Embed Size (px)
Citation preview

Toxicology and Applied Pharmacology 256 (2011) 114–121
Contents lists available at SciVerse ScienceDirect
Toxicology and Applied Pharmacology
j ourna l homepage: www.e lsev ie r.com/ locate /ytaap
Reactive oxygen species mediate arsenic induced cell transformation andtumorigenesis through Wnt/β-catenin pathway in human colorectaladenocarcinoma DLD1 cells
Zhuo Zhang b, Xin Wang a, Senping Cheng a, Lijuan Sun a, Young-Ok Son a, Hua Yao c, Wenqi Li b,Amit Budhraja a, Li Li d, Brent J. Shelton e, Thomas Tucker e, Susanne M. Arnold f, Xianglin Shi a,⁎a Graduate Center for Toxicology, University of Kentucky, 1095 VA Drive, Lexington, KY 40536, USAb Department of Preventive Medicine and Environmental Health, University of Kentucky, 121 Washington Avenue, Lexington, KY 40536, USAc Department of Stomatology, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310003, Chinad Department of Family Medicine, Case Western Reserve University, Cleveland, OH 44106, USAe Markey Cancer Control Program, University of Kentucky, 2365 Harrodsburg Rd, Lexington, KY 40504, USAf Markey Cancer Center, University of Kentucky, 800 Rose street, Lexington, KY 40536, USA
⁎ Corresponding author. Fax: +1 859 323 1059.E-mail address: [email protected] (X. Shi).
0041-008X/$ – see front matter © 2011 Published by Edoi:10.1016/j.taap.2011.07.016
a b s t r a c t
a r t i c l e i n f oArticle history:Received 1 July 2011Revised 27 July 2011Accepted 28 July 2011Available online 11 August 2011
Keywords:ArsenicReactive oxygen speciesNADPH oxidaseβ-cateninCell transformationTumorigenesis
Long term exposure to arsenic can increase incidence of human cancers, such as skin, lung, and colon rectum.The mechanism of arsenic induced carcinogenesis is still unclear. It is generally believed that reactive oxygenspecies (ROS) may play an important role in this process. In the present study, we investigate the possiblelinkage between ROS, β-catenin and arsenic induced transformation and tumorigenesis in human colorectaladenocarcinoma cell line, DLD1 cells. Our results show that arsenic was able to activate p47phox and p67phox,two key proteins for activation of NADPH oxidase. Arsenic was also able to generate ROS in DLD1 cells. Arsenicincreased β-catenin expression level and its promoter activity. ROS played a major role in arsenic-inducedβ-catenin activation. Treatment of DLD1 cells by arsenic enhanced both transformation and tumorigenesis ofthese cells. The tumor volumes of arsenic treated group were much larger than those without arsenictreatment. Addition of either superoxide dismutase (SOD) or catalase reduced arsenic induced celltransformation and tumor formation. The results indicate that ROS are involved in arsenic induced celltransformation and tumor formation possible through Wnt/β-catenin pathway in human colorectaladenocarcinoma cell line DLD1 cells.
lsevier Inc.
© 2011 Published by Elsevier Inc.
Introduction
Arsenic is a metalloid element that is widely distributed in theenvironment and a variety of occupational settings (Bode and Dong,2002). Long term exposure to this metal is associated with increasedincidence of cancer in lung, liver, kidney, skin, bladder, and colon(Stevens et al., 2010; Tchounwou et al., 2003). The exact molecularmechanisms of arsenic carcinogenesis are not well understood. It isgenerally acknowledged that arsenic does not act via a classic genotoxicor mutagenic mechanism, because it is not a direct mutagen (Huff et al.,2000; Kitchin andWallace, 2008). Arsenicwas thought to act principallyas a co-mutagen and co-carcinogen. Other potential carcinogenicactions include oxidative stress (Ding et al., 2005; Shi et al., 2004a),genotoxic damage, DNA repair inhibition (Ding et al., 2008), tumorpromotion, cell proliferation, chromosomal aberrations, and activationof certain signal transduction pathways leading to changes in gene
expression (Benbrahim-Tallaa andWaalkes, 2008; Galanis et al., 2009).It is probable that thesemechanisms do not act in isolation, but overlap,and contribute to the complexnature of arsenic-induced carcinogenesis.
Wnt ligands are a large family of highly conserved secretedproteins that are important for normal cell development. Wnts bindto receptors at the plasma membrane and initiate intracellularsignaling cascades to control a wide variety of processes in embryonicdevelopment and adult homeostasis. Wnts direct programs ofproliferation, stem cell self-renewal, cell fate decisions, cell polarity,and convergent extension behaviors of migrating cells (Klaus andBirchmeier, 2008; MacDonald et al., 2009; Reya and Clevers, 2005).Each of theseWnt-directed outcomes is critical for proper development ofthe intestine (vander Flier andClevers, 2009).Many components of theβ-catenin-dependent pathway are often differentially regulated betweennormal tissue and its cancerous counterpart. In particular, melanoma,hepatocellular carcinoma (HCC), prostate, colon, thyroid, and ovariancancers, as well as certain subsets of breast cancers, harbor β-catenin-stabilizingmutations (Polakis, 2000). Suchmutations result in high levelsof β-catenin, leading to constitutively activeWnt signaling (Korinek et al.,1997;Morin et al., 1997).While the regulation of β-catenin appears to be

115Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
one of the most significant events linking Wnt signaling to cancer,modulation of other pathway components, both upstream and down-stream of β-catenin, has also been shown to play a role.
Although the molecular mechanism of arsenic carcinogenesisremains to be investigated, it is generally believed that reactive oxygenspecies (ROS) play an important role in the initiation of arsenic-inducedcellular injury which can lead to cancer development (Harris and Shi,2003; Huang et al., 2004; Leonard et al., 2004;Qian et al., 2005; Shi et al.,2004b). ROS can induce direct cellular injury, which may trigger acascade of radical reactions enhancing secondary ROS generation.Furthermore, excessive generation of ROSmay lead to the stimulation ofinflammatory processes involving secretion of chemotactic factors,growth factors, proteolytic enzymes, lipoxygenases, and cyclooxygen-ase, inactivation of anti-proteolytic enzymes, and the release ofsignaling proteins (Chen and Shi, 2002a, b; Harris and Shi, 2003). Animportant physiological system for ROS production is the NADPHoxidase (Nox) complex. The Nox system has been known as a source ofROS in phagocytes. NOX consists of three cytosolic components(p67phox, p47phox and p40phox), two membrane-bound elements(gp91phox and p22phox), and a low-molecular-weight G protein. Nox isdormant in the resting cells. Upon stimulation, NOX is rapidly activated.Activation of Nox is caused by translocation of the cytosolic componentsto the cell membrane so that the complete oxidase can be assembled.Activations of both p47 and p67 are required for the complete functionof Nox. In the past several years, different Nox complexes have beenidentified as a key machinery to regulate cellular ROS production bydifferent signaling pathways (Brown and Griendling, 2009). Somestudies indicated an accumulation of ROS in the cells treated witharsenic (Bower et al., 2006; Chou et al., 2004; Ding et al., 2005; Shi et al.,2004a). Arsenic is highly capable of activating NADPH oxidase anddestabilizing the out-membrane of the mitochondria, leading to thegeneration of superoxide radical, hydroxyl radical and hydrogenperoxide (Chou et al., 2004). It has been generally accepted that ROSare critical regulators for a wide range of cellular responses, from kinaseactivation, gene expression, DNA damage, cell proliferation, to cellmigration in the arsenic treated cells.
Aberrant activation ofWnt signaling, arising fromgenetic defects inthe tumor suppressor genes adenomatous polyposis coli (APC) oractivating mutations in β-catenin, is common in colorectal cancer(Barker and Clevers, 2006; Segditsas and Tomlinson, 2006). Aprimary consequence of Wnt signaling activation is the stabilizationofβ-catenin in the cytoplasm, resulting in an increased translocation ofβ-catenin to the nucleus. Nuclear β-catenin forms a complex with theTCF/LEF transcription factor leading to activation of Wnt target geneexpression (Li et al., 2011). A number of recent publications indicatethat the WNT/β-catenin pathway is regulated by ROS (Ladelfa et al.,2011). However, it is still unclear if arsenic induced ROS generationis responsible for the cell transformation and tumorigenesis throughβ-catenin pathway in colon cancer cells.
Materials and methods
Cells and chemicals. Colorectal adenocarcinoma cell line DLD1 cellswere cultured inmonolayer at 37 °C, 5% CO2 usingDulbecco'smodifiedEagle's medium (DMEM) containing 10% fetal bovine serum (FBS),2 mM L-glutamine, and 25 μg gentamicin/ml. Arsenic trichloride(AsCl3) was purchased from Sigma (St. Louis, MO). All antibodiesused in Western blotting were purchased from Santa Cruz Biotech-nology (Santa Cruz, CA). 2′,7′-dichlorodihydrofluorescein diacetateethyl ester (DCFDA) was from Molecular Probes (Eugene, OR).
Western blot. The cells were solubilized in lysis buffer containing 10mMTris–HCl, pH 7.4, 150mM NaCl, 1 mM EDTA, 1% Triton X-100, proteaseinhibitormixture, including 5 μg/ml aprotinin, 1 mMphenylmethylsulfonylfluoride, and 1 mM sodium orthovanadate (Sigma), for 30 min at 4 °C.After centrifugation at 10,000×g for 20 min at 4 °C, the supernatants
were transferred, and the protein contentwasmeasured. Lysates (30 μg)were separated by 10% SDS-PAGE, followed by transfer to Immobilon-Pmembrane (Millipore, MA). Membranes were blocked and probed withthe appropriate antibodies at a concentration of 1 μg/ml. Primaryantibodies were suspended in 5% milk in 10 mM Tris–HCl, pH 7.5,100 mM NaCl, and 0.1% Tween 20. After washes, blots were incubatedwith secondary antibodies conjugated to horseradish peroxidase.Immunoreactive bands were detected by the enhanced chemilumines-cence reagent (Pierce).
Measurement of intracellular ROS. Detection of ROS was performedusing the fluorescent dye 2′,7′-dichlorodihydrofluoresceine acetate(DCFDA). DCFDA is oxidized into fluorescent 2′,7′-dichlorofluorescein(DCF) in the presence of hydrogen peroxide (H2O2). The intensity offluorescence represents the generation of H2O2. The cells were washedonce with PBS and incubated with 10 μM DCFDA in PBS for 30 min.Then, the cells were washed with PBS and incubated in culture mediafor 30 min. After incubation, cells were trypsinized, washed twice withcold PBS, and analyzed by using a FACS Calibur flow cytometry. Thefluorescence intensity of DCF was measured at an excitation wave-length of 492 nm and an emission wavelength of 517 nm.
Electron spin resonance (ESR) assay. All ESR measurements wereconducted using a Bruker EMX spectrometer (Bruker Instruments,Billerica, MA) and a flat cell assembly. Since hydroxyl radical isreactive and has a very short life-time, direct ESR measurementcannot be used for its detection. Instead, ESR spin trapping or radicaltrapping, an indirect method, was used. This technique involves theaddition-type reaction of a nonradical compound (spin or radicaltrap) to form a relatively long-lived free radical product (spin orradical adduct), which can then be studied using ESR. The intensity ofthe ESR signal is used to measure the amount of short-lived hydroxylradical trapped and the hyperfine couplings of the spin adduct aregenerally characteristic of the original trapped radicals. The spintrapping is a method of choice for detection and identification ofhydroxyl radical generation due to its specificity and sensitivity.5,5-dimethyl-1-pyrroline 1-oxide (DMPO) was used as spin or radicaltrap. DMPO was charcoal purified and distilled to remove all ESRdetectable impurities before use. Hyperfine couplings were measured(to 0.1 G) directly from magnetic field separation using potassiumtetraperoxochromate (K3CrO8) and 1,1-diphenyl-2-picrylhydrazyl(DPPH) as reference standards. The Acquisit program was used fordata acquisitions and analyses (Bruker Instruments). Reactants weremixed in test tubes to a total final volume of 0.5 ml. The reactionmixture was then transferred to a flat cell for ESR measurement.
Luciferase assay. β-catenin activity was measured using luciferasereporter assay. The luciferase reporter contains eight optimal copies ofthe LEF/TCF binding site upstream of a minimal thymidine kinasepromoter directing transcription of a luciferase gene. The LEF/TCFReporter is designed to monitor the activity of Wnt signal transductionpathways in cultured cells. The Wnt signaling pathway leads to thedephosphorylation, stabilization, and nuclear translocation of β-catenin.The stabilized β-catenin complexes with the TCF/LEF transcriptionfactors, leading to the activation of Wnt-responsive genes. Therefore,the activity of LEF/TCF luciferase represents β-catenin activity. The DLD1cells were seeded in 12-well plates. After 80% confluence, the cells weretransfectedwith LEF/TCF luciferase reporter plasmid in combinationwithcatalase plasmid (Origene Inc). After 4 h of transfection, the cells weretreated with 10 μM arsenic for 24 h. The cells were harvested. Luciferaseassay was conducted by luciferase assay reagent (Promega) andmeasured in Luminometer (Promega). The relative luciferase activitywas normalized by protein concentration.
Cell transformation assay. Arsenic induced cell transformation wasassayed in soft agar. The cells were split to 10 cm culture dishes after
116 Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
90% confluence, 0.5 μM of arsenic, either 100 U/ml SOD or 500 U/mlcatalase was added individually or combined according to experi-mental design. The cells were repeatedly treated with arsenic, SODand catalase for up to 2 weeks. Those cells were used for celltransformation assay. Briefly, 2 ml of 1% agar mixturewith DMEMwasadded into 6-well plate. The 2500 cells were mixed with 0.7% agar,aliquot 1 ml of the cell mixture on to the base agar. Every 3 days, 1 mlof fresh DMEMmedium was added to the each well of plate. The cellswere grown in the incubator for 2 weeks. After 2 weeks, crystal violetwas added to make the cells visible. The colonies were counted.
Tumorigenicity assay. 6 week old male athymic nude mice werepurchased from Charles River Laboratory. DLD1 cells were treated witharsenic, SOD and catalase for up to 2 weeks as described in the “Celltransformation assay” section. The cells were used to test theirtumorigenicity in nude mice. A non-treated same passage cells will beused as control. The cells were harvested and adjusted to cell densities of107 cells/ml in cell culture medium. One site of the axillary region of thenudemicewill be subcutaneously injectedwith 0.2 ml of cell suspension(2×106 cells/site). Nude mice were fed with regular food. In order todetermine tumor volume by external caliper, the greatest longitudinaldiameter (length) and the greatest transverse diameter (width) weredetermined. Tumor volume based on caliper measurements wascalculated by the modified ellipsoidal formula (Euhus et al., 1986;Tomayko and Reynolds, 1989). Tumor volume=1/2(length×width2).Tumor volume (inch3) was measured 10 days after injection.
Results
Arsenic induces activations of p67phox and p47phox and ROS generation
The hypothesis of arsenic-induced ROS generation in the presentstudy is that arsenic induces phosphorylation of p47phox, transloca-tion of p47phox and p67phox, and activation of NADPH oxidase. In ourstudy, we examined two important proteins involved in the activation
A
C
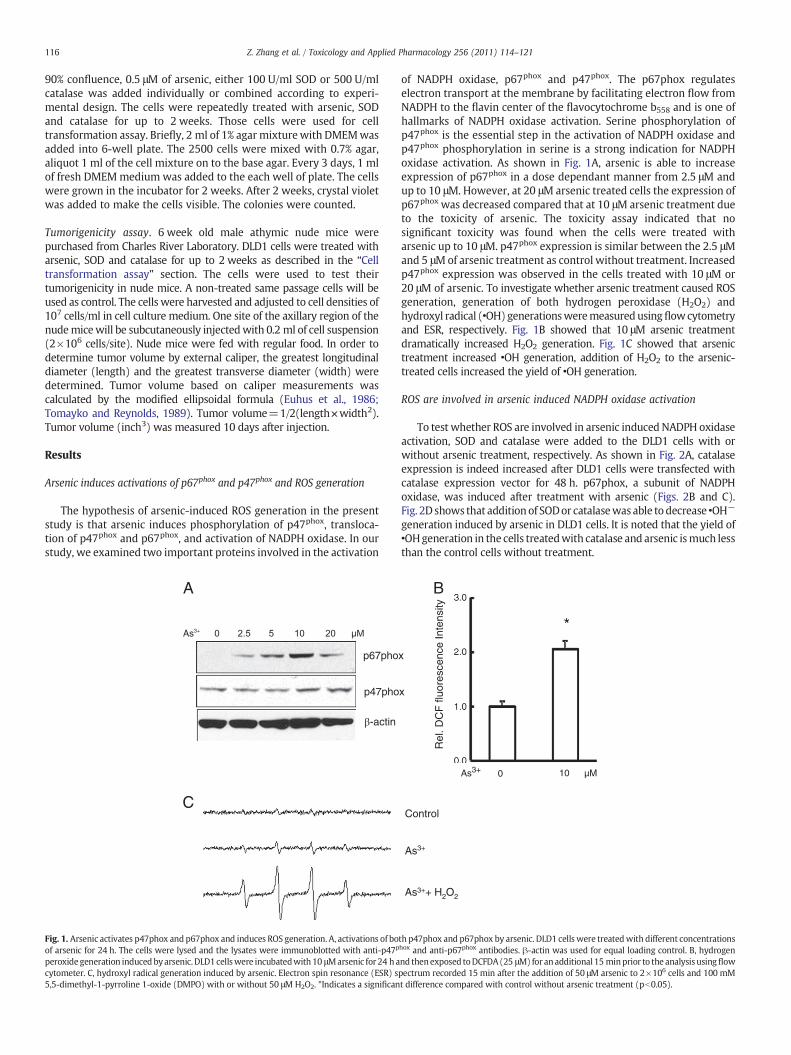
Fig. 1. Arsenic activates p47phox and p67phox and induces ROS generation. A, activations of boof arsenic for 24 h. The cells were lysed and the lysates were immunoblotted with anti-p47p
peroxidegeneration inducedbyarsenic.DLD1cellswere incubatedwith10 μMarsenic for 24 hacytometer. C, hydroxyl radical generation induced by arsenic. Electron spin resonance (ESR) s5,5-dimethyl-1-pyrroline 1-oxide (DMPO) with or without 50 μM H2O2. *Indicates a significan
of NADPH oxidase, p67phox and p47phox. The p67phox regulateselectron transport at the membrane by facilitating electron flow fromNADPH to the flavin center of the flavocytochrome b558 and is one ofhallmarks of NADPH oxidase activation. Serine phosphorylation ofp47phox is the essential step in the activation of NADPH oxidase andp47phox phosphorylation in serine is a strong indication for NADPHoxidase activation. As shown in Fig. 1A, arsenic is able to increaseexpression of p67phox in a dose dependant manner from 2.5 μM andup to 10 μM. However, at 20 μM arsenic treated cells the expression ofp67phox was decreased compared that at 10 μM arsenic treatment dueto the toxicity of arsenic. The toxicity assay indicated that nosignificant toxicity was found when the cells were treated witharsenic up to 10 μM. p47phox expression is similar between the 2.5 μMand 5 μMof arsenic treatment as control without treatment. Increasedp47phox expression was observed in the cells treated with 10 μM or20 μM of arsenic. To investigate whether arsenic treatment caused ROSgeneration, generation of both hydrogen peroxidase (H2O2) andhydroxyl radical (•OH)generationsweremeasuredusingflowcytometryand ESR, respectively. Fig. 1B showed that 10 μM arsenic treatmentdramatically increased H2O2 generation. Fig. 1C showed that arsenictreatment increased •OH generation, addition of H2O2 to the arsenic-treated cells increased the yield of •OH generation.
ROS are involved in arsenic induced NADPH oxidase activation
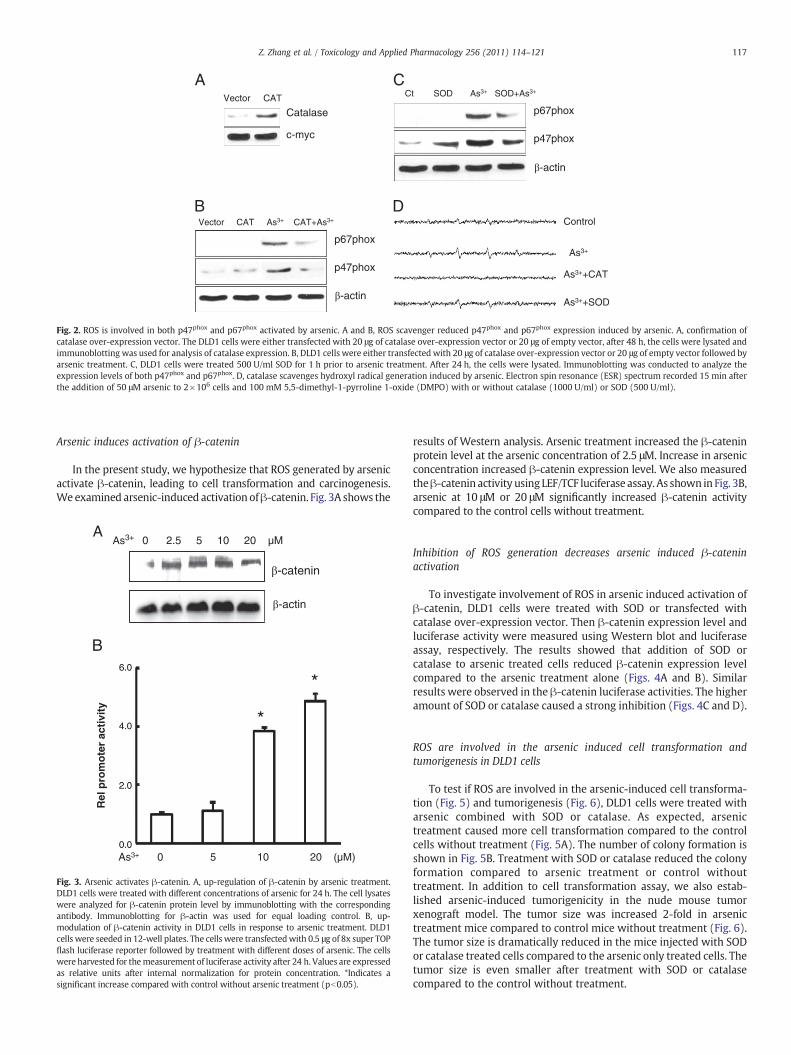
To test whether ROS are involved in arsenic induced NADPH oxidaseactivation, SOD and catalase were added to the DLD1 cells with orwithout arsenic treatment, respectively. As shown in Fig. 2A, catalaseexpression is indeed increased after DLD1 cells were transfected withcatalase expression vector for 48 h. p67phox, a subunit of NADPHoxidase, was induced after treatment with arsenic (Figs. 2B and C).Fig. 2D shows that addition of SODor catalasewas able to decrease •OH−
generation induced by arsenic in DLD1 cells. It is noted that the yield of•OHgeneration in the cells treatedwith catalase and arsenic ismuch lessthan the control cells without treatment.
B
th p47phox and p67phox by arsenic. DLD1 cells were treatedwith different concentrationshox and anti-p67phox antibodies. β-actin was used for equal loading control. B, hydrogennd thenexposed toDCFDA(25 μM) for anadditional 15 minprior to the analysis usingflowpectrum recorded 15 min after the addition of 50 μM arsenic to 2×106 cells and 100 mMt difference compared with control without arsenic treatment (pb0.05).
A
B
C
D
Fig. 2. ROS is involved in both p47phox and p67phox activated by arsenic. A and B, ROS scavenger reduced p47phox and p67phox expression induced by arsenic. A, confirmation ofcatalase over-expression vector. The DLD1 cells were either transfected with 20 μg of catalase over-expression vector or 20 μg of empty vector, after 48 h, the cells were lysated andimmunoblotting was used for analysis of catalase expression. B, DLD1 cells were either transfected with 20 μg of catalase over-expression vector or 20 μg of empty vector followed byarsenic treatment. C, DLD1 cells were treated 500 U/ml SOD for 1 h prior to arsenic treatment. After 24 h, the cells were lysated. Immunoblotting was conducted to analyze theexpression levels of both p47phox and p67phox. D, catalase scavenges hydroxyl radical generation induced by arsenic. Electron spin resonance (ESR) spectrum recorded 15 min afterthe addition of 50 μM arsenic to 2×106 cells and 100 mM 5,5-dimethyl-1-pyrroline 1-oxide (DMPO) with or without catalase (1000 U/ml) or SOD (500 U/ml).
117Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
Arsenic induces activation of β-catenin
In the present study, we hypothesize that ROS generated by arsenicactivate β-catenin, leading to cell transformation and carcinogenesis.We examined arsenic-induced activation ofβ-catenin. Fig. 3A shows the
Rel
pro
mo
ter
acti
vity
A
B
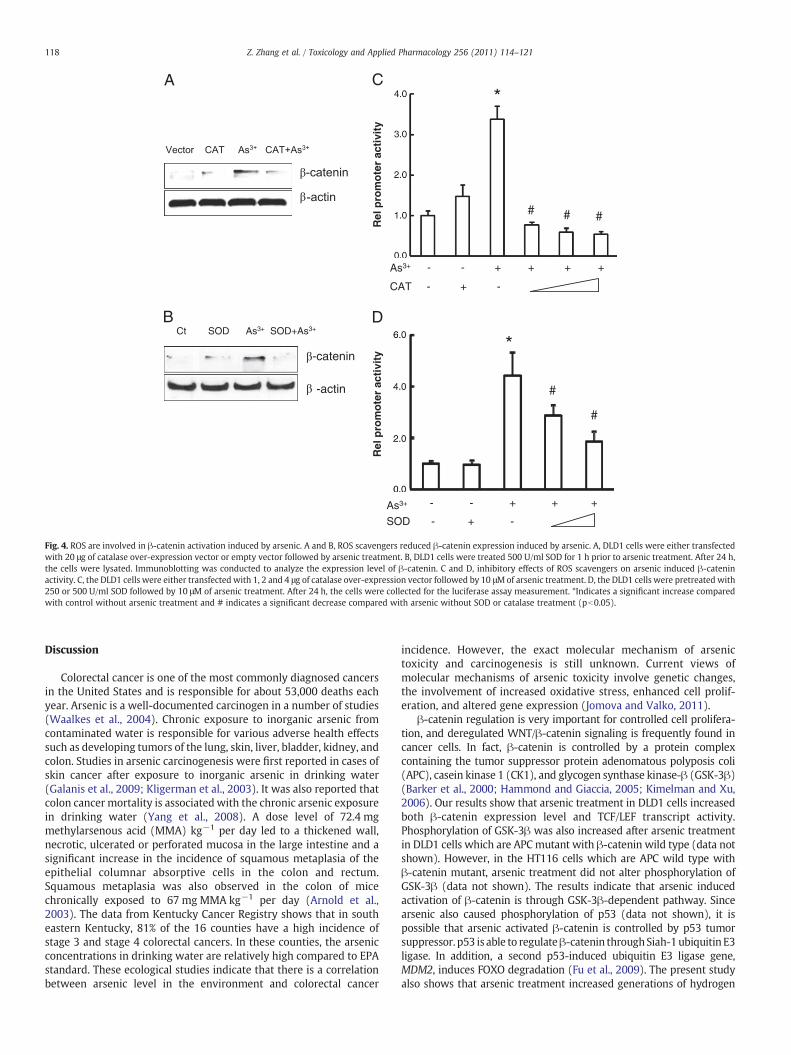
Fig. 3. Arsenic activates β-catenin. A, up-regulation of β-catenin by arsenic treatment.DLD1 cells were treated with different concentrations of arsenic for 24 h. The cell lysateswere analyzed for β-catenin protein level by immunoblotting with the correspondingantibody. Immunoblotting for β-actin was used for equal loading control. B, up-modulation of β-catenin activity in DLD1 cells in response to arsenic treatment. DLD1cells were seeded in 12-well plates. The cells were transfectedwith 0.5 μg of 8x super TOPflash luciferase reporter followed by treatment with different doses of arsenic. The cellswere harvested for themeasurement of luciferase activity after 24 h. Values are expressedas relative units after internal normalization for protein concentration. *Indicates asignificant increase compared with control without arsenic treatment (pb0.05).
results of Western analysis. Arsenic treatment increased the β-cateninprotein level at the arsenic concentration of 2.5 μM. Increase in arsenicconcentration increased β-catenin expression level. We also measuredtheβ-catenin activity using LEF/TCF luciferase assay. As shown in Fig. 3B,arsenic at 10 μM or 20 μM significantly increased β-catenin activitycompared to the control cells without treatment.
Inhibition of ROS generation decreases arsenic induced β-cateninactivation
To investigate involvement of ROS in arsenic induced activation ofβ-catenin, DLD1 cells were treated with SOD or transfected withcatalase over-expression vector. Then β-catenin expression level andluciferase activity were measured using Western blot and luciferaseassay, respectively. The results showed that addition of SOD orcatalase to arsenic treated cells reduced β-catenin expression levelcompared to the arsenic treatment alone (Figs. 4A and B). Similarresults were observed in the β-catenin luciferase activities. The higheramount of SOD or catalase caused a strong inhibition (Figs. 4C and D).
ROS are involved in the arsenic induced cell transformation andtumorigenesis in DLD1 cells
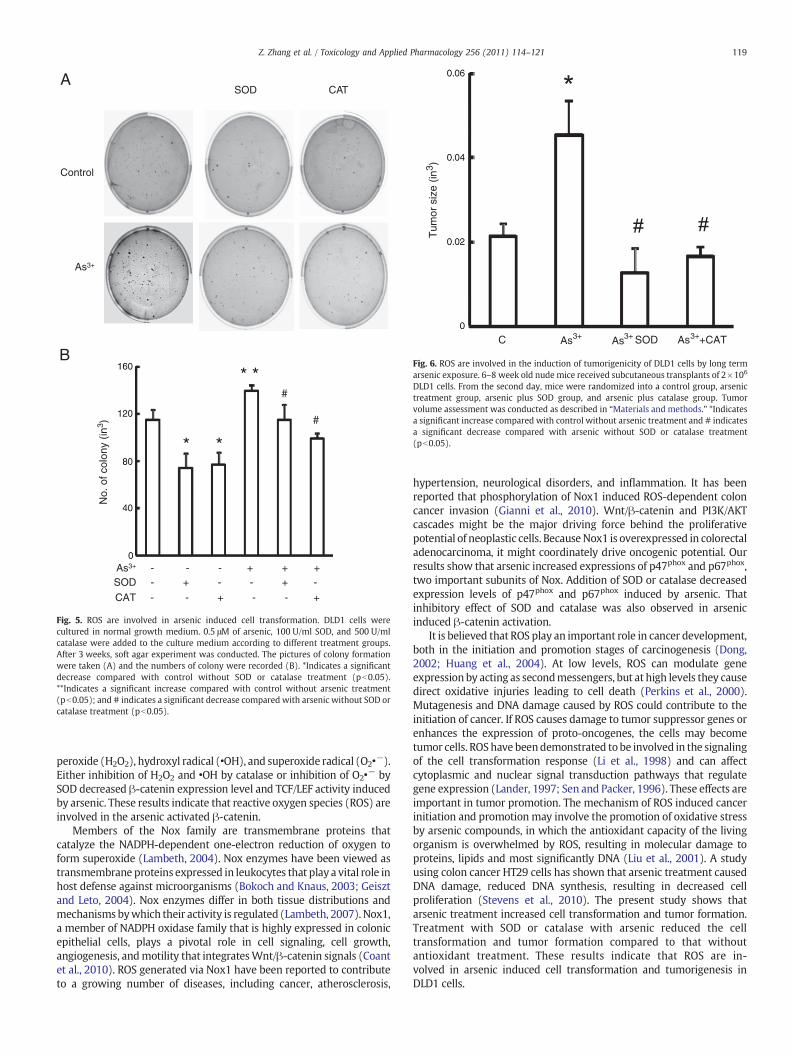
To test if ROS are involved in the arsenic-induced cell transforma-tion (Fig. 5) and tumorigenesis (Fig. 6), DLD1 cells were treated witharsenic combined with SOD or catalase. As expected, arsenictreatment caused more cell transformation compared to the controlcells without treatment (Fig. 5A). The number of colony formation isshown in Fig. 5B. Treatment with SOD or catalase reduced the colonyformation compared to arsenic treatment or control withouttreatment. In addition to cell transformation assay, we also estab-lished arsenic-induced tumorigenicity in the nude mouse tumorxenograft model. The tumor size was increased 2-fold in arsenictreatment mice compared to control mice without treatment (Fig. 6).The tumor size is dramatically reduced in the mice injected with SODor catalase treated cells compared to the arsenic only treated cells. Thetumor size is even smaller after treatment with SOD or catalasecompared to the control without treatment.
A
B
Rel
pro
mo
ter
acti
vity
R
el p
rom
ote
r ac
tivi
ty
C
D
# # #
#
#
*
*
Fig. 4. ROS are involved in β-catenin activation induced by arsenic. A and B, ROS scavengers reduced β-catenin expression induced by arsenic. A, DLD1 cells were either transfectedwith 20 μg of catalase over-expression vector or empty vector followed by arsenic treatment. B, DLD1 cells were treated 500 U/ml SOD for 1 h prior to arsenic treatment. After 24 h,the cells were lysated. Immunoblotting was conducted to analyze the expression level of β-catenin. C and D, inhibitory effects of ROS scavengers on arsenic induced β-cateninactivity. C, the DLD1 cells were either transfected with 1, 2 and 4 μg of catalase over-expression vector followed by 10 μMof arsenic treatment. D, the DLD1 cells were pretreated with250 or 500 U/ml SOD followed by 10 μM of arsenic treatment. After 24 h, the cells were collected for the luciferase assay measurement. *Indicates a significant increase comparedwith control without arsenic treatment and # indicates a significant decrease compared with arsenic without SOD or catalase treatment (pb0.05).
118 Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
Discussion
Colorectal cancer is one of the most commonly diagnosed cancersin the United States and is responsible for about 53,000 deaths eachyear. Arsenic is a well-documented carcinogen in a number of studies(Waalkes et al., 2004). Chronic exposure to inorganic arsenic fromcontaminated water is responsible for various adverse health effectssuch as developing tumors of the lung, skin, liver, bladder, kidney, andcolon. Studies in arsenic carcinogenesis were first reported in cases ofskin cancer after exposure to inorganic arsenic in drinking water(Galanis et al., 2009; Kligerman et al., 2003). It was also reported thatcolon cancer mortality is associated with the chronic arsenic exposurein drinking water (Yang et al., 2008). A dose level of 72.4 mgmethylarsenous acid (MMA) kg−1 per day led to a thickened wall,necrotic, ulcerated or perforated mucosa in the large intestine and asignificant increase in the incidence of squamous metaplasia of theepithelial columnar absorptive cells in the colon and rectum.Squamous metaplasia was also observed in the colon of micechronically exposed to 67 mg MMA kg−1 per day (Arnold et al.,2003). The data from Kentucky Cancer Registry shows that in southeastern Kentucky, 81% of the 16 counties have a high incidence ofstage 3 and stage 4 colorectal cancers. In these counties, the arsenicconcentrations in drinking water are relatively high compared to EPAstandard. These ecological studies indicate that there is a correlationbetween arsenic level in the environment and colorectal cancer
incidence. However, the exact molecular mechanism of arsenictoxicity and carcinogenesis is still unknown. Current views ofmolecular mechanisms of arsenic toxicity involve genetic changes,the involvement of increased oxidative stress, enhanced cell prolif-eration, and altered gene expression (Jomova and Valko, 2011).
β-catenin regulation is very important for controlled cell prolifera-tion, and deregulated WNT/β-catenin signaling is frequently found incancer cells. In fact, β-catenin is controlled by a protein complexcontaining the tumor suppressor protein adenomatous polyposis coli(APC), casein kinase 1 (CK1), and glycogen synthase kinase-β (GSK-3β)(Barker et al., 2000; Hammond and Giaccia, 2005; Kimelman and Xu,2006). Our results show that arsenic treatment in DLD1 cells increasedboth β-catenin expression level and TCF/LEF transcript activity.Phosphorylation of GSK-3β was also increased after arsenic treatmentin DLD1 cells which are APCmutant with β-catenin wild type (data notshown). However, in the HT116 cells which are APC wild type withβ-catenin mutant, arsenic treatment did not alter phosphorylation ofGSK-3β (data not shown). The results indicate that arsenic inducedactivation of β-catenin is through GSK-3β-dependent pathway. Sincearsenic also caused phosphorylation of p53 (data not shown), it ispossible that arsenic activated β-catenin is controlled by p53 tumorsuppressor. p53 is able to regulateβ-catenin throughSiah-1ubiquitin E3ligase. In addition, a second p53-induced ubiquitin E3 ligase gene,MDM2, induces FOXO degradation (Fu et al., 2009). The present studyalso shows that arsenic treatment increased generations of hydrogen
SOD CAT
B
A
* *
* #
#
*
Fig. 5. ROS are involved in arsenic induced cell transformation. DLD1 cells werecultured in normal growth medium. 0.5 μM of arsenic, 100 U/ml SOD, and 500 U/mlcatalase were added to the culture medium according to different treatment groups.After 3 weeks, soft agar experiment was conducted. The pictures of colony formationwere taken (A) and the numbers of colony were recorded (B). *Indicates a significantdecrease compared with control without SOD or catalase treatment (pb0.05).**Indicates a significant increase compared with control without arsenic treatment(pb0.05); and # indicates a significant decrease compared with arsenic without SOD orcatalase treatment (pb0.05).
Fig. 6. ROS are involved in the induction of tumorigenicity of DLD1 cells by long termarsenic exposure. 6–8 week old nude mice received subcutaneous transplants of 2×106
DLD1 cells. From the second day, mice were randomized into a control group, arsenictreatment group, arsenic plus SOD group, and arsenic plus catalase group. Tumorvolume assessment was conducted as described in “Materials and methods.” *Indicatesa significant increase compared with control without arsenic treatment and # indicatesa significant decrease compared with arsenic without SOD or catalase treatment(pb0.05).
119Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
peroxide (H2O2), hydroxyl radical (•OH), and superoxide radical (O2•−).
Either inhibition of H2O2 and •OH by catalase or inhibition of O2•− by
SOD decreased β-catenin expression level and TCF/LEF activity inducedby arsenic. These results indicate that reactive oxygen species (ROS) areinvolved in the arsenic activated β-catenin.
Members of the Nox family are transmembrane proteins thatcatalyze the NADPH-dependent one-electron reduction of oxygen toform superoxide (Lambeth, 2004). Nox enzymes have been viewed astransmembrane proteins expressed in leukocytes that play a vital role inhost defense against microorganisms (Bokoch and Knaus, 2003; Geisztand Leto, 2004). Nox enzymes differ in both tissue distributions andmechanisms bywhich their activity is regulated (Lambeth, 2007). Nox1,a member of NADPH oxidase family that is highly expressed in colonicepithelial cells, plays a pivotal role in cell signaling, cell growth,angiogenesis, andmotility that integratesWnt/β-catenin signals (Coantet al., 2010). ROS generated via Nox1 have been reported to contributeto a growing number of diseases, including cancer, atherosclerosis,
hypertension, neurological disorders, and inflammation. It has beenreported that phosphorylation of Nox1 induced ROS-dependent coloncancer invasion (Gianni et al., 2010). Wnt/β-catenin and PI3K/AKTcascades might be the major driving force behind the proliferativepotential of neoplastic cells. BecauseNox1 is overexpressed in colorectaladenocarcinoma, it might coordinately drive oncogenic potential. Ourresults show that arsenic increased expressions of p47phox and p67phox,two important subunits of Nox. Addition of SOD or catalase decreasedexpression levels of p47phox and p67phox induced by arsenic. Thatinhibitory effect of SOD and catalase was also observed in arsenicinduced β-catenin activation.
It is believed that ROS play an important role in cancer development,both in the initiation and promotion stages of carcinogenesis (Dong,2002; Huang et al., 2004). At low levels, ROS can modulate geneexpression by acting as secondmessengers, but at high levels they causedirect oxidative injuries leading to cell death (Perkins et al., 2000).Mutagenesis and DNA damage caused by ROS could contribute to theinitiation of cancer. If ROS causes damage to tumor suppressor genes orenhances the expression of proto-oncogenes, the cells may becometumor cells. ROShave beendemonstrated to be involved in the signalingof the cell transformation response (Li et al., 1998) and can affectcytoplasmic and nuclear signal transduction pathways that regulategene expression (Lander, 1997; Sen and Packer, 1996). These effects areimportant in tumor promotion. The mechanism of ROS induced cancerinitiation and promotion may involve the promotion of oxidative stressby arsenic compounds, in which the antioxidant capacity of the livingorganism is overwhelmed by ROS, resulting in molecular damage toproteins, lipids and most significantly DNA (Liu et al., 2001). A studyusing colon cancer HT29 cells has shown that arsenic treatment causedDNA damage, reduced DNA synthesis, resulting in decreased cellproliferation (Stevens et al., 2010). The present study shows thatarsenic treatment increased cell transformation and tumor formation.Treatment with SOD or catalase with arsenic reduced the celltransformation and tumor formation compared to that withoutantioxidant treatment. These results indicate that ROS are in-volved in arsenic induced cell transformation and tumorigenesis inDLD1 cells.

120 Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
It has been reported that a low dose of H2O2 treatment inducedrapid stabilization of β-catenin and a concomitant increase in theexpression of endogenous Wnt target genes (Funato et al., 2006).Paradoxically, it has also been reported that prolonged exposure toROS impairs Wnt/β-catenin signaling (Korswagen, 2006). There is atimeframe difference in those two experiments: H2O2 induces a rapidincrease in Wnt signaling that peaks around 20 min after stimulation,and it is reduced when cells are analyzed several hours later.Oxidative stress modulates the function of β-catenin. Low levels ofoxidative stress stimulate the interaction of β-catenin with FOXOtranscription factors (Essers et al., 2005). Binding of β-catenin toFOXO strongly enhances activation of these transcription factors,leading to a protective response that inhibits cell cycle progressionand allows the cell to deal effectively with the oxidative damage.Increased level of ROS can also stimulate cell proliferation andtransformation (Arnold et al., 2001). This enhanced proliferation is inpart mediated by releasing the NRX dependent inhibition of Wnt/β-catenin signaling. β-catenin may thus be a key regulator thatdetermines whether the cell will proliferate or arrest to repair theoxidative damage. The results from the present study indicate thatROS mediated β-catenin activation contributes to arsenic induced celltransformation and tumorigenesis.
In conclusion, the present study indicates that ROS mediated β-catenin play important role in arsenic induced cell transformation andtumorigenesis in DLD1 cell. Nox is involved in the arsenic inducedcarcinogenesis through generation of ROS.
Conflict of interest
This manuscript will not be submitted to any other scientificjournal prior to the decision by Toxicology and Applied Pharmacology.Each listed author on the manuscript is aware of and agrees to thecontents of the manuscript, including the authorship. None of thelisted authors has any financial or other interests that could be ofconflict.
Acknowledgments
This research is supported by NIH grants R01CA119028,R01CA116697, R01ES015375, and R01ES015518.
References
Arnold, R.S., Shi, J., Murad, E., Whalen, A.M., Sun, C.Q., Polavarapu, R., et al., 2001.Hydrogen peroxide mediates the cell growth and transformation caused by themitogenic oxidase Nox1. Proc. Natl. Acad. Sci. U. S. A. 98 (10), 5550–5555.
Arnold, L.L., Eldan, M., van Gemert, M., Capen, C.C., Cohen, S.M., 2003. Chronic studiesevaluating the carcinogenicity of monomethylarsonic acid in rats and mice.Toxicology 190 (3), 197–219.
Barker, N., Clevers, H., 2006. Mining the Wnt pathway for cancer therapeutics. Nat. Rev.Drug Discov. 5 (12), 997–1014.
Barker, N., Morin, P.J., Clevers, H., 2000. The Yin–Yang of TCF/beta-catenin signaling.Adv. Cancer Res. 77, 1–24.
Benbrahim-Tallaa, L., Waalkes, M.P., 2008. Inorganic arsenic and human prostatecancer. Environ. Health Perspect. 116 (2), 158–164.
Bode, A.M., Dong, Z., 2002. The paradox of arsenic: molecular mechanisms of celltransformation and chemotherapeutic effects. Crit. Rev. Oncol. Hematol. 42 (1),5–24.
Bokoch, G.M., Knaus, U.G., 2003. NADPH oxidases: not just for leukocytes anymore!Trends Biochem. Sci. 28 (9), 502–508.
Bower, J.J., Leonard, S.S., Chen, F., Shi, X., 2006. As(III) transcriptionally activates thegadd45a gene via the formation of H2O2. Free Radic. Biol. Med. 41 (2),285–294.
Brown, D.I., Griendling, K.K., 2009. Nox proteins in signal transduction. Free Radic. Biol.Med. 47 (9), 1239–1253.
Chen, F., Shi, X., 2002a. Signaling from toxic metals to NF-kappaB and beyond: not just amatter of reactive oxygen species. Environ. Health Perspect. 110 (Suppl 5),807–811.
Chen, F., Shi, X., 2002b. Intracellular signal transduction of cells in response tocarcinogenic metals. Crit. Rev. Oncol. Hematol. 42 (1), 105–121.
Chou, W.C., Jie, C., Kenedy, A.A., Jones, R.J., Trush, M.A., Dang, C.V., 2004. Role of NADPHoxidase in arsenic-induced reactive oxygen species formation and cytotoxicity inmyeloid leukemia cells. Proc. Natl. Acad. Sci. U. S. A. 101 (13), 4578–4583.
Coant, N., Ben Mkaddem, S., Pedruzzi, E., Guichard, C., Treton, X., Ducroc, R., et al., 2010.NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate ofproliferative progenitor cells in the colon. Mol. Cell. Biol. 30 (11), 2636–2650.
Ding, W., Hudson, L.G., Liu, K.J., 2005. Inorganic arsenic compounds cause oxidativedamage to DNA and protein by inducing ROS and RNS generation in humankeratinocytes. Mol. Cell. Biochem. 279 (1–2), 105–112.
Ding, W., Hudson, L.G., Sun, X., Feng, C., Liu, K.J., 2008. As(III) inhibits ultravioletradiation-induced cyclobutane pyrimidine dimer repair via generation of nitricoxide in human keratinocytes. Free Radic. Biol. Med. 45 (8), 1065–1072.
Dong, Z., 2002. The molecular mechanisms of arsenic-induced cell transformation andapoptosis. Environ. Health Perspect. 110 (Suppl 5), 757–759.
Essers, M.A., de Vries-Smits, L.M., Barker, N., Polderman, P.E., Burgering, B.M.,Korswagen, H.C., 2005. Functional interaction between beta-catenin and FOXO inoxidative stress signaling. Science 308 (5725), 1181–1184.
Euhus, D.M., Hudd, C., LaRegina, M.C., Johnson, F.E., 1986. Tumor measurement in thenude mouse. J. Surg. Oncol. 31 (4), 229–234.
Fu, W., Ma, Q., Chen, L., Li, P., Zhang, M., Ramamoorthy, S., et al., 2009. MDM2 actsdownstream of p53 as an E3 ligase to promote FOXO ubiquitination anddegradation. J. Biol. Chem. 284 (21), 13987–14000.
Funato, Y., Michiue, T., Asashima, M., Miki, H., 2006. The thioredoxin-related redox-regulating protein nucleoredoxin inhibits Wnt-beta-catenin signalling throughdishevelled. Nat. Cell Biol. 8 (5), 501–508.
Galanis, A., Karapetsas, A., Sandaltzopoulos, R., 2009. Metal-induced carcinogenesis,oxidative stress and hypoxia signalling. Mutat. Res. 674 (1–2), 31–35.
Geiszt, M., Leto, T.L., 2004. The Nox family of NAD(P)H oxidases: host defense andbeyond. J. Biol. Chem. 279 (50), 51715–51718.
Gianni, D., Taulet, N., DerMardirossian, C., Bokoch, G.M., 2010. c-Src-mediatedphosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)-dependent formation of functional invadopodia in human colon cancer cells. Mol.Biol. Cell 21 (23), 4287–4298.
Hammond, E.M., Giaccia, A.J., 2005. The role of p53 in hypoxia-induced apoptosis.Biochem. Biophys. Res. Commun. 331 (3), 718–725.
Harris, G.K., Shi, X., 2003. Signaling by carcinogenic metals and metal-induced reactiveoxygen species. Mutat. Res. 533 (1–2), 183–200.
Huang, C., Ke, Q., Costa, M., Shi, X., 2004. Molecular mechanisms of arseniccarcinogenesis. Mol. Cell. Biochem. 255 (1–2), 57–66.
Huff, J., Chan, P., Nyska, A., 2000. Is the human carcinogen arsenic carcinogenic tolaboratory animals? Toxicol. Sci. 55 (1), 17–23.
Jomova, K., Valko, M., 2011. Advances in metal-induced oxidative stress and humandisease. Toxicology 283 (2–3), 65–87.
Kimelman, D., Xu, W., 2006. beta-Catenin destruction complex: insights and questionsfrom a structural perspective. Oncogene 25 (57), 7482–7491.
Kitchin, K.T., Wallace, K., 2008. The role of protein binding of trivalent arsenicals inarsenic carcinogenesis and toxicity. J. Inorg. Biochem. 102 (3), 532–539.
Klaus, A., Birchmeier, W., 2008. Wnt signalling and its impact on development andcancer. Nat. Rev. Cancer 8 (5), 387–398.
Kligerman, A.D., Doerr, C.L., Tennant, A.H., Harrington-Brock, K., Allen, J.W., Winkfield,E., et al., 2003. Methylated trivalent arsenicals as candidate ultimate genotoxicforms of arsenic: induction of chromosomal mutations but not gene mutations.Environ. Mol. Mutagen. 42 (3), 192–205.
Korinek, V., Barker, N., Morin, P.J., vanWichen, D., deWeger, R., Kinzler, K.W., et al., 1997.Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/−colon carcinoma. Science 275 (5307), 1784–1787.
Korswagen, H.C., 2006. Regulation of theWnt/beta-catenin pathway by redox signaling.Dev. Cell 10 (6), 687–688.
Ladelfa, M.F., Toledo, M.F., Laiseca, J.E., Monte, M., 2011. Interaction of p53 with tumorsuppressive and oncogenic signaling pathways to control cellular reactive oxygenspecies production. Antioxid. Redox Signal.
Lambeth, J.D., 2004. NOX enzymes and the biology of reactive oxygen. Nat. Rev.Immunol. 4 (3), 181–189.
Lambeth, J.D., 2007. Nox enzymes, ROS, and chronic disease: an example of antagonisticpleiotropy. Free Radic. Biol. Med. 43 (3), 332–347.
Lander, H.M., 1997. An essential role for free radicals and derived species in signaltransduction. FASEB J. 11 (2), 118–124.
Leonard, S.S., Harris, G.K., Shi, X., 2004. Metal-induced oxidative stress and signaltransduction. Free Radic. Biol. Med. 37 (12), 1921–1942.
Li, J.J., Oberley, L.W., Fan, M., Colburn, N.H., 1998. Inhibition of AP-1 and NF-kappaB bymanganese-containing superoxide dismutase in human breast cancer cells. FASEB J.12 (15), 1713–1723.
Li, Y., Bavarva, J.H., Wang, Z., Guo, J., Qian, C., Thibodeau, S.N., et al., 2011. HEF1, a noveltarget of Wnt signaling, promotes colonic cell migration and cancer progression.Oncogene.
Liu, S.X., Athar, M., Lippai, I., Waldren, C., Hei, T.K., 2001. Induction of oxyradicals byarsenic: implication for mechanism if genotoxicity. Proc. Natl. Acad. Sci. U.S.A. 98(4), 1643–1648.
MacDonald, B.T., Tamai, K., He, X., 2009. Wnt/beta-catenin signaling: components,mechanisms, and diseases. Dev. Cell 17 (1), 9–26.
Morin, P.J., Sparks, A.B., Korinek, V., Barker, N., Clevers, H., Vogelstein, B., et al., 1997.Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275 (5307), 1787–1790.
Perkins, C., Kim, C.N., Fang, G., Bhalla, K.N., 2000. Arsenic induces apoptosis ofmultidrug-resistant human myeloid leukemia cells that express Bcr-Abl oroverexpress MDR, MRP, Bcl-2, or Bcl-x(L). Blood 95 (3), 1014–1022.

121Z. Zhang et al. / Toxicology and Applied Pharmacology 256 (2011) 114–121
Polakis, P., 2000. Wnt signaling and cancer. Genes Dev. 14 (15), 1837–1851.Qian, Y., Liu, K.J., Chen, Y., Flynn, D.C., Castranova, V., Shi, X., 2005. Cdc42 regulates
arsenic-induced NADPH oxidase activation and cell migration through actinfilament reorganization. J. Biol. Chem. 280 (5), 3875–3884.
Reya, T., Clevers, H., 2005. Wnt signalling in stem cells and cancer. Nature 434 (7035),843–850.
Segditsas, S., Tomlinson, I., 2006. Colorectal cancer and genetic alterations in the Wntpathway. Oncogene 25 (57), 7531–7537.
Sen, C.K., Packer, L., 1996. Antioxidant and redox regulation of gene transcription.FASEB J. 10 (7), 709–720.
Shi, H., Hudson, L.G., Liu, K.J., 2004a. Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic. Biol. Med. 37 (5), 582–593.
Shi, H., Hudson, L.G., Ding,W.,Wang, S., Cooper, K.L., Liu, S., et al., 2004b. Arsenite causesDNA damage in keratinocytes via generation of hydroxyl radicals. Chem. Res.Toxicol. 17 (7), 871–878.
Stevens, J.J., Graham, B., Walker, A.M., Tchounwou, P.B., Rogers, C., 2010. The effects ofarsenic trioxide on DNA synthesis and genotoxicity in human colon cancer cells. Int.J. Environ. Res. Public Health 7 (5), 2018–2032.
Tchounwou, P.B., Patlolla, A.K., Centeno, J.A., 2003. Carcinogenic and systemic healtheffects associated with arsenic exposure—a critical review. Toxicol. Pathol. 31 (6),575–588.
Tomayko, M.M., Reynolds, C.P., 1989. Determination of subcutaneous tumor size inathymic (nude) mice. Cancer Chemother. Pharmacol. 24 (3), 148–154.
van der Flier, L.G., Clevers, H., 2009. Stem cells, self-renewal, and differentiation in theintestinal epithelium. Annu. Rev. Physiol. 71, 241–260.
Waalkes, M.P., Liu, J., Ward, J.M., Diwan, B.A., 2004. Mechanisms underlying arseniccarcinogenesis: hypersensitivity of mice exposed to inorganic arsenic duringgestation. Toxicology 198 (1–3), 31–38.
Yang, C.Y., Chang, C.C., Ho, S.C., Chiu, H.F., 2008. Is colon cancer mortality related toarsenic exposure? J. Toxicol. Environ. Health A 71 (8), 533–538.





























