Embed Size (px)
Citation preview

Motor Neuron Disease / Amyotrophic Lateral Sclerosis (ALS)



Motor Neuron Disease / ALSAetiology :
o The cause of motor neuron disease is unknown. Several possibilities have been suggested:
o Gentic – Mutations in the SOD1 gene (responsible for producing the enzyme superoxide dismutase) are found in 20% of familial cases of ALS. Superoxide dismutase is important in removing toxic superoxide radicals and converting them into non-harmful substances. Defects in the enzyme lead to accumulation and anterior horn cell death

o Viruses – Chronic virus infection has been suggested. Polio virus will acutely damage the anterior horn cell and chronic polio infection could theoretically produce motor neuron disease. Some claim that motor neuron disease follows acute poliomyelitis; however, when this occurs the clinical picture is not typical and may resemble more closely Spinal Muscular Atrophy. Polio antibody titres remain normal. Virus-like particles have been reported in some patients with MND, but transmission to non-human primates has been unsuccessful, HIV-associated ALS like syndromes with similar pathological evidence of anterior horn cell loss have raised the possibility that ALS could be due to retroviral infection (retroviruses persist by integration into the host’s DNA and would be ideal suspects.

o Toxins – Cetain metals, lead, selenium, mercury and managanese have been incriminated, but again evidence is inconclusive
o Minerals – Clinical similarities between MND and neurological involvement in hyperparathyroidism and phosphate deficiency suggest a relationship with chronic calcium deficiency

o The final common pathway of anterior horn cell death, irrespective of what actually triggers the process, is a complex interaction of genetic factors, oxidative stress and glutamate excess (excitatory injury). Abnormal clumps of proteins (neurofilaments) can be found in motor neurons that may themselves be toxic or by-products of overwhelming cell injury. Calcium channel antibodies have been isolated and these further accelerate injury by stimulating additional glutamate release and intracellular calcium influx activating enzymes that promote additional cell damage.

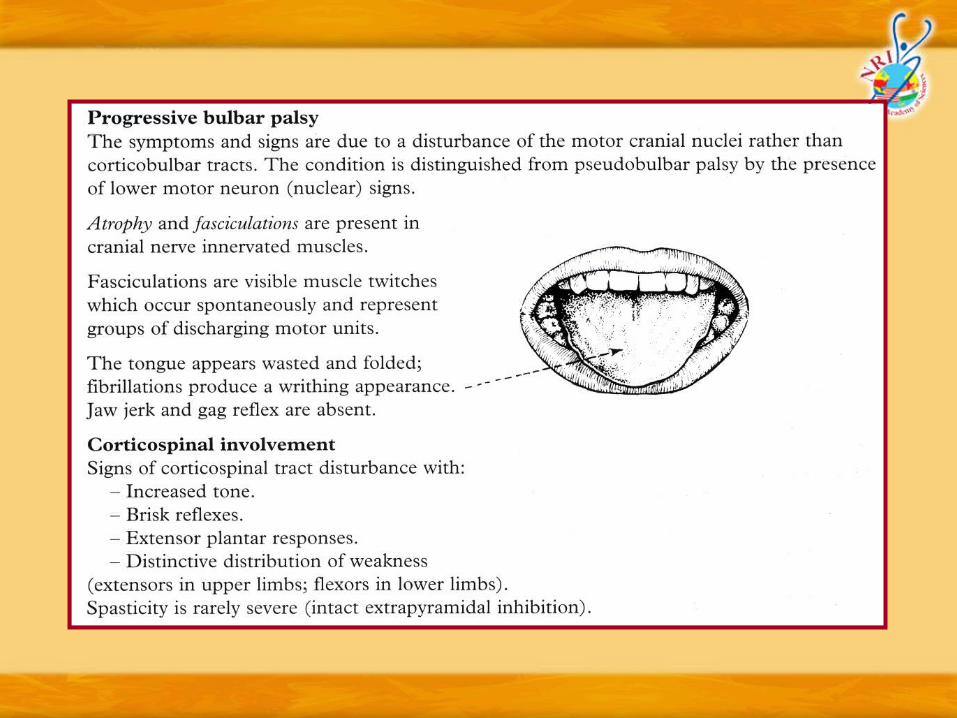
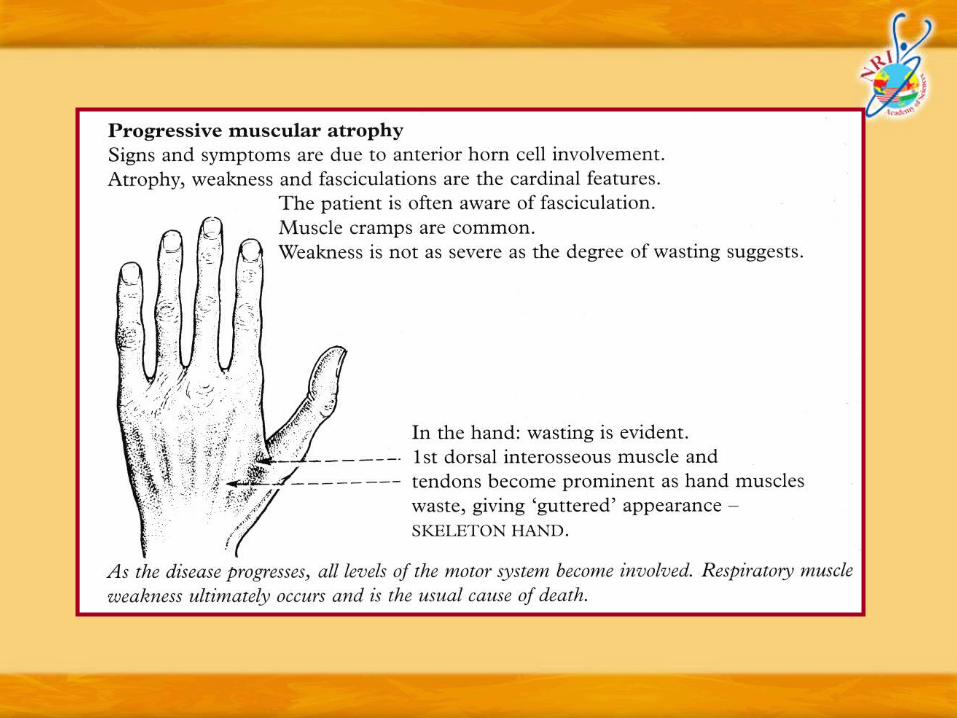


Diagnostic criteria (EI Escorial criteria for MND/ALS – World Federation of Neurology)
Presence of –
o LMN signs in at least 2 limbs
o UMN signs in at least 1 region (bulbar/ cervical/ lumbosacral)
o Progression of disease

Absence of –
o Sensory signs
o Neurogenic sphincter disturbance
o Other clinically evident CNS/PNS disease
o Exclusion of ALS-like syndromes

Treatment :
o Treatment is primarily that of managing symptoms and supporting both patient and family as these progress and their needs change
o Counselling is essential to a full understanding of the illness and its natural history. Support from a Nurse Specialist is invaluable to meeting the challenges of each phase of illness and issues of feeding and methods of ventilatory support are best discussed well in advance so that informed decisions can be made. The comprehensive care of patients is challenging with medical, legal and ethical considerations

Symptomatic treatment :
o Anarthria and dysarthria – Speech assessment and communicaiton aids when indicated.
o Dysphagia and aspiration - Percutaneous endoscopic gastrostomy (PEG)
o Nutrition – Estimate calorific content and supplement diet with vitamins
o Muscle weakness – Physiotherapy, walking aids. Splints, etc

o Respiratory failure – As vital capacity drops respiratory failure becomes inevitable. Non-invasive ventilatory assistance should be considered, if requested, when this falls below 70%. Mechanical ventilation should only be provided in those who insist upon it. Rarely ALS can present with early respiratory failure before treatment issues have been discussed. This creates a major management dilemma

Disease-modifying treatment :
o Riluzole is a drug with energy buffering and anti-glutamate properties. It is the only approved treatment and in a dose of 100 mg daily is safe with a marginal effect in prolonging survival by 2 months
o Recombinant human insulin-like growth factor requires further evaluation. Anti retroviral treatment (Indinavir) is currently under trial. Recent research suggests that anti-oxidant drugs may be beneficial but a greater understanding of the molecular basis of this illness is required to design more promising treatments.
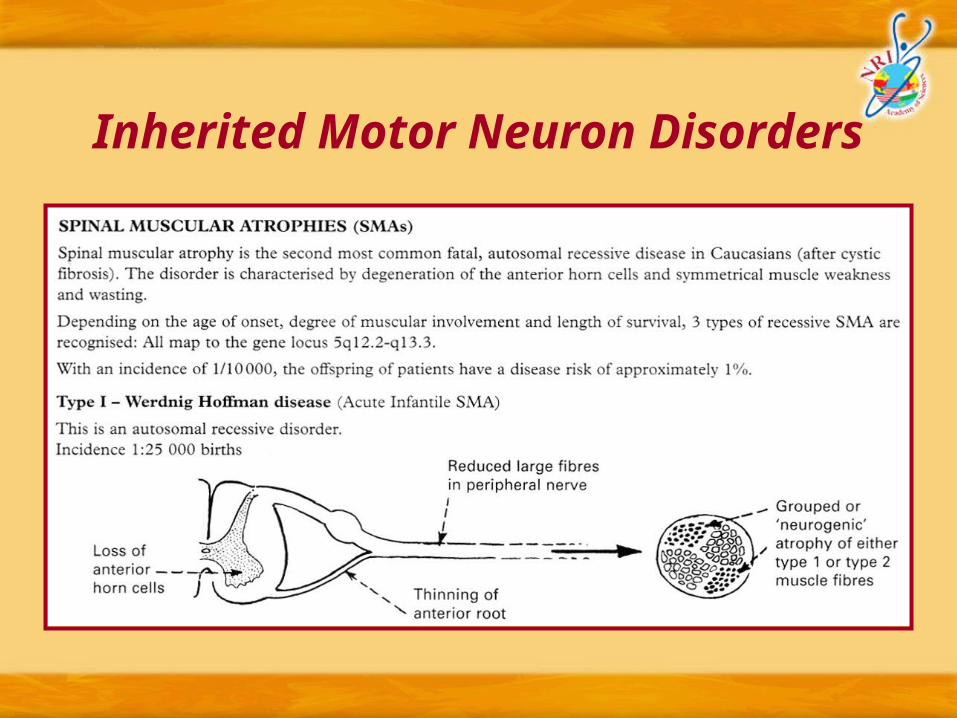
Inherited Motor Neuron Disorders


Type II – Kugelberg Welander Disease : (Late infantile or juvenile SMA)
o Pathological features similar to Werdnig Hoffman disease
Clinical features :
o Limb girdle muscles affected
o It is slowly progressive with great variability even within the same family. Median age at death 12 years. Survival to adulthood occurs in the dominant form

Type III (Adult onset SMA)
o Onset between 2nd and 5th decade with progressive limb girdle weakness.
o Distinction from progressive muscular atrophy form of ALS is difficult. A benign course supports the former
Distal and scapuloperoneal forms :
o Differentiation from HMSN type I and II and scapuloperoneal dystrophy is clinically difficult and separation may only be possible in histological and neurphysiological grounds

o Spinal and bulbar muscular atrophy (Kennedy’s syndrome)
o X-linked adult-onset neurogenic muscular atrophy with late distal and bulbar involvement (Gene Locus:Xq11-q12)
o Onset of fasciculations followed by muscle weakness and wasting occur at approximately 40 years of age. Bulbar signs and facial fasciculations are characteristic.
o Babinski sign is negative. The disorder is compatible with long life
Management of spinal muscular atrophies :
o There is no specific treatment
o Care is supportive
o Genetic counselling is essenttial

Management of spinal muscular atrophies :
o There is no specific treatment
o Care is supportive
o Genetic counselling is essenttial























![NFL Football & Amyotrophic Lateral Sclerosis [ALS]](https://img.pdfslide.net/doc/110x75/559430511a28ab4c3d8b4747/nfl-football-amyotrophic-lateral-sclerosis-als.jpg)





