion of particles of the peptidecompound 27 in alkanes or in the mixture of polar solventssuch as water/methanol (with the ratio of 1 : 4) can undergo aprecipitate–gel transition when treated with ultrasound at 20 1Cfor 1–4 min.68 Interestingly, the compound can selectively gelalkanes in water–alkane mixtures when exposed to ultrasoundwithout heating at room temperature, which might be moremeaningful for potential applications of the gel in industry, forexample, in the collection of petroleum from water.
Sonication not only decreases the CGC of the gelationprocess but in some cases can also expand the range ofapplicable gelation solvents to a large extent. The terminallyprotected tripeptide 28 was observed to form a very weak gelonly in 1,2-dichlorobenzene by a typical heating–cooling pro-cess.69 However, by applying sonication for over 40 s, thetripeptide could gel in many organic solvents, such as hexane,cyclohexane, petrol, benzene, toluene, xylene and ethanol.Some peptide-based gelators can not only gelate organicsolvents but can also form hydrogels in water under ultrasonictreatment, which makes the gels more applicable in the field ofdrug delivery. For example, the L-glutamic acid-based com-pound NBGE 29 reported by M. H. Liu could form stable gelscomposed of a nanofibre network by the sol–gel and precipitate–gel processes in hexane, toluene and even water when treatedwith ultrasound, displaying multiple transitions among different
Fig. 13 Chemical structures of 26–30 containing a peptide.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5358 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
phases triggered by sonication and heating.70 Heinz-BernhardKraatz and co-workers recently investigated the sonication-induced gelation properties and coiled fibrous architectures ina short peptide of Boc-L-Phe-L-Lys(Z)-OMe (30).71 The peptidedisplayed instant gelation in aliphatic and achromatic hydro-carbons starting from a suspension state within 10 s whensubjected to sonication. Additionally, the sonogel of thedipeptide was also observed to exhibit thixotropic and self-healing characteristics, as well as thermal chiroptical switch-ing behaviour.
As one example of cyclopeptides, compound 31 with anexposed –NH2 group dissolves only in highly polar solvents(Fig. 14), and gelation has been observed in some of thesesolvents at low concentrations (usually in the range 0.5–2.0 wt%)at room temperature.72 In fact, the preparation of 31 in DMF led
invariably to instant gelation at a very low concentration(0.3 wt%). Although 31 precipitates from water under ambientconditions, ultrasound irradiation promotes the formation of aweak gel instead of precipitation during the cooling process.This hydrogel and precipitate returned to the soluble phaseupon heating, and this cycle could be repeated many times.These results demonstrate that ultrasound induces and accel-erates efficient primary nucleation that favours fibrillar aggrega-tion. It should be noted that hydrogelation occurred even at56 1C under sonication at a concentration of 1.5 wt%. TheN-acylated derivatives 32a–32g lack the capacity to gel in DMFand DMSO, even at relatively high concentrations.
In the molecular design of peptide-based organogelators,the pendant groups of the peptides highly affect the gel pro-perties. K. Das et al. observed that peptide-appended bola-amphiphiles 33a and 33b were capable of forming stable,transparent, self-supporting hydrogels upon sonication at roomtemperature at a final pH of 5.4, whereas bolaamphiphile 33cwas unable to form a gel under similar conditions (Fig. 15).73
These gelation experiments reveal that 33a is a more efficientgelator than 33b upon sonication. After only 15 s of sonication,33a forms a hydrogel instantaneously, whereas compound 33brequires 1 h to gel. All of these hydrogels are exceptionallystable over a wide range of pH levels (pH 4–8) for more than3 months at room temperature. The polypeptide 34 could gelin mixed solvents of THF/water (1 : 1) after sonication for10–30 min, exhibiting a fibrillar morphology.74 In particular, whenincubated at 60 1C, the gel underwent a reversible and in situ fibre–vesicle transition tuned by heat and sonication stimuli. It wasconcluded that the lysine residue played an important role in thegelation from a molecular structure point of view.Fig. 14 Chemical structures of 31–32.
Fig. 15 Chemical structures of 33–34.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5359
Compared with the number of reports dedicated to sono-gels, there are fewer reports about peptide-based thixotropicorganogels. As a typical example, the peptide compoundNap-GFFYGGKOGEOGKOGSO (35) reported by Z. M. Yangcould self-assemble into a hydrogel by heating–cooling cyclesat a concentration above 0.8 wt% (Fig. 16).75 The gel wasthixotropic and could be converted to a viscous solution byvortexing, shaking or pipetting; the viscous solution could thenbe reverted to a gel after sitting at room temperature for acertain period of time. This thixotropic property of gels formedby 35 can facilitate cell harvesting post-culture. Further workhas shown that the hydrogel surface can slow down the spreadof EBs and the proliferation of murine ES cells compared withthat of gelatin. Moreover, the hydrogel can selectively improveflk1 expression in different ES cells.
Gelator 36 is an unusual example of a peptidedithienylcyclopentene-based gelator that shows multi-responsivecharacteristics.76 36 forms an opaque gel in THF, acetone andacetonitrile, in which the formation of anti-parallel b-sheets of thebiomimetic tetrapeptides is deemed to be the key driving force forgelation. The organogel is responsive to various external stimuli,including temperature, light, chemicals and mechanical force. Inthe presence of catechol, the gel strength is enhanced, but the gelretains thixotropic properties.
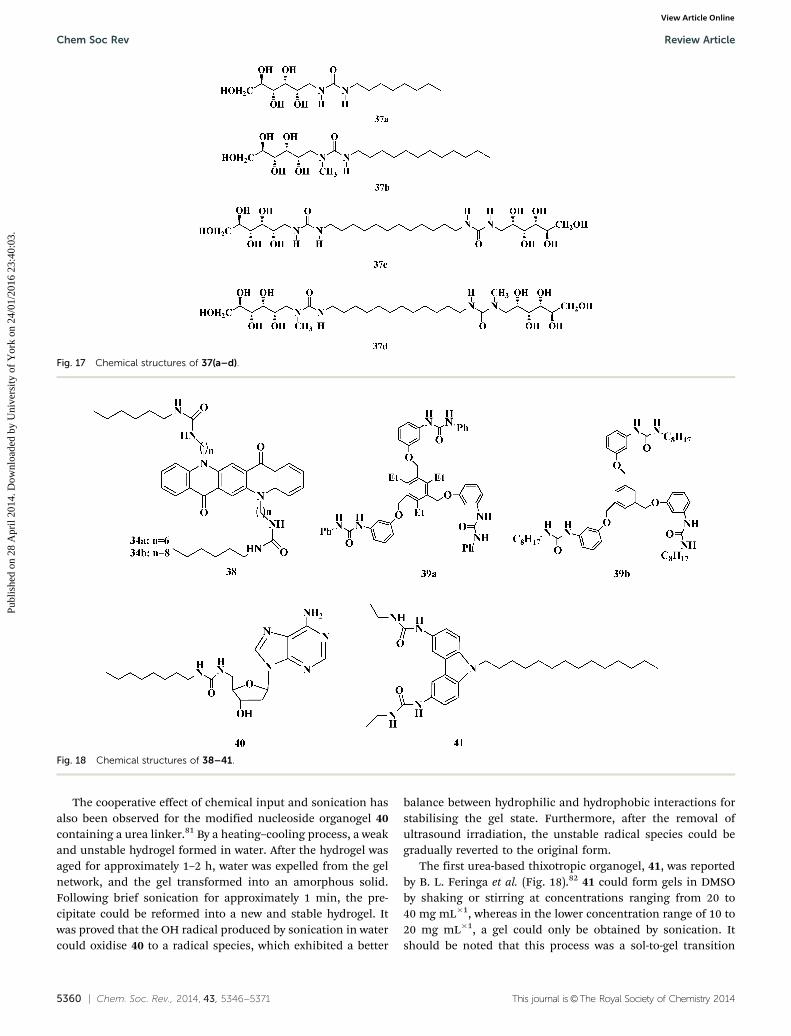
2.4.2 Urea-based organogels. It was demonstrated morethan five decades ago that urea-type hydrogen bonds werecapable of stabilising the assemblies of gelators. The self-association of urea involving two hydrogen bonds is muchstronger than that of amides or urethanes, thus decreasing thesolubility of the materials resulting in gelation or even precipi-tates. A family of ureido amphiphiles and bolaamphiphilesderived from chiral aminopolyols (namely D-glucamine andN-methyl-D-glucamine, 37a–37d) have been observed to be excel-lent hydrogelators (Fig. 17).77 Such amphiphiles can be generatedfrom readily available unprotected aminosugars and hydro-phobic isocyanates by one-step procedures in aqueous media.
These gelators can form hydrogels at room temperature aftersonication at low concentrations (1.0–3.0 wt%). It is noteworthythat some derivatives (37b–37d) also form lyotropic liquidcrystals with lamellar or hexagonal morphologies.
Some urea-based compounds have poor solubility and pre-cipitate from solvents by the typical heating–cooling process. Incases in which the direct transformation of these compoundsfrom precipitate to gel triggered by sonication at room tem-perature hardly occurs due to the strong interaction betweengelator molecules, heat, as a favourable secondary stimulus,can be used in conjunction with sonication to produce sono-gels. For example, compound 38 reported by Dou C. et al. wasdissolved in dimethylformamide (DMF) and dimethylsulfoxide(DMSO) at elevated temperatures (Fig. 18).78 Upon slowlycooling the hot solution to room temperature for over 15 min,a large amount of ordinary precipitate was generated. To obtaingels, the hot solution was stimulated by sonication in a waterbath at 25 1C for 10 s, and this process successfully prevented asol–precipitate transition. However, during this process, thetemperature difference played an important role in determin-ing the gelation and physical properties of the gel, which issimilar to the phenomenon observed in the sonogel process of11a and 11b.
With a reasonable design, sonogels obtained by sonicationcan also respond to other stimuli resulting in phase changesand can then be reversed by a second stimulus without heat. Asuspension of tris-urea compound 39a could gel in organicsolvents, including acetone, MeOH, THF and diethyl phthalate,after brief sonication.79 These gels were observed to be stablefor several months. Addition of Bu4N+X� (but not that ofBu4N+BF4
�) caused a gel–sol transition. In another study, theLewis acid BF3�OEt2 or ZnBr2, acting as a positive stimulus,could re-gel the corresponding sol upon ultrasound treatment.Thus, this gel was regarded as a smart switch stimulatedcooperatively by salt and ultrasound. Similar transitions havealso been achieved for 39b.80
Fig. 16 Chemical structures of 35–36.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online
5360 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
The cooperative effect of chemical input and sonication hasalso been observed for the modified nucleoside organogel 40containing a urea linker.81 By a heating–cooling process, a weakand unstable hydrogel formed in water. After the hydrogel wasaged for approximately 1–2 h, water was expelled from the gelnetwork, and the gel transformed into an amorphous solid.Following brief sonication for approximately 1 min, the pre-cipitate could be reformed into a new and stable hydrogel. Itwas proved that the OH radical produced by sonication in watercould oxidise 40 to a radical species, which exhibited a better
balance between hydrophilic and hydrophobic interactions forstabilising the gel state. Furthermore, after the removal ofultrasound irradiation, the unstable radical species could begradually reverted to the original form.
The first urea-based thixotropic organogel, 41, was reportedby B. L. Feringa et al. (Fig. 18).82 41 could form gels in DMSOby shaking or stirring at concentrations ranging from 20 to40 mg mL�1, whereas in the lower concentration range of 10 to20 mg mL�1, a gel could only be obtained by sonication. Itshould be noted that this process was a sol-to-gel transition
Fig. 17 Chemical structures of 37(a–d).
Fig. 18 Chemical structures of 38–41.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5361
triggered by shaking or stirring, which was different from theschemes reported by our group50 or Y. Fang,53 in which pre-existing fibrils are broken by mechanical force. The roomtemperature gels formed by mechanical stress usually exhibiteda lower Tg o 45 1C; therefore, it was concluded that the precipitate-to-gel transition induced by shaking may be composed ofprecipitate-to-sol and sol-to-gel transitions rather than a directprecipitate-to-gel transition. The mechanism is such thatmechanically breaking fibrils would yield significantly morefibril fragments as nucleation points. These fragments wouldgrow again rapidly into fibrils of sufficient size and aggregate toform a fibrous gel network.
2.5 Other types of organogels responsive to ultrasound andmechanical stress
In addition to the above-mentioned gelator moieties, which areclassic and extensively used to construct organogels, there arealso many other types of organogelators discovered by seren-dipity that can respond to sonication or mechanical force. As atypical example, Weiss et al. reported a series of organogelatorsbased on hydroxystearic acid (HSA) and stearic acid (SA) with-out any p units (Fig. 19).83 Some of these organogels recovertheir viscoelasticity very rapidly after being destroyed by shear.For example, gels of 42a in silicone oil (2 wt%) can recoverapproximately 90% of their original viscoelasticity within 10 safter the cessation of destructive shear, and several other gels,
including HAS, 42b, 43d and 43f, show reduced recovery ofviscoelasticity but are equally fast in doing so.
Due to the competition of multiple noncovalent interactionsbetween gelators and gelator–solvent systems, LMOGs alwaysexhibit weak mechanical behaviour. Recently, multiple com-plementary hydrogen bonding motifs or cooperative multiplenoncovalent interactions have been observed for achievingstrong binding in organic solvents or water. These strong yetreversible noncovalent interactions lead to thermodynamicallycontrolled supramolecular polymerisation, thus resulting inself-supporting gels that can be moulded into different shapeswithout cracking. As smart and adaptable materials, these gelsalso show outstanding self-healing properties. For example,Araki et al. reported the first nucleoside derived supragelatorcontaining alkylsilyl (48, Fig. 20), which could be used toconstruct a flexible, self-supporting film from an ethyl acetatesolution (5% wt/wt).84
The sugar-based organogels 49a and 49b reported by A.Vidyasagar and co-workers are also examples of self-supporting gels that can gel in hydrocarbon solvents at con-centrations as low as 0.2 wt% (Fig. 21).85 These alkane gelsshow high gel–sol transition temperatures indicative of thehigh strength of the gels, which could be moulded into anyself-supporting geometrical shape. These gels also showedremarkable self-healing properties. When a block of gel wascut into two pieces and then joined together, the pieces merged
Fig. 19 Chemical structures of 42–47.
Fig. 20 Chemical structure of compound 48 and the image of the self-supported gel film. (Reprinted with permission from ref. 84. Copyright 2004,Wiley-VCH Verlag GmbH & Co. KGaA.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5362 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
into a continuous block due to a dynamic equilibrium betweenfree dissolved gelators and self-assembled gelators (fibres) inthe gel. Such movement of free gelator molecules across theinterface allowed for the growth of fibres across the fusioninterface or the connection of broken fibres across the inter-face, thus facilitating the self-healing process. These gels alsodisplayed remarkably high transparency in the visible regionand weak transmittance in the ultraviolet region. Based on thisresult, the gel may be placed in paraffin oil and moulded intodifferent optical components, such as lenses, prisms, gel platesand tubes.
2.6 Two-component organogels
Two-component systems often display multiple functions insurface patterns, optical properties, multi-response, etc. Theproperties and functionality of these two-component systemscan be more precisely tuned and controlled in an intelligentmode. However, in a two-component system, phase separationis inevitable because of the competition between congenericmolecules and heterogeneous components. As a result, the self-assembly of congeneric molecules themselves rather than hetero-geneous components may be generated in two-component systems.As an intense high-frequency compressional wave, ultrasoundmay cleave and tune inter- or intramolecular interactions,including hydrogen bonding, p-stacking and hydrophobicinteractions, in the self-assembly process of gel formation;thus, ultrasound treatment may promote intermolecular inter-actions between heterogeneous molecules to produce a newhomogeneous gel system that has the functions of two compo-nents combined in one system. To form such a system, hydro-gen bonding, electrostatic, coordination as well as p-stackinginteractions have been used to induce host–guest interactionsin organogels. A few typical examples are presented in thefollowing.
The smallest host–guest robust hydrogels formed by multi-ple hydrogen bond interactions among the rigid melamine (50)and uric acid (51) were reported by J. W. Steed (Fig. 22).86
Neither of these compounds is soluble in water, but whensubjected to sonication and shaking over a period of approxi-mately 5 minutes, the compounds could gel in water in a ratioof 1 : 1 at concentrations of 0.8 wt% and above. Theoreticalcalculations and 13C MAS-NMR and PXRD data revealed thatthe rigid and planar assembly formed by strong p–p stackingand infinite hydrogen bonding assisted the gel formation.
It is reasonable to expect that ultrasound could be favour-able in strengthening the hydrogen bonding among con-genic molecules. Recently, we reported the ultrasound-assistedco-aggregation of a gel (52) and a micro-crystal (53) system featur-ing an amino acid and imidazole-based derivative.87 Circulardichroism (CD), XRD, IR data and optimised molecular geometrycalculations indicated that sonication weakened the inter-molecular interaction between congeneric molecules of 52, thusproviding the opportunity for the naked –COOH from 52 to bindwith the N-imidazole of 53 by hydrogen bonding. The sonication-triggered co-aggregation was thermally stable and could not bedestroyed by repeated heating–cooling cycles. In a further study,fibres with multicolour emission were obtained by tuning theratio of 52 to 53. The two-component sonogels are sensitive toacid and metal ions, with the morphology transformed from thatof a fibre structure to that of vesicles. Moreover, both the gelstate and the morphology can be reversed by further addition ofalkali.
By virtue of a characteristic structure with a hydrophilicexterior surface and a hydrophobic interior cavity, which canaccommodate a wide range of molecules as guests, cyclo-dextrins (CDs) have been widely used as hosts in supramole-cular chemistry for fabricating various nanostructures.88,89
Among the varieties of guest molecules used, those containingan adamantane group (AD) have been extensively used becauseof the high-complexation ability between AD and CD, especiallyb-CD.90,91 To investigate the aggregation properties of thehost–guest complex, we designed and synthesised a gelatorcontaining an AD group (54), whose gelation behaviour can becontrolled by sonication (Fig. 23).92 It is surprising that the self-assembly process of 54 can also be tuned by host–guest inter-actions via addition of b-CD under sonication. A structuralevolution from nanoribbons to ordered nanothreads wasobserved by manipulation of the self-assembly between thisadamantine-based gelator and b-CD.93
Fig. 21 Chemical structures of 49.
Fig. 22 Chemical structures of 50–53.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5363
The two-component gels formed by host–guest interactioncan also be thixotropic. Recently, Liu and coworkers reportedtwo-component hydrogels composed of bolaamphiphilic L-histidine(55) and benzene dicarboxylic acids (OPA, IPA or TPA) with thecapacity for noncovalent polymer formation after a traditionalheating–cooling process (Fig. 24). The structure of the dicarb-oxylic acids was proved to be an important factor for thedifferent behaviours of the gels. Hydrogels formed usingOPA–55 and IPA–55 showed thixotropic properties. When thegels were shaken by hand, the hydrogels could transform intosols without any phase separation, and the sols could againtransform into gels after being rested at room temperature forseveral hours. However, thixotropy cannot be achieved byTPA–55 due to the linear molecular packing and orderedhydrogen bonds among complexes in the compound.94
Using electrostatic interaction to construct host–guest func-tional organogels is also an effective way to obtain multi-responsive two-component gels. For example, Shinkai et al.reported an oligothiophene-based organogelator 56 that couldgel in mixed solvents of EtOH and CH3Cl with a volume ratio of1 : 1 and exhibited thixotropic behaviour.95 The addition of1,2-bisammonium ethylene compounds as the guests of 56 couldshorten the recovery time after being mechanically broken andmade the gel more transparent without phase separationbecause the host–guest interactions served as junction points.The recovery time could be tuned by the concentration of theguest (from 10 mg L�1 to 1.2 g L�1) from minutes to days. Themost interesting result obtained was the chiral memory switch-ing displayed by the host–guest interaction. By a heating–cooling process, a gel with R,R-1,2-bisammonium cyclohexaneor S,S-1,2-bisammonium cyclohexane showed remarkable
induced CD spectra. By a shaking–rest process, the recoveredgel was CD-silent; then, after a further heating–cooling cycle,the CD signal could be recovered.
In a thixotropic gel system, emission can also be triggeredby mechanical force, just as in sonogels. Recently, ThomasBaumgartner et al. reported an interesting mechanically driventransition in emission colour controlled by heat and mechanicalforce.96 In a homogeneous gel system, the electrostatic repulsionprevented strong intermolecular interactions between 57 and 58(57 as the emission donor, 58 as the acceptor, in a ratio of100 : 1), thus leading to less energy transfer and the blue emis-sion of 57; after shaking, changes in intramolecular conforma-tion and phase separation decreased the intermolecular distancebetween 57 and 58, resulting in an efficient energy transferprocess and thus orange emission. By further treating with heat,the blue emission and gel state could be restored.
3. Mechanism of gelation controlledby ultrasound and mechanical stress
As indicated in the examples described above, it is intriguingthat physical stimuli such as sonication and shearing stress donot always serve as inhibitors of the gelation process; thestimuli can also be favourable for gelation events. Unlike gelscontrolled by chemical stimuli, whose structure is more pre-dictable, most gel systems and their amazing physical pro-perties, which are sensitive to physical stimuli, have beendiscovered by serendipity. However, with the aid of techniquessuch as X-ray diffraction (XRD), small angle X-ray scattering(SAXS), NMR, SEM, TEM, confocal laser scanning microscopy,
Fig. 23 (A) The structure of 54; the morphology of the DMF gel of (B) 54 and (C) 54 + b-CD triggered by sonication. (Reprinted with permission fromref. 93. Copyright 2013, Royal Society of Chemistry.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online
5364 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
circular dichroism (CD) and absorption and fluorescentspectroscopy, general rules that can guide us in designingand studying new molecular systems have been developed thatmake gelation predicable. These rules are associated with theintrinsic nature of gelation and the corresponding responseprocesses.
3.1 Effect of ultrasound and mechanical stress expressed atthe molecular level of gelators
From the above results, it is indicated that the mechanical forceand moderate sonication generally do not change or destroy thechemical structure of a molecule in gel tissues.9,23 Sonicationserves two functions in the gelation process. First, sonicationcan transform intramolecular interactions into intermolecularinteractions to assist the gelation process in case that the
energy difference between formation of the two kinds of inter-actions is in a certain range.34–36,45,46 The instant sol–geltransition afforded by ultrasound is often attributed to theconversion of the mode of molecular hydrogen bonding or p–pbonding from intramolecular interaction to intermolecularinteraction. By applying a heating–cooling process, a gel canreform into the original sol; thus, the process can be regardedas a switch in the structure and rheology tuned by heat andultrasound. For example, the sol–gel transition of compoundanti-1 is triggered by structural transformation from self-lockedp–p stacking to inter-locked p–p stacking.34 The intramolecularhydrogen bonding between the vicinal –NH and –Cl in Pd-basedpeptide gelator 2a/2b can be disrupted when subjected toultrasound.35 As a result, aggregates of the molecules can bereorganised by intermolecular hydrogen bonding to form a
Fig. 24 Chemical structures of 55–58.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5365
belt-like network with a b-sheet structure to trap solvent moleculesand thereby form a gel. In an ALS system such as 10b, we have alsoobserved a conformational transition from intramolecular hydro-gen bonding between the –CQO bond of naphthalic anhydrideand –NH (ethyl imide) to intermolecular hydrogen bondingaccompanied by vesicle–fibre morphological changes.45
The other function that sonication may serve is the optimi-sation of the sequence or strength of intermolecular inter-actions in systems featuring multiple noncovalent interactionsto promote gelation.46,47 Therefore, a supramolecular systemwith multiple hydrogen bonding interactions is prone to be asonication-sensitive organogel. For example, with the aid ofsonication, compound 59 can form weak opaque gels in amixed solvent composed of CH2Cl2–CH3OH or CH3Cl3–CH3OH.This finding indicates that there is a transition from dimericcapsules to a cross-linked hydrogen bonding mode amongmolecules that was favourable for gelation (Fig. 25).97 In somecases, to optimise intermolecular interactions, sonication mayalso trigger the transition of molecular conformations to adynamic mode. Complex 4 is a typical example in whicha sonication-induced transformation of the conformation froma perpendicular mode to a coplanar mode of 1,8-naphthalimideand the tBu3tpy ring has been suggested.37 Sonication may thusbecome an effective method for generating two-componentmulti-functional gel systems or other nano/micro materialssuch as in the co-aggregate state of 52 + 53, with the trans-formation of hydrogen bonds among congeneric molecules intointeractions among heterogeneous components. In summary,conformational changes in molecules toward intramolecular orintermolecular interactions can be adjusted via sonication,which makes the gelation process or gels themselves moretuneable, easier to implement and intelligent systems for furtherpotential applications.
3.2 Effect of ultrasound and mechanical stress expressed asmacro aggregation of self-assemblies
The macroscopic effects of ultrasound and mechanical stress on gelsystems can provisionally be divided into four types: (a) sol-to-gel; (b)gel-to-gel; (c) precipitate-to-gel; and (d) gel-to-sol transformations.
The sol-to-gel transformation is generally caused by a moleculartransformation of intramolecular interactions into inter-molecular interactions by a stimulus, as previously discussed.The other three situations or transformations are more com-plicated. It should be noted that among most sonication-responsive or thixotropic gels, morphological transformationplays an important role in determining macro state changes.For example, in the sonication-induced direct gel-to-gel processof compound 11b in CH3CN, the sonication triggered thetransformation of a core–shell structure into nanospheres,and the nanospheres regenerated in cross-linked nanofibreswere then observed by a confocal laser microscope (CLSM).47
The observation of a micro-phase transformation from a gel toa partial gel and then back to a gel is in accordance with thisresult. However, XRD experiments did not provide any datacontradicting the conformation change. In toluene, sonicationdisplayed a cutting effect on the morphology; fibres measuring100 nm in width were cut into spindly fibres measuring20–40 nm in width. A similar effect was observed for compound13a in toluene; specifically, 2D pattern changes in which largecross-linked rings were transformed into smaller rings undersonication were observed. In all of these processes, destructionand regeneration coexist; thus, the gel state is retained through-out the entire processes.
The sonication-triggered precipitate-to-gel transition is simi-lar to the gel-to-gel transition, with the entangled fibrillarstructure as the terminal state in most cases. For example,separated spherulites were obtained from precipitation particlesof a nongelled sample of 3 wt% 26–octanol.67 Each spheruliteshowed a porous structure formed by fibre arrays in which thefibre arms grew radially from the centre. In contrast, homo-geneous 3D interconnected fibre networks were obtained fromthe ultrasound-induced gel phase. In such networks, nanometre-sized branched fibres can extend from one network to theadjacent one via mutual interpenetration and entanglement.The precipitate of 11a in CH3CN also undergoes a precipitate-to-gel transition under sonication over a temperature range of50–70 1C, accompanied by a morphological transition fromirregular nanomicelles to entangled fibres.
Fig. 25 Possibility of the tris(urea) 59 related to the transformation between dimeric capsules and supramolecular gels by sonication. (Reprinted withpermission from ref. 97. Copyright 2012, Royal Society of Chemistry.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5366 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
Amazingly, sonication-triggered morphological changes ingel formation are mostly reversible. When a sonogel is heatedto form a sol and then cooled, the initial state before sonicationcan be repaired by heat stimulus without fatigue. There-fore, switches in morphology or phase, even surfaces, can beeasily obtained by sonication and heat. For example, Yao andco-workers reported a direct morphological switch betweenvesicles in solution and fibres in a sonogel based on poly-peptide organogelator 34.74 This morphological transition wasreversible and repeatable without obvious fatigue effects andwas accompanied by a switch in macroscopic properties betweenthose of a transparent solution and those of an opaque gel. Areversible conversion between vesicles and three-dimensionalgel networks by self-assembly in response to heating and sonica-tion over a certain concentration range was also observed in ourALS structure (54). We were able to use this special property totransform the ethanol sol of 54 into a gel state or vesicles over aconcentration range of 1.7–4.4 wt% by sonication or a heating–cooling process (Fig. 26).92
Generally, simple mechanical stress generated by shaking orshear stress cannot affect molecular conformation; therefore,the destruction and self-healing process in a thixotropic gel ishighly dependent on the transformation of aggregates and thespontaneous balance between the aggregates and solvent mole-cules. Fibrillar or porous networks are inclined to trap solventmolecules to support a gel state. When the primary junctionsare destroyed, the gel transforms into a disordered aggregationor other phase state such as a sol, precipitate or partial gel. Thetransformation from a spherical structure to fibres has beenobserved in self-healing processes after the removal of theexternal source of stress. For example, the dense packing ofNMOPs (nanoscale metal–organic particles) measuring approxi-mately 50–300 nm in diameter was observed to support the gelstructure of 8 + CuX.41 When exposed to shaking, the gelaccumulation was separated with the destruction of the sec-ondary structure; then, after removing the stress, the sphericalparticles were cross-linked into nanofibres within 6–12 weeks(Fig. 27). The conversion was completed after approximatelyone year. For this slow transition, an intermediate pearl-necklace-like structure was clearly observed. Recently, Shinkaiet al. also reported a similar morphological evolution ofisolated disc-like aggregates into one-dimensional cross-linked assemblies. It is interesting that in many sonogels or
thixotropic gels, a fibrillar structure or fibril network is theterminal state.
By shaking and resting, transformations in phase andmorphology, and even chirality, can be obtained in thixotropicgels by internal stimuli. Such materials are regarded as third-generation self-healing materials. However, as self-healing softmaterials, the repaired structure of thixotropic gels is reason-ably altered in some respect compared with the original struc-ture. This phenomenon may be compared to the self-healing ofnatural materials such as skin and bone, whose recovered stateshows a slight variation from the original, indicating somedegree of destruction. In some thixotropic gel systems, thetransition from a gel to a sol then to a self-healed gel is notalways infinite due to the dynamic evolution of the instablestructures. For example, the organogel of CNC 12b in an alkanesolvent demonstrated the importance of morphology in deter-mining the thixotropic properties.48 At a gelation temperaturer28 1C, the gel formed with highly branched, small fibres inspherulitic aggregates, which was thixotropic. After severalcycles, the system exhibited fatigue, and the structure wasincreasingly fibre-like. In contrast, at a temperature 428 1C,only long fibres were observed, and self-healing could not occur.
Fig. 26 SEM images of compound 54 from ethanol (2.4 wt%): (a) precipitate from a heating–cooling process; (b) partial gel after sonication for 5 min; (c)S-gel after sonication for 10 min; scale bars for a, b and c are 5, 5 and 1 mm, respectively. (Reprinted with permission from ref. 92. Copyright 2012, RoyalSociety of Chemistry.)
Fig. 27 TEM micrographs of a hydrogel 41: (a) fresh gel upon standing(44 h), (b) sol after shaking (5 min), (c) transition of NMOPs of reformed gelinto ‘‘pearl-necklace’’ structures (in red) (3–6 weeks), (d) pearl-necklacenanostructures (red rectangle) and 1D nanobundles (red arrow) (6–12 weeks),(e) nanobundles and nanospheres (6–12 weeks), and (f) nanobundles in agedgels (about a year). (Reprinted with permission from ref. 41. Copyright 2011,American Chemical Society.)
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5367
The self-healing ability was ultimately attributed to the constantgrowth of spherulitic aggregates into fibres throughout theentire evolution process.
4. Application of LMOGs respondingto ultrasound and mechanical stress
In addition to the physical and gelation properties of LMOGs,as well as the mechanisms that are important for designing andstudying new organogelators, the extensive applications ofthese organogels are also of great importance. Physically respon-sive organogels have found applications in many fields, such aswettability switches, hybrid materials and pollutant removal.
4.1 Surface switches constructed by sonogels
Wettability, which is usually determined by multiple factorssuch as chemical composition and the micro- or nanostructureon surfaces, is one of the most important properties of solidsurfaces and has attracted increasing attention in recent yearsfor biomimetic applications and self-cleaning materials. Thesurface morphology of physically responsive gels can be trans-formed on the micro- or macroscale due to molecular trans-formation, molecular aggregation mode, etc., when subjected toexternal stimulation, thus resulting in the transformation of
the surface of xerogels. The first inspiring example of control-lable surface wettability adjusted by sonication in an organogelsystem was reported by our group.45,46 After a heating–coolingprocess, a xerogel film of 10a showed a superhydrophobicsurface with a vesicle morphology (composed of a fibrousnetwork with pores on the surface) (Fig. 28). After being treatedwith sonication, the sonication-induced S-xerogel had a surfacewith a water contact angle (CA) of 104.41; then, at applying aheating–cooling process to the S-gel, the contact angle could berecovered. However, with a similar conformation transforma-tion from a spherulitic structure to a fibre network stimulatedby sonication, the opposite result was obtained for sonogel 4:the precipitate generated by a heating–cooling process had amore hydrophilic surface with an average CA of 951, whereasthe gel formed by ultrasound exhibited a hydrophobic surface(CA = 1441).
4.2 Hybrid functional materials constructed by sonogels
One of the most important properties of responsive gels is thefacile tuneability of the structure of the gelators responsiblefor the observed properties. To obtain complex functionalmaterials, one strategy is to generate new composites by theassembly of gels with other systems.98–103 David Bardelang andcoworkers incorporated QDs (quantum dots) into an organogelnetwork via ultrasound for the first time.104–106 Morphologicalstudies suggested that large QDs were incorporated into thesurfaces of gel fibres, whereas the gelation properties and thefluorescence of the QDs were not significantly affected. More-over, a hybrid gel or xerogel with TEMPO-bound QDs couldsense the decomposition product of azobisisobutyronitrile(AIBN) with apparent changes in fluorescence.
Our group also prepared a hybrid gel featuring peptide-based derivatives (60, Fig. 29A) and up-conversion nano-phosphors (UCNPs) that could exhibit multi-colour emissionwhen excited by near-infrared light.107 UCNPs (NaYF4) weresuccessfully transformed into a self-assembled LMOG networkthrough physical interactions in a polar solvent via sonication.As a result, the UCNPs are dispersed in a gel matrix, and theindividually dispersed UCNPs acted as physical cross-linksbetween the supramolecular fibres, thereby reinforcing the gelstructure (Fig. 29B). This new strategy for producing UCNP–peptidehybrid multi-colour gels allows the UCNPs to retain their nano-phosphor properties in the gel state. These UCNP nano-composite
Fig. 28 SEM images of the xerogel of T-gel (a), S-gel (b), R-gel (c) of 10afrom p-xylene (15 mg mL�1) and the photographs of the water droplet onglass slices, spin-coated with the T-gel (d), S-gel (e), and R-gel (f).(Reprinted with permission from ref. 84. Copyright 2004, Wiley-VCHVerlag GmbH & Co. KGaA.)
Fig. 29 (A) Chemical structures of 60a and 60b. (B) Self-assembly of 60b + UCNPs and fluorescent images of 60b + UCNP gels in n-hexanol ([60b] =2.3 � 10�2 M, 1.5 wt% of UCNPs); images from left to right: 60b + UCNPs-Red, 60b + [UCNPs-Red/UCNPs-Green (1/1 wt)], 60b + UCNPs-Green, 60b +[UCNPs-Green/UCNPs-Blue (1/1 wt)], 60b + UCNPs-Blue, 60b + [UCNPs-Blue/UCNPs-Red (1/1 wt)] (lex = 980 nm). (Reprinted with permission fromref. 107. Copyright 2009, Royal Society of Chemistry.)
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5368 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
gels may fulfil the requirements for colour displays and NIR lightabsorption materials for generating power or may be modified tofunction as biological labels.
4.3 Thixotropic gels for oil collection and pollutant removal
Notably, the phase-selective gelation between water and oiltriggered by mechanical force at room temperature indicatesthe feasibility of removing oil or pollutants from water sources.108
The first example of phase-selective gelation was demonstratedby Bhattacharya and Krishnan-Ghosh with amino acid amphi-philes.109 Molecular gelators based on closed-chain and open-chainsugars have been studied recently, which exhibit phase-selective gelation and can gel oil in the presence of waterat room temperature.110,111 After the phase-selective gelationof the oil in these mixtures, both the oil and gelator can berecovered and reused.
Thixotropic gels can also be used for pollutant removal. Forexample, the compound N-lauroyl-L-glutamic-a,g-dibutylamidecan gel in many organic solvents and exhibit thixotropic proper-ties. By combining the gelator and an appropriate extractant,such as thiophene for mercury chelators or 4-(2-pyridylazo)-resorcinol (PAR) for nickel ions, with an organic hydrocarbonsolvent to form a thixotropic gel, a solid–liquid system forextraction was constructed in a previous study. Experimentalresults revealed that the system required only 10 min ofshaking to extract metal from water with high selectivity.112
4.4 Rheology switching in thixotropic gels
The control of the rheological properties of fluids during self-healing processes is of practical importance for industrial applica-tions, biomaterials and smart systems.20,21,50 Switches in rheologymay control the motion of fluid molecules toward damaged areasto realise self-healing. Some thixotropic organogels are verysensitive to mechanical stress and show switchable rheologicalproperties. These gels hold potential applications in, for example,art cleaning and protection113 and blood and cell conservation.114
5. Conclusions and outlook
In conclusion, the history, types, mechanisms, as well asapplications of ultrasound and mechanical stress driven/responsive organogels were examined and summarised in thisreview. These gels show distinct gelation properties such asin situ and instant gelation, phase-selective gelation and self-healing properties. The structures of the molecules in thesecompounds greatly determine the resulting gelation properties.Alkane chain length, the position of chirality units, substitutiongroups, peptide sequences as well as the hydrogen bondingmode or hydrogen bond numbers can all cause the responsiveproperties of gels to vary. The reported cholesterol-based organo-gels with the structure of an ALS2 system are often bothsonication-responsive and thixotropic and exhibit gelation pro-perties at room temperature; phenyl alkane ester-based organo-gelators exhibit properties similar to those of cholesterol-basedorganogelators; metallogels often exhibit reversible and facile gel
colour or emission control due to the adjustment of p–p inter-actions; and amide-based organogels, including peptide-basedorganogels, exhibit low toxicity, which makes the gels moreinjectable and practical in the field of drug delivery.
The gelation and stimulus-responsive mechanisms of theaforementioned gels have been thoroughly studied and ana-lysed. The physically driven gelation process depends on manyfactors, such as the structural adjustment of gelator molecules,solvent effects, concentration, temperature, sonication and shearstress. A tiny change in these factors greatly impacts the gelationproperties of gels due to the precise balance between themolecular assembly of gelator molecules, solvent–gelator inter-actions and the applied external stimulus. It has been deter-mined that conformational changes in molecular structure mayoccur either in sonogels or in thixotropic gels during the sol–gelphase-transition process; however, it is still difficult to studythese changes using the present techniques without crystallisa-tion data. Sonication- or shear-stress-induced dissociation or thecross-linking effect on aggregates of gelators often results infibrillisation phenomena, which offer schemes for mouldingfibres into living tissues. Therefore, the study of dynamic assem-blies stimulated by mechanical stimuli in supramolecular geltissues holds great significance in the field of biomimetics.
The extensive applications of organogels are of great impor-tance in the design and study of new organogelators, helping us toaddress open issues such as characterising gels and determiningpractical applications for gels. In reality, most LMOGs are far fromfinding applications due to the poor robustness of non-covalentsystems. However, some gels with outstanding mechanical andoptical properties would be more useful for practical application,and great efforts have been undertaken in this field by severalresearchers. The reversible control of organogel properties by theapplication of heat and mechanical stimuli is often accompaniedby transformations in phase, morphological structure and rheolo-gical and spectroscopic properties, which make these gels suitablefor application in the field of drug delivery, switchable devices,adaptive materials and self-healing materials, among others.
Acknowledgements
The authors acknowledge financial support from the NationalBasic Research Program of China (2013CB733700), the ChinaNational Funds for Distinguished Young Scientists (21125104),the National Natural Science Foundation of China (51373039,91022021, and 21301047), the Program for Innovative ResearchTeam in University (IRT1117), the Program of Shanghai SubjectChief Scientist (12XD1405900), and the Shanghai Leading Aca-demic Discipline Project (B108).
References
1 D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12,1237–1247.
2 N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34,821–836.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5369
3 B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012,44, 544–554.
4 P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159.5 J. H. Jung, J. H. Lee, J. R. Silverman and G. John, Chem. Soc.
Rev., 2013, 42, 924–936.6 M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and
B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098.7 M. Moniruzzaman, A. Sahin and K. I. Winey, Carbon, 2009,
47, 645–650.8 T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335,
813–817.9 R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer,
J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe andE. W. Meijer, Science, 1997, 278, 1601–1604.
10 S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida,S. Kiyonaka and I. Hamachi, Chem. – Eur. J., 2005, 11,1130–1136.
11 P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler,Nature, 2008, 451, 977–980.
12 L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104,1201–1218.
13 K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew.Chem., Int. Ed., 2003, 42, 980–999.
14 J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160.15 F. Fages, Low Molecular Mass Gelators, Top. Curr. Chem.,
2005, 256, 283.16 T. Ishi-i, S. Shinkai and F. Wurthner, Supramolecular Dye
Chemistry, Top. Curr. Chem., 2005, 258, 119–160.17 D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12,
1237–1247.18 J. H. Van Esch and B. L. Feringa, Angew. Chem., Int. Ed.,
2000, 39, 2263–2266.19 S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu
and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139.20 G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697.21 J. M. J. Paulusse and R. P. Sijbesma, Angew. Chem., Int. Ed.,
2006, 45, 2334–2337.22 V. F. Humphrey, Prog. Biophys. Mol. Biol., 2007, 93,
195–211; C. Gong and D. P. Hart, J. Acoust. Soc. Am.,1998, 104, 1–16.
23 G. Cravotto and P. Cintas, Chem. Sci., 2012, 3, 295–307;K. S. Suslick, Sci. Am., 1989, 260, 80–86.
24 W. T. Richards and A. L. Loomis, J. Am. Chem. Soc., 1927,49, 3086–3100.
25 D. J. Donaldson, M. D. Farrlngton and P. Kruus, J. Phys.Chem., 1979, 83, 3130–3135.
26 C. Petrier, A. Jeunet, J.-L. Luche and G. Reverdy, J. Am.Chem. Soc., 1992, 114, 3148–3150.
27 T. Kitazume and N. Ishikawa, J. Am. Chem. Soc., 1985, 107,5186–5191.
28 J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Science,2011, 333, 1606–1609.
29 D. Bardelang, Soft Matter, 2009, 5, 1969–1971.30 M. Lescanne, P. Grondin, A. d’Aleo, F. Fages, J. Pozzo,
O. Monval, P. Reinheimer and A. Colin, Langmuir, 2004, 20,3032–3041.
31 S. Burattini, B. W. Greenland, D. H. Merino, W. Weng,J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay,I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132,12051–12058.
32 A. B. W. Brochu, S. L. Craig and W. M. Reichert, J. Biomed.Mater. Res., Part A, 2011, 96, 492–506.
33 M. D. Hager, P. Greil, C. Leyens, S. Zwaag andU. S. Schubert, Adv. Mater., 2010, 22, 5424–5430.
34 T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127,9324–9325.
35 K. Isozaki, H. Takaya and T. Naota, Angew. Chem., Int. Ed.,2007, 46, 2855–2857.
36 N. Komiya, T. Muraoka, M. Lida, M. Miyanaga,T. Talahashi and T. Naota, J. Am. Chem. Soc., 2011, 133,16054–16061.
37 K. Liu, L. Meng, S. Mo, M. Zhang, Y. Mao, X. Cao, C. Huangand T. Yi, J. Mater. Chem. C, 2013, 1, 1753–1762.
38 Y. Li, E. S.-H. Lam, A. Y.-Y. Tam, K. M.-C. Wong, W. H.Lam, L. Wu and V. W.-W. Yam, Chem. – Eur. J., 2013, 19,9987–9994.
39 J. Liu, P. He, J. Yan, X. Fang, J. Peng, K. Liu and Y. Fang,Adv. Mater., 2008, 20, 2508–2511.
40 W. Weng, J. B. Beck, A. M. Jamieson and S. J. Rowan, J. Am.Chem. Soc., 2006, 128, 11663–11672.
41 T. D. Hamilton, D.-K. Bucar, J. Baltrusaitis, D. R. Flanagan,Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am.Chem. Soc., 2011, 133, 3365–3371.
42 M. Zinic, F. Votle and F. Fages, Cholesterol-based gelators,Top. Curr. Chem., 2005, 256, 39–76.
43 Y. Lin and R. G. Weiss, Macromolecules, 1987, 20, 414–417.44 Y. Lin, B. Kachar and R. G. Weiss, J. Am. Chem. Soc., 1989,
111, 5542–5551.45 J. Wu, T. Yi, T. Shu, M. Yu, Z. Zhou, M. Xu, Y. Zhou,
H. Zhang, J. Han, F. Li and C. Huang, Angew. Chem., Int.Ed., 2008, 47, 1063–1067.
46 J. Wu, T. Yi, Q. Xia, Y. Zou, F. Liu, J. Dong, T. Shu, F. Li andC. Huang, Chem. – Eur. J., 2009, 15, 6234–6243.
47 X. D. Yu, Q. Liu, J. C. Wu, M. Zhang, X. Cao, S. Zhang,Q. Wang, L. Chen and T. Yi, Chem. – Eur. J., 2010, 16,9099–9106.
48 X. Huang, S. R. Raghavan, P. Terech and R. G. Weiss, J. Am.Chem. Soc., 2006, 128, 15341–15352.
49 X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am.Chem. Soc., 2005, 127, 4336–4344.
50 X. Yu, X. Cao, L. Chen, H. Lan, B. Liu and T. Yi, Soft Matter,2012, 8, 3329–3334.
51 J. Peng, K. Liu, J. Liu, Q. Zhang, X. Feng and Y. Fang,Langmuir, 2008, 24, 2992–3000.
52 M. Xue, D. Gao, K. Liu, J. Peng and Y. Fang, Tetrahedron,2009, 65, 3369–3377.
53 X. Y. Hou, D. Gao, J. Yan, Y. Ma, K. Liu and Y. Fang,Langmuir, 2011, 27, 12156–12163.
54 X. Q. Cai, K. Q. Liu, J. Yan, H. Zhang, X. Hou, Z. Liu andY. Fang, Soft Matter, 2012, 8, 3756–3761.
55 X. Cai, Y. Wu, L. Wang, N. Yan, J. Liu, X. Fang and Y. Fang,Soft Matter, 2013, 9, 5807–5814.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

5370 | Chem. Soc. Rev., 2014, 43, 5346--5371 This journal is©The Royal Society of Chemistry 2014
56 V. Percec, M. Peterca, M. E. Yurchenko, J. G. Rudick andP. A. Heiney, Chem. – Eur. J., 2008, 14, 909–918.
57 Z.-X. Liu, Y. Feng, Z.-C. Yan, Y.-M. He, C.-Y. Liu andQ.-H. Fan, Chem. Mater., 2012, 24, 3751–3757.
58 P. Mukhopadhyay, N. Fujita, A. Takada, T. Kishida,M. Shirakawa and S. Shinkai, Angew. Chem., Int. Ed.,2010, 49, 6338–6342.
59 A. Dawn, T. Shiraki, H. Ichikawa, A. Takada, Y. Takahashi,Y. Tsuchiya, L. T. N. Lien and S. Shinkai, J. Am. Chem. Soc.,2012, 134, 2161–2171.
60 X. F. Zhu, P. F. Duan, L. Zhang and M. H. Liu, Chem. – Eur.J., 2011, 17, 3429.
61 G. M. Whitesides, J. P. Mathias and C. T. Seto, Science,1991, 254, 1312.
62 T. Ishi-i, R. Kuwahara, A. Takata, Y. Jeong, K. Sakurai andS. Mataka, Chem. – Eur. J., 2006, 12, 763.
63 A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith,Angew. Chem., Int. Ed., 2008, 47, 8002.
64 S. Yagai, H. Aonuma, Y. Kikkawa, S. Kubota, T. Karatsu,A. Kitamura, S. Mahesh and A. Ajayaghosh, Chem. – Eur. J.,2010, 16, 8652.
65 J. L. Bada, Nature, 1995, 374, 594; P. K. Vemula andG. John, Acc. Chem. Res., 2008, 41, 769.
66 C. L. Zhan, P. Gao and M. H. Liu, Chem. Commun., 2005,462.
67 R. Wang, X. Liu and J. Li, Cryst. Growth Des., 2009, 9,3286–3291.
68 D. Bardelang, F. Camerel, J. C. Margeson, D. Leek,M. Schmutz, M. B. Zaman, K. Yu, D. V. Soldatov,R. Ziessel, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem.Soc., 2008, 130, 3313–3315.
69 S. Maity, P. Kumar and D. Haldar, Soft Matter, 2011, 7,5239–5245.
70 Y. G. Li, T. Wang and M. H. Liu, Tetrahedron, 2007, 63,7468–7473.
71 R. Afrasiabi and H. Kraatz, Chem. – Eur. J., 2013, 19,1769–1777.
72 Z. Xie, A. Zhang, L. Ye and Z.-G. Feng, Soft Matter, 2009, 5,1474–1482.
73 I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8,5301–5308.
74 D. Ke, C. Zhan, A. D. Q. Li and J. Yao, Angew. Chem., Int.Ed., 2011, 50, 3715–3719.
75 H. Liu, Y. Hu, H. Wang, J. Wang, D. Kong, L. Wang,L. Chen and Z. Yang, Soft Matter, 2011, 7, 5430–5436.
76 Y. Jiang, F. Zeng, R. Gong, Z. Guo, C. Chen and X. Wan, SoftMatter, 2013, 9, 7538–7544.
77 M. Avalos, R. Babiano, P. Cintas, A. Gomez, J. L. Jimenez,M. Lozano, A. L. Ortiz, J. C. Palacios and A. Pinazo,Chem. – Eur. J., 2008, 14, 5656–5669.
78 C. D. Dou, C. G. Wang, H. Y. Zhang, H. Z. Gao andY. Wang, Chem. – Eur. J., 2010, 16, 10744–10751.
79 M. Yamanaka, T. Nakamura, T. Nakagawa and H. Itagaki,Tetrahedron Lett., 2007, 48, 8990–8993.
80 M. Yamanaka and H. Fujii, J. Org. Chem., 2009, 74,5390–5394.
81 S. M. Park and B. H. Kim, Soft Matter, 2008, 4, 1995–1997.82 J. T. V. Herpt, M. C. A. Stuart, W. R. Browne and B. L.
Feringa, Langmuir, 2013, 29, 8763–8767.83 V. A. Mallia, M. George, D. L. Blair and R. G. Weiss,
Langmuir, 2009, 25, 8615–8625.84 I. Yoshikawa, J. Li, Y. Sakata and K. Araki, Angew. Chem.,
Int. Ed., 2004, 43, 100–103.85 A. Vidyasagar, K. Handore and K. M. Sureshan, Angew.
Chem., Int. Ed., 2011, 50, 8021–8024.86 K. M. Anderson, G. M. Day, M. J. Paterson, P. Byrne,
N. Clarke and J. W. Steed, Angew. Chem., Int. Ed., 2008,47, 1058–1062.
87 X. Yu, Q. Liu, X. Xu, H. Lan, X. Cao, L. Chen, B. Liu andT. Yi, Acta Chim. Sin., 2012, 70, 2016–2023.
88 K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim andJ. Kim, Chem. Soc. Rev., 2006, 36, 267–279.
89 Y. Liu and Y. Chen, Acc. Chem. Res., 2006, 39, 681–691.90 M. Guo and M. Jiang, Soft Matter, 2009, 5, 495–500.91 M. Escalante, Y. Zhao, M. J. W. Ludden, R. Vermeij,
J. D. Olsen, E. Berenschot, C. N. Hunter, J. Huskens,V. Subramaniam and C. Otto, J. Am. Chem. Soc., 2008,130, 8892–8893.
92 M. Zhang, L. Meng, X. Cao, M. Jiang and T. Yi, Soft Matter,2012, 8, 4494–4498.
93 M. Zhang, M. Jiang, L. Meng, K. Liu, Y. Mao and T. Yi, SoftMatter, 2013, 9, 9449–9454.
94 Y. Liu, T. Wang and M. Liu, Chem. – Eur. J., 2012, 18,14650–14659.
95 A. A. Sobczuk, Y. Tsuchiya, T. Shiraki, S. Tamaru andS. Shinkai, Chem. – Eur. J., 2012, 18, 2832–2838.
96 Y. Ren, W. H. Kan, V. Thangadurai and T. Baumgartner,Angew. Chem., Int. Ed., 2012, 51, 3964–3968.
97 C. Deng, R. Fang, Y. Guan, J. Jiang, C. Lin and L. Wang,Chem. Commun., 2012, 48, 7973–7975.
98 W. C. Wang, C. Du, H. Bi, Y. H. Sun, Y. Wang, C. Mauser,E. D. Como, H. Fuchs and L. F. Chi, Adv. Mater., 2010, 22,2764–2769.
99 S. K. M. Nalluri and B. J. Ravoo, Angew. Chem., Int. Ed.,2010, 49, 5371–5374.
100 S. Mahesh, R. Thirumalai, S. Yagai, A. Kitamura andA. Ajayaghosh, Chem. Commun., 2009, 5984–5986.
101 Q. Chen, D. Zhang, G. Zhang, X. Yang, Y. Feng, Q. Fangand D. B. Zhu, Adv. Funct. Mater., 2010, 20, 3244–3251.
102 J. L. Li and X. Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216.103 C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang,
R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132,3092–3096.
104 D. Bardelang, M. Z. Zaman, I. L. Moudrakovski, S. Pawsey,J. C. Margeson, D. Wang, X. Wu, J. A. Ripmeester,C. I. Ratcliffe and K. Yu, Adv. Mater., 2008, 20, 4517.
105 X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater.,2008, 20, 1522.
106 N. Gaponik, A. Wolf, R. Marx, V. Lesnyak and K. Schilling,Adv. Mater., 2008, 20, 4257.
107 J. Wu, Q. Tian, H. Hu, Q. Xia, Y. Zou, F. Li, T. Yi andC. Huang, Chem. Commun., 2009, 4100–4102.
Chem Soc Rev Review Article
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online

This journal is©The Royal Society of Chemistry 2014 Chem. Soc. Rev., 2014, 43, 5346--5371 | 5371
108 P. K. Vemula and G. John, Acc. Chem. Res., 2008, 41, 769–782.109 S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun.,
2001, 185–186.110 S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and
G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698.111 S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2,
2270–2273.
112 S. Abe, M. Endo, M. Sato and K. Awano, Anal. Commun.,1996, l33, 137–138.
113 E. Carretti, M. Bonini, L. Dei, B. H. Berrie, L. V. Angelova,P. Baglioni and R. G. Weiss, Acc. Chem. Res., 2010, 43,751–760.
114 K. Sun, H. Oh, J. F. Emerson and S. R. Raghavan, J. Mater.Chem., 2012, 22, 2378–2382.
Review Article Chem Soc Rev
Publ
ishe
d on
28
Apr
il 20
14. D
ownl
oade
d by
Uni
vers
ity o
f Y
ork
on 2
4/01
/201
6 23
:40:
03.
View Article Online