Embed Size (px)
Citation preview

lable at ScienceDirect
Progress in Biophysics and Molecular Biology xxx (2014) 1e9
Contents lists avai
Progress in Biophysics and Molecular Biology
journal homepage: www.elsevier .com/locate/pbiomolbio
Original research
Hot spots in proteineprotein interfaces: Towards drug discovery
Engin Cukuroglu, H. Billur Engin, Attila Gursoy, Ozlem Keskin*
Center for Computational Biology and Bioinformatics and College of Engineering, Koc University, Istanbul, Turkey
a r t i c l e i n f o
Article history:Available online xxx
Keywords:Hot spotProteineprotein interfaceDrug designProteineprotein interaction
* Corresponding author.E-mail address: [email protected] (O. Keskin).
http://dx.doi.org/10.1016/j.pbiomolbio.2014.06.0030079-6107/© 2014 Published by Elsevier Ltd.
Please cite this article in press as: Cukurogluand Molecular Biology (2014), http://dx.doi.
a b s t r a c t
Identification of drug-like small molecules that alter proteineprotein interactions might be a key step indrug discovery. However, it is very challenging to find such molecules that target interface regions inprotein complexes. Recent findings indicate that such molecules usually target specifically energeticallyfavored residues (hot spots) in proteineprotein interfaces. These residues contribute to the stability ofproteineprotein complexes. Computational prediction of hot spots on bound and unbound structuresmight be useful to find druggable sites on target interfaces. We review the recent advances in compu-tational hot spot prediction methods in the first part of the review and then provide examples on howhot spots might be crucial in drug design.
© 2014 Published by Elsevier Ltd.
1. Introduction
Proteineprotein interactions play crucial roles in regulatingbiological processes, cellular and signaling pathways. Proteineprotein binding sites are called interfaces. Residue properties ininterfaces are the key elements in proteineprotein recognition,binding and affinity. Residue based analysis can help revealingproteineprotein binding mechanisms. Alterations in native pro-teineprotein interactions may lead to several diseases. Therefore,targeting the interfaces between proteins has an enormous po-tential in drug discovery (Kar et al., 2012; Thangudu et al., 2012;Wells and McClendon, 2007). Drugs targeting proteineprotein in-teractions should ultimately bind to protein interfaces if not toallosteric sites. However, targeting interfaces is more challengingthan targeting active sites of enzymes or G protein-coupled re-ceptors in drug discovery since interfaces are relatively large, oftenflat without specific ligand binding pockets.
Residues in proteineprotein interfaces do not equally contributeto the binding energies. There are critical residues called hot spotswhich contribute most to the binding energy (Bogan and Thorn,1998; Clackson and Wells, 1995). Hot spot residues can be detec-ted by alanine scanning mutagenesis experiments (Clackson andWells, 1995). If the binding energy difference is more than 2 kcal/mol aftermutating a residue to an alanine, it is labeled as a hot spot.Bogan and Thorn (Bogan and Thorn, 1998) analyzed amino acidcompositions of hot spots and concluded that some residues are
, E., et al., Hot spots in proteiorg/10.1016/j.pbiomolbio.201
more favorable as hot spots in proteineprotein interfaces. Tyr, Argand Trp are the most frequent ones which are critical due to theirsize and conformation. They also reported that hot spots are sur-rounded by a set of residues, that are energetically less importantresembling to an O-ring in a pipe fitting, to occlude hot spots fromwater molecules. There is a correlation between change in theaccessible surface area and energy contribution of residues(Guharoy and Chakrabarti, 2005). Moreira et al. (2007a) also sup-ported O-ring hypothesis using Molecular Dynamic (MD) simula-tions. Further, these hot spots are not randomly distributed in theinterfaces but rather clustered. Hot spots are assembled withindensely packed regions. These modular assembly regions are calledhot regions (Keskin et al., 2005). This binding site organizationjustify how a given protein molecule may bind to different proteinpartners. Kleanthous and coworkers (Meenan et al., 2010) showedthat a limited number of mutations at the interface of cognatecomplex of colicin E9 endonuclease and immunity protein 9 pro-vide high-affinity binding of E9 to immunity protein 2, although ata weaker affinity compared to the cognate complex. These experi-mental findings were also studied by computational hot spot or-ganizations (Cukuroglu et al., 2012). The organization of hot spotresidues provides a mechanism to obtain binding affinity andspecificity to different partners. Therefore, cooperativity of theseresidues can reveal the complex binding organizations in specificity(Cukuroglu et al., 2010; Shulman-Peleg et al., 2007).
Hot spots might also be important to find kinetic behavior ofproteineprotein complexes. Agius et al. (2013) made use of hot spotenergetics and architectures of hot spots/hot regions to predictchanges in dissociation rates upon a mutation. They used a set ofbiophysical and statistical descriptors to estimate hot spot energies.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e92
These descriptors are then used as features to estimate successfullyoff-rate changes of single e or multi-point mutations.
Determining hot spot residues by experimental techniques iscostly and time consuming, so computational methods have beendeveloped to predict hot spot residues in bound and unboundprotein structures (Cho et al., 2009; Darnell et al., 2007; del Sol andO'Meara, 2005; Grosdidier and Fernandez-Recio, 2008; Gueroiset al., 2002; Huo et al., 2002; Kortemme and Baker, 2002; Li and Liu,2009; Moreira et al., 2007a; Ofran and Rost, 2007; Tuncbag et al.,2009). Prediction results show that prediction accuracies areimproving over the years. Prediction methods will be a comple-ment to experimental studies to find hot spot residues which havean important role on binding affinity and specificity. In addition,computational methods can guide experimental mutational studiesand explain functional and mechanistic aspects of protein binding(Fern�andez-Recio, 2011).
Last but not least, there is an increasing number of studiesshowing that hot spots may be important in drug design (Fry, 2012;Jubb et al., 2012; Thangudu et al., 2012; Wells and McClendon,2007). Drug-like small molecules prefer to bind to hot spots atproteineprotein interfaces (Arkin and Wells, 2004). Below, we willreview first the recent advances in computational hot spot pre-diction and then protein-drug interactions and how hot spots arecrucial in drug design.
2. Predicting hot spot residues
Hot spot prediction methods can be classified into two cate-gories: based on unbound, monomeric structures and proteine-protein complex structures. Previous studies used different datasets and thresholds to predict hot spot residues. A complete list ofthe available tools and their properties are tabulated inSupplementaryFile_1.
3. Hot spot predictions on bound protein structures
Most of the hot spot prediction tools concentrated on boundproteineprotein interactions in order to detect hot spot residues inprotein interfaces. The analysis of hot spot residues detected byexperimental techniques is limited to a small number of complexes.Information on experimentally determined hot spot residues hasbeen deposited in databases. ASEdb is the first alanine mutationdatabase developed by Thorn and Bogan (2001) and later BID wasformed by Fischer et al. (2003). The cost and difficulty of deter-mining hot spot residues experimentally led to prediction of theseresidues by computational approaches. Hence, Kortemme andBaker (Kortemme and Baker, 2002; Kortemme et al., 2004) pro-posed a computational alanine scanning method that uses energiesof packing interactions, hydrogen bonds and solvation. Gueroiset al. (2002) used FOLD-X energies to predict the hot spot resi-dues. Another energy based method was developed by Gao et al.(2004) using hydrogen bond, hydrophobic and VdW interactions(three major non-covalent interactions) in order to estimate theindividual contribution of each interfacial residue to the bindingenergy. The calculated energy changes of mutations werecompatible with experimental results.
MD simulations are suitable for detailed analysis of proteine-protein interactions at the atomic level and they can be used forprediction of hot spot residues (Gonzalez-Ruiz and Gohlke, 2006;Grosdidier and Fernandez-Recio, 2008; Huo et al., 2002; Landonet al., 2007;Moreira et al., 2007a; Rajamani et al., 2004;Wang et al.,2013; Yogurtcu et al., 2008). Both energy and MD based hot spotprediction methods have high accuracy rates, but they arecomputationally expensive and difficult to apply on large scalestudies.
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
Knowledge-based methods form another approach to predicthot spot residues. They usually use machine learning methods tolearn from known hot spot data. The major advantage ofknowledge-based methods is their computational efficiency.However, they are very sensitive to the selection of features such asresidue type, size, hydrophobicity, accessible surface areas, etc. tocharacterize hot spot residues, and it is hard to find the best featurecombination. Most of the studies use diverse features in order toincrease their prediction accuracies even if they use similar ma-chine learning algorithms such as support vector machine (SVM),linear regression, neural networks, Bayesian networks, and randomforest models. Assi et al. (2010) used sequence conservation, energyscores and contact number information to predict hot spot resi-dues. Lise et al. built up a prediction method based on machinelearning (Lise et al., 2009) but their method was not working wellon Arg and Glu, so they improved their approach adding twoadditional classifiers specific for these two amino acids (Lise et al.,2011). They mainly used van der Waals potentials, desolvation,hydrogen bonds and electrostatistics energies in order to predicthot spot residues. Koes and Camacho (Koes and Camacho, 2012)usedmachine learning approachwith accessible surface area (ASA),relative ASA (RASA), evolutionary rate, conservation score, freeenergy of complexation and change in free energy of the alaninemutation values. They reported that ASA, RASA and per residuesestimate of the free energy values were the most informative fea-tures and had good classification accuracies. Also, Xia et al. (2010)exhaustively searched different features of protein structures inorder to increase the hot spot prediction accuracy and theyconcluded that ASA related features showed better discriminativepower as suggested by Cho et al. (2009). According to the work ofXia et al. (2010) protrusion index was also a good discriminator ofhot spots which was also shown in Li et al.'s work (Li et al., 2004).Unlike these features, Cho et al. (2009) found that weighted atomicpacking density and weighted hydrophobicity had a discriminativepower on hot spot residue predictions. Mitchell and her colleaguesproposed two distinct methods: KFC (Darnell et al., 2007) whichused shape specificity and biochemical contact features of theinterface residues and they updated their approach with KFC2 (Zhuand Mitchell, 2011) which used interface solvation, atomic densityand plasticity features. They concluded that lack of plasticity wasstrongly indicative of a hot spot residue but it was not a require-ment. Wang et al. (2012a) used mass, polarizability and isoelectricpoint of residues, the relative side-chain accessible surface area andthe average depth index to predict hot spot residues. As shown inprevious studies, using accessible surface area, energy, atomicpacking density and plasticity related features in a suitable com-bination increases the hot spot prediction accuracy.
Empirical formula based methods are also used instead of ma-chine learning algorithms in order to predict hot spot residues.Pavelka et al. (2009) used only conservation scores to identify hotspot residues. Guney et al. (2008) used conservation score and ASAvalues with an empirical formula. Tuncbag et al. (2009) showedthat RASA and pair wise potentials are much more discriminativethan conservation scores for predictions. Indeed, it should be thefamily of proteins that determine whether conservation isdiscriminative. For antibody/antigen complexes, conservation isnot a good feature (Assi et al., 2010). Kruger and Gohlke (2010) usedpair wise potentials with degree of buriedness to predict hot spotresidues. Geppert et al. (2011) used pair wise potentials, atom typesand residue properties to generate an empirical formula with anadditional voting system in order to find the functional hot spotresidues. Shulman-Peleg et al. (2007) performed structural align-ment of functionally similar proteineprotein complexes in order tofind spatial chemical conservation of the residues which corre-spond to hot spot residues. Hot regions in proteineprotein
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e9 3
interfaces are functionally important and previous studies usedsequence and structural information to find these regions (Armonet al., 2001; Cukuroglu et al., 2012; Hsu et al., 2007).
Network topology properties came into prominence in order topredict hot spot residues. Del Sol and O'Meara (del Sol and O'Meara,2005) applied network topology to the residue networks of proteincomplexes in order to find hot spot residues. They stated that highlycentral residues in the network were most probably hot spot resi-dues. Li and Liu (2009) generated a bipartite graph from twomonomers of the complexes and located biclique patterns from themaximal biclique subgraphs in order to identify hot spot residues.Tuncbag et al. (2010b) interpreted the residue contributions toseveral mincuts in minimum cut trees to define hot spot residues.
4. Hot spot predictions on unbound protein structures
There are three studies basedonunboundprotein structures. ISIS(Ofran and Rost, 2007) is the first prediction tool which uses aminoacid sequences to predict the hot spot residues on unbound proteinstructures. pyDockNIP (Grosdidier and Fernandez-Recio, 2008) usednormalized interface propensity values derived from rigid bodydockingwith electrostatics and desolvation scoring for prediction ofinteraction hot spots. In this work, they used complex forms of theproteins to derive their prediction function; however, this functioncan be used to find hot spot residues in unbound form. Ozbek et al.(2013) used dynamic fluctuations in high frequency modes ob-tained fromGaussian Network Model (GNM) in order to predict hotspot residues onunbound structures. If the structure of the unboundform is present GNM (Haliloglu and Erman, 2009; Ozbek et al., 2013)performs better, but if the only sequence information is present ISIS(Ofran andRost, 2007) has the advantage. There is limitednumberofstudies for hot spot prediction of the residues on unbound struc-tures. Much work needs to be done to predict the hot spots onprotein interfaces with high precision and specificity.
5. Multi-interface binding and hot spot residues
Some proteins can interact with multiple partners using thesame or different interfaces on their surfaces. It is impossible for
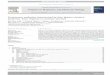
Fig. 1. Actin monomer is shown in blue cartoon representation. The other colors are the irepresentations show the interface residues and big red ball representations show the residshown in blue cartoon representation. (VMD (Humphrey et al., 1996) was used to prepare
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
these proteins to interact with many partners using different in-terfaces, because they have limited surface area for binding. Hence,they use some binding sites repeatedly (Keskin and Nussinov, 2007;Kim et al., 2006). Thangudu et al. (2012) showed that this multiplebinding sites have at least one hot spot residue and these sitesusually overlap. The affinity and specificity of multiple interactionscan originate from cooperativity of different hot spot residues(Cukuroglu et al., 2012). An experimental study also highlightedthat the mutations of different hot spot residues of the hub proteinSMAD3 changed the affinity of the interactions with its partners(Schiro et al., 2011). Hot spot residue analysis can reveal the com-plex binding strategies of the proteins withmulti-interface binding.Cukuroglu et al. (2014) extracted all possible proteins with multi-interface binding strategies in PDB (Berman et al., 2000). Thesestructures were all from experimental non-redundant structures.According to the results of an analysis, actin used 40 differentbinding modes while interacting with another actin monomer asshown in Fig. 1 (40 different complexes are superimposed). Theblue monomer on the center of the Fig. 1 is the actin monomer incommon and the other actin monomers represented with differentcolors are the interaction partners. When the hot spot residues ofthe complexes are analyzed (hot spots and hot regions are found byHot region (Cukuroglu et al., 2012)), only two interfaces did nothave any computational hot spots. 35 interfaces used 67 differenthot spot residues on the actin surface (colored blue in Fig. 1) andnone of the hot spot residue combinations were repeated in anyinteraction which generates the binding affinity and specificity. Inthree complexes, the actin monomer (colored blue in Fig. 1) did nothave any hot spot residues even if their partner monomers had hotspot residues (Supplementary Table 1). Ubiquitin used 22 differentbinding strategies while interacting with another ubiquitinmonomer as shown in Fig. 2 (22 different complexes are super-imposed). The computational hot spots were tabulated inSupplementary Table 2 and the complexes with computational hotspots are shown separately in Fig. 3. The ubiquitin monomer(colored blue in Fig. 2) used 17 different residues as hot spots andthese hot spot residues were used in 13 complexes. In four com-plexes, the ubiquitin monomer (colored blue in Fig. 2) did not haveany hot spot residues even if their partner monomers had hot spot
nteraction partners of the Actin monomer (Actin e actin interactions). Ball and stickues which are used as a hot spot at least in one of the interaction by actin monomer asthe figure).
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

Fig. 2. Ubiquitin monomer is shown in blue cartoon representation. The other colors are the interaction partners of the Ubiquitin monomer (UbiquitineUbiquitin interactions). Balland stick representations show the interface residues and big red ball representations show the residues which are used as a hot spot at least in one of the interaction by ubiquitinmonomer as shown in blue cartoon representation. (VMD (Humphrey et al., 1996) was used to prepare the figure).
E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e94
residues. Only five complexes did not have any computational hotspots ant the other 17 complexes with hot spots residues are shownin Fig. 3. The coloring of the Fig. 3 is the same as Fig. 2. The bluecolored ubiquitin monomer used different hot spot residue com-binations with their partners in order to provide binding affinityand specificity. For these two examples, the hot spot residues in theinterfaces are formed mostly a hot region. For actin complexes, hotspot residues in 27 complexes out of 32 complexes that have hotspot residues both on the target and the partner proteins makecontacts with each other, also in the ubiquitin example this caseoccurred in 10 complexes out of 11 complexes. These comple-mentary hot spot interactions will be a promising target for drugstudies. However, even if hot spots do not have any complementaryhot spot residues in partner protein they will also be a promisingtarget for inhibiting proteineprotein interactions.
6. Proteinedrug interactions and hot spots
Until very recently the druggable genome was believed toconsist of only a limited number of protein families, such as Gprotein coupled receptors (GPCRs), serine/threonine and tyrosineprotein kinases, zinc metallopeptidases, serine proteases, nuclearhormone receptors and phosphodiesterase (Hopkins and Groom,2002). These drug target groups are mostly enzymes, ion chan-nels or receptors, with well-defined and preformed concave bind-ing sites (Jubb et al., 2012). However the structural and sequencesimilarity among the members of the same protein familyincreased the possibility of drug promiscuity (Engin et al., 2014).Besides the researchers realized that the target based drug designapproaches that aimed to produce high affinity drugs yielded lowselectivity (Jubb et al., 2012). The increasing awareness of thedamage (side effects) caused by drug promiscuity lead to searchesfor new strategies in drug discovery.
Since the discovery of the important role of hot spots in stabi-lizing the proteineprotein interactions (PPIs) (Bogan and Thorn,
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
1998; Clackson and Wells, 1995; DeLano, 2002), inhibition of PPIswith small molecules targeting hot spots have been extensivelystudied (Jubb et al., 2012; London et al., 2013). As the biologicalfunctions aremediated through proteineprotein interactions (PPIs),they became attractive therapeutic targets. In the former years thePPI interfaces were believed to be undruggable, since they lackpockets and are generally flat, large and featureless. However, thisimpression vanished with the successful PPI inhibitors and thedruggable genome expanded with these additional targets. TheBenzodiazepines (Chung et al., 2011; Filippakopoulos et al., 2010;Nicodeme et al., 2010), the ICG-001 that inhibits the interactionbetween b-catenin and CBP (Emami et al., 2004), the inhibitors of IL-2 (Braisted et al., 2003),MDM2 (Canner et al., 2009; Vu and Vassilev,2011), BCL-2/BCL-XL (Tse et al., 2008), XIAP (Straub, 2011) andVLA-4(Straub, 2011) may be listed as examples of PPI targeting drugs. Inorder to better explain the concept of small molecules that blockPPIs, hindrance of IL2 e IL2R interaction by a small molecule isvisualized in Fig. 4 and there are further examples of known PPIinhibitors in the literature (Basse et al., 2013; Cesa et al., 2013; Fry,2012; Thangudu et al., 2012; Villoutreix et al., 2012; Wells andMcClendon, 2007). The major characteristics of druggable hotspots are about their location preferences: hot spots generally arenot observed all over the interface but are clustered together (Keskinet al., 2005; Reichmann et al., 2005) and druggable hot spots have atendency to be located in complemented pockets (pockets thatdisappear after proteineprotein binding) (Li et al., 2004).
Allosteric drugs, the drugs that bind away from the PPI interface,are also capable of altering the PPIs. Ma et al. suggested thatdesigning drugs that target the allosteric hot spots may enlarge thedruggable genome in a similar way druggable hot spots did; anddeveloping methods to identify allosteric hot spots may make theallosteric drug design more efficient (Ma and Nussinov, 2014). Theyobserved hot spots in the allosteric region of the Ras protein andclaimed that the allosteric regulation of Ras protein conformationmay be a useful approach for targeting Ras pathways.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

Fig. 3. UbiquitineUbiquitin interactions with hot spot residues are representedseparately. The hot spot residues are shown in Supplementary Table 2. The colors ofthe cartoon representations are the same as Fig. 2. (VMD (Humphrey et al., 1996) wasused to prepare the figure).
E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e9 5
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
7. Hot spots and drug discovery
A small molecule needs to submerge a portion of its surface intothe target protein in order to perform a high affinity binding.Generally this is done via binding to the cavities of the targetprotein (Wells and McClendon, 2007). However proteineproteininteractions (PPIs), which are interesting targets for therapeuticapplications (Arkin and Wells, 2004; Berg, 2003; Domling, 2008;Fry, 2006; Wells and McClendon, 2007) lack such cavities. OnPPIs the binding energy is dominated by hot spots and they areburied on the surface by the other residues (Moreira et al., 2007b).This fact forms the basis for a promising protein drug designstrategy named hot spot grafting.
Hot spot grafting is to hunt for suitable scaffolds from a librarythat contain the hot spots and submerge these hot spots on thesurface (Zhang and Lai, 2011). Graph theory-based pattern match-ing approaches or set reduction algorithms may be employed inspotting scaffolds that can accommodate desired hot spot patterns(Liang et al., 2000). Lai and his coworkers developed such algo-rithms (Liang et al., 2000; Zhang and Lai, 2012) and they success-fully designed a protein to bind the human EPO receptor (Liu et al.,2007). Fleishman et al. (Fleishman et al., 2011) suggested anotherapproach to design protein drugs with predetermined hot spotpatterns. In their method, first a scaffold protein was docked to thetarget protein to find good conformations with shape comple-mentary, and then the hot spot patterns were used to fix theconfiguration. Theywere able to design two proteins that target theconserved stem region on influenza hemagglutinin.
Another hot spot guided structure based approach, HS-Pharm,for biologically active compound design was introduced byBarillari et al. (2008). HS-Pharm is a fast knowledge-basedapproach that allows the prioritization of cavity atoms thatshould be targeted for ligand binding, by training machine learningalgorithms with known ligand-binding pockets. Here, the hot spotsare utilized for focusing structure-based pharmacophore models.
Designing drugs that target hot spots is a good strategy to breakPPIs, since they have a major contribution in the stability. Plus, thehot spots of PPIs help detecting the regions which a ligand maybind with high affinity. The relationship between PPI hot spots andligand binding hot spots is still being questioned but there are somestudies (Zerbe et al., 2012) pointing a correlation between them.Inspired by this fact, Koes and Camacho (2012) proposed a struc-tural bioinformatics approach to detect starting points for rationalPPI targeting small-molecule design. They identified clusters ofinterface residues that they refer as Small-Molecule InhibitorStarting Points (SMISPs). These regions are bigger than hot spots,smaller than the whole interface.
Seco et al. (2009) merged the identification of hot spots andbinding affinity calculations to produce a druggability index todiscover druggable sites. Their method is independent of surfacedescriptors or training sets and it works on PPIs as well as allostericbinding sites. They tested their method on five pharmacologicaltargets including protein phosphatase 1B (PTP-1B) which is a verychallenging target. They successfully identified the druggable siteson this protein and discovered that the most potent druggable sitewas overlapping with the PTP-1B and insulin receptor tyrosinekinase (IRK) interface. Li et al. (2011) described a novel drug dis-covery and drug repositioning method using multiple ligandsimultaneous docking (MLSD) and hot spots. They have used STAT3,a cancer target, as the test case. First they docked multiple drugscaffolds into the hot spots of STAT3 and produced virtual templatecompounds. Next they searched drug databases for similar hits andidentified celecoxib as a novel inhibitor of STAT3. Besides, theymodeled two novel lead inhibitors based on one of the lead tem-plates and celecoxib.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

Fig. 4. A small molecule breaking the PPI between IL2 and IL2R. In the right figure, the complex between IL2 and IL2R (PDB ID: 1Z92) and the drugeprotein interaction between asmall molecule and IL2 (PDB ID: 1M49) are superimposed. This small molecule blocks the interaction between IL2 and its receptor (Arkin et al., 2003). As can be observed from thefigure on the left, the small molecule binds around some of the IL2-IL2R interface hot spots shown in red. Hot spot of the IL2-IL2R interface is obtained via Hotpoint (Tuncbag et al.,2010a) server. (VMD (Humphrey et al., 1996) was used to prepare the figure).
E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e96
Hot spots are of crucial importance for fragment based drugdiscovery as well. It is known that different probes only clusteraround hot spots (Hall et al., 2012). Hadjuk et al. experimented overfragment-sized compounds (Hajduk et al., 2005) and Liepinsh andOtting (1997) investigated organic solvents in aqueous solutionsby using nuclear magnetic resonance method. They observed thatboth of themolecules bind to hot spotswith a hit rate of 90% ormorethan 90%. Many other experiments had also resulted in similarconclusions (English et al., 1999; Mattos and Ringe, 1996) andconfirmed that hot spots are capable of binding a range of differentkinds of small molecules. FTMap (Hall et al., 2012) server finds thebinding hot spots and provides complementary information forfragment based drug discovery. Kozakov et al. (201) proposed afragment based method that works even when the proteins are intheir unbound states to discover regions on a proteineproteininterface capable of binding to drug-size ligands based on hot spots.
Sequence and structure similarity within a protein family is oneof the biggest challenges in drug discovery. In order to preventincreased risk of toxicology, targeting conserved interfaces shouldbe avoided.
To sum up, the hot spots differentiate the PPI binding sites fromthe rest of the protein surface and as mentioned earlier theyconstitute the majority of the binding energy of the PPIs. Since themodulation of hot spots became an important strategy for blockingPPIs, this discovery had a great impact in the drug discovery. It ledto the enlargement of druggable genome and the emergence ofnew drug development techniques which are discussed above.
8. Hot spots and disease causing mutations
Within the last decade a vast amount of sequence data havebeen produced thanks to international efforts put on large-scaleprojects such as 1000genomes (Genomes Project et al., 2012),TCGA (Cancer Genome Atlas Research et al., 2013), HAPmap(International HapMap et al., 2010) and many others. Meanwhile,the accumulation of structural data in the Protein Databank(Berman et al., 2007) continued in an increasing pace. Meltingprotein structure and genetic variations together in one potpermitted the researchers to obtain a broader understanding on thecomplex disease mechanisms. In 2008, a curated set of PPI relatedmutations was published (Schuster-Bockler and Bateman, 2008).Afterward, Zhong et al. (2009) published a pioneering study on thedistinct outcomes of different classes of disease mutations (node
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
removals and edgetic perturbations). Subsequently, David et al.(2012) claimed that disease-causing mutations prefer to happenin the interface region of a given protein, if they are not located inthe core. It was understood that disease-causing mutations tend tomodify the function of a target protein either by destabilizing it viacore mutations or altering its interactions via interface mutations.Likewise, Wang et al. (2012b) observed that in-framemutations areenriched in the interaction interface region of proteins linked withthe corresponding disease.
Destabilizing effects of mutated hot spots will most likely followsuch discoveries linking disease-causing mutations with PPIs. Eventhough currently there isn't a large-scale study focusing on thistopic, some researchers published relevant case studies. Engin et al.(2013), modeled the interaction between ELANE and CSF3. Theyclaimed that the variants of 101 residue, which is an interaction hotspot on ELANE, may be altering the affinity of CSF3-ELANE bindingenergy. Plus, they mentioned that these variants are susceptible tobe linked with the metastasis progression in breast cancer patients.Acuner Ozbabacan et al. (2014), investigated the distribution ofoncogenic mutations and SNPs over the IL-1 signaling pathwayproteins. Theywere able tomap some of these genetic variations onto the computational hot spots of the interactions in IL-1 signalingpathway. Later, a similar study was performed on the structuralinteractions of IL-10 with other signaling molecules, in order tounderstand the impact of oncogenic mutations on the mechanismsthat leads to cancer and inflammation (Acuner-Ozbabacan et al.).
9. Hot spots and post-translational modifications
Post-translational modifications (PTMs) assist the PPIs while thecell is responding to a variety of dynamic signals. The PTM eventsthat take place at the functional sites are capable of creating newbinding sites (Schaller and Parsons, 1995), increasing the interac-tion partner number of the protein and accordingly diversifyingfunctionality. Meanwhile a PTM may cause disassociation of abinding partner as well (Nussinov et al., 2012). It has been shownthat phosphorylation sites are more likely to happen on bindinginterfaces in hetero-oligomeric and weak transient homo-oligomeric complexes, whereas binding hot spots tend to bephosphorylated in hetero-oligomers. Moreover, phosphorylationwas claimed to be capable of causing significant changes in thebinding energy of some complexes (Nishi et al., 2011). Anotherconclusion of the same study was that compared to the rest of the
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e9 7
residues in a hetero-oligomer interface, interface phosphositeswere twice as likely to be binding hot spots.
10. Conclusion
The binding affinity and the specificity of the proteineproteininteractions are provided by energetically important residues.Mutations of these residues cause dissociation of the proteins orforce them to change their binding modes. These energeticallyfavored residues (hot spots) are also the residues where drug-likemolecules at proteineprotein interfaces. For drug developmentstudies, assigning hot spot residues as the target sites will guidedesigning small molecules for these specific residues. This strategymight prevent possible side effects of the drugs. Finding these hotspot residues experimentally is a costly process which has led theresearchers to develop computational algorithms to predict hotspots efficiently. Improvements in hot spot predictions in the lastdecade are promising and relating these studies with drug dis-covery is promising for future.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.pbiomolbio.2014.06.003.
References
Acuner-Ozbabacan, S.E., Engin, H.B., Guven-Maiorov, E., Kuzu, G., Muratcioglu, S.,Baspinar, A., Chen, Z., Van Waes, C., Gursoy, A., Keskin, O. and Nussinov, R. TheNetwork of Interleukin-10 Protein Structural Interactions with Other SignalingMolecules and Its Implications in Inflammation and Cancer, BMC Genomics.
Acuner Ozbabacan, S.E., Gursoy, A., Nussinov, R., Keskin, O., 2014. The structuralpathway of interleukin 1 (IL-1) initiated signaling reveals mechanisms ofoncogenic mutations and SNPs in inflammation and cancer. PLoS Comput Biol.10, e1003470.
Agius, R., Torchala, M., Moal, I.H., Fernandez-Recio, J., Bates, P.A., 2013. Character-izing changes in the rate of protein-protein dissociation upon interface muta-tion using hotspot energy and organization. PLoS Comput Biol. 9, e1003216.
Arkin, M.R., Randal, M., DeLano, W.L., Hyde, J., Luong, T.N., Oslob, J.D., Raphael, D.R.,Taylor, L., Wang, J., McDowell, R.S., Wells, J.A., Braisted, A.C., 2003. Binding ofsmall molecules to an adaptive protein-protein interface. Proc. Natl. Acad. Sci. U.S. A. 100, 1603e1608.
Arkin, M.R., Wells, J.A., 2004. Small-molecule inhibitors of protein-protein in-teractions: progressing towards the dream. Nat. Rev. Drug. Discov. 3, 301e317.
Armon, A., Graur, D., Ben-Tal, N., 2001. ConSurf: an algorithmic tool for the iden-tification of functional regions in proteins by surface mapping of phylogeneticinformation. J. Mol. Biol. 307, 447e463.
Assi, S.A., Tanaka, T., Rabbitts, T.H., Fernandez-Fuentes, N., 2010. PCRPi: presagingcritical residues in protein interfaces, a new computational tool to chart hotspots in protein interfaces. Nucleic Acids Res. 38, e86.
Barillari, C., Marcou, G., Rognan, D., 2008. Hot-spots-guided receptor-based phar-macophores (HS-Pharm): a knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmaco-phores. J. Chem. inf. Model. 48, 1396e1410.
Basse, M.J., Betzi, S., Bourgeas, R., Bouzidi, S., Chetrit, B., Hamon, V., Morelli, X.,Roche, P., 2013. 2P2Idb: a structural database dedicated to orthosteric modu-lation of protein-protein interactions. Nucleic acids Res. 41, D824eD827.
Berg, T., 2003. Modulation of protein-protein interactions with small organicmolecules. Angew. Chem. 42, 2462e2481.
Berman, H., Henrick, K., Nakamura, H., Markley, J.L., 2007. The worldwide proteinData Bank (wwPDB): ensuring a single, uniform archive of PDB data. NucleicAcids Res. 35, D301eD303.
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H.,Shindyalov, I.N., Bourne, P.E., 2000. The protein data bank. Nucleic Acids Res. 28,235e242.
Bogan, A.A., Thorn, K.S., 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol.280, 1e9.
Braisted, A.C., Oslob, J.D., Delano, W.L., Hyde, J., McDowell, R.S., Waal, N., Yu, C.,Arkin, M.R., Raimundo, B.C., 2003. Discovery of a potent small molecule IL-2inhibitor through fragment assembly. J. Am. Chem. Soc. 125, 3714e3715.
Cancer Genome Atlas Research, N, Weinstein, J.N., Collisson, E.A., Mills, G.B.,Shaw, K.R., Ozenberger, B.A., Ellrott, K., Shmulevich, I., Sander, C., Stuart, J.M.,2013. The Cancer genome atlas Pan-Cancer analysis project. Nat. Genet. 45,1113e1120.
Canner, J.A., Sobo, M., Ball, S., Hutzen, B., DeAngelis, S., Willis, W., Studebaker, A.W.,Ding, K., Wang, S., Yang, D., Lin, J., 2009. MI-63: a novel small-molecule inhibitor
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
targets MDM2 and induces apoptosis in embryonal and alveolar rhabdomyo-sarcoma cells with wild-type p53. Br. J. Cancer 101, 774e781.
Cesa, L.C., Patury, S., Komiyama, T., Ahmad, A., Zuiderweg, E.R., Gestwicki, J.E., 2013.Inhibitors of difficult protein-protein interactions identified by high-throughput screening of multiprotein complexes. ACS Chem. Biol. 8, 1988e1997.
Cho, K.I., Kim, D., Lee, D., 2009. A feature-based approach to modeling protein-protein interaction hot spots. Nucleic Acids Res. 37, 2672e2687.
Chung, C.W., Coste, H., White, J.H., Mirguet, O., Wilde, J., Gosmini, R.L., Delves, C.,Magny, S.M., Woodward, R., Hughes, S.A., Boursier, E.V., Flynn, H., Bouillot, A.M.,Bamborough, P., Brusq, J.M., Gellibert, F.J., Jones, E.J., Riou, A.M., Homes, P.,Martin, S.L., Uings, I.J., Toum, J., Clement, C.A., Boullay, A.B., Grimley, R.L.,Blandel, F.M., Prinjha, R.K., Lee, K., Kirilovsky, J., Nicodeme, E., 2011. Discoveryand characterization of small molecule inhibitors of the BET family bromodo-mains. J. Med. Chem. 54, 3827e3838.
Clackson, T., Wells, J.A., 1995. A hot spot of binding energy in a hormone-receptorinterface. Science 267, 383e386.
Cukuroglu, E., Gursoy, A., Keskin, O., 2010. Analysis of hot region organization in hubproteins. Ann. Biomed. Eng. 38, 2068e2078.
Cukuroglu, E., Gursoy, A., Keskin, O., 2012. HotRegion: a database of predicted hotspot clusters. Nucleic Acids Res. 40, D829eD833.
Cukuroglu, E., Gursoy, A., Nussinov, R., Keskin, O., 2014. Non-redundant uniqueinterface structures as templates for modeling protein interactions. PLoS One 9,e86738.
Darnell, S.J., Page, D., Mitchell, J.C., 2007. An automated decision-tree approach topredicting protein interaction hot spots. Proteins 68, 813e823.
David, A., Razali, R., Wass, M.N., Sternberg, M.J., 2012. Protein-protein interactionsites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat.33, 359e363.
del Sol, A., O'Meara, P., 2005. Small-world network approach to identify key resi-dues in protein-protein interaction. Proteins 58, 672e682.
DeLano, W.L., 2002. Unraveling hot spots in binding interfaces: progress andchallenges. Curr. Opin. Struct. biol. 12, 14e20.
Domling, A., 2008. Small molecular weight protein-protein interaction antagonists:an insurmountable challenge? Curr. Opin. Chem. biol. 12, 281e291.
Emami, K.H., Nguyen, C., Ma, H., Kim, D.H., Jeong, K.W., Eguchi, M., Moon, R.T.,Teo, J.L., Kim, H.Y., Moon, S.H., Ha, J.R., Kahn, M., 2004. A small molecule in-hibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc.Natl. Acad. Sci. U. S. A 101, 12682e12687.
Engin, H.B., Guney, E., Keskin, O., Oliva, B., Gursoy, A., 2013. Integrating structure toprotein-protein interaction networks that drive metastasis to brain and lung inbreast cancer. PLoS One 8, e81035.
Engin, H.B., Gursoy, A., Nussinov, R., Keskin, O., 2014. Network-based strategies canhelp mono- and poly-pharmacology drug discovery: a systems biology view.Curr Pharm Des 20, 1201e1207.
English, A.C., Done, S.H., Caves, L.S., Groom, C.R., Hubbard, R.E., 1999. Locatinginteraction sites on proteins: the crystal structure of thermolysin soaked in 2%to 100% isopropanol. Proteins 37, 628e640.
Fern�andez-Recio, J., 2011. Prediction of protein binding sites and hot spots. WIREsComput Mol. Sci. 1, 18.
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W.B., Fedorov, O., Morse, E.M.,Keates, T., Hickman, T.T., Felletar, I., Philpott, M., Munro, S., McKeown, M.R.,Wang, Y., Christie, A.L., West, N., Cameron, M.J., Schwartz, B., Heightman, T.D., LaThangue, N., French, C.A., Wiest, O., Kung, A.L., Knapp, S., Bradner, J.E., 2010.Selective inhibition of BET bromodomains. Nature 468, 1067e1073.
Fischer, T.B., Arunachalam, K.V., Bailey, D., Mangual, V., Bakhru, S., Russo, R.,Huang, D., Paczkowski, M., Lalchandani, V., Ramachandra, C., Ellison, B., Galer, S.,Shapley, J., Fuentes, E., Tsai, J., 2003. The binding interface database (BID): acompilation of amino acid hot spots in protein interfaces. Bioinformatics 19,1453e1454.
Fleishman, S.J., Whitehead, T.A., Ekiert, D.C., Dreyfus, C., Corn, J.E., Strauch, E.M.,Wilson, I.A., Baker, D., 2011. Computational design of proteins targeting theconserved stem region of influenza hemagglutinin. Science 332, 816e821.
Fry, D.C., 2006. Protein-protein interactions as targets for small molecule drugdiscovery. Biopolymers 84, 535e552.
Fry, D.C., 2012. Small-molecule inhibitors of protein-protein interactions: how tomimic a protein partner. Curr. Pharm. Des. 18, 4679e4684.
Gao, Y., Wang, R., Lai, L., 2004. Structure-based method for analyzing protein-protein interfaces. J. Mol. Model 10, 44e54.
Genomes Project, C., Abecasis, G.R., Auton, A., Brooks, L.D., DePristo, M.A.,Durbin, R.M., Handsaker, R.E., Kang, H.M., Marth, G.T., McVean, G.A., 2012. Anintegrated map of genetic variation from 1092 human genomes. Nature 491,56e65.
Geppert, T., Hoy, B., Wessler, S., Schneider, G., 2011. Context-based identification ofprotein-protein interfaces and “hot-spot” residues. Chem. Biol. 18, 344e353.
Gonzalez-Ruiz, D., Gohlke, H., 2006. Targeting protein-protein interactions withsmall molecules: challenges and perspectives for computational bindingepitope detection and ligand finding. Curr. Med. Chem. 13, 2607e2625.
Grosdidier, S., Fernandez-Recio, J., 2008. Identification of hot-spot residues inprotein-protein interactions by computational docking. BMC Bioinfo. 9, 447.
Guerois, R., Nielsen, J.E., Serrano, L., 2002. Predicting changes in the stability ofproteins and protein complexes: a study of more than 1000 mutations. J. Mol.Biol. 320, 369e387.
Guharoy, M., Chakrabarti, P., 2005. Conservation and relative importance of residuesacross protein-protein interfaces. Proc. Natl. Acad. Sci. U. S. A. 102,15447e15452.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e98
Guney, E., Tuncbag, N., Keskin, O., Gursoy, A., 2008. HotSprint: database ofcomputational hot spots in protein interfaces. Nucleic Acids Res. 36,D662eD666.
Hajduk, P.J., Huth, J.R., Fesik, S.W., 2005. Druggability indices for protein tar-gets derived from NMR-based screening data. J. Med. Chem. 48,2518e2525.
Haliloglu, T., Erman, B., 2009. Analysis of correlations between energy and residuefluctuations in native proteins and determination of specific sites for binding.Phys. Rev. Lett. 102, 088103.
Hall, D.R., Ngan, C.H., Zerbe, B.S., Kozakov, D., Vajda, S., 2012. Hot spot analysis fordriving the development of hits into leads in fragment-based drug discovery.J. Chem. inf. Model. 52, 199e209.
Hopkins, A.L., Groom, C.R., 2002. The druggable genome. Nat. Rev. Drug. Discov. 1,727e730.
Hsu, C.M., Chen, C.Y., Liu, B.J., Huang, C.C., Laio, M.H., Lin, C.C., Wu, T.L., 2007.Identification of hot regions in protein-protein interactions by sequentialpattern mining. BMC Bioinforma. 8 (Suppl. 5), S8.
Humphrey, W., Dalke, A., Schulten, K., 1996. VMD: visual molecular dynamics.J. Mol. Graph 14, 33e38, 27e28.
Huo, S., Massova, I., Kollman, P.A., 2002. Computational alanine scanning of the 1:1human growth hormone-receptor complex. J. Comput Chem. 23, 15e27.
International HapMap, C., Altshuler, D.M., Gibbs, R.A., Peltonen, L., Altshuler, D.M.,Gibbs, R.A., Peltonen, L., Dermitzakis, E., Schaffner, S.F., Yu, F., Peltonen, L.,Dermitzakis, E., Bonnen, P.E., Altshuler, D.M., Gibbs, R.A., de Bakker, P.I.,Deloukas, P., Gabriel, S.B., Gwilliam, R., Hunt, S., Inouye, M., Jia, X., Palotie, A.,Parkin, M., Whittaker, P., Yu, F., Chang, K., Hawes, A., Lewis, L.R., Ren, Y.,Wheeler, D., Gibbs, R.A., Muzny, D.M., Barnes, C., Darvishi, K., Hurles, M.,Korn, J.M., Kristiansson, K., Lee, C., McCarrol, S.A., Nemesh, J., Dermitzakis, E.,Keinan, A., Montgomery, S.B., Pollack, S., Price, A.L., Soranzo, N., Bonnen, P.E.,Gibbs, R.A., Gonzaga-Jauregui, C., Keinan, A., Price, A.L., Yu, F., Anttila, V.,Brodeur, W., Daly, M.J., Leslie, S., McVean, G., Moutsianas, L., Nguyen, H.,Schaffner, S.F., Zhang, Q., Ghori, M.J., McGinnis, R., McLaren, W., Pollack, S.,Price, A.L., Schaffner, S.F., Takeuchi, F., Grossman, S.R., Shlyakhter, I.,Hostetter, E.B., Sabeti, P.C., Adebamowo, C.A., Foster, M.W., Gordon, D.R.,Licinio, J., Manca, M.C., Marshall, P.A., Matsuda, I., Ngare, D., Wang, V.O.,Reddy, D., Rotimi, C.N., Royal, C.D., Sharp, R.R., Zeng, C., Brooks, L.D.,McEwen, J.E., 2010. Integrating common and rare genetic variation in diversehuman populations. Nature 467, 52e58.
Jubb, H., Higueruelo, A.P., Winter, A., Blundell, T.L., 2012. Structural biology and drugdiscovery for protein-protein interactions. Trends Pharmacol. Sci. 33, 241e248.
Kar, G., Kuzu, G., Keskin, O., Gursoy, A., 2012. Protein-protein interfaces integratedinto interaction networks: implications on drug design. Curr. Pharm. Des. 18,4697e4705.
Keskin, O., Ma, B., Nussinov, R., 2005. Hot regions in proteineprotein interactions:the organization and contribution of structurally conserved hot spot residues.J. Mol. Biol. 345, 1281e1294.
Keskin, O., Nussinov, R., 2007. Similar binding sites and different partners: impli-cations to shared proteins in cellular pathways. Structure 15, 341e354.
Kim, P.M., Lu, L.J., Xia, Y., Gerstein, M.B., 2006. Relating three-dimensional structuresto protein networks provides evolutionary insights. Science 314, 1938e1941.
Koes, D.R., Camacho, C.J., 2012. Small-molecule inhibitor starting points learnedfrom protein-protein interaction inhibitor structure. Bioinformatics 28,784e791.
Kortemme, T., Baker, D., 2002. A simple physical model for binding energy hot spotsin protein-protein complexes. Proc. Natl. Acad. Sci. U. S. A. 99, 14116e14121.
Kortemme, T., Kim, D.E., Baker, D., 2004. Computational alanine scanning of protein-protein interfaces. Sci. STKE 2004, l2.
Kozakov, D., Hall, D.R., Chuang, G.Y., Cencic, R., Brenke, R., Grove, L.E., Beglov, D.,Pelletier, J., Whitty, A., Vajda, S., 2011. Structural conservation of druggable hotspots in protein-protein interfaces. Proc. Natl. Acad. Sci. U. S. A 108,13528e13533.
Kruger, D.M., Gohlke, H., 2010. DrugScorePPI webserver: fast and accurate in silicoalanine scanning for scoring protein-protein interactions. Nucleic Acids Res. 38,W480eW486.
Landon, M.R., Lancia Jr., D.R., Yu, J., Thiel, S.C., Vajda, S., 2007. Identification of hotspots within druggable binding regions by computational solvent mapping ofproteins. J. Med. Chem. 50, 1231e1240.
Li, H., Liu, A., Zhao, Z., Xu, Y., Lin, J., Jou, D., Li, C., 2011. Fragment-based drug designand drug repositioning using multiple ligand simultaneous docking (MLSD):identifying celecoxib and template compounds as novel inhibitors of signaltransducer and activator of transcription 3 (STAT3). J. Med. Chem. 54,5592e5596.
Li, J., Liu, Q., 2009. 'Double water exclusion': a hypothesis refining the O-ring theoryfor the hot spots at protein interfaces. Bioinformatics 25, 743e750.
Li, X., Keskin, O., Ma, B., Nussinov, R., Liang, J., 2004. Protein-protein interactions:hot spots and structurally conserved residues often locate in complementedpockets that pre-organized in the unbound states: implications for docking.J. Mol. Biol. 344, 781e795.
Liang, S., Liu, Z., Li, W., Ni, L., Lai, L., 2000. Construction of protein binding sites inscaffold structures. Biopolymers 54, 515e523.
Liepinsh, E., Otting, G., 1997. Organic solvents identify specific ligand binding siteson protein surfaces. Nat. Biotechnol. 15, 264e268.
Lise, S., Archambeau, C., Pontil, M., Jones, D.T., 2009. Prediction of hot spot residuesat protein-protein interfaces by combining machine learning and energy-basedmethods. BMC Bioinforma. 10, 365.
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
Lise, S., Buchan, D., Pontil, M., Jones, D.T., 2011. Predictions of hot spot residues atprotein-protein interfaces using support vector machines. PLoS One 6, e16774.
Liu, S., Zhu, X., Liang, H., Cao, A., Chang, Z., Lai, L., 2007. Nonnatural protein-proteininteraction-pair design by key residues grafting. Proc. Natl. Acad. Sci. U. S. A.104, 5330e5335.
London, N., Raveh, B., Schueler-Furman, O., 2013. Druggable protein-protein in-teractions - from hot spots to hot segments. Curr. Opin. Chem. biol. 17, 952e959.
Ma, B., Nussinov, R., 2014. Druggable orthosteric and allosteric hot spots to targetprotein-protein interactions. Curr. Pharm. Des. 20, 1293e1301.
Mattos, C., Ringe, D., 1996. Locating and characterizing binding sites on proteins.Nat. Biotechnol. 14, 595e599.
Meenan, N.A., Sharma, A., Fleishman, S.J., Macdonald, C.J., Morel, B., Boetzel, R.,Moore, G.R., Baker, D., Kleanthous, C., 2010. The structural and energetic basisfor high selectivity in a high-affinity protein-protein interaction. Proc. Natl.Acad. Sci. U. S. A. 107, 10080e10085.
Moreira, I.S., Fernandes, P.A., Ramos, M.J., 2007a. Computational alanine scanningmutagenesisean improved methodological approach. J. Comput Chem. 28,644e654.
Moreira, I.S., Fernandes, P.A., Ramos, M.J., 2007b. Hot spotsea review of the protein-protein interface determinant amino-acid residues. Proteins 68, 803e812.
Nicodeme, E., Jeffrey, K.L., Schaefer, U., Beinke, S., Dewell, S., Chung, C.W.,Chandwani, R., Marazzi, I., Wilson, P., Coste, H., White, J., Kirilovsky, J., Rice, C.M.,Lora, J.M., Prinjha, R.K., Lee, K., Tarakhovsky, A., 2010. Suppression of inflam-mation by a synthetic histone mimic. Nature 468, 1119e1123.
Nishi, H., Hashimoto, K., Panchenko, A.R., 2011. Phosphorylation in protein-proteinbinding: effect on stability and function. Structure 19, 1807e1815.
Nussinov, R., Tsai, C.J., Xin, F., Radivojac, P., 2012. Allosteric post-translationalmodification codes. Trends Biochem Sci. 37, 447e455.
Ofran, Y., Rost, B., 2007. Protein-protein interaction hotspots carved into sequences.PLoS Comput Biol. 3, e119.
Ozbek, P., Soner, S., Haliloglu, T., 2013. Hot spots in a network of functional sites.PLoS One 8, e74320.
Pavelka, A., Chovancova, E., Damborsky, J., 2009. HotSpot Wizard: a web server foridentification of hot spots in protein engineering. Nucleic Acids Res. 37,W376eW383.
Rajamani, D., Thiel, S., Vajda, S., Camacho, C.J., 2004. Anchor residues in protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 101, 11287e11292.
Reichmann, D., Rahat, O., Albeck, S., Meged, R., Dym, O., Schreiber, G., 2005. Themodular architecture of protein-protein binding interfaces. Proc. Natl. Acad. Sci.U. S. A 102, 57e62.
Schaller, M.D., Parsons, J.T., 1995. pp125FAK-dependent tyrosine phosphorylation ofpaxillin creates a high-affinity binding site for Crk. Mol. Cell. Biol. 15,2635e2645.
Schiro, M.M., Stauber, S.E., Peterson, T.L., Krueger, C., Darnell, S.J., Satyshur, K.A.,Drinkwater, N.R., Newton, M.A., Hoffmann, F.M., 2011. Mutations in protein-binding hot-spots on the hub protein Smad3 differentially affect its proteininteractions and Smad3-regulated gene expression. PLoS One 6, e25021.
Schuster-Bockler, B., Bateman, A., 2008. Protein interactions in human geneticdiseases. Genome Biol. 9, R9.
Seco, J., Luque, F.J., Barril, X., 2009. Binding site detection and druggability indexfrom first principles. J. Med. Chem. 52, 2363e2371.
Shulman-Peleg, A., Shatsky, M., Nussinov, R., Wolfson, H.J., 2007. Spatial chemicalconservation of hot spot interactions in protein-protein complexes. BMC Biol. 5,43.
Straub, C.S., 2011. Targeting IAPs as an approach to anti-cancer therapy. Curr. Top.Med. Chem. 11, 291e316.
Thangudu, R.R., Bryant, S.H., Panchenko, A.R., Madej, T., 2012. Modulating protein-protein interactions with small molecules: the importance of binding hotspots.J. Mol. Biol. 415, 443e453.
Thorn, K.S., Bogan, A.A., 2001. ASEdb: a database of alanine mutations and theireffects on the free energy of binding in protein interactions. Bioinformatics 17,284e285.
Tse, C., Shoemaker, A.R., Adickes, J., Anderson, M.G., Chen, J., Jin, S., Johnson, E.F.,Marsh, K.C., Mitten, M.J., Nimmer, P., Roberts, L., Tahir, S.K., Xiao, Y., Yang, X.,Zhang, H., Fesik, S., Rosenberg, S.H., Elmore, S.W., 2008. ABT-263: a potent andorally bioavailable Bcl-2 family inhibitor. Cancer Res. 68, 3421e3428.
Tuncbag, N., Gursoy, A., Keskin, O., 2009. Identification of computational hot spotsin protein interfaces: combining solvent accessibility and inter-residue poten-tials improves the accuracy. Bioinformatics 25, 1513e1520.
Tuncbag, N., Keskin, O., Gursoy, A., 2010a. HotPoint: hot spot prediction server forprotein interfaces. Nucleic acids Res. 38, W402eW406.
Tuncbag, N., Salman, F.S., Keskin, O., Gursoy, A., 2010b. Analysis and network rep-resentation of hotspots in protein interfaces using minimum cut trees. Proteins78, 2283e2294.
Villoutreix, B.O., Labbe, C.M., Lagorce, D., Laconde, G., Sperandio, O., 2012. A leapinto the chemical space of protein-protein interaction inhibitors. Curr. Pharm.Des. 18, 4648e4667.
Vu, B.T., Vassilev, L., 2011. Small-molecule inhibitors of the p53-MDM2 interaction.Curr. Top. Microbiol. Immunol. 348, 151e172.
Wang, L., Hou, Y., Quan, H., Xu, W., Bao, Y., Li, Y., Fu, Y., Zou, S., 2013. A compound-based computational approach for the accurate determination of hot spots.Protein Sci. 22, 1060e1070.
Wang, L., Liu, Z.P., Zhang, X.S., Chen, L., 2012a. Prediction of hot spots in proteininterfaces using a random forest model with hybrid features. Protein Eng. Des.Sel. 25, 119e126.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003

E. Cukuroglu et al. / Progress in Biophysics and Molecular Biology xxx (2014) 1e9 9
Wang, X., Wei, X., Thijssen, B., Das, J., Lipkin, S.M., Yu, H., 2012b. Three-dimensionalreconstruction of protein networks provides insight into human genetic dis-ease. Nat. Biotechnol. 30, 159e164.
Wells, J.A., McClendon, C.L., 2007. Reaching for high-hanging fruit in drug discoveryat protein-protein interfaces. Nature 450, 1001e1009.
Xia, J.F., Zhao, X.M., Song, J., Huang, D.S., 2010. APIS: accurate prediction of hot spotsin protein interfaces by combining protrusion index with solvent accessibility.BMC Bioinforma. 11, 174.
Yogurtcu, O.N., Erdemli, S.B., Nussinov, R., Turkay, M., Keskin, O., 2008. Restrictedmobility of conserved residues in protein-protein interfaces in molecular sim-ulations. Biophys. J. 94, 3475e3485.
Zerbe, B.S., Hall, D.R., Vajda, S., Whitty, A., Kozakov, D., 2012. Relationship betweenhot spot residues and ligand binding hot spots in protein-protein interfaces.J. Chem. inf. Model. 52, 2236e2244.
Please cite this article in press as: Cukuroglu, E., et al., Hot spots in proteiand Molecular Biology (2014), http://dx.doi.org/10.1016/j.pbiomolbio.201
Zhang, C., Lai, L., 2011. Towards structure-based protein drug design. Biochem. Soc.Trans. 39 (Suppl. 1), 1382e1386 following 1386.
Zhang, C., Lai, L., 2012. AutoMatch: target-binding protein design and enzymedesign by automatic pinpointing potential active sites in available proteinscaffolds. Proteins 80, 1078e1094.
Zhong, Q., Simonis, N., Li, Q.R., Charloteaux, B., Heuze, F., Klitgord, N., Tam, S., Yu, H.,Venkatesan, K., Mou, D., Swearingen, V., Yildirim, M.A., Yan, H., Dricot, A.,Szeto, D., Lin, C., Hao, T., Fan, C., Milstein, S., Dupuy, D., Brasseur, R., Hill, D.E.,Cusick, M.E., Vidal, M., 2009. Edgetic perturbation models of human inheriteddisorders. Mol. Syst. Biol. 5, 321.
Zhu, X., Mitchell, J.C., 2011. KFC2: a knowledge-based hot spot prediction methodbased on interface solvation, atomic density, and plasticity features. Proteins 79,2671e2683.
neprotein interfaces: Towards drug discovery, Progress in Biophysics4.06.003