Embed Size (px)
Citation preview

Novel Small Molecule Inhibitors
for the Human Kinesins
Mklp2 and Kif18A
DISSERTATION
zur Erlangung des Akademischen Grades
des Doktors der Naturwissenschaften
(Dr. rer. nat.)
an der Universität Konstanz
Mathematisch‐Naturwissenschaftliche Sektion
Fachbereich Chemie
vorgelegt von
Joachim Braun
aus Bessenbach
2015
Tag der mündlichen Prüfung: 17. April 2015
1. Referent: Prof. Dr. Andreas Marx
2. Referent: Prof. Dr. Thomas U. Mayer
Konstanzer Online-Publikations-System (KOPS) URL: http://nbn-resolving.de/urn:nbn:de:bsz:352-0-287464




„Viele Menschen treten in dein Leben ein, aber nur ein paar besondere Menschen hinterlassen auch Spuren in
deinem Herzen.“
Autor unbekannt.
Gewidmet Alfred und Anni Prößler


Danksagung
An erster Stelle möchte ich dem kürzlich verstorbenen Prof Dr. Ulrich Groth für die
Aufnahme in seine Arbeitsgruppe und die jahrelange Finanzierung danken.
Des Weiteren gilt mein besonderer Dank Prof. Dr. Thomas U. Mayer für die
Überlassung des sehr interessanten und interdisziplinären Themas sowie die
Aufnahme in seine Arbeitsgruppe, viele wissenschaftliche Diskussionen, die
Übernahme des Zweitgutachtens und die Möglichkeit als Chemiker molekular- und
zellbiologisches Arbeiten erlernen zu können.
Außerdem danke ich Prof. Dr. Andreas Marx für die Übernahme des Erstgutachtens
und zahlreiche Diskussionen in meinem Thesis Committee. Prof Dr. Müller danke ich
für die Übernahme des Prüfungsvorsitzes.
Besonders möchte ich mich bei Johanna Kastl und Martin Möckel bedanken für die
fruchtbare Zusammenarbeit, viele Diskussionen und die Hilfe bei den
Versuchsdurchführungen.
Meinen drei Bachelor möchte ich für die schöne Zeit und gute Zusammenarbeit
danken, sowie den zahlreichen Mitarbeiterpraktikanten.
Allen ehemaligen sowie übrig gebliebenen Mitgliedern der AG Groth danke ich für
zahlreiche wissenschaftliche Diskussionen, die gute Arbeitsatmosphäre und vieles
mehr. Besonderen Dank gebührt dem „Wölfsche“ für das „Käffsche“ um 15 Uhr bei
dem sowohl wissenschaftliches als auch anderes diskutiert und in humoristischer Art
verarbeitet wurde. Auch meinen zwei rumänischen Labormitbewohnerinnen Dana
und Carmen ein herzliches Dankeschön für die unvergessliche Zeit mit euch beiden!
Der ganzen AG Mayer danke ich für ihre Hilfsbereitschaft, viele Diskussionen und das
Geraderücken, wenn der Chemiker mal wieder etwas unsicher war und eine super
Arbeitsatmosphäre.
Malin Bein danke ich für die Durchführung der Zytotoxizitätsstudien.
Für das Korrekturlesen dieser Arbeit danke ich Hüsnü Topal, Tobias Strittmatter,
Johannes Drexler, Juliane Leutzow, Martin Möckel und Holger Bußkamp.
Besonders danken möchte ich all meinen Freunden, die meine Zeit hier in Konstanz
zu einer unvergesslichen, wunderbaren Erfahrung gemacht haben.
Zuletzt gilt mein größter Dank meinen Eltern und meinen Geschwistern, die in
jeglicher Lage zu mir gestanden und mich bedingungslos während der gesamten
Studienzeit unterstütz haben. Ein sicherer Hafen in dem man sich wohlfühlen und
zurückziehen kann, während alles andere seine Bedeutung verliert, ist unbezahlbar.

Die vorliegende Arbeit entstand in der Zeit von März 2010 bis Dezember 2014 in den Arbeitsgruppen von Prof. Dr. Ulrich Groth am Lehrstuhl für Organische Chemie im Fachbereich Chemie und Prof. Dr. Thomas U. Mayer am Lehrstuhl für Molekulare Genetik im Fachbereich Biologie an der Universität Konstanz.

Publikationen
Teile dieser Arbeit sind veröffentlicht in:
J. Braun, M. M. Möckel, T. Strittmatter, A. Marx, U. Groth, and T. U. Mayer
“Synthesis and Biological Evaluation of Optimized Inhibitors of the Mitotic Kinesin
Kif18A”
ACS Chem. Biol. 2015, 10, 554–560
Weitere Publikationen:
H. Strobelt, E. Bertini, J. Braun, O. Deussen, U. Groth, T.U. Mayer, D. Merhof
“HiTSEE KNIME: a visualization tool for hit selection and analysis in high-throughput
screening experiments for the KNIME platform”
BMC Bioinformatics 2012, 13 (Suppl 8):S4
J. Kastl, J. Braun, A. Prestel, H. Möller, T. Huhn, and T. U. Mayer
“M2I-1: A Small protein-protein interaction inhibitor targeting the mitotic spindle
assembly checkpoint”
ACS Chem. Biol. 2015, submitted

Table of content
1. Introduction ......................................................................................................................... 1
1.1 Cell Cycle ........................................................................................................................ 1
1.2 Kinesins in Mitosis and Cytokinesis .............................................................................. 2
1.2.1 Structure of Kinesins ............................................................................................... 2
1.2.2 Mitosis and Cytokinesis ........................................................................................... 3
1.3 Relevance of Kinesins in Cancer .................................................................................... 7
1.4 Screening for Kinesin Inhibitors .................................................................................... 8
1.5 Chemical Genetics ........................................................................................................ 10
2. Aim of the Work ................................................................................................................ 12
3. Results and Discussion ....................................................................................................... 14
3.1 Mklp2 and its Inhibitors SH1 and Paprotrain ............................................................. 14
3.1.1 Synthesis of Substituted Quinoxalines .................................................................. 15
3.1.2 Design of SH1 Analogs ........................................................................................... 15
3.1.3 Synthesis of Substituted 2-Phenylquinoxalines .................................................... 16
3.1.4 Synthesis of 2-Thiophenquinoxalines .................................................................... 19
3.1.5. Prodrug Approach ................................................................................................ 20
3.1.6 Effects on Cells ...................................................................................................... 22
3.1.7 Enzyme Coupled Assay .......................................................................................... 23
3.1.8 Malachite Green Assay .......................................................................................... 25
3.1.9 Summary and Outlook ........................................................................................... 26
3.2 Kif18A and its Inhibitor BTB-1 ..................................................................................... 29
3.2.1 Synthesis of BTB-1 ................................................................................................. 29
3.2.2 Design of BTB-1 Analogs ........................................................................................ 30
3.2.3 Synthesis of BTB-1 Analogs ................................................................................... 31
3.2.4 Screening for Kif18A Inhibition and IC50 Determination ....................................... 34
3.2.5 Mode of Inhibition and Selectivity ........................................................................ 36
3.2.6 Cellular Toxicity Studies ......................................................................................... 39
3.2.7 Live Cell Imaging .................................................................................................... 42
3.2.8 Cellular Thermal Shift Assay .................................................................................. 43
3.2.9 Immunofluorescence Imaging for Kif18A Localization .......................................... 45
3.2.10 Tubulin Polymerization Assay and IC50 Determination ....................................... 48
3.2.11 Structure Activity Relationships and Outlook ..................................................... 50
4. Summary ............................................................................................................................ 54
5. Zusammenfassung ............................................................................................................. 56

6. Materials and Methods ..................................................................................................... 58
6.1 General ........................................................................................................................ 58
6.2 Synthesis of SH1 Analogs ............................................................................................ 59
General Methods for Glyoxal Synthesis (Method A) ..................................................... 59
General Method for Preparation of Substituted Quinoxalines (Method B) .................. 59
General Method for Preparation of Amide and Ester Analogs of SH1 (Method C) ....... 59
3,4-Dichlorophenylglyoxal (1) ........................................................................................ 60
3,4-Difluorophenylglyoxal (2) ........................................................................................ 60
Phenylglyoxal (3) ............................................................................................................ 61
4-Bromophenylglyoxal (4) .............................................................................................. 61
4-Nitrophenylglyoxal (5) ................................................................................................ 61
1-(5-Chlorothiophen-2-yl)-2,2-dihydroxyethan-1-one (6) ............................................. 62
5-Chloro-2-phenyl-7-(trifluoromethyl)quinoxaline (13) ................................................ 62
2-(3,4-Dichlorophenyl)quinoxaline-6-carboxylic acid (14) ............................................. 62
2-(3,4-Difluorophenyl)quinoxaline-6-carboxylic acid (15) ............................................. 63
2-Phenylquinoxaline (16) ............................................................................................... 63
2-(4-Bromophenyl)-5-chloro-7-(trifluoromethyl)quinoxaline (17) ................................ 64
2-(5-chlorothiophen-2-yl)quinoxaline (18) .................................................................... 64
5-chloro-2-(5-chlorothiophen-2-yl)-7-(trifluoromethyl)quinoxaline (19) ...................... 65
Ethyl 2-(3,4-dichlorophenyl)quinoxaline-6-carboxylate (20) ......................................... 65
2-(3,4-Dichlorophenyl)-N,N-diethylquinoxaline-6-carboxamide (21)............................ 66
(2-(3,4-dichlorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone (22) ........ 67
(2-(3,4-Dichlorophenyl)quinoxalin-6-yl)(morpholino)methanone (23) ......................... 67
2-(3,4-Difluorophenyl)-N,N-diethylquinoxaline-6-carboxamide (24) ............................ 68
(2-(3,4-difluorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone (25) ........ 69
(2-(4-Fluoro-3-morpholinophenyl)quinoxalin-6-yl)(morpholino)methanone (26) ........ 69
6.3 BTB-1 Analogs Synthesis ............................................................................................. 70
General Methods for the Preparation of Sulfones and Sulfoxides ................................ 70
1-Chloro-2-nitro-4-(phenylsulfonyl)benzene (70) ......................................................... 71
2,4-Dinitro-1-(phenylsulfonyl)benzene (52) .................................................................. 72
2,4-Dinitro-1-(phenylsulfinyl)benzene (64) ................................................................... 72
4-Fluoro-2-nitro-1-(phenylsulfonyl)benzene (53) .......................................................... 73
2-Nitro-1-(phenylsulfonyl)-4-(trifluoromethyl)benzene (54) ......................................... 74
2-((4-Chloro-2-nitrophenyl)sulfonyl)thiophene (55) ..................................................... 74
2-((4-Chloro-2-nitrophenyl)sulfonyl)naphthalene (56) .................................................. 75
2-((4-Chloro-2-nitrophenyl)sulfinyl)naphthalene (65) ................................................... 76

4-Chloro-2-nitro-1-tosylbenzene (57) ............................................................................ 76
4-Chloro-1-((4-methoxyphenyl)sulfonyl)-2-nitrobenzene (58) ...................................... 77
4-Chloro-1-((4-methoxyphenyl)sulfinyl)-2-nitrobenzene (66) ....................................... 78
1-Nitro-2-(phenylsulfonyl)benzene (59) ......................................................................... 78
1-Nitro-2-(phenylsulfinyl)benzene (67) .......................................................................... 79
1-((4-Methoxyphenyl)sulfonyl)-2-nitrobenzene (60) ..................................................... 79
1-((4-Methoxyphenyl)sulfinyl)-2-nitrobenzene (68) ...................................................... 80
2,4-Dinitro-1-tosylbenzene (61) ..................................................................................... 81
2,4-Dinitro-1-(p-tolylsulfinyl)benzene (69) .................................................................... 81
4-Chloro-1-((4-chlorophenyl)sulfonyl)-2-nitrobenzene (62) .......................................... 82
1-((4-Chlorophenyl)sulfonyl)-2,4-dinitrobenzene (63) ................................................... 83
4-Chloro-2-nitro-1-phenoxybenzene (71) ...................................................................... 83
4-Chloro-1-iodo-2-nitrobenzene (72) ............................................................................. 84
4-Chloro-2-nitrobenzenediazonium (74) ........................................................................ 85
6.4 Biochemical and Cellular Assays ................................................................................. 85
Protein Expression and Purification from Bacteria via Poly-histidine (His) Tag ............. 85
Polymerization of Taxol Stabilized Microtubules ........................................................... 86
Malachite Green Assay ................................................................................................... 86
Enzyme Coupled Assay ................................................................................................... 87
Enzyme Coupled Assay SH1 Analogs .............................................................................. 87
Enzyme Coupled Assay SH1 Titration with Triton X-100 ................................................ 87
Enzyme Coupled Assay BTB-1 Analogs ........................................................................... 88
IC50 (Kif18A) .................................................................................................................... 88
Basal Kif18A ATPase Activity .......................................................................................... 88
Inhibition Mode of 59 ..................................................................................................... 88
Cellular Thermal Shift Assay (CETSA) ............................................................................. 88
Western Blot Analysis ..................................................................................................... 89
Tubulin Polymerization Assay ........................................................................................ 89
IC50 (Tub.Polym.) ............................................................................................................. 90
Cell Culture ..................................................................................................................... 90
Immunofluorescence ..................................................................................................... 90
Live Cell Imaging ............................................................................................................. 91
Alamar Blue Assay and EC50 Values ................................................................................ 91
7. References ......................................................................................................................... 93
8. Appendix .......................................................................................................................... 107
8.1 Appreviations ............................................................................................................. 107



Introduction
1
1. Introduction
1.1 Cell Cycle
Cell reproduction is an essential process in all higher eukaryotes in order to develop
a multicellular organism and replace cells that died due to natural causes or
environmental damage.1 The cell cycle can be divided into different distinct phases:
gap1 phase (G1), synthesis phase (S phase), gap2 phase (G2) and the mitotic phase
(M phase) (Figure 1). In the G1 phase the cell grows and verifies, if the conditions are
ideal for proliferation or if there are any inhibitory signals. As a result of this, the cell
enters a quiescent state or progress in the cell cycle. In the S phase the
deoxyribonucleic acid (DNA) is replicated and the chromosomes as well as the
centrioles are duplicated. To ensure an error free duplication of the DNA, the G2
phase includes a DNA damage checkpoint. G1, S and G2 phase are also referred to as
interphase.
Figure 1: Schematic representation of the cell cycle and the different phases.
In the M phase the duplicated chromosomes are separated by the mitotic spindle
machinery and equally distributed into two daughter cells. To ensure the correct
distribution of the genetic material to the daughter cells, the spindle assembly
checkpoint (SAC) monitors the correct attachment of chromosomes to the mitotic
spindle.2 The final physical separation of the daughter cells is achieved through
cytokinesis. All these events are orchestrated by different mechanisms in order to
sustain genomic integrity. One important class of motor proteins involved in these
processes is the kinesin superfamily.
Introduction
2
1.2 Kinesins in Mitosis and Cytokinesis
1.2.1 Structure of Kinesins
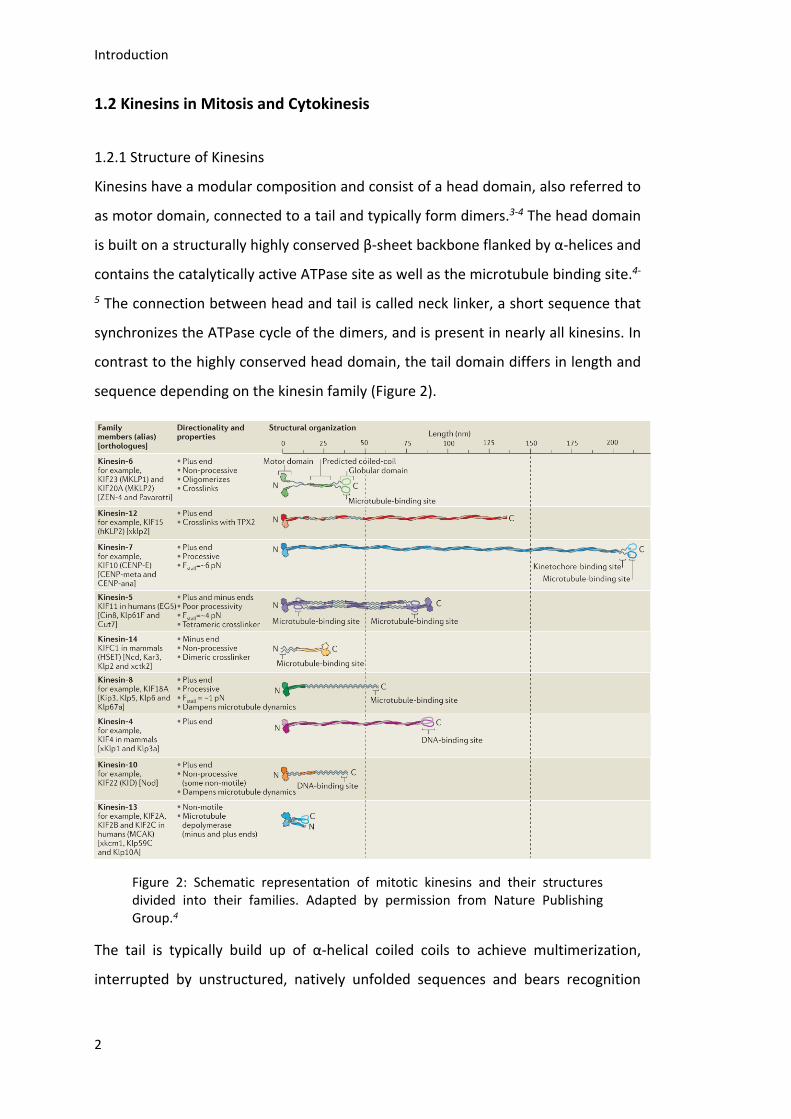
Kinesins have a modular composition and consist of a head domain, also referred to
as motor domain, connected to a tail and typically form dimers.3-4 The head domain
is built on a structurally highly conserved β-sheet backbone flanked by α-helices and
contains the catalytically active ATPase site as well as the microtubule binding site.4-
5 The connection between head and tail is called neck linker, a short sequence that
synchronizes the ATPase cycle of the dimers, and is present in nearly all kinesins. In
contrast to the highly conserved head domain, the tail domain differs in length and
sequence depending on the kinesin family (Figure 2).
Figure 2: Schematic representation of mitotic kinesins and their structures divided into their families. Adapted by permission from Nature Publishing Group.4
The tail is typically build up of α-helical coiled coils to achieve multimerization,
interrupted by unstructured, natively unfolded sequences and bears recognition

Introduction
3
sequences for co-proteins, regulatory kinases, cargo and in some cases another
microtubule binding site.3-4, 6-7
1.2.2 Mitosis and Cytokinesis
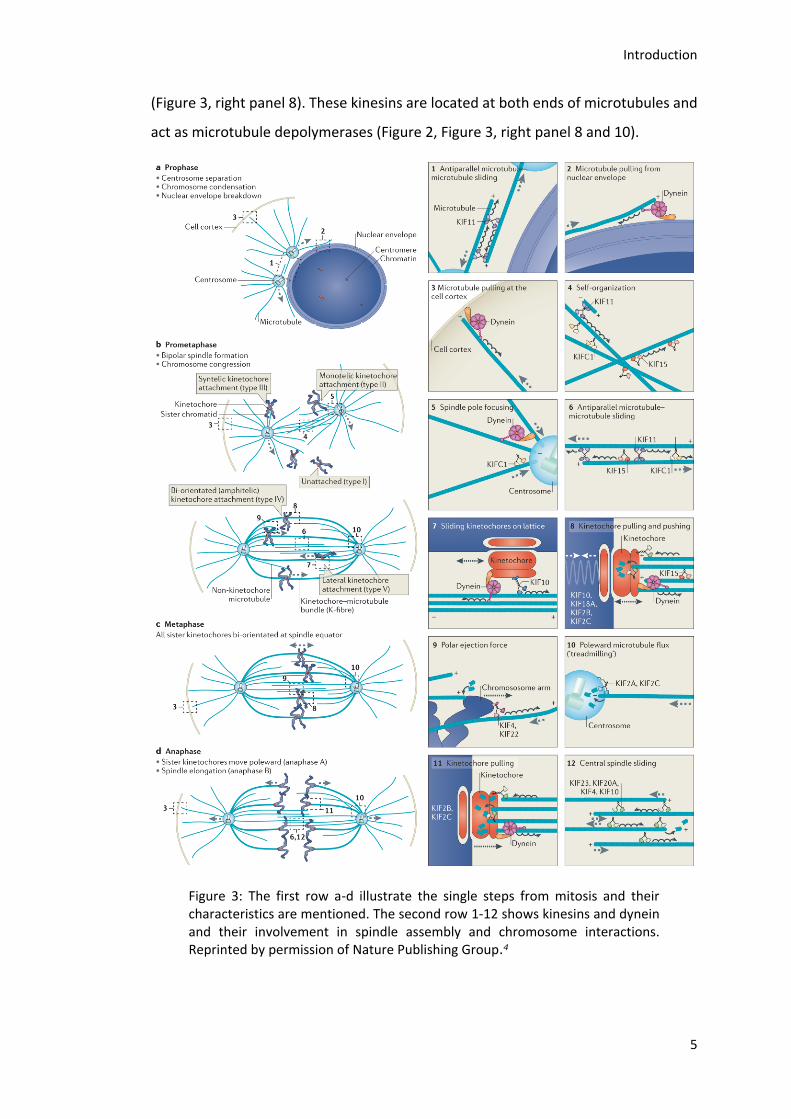
Mitosis is divided into different distinct phases: Prophase, prometaphase,
metaphase, anaphase and telophase.
The main events during prophase are centrosome separation, which is a motor-
dependent event, chromosome condensation and nuclear envelope break down
(NEBD) (Figure 3, a). Kif11, also known as Eg5, is the only member of the human
kinesin-5 family and essential for centrosome separation.4, 8-11 It consists of two
heterodimers forming a tetramer with two heads at each end functioning as a
microtubule cross-linker (Figure 2 and Figure 3, right panel 1).8, 12-13 Besides, Eg5 has
also microtubule binding sites in the tail (Figure 2) and moves towards the plus end
of microtubules, if attached to antiparallel microtubules.8, 14-16 Therefore, Eg5 enables
the separation of the centrosomes by sliding antiparallel microtubules apart, which
originate from different centrosomes (Figure 3, right panel 1).8, 14 The Eg5-driven
microtubule sliding resists forces that tend to speed up sliding, like pulling forces of
dynein on the centrosomal microtubules (Figure 3 right panel 2 and 3).17
The end of prophase in mammalian cells is reached with NEBD and followed by
prometaphase, in which a bipolar mitotic spindle is formed and chromosome
congression takes place (Figure 3, b). The mitotic spindle self-organizes through the
dynamics of microtubules, which are influenced by motor- and microtubule binding-
proteins in order to nucleate, capture, slide and reorient microtubules from both
asters.18 Microtubules originating from opposite poles can either form antiparallel
overlaps or capture kinetochores forming parallel bundles known as kinetochore
microtubules (K-fibers). The major difference between these kinds of microtubules is
their dynamic behavior: non kinetochore microtubules have a half-life of
approximately 10 seconds compared to K-fibers with a half-life of several minutes.19
At the beginning of spindle assembly the activity of Eg5 as well as pulling from cortical
myosin and pushing forces from kinetochores are required, and the continued
complete separation of the centrosomes take place (Figure 3, right panel 3-5).20-22 It
was found that Kif15 (KLP2) overexpression compensates the loss of Eg5, suggesting

Introduction
4
that both kinesins are able to slide away overlapping antiparallel microtubules (Figure
3, right panel 6).23-24 Recent studies revealed that Kif15 preferably associates with
parallel microtubules in K-fibers counteracting the forces generated by Eg5.23, 25 After
assembly of a bipolar spindle its maintenance requires outward sliding forces
generated by Eg5 and/or Kif15 and inward forces generated by KifC1, also known as
HSET, a member of kinesin-14 family (Figure 3, right panel 4-6).24, 26-27 As shown in
Figure 2 kinesin-14 family members have their motor domain at the C terminus and
are microtubule minus end directed motors (Figure 3, right panel 5). The HSET
analogs in Drosophila melanogaster (Ncd) and Schizosaccharomyces pombe (Klp2)
are able to stabilize parallel and slide apart antiparallel microtubules, in the opposite
direction compared to Eg5.28-30 These findings highlight the antagonistic activity of
Eg5 and HSET during bipolar spindle formation.26, 31-33 In parallel to bipolar spindle
formation the chromosomes have to be attached to microtubules and aligned in the
metaphase plate.34 Multiple kinesins like CENP-E (Kif10), Kif18A, Kif2B and C (MCAK)
are necessary for this process (Figure 3, right panel 7 and 8). CENP-E, for example, is
a plus end-directed motor, which is able to transport kinetochores, and therefore,
the whole chromosome, to the plus ends of microtubules (Figure 3, right panel 7).35-39
When sister kinetochores are attached to microtubules originating from opposite
poles they are able to oscillate between the poles by utilizing microtubule dynamics.
This is achieved by the regulation of microtubules growth and shrinkage and
switching between them by kinetochores with the help of kinesins, like Kif18A.
Kif18A, member of the kinesin-8 family, is a plus end-directed motor involved in
modulating microtubule plus end dynamics and is localized at kinetochores
(Figure 2, Figure 3 right panel 8).40-49 It is under debate, if Kif18Aposseses a
microtubule depolymerization activity. One model suggests that newly arriving
Kif18A molecules “bounce” into already plus-tip localized Kif18A ,which than falls off
together with one tubulin dimer. The ability to depolymerize microtubules is
consistent with the observed higher oscillation amplitude of kinetochores following
Kif18A depletion and the resulting severe chromosome congression defect.42, 46, 50-51
The processivity of Kif18A is increased by a C-terminal microtubule binding site, which
is also required for the mitotic function of the kinesin.43-45 Kif2A, Kif2B and MCAK
(Kif2C), like Kif18A, are important regulators of kinetochore oscillation
Introduction
5
(Figure 3, right panel 8). These kinesins are located at both ends of microtubules and
act as microtubule depolymerases (Figure 2, Figure 3, right panel 8 and 10).
Figure 3: The first row a-d illustrate the single steps from mitosis and their characteristics are mentioned. The second row 1-12 shows kinesins and dynein and their involvement in spindle assembly and chromosome interactions. Reprinted by permission of Nature Publishing Group.4

Introduction
6
At the minus end of K-fibers, they generate a pulling force on kinetochores whereas
at the plus end of microtubules, they seem to be involved in correcting erroneous
kinetochore attachments and regulating the speed of kinetochore motility.50, 52-55
When the chromosomes are properly aligned metaphase is reached, which is
followed by anaphase (Figure 3 c and d). In anaphase, the two sister chromatids are
separated and pulled to opposite poles. The motor requirements in human cells for
anaphase are still unknown.4 In telophase in mammalian cells, the chromosomes
decondens and new nuclei are formed.
Afterwards, cytokinesis as the final physical division takes place. A main component
of cytokinesis is the contractile ring, consisting of actin and myosin.56-58 It assembles
through actin filament polymerization by GTPase RhoA and myosin motor
activity.59-62 Another key component is the central spindle, which forms during
anaphase progression. It consists of bundled antiparallel microtubules and serves as
a signaling platform for the positioning of the cleavage furrow.4, 56, 63-64 For the
assembly of the central spindle activity of different microtubule-associated proteins
(MAPs) like protein regulator of cytokinesis (PRC1), the centralspindlin complex and
the chromosomal passenger complex (CPC) are required. Additionally, members of
the kinesin-6 family like mitotic kinesin-like protein 2 (Mklp2, Kif20A) and M-phase
phosphoprotein 1 (MPP1, Kif20B) contribute to central spindle assembly
(Figure 3, right panel 12). PRC1 is localized at the central spindle and contains a
conserved central domain, which induces microtubule bundling.65-66 Further, the N-
terminus of PRC1 comprises an oligomerization and a Kif4A binding domain that
enhances the localization to the central region.65, 67 Despite its interaction with PRC1,
Kif4A has a key role in the regulation of microtubule dynamics and therefore
controlling the size of the central spindle (Figure 3, right panel 12).68-69 A dimer of
Mklp1 (Kif23) together with a dimer of Cyk4 - a Rho family GTPase-activating protein
(GAP) - forms the tetrameric centralspindlin complex and localizes to the center of
the central spindle.70-71 It supports microtubule bundling, RhoA regulation and
recruits regulators of abscission.60, 72 The third important multi protein complex for
central spindle assembly, the CPC, consists of Aurora B, survivin, borealin and inner
centromere protein (INCENP) and is not only active at the central spindle during

Introduction
7
anaphase, but throughout mitosis. Its activity is not only constricted by activation and
localization of Aurora B and, related to that phosphorylation of central spindle
components, it is also suggested to be involved in microtubule bundling.73-76 The
accumulation of Aurora B and polo-like kinase 1 (PLK1) is achieved by Mklp2.77-78
MPP1 as well as Mklp2 are essential in the late steps of cytokinesis.79-80
In summary, kinesins have many talents and functions ranging from their importance
for the correct distribution of the genetic material during mitosis to the precise
abscission of the daughter cells.
1.3 Relevance of Kinesins in Cancer
Due to the rapid development of resistance in cancer cells to antimitotic agents like
docetaxol and other taxol derivatives, and severe side effects of those, there is an
emergent need for new antimitotic drugs and new targets beside the mitotic spindle.
In that context and as a result of their functions during cell division, kinesins have
emerged as targets for cancer therapy.81-84 For example, several inhibitors for Eg5
and CENP-E have entered clinical trials.81 For other kinesins so far no inhibitors are
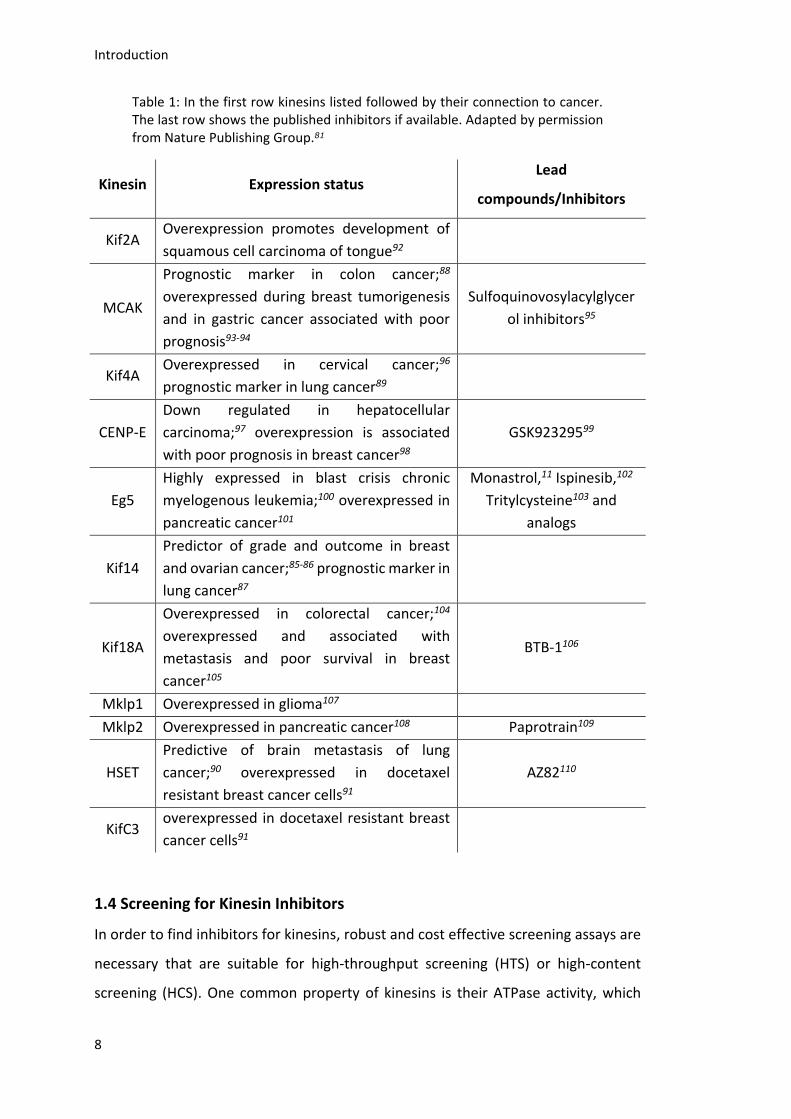
available albeit they could be used as promising targets. In Table 1 some examples of
kinesins, their connection to different cancer types and their published inhibitors are
shown. Kinesins are not only used as drug targets, but also serve as cancer and
prognostic markers. 85-90 Furthermore, in some cases overexpression of kinesins are
involved in the development of drug resistance.91 It is obvious that kinesins can serve
as targets for cancer chemotherapy but therefore their cellular functions have to be
fully understood and selective inhibitors have to be developed.
Introduction
8
Table 1: In the first row kinesins listed followed by their connection to cancer. The last row shows the published inhibitors if available. Adapted by permission from Nature Publishing Group.81
Kinesin Expression status Lead
compounds/Inhibitors
Kif2A Overexpression promotes development of
squamous cell carcinoma of tongue92
MCAK
Prognostic marker in colon cancer;88
overexpressed during breast tumorigenesis
and in gastric cancer associated with poor
prognosis93-94
Sulfoquinovosylacylglycer
ol inhibitors95
Kif4A Overexpressed in cervical cancer;96
prognostic marker in lung cancer89
CENP-E
Down regulated in hepatocellular
carcinoma;97 overexpression is associated
with poor prognosis in breast cancer98
GSK92329599
Eg5
Highly expressed in blast crisis chronic
myelogenous leukemia;100 overexpressed in
pancreatic cancer101
Monastrol,11 Ispinesib,102
Tritylcysteine103 and
analogs
Kif14
Predictor of grade and outcome in breast
and ovarian cancer;85-86 prognostic marker in
lung cancer87
Kif18A
Overexpressed in colorectal cancer;104
overexpressed and associated with
metastasis and poor survival in breast
cancer105
BTB-1106
Mklp1 Overexpressed in glioma107
Mklp2 Overexpressed in pancreatic cancer108 Paprotrain109
HSET
Predictive of brain metastasis of lung
cancer;90 overexpressed in docetaxel
resistant breast cancer cells91
AZ82110
KifC3 overexpressed in docetaxel resistant breast
cancer cells91
1.4 Screening for Kinesin Inhibitors
In order to find inhibitors for kinesins, robust and cost effective screening assays are
necessary that are suitable for high-throughput screening (HTS) or high-content
screening (HCS). One common property of kinesins is their ATPase activity, which

Introduction
9
could be used as readout. Therefore the released phosphate has to be detected in a
quantitative manner.
In a malachite green assay (MGA) the released phosphate forms a complex with
molybdate of the stoichiometry PMo12O403-(Scheme 1).111-113 The molybdate anion
forms another complex with malachite green exposing its typically green color
(Scheme 1). The exact nature of the complex is not described so far. The color change
can be detected by measuring the absorption at 650 nm.
Scheme 1: Formation of molybdate and phosphate complex and further reaction to green colored complex with malachite green.
Another spectroscopic approach is the enzyme-coupled assay (ECA) (Figure 4).114 This
assay utilizes the regeneration of ATP by pyruvate kinase and phosphoenolpyruvate
(PEP). The resulting pyruvate is reduced to lactate under consumption of NADH by
lactate dehydrogenase (Figure 4). The decrease of NADH can be monitored using its
absorption at 340 nm.
Figure 4: Schematic representation of the ECA. The kinesin-consumed ATP is regenerated by an enzyme cascade using NADH. Therefore, the decrease of absorbance at 340 nm can be measured to quantify NADH consumption.
Introduction
10
1.5 Chemical Genetics
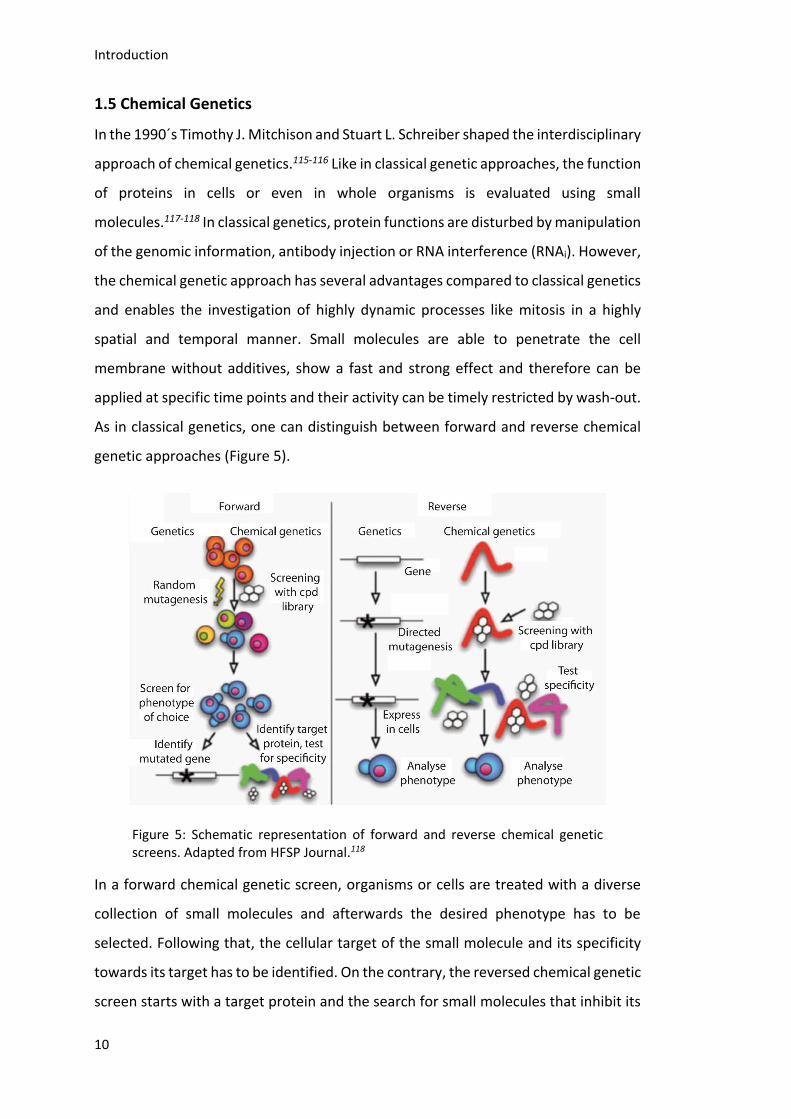
In the 1990´s Timothy J. Mitchison and Stuart L. Schreiber shaped the interdisciplinary
approach of chemical genetics.115-116 Like in classical genetic approaches, the function
of proteins in cells or even in whole organisms is evaluated using small
molecules.117-118 In classical genetics, protein functions are disturbed by manipulation
of the genomic information, antibody injection or RNA interference (RNAi). However,
the chemical genetic approach has several advantages compared to classical genetics
and enables the investigation of highly dynamic processes like mitosis in a highly
spatial and temporal manner. Small molecules are able to penetrate the cell
membrane without additives, show a fast and strong effect and therefore can be
applied at specific time points and their activity can be timely restricted by wash-out.
As in classical genetics, one can distinguish between forward and reverse chemical
genetic approaches (Figure 5).
Figure 5: Schematic representation of forward and reverse chemical genetic screens. Adapted from HFSP Journal.118
In a forward chemical genetic screen, organisms or cells are treated with a diverse
collection of small molecules and afterwards the desired phenotype has to be
selected. Following that, the cellular target of the small molecule and its specificity
towards its target has to be identified. On the contrary, the reversed chemical genetic
screen starts with a target protein and the search for small molecules that inhibit its

Introduction
11
activity. After elucidation of the specificity, cells or organisms are treated with the
small molecule and the received phenotype is examined. Both methods include
critical steps. For the forward approach the identification of the cellular target is
often challenging, because the received phenotype could be a consequence of
perturbing multiple protein activities rather than that of a single one.119 For the
reverse approach the identification of the phenotype is the bottle neck. Since the
identified compound is so far only tested for selectivity in vitro it could act with
different targets in a cellular environment.Nevertheless, chemical genetics is a
powerful tool to dissect highly dynamic cellular processes. As a result of the rapid
development in organic and combinatorial chemistry, compound libraries consisting
of natural and synthetic products are readily available.120-121 Hence, the discovered
molecular tools from chemical genetic screens are not only of great value for basic
science but also may open up novel avenues for the treatment of diseases.
The aim of this thesis was the development and biological evaluation of inhibitors
targeting the mitotic kinesins Mklp2 and Kif18A.

Aim of the Work
12
2. Aim of the Work
Kinesins are essential for the correct distribution of the genetic material into the
daughter cells during mitosis and meiosis.4 Different kinesins can have multiple
functions at different time points during the cell cycle. Specific small molecule
inhibitors are therefore ideal tools to dissect the different functions of kinesins,
because they act in a very rapid and often reversible manner. For some kinesins
inhibitors are already available as described in 1.3.
For the kinesin Mklp2, Paprotrain was discovered in 2010 by Tcherniuk et al.109,
whereas a small molecule named SH1 was identified and characterized in the
laboratory of Prof. Thomas U. Mayer in 2009 122, which served as lead compound for
the SH1 project. SH1 showed a low water solubility, which complicated its
characterization. Thus, the aim of this part of the thesis was to synthesize polar SH1
derivatives in order to enhance water solubility and establish structure activity
relationships. Therefore, the synthesized small molecules should be studied for their
ability to inhibit Mklp2 in vitro as well as their effect on cells. To achieve this, different
synthetic approaches, biochemical and cellular assays were performed.
For the kinesin Kif18A, a small molecule named BTB-1 was identified as potent
inhibitor by Catarinella et al. in 2009 106. BTB-1 shows the desired inhibition of the
ATPase activity of Kif18A in vitro and led to an increased percentage of cells in mitosis
upon cellular treatment. Nevertheless, cells treated with high concentration of BTB-1
did not show elongated spindles that are characteristic for the depletion of Kif18A.
Here, the aim was to establish a synthetic pathway in order to synthesize a small
molecule collection of BTB-1 analogs. The small compound collection should be
investigated for their inhibitory activity and selectivity towards Kif18A. Further,
structure activity relationships should be established for a better understanding of
the mode of action of BTB-1. Next, the small compound library should be studied for
their effect on the cellular level. Different biochemical and cellular assays were
carried out to characterize the new small molecule collection.

Aim of the work
13
In addition to their undeniable value for molecular biology, small molecules inhibitors
of Kif18A and Mklp2 could serve as starting point for the development of novel drugs
for mitosis-related diseases, such as tumors that overexpress these kinesins104-105, 108.

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
14
3. Results and Discussion
3.1 Mklp2 and its Inhibitors SH1 and Paprotrain
As described in 1.2 Mklp2 is indispensable for cytokinesis. In the group of Prof.
Thomas U. Mayer an inhibitor for Mklp2, called SH1, was identified in a reversed
chemical genetic screen (Scheme 2, a).122 The treatment of HeLa cells with SH1
caused a cytokinesis defect resulting in binucleated cells.122 SH1 was successfully
resynthesized in my diploma thesis by condensation of ortho-phenylenediamines
with glyoxales and the biological activity on HeLa cells were reproduced
(Scheme 2, a).123 The main problem of SH1 was its poor water solubility.
Scheme 2: The structure of SH1 and Paprotrain as well as their synthesis are shown. a) SH1 was synthesized using phenylglyoxal and phenylenediamines receiving the desired quinoxaline. b) Literature described synthesis of Paprotrain by aldol condensation of 3-pyridinecarboxaldeyde and 3-indolylacetonitrile.
In 2010 Tcherniuk et al. published Paprotrain as a selective Mklp2 inhibitor with a
half-maximal inhibitory concentration (IC50) of 0.83±0.1 µM.109 It was synthesized
using an aldol reaction of 3-indolylacetonitrile with 3-pyridinecarboxaldehyde
(Scheme 2, b). The treatment of cells with Paprotrain resulted in a binucleated
cellular phenotype similar to that received from RNAi-mediated depletion of Mklp2.
Paprotrain compared to SH1 shows a higher solubility in aqueous media but is
sensitive to light induced isomerization due to its double bond. In the following, the
focus will be on SH1 and the synthesis of analogs while Paprotrain serves as point of
comparison. In order to establish structure activity relationships (SARs) and enhance
water solubility new compounds based on SH1 should be synthesized in fast and
efficient manner.

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
15
3.1.1 Synthesis of Substituted Quinoxalines
Quinoxalines are readily available by the reaction of 1,2-arylenediamins and various
two carbon unit donors like α-diketones, α-halocarbonyl compounds, oxalic acid
derivatives, epoxides and dihalides to mention some.124 All these reactions are
restricted to symmetrical diamine building blocks, because otherwise regioisomeric
mixtures of quinoxalines are obtained. To achieve regioselectivity addition of
different catalysts is described in literature.125-130 As described in my diploma thesis
SH1 can be synthesized in a regioselective manner using glyoxal hydrate,123 which
served as the synthetic approach for further synthesis of SH1 analogs (Scheme 2, a).
3.1.2 Design of SH1 Analogs
In order to improve the water solubility of SH1 and establish SARs, different
approaches were used (Scheme 3). One approach was to introduce different polar
substituents at the phenyl or quinoxaline ring, which was started in my diploma thesis
and followed up (Scheme 3, a).123 The second approach was to isosterically exchange
the phenyl moiety to a thiophene (Scheme 3, b). This exchange should improve the
solubility remarkably since heterocycles, like quinoxalines and thiophenes, are
readily water soluble compared to the aromatic phenyl moiety. The last approach
was to enhance the solubility by conjugation to an enzymatic cleavable carbohydrate,
so-called prodrug approach (Scheme 3, c). As linker between the carbohydrate and
the bioactive quinoxaline ethylene glycol scaffolds of different length were chosen.
Despite their enhancement of water solubility, they also show a high flexible
backbone, which could be beneficial for enzymatic cleavage of the ester bond or if
not cleaved for binding of the quinoxaline to its target. A second effect is, that the
compounds uptake is not restricted to passive diffusion, because carbohydrates are
actively transported into cells. All approaches are based on the initial reaction of
phenylenediamines with glyoxales. In the next section the approach a will be
described.

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
16
Scheme 3: Different approaches for enhancement of water solubility. a) Introduction of polar groups at the phenyl or quinoxaline ring. b) Isosterical exchange of the phenyl moiety to more water soluble thiophene. c) Conjugation of SH1 analog to propargyl-2-acetamido-2-deoxy-α-D-glucoside.
3.1.3 Synthesis of Substituted 2-Phenylquinoxalines
First, analog to methods described in literature, differently substituted 2-phenyl-
glyoxal hydrates (compound 1-5) were synthesized as well as one thiophene analog
6.123, 131-133 Therefore, the selective oxidation of activated methyl groups of
acetophenones with selenium (IV) oxide (SeO2) was applied (Scheme 4, compounds
1-5). For the thiophene glyoxal hydrate 6 no yield is given, because the crude product
was directly used for condensation. The next step consists of the condensation of
phenylenediamines with the received glyoxal hydrates (Table 2). Yields marked
with * correspond to small molecules synthesized during my diploma thesis using
phenacylbromids instead of glyoxales and are listed for completeness
(Table 2, SH1 and compounds 7-12).123

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
17
Scheme 4: Synthesis of glyoxal hydrates by SeO2 mediated oxidation of activated methyl groups. The yields are shown except for the thiophene glyoxal, which was directly used for further synthesis due to instability.
Compound 7 and 8 were regioisomeric mixtures. Compared to the previously used
phenacylbromid approach the yields were improved. As mentioned above, the
thiophene glyoxal was used directly without further purification and converted to the
corresponding quinoxalines. Two examples 18 and 19 are given in Table 3 and this
compound class is discussed in chapter 3.1.5 in further detail.
Table 2: Reaction scheme for the synthesis of substituted 2-phenylquinoxalines. The substitution pattern and yields are given in the table. Yields marked with * correspond to small molecules synthesized during my diploma thesis.
Cpd. R1 R2 R3 R4 R5 Yield
SH1 Cl Cl Cl H CF3 83%*
7 Cl Cl Cl H CF3
33%
regioisomers*
8 F F Cl H CF3
28%
regioisomers*
9 Cl Cl H H H 47%*
10 F F H H H 35%*
11 F F H COOEt H 14%*
12 OMe OMe H H H 71%*
13 H H Cl H CF3 65%
14 Cl Cl H COOH H 94%
15 F F H COOH H 75%
16 H H H H H 97%
17 H Br Cl H CF3 72%

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
18
Table 3: The reaction scheme depicts the synthesis of 2-thiophenequinoxalines and the corresponding compounds and yields are given in the table.
Cpd. R3 R5 Yield
18 H H 51% 19 Cl CF3 50%
To enhance cell membrane permeability the free acids were converted into amides
or ethyl esters. N-Methylpiperazine and morpholine were selected as suitable
amines, because they bear another hetero atom, which enhances the solubility for
polar solvents, and diethylamine as a less bulky substituent. Further, the sterical
demand of this position for biological activity was addressed. Therefore, the carbonic
acid was transformed into an acid chloride to enhance reactivity and reacted with the
corresponding nucleophile. In a first attempt to synthesize the morpholide 26, the
base was used as solvent and the reaction was heated, which resulted in a
nucleophilic aromatic substitution (SNAr) where morpholine replaced one fluoride of
the phenyl ring (Table 4, 27). To circumvent this problem the corresponding amine
was used in a small excess (1.1 equiv.) and triethylamine (NEt3) was added as acid
scavenger (Table 4). Interestingly, all difluoro substituted compounds 24-26 were
received in lower yields compared to their dichloro analogs 21-23. This could be
explained by the altered electronegativity through the two fluoride substituents.

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
19
Table 4: Reaction pathway to amides and ethyl ester are shown. The corresponding acid is first converted into its acid chloride followed by conversion to the desired amid/ester. The yields and different products are listed in the table.
Cpd. R1 R2 R4 Yield
20 Cl Cl COOEt 59%
21 Cl Cl diethylamide 74%
22 Cl Cl N‐methylpiperazide 53%
23 Cl Cl morpholide 68%
24 F F diethylamide 53%
25 F F N‐methylpiperazide 37%
26 morpholine F morpholide 24%
3.1.4 Synthesis of 2-Thiophenquinoxalines134
The synthesis of 27-40 described in this part was carried out by Kim M. Leitner during
her bachelor thesis. First, differently chlorinated 2-acetylthiophenes were
synthesized according to literature procedures applying the swamping catalyst effect
(Scheme 5).135-137 The swamping catalyst effect, which is an excess of AlCl3, leads to
the complexation of the carbonyl group and prevents α-halogenation of it.
Scheme 5: Synthesis of differentially chlorinated 2-acetothiophenes by temperature control and excess of 3 equiv. AlCl3.
The regioselectivity is achieved by temperature control. This was done to receive
electron poor thiophenes comparable to dichloroacetophenones used for the
Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
20
synthesis of SH1 analogs. For the formation of 2-thiophenequinoxalines the synthetic
procedures described in 3.1.4 were used. The acetylthiophenes were oxidized to
glyoxales followed by condensation to phenylenediamines. The amides and esters
were synthesized using the corresponding acid chloride as described in 3.1.4. Table 5
shows the received compounds and achieved yields. For further details and exact
synthetic procedures see bachelor thesis of Kim M. Leitner.134
Table 5: Synthesized 2-thiophenequinoxalines and yields. For the amides/esters the yields correspond to the amidation/esterification via the acid chloride pathway.
Cpd. R1 R2 R3 R4 R5 Overall yield
27 H H H H H 51%
28 Cl Cl Cl H CF3 50%
29 Cl H Cl H CF3 13%
30 H Cl Cl H CF3 11%
31 Cl H H H H 19%
32 H Cl H H H 16%
33 Cl H H COOH H 11%
34 Cl Cl H COOH H 54%
35 H Cl H COOH H 30%
36 Cl H H diethylamide H 30% (in respect to the
acid)
37 Cl H H morpholide H 18% (in respect to the
acid)
38 Cl H H COOMe H 32% (in respect to the
acid)
39 H Cl H COOMe H 33% (in respect to the
acid)
40 Cl H H COOEt H 22% (in respect to the
acid)
3.1.5. Prodrug Approach138
The work presented in this section was carried out by Sabrina Batke during her
bachelor thesis. The main reaction used herein was the Huisgen 1,3 dipolar

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
21
cycloaddition of alkines and azides.139 Beforehand, the quinoxaline structures were
synthesized as described in 3.1.4. The acid chlorides were esterified with ethylene
glycol linkers of different lengths bearing a terminal azide group (Table 6). With the
azide in hand the next step was the copper (I) catalyzed 1,3 dipolar cycloaddition.
Table 6: Schematic representation of the synthesis of azide bearing ethylene glycol linker and the corresponding yields are given in the table.
Cpd. R1 R2 Yield
41 F OCH2CH
2N
3 76%
42 F OCH2CH
2OCH
2CH
2N
3 59%
43 F O(CH2CH
2O)
3CH
2CH
2N
3 76%
44 Cl OCH2CH
2N
3 64%
45 Cl OCH2CH
2OCH
2CH
2N
3 64%
46 Cl O(CH2CH
2O)
3CH
2CH
2N
3 65%
Table 7: Scheme of the copper (I) mediated 1,3 dipolar cycloaddition of azides and alkine modified carbohydrates. The yields of the synthesized compounds are shown in the table.
Cpd. R n Yield
47 Cl 1 35%
48 F 1 31%
49 Cl 2 60%
50 F 2 34%
51 F 4 36%

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
22
Therefore, the reaction was carried out with propargyl-2-acetamido-2-deoxy-α-D-
glucoside under microwave irradiation and copper catalysis.140-142 The results are
summarized in Table 7. For further details of synthetic procedures see bachelor thesis
of Sabrina Batke.138
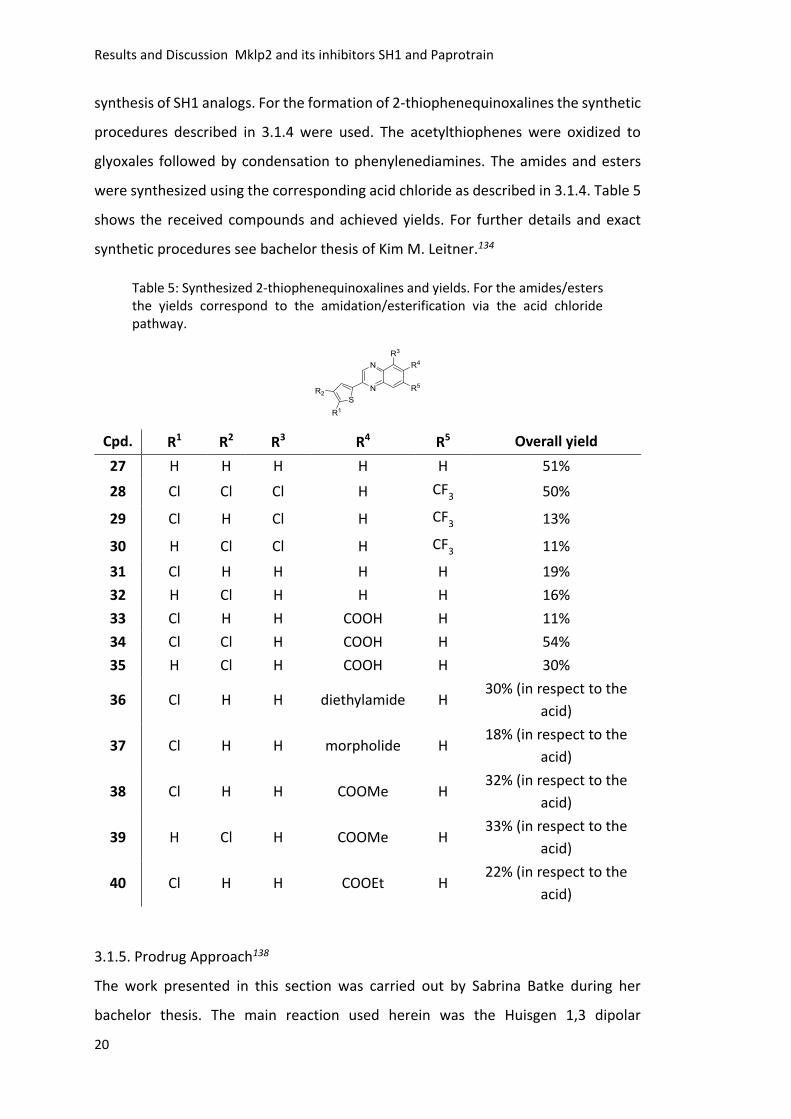
3.1.6 Effects on Cells
First, the cellular effect of selected compounds was investigated and the results
received during my diploma thesis were completed using the same experimental
setup. As described in the PhD thesis of Stefan Hümmer, the inhibition of Mklp2
should result in a failure of cytokinesis and therefore lead to binucleated cells.122-123
S-phase synchronized HeLa cells were treated with 100 µM compound or 1% DMSO
as solvent control and imaged for 18 h using live cell imaging (Figure 6). It was
distinguished between mononucleated, binucleated and undefined cells. All
compounds marked with “*” showed crystallization during the experiments.
Compounds marked with + were only tested once. Compounds 12, 15 and 20-26 did
not show any significant effect on cells. The resynthesized and commercial SH1
showed similar activity in contrast to the regioisomeric mixture of SH1 7, which was
less active. Small molecules 9 and 11 showed an activity comparable to SH1.
Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
23
Figure 6: Schematic representation of assay setup. S-phase synchronized HeLa cells were treated with 100 µM compound or 1% DMSO as solvent control and observed for 18 h. It was distinguished between mononucleated and binucleated cells. Compound marked with * showed crystallization during the experiment, whereas compounds marked with + were only tested once. For all other small molecules the mean of three independent experiments and standard deviations are shown.
These results suggest that the substitution pattern of the phenyl moiety is essential
for activity and that small changes at the quinoxaline are tolerated (9, 11), but bulkier
substituents diminish activity (21-26). It is noticeable that all active small molecules
showed crystallization during the assay and thus, the exact active concentration of
the corresponding small molecule is unknown. With the first hints for bioactivity, the
next step was to confirm Mklp2 as the target in vitro.
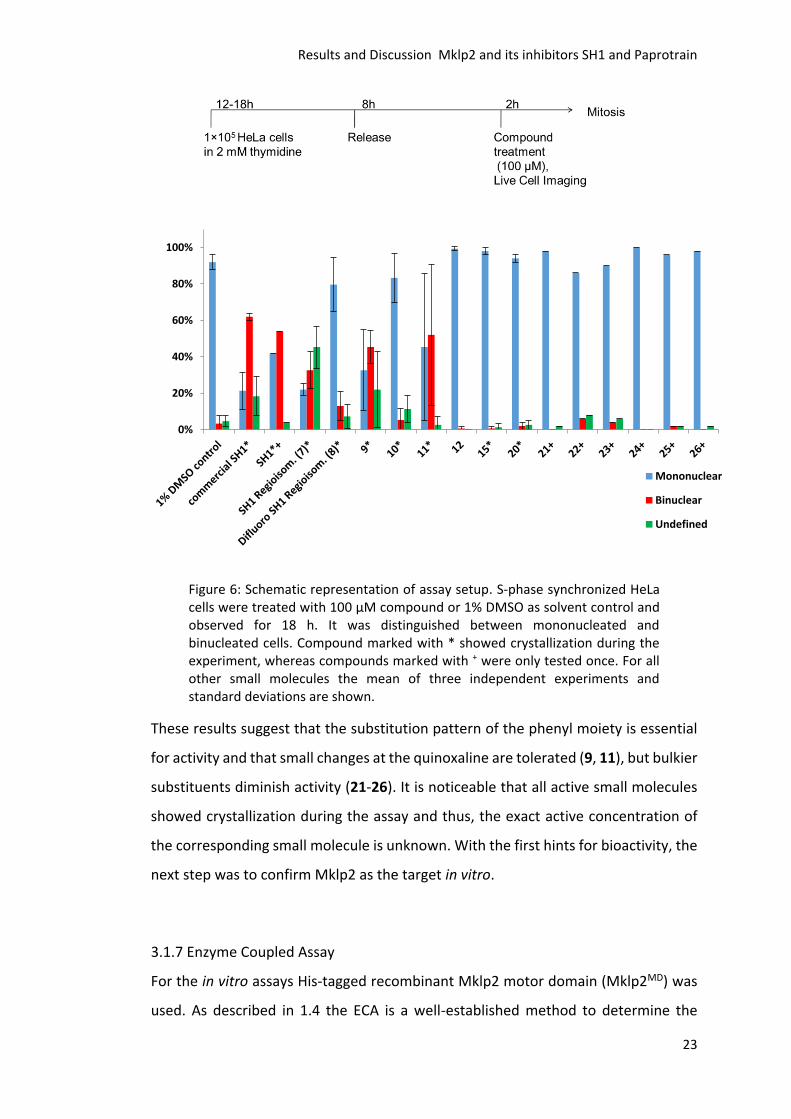
3.1.7 Enzyme Coupled Assay
For the in vitro assays His-tagged recombinant Mklp2 motor domain (Mklp2MD) was
used. As described in 1.4 the ECA is a well-established method to determine the
0%
20%
40%
60%
80%
100%
Mononuclear
Binuclear
Undefined
Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
24
ATPase activity of kinesins and was used to investigate, if the synthesized small
molecules are able to inhibit the ATPase activity of Mklp2MD. First, the assay condition
were evaluated by titration of Mklp2MD, taxol stabilized microtubules and ATP. After
that a compound collection was applied at a concentration of 50 µM. DMSO was used
as solvent control and the ATPase activity was set to 100%, whereas 50 µM Paprotrain
was used as a positive control.
Figure 7: The ATPase activity of Mklp2MD is shown relative to the DMSO control. Small molecules were used in a concentration of 50 µM. All compounds were tested with two different end concentrations of DMSO. The first bar shows the activity with a final concentration of 5% and the second bar of 10% DMSO. Bars colored in red mark compounds, which showed no linear decrease of absorption whereas yellow bars indicate that linear ranges were observable. Green bars point to linear absorbance decrease during the whole assay. Average of three independent experiments and standard deviations are shown.
Due to the solubility problems experienced in the cellular assays two different DMSO
concentrations (5% and 10%) were used, in order to enhance the solubility of the
small molecules in polar media. The screening results are depicted in Figure 7.
Despite the increased concentration of DMSO, crystallization of some compounds
was observed. Owing to the high absorbance of SH1 at 341 nm the crystallization
resulted in a nonlinear absorption course resulting in a more difficult determination
of the ATPase activity. All compounds showing that behavior are colored in red in
1. Bar ATPase activity 5% DMSO
2. Bar ATPase activity 10% DMSO
0%
20%
40%
60%
80%
100%
120%
140%
ATP
ase
act
ivit
y re
l. t
o D
MSO
co
ntr
ol
Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
25
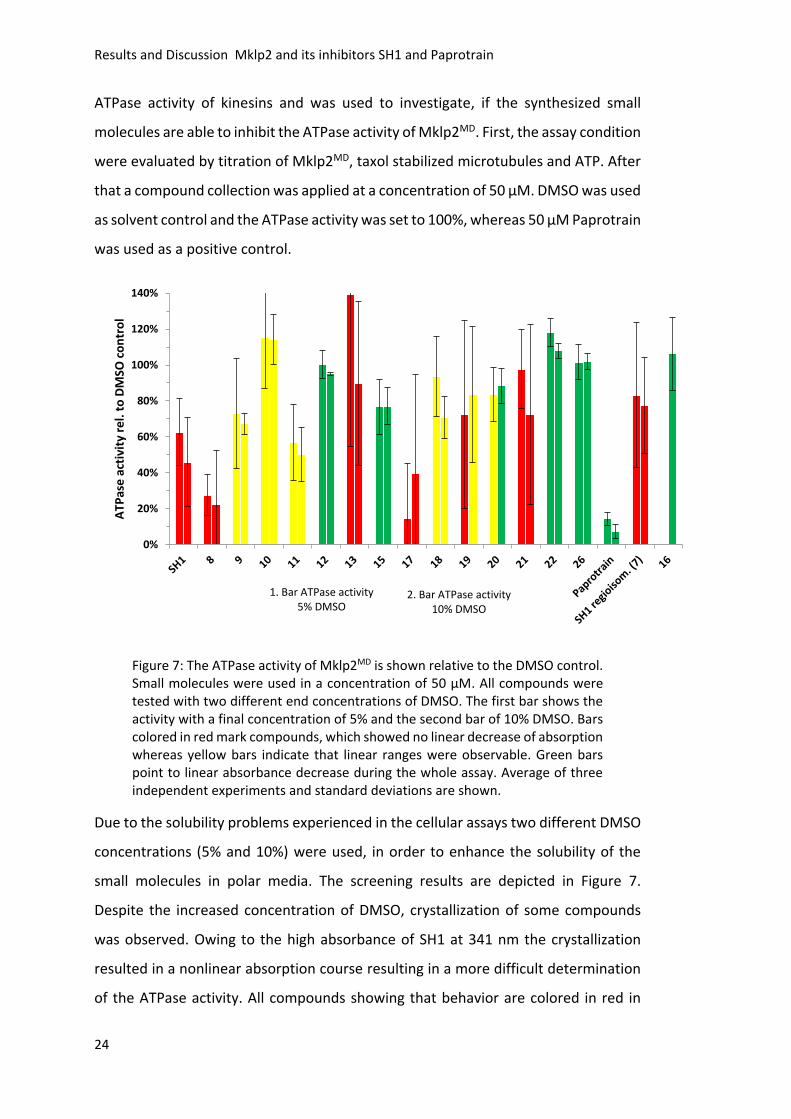
Figure 7 (SH1, 8, 13, 17, 19, 21 and 7). Yellow bars indicate that a linear range is
observable but not for the whole assay and green bars show a linear behavior for the
whole time. For 19 and 20 the solubility was enhanced as a result of the higher DMSO
concentration, leading to better assay results. Paprotrain, for example, showed a
linear decrease of absorption at 340 nm independent of the DMSO concentration and
showed the highest inhibitory activity. In a second approach Triton X-100 was added
additionally to higher DMSO concentration as detergent in a final concentration of
0.1%, which resulted in crystallization inhibition but also in a complete loss of
inhibitory activity of SH1 (Figure 8). Furthermore, two different microtubule (MT)
concentrations were used, but they did not show any influence on the ATPase
activity. As a consequence of the readout problem caused by the absorption of SH1,
the malachite green assay was used as an alternative approach.
Figure 8: The ATPase activity of Mklp2MD in presence of 0.1% Triton X-100 is shown relative to the DMSO control. SH1 was applied in different concentrations ranging from 100-25 µM. Blue bars indicate the ATPase activity at 2 µM microtubules (MT) and orange at 400 nm MT´s. No inhibition of SH1 and no significant difference between the applied MT concentrations was observed. Average of three independent experiments and standard deviations are shown.
3.1.8 Malachite Green Assay
Since the readout of the MGA is absorbance measurements at 650 nm SH1 should
not interfere with the assay. In the beginning, the assay conditions were determined
by titration of Mklp2MD. In order to enhance the solubility of SH1 Triton X-100 was
0%
20%
40%
60%
80%
100%
120%
140%
160%
25 µM SH1 50 µM SH1 75 µM SH1 100 µM SH1
ATP
ase
act
ivit
y re
l. t
o D
MSO
co
ntr
ol
2 µM MT 400 nM MT
Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
26
added in a final concentration of 0.1%. Mklp2MD showed no sensitivity towards Triton
X-100 in the assay, excluding interactions of the additive with the kinesin or other
assay components. First, different concentrations of SH1 were used to elucidate if
SH1 inhibits Mklp2MD in a concentration dependent manner. As described above,
DMSO was used as solvent control and the activity was set to 100%. Unexpectedly,
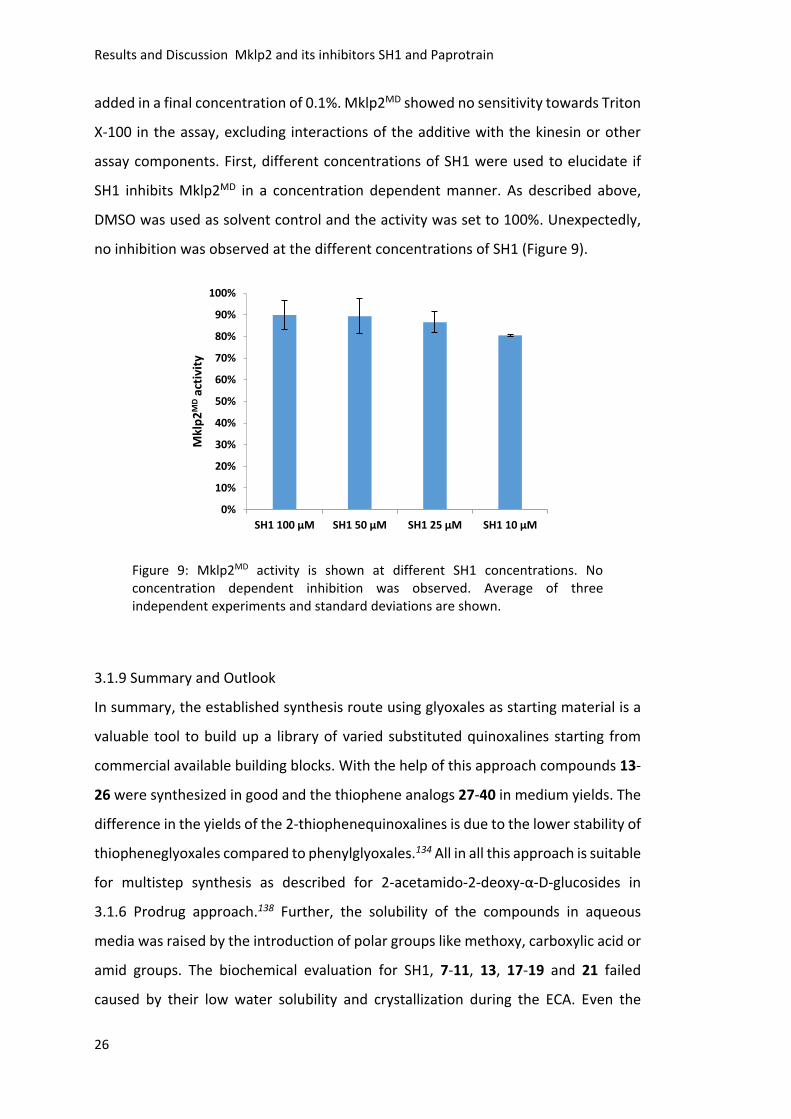
no inhibition was observed at the different concentrations of SH1 (Figure 9).
Figure 9: Mklp2MD activity is shown at different SH1 concentrations. No concentration dependent inhibition was observed. Average of three independent experiments and standard deviations are shown.
3.1.9 Summary and Outlook
In summary, the established synthesis route using glyoxales as starting material is a
valuable tool to build up a library of varied substituted quinoxalines starting from
commercial available building blocks. With the help of this approach compounds 13-
26 were synthesized in good and the thiophene analogs 27-40 in medium yields. The
difference in the yields of the 2-thiophenequinoxalines is due to the lower stability of
thiopheneglyoxales compared to phenylglyoxales.134 All in all this approach is suitable
for multistep synthesis as described for 2-acetamido-2-deoxy-α-D-glucosides in
3.1.6 Prodrug approach.138 Further, the solubility of the compounds in aqueous
media was raised by the introduction of polar groups like methoxy, carboxylic acid or
amid groups. The biochemical evaluation for SH1, 7-11, 13, 17-19 and 21 failed
caused by their low water solubility and crystallization during the ECA. Even the
0%
10%
20%
30%
40%
50%
60%
70%
80%
90%
100%
SH1 100 µM SH1 50 µM SH1 25 µM SH1 10 µM
Mkl
p2
MD
acti
vity

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
27
attempt to enhance solubility with a higher DMSO concentration was not sufficient
to prevent crystallization. Hence, SH1 serves as point of reference it was titrated
using Triton X-100 as additive for solubility enhancement. As a result the
crystallization of SH1 was avoided but no inhibitory activity was detectable. To avoid
the interference of SH1 absorption at 341 nm with the assay readout the MGA was
applied. As described before Triton X-100 was added as detergent and SH1 was used
in different concentrations. In accordance with the ECA results addition of
Triton X-100 resulted in a complete loss of inhibitory activity of SH1. It seems that the
low solubility of SH1 is necessary for its activity. In the cellular assay it was also
proven, that only small molecules that crystallized during the assay showed
bioactivity. Therefore, the idea is that the low solubility of SH1 is necessary in order
to develop its bioactivity and that SH1 acts as an aggregation based inhibitor. In
literature different approaches are described to detect aggregation based inhibitors
and β-lactamase was used as model system.143-147 The general approach was to
diminish inhibition by addition of Triton X-100. As described above SH1 shows this
effect and could be termed as detergent dependent inhibitor. To identify SH1 as an
aggregation based inhibitor the proof of the inhibitory activity without detergence
has to be shown. The ECA could not be used for this, but the MGA as well as other
assays suitable for detection of ATPase activity like using radioactive labeled ATP or
Förster resonance energy transfer (FRET) based ATP probes can be applied.148-149
Another possibility would be to alter the molecular nature of SH1 in a way that the
inhibition of Mklp2 remains and the water solubility is enhanced. Looking at the
structure of Paprotrain the pyridine moiety, as a nitrogen containing aromatic
heterocycle, could be replaced by the substituted quinoxaline scaffold of SH1,
resulting in a fusion of SH1 and Paprotrain (Scheme 6). Further, the
3,4-dichlorophenyl residue could be replaced by methylindol as described by Finlay
et al. (Scheme 6).150 In their work they described the exchange of a dichlorophenyl
by a methylindol moiety resulting in a higher efficacy of the small molecule as IKur
inhibitor. For the synthesis of these indole substituted quinoxalines the glyoxal
approach can be used. The synthesis of 5-chloro-2-(1-methyl-1H-indol-3-yl)-7-
(trifluoromethyl)quinoxaline is under current investigation.

Results and Discussion Mklp2 and its inhibitors SH1 and Paprotrain
28
Scheme 6: Synthetic approaches to enhance solubility in polar media by combination of SH1 and Paprotrain (upper panel) or exchange of dichlorophenyl by methylindole (lower panel).
The last mentioned approach to modify the structure of SH1 has the benefit that a
soluble inhibitor is in general a better analyzable small molecule concerning its
bioactivity and could be used as a tool in molecular biology. Furthermore, the newly
developed inhibitor could serve as starting point for additional synthesis of selective
Mklp2 inhibitors and could open up new avenues for treatment of cytokinesis related
diseases like Mklp2 overexpressing tumors.108

Results and Discussion Kif18A and its inhibitor BTB-1
29
3.2 Kif18A and its Inhibitor BTB-1
The unique mitotic kinesin Kif18A integrates plus-end directed motility with the
ability to affect microtubule dynamics. It is essential for the accurate architecture of
the mitotic spindle and proper alignment of chromosomes in mammalian cells.42-47
Cells depleted of Kif18A by RNAi display elongated spindles with hyperstable
microtubules and the majority of chromosomes is scattered throughout the
spindle.42-46 Since multiple cancer types show an elevated level of Kif18A, its function
might be beneficial for the survival and development of these cells.104-105 So far BTB-1
is the only known inhibitor of Kif18A. As shown by Catarinella et al. in 2009106, BTB-1
inhibited the microtubule-stimulated ATPase activity of Kif18A with an IC50 value of
1.7 µM in a reversible manner. Treatment of HeLa cells with high concentrations of
BTB-1 did not result in a phenotype with elongated spindles reminiscent of Kif18A
depletion, but resulted in an increase of the mitotic index. This result showed that
BTB-1 is bioactive in tissue culture cells. Based on the different phenotypes, it was
speculated that Kif18A might not be the only or relevant binding partner of BTB-1 in
cells. Therefore, a small molecule collection of structurally near analogs of BTB-1
should be synthesized in order to establish SARs and to get a better understanding of
the inhibition mode of BTB-1. Further, their biological activity on cultured tissue cells
and in vitro should be evaluated.
3.2.1 Synthesis of BTB-1
BTB-1 was synthesized according to known literature procedures
(Scheme 7).106, 151-152 The first step consists of a nucleophilic aromatic substitution
(SNAr) under basic conditions to displace the activated chloride at the aromatic core
by a thiophenolate. Subsequent oxidation of the resulting sulfide by H2O2 yields
BTB-1. This general approach was used to build up the small library of BTB-1 analogs
as described in 3.2.3. The synthetic pathway starts from commercially available
building blocks and represents a robust and cost-efficient method to assemble a small
scale compound collection in a fast manner.

Results and Discussion Kif18A and its inhibitor BTB-1
30
Scheme 7: BTB-1 was synthesized using a nucleophilic aromatic substitution under basic conditions followed by oxidation to the corresponding sulfone with H2O2 in an overall yield of 74%.
3.2.2 Design of BTB-1 Analogs
A subset of closely related analogs of BTB-1 were synthesized to get a better
understanding, which structural motifs of BTB-1 are necessary for its inhibitory
activity against its target Kif18A, and to potentially find more effective inhibitors. To
this end, the unsymmetrically substituted diphenylsulphone BTB-1 was divided into
three scaffolds: the two phenyl moieties I, II and a linker between them (Figure 10).
To enhance lipophilicity and cell membrane permeability of BTB-1, lipophilic groups
like fluorine and trifluoromethyl were introduced at phenyl moiety I in para position
to the linker. Additionally, the electron withdrawing group NO2 was introduced to
elucidate how altered electronegativity and sterical demand at this position affect
the inhibitory activity towards Kif18A. In order to investigate the importance of the
NO2 group in ortho position at phenyl moiety I for Kif18A inhibition, it was shifted to
the meta position to the linker. The phenyl moiety II was isosterically exchanged to
thiophene and the aromatic system was expanded to naphthalene. Various
substituents (Cl, OMe, Me) were introduced in para position to the linker
(phenyl moiety II). Finally, the sulfone linker was changed to an ether or sulfoxide to
analyze the effect of the geometry of the linker on the inhibition of Kif18A.
Figure 10: Structure of BTB-1 and its division into scaffolds I, II and linker.

Results and Discussion Kif18A and its inhibitor BTB-1
31
3.2.3 Synthesis of BTB-1 Analogs
The synthetic approach for BTB-1, as described in 3.2.1, was used in order to
synthesize differently substituted sulfones. Starting with a nucleophilic aromatic
substitution under basic conditions followed by H2O2 oxidation the sulfones 52-63
were received in low to good yields (Table 8).
Table 8: Reaction scheme for the synthesis of substituted diphenylsulfones. The substitution pattern and yields over two steps are given in the table. For BTB-1, the literature yield is given for comparison. Compounds marked with * were synthesized using sodium thiophenolate and 0.3 equiv. NaOH.
Cpd. R1 R2 Overall yield
BTB-1 Cl Ph 74%106
52* NO2 Ph 25%
53* F Ph 12%
54* CF3 Ph 26%
55 Cl 2-thiophene 68%
56 Cl 2-naphthalene 6%
57 Cl 4-methylbenzene 7%
58 Cl 4-methoxybenzene 12%
59 H Ph 3%
60 H 4-methoxybenzene 5%
61 NO2 4-methylbenzene 18%
62 Cl 4-chlorobenzene 55%
63 NO2 4-chlorobenzene 77%
The racemic sulfoxides 64-69 were synthesized by changing the oxidant to
meta-chloroperoxybenzoic acid (m-CPBA) and using it in stoichiometric quantities to
receive a single oxidation process to the sulfoxides (Table 9).

Results and Discussion Kif18A and its inhibitor BTB-1
32
Table 9: Synthetic route to the racemic sulfoxides. The yields and substitution patterns are given in the table.
Cpd. R1 R2 Yield
64 NO2 Ph 38%
65 Cl 2-naphthalene 66%
66 Cl 4-methoxybenzene 72%
67 H Ph 13%
68 H 4-methoxybenzene 2%
69 NO2 4-methylbenzene 20%
The NO2 substituent in ortho position to the linker was shifted to meta position.
Sulfone 70 was synthesized according to Moore et al. by an AlCl3 mediated reaction
of benzene and the corresponding sulfonyl chloride in 46% yield (Scheme 8).153
Scheme 8: AlCl3 mediated reaction of benzene and 4-chloro-3-nitrobenzenesulfonyl chloride to 70.
Further, the linker was changed from sulfone or sulfoxide to an ether by synthesizing
71 in 96% yield applying a SNAr of 1,4-dichloro-2-nitrobenzene with phenol
(Scheme 9).
Scheme 9: Synthesis of the biphenylether 71 by nucleophilic aromatic substitution under basic conditions.

Results and Discussion Kif18A and its inhibitor BTB-1
33
Another effort to diversify the linker was to introduce a phosphinic acid residue. 4-
chloro-2-nitroaniline was converted into the iodide 72 using a diazetation-iodination
as described in literature (Scheme 10).154 The following copper mediated reaction of
the iodide with phenylphosphinic acid did not yield the desired small molecule 73
(Scheme 10).155 Only decomposition of the starting material was observed.
Scheme 10: Synthesis of iodide 72 in a one-pot procedure and the subsequent copper mediated reaction to 73.
In another attempt to synthesize 73 according to literature procedures 4-chloro-2-
nitroaniline was first converted into its diazonium salt and afterwards the copper
mediated reaction with dichloro(phenyl)phosphane was carried out
(Scheme 11).156-157 As well as the before mentioned approach this attempt did not
lead to the desired product but only to decomposition of the starting material. Since
at this time point in vitro studies revealed that the sulfone linker is essential for Kif18A
inhibition, as discussed later on (chapter 3.2.4 and 3.2.11), no further attempts to
synthesize 73 were carried out.
Scheme 11: Proposed synthetic rout to 73. The diazonium compound 74 was readily available through diazetation, but the copper mediated reaction to 73 lead only to decomposition of the starting material.

Results and Discussion Kif18A and its inhibitor BTB-1
34
The main focus for the synthesis was not to optimize the synthetic procedures, but
to yield pure substances in a fast manner for evaluation of their biological activity.
Therefore, all compounds were purified by column chromatography and/ or
successive recrystallization resulting in lower yields.
3.2.4 Screening for Kif18A Inhibition and IC50 Determination
With the synthesized BTB-1 analogs 52-71 in hand the next step was to evaluate their
ability to inhibit the ATPase activity of Kif18A. The motor-domain of Kif18A fused to
a His-Tag (Kif18AMD) was used in an enzyme coupled assay to evaluate the potency of
the respective analogs. The ECA conditions were adjusted to obtain 50% inhibition at
5 µM BTB-1 and the ATPase activity in the presence of DMSO was normalized to 100%
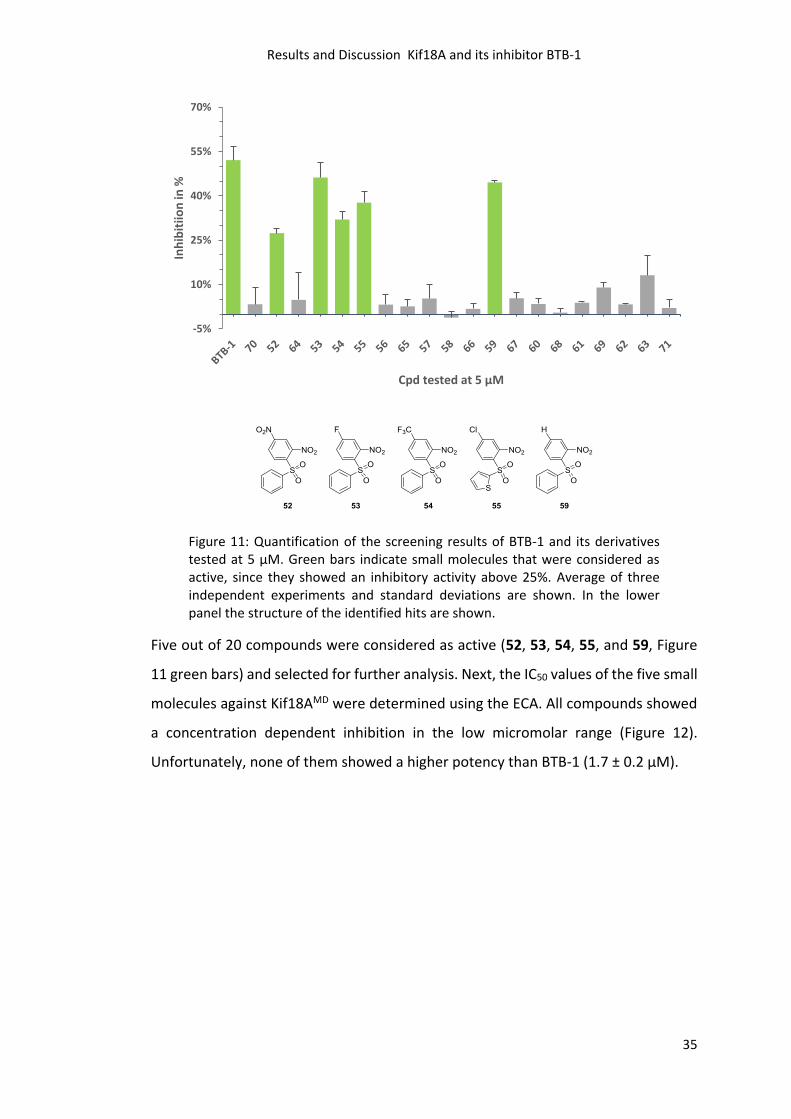
(Figure 11). All compounds that showed higher inhibition than 25% were considered
as active. This selection criteria was chosen in order to identify inhibitors with
comparable properties like BTB-1.
Results and Discussion Kif18A and its inhibitor BTB-1
35
Figure 11: Quantification of the screening results of BTB-1 and its derivatives tested at 5 µM. Green bars indicate small molecules that were considered as active, since they showed an inhibitory activity above 25%. Average of three independent experiments and standard deviations are shown. In the lower panel the structure of the identified hits are shown.
Five out of 20 compounds were considered as active (52, 53, 54, 55, and 59, Figure
11 green bars) and selected for further analysis. Next, the IC50 values of the five small
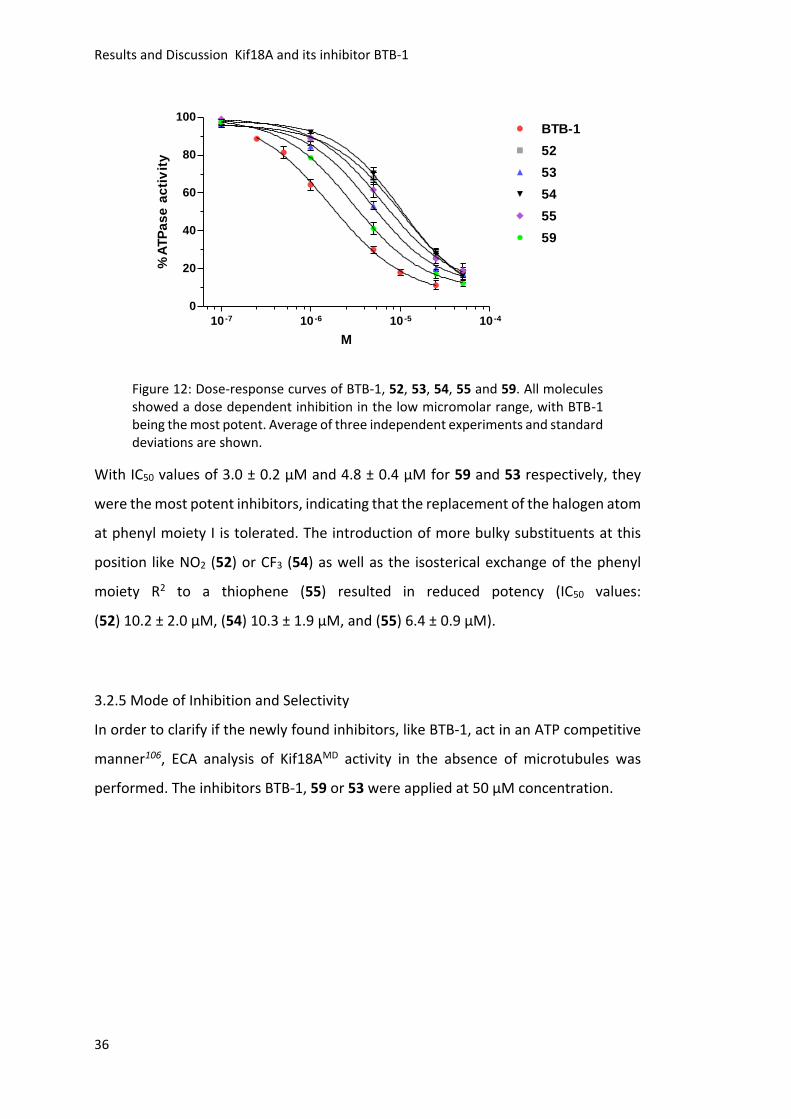
molecules against Kif18AMD were determined using the ECA. All compounds showed
a concentration dependent inhibition in the low micromolar range (Figure 12).
Unfortunately, none of them showed a higher potency than BTB-1 (1.7 ± 0.2 μM).
-5%
10%
25%
40%
55%
70%
Inh
ibit
iion
in %
Cpd tested at 5 µM
Results and Discussion Kif18A and its inhibitor BTB-1
36
10 -7 10 -6 10 -5 10 -4
0
20
40
60
80
100BTB-1
52
53
54
55
59
M
% A
TP
as
e a
cti
vit
y
Figure 12: Dose-response curves of BTB-1, 52, 53, 54, 55 and 59. All molecules showed a dose dependent inhibition in the low micromolar range, with BTB-1 being the most potent. Average of three independent experiments and standard deviations are shown.
With IC50 values of 3.0 ± 0.2 μM and 4.8 ± 0.4 μM for 59 and 53 respectively, they
were the most potent inhibitors, indicating that the replacement of the halogen atom
at phenyl moiety I is tolerated. The introduction of more bulky substituents at this
position like NO2 (52) or CF3 (54) as well as the isosterical exchange of the phenyl
moiety R2 to a thiophene (55) resulted in reduced potency (IC50 values:
(52) 10.2 ± 2.0 μM, (54) 10.3 ± 1.9 μM, and (55) 6.4 ± 0.9 μM).
3.2.5 Mode of Inhibition and Selectivity
In order to clarify if the newly found inhibitors, like BTB-1, act in an ATP competitive
manner106, ECA analysis of Kif18AMD activity in the absence of microtubules was
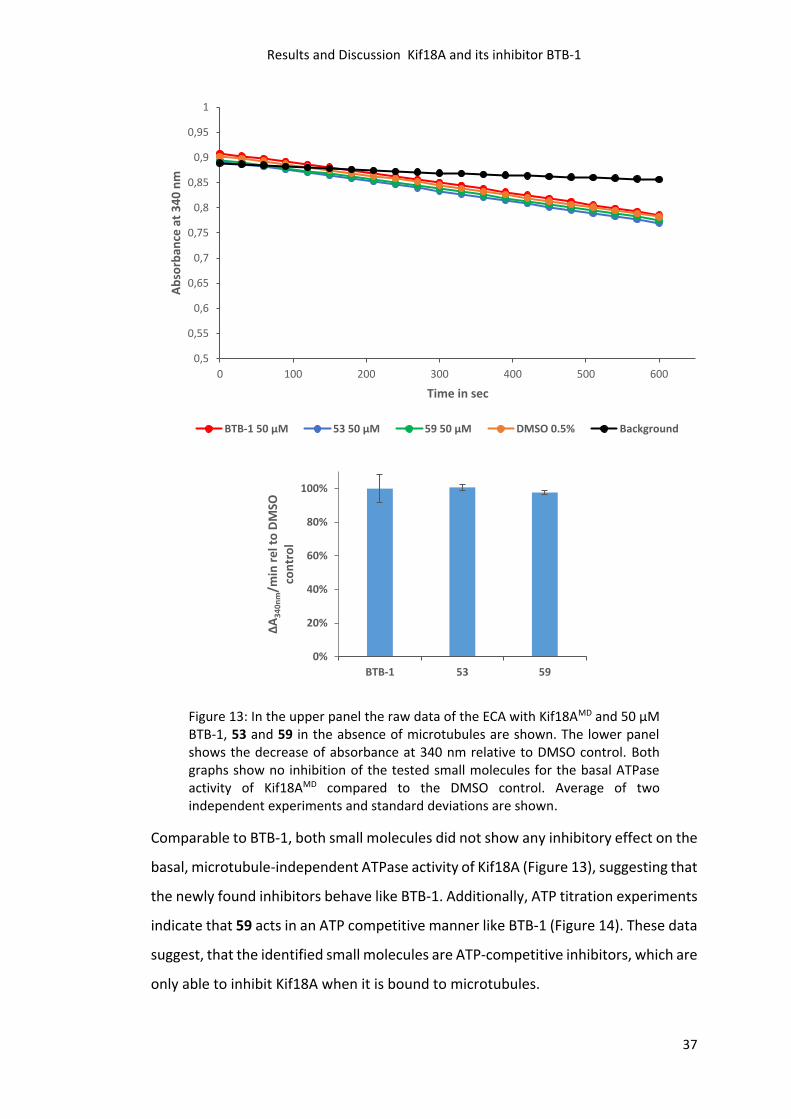
performed. The inhibitors BTB-1, 59 or 53 were applied at 50 µM concentration.
Results and Discussion Kif18A and its inhibitor BTB-1
37
Figure 13: In the upper panel the raw data of the ECA with Kif18AMD and 50 µM BTB-1, 53 and 59 in the absence of microtubules are shown. The lower panel shows the decrease of absorbance at 340 nm relative to DMSO control. Both graphs show no inhibition of the tested small molecules for the basal ATPase activity of Kif18AMD compared to the DMSO control. Average of two independent experiments and standard deviations are shown.
Comparable to BTB-1, both small molecules did not show any inhibitory effect on the
basal, microtubule-independent ATPase activity of Kif18A (Figure 13), suggesting that
the newly found inhibitors behave like BTB-1. Additionally, ATP titration experiments
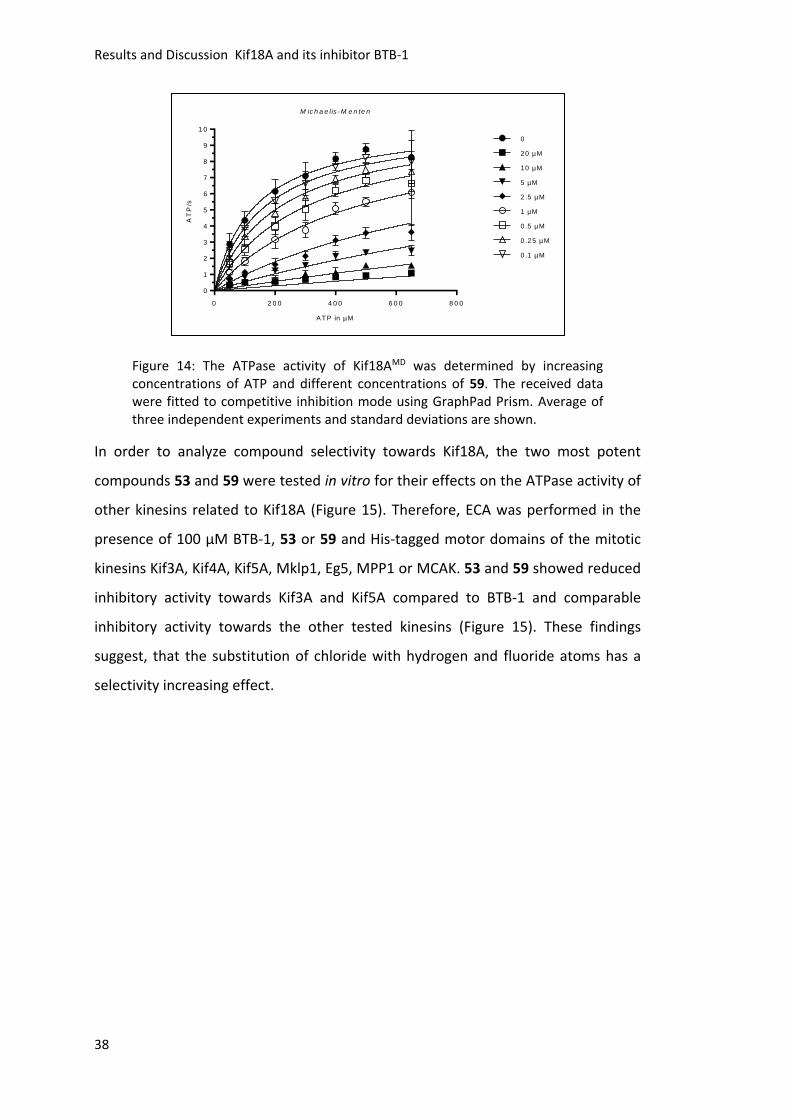
indicate that 59 acts in an ATP competitive manner like BTB-1 (Figure 14). These data
suggest, that the identified small molecules are ATP-competitive inhibitors, which are
only able to inhibit Kif18A when it is bound to microtubules.
0,5
0,55
0,6
0,65
0,7
0,75
0,8
0,85
0,9
0,95
1
0 100 200 300 400 500 600
Ab
sorb
ance
at
34
0 n
m
Time in sec
BTB-1 50 µM 53 50 µM 59 50 µM DMSO 0.5% Background
0%
20%
40%
60%
80%
100%
BTB-1 53 59
ΔA
34
0n
m/m
in r
el t
o D
MSO
co
ntr
ol
Results and Discussion Kif18A and its inhibitor BTB-1
38
0 2 0 0 4 0 0 6 0 0 8 0 0
0
1
2
3
4
5
6
7
8
9
1 0
M ic h a e lis -M e n te n
A TP in µM
AT
P/s
0
20 µM
10 µM
5 µM
2 .5 µM
1 µM
0 .5 µM
0 .2 5 µM
0 .1 µM
Figure 14: The ATPase activity of Kif18AMD was determined by increasing concentrations of ATP and different concentrations of 59. The received data were fitted to competitive inhibition mode using GraphPad Prism. Average of three independent experiments and standard deviations are shown.
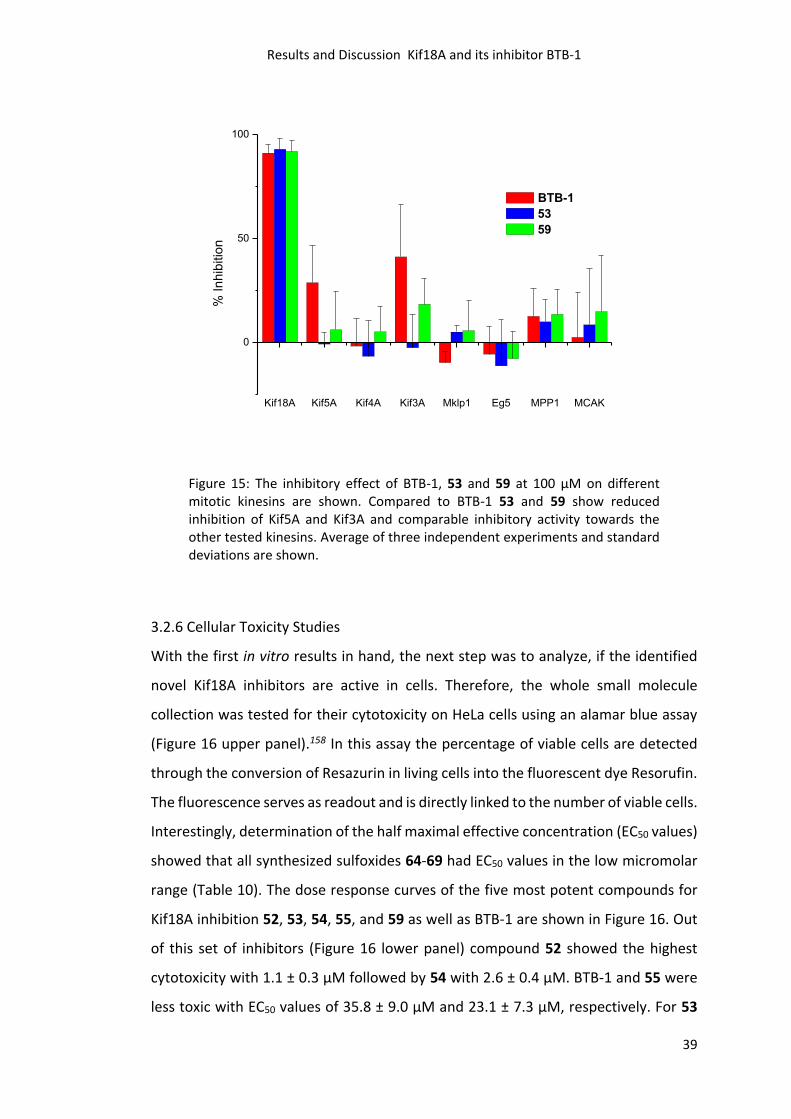
In order to analyze compound selectivity towards Kif18A, the two most potent
compounds 53 and 59 were tested in vitro for their effects on the ATPase activity of
other kinesins related to Kif18A (Figure 15). Therefore, ECA was performed in the
presence of 100 μM BTB-1, 53 or 59 and His-tagged motor domains of the mitotic
kinesins Kif3A, Kif4A, Kif5A, Mklp1, Eg5, MPP1 or MCAK. 53 and 59 showed reduced
inhibitory activity towards Kif3A and Kif5A compared to BTB-1 and comparable
inhibitory activity towards the other tested kinesins (Figure 15). These findings
suggest, that the substitution of chloride with hydrogen and fluoride atoms has a
selectivity increasing effect.
Results and Discussion Kif18A and its inhibitor BTB-1
39
Kif18A Kif5A Kif4A Kif3A Mklp1 Eg5 MPP1 MCAK
0
50
100
% In
hib
itio
n
BTB-1
53
59
Figure 15: The inhibitory effect of BTB-1, 53 and 59 at 100 µM on different mitotic kinesins are shown. Compared to BTB-1 53 and 59 show reduced inhibition of Kif5A and Kif3A and comparable inhibitory activity towards the other tested kinesins. Average of three independent experiments and standard deviations are shown.
3.2.6 Cellular Toxicity Studies
With the first in vitro results in hand, the next step was to analyze, if the identified
novel Kif18A inhibitors are active in cells. Therefore, the whole small molecule
collection was tested for their cytotoxicity on HeLa cells using an alamar blue assay
(Figure 16 upper panel).158 In this assay the percentage of viable cells are detected
through the conversion of Resazurin in living cells into the fluorescent dye Resorufin.
The fluorescence serves as readout and is directly linked to the number of viable cells.
Interestingly, determination of the half maximal effective concentration (EC50 values)
showed that all synthesized sulfoxides 64-69 had EC50 values in the low micromolar
range (Table 10). The dose response curves of the five most potent compounds for
Kif18A inhibition 52, 53, 54, 55, and 59 as well as BTB-1 are shown in Figure 16. Out
of this set of inhibitors (Figure 16 lower panel) compound 52 showed the highest
cytotoxicity with 1.1 ± 0.3 μM followed by 54 with 2.6 ± 0.4 μM. BTB-1 and 55 were
less toxic with EC50 values of 35.8 ± 9.0 μM and 23.1 ± 7.3 μM, respectively. For 53
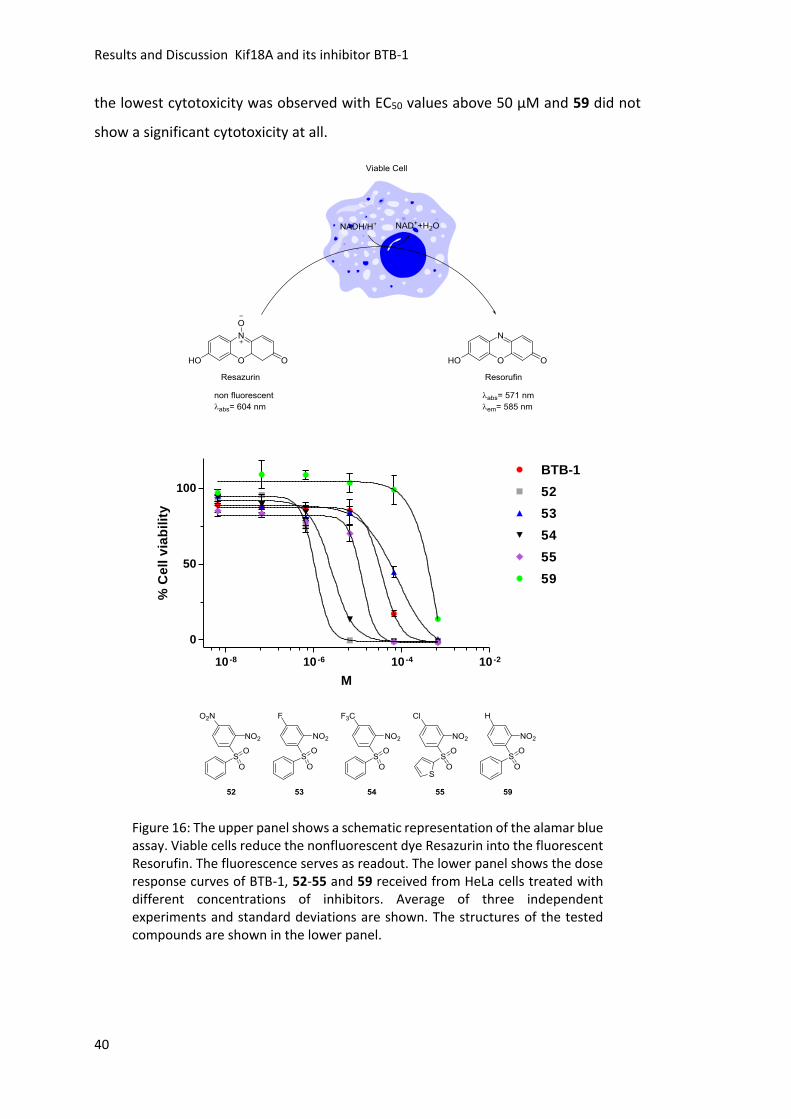
Results and Discussion Kif18A and its inhibitor BTB-1
40
the lowest cytotoxicity was observed with EC50 values above 50 μM and 59 did not
show a significant cytotoxicity at all.
10 -8 10 -6 10 -4 10 -2
0
50
100
BTB-1
52
53
54
55
59
M
% C
ell
via
bil
ity
Figure 16: The upper panel shows a schematic representation of the alamar blue assay. Viable cells reduce the nonfluorescent dye Resazurin into the fluorescent Resorufin. The fluorescence serves as readout. The lower panel shows the dose response curves of BTB-1, 52-55 and 59 received from HeLa cells treated with different concentrations of inhibitors. Average of three independent experiments and standard deviations are shown. The structures of the tested compounds are shown in the lower panel.

Results and Discussion Kif18A and its inhibitor BTB-1
41
Table 10: EC50 values in µM of the BTB-1 analogs received from the alamar blue assay. No cytotoxicity or values above 60 µM are described with “ “.
Cpd. EC50 (µM)
BTB-1 35.8 ± 9
52 1.1 ± 0.3
53 54.7 ± 7.8
54 2.6 ± 0.4
55 23.1 ± 7.3
56 13.8 ± 2.9
57 -
58 36.2 ± 9.3
59 -
60 -
61 2.6 ± 0.4
62 20.8 ± 6.3
63 -
64 1.9 ± 0.2
65 5.5 ± 1.2
66 9.3 ± 2.1
67 20.8 ± 3.0
68 30.1 ± 4.6
69 1.8 ± 0.3
70 9.6 ± 1.3
71 -
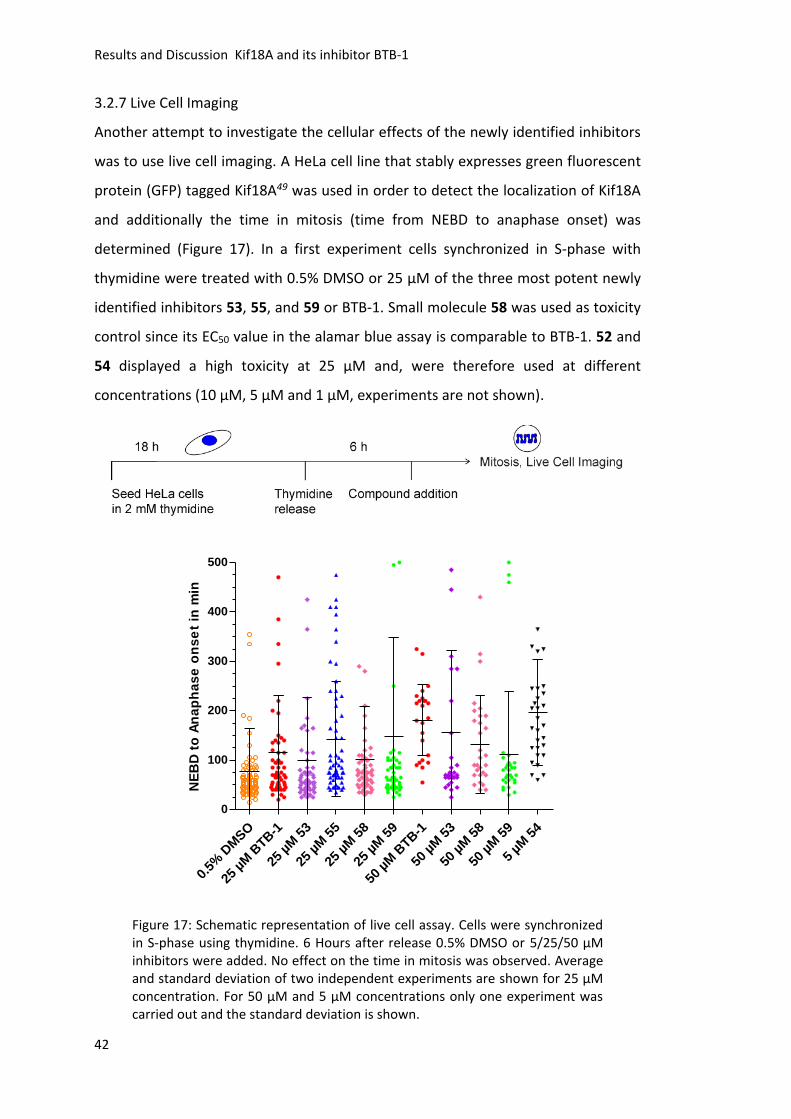
Results and Discussion Kif18A and its inhibitor BTB-1
42
3.2.7 Live Cell Imaging
Another attempt to investigate the cellular effects of the newly identified inhibitors
was to use live cell imaging. A HeLa cell line that stably expresses green fluorescent
protein (GFP) tagged Kif18A49 was used in order to detect the localization of Kif18A
and additionally the time in mitosis (time from NEBD to anaphase onset) was
determined (Figure 17). In a first experiment cells synchronized in S-phase with
thymidine were treated with 0.5% DMSO or 25 µM of the three most potent newly
identified inhibitors 53, 55, and 59 or BTB-1. Small molecule 58 was used as toxicity
control since its EC50 value in the alamar blue assay is comparable to BTB-1. 52 and
54 displayed a high toxicity at 25 µM and, were therefore used at different
concentrations (10 µM, 5 µM and 1 µM, experiments are not shown).
0.5%
DM
SO
25 µ
M B
TB-1
25 µ
M 5
3
25 µ
M 5
5
25 µ
M 5
8
25 µ
M 5
9
50 µ
M B
TB-1
50 µ
M 5
3
50 µ
M 5
8
50 µ
M 5
9
5 µM
54
0
100
200
300
400
500
NE
BD
to
An
ap
ha
se
on
se
t in
min
Figure 17: Schematic representation of live cell assay. Cells were synchronized in S-phase using thymidine. 6 Hours after release 0.5% DMSO or 5/25/50 µM inhibitors were added. No effect on the time in mitosis was observed. Average and standard deviation of two independent experiments are shown for 25 µM concentration. For 50 µM and 5 µM concentrations only one experiment was carried out and the standard deviation is shown.

Results and Discussion Kif18A and its inhibitor BTB-1
43
Nevertheless, none of the small molecules interfered with the localization of Kif18A
and no significant increase in time in mitosis compared to the DMSO control was
detected (Figure 17). The experiment was repeated using 50 µM inhibitor
concentration, except for 54 and 52, where 5 µM was applied. Due to toxic effects of
55 at 50 µM and 52 at 5 µM concentration both molecules were excluded from
analysis. Even the higher concentrations of the inhibitors did not result in a
detectable delocalization of Kif18A. Despite that, 54 led to an increased time in
mitosis at 5 µM concentration compared to the DMSO control (Figure 17). BTB-1
showed a slight increase in mitotic timing (Figure 17). Since no effect on the Kif18A
localization was observed, this experiment was only carried out once. In summary,
the inhibitors are not efficient enough to inhibit Kif18A in cells, since the localization
of Kif18A was not altered upon compound treatment.
3.2.8 Cellular Thermal Shift Assay
As another approach to elucidate the cellular effects of BTB-1 and the most promising
inhibitor 53 a cellular thermal shift assay (CETSA) was performed. In this assay cell
lysate is applied and therefore the small molecules do not have to pass the cell
membrane.159 This assays relies on the thermal stabilization of the target protein
upon Inhibitor binding. Mitotic HeLa cell extract was prepared and treated with 0.5%
DMSO as solvent control or compounds. After incubation and thermal treatment the
samples were analyzed by SDS-PAGE followed by western blot analysis. First, the
temperature at which Kif18A denatures was determined in the presence of 100 µM
BTB-1, 53 or 0.5% DMSO. Unfortunately, no stabilization of Kif18A due to inhibitor
addition was observed. No intensity differences of the samples were detectable
(Figure 18). The DMSO control revealed that Kif18A is destabilized at temperatures
between 50°-54°C (Intensity decrease, Figure 18) and therefore this temperature
range was used to evaluate if higher inhibitor concentrations are able to stabilize
Kif18A.

Results and Discussion Kif18A and its inhibitor BTB-1
44
Figure 18: Western blot analysis of temperature gradient from 46°C to 62°C in the presence of 0.5% DMSO, 100 µM BTB-1 or 53. In line 10 (DMSO control 52°C) was less lysate loaded. No stabilization of Kif18A was observed. CycB, Cdc27 and tubulin served as mitotic markers and loading controls.
BTB-1 and 53 were applied in 500 µM, 200 µM, 100 µM and 50 µM concentration,
but still no stabilization of Kif18A was observed (Figure 19). Since BTB-1 as well as 53
are not able to stabilize Kif18A even at concentrations up to 500 µM, it is possible
that these inhibitors are not able to bind efficiently to Kif18A in a cellular
environment.
Figure 19: Western blot analysis of compound titration of BTB-1 and 53 from 50°C to 54°C. The first and last line was loaded with random lysate. No stabilization of Kif18A was observed. CycB, Cdc27 and tubulin served as mitotic markers and loading control.

Results and Discussion Kif18A and its inhibitor BTB-1
45
3.2.9 Immunofluorescence Imaging for Kif18A Localization
In order to analyze in detail the effect of BTB-1 and derivatives 53, 54, and 59 on cells
and the architecture of the mitotic spindle, immunofluorescence imaging was
performed. The same HeLa cell line as described for live cell imaging (chapter 3.2.7)
was used.49 Cells synchronized in S-phase using a high concentration of thymidine
were released after nine hours and then treated with the solvent control DMSO or
30/50 µM of BTB-1 or derivatives 53, 54, and 59 (Figure 20). Thirty minutes after
compound addition, cells were chemically fixed and tubulin (red) and DNA (blue)
structures were visualized by immunostaining and treatment with the dye Hoechst
33342, respectively. Since initial immunofluorescence analysis of derivative 52
revealed that this small molecule caused unspecific cytotoxic effects on both dividing
and non-dividing cells, it was excluded from further microscopic studies. The
immunofluorescence images revealed that neither 53 nor 59 significantly affected
spindle structures or alignment of chromosomes at 30/50 µM concentration
(Figure 20, only 50 µM is shown). Additionally, the plus-end accumulation of
GFP-Kif18A at microtubules – which as shown previously depends on the motor
activity of Kif18A44-45 – was not affected by the treatment with 53 or 59.
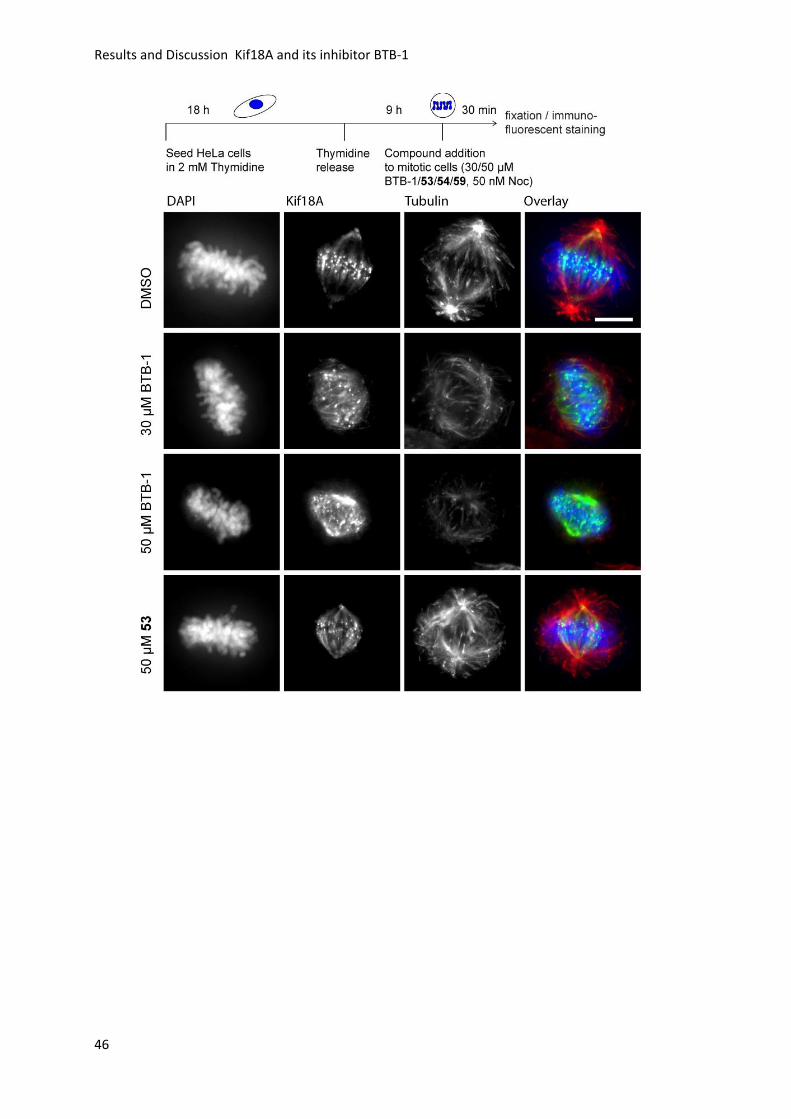
Results and Discussion Kif18A and its inhibitor BTB-1
46
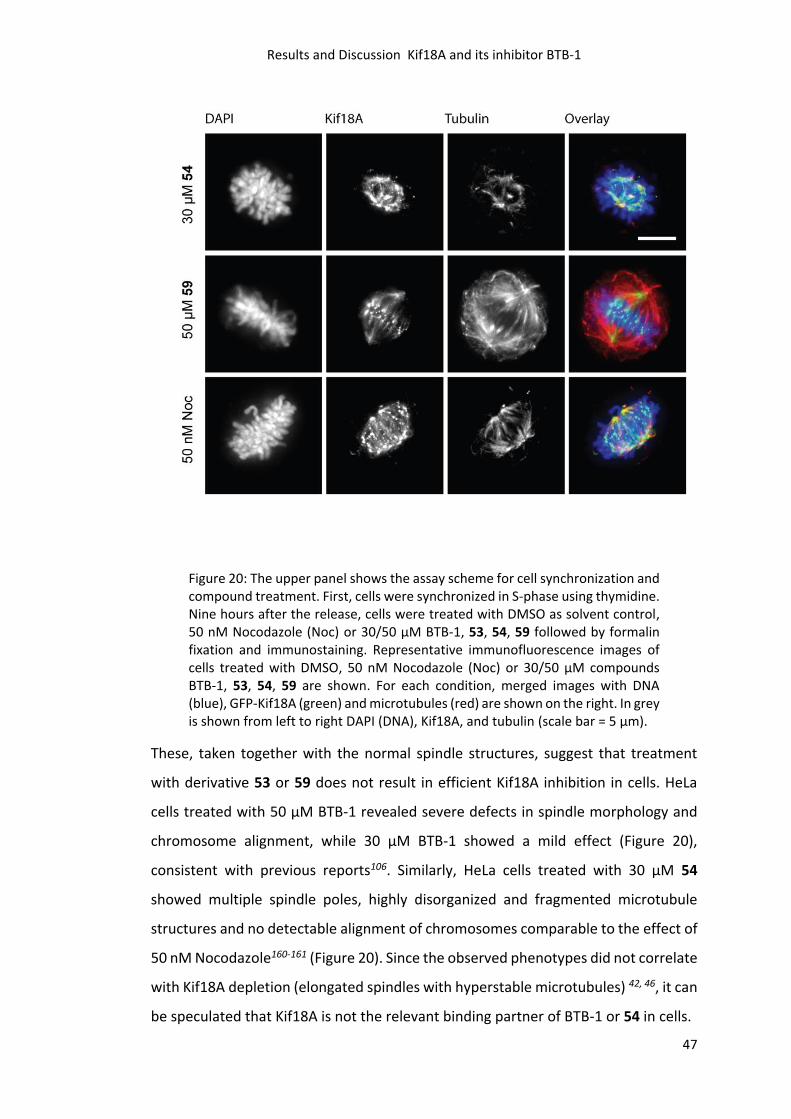
Results and Discussion Kif18A and its inhibitor BTB-1
47
Figure 20: The upper panel shows the assay scheme for cell synchronization and compound treatment. First, cells were synchronized in S-phase using thymidine. Nine hours after the release, cells were treated with DMSO as solvent control, 50 nM Nocodazole (Noc) or 30/50 µM BTB-1, 53, 54, 59 followed by formalin fixation and immunostaining. Representative immunofluorescence images of cells treated with DMSO, 50 nM Nocodazole (Noc) or 30/50 µM compounds BTB-1, 53, 54, 59 are shown. For each condition, merged images with DNA (blue), GFP-Kif18A (green) and microtubules (red) are shown on the right. In grey is shown from left to right DAPI (DNA), Kif18A, and tubulin (scale bar = 5 µm).
These, taken together with the normal spindle structures, suggest that treatment
with derivative 53 or 59 does not result in efficient Kif18A inhibition in cells. HeLa
cells treated with 50 µM BTB-1 revealed severe defects in spindle morphology and
chromosome alignment, while 30 µM BTB-1 showed a mild effect (Figure 20),
consistent with previous reports106. Similarly, HeLa cells treated with 30 µM 54
showed multiple spindle poles, highly disorganized and fragmented microtubule
structures and no detectable alignment of chromosomes comparable to the effect of
50 nM Nocodazole160-161 (Figure 20). Since the observed phenotypes did not correlate
with Kif18A depletion (elongated spindles with hyperstable microtubules) 42, 46, it can
be speculated that Kif18A is not the relevant binding partner of BTB-1 or 54 in cells.

Results and Discussion Kif18A and its inhibitor BTB-1
48
3.2.10 Tubulin Polymerization Assay and IC50 Determination
The spindle phenotype induced by 54 was reminiscent of the phenotype caused by
low doses of Nocodazole, a microtubule destabilizing compound (Figure 20). To
analyze if the small molecule collection or BTB-1 target microtubules, a
turbidity-based in vitro microtubule polymerization assay was performed.162-163 The
ability of tubulin to polymerize in the presence of GTP at elevated temperatures in a
glutamate buffer and a turbidity based readout was used (Figure 21).
Figure 21: Schematic representation of tubulin polymerization. α-(light green) and β-(dark green or brown) tubulin heterodimers polymerize to microtubules. After GTP hydrolysis the destabilized plus-end depolymerizes. Upon exchange of GDP to GTP in the tubulin dimers repolymerization occurs.
α- and β- tubulin heterodimers are able to polymerize in the presence of GTP. The
GTP cap consists of tubulin dimers in the GTP bound state and stabilizes the highly
dynamic plus-ends of microtubules.164-166 After GTP hydrolysis, depolymerization,
often called catastrophe, takes place. This process can be stopped by addition of
GTP-bound tubulin dimers. After a certain time, the polymerization and
depolymerization processes reach a dynamic equilibrium as long as enough GTP is
present. The microtubule polymerization efficiency in the presence of the solvent
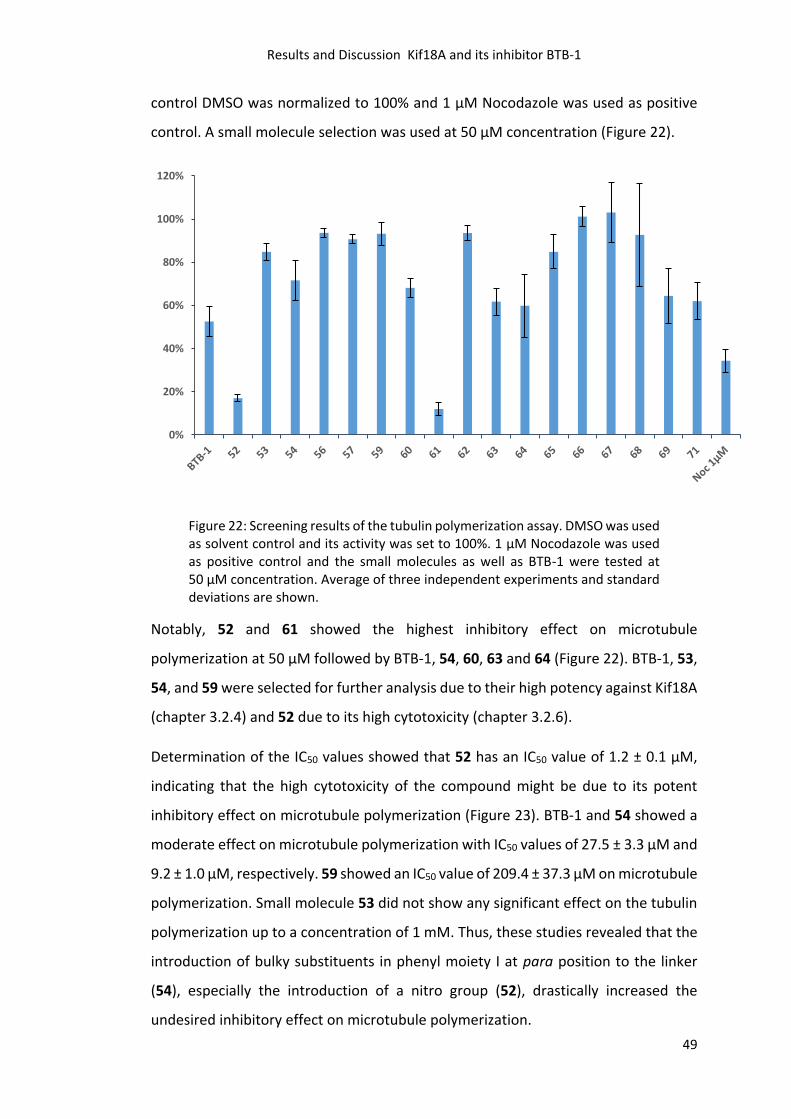
Results and Discussion Kif18A and its inhibitor BTB-1
49
control DMSO was normalized to 100% and 1 µM Nocodazole was used as positive
control. A small molecule selection was used at 50 µM concentration (Figure 22).
Figure 22: Screening results of the tubulin polymerization assay. DMSO was used as solvent control and its activity was set to 100%. 1 µM Nocodazole was used as positive control and the small molecules as well as BTB-1 were tested at 50 µM concentration. Average of three independent experiments and standard deviations are shown.
Notably, 52 and 61 showed the highest inhibitory effect on microtubule
polymerization at 50 µM followed by BTB-1, 54, 60, 63 and 64 (Figure 22). BTB-1, 53,
54, and 59 were selected for further analysis due to their high potency against Kif18A
(chapter 3.2.4) and 52 due to its high cytotoxicity (chapter 3.2.6).
Determination of the IC50 values showed that 52 has an IC50 value of 1.2 ± 0.1 µM,
indicating that the high cytotoxicity of the compound might be due to its potent
inhibitory effect on microtubule polymerization (Figure 23). BTB-1 and 54 showed a
moderate effect on microtubule polymerization with IC50 values of 27.5 ± 3.3 µM and
9.2 ± 1.0 µM, respectively. 59 showed an IC50 value of 209.4 ± 37.3 µM on microtubule
polymerization. Small molecule 53 did not show any significant effect on the tubulin
polymerization up to a concentration of 1 mM. Thus, these studies revealed that the
introduction of bulky substituents in phenyl moiety I at para position to the linker
(54), especially the introduction of a nitro group (52), drastically increased the
undesired inhibitory effect on microtubule polymerization.
0%
20%
40%
60%
80%
100%
120%
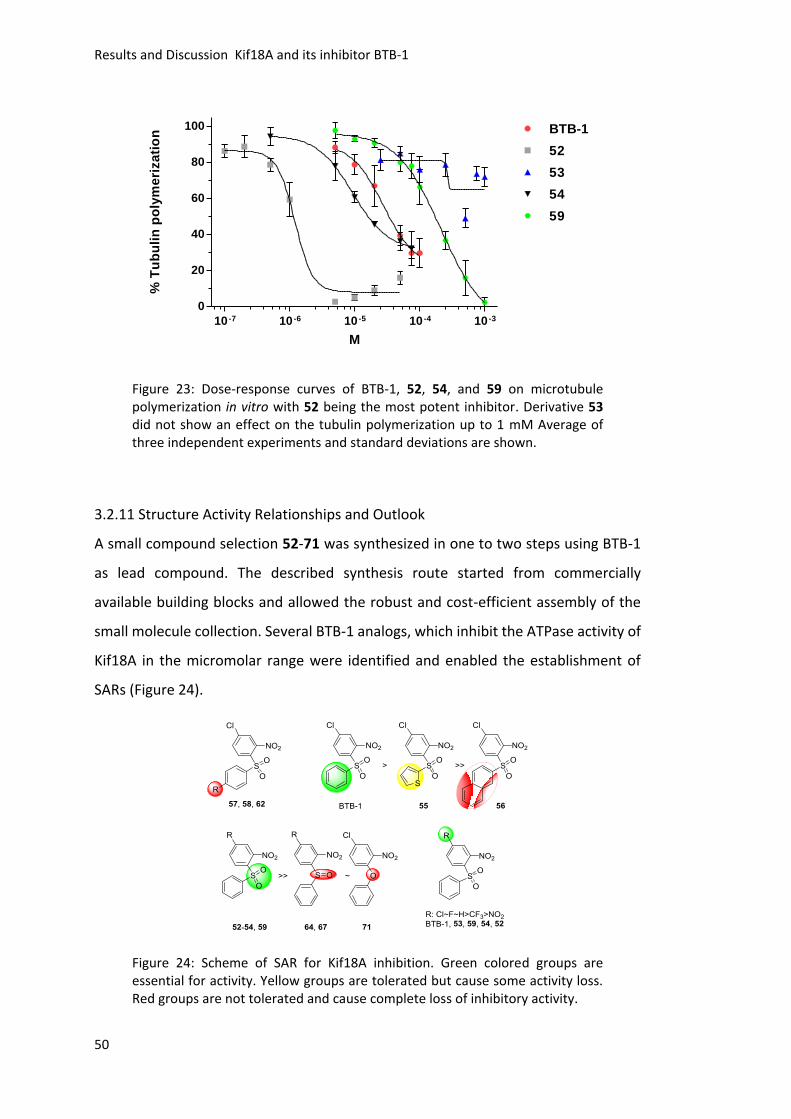
Results and Discussion Kif18A and its inhibitor BTB-1
50
10 -7 10 -6 10 -5 10 -4 10 -3
0
20
40
60
80
100 BTB-1
52
53
54
59
M
% T
ub
uli
n p
oly
me
riza
tio
n
Figure 23: Dose-response curves of BTB-1, 52, 54, and 59 on microtubule polymerization in vitro with 52 being the most potent inhibitor. Derivative 53 did not show an effect on the tubulin polymerization up to 1 mM Average of three independent experiments and standard deviations are shown.
3.2.11 Structure Activity Relationships and Outlook
A small compound selection 52-71 was synthesized in one to two steps using BTB-1
as lead compound. The described synthesis route started from commercially
available building blocks and allowed the robust and cost-efficient assembly of the
small molecule collection. Several BTB-1 analogs, which inhibit the ATPase activity of
Kif18A in the micromolar range were identified and enabled the establishment of
SARs (Figure 24).
Figure 24: Scheme of SAR for Kif18A inhibition. Green colored groups are essential for activity. Yellow groups are tolerated but cause some activity loss. Red groups are not tolerated and cause complete loss of inhibitory activity.

Results and Discussion Kif18A and its inhibitor BTB-1
51
Comparison of identically substituted sulfone (52 and 59) and sulfoxide (64 and 67)
derivatives highlight the importance of the sulfone functionality for the inhibitory
activity of the compounds against Kif18A (Figure 24). Consistently, the replacement
of the sulfone linker of BTB-1 with an ether linkage (71) resulted in an almost
complete loss of inhibitory activity (Figure 24). Additional SAR studies revealed that
the introduction of substituents in para position to the linker at phenyl moiety II was
not tolerated whether they were electron withdrawing (62 and 63) or donating
(57, 58, 66, 60, 61, 68 and 69). Likewise, the sterical expansion of the aromatic
phenyl moiety II by replacing it with naphthalene (56) resulted in a complete loss of
activity (Figure 24). Shifting the NO2 group from ortho (BTB-1) to meta (70) position
to the sulfone linker within phenyl moiety I caused compound inactivity. In contrast,
the replacement of chloride (BTB-1) at R1 of phenyl moiety I with hydrogen (59) or
different electron withdrawing groups like fluoride (53), NO2 (52), or CF3 (54) showed
no drastic effect on compound activity (Figure 24). Since 53 and 59 – the most potent
derivatives – did not interfere with the localization of Kif18A to the plus-ends of
microtubules in mitotic cells, it might be that treatment with these small molecule
inhibitors did not result in efficient Kif18A inhibition. One possible explanation could
be that the compounds 53 and 59 are not cell membrane permeable, but the cellular
effects received from structurally related compounds BTB-1 and 54 argues against
this idea. Another possibility could be that association of Kif18A with binding partners
or posttranslational modifications render Kif18A resistant to the binding of 53 and
59. Remarkably, BTB-1 and 54 revealed a cellular phenotype reminiscent of low doses
of Nocodazole leading to the conclusion that they might interact with tubulin.
Additional in vitro studies confirmed that these compounds potently inhibited
microtubule polymerization (Figure 25). For BTB-1 and 54 it is difficult to determine
whether the altered Kif18A localization in cells treated with these compounds is due
to their direct interaction with Kif18A or caused by the undesired effect on
microtubule dynamics, which per se has an influence on Kif18A plus–end
accumulation at microtubules45. The in vitro microtubule polymerization assays
revealed 52 as the most potent inhibitor (Figure 25). Therefore, it is likely that the
high cytotoxicity of 52 is caused by its severe effect on microtubule polymerization.

Results and Discussion Kif18A and its inhibitor BTB-1
52
Figure 25: Results of the SAR studies. The upper panel shows molecules sorted based on their inhibitory effect on microtubule polymerization, decreasing activity from left to right with 52 being the most potent inhibitor for microtubule polymerization. In the lower panel, compounds are sorted based on their inhibitory activity towards Kif18A with the most potent compound BTB-1 shown on the right side.
All these data suggest the introduction of electron withdrawing sterically demanding
groups like NO2 (52) or CF3 (54) at R1 of phenyl moiety I shift the inhibitory activity
towards microtubules as well as the combination of a NO2 group at R1 and a methyl
group at the para position of phenyl moiety II (61). In contrast to that the introduction
of small substituents at R1 like hydrogen (59) or fluoride (53) influence the activity in
favor of Kif18A inhibition (Figure 25).
The insights of the SAR studies might provide an important guideline for future design
and synthesis of more potent Kif18A inhibitors. In order to establish even more
powerful SARs the exact binding of BTB-1 and its analogs to Kif18A has to be resolved.
One possibility to achieve this would be the crystallization of Kif18A with bound
inhibitor. The structure would reveal the interactions of the small molecules with the
amino acids of Kif18A and allow the design of inhibitors that fit to the binding surface.
Such compounds would be invaluable tools for basic research, because Kif18A seems
to have different functions at different times during mitosis and small molecules
could be applied at specific time points. Further, the inhibitor would have the

Results and Discussion Kif18A and its inhibitor BTB-1
53
potential to open up new strategies in the treatment of mitosis-related diseases such
as Kif18A overexpressing tumors.

Summary
54
4. Summary
Kinesins are essential for the correct distribution of the genetic material into the
daughter cells during mitosis.4 The functional dissection of individual kinesins
involved in chromosome segregation is a challenging task, because of the complexity
and dynamicity of mitosis. Due to their fast and often reversible mode of action,
membrane permeable small molecules inhibitors are ideal tools to dissect the
different functions of kinesins.
Paprotrain was discovered in 2010 by Tcherniuk et al.109 as inhibitor for the kinesin
Mklp2, whereas a small molecule named SH1, a 5-chloro-2-(3,4-dichlorophenyl)-7-
(trifluoromethyl)quinoxaline, was identified and characterized in the laboratory of
Prof. Thomas U. Mayer in 2009 122. For the design of new Mklp2 inhibitors, SH1 served
as the lead compound. The main drawback of SH1 was its low solubility in aqueous
media. Thus, the aim was to develop new derivatives with enhanced solubility. After
the establishment of a synthetic pathway towards SH1 using glyoxales, several
compounds bearing polar substituents (7-26) as well as their precursors (1-6) were
synthesized (chapter 3.1.3). In a second approach to enhance the water solubility the
phenyl moiety of SH1 was exchanged to a thiophene (27-40, chapter 3.1.4).134
Moreover, SH1 analogs linked to an enzymatic cleavable carbohydrate residue were
synthesized (47-51, chapter 3.1.5).138 All different synthetic approaches to enhance
the water solubility of SH1 derivatives were successful. Characterization of the
biological activity of the synthesized compounds revealed that only compounds
barely soluble in aqueous media were active (chapter 3.1.6). Furthermore, addition
of the detergent Triton X-100 suppressed the inhibitory activity of SH1 towards Mklp2
in vitro. Collectively, these results suggest, that aggregation of SH1 and its derivatives
is necessary for their inhibitory activity (chapter 3.1.7 and 3.1.8).
For the kinesin Kif18A, a small molecule named BTB-1 was identified by Catarinella et
al. in 2009 106 as a potent inhibitor. BTB-1 inhibited the ATPase activity of Kif18A in
vitro and led to an increased percentage of cells in mitosis upon cellular treatment.
Nevertheless, cells treated with high concentration of BTB-1 did not show elongated
spindles that are characteristic for the depletion of Kif18A. Here, the aim was to
establish a synthetic pathway in order to synthesize a small molecule collection of

Summary
55
BTB-1 analogs. For the synthesis of the small molecule collection, the main reaction
utilized was a nucleophilic aromatic substitution under basic conditions followed by
an oxidation (52-63 and 64-69, chapter 3.2.3). The screening for Kif18A inhibition
identified five active compounds (52-55 and 59), which showed IC50 values in the low
micromolar range (<10 µM, chapter 3.2.4). Further, the selectivity of the newly found
two most potent inhibitors (53 and 59) towards Kif18A was investigated. Both
showed a higher selectivity towards Kif18A compared to BTB-1 (chapter 3.2.5). Next,
the cellular effects of the novel small molecules were investigated. The cellular
cytotoxicity was determined, followed by studies to detect Kif18A localization in cells
(chapters 3.2.6-3.2.8). Fluorescence imaging revealed that the newly identified
inhibitors did not alter the localization of Kif18A to the plus-ends of microtubules.
They might be not efficient enough to inhibit Kif18A in cells, due to binding partners
or posttranslational modifications of Kif18A. Instead some compounds showed a
severe effect on the microtubule network comparable to low doses of the
microtubule poison Nocodazole (BTB-1 and 54, chapter 3.2.9). An in vitro microtubule
polymerization assay confirmed that some compounds interfered with microtubule
polymerization (BTB-1, 52 and 54, chapter 3.2.10). Most importantly, the undesired
effect on microtubule polymerization was separated from the desired Kif18A
inhibition with the help of in-depth SAR studies (chapter 3.2.11). The insights of the
SAR studies provide a powerful guideline for future design and synthesis of highly
potent Kif18A inhibitors, which would be invaluable tools for basic research and could
open up new strategies in the development of novel drugs for mitosis-related
diseases, like Kif18A overexpressing tumors.

Zusammenfassung
56
5. Zusammenfassung
Mitotische Kinesine sind wichtig für die korrekte Verteilung des genetischen
Materials in die beiden Tochterzellen während der Mitose.4 Sie haben verschiedene
Funktionen zu unterschiedlichen Zeitpunkten des Zellzyklus. Spezifische
niedermolekulare Inhibitoren sind daher ideale Werkzeuge, um die verschiedenen
Funktionen der Kinesine zu analysieren, da sie in einer sehr schnellen und oft
reversiblen Art wirken.
Paprotrain wurde 2010 von Tcherniuk et al.109 als Inhibitor für das Kinesin Mklp2
entdeckt, während die niedermolekulare Substanz SH1, ein 5-Chlor-2-(3,4-
dichlorphenyl)-7-(trifluormethyl)chinoxalin, im Labor von Prof. Thomas U. Mayer im
Jahr 2009122 identifiziert und charakterisiert wurde. Für das Design neuer Mklp2
Inhibitoren diente SH1 als Leitstruktur. Der größte Nachteil von SH1 liegt in seiner
schlechten Wasserlöslichkeit, weshalb neue Inhibitoren mit erhöhter
Wasserlöslichkeit synthetisiert werden sollten. Nach der Etablierung eines
Synthesewegs zur Darstellung von SH1 mittels Glyoxalen, wurden verschiedene
Verbindungen mit polaren Substituenten (7-26) sowie deren Vorstufen (1-6)
hergestellt (Kapitel 3.1.3). In einem weiteren Ansatz, um die Löslichkeit zu erhöhen,
wurde der Phenylrest von SH1 durch Thiophen ersetzt (27-40, Kapitel 3.1.4).134
Außerdem wurden SH1 Analoga verbunden mit einem enzymatisch spaltbaren
Kohlenhydratrest synthetisiert (47-51, Kapitel 3.1.5).138 Die verschiedenen
strukturellen Modifikationen von SH1 führten erfolgreich zur Erhöhung der
Wasserlöslichkeit. Die Charakterisierung der biologischen Aktivität zeigte jedoch,
dass nur in wässrigen Medien schwer lösliche Verbindungen, aktiv waren
(Kapitel 3.1.6). In in vitro Untersuchungen mit Triton X-100 wies SH1 keine Inhibition
von Mklp2 auf, was zu dem Schluss führt, dass die Aggregation von SH1 notwendig
für seine hemmende Aktivität ist (Kapitel 3.1.7 und 3.1.8).
Für das Kinesin Kif18A wurde ein niedermolekulares Molekül namens BTB-1 als
potenter Inhibitor von Catarinella et al. 2009106 identifiziert. BTB-1 inhibierte die
ATPase-Aktivität von Kif18A in vitro und führte bei zellulärer Anwendung zu einer
Erhöhung der Anzahl mitotischer Zellen.106 Dennoch wiesen Zellen, die mit einer
hohen Konzentration von BTB-1 behandelt wurden, keine verlängerten Spindeln auf,

Zusammenfassung
57
die charakteristisch für die Depletion von Kif18A sind. Daher sollte ein Syntheseweg
etabliert werden, um eine Sammlung von BTB-1-Analoga zu synthetisieren und
Struktur-Aktivitäts-Beziehungen aufzustellen. Zur Synthese der BTB-1 Analoga wurde
als Hauptreaktion eine nukleophile aromatische Substitution unter basischen
Bedingungen verwendet, gefolgt von einer Oxidation (52-63 und 64-69,
Kapitel 3.2.3). Im Screening zur Kif18A Inhibition wurden fünf aktive Verbindungen
(52-55 und 59) identifiziert, die IC50-Werte im niedrigen mikromolaren Bereich
zeigten (<10 µM, Kapitel 3.2.4). Ferner wurde die Selektivität der zwei potentesten
Inhibitoren (54 und 60) gegenüber Kif18A im Vergleich zu anderen mitotischen
Kinesinen untersucht und beide zeigten eine erhöhte Selektivität gegenüber Kif18A
im Vergleich zu BTB-1 (Kapitel 3.2.5). Als nächstes wurden die zellulären Effekte der
neuen Inhibitoren untersucht. Zuerst wurde die Zytotoxizität bestimmt, gefolgt von
Studien zur Lokalisation von Kif18A in Zellen (Kapitel 3.2.6-3.2.8).
Fluoreszenz-Bildgebung zeigte, dass die neu identifizierten Inhibitoren nicht die
Lokalisierung von Kif18A zu den Plus-Enden der Mikrotubuli veränderten. Trotz der
Inhibition der ATPase-Aktivität von Kif18A in vitro sind die neu identifizierten
Inhibitoren möglicherweise nicht effizient genug, um Kif18A in Zellen zu hemmen.
Dies könnte durch mögliche Bindungspartner oder posttranslationale Modifikationen
von Kif18A verursacht werden. Stattdessen zeigten einige Verbindungen eine starke
Wirkung auf das Mikrotubuli-Netzwerk, vergleichbar mit niedrigen Dosen des
Spindelgifts Nocodazol (BTB-1 und 54, Kapitel 3.2.9). Ein in vitro
Mikrotubuli-Polymerisationsversuch bestätigte, dass einige Verbindungen die
Mikrotubuli-Polymerisation störten (BTB-1, 52 und 54, Kapitel 3.2.10). Jedoch wurde
mit Hilfe intensiver Struktur-Aktivitäts-Studien die unerwünschte Wirkung auf die
Mikrotubuli-Polymerisation von der gewünschten Kif18A Inhibition getrennt (Kapitel
3.2.11). Die Erkenntnisse der Struktur-Aktivitäts-Studien bieten einen sehr guten
Leitfaden für das zukünftige Design und die Synthese von hochwirksamen
Kif18A Inhibitoren, die nicht nur wertvolle Werkzeuge für die Grundlagenforschung
wären, sondern auch neue Strategien in der Entwicklung innovativer Medikamente
für Mitose-Erkrankungen, wie Kif18A überexprimierende Tumoren, darstellen
könnten.104-105

Materials and Methods
58
6. Materials and Methods
6.1 General
Chemicals, reagents and solvents
All solvents, reagents and fine chemicals are commercially available (Sigma‐Aldrich,
Acros, Merck, Fluka, Roth, TCI, MCAT, ABCR, or Fluorochem) and are used without
further purification. The used petroleum ether (PE) had a boiling point range of
35-80°C.
NMR‐Spectroscopy
1H‐, 13C‐ and 19F‐NMR spectra were recorded either on a Bruker Avance III 400 or
Bruker Avance 600 spectrometer in the NMR facility of the University of Konstanz.
Chemical shifts are given in the δ‐scale and referenced to residual non deuterated
solvent. A BBFOplus probe with actively shielded z‐gradient was used with its inner
(BB‐) coil tuned to 19F. The following abbreviations were used for spin‐systems:
s (singlet); d (doublet); t (triplet); q (quartet). In case of multiple splittings, identifiers
in the fashion of dt (doublet of a triplet) were used or m for undistinguishable
multiplets. The spectra were processed using MestReNova.
Elementary Analysis
The elementary analysis was performed by the University of Konstanz micro
analytical laboratory.
Melting Points
Melting points were measured on a Gallenkamp Melting Point Apparatus and are
uncorrected.
Chromatography
For TLC, pre‐coated plastic sheets with fluorescence indicator Polygram Sil G/UV254
by Macherey‐Nagel with a coating thickness of 0.2 mm were used. The substances
were detected under 254 or 366 nm light. Flash column chromatography
(200 - 300 mbar) was performed on Macherey‐Nagel silica gel 60 M (particle size:
40 - 63 μm). Technical grade solvents were distilled prior to use.

Materials and Methods
59
6.2 Synthesis of SH1 Analogs
General Methods for Glyoxal Synthesis (Method A)
The glyoxales were prepaired according to literature procedures. 131-133 The
corresponding acetophenone and 1.5 equivalents (equiv.) of selenium (IV) oxide
(SeO2) were heated to reflux in dioxane / water mixture (1 / 0.03) overnight. The
formed black Se precipitate was removed by filtration and the solution was
evaporated to dryness. The received solid was suspended in water, heated to reflux
and the solvent was removed. The subsequent purification is described for each
compound.
General Method for Preparation of Substituted Quinoxalines (Method B)
The corresponding phenylenediamine and glyoxal in equimolar stoichiometry were
dissolved in EtOH and heated to reflux. Upon cooling the formed precipitate was
collected and further purified as indicated.
General Method for Preparation of Amide and Ester Analogs of SH1 (Method C)
First, the corresponding quinoxaline-6-carboxylic acid is converted into its acid
chloride by dissolving it in an excess of thionylchloride (25 mL) and heating to reflux.
After removing the solvent, the residue was suspended in CH2Cl2 (25 mL) and cooled
to 0°C. First 1.2 equiv. Et3N followed by slow addition of 1.1 equiv. amine/alcohol.
The reaction mixture was allowed to warm to ambient temperature after complete
addition. If not otherwise mentioned water (20 mL) was added and the aqueous
phase was extracted two times with CH2Cl2 (20 mL). The combined organic layers
were washed with water, dried over MgSO4 and the solvent was removed. Further
purification was carried out as mentioned for the corresponding compound.

Materials and Methods
60
3,4-Dichlorophenylglyoxal (1)
C8H6Cl2O3
M: 221.04 g mol-1
3,4-Dichloroacetophenone (2.0 g, 10.6 mmol) was converted into the glyoxal
according to Method A. After evaporation of the water the white precipitate was
recrystallized from benzene to yield 3,4-dichlorophenylglyoxal monohydrate as white
crystals (1.28 g, 40.4 mmol, 55% yield).
1H NMR (400 MHz, DMSO-d6) δ: 8.18 (d, J = 1.9 Hz, 1H), 7.96 (dd, J = 8.4, 1.9 Hz, 1H),
7.79 (d, J = 8.4 Hz, 1H), 5.92 (d, J = 5.8 Hz, 1H).
3,4-Difluorophenylglyoxal (2)
C8H6F2O3
M: 188.13 g mol-1
3,4-Difluoroacetophenone (10.0 g, 64 mmol) was converted into the glyoxal
according to Method A. Instead of water the yellow oil was dissolved in CH2Cl2 and
heated to reflux. Upon cooling a precipitate was formed and removed by filtration.
The solvent was removed and water was added. After heating to reflux the water was
removed and the received solid was purified by column chromatography
(EtOAc/n-hexane : 7/3). After recrystallization from benzene/n-hexane
3,4-difluorophenylglyoxal hydrate was received as white crystals (7.29 g, 39 mmol,
61% yield).
1H NMR (400 MHz, Chloroform-d) δ: 8.04 – 7.91 (m, 2H), 7.29 (q, J = 8.9 Hz, 1H), 6.27
(d, J = 10.2 Hz, 1H), 5.00 (d, J = 10.2 Hz, 1H).
19F NMR (376 MHz, Chloroform-d) δ: -125.87 (d, J = 20.8 Hz), -134.93 (d, J = 20.9 Hz).

Materials and Methods
61
Phenylglyoxal (3)
C8H8O3
M: 152.15 g mol-1
Acetophenone (5.0 g, 42 mmol) was converted into the glyoxal according to Method
A. The resulting oil was suspended in water and heated to reflux. Afterwards the
solvent was removed and the received solid was purified by recrystallization from
benzene/PE to yield the phenylglyoxal hydrate (2.7 g, 18 mmol, 43% yield).
1H NMR (400 MHz, DMSO-d6): 8.07 (dt, J = 7.2, 1.4 Hz, 2H), 7.68 – 7.58 (m, 1H), 7.52
(dd, J = 8.4, 7.0 Hz, 2H), 6.72 (s, 1H), 5.68 (t, J = 7.1 Hz, 1H).
4-Bromophenylglyoxal (4)
C8H7BrO3
M: 231.05 g mol-1
Bromoacetophenone (1.0 g, 5.0 mmol) was converted into the glyoxal according to
Method A. The received solid was recrystallized from H2O to yield
4-bromophenylglyoxal hydrate as colorless solid (0.3 g, 1.3 mmol. 26% yield).
1H NMR (400 MHz, DMSO-d6) δ: 8.06 – 7.95 (m, 2H), 7.78 – 7.69 (m, 2H), 6.84 (d,
J = 7.0 Hz, 2H), 5.62 (t, J = 7.0 Hz, 1H).
4-Nitrophenylglyoxal (5)
C8H7NO5
M: 197.03 g mol-1
4-Nitroacetophenone (1.0 g, 6 mmol) was converted into the glyoxal according to
Method A. After removal of the solvent, the received yellow solid was purified with
column chromatography (EtOAc/PE : 1/1) to yield 4-nitrophenylglyoxal hydrate
(1.01 g, 5.1 mmol, 85%)

Materials and Methods
62
1H NMR (400 MHz, DMSO-d6) δ: 8.43 – 8.24 (m, 4H), 7.04 (d, J = 6.7 Hz, 2H), 5.66 (s,
1H).
1-(5-Chlorothiophen-2-yl)-2,2-dihydroxyethan-1-one (6)
C6H5ClO3S
M: 192.61 g mol-1
5-Chloro-1-acetylthiophene (0.5 g, 3.2 mmol) was converted into the glyoxal
according to Method A. After filtration the solvent was removed and no water was
added. The crude product was directly used without further purification.
5-Chloro-2-phenyl-7-(trifluoromethyl)quinoxaline (13)
C15H8ClF3N2
M: 308.69 g mol-1
3-Chloro-5-(trifluoromethyl)benzene-1,2-diamine (0.5 g, 3.2 mmol) was converted to
5-Chloro-2-phenyl-7-(trifluoromethyl)quinoxaline according to Method B. After
excessive washing of the received solid with EtOH 5-chloro-2-phenyl-7-
(trifluoromethyl)quinoxaline was received as pink solid (0.64 g, 2.1 mmol, 65% yield).
1H NMR (400 MHz, DMSO-d6) δ: 9.47 (s, 1H), 8.31 – 8.30 (m, 3H), 8.05 (d, J = 1.8 Hz,
1H), 7.57 – 7.62 (m, 3H).
19F NMR (376 MHz, Chloroform-d) δ: –62.74 (s).
CHN (calc/found %): C(58.36/58.05), H(2.61/2.67), N(9.08/9.14).
2-(3,4-Dichlorophenyl)quinoxaline-6-carboxylic acid (14)
C15H8Cl2N2O2
M: 319.14 g mol-1

Materials and Methods
63
3,4-Diaminobenzoic acid (5.0 g, 22.6 mmol) was converted into
2-(3,4-dichlorophenyl)quinoxaline-6-carboxylic acid using Method B. The crude
product was recrystallized from dioxane yielding 2-(3,4-dichlorophenyl)quinoxaline-
6-carboxylic acid as off-white solid (6.8 g, 21.2 mmol, 94% yield).
1H NMR (400 MHz, DMSO-d6, 72°C) δ: 13.19 (s, 1H), 9.65 (s, 1H), 8.72 – 8.46 (m, 2H),
8.39 – 8.12 (m, 3H), 7.83 (d, J = 8.3 Hz, 1H).
2-(3,4-Difluorophenyl)quinoxaline-6-carboxylic acid (15)
C15H8F2N2O2
M: 286.24 g mol-1
3,4-Diaminobenzoic acid (578 mg, 3.4 mmol) was converted into
2-(3,4-difluorophenyl)quinoxaline-6-carboxylic acid using Method B without heating.
The resulting solid was washed with EtOH to yield 2-(3,4-difluorophenyl)quinoxaline-
6-carboxylic acid as off-white solid (680 mg, 2.5 mmol, 75% yield).
1H NMR (400 MHz, DMSO-d6) δ: 13.36 (s, 1H), 9.65 (s, 1H), 8.57 (s, 1H), 8.33 (t, J =
10.0 Hz, 1H), 8.28 – 8.16 (m, 3H), 7.66 (q, J = 10.0, 8.6 Hz, 1H).
19F NMR (376 MHz, DMSO-d6) δ: –135.22 (d, J = 22.2 Hz), –137.29 (d, J = 22.2 Hz).
2-Phenylquinoxaline (16)
C14H10N2
M: 206.25 g mol-1
1,2-Phenylenediamine (0.3 g, 2.0 mmol) was converted into 2-phenylquinoxaline
using Method B. After cooling to ambient temperature the resulting black precipitate
was removed. The filtrate was concentrated and 2-phenylquinoxaline was received
as off-white solid (0.4 g, 2.0 mmol, 97% yield).

Materials and Methods
64
1H NMR (400 MHz, DMSO-d6) δ: 9.58 (s, 1H), 8.35 – 8.33 (m, 2H), 8.13 (td, J = 8.3, 1.7
Hz, 2H), 7.63– 7.55 (m, 3H), 7.89 – 7.84 (m, 2H).
CHN (calc/found %): C(81.53/81.35), H(4.89/4.78), N(13.58/13.73).
2-(4-Bromophenyl)-5-chloro-7-(trifluoromethyl)quinoxaline (17)
C15H7BrClF3N2
M: 387.58 g mol-1
3-Chloro-5-(trifluoromethyl)benzene-1,2-diamine (0.18 g, 0.9 mmol) was converted
to 2-(4-bromophenyl)-5-chloro-7-(trifluoromethyl)quinoxaline according to Method
B. After excessive washing with EtOH 2-(4-bromophenyl)-5-chloro-7-
(trifluoromethyl)quinoxaline was received as colorless solid (0.24 g, 0.62 mmol, 72%
yield).
1H NMR (400 MHz, DMSO-d6) δ: 9.78 (s, 1H), 8.44 – 8.42 (m, 1H), 8.36 – 8.34 (m, 3H),
7.83 – 7.81 (m, 2H), 8.28 – 8.16 (m, 3H), 7.66 (q, J = 10.0, 8.6 Hz, 1H).
19F NMR (376 MHz, DMSO-d6) δ: –61.15 (s).
CHN (calc/found %): C(46.48/46.54), H(1.82/1.75), N(7.23/7.57).
2-(5-chlorothiophen-2-yl)quinoxaline (18)
C12H7ClN2S
M: 246.71 g mol-1
1,2-Phenylenediamine (0.34 g, 3.2 mmol) was converted into 2-phenylquinoxaline
and the crude 1-(5-Chlorothiophen-2-yl)-2,2-dihydroxyethan-1-one (6) using Method
B. The pure product was received after excessive washing with EtOH (0.40 g, 1.6
mmol, 51% yield).

Materials and Methods
65
1H NMR (400 MHz, Chloroform-d) δ: 9.53 (s, 1H), 8.10 (d, J = 4.1 Hz, 1H), 8.08-8.04
(m, 1H), 8.02-7.97 (m, 1H), 7.87-7.75 (m, 2H), 7.30 (d, J = 4.1 Hz, 1H)
CHN (calc/found %): C(58.42/58.21), H(2.86/2.82), N(11.35/11.30).
5-chloro-2-(5-chlorothiophen-2-yl)-7-(trifluoromethyl)quinoxaline (19)
C13H5Cl2F3N2S
M: 349.15 g mol-1
3-Chloro-5-(trifluoromethyl)benzene-1,2-diamine (0.67 g, 3.2 mmol) was converted
to 5-chloro-2-(5-chlorothiophen-2-yl)-7-(trifluoromethyl)quinoxaline using crude 1-
(5-Chlorothiophen-2-yl)-2,2-dihydroxyethan-1-one (6) according to Method B. The
crude product was recrystallized from EtOH and received as crystals (0.55 g, 1.6
mmol).
1H NMR (400 MHz, DMSO-d6) δ: 9.63 (s, 1H), 8.34 (m, 1H), 8.21 (d, J = 1.9 Hz, 1H),
8.17 (d, J = 4.1 Hz, 1H), 7.31 (dd, J = 0.5, 4.1 Hz, 1H).
19F NMR (376 MHz, DMSO-d6) δ: –61.27 (s).
CHN (calc/found %): C(44.72/44.64), H(1.44/1.46), N(8.02/8.18).
Ethyl 2-(3,4-dichlorophenyl)quinoxaline-6-carboxylate (20)
C17H12Cl2N2O2
M: 347.20 g mol-1
2-(3,4-dichlorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.6 mmol) was converted
into ethyl 2-(3,4-dichlorophenyl)quinoxaline-6-carboxylate using Method C. The
crude product was recrystallized from toluene and washed with hexane to yield ethyl

Materials and Methods
66
2-(3,4-dichlorophenyl)quinoxaline-6-carboxylate as white crystal needles (0.32 g,
0.9 mmol, 59% yield).
1H NMR (400 MHz, Chloroform-d) δ: 9.36 (s, 1H), 8.84 (d, J = 1.8 Hz, 1H), 8.45 – 8.33
(m, 2H), 8.18 (d, J = 8.7 Hz, 1H), 8.07 (dd, J = 8.4, 2.1 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H),
4.49 (q, J = 7.2 Hz, 2H), 1.47 (t, J = 7.2 Hz, 3H).
2-(3,4-Dichlorophenyl)-N,N-diethylquinoxaline-6-carboxamide (21)
C19H17Cl2N3O
M: 374.27 g mol-1
2-(3,4-dichlorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.6 mmol) was converted
into 2-(3,4-dichlorophenyl)-N,N-diethylquinoxaline-6-carboxamide using Method C.
After warming to ambient temperature no water was added. Instead, the solvent was
removed and the crude product was dissolved in EtOAc and filtered over a SiO2-pad.
The solvent was removed and the product was washed wit n-hexane yielding
2-(3,4-dichlorophenyl)-N,N-diethylquinoxaline-6-carboxamide as white solid (0,45 g,
1.2 mmol, 74% yield).
1H NMR (400 MHz, DMSO-d6) δ: 9.67 (s, 1H), 8.58 (d, J = 2.1 Hz, 1H), 8.34 (dd, J = 8.5,
2.1 Hz, 1H), 8.20 (d, J = 8.6 Hz, 1H), 8.05 (d, J = 1.7 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H),
7.84 (dd, J = 8.6, 1.8 Hz, 1H), 3.38 (broad, 4H), 1.15 (broad, 6H).
13C NMR (101 MHz, DMSO-d6) δ: 168.55, 149.23, 144.44, 141.00, 140.77, 138.83,
136.36, 133.44, 132.12, 131.33, 129.76, 129.17, 128.94, 127.56, 125.87, 99.50, 42.93,
14.03, 12.78.
CHN (calc/found %): C(60.97/61.10), H(4.58/4.70), N(11.23/11.33).

Materials and Methods
67
(2-(3,4-dichlorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone (22)
C20H18Cl2N4O
M: 401.29 g mol-1
2-(3,4-dichlorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.6 mmol) was converted
into (2-(3,4-dichlorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone
using Method C. The combined organic layers were additionally washed with 0.1 M
aq. HCl and finally with water. After drying over MgSO4 the solvent was removed, the
crude product was washed with EtOAc and n-hexane to yield the desired product as
off-white solid (0.42 g, 1.1 mmol, 53% yield).
1H NMR (400 MHz, Chloroform-d) δ: 9.34 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 8.20 (d, J =
8.6 Hz, 1H), 8.15 (d, J = 1.8 Hz, 1H), 8.06 (dd, J = 8.4, 2.1 Hz, 1H), 7.85 (dd, J = 8.6, 1.9
Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 3.75 (broad, 4H), 2.53 (broad, 4H), 2.39 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 168.92, 150.37, 143.59, 142.54, 141.34, 137.21,
136.33, 135.27, 133.96, 131.35, 130.51, 129.62, 129.57, 127.91, 126.65, 55.18, 46.00.
CHN (calc/found %): C(59.86/58.19), H(4.52/4.44), N(13.96/13.65).
(2-(3,4-Dichlorophenyl)quinoxalin-6-yl)(morpholino)methanone (23)
C19H15Cl2N3O2
M: 388.25 g mol-1
2-(3,4-Dichlorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.6 mmol) was converted
into (2-(3,4-dichlorophenyl)quinoxalin-6-yl)(morpholino)methanone using Method
C. The combined organic layers were additionally washed with 0.1 M aq. HCl and
finally with water. After drying over MgSO4 the solvent was removed, the crude
product was washed with n-hexane to yield the desired product as off-white solid
(0.41 g, 1.1 mmol, 68% yield).

Materials and Methods
68
1H NMR (400 MHz, Chloroform-d) δ: 9.34 (s, 1H), 8.38 (d, J = 2.1 Hz, 1H), 8.21 (d, J =
8.6 Hz, 1H), 8.15 (d, J = 1.8 Hz, 1H), 8.06 (dd, J = 8.4, 2.1 Hz, 1H), 7.85 (dd, J = 8.6, 1.9
Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 3.67 (broad, 8H).
13C NMR (101 MHz, Chloroform-d) δ: 169.02, 150.44, 143.61, 142.57, 141.30, 136.84,
136.28, 135.31, 133.97, 131.36, 130.60, 129.62, 129.57, 128.00, 126.65, 67.03.
CHN (calc/found %): C(58.78/58.62), H(3.89/4.07), N(10.82/10.77).
2-(3,4-Difluorophenyl)-N,N-diethylquinoxaline-6-carboxamide (24)
C19H17F2N3O
M: 341.36 g mol-1
2-(3,4-Difluorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.7 mmol) was converted
into 2-(3,4-difluorophenyl)-N,N-diethylquinoxaline-6-carboxamide using Method C.
The crude product was dissolved in EtOAc and filtered through a short SiO2 pad.
Afterwards the solvent was removed to yield the desired product (0.30 g, 0.9 mmol,
53% yield).
1H NMR (400 MHz, DMSO-d6) δ: 9.63 (s, 1H), 8.39 (ddd, J = 11.8, 7.8, 1.9 Hz, 2H), 8.26
– 8.20 (m, 1H), 8.16 (d, J = 8.5 Hz, 1H), 8.04 (d, J = 1.4 Hz, 1H), 7.82 (dd, J = 8.6, 1.7 Hz,
1H), 7.72 – 7.61 (m, 1H), 3.51 (broad, 4H), 3.25 (broad, 6H).
13C NMR (101 MHz, DMSO-d6) δ: 168.58 , 151.07 (dd, J = 250.5, 12.6 Hz), 149.93 (dd,
J = 246.1, 12.9 Hz), 149.45 , 144.35 , 140.97 , 140.58 , 138.64 , 133.42 (dd, J = 6.0, 3.5
Hz), 129.67 , 128.89 , 125.87 , 124.77 (dd, J = 7.0, 3.3 Hz), 118.33 (d, J = 17.5 Hz),
116.57 (d, J = 18.7 Hz), 42.91 , 14.02 , 12.78 .
19F NMR (376 MHz, DMSO-d6) δ: -135.76 (d, J = 22.3 Hz), -137.42 (d, J = 22.3 Hz).
CHN (calc/found %): C(66.85/66.61), H(5.02/4.80), N(12.31/12.26).

Materials and Methods
69
(2-(3,4-difluorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone (25)
C20H18F2N4O
M: 368.39 g mol-1
2-(3,4-Difluorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.7 mmol) was converted
into (2-(3,4-difluorophenyl)quinoxalin-6-yl)(4-methylpiperazin-1-yl)methanone using
Method C. The crude product was dissolved in EtOAc and filtered through a short SiO2
pad. Afterwards the solvent was removed to yield the desired product (0.23 g,
0.6 mmol, 37% yield).
1H NMR (400 MHz, DMSO-d6) δ: 9.65 (s, 1H), 8.61 – 8.35 (m, 2H), 8.33 – 8.12 (m, 3H),
8.08 (d, J = 2.3 Hz, 1H), 7.84 (ddd, J = 13.2, 8.5, 1.9 Hz, 1H), 7.75 – 7.60 (m, 1H), 3.69
(broad, 4H), 2.36 (broad, 4H), 2.21 (s, 3H).
13C NMR (151 MHz, DMSO-d6) δ: 167.62 , 151.13 (dd, J = 250.6, 12.7 Hz), 149.96 (dd,
J = 245.9, 12.5 Hz), 149.60 , 144.40 , 141.24 , 137.40 , 133.40 (dt, J = 5.7, 2.9 Hz),
129.71 , 129.31 , 127.15 , 126.93 , 124.89 – 124.78 (m), 118.39 (d, J = 17.5 Hz), 116.64
(d, J = 18.7 Hz), 47.08 , 45.57 , 41.53 .
19F NMR (376 MHz, DMSO-d6) δ: -135.65 (d, J = 22.5 Hz), -137.38 (d, J = 22.1 Hz).
(2-(4-Fluoro-3-morpholinophenyl)quinoxalin-6-yl)(morpholino)methanone (26)
C23H23FN4O3
M: 422.46 g mol-1
2-(3,4-Difluorophenyl)quinoxaline-6-carboxylic acid (0.5 g, 1.7 mmol) was converted
into the acid chloride as described in Method C. Afterwards an excess of morpholine
(25 mL) was added and the reaction mixture was heated to reflux. After 3h the
solvent was removed and the residue was dissolved in CH2Cl2. Unsoluble solid was

Materials and Methods
70
removed by filtration and the filtrate was washed once with 1 M aq HCl and twice
with H2O, dried over MgSO4 and the solvent was removed. The crude product was
dissolved in EtOAc and filtered through a SiO2 pad. The solvent was removed and the
resulting solid was washed with n-hexane to yield (2-(4-fluoro-3-
morpholinophenyl)quinoxalin-6-yl)(morpholino)methanone as yellow solid (0.17 g,
0.4 mmol, 24% yield).
1H NMR (400 MHz, Chloroform-d) δ: 9.31 (s, 1H), 8.15 (dd, J = 8.5, 2.4 Hz, 1H), 8.10
(d, J = 1.6 Hz, 1H), 8.04 – 7.87 (m, 3H), 7.78 (ddd, J = 21.6, 8.6, 1.8 Hz, 1H), 7.07 (t, J =
8.6 Hz, 1H), 3.94 – 3.87 (m, 4H), 3.66 (broad, 8H), 3.30 – 3.13 (m, 4H).
13C NMR (101 MHz, Chloroform-d) δ: 169.22, 155.77 (d, J = 246.9 Hz), 151.32 (d, J =
2.4 Hz), 143.77 , 142.74 , 140.76 , 137.13 , 135.94 , 130.30 , 129.22 , 123.93 (d, J = 3.0
Hz), 118.75 (d, J = 3.7 Hz), 115.52 (d, J = 22.9 Hz), 66.98 (d, J = 5.6 Hz), 50.56 (d, J =
4.0 Hz).
CHN (calc/found %): C(65.39/65.12), H(5.49/5.50), N(13.26/13.58).
6.3 BTB-1 Analogs Synthesis
General Methods for the Preparation of Sulfones and Sulfoxides
The sulfones and sulfoxides were synthesized according to known literature
procedures.106, 151-152 To a solution of the corresponding phenol, thiophenol or
thiophenolate and substituted nitrobenzene, an equimolar amount of base for the
thiophenol or 0.3 equiv. for the thiophenolate was added and heated to reflux. EtOH
and 0.1 M aqueous NaOH was used as the solvent if not otherwise mentioned. After
complete consumption of the starting material the solvent was removed and a 1/1
mixture of water and EtOAc was added. The aqueous phase was extracted twice with
EtOAc. The combined organic layers were dried over MgSO4 and the solvent was
removed in vacuum to yield the crude sulfide.
Method D The sulfide was dissolved in 5 mL acetic acid and an excess of 0.7 mL
30% H2O2 was added dropwise if not otherwise mentioned. After
complete addition the reaction mixture was heated to reflux. If

Materials and Methods
71
necessary another equivalent of H2O2 was added. After complete
reaction the solvent was removed in vacuum.
Method E The sulfide was dissolved in CH2Cl2 cooled to 0°C and 1 equiv. of m-
CPBA solution in CH2Cl2 was added drop wise. Saturated NaHCO3
solution was added for neutralization and the organic phase was
separated and the solvent removed to yield the crude sulfoxide.
1-Chloro-2-nitro-4-(phenylsulfonyl)benzene167 (70)
C12H8ClNO4S
M: 297.71 g mol-1
AlCl3 (1.56 g, 11.7 mmol) was suspended in benzene (5 mL) and 4-chloro-3-
nitrobenzenesulfonyl chloride (2.5 g, 9.8 mmol) was added. The mixture was stirred
for 24 h at ambient temperature and hydrolyzed with water (10 mL). The mixture was
extracted three times with EtOAc (15 mL). The organic layer was washed two times
with 1 M NaOH (5 mL) and with brine (5 mL). After drying over MgSO4 the solvent
was removed followed by column chromatographic purification (PE/EtOAc : 4/1) and
recrystallization from EtOAc/n-hexane to yield 1-chloro-2-nitro-4-
(phenylsulfonyl)benzene as white crystals (1.36 g, 4.5 mmol, 46% yield).
m.p: 124°C.
1H NMR (400 MHz, Chloroform-d) δ: 8.41 (d, J = 2.1 Hz, 1H), 8.05 (dd, J = 8.4, 2.1 Hz,
1H), 7.96 (m, 2H), 7.71 (d, J = 8.5 Hz, 1H), 7.68 – 7.61 (m, 1H), 7.61 – 7.53 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 148.14, 142.16, 139.98, 134.40, 133.37, 132.48,
131.67, 129.95, 128.11, 125.07.
CHN (calc/found %): C(48.41/48.42), H(2.71/2.73), N(4.70/4.62).

Materials and Methods
72
2,4-Dinitro-1-(phenylsulfonyl)benzene (52)
C12H8N2O6S
M: 308.26 g mol-1
2,4-Dinitro-1-(phenylsulfonyl)benzene was synthesized according to Method D using
2,4-dinitrochlorobenzene (288 mg, 1.4 mmol) and sodium thiophenolate (188 mg, 1.4
mmol). After pruficiation by column chromatography (PE/EtOAc : 3/1) and
recrystallization from EtOAc/hexane 2,4-dinitro-1-(phenylsulfonyl)benzene was
received as off-white crystalls (110 mg, 0.3 mmol , 25% yield).
m.p.: 153°C
1H NMR (400 MHz, Chloroform-d) δ: 8.58 (d, J = 1.3 Hz, 2H), 8.53 (t, J = 1.3 Hz, 1H),
8.07 – 7.94 (m, 2H), 7.75 – 7.66 (m, 1H), 7.65 – 7.56 (m, 2H)
13C NMR (101 MHz, Chloroform-d) δ 150.57, 148.88, 140.10, 139.15, 134.86,
133.47, 129.63, 128.84, 126.93, 120.31.
CHN (calc/found %): C(46.75/46.78), H(2.62/2.77), N(9.09/8.88).
2,4-Dinitro-1-(phenylsulfinyl)benzene (64)
C12H8N2O5S
M: 292.27 g mol-1
2,4-Dinitro-1-(phenylsulfinyl)benzene was prepared according to Method E using
thiophenole (0.4 mL, 3.4 mmol) and 2,4-dinitrochlorobenzene (705 mg, 3.4 mmol).
The crude sulfoxide was purified by column chromatography (PE/EtOAc : 9/1)
followed by recrystallization from EtOH to yield 2,4-dinitro-1-(phenylsulfinyl)benzene
as yellow crystalles (370 mg, 1.3 mmol, 38% yield).
m.p.: 108°C

Materials and Methods
73
1H NMR (400 MHz, Chloroform-d) δ: 9.04 (d, J = 1.9 Hz, 1H), 8.86 – 8.76 (m, 2H),
7.77 – 7.68 (m, 2H), 7.51 – 7.40 (m, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 151.24, 149.58, 144.96, 144.06, 132.41,
129.78, 129.31, 128.24, 126.94, 120.81.
CHN (calc/found %): C(49.31/49.23), H(2.76/2.85), N(9.58/9.44).
4-Fluoro-2-nitro-1-(phenylsulfonyl)benzene (53)
C12H8FNO4S
M: 281.26 g mol-1
4-Fluoro-2-nitro-1-(phenylsulfonyl)benzene was synthesized according to Method D
using 1-chloro-4-fluoro-2-nitrobenzene (500 mg, 2.8 mmol) and sodium
thiophenolate (380 mg, 2.8 mmol). After column chromatography (n-hexane/EtOAc :
3/1) 4-fluoro-2-nitro-1-(phenylsulfonyl)benzene was received as a white solid (50 mg,
0.2 mmol, 12% yield).
m.p.: 126°C
1H NMR (400 MHz, Chloroform-d) δ 8.44 – 8.37 (m, 1H), 7.99 – 7.92 (m, 2H), 7.69 –
7.62 (m, 1H), 7.61 – 7.54 (m, 2H), 7.51 – 7.43 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ: -99.24.
13C NMR (151 MHz, Chloroform-d) δ: 164.92 (d, J = 261.8 Hz), 149.77 (d, J = 8.8 Hz),
140.26 , 134.21 (d, J = 9.4 Hz), 133.96 , 130.94 (d, J = 4.1 Hz), 129.20 , 128.24 ,
119.55 (d, J = 21.4 Hz), 113.08 (d, J = 27.2 Hz).
CHN (calc/found %): C(51.24/51.05), H(2.87/2.99), N(4.98/5.16).

Materials and Methods
74
2-Nitro-1-(phenylsulfonyl)-4-(trifluoromethyl)benzene (54)
C13H8F3NO4S
M: 331.27 g mol-1
2-Nitro-1-(phenylsulfonyl)-4-(trifluoromethyl)benzene was prepared following
Methode D using 1-chloro-2-nitro-4-(trifluoromethyl)benzene (321 mg, 1.4 mmol)
and sodium thiophenolate (188 mg, 1.4 mmol). After column chromatography
(PE/EtOAc : 3/1) 2-nitro-1-(phenylsulfonyl)-4-(trifluoromethyl)benzene was
recrystallized from EtOAc/n-hexane and recieved as white crystalls (120 mg, 0.3
mmol, 26% yield).
m.p.: 142°C
1H NMR (400 MHz, Chloroform-d) δ 8.52 (d, J = 8.2 Hz, 1H), 8.08 – 8.01 (m, 1H), 8.01
– 7.95 (m, 3H), 7.73 – 7.65 (m, 1H), 7.64 – 7.55 (m, 2H).
19F NMR (376 MHz, Chloroform-d) δ: -63.33.
13C NMR (101 MHz, Chloroform-d) δ: 148.73 , 139.61 , 138.23 , 136.63 (q, J = 34.9
Hz), 134.54 , 132.77 , 129.50 , 129.70 – 129.26 (m), 128.69 , 122.26 (q, J = 3.7 Hz),
122.03 (q, J = 273.8 Hz).
CHN (calc/found %): C(47.13/46.89), H(2.43/2.63), N(4.23/4.46).
2-((4-Chloro-2-nitrophenyl)sulfonyl)thiophene (55)
C10H6ClNO4S2
M: 303.73 g mol-1
2-((4-Chloro-2-nitrophenyl)sulfonyl)thiophene was synthesized according to Method
D using thiophene-2-thiol (163 mg, 1.4 mmol) and 1,4-dichloro-2-nitrobenzene (269
mg, 1.4 mmol) and 1 M aq. NaOH as base. After column chromatography (PE/EtOAc
: 3/1) 2-((4-Chloro-2-nitrophenyl)sulfonyl)thiophene was received as off-white
crystalls (290 mg, 0.95 mmol, 68% yield).

Materials and Methods
75
m.p.: 134°C
1H NMR (400 MHz, Chloroform-d) δ: 8.21 (d, J = 8.4 Hz, 1H), 7.92 (dd, J = 3.9, 1.3 Hz,
1H), 7.78 (dd, J = 5.0, 1.3 Hz, 1H), 7.71 (dd, J = 8.4, 2.0 Hz, 1H), 7.69 (d, J = 1.9 Hz,
1H), 7.17 (dd, J = 4.9, 3.9 Hz, 1H).
13C NMR (101 MHz, Chloroform-d) δ: 148.77, 141.12, 140.66, 136.27, 135.76,
133.58, 132.70, 132.46, 128.15, 124.95.
CHN (calc/found %): C(39.54/39.44), H(1.99/2.14), N(4.61/4.85).
2-((4-Chloro-2-nitrophenyl)sulfonyl)naphthalene (56)
C16H10ClNO4S
M: 347.77 g mol-1
2-((4-Chloro-2-nitrophenyl)sulfonyl)naphthalene was prepared according to Method
D using 2-thionaphthalene (224 mg, 1.4 mmol) and 1,4-dichloro-2-nitrobenzene (269
mg, 1.4 mmol). The crude sulfone was purified by column chromatography (n-
hexane/EtOAc : 3/1) followed by recrystallization from EtOAc/ n-hexane to yield 2-
((4-chloro-2-nitrophenyl)sulfonyl)naphthalene as off-white solid (30 mg, 0.08 mmol,
6% yield).
m.p.: 155°C
1H NMR (400 MHz, Chloroform-d) δ: 8.60 (d, J = 8.5 Hz, 1H), 8.35 (s, 1H), 8.23 (d, J =
2.0 Hz, 1H), 7.99 (dd, J = 8.5, 2.0 Hz, 1H), 7.95 – 7.89 (m, 1H), 7.88 – 7.78 (m, 2H),
7.65 – 7.52 (m, 3H), 1.55 (s, 2H, H2O).
13C NMR (101 MHz, Chloroform-d) δ: 142.70, 141.54, 138.07, 135.54, 134.68,
132.71, 129.97, 129.08, 128.55, 128.34, 128.06, 127.91, 127.55, 125.59, 121.66.
CHN (calc/found %): C(55.26/54.76), H(2.90/3.07), N(4.03/4.10).

Materials and Methods
76
2-((4-Chloro-2-nitrophenyl)sulfinyl)naphthalene (65)
C16H10ClNO3S
M: 331.77 g mol-1
2-((4-Chloro-2-nitrophenyl)sulfinyl)naphthalene was synthesized following Methode
E using 2-thionaphthalene (224 mg, 1.4 mmol) and 1,4-dichloro-2-nitrobenzene (269
mg, 1.4 mmol). The crude sulfoxide was received as yellow oil which crystallizes upon
cooling. The yellow crystalls were washed with n-hexane to yield the pure 2-((4-
chloro-2-nitrophenyl)sulfinyl)naphthalene (306 mg, 0.9 mmol, 66% yield).
m.p.: 126°C
1H NMR (600 MHz, Chloroform-d) δ: 8.59 (d, J = 8.5 Hz, 1H), 8.35 (d, J = 1.8 Hz, 1H),
8.21 (d, J = 2.1 Hz, 1H), 7.98 (dd, J = 8.5, 2.1 Hz, 1H), 7.93 – 7.88 (m, 1H), 7.85 (d, J =
8.7 Hz, 1H), 7.83 – 7.79 (m, 1H), 7.59 (dd, J = 8.7, 1.9 Hz, 1H), 7.58 – 7.54 (m, 2H).
13C NMR (151 MHz, Chloroform-d) δ: 145.11, 142.62, 141.47, 138.00, 135.50,
134.60, 132.64, 129.92, 129.03, 128.50, 128.30, 128.01, 127.85, 127.50, 125.53,
121.63.
CHN (calc/found %): C(57.92/57.97), H(3.04/3.11), N(4.22/4.43).
4-Chloro-2-nitro-1-tosylbenzene (57)
C13H10ClNO4S
M: 311.74 g mol-1
4-Chloro-2-nitro-1-tosylbenzene was synthesized according to Method D using 4-
methylbenzenethiol (174 mg, 1.4 mmol) and 1,4-dichloro-2-nitrobenzene (269 mg,
1.4 mmol). After column chromatography (n-hexane/EtOAc : 3/1) the product was
received as light brown solid (30 mg, 0.09 mmol, 7%).

Materials and Methods
77
m.p.: 117°C
1H NMR (400 MHz, Chloroform-d) δ: 8.26 (d, J = 8.5 Hz, 1H), 7.88 – 7.81 (m, 2H),
7.71 (dd, J = 8.5, 2.1 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 7.40 – 7.32 (m, 2H), 2.44 (s,
3H).
13C NMR (101 MHz, Chloroform-d) δ: 148.92, 145.47, 140.89, 137.20, 133.58,
132.78, 132.58, 130.00, 128.58, 125.01, 21.85.
CHN (calc/found %): C(50.09/50.05), H(3.23/3.53), N(4.49/4.53).
4-Chloro-1-((4-methoxyphenyl)sulfonyl)-2-nitrobenzene (58)
C13H10ClNO5S
M: 327.74 g mol-1
4-Chloro-1-((4-methoxyphenyl)sulfonyl)-2-nitrobenzene was synthesized according
to Methode D using 4-methoxybenzenethiol (196 mg, 1.4 mmol) and 1,4-dichloro-2-
nitrobenzene (269 mg, 1.4 mmol). After column chromatography (PE/EtOAc : 3/1) the
product was received as off-white solid (54 mg, 0.16 mmol, 12%).
m.p.: 104°C
1H NMR (400 MHz, Chloroform-d) δ: 8.23 (d, J = 8.5 Hz, 1H), 7.95 – 7.86 (m, 2H),
7.70 (dd, J = 8.5, 2.1 Hz, 1H), 7.66 (d, J = 2.0 Hz, 1H), 7.08 – 6.95 (m, 2H), 3.88 (s,
3H).
13C NMR (101 MHz, Chloroform-d) δ: 164.27, 148.84, 140.68, 133.95, 132.59,
132.52, 131.40, 131.06, 124.93, 114.62, 55.91.
CHN (calc/found %): C(47.64/47.65), H(3.08/3.16), N(4.27/4.38).

Materials and Methods
78
4-Chloro-1-((4-methoxyphenyl)sulfinyl)-2-nitrobenzene (66)
C13H10ClNO4S
M: 311.74 g mol-1
4-Chloro-1-((4-methoxyphenyl)sulfinyl)-2-nitrobenzene was synthesized according
to Method E using 4-methoxybenzenethiol (350 mg, 2.5 mmol) and 1,4-dichloro-2-
nitrobenzene (480 mg, 2.5 mmol). The crude sulfoxide was crystallized from
EtOAc/PE. Due to traces of m-chlorbenzoic acid the crude product was dissolved in
CH2Cl2, washed with sat. K2CO3 solution and dried over MgSO4. After removal of the
solvent 4-chloro-1-((4-methoxyphenyl)sulfinyl)-2-nitrobenzene was received as
yellow solid (560 mg, 1.8 mmol, 72% yield).
m.p.: 118°C
1H NMR (400 MHz, Chloroform-d) δ: 8.53 (d, J = 8.5 Hz, 1H), 8.22 (d, J = 2.1 Hz, 1H),
7.95 (dd, J = 8.5, 2.1 Hz, 1H), 7.69 – 7.52 (m, 2H), 6.94 – 6.85 (m, 2H), 3.80 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 162.39, 145.04, 143.01, 137.78, 135.97,
135.35, 128.90, 127.71, 125.54, 114.86, 55.66.
CHN (calc/found %): C(50.09/49.94), H(3.23/3.32), N(4.49/4.63).
1-Nitro-2-(phenylsulfonyl)benzene (59)
C12H9NO4S
M: 263.27 g mol-1
According to Methode D thiophenol (1.47 g, 13.3 mmol) and 1-chloro-2-nitrobenzene
(2.1 g 13.3 mmol) were converted to 1-nitro-2-(phenylsulfonyl)benzene. The crude
sulfide was recrystallized from water. After oxidation the crude sulfone was purified
with column chromatography (PE/EtOAc : 1/1) and recrystalized from EtOH to yield
1-nitro-2-(phenylsulfonyl)benzene as white solid (120 mg, 0.4 mmol, 3% yield).
m.p.: 145°C

Materials and Methods
79
1H NMR (400 MHz, Chloroform-d) δ: 8.41 – 8.33 (m, 1H), 8.03 – 7.94 (m, 2H), 7.84 –
7.70 (m, 3H), 7.68 – 7.60 (m, 1H), 7.60 – 7.52 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 148.68, 140.61, 134.78, 134.70, 133.94,
132.63, 131.75, 129.25, 128.39, 124.91.
CHN (calc/found %): C(54.75/54.64), H(3.45/3.52), N(5.32/5.45).
1-Nitro-2-(phenylsulfinyl)benzene (67)
C12H9NO3S
M: 247.27 g mol-1
According to Method E thiophenol (1.47 g, 13.3 mmol) and 1-chloro-2-nitrobenzene
(2.1 g 13.3 mmol) were converted to 1-nitro-2-(phenylsulfinyl)benzene. The crude
sulfide was recrystallized from water. After oxidation the crude sulfoxide was purified
with column chromatography (EtOAc/PE : 1/1) and recrystallized from EtOH to yield
1-nitro-2-(phenylsulfinyl)benzene as yellow crystalls(1.7 mmol, 427 mg, 13% yield).
m.p.: 88°C
1H NMR (400 MHz, Chloroform-d) δ: 8.59 (dd, J = 7.9, 1.4 Hz, 1H), 8.26 (dd, J = 8.1,
1.2 Hz, 1H), 8.01 (m), 7.78 – 7.65 (m, 3H), 7.49 – 7.36 (m, 3H).
13C NMR (101 MHz, Chloroform-d) δ: 145.29, 144.76, 144.05, 135.52, 131.69,
131.57, 129.41, 126.92, 126.45, 125.46.
CHN (calc/found %): C(58.29/58.20), H(3.67/3.65), N(5.66/5.86).
1-((4-Methoxyphenyl)sulfonyl)-2-nitrobenzene (60)
C13H11NO5S
M: 293.29 g mol-1

Materials and Methods
80
4-Methoxybenzenethiol (0.89 g, 6.3 mmol) and 1-chloro-2-nitrobenzene (1.00 g, 6.3
mmol) were dissolved in EtOH. 0.8 g KOH was dissolved in EtOH and was added to
the reaction mixture followed by heating to 70°C. After filtration water was added to
the filtrate and extracted twice with 30 mL CH2Cl2. The organic layer was washed with
30 mL of 2M aq. KOH and dried over Na2SO4. After removal of the solvent the crude
sulfide was oxidized to the corresponding sulfone following Method D. The crude
product was washed with EtOAc yielding 1-((4-methoxyphenyl)sulfonyl)-2-
nitrobenzene as a white solid (100 mg, 0.34 mmol, 5% yield).
m.p: 148°C
1H NMR (400 MHz, Chloroform-d) δ: 8.35 – 8.25 (m, 1H), 7.99 – 7.87 (m, 2H), 7.78 –
7.64 (m, 3H), 7.09 – 6.94 (m, 2H), 3.87 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 164.08, 148.54, 135.46, 134.32, 132.47,
131.81, 131.40, 131.01, 124.69, 114.51, 55.87.
CHN (calc/found %): C(53.24/53.26), H(3.78/3.72), N(4.78/4.94).
1-((4-Methoxyphenyl)sulfinyl)-2-nitrobenzene (68)
C13H11NO4S
M: 277.29 g mol-1
4-Methoxybenzenethiol (1.78 g, 12.7 mmol) and 1-chloro-2-nitrobenzene (2.00 g,
12.7 mmol) were dissolved in EtOH. 0.8 g KOH was dissolved in EtOH and was added
to the reaction mixture followed by heating to 70°C. After filtration water was added
to the filtrate and extracted twice with 30 mL CH2Cl2. The organic layer was washed
with 30 mL of 2 M aq. KOH and dried over Na2SO4. After removal of the solvent the
crude sulfide was oxidized to the corresponding sulfoxide following Method E. The
sulfoxide was purified via cholumn chromatography (EtOAc/PE : 2/1) yielding 1-((4-
methoxyphenyl)sulfinyl)-2-nitrobenzene as yellow crystals (70 mg, 0.25 mmol, 2%).
m.p: 94°C

Materials and Methods
81
1H NMR (400 MHz, Chloroform-d) δ: 8.57 (dd, J = 7.9, 1.2 Hz, 1H), 8.24 (dd, J = 8.1,
1.0 Hz, 1H), 8.05 – 7.95 (m, 1H), 7.70 – 7.64 (m, 1H), 7.63 – 7.57 (m, 2H), 6.87 (dd, J
= 9.4, 2.5 Hz, 2H), 3.77 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 162.29, 144.80, 144.49, 136.49, 135.49,
131.47, 129.04, 126.44, 125.58, 114.83, 55.70.
CHN (calc/found %): C(56.31/56.32), H(4.00/4.00), N(5.05/5.16).
2,4-Dinitro-1-tosylbenzene (61)
C13H10N2O6S
M: 322.29 g mol-1
2,4-Dinitro-1-tosylbenzene was synthesized according to Method E using 1-chloro-
2,4-dinitrobenzene (910 mg, 4.5 mmol) and 4-methylbenzenethiol (558 mg, 4.5
mmol). After purification by column chromatography (EtOAc/PE : 1/1) 2,4-dinitro-1-
tosylbenzene was received as an off-white solid (241 mg, 0.8 mmol, 18%).
m.p: 181°C
1H NMR (400 MHz, Chloroform-d) δ: 8.59 – 8.47 (m, 3H), 7.93 – 7.77 (m, 2H), 7.39
(d, J = 8.1 Hz, 2H), 2.46 (s, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 150.42, 148.79, 146.36, 140.44, 136.07,
133.31, 130.28, 128.99, 126.84, 120.19, 21.92.
CHN (calc/found %): C(48.45/48.31), H(3.13/3.25), N(8.69/8.81).
2,4-Dinitro-1-(p-tolylsulfinyl)benzene (69)
C13H10N2O5S
M: 306.29 g mol-1

Materials and Methods
82
2,4-Dinitro-1-(p-tolylsulfinyl)benzene was synthesized according to Method E using
1-chloro-2,4-dinitrobenzene (910 mg, 4.5 mmol) and 4-methylbenzenethiol (558 mg,
4.5 mmol). After column chromatographic purification (EtOAc/PE : 1/2) and
recrystallization from EtOAc/n-hexane 4-dinitro-1-(p-tolylsulfinyl)benzene was
received as yellow crystals (260 mg, 0.9 mmol, 20%).
m.p: 134°C
1H NMR (400 MHz, Chloroform-d) δ: 9.03 (d, J = 1.9 Hz, 1H), 8.81 (d, J = 8.6 Hz, 1H),
8.78 (dd, J = 8.6, 1.9 Hz, 1H), 7.61 – 7.55 (m, 2H), 7.25 – 7.20 (m, 2H), 2.35 (s, 3H).
13C NMR (101 MHz, Chloroform-d) δ 151.49, 149.50, 144.93, 143.27, 140.87,
130.41, 129.23, 128.22, 127.00, 120.79, 21.58.
CHN (calc/found %): C(50.98/50.90), H(3.29/3.36), N(9.15/9.22).
4-Chloro-1-((4-chlorophenyl)sulfonyl)-2-nitrobenzene (62)
C12H7Cl2NO4S
M: 332.15 g mol-1
4-Chloro-1-((4-chlorophenyl)sulfonyl)-2-nitrobenzene was synthesized according to
Method D using 4-chlorobenzenethiol (750 mg, 5.18 mmol), 1,4-dichloro-2-
nitrobenzene (1.00 g, 5.18 mmol), MeOH as the solvent and 1 M aqueous NaOH as
the base. The crude reaction mixture was evaporated to dryness and the residue
dissolved in CH2Cl2 and washed with H2O. The aqueous layer was extracted twice with
CH2Cl2. The combined organic layers were dried over MgSO4 and the solvent was
removed in order to recieve the crude sulfide as a yellow solid. After oxidation the
crude product was purified by column chromatography (n-hexane/EtOAc : 3/1)
yielding 4-chloro-1-((4-chlorophenyl)sulfonyl)-2-nitrobenzene as a white powder
(850 mg, 2.83 mmol, 55% yield).
m.p.: 134°C

Materials and Methods
83
1H NMR (400 MHz, Chloroform-d) δ: 8.29 (d, J = 8.3 Hz, 1H), 7.94 – 7.85 (m, 2H),
7.79 – 7.71 (m, 2H), 7.59 – 7.49 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 148.97, 141.48, 141.05, 138.70, 132.92,
132.82, 129.96, 129.79, 129.70, 125.30.
CHN (calc/found %): C(43.39/43.34), H(2.12/2.28), N(4.22/4.29).
1-((4-Chlorophenyl)sulfonyl)-2,4-dinitrobenzene (63)
C12H7ClN2O6S
M: 342.71 g mol-1
1-((4-Chlorophenyl)sulfonyl)-2,4-dinitrobenzene was synthesized according to
Method D using 1-chloro-2,4-dinitrobenzene (284 mg, 1.4 mmol) and 4-
chlorobenzenethiol (202 mg, 4.5 mmol). Upon cooling after the oxidation with H2O2
1-((4-chlorophenyl)sulfonyl)-2,4-dinitrobenzene crystallizes from glacial acetic acid
(368 mg, 1.1 mmol, 77% yield).
m.p.: 166°C
1H NMR (400 MHz, Chloroform-d) δ: 8.55 – 8.49 (m, 2H), 8.48 (dd, J = 2.0, 0.8 Hz,
1H), 7.92 – 7.78 (m, 2H), 7.53 – 7.47 (m, 2H).
13C NMR (101 MHz, Chloroform-d) δ: 150.72, 148.92, 141.91, 139.79, 137.58,
133.49, 130.36, 130.00, 127.07, 120.44.
CHN (calc/found %): C(42.06/41.90), H(2.06/2.34), N(8.17/7.99).
4-Chloro-2-nitro-1-phenoxybenzene (71)168
C12H8ClNO3
M: 249.65 g mol-1

Materials and Methods
84
To a solution of phenol (508 mg, 5.4 mmol) in DMSO (30 mL) was added KOH (303
mg, 5.4 mmol) and the reaction mixture was heated to 50°C for 30 min. After cooling
to ambient temperature a solution of 1,4-dichloro-2-nitrobenzene (1.037 g, 5.4
mmol) in DMSO (7 mL) was added and heated to 90°C for 16 h. After addition of H2O
(15 mL) the aqueous phase was extracted three times with EtO2 (50 mL). The organic
layer was washed with H2O and dried over MgSO4. After removal of the solvent 4-
chloro-2-nitro-1-phenoxybenzene was received as brown oil (1.300 g, 5.2 mmol, 96%
yield).
1H NMR (400 MHz, Chloroform-d) δ: 7.95 (d, J = 2.6 Hz, 1H), 7.45 (dd, J = 8.9, 2.6 Hz,
1H), 7.43 – 7.36 (m, 2H), 7.24 – 7.18 (m, 1H), 7.07 – 7.02 (m, 2H), 6.96 (d, J = 8.9 Hz,
1H).
13C NMR (151 MHz, Chloroform-d) δ: 155.53, 149.68, 141.42, 134.22, 130.35,
128.23, 125.74, 125.12, 121.62, 119.43.
CHN (calc/found %): C(57.37/57.48), H(3.23/3.35), N(5.61/5.72).
4-Chloro-1-iodo-2-nitrobenzene (72)154
C6H3ClINO2
M: 283.45 g mol-1
Para-toluenesulfonic acid (1.7 g, 9 mmol) was dissolved in MeCN (12 mL). Upon
addition of 4-chloro-2-nitroaniline (0.52 g, 3 mmol) a yellow salt was formed. The
reaction mixture was cooled to 0°C and a solution of NaNO2 (0.41 g, 6 mmol) and KI
(1.2 g, 7.5 mmol) in water (1.8 mL) was added dropwise. After 10 min the reaction
mixture was allowed to warm to ambient temperature and was stirred overnight. H2O
(50 mL) was added and the pH was brought to 9 by addition of saturated aq. Na2CO3
solution. The aqueouse phase was extracted three times with Et2O (16 mL). The
combined organic phases were washed with saturated aq. NaCl solution, dried over
MgSO4 and the solvent was removed. The crude oil was purified by column
chromatography (n-hexane/CH2Cl2 = 3/1) to yield 4-chloro-1-iodo-2-nitrobenzene as
yellow solid (0.58 g, 2 mmol, 68% yield).

Materials and Methods
85
1H NMR (400 MHz, DMSO-d6) δ: 8.12 (d, J = 2.4 Hz, 1H), 8.10 (d, J = 8.5 Hz, 1H), 7.52
(dd, J = 8.5, 2.4 Hz, 1H).
13C NMR (101 MHz, DMSO-d6) δ: 154.18, 142.38, 133.88, 133.57, 124.78, 86.33.
4-Chloro-2-nitrobenzenediazonium (74)156
C6H3ClN3O2+
M: 184.56 g mol-1
4-Chloro-2-nitroaniline (8.6 g, 50 mmol) was suspended in 50% aqueous HBF4-
solution (70 mL) and diazotized at 0°C with a aq. NaNO2 solution (3.4 g, 50 mmol).
After 15 min the precipitate was separated and washed with EtOH and Et2O.
Afterwards, the salt was dissolved in MeCN and precipitated upon addition of Et2O to
yield 4-chloro-2-nitrobenzenediazonium tetrafluoroborate as off-white salt (5.2 g, 19
mmol, 38% yield).
6.4 Biochemical and Cellular Assays
Protein Expression and Purification from Bacteria via Poly-histidine (His) Tag
All kinesin motor domains were expressed and purified as described previously.106
Bacteria of the E.coli strain BL21RIL were transformed with the plasmids coding for
poly-histidine-tagged motor proteins in the presence of selective antibiotics
(ampicillin 100 μg mL-1 and chloramphenicol 34 μg mL-1) and incubated over night at
37°C. Multiple colonies were used to inoculate 25 to 100 mL culture of LB medium,
grown over night at 37°C including antibiotics as above. 10 mL per liter of pre-
inoculum were used to inoculate a new liquid culture (2 to 4 liters) plus antibiotics as
above, which was grown at 37°C until OD600 0.6 units was reached. Afterwards the
expression of the recombinant protein was induced with 0.5 mM IPTG and allowed
for 18 h at 18°C and the bacteria were harvested by centrifugation (4500 x g, 15 min,
4°C). The pellet was frozen (-80°C, 20 min) and thawe, resuspended with 10 mL lysis
buffer (20 mM Tris pH 8.0, 300 mM NaCl, 5 mM imidazole, 0.1% Triton X 100,

Materials and Methods
86
Complete protease inhibitor Roche) per liter of original culture. The cells were lysed
by high-pressure treatment using the EmulsiFlex-C5 or C3 homogenizer (Avestin) and
then cleared by centrifugation (40000 x g, 30 min, 4°C). The lysate was incubated with
Ni-NTA beads (Qiagen) for 2 h at 4°C on a rotating wheel (0.25 to 0.5 mL resin per
liter of original culture), washed 3 times in batch with 20 mL washing buffer (20 mM
Tris pH 8.0, 300 mM NaCl, 20 mM imidazole, +/- 1 mM NaATP and 1 mM MgCl2) and
finally eluted over a Poly- Prep Chromatography Column (Bio-Rad) in 0.5 to 1 mL
fractions (elution buffer: 20 mM Tris pH 8.0, 300 mM NaCl, 200 mM imidazole). The
fractions containing the recombinant protein were pooled together and dialyzed o/n
at 4°C (dialysis buffer: 10 mM β-mercaptoethanol, 10% glycerol, 25 mM Tris pH 7.4,
300 mM NaCl) then aliquoted, snap-frozen and stored at -80°C.
Polymerization of Taxol Stabilized Microtubules
1 mL of pig brain purified tubulin dimers (10 mg mL-1) was precleared by
ultracentrifugation (186000 g, 4°C, 10 min) and supplemented with 25% glycerol, 1.5
mM GTP and 668 µl BRB80. The mixture was incubated at 37°C for 30 min and
afterwards taxol was added to a final concentration of 40 µM to the mix and the
microtubules were allowed to polymerize for additional 30 min at 37°C. Finally the
microtubules were pelleted by ultra-centrifugation (186000 g, 30 min, 35°C) and the
pellet was resuspended in BRB80 containing 25 μM taxol. The concentration of
polymerized microtubules was determined by measuring the absorbance at 280 nm
of tubulin dimers (resuspended in 6 M guanidine-HCl).
Malachite Green Assay
Mklp2MD (80 nM), taxol stabilized microtubules (1 µM), Triton X-100 (0.1%), MgATP
(400 µM) and SH-1 (10 µM, 25 µM, 50 µM and 100 µM) were incubated in buffer
containing 20 mM PIPES (pH 6.8), 1 mM MgCl2, 1 mM EGTA, 0.1 mg mL-1 BSA and 0.1
μM taxol at room temperature. In 6 minute intervals 10 µL of reaction mixture were
quenched by addition of 40 μl 1 M HClO4. 50 μl malachite green solution (1.3 M HCl,

Materials and Methods
87
8.5 mM ammonium molybdate, 363 nM malachite green) were added. After 20
minutes at room temperature absorbance at 650 nm was measured. Relative
absorbance was calculated by subtracting the absorbance after 0 min.
Enzyme Coupled Assay
A mix of MgATP, taxol stabilized microtubules, 200 µM NADH, 5 mM PEP (pH 7), 4 U
mL-1 Pyruvate kinase, 8 U mL-1 Lactate dehydrogenase and assay buffer (10 mM
Imidazole/acetate pH 7.2, 5 mM Mg-acetate, 2 mM EGTA, 20 µM taxol) was
supplemented with DMSO or compounds. The reaction was started by addition of
kinesin motor domain and absorbance was measured every 30 seconds at 340 nm for
10 minutes using the Tecan 500 plate reader. DMSO was used as solvent control and
the activity of the kinesin motor domain was set to 100%. The ATPase activity was
calculated using following equation
[(-∆A340nm min-1/(εNADH × d)]/c(Kif18A)/(60s × min-1)
Enzyme Coupled Assay SH1 Analogs
A mix of 1 mM MgATP, 2µM taxol stabilized microtubules, 40 nM Mklp2MD and the
standard conditions as described above were used. DMSO was applied in 5% or 10%
and the SH-1 analogs at 50 µm.
Enzyme Coupled Assay SH1 Titration with Triton X-100
A mix of 1 µM MgATP, 2 µM or 400 nM taxol stabilized microtubules, 40 nM Mklp2MD
and the standard conditions as described above were used. DMSO was applied at 5%
and SH1 in 25 µM, 50 µM 75 µM and 100 µM concentration.

Materials and Methods
88
Enzyme Coupled Assay BTB-1 Analogs
A mix of 650 µM MgATP, 3 µM taxol stabilized microtubules, Kif18AMD (20 nM) and
the standard conditions as described above were used.The mix was supplemented
with DMSO (0.5%) or BTB-1 and analogs (5µM).
IC50 (Kif18A)
Increasing concentrations of BTB-1 analogs were used to determine the residual
activity of 20 nM His-Kif18AMD in the presence of 650 μM ATP and 3 μM taxol
stabilized microtubules by enzyme-coupled assay (see above). DMSO was used as
solvent control and the activity of the protein in its presence was set to 100%. The
assays were performed in 384 well plates and the oxidation of NADH over time was
monitored using a plate reader as reported above.
Basal Kif18A ATPase Activity
The ATPase activity of 300 nM Kif18AMD without microtubules was determined using
the enzyme coupled assay (as described above) in the presence of 50 µM BTB-1, 53
and 59 or 0.5% DMSO as solvent control.
Inhibition Mode of 59
Increasing concentrations of 59 and ATP were used in an enzym coupled assay (as
described above) to elucidate the inhibition mode. According to the Michaelis-
Menten kinetics each data set was fit to competitive inhibitory model using the
software Prism 5 (GraphPad).
Cellular Thermal Shift Assay (CETSA)159
In order to prepare a mitotic extract HeLa cells were synchronized using 2mM
thymidine (24 h) and released into 0.5 µM Nocodazole (14 h). The cells were shook

Materials and Methods
89
off, washed three times with PBS and released in fresh media for 30 min. After
harvesting, the cell suspension was washed three times with PBS supplemented with
Complete protease inhibitor (Roche) and snap frozen in liquid nitrogen. The cell
suspension was freeze-thawed three times and cleared by centrifugation (20000 g,
20 min at 4°C). The supernatant was diluted with PBS containing Complete protease
inhibitor (Roche) and divided into three aliquots, with two aliquots treated with
compounds and one aliquot used as DMSO control. The samples were incubated for
30 min at room temperature and divided into smaller aliquots (25 µl), which were
heated individually at different temperatures (46-62°C in 2°C steps) using the Veriti
thermal cycler followed by cooling to room temperature for 3 min. The treated
lysates were cleared by centrifugation (20000 g, 20 min at 4°C) and analyzed by SDS-
PAGE followed by western blot analysis.
Western Blot Analysis
SDS page electrophoresis and western blot analysis were performed according to
standard protocols.169 The detection of the protein of interest was performed using
specific primary antibodies, commercially available HRP-conjugated secondary
antibodies (Bio-Rad and Dianova) and ECL (100 mM Tris-HCl pH 8.5, 1.25 mM luminol,
225 μM coumaric acid, 0.015% v/v H2O2) reagent.
Antigen Organism Specification/Antibody number
Kif18A (human) Rabbit M. Mayr42
Tubulin Mouse FITC conjugated (SIGMA)
CycB Mouse TUM359
Cdc27 Rabbit TUM 229
Tubulin Polymerization Assay
A mix of 0.8 µM glutamate (pH 6.6), 100 µM MgCl2 and 10 µM tubulin was
supplemented with DMSO (0.5%), compound (50µM) or Nocodazole, which was used
as a control tubulin polymerization inhibitor, and incubated for 15 min at ambient

Materials and Methods
90
temperature. Afterwards, the mixtures were cooled on an ice bath for 5 min and the
polymerization was started by addition of GTP (final conc. 0.4 mM) and incubated for
30 sec at 30°C. The assays were performed in 384 well plates and the polymerization
was measured turbidimetrically at 350 nm every 20 sec over a time period of 30 min
using the plate VARIOSKAN FLASH from Thermo Scientific. The tubulin polymerization
velocity was determined by linear regression and compared to the DMSO control
which was set to 100%.
IC50 (Tub.Polym.)
The experiments were performed using the conditions described above in quartz
cuvettes with the final assay volume of 150 µL with increasing concentrations of BTB-
1 and its analogs. The polymerization was measured with VarianInc Cary100bio
spectrophotometer for 20 min at 30°C and the depolymerization for 15 min at 15°C.
The tubulin polymerization velocity was determined by linear regression and
compared to the DMSO control which was set to 100%.
Cell Culture
HeLa cells were cultured in Dulbecco’s modified Eagle medium (DMEM)
supplemented with 10% fetal calf serum (Gibco), penicillin (100 U mL-1) and
streptomycin (100 μg mL-1) at 37°C in a humid atmosphere with 5% CO2.
Immunofluorescence
A DeltaVision Core system (Applied Precision) mounted on a IX-71 inverted
microscope (Olympus) equipped with a LED-system, a CoolSnap HQ-2 camera
(Photometrics) and a 40 x 1.35 NA UApo/340 (Olympus) oil objective was used for
image acquistion.
HeLa cells stably expressing GFP labeled Kif18A, as previously described in Häfner et
al.49 were synchronized using 2 mM thymidine (18 hours) and plated on coverslips in
6 well plates (200,000 cells/well). The coverslips were treated with different
concentrations of BTB-1 or its analogs or DMSO as solvent control for 30 min. The

Materials and Methods
91
cells were then treated and immunostained as described previously to visualize
microtubules and DNA.42
Live Cell Imaging
A 20 x 0.4 NA LD Plan Neo (Zeiss) objective on a Axio Observer Z1 (Zeiss) equipped
with an environmental chamber (Zeiss), Colibri LED Modules and a CoolSnap-ES2
camera (Photometrics) controlled by MetaMorph Software were used for image
acquisition.
SH1 Analogs
For live cell imaging a stably GFP-H2B expressing HeLa cell line was used. The cells
were seeded in a 12 well plate 24 h prior to use in a density of 100000 mL-1 in 2mM
thymidine. After 16 h the cells were released from the thymidine block and after 8 h
the CO2 independent live cell media premixed with compound solution was applied
to the cells. The cells were subsequently imaged for about 18 hours. Five random
positions were determined per well and every 10 minutes a picture was taken of
every position using bright field and the 470 nm (for GFP) fluorescence channel.
BTB-1 Analogs
HeLa cells stably expressing GFP labeled Kif18A, as previously described in Häfner et
al.49 were synchronized using 2 mM thymidine in a density of 200000 mL-1 (18 hours).
The cells were released from the thymidine block and after 6 h the CO2 independent
live cell media premixed with compound solution was applied to the cells. The cells
were subsequently imaged for about 18 hours. Five random positions were
determined per well and every 10 minutes a picture was taken of every position using
bright field and the 470 nm (for GFP) fluorescence channel.
Alamar Blue Assay158 and EC50 Values
HeLa cells were seeded in 96-well plates (4,000 cells/well) and allowed to attach for
24 h. Compounds to be tested were dissolved in a suitable amount of DMSO and
different concentrations were prepared to give final concentrations with a maximum

Materials and Methods
92
DMSO content of 1%. The cells were incubated for 48 h with different concentrations
of the compound. AlamarBlue (10 µL) was added and the cells were incubated for
another hour. After excitation at 530 nm, fluorescence at 590 nm was measured using
a FL600 Fluorescence Microplate Reader (Bio-TEK). Cell viability is expressed in
percent with respect to a control containing 1% DMSO.

References
93
7. References
(1) Morgan, D. O. (2007) The Cell Cycle Principles of Control, Oxford University Press.
(2) Musacchio, A., and Salmon, E. D. (2007) The spindle-assembly checkpoint in space and time, Nat. Rev. Mol. Cell Biol. 8, 379-393.
(3) Woehlke, G., and Schliwa, M. (2000) Walking on two heads: the many talents of kinesin, Nat. Rev. Mol. Cell Biol. 1, 50-58.
(4) Cross, R. A., and McAinsh, A. (2014) Prime movers: the mechanochemistry of mitotic kinesins, Nat. Rev. Mol. Cell Biol. 15, 257-271.
(5) Gigant, B., Wang, W., Dreier, B., Jiang, Q., Pecqueur, L., Plückthun, A., Wang, C., and Knossow, M. (2013) Structure of a kinesin–tubulin complex and implications for kinesin motility, Nat. Struct. Mol. Biol. 20, 1001-1007.
(6) Seeger, M. A., Zhang, Y., and Rice, S. E. (2012) Kinesin tail domains are intrinsically disordered, Proteins: Struct., Funct., Bioinf. 80, 2437-2446.
(7) Verhey, K. J., and Hammond, J. W. (2009) Traffic control: regulation of kinesin motors, Nat. Rev. Mol. Cell Biol. 10, 765-777.
(8) Kapitein, L. C., Peterman, E. J., Kwok, B. H., Kim, J. H., Kapoor, T. M., and Schmidt, C. F. (2005) The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks, Nature 435, 114-118.
(9) Blangy, A. (1995) Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo, Cell 83, 1159-1169.
(10) Tanenbaum, M. E., and Medema, R. H. (2010) Mechanisms of centrosome separation and bipolar spindle assembly, Dev. Cell 19, 797-806.
(11) Mayer, T. U., Kapoor, T. M., Haggarty, S. J., King, R. W., Schreiber, S. L., and Mitchison, T. J. (1999) Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen, Science 286, 971-974.
(12) Kashina, A. S., Baskin, R. J., Cole, D. G., Wedaman, K. P., Saxton, W. M., and Scholey, J. M. (1996) A bipolar kinesin, Nature 379, 270-272.
(13) Acar, S., Carlson, D. B., Budamagunta, M. S., Yarov-Yarovoy, V., Correia, J. J., Ninonuevo, M. R., Jia, W., Tao, L., Leary, J. A., Voss, J. C., Evans, J. E., and Scholey, J. M. (2013) The bipolar assembly domain of the mitotic motor kinesin-5, Nat Commun 4, 1343.

References
94
(14) Kapitein, L. C., Kwok, B. H., Weinger, J. S., Schmidt, C. F., Kapoor, T. M., and Peterman, E. J. (2008) Microtubule cross-linking triggers the directional motility of kinesin-5, J. Cell Biol. 182, 421-428.
(15) Weinger, J. S., Qiu, M., Yang, G., and Kapoor, T. M. (2011) A nonmotor microtubule binding site in kinesin-5 is required for filament crosslinking and sliding, Curr. Biol. 21, 154-160.
(16) Kwok, B. H., Kapitein, L. C., Kim, J. H., Peterman, E. J., Schmidt, C. F., and Kapoor, T. M. (2006) Allosteric inhibition of kinesin-5 modulates its processive directional motility, Nat. Chem. Biol. 2, 480-485.
(17) Kwok, B. H., Yang, J. G., and Kapoor, T. M. (2004) The rate of bipolar spindle assembly depends on the microtubule-gliding velocity of the mitotic kinesin Eg5, Curr. Biol. 14, 1783-1788.
(18) Walczak, C. E., and Heald, R. (2008) Mechanisms of mitotic spindle assembly and function, Int. Rev. Cytol. 265, 111-158.
(19) Zhai, Y., Kronebusch, P. J., and Borisy, G. G. (1995) Kinetochore microtubule dynamics and the metaphase-anaphase transition, J. Cell Biol. 131, 721-734.
(20) Rosenblatt, J., Cramer, L. P., Baum, B., and McGee, K. M. (2004) Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly, Cell 117, 361-372.
(21) Toso, A., Winter, J. R., Garrod, A. J., Amaro, A. C., Meraldi, P., and McAinsh, A. D. (2009) Kinetochore-generated pushing forces separate centrosomes during bipolar spindle assembly, J. Cell Biol. 184, 365-372.
(22) Whitehead, C. M., and Rattner, J. B. (1998) Expanding the role of HsEg5 within the mitotic and post-mitotic phases of the cell cycle, J. Cell Sci. 111 ( Pt 17), 2551-2561.
(23) Sturgill, E. G., and Ohi, R. (2013) Kinesin-12 differentially affects spindle assembly depending on its microtubule substrate, Curr. Biol. 23, 1280-1290.
(24) Tanenbaum, M. E., Macurek, L., Janssen, A., Geers, E. F., Alvarez-Fernandez, M., and Medema, R. H. (2009) Kif15 cooperates with eg5 to promote bipolar spindle assembly, Curr. Biol. 19, 1703-1711.
(25) Vladimirou, E., McHedlishvili, N., Gasic, I., Armond, J. W., Samora, C. P., Meraldi, P., and McAinsh, A. D. (2013) Nonautonomous movement of chromosomes in mitosis, Dev. Cell 27, 60-71.
(26) Mountain, V., Simerly, C., Howard, L., Ando, A., Schatten, G., and Compton, D. A. (1999) The kinesin-related protein, HSET, opposes the activity of Eg5 and cross-links microtubules in the mammalian mitotic spindle, J. Cell Biol. 147, 351-366.

References
95
(27) Vanneste, D., Takagi, M., Imamoto, N., and Vernos, I. (2009) The role of Hklp2 in the stabilization and maintenance of spindle bipolarity, Curr. Biol. 19, 1712-1717.
(28) Braun, M., Drummond, D. R., Cross, R. A., and McAinsh, A. D. (2009) The kinesin-14 Klp2 organizes microtubules into parallel bundles by an ATP-dependent sorting mechanism, Nat. Cell Biol. 11, 724-730.
(29) Braun, M., Lansky, Z., Fink, G., Ruhnow, F., Diez, S., and Janson, M. E. (2011) Adaptive braking by Ase1 prevents overlapping microtubules from sliding completely apart, Nat. Cell Biol. 13, 1259-1264.
(30) Fink, G., Hajdo, L., Skowronek, K. J., Reuther, C., Kasprzak, A. A., and Diez, S. (2009) The mitotic kinesin-14 Ncd drives directional microtubule-microtubule sliding, Nat. Cell Biol. 11, 717-723.
(31) Saunders, W., Lengyel, V., and Hoyt, M. A. (1997) Mitotic spindle function in Saccharomyces cerevisiae requires a balance between different types of kinesin-related motors, Mol. Biol. Cell 8, 1025-1033.
(32) Sharp, D. J., Rogers, G. C., and Scholey, J. M. (2000) Microtubule motors in mitosis, Nature 407, 41-47.
(33) Hentrich, C., and Surrey, T. (2010) Microtubule organization by the antagonistic mitotic motors kinesin-5 and kinesin-14, J. Cell Biol. 189, 465-480.
(34) Kops, G. J., Saurin, A. T., and Meraldi, P. (2010) Finding the middle ground: how kinetochores power chromosome congression, Cell. Mol. Life Sci. 67, 2145-2161.
(35) Kapoor, T. M., Lampson, M. A., Hergert, P., Cameron, L., Cimini, D., Salmon, E. D., McEwen, B. F., and Khodjakov, A. (2006) Chromosomes can congress to the metaphase plate before biorientation, Science 311, 388-391.
(36) Wood, K. W., Sakowicz, R., Goldstein, L. S., and Cleveland, D. W. (1997) CENP-E is a plus end-directed kinetochore motor required for metaphase chromosome alignment, Cell 91, 357-366.
(37) Schaar, B. T., Chan, G. K., Maddox, P., Salmon, E. D., and Yen, T. J. (1997) CENP-E function at kinetochores is essential for chromosome alignment, J. Cell Biol. 139, 1373-1382.
(38) Yardimci, H., van Duffelen, M., Mao, Y., Rosenfeld, S. S., and Selvin, P. R. (2008) The mitotic kinesin CENP-E is a processive transport motor, Proc. Natl. Acad. Sci. U. S. A. 105, 6016-6021.
(39) Sardar, H. S., and Gilbert, S. P. (2012) Microtubule capture by mitotic kinesin centromere protein E (CENP-E), J. Biol. Chem. 287, 24894-24904.

References
96
(40) Roostalu, J., and Surrey, T. (2013) The multiple talents of kinesin-8, Nat. Cell Biol. 15, 889-891.
(41) Su, X., Arellano-Santoyo, H., Portran, D., Gaillard, J., Vantard, M., Thery, M., and Pellman, D. (2013) Microtubule-sliding activity of a kinesin-8 promotes spindle assembly and spindle-length control, Nat. Cell Biol. 15, 948-957.
(42) Mayr, M. I., Hummer, S., Bormann, J., Gruner, T., Adio, S., Woehlke, G., and Mayer, T. U. (2007) The human kinesin Kif18A is a motile microtubule depolymerase essential for chromosome congression, Curr. Biol. 17, 488-498.
(43) Mayr, M. I., Storch, M., Howard, J., and Mayer, T. U. (2011) A non-motor microtubule binding site is essential for the high processivity and mitotic function of kinesin-8 Kif18A, PLoS One 6, e27471.
(44) Weaver, L. N., Ems-McClung, S. C., Stout, J. R., LeBlanc, C., Shaw, S. L., Gardner, M. K., and Walczak, C. E. (2011) Kif18A uses a microtubule binding site in the tail for plus-end localization and spindle length regulation, Curr. Biol. 21, 1500-1506.
(45) Stumpff, J., Du, Y., English, C. A., Maliga, Z., Wagenbach, M., Asbury, C. L., Wordeman, L., and Ohi, R. (2011) A tethering mechanism controls the processivity and kinetochore-microtubule plus-end enrichment of the kinesin-8 Kif18A, Mol. Cell 43, 764-775.
(46) Stumpff, J., von Dassow, G., Wagenbach, M., Asbury, C., and Wordeman, L. (2008) The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment, Dev. Cell 14, 252-262.
(47) Du, Y., English, C. A., and Ohi, R. (2010) The kinesin-8 Kif18A dampens microtubule plus-end dynamics, Curr. Biol. 20, 374-380.
(48) Stumpff, J., Wagenbach, M., Franck, A., Asbury, C. L., and Wordeman, L. (2012) Kif18A and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension, Dev. Cell 22, 1017-1029.
(49) Häfner, J., Mayr, M. I., Möckel, M. M., and Mayer, T. U. (2014) Pre-anaphase chromosome oscillations are regulated by the antagonistic activities of Cdk1 and PP1 on Kif18A, Nat Commun 5, 4397.
(50) Jaqaman, K., King, E. M., Amaro, A. C., Winter, J. R., Dorn, J. F., Elliott, H. L., McHedlishvili, N., McClelland, S. E., Porter, I. M., Posch, M., Toso, A., Danuser, G., McAinsh, A. D., Meraldi, P., and Swedlow, J. R. (2010) Kinetochore alignment within the metaphase plate is regulated by centromere stiffness and microtubule depolymerases, J. Cell Biol. 188, 665-679.

References
97
(51) Peters, C., Brejc, K., Belmont, L., Bodey, A. J., Lee, Y., Yu, M., Guo, J., Sakowicz, R., Hartman, J., and Moores, C. A. (2010) Insight into the molecular mechanism of the multitasking kinesin-8 motor, EMBO J. 29, 3437-3447.
(52) Waters, J. C., Mitchison, T. J., Rieder, C. L., and Salmon, E. D. (1996) The kinetochore microtubule minus-end disassembly associated with poleward flux produces a force that can do work, Mol. Biol. Cell 7, 1547-1558.
(53) Ganem, N. J., Upton, K., and Compton, D. A. (2005) Efficient mitosis in human cells lacking poleward microtubule flux, Curr. Biol. 15, 1827-1832.
(54) Wordeman, L., Wagenbach, M., and von Dassow, G. (2007) MCAK facilitates chromosome movement by promoting kinetochore microtubule turnover, J. Cell Biol. 179, 869-879.
(55) Bakhoum, S. F., Thompson, S. L., Manning, A. L., and Compton, D. A. (2009) Genome stability is ensured by temporal control of kinetochore-microtubule dynamics, Nat. Cell Biol. 11, 27-35.
(56) Glotzer, M. (2009) The 3Ms of central spindle assembly: microtubules, motors and MAPs, Nature Rev. Mol. Cell Biol. 10, 9-20.
(57) Schroeder, T. E. (1968) Cytokinesis: filaments in the cleavage furrow, Exp. Cell Res. 53, 272-276.
(58) Schroeder, T. E. (1973) Actin in dividing cells: contractile ring filaments bind heavy meromyosin, Proc. Natl. Acad. Sci. U. S. A. 70, 1688-1692.
(59) Eggert, U. S., Mitchison, T. J., and Field, C. M. (2006) Animal cytokinesis: from parts list to mechanisms, Annu. Rev. Biochem. 75, 543-566.
(60) Barr, F. A., and Gruneberg, U. (2007) Cytokinesis: placing and making the final cut, Cell 131, 847-860.
(61) Piekny, A., Werner, M., and Glotzer, M. (2005) Cytokinesis: welcome to the Rho zone, Trends Cell Biol 15, 651-658.
(62) Werner, M., and Glotzer, M. (2008) Control of cortical contractility during cytokinesis, Biochem. Soc. Trans. 36, 371-377.
(63) Glotzer, M. (2004) Cleavage furrow positioning, J. Cell Biol. 164, 347-351.
(64) Somers, W. G., and Saint, R. (2003) A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis, Dev. Cell 4, 29-39.
(65) Zhu, C., and Jiang, W. (2005) Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis, Proc. Natl. Acad. Sci. U. S. A. 102, 343-348.

References
98
(66) Mollinari, C., Kleman, J. P., Jiang, W., Schoehn, G., Hunter, T., and Margolis, R. L. (2002) PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone, J. Cell Biol. 157, 1175-1186.
(67) Kurasawa, Y., Earnshaw, W. C., Mochizuki, Y., Dohmae, N., and Todokoro, K. (2004) Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation, EMBO J. 23, 3237-3248.
(68) Bieling, P., Telley, I. A., and Surrey, T. (2010) A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps, Cell 142, 420-432.
(69) Hu, C. K., Coughlin, M., Field, C. M., and Mitchison, T. J. (2011) KIF4 regulates midzone length during cytokinesis, Curr. Biol. 21, 815-824.
(70) Mishima, M., Kaitna, S., and Glotzer, M. (2002) Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity, Dev. Cell 2, 41-54.
(71) Pavicic-Kaltenbrunner, V., Mishima, M., and Glotzer, M. (2007) Cooperative assembly of CYK-4/MgcRacGAP and ZEN-4/MKLP1 to form the centralspindlin complex, Mol. Biol. Cell 18, 4992-5003.
(72) Glotzer, M. (2005) The molecular requirements for cytokinesis, Science 307, 1735-1739.
(73) Ban, R., Irino, Y., Fukami, K., and Tanaka, H. (2004) Human mitotic spindle-associated protein PRC1 inhibits MgcRacGAP activity toward Cdc42 during the metaphase, J. Biol. Chem. 279, 16394-16402.
(74) Guse, A., Mishima, M., and Glotzer, M. (2005) Phosphorylation of ZEN-4/MKLP1 by Aurora B regulates completion of cytokinesis, Curr. Biol. 15, 778-786.
(75) Neef, R., Klein, U. R., Kopajtich, R., and Barr, F. A. (2006) Cooperation between mitotic kinesins controls the late stages of cytokinesis, Curr. Biol. 16, 301-307.
(76) Wheatley, S. P., Carvalho, A., Vagnarelli, P., and Earnshaw, W. C. (2001) INCENP is required for proper targeting of Survivin to the centromeres and the anaphase spindle during mitosis, Curr. Biol. 11, 886-890.
(77) Gruneberg, U., Neef, R., Honda, R., Nigg, E. A., and Barr, F. A. (2004) Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2, J. Cell Biol. 166, 167-172.
(78) Neef, R., Gruneberg, U., Kopajtich, R., Li, X., Nigg, E. A., Sillje, H., and Barr, F. A. (2007) Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1, Nat. Cell Biol. 9, 436-444.

References
99
(79) Neef, R. (2003) Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis, J. Cell Biol. 162, 863-875.
(80) Abaza, A., Soleilhac, J. M., Westendorf, J., Piel, M., Crevel, I., Roux, A., and Pirollet, F. (2003) M phase phosphoprotein 1 is a human plus-end-directed kinesin-related protein required for cytokinesis, J. Biol. Chem. 278, 27844-27852.
(81) Rath, O., and Kozielski, F. (2012) Kinesins and cancer, Nat. Rev. Cancer 12, 527-539.
(82) Sarli, V., and Giannis, A. (2006) Inhibitors of mitotic kinesins: next-generation antimitotics, ChemMedChem 1, 293-298.
(83) Yu, Y., and Feng, Y. M. (2010) The role of kinesin family proteins in tumorigenesis and progression: potential biomarkers and molecular targets for cancer therapy, Cancer 116, 5150-5160.
(84) Liu, X., Gong, H., and Huang, K. (2013) Oncogenic role of kinesin proteins and targeting kinesin therapy, Cancer Sci. 104, 651-656.
(85) Corson, T. W., and Gallie, B. L. (2006) KIF14 mRNA expression is a predictor of grade and outcome in breast cancer, Int. J. Cancer 119, 1088-1094.
(86) Theriault, B. L., Pajovic, S., Bernardini, M. Q., Shaw, P. A., and Gallie, B. L. (2012) Kinesin family member 14: an independent prognostic marker and potential therapeutic target for ovarian cancer, Int. J. Cancer 130, 1844-1854.
(87) Corson, T. W., Zhu, C. Q., Lau, S. K., Shepherd, F. A., Tsao, M. S., and Gallie, B. L. (2007) KIF14 messenger RNA expression is independently prognostic for outcome in lung cancer, Clin Cancer Res 13, 3229-3234.
(88) Ishikawa, K., Kamohara, Y., Tanaka, F., Haraguchi, N., Mimori, K., Inoue, H., and Mori, M. (2008) Mitotic centromere-associated kinesin is a novel marker for prognosis and lymph node metastasis in colorectal cancer, Br. J. Cancer 98, 1824-1829.
(89) Taniwaki, M., Takano, A., Ishikawa, N., Yasui, W., Inai, K., Nishimura, H., Tsuchiya, E., Kohno, N., Nakamura, Y., and Daigo, Y. (2007) Activation of KIF4A as a prognostic biomarker and therapeutic target for lung cancer, Clin Cancer Res 13, 6624-6631.
(90) Grinberg-Rashi, H., Ofek, E., Perelman, M., Skarda, J., Yaron, P., Hajduch, M., Jacob-Hirsch, J., Amariglio, N., Krupsky, M., Simansky, D. A., Ram, Z., Pfeffer, R., Galernter, I., Steinberg, D. M., Ben-Dov, I., Rechavi, G., and Izraeli, S. (2009) The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain, Clin Cancer Res 15, 1755-1761.

References
100
(91) De, S., Cipriano, R., Jackson, M. W., and Stark, G. R. (2009) Overexpression of kinesins mediates docetaxel resistance in breast cancer cells, Cancer Res. 69, 8035-8042.
(92) Wang, C. Q., Qu, X., Zhang, X. Y., Zhou, C. J., Liu, G. X., Dong, Z. Q., Wei, F. C., and Sun, S. Z. (2010) Overexpression of Kif2a promotes the progression and metastasis of squamous cell carcinoma of the oral tongue, Oral Oncol 46, 65-69.
(93) Shimo, A., Tanikawa, C., Nishidate, T., Lin, M. L., Matsuda, K., Park, J. H., Ueki, T., Ohta, T., Hirata, K., Fukuda, M., Nakamura, Y., and Katagiri, T. (2008) Involvement of kinesin family member 2C/mitotic centromere-associated kinesin overexpression in mammary carcinogenesis, Cancer Sci. 99, 62-70.
(94) Nakamura, Y., Tanaka, F., Haraguchi, N., Mimori, K., Matsumoto, T., Inoue, H., Yanaga, K., and Mori, M. (2007) Clinicopathological and biological significance of mitotic centromere-associated kinesin overexpression in human gastric cancer, Br. J. Cancer 97, 543-549.
(95) Aoki, S., Ohta, K., Yamazaki, T., Sugawara, F., and Sakaguchi, K. (2005) Mammalian mitotic centromere-associated kinesin (MCAK): a new molecular target of sulfoquinovosylacylglycerols novel antitumor and immunosuppressive agents, FEBS J. 272, 2132-2140.
(96) Narayan, G., Bourdon, V., Chaganti, S., Arias-Pulido, H., Nandula, S. V., Rao, P. H., Gissmann, L., Durst, M., Schneider, A., Pothuri, B., Mansukhani, M., Basso, K., Chaganti, R. S., and Murty, V. V. (2007) Gene dosage alterations revealed by cDNA microarray analysis in cervical cancer: identification of candidate amplified and overexpressed genes, Genes, Chromosomes Cancer 46, 373-384.
(97) Liu, Z., Ling, K., Wu, X., Cao, J., Liu, B., Li, S., Si, Q., Cai, Y., Yan, C., Zhang, Y., and Weng, Y. (2009) Reduced expression of cenp-e in human hepatocellular carcinoma, J Exp Clin Cancer Res 28, 156.
(98) Agarwal, R., Gonzalez-Angulo, A. M., Myhre, S., Carey, M., Lee, J. S., Overgaard, J., Alsner, J., Stemke-Hale, K., Lluch, A., Neve, R. M., Kuo, W. L., Sorlie, T., Sahin, A., Valero, V., Keyomarsi, K., Gray, J. W., Borresen-Dale, A. L., Mills, G. B., and Hennessy, B. T. (2009) Integrative analysis of cyclin protein levels identifies cyclin b1 as a classifier and predictor of outcomes in breast cancer, Clin Cancer Res 15, 3654-3662.
(99) Wood, K. W., Lad, L., Luo, L., Qian, X., Knight, S. D., Nevins, N., Brejc, K., Sutton, D., Gilmartin, A. G., Chua, P. R., Desai, R., Schauer, S. P., McNulty, D. E., Annan, R. S., Belmont, L. D., Garcia, C., Lee, Y., Diamond, M. A., Faucette, L. F., Giardiniere, M., Zhang, S., Sun, C. M., Vidal, J. D., Lichtsteiner, S., Cornwell, W. D., Greshock, J. D., Wooster, R. F., Finer, J. T., Copeland, R. A., Huang, P. S., Morgans, D. J., Jr., Dhanak, D., Bergnes, G., Sakowicz, R., and

References
101
Jackson, J. R. (2010) Antitumor activity of an allosteric inhibitor of centromere-associated protein-E, Proc. Natl. Acad. Sci. U. S. A. 107, 5839-5844.
(100) Nowicki, M. O., Pawlowski, P., Fischer, T., Hess, G., Pawlowski, T., and Skorski, T. (2003) Chronic myelogenous leukemia molecular signature, Oncogene 22, 3952-3963.
(101) Liu, M., Wang, X., Yang, Y., Li, D., Ren, H., Zhu, Q., Chen, Q., Han, S., Hao, J., and Zhou, J. (2010) Ectopic expression of the microtubule-dependent motor protein Eg5 promotes pancreatic tumourigenesis, J Pathol 221, 221-228.
(102) Lad, L., Luo, L., Carson, J. D., Wood, K. W., Hartman, J. J., Copeland, R. A., and Sakowicz, R. (2008) Mechanism of inhibition of human KSP by ispinesib, Biochemistry 47, 3576-3585.
(103) Skoufias, D. A., DeBonis, S., Saoudi, Y., Lebeau, L., Crevel, I., Cross, R., Wade, R. H., Hackney, D., and Kozielski, F. (2006) S-Trityl-L-cysteine Is a Reversible, Tight Binding Inhibitor of the Human Kinesin Eg5 That Specifically Blocks Mitotic Progression, J. Biol. Chem. 281, 17559-17569.
(104) Nagahara, M., Nishida, N., Iwatsuki, M., Ishimaru, S., Mimori, K., Tanaka, F., Nakagawa, T., Sato, T., Sugihara, K., Hoon, D. S., and Mori, M. (2011) Kinesin 18A expression: clinical relevance to colorectal cancer progression, Int. J. Cancer 129, 2543-2552.
(105) Zhang, C., Zhu, C., Chen, H., Li, L., Guo, L., Jiang, W., and Lu, S. H. (2010) Kif18A is involved in human breast carcinogenesis, Carcinogenesis 31, 1676-1684.
(106) Catarinella, M., Gruner, T., Strittmatter, T., Marx, A., and Mayer, T. U. (2009) BTB-1: A Small Molecule Inhibitor of the Mitotic Motor Protein Kif18A, Angewandte Chemie-International Edition 48, 9072-9076.
(107) Takahashi, S., Fusaki, N., Ohta, S., Iwahori, Y., Iizuka, Y., Inagawa, K., Kawakami, Y., Yoshida, K., and Toda, M. (2012) Downregulation of KIF23 suppresses glioma proliferation, J Neurooncol 106, 519-529.
(108) Imai, K., Hirata, S., Irie, A., Senju, S., Ikuta, Y., Yokomine, K., Harao, M., Inoue, M., Tomita, Y., Tsunoda, T., Nakagawa, H., Nakamura, Y., Baba, H., and Nishimura, Y. (2011) Identification of HLA-A2-restricted CTL epitopes of a novel tumour-associated antigen, KIF20A, overexpressed in pancreatic cancer, Br. J. Cancer 104, 300-307.
(109) Tcherniuk, S., Skoufias, D. A., Labriere, C., Rath, O., Gueritte, F., Guillou, C., and Kozielski, F. (2010) Relocation of Aurora B and survivin from centromeres to the central spindle impaired by a kinesin-specific MKLP-2 inhibitor, Angew Chem Int Ed Engl 49, 8228-8231.

References
102
(110) Wu, J. Q., Mikule, K., Wang, W. X., Su, N., Petteruti, P., Gharandaghi, F., Code, E., Zhu, X. H., Jacques, K., Lai, Z. W., Yang, B., Lamb, M. L., Chuaqui, C., Keen, N., and Chen, H. W. (2013) Discovery and Mechanistic Study of a Small Molecule Inhibitor for Motor Protein KIFC1, ACS Chem. Biol. 8, 2201-2208.
(111) Bell, R. D., and Doisy, E. A. (1920) Rapid colorimetric methods for the determination of phosphorus in urine and blood., J. Biol. Chem. 44, 55-67.
(112) Baykov, A. A., Evtushenko, O. A., and Avaeva, S. M. (1988) A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay, Anal. Biochem. 171, 266-270.
(113) Geladopoulos, T. P., Sotiroudis, T. G., and Evangelopoulos, A. E. (1991) A malachite green colorimetric assay for protein phosphatase activity, Anal. Biochem. 192, 112-116.
(114) Huang, T. G., and Hackney, D. D. (1994) Drosophila kinesin minimal motor domain expressed in Escherichia coli. Purification and kinetic characterization, J. Biol. Chem. 269, 16493-16501.
(115) Mitchison, T. J. (1994) Towards a pharmacological genetics, Chem. Biol. 1, 3-6.
(116) Schreiber, S. L. (1998) Chemical genetics resulting from a passion for synthetic organic chemistry, Bioorg. Med. Chem. 6, 1127-1152.
(117) Mayer, T. U. (2003) Chemical genetics: tailoring tools for cell biology, Trends Cell Biol 13, 270-277.
(118) Florian, S., Hummer, S., Catarinella, M., and Mayer, T. U. (2007) Chemical genetics: reshaping biology through chemistry, HFSP J 1, 104-114.
(119) Schenone, M., Dancik, V., Wagner, B. K., and Clemons, P. A. (2013) Target identification and mechanism of action in chemical biology and drug discovery, Nat. Chem. Biol. 9, 232-240.
(120) Wetzel, S., Bon, R. S., Kumar, K., and Waldmann, H. (2011) Biology-Oriented Synthesis, Angewandte Chemie-International Edition 50, 10800-10826.
(121) Dolle, R. E. (2002) Comprehensive survey of combinatorial library synthesis: 2001, J. Comb. Chem. 4, 369-418.
(122) Hümmer, S. (2009) Functional characterization of the mitotic kinesin-like protein Mklp2, Ludwig-Maximilians-Universität München.
(123) Braun, J. (2009) Synthesis and Biological Evaluation of Cytokinesis Inhibitors, University of Konstanz.
(124) Saifina, D. F., and Mamedov, V. A. (2010) New and modified classical methods for the synthesis of quinoxalines, Russ. Chem. Rev. 79, 351-370.

References
103
(125) Darabi, H. R., Mohandessi, S., Aghapoor, K., and Mohsenzadeh, F. (2007) A recyclable and highly effective sulfamic acid/MeOH catalytic system for the synthesis of quinoxalines at room temperature, Catal. Commun. 8, 389-392.
(126) Darabi, H. R., Tahoori, F., Aghapoor, K., Taala, F., and Mohsenzadeh, F. (2008) NH4Cl-CH3OH: an Efficient, Acid- and Metal-Free Catalyst System for the Synthesis of Quinoxalines, J. Braz. Chem. Soc. 19, 1646-1652.
(127) Cho, C. S., and Oh, S. G. (2007) Copper-catalyzed oxidative cyclization of alpha-hydroxyketones with o-phenylenediamines leading to quinoxalines, Journal of Molecular Catalysis a-Chemical 276, 205-210.
(128) Zall, A., Bensinger, D., and Schmidt, B. (2012) Oxidative Homologation of Aldehydes to a-Ketoaldehydes by using Iodoform, o-Iodoxybenzoic Acid, and Dimethyl Sulf-oxide, Eur. J. Org. Chem., 1439-1447.
(129) Meshram, H. M., Kumar, G. S., Ramesh, P., and Reddy, B. C. (2010) A mild and convenient synthesis of quinoxalines via cyclization-oxidation process using DABCO as catalyst, Tetrahedron Lett. 51, 2580-2585.
(130) Pan, F., Chen, T.-M., Cao, J.-J., Zou, J.-P., and Zhang, W. (2012) Ga(ClO4)3-catalyzed synthesis of quinoxalines by cycloaddition of α-hydroxyketones and o-phenylenediamines, Tetrahedron Lett. 53, 2508-2510.
(131) Kornblum, N., Powers, J. W., Anderson, G. J., Jones, W. J., Larson, H. O., Levand, O., and Weaver, W. M. (1957) A New and Selective Method of Oxidation, J. Am. Chem. Soc. 79, 6562-6562.
(132) Baldwin, J. J., Engelhardt, E. L., Hirschmann, R., Lundell, G. F., Ponticello, G. S., Ludden, C. T., Sweet, C. S., Scriabine, A., Share, N. N., and Hall, R. (1979) beta-Adrenergic blocking agents with acute antihypertensive activity, J. Med. Chem. 22, 687-694.
(133) Saggiomo, A. J., Kano, S., Kikuchi, T., Okubo, K., and Shinbo, M. (1972) Antimalarial potency of 2-benzoyl-4-quinolinemethanols, J. Med. Chem. 15, 989-994.
(134) Leitner, K. M. (2010) Synthese und Charakterisierung von 2-Thiophenchinoxalinen, Universität Konstanz.
(135) Pearson, D. E., Pope, H. W., Hargrove, W. W., and Stamper, W. E. (1958) The Swamping Catalyst Effect .2. Nuclear Halogenation of Aromatic Aldehydes and Ketones, J. Org. Chem. 23, 1412-1419.
(136) Pearson, D. E., Stamper, W. E., and Suthers, B. R. (1963) Swamping Catalyst Effect .5. Halogenation of Aromatic Acid Derivatives, J. Org. Chem. 28, 3147-&.
(137) Stanetty, P., Puschautz, E., and Friedbacher, G. (1989) Herbicidal Thienylureas .1., Monatsh. Chem. 120, 53-63.

References
104
(138) Batke, S. (2011) Synthese und biologische Evaluation von wasserlöslichen 2-Phenylchinoxalinen als Zytokinese-Inhibitoren, Universität Konstanz.
(139) Huisgen, R. (1963) Kinetik Und Mechanismus 1.3-Dipolarer Cycloadditionen, Angewandte Chemie-International Edition 75, 742-&.
(140) Beckmann, H. S. G. (2006) Entwicklung einer Eintopfreaktion aus Diazotransfer und Azid-Alkin-Cycloaddition zur Synthese von Neoglycokonjugaten, Universität Konstanz.
(141) Beckmann, H. S. G., and Wittmann, V. (2007) One-pot procedure for diazo transfer and azide-alkyne cycloaddition: Triazole linkages from amines, Org. Lett. 9, 1-4.
(142) Rostovtsev, V. V., Green, L. G., Fokin, V. V., and Sharpless, K. B. (2002) A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes, Angewandte Chemie-International Edition 41, 2596-+.
(143) Feng, B. Y., Shelat, A., Doman, T. N., Guy, R. K., and Shoichet, B. K. (2005) High-throughput assays for promiscuous inhibitors, Nat. Chem. Biol. 1, 146-148.
(144) Feng, B. Y., and Shoichet, B. K. (2006) A detergent-based assay for the detection of promiscuous inhibitors, Nat. Protoc. 1, 550-553.
(145) Feng, B. Y., Simeonov, A., Jadhav, A., Babaoglu, K., Inglese, J., Shoichet, B. K., and Austin, C. P. (2007) A high-throughput screen for aggregation-based inhibition in a large compound library, J. Med. Chem. 50, 2385-2390.
(146) McGovern, S. L., Helfand, B. T., Feng, B., and Shoichet, B. K. (2003) A specific mechanism of nonspecific inhibition, J. Med. Chem. 46, 4265-4272.
(147) Seidler, J., McGovern, S. L., Doman, T. N., and Shoichet, B. K. (2003) Identification and prediction of promiscuous aggregating inhibitors among known drugs, J. Med. Chem. 46, 4477-4486.
(148) Hacker, S. M., Pagliarini, D., Tischer, T., Hardt, N., Schneider, D., Mex, M., Mayer, T. U., Scheffner, M., and Marx, A. (2013) Fluorogenic ATP analogues for online monitoring of ATP consumption: observing ubiquitin activation in real time, Angew Chem Int Ed Engl 52, 11916-11919.
(149) Hardt, N., Hacker, S. M., and Marx, A. (2013) Synthesis and fluorescence characteristics of ATP-based FRET probes, Org. Biomol. Chem. 11, 8298-8305.
(150) Finlay, H. J., Lloyd, J., Vaccaro, W., Kover, A., Yan, L., Bhave, G., Prol, J., Huynh, T., Bhandaru, R., Caringal, Y., DiMarco, J., Gan, J., Harper, T., Huang, C., Conder, M. L., Sun, H., Levesque, P., Blanar, M., Atwal, K., and Wexler, R. (2012) Discovery of ((S)-5-(Methoxymethyl)-7-(1-methyl-1H-indol-2-yl)-2-

References
105
(trifluoromethyl)-4,7-dihydropyrazolo[1,5-a]pyrimidin-6-yl)((S)-2-(3-methylisoxazol-5-yl)pyrrolidin-1-yl)methanone As a Potent and Selective IKur Inhibitor, J. Med. Chem. 55, 3036-3048.
(151) Grundon, M. F., Johnston, B. T., and Matier, W. L. (1966) Proximity effects in diaryl derivatives. Part IV. Base-catalysed reactions of 2,2[prime or minute]-di(hydroxyamino)diaryl sulphones and of 2-(hydroxyamino)aryl phenyl sulphones, Journal of the Chemical Society B: Physical Organic, 260-266.
(152) Parveen, S., Khan, M. O. F., Austin, S. E., Croft, S. L., Yardley, V., Rock, P., and Douglas, K. T. (2005) Antitrypanosomal, Antileishmanial, and Antimalarial Activities of Quaternary Arylalkylammonium 2-Amino-4-Chlorophenyl Phenyl Sulfides, a New Class of Trypanothione Reductase Inhibitor, and of N-Acyl Derivatives of 2-Amino-4-Chlorophenyl Phenyl Sulfide, J. Med. Chem. 48, 8087-8097.
(153) Moore, W. J., Kern, J. C., Bhat, R., Commons, T. J., Fukayama, S., Goljer, I., Krishnamurthy, G., Magolda, R. L., Nogle, L., Pitts, K., Stauffer, B., Trybulski, E. J., Welmaker, G. S., Wilson, M., and Bodine, P. V. N. (2008) Modulation of Wnt Signaling Through Inhibition of Secreted Frizzled-Related Protein I (sFRP-1) with N-Substituted Piperidinyl Diphenylsulfonyl Sulfonamides†, J. Med. Chem. 52, 105-116.
(154) Krasnokutskaya, E. A., Semenischeva, N. I., Filimonov, V. D., and Knochel, P. (2007) A new, one-step, effective protocol for the iodination of aromatic and heterocyclic compounds via aprotic diazotization of amines, Synthesis-Stuttgart, 81-84.
(155) Rao, H., Jin, Y., Fu, H., Jiang, Y., and Zhao, Y. (2006) A Versatile and Efficient Ligand for Copper-CPyrrolidine-2-Phosphonic Acid Phenyl Monoester, Chemistry – A European Journal 12, 3636-3646.
(156) Pikulik, I. I., Weber, R. U., and Zollinger, H. (1981) Nucleophilic Aromatic Substitutions. Part XIV.. Investigation of the mechanism of hydroxy-denitration of 4,2-and 2,4-chloronitrobenzenediazonium ions as a function of pH, Helv. Chim. Acta 64, 1777-1789.
(157) Doak, G. O., and Freedman, L. D. (1951) The Synthesis of Arylphosphonic and Diarylphosphinic Acids by the Diazo Reaction1, J. Am. Chem. Soc. 73, 5658-5660.
(158) Hamid, R., Rotshteyn, Y., Rabadi, L., Parikh, R., and Bullock, P. (2004) Comparison of alamar blue and MTT assays for high through-put screening, Toxicol. in Vitro 18, 703-710.
(159) Martinez Molina, D., Jafari, R., Ignatushchenko, M., Seki, T., Larsson, E. A., Dan, C., Sreekumar, L., Cao, Y., and Nordlund, P. (2013) Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay, Science 341, 84-87.

References
106
(160) Jordan, A., Hadfield, J. A., Lawrence, N. J., and McGown, A. T. (1998) Tubulin as a target for anticancer drugs: agents which interact with the mitotic spindle, Med. Res. Rev. 18, 259-296.
(161) Van den Bossche, H., Rochette, F., and Horig, C. (1982) Mebendazole and related anthelmintics, Advances in pharmacology and chemotherapy 19, 67-128.
(162) Dong, H., Li, Y. Z., and Hu, W. (2004) Analysis of purified tubulin in high concentration of glutamate for application in high throughput screening for microtubule-stabilizing agents, Assay Drug Dev. Technol. 2, 621-628.
(163) Hamel, E. (2003) Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin, Cell Biochem. Biophys. 38, 1-22.
(164) Jordan, M. A., and Wilson, L. (2004) Microtubules as a target for anticancer drugs, Nat. Rev. Cancer 4, 253-265.
(165) Amos, L. A. (2004) Microtubule structure and its stabilisation, Org. Biomol. Chem. 2, 2153-2160.
(166) Desai, A., and Mitchison, T. J. (1997) Microtubule polymerization dynamics, Annu. Rev. Cell Dev. Biol. 13, 83-117.
(167) Gopalsamy, A., Shi, M., Stauffer, B., Bahat, R., Billiard, J., Ponce-de-Leon, H., Seestaller-Wehr, L., Fukayama, S., Mangine, A., Moran, R., Krishnamurthy, G., and Bodine, P. (2008) Identification of Diarylsulfone Sulfonamides as Secreted Frizzled Related Protein-1 (sFRP-1) Inhibitors, J. Med. Chem. 51, 7670-7672.
(168) Wang, C., Ma, Z., Sun, X.-L., Gao, Y., Guo, Y.-H., Tang, Y., and Shi, L.-P. (2006) Synthesis and Characterization of Titanium(IV) Complexes Bearing Monoanionic [O-NX] (X = O, S, Se) Tridentate Ligands and Their Behaviors in Ethylene Homo- and Copolymerizaton with 1-Hexene, Organometallics 25, 3259-3266.
(169) Burnette, W. N. (1981) "Western blotting": electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A, Anal. Biochem. 112, 195-203.

Appendix
107
8. Appendix
8.1 Appreviations
ADP adenosine diphosphate
aq. aqueous
ATP adenosine triphosphate
CETSA cellular thermal shift assay
DMAP 4-dimethylaminopyridine
DMF dimethylformamide
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
EC50 half maximal effective concentration
ECA enzyme coupled assay
equiv. equivalents
EtOAc ethyl acetate
EtOH ethanol
FRET Förster resonance energy transfer
G1-phase gap1 phase
G2-phase gap2 phase
GAP GTPase-activating protein
GDP guanosine diphosphate
GFP green fluorescent protein
GTP guanosine triphosphate
HCS high content screening
HTS high throughput screening
IC50 half maximal inhibitory concentration
INCENP inner centromere protein
K-fibers kinetochore microtubules
MAPs microtubule-associated proteins
m-CPBA meta-chloroperoxybenzoic acid

Appendix
108
MeCN acetonitrile
MGA malachite green assay
Mklp2 mitotic kinesin-like protein 2
M phase mitotic phase
MPP1 M-phase phosphoprotein 1
NADH nicotinamide adenine dinucleotide
NEBD nuclear envelope breakdown
PE petroleum ether
PEP phosphoenolpyruvate
PLK1 polo-like kinase 1
PRC1 protein regulator of cytokinesis
p-TsOH para-toluenesulfonic acid
RNAi ribonucleic acid interference
SAC spindle assembly checkpoint
SAR structure activity relationship
SNAr nucleophilic aromatic substitution
S phase synthesis phase
TMEDA Tetramethylethylenediamine





























