Embed Size (px)
Citation preview


METABOLĠZMA ANABOLĠZMA +
KATABOLĠZMA

Metabolik hastalıklarda protein, karbonhidrat ve lipid sentezinde veya yıkılmasında defekt söz konusudur.

Yapısı bozulan protein genellikle bir enzimdir. Ancak bir reseptör, taĢıyıcı protein, membran pompası veya yapısal element de olabilir.

Kilo alamama
Persistan kusma
Konvülsiyon
Tonus /
Letarji, koma
RDS / apne
Kardiyomiyopati
Renal kistler
Ġshal
Sarılık
Hepatomegali
Vücutta değiĢik koku
Kaba yüz görünümü
Katarakt
Lens dislokasyonu
Persistan egzema

Saç değiĢiklikleri
Hipoglisemi
Metabolik asidoz
Ġdrarda redüktan madde
Ġdrar ve serumda aminoasit veya amonyağın artması

DOĞUMDA NORMAL OLUP DAHA SONRA KLĠNĠK BOZULMA GÖRÜLMESĠ METABOLĠK HASTALIKLAR YÖNÜNDEN ĠPUCUDUR!!

ÇOĞU OTOZOMAL RESESĠF GEÇER. BU YÜZDEN AĠLE ÖYKÜSÜ (KARDEġ ÖLÜMÜ GĠBĠ) TANI ġÜPHESĠNĠ ARTTIRIR!!!
ÖZEL KOKULAR YARDIMCI OLABĠLĠR!!

KLĠNĠK BULGULAR NONSPESĠFĠKTĠR; SEPSĠSLE
KARIġIR!! DAHA HAFĠF VARYANTLAR DAHA GEÇ YAġTA
ORTAYA ÇIKAR. ANCAK ÇOĞU HAYATIN ĠLK BĠR KAÇ GÜNÜNDE SEMPTOM VERĠR.

AMONYAK, HCO3 VE PH DEĞERLERĠ AYIRICI TANIDA ÖNEMLĠDĠR.


KAN AMONYAK DÜZEYĠ ÜRE SĠKLUS DEFEKTLERĠNDE VE ORGANĠK ASĠDEMĠLERDE ANCAK ÜRE SĠKLUS DEFEKTLERĠNDE ASĠDOZ YOKKEN, ORGANĠK ASĠDEMĠLERDE ASĠDOZ VARDIR. HEPSĠ NORMALSE AMĠNOASĠDOPATĠLER VEYA GALAKTOZEMĠ DÜġÜNÜLMELĠDĠR.

Glutarik asidemi (tip2)...........Terli ayak
Ġzovalerik asidemi..................Terli ayak
FKU..........................Küflü veya faremsi
MSUD........................................Çemen
Hawkinsüri......................Yüzme havuzu

Metionin malabsorbsiyonu..........Lahana
Tirozinemi...................................Lahana
Multipl karboksilaz eks...........Kedi idrarı
Trimetilaminüri...................KokmuĢ balık

MENTAL MOTOR RETARDASYON, KONVÜLSĠYONLAR VEYA ENFEKSĠYON GĠBĠ BĠR STRESĠN TETĠKLEDĠĞĠ AÇIKLANAMAYAN KUSMA, ASĠDOZ, KOMA TABLOSU DA ĠPUCU OLABĠLĠR!!

Açıklanmayan MMR, konvülsiyonlar
Özellikle akut hastalık sırasında olağan olmayan koku
Açıklanamayan tekrarlayıcı kusma, asidoz, mental bozulma ve koma atakları
Hepatomegali
Renal taĢlar olması halinde metabolik hastalıklar akla gelmelidir!!


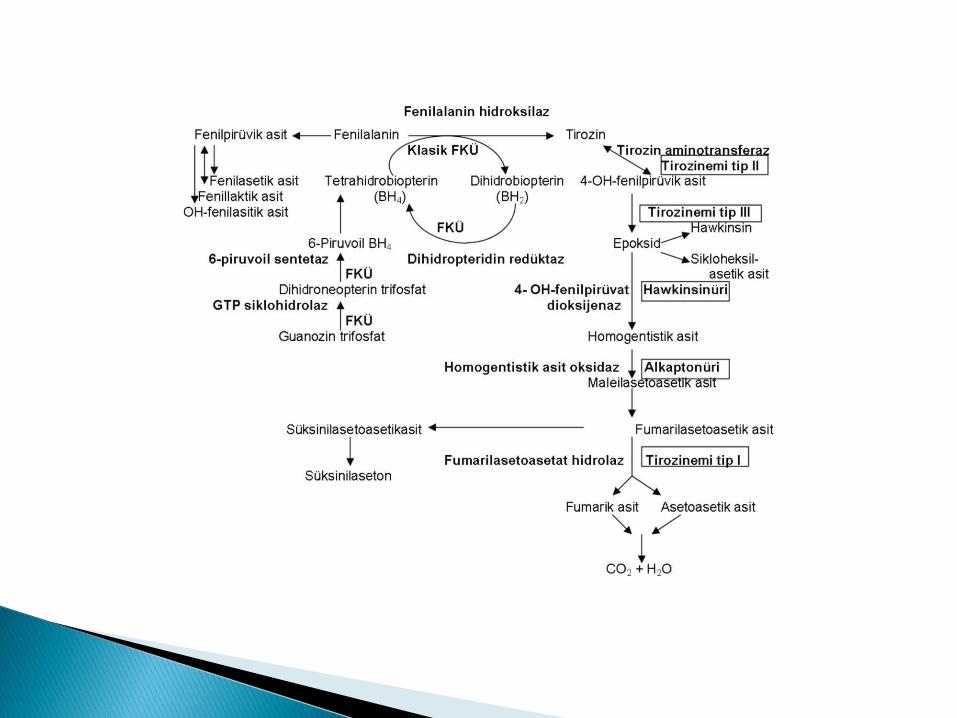

Klasik fenilketonüri (En sık, benign)
BH4 eksikliğine bağlı hiperfenilalaninemi (malign)
Benign hiperfenilalaninemi
Geçici hiperfenilalaninemi

Fenilalanin hidroksilaz enzimi eksiktir.
Fenilalanin tirozine çevrilemez.
OR geçer.
Biriken fenil alanin toksik metabilitlere (fenilpirüvik asit, feniletilenamin) dönüĢerek beyin harabiyetine yol açar.

Klinik: ◦ Doğumda normaldirler.
◦ Giderek artan Ģekilde MR geliĢir.
◦ Ġnatçı kusmalar (PĠLOR STENOZU GĠBĠ)
◦ Amaçsız hareketler, hiperaktivite, atetoz

Fizik muayene: ◦ SarıĢın ve mavi gözlüdürler.
◦ Seboreik veya ekzamatoid cilt raĢları
◦ Ġdrarda FARE VEYA KÜF KOKUSU (fenilasetik asite bağlı)
◦ Hipertonisite, DTR’ ler
◦ Konvülsiyon, EEG bozuklukları, mikrosefali

Tanı: ◦ Guthrie testi :
Doğumdan 48-72 saat sonra tarama testi olarak yapılır. Kanda artan pha BACILLUS SUBTILISI’i inhibe eder. Bu test (+) ise pha ve tirozin düzeyi ölçülür. False (-) sonuç olasılığını azaltmak için proteinle beslemeden sonra bakılır.

Tanı: ◦ Ġdrarda %10’luk FeCl3 testi:
En basit yöntemdir.6 damla idrar üzerine 3 damla FeCl3 damlatıldığında yeĢil renk oluĢur. YeĢil renk 1-2 dk’da kaybolur. (+) sonuç alındığında serumda pha düzeyi ölçülerek tanı doğrulanır. Ġdrardaki fenilpirüvik asit hayatın ilk günlerinde (-) olabileceğinden test false (-) olabilir!!

TANI KRĠTERLERĠ: ◦ Kan pha düzeyi > 20 mg/dl olması
◦ Plazma tirozin düzeyininin N olması
◦ Ġdrarda pha metabolitlerinin artması
◦ BH4’ün N olması
◦ TANI KRĠTERLERĠ ĠÇĠNDE ENZĠM DÜZEYĠ YOK!!

TEDAVĠ: ◦ Amaç, vücut sıvılarında phe ve metabolitlerini
azaltmaktır.
◦ Diyetten phe kısıtlanır. Süresi??
◦ 6 yaĢından sonra diyet biraz gevĢetilebilir.
◦ Kan pha düzeyi 3-15 (5-7) mg/dl arasında tutulmaya çalıĢılır.
◦ Yeterli kalori ve vitamin sağlanır.

FKU’LU ANNE BEBEĞĠ: ◦ Abortus riski, DDA
◦ Mikrosefali
◦ Kalp anomalisi
◦ Korpus kallosum hipoplazisi
◦ MR
◦ Maternal kan pha düzeyi 5 mg/dl’nin altında tutulur.

Malign hiperfenilalaninemi de denir. BH4 SENTEZĠNDE ROL ALAN DĠHĠDROPTERĠDĠN
REDÜKTAZ, 6-PĠRUVOĠL SENTETAZ (EN SIK), GTP SĠKLOHĠDROLAZ,KARBĠNOLAMĠN DEHĠDRATAZ ENZĠMLERĠNDEN BĠRĠ EKSĠKTĠR.

BH4 FENĠLALANĠN, TĠROZĠN VE TRĠPTOFAN HĠDROKSĠLAZ ENZĠMLERĠNĠN KOFAKTÖRÜDÜR!!
BH4 NO SENTAZIN KOFAKTÖRÜDÜR.
Tirozin ve triptofan hidroksilaz dopamin ve seronin sentezinde esansiyeldir.

KLĠNĠK BULGULAR KLASĠK TĠPLE AYNIDIR.
ANCAK DĠYETE RAĞMEN NÖROLOJĠK BULGULAR ĠLERLER!!

TANI: ◦ Ġdrarda neopterin ve biopterin tayini
◦ BH4 yükleme testi: 2-10 mg/kg BH4 p.o/ i.v
◦ Enzim tayini
◦ Gen çalıĢmaları

TEDAVĠ: ◦ DüĢük pha li diyet
◦ Nörotransmitter prokürsörleri (L-Dopa, 5-0H Trp gibi)
◦ BH4 20-40 mg/kg/gün p.o.

Fenilalanin hidroksilaz kısmen eksiktir.
Kan pha düzeyi <20 mg/dl
Asemptomatiktirler.
Tarama testlerinde tanı koyulur.
Diyetsiz da hastaların geliĢimi normaldir.
Diyette protein kısıtlanır.

Yenidoğanın geçici tirozinemisinde pha düzeyi de orta oranda yükselir.
Ġnfantın tirozini okside etme özelliği mature olunca artmıĢ olan tirozin ve pha düzeyleri de normale iner.


Tirozin metabolizma bozuklukları: ◦ Tirozinemi tip 1,2,3
◦ Yenidoğanın geçici tirozinemisi
◦ Hawkinsüri
◦ Alkaptonüri
◦ Albinizm

Tirozin, protein, dopamin, norepinefrin, epinefrin, melanin ve tiroksin sentezinde rol oynar.

Hipertirozinemi; ◦ Tirozinemi tip 1,2,3
◦ Ağır hepatosellüler disfonksiyon
◦ Skorbüt (Vitamin C 4-HPPD’nin kofaktörü)
◦ Hipertiroidi

Herediter tirozinemi, hepatorenal tirozinemi
OR geçer.
FUMARĠLASETOASETAT HĠDROLAZ ENZĠMĠ EKSĠKTĠR.

Tirozin metabolitleri özellikle SÜKSĠNĠLASETON birikir.
KC, BÖBREKLER VE SSS’Ġ ETKĠLENĠR.

KLĠNĠK: ◦ Akut:
Ġlk 6 ayda semptomatik hale gelir.
BGG, irritabilite, kusma, diyare, ateĢ, HM, sarılık, hipoglisemi, kanama eğilimi
ÇÜRÜK LAHANA KOKUSU vardır. (metionin!!)
2 yaĢından önce kc yetmezliğinden ölüm görülür. Daha büyüklerde kc ca geliĢebilir.

KLĠNĠK: ◦ Kronik:
1 yaĢından sonra bulgu verir.
BGG, siroz, FANKONĠ SENDROMU, VĠTAMĠN D DĠRENÇLĠ RĠKETS GÖRÜLEBĠLĠR. USG’de nefromegali ve nefrokalsinoz
Akut polinöropati (ayak ve karın ağrısı, paralitik ileus, self mutilasyon, kusma)
10 yaĢ civarında kc yetmezliği ve hepatoma nedeniyle ex olurlar.

LAB: ◦ NN anemi
◦ Serum bilirubinleri ve KCFT
◦ AFP
◦ Plazmada tirozin ve metionin
◦ Generalize aminoasidüri
◦ Ġdrarda gama aminolevülinik asit

SERUM VE ĠDRARDA SUKSINIL ASETOASETAT VE SUKSINIL ASETON ARTIġI TANISALDIR!!

TANI: ◦ Enzim analizi
◦ BEBEKLERDE HEPATĠT VE HEPATĠK YETMEZLĠK YAPAN DURUMLARDAN AYIRT EDĠLMELĠDĠR!!

TEDAVĠ: ◦ TĠROZĠN, METĠONĠN VE PHA’DEN KISITLI DĠYET
◦ KARACĠĞER TRANSPLANTASYONU

Richner – Hanhart Sendromu
Okulokutanöz tirozinemi
OR geçer.
Ciddi hipertirozinemi ve tirozinüri vardır.
TĠROZĠN TRANSAMĠNAZ (AMĠNOTRANSFERAZ) EKSĠKTĠR!!

KLĠNĠK: ◦ Mental retardasyon
◦ Palmar ve plantar punktat hiperkeratoz
◦ Herpetiform korneal ülserler
◦ Gözlerde aĢırı sulanma, kızarıklık, ağrı ve fotofobi

TĠROZĠNEMĠ TĠP 1 ‘DEN FARKLI OLARAK KC, BÖBREK FONKSĠYONLARI VE DĠĞER AMĠNOASĠTLERĠN SERUM KONSANTRASYONLARI NORMALDĠR!!

TEDAVĠ ◦ TĠROZĠN VE FENĠLALANĠNDEN KISITLI DĠYET VERĠLĠR.

4-HPPD (4-0H fenilpirüvat dioksijenaz) aktivitesi düĢüktür.
Nörolojik anomaliler vardır. Karaciğer ve böbrek anormalliği yoktur.
DüĢük tirozin ve fenilalaninli diyetle birlikte vitamin C verilir.

Prematürelerde ve yüksek proteinli diyetle beslenenlerde görülür.
Çoğu asemptomatiktir.
GUTRĠE TARAMA TESTĠNDE TANI ALIRLAR.

Kendiliğinden düzelir.
Etiyolojide 4-HPPD enzim maturasyonunun gecikmesi rol oynar.
Diyette protein kısıtlanır ve vitamin C verilir.

4-HPPD enzim kompleksinden biri eksiktir.
OD geçer.
Sadece süt çocukluğunda semptomatiktir.

KLĠNĠK: ◦ Ciddi metabolik asidoz, ketozis
◦ BGG
◦ Hepatomegali
◦ YÜZMA HAVUZU KOKUSU
◦ Mental geliĢim normaldir.

TANI: ◦ Ġdrarda 4-OH-fenilpirüvik asit, 4-OH fenil laktik
asit, 4-OH siklohekzil laktik asit ve HAWKĠNSĠN artar.

TEDAVĠ: ◦ Proteinden, tirozinden ve fenilalaninden kısıtlı diyet
◦ Yüksek doz vitamin C
◦ Bir yaĢından sonra tedavi gerekmez.

HOMOGENTĠSĠK ASĠT OKSĠDAZ EKSĠKTĠR.(!!)
VÜCUTTA HOMOGENTĠSĠK ASĠT BĠRĠKĠR VE ĠDRARLA ATILIR. (!!)
OD geçer.

KLĠNĠK: ◦ ĠDRAR BEKLETĠLĠNCE SĠYAHLAġIR. (!!)
◦ OCHRONOSĠS (Özellikle kıkırdak ve mezenĢimal dokuların renginin koyulaĢması)
◦ ARTRĠT
◦ Kalp kapakçık hastalıkları olabilir.

TANI: ◦ ĠDRARDA HOMOGENTĠSĠK ASĠT ÖLÇÜLÜR. ◦ HOMOGENTĠSĠK ASĠT FEHLĠNG VE BENEDĠCT
AYIRAÇLARINI ĠNDĠRGER. ◦ ETKĠLĠ BĠR TEDAVĠSĠ YOKTUR. YÜKSEK DOZLARDA
VĠTAMĠN C ARTĠRĠTĠK DEĞĠġĠKLERĠN GELĠġMESĠNĠ GECĠKTĠREBĠLĠR.

MELANĠNĠN SENTEZ VE DAĞILIMI BOZUKTUR.
MELANĠN TĠROZĠNDEN SENTEZLENĠR


KLĠNĠK: ◦ Cilt, iris ve retinada depigmentasyon
◦ Nistagmus, strabismus, fotofobi, görme keskinliğinde azalma
◦ Geç dönemde körlük ve cilt kanseri (squamöz tip)

OR geçer.
Ġki genetik formu vardır: OKA1 ve OKA2 ◦ OKA1 tirozinaz (-) ve OKA2 tirozinaz (+) (!!)
◦ Pigmentasyon kaybı OKA1’de OKA2’den daha fazladır.

OKA2(Tirozinaz (+) albinizm): ◦ EN SIK RASTLANAN FORMUDUR.
◦ Tirozinin melanozom membranından geçiĢi bozuktur (p proteini).
◦ 15. kromozomda del. Vardır.
◦ Bu kromozomdaki p geni p proteinini kodlar.

PRADER WĠLLĠ VE ANGELMAN SENDROMUNDA DA 15. KROMOZOMDA DELESYON VARDIR VE BU TĠP ALBĠNĠZM GÖRÜLÜR!!

HERMANSKY-PUDLAK SENDROMU ◦ Tirozinaz (+), generalize albinizm görülür.
◦ Platelletlerde disfonksiyon vardır.
◦ Kanama eğilimi ve kanama zamanında uzama görülür.
◦ Dokularda ceroid like materyal birikir.

CHEDĠAK HĠGASHĠ SENDROMU ◦ Tirozinaz (+) parsiyel albinizm vardır
◦ OR geçer.
◦ Lökositlerde anormal granülasyon vardır.
◦ Enfeksiyonlara eğilim artmıĢtır.
◦ Lenfofolliküler maligniteler sıktır.
◦ Melanosom sayısı azalmıĢ büyüklükleri artmıĢtır.

CHEDĠAK HĠGASHĠ SENDROMU: ◦ Tedavide YÜKSEK DOZ VĠTAMĠN C verilir.
◦ KĠT de diğer bir alternatif tedavi seçeneğidir.

OKA1Tirozinaz (-) albinizm: ◦ Ġkinci sıklıkta görülür.
◦ Tirozinaz enzimi eksiktir.
◦ OR geçer.

OKÜLER ALBĠNĠZM: ◦ Albinizm gözle sınırlıdır.
◦ Saçta Tirozinaz (+) tir.
◦ OD,OR ve X’e bağlı formları vardır.
◦ OA1, OA2 ve 0A3 olmak üzere 3 tipi vardır.
◦ OA1 (Nettleship Falls tipi)

PARSĠYEL ALBĠNĠZM: ◦ Piebaldizm de denir.
◦ Cilt ve saçlarda lokalize depigmentasyon vardır.
◦ OD geçer.
◦ Beyaz perçem, cilt yamaları tek bulgu olabilir.
◦ Hasarlı bölgeler melanositten yoksundur.

WAARDENBURG SENDROMU: ◦ OD geçer.
◦ Sensorinöral sağırlık
◦ Beyaz perçem
◦ Anormal yüz görünümü


Metionin metabolizma bozuklukları: ◦ Homosistinüri
◦ Sistationinemi
◦ Hipermetioninemi


3 tipi vardır: ◦ TĠP 1: Sistationin sentetaz eksikliğine bağlı (klasik)
EN SIK!!
◦ TĠP 2: Metilkobalamin oluĢumundaki defekte bağlı
◦ TĠP 3: Metilentetrahidrofolat redüktaz eksikliğine bağlı

KLĠNĠK: ◦ Büyüme geliĢme geriliği
◦ LENS SUBLUKSASYONU (TANI!!)
◦ Ġleri dönemde astigmatizma, glokom, katarakt, retina dekolmanı, optik atrofi
◦ PROGRESĠF MENTAL RETARDASYON
◦ Konvülsiyon

KLĠNĠK 2: ◦ iskelet bozuklukları (skolyoz, pectus ex, pectus
carinatum,genu valgum vs)
◦ Zayıf ve uzun boylu, uzun ekstremitelilerdir.
◦ ARAKNODAKTĠLĠLERĠ VARDIR.
◦ MARFAN SEND’U ĠLE KARIġABĠLĠR!!

KLĠNĠK 3: ◦ Sarı saçlı ve mavi gözlüdürler. FKU GĠBĠ!!
◦ Generalize osteoporoz olabilir.
◦ Tromboemboliler sonucu; optik atrofi, paralizi, nöbetler, kor pulmonale, ciddi HT)

HOMOSĠSTĠNĠN VASKÜLER ENDOTELDE OLUġTURDUĞU HARABĠYET TROMBOEMBOLĠYE NEDEN OLUR!!
ARTMIġ HOMOSĠSTĠN DÜZEYLERĠ TROM ADEZĠVĠTESĠNĠ ARTTIRIR!!
ÖZELLĠKLE SEREBRAL TROMBOEMBOLĠYE BAĞLI PARALĠZĠLER GELĠġEBĠLĠR!!

TANI: ◦ Vücut sıvılarında metionin ve homosistein artmıĢtır.
(TANISAL)
◦ Plazma sistin seviyesi düĢüktür.
◦ Enzim eksikliği gösterilebilir.
◦ Prenatal tanısı mümkündür.

TEDAVĠ: ◦ YÜKSEK DOZ B6
◦ Folik asit
◦ Metionin kısıtlanır, sistein eklenir.
◦ Metil grup donörü olarak betain (trimetil glisin) kullanılarak homosisteinin metionine remetilasyonu sağlanır.

Metilkobalamin metionin sentazın kofaktörüdür.
Metilkobalamin oluĢumunda defekt vardır.
Metilmalonik asidüri görülebilir.

TANI: ◦ Megaloblastik anemi
◦ Homosistinüri
◦ Hipometioninemi
◦ Metilkobalamin düzeyi

TEDAVĠ: ◦ Yüksek doz vitamin B12

Metilentetrahidrofolat redüktaz eksiktir.
5-metil THF homosisteinin metionine remetilasyonu için gereklidir.

TANI: ◦ Orta derecede homosistinemi ve homosistinüri
◦ Metionin azalmıĢ veya N
◦ Megaloblastik anemi yok
◦ Tromboembolik olaylar görülebilir.
◦ Enzim tayini

TEDAVĠ: ◦ Folik asit
◦ Metionin
◦ B6
◦ B12
◦ Betain

Sistationin SĠSTATĠONĠNAZ ile sistein ve homoserine yıkılır.
Bu enzimin kofaktörü B6’dır.

Fetus ve yenidoğanda sistationinaz enzimi bulunmadığından SĠSTEĠN YENĠDOĞANLARDA ÖZELLĠKLE PREMATÜRELERDE ESANSĠYELDĠR!
TEDAVĠDE VĠTAMĠN B6 KULLANILIR.

SĠSTATĠONÜRĠ: ◦ B6 eksikliği
◦ B12 eksikliği
◦ Karaciğer hastalıkları özellikle galaktozemi
◦ Tirotoksikoz
◦ Ganglioblastoma, nöroblastoma, hepatoblastom
◦ Homosistinüri tip 2 ve 3
◦ Sistatioinaz eks

HĠPERMETĠONĠNEMĠ: ◦ Homosistinemi tip 1
◦ Tirozinemi tip 1
◦ Karaciğer hastalığı
◦ Prematüreler
◦ Metionin adenozil transferaz olgunlaĢmasında gecikme olan bazı yenidoğanlar
◦ Primer hipermetioninemi

ĠDRARDA ÇÜRÜK LAHANA KOKUSU OLABĠLĠR.
NÖROLOJĠK ANORMALLĠK TANIMLANAN VAKALAR BĠLDĠRĠLMĠġTĠR.
TEDAVĠDE PROTEĠN KISITLANIR.


Sistein METĠONĠNDEN sentezlenir, sistine oksitlenir.
Sistein metabolizma bozuklukları: ◦ Sistinüri
◦ Sistinozis
◦ Sülfit oksidaz eksikliği

DĠBAZĠK AMĠNOASĠTLERĠN (SĠSTĠN, ORNĠTĠN,
ARGĠNĠN, LĠZĠN) TRANSPORTUNDAKĠ DOĞUMSAL DEFEKT ĠDRARLA ATILMALARINA NEDEN OLUR!!
Radyoopak RENAL TAġLAR oluĢur. (TEK KOMPLĠKASYON!!)

OR Dalak, karaciğer, lenf düğümleri, kemik iliği,
böbrekler, kornea, konjunktiva, tiroid, pn, barsak ve beyin lizozomlarında SĠSTĠN birikir.
MEMBRAN TRANSPORT SĠSTEMĠNDEKĠ BĠR DEFEKT SĠSTĠNĠ LĠZOZOM ĠÇĠNDE TUTAR.!!

HEREDĠTER FANKONĠ SENDROMUNUN EN SIK NEDENĠ SĠSTĠNOZĠSTĠR!!
SĠSTĠNOZĠSLĠ FANKONĠ SENDROMUNA LĠGNAC SENDROMU DENĠR.

KLĠNĠK:
INFANTIL FORM (NEFROPATĠK): ◦ Fankoni sendromu , 1. Dekatta KBY geliĢir.
◦ Ağır geliĢme geriliği
◦ Hipotiroidi
◦ Sarı saçlar (melaninde defekt), fotofobi
ADÖLESAN ve ERĠġKĠN FORM

TANI: ◦ GÖZDE, KEMĠK ĠLĠĞĠNDE VEYA LENF DÜĞÜMLERĠNDE
SĠSTĠN KRĠSTALLERĠ ◦ Lökosit sistin içeriğinin ölçülmesi
TEDAVĠ: ◦ SĠSTEAMĠN (Sülfidril bağlayıcısı, hücre içi sistin
miktarını azaltır) ◦ KBY’de
Hemodiyaliz
Renal transplantasyon

Sülfit, SÜLFĠT OKSĠDAZLA sülfata oksitlenir.
ENZĠMĠN KOFAKTÖRÜ MOLĠBDENDĠR.
MOLĠBDEN AYRICA KSANTĠN DEHĠDROGENAZIN VE ALDEHĠT OKSĠDAZIN DA KOFAKTÖRÜDÜR!!

KLĠNĠK: ◦ Beslenmeyi reddetme, kusma
◦ MR
◦ Dirençli konvülsiyon (tonik, klonik, myoklonik)
◦ BĠLATERAL LENS DĠSLOKASYONU

TANI: ◦ Ġdrarlarında aĢırı miktarda sülfit, tiosülfat, s-
sülfosistein, ksantin ve hipoksantin bulunur. ◦ Kan ve idrarda sülfat ve ürik asit düzeyleri
düĢüktür. ◦ Sülfit oksidaz ve molibden tayini ◦ Tedavisi yoktur.


TRĠPTOFAN NĠKOTĠNĠK ASĠDĠN VE SEROTONĠNĠN PREKÜRSÖRÜDÜR!!

TRP metabolizma bozuklukları: ◦ Hartnup hastalığı
◦ Serotonin eksikliği
◦ Ġndikanüri
◦ Triptofanemi


OR
NÖTRAL AMĠNOASĠTLERĠN ĠNTESTĠNAL MUKOZA VE RENAL TUBULUSLARDAN TRANSPORTUNDA DEFEKT VARDIR.

KLĠNĠK: ◦ Asemptomatik
◦ Fotosensitivite
◦ Pellegra benzeri raĢ
◦ Ġntermittan ataksi, MR
◦ Epizodik psikolojik değiĢiklikler

TANI: ◦ AMĠNOASĠDÜRĠ (SADECE NÖTRAL
OLANLAR)****FANKONĠDE GENERALĠZE AMĠNOASĠDÜRĠ OLUR!!
◦ Nötral aminoasitlerin plazma konsantrasyonu normaldir.
◦ Ġndol derivelerinin özellikle indikanın atılımı artmıĢtır.

TEDAVĠ: ◦ Nikotinik asit
◦ Nikotinamid
◦ YÜKSEK !! konsantrasyonda protein içeren diyet

Ġlk basamak triptofanın triptofan hidroksilazla hidroksilasyonudur.
Enzimin kofaktörü BH4’tür.

Ġntestinal sistemde emilemeyen trp bakterilerce ĠNDOLE parçalanır ve ĠNDĠKAN olarak atılır.
Ġdrar havayla temas edince indikan ĠNDĠGO mavisine dönüĢür.

Ġndikanüri: ◦ Kör lup sendromu STAZ
◦ Konstipasyon STAZ
◦ Hartnup hastalığı
◦ FKU
◦ Bue diaper sendromu

Blue diaper sendromu: ◦ Ailesel
◦ Hiperkalsemi
◦ Nefrokalsinozis
◦ Ġndikanüri (Trp met bozukluğu)


BU AMĠNOASĠTLERĠN (DALLI ZĠNCĠRLĠ AMĠNOASĠTLER) YIKILIMI SIRASINDA ARA ÜRÜN OLARAK ORGANĠK ASĠTLER OLUġUR. BU DA ASĠDOZA NEDEN OLUR!!



DALLI ZĠNCĠRLĠ -KETOASĠT DEHĠDROGENAZ EKSĠKTĠR!!
ENZĠMĠN KOFAKTÖRÜ TĠAMĠN PĠROFOSFATTIR!!
HASTALARDAKĠ AKÇAAĞACI ġURUBU (ÇEMEN) KOKUSU TĠPĠKTĠR!!

KLĠNĠK: ◦ Beslenme bozukluğu, kusma, zayıf emme
◦ Letarji, koma
◦ Hipertonisite, rijidite, opistotonus, MMR
◦ Konvülsiyon, hipoglisemi
◦ SEPSĠS, MENENJĠT VE NEONATAL TETANOZ ĠLE KARIġIR.!!

TANI: ◦ Ciddi asidoz
◦ ÇEMEN KOKUSU
◦ KANDA VALĠN,LÖSĠN. ĠZOLÖSĠN ARTAR. ALANĠN AZALIR.
◦ ĠDRARDA VALĠN, LÖSĠN, ĠZOLÖSĠN VE KETOASĠTLER ARTAR.

ĠDRARDAKĠ KETOASĠTLER 2,4 DĠNĠTROFENĠLHĠDRAZĠN TESTĠ ĠLE GÖSTERĠLEBĠLĠR!!
Enzim analizi yapılabilir.
MSUD’li anne bebeklerinde bu güne değin bildirilmiĢ bir anomali yoktur!!

TEDAVĠ: ◦ PERĠTON DĠYALĠZĠ
◦ Yeterli kalori desteği
◦ Dallı zincirli a.a’lerden fakir diyet ömür boyu uygulanır.
◦ TĠAMĠN !!

Plazma izolösin konsantrasyonu çok düĢerse akrodermatitis enteropatikaya benzer bir klinik tablo oluĢabilir.

Lösin metabolizma bozukluğudur.
ĠZOVALERĠL CoA DEHĠDROGENAZ eksiktir.

KLĠNĠK: ◦ Kusma (pilor stenozu gibi)
◦ Ciddi asidoz !!
◦ Letarji, konvülsiyon, koma
◦ TERLĠ AYAK KOKUSU

TANI: ◦ Ġdrarda ve kanda izovalerik asit ve metabolitleri
artar.
◦ Ketoasidoz
◦ Pansitopeni
◦ Hiperamonemi (Üre siklus bozukluğu ile karıĢabilir)
◦ Hiperglisemi
◦ Hipokalsemi

TANI: ◦ Enzim tayini
◦ A. Pankreatit tablosu sıktır.

TEDAVĠ: ◦ Hidrasyon düzeltilir.
◦ NaHCO3 verilir.
◦ GLĠSĠN VE KARNĠTĠN VERĠLĠR. (Ġzovalerik asitin idrarla atılımını arttırmak için)
◦ Yeterli kalori sağlanır.(Katabolizmayı durdurmak için)
◦ Kan değiĢimi veya periton diyalizi
◦ Proteinden kısıtlı diyet

BĠYOTĠN VÜCUTTAKĠ TÜM KARBOKSĠLAZLARIN KOFAKTÖRÜDÜR!!

BĠOTĠNĠDAZ, biyotinin barsaklardan emiliminde ve karboksilazlara bağlı biyotinin ayrılarak yeniden kullanılmasında görev alır.
HOLOKARBOKSĠLAZ, biyotinin aktive edeceği karboksilaza bağlanmasını sağlar.

Biotinidaz ve holokarboksilazdaki eksiklikler bütün karboksilazların disfonksiyonuna ve organik asidemilere neden olur.

KLĠNĠK: ◦ Solunum problemi (takipne, apne)
◦ Kusma
◦ Hipotoni !!
◦ Konvülsiyon
◦ GeliĢme geriliği
◦ Ġmmün yetmezlik

KLĠNĠK: ◦ ERKEK KEDĠ ĠDRARI KOKUSU
◦ Cilt bulguları (raĢ, eksfolyasyon, alopesi totalis)
◦ CĠLT BULGULARI BU HASTALIĞI DĠĞER ORGANĠK ASĠDEMĠLERDEN ÖZELLĠKLE PROPĠYONĠK ASĠDEMĠDEN AYIRIR!!

TANI: ◦ Metabolik asidoz, ketozis
◦ Ġdrarda organik asit artıĢı
◦ Hiperamonemi
◦ Enzim tayini

TEDAVĠ: ◦ BĠOTĠN 10 mg/gün

Tanı enzim analizi ile konur.
Ġdrar organik asit profili ve tedavi infantil formla aynıdır.

***Atopik / seboreik dermatit
***Alopesi
Ataksi
Myoklonik nöbet
Hipotoni
GeliĢme geriliği
ĠĢitme kaybı
Ġmmün yetmezlik
Metabolik asidoz

Biotin içermeyen TPN
Uzun süre antikonvülzan kullanımı
DüĢük proteinli mama
Kısa barsak sendromu veya kronik ishal
ÇĠĞ YUMURTA ĠLE BESLENME hallerinde görülür.

Asetoasetil CoA tiolaz eksikliği
Asemptomatik ......>>>> Ciddi asidoz
Asidoz, ketoz, hiperamonemi, koma ve ölüm görülebilir.
Asidoz atakları araya giren enfeksiyon ile provake olur.

Ataklar arasında tamamen normaldir.
Mental geliĢim genelde normaldir.
ATAKLAR ASPRĠN ENTOKSĠKASYONU ĠLE KARIġABĠLĠR!!
Salisilat için kullanılan asetoasetat testi (+)’tir.

TANI: Enzim analizi ile koyulur.

TEDAVĠ: ◦ Hidrasyon, NaHCO3, %10 glukoz
◦ Uygun sıvı ve elektrolit
◦ Hiperamonemi tedavisi
◦ Periton diyalizi
◦ Protein kısıtlı diyet
◦ Sekonder karnitin eksikliği geliĢtiği için karnitin verilir.

Propiyonik asiti metil malonik aside çeviren PROPĠYONĠL CoA KARBOKSĠLAZ eksiktir.
Enzimin kofaktörü BĠYOTĠN’dir.

KLĠNĠK: ◦ Beslenme bozukluğu, kusma, hipotoni, letarji,
dehidratasyon, konvülsiyon
◦ Ketoasidoz atakları !! Enfeksiyonlarla tetiklenir!!

TANI: ◦ Metabolik asidoz, ketozis!!
◦ Pansitopeni!!
◦ Hipoglisemi
◦ Hiperamonemi!!
◦ Hiperglisinemi

TANI: ◦ Plazmada ve idrarda propionik asit ve metilsitrik
asit artar.
◦ Enzim analizi

PA, multipl karboksilaz eksikliklerinden ve üre siklusu defektlerinden ayrılmalıdır!!

TEDAVĠ: ◦ Rehidratasyon, NaHCO3, yeterli kalori
◦ DüĢük proteinli diyet
◦ Neomisin, metronidazol p.o.
◦ Karnitin
◦ Hiperamonemi tedavisi

TEDAVĠ: ◦ Periton diyalizi, hemodiyaliz
◦ Kesin tanı konulana kadar biyotin baĢlanabiir.
◦ Uzun dönemde; düĢük proteinli diyet, karnitin, kronik alkali ve kronik hiperamonemi tedavisi

Ketotik hiperglisinemiler: ◦ PA
◦ MMA
◦ Biriken organik asitler glisini yıkan enzimi inhibe eder.

MMA, metilmalonil CoA mutaz / rasemaz ile süksinik aside dönüĢür.
MMA CoA mutazın koenzimi adenozil kobalamindir.
MMA CoA MUTAZIN VEYA KOENZĠMĠNĠN EKSĠKLĠĞĠNDE VÜCUTTA MMA VE PREKÜRSÖRLERĠ BĠRĠKĠR!!

RASEMAZIN DA MUTAZIN DA EKSĠKLĠĞĠ MMA’ ‘YA NEDEN OLUR. ANCAK RASEMAZ EKSĠKLĠĞĠ GÖSTERĠLMEMĠġTĠR!!

KLĠNĠK: ◦ Enfeksiyonlarla tetiklenen ketoasidoz tablosu, koma
◦ Ġleri dönemde hipotoni, BGG
◦ Bazı MMA’lılarda tipik yüz görünümü vardır.

TANI: ◦ Ketozis,asidoz
◦ Pansitopeni
◦ Hiperglisinemi
◦ Hiperamonemi
◦ Hipoglisemi
◦ Vücut sıvılarında MMA artar.

TEDAVĠ: ◦ Akut atakta yüksek doz vitamin B12
◦ Kronik tedavide
Protein kısıtlanır.
L-karnitin
Vitamin B12
Kronik alkali tedavisi

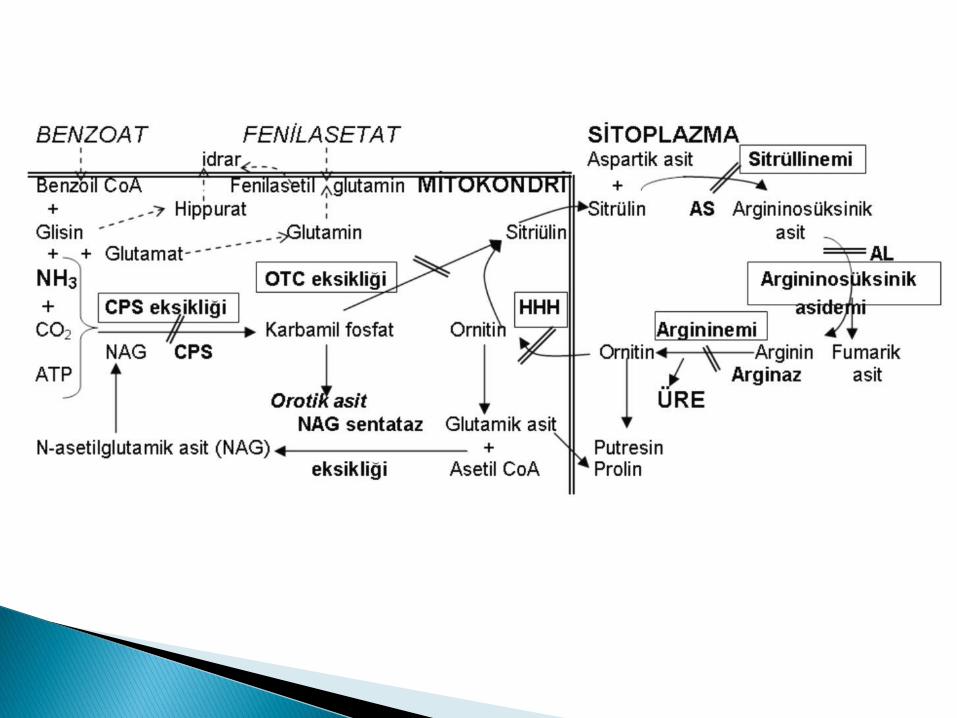

Üre sentezinde görev alan enzimler: ◦ Karbamil fosfat sentetaz (CPS): OR
◦ Ornitin transkarbamilaz (OTC): XD (En sık)
◦ Arjinino süksinat sentetaz (AS): 0R
◦ Arjininosüksinat liyaz (AL): OR
◦ Arjininaz: OR
◦ N-asetilglutamat (NAG) sentetaz

AS..............>Sitrülinemi
AL..............>Arjininosüksinik asidemi
Arjinaz.......>Arjininemi
Hiperamonemili hastada kanda spesifik aa’ler artmıĢsa sitrülinemi,arjininemi ve arjininosüksinik asidemi düĢünülür.


Üre siklusu defektleri (EN SIK!!) Organik asidemiler Lizinürik protein intoleransı YD’nın geçici hiperamonemisi Hperornitin-hiperamonemi-homositrülinemi
(HHH) sendromu Konj hiperinsülinizm ile birlikte
hiperamonemi

KLĠNĠK: ◦ Bebekler doğumda normaldir.
◦ PROTEĠN beslenmeyi takiben ortaya çıkan emmeme, kusma, takipne , letarji ve koma
◦ Konvülsiyon
◦ Hepatomagali
◦ SEPSĠSLE KARIġIR!!

LAB: ◦ Hiperamonemi (>200µM)
◦ BUN ve üre genelde düĢüktür.
◦ ASĠDOZ YOKTUR!!

CPS ve OTC eksikliğinde kanda glutamat, aspartat ve alanin artar.
OTC eksikliğinde idrarda orotik asit artar.

Oral karbamil glutamat verilmesinden sonra kliniğin düzelmesi NAG sentetaz eksikliğini destekler!!
Karbamilfosfat sentaz eksikliğini NAG sentetaz eksikliğinden ayırt etmek için enzim analizi gerekir.

TEDAVĠ: ◦ Yeterli sıvı, kalori ve elektrolit sağlanır. ◦ Ġ.v. Lipid 1g / kg /gün ◦ Ġ.v. Protein 0,25 g / kg /gün. Sonra N/G ile 0,5-1,0
g / kg / gün protein ◦ Renal atılımı arttıranlar:
Sodyum benzoat
Fenilasetat
Fenilbütirat

TEDAVĠ ◦ Arjinin (arjininaz eksikliği olanlar hariç)
◦ OTC eksikliği olanlarda sitrülin
◦ Hemodiyaliz (EN ETKĠLĠ)
◦ Periton diyalizi (EN PRATĠK VE EN HIZLI)



GLĠSĠN SERĠN VE TREONĠNDEN SENTEZLENEN NONESANSĠYEL BĠR AMĠNOASĠTTĠR.

Hiperglisinemi: ◦ PA ( Organik asidemiler glisin ayırma enzim
sistemini inhibe ederler)
◦ MMA
◦ Nonketotik hiperglisinemi (Glisin ayırma enzim sisteminde genetik defekt vardır!!)

NEONATAL FORM: ◦ EN SIK!!! ◦ KLĠNĠK:
Emmeme
Beslenme güçlüğü
Ağıt hipotoni, koma, apne, ölüm
MYOKLONĠK NÖBETLER !!
HIÇKIRIK!!
MMR, epilepsi

TANI: ◦ Hiperglisinemi
◦ Hiperglisinüri
◦ SPĠNAL SIVIDA PLAZMADAN 15-30 KAT DAHA FAZLA GLĠSĠN BULUNMASI TANISALDIR!!
◦ Enzim analizi

TEDAVĠ: ◦ Kan değiĢimi
◦ Glisin alımının kısıtlanması
◦ Sodyum benzoat veya folat ilavesi
◦ Striknin, diazepam ve dextrametorfan

OKSALĠK ASĠT BÜYÜK ÇOĞUNLUKLA GLĠOKSĠLĠK ASĠTĠN AZ ORANDA ĠSE ASKORBĠK ASĠDĠN OKSĠDASYONUNDAN TÜRER.!!!!

Sekonder hiperokzalüri: ◦ *****Pridoksin eksikliği
◦ *****Vitamin C intoksikasyonu
◦ ĠBH
◦ Etilen glikol alımı
◦ Metoksifluran anestezisi
◦ AĢırı barsak rezeksiyonu

PRĠDOKSĠN ALANĠN-GLĠOKSALAT AMĠNOTRANSFERAZIN KOFAKTÖRÜDÜR!!
Kuzukulağı gibi bitkilerin alınmasından sonra fatal hiperoksalüri geliĢebilir!!!!

Alanin:glioksalat aminotransferaz eksiktir.
Enzimin kofaktörü pridoksindir.
5 yaĢından önce semptomatiktir.

KLĠNĠK: ◦ Böbrek taĢı
◦ Nefrokalsinozis
◦ Renal kolik
◦ Asemptomatik hematüri
◦ BFT bozukluğu, geliĢme geriliği, renal yetmezlik
◦ Akut artrit

ÇOĞU HASTA <20 YAġTA RENAL YETMEZLĠK
NEDENĠYLE ÖLÜR!! ÜRĠK ASĠT YÜKSEK OLDUĞUNDAN GUT ĠLE
KARIġABĠLĠR!!!!

TANI: ◦ OKZALÜRĠ!!
◦ ĠDRARDA GLĠKOLĠK VE GLĠOKSĠLĠK ASĠT ARTIMI ARTMIġTIR!!
◦ ENZĠM TAYĠNĠ

TEDAVĠ ◦ Yüksek doz pridoksin
◦ RENAL TP ETKĠSĠZDĠR.!!!!
◦ KOMBĠNE KARACĠĞER VE BÖBREK TP’U EN ETKĠLĠ YÖNTEMDĠR!!

L-gliserik asidüri
D-gliserat dehidrogenaz/glioksilat redüktaz eksiktir
Klinik tip1 ile aynıdır.
Renal taĢlar 2 yaĢından önce geliĢebilir.
RENAL YETMEZLĠK GÖRÜLMEZ!!

TANI: ◦ Ġdrarda oksalat ve L-gliserik asit artmıĢtır.
◦ GLĠKOLĠK VE GLĠOKSĠLĠK ASĠT YOKKEN ĠDRARDA L-GLĠSERĠK ASĠT BULUNMASI TĠP 1’DEN AYIRT EDĠLMESĠNĠ SAĞLAR!!
◦ Etkili bir tedavisi yoktur.


ASPARTOAÇĠLAZ ENZĠMĠ EKSĠKTĠR.
Asetil aspartik asit beyaz cevherde birikir.
Bir tür lökodistrofidir.
Bilinen bir tedavisi yoktur.


Glutamik asit metabolizma bozuklukları: ◦ Glutatyon sentetaz eksikliği
◦ GABA metabolizma bozukluğu
Pridoksin eksikliği
GAB Asidemi


Lizin metabolizma bozuklukları: ◦ Hiperlizinemi
◦ Glutarik asidüri tip 1
◦ Lizinürik protein intoleransı


Ailesel iminoglisinüri PROLĠN metabolizma bozukluğudur.


Normalde kan laktik asit düzeyi<18 mg/dl
Enzimatik defekte bağlı olmayan hiperlaktikasidemi hipoksemide görülür. Bu durumda pirüvik asit düzeyi normaldir. Enzimatik defektte ise pirüvat düzeyi de yüksektir.

Hiperlaktik asidemi pirüvatın ya glukoneognez yolunda glukoza ya da TCA’da CO2 ve H20’ya çevrilmesindeki defekt sonucu meydana gelir.
Anyon açığı >16 Mm olan açıklanamayan metabolik asidozda laktat düzeyi ölçülmelidir.

Laktik asidoz olmayan defektler: ◦ Galaktozemi
◦ Benign fruktozüri
◦ Herediter fruktoz intoleransı
◦ Kas fosfogliserat mutaz eksikliği
◦ Kas tipi laktat dehidrogenaz eksikliği

Laktik asidoz olan defektler: ◦ Glukoz 6-fosfataz eksikliği
◦ Fruktoz 1-6 difosfataz eksikliği
◦ Pirüvat dehidrogenaz eksikliği
◦ Pirüvat karboksilaz eksiklği
◦ Karnitin eksikliği
◦ Konjenital idiyopatik laktik asidoz
◦ Pirüvat dehidrogenaz fosfataz eksikliği
◦ LEĠGH subakut nekrotizan ensefalopatisi




3 enzim defekti tanımlanmıĢtır: ◦ Galaktokinaz eksikliği
◦ Galaktoz 1-fosfat üridil transferaz eksikliği
◦ Epimeraz eksikliği

OR geçer.
Galaktozemi, galaktozüri ve katarakt ile karakterizedir.
MR ve aminoasidüri yoktur.
ĠRM (+)’tir.
Galaktozsuz diyetle katarakt önlenebilir.
Prognoz iyidir.

GALAKTOZ 1-FOSFAT ÜRĠDĠL TRANSFERAZ EKSĠKTĠR!!
Galaktoz 1 fosfat karaciğer, böbrek ve beyinde birikir

Sarılık
Hepatomegali
Kusma
Hipoglisemi
Konvülsiyon
Letarji
Ġrritabilite
Beslenme güçlüğü
Kilo alamama
Aminoasidüri
Katarakt
Siroz
Asit
Splenomegali
Mental retardasyon
Amenore

KATARAKT GALAKTĠLOL BĠRĠKĠMĠNE;
DĠĞERLERĠ GALAKTOZ 1 FOSFAT BĠRĠKĠMĠNE BAĞLIDIR!!!!

TEDAVĠDE GALAKTOZ VE GALAKTOZ ĠÇEREN BESĠNLER (ANNE SÜTÜ GĠBĠ) KISITLANIR !!



FRUKTOKĠNAZ EKSĠKTĠR!!
Asemptomatiktirler.
ĠRM (+)’tir.

ALDOLAZ B (fosfofruktoaldolaz) eksiktir.
Fruktoz veya sükroz içeren gıdaların (meyveler) alımıyla ortaya çıkar.

KLĠNĠK: ◦ Sarılık ◦ Hepatomegali ◦ Kusma ◦ Letarji ◦ Ġrritabilite ◦ Konvülsiyon ◦ Hipoglisemi

TANI: ◦ ĠRM(+)
◦ ġeker kromotografisinde fruktoz saptanır.
Tedavi diyetten fruktozun tamamen eliminasyonunu gerektirir.


EGZERSĠZ SONRASI MĠYOGLOBĠNÜRĠ VE KAS KRAMPLARI ORTAYA ÇIKAR.
ĠSKEMĠK EGZERSĠZĠ TAKĠBEN KAN LAKTĠK ASĠT KONSANTRASYONU ARTTIRILAMAZ.

GLĠKOJENOZĠSLER ĠÇĠNDE AĞIR LAKTĠK ASĠDOZLA SEYREDEN TEK GLĠKOJEN DEPO HASTALIĞIDIR!!

Fruktoz 1,6 difosfataz glikoneogenezin anahtar enzimlerinden biridir.
Fruktoz veya sükroz alımını takiben hipoglisemi, Ģok, koma, konvülsiyon ve hiperlaktik asidemiye bağlı metabolik asidoz geliĢtirirler.
Asemptomatik dönemde HM hariç FM normaldir.


Tip 1,3,4,6,9,glikojen sentetaz eksikliği ve GLUT-2 defektinde AÇLIK HĠPOGLĠSEMĠSĠ VE HEPATOMEGALĠ olur.
TĠP 3 ve 4’te siroz görülür.
TĠP 1’de renal disfonksiyon olur.
TĠP 3 ve 4’e miyopati eĢlik

Von Gierke hastalığı, hepatorenal hastalık GLĠKOZ-6 FOSFATAZ EKSĠKTĠR. Karaciğer , böbrek ve barsaklarda glikojen
birikir. Kasta glukoz 6 fosfataz bulunmadığından
kaslar etkilenmez.

KLĠNĠK : ◦ Hipoglisemi
◦ Laktik asidoz
◦ Hepatomegali
◦ Hipoglisemik konvülsiyon
◦ TaĢ bebek yüzü- Doll face

KLĠNĠK : ◦ Hiperlipidemi
◦ Hiperürikasidemi
◦ Ġntermittan diyare
◦ Kanama
◦ KCFT yüksekliği
◦ Nefromegali

Hepatik adenom geliĢebilir, nadiren malign
Pulmoner HT ve osteoporoz olabilir.

Renal hastalık geç komplikasyondur.
FANKONĠ SENDROMU GÖRÜLEBĠLĠR!!
Glukoz ve fruktoz verilmesi kan glukozunu yükseltmez. Glukagon verildiğinde de beklenen glukoz artıĢı olmaz, tip 3’te olması ayırt edicidir.

TEDAVĠ: ◦ HĠPOGLĠSEMĠ ÖNLENMEYE ÇALIġILIR!!
◦ Sürekli N/ G glukoz infüzyonu
◦ Oral piĢmemiĢ mısır niĢastası
◦ K.H içeriği yüksek sık beslenme

PSEUDO GDH1 (GDH 1b) ◦ Klinik tip 1a ile aynıdır.
◦ NÖTROPENĠ görülür.
◦ Glikoz 6 fosfatın mikrozomal membrandan transportunda (translokaz) defekt vardır.

Amilo-1,6 glukozidaz (debrancher enzim) eksiktir.
CORĠ, FORBES HASTALIĞI da denir.

KLĠNĠK: ◦ Hepatosplenomegali
◦ Hipoglisemi
◦ Hiperlipidemi
◦ Büyüme geriliği
◦ NEFROMEGALĠ YOKTUR!!!

HM ve hapatik semptomlar yaĢla hafifler ve puberteden sonra genellikle kaybolur.
Siroz görülebilir.
KMP ve myopati olabilir.
KCFT yüksek, açlık ketozu belirgin, ürik asit ve laktat normaldir.
Tedavide yüksek proteinli diyet uygulanır.

Amilo-1,4-1,6 transglukozidaz (brancher enzim) eksiktir.
ANDERSEN HASTALIĞI da denir.

KLĠNĠK: ◦ Hepatosplenomegali
◦ Siroz ve kc yetmezliği 5 yaĢından önce ölüme yol açar.
TEDAVĠ:
◦ Steroid
◦ Karaciğer transplantasyonu

Karaciğer fosforilaz enzimi eksiktir.
HERS HASTALIĞI da denir.
Hepatomegali ve BGG olur.
Hipoglisemi, hiperlipidemi ve hiperketozis olursa genellikle hafiftir.
Laktik asit ve ürik asit normaldir.
Puberteden sonra düzelir.

Karaciğer fosforilaz kinazı eksiktir.

Hipoglisemik konvülsiyonlar görülür.
Hiperketonemi vardır.
Hipoglisemi açlıkta görülür ve glukagona yanıt vermez.
Glukoz verildikten sonra kan Ģekeri uzun süre yüksek kalır.
Sık arayla proteinden zengin beslenme hipoglisemiyi ve MR geliĢimini önleyebilir.

FANCONĠ-BICKEL sendromu da denir.
Renal fanconi sendromu ile birlikte hepatik glikojenoz vardır.
GLUT-2’de defekt vardır.
OR geçer.


1. Grup: progresif kas güçsüzlüğü, atrofi veya KMP (TĠP 2)
2. Grup: kas ağrısı, egzersiz intoleransı, myoglobinüri (TĠP 5, 7)

Lizozomal asit -glukosidaz (asit maltaz) eksiktir.
GLĠKOJENOZLAR ĠÇĠNDE TEK LĠZOZOMAL HASTALIK GDH 2’DĠR!!
POMPE HASTALIĞI (kardiyak glikojenoz, generalize glikojenoz da denir)
Bebekleri 2a ve büyük çocukları ve eriĢkinleri etkileyen 2b olmak üzere iki tipi vardır.

Klasik formdur.
Ġlk 2 yılda ölümle sonuçlanır.
Ġleri derecede hipotoni, kardiyomegali ve orta derecede HM geliĢir.
Kısa PR görülebilir.
Mental geliĢim normaldir.

Emme zayıftır, solunum yüzeyeldir ve MAKROGLOSSĠYE bağlı hava açlığı vardır.
Aspirasyon pnömonisi sıktır.
Ölüm solunum kaslarının yetmezlği nedeniyle olur.
KAN GLUKOZ DÜZEYĠ NORMALDĠR!!

Ġskelet kaslarındaki zayıflık daha geç baĢlar.
YaĢam süresi normal olabilir.
Solunum yetmezliğine bağlı ölüm görülebilir.
Kardiyomegali ve EKG anomalileri yoktur.

Kas fosforilazı eksiktir.
MC ARDLLE SENDROMU da denir.
Rekürren miyoglobinüri, rabdomyoliz atakları ve devamlı kas ağrısı olabileceği gibi asemptomatik de olabilir.

Tanı iskemik egzersiz testi ile konulabilir.
Fosforilaz aktivitesi iskelet kasında yoktur, karaciğer ve düz kasta normaldir.
Tedavide yüksek proteinli diyet önerilir.

Fosfofruktokinaz enzimi eksiktir.
TARUI HASTALIĞI da denir.
Semptomlar tip 5’e benzer ama daha hafiftir.
Enzim iskelet kasında eksik, karaciğerde normaldir.

Hiperürisemi sıktır.
Kompanse hemolitik anemi görülür.

Progresif beyin hastalığı vardır.
HM görülür.
Yutma güçlüğü nedeniyle aspirasyon pnömonisi sıktır.
Enzim eksikliği gösterilememiĢtir, fosforilaz aktivasyon sisteminde bozukluk vardır.

CAMP bağımlı kinaz eksiktir.
Belirgin HM dıĢında hastalar normaldir.

Glikojen depo hastalıklarından GDH 9b X’e bağlı diğerleri OR geçer.
GDH 1’in prenatal tanısı yoktur.
Prenatal tanının en yararlı olduğu hastalık Pompe hastalığıdır.



MPS’ler yetersiz parçalanır ve depolanır.
Klinik bulgular bu depolanmaya bağlıdır.
Tamamında spesifik lizozomal parçalayıcı enzim defektleri saptanmıĢtır.

MPS patogenezinde yer alan majör GAG’lar; dermatan sülfat, heparan sülfat ve keratan sülfattır.
MPS’lerin özelliği kemik değiĢiklikleridir.
Ġskelet değiĢikliklerine genel olarak disostozis multiplex denir.
Progresif MR olur.

KVS, karaciğer, dalak, tendonlar, eklemler ve cilt tutulabilir.
X’e bağlı geçen Hunter sendromu hariç tümü OR geçer.
Klinik ve radyolojik bulgularla hastalıklardan Ģüphelenilir.
GAG’ların idrar atılımındaki artıĢ ve enzim analizi ile tanı doğrulanır.


EN AĞIR MPS FORMUDUR.
-L-idüronidaz eksiktir.
Dokularda dermatan ve heparan sülfat birikir, idrarla atılır.
Gargoyle hücreler görülür.

KLĠNĠK: ◦ HSM
◦ Persistan nazal akıntı, gürültülü solunum
◦ Kaba yüz görünümü
◦ BaĢ büyük ve dolikosefalik
◦ Korneal bulanıklık
◦ Umblikal ve inguinal heni
◦ BBG, MR
◦ Eklemlerde ilerleyici sertlik ve kontraktür

RÖNTGEN: ◦ DĠSOSTOZĠS MULTĠPLEX
Dolikosefalik kafa ve kalınlaĢmıĢ kemikler
J Ģeklinde sella tursika
Klavikulanın mediyal 1/3’ünde kalınlaĢma
Vertebra gövdelerinde gagalaĢma
Gibbus deformitesi
Kostalar spatula gibi

RÖNTGEN ◦ DĠSOSTOZĠS MULTĠPLEX
Elde terminal falankslarda incelme
Metakarp prok incelme, distalde geniĢleme
Uzun kemiklerin korteksinde incelme, medullada geniĢleme
Radiusun ulnaya deviasyonu sonucu V oluĢumu



MPS’lerin en hafif formudur.
Zeka normaldir.
-L-idüronidaz eksiktir.
Heparan ve dermatan sülfat birikir.

Ara formdur.
Zeka normaldir.

MPS’ler arasında tek X’e bağlı olandır.
Ġduronat sülfataz eksiktir.
Dermatan ve heparan sülfat birikir.
Klinik Hurler’den hafiftir.

TĠP A: ◦ KORNEAL BULANIKLIK YOKTUR.!!

TĠP B: ◦ TRAKEA VE BRONġTA GAG BĠRĠKĠMĠNE BAĞLI HAVA
YOLU OBSTRUKSĠYONU BU TĠPĠN ÖZELLĠĞĠDĠR!!!!

AĢağıdaki enzimlerden biri eksiktir: ◦ Heparan sülfamidaz
◦ N-asetilglukozaminidaz
◦ N-asetil transferaz
◦ N-asetilglukozamin-6-sülfataz

Heparan sülfat birikir ve idrarla atılır.
Hurler ve Hunter’dan daha hafiftir.
GeliĢim basamaklarında gecikme ve HĠPERAKTĠVĠTE vardır. Nörodejenerasyon sonucu yatalak hale gelirler.

Keratan sülfatüri ve iskelet displazisi ile karakterizedir. Keratan ve kondroidin sülfat birikir.
Tip A’da N- asetilgalaktozamin-6-sülfat sülfataz, tip B’de beta- galaktozidaz eksiktir. (GM1 gangliosidozda da aynı enzim eksiktir.
Ağır somatik bulgular vardır, MR yoktur. Eklem laksitesi, boy kısalığı, iskelet
anomalileri, korneal bulanıklık, HSM görülebilir.

Sürekli sırıtıyor gibi görünürler.
Solunum yetmezliğine bağlı kor pulmonaleden ölürler.

MR olmaz.
N-asetilglukozamin-4-sülfat sülfataz (aril sülfataz B eksiktir.
Ġdrarla dermatan sülfat atılır.
Diğer bulgular Hurler’a benzer.

Dermatan ve heparan sülfat birikir.
Klinik Hurler’e benzer.
Sly sendromu da denir.



Özellikle kh rezervinin kalmadığı açlık durumlarında yağ asidi oksidasyonu ile enerji sağlanır.
Yağ asitleri egzersiz yapan iskelet kasları ve kalp için de önemli yakıttır. Buralarda CO2 ve H20’ya kadar yıkılırlar.
Yağ asitlerinin karaciğerde oksidasyon son ürünleri ketonlardır.

KLĠNĠK: ◦ Koma ve hipoglisemi
◦ Progresif kas zayıflığı ve KMP
◦ HĠPOGLĠSEMĠYE KETOZĠS EġLĠK ETMEZ.
◦ UZUN SÜRE AÇ KALINMADIKÇA BULGU VERMEZ.
Ataklar yanlıĢlıkla Reye sendromu veya ani bebek ölümü sanılabilir.

Yağ asitleri sitoplazmada CoA ile aktive edildikten sonra karnitin ile mitokondri iç zarından geçer. Mitokondri matriksinde yağ asidi beta-oksidasyonu ile asetil CoA ‘ya parçalanırken FADH2 ve NADH oluĢur. Bunlar da ETS’ye girerek ATP yaparlar.


Uzun süreli açlıkta hipoketotik hipoglisemi, dikarboksilik asidüri ve diğer organik asidüriler ve sekonder karnitin eksikliği görülür.

En sık karĢılaĢılan yağ asidi oksidasyon defektidir.
Uzun süreli açlık olmadıkça sağlıklı görülürler.
Ġlk atak 6-24 ayda görülür.
Ataklar genelde yaĢamı tehdit eder.

KLĠNĠK: ◦ Kusma
◦ Letarji, koma
◦ Konvülsiyon
◦ Hepatomegali yoktur veya hafiftir.

LAB: ◦ Hipoketotik hipoglisemi
◦ Hafif asidoz
◦ Üre, amonyak, ürik asit artar.
◦ AST, ALT artar. PT ve PTT uzar.
◦ Ġdrarda orta zincirli dikarboksilik asit düzeyleri artar.
◦ Sekonder karnitin eksikliği

TEDAVĠ: ◦ Akut atakta %10 dx i.v
◦ Kronik tedavide hastalarının asla 10-12 saatten fazla aç kalmamaları ile sağlanır.
◦ Prognoz mükemmeldir, kas zayıflığı veya KMP görülmez.

Daha ağır seyreder.
Ġlk 1 yılda hipoglisemi ve koma görülür.
Kas zayıflığı ve hipertrofik KMP vardır.

Multiple açil CoA dehidrojenasyon defektleri, glutarik asidüri tip 2
Bu iki enzim yağ asidi oksidasyonunda ve dallı zincirli aa’lerin ve glutarik asidin oksidasyonunda görev alır.
ETF: Elektron transfer flavoprotein
ETF-DH: ETF dehidrogenaz

ETF veya ETF-DH’nin tam yokluğunda YD döneminde asidoz, hipoglisemi, koma ve hipotoni geliĢir.
Ġdrar organik asit profili tanıyı koydurur.

Parsiyel eksikliklerde tablo MCAD veya LCAD eksikliğine benzer.
Bu hastalarda da sekonder karnitin eksikliği geliĢir.
Tedavi uzun süre aç kalmamayı gerektirir.
Bazı hastalar B2’den fayda görür.

Altta yatan defekt plazma membran karnitin transport sistemindedir.
2-4 yaĢında baĢlayıp sürekli ilerleyen KMP ve iskelet kas zayıflığı vardır.
Hipoketotik hipoglisemi atakları görülebilir. Tedavide yüksek doz p.o karnitin verilir.

Açlıkta hipoketotik hipoglisemi olur.
Kas zayıflığı veya KMP yoktur.

Egzersiz sonrası kas ağrısı, rabdomiyoliz ve miyoglobinüri olur.
CPT eksikliklerinde karnitin düzeyleri ve idrar organik asit atılımı normaldir.




























