Embed Size (px)
Citation preview

Physical Chemistry I I. Introduction------------------------------------------------------------------------------------------------------------
1
Physical Chemistry I
Prof. Dr. W.S. Veeman
Bachelor CourseWater: Chemistry, Analysis and Microbiology
WS 2002/2003
Literature: P.W. Atkins Physical Chemistry / Physikalische ChemieT. Engel and P. Reid Physical Chemistry / Physikalische Chemie
G.K. Vemulapalli Physical Chemistry
I. Introduction
In the universe we find all kinds of „objects“, very large objects like the sun, earth and moon, medium-size objects like a chair, an engine or an animal, small objects like bacteria andviruses and very small objects like electrons, atoms and molecules. All larger objects arecomposed of atomic particles. On a microscopic scale these atomic particles interact witheach other and can give rise to something like a chemical bond in a molecule or a hydrogenbond between water molecules. In addition to atomic particles electric and magnetic fieldsexist which can interact with these atomic particles. Radiation like visible light or X-rays, forinstance, can be described as rapidly varying electric and magnetic fields (electromagneticwaves) which propagate in space with a very high speed, the light velocity c, ca. 300.000km/s (this speed is dependent on the medium in which the wave propagates, the fastest speedis found when the wave propagates in vacuum).
Physicists, chemists and biologists have developed a large number of theories to describeproperties of living and dead objects and the process in which these objects are involved.From a physical point of view there exist however a few almost universal theories on whichall other theories must be based.
- Classical or Newtonian mechanics describes the motion of large objects, like for instanceplanets or bullets, extremely well.
- Quantum mechanics is needed to describe the properties of atomic particles and ofprocesses in which such particles are involved. On such a microscopical scale classicalmechanics does not work. Quantum mechanics was developed in 1920-1930 and functionswell for the description of the behaviour of atomic particles.
- Relativity Theory describes processes in which particles that move at very high speeds(comparable to the speed of light) are involved. Although this seems very far from dailylife, do not forget that electrons in atoms seem to move at such speeds. The quantummechanics description of molecules therefore sometimes needs a relativistic correction.
- The electromagnetic theory (EM theory) describes the interaction of electric and magneticfields with microscopic objects( electrons, atoms, molecules,...) and with macroscopicobjects (radio or radar wave antennas). Since light is an electromagnetic wave ( apropagation of periodic electric and magnetic fields) the EM theory in combination with

Physical Chemistry I I. Introduction------------------------------------------------------------------------------------------------------------
2
quantum mechanics is also needed to describe phenomena like the absorption, emission ordiffraction of light by matter.
- Thermodynamics was originally the theory for the description of the flow of heat andenergy between macroscopic machines and their surrounding (especially useful for thedevelopment of the steam engine in 18....) but it has been developed into a general theoryfor the description of physical, chemical and biological processes. Classical orphenomenological thermodynamics describes macroscopical objects without referring totheir atomic or molecular structure (the earlier investigators probably had no notion ofmolecules) and is based on several, often very ingenious, experiments from which generalaxiomatic laws were derived. In later times a thermodynamic theory was developed whichdescribes matter as an ensemble of very many microscopical particles, each of whichfollows a quantummechanical description. In view of the large number of microscopicalparticles in an ensemble statistical methods are applied to deal with such large ensembles,therefore this branch of thermodynamics is called Statistical Thermodynamics.
Roughly speaking physical chemistry (the „physics of molecules“) can be divided into a description of the molecular structure of matter and the consequences thereof for the chemicalor biological processes in which the molecules are involved, and into a macroscopicaldescription of matter for which the knowledge of the precise molecular structure is not veryrelevant. The first approach would need quantum mechanics, the second approach is theapproach of classical thermodynamics.In this lecture we will start with the second approach, but we will try to make a relation to thefirst approach whenever possible.
Classical thermodynamics is based on a number of axioms (thermodynamic „laws“) which result from very many laboratory and „Gedanken“-experiments. The strength and the beautyof classical thermodynamics is that we can describe certain processes without knowing thedetails of the process and the structure of the components that take part in these processes. Asan example, consider the daily life experience that when you put cheese and bread in arefrigerator, everyone knows that after a while both have attained the same temperature,although the molecular structures of cheese and bread are very different.
Thermodynamics is built up like mathematics with strict rules and definitions (for instance,we have to define what the meaning of temperature is before we can for instance say that thetemperature of cheese and bread is the same). We therefore start with a list of definitions.

Physical Chemistry I II. Definitions------------------------------------------------------------------------------------------------------------
3
II. Definitions
As described above, thermodynamics tries to describe macroscopical objects or structures ororganizations almost without knowledge about the details of the internal structure orcomposition. Such objects are called systems. Because the notion of a system is so general,also the definition has to be very general and, at first look, the definition seems to beunnecessary abstract. The advantage of this general definition is that you can applythermodynamic theory to a large number of phenomena. Important is that the abstractdefinition of a system automatically implies the definition of the surrounding of the system.Everything in the universe that does not belong to the system is called the surrounding.
System = Part of the universe that one wants to describe and that can be separated from itsenvironment.
A system could be a reaction vessel or a biological cell or an engine or the earth or a containerwith gas. For the discussion in the first chapters it is easy to think of the system as a cylinderfilled with gas and closed by a piston. Properties of the gas like the volume can then bechanged by exerting pressure on the piston.The system and its surroundings may exchange energy and/or matter.To visualize the exchange of energy between the system and its surroundings think of the gascylinder. By applying pressure to the piston we (we = part of the surrounding) do work on thesystem. As a result of the work done on the system the energy of the system must have beenincreased.For the exchange of matter take the example of a biological cell. The cell wall separates thecell from its surroundings but this wall is open for certain substances to enter or leave the cell.
The following general situations can be distinguished:
Closed system : no exchange of energy or matter between system andsurroundings
Open system : exchange of energy and matter between system andsurroundings is possible
Isolated system : no exchange of energy between system and surroundingsThermally insulated system : no heat exchange between system and surroundings, an
isolated system is also a thermally insulated system, the reverseis not true.
The state of a system: state variables.
The state of a system is characterized by values of certain properties. Think again of ourcylinder with gas as the system. The gas has a certain volume V (in m3), temperature T (forthe unit see below) and pressure p in Pascal (1 Pa = 1 N/m2, 1 bar = 105 Pa, 1 atm = 101325Pa). These properties describe the state of the system and are called state variables. One candistinguish intensive variables whose values do not change when the system is enlarged:
intensive variables : p,T, density ,...... (the density is the mass per volume in units kg/m)
and extensive variables, whose values change with the size of the system:

Physical Chemistry I II. Definitions------------------------------------------------------------------------------------------------------------
4
extensive variables : V, the energy of the system E (in units Joule), the amount of thesubstance n expressed in mol, ….
Most systems contain more than one substance and a mol number ni can be defined for everysubstance i.
There is always a relation between the temperature, the volume, the pressure and the amountof substance, this relation F(p,V,T,n) = 0 is called the equation of state. As we will see later,this relation is particularly simple for gases at low pressure (ideal gases).
A system is in an equilibrium state when all the state variables are independent of time. Asystem usually is not in an equilibrium state, which means that one or more variables arechanging in time. Sometimes it looks as if a system is in equilibrium, while many variablesare constant in the time. In winter the room may be heated and the room temperature may beconstant in time but higher than the outside temperature. This is called a stationary state. Tocheck whether a system is in a stationary or in an equilibrium state, make it a closed system(“disconnect” the system from its surroundings). When the system variables stay constant in time, the system was in an equilibrium state, if not it was in a stationary state.
Process
A system may undergo processes that cause some state variables to change with time. Forinstance we can take our gas in the cylinder and raise the outside pressure on the piston. Thestate of the gas will change in that the internal pressure of the gas will increase and thevolume decrease. An important difference is made between reversible and irreversibleprocesses. This difference will be discussed later.
Temperature
The concept of temperature follows from the zero’th law of thermodynamics. This law states that when a system A is in thermal equilibrium with a system B (that means that when webring A in thermal contact with B both A and B do not change) and B is in thermalequilibrium with C, that then also A is in thermal equilibrium with C.
A, B and C can be very different systems, for instance a cylinder with gas, a mercurythermometer and a block of ice. The fact that when the systems are brought into thermalcontact, the state of either of the systems does not change, in spite of the fact that thecomposition of each system is very different, means that they have some property in common.This property is called the temperature. Although the precise definition of temperature is nottrivial, we will not discuss it here. We assume that everyone has a practical feeling for theconcept of temperature. The assignment of a value to the temperature of a certain system israther arbitrary. Several temperature scales exist , the familiar Celcius scale compares thelength of a mercury column when it is in thermal contact with melting ice (0o) with the
A B C

Physical Chemistry I II. Definitions------------------------------------------------------------------------------------------------------------
5
expanded length when it is in contact with boiling water (100o C). In the SI unit system(SI=Système Internationale) the temperature is expressed in Kelvins (K, not “degree” Kelvin), the absolute temperature. The absolute temperature can be expressed in the temperatureaccording to the Celcius scale by the equation:
T (in K) = t (in C) +273.15
The absolute lowest temperature that can be reached is 0 K.
The states of matter
We spoke already about a gas in a cilinder. In this case the system is a gas in which theparticles are more or less free to move in every direction. Therefore a gas, if it is contained ina container, will completely fill the container. Usually the density of a gas is relatively lowcompared to the other forms in which matter can be found: liquids and solids. A liquid in acontainer does not have to fill the container completely. When not, there always is a surfacebetween the liquid and the gas or vapour above it. The particles which form the liquid, usuallymolecules, can also move freely but now the density is so much higher than in a gas thatcollisions with neighboring particles occur every few ps (1 ps = 10-12s). The interparticleinteractions are therefore very strong. Gas and liquids have in common that the long-rangeorder is very low. We will see later that, for instance, for water there is a short-lived short-range order. Also in solids the interparticle interactions usually are very strong. Solids can befound in a highly ordered state, crystals, or in an unordered state, the amorphous state.Usually the translational mobility of the particles, atoms or molecules, in solids is very low. Inmany solids built of molecules, organic solids, however, the molecules show varying degreesof rotational freedom. In addition, certain functional groups may show a high rotationalmobility, e.g. it is known that methyl groups rotate fast about their threefold symmetry axiseven at low temperatures (20 K).
Because of the very weak interparticle interactions gases are much easier to describe thanliquids or solids. For that reason we start with a description of the properties of gases.
Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
6
III. The properties of gases
III. a. Perfect gases
The most simple state of matter is a gas. A gas consists of particles (atoms or molecules)which are free to move, except that the particles may collide with each other and with the wallif the gas is in a container. Between the particles there is empty space! The pressure on thewalls of the container arises from the collisions of the gas particles with the wall. This impliesthat the pressure must be fluctuating in time, but because there are so many particles, 1 molcontains 6.02208 x 1023 particles, from a macroscopical point of view the pressure p isconstant in time as long as the state of the gas does not change.
For a gas consisting of one type of particles (for instance H2 for a hydrogen gas) the statevariables are: volume V, temperature T, pressure p and amount (=number of moles) n. Asmentioned above there must be a relation between these state variables, the equation of state.For gases there exist several different equations of state, depending on how accurate onewants to describe the gas. The most simple form is:
nRTpV (1)
where R= 8.31451 JK-1mol-1 is a constant equal for all gases (the gas constant) and T has tobe expressed in Kelvin (K).
This equation of state describes gases when the pressure is so low that collisions between theparticles are very unlikely. Such a gas, where the particles do not interact, is called a perfector ideal gas.
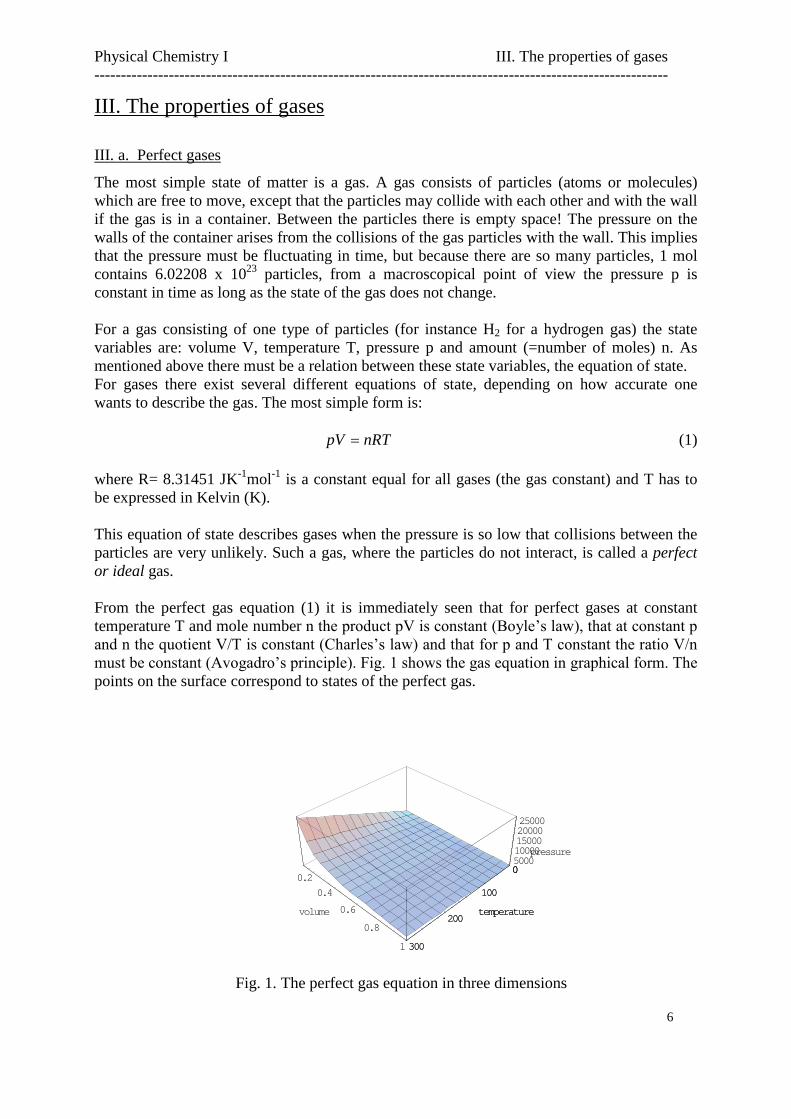
From the perfect gas equation (1) it is immediately seen that for perfect gases at constanttemperature T and mole numbern the product pV is constant (Boyle’s law), that at constant p and n the quotient V/T is constant (Charles’s law) and that for p and T constant the ratio V/n must be constant (Avogadro’s principle). Fig. 1 shows the gas equation in graphical form. The points on the surface correspond to states of the perfect gas.
0
100
200
300
temperature
0.20.4
0.6
0.8
1
volume
0500010000150002000025000
pressure
0
100
200
300
temperature
Fig. 1. The perfect gas equation in three dimensions
Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
7
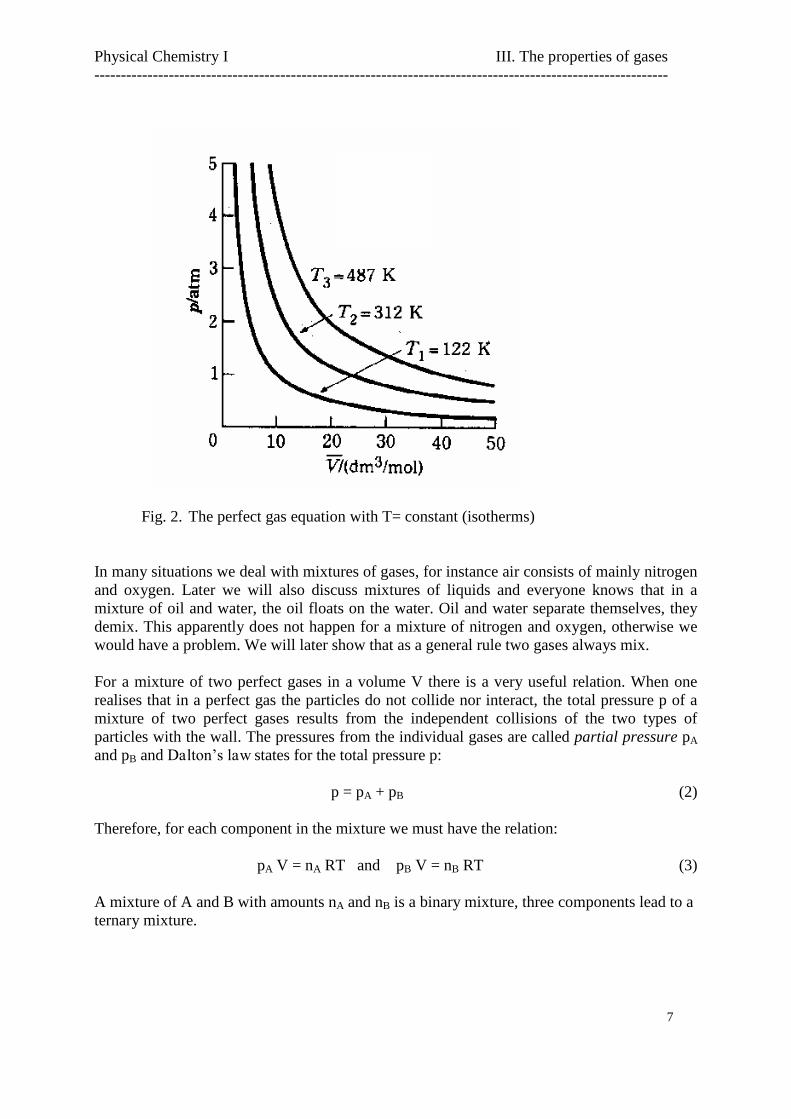
Fig. 2. The perfect gas equation with T= constant (isotherms)
In many situations we deal with mixtures of gases, for instance air consists of mainly nitrogenand oxygen. Later we will also discuss mixtures of liquids and everyone knows that in amixture of oil and water, the oil floats on the water. Oil and water separate themselves, theydemix. This apparently does not happen for a mixture of nitrogen and oxygen, otherwise wewould have a problem. We will later show that as a general rule two gases always mix.
For a mixture of two perfect gases in a volume V there is a very useful relation. When onerealises that in a perfect gas the particles do not collide nor interact, the total pressure p of amixture of two perfect gases results from the independent collisions of the two types ofparticles with the wall. The pressures from the individual gases are called partial pressure pA
and pB andDalton’s lawstates for the total pressure p:
p = pA + pB (2)
Therefore, for each component in the mixture we must have the relation:
pA V = nA RT and pB V = nB RT (3)
A mixture of A and B with amounts nA and nB is a binary mixture, three components lead to aternary mixture.

Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
8
III. b. Motion of gas molecules
As mentioned already in a gas the particles (atoms or molecules) are very free to move andthey will do so because one can proof (statistical thermodynamics) that the average energy perparticle available for translational motions (kinetic energy = ½mv2) is (3/2) kT, where k is theBoltzmann constant (k = 1.38066 x 10-23 JK-1). This shows that one can expect that theaverage speed of the particles in a gas increases with temperature.The average kinetic energy of the particles can be expressed as:
kTvmmv 232
212
21
or: TkNvmN AA 232
21
or: RTvM 232
21
or:
MRT
v32 (4)
Here the left hand side of the equation is the root mean square velocity (<v2> means the(velocity)2, averaged over all particles). M in eq. (4) is the molar mass, the mass of one moleof the gas particles. For CO2 (M=44x10-3 kg/mol) at 298 K the average speed according to eq.(4) is 411 ms-1.Eq. (4) is an expression for the average speed of the particles, some are faster, others areslower. The division between slow and fast is temporary, a fast particle may slow downbecause of collisions with other particles. There really is a distribution of speeds.The distribution function can be calculated (Maxwell distribution):
RTMv
vRT
Mvf
2exp
24)(
222
3
(5)
The function is plotted in fig. 3 for different temperatures, the higher the temperature thehigher the average speed and the broader the distribution. The area under the curves mustremain constant.
.
500 1000 1500 2000m2????????s
0.00005
0.0001
0.00015
0.0002
0.00025
fraction
Fig. 3. The distribution of molecular speeds for N2 (molar mass 28.02 x 10-3 kg/mol)at 100, 273 and 500 K.

Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
9
III. c. Real Gases
The difference between perfect gases and real gases is that for real gases the particles caninteract with each other when their interparticle distance becomes of the order of magnitudeof their radii. Depending on their distance they attract or repulse each other. Attractive forceswork at relatively large distance, repulsive forces act only when the particles come so closethat their electron clouds interpenetrate. These interactions are particularly important for highpressures. A complete understanding of the forces between molecules (or atoms) is part of thequantummechanical description of matter. In this thermodynamic picture it suffices to knowthat atomic particles are composed of electrically charged particles (negatively chargedelectrons and positively charged atomic nuclei). When two atomic particles approach eachother, these charges interact with each other. At first they usually attract but when they comevery close, so that the electron clouds of different atoms almost penetrate, the repulsive forcebecomes very strong, much stronger than the attractive forces. An approximate equationwhich describes the potential energy for the attraction and repulsion is the Lennard-Jonesequation:
6
012
04rr
rr
V (6)
The distance dependence of this energy is shown in fig. 4. The parameters α and r0 representthe depth of the energy well (where V has a minimum) and the distance where V=0,respectively.
0.5 1 1.5 2 2.5
r????????r0
-2?10- 2 2
-1.5?10-2 2
-1?10- 2 2
-5?10- 2 3
5?10- 23
1?10- 22
1.5?10- 2 2
2?10- 22
energy
Fig. 4. The potential energy of two atoms as a function of their distance, according toeq. (6)
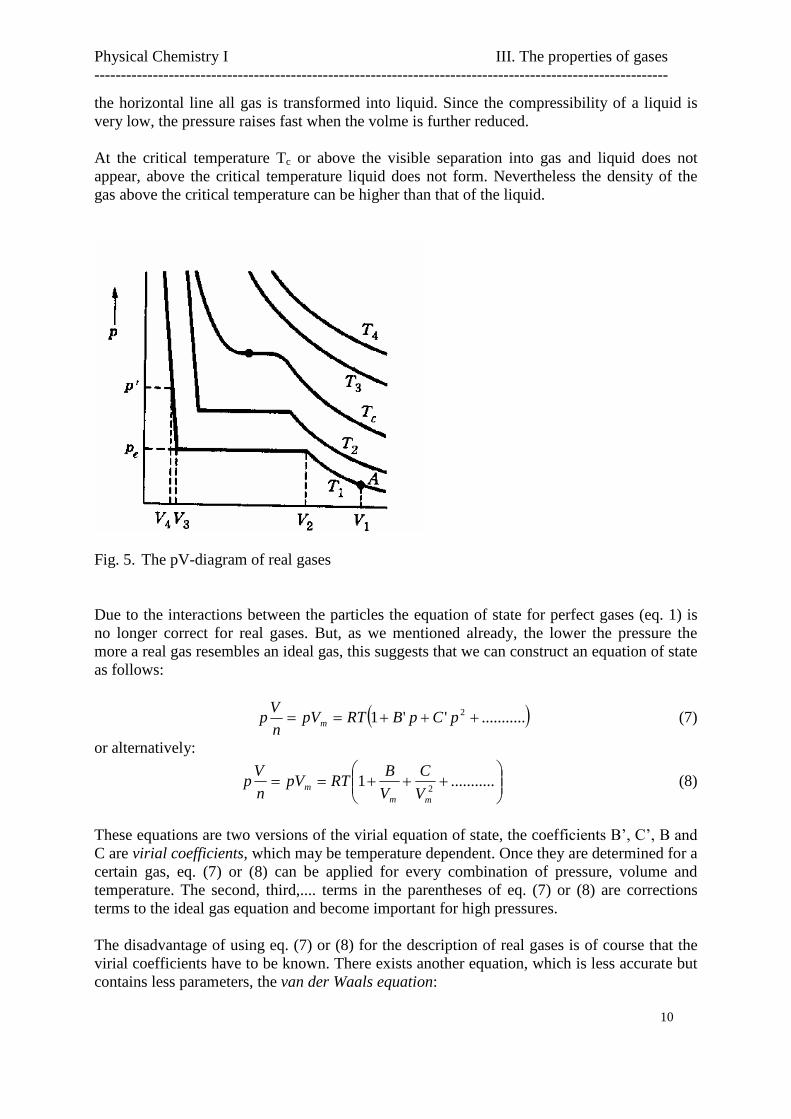
At this point it is good to look at the behaviour of a real gas when we increase the pressure onthe piston of our gas cilinder. In fig. 5 the pV-diagram (isotherms) of a certain gas is shown.At relatively low temperatures at a certain volume of the gas the pressure does not raise uponfurther compression. This is represented in fig. 5 by a horizontal line. What happens is thatthe gas liquefies, liquid and gas coexist in the cylinder in equilibrium and are separated by aclear surface. By lowering the volume more and more liquid is produced until at the end of
Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
10
the horizontal line all gas is transformed into liquid. Since the compressibility of a liquid isvery low, the pressure raises fast when the volme is further reduced.
At the critical temperature Tc or above the visible separation into gas and liquid does notappear, above the critical temperature liquid does not form. Nevertheless the density of thegas above the critical temperature can be higher than that of the liquid.
Fig. 5. The pV-diagram of real gases
Due to the interactions between the particles the equation of state for perfect gases (eq. 1) isno longer correct for real gases. But, as we mentioned already, the lower the pressure themore a real gas resembles an ideal gas, this suggests that we can construct an equation of stateas follows:
...........''1 2 pCpBRTpVnV
p m (7)
or alternatively:
...........1 2
mmm V
CVB
RTpVnV
p (8)
These equations are two versions of the virial equation of state, the coefficients B’, C’, B and C are virial coefficients, which may be temperature dependent. Once they are determined for acertain gas, eq. (7) or (8) can be applied for every combination of pressure, volume andtemperature. The second, third,.... terms in the parentheses of eq. (7) or (8) are correctionsterms to the ideal gas equation and become important for high pressures.
The disadvantage of using eq. (7) or (8) for the description of real gases is of course that thevirial coefficients have to be known. There exists another equation, which is less accurate butcontains less parameters, the van der Waals equation:

Physical Chemistry I III. The properties of gases------------------------------------------------------------------------------------------------------------
11
2
Vn
anbV
nRTp (9)
A comparison of this equation and the ideal gas equation (1) shows that (i) the volumeavailable to the molecules in (9) is smaller than V by the volume of the molecules themselves(nb) and (ii) due to the attracting forces between the molecules the pressure is lowered by anamount proportional to a.

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
12
IV. The first Law of Thermodynamics
IV. a. The first Law
As defined above we separate the universe into a system and its surroundings. The system hasproperties which are described by the state variables. A very important property of the systemis its total energy (expressed for instance in Joule/mol) , which we call U. This internal energyof the system can be changed by the transfer of energy between system and surroundings, forinstance because of the transfer of energy in the form of heat q or by applying work to thesystem: to compress the gas in a cilinder we (the surrounding) have to perform work on thesystem. Due to this work w the internal energy of the system is increased. Experience frommany experiments during many centuries has shown that energy does not disappear.Therefore the exact amount of energy that is transferred from the surrounding into the system,must exactly equal to the change ΔU of the internal energy of the system:
wqU (10)
This is the first law of thermodynamics and it is important to realise that this law is based onexperience, it is an axiom. We assume that this law is always true and that means that eq. (10)is also true when the amount of exchanged energy is very small, infinitely small. Suchinfinitely small amounts in mathematics are denoted by the differential sign d, therefore eq.(10) can also be written in a differential form as:
dwdqdU (11)
Eq. (10) and (11) also express that heat and work are equivalent forms of energy. When anengine does work w, the system = engine must receive energy from the outside (q) and/or itsinternal energy decreases by ΔU. No engine can do work without consuming some form of energy (no perpetuum mobile of the first kind).
Equations (10) and (11) allow the following conclusions:
(i) The internal energy of a closed system remains constant(ii) When the system is thermally isolated (q or dq=0), the internal energy can only
change by work (dU = dw).
By the first law three important state variables have been introduced, U, q and w.
IV. b. The internal energy U
When we assume that we mainly consider systems composed of molecules the internal energyrepresent the energy of all these molecules. Molecules can store energy as they have differentdegrees of freedom: they can translate in space (translational energy), the molecule as awhole or some part of the molecules (e.g. a methyl group) can rotate (rotational energy), theelectrons in the molecule can be excited into higher orbitals (electron energy), the nuclei in amolecule can execute vibrations (vibrational energy) and the interaction energy betweenmolecules can be changed. All these energies together form the internal energy U.

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
13
IV. c. Work w
Work can be applied to a system in various ways. For our discussion the work done tocompress or expand a gas (“volume” work) is important, this is the work that has to be doneon a system when we change the volume of the system. Again our cylinder with piston is anice example, for the compression of the gas in the cylinder we have to apply work. Theamount of work certainly depends on the pressure p we have to exert on the piston and on thechange of the volume ΔV (or in differential form dV) we accomplish:
pdVdworVpw (12)
The minus sign expresses that for a decrease of the volume (ΔV and dV negative, compression ) we have to do positive work.
The two expressions (12) are related, while:
II
I
II
IpdVdww (13)
Volume work is certainly not the only form of work, one can distinguish mechanical andelectric work. To change the surface of a droplet or of a film from area A to A + dA work hasto be done against the surface tensionγ:
dw = γdA (14)
Mechanical work is always the product of a force K and a displacement ds:
To displace a mass over a distance ds: dw = KdsTo raise a mass over a height dh: dw = Kdh
Electrical work is done when electrical charges are moved or changed in an electric potentialfield:
To change the charge of a particle in a potential field φ from Q to Q + dQ: φdQdw (15)To move an electrical charge Q from a potential φ to φ + dφ: dw = Qdφ (16)
IV. d. Heat q
Heat is one of the forms of energy and it can flow into or out of a system. A copper rod, forinstance, can be heated on one side by a flame. This input of heat raises the temperature of thesystem. The temperature raise dT for a given amount of heat dq depends on the heat capacityC of the system:
CdTdq
The heat dq in general is also used to make the system expand (the copper rod becomeslonger, actually the volume increases). So heat can be used to raise the temperature (increaseof the internal energy U) and/or increase the volume (expansion work).

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
14
Two special situations can occur:a. All of the heat is used to raise the temperature by keeping the volume constant:
dTCdq Vb. When the pressure of the system is kept constant, the applied heat is used for expansion
and temperature raise: dTCdq p
When the same system is heated with the same amount dq, once at constant volume and onceat constant pressure, the resulting temperature raises (dT)V and (dT)p, respectively, are notequal:
(dT)v > (dT)p
while at constant volume all energy is used to raise the temperature and at constant pressureonly part of the energy is used for expansion. With:
Cv (dT)v = Cp (dT)p
Cv < Cp (17)
We can now derive a useful relation between CV and the internal energy U:
dU = dq + dw = dq - pdV when only volume work is considereddU = dq = Cv dT if we assume dV = 0 (constant volume)
and then:
V
V TU
C
(18)
Equation (18) can be considered as the definition of the heat capacity CV.
IV. e. State functions
So far we have seen many state variables, p, V, T, U...... which describe the state of thesystem. An important general distinction between state variables can be made: the value ofsome variables depend only on the present state of the system and of others on the path thesystem has reached the present state. The temperature of a system, for instance, describes thestate of the system and is independent of the path over which the system has reached thisstate. From one temperature measurement of a system it can not be determined if the systemwas before at a lower, the same or a higher temperature. The energy therefore is a statefunction. When we look, however, at our definition of volume work, eq. (13):
II
I
II
IpdVdww (13)
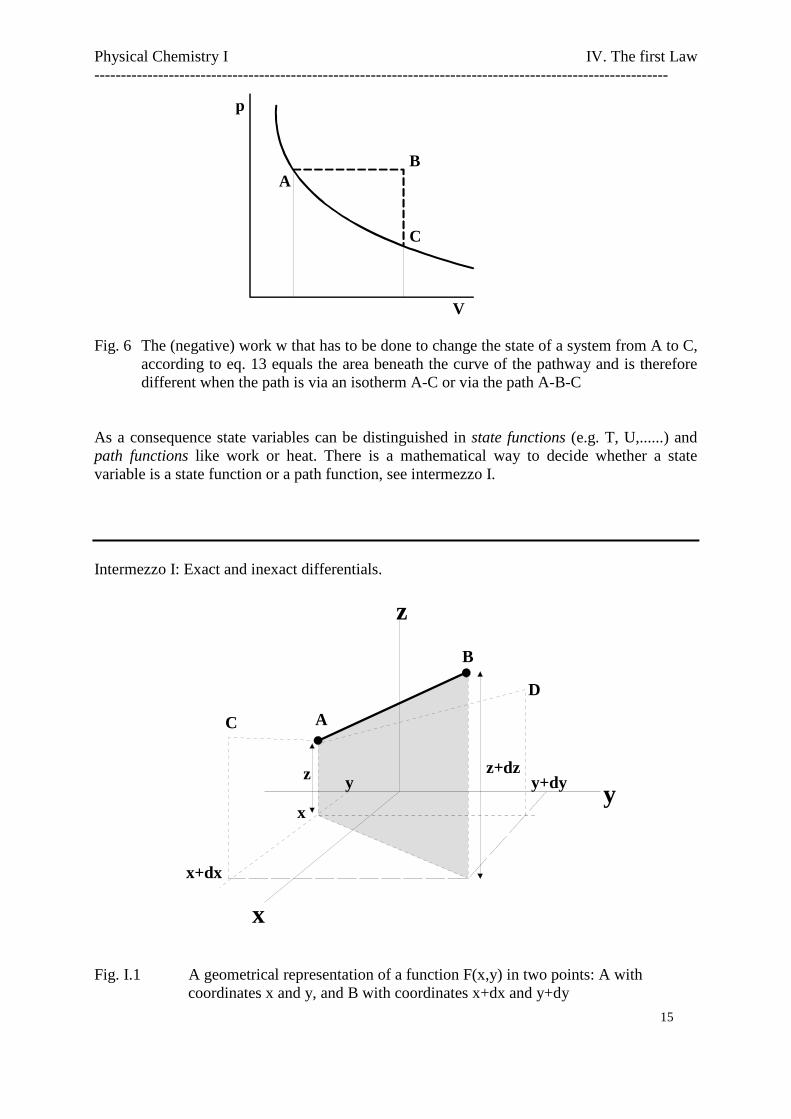
it is immediately clear from fig. 6 that the work w depends on the path the system has gonebetween state I and state II, while the integral is equal to the area beneath the curve in the p-Vdiagram.
Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
15
AB
C
V
p
Fig. 6 The (negative) work w that has to be done to change the state of a system from A to C,according to eq. 13 equals the area beneath the curve of the pathway and is thereforedifferent when the path is via an isotherm A-C or via the path A-B-C
As a consequence state variables can be distinguished in state functions (e.g. T, U,......) andpath functions like work or heat. There is a mathematical way to decide whether a statevariable is a state function or a path function, see intermezzo I.
Intermezzo I: Exact and inexact differentials.
z
x
yz z+dz
A
B
C
D
x
x+dx
y y+dy
Fig. I.1 A geometrical representation of a function F(x,y) in two points: A withcoordinates x and y, and B with coordinates x+dx and y+dy

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
16
Let us consider a function of two variables: z=F(x,y), this function represent a surface in the(x,y,z)-coordinate system (fig. 7). In point A with coordinates x,y,z the value of the functionis z; in point B with coordinates x+dx,y+dy,z+dz the function has the value z+dz. When dxand dy are both infinitesimally small, then also dz is infinitesimally small. In that case thesurface between A and B can be considered to be a straight plane and the increase in height,when going from A to B, is the sum of the increase in height (dz)y when going from A to C,parallel to the x-axis, and the increase of z going from A to D, (dz)x:
dz = (dz)y + (dz)x
The increases in z along the x and y axes are: dxxz
dzy
y
and dyyz
dzx
x
,
and therefore: dyyz
dxxz
dzxy
(I.1)
We now investigate the integral D
A
dz for two different path ways in the x,y-plane (fig. 8):
A-B-D and A-C-D.
A
B D
C
X
Y
x x+dx
y
y+dy
Fig. I.2
Path 1:
D
A dyyx
D
B dyy
B
A x
D
A xy
D
A
dxxz
dyyz
dxxz
dyyz
dyyz
dxxz
dz
Path 2:
D
A dxxy
D
C dxx
C
A y
D
A xy
D
A
dyyz
dxxz
dyyz
dxxz
dyyz
dxxz
dz
The two integrals are identical when: dxxz
dyyz
dyyx
= dyyz
dxxz
dxxy
,

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
17
or when: dxxz
xz
dyyz
yz
ydyyxdxx
. (I.2)
With the definitions for differentiation:
yxz
yz
xdx
yz
yz
xdxx
2
andxyz
xz
ydy
xz
xz
ydyy
2
,
eq. (I.2) changes into:xyz
yxz
22
(I.3)
In general one can prove, that the integral D
A
dz is independent of the path A–D when dz can
be written as:
dyyz
dxxz
dzxy
(I.1)
and the condition (I.3) is fulfilled.
In that case dz in expression (I.1) is called the exact or complete differential and inthermodynamics z is a state function.When condition (I.3) is not fulfilled, dz in (I.1) is the inexact or incomplete integral and z willbe a path function.
For an exact differential the cyclic integral: dz must be zero!
IV. f. Enthalpy
In IV. d. we found that CUTV
V
and we also saw that there is a heat capacity Cp. We can
ask ourselves would there be a variable H with which we can make a definition for Cp as:
P
P TH
C
(19)
When we define H = U + pV ,
we find by differentiation

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
18
dH = dU + pdV + Vdp = (if we consider only volume work)= dq - pdV + pdV + Vdp = dq + Vdp
dH = dq , when dp = 0with (dq)p = Cp dT
dH = Cp dT (20)
orp
TH
Cp
H = U + pV Enthalpy
The enthalpy is a form of energy and therefore an extensive variable. Is H also a statefunction? Using the recipe of Intermezzo I we find that the product pV is a state function andwhile U also is a state function, H is a state function.
The enthalpy plays an important role in chemistry while many chemical processes run at aconstant pressure. We now have two equivalent expressions for the first law:
dU = dq - pdV dH = dq + Vdp
The usefulness of the second expression becomes clear when we look at how the expressionschange at constant pressure :
dU dq for p = constant dH = dq for dp = 0
IV. g. Thermochemistry
At constant pressure the heat input into the system equals the change in enthalpy ΔH of the system and not the change of the internal energy ΔU. Therefore, for every process at constant pressure where heat is supplied to the system or furnished by the system the amount of thisenergy can be expressed as an enthalpy change. There are many physical or chemical changesof a system which can be described by an enthalpy change. To mention a few:
Melting melt enthalpy ΔmHSublimation:. sublimation enthalpy ΔsHChemical reaction: reaction enthalpy ΔrHAdsorption: adsorption enthalpy ΔaH
d (pV) = pdV + Vdp
pp
VV
1 1

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
19
Combustion: combustion enthalpy ΔcH
Usually enthalpy changes are reported for processes under a set of standard conditions. Thestandard enthalpy change ΔHØ(T) is defined for a process where all species are in theirstandard state:
The standard state of a substance is the thermodynamically most stable state at the specifiedtemperature T and at a pressure of 1 bar = 105 Pascal.
Example: C(solid, graphite) + O2 (gas)→ CO2 (gas) ΔrHØ(298 K) = - 393.5 kJ/mol
At 298 K the stable form of carbon is graphite and for O2 and CO2 the gasstate.
For a chemical reaction the standard reaction enthalpy can be positive or negative:
Endothermal reaction : r H > 0 The reaction needs energy:If isothermal process Heat flows into the systemIf adiabatic process The temperature decreases
Exothermal reaction : r H < 0 The reaction produces energy:Isothermal process The reaction produces heatAdiabatic process The temperature increases
A reaction enthalpy can be calculated from the formation enthalpies of the compoundsinvolved. The formation enthalpy is the enthalpy needed to build a compound from itselements:
Example: 2HN3 (l) + 2 NO(g)→ H2O2 (l) + 4N2(g)
The compounds consist of the following elements: H, N, O. The standard formationenthalpies at T=298 K to build these compounds from the elements are:
HN3(l) 264.0 kJ/molNO(g) 90.25H2O2 (l) -187.84N2(g) 0
We can construct the following triangle:
ΔrH2HN3 (l) + 2 NO(g) H2O2 (l) + 4N2(g)
2* 264.0 + 2* 90.25 1*–187.8 + 4* 0
H , N, O
Since energy is a state function, independent of the path, we have the following equality:2*264.0 +2*90.25 +ΔrHØ(298 K) =– 187.8 + 4*0 or ΔrHØ(298 K) = -896.3 kJ/mol.

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
20
A comparable situation is found when an overall reaction can be written as a series ofreactions. Hess‘ law states that the overall reaction enthalpy is the sum of the reactionenthalpies of the individual reactions. This is also a consequence of the first law ofthermodynamics.
ΔrH in kJ/molNa (s) + H2O (l) → NaOH (s) + ½H2(g) -139,78½H2(g) + ½Cl2(g) → HCl(g) -92,31HCl(g) + NaOH(s) → Nacl(s) + H2O(l) -179,06
Na(s) + ½Cl2(g) → NaCl(s) ΔrH = -411,15 kJ/mol
Na+(g) + e-(g) + Cl(g)___________________
Na+(g) + e-(g) + ½Cl2(g) +121____________________________
-351
Na+(g) + Cl-(g)+498 ______________________
Na(g) + ½Cl2(g)_________________
+107 -xNa(s) + ½Cl2(g)__________________
+407NaCl(aq)
____________________________
Fig. 7 A thermochemical cycle to determine the standard hydration enthalpy of Na+(g) and Cl-
(g) in water.Related to this law are the so called thermochemical cycles.
Let us suppose we want to know the standard enthalpy of hydration ΔhydrHØ(298 K) of Na+
and Cl- ions in water. The cycle can be built from individual steps, as shown in fig. 7:
The cycle is built from the following individual reactions:
(1) Na(s) + ½ Cl2(g)→ NaCl(aq) ΔrHØ(298 K) = - 407 kJ/mol(2) Na(s)→ Na(g) ΔsHØ(298 K) = 107 kJ/mol(3) Na(g)→ Na+(g) + e-(g) ΔiHØ(298 K) = 498 kJ/mol(4) ½ Cl2(g)→ Cl(g) ΔdisHØ(298 K) = 121 kJ/mol(5) Cl(g) + e-(g)→ Cl-(g) ΔegHØ(298 K) = -351 kJ/mol

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
21
The hydration enthalpy can then be calculated since energy is conserved:ΔhydrHØ(298 K)= -407-107-498-121+351=-782 kJ/mol
The combustion enthalpy ΔcH is the enthalpy for the oxidation of a substance to CO2 andH2O. For instance:
Glucose: C6H12O6 (s) + 6O2 (g) 6CO2 (g) + 6H2O (l) ΔcH(298 K)= -2808 kJ/mol
The anaerobic fermentation of glucose brings much less energy:
enzymesC6H12O6 (s) 2CH3CH(OH)COOH (s) ΔcH(298 K)= -120 kJ/mol(glucose) (lactic acid)
In general reaction enthalpies are temperature dependent. Therefore if we find values forenthalpy changes in a handbook at a given temperature T1, we must be able to calculate thesevalues for the wanted temperature T2.
Since reaction enthalpies usually are reported for a single temperature, we must be able tocalculate the reaction enthalpies at an other temperature.To find the temperature dependence of the enthalpy integrate eq. (20):
dH = Cp dT (20)
2
1
)()( 12
T
TpdTCTHTH
When the temperature change T2-T1 is not large, we often can make the approximation thatthe Cp’s are independent of the temperature. Then the integral can be easily solved and we find:
1212 )()( TTCTHTH p (21)
When eq. (21) is correct, it is correct for the formation enthalpy of each component involvedin a reaction:
aA + bB→ cC + dD
)()()()()( 11111 THdTHcTHbTHaTH fDfCfBfAr
)()()()()( 22222 THdTHcTHbTHaTH fDfCfBfAr
)()()()(
)()()()()()(
1212
121212
THTHdTHTHc
THTHbTHTHaTHTH
fDfDfCfC
fBfBfAfArr
Using eq. (21):

Physical Chemistry I IV. The first Law------------------------------------------------------------------------------------------------------------
22
)()(
)()()()()(
1212
12121212
TTCTTCd
TTCcTTCbTTCaTHTH
prpD
pCpBpArr
where pDpCpBpApr dCcCbCaCC
For a general reaction, written as:
J
J J0 (the stoichiometric coefficients νJ are negative for educts and
positive for products)
J
pJJpr CC (22)
Also the energy involved at phase transitions at constant pressure (melting, vaporization,sublimation) can be expressed in enthalpy:
H2O (l) H2O (g) ΔHvap(298 K)= 44 kJ/mol
ΔHvap(373 K)= 40.7 kJ/mol
Above we calculated the change in enthalpy, what would be the change of the internalenergy? At 100oC and 1 bar the volume of 1 mol H2O(l) is 18 cm3, of the gas it is 30.000 cm3.Then:
ΔUvap(373 K)= ΔHvap
(373 K)–Δ(pV)= ΔHvap(373 K)–Δ(nRT)=
= ΔHvap(373 K)–RTΔ(n)=40.7-3.1=37.6 kJ/mol
Here we neglected the molar volume of the liquid and assumed that the vapour is an ideal gas.

Physical Chemistry I V. The Second Law------------------------------------------------------------------------------------------------------------
23
V. The Second Law of Thermodynamics
The second law, which, just as the First Law, is based on many experiments in the past andmust be considered as an axiom, can be formulated in several ways. Traditionally thefollowing two formulations are used.
1. (After Kelvin) : It is impossible to completely transform heat from a reservoir intomechanical work. Schematically:
is not possibleq = heatw= work
(As before, we use the sign convention, that when the internal energy of a system increases byheat input q or work w done on the system, this heat or work is taken positive. If the internalenergy decreases, the energy input is negative).
2. (After Clausius): Without changing the surrounding it is impossible to transfer heat(energy) from a reservoir with temperature T2 to a reservoir with temperature T1 if T2<T1.
Schematically:
is not possible without changing the environment.
These two formulations lead to a formulation, which is most important for chemicalthermodynamics:
3. a. There exists a state function S, called the entropy.b. When a system changes through a process from state I to II, the relation between thechange of the entropy, ΔS or dS, ofthis system and the heat exchanged between thesystem and its environment depends on the type of process:
I II reversibel: II
I
II
I
reversibelSystem T
dqdSΔS , (23)
I II irreversibel: II
I
II
I
elirreversibSystem T
dqdSΔS , (24)
or combined: II
ISystem T
dqΔS or in differential form
Tdq
dS (25)
II
I Tdq
ΔS ; when T = constant: II
I Tq
dqT1
ΔS (26)
T Sq w= -q
T2 T1
q

Physical Chemistry I V. The Second Law------------------------------------------------------------------------------------------------------------
24
At first sight, this inequality (26), called after Clausius, seems to be in contradiction with theformulation 3. a, which implies that the entropy, being a state function, should be independentof the path or type of process (reversible or irreversible):
leirreversibSystem
reversibleSystem ΔSΔS (27)
It is important to realize, however, that the amount of involved heat dq in eqs. (23) and (24) isnot the same for reversible and irreversible processes.
To get a better understanding what entropy really is, let us look at the following twoprocesses:
Process 1.
T=100o C T=0o C
Process 2.
gas
In the first example two heat reservoirs, at different temperatures, are brought into thermalcontact. Everyone knows that after a while the two reservoirs reach an equilibrium at the sametemperature, depending on the heat capacities, somewhere between 0o and 100o C.In the second example at first a gas fills the left half of a volume and the right half is empty(vacuum). After opening a valve between the two halves the gas fills both halves at the samepressure. Both processes have in common that:
- the process is spontaneous in one direction; the opposite process will never be observed,- the total energy change is zero (the same processes will take place, when the two heat
reservoirs or the total volume would be a closed system, for which the first law tells that theenergy must be conserved).
The question is, what force drives these spontaneous processes? Clearly, it is not the energy,there must be another variable: the entropy. By looking at the two processes discussed above,we see that in the first example energy that at first was concentrated in the high temperaturereservoir, spontaneously distributes itself over both reservoirs. The second process shows thatmatter concentrated in a relative small volume has the tendency to distribute itself over alarger volume (when it can!). Entropy describes that energy and matter tend to distribute overas many degrees of freedom as is possible. Entropy is often said to be a measure of disorder,the higher the disorder, the higher the entropy. Order and disorder do not have a meaning in

Physical Chemistry I V. The Second Law------------------------------------------------------------------------------------------------------------
25
thermodynamics. In statistical thermodynamics it can be shown that the entropy is related tothe number of states accessible to the system. In our above example of the gas that distributesspontaneously over the total volume when a valve between the two halves is opened, the gasmolecules get access to positions in the empty half volume.
Entropy is a measure of disorder when disorder is somehow defined as the number of statesavailable to the system. This idea can be further clarified by the following often mentionedexample. Let us suppose there are two professors A and B. In the office of professor A booksand papers are lying in a seemingly random way all over the place. In professor B’s office allbooks are in book shelves and papers are stored in an orderly way. At first sight you wouldsay that the disorder in Prof. A’s office is much higher than in the office of Prof. B.However,when you visit both offices regularly and find Prof. B rearranging his books and papers all thetime, while the position of books and papers in the office of Prof. A do not change with time,the entropy of the office of Prof. B is higher than that of the office of Prof. A, while in Prof.B’s office the books and papers sample more positions than in Prof. A’s office. In the sense of entropy the disorder in Prof. B’s office is higher than in Prof. A’s office!
For some very simple systems and simple process we can calculate how much the entropychanges, but first we will discuss some consequences of eqs. (25) and (26):
II
ISystem T
dqΔS
For a closed system dq =0 and ΔSSystem ? 0: the entropy of a closed system increases when inthe system an irreversible process takes place. The entropy of a closed system can neverdecrease!A system and its surroundings form together a closed system (according to the definition ofsurrounding, outside the surroundings there is nothing!). Consequently:
Reversible process in the system: ΔSsystem + ΔSsurrounding = 0 or ΔSsurrounding = -ΔSsystem
Irreversible process : ΔSsystem + ΔSsurrounding > 0 or ΔSsurrounding > -ΔSsystem
The change of the entropy of the system is independent of the type of process (while S is astate function, see also eq. (27)), the change in entropy of the surroundings, however, dependson the process.
Entropy is the only variable that can be used to decide in what direction time changes. Whenwe agree that the universe is a closed system, the entropy of the universe steadily increases.By measuring the entropy of the universe at two different times, we can in principle decide atwhich time the universe is older.

Physical Chemistry I VI. Changes of the entropy------------------------------------------------------------------------------------------------------------
26
VI. Changes of the entropy
VI. 1. Isothermal expansion of an ideal gas
pa
We consider a gas in a cylinder closed by a pistonunder pressure p. When we only consider volume work,the first law states:
gas qdVpdqdU a pi
When the gas is ideal, the atoms or molecules do not interact(Ch. III. a.) and the internal energy does not change when we compressor expand the gas: dU=0. Further we expand the gas underreversible conditions, i.e. in such a way that always pa = pi.
dVpdq a
1
2ln2
1
2
1
2
1
2
1VV
nRTVdV
nRTdVV
nRTdVpdVpq
V
V
V
V
V
Vi
V
Varev
where we used the fact that the gas is ideal and the temperature is constant.
1
2lnVV
nRT
qS rev
gas (28)
Eq. (28) shows that the entropy of a gas increases when it is expanded.Since S is a state function, the above equation is also correct when the expansion would be viaan irreversible path. Conclusion:
Reversible expansion : Sgas + Ssurrounding = 0 ; Ssurrounding =
1
2lnVV
nR
Irreversibel : Sgas + Ssurrounding > 0 ; Ssurrounding >
1
2lnVV
nR
As already discussed in the previous chapter, the entropy change of the system is independentof the path of the process, the entropy change of the surrounding depends on the path!
VI. 2. The entropy change when a substance is heated
When a substance with heat capacity Cp is heated at constant pressure, the change intemperaure is proportional to the heat input (Chapter IV. d):
dTCdq p
The change of the entropy of the system does not depend on the reversibility or irreversibility
Physical Chemistry I VI. Changes of the entropy------------------------------------------------------------------------------------------------------------
27
of the process, therefore we can assume that it is reversible. Then:
2
1
2
1
2
1
2
1 1
2lnT
Tp
T
Tp
pT
T
revT
T TT
CTdT
CT
dTC
Tdq
dSS (29)
where it has been assumed that over the temperature range T2 –T1 the heat capacity isindependent of the temperature. We can change eq. (29) into:
T
p
T
dTCSTS
0
)0()( (30)
where S(0) is the entropy at the temperature 0 K. When S(0) would be known, eq. (30) wouldallow the calculation of the entropy at a higher temperature T. The entropy at 0 K will bediscussed in one of the following chapters. First we calculate what happens when between 0K and temperature T a phase transition occurs.
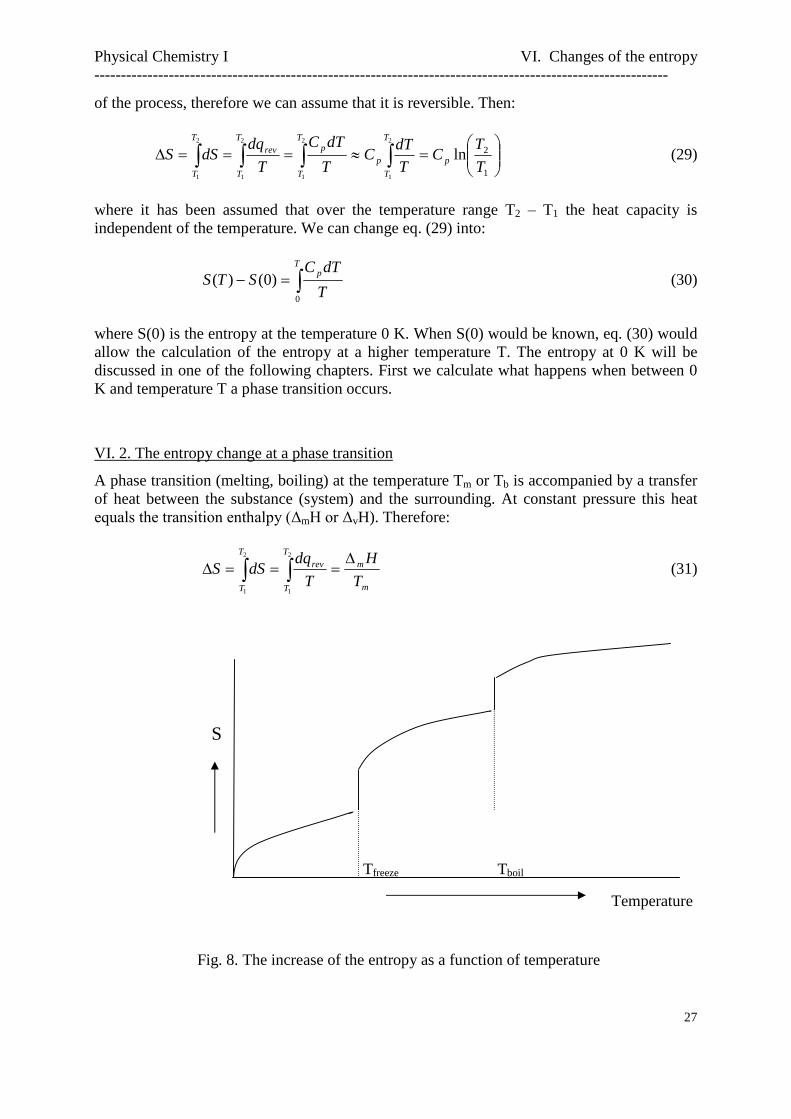
VI. 2. The entropy change at a phase transition
A phase transition (melting, boiling) at the temperature Tm or Tb is accompanied by a transferof heat between the substance (system) and the surrounding. At constant pressure this heatequals the transition enthalpy (ΔmH or ΔvH). Therefore:
m
mT
T
revT
T TH
Tdq
dSS
2
1
2
1
(31)
Fig. 8. The increase of the entropy as a function of temperature
Temperature
S
Tfreeze Tboil

Physical Chemistry I VI. Changes of the entropy------------------------------------------------------------------------------------------------------------
28
By combining eqs. (30) and (31) we are able the calculate the entropy difference S(T)–S(0)of a substance at an arbitrary temperature T, at least when the heat capacity at everytemperature is known:
T
T
p
m
vT
T
p
m
mT
p
v
v
m
m
T
dTC
TH
T
dTC
TH
T
dTCSTS
0
)0()( (32)
The change of the entropy with temperature is sketched in fig. 8.

Physical Chemistry I VII. The third law------------------------------------------------------------------------------------------------------------
29
VII. The third law
We saw that to calculate the entropy at an arbitrary temperature T (eq. 32) we need to knowthe entropy at 0 Kelvin. Without going into too much detail, we simply use the followinginterpretation of the third law:
The entropy of a perfect crystal at 0 Kelvin is zero.
Actually, the third law only claims that the entropies of all perfect crystals at T=0 are equal,but it is convenient to take the value S(0)=0. The difficult part of the above statement is thenotion of a perfect crystal. When we would have a crystal with defects (a defect could be thatat some lattice points an atom is missing). This “disorder” (see the discussion on entropy and disorder) raises the entropy and therefore the entropy of an imperfect crystal at 0 K can not beequal to 0. In this respect it is of interest to look at the crystal structure of ice: fig. 9.
Fig. 9 The structure of ice. Each oxygen is surrounded by four other oxygens in a regulartetrahedron. Each oxygen bonds to two hydrogens by short hydrogen bonds and to twoother hydrogens by long hydrogen bonds (from: L. Pauling, General Chemistry).
In the structure of ice (we will see later that at high pressures there are other ice structures,this structure is that of ice at normal pressure) each oxygen bonds to four other oxygens byfour hydrogen bonds. Two of these hydrogen bonds are short, two are long. Even at 0 K it isnot determined which hydrogen bonds are short and which are long. This „disorder“ contributes to a residual entropy S(0) of 3.4 J/Kmol.
Just as for the other thermodynamic variables, one defines standard entropies:
S(T) = standard entropy at standard conditions at temperature T.

Physical Chemistry I VII. The third law------------------------------------------------------------------------------------------------------------
30
r S= standard reaction entropy
Chemical reaction : 0 = J
JJν
J = educts and products, J positive for products, negative für educts.
r S=
J JSJν
Example: H2 (g) +12
O2 (g) H2O (l)
0 H2O (l) - H2 (g) -12
O2 (g)
r S= 69,9 - 130,7 -12
205,1 = - 163,4 J K-1 mol-1
Although r S< 0 the reaction is spontaneous because the entropy of the surroundingincreases strongly due to the highly exothermal reaction:
:
Ssurrounding = rH/T=2220000/298=7449 J/mol.Kand :
S total = r S+ S surrounding =-163+7449=7286 J/mol.K >0

Physical Chemistry I VIII. Free Energy and Free Enthalpy------------------------------------------------------------------------------------------------------------
33
VIII. Free Energy and Free Enthalpy
VIII. 1 Introduction of F and G
In the previous chapters we have seen that we can predict whether a chemical reaction runsspontaneously or not. The recipe was to calculate the entropy change of the system and of thesurrounding. When the total entropy change was positive the reaction would spontaneouslygo. In this chapter we introduce two new state functions with which it is somewhat easier todecide whether a reaction goes spontaneously or not. We do this in the following schematicway:
Assumption: only volume work
dU = dq - p dV first law dH = dq + V dpwith dV = 0 with dp = 0dU = dq dH = dq
Clausius´ inequality dSSystem dqT
dS dUT
dS dHT
T dS dU T dS dHdU - T dS 0 dH - T dS 0
d (U - T S) 0 if T = constant d (H - T S) 0(dF) V,T 0 (33) (dG) p,T 0 (34)
F =U-TS= Free Energy G = H-TS = Free Enthalpy(IUPAC : A = Helmholz Energy) (IUPAC: G = Gibbs-Energy)
F and G are functions of state, because U, H and (TS) are functions of state.
Eq. (33) shows that at constant volume V and temperature T the change in free energy of thesystem is negative for irreversible (spontaneous) processes and = 0 for reversible processes.Eq. (34) shows that at constant pressure p and temperature T the change in free enthalpy ofthe system is negative for irreversible processes and = 0 for reversible processes.
Now we can use the free energy F and the free enthalpy G of the system to decide whether aprocess goes spontaneously or not and we do not have to consider the entropy change of thesystem + surrounding. However, it is important to realize that the eqs. (33) and (34) are onlyvalid for V, T constant, respectively p, T constant. In case of the entropy we did not have sucha limitation.
According to the definition of G we can write:
ΔG = ΔH - T ΔS if T = constant (35)
energy- entropy-contribution contribution

Physical Chemistry I VIII. Free Energy and Free Enthalpy------------------------------------------------------------------------------------------------------------
34
and this shows that a spontaneous process for which ΔG < 0 not only can occur when H < 0(exothermic) and S > 0, but also when:
S < 0 and ΔH < T ΔS (highly exothermic process)or H > 0 and T ΔS > ΔH (spontaneous endothermic reaction!)
VIII. 2 Free Energy, Free Enthalpy and work
In the same schematic way as before we can derive very useful relations between F and G andthe maximum amount of work a system can deliver.
dU = dq + dwdw = dU–dqT dS dqdw dU–T dS–dw T dS–dUdw= work done on the systemdw´=–dw = work done by the systemdw’T dS–dUdw’–dF (T= constant)
The maximum amount of work a system candeliver, is obtained for a reversible processand is equal to -dF (T= constant)
dw’max = - dU + TdS
The internal energy U can not completely beused for work when dS is positive
dU „ordered“ stored energyTd S „disordered“, non usable energy
dH = dU + p dV + V dpdH = dq + dw + p dV + V dpT dS dqdH T dS + dw + p dV + V dpdw = dwa - p dVdwa = all work that is not volume workdH T dS + dwa + V dpdH–T dS dwa + V dpp,T = constantdG dwa
dwa’ = –dwa = work done by the systemdwa’ –dG
The maximum non-volume work that asystem can deliver is obtained for areversible process and is equal to–dG(p,T = constant)

Physical Chemistry I VIII. Free Energy and Free Enthalpy------------------------------------------------------------------------------------------------------------
35
VIII. 3 Free Energy and rubber elasticity
Maybe entropy is still an abstract idea, therefore a little “experiment” to show that entropy is so real you can feel it! We first make a combination of the first and second law:
dU = dq + dw = TdS +dw (is true if the process is reversible)dU = TdS +dw (must be true also for irreversible processes while U is a
function of state).
When we stretch a rubber band from length l to a length l+dl by applying a force K we haveto do stretching work Kdl and possibly volume work -pdV. The internal energy U changes:
dU=TdS+Kdl-pdV
In good approximation the volume of a rubber band does not change upon stretching, dV=0.With:
dF=dU-TdS (if T = constant)
one finds:
dF=Kdl,
or: KTV,l
F
polymer chain:
The above equation means that the force K we have to apply to stretch the rubber band atconstant temperature and volume equals the derivative of the Free Energy with respect to thelength of the band.
KTV,l
ST
TV,lU
TV,lF
(36)
Energy Entropyelasticity elasticity
Eq. (36) shows that part of the force K we have to apply to stretch a rubber band is due to thechange of the entropy with the length of the band (for most rubbery systems the entropy partis the main part, rubbers for which the energetic part is zero are called ideal rubbers). Thischange of the entropy comes about while upon stretching the polymer molecules are forced tochange their form from a more chaotic (coiled form) to a more regular stretched form.
Let us consider the experiment when a weight hangs on a rubber band and the temperature ofthe rubber is increased. What will happen to the length of the rubber band?

Physical Chemistry I VIII. Free Energy and Free Enthalpy------------------------------------------------------------------------------------------------------------
36
The right side of eq. (36), the force K, does not change with temperature because it is equal tothe mass of the weight multiplied with the gravity factor g. Where we are looking for is thepartial derivative:
KTl
To derive the sign of this derivative we use two mathematical relations:
a. For a function z, which depends on two independent variables x and y, z = z (x,y), one hasthe cyclic rule (“Kettenregel”):
1
yxz xz
zy
yx
For the function l = l (K,T) this implies:
1
TKl Kl
lT
TK
orTl
K
K Kl
TK
lTT
l
1
(37)
We immediately see that the second term on the right sideTK
l
>0, the length at constant
temperature will increase with increasing force K.
b. The derivativelT
K
in (37) can be rewritten using the fact that the free energy F is a state
function:dF=dU-TdS-SdT=TdS+Kdl-TdS-SdT=Kdl-SdT
and
Tl lS
TK
(Schwarz equation) (38)
The entropy will decrease with increasing length l because the molecules become more
ordered when the band is stretched, thereforelT
K
in eq. (38) is positive, which means that
KTl
in eq. (37) must be negative, the length of the rubber band will decrease at increasing
temperature.

Physical Chemistry I IX. The fundamental equations and the chemical potential------------------------------------------------------------------------------------------------------------
37
I. IX. The fundamental equations and the chemical potentialIX.1 The fundamental equations
When the only work we consider is volume work, we can write the following equations, thefundamental equations, which combine the statements of the First and Second Law ofThermodynamics.
dU = dq-pdV dU=TdS-pdV (38)
(the = sign instead of the ? sign can be used here because U is a state function, whose valueshould be independent of the path, reversible or irreversible).
Then:dH=dU+pdV+Vdp dH=TdS+Vdp (39)
dF=dU-TdS-SdT dF=-SdT-pdV (40)
dG=dH-TdS=SdT dG=Vdp-SdT (41)
IX.2 The chemical potential
U, H, F and G are extensive variables, this implies that their values should also depend on theamounts (mols) of the substances in the system:
G = G(p, T, nA, nB,………….nN)Or:
N
nTpNA
nTpAnpnT
dnnG
dnnG
dTTG
dppG
dGiiii ,,,,,,
......
(42)
(in the partial derivatives ni means that all n are to be kept constant, except the n involved inthe differentiation)
This can also be written as:
j
j
nTpjnpnT
dnnG
dTTG
dppG
dGi
ii ,,,,
(43)
A comparison between eqs. (41) and (43) shows that the coefficients of dp and dT in eq. (43)must be:
VpG
inT
,
STG
inp
,
The coefficients of the differential dnj in eq. (43) are defined as the chemical potential μj ofthe substance j:

Physical Chemistry I IX. The fundamental equations and the chemical potential------------------------------------------------------------------------------------------------------------
38
j
nTpji
nG
,,
(44)
Combining eqs. (43) and (44) we find an important equation, which is often called the
fundamental equation of chemical thermodynamics:
j
jj dnSdTVdpdG (45)
The fundamental equations (38)–(40) can also be written in the form of eq. (45), but we willnot need them. The fundamental equations (38)-(41) are correct as long as the amounts of thesubstances are constant: dnj=0.
The chemical potentialμ for a single component system with n mol substance is very simple, while:
GnG and ,,
GnG
Tp
(46)
where G is the Free Enthalpy of 1 mol of the substance, the molar Free Enthalpy.
To show why the chemical potential is so important, we consider a N-component system. Wesuppose that in this system the chemical potential of any component is the same everywherein the system, except for component I, which differs at site 1 and 2: μi(1) > μi(2)
When we bring at constant p and T an amount dni of the substance i from site 1 to site 2 theresulting change of the free enthalpy is negative:
0)1()2()2()1( iiiiiii dndndndG
Therefore such a transport of substance i from a site with high chemical potential to a sitewith lower chemical potential is a spontaneous process! This process will continue until inequilibrium the chemical potential for each component of the system is the same everywherein the system. Thermodynamics does not say, however, how fast this process goes. It couldtake an infinitely long time.
1 2

Physical Chemistry I IX. The fundamental equations and the chemical potential------------------------------------------------------------------------------------------------------------
39
The conclusion is:
For a system in equilibrium the chemical potential for every substance is the sameeverywhere in the system, independent of the phase (the definition of phase comeslater).
For instance when at ca. 273 K and 1 bar ice and water are in equilibrium, then the chemicalpotential of H2O is the same in ice and water. At temperatures below 273K at 1 bar thechemical potential of ice is lower than that of water, therefore at this temperature water shouldfreeze. At room temperature and 1 bar the chemical potential of graphite is lower than that ofdiamond. Graphite at these conditions is thermodynamically more stable than diamond. Thetransition from diamond to graphite, however, is infinitely slow and therefore will not beobservable. So thermodynamics tells in what direction a process should go but not how fast!
IX. 3 The chemical potential of an ideal gas
By comparing eqs. (41) and (46) we find for a constant temperature:
dpVd By using the equation of state for an ideal gas RTVp this can be rewritten as:
dpp
RTd
which after integration from pressure pA to pE gives:
A
Ep
pAE p
pRT
pdp
RTppE
A
ln)()(
By choosing a special starting pressure (standard pressure) pA=p=1 bar=105 Pa (Pascal), theabove expression becomes:
pp
RTTTp ln)(),( (47)

Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
40
II. X. MixturesX.1 General
In most real cases a system consists of many different substances and can be considered to bemixtures of many components. Mixtures can also consist of more than one phase, for instancethe mixture in a bottle of beer consists of a liquid phase and a gas phase. The liquid phasecontains at least two components, water and ethanol, the gas phase is also a mixture of watervapour and ethanol vapour. The thermodynamics of the previous chapter can be perfectly wellapplied to the description of mixtures. To keep things simple we discuss only mixtures of twocomponents A and B, binary mixtures. The composition of a binary mixture is defined by themole numbers nA and nB. We frequently will work with the mole fractions xA and xB:
1
BABABA
BB
BA
AA xxnnn
nnn
xnn
nx
Another definition we need is the volume of 1 mole of A and B (molar volume), thesevolumes in general are not equal:
Molar volume of A = AV ; molar volume of B =
BV
The cross indicates that it is the volume of pure A or B.
X. 2. Partial molar quantities
As an introduction to the description of the free enthalpy of a mixture we first describe whathappens to the total volume V of a mixture when we add nA moles of A (volume =
AAVn ) tonB moles of B (volume =
BBVn ):
BBAA VnVnV
The reason is that the interaction between A and B molecules may differ from the interactionsbetween A molecules or the interaction between B molecules. As an example let us add 1mole water to a very large amount (say 1000 moles) of water and to a large amount of ethanol(also 1000 moles ethanol). In both cases the volume of the mixture increases by V:
1 mole water to 1000 moles of water V=18 cm3
1 mole water to 1000 moles of ethanol V=14 cm3
The reason for the difference is that in the first case every added water molecule is surroundedby other, identical water molecules. In the second case statistically the added water moleculeis surrounded by ethanol molecule sonly. Due to a difference in the intermolecular interactionwater–ethanol the density around the water molecules in the ethanol–water mixture ishigher than in the water–water mixture.
Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
41
This means that the volume of 1 mole water depends on the type and amount of the othercomponent in the mixture. In the pure liquid 1 mole of A has a volume equal to the molarvolume
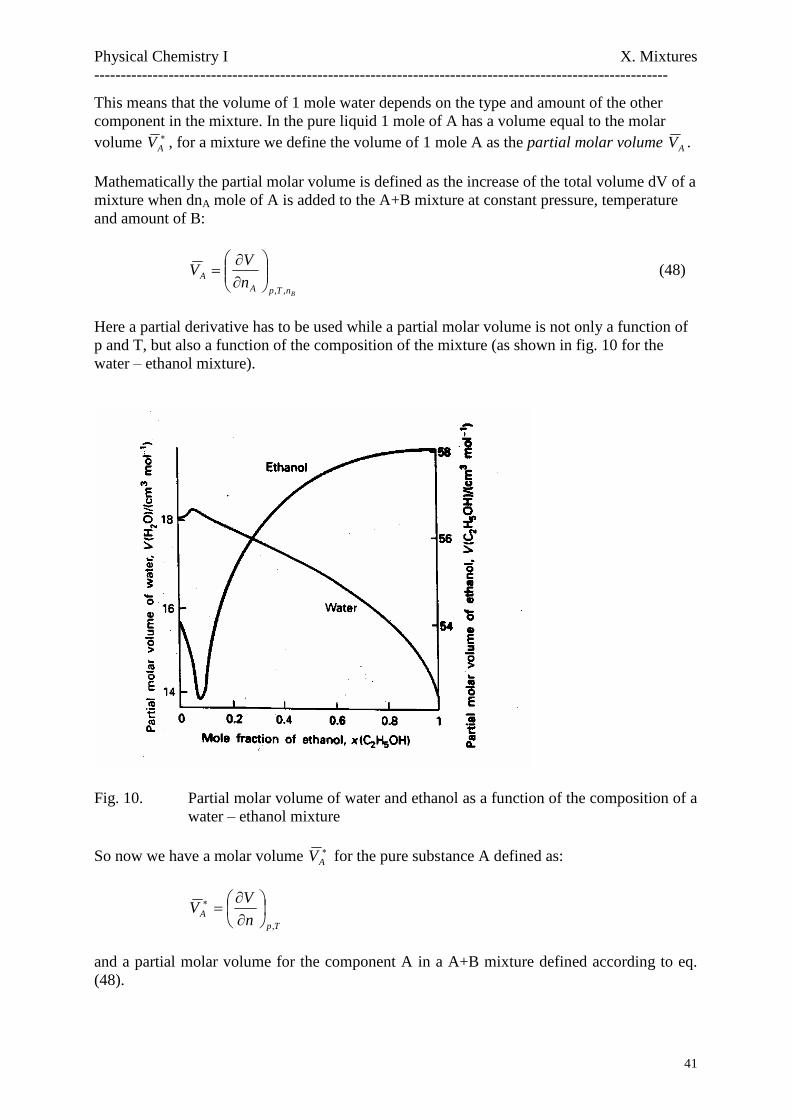
AV , for a mixture we define the volume of 1 mole A as the partial molar volume AV .
Mathematically the partial molar volume is defined as the increase of the total volume dV of amixture when dnA mole of A is added to the A+B mixture at constant pressure, temperatureand amount of B:
BnTpAA n
VV
,,
(48)
Here a partial derivative has to be used while a partial molar volume is not only a function ofp and T, but also a function of the composition of the mixture (as shown in fig. 10 for thewater–ethanol mixture).
Fig. 10. Partial molar volume of water and ethanol as a function of the composition of awater–ethanol mixture
So now we have a molar volume AV for the pure substance A defined as:
TpA n
VV
,
and a partial molar volume for the component A in a A+B mixture defined according to eq.(48).

Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
42
When we consider the total volume V of a mixture as a function of nA and nB, we can writefor the differential dV:
BBAAA
nTpBA
nTpA
dnVdnVdnnV
dnnV
dVAB
,,,,
(49)
Eq. (49) is a differential equation that can be integrated:
BBAA VnVnV (50)
The result eq. (50) is surprising because it looks as if one can integrate eq. (49) by assumingthat AV and BV are independent of nA and nB, which is certainly not the case (see for instancefig. 10). The proof that eq. (50) can be obtained by integration of eq. (49) will be given inAppendix I.
For the free enthalpy G we have a similar expression:
BBAABAAAB
nTpBA
nTpA
dndndnGdnGdnnG
dnnG
dGAB
,,,,
(51)
which we can integrate to:
BBAA dndnG (p,T=constant) (52)
Eq. (51) shows that the chemical potential can also be viewed as a partial molar entity.Remember that just as AV and BV also μA und μB are functions of nA and nB, or better xA!
X. 3. Binary mixture in two phases.
Before we can describe the chemical potential in a liquid mixture, we must have anexpression for the chemical potential of a component in a liquid phase. We can obtain such arelation by considering a pure liquid A in equilibrium with its vapour in a closed system (fig.11).
Fig. 11
A vapour
A liquid

Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
43
We assume that the vapour can be described as an ideal gas with pressure Ap . Applying eq.
(47) we have for the chemical potential of the gas:
pp
RTg AA ln)(
Now we use the fact that in equilibrium the chemical potential of A in the liquid state equalsthe chemical potential of A in the gas phase:
pp
RTgl AAA ln)()( (53)
Now we look at the binary mixture A+B in two phases, fig. 12:
Fig.12
A+B
A+B
We can apply eq. (53) also to this situation when we replace Ap by the partial pressure of A
in the gas mixture pA (we remove the star while now we discuss a mixture!):
pp
RTg AA ln)( (54)
By combining eqs. (53) and (54) we can write:
A
AAA p
pRTll ln)()( (55)
With equation (55) we got an expression for the chemical potential of a component in a fluidmixture and it shows that the presence of a second component A in the mixture A+B lowersthe chemical potential of A!Eq. (55) for A and a similar expression for B is still not so convenient for a liquid mixturewhile it depends on the partial pressure in the corresponding vapour. We would like to havean expression without parameters with depend on the vapour composition of the mixture.The following equations are correct for every possible composition of the mixture:
A
AAA p
pRTll ln)()(
B
BBB p
pRTll ln)()( (55)

Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
44
Now we first consider some special mixtures:
A. the mixture is ideal, that means that the molecular interaction between A molecules,A-A, is equal to that interaction between B molecules, B-B, and between A and Bmolecules, A-B. In an ideal gas these interactions are zero, in an ideal liquid mixturethese interactions of course can not be zero but are equal. An ideal mixture is formedonly when the molecules are very similar, for instance benzene and toluene. In thecase of an ideal mixture:
AAA pxp and BBB pxp (Raoult’s law) (56)
which changes eq. (55) into:
AAA xRTll ln)()( BBB xRTll ln)()( (57)
B. A special situation for two dissimilar molecules which do not form an ideal mixture(or solution) is found when the solution is very dilute. In such an ideal-dilute solution(N.B. an ideal-dilute solution is less ideal than an ideal solution!) both the solventmolecules A and the solute molecules B statistically are surrounded exclusively by Amolecules, the probability that two neighbouring molecules are both of the B type isvery small. For the A molecules this means that the situation is like in an idealmixture:
AAA xRTll ln)()( (Raoult’s law) (58)
For the solute molecules B this is different, here we writeB
BB K
px (Henry’s law)
and:
B
BBBB p
xKRTll ln)()( = BBB
B
BB xRTxRT
pK
RTl lnlnln)(
(59)
Here KB, the Henry constant, is a constant with the dimension of pressure and whichdepends on B only;
B is the chemical potential of B in a (hypothetical) mixture withfor which the partial pressure is KB.
C. For a real solution, not ideally diluted, only equations (55) are correct and here theexpressions for the chemical potentials are rewritten by replacing xA and xB in eqs.(58) and (59) by the activities aA and aB:
AAA aRTll ln)()( and BBB aRTll ln)()(
These activities are numbers like a mole fraction. The activities can also be introducedvia the molality (mole/kg solvent).
X. 4. A mixture of two liquids
We consider a binary mixture of nA mole of A and nB mole of B, n=nA+nB. Using eq, (52):

Physical Chemistry I X. Mixtures------------------------------------------------------------------------------------------------------------
45
BBAA dndnG
we can now calculate the total Free Enthalpy GB before (pure substances A and B) and aftermixing GM:
BBAAM
BBAAB
nnG
nnG
(60)
The difference is the Free Enthalpy of mixingΔMG and for an ideal mixture we can apply eq.(57):
BBAABBAABMM xxxxnRTxRTnxRTnGGG lnlnlnln (61)
For a mixture xA and xB are both smaller than 1, which makes ΔMG negative. This implies thatthe mixing process for an ideal mixture, at constant p and T, is always spontaneous for everycomposition! Components which are so similar that they form an ideal mixture, always mix.As the mixing Free Enthalpy is the sum of an energy and an entropy term:
STHG MMM (at constant temperature)
From eq. (45) we have:
BA nnpTG
S,,
and consequently also
BA xxp
MM T
GS
,,
; we find:
BBAAM xxxxnRS lnln (62)
The mixing entropy is positive, which also shows that the mixing process must bespontaneous. Combining eqs. (61) and (62) shows that for an ideal mixing process ΔMH=0(that is while all intermolecular interactions are equal). The mixing of an ideal mixture iscompletely driven by the entropy!
For many mixtures the mixing free enthalpy is not negative (or the mixing entropy is notpositive). Then also ΔMH is not equal to zero:
BBAAEM
BBAAEM
EM
xxxxnRTGGxxxxnRSS
HH
lnlnlnln
where the new parameters are the excess parameters, whose values indicate how much thereal mixture deviate from an ideal mixture. When ΔMG>0 the two components do not mix (forinstance oil and water for most compositions). When they have been somehow broughttogether, they spontaneously demix. It is found that for most substances the question whetherthey mix, depends on the relative concentrations and p and T. This can be indicated in aphase diagram, as will be discussed in Chapter XII.
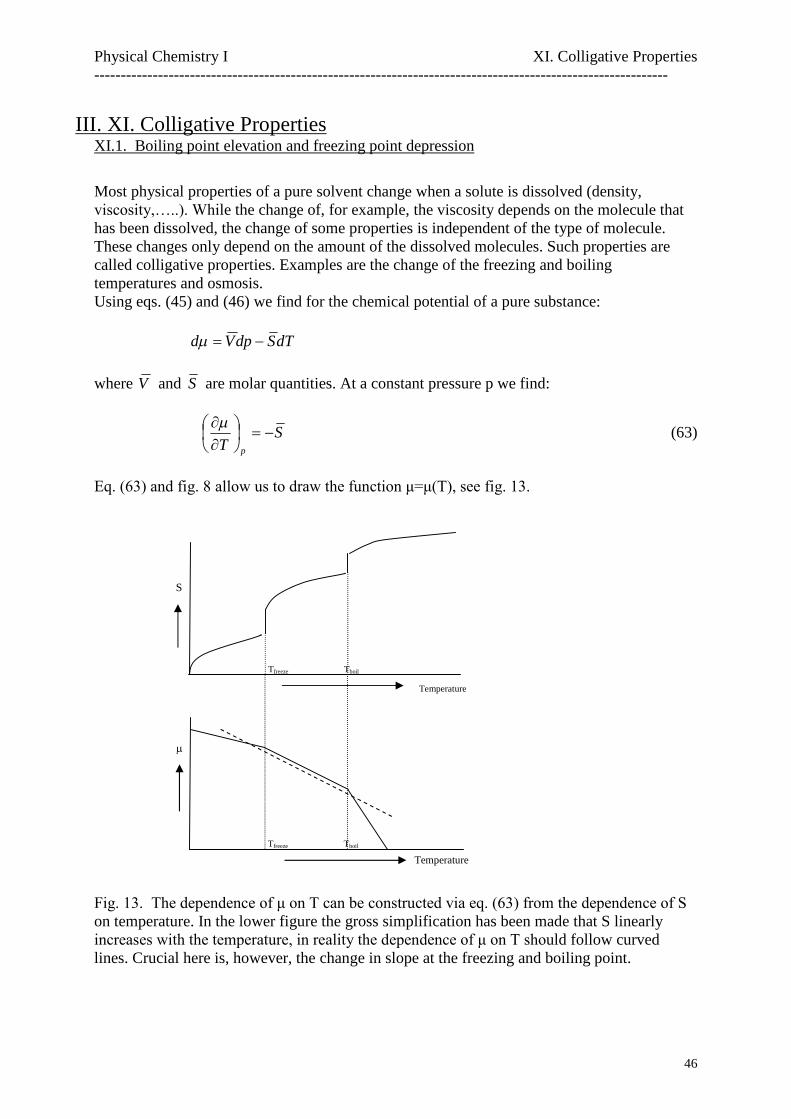
Physical Chemistry I XI. Colligative Properties------------------------------------------------------------------------------------------------------------
46
III. XI. Colligative PropertiesXI.1. Boiling point elevation and freezing point depression
Most physical properties of a pure solvent change when a solute is dissolved (density,viscosity,…..). While the change of, for example, the viscosity depends on the molecule thathas been dissolved, the change of some properties is independent of the type of molecule.These changes only depend on the amount of the dissolved molecules. Such properties arecalled colligative properties. Examples are the change of the freezing and boilingtemperatures and osmosis.Using eqs. (45) and (46) we find for the chemical potential of a pure substance:
dTSdpVd
where V and S are molar quantities. At a constant pressure p we find:
ST p
(63)
Eq. (63) and fig. 8 allow us to draw the function μ=μ(T), see fig. 13.
Fig. 13. The dependence of μ on T can be constructed via eq. (63) from the dependence of S on temperature. In the lower figure the gross simplification has been made that S linearlyincreases with the temperature, in reality the dependence of μ on T should follow curvedlines. Crucial here is, however, the change in slope at the freezing and boiling point.
Temperature
S
Tfreeze Tboil
Tfreeze Tboil
Temperature

Physical Chemistry I XI. Colligative Properties------------------------------------------------------------------------------------------------------------
47
XI. 2 Boiling point elevation
When a compound B is dissolved in the solvent A the chemical potential of the solventchanges:
AAA xRTll ln)()( (assumption the solution is ideal or ideally diluted)
Since xA=1-xB<1 the chemical potential of A has decreased relative to that of the pure liquid.When we assume for simplicity that the partial pressure of the solute is zero and that thesolute and solvent do not mix in the solid state, then only the curve of μ in the liquid phase changes, as indicated in fig. 13 by the dashed line. From this figure it is immediately clear thatthe boiling temperature has increased and the freezing point depressed.
The changes in freezing and boiling point can be calculated using expressions for thechemical potential. For the equilibrium of the vapour A (the vapour consists only of A whenwe assume that B has no vapour pressure) with the mixture A+B:
)()( lg AA and
AAA xRTll ln)()( follows:
RTTG
RTlGgG
RTlg
x AAAAAA
)()()()()(ln
(64)
where AG is the vaporisation free enthalpy of A.By differentiation of eq. (64) we get:
TTG
dTd
Rx
dTd A
A
)(1ln (65)
The reason to write eq. (64) in the clumsy form of eq. (65) is that we know how todifferentiate the terms in eq. (65).
Intermezzo: the Gibbs-Helmholtz equation.
SdtVdpdG (41)
STG
p
(66)
THG
STSHG
THG
TG
p

Physical Chemistry I XI. Colligative Properties------------------------------------------------------------------------------------------------------------
48
222
1TH
TG
THG
TT
GTG
T
TG
T
(67)
Eq. (67) is correct for any free enthalpy, therefore also for the vaporisation enthalpy:
222
)()()()(TH
TlH
TgH
TlGgG
TTG
TAAAAAA
(68)
By insertion of eq. (68) into eq. (65) we find:
dTRTH
xd AA 2ln
and integrating:
boil
boil
A T
T
Ax
A dTRT
Hxd 2
1
ln (69)
Here T*boil is the boiling temperature of the pure liquid A and Tboil the boiling temperature ofthe solution (Tboil>T*boil). When we assume that the vaporisation enthalpy AH does notdepend on the temperature over the temperature range Tboil - T*
boil= δTboil<<T*boil, then:
22
11ln
boil
boilA
boilboil
boilboilA
boilboil
AT
T
AA T
TRH
TTTT
RH
TTRH
TdT
RH
xboil
boil
(70)
The last step we have to make is to relate xA to the amount of dissolved species xB:
BBA xxx )1ln(ln when xB<<1.
The change in boiling temperature of the solvent due to dissolving B is then:
BA
boilboil x
HRT
T
2
(71)
The change in boiling temperature of A only depends on the amount B, not on the type ofmolecule B and further on the vaporisation enthalpy of the solvent.
XI. 3. Freezing point depression
In a completely analogous way the freezing temperature depression can be calculated, withthe result:

Physical Chemistry I XI. Colligative Properties------------------------------------------------------------------------------------------------------------
49
BA
freezefreeze x
H
RTT
2
(72)
XI. 4. Osmosis
Osmosis is found when the transport of solvent and solute molecules into a certain enclosedvolume is different. Many osmosis effects are found in biology, for instance for a cell forwhich the cell wall acts as a selective membrane. To describe the effect in a thermodynamicway we consider a volume with solvent A and solute B separated from the pure solvent A bya semi-permeable membrane, see fig.14. Experimentally it is found that the solution in thecompartment raises to a higher level than the level of the pure solvent. This causes an extrapressure, the osmotic pressure, on the solution of:
gh
where ρ is the density of the solution, g the acceleration due to gravity and h the difference in liquid levels..
Fig. 14. Osmosis; the semi-permeable membrane (dashed line) allows the transport of solventmolecules A but not of the solute B.
In equilibrium the chemical potential of A at the same height is the same in the pure solvent atpressure p and in the solution at pressure p+π:
AAA xRTppp ln)()( (73)
p
pAAA dpVpp )()( (74)
where AV is the molar volume of A.
By combining the eqs. (73) and (74) we find:
p
pAA dpVxRT ln
A+B
A
hp

Physical Chemistry I XI. Colligative Properties------------------------------------------------------------------------------------------------------------
50
or: AB VxRT (75)
obtained by using:
)(
)1()1ln(ln
pandpbetweenpressureoftindependenVifVdpV
xxRTxRTxRT
AA
p
pA
BBBA
In the compartment with volume V we have nA mole A and nB mole B; with nB<<nA we havethe following equations:
VVn
nn
x
AA
A
BB
(76)
By combination of the eqs. (75) and (76) we find the van ‘t Hoff equation for ideal solutions:
RTnV Bor:
RTB
where [B] is the concentration of B in mol per liter. For real solutions a virial equation can beassumed to exist:
..............1 2 BCBBRTB
Also the osmotic pressure π is not dependent on the type of molecule B, it only depends on the amount of B!

Physical Chemistry I VIII. Free Energy and Free Enthalpy___________________________________________________________________________
51





























