Embed Size (px)
Citation preview

Tumori surrenaliciin età pediatrica
Prof. Claudio GambiniDirettore
U.O. di Anatomia PatologiaIstituto G. Gaslini - Genova

Tumori surrenaliciI tumori surrenalici si differenziano dalle
altre neoplasie in quanto all’aspetto prettamente oncologico (tumori benigni o
maligni) può associarsi un aspetto funzionale endocrino (tumori
iperfunzionanti o non funzionanti).
Tumori della corticaleTumori della midollare

TUMORI DELLA
CORTI
CALE

GeneralitàRari in età pediatrica < 18 anni (in una revisione di 20 anni su 141 casi: 130 neuroblastomi, 8 neoplasie cortico-surrenaliche e 3 feocromocitomi).
Età media alla diagnosi di 4.6 anni.
Distribuzione bimodale con un picco nella prima infanzia e con un gruppo distribuito più omogeneamente nell’adolescenza. La metà dei pazienti è diagnosticata nei primi 4 anni di vita.
F:M = 2-3:1.
I tumori pediatrici sono spesso funzionanti mostrando segni e sintomi di anomalie endocrine. In una serie di 200 tumori corticali solo il 7,5% era non funzionante.La maggior parte si presenta con virilizzazione seguita da sindrome di Cushing o con una sindrome endocrina mista che include virilizzazione. Rara la sindrome di Cushing da sola; insoliti femminilizzazione e iperaldosteronismo primario.

1. Iperfunzione endocrina (ipercorticalismo)Ipercortisolismo (sindrome di Cushing)Iperaldosteronismo (sindrome di Conn)Virilizzazione FemminilizzazioneSindromi endocrine miste
2. Non funzionali o non iperfunzionali(difetto enzimatico con produzione e liberazione di deidroepiandrosterone o 11-deossi-cortisolo)
3. Status funzionale sconosciuto

Sindromi ereditarie associate atumori cortico-surrenalici
(piccola percentuale di casi)
Sindrome di Li FraumeniSindrome di Beckwith e WiedemannComplesso di Carney

I tumori cortico-surrenalici includono classicamente adenomi e carcinomi
Criteri patologici derivati dalla più ampia casistica dell’adulto.
Così sintetizzabili

Adenoma
Generalità:M:F = 1:1,5età media = 5 aa.
Trattamento chirurgico
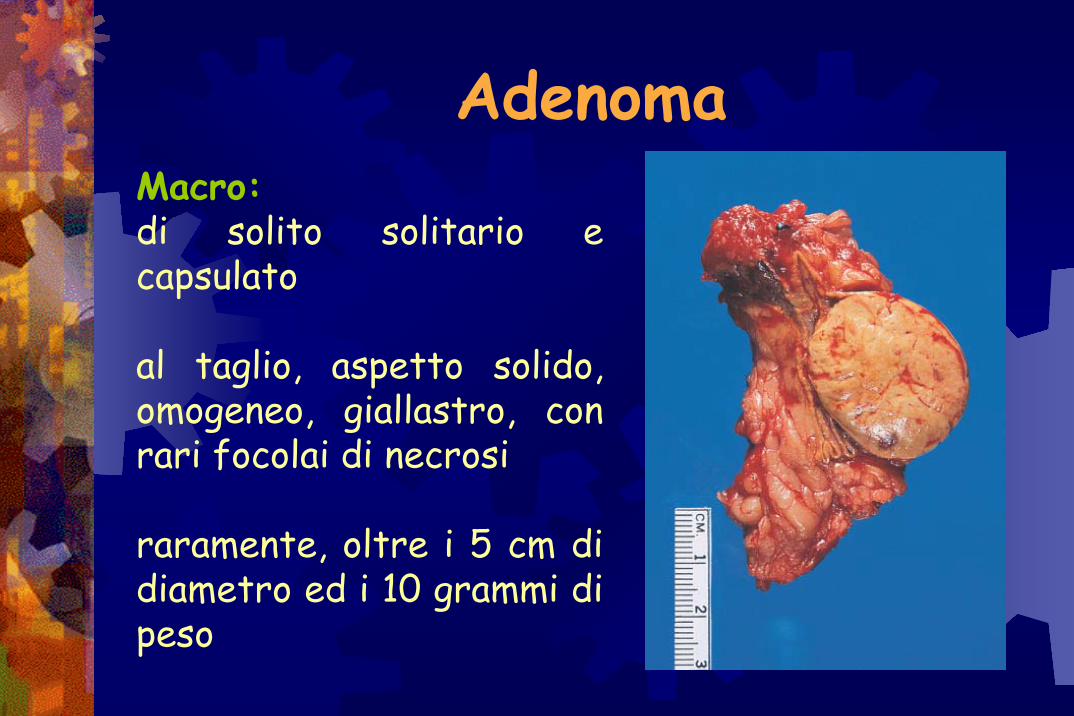
AdenomaMacro:di solito solitario e capsulato
al taglio, aspetto solido, omogeneo, giallastro, con rari focolai di necrosi
raramente, oltre i 5 cm di diametro ed i 10 grammi di peso
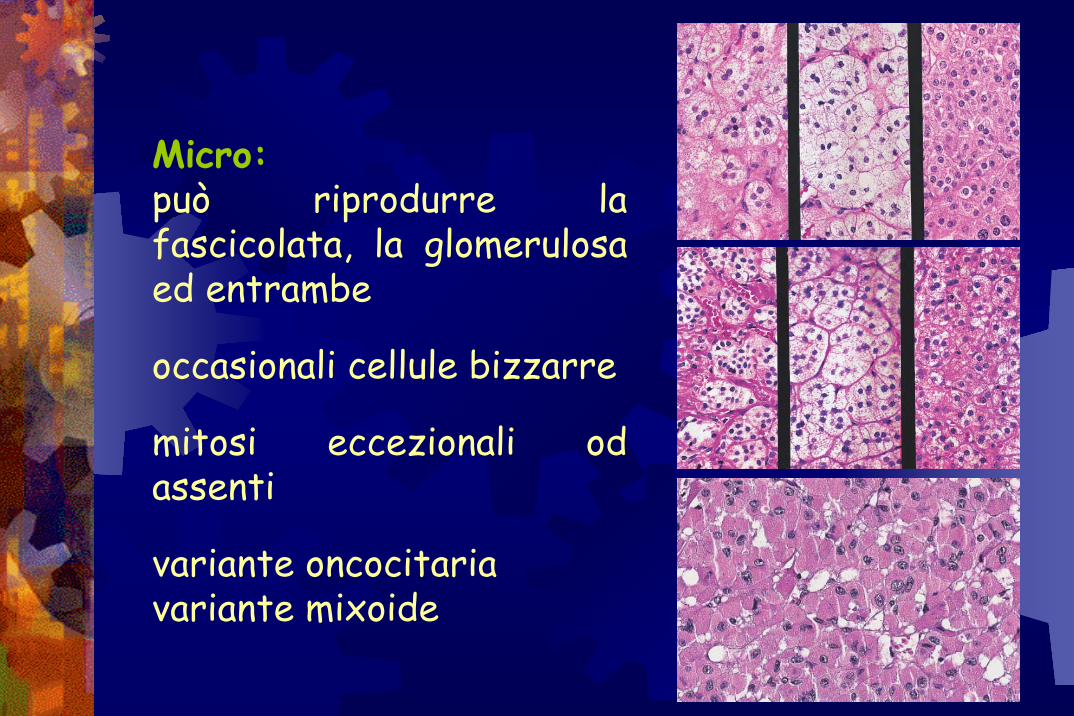
Micro:può riprodurre la fascicolata, la glomerulosa ed entrambe
occasionali cellule bizzarre
mitosi eccezionali od assenti
variante oncocitariavariante mixoide

Immunoistochimica:
forte positività per le citocheratine a basso peso molecolare e debole espressione della vimentina (al contrario del carcinoma cortico-surrenalico)
negatività per EMA e isoantigeni del gruppo sanguigno Lewis (al contrario del carcinoma renale)
positività per diversi ormoni steroidei
Analisi DNA: alterazioni della ploidia = 20% (vs. circa 70% nel carcinoma)

CarcinomaGeneralità: M/F = 1/1
età media = 10 aa.
Comportamento maligno, con metastasi nel 50% dei casi alla diagnosi [fegato (60%), linfonodi loco-regionali (40%), polmone (40%), sierose, ossa e cute]
Trattamento chirurgico
Sopravvivenza: 50% a 2 aa., 20-35% a 5 aa.
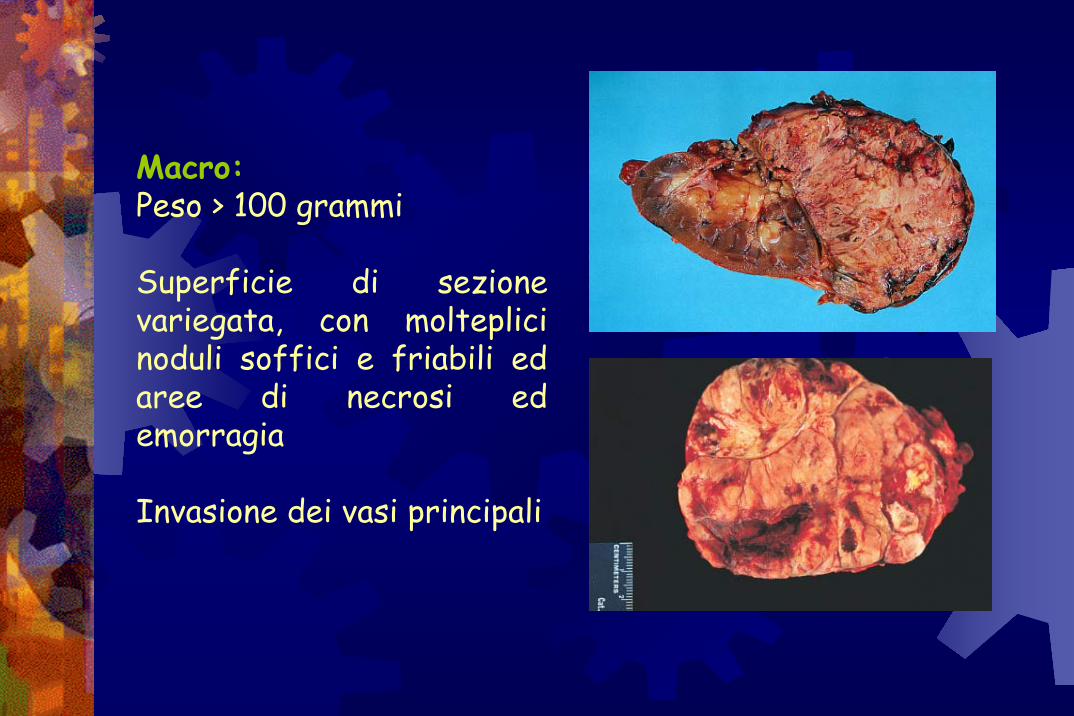
Macro:Peso > 100 grammi
Superficie di sezione variegata, con molteplici noduli soffici e friabili ed aree di necrosi ed emorragia
Invasione dei vasi principali
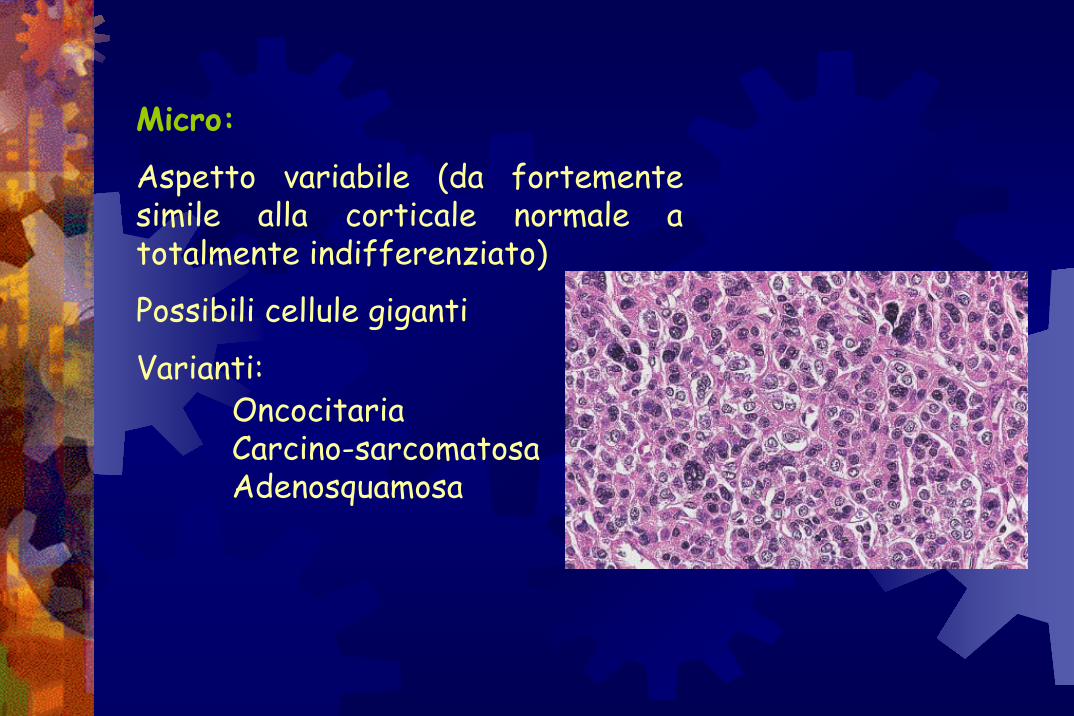
Micro:
Aspetto variabile (da fortemente simile alla corticale normale a totalmente indifferenziato)
Possibili cellule giganti
Varianti:Oncocitaria Carcino-sarcomatosaAdenosquamosa

Immunoistochimica
Vimentina +Cheratine – (/+)EMA –CEA –IGFR + e EGFR +ormoni steroidei – (/+)
Biologia molecolare
Sovraespressione di TRF-1 (telomeric-repeat binding factor-1), LOH dei geni p53 e Rb.

Di grande rilevanza è che a differenza dall’adulto i criteri patologici per la malignità nei casi pediatrici rimangono incerti.In un recente lavoro di J. Wieneke e Thompson (Am. J. Surg. Path. 2003; 27 (7): 867-881) sono stati esaminati aspetti clinici e patologici macro e microscopici che hanno individuato alcune caratteristiche associate ad aumentata probabilità di comportamento clinico maligno.

Criteri macroscopici e microscopici di malignità delle neoplasie cortico-surrenaliche in pazienti pediatrici
Peso del tumore > 400 grammiDimensioni del tumore > 10,5 cmEstensione ai tessuti molli perirenali e/o agli
organi adiacentiInvasione venosaInvasione capsularePresenza di necrosi> 15 mitosi per 20 HPFPresenza di figure mitotiche atipiche

L’invasione della vena cava, la necrosi e l’incremento dell’attivitàmitotica ( > 15 f.m. su 20 HPF) si sono dimostrati ad una analisi multivariata quali criteri che, indipendentemente, sono suggestivi di comportamento clinico maligno.

L’analisi dell’assetto cromosomico tumorale ha consentito di evidenziare numerose alterazioni cromosomiche, sia nei tumori surrenalici benigni sia in quelli maligni. In particolare, in ¼ degli adenomi, vi è un aumento delle copie di DNA del braccio corto del cromosoma 4, mentre nei tumori maligni, oltre ad un aumento di copie nei cromosomi 4, 5, 9 e 12, vi può essere una perdita di frammenti genici sui cromosomi 1, 2, 11 e 17.Il gruppo di geni più studiati nel carcinoma surrenalico è situato sul braccio corto del cromosoma 11.

Oncogeni, geni onco-soppressori e fattori di crescita implicati nella tumorigenesi
delle neoplasie cortico-surrenalicheOncogeni
Proteina G (gas) RASAttività proteina kinasi C calcio-dipendente
Geni soppressori tumoraliTP53MEN1p57Kip2 H19
Fattori di crescitaSovraespressione di IGF II

Bornstein SR and Hornsby PJ: What can we learn from gene expression profiling for adrenal tumour management?J. Clin. Endocrinol Metab 90:1900-2, 2005
Il carcinoma mostra uno specifico profilo:• sovra-espressione di IGF 2;• sovra-espressione di ciclina E;• sovra-espressione di GATA 4 (diffusione metastatica)
• ipo-espressione di 14 geni correlati alla steoidogenesi;• ipo-espressione del genotipo e fenotipo MHC class II (perdita di controllo dei meccanismi immunitari);• ipo-espressione del sistema FAS/FAS-L (idem);• ipo-espressione di AKR1B1 (cAMP regulated aldose reductase)

TUMORI DELLA MIDOLLARE

FeocromocitomaGeneralità Età: 3-81 anniIncidenza: 6,5% di tutti i tumori surrenaliciTumore del 10%: 10% bilaterale, 10% extra-surrenalico, 10% in età pediatrica, 10% maligno
Familiarità (associazione con neurofibromatosi, displasia dell’a. renale, s. di von Hippel-Lindau, sindrome di Turner, MEN IIA/IIB).

ClinicaI segni derivano dalla liberazione di nor-epinefrina, epinefrina (surrene) od entrambe;
Attacchi ipertensivi intermittenti, scatenati da farmaci, parto, intervento chirurgico o massaggio del tumore;
Triade: eccessi di sudorazionetachicardiacefalea
Conferma: catecolamine urinarie + TAC
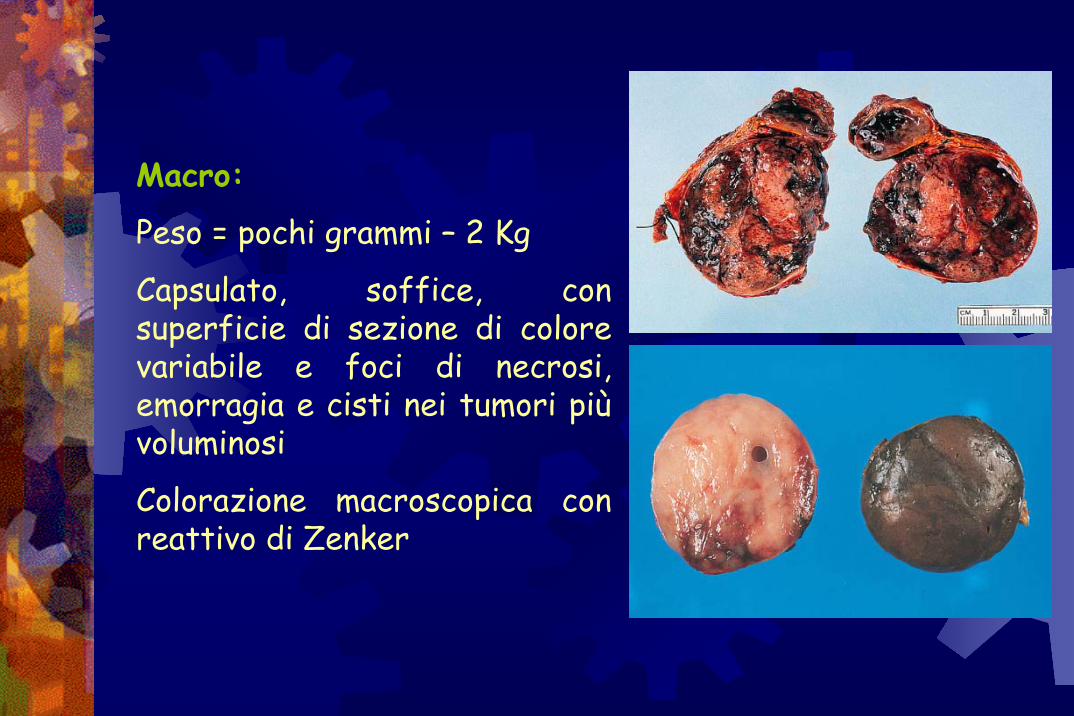
Macro:
Peso = pochi grammi – 2 Kg
Capsulato, soffice, con superficie di sezione di colore variabile e foci di necrosi, emorragia e cisti nei tumori più voluminosi
Colorazione macroscopica con reattivo di Zenker
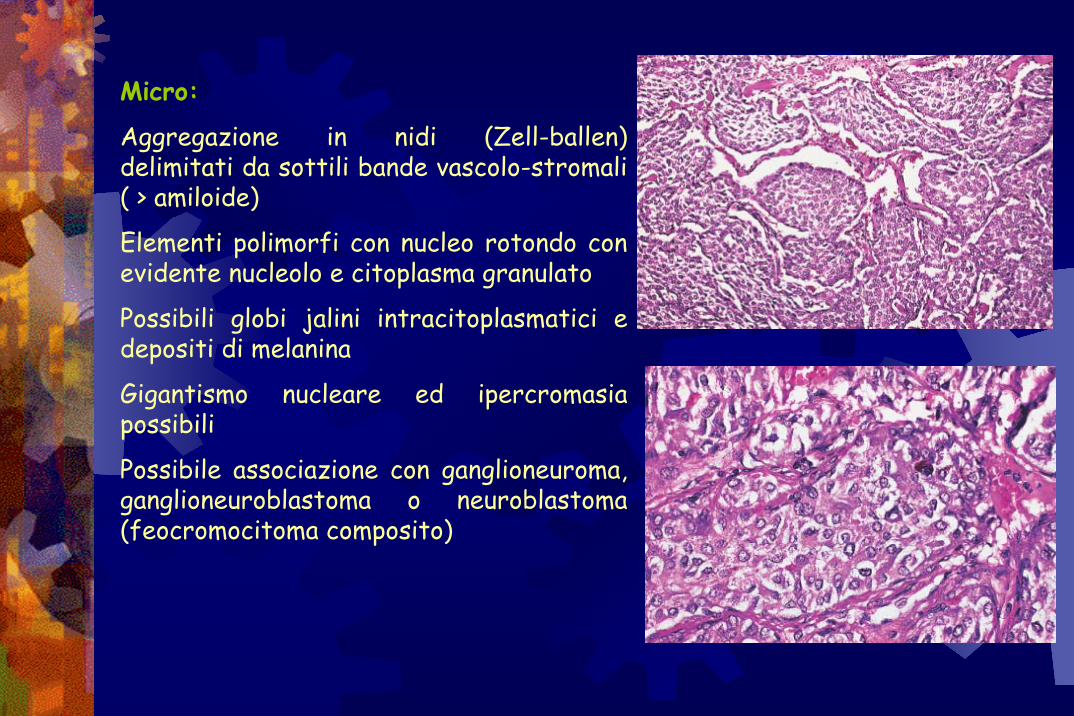
Micro:
Aggregazione in nidi (Zell-ballen) delimitati da sottili bande vascolo-stromali ( > amiloide)
Elementi polimorfi con nucleo rotondo con evidente nucleolo e citoplasma granulato
Possibili globi jalini intracitoplasmatici e depositi di melanina
Gigantismo nucleare ed ipercromasiapossibili
Possibile associazione con ganglioneuroma, ganglioneuroblastoma o neuroblastoma (feocromocitoma composito)
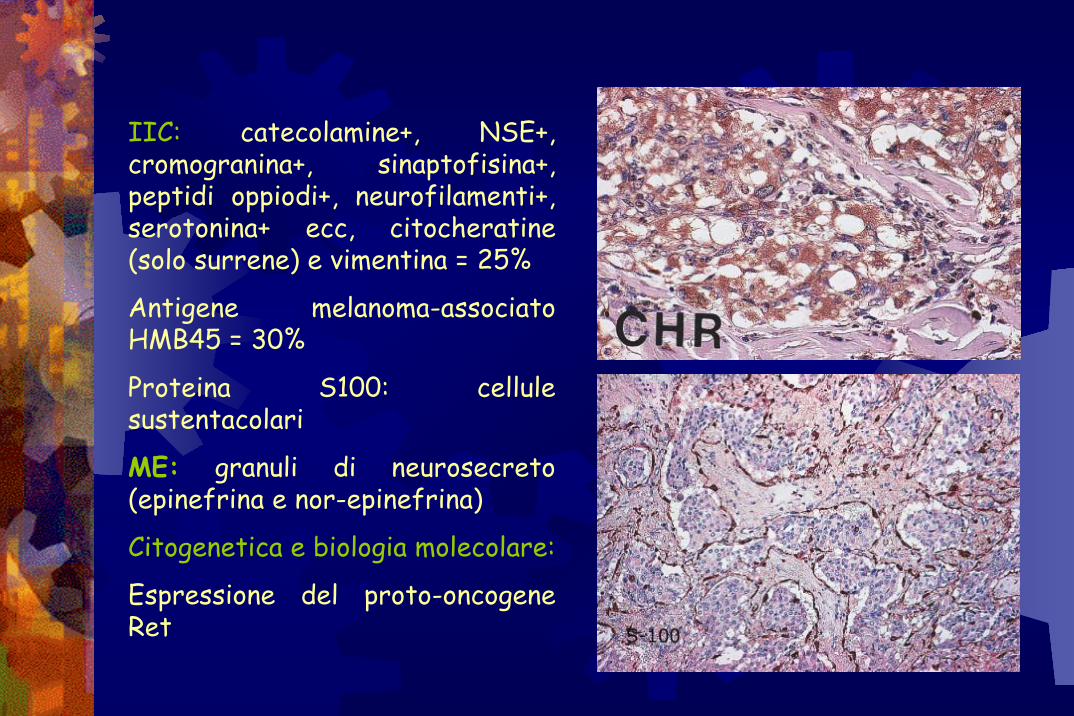
IIC: catecolamine+, NSE+, cromogranina+, sinaptofisina+, peptidi oppiodi+, neurofilamenti+, serotonina+ ecc, citocheratine(solo surrene) e vimentina = 25%
Antigene melanoma-associato HMB45 = 30%
Proteina S100: cellule sustentacolari
ME: granuli di neurosecreto(epinefrina e nor-epinefrina)
Citogenetica e biologia molecolare:
Espressione del proto-oncogene Ret

Malignità: l’unico criterio certo di malignità è la presenza di metastasi (presenza di malattia in sedi dove normalmente non è presente tessuto cromaffine).
Possibili indicatori di comportamento aggressivo: criteri microscopici riduzione o assenza di cellule sustentacolari alto indice di marcatura per Ki-67alterazioni della ploidiaespressione del gene hTERTperdita della proteina del Rb
Metastasi: scheletro (coste e colonna), polmone, fegato e linfonodi.
Prognosi: generalmente, i pazienti con metastasi muoiono entro un anno; si hanno talora metastasi tardive

Criteri microscopici da valutare1. invasione capsulare2. invasione vascolare3. estensione al tessuto adiposo perisurrenalico4. presenza di grossi nidi confluenti5. crescita diffusa6. necrosi7. incremento di cellularità8. presenza di cellule fusate9. marcato pleomorfismo cellulare e nucleare10. monotonia cellulare11. ipercromasia nucleare12. macronucleoli13. aumento di figure mitotiche (> 3x10 HPF)14. figure mitotiche atipiche15. assenza di globuli jalini

PASS (Pheochromocytoma of the
Adrenal Gland Scaled Score)Sistema di valutazione a score del feocromocitoma della ghiandola surrenale secondo Lester D.R. ThompsonThe American Journal of Surgical Pathology 26(5): 551-566, 2002
Invasione vascolareInvasione capsulareMarcato pleomorfismo nucleareIpercromasia nucleare
Tutti gli altri criteri 2 punti
Un punteggio uguale o superiore a 4 suggerisce comportamento biologico potenzialmente maligno
1 punto

August C et al CGH and CD44/MIB1 immunohistochemistry are helpful to distinguish metastasized from non metastasized sporadic pheochromocytomas. Mod. Pathol., 17: 1119-8, 2004
CGH = guadagni in 17q.
Ohta S. et al Down-regulation of metastasis suppressor genes in malignant pheochromocytoma. Int. J. Cancer, 114: 139-43, 2005
Sei geni: nm23-H1, TIMP-4, BRMS-1 TXNIP, CRSP-3, E-Cad

Tumori del sistema
simpatico-adrenergico
Oltre che nella midollare del surrene, possono occorrere a livello del collo, mediastino e retroperitoneale.
1. Neuroblastoma
2. Ganglioneuroblastoma nodulare
3. Ganglioneuroblastoma intermixed
4. Ganglioneuroma
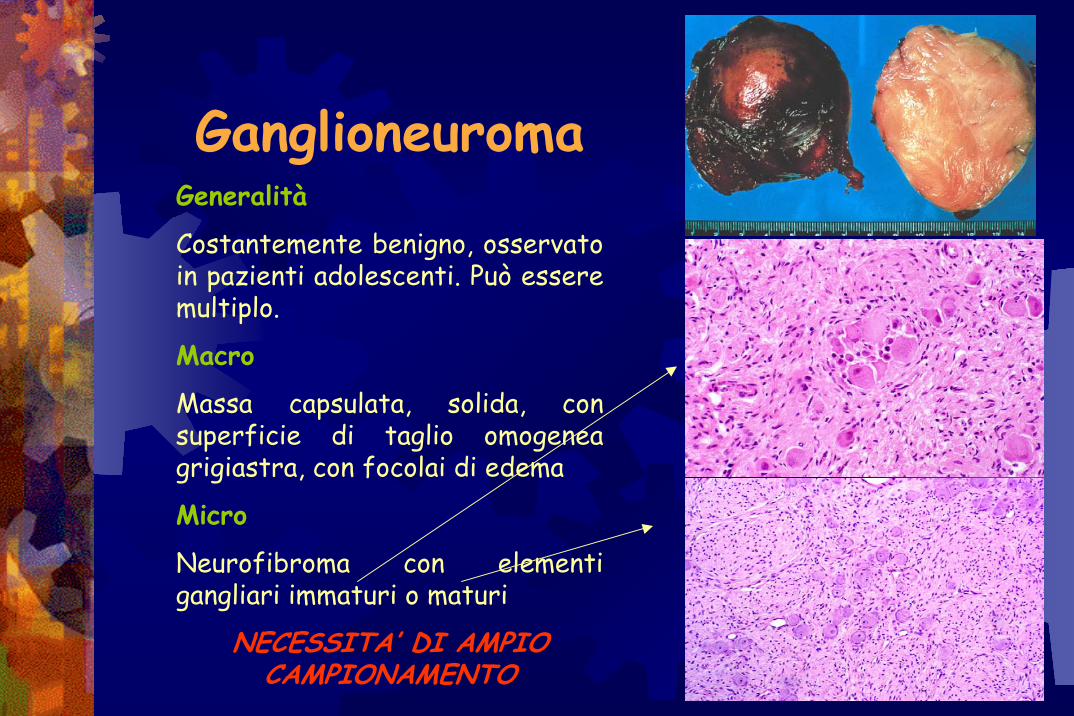
GanglioneuromaGeneralità
Costantemente benigno, osservato in pazienti adolescenti. Può essere multiplo.
Macro
Massa capsulata, solida, con superficie di taglio omogenea grigiastra, con focolai di edema
Micro
Neurofibroma con elementi gangliari immaturi o maturi
NECESSITA’ DI AMPIO CAMPIONAMENTO

Altre neoplasie surrenaliche
• Mielolipoma• Tumori benigni mesenchimali• Sarcomi• Linfomi• Melanoma maligno• Metastasi: polmone
mammellacute (m.m.)rene

GeneralitàTumore benigno, raro in età pediatrica (15-18 anni), raramente bilaterale. F=M. Dimensioni molto variabili da piccole a masse di notevoli dimensioni. Lesioni con poco tessuto adiposo difficili da distinguere radiologicamente da altri tumori surrenalici. 50% dei casi asintomatico; 50%: dolore addominale o al fianco, ematuria, ipertensione, massa palpabile, raramente emorragia retroperitoneale.
Possibile associazione con disturbi endocrini: m. di Cushing, m. di Addison, virilismo, pseudoermafroditismo
Mielolipoma
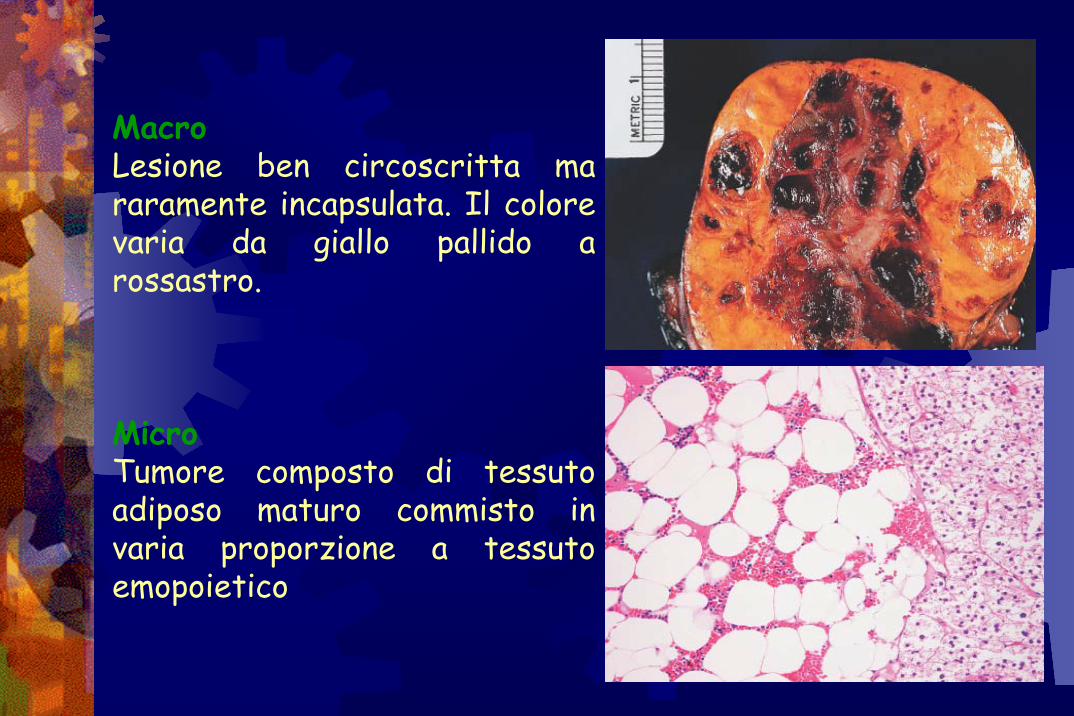
MacroLesione ben circoscritta ma raramente incapsulata. Il colore varia da giallo pallido a rossastro.
MicroTumore composto di tessuto adiposo maturo commisto in varia proporzione a tessuto emopoietico


Tumori della corticale: correlazioni clinico-patologiche
Lesioni non funzionanti:Difetto enzimatico con produzione e liberazione di deidroepiandrosterone o 11-deossi-cortisoloCarcinoma
Iper-aldosteronismo (s. di Conn):Perdita urinaria di potassio, ritenzione di sodio, soppressa produzione di renina, ipertensione e debolezza muscolare; 70% = adenoma; 30% = iperplasia; rarissimo = carcinoma

Tumori della corticale: correlazioni clinico-patologiche
Sindrome di Cushing:Iperproduzione di cortisolo (femmine adulte = 80%)20% = causa surrenalica (iperplasia > adenoma > carcinoma)80% = adenoma ipofisario, microcitoma, carcinoide, feocromocitoma, tumore ovarico
Sindrome adreno-genitale:Mascolinizzazione; 50% dei casi prima della pubertà; 80% dei pazienti di sesso femminile;Iperplasia surrenalica congenita > carcinoma (adulti)





























