Embed Size (px)
Citation preview

PredictingRNA Structure and Function
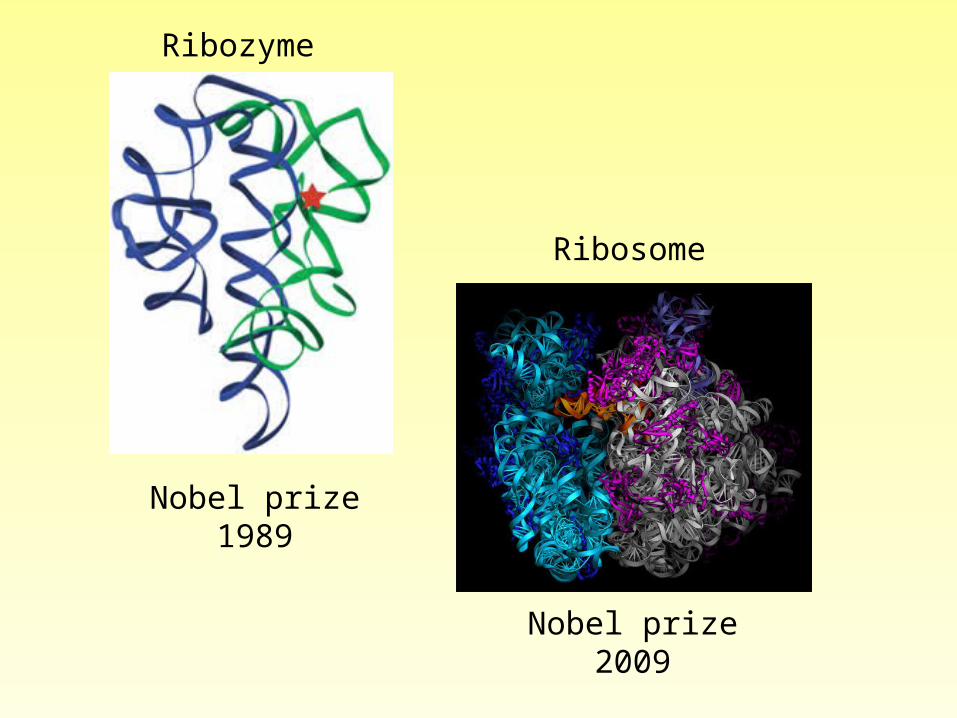
Nobel prize1989
Nobel prize2009
Ribozyme
Ribosome

RNA has many biological functions
• Enzymatic reaction (protein synthesis)
• Control of mRNA stability (UTR)
• Control of splicing (snRNP)
• Control of translation (microRNA)
The function of the RNA molecule depends on its folded structure

Protein structures RNA structures
Total 65000 Total 700

RNA Structural levels
tRNA
Secondary Structure Tertiary Structure

RNA Secondary Structure
U U
C G U A A UG C
5’ 3’
5’G A U C U U G A U C
3’
STEM
LOOP• The RNA molecule folds on itself. • The base pairing is as follows: G C A U G U hydrogen bond.
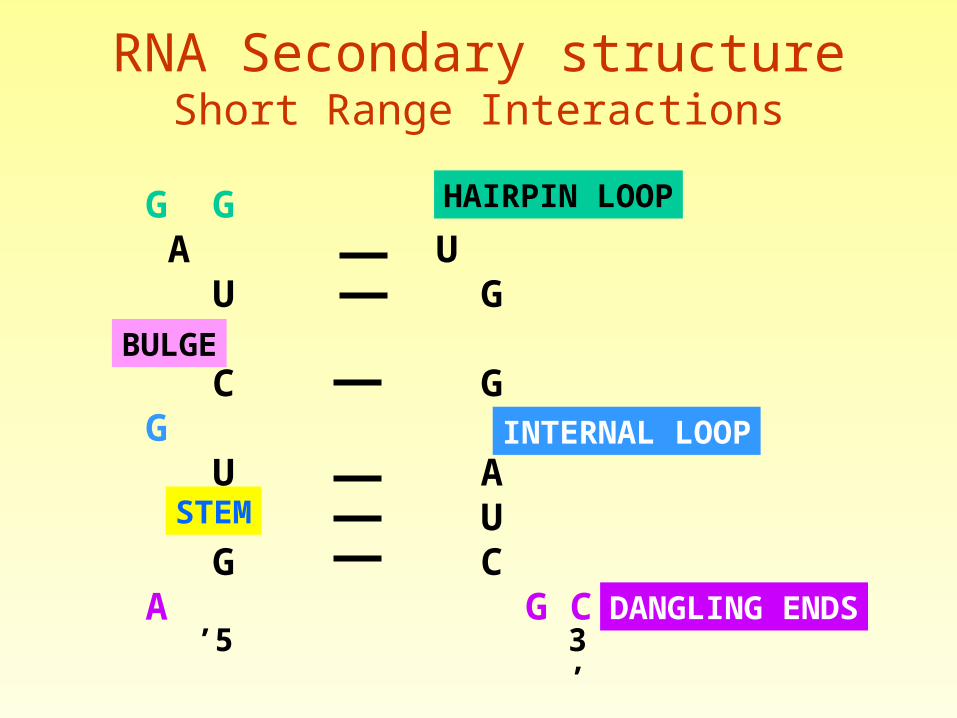
RNA Secondary structureShort Range Interactions
G G A U
U GC C GG A U A A U G CA G C U U
INTERNAL LOOP
HAIRPIN LOOP
BULGE
STEM
DANGLING ENDS5’ 3’

The function of the RNA molecule depends on its folded structure
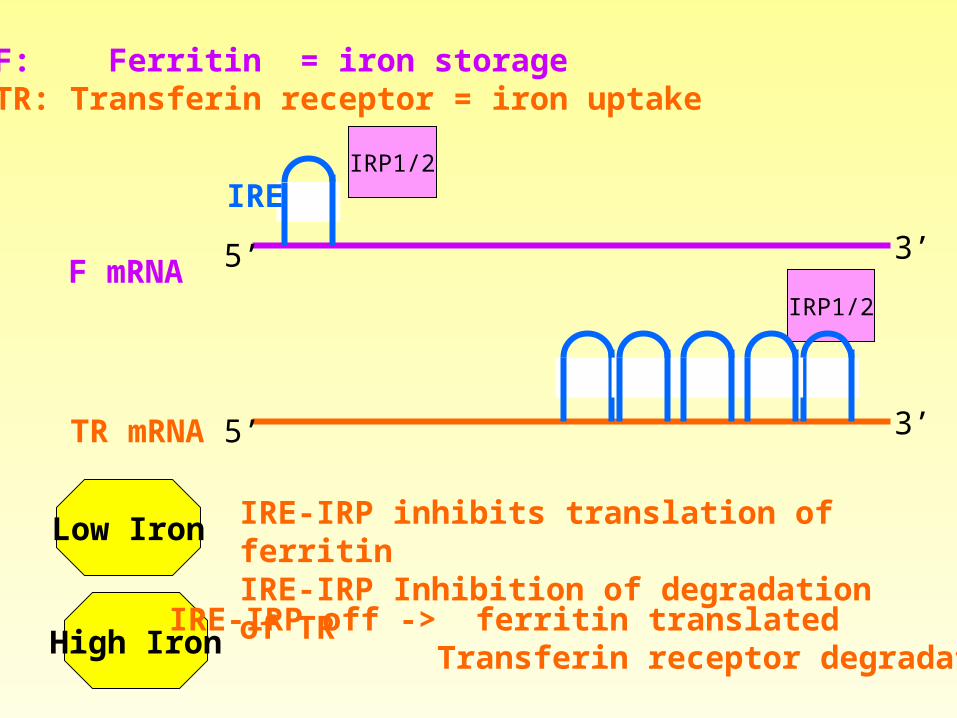
Example: mRNA structure involved in control of Iron levels
G U A GC N N N’ N N’ N N’ N N’C N N’ N N’ N N’ N N’ N N’ 5’ 3’
conserved
Iron Responsive ElementIRE
Recognized byIRP1, IRP2
IRP1/2
5’ 3’F mRNA
5’ 3’TR mRNA
IRP1/2
F: Ferritin = iron storageTR: Transferin receptor = iron uptake
IRE
Low Iron IRE-IRP inhibits translation of ferritinIRE-IRP Inhibition of degradation of TR
High IronIRE-IRP off -> ferritin translated
Transferin receptor degradated

Predicting RNA secondary Structure
Most common approach:
Search for a RNA structure with a
Minimal Free Energy (MFE)
Fold, Mfold Zucker & Stiegler (1981) Nuc. Acids Res. 9:133-148Zucker (1989) Science 244:48-52
RNAfoldVienna RNA secondary structure serverHofacker (2003) Nuc. Acids Res. 31:3429-3431
Free energy model
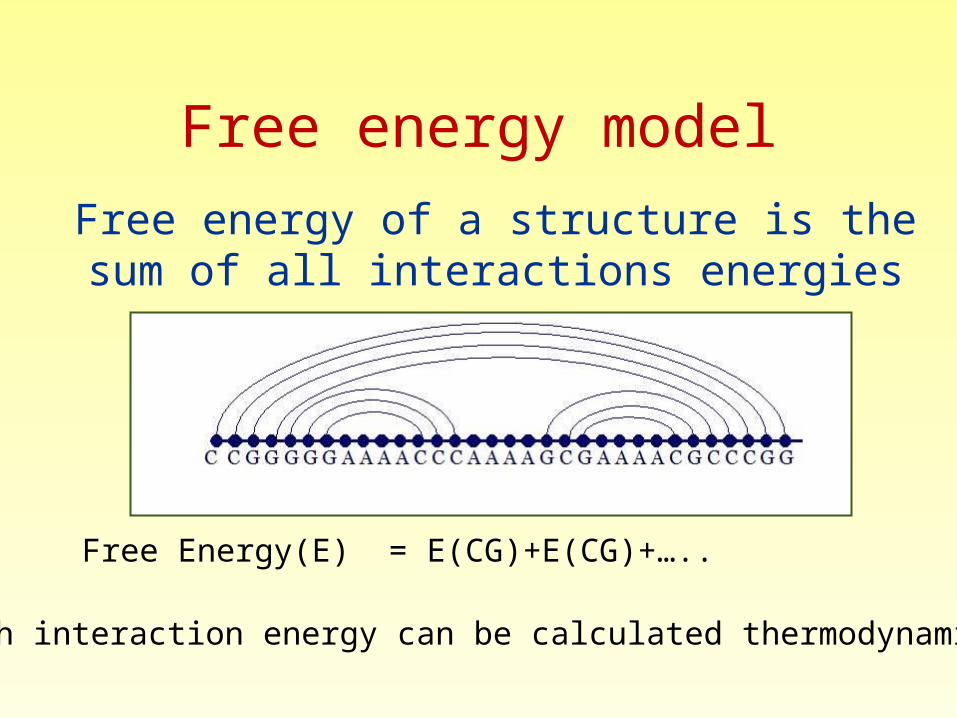
Free energy of a structure is the sum of all interactions energies
Each interaction energy can be calculated thermodynamicly
Free Energy(E) = E(CG)+E(CG)+…..

Why is MFE secondary structure prediction hard?
• MFE structure can be found by calculating free energy of all possible structures
• BUT the number of potential structures grows exponentially with the number, n, of bases
RNA folding with Dynamic programming (Zucker and Steigler)
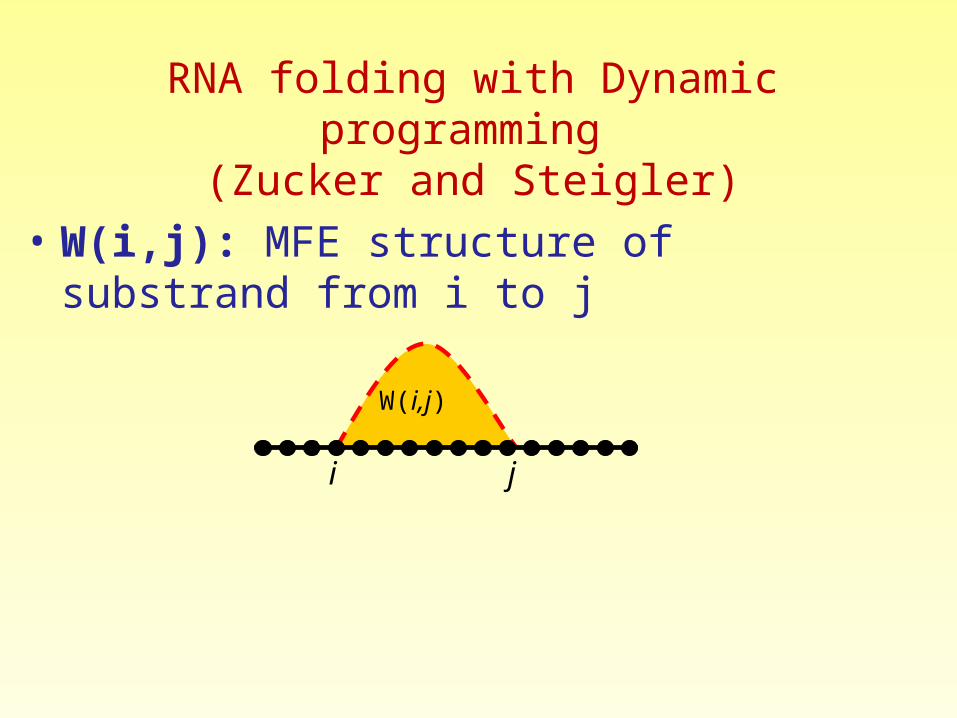
• W(i,j): MFE structure of substrand from i to j
i j
W(i,j)
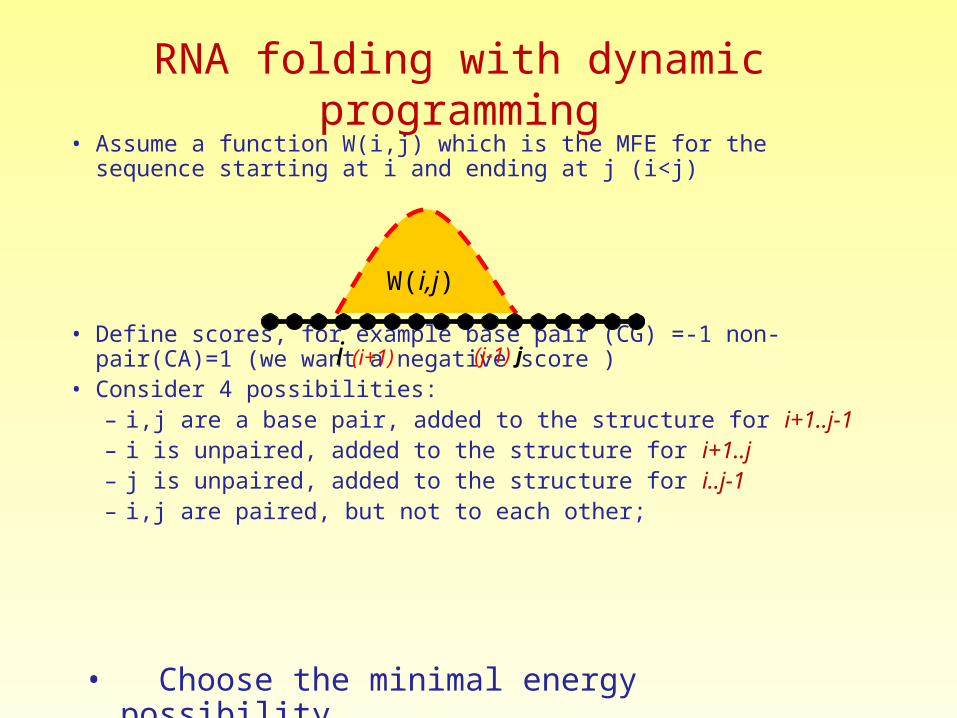
RNA folding with dynamic programming• Assume a function W(i,j) which is the MFE for the sequence starting at i
and ending at j (i<j)
• Define scores, for example base pair (CG) =-1 non-pair(CA)=1 (we want a negative score )
• Consider 4 possibilities:– i,j are a base pair, added to the structure for i+1..j-1– i is unpaired, added to the structure for i+1..j– j is unpaired, added to the structure for i..j-1– i,j are paired, but not to each other;
• Choose the minimal energy possibility
i (i+1)
W(i,j)
(j-1) j

Simplifying Assumptions for Structure Prediction
• RNA folds into one minimum free-energy structure.
• The energy of a particular base can be calculated independently– Neighbors do not influence the energy.

Sequence dependent free-energy Nearest Neighbor Model
U U
C G G C A UG CA UCGAC 3’5’
U U
C G U A A UG CA UCGAC 3’5’
Energy is influenced by the previous base pair (not by the base pairs further down).

Sequence dependent free-energy values of the base pairs
(nearest neighbor model) U U
C G G C A UG CA UCGAC 3’5’
U U
C G U A A UG CA UCGAC 3’5’
Example values:GC GC GC GCAU GC CG UA -2.3 -2.9 -3.4 -2.1
These energies are estimated experimentally from small synthetic RNAs.

Mfold :Adding Complexity to Energy Calculations
• Positive energy - added for destabilizing regions such as bulges, loops, etc.
• More than one structure can be predicted

Free energy computation
U UA A G C G C A G C U A A U C G A U A 3’A5’
-0.3
-0.3
-1.1 mismatch of hairpin-2.9 stacking
+3.3 1nt bulge -2.9 stacking
-1.8 stacking
5’ dangling
-0.9 stacking-1.8 stacking
-2.1 stacking
G= -4.6 KCAL/MOL
+5.9 4 nt loop

Mfold :Adding Complexity to Energy Calculations
• Positive energy - added for destabilizing regions such as bulges, loops, etc.
• More than one structure can be predicted

Frey U H et al. Clin Cancer Res 2005;11:5071-5077
©2005 by American Association for Cancer Research
More than one structure can be predicted for the same RNA

RNA fold prediction based on Multiple Alignment
Information from multiple sequence alignment (MSA) can help to predict the probability of positions i,j to be base-paired.
G C C U U C G G G CG A C U U C G G U CG G C U U C G G C C

Compensatory Substitutions
U U
C G U A A UG CA UCGAC 3’
G C
5’
Mutations that maintain the secondary structure
can help predict the fold
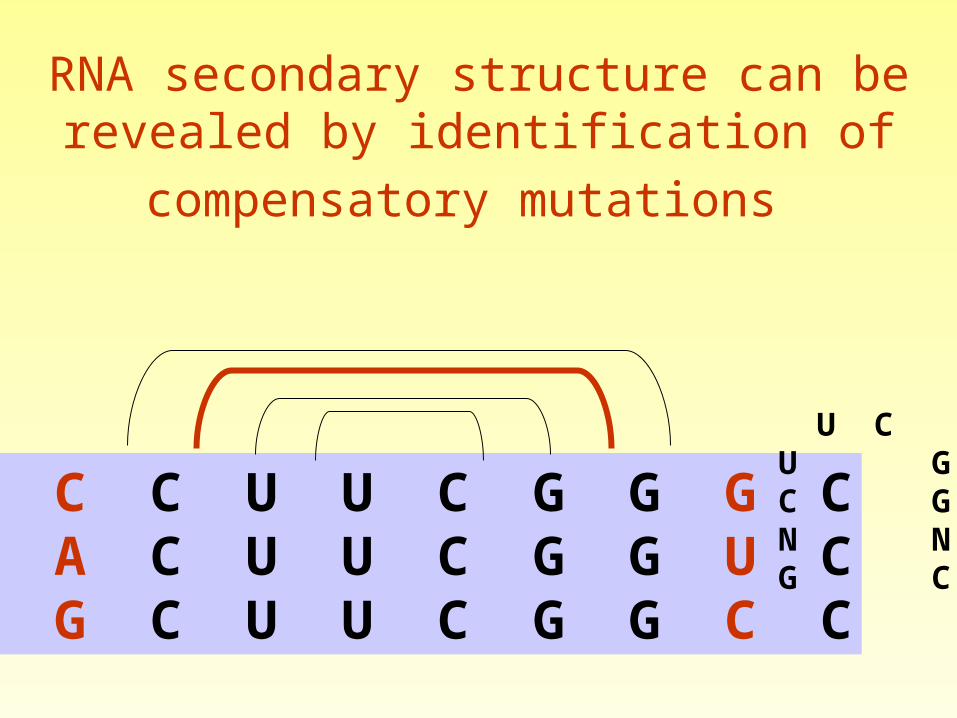
RNA secondary structure can be revealed by
identification of compensatory mutations
G C C U U C G G G CG A C U U C G G U CG G C U U C G G C C
U CU GC GN N’G C

Insight from Multiple Alignment
Information from multiple sequence alignment (MSA) can help to predict theprobability of positions i,j to be base-paired.
•Conservation – no additional information•Consistent mutations (GC GU) – support stem•Inconsistent mutations – does not support stem.•Compensatory mutations – support stem.

RNAalifold (Hofacker 2002)From the vienna RNA package
Predicts the consensus secondarystructure for a set of aligned RNA sequences by using modified dynamic programming algorithm that addalignment information to the standardenergy model
Improvement in prediction accuracy

Other related programs
• COVE
RNA structure analysis using the covariance model (implementation of the stochastic free grammar method)
• QRNA (Rivas and Eddy 2001)
Searching for conserved RNA structures
• tRNAscan-SE tRNA detection in genome sequences
Sean Eddy’s Lab WUhttp://www.genetics.wustl.edu/eddy

RNA families
• Rfam : General non-coding RNA database
(most of the data is taken from specific databases)
http://www.sanger.ac.uk/Software/Rfam/
Includes many families of non coding RNAs and functionalmotifs, as well as their alignment and their secondary structures

An example of an RNA family miR-1 MicroRNAs
mir-1 microRNA precursor family This family represents the microRNA (miRNA) mir-1 family. miRNAs are transcribed as ~70nt precursors (modelled here) and subsequently processed by the Dicer enzyme to give a ~22nt product. The products are thought to have regulatory roles through complementarity to mRNA.
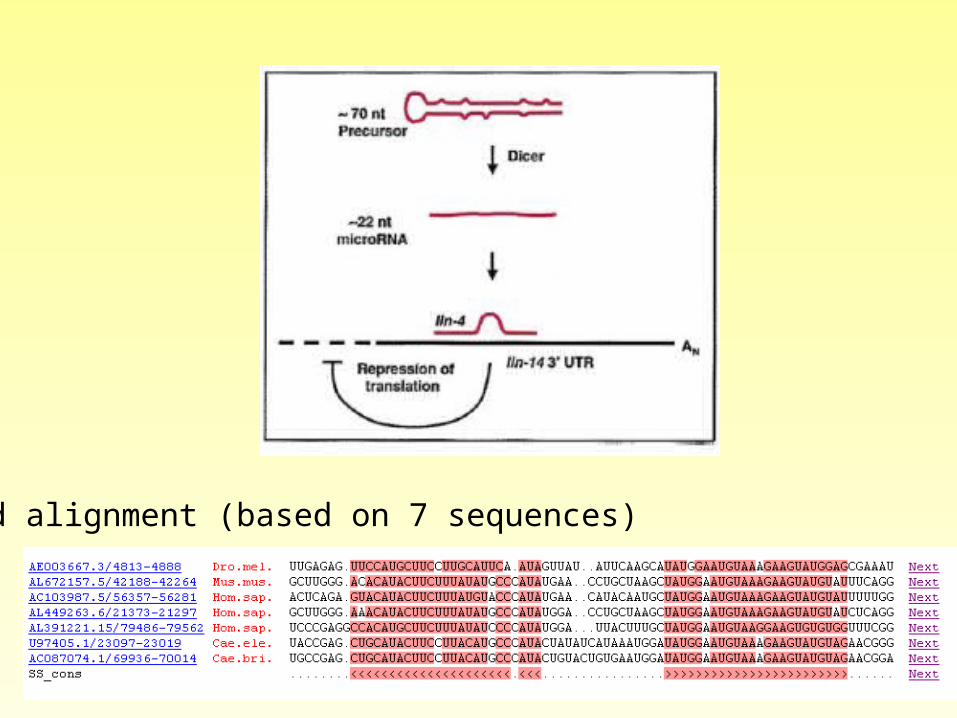
Seed alignment (based on 7 sequences)

Predicting microRNA target

Predicting microRNA target genes
• Why is it hard??– Lots of known miRNAs with similar seeds– Base pairing is required only for seed (7 nt)
Very High probability to find by chance
• Initial methods– Look at conserved miRNAs– Look for conserved target sites– Consider the RNA fold

TargetScan Algorithm by Lewis et al 2003
The Goal – find miRNA candidate target genes of a given miRNA
• Stage 1: Select only the 3’UTR of all genes
– Search for 7nts which are complementary to bases 2-8 from miRNA (miRNA seed”) in 5’UTRs

TargetScan Algorithm
• Stage 2: Extend seed matches in both directions
– Allow G-U (wobble) pairs

TargetScan Algorithm
• Stage 3: Optimize base-pairing
in remaining 3’ region of miRNA
(not applied in the later versions)

• Stage 4: Calculate the folding free energy (G) assigned to each putative miRNA:target interaction using RNAfold
Low energy get high Score
• Stage 5: Calculate a final score for a UTR to be a target
adding evolutionary conservation (by doing the same steps on UTR from other species)
TargetScan Algorithm

Predicting targets of RNA-binding protein (RBP)

Predicting RBPs target• Different types of RBPs
– Proteins that regulate RNA stability (bind usually at the 3’UTR)
– Splicing Factors (bind exonic and intronic regions) – ……
• Why is it hard– Most RBP sites are short and degenerative (e.g.
CTCTCT )

Predicting Exon Splicing Enhancers ESE-finder
1. Built PSSM for ESE, based on experimental data (SELEX)

ESE-finder
2. A given sequence is tested against5 PSSM in overlapping windows
3. Each position in the sequence is given a score
4. Position which fit a PSSM (score above a cutoff) are predicted asESEs
http://rulai.cshl.edu/

http://sfmap.technion.ac.il

Predicting RBPs target• RNA binding sites can be predicted by
general motif finders
- MEME http://metameme.sdsc.edu/
- DRIM http://bioinfo.cs.technion.ac.il/drim/