Embed Size (px)
Citation preview

Sequence Comparison
Intragenic - self to self.-find internal repeating units.
Intergenic -compare two different sequences.
Dotplot - visual alignment of two sequences
Multiple Sequence Alignment -Two or more sequences

OverviewOverview
Why compare sequencesWhy compare sequences Homology vs. identity/similarityHomology vs. identity/similarity DotPlotsDotPlots ScoringScoring
MatchMatch MismatchMismatch Gap penalityGap penality
Global vs. local alignmentGlobal vs. local alignment Do the results make biological sense?Do the results make biological sense?

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences Identify elements that repeat in a single Identify elements that repeat in a single
sequence.sequence.

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences Identify elements that repeat in a single Identify elements that repeat in a single
sequence.sequence. Identify elements conserved between genes.Identify elements conserved between genes.

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences Identify elements that repeat in a single Identify elements that repeat in a single
sequence.sequence. Identify elements conserved between genes.Identify elements conserved between genes. Identify elements conserved between species.Identify elements conserved between species.

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences Identify elements that repeat in a single Identify elements that repeat in a single
sequence.sequence. Identify elements conserved between genes.Identify elements conserved between genes. Identify elements conserved between species.Identify elements conserved between species.
• Regulatory elementsRegulatory elements

Why Align SequencesWhy Align Sequences
Identify conserved sequencesIdentify conserved sequences Identify elements that repeat in a single Identify elements that repeat in a single
sequence.sequence. Identify elements conserved between genes.Identify elements conserved between genes. Identify elements conserved between species.Identify elements conserved between species.
• Regulatory elementsRegulatory elements• Functional elementsFunctional elements

Underlying Underlying Hypothesis?Hypothesis?

Underlying Underlying Hypothesis?Hypothesis?
EVOLUTIONEVOLUTION

Underlying Underlying Hypothesis?Hypothesis?
EVOLUTIONEVOLUTION
Based upon conservation of Based upon conservation of sequence during evolution we can sequence during evolution we can
infer function.infer function.

Basic terms:Basic terms:
SimilaritySimilarity - measurable quantity. - measurable quantity. Similarity- applied to proteins using concept of Similarity- applied to proteins using concept of
conservative substitutionsconservative substitutions IdentityIdentity percentagepercentage
HomologyHomology-specific term indicating -specific term indicating relationship by evolutionrelationship by evolution

Basic terms:Basic terms:
Orthologs: homologous sequences found Orthologs: homologous sequences found in in two or moretwo or more species, that have the species, that have the same function (i.e. alpha- hemoglobin).same function (i.e. alpha- hemoglobin).

Basic terms:Basic terms:
Orthologs: homologous sequences found Orthologs: homologous sequences found it it two or moretwo or more species, that have the species, that have the same function (i.e. alpha- hemoglobin).same function (i.e. alpha- hemoglobin).
Paralogs: homologous sequences found in Paralogs: homologous sequences found in the the samesame species that arose by gene species that arose by gene duplication. ( alpha and beta hemoglobin).duplication. ( alpha and beta hemoglobin).

Pairwise comparisonPairwise comparison
DotplotDotplot All against all comparison.All against all comparison.
• Every position is compared with every other Every position is compared with every other position.position.

Pairwise comparisonPairwise comparison
DotplotDotplot All against all comparison.All against all comparison.
• Every position is compared with every other Every position is compared with every other position.position.
• Nucleic acids and proteins have polarity.Nucleic acids and proteins have polarity.

Pairwise comparisonPairwise comparison
DotplotDotplot All against all comparison.All against all comparison.
• Every position is compared with every other Every position is compared with every other position.position.
• Nucleic acids and proteins have polarity.Nucleic acids and proteins have polarity.• Typically only one direction makes biological Typically only one direction makes biological
sense. sense.

Pairwise comparisonPairwise comparison
DotplotDotplot All against all comparison.All against all comparison.
• Every position is compared with every other Every position is compared with every other position.position.
• Nucleic acids and proteins have polarity.Nucleic acids and proteins have polarity.• Typically only one direction makes biological Typically only one direction makes biological
sense. sense. 5’ to 3’ or amino terminus to carboxyl terminus.5’ to 3’ or amino terminus to carboxyl terminus.

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT
.

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT
..

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT
... .

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT
... ..

DotPlotDotPlot
Dotplot- matrix, with one sequence across Dotplot- matrix, with one sequence across top, other down side. Put a dot, or 1, top, other down side. Put a dot, or 1, where ever there is identity.where ever there is identity.
G A T C T
GATCT
... ... .

G A T A C T G C G A T A C T G C G C AG 1 1 1 1 1A 1 1 1 1 1T 1 1 1 1 1A 1 1 1 1C 1 1 1 1 1T 1 1 1 1G 1 1 1 1C 1 1 1 1G 1 1 1A 1 1 1T 1 1A 1C 1 1 1T 1G 1 1C 1 1G 1C 1A 1

Simple plotSimple plot
Window: size of sequence block used for Window: size of sequence block used for comparison. In previous example:comparison. In previous example: window = 1window = 1
Stringency = Number of matches required Stringency = Number of matches required to score positive. In previous example:to score positive. In previous example: stringency = 1 (required exact match)stringency = 1 (required exact match)
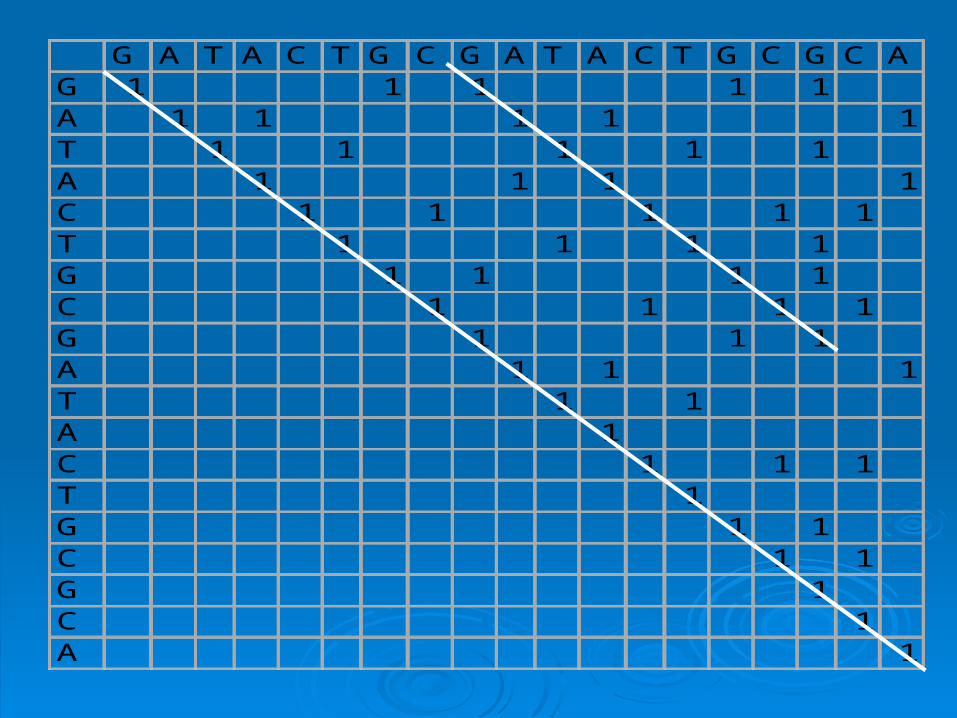
G A T A C T G C G A T A C T G C G C AG 1 1 1 1 1A 1 1 1 1 1T 1 1 1 1 1A 1 1 1 1C 1 1 1 1 1T 1 1 1 1G 1 1 1 1C 1 1 1 1G 1 1 1A 1 1 1T 1 1A 1C 1 1 1T 1G 1 1C 1 1G 1C 1A 1

G A T A C T G C A T C G T C A C T C AG 1 1 1A 1 1 1 1 1T 1 1 1 1 1A 1 1 1 1C 1 1 1 1 1 1T 1 1 1 1G 1 1C 1 1 1 1 1A 1 1 1T 1 1 1C 1 1 1 1G 1T 1 1C 1 1 1A 1 1C 1 1T 1C 1A 1
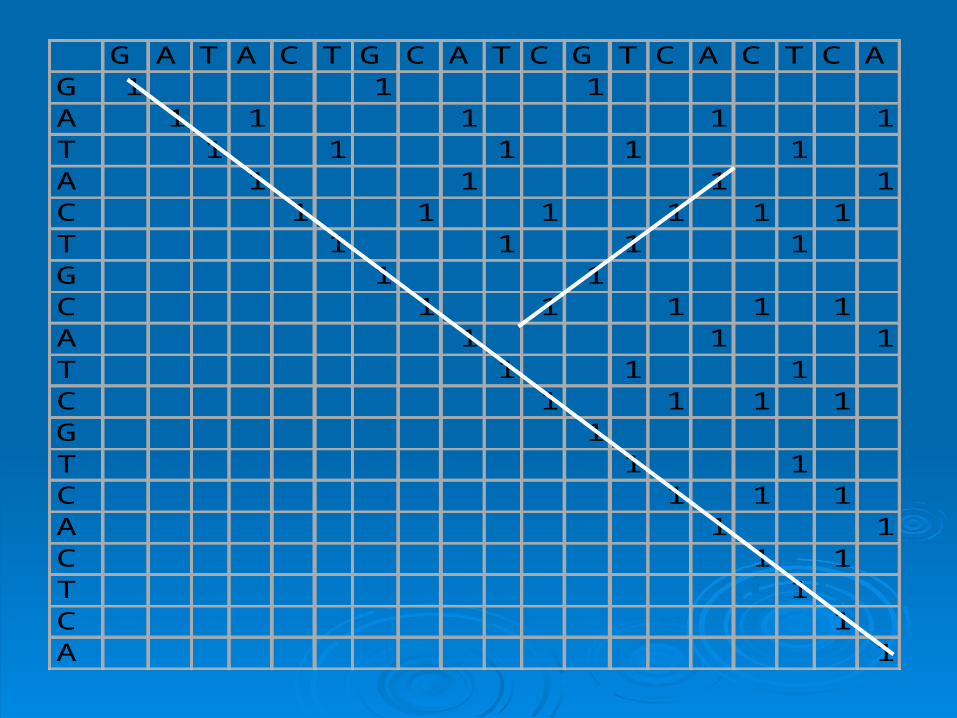
G A T A C T G C A T C G T C A C T C AG 1 1 1A 1 1 1 1 1T 1 1 1 1 1A 1 1 1 1C 1 1 1 1 1 1T 1 1 1 1G 1 1C 1 1 1 1 1A 1 1 1T 1 1 1C 1 1 1 1G 1T 1 1C 1 1 1A 1 1C 1 1T 1C 1A 1

Dot PlotDot Plot
Compare two sequences in every Compare two sequences in every register.register.
Vary size of window and stringency Vary size of window and stringency depending upon sequences being depending upon sequences being compared.compared.
For nucleotide sequences typically start For nucleotide sequences typically start with window = 21; stringency = 14with window = 21; stringency = 14

GATCGTACCATGGAATCGTCCAGATCAGATC + (4/4)
GATCGATC
GATC - (0/4)
- (0/4)+ (2/4)
WINDOW = 4; STRINGENCY = 2
DotPlot

G A T C G T A C C A T G G A T C G T C A G A TG * * * * * * *
A * * * * * *T * * * *C *G *T *A *C *C *A *T *G *G *A *T *C *G *T *C *A *G *A *T *
This “match” from G and C out of the four
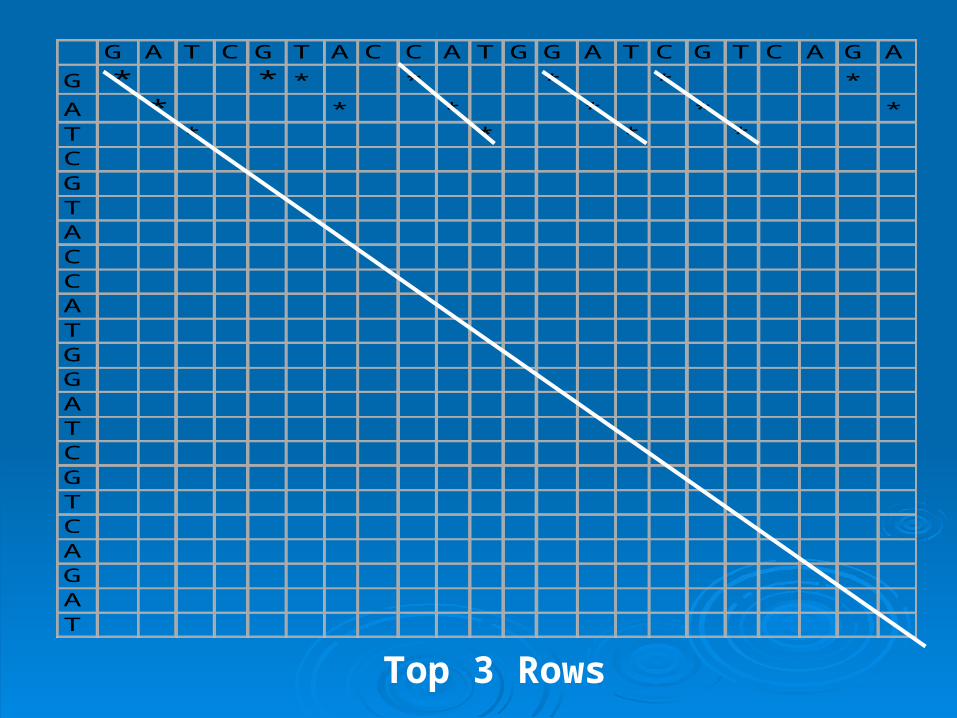
G A T C G T A C C A T G G A T C G T C A G A
G * * * * * * *
A * * * * * *T * * * *CGTACCATGGATCGTCAGAT
Top 3 Rows

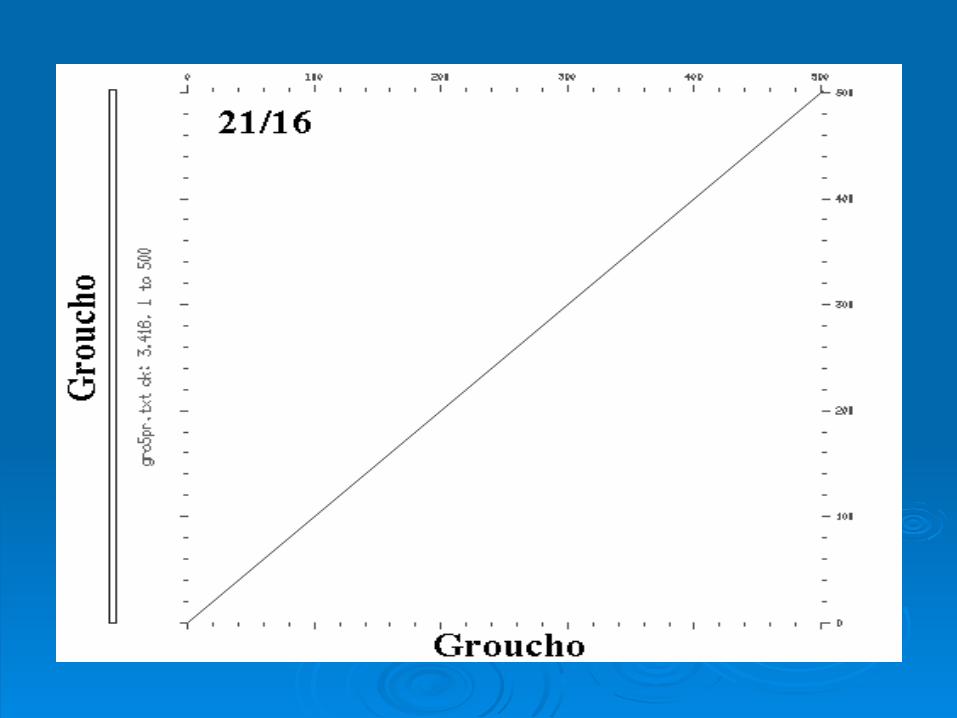
Intragenic ComparisonIntragenic Comparison
Rat Groucho Gene Rat Groucho Gene
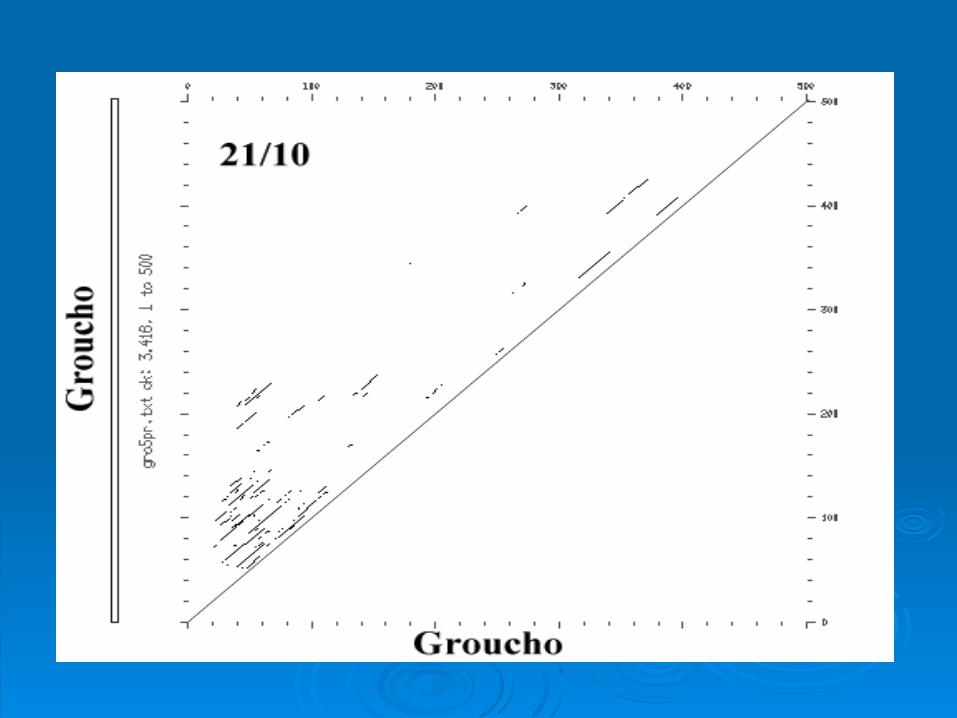
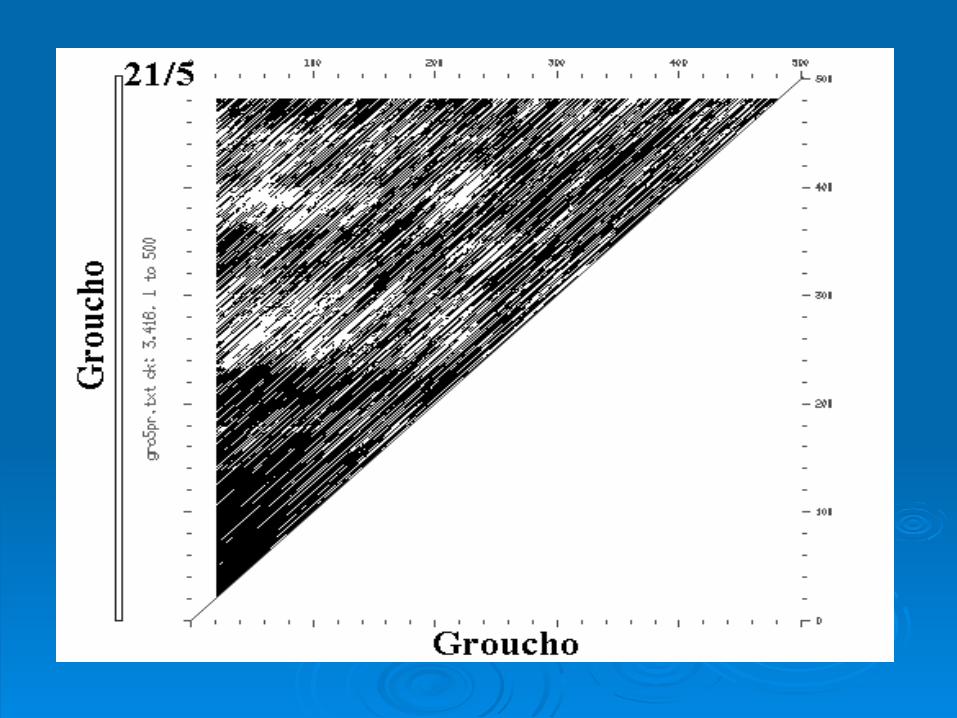

Intergenic ComparisonIntergenic Comparison
Rat and Drosophila Groucho Rat and Drosophila Groucho GeneGene
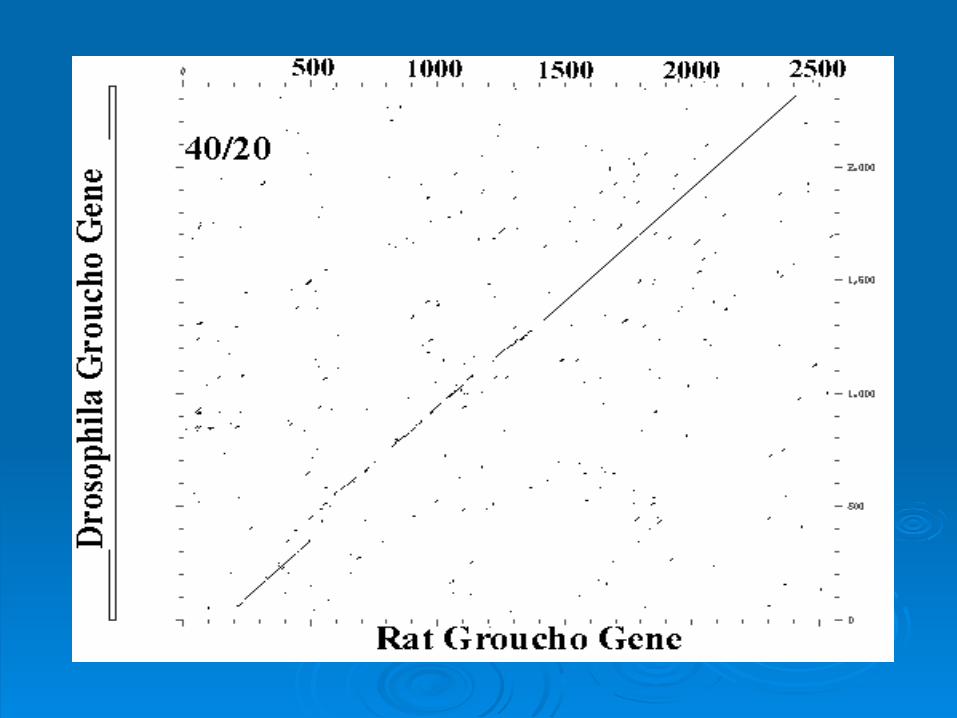
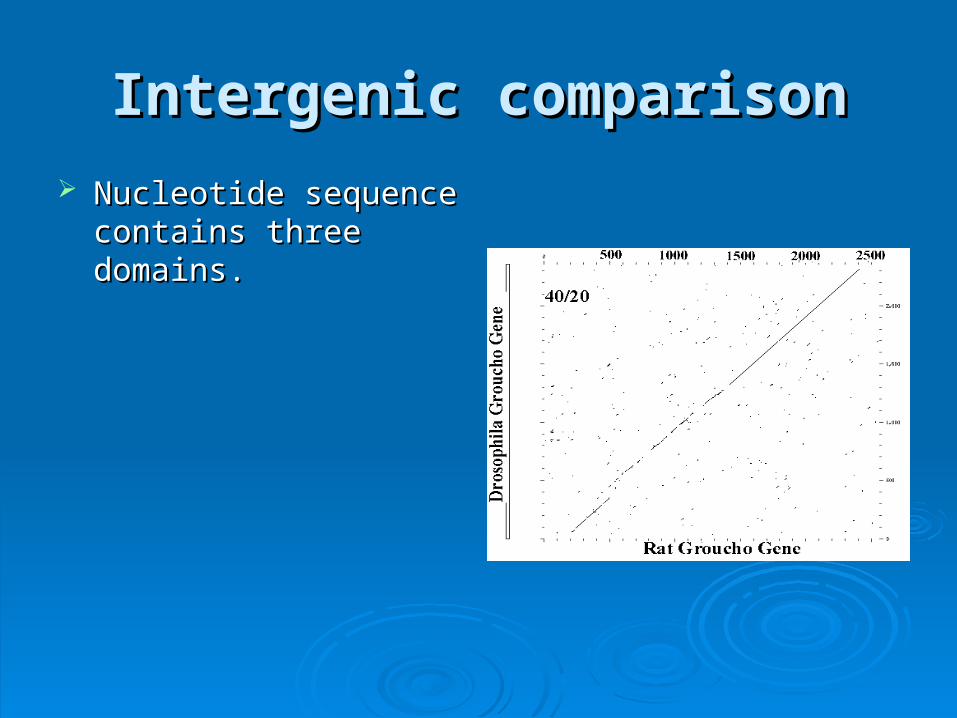
Intergenic comparisonIntergenic comparison
Nucleotide sequence Nucleotide sequence contains three domains.contains three domains.
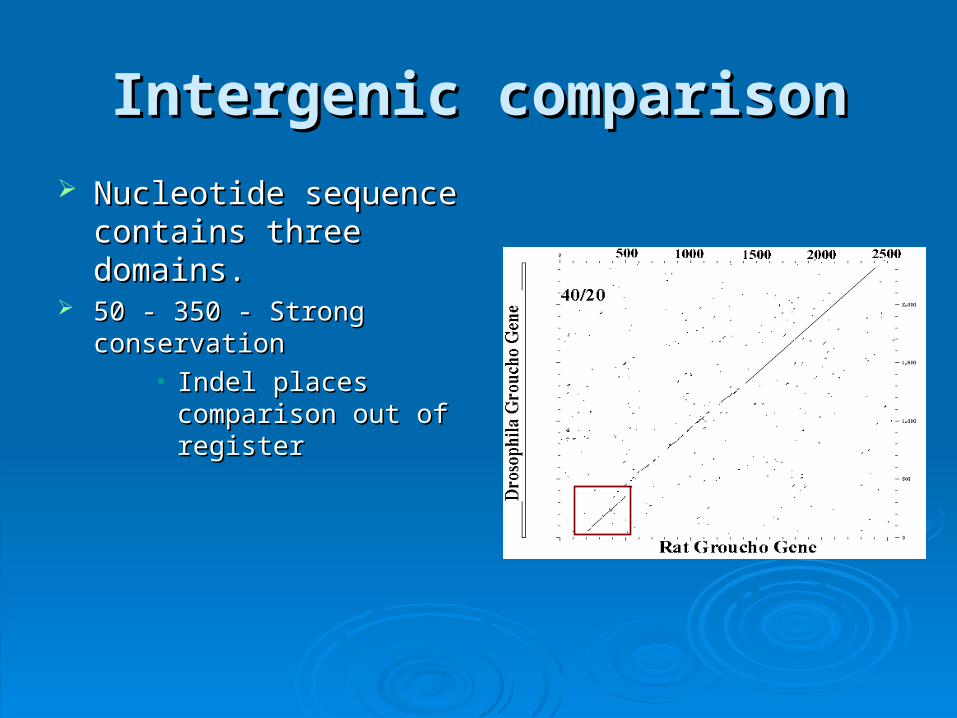
Intergenic comparisonIntergenic comparison
Nucleotide sequence Nucleotide sequence contains three domains.contains three domains.
50 - 350 - Strong conservation50 - 350 - Strong conservation• Indel places comparison Indel places comparison
out of registerout of register
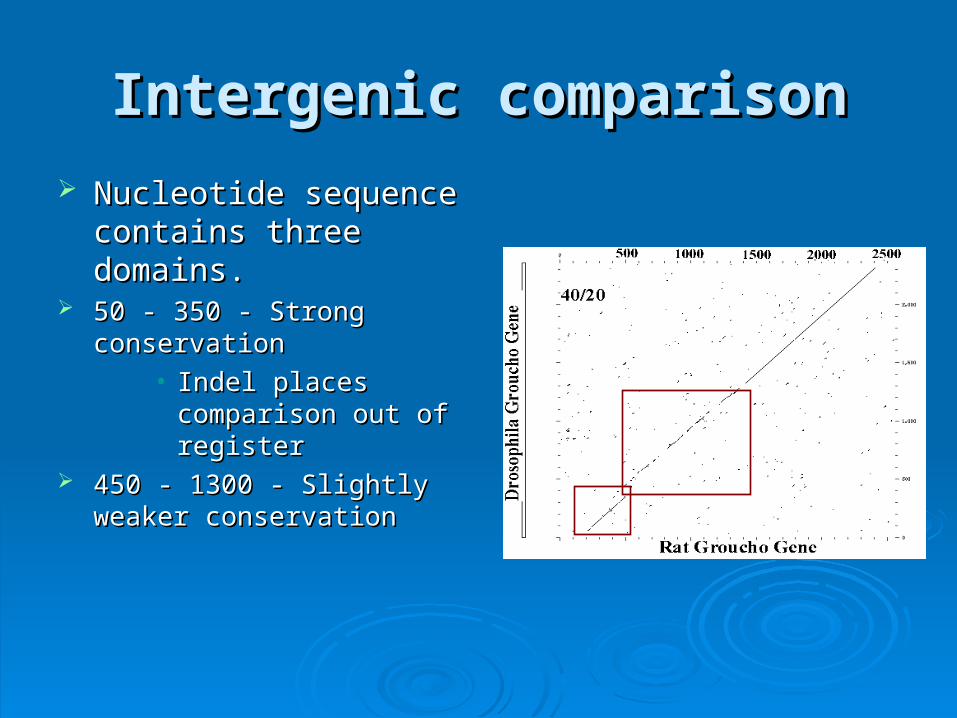
Intergenic comparisonIntergenic comparison
Nucleotide sequence Nucleotide sequence contains three domains.contains three domains.
50 - 350 - Strong conservation50 - 350 - Strong conservation• Indel places comparison Indel places comparison
out of registerout of register 450 - 1300 - Slightly weaker 450 - 1300 - Slightly weaker
conservationconservation

Intergenic comparisonIntergenic comparison
Nucleotide sequence Nucleotide sequence contains three domains.contains three domains.
50 - 350 - Strong conservation50 - 350 - Strong conservation• Indel places comparison Indel places comparison
out of registerout of register 450 - 1300 - Slightly weaker 450 - 1300 - Slightly weaker
conservationconservation 1300 - 2400 - Strong 1300 - 2400 - Strong
conservationconservation

GrouchoGroucho
These three coding regions correspond to These three coding regions correspond to apparent functional domains of the apparent functional domains of the encoded proteinencoded protein

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: : Score x for match, -y for mismatch; Score x for match, -y for mismatch;

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: : Score x for match, -y for mismatch; Score x for match, -y for mismatch;
• Penalty for:Penalty for: Creating GapCreating Gap Extending a gapExtending a gap

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: :
QualityQuality = [10(match)] = [10(match)]

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: :
QualityQuality = [10(match)] + [-1(mismatch)] = [10(match)] + [-1(mismatch)]

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: :
QualityQuality = [10(match)] + [-1(mismatch)] - = [10(match)] + [-1(mismatch)] -
[(Gap Creation Penalty)(#of Gaps)[(Gap Creation Penalty)(#of Gaps)

Scoring AlignmentsScoring Alignments
Quality ScoreQuality Score: :
QualityQuality = [10(match)] + [-1(mismatch)] - = [10(match)] + [-1(mismatch)] -
[(Gap Creation Penalty)(#of Gaps) +(Gap Ext. Pen.)(Total [(Gap Creation Penalty)(#of Gaps) +(Gap Ext. Pen.)(Total length of Gaps)]length of Gaps)]

Z Score (standardized score)Z Score (standardized score)
Z = (ScoreZ = (Scorealignmentalignment - Average Score - Average Scorerandomrandom))
Standard Deviationrandom

Quality Score:Randomization•Program takes sequence and randomizes it X times (user select).•Determines average quality score and standard
deviation with randomized sequences•Compare randomized scores with Quality score to help determine if alignment is potentially significant.

RandomizationRandomization It has become clear thatIt has become clear that
Sequences appear to evolve in a Sequences appear to evolve in a “word” like fashion.“word” like fashion.• 26 letters of the alphabet--combined to 26 letters of the alphabet--combined to
make words. make words. • Words actually communicate information.Words actually communicate information.
Randomization should actually occur at Randomization should actually occur at the level of strings of nucleotides (2-4). the level of strings of nucleotides (2-4).

Global AlignmentGlobal Alignment
Global - Compares all possible Global - Compares all possible alignments of two sequences and alignments of two sequences and presents the presents the one with the greatest one with the greatest number of matches and the fewest number of matches and the fewest gapsgaps. .

Global AlignmentGlobal Alignment
Global - Compares all possible Global - Compares all possible alignments of two sequences and alignments of two sequences and presents the presents the one with the greatest one with the greatest number of matches and the fewest number of matches and the fewest gapsgaps..
Alignment will “run” from one end of the Alignment will “run” from one end of the longest sequence, to the other end. longest sequence, to the other end.

Global AlignmentGlobal Alignment
Global - Compares all possible Global - Compares all possible alignments of two sequences and alignments of two sequences and presents the presents the one with the greatest one with the greatest number of matches and the fewest number of matches and the fewest gapsgaps..
Alignment will “run” from one end of the Alignment will “run” from one end of the longest sequence, to the other end. longest sequence, to the other end.
Best for closely related sequences.Best for closely related sequences.

Global AlignmentGlobal Alignment
Global - Compares all possible alignments of Global - Compares all possible alignments of two sequences and presents the two sequences and presents the one with the one with the greatest number of matches and the fewest greatest number of matches and the fewest gapsgaps..
Alignment will “run” from one end of the Alignment will “run” from one end of the longest sequence, to the other end. longest sequence, to the other end.
Best for closely related sequences.Best for closely related sequences. Can miss short regions of strongly conserved Can miss short regions of strongly conserved
sequence. sequence.

Local AlignmentLocal Alignment
Identifies segments of alignment with the Identifies segments of alignment with the highest possible score.highest possible score.

Local AlignmentLocal Alignment
Identifies segments of alignment with the Identifies segments of alignment with the highest possible score.highest possible score.
Align sequences, extends aligned regions in Align sequences, extends aligned regions in both directions until score falls to zero.both directions until score falls to zero.

Local AlignmentLocal Alignment
Identifies segments of alignment with the highest Identifies segments of alignment with the highest possible score.possible score.
Align sequences, extends aligned regions in both Align sequences, extends aligned regions in both directions until score falls to zerodirections until score falls to zero..
Best for comparing sequences whose relationship is Best for comparing sequences whose relationship is unknown.unknown.

Global Alignment:
Local Alignment:

Blast 2
Basic Local Alignment Search Tool
E (expect) valueE (expect) value: number of hits expected by randomchance in a database of same size.
Larger numerical value = lower significance
HIV sequence

Both Global (Gap) and Local (Bestfit) tools will Both Global (Gap) and Local (Bestfit) tools will (almost) (almost) alwaysalways give a match. give a match.

Both Global (Gap) and Local (Bestfit) tools will Both Global (Gap) and Local (Bestfit) tools will (almost) (almost) alwaysalways give a match. give a match.
It is important to determine if the match is It is important to determine if the match is biologically relevant.biologically relevant.

Both Global (Gap) and Local (Bestfit) tools will Both Global (Gap) and Local (Bestfit) tools will (almost) (almost) alwaysalways give a match. give a match.
It is important to determine if the match is It is important to determine if the match is biologically relevant.biologically relevant.
Not necessarily relevant: Low complexity Not necessarily relevant: Low complexity regions.regions. Sequence repeats (glutamine runs)Sequence repeats (glutamine runs)

Both Global (Gap) and Local (Bestfit) tools will Both Global (Gap) and Local (Bestfit) tools will (almost) (almost) alwaysalways give a match. give a match.
It is important to determine if the match is It is important to determine if the match is biologically relevant.biologically relevant.
Not necessarily relevant: Low complexity Not necessarily relevant: Low complexity regions.regions. Sequence repeats (glutamine runs)Sequence repeats (glutamine runs) Transmembrane regions (high in hydrophobes)Transmembrane regions (high in hydrophobes)

Both Global (Gap) and Local (Bestfit) tools will Both Global (Gap) and Local (Bestfit) tools will (almost) (almost) alwaysalways give a match. give a match.
It is important to determine if the match is It is important to determine if the match is biologically relevant.biologically relevant.
Not necessarily relevant: Low complexity Not necessarily relevant: Low complexity regions.regions. Sequence repeats (glutamine runs)Sequence repeats (glutamine runs) Transmembrane regions (high in hydrophobes)Transmembrane regions (high in hydrophobes)
If working with coding regions, you are If working with coding regions, you are typically better off typically better off comparing proteincomparing protein sequencessequences. Greater information content.. Greater information content.





























