Embed Size (px)
Citation preview

Ministério da Saúde Instituto Nacional de Câncer Coordenação de Pós-graduação
INSTITUTO NACIONAL DE CÂNCER Pós-Graduação em Oncologia
SHEILA COELHO SOARES LIMA
ALTERAÇÕES EPIGENÉTICAS E SUAS CONSEQUÊNCIAS FUNCIONAIS EM CARCINOMA
EPIDERMÓIDE DE ESÔFAGO
Orientador(es): Prof. Dr. Luis Felipe Ribeiro Pinto Prof. Dr. Zdenko Herceg
RIO DE JANEIRO 2012

ii
Ministério da Saúde Instituto Nacional de Câncer Coordenação de Pós-graduação
INSTITUTO NACIONAL DE CÂNCER Pós-Graduação em Oncologia
SHEILA COELHO SOARES LIMA
ALTERAÇÕES EPIGENÉTICAS E SUAS CONSEQUÊNCIAS FUNCIONAIS EM CARCINOMA
EPIDERMÓIDE DE ESÔFAGO
Tese apresentada ao Instituto Nacional de Câncer como parte dos requisitos para obtenção do título de Doutor em Oncologia.
Orientador(es): Prof. Dr. Luis Felipe Ribeiro Pinto Prof. Dr. Zdenko Herceg
RIO DE JANEIRO 2012

iii
Ficha Catalográfica
L732a Lima, Sheila Coelho Soares
Alterações epigenéticas e suas consequências funcionais em carcinoma epidermóide de esôfago /
Sheila Coelho Soares Lima. - Rio de Janeiro, 2012.
de . -
xvii, 442f. : il.
Tese (Doutorado em Oncologia) – Instituto Nacional de Câncer José Alencar Gomes da Silva – INCA,
2012.
Orientadores: Luis Felipe Ribeiro Pinto.
Zdenko Herceg.
1.Neoplasias esofágicas. 2. Carcinoma de células escamosas. 3. Metilação de DNA. 4. Diferenciação
celular. 5. Dissertações acadêmicas. I. Pinto, Luis Felipe Ribeiro (Orient.). II. Herceg, Zdenko (Orient.).
III. Instituto Nacional de Câncer José Alencar Gomes da Silva. IV. Título.
CDD-616.99432
CDD-616.994

iv
Ministério da Saúde Instituto Nacional de Câncer Coordenação de Pós-graduação
INSTITUTO NACIONAL DE CÂNCER Pós-Graduação em Oncologia
SHEILA COELHO SOARES LIMA
ALTERAÇÕES EPIGENÉTICAS E SUAS CONSEQUÊNCIAS FUNCIONAIS EM CARCINOMA
EPIDERMÓIDE DE ESÔFAGO
Orientador(es): Prof. Dr. Luis Felipe Ribeiro Pinto Prof. Dr. Zdenko Herceg
Aprovada em: 06/03/2012
EXAMINADORES: Prof. Dr. João Paulo de Biaso Viola - Presidente Prof. Dr. Miguel Ângelo Martins Moreira Prof. Dr. Milton Ozório Moraes Prof. Dr. Verônica Maria Morando da Silva Prof. Dr. Hector Nicolas Seuánez Abreu - Suplente I Prof. Dr. Thereza Christina Barja Fidalgo - Suplente II
RIO DE JANEIRO 2012

v
À minha avó e à minha mãe,
as mulheres mais guerreiras que já conheci.

vi
Agradecimentos
- Ao Prof. Dr. Luis Felipe Ribeiro Pinto, por ter sido fundamental em minha formação científica, tendo estado ao meu lado desde a iniciação até o doutorado. Sob sua orientação, aprendi não só o significado de ser pesquisador, como também tirei lições para a vida inteira! Muito obrigada! - To Dr. Zdenko Herceg, for receiving me so well in his lab and for making possible my PhD. I learned a lot during these two years and a half and I hope the collaboration will continue. Thank you! To Dr Héctor Hernandez-Vargas, thank you for all the discussion, the supervision and, of course, the beers! I’m sure my thesis wouldn’t have been the same without you! Thank you! - Aos Prof. Dr. Ana Rossini e Rodolpho Albano por estarem sempre dispostos a ajudar. - À Dra. Tatiana de Almeida Simão (Tatí, para os íntimos), por ter estado por perto desde o início de tudo, me ajudando desde a fazer uma reação de PCR funcionar, passando pelo PCR em tempo real, até a era dos microarranjos. Com certeza, tudo ficou mais fácil e mais divertido com você ao meu lado! Porém queria agradecer acima de tudo pela amizade, nada é mais importante nesse meio tão competitivo! - À Dra Nathalia Meireles da Costa (Nath), por ter sido fundamental principalmente durante a minha estadia em Lyon. Sua ajuda em TUDO e o início de uma grande amizade foram muito importantes para mim! Obrigada, gata! - Ao Dr Davy Carlos Mendes Rapozo, por ter sido, sem sombra de dúvida, a pessoa que mais me ligou quando eu estava em Lyon! Brincadeiras à parte, seu apoio e ajuda desde os primeiros momentos no laboratório, assim como a sua amizade, me ajudaram a chegar onde estou hoje. Muito obrigada! - A todos os membros do meu laboratório no Brasil: Adilson, Alda, Andreia, Carolina, Cíntia, Claudia, Daniel, Diego, Elisa, Ester, Isabela, Janaina, Juliana, Marina, Monique, Nina, Paulo Thiago, Pedro Bello, Pedro Nicolau, Vagner, Wagner e Vanessa. Muito obrigada pela ajuda e pelo apoio mesmo de longe! - To everybody from my lab in Lyon: Anastas, Anupam, Asiedua, Carla, Clément, Cyrille, Gabriel, Ho-Sun, Karen, Maria, Marie-Pierre Cros, Marie-Pierre Lambert, Marion, Meabh, Pushpinder, Samson, Thomas, Vasily, and Vladimir. Thank you for all the help and the good moments we spent together! - A special thanks to Cyrille, Maria, Marie-Pierre Cros, Marie-Pierre Lambert, Marion and Meabh for helping me in a day-by-day basis in all experiments and discussions, sharing all the good and bad moments. - A todos os membros da banca avaliadora desta tese por aceitarem o convite de avaliar este trabalho.

vii
- Aos médicos Adami Andreollo (GASTC/UNICAMP), Rodolfo Acatauassu (HUPE/UERJ) e Kleber Kruel (HC/UFRS) pela coleta das biópsias. Sem este material o trabalho não teria sido realizado. - A todos os pacientes e suas famílias por aceitarem fazer parte deste trabalho, mesmo em um momento tão difícil. - Ao Dr Kunitada Shimotohno da Kyoto University pela doação do vetor de expressão contendo o gene BCL3. - To Dr Florence Le Calvez-Kelm and Geoffroy Durand from the Genetic Cancer Susceptibility Group – IARC for helping in the bead array analysis. - Aos meus pais, por me apoiarem SEMPRE, mesmo quando as dificuldades foram grandes. Estar longe de vocês não foi fácil, mas saber que vocês estariam sempre ao meu lado, me ajudou a superar as dificuldades. - Ao meu irmão não só por me apoiar 100% nas minhas escolhas, mas também por torná-las possíveis. A segurança que você me ofereceu no período que passei fora foi fundamental para que meu trabalho pudesse ser realizado! - À minha grande amiga e irmã Rê. Seu apoio, sua confiança em meu potencial e sua amizade são essenciais em minha vida. Saber que você está sempre ao meu lado e que nossa amizade não se enfraqueceria foram muito importantes para o desafio que enfrentei durante meu doutorado. Obrigada por TUDO! - À minha grande amiga Lu. As horas no telefone para contar fofocas, treinar apresentações e, principalmente, desabafar foram fundamentais durante o tempo que passei em Lyon. Sem você, amiga linda, não sei como suportaria a distância de todos. Obrigada por estar sempre por perto, mesmo longe! - To Marion for being such a good friend, helping me in all aspects from French burocratic stuff to home-sickness. I’m glad I had you by my side during the period I stayed in Lyon and I hope our friendship will last for life! - À pessoa que mais me apoiou, mesmo quando isso significou colocar nosso relacionamento em risco. Pablinho, você foi fundamental durante o meu doutorado, assim como é na minha vida! Ter seu apoio, sua compreensão e seu amor me ajudam a seguir adiante. Amo você demais da conta!

viii
Índice
Página
Lista de quadros..........................................................................................................xi Lista de figuras................................................................................. ...........................xii Lista de siglas e abreviaturas....................................................................................xiv Resumo..................................................................................................... .................xvi Abstract.....................................................................................................................xvii INTRODUÇÃO GERAL................................................................................................1 1. Câncer................................................................................................................1 2. Epigenética.........................................................................................................5 2.1. Metilação do DNA.......................................................................................5 2.2. Modificações de Histonas...........................................................................7 2.3. miRNAs.......................................................................................................8 OBJETIVOS...............................................................................................................11 CAPÍTULO I: Alterações em metilação do DNA em carcinoma epidermóide de esôfago......................................................................................................................12 INTRODUÇÃO...........................................................................................................12 1. Câncer de esôfago................................................................................................12
1.1. Alterações Genéticas em CEE.......................................................................13 1.2. Alterações Epigenéticas em CEE...................................................................15
ARTIGO: Identification of a DNA Methylome Signature of Esophageal Squamous Cell Carcinoma and Potential Epigenetic Biomarkers......................17 DISCUSSÃO..............................................................................................................33 CAPÍTULO II: Alterações epigenéticas no gene BCL3 e suas consequências funcionais.................................................................................................................39 INTRODUÇÃO...........................................................................................................39
1. BCL3: um membro diferenciado da família IB...............................................39 2. BCL3, proliferação e sobrevivência celular.....................................................40 3. Estrutura e regulação de BCL3.......................................................................42
MATERIAL E MÉTODOS..........................................................................................44 1. Linhagens celulares..............................................................................................44
1.1. Ensaio de proliferação celular........................................................................44 1.2. Ensaio de wound healing……………….……….………………………………..44 1.3. Tratamento com Interleucina 6 (IL6)..............................................................45 1.4. Extração de DNA a partir de linhagens celulares...........................................45
1.4.1. Quantificação do DNA...........................................................................45 1.4.2. Tratamento com Bissulfito de Sódio.....................................................45 1.4.3. Pirosequenciamento.............................................................................46
1.5. Extração de RNA a partir de linhagens celulares...........................................49 1.5.1. Reação de transcrição reversa (RT) com a enzima MML V-RT...........49 1.5.2. Reação em cadeia da polimerase quantitativa (PCRq)........................50
1.6. Imunofluorescência de células cultivadas em monocamada.........................52

ix
1.7. Transfecção com plasmídeo e/ou esiRNA.....................................................52 2. Cultivo de células em matrigel..............................................................................53
2.1. Extração de RNA de células cultivadas em matrigel......................................53 2.2. Imunofluorescência de células cultivadas em matrigel..................................54 2.3. Inclusão das células cultivadas em matrigel em parafina..............................55
3. Análises estatísticas.............................................................................................55 RESULTADOS E DISCUSSÃO.................................................................................56 1. Regulação da expressão de BCL3 por IL6...........................................................56 2. Avaliação do perfil de metilação do gene BCL3 em amostras humanas e em
linhagens celulares após o tratamento com IL6...................................................57 3. Avaliação da expressão de transcritos alternativos do gene BCL3 em linhagens
celulares esofágicas tratadas com IL6..................................................................62 4. Estabelecimento do modelo de diferenciação celular em matrigel utilizando
células esofágicas.................................................................................................64 4.1. Escolha das linhagens a serem usadas no modelo de diferenciação celular
em matrigel.....................................................................................................65 4.1.1. Caracterização de linhagens celulares tumorais esofágicas cultivadas
em monocamadas....................................................................................65 4.1.2. Estabelecimento do modelo de diferenciação celular em matrigel.......67
5. Caracterização do modelo de diferenciação celular em matrigel utilizando a linhagem TE-11.....................................................................................................71 5.1. Avaliação da presença de caspase 3 clivada por imunohistoquímica...........71 5.2. Avaliação da expressão por RT-PCRq de genes envolvidos no processo de
diferenciação e genes potencialmente importantes para o processo de carcinogênese esofágica................................................................................72
6. Efeito da IL6 sobre a diferenciação celular em matrigel.......................................75 6.1. Análise da expressão do gene BCL3 em esferas obtidas a partir de células
TE-11 cultivadas em matrigel na ausência ou presença de IL6.....................78 7. Modulação do gene BCL3 em células TE-11.......................................................79
7.1. Modulação do gene BCL3 em células TE-11 cultivadas em monocamada..................................................................................................79
7.2. Modulação do gene BCL3 em células TE-11 cultivadas em matrigel............81 DISCUSSÃO GERAL.................................................................................................83 CONCLUSÕES..........................................................................................................87 PERSPECTIVAS........................................................................................................88 REFERÊNCIAS BIBLIOGRÁFICAS..........................................................................89 ANEXO I - The Effects of Diet on Epigenetic Processes....................................108 ANEXO II - Epigenetic Signatures in Cancer: Implications for the Control of Cancer in the Clinic................................................................................................119

x
ANEXO III - Aberrant DNA methylation of cancer-associated genes in gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC–EURGAST)...................................................................................................129 ANEXO IV: DNA methylation changes associated with risk factors in tumours of the upper aerodigestive tract............................................................................141

xi
Lista de quadros
Página Quadro 2.1: Principais características das linhagens utilizadas neste estudo……....44 Quadro 2.2: Sequências dos primers utilizados nas análises por pirosequenciamento...................................................................................................46 Quadro 2.3: Sequências dos primers utilizados nas análises de expressão por PCRq………....................................................................................................………51

xii
Lista de figuras
Página
Figura 1: Modelo Genético Clonal................................................................................2 Figura 2: Modelo do Progenitor Epigenético................................................................4 Figura 3: Metilação do DNA e Câncer..........................................................................6 Figura 4: Mapa de modificações de histonas de um cromossomo típico em células normais e células tumorais..........................................................................................8 Figura 5: Biogênese de miRNAs..................................................................................9 Figura 2.1: Organização do gene BCL3 no genoma e suas potencias regiões de controle..................................................................................................................... ..42 Figura 2.2: Esquema geral da técnica de pirosequenciamento.................................48 Figura 2.3: Exemplo de pirograma.............................................................................49 Figura 2.4: Expressão do RNAm de BCL3 em diferentes linhagens esofágicas após o tratamento com IL6 em diferentes doses (10, 20 e 100 ng/mL) por diferentes períodos de tempo (30 minutos, 24 e 72 horas)........................................................57 Figura 2.5: Perfil de metilação da ilhota CpG 2 do gene BCL3 em amostras esofágicas de indivíduos saudáveis e pacientes com CEE (tecido normal adjacente e tumor)....................................................................................................................... ..59 Figura 2.6: Perfil de metilação da ilhota CpG 3 do gene BCL3 em amostras esofágicas de indivíduos saudáveis e pacientes com CEE (tecido normal adjacente e tumor)........................................................................................................... ..............60 Figura 2.7: Perfil de metilação da ilhota CpG 3 do gene BCL3 em linhagens celulares esofágicas HET-1A e TE-11 tratadas com IL6...........................................................61 Figura 2.8: Transcritos do gene BCL3. O transcrito BCL3-001 representa o transcrito “clássico”, enquanto os outros seriam os transcritos alternativos..............................62 Figura 2.9: Análise da expressão dos transcritos alternativos de BCL3 por PCRq nas linhagens celulares HET-1A e TE-11 após tratamento com IL6................................63 Figura 2.10: Ensaio de proliferação celular das linhagens celulares de CEE TE-1, TE-11 e TE-13........................................................................................................... .65 Figura 2.11: Ensaio de wound healing utilizando diferentes linhagens celulares.................................................................................................................... .66

xiii
Figura 2.12: Esquema do modelo de diferenciação celular em matrigel..................................................................................................................... ..67 Figura 2.13: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-1..................................................................................................................................68 Figura 2.14: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-11................................................................................................................................69 Figura 2.15: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-13................................................................................................................. ...............70 Figura 2.16: Avaliação da presença de caspase 3 clivada por imunohistoquímica em células TE-11 cultivadas em matrigel por nove dias..................................................72 Figura 2.17: Análise da expressão do RNAm de genes correlacionados ao processo de diferenciação celular e genes encontrados diferencialmente metilados no presente estudo em células TE-11 cultivadas em diferentes condições....................74 Figura 2.18: Classificação das esferas obtidas a partir de células TE-11 cultivadas em matrigel por quinze dias.......................................................................................76 Figura 2.19: Quantificação de estruturas verdadeiras obtidas a partir de células TE-11 cultivadas em matrigel na ausência e na presença de IL6 50 ng/mL por quinze dias......................................................................................................................... ....77 Figura 2.20: Fotografias representativas das esferas obtidas a partir de células TE-11 cultivadas em matrigel na ausência ou na presença de IL6 50 ng/mL.................78 Figura 2.21: Análise da expressão do RNAm do gene BCL3 em células TE-11 cultivadas em matrigel na ausência ou presença de IL6 50 ng/mL por diferentes períodos de tempo.....................................................................................................79 Figura 2.22: Análise da expressão do RNAm de BCL3 por RT-PCRq em células TE-11 transfectadas com esiRNA contra BCL3 e/ou vetor de expressão contendo este gene......................................................................................................................... ..80 Figura 2.23: Avaliação da expressão de BCL3 por imunofluorescência em células TE-11 transfectadas com (A) RNA de interferência contra BCL3 (esiRNA) ou (B) vetor de expressão contendo BCL3...........................................................................81 Figura 2.24: Quantificação de estruturas verdadeiras obtidas a partir de células TE-11 cultivadas em matrigel...........................................................................................82

xiv
Lista de siglas e abreviaturas
ADE – adenocarcinoma de esôfago APC - “adenomatosis polyposis coli” APS - adenosina 5’ fosfosulfato ASO - oligonucleotídeos alelo-específicos ATP – adenosina trifosfato BCL3 – “B-cell CLL/lymphoma 3” CD2 – molécula CD2 CD34 – molécula CD34 CDX2 – “caudal type homeobox 2” CEE - carcinoma epidermóide de esôfago CpG - dinucleotídeos 5’-CpG-3’ DES - desmina DNA – ácido desoxirribonucléico DNMT3a – DNA metil-transferase 3a DNMTs – DNA metil-transferases dNTP – desoxirribonucleotídeo trifosfato DSC2 – desmocolina 2 DSG1 – desmogleína 1 EDTA – etilenodiamina tetracetato EGF - fator de crescimento epidérmico EGFR – receptor do fator de crescimento epidérmico EUA – Estados Unidos da América FHIT – “fragile histidine triad” GABRA5 – “gamma-aminobutyric acid (GABA) A receptor, alpha 5” GPT - genes progenitores de tumores GST - genes supressores de tumor GSTP1- glutationa S-transferase pi GSTT1 – glutationa S-transferase theta H3K27 - lisina 27 da histona H3 H3K4 - lisina 4 da histona H3 H3K9 - lisina 9 da histona H3 H4K20 - lisina 20 da histona H4 HATs – histona acetil-transferases HCl – ácido clorídrico HCPA - Hospital das Clínicas de Porto Alegre HDACs – histona desacetilases HIF1α – “hypoxia inducible factor 1” HLA-DPA1 – “major histocompatibility complex, class II, DP alpha 1” HLA-DPB1 – “major histocompatibility complex, class II, DP beta 1” hMLH1 – “mutL homolog 1” humano HMTs – histona metil-transferases HPV – papilomavirus humano HUPE - Hospital Universitário Pedro Ernesto IFNG – interferon gamma IL10 – interleucina 10 IL16 – interleucina 16 IL1α – interleucina 1α

xv
IL1β – interleucina 1β IL6 – interleucina 6 Ile – aminoácido interleucina INCA - Instituto Nacional de Câncer IRAK1 – “interleukin-1 receptor-associated kinase 1” KRAS – “Kirsten rat sarcoma-2 viral oncogene homologue” KRT13 – queratina 13 KRT5 – queratina 5 LAT – “linker for activation of T cells” LINEs - “long interspersed nuclear elements” LSO - oligonucleotídeos locus-específicos MAP3K8 – “mitogen-activated protein kinase kinase kinase 8” MBDs - proteínas que se ligam ao DNA metilado MECP2 - methyl CpG binding protein 2” MGK - mutação “gate-keeper” MGMT - O6-metilguanina-DNA metiltransferase miRNAs – microRNAs mRNA – RNA mensageiro OMS - Organização Mundial de Saúde ONC – oncogenes ORFs – “open reading frames” p16 – também conhecido como CDKN2A, “cyclin-dependent kinase inhibitor 2A” PCR – reação em cadeia da polimerase PECAM1 – “platelet/endothelial cell adhesion molecule” PPi – pirofosfato pRb - proteína retinoblastoma RAR-β – receptor do ácido retinóico β RASSF1A – “Ras association (RalGDS/AF-6) domain family member 1” RISC - complexo de silenciamento induzido por RNA RJ – Rio de Janeiro RNA – ácido ribonucléico rpm – rotações por minuto RS – Rio Grande do Sul SDS – dodecil sulfato de sódio SINEs - “short interspersed nuclear elements” SMARCB1 – “SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1” SP – São Paulo SPP1 – “secreted phosphoprotein 1”, também conhecida como osteopontina TFF1 – “trefoil factor 1” TFFs - “trefoil factors” TIMP1 – “TIMP metallopeptidase inhibitor 1” TP53 - “tumour protein 53” Tris – trihidroximetil aminometano UERJ – Universidade do Estado do Rio de Janeiro Val - aminoácido valina

xvi
Resumo
O carcinoma epidermóide de esôfago (CEE) é um dos dez tipos de tumores mais frequentes no mundo e apresenta baixa sobrevida, consequência principalmente de um diagnóstico tardio e da falta de um tratamento eficaz. Atualmente, um grande enfoque tem sido dado às alterações epigenéticas como potenciais biomarcadores em câncer. Entretanto, para CEE, muito pouco é conhecido em termos de tais alterações Desta forma, nosso estudo procurou estabelecer uma assinatura de metilação do DNA para CEE e investigar as consequências funcionais de tais alterações. Para isso, foram incluídos no estudo 10 pacientes com CEE (tumor e tecido normal adjacente) e 4 indivíduos saudáveis que doaram uma biópsia de tecido esofágico. Tais amostras foram submetidas à análise pela plataforma GoldenGate (Illumina) que investiga o perfil de metilação do promotor de cerca de 800 genes relacionados a câncer. A comparação de classes pareada identificou 37 sítios CpG diferencialmente metilados, correspondentes a 34 genes, entre tumor e tecido normal adjacente. Com base nestes genes, o componente inflamatório e a adesão celular foram apontados como as principais vias celulares alteradas. A análise dos tumores e tecido normal adjacente em comparação ao tecido esofágico de indivíduos saudáveis nos mostrou ainda que a hipermetilação de TFF1 e a hipometilação de BCL3 já estavam presentes na mucosa adjacente, o que pode representar alterações precoces. Para validar os resultados obtidos com a plataforma GoldenGate, avaliamos por pirosequenciamento o perfil de metilação de três genes encontrados hipermetilados nos tumores, dois genes hipometilados, dois genes para os quais não foram encontradas alterações e do gene TFF1 em uma nova série de amostras. Nesta mesma série, avaliamos ainda o perfil de metilação dos elementos repetitivos LINE1 por pirosequenciamento, o que nos mostrou que os tumores apresentam uma hipometilação global em comparação ao tecido não tumoral.
Neste trabalho também investigamos as consequências funcionais das alterações encontradas no gene BCL3. Este gene é extremamente interessante pois possui três ilhotas CpG, é sabidamente induzido por IL6 (gene encontrado hipometilado nas nossas amostras) e é considerado um proto-oncogene. A avaliação das duas ilhotas situadas no corpo do gene nos mostrou que em uma delas (ilhota CpG 3) os tumores apresentam uma hipermetilação em comparação ao tecido não tumoral, perfil semelhante ao de linhagens celulares tratadas com IL6 por apenas trinta minutos. Também avaliamos a expressão em linhagens tratadas com IL6 de quatro transcritos alternativos descritos para BCL3. Entretanto, todos tiveram sua expressão induzida após o tratamento, sugerindo que a ilhota CpG 3 não seja um promotor alternativo para tais transcritos. Além disso, a expressão de BCL3 induzida por IL6 perturba o processo de diferenciação em um modelo in vitro. Tais resultados sugerem que as alterações encontradas em BCL3 podem ser induzidas pelo processo inflamatório e a superexpressão deste gene pode desregular a diferenciação celular em estágios precoces do processo de carcinogênese. Sendo assim, este estudo foi o primeiro a determinar uma assinatura de metilação do DNA para CEE, identificando não só importantes vias de sinalização celular alteradas, mas também potencias alterações precoces e consequências funcionais.

xvii
Abstract
Esophageal cancer (EC) is one of the ten most incident tumors worldwide. The mortality rates for this type of tumor are very similar to the incidence rates, which is a direct consequence of a late diagnosis and a poor treatment. Esophageal squamous cell carcinoma (ESCC) is believed to arise from esophageal mucosa through accumulation of both genetic and epigenetic changes. Alterations on DNA methylation have been pointed as early changes during tumor development. The aim of this study was to examine the global deregulation of methylation states in ESCC and identify DNA methylation signatures associated with different stages of the disease. We performed a bead array analysis of more than 800 cancer-related genes in a series of 10 ESCC samples, 10 matched surrounding tissues, and 4 healthy esophageal mucosa. The comparison between tumors and matched surrounding tissue revealed 37 differentially methylated CpG sites, corresponding to 34 genes, and these CpG sites were significantly enriched in genes related to several pathways including the cell communication pathway and the inflammatory component. In addition, by comparing the samples from ESCC patients with healthy esophageal mucosa, we identified TFF1 hypermethylation and BCL3 hypomethylation as potential early markers of ESCC. Pyrosequencing was used for validation of DNA methylation changes as well as for the assessment of global methylation by LINE1 assay. This analysis revealed a global hypomethylation in esophageal tumors, what is accordance with previous reports for other cancer types. We also investigated the functional consequences of BCL3 hypomethylation, a gene supposed to be involved in cell proliferation and apoptosis control. Very interestingly, this gene possesses three CpG islands and it is induced by IL6 (a gene found hypomethylated in our tumor samples). The role of these putative control regions is not clear and we decided not only to assess their methylation profile, but also their functional role. The third CpG island showed a hypermethylation in tumors and normal surrounding tissue in comparison with mucosa from healthy individuals as well as in cell lines treated with IL6, leading us to suspect of the presence of an alternative promoter. We then evaluated the expression of four alternative transcripts reported for BCL3 in cell lines treated with IL6, but no inverse correlation was found between the expression and the methylation status. This suggests that the region analysed doesn’t encompass a promoter for these alternative transcripts. We then investigated the functional role of BCL3 in a cell differentiation model in matrigel. Interestingly, we showed that IL6 treatment impairs proper differentiation and this effect is most likely dependent, at least in part, of BCL3 induction. Our results suggest that BCL3 expression may be induced by the inflammatory component and this may be mediated by epigenetic alterations. Besides, the overexpression of this gene may result in increased proliferation and apoptosis blockade, impairing differentiation in early stages of esophageal carcinogenesis. This was the first study to investigate the changes on DNA methylation in large scale in ESCC and provided evidences that important cellular pathways are altered in this type of tumor. Besides, we also identified possible early alterations in ESCC and provided hints on functional consequences of those changes.

1
Introdução Geral
1. Câncer
O câncer foi responsável por 13% das mortes no mundo em 2002, sendo que
70% destas ocorreram em países em desenvolvimento. Segundo as estimativas da
Organização Mundial de Saúde (OMS), a tendência é de que haja um aumento de
50% no número de novos casos e de 100% na mortalidade decorrente de neoplasias
até 2020. No Brasil, o câncer representa atualmente a segunda causa de morte,
acometendo por volta de 500 mil pessoas e levando à morte cerca de 150 mil
pessoas a cada ano. Este cenário ressalta a importância da doença não só no
Brasil, mas também no mundo e nos remete à urgência em se conhecer melhor a
biologia de tumores.
O câncer já foi definido de muitas maneiras diferentes. Tudo teve início com as
observações de Hipócrates, o qual sugeriu o nome “câncer” à doença devido aos
vasos sanguíneos que alimentam os tumores e se assemelham às garras de
caranguejos (Feinberg, Ohlsson et al., 2006). Além disso, há muito tempo os
patologistas viram o câncer como células que adquirem determinadas características
de maneira inapropriada, resultando no desenvolvimento de um tumor (Pitot, 1986).
Entretanto, no último século, a idéia do câncer como uma doença genética tem
predominado, sendo que Boveri e Boveri foram os primeiros a sugerir uma
associação entre tumores e cromossomos aberrantes (Boveri e Boveri, 1929). Hoje,
a visão clássica do câncer sugere que a doença surge a partir de uma única célula
que, por meio da aquisição de uma série de mutações, sofre ondas de replicação
clonal e leva ao surgimento de um tumor (Fig. 1) (Iacobuzio-Donahue, 2009). Cada
mutação leva ao crescimento seletivo de uma sub-população de células tumorais e
cada uma dessas sub-populações contém uma ou mais propriedades relacionadas
ao potencial de invasão, à habilidade metastática e à resistência terapêutica
(Iacobuzio-Donahue, 2009). Este chamado modelo genético clonal ganhou força
com a descoberta dos genes supressores de tumor e oncogenes, os quais são
peças fundamentais no processo de tumorigênese (Feinberg, Ohlsson et al., 2006).
Neste contexto, diversas mutações já foram descritas em diferentes tipos de tumores
e, para alguns, a sequência de eventos já é bem descrita, como no caso do câncer
coloretal. Este processo de tumorigênese envolve uma série de alterações em

2
oncogenes e genes supressores de tumor, incluindo “adenomatosis polyposis coli”
(APC), “Kirsten rat sarcoma-2 viral oncogene homologue” (KRAS) e “tumour protein
53” (TP53). A aquisição sequencial de alterações nestes genes se dá de acordo com
o estágio da doença, progredindo de pequenos adenomas benignos até tumores
metastáticos avançados (Feinberg, Ohlsson et al., 2006).
Figura 1: Modelo Genético Clonal. A visão clássica do câncer sugere que esta doença surge a partir de uma série de mutações tanto em oncogenes (ONC) quanto em genes supressores de tumor (GST) que levam ao crescimento seletivo de uma sub-população de células. Estas mutações seriam as responsáveis pelas propriedades do tumor, tais quais potencial de invasão, metástase e resistência a drogas. Adaptado de Feinberg et al., 2006.
Apesar da relevância deste modelo, avanços na pesquisa básica do câncer
têm demonstrado que as alterações genéticas não parecem ser as únicas a
contribuir para o processo de carcinogênese. Isto se baseia, por exemplo, na
observação de que algumas alterações de fenótipo nas células tumorais não são
relacionadas ao aparecimento de mutações. Este é o caso da aquisição de potencial
metastático pelas células de tumores coloretais. Apesar da pesquisa massiva na
área, mutações não foram relacionadas a este potencial, enquanto modificações
específicas de expressão gênica parecem estar envolvidas. Isto sugere que outros
mecanismos participam da desregulação de vias celulares durante a tumorigênese
(Feinberg, Ohlsson et al., 2006). Além disso, o modelo genético clonal se baseia na
aquisição de múltiplas mutações, o que requereria muito tempo. A aquisição de
mutações em genes específicos é um processo ineficiente devido à complexidade e
eficácia dos sistemas de monitoramento e reparo de DNA, portanto, pouco provável

3
de ocorrer repetidamente mesmo em uma sub-população de células (Hanahan e
Weinberg, 2000). Desta maneira, se décadas são necessárias para o surgimento de
um tumor, outras décadas seriam necessárias para a progressão do mesmo, já que
a maior parte dos tumores sólidos não apresenta um potencial replicativo maior do
que o de uma célula normal (Feinberg, Ohlsson et al., 2006). Tudo isto levou a um
enfraquecimento do modelo clonal genético.
Sabe-se hoje que os mecanismos epigenéticos de regulação da expressão
gênica desempenham um papel fundamental durante o desenvolvimento e no
processo de diferenciação celular, quando as células precisam ligar ou desligar
genes específicos. Esta observação associada às alterações epigenéticas
encontradas em células tumorais, especialmente em estágios iniciais, levaram à
proposição de um novo modelo de desenvolvimento do câncer, o chamado modelo
do progenitor epigenético (Fig. 2). Neste modelo, a primeira etapa consiste de
alterações epigenéticas que levariam à desregulação de células progenitoras em um
determinado tecido, tornando essas células hábeis a se tornar neoplásicas. Uma
evidência desta primeira etapa seria a identificação de alterações epigenéticas
características de tumores já no tecido normal adjacente (Holst, Nuovo et al., 2003).
Além disso, todos os tumores até então investigados apresentam uma hipometilação
global do DNA. Já que as modificações genéticas também são clonais, esses dois
eventos deveriam ocorrer simultaneamente e de maneira universal, o que seria
pouco provável. Sendo assim, alterações epigenéticas parecem preceder as
mutações e outras modificações genéticas (Feinberg, Ohlsson et al., 2006).

4
Figura 2: Modelo do Progenitor Epigenético. De acordo com este modelo, o câncer surge em três etapas. Na primeira delas, alterações epigenéticas em “genes progenitores de tumores” (GPT) tornariam as células afetadas hábeis a se tornar neoplásicas. Em seguida, uma mutação “gate-keeper” (MGK)/iniciadora em oncogenes (ONC) ou genes supressores de tumor (GST) surgiria ainda nos estágios iniciais do processo de tumorigênese. Por fim, a plasticidade genética e/ou epigenética levaria a uma evolução acelerada do tumor. Adaptado de Feinberg et al., 2006.
A segunda etapa deste modelo envolve uma mutação iniciadora que
apareceria nos estágios iniciais do desenvolvimento de uma neoplasia (Feinberg,
Ohlsson et al., 2006). Estas mutações poderiam levar à ativação de oncogenes ou
ao silenciamento de genes supressores de tumor e, por terem sido anteriormente
apontadas como o primeiro passo do processo de carcinogênese, já foram
extensivamente estudadas em diversos tipos de câncer. Em tumores coloretais, por
exemplo, essa mutação iniciadora ocorreria no gene APC, enquanto em leucemia
mielóide crônica os rearranjos BCR–ABL são bem descritos (Lengauer, Kinzler et al.,
1998; Futreal, Coin et al., 2004).
Por fim, o terceiro passo do modelo do progenitor epigenético envolveria a
plasticidade genética e epigenética das células que já adquiriram a mutação
iniciadora. Esta plasticidade conferiria a estas células uma maior habilidade de
modificação do seu fenótipo, o que poderia resultar na aquisição de novas
características favoráveis não só para o estabelecimento do tumor em si, mas
também para a aquisição de poder metastático, invasão e resistência a
quimioterápicos (Feinberg, Ohlsson et al., 2006).
Este modelo proposto recentemente tem encontrado fortes alicerces em
trabalhos já consolidados e também na série de estudos sobre alterações
epigenéticas em diversos tipos de câncer que têm sido desenvolvidos nos últimos
anos. Isto nos remete à importância dos mecanismos epigenéticos de regulação

5
gênica para o desenvolvimento desta doença e, por essa razão, este é o foco do
nosso estudo.
2. Epigenética
O termo “epigenética” foi primeiramente introduzido em 1940 por um
embriologista e geneticista britânico chamado Conrad Hal Waddington para
descrever o estudo das causas do desenvolvimento (Slack, 2002). Hoje, este mesmo
termo se refere a modificações herdáveis que resultam em alterações de expressão
gênica sem modificar a sequência de bases do DNA. Três mecanismos epigenéticos
foram até então descritos: metilação do DNA, modificações de histonas e
microRNAs. Estes mecanismos desempenham um papel fundamental no
funcionamento de células normais e, portanto, sua desregulação pode levar ao
aparecimento de doenças, como o câncer.
2.1. Metilação do DNA
Atualmente, o mecanismo epigenético mais amplamente estudado é a metilação
do DNA. Este se baseia na adição de grupamentos metil a citosinas posicionadas 5’
em relação a guaninas nos chamados dinucleotídeos CpG, reação catalisada por
enzimas DNA metil-tranferases (DNMTs) (Adams, Mckay et al., 1979; Herceg, 2007).
Os dinucleotídeos CpG compõem 2-5% do genoma, mas não são igualmente
distribuídos (Lopez, Percharde et al., 2009). Eles são encontrados principalmente em
sequências repetitivas, como nos SINEs e LINEs (“short interspersed nuclear
elements” e “long interspersed nuclear elements”), e nas regiões promotoras dos
genes, formando as chamadas ilhotas CpG (encontradas em cerca de 60% dos
genes) (Bird, 2002; Lopez, Percharde et al., 2009).
Em células normais, os dinucleotídeos CpG localizados em sequências
repetitivas geralmente encontram-se metilados, o que pode prevenir a expressão de
retrotransposons e confere estabilidade ao genoma (Lopez, Percharde et al., 2009).
Já as ilhotas CpG localizadas em regiões promotoras encontram-se normalmente
desmetiladas, excetuando-se aquelas associadas a genes que sofrem “imprinting” e
genes localizados no cromossomo X inativo (Laird, 2003).
Células tumorais normalmente apresentam um perfil de metilação aberrante: a
hipometilação global é comum, enquanto promotores de genes específicos
encontram-se hipermetilados (Fig. 3). A hipometilação normalmente afeta
sequências repetitivas e retrotransposons, o que tem sido correlacionado à

6
instabilidade genômica, característica comum em tumores (Hinshelwood e Clark,
2008). Além disso, a hipometilação também pode afetar promotores específicos,
induzindo a expressão de genes que deveriam estar silenciados, como oncogenes e
genes que sofrem “imprinting”. Já a hipermetilação frequentemente afeta promotores
de genes importantes para a homeostase celular, como os genes supressores de
tumor. Este estado normalmente é associado a uma expressão gênica reduzida e
poderia explicar como a função destes genes é perdida na ausência de mutações
(Jones e Baylin, 2002).
Figura 3: Metilação do DNA e Câncer. Células tumorais apresentam um perfil de metilação alterado em comparação a células normais, caracterizado por uma hipometilação global associada à hipermetilação de promotores específicos. Isto pode resultar em: (A) hipermetilação e consequente silenciamento de genes supressores de tumor (GSTs); (B) perda do “imprinting” e ativação de alelos antes silenciados; (C) hipometilação de sequências repetitivas, possível ativação de retrotransposons e consequente instabilidade genômica.
O silenciamento gênico induzido por hipermetilação de promotores parece
ocorrer por dois mecanismos. O primeiro deles sugere que os grupamentos metil
bloqueiam a ligação de fatores de transcrição e a RNA polimerase é impossibilitada

7
de transcrever o gene (Molloy e Watt, 1990). Além disso, citosinas metiladas
recrutam proteínas específicas, como as proteínas que se ligam ao DNA metilado
(MBDs) e histona desacetilases (HDACs, mais detalhes a seguir), as quais
competem com os fatores de transcrição por seus sítios de ligação no DNA e tornam
a cromatina menos acessível (Nan, Cross et al., 1998). Estes dois mecanismos
parecem interagir na regulação da expressão de diversos genes.
2.2. Modificações de Histonas
Outro mecanismo epigenético de regulação gênica inclui as modificações de
histonas. Estas proteínas empacotam o DNA em estruturas chamadas
nucleossomos e regulam a conformação da cromatina (Loizou, Murr et al., 2006;
Esteller, 2007). Esta regulação se dá por meio da adição de modificações às caudas
das histonas, as quais interferem na afinidade destas proteínas pelo DNA. Dentre
tais modificações, temos acetilação, metilação, fosforilação, ubiquitinação e
sumoilação, porém a adição de grupamentos acetil e metil tem sido mais
amplamente estudada (Wang, Fischle et al., 2004).
A acetilação de histonas consiste na adição de grupamentos acetil a lisinas
localizadas principalmente na cauda N-terminal das histonas, reação catalisada por
enzimas chamadas histona acetil-transferases (HATs) (Lafon-Hughes, Di Tomaso et
al., 2008). A acetilação induz um afrouxamento da cromatina, que se torna mais
permissiva devido à neutralização das cargas positivas das lisinas, responsáveis
pela interação com o DNA (negativamente carregado) (Garcia-Ramirez, Rocchini et
al., 1995). Dessa forma, histonas acetiladas são associadas à eucromatina e a uma
transcrição ativa. O processo inverso, catalizado por histona desacetilases (HDACs),
resulta na formação de heterocromatina associada à repressão transcricional
(Lopez, Percharde et al., 2009).
Outra modificação bastante estudada é a adição de grupamentos metil às
histonas. Estas proteínas podem ser mono-, di- ou trimetiladas em resíduos de lisina
ou arginina por enzimas chamadas histona metil-transferases (HMTs), enquanto a
desmetilação é catalisada por histona desmetilases (Ellis, Atadja et al., 2009). Ao
contrário da acetilação de histonas, a metilação pode ser associada tanto a um
estado permissivo, quanto ao silenciamento de genes, dependendo do resíduo
modificado (Bernstein, Meissner et al., 2007).
Em comparação à metilação do DNA, o conhecimento acerca do papel das
modificações de histonas no câncer é bem menos claro. De uma maneira geral, a

8
desacetilação das histonas H3 e H4, a perda da trimetilação da lisina 4 da histona
H3 (H3K4) e a metilação H3K9 assim como a trimetilação H3K27 têm sido
associadas à hipermetilação do DNA e ao silenciamento gênico, especialmente de
GSTs (Fig. 4) (Fahrner, Eguchi et al., 2002; Esteller, 2007). Já em regiões
repetitivas, as modificações associadas a uma cromatina mais permissiva são mais
comuns (Fig. 4) (Esteller, 2007). Entretanto, a regulação destes processos e como
eles são perturbados no processo de carcinogênese ainda não são conhecidos em
maiores detalhes.
Figura 4: Mapa de modificações de histonas de um cromossomo típico em células normais e células tumorais. Em células normais, regiões que incluem promotores de GSTs são enriquecidas em modificações de histonas associadas a uma transcrição ativa, como a acetilação das histonas H3 e H4 e a trimetilação da lisina 4 da histona H3. Nas mesmas células, regiões repetitivas e outras regiões de heterocromatina são caracterizadas por marcas repressivas como a trimetilação H3K27, dimetilação H3K9 e trimetilação H4K20. Em células tumorais este perfil se inverte, sendo que as marcas repressivas encontram-se associadas aos GSTs, enquanto as modificações de histonas relacionadas a uma transcrição ativa são prevalentes em regiões repetitivas do DNA. Adaptado de Esteller, 2007.
2.3. miRNAs
A expressão gênica também pode ser regulada pelos microRNAs (miRNAs),
pequenos (20-22 nucleotídeos) RNAs regulatórios que podem ser transcritos a partir
de sequências codificantes e não codificantes. As “open reading frames” (ORFs) a
partir das quais estas moléculas são transcritas estão localizadas tanto em íntrons
ou exons de genes que codificam proteínas (70%), quanto em regiões intergênicas

9
(30%), sendo que a RNA polimerase II é responsável pela transcrição (Lee, Kim et
al., 2004; Rodriguez, Griffiths-Jones et al., 2004). Este transcrito primário, chamado
pri-miRNA, pode conter até várias quilobases e é processado ainda no núcleo pela
RNase III Drosha, formando o pré-miRNA (um “hairpin” de cerca de 70 nucleotídeos)
(Lee, Ahn et al., 2003). O pré-miRNA é então exportado para o citoplasma onde é
processado por outra RNase III chamada Dicer (Hutvagner, Mclachlan et al., 2001;
Yi, Qin et al., 2003). O produto é um RNA de fita dupla de aproximadamente 20-22
nucleotídeos que se liga ao complexo de silenciamento induzido por RNA (RISC)
(Chendrimada, Gregory et al., 2005). Neste complexo, uma das fitas de RNA é
selecionada (fita guia) e o RISC maduro é formado, sendo capaz de silenciar genes
(Bartel, 2004).
Figura 5: Biogênese de miRNAs. (1) Os miRNAs são transcritos pela RNA polimerase II em um transcrito primário (pri-miRNA) que é clivado no núcleo pela RNase III Drosha, formando um “hairpin” chamado pré-miRNA. (2) O pré-miRNA é exportado para o citoplasma pela exportina 5 e é posteriormente processado por outra RNase III chamada Dicer, formando um miRNA de fita dupla (3). Este miRNA se liga ao complexo de silenciamento induzido por RNA (RISC), onde uma das fitas é selecionada. (4) O RISC maduro é então capaz de silenciar genes pela degradação do mRNA ou por inibir a tradução, dependendo do grau de complementaridade entre o miRNA e o mRNA alvo. Adaptado de Garzon et al., 2006.

10
Cerca de 30% dos genes humanos parecem ser regulados por miRNAs
(Bartel, 2004). Esta regulação é mediada pela interação direta entre os miRNAs e os
mRNAs e pode se dar por dois mecanismos, repressão traducional ou degradação
do mRNA (Fig. 5). Estes dois mecanismos são dirigidos pela complementaridade
entre o miRNA e o mRNA alvo e ambos resultam em silenciamento gênico (Molnar,
Tamasi et al., 2008).
Nos últimos anos, o conhecimento acerca dos miRNAs em câncer tem
avançado consideravelmente. De maneira interessante, cerca de 50% dos miRNAs
conhecidos estão localizados em regiões do genoma frequentemente afetadas em
tumores, como sítios frágeis, regiões de perda de heterozigosidade e amplificação
(Calin e Croce, 2006). Algumas destas regiões já têm sido correlacionadas ao
processo de carcinogênese, mas os genes responsáveis ainda não eram
conhecidos. Sabe-se hoje que isto pode ser atribuído aos miRNAs.
De uma maneira geral, apesar da expressão de alguns miRNAs estar
aumentada em tumores, a maioria deles é silenciada em células tumorais (Lu, Getz
et al., 2005). Entretanto, o tipo e não a quantidade total de miRNAs alterados parece
ser importante para o processo de carcinogênese (Molnar, Tamasi et al., 2008).
Estas moléculas têm sido classificadas como supressores de tumor e oncogenes. No
primeiro caso, a perda da função do miRNA pode levar à ativação de oncogenes e
iniciar ou contribuir para a transformação de uma célula normal (Garzon, Calin et al.,
2009). Já os miRNAs oncogênicos geralmente são superexpressos em neoplasias e
agem silenciando genes supressores de tumor, promovendo o crescimento tumoral
(Molnar, Tamasi et al., 2008).
Alterações em cada um dos mecanismos epigenéticos de regulação gênica já
foram identificadas em diferentes tumores e, cada vez mais, estas alterações têm
sido apontadas como biomarcadores de diagnóstico, prognóstico e resposta a
tratamento, além de representarem potenciais alvos terapêuticos devido à sua
reversibilidade. Entretanto, para alguns tumores, como o carcinoma epidermóide de
esôfago, estes estudos ainda são escassos. Por isso, esta tese procura
compreender o papel de alterações epigenéticas neste tipo de tumor.

11
Objetivos
Conforme mencionado anteriormente, o carcinoma epidermóide de esôfago
ainda é uma doença pouco estudada e, portanto, o conhecimento acerca da sua
biologia ainda é escasso. Além disso, os mecanismos epigenéticos que contribuem
para o seu desenvolvimento foram estudados apenas superficialmente até então.
Sendo assim, essa tese tem como objetivo principal identificar as alterações em
metilação do DNA que contribuem para o desenvolvimento do CEE e de que
maneira tais modificações afetam o funcionamento da célula esofágica normal. Para
isso, objetivos específicos foram traçados:
- Identificar os genes diferencialmente metilados em CEE.
- Identificar potenciais biomarcadores para o diagnóstico precoce de CEE
- Realizar ensaios funcionais utilizando linhagens celulares para melhor
compreender as consequências das diferentes alterações em metilação do
DNA encontradas.

12
Capítulo I: Alterações em metilação do DNA em
carcinoma epidermóide de esôfago
Introdução
1. Câncer de Esôfago
O câncer de esôfago é um dos dez tipos de neoplasia mais frequentes no
mundo. De acordo com os dados da GLOBOCAN (2008), este tipo de câncer é o
sexto mais frequente e o quinto em mortalidade entre homens e o décimo primeiro
mais frequente e o oitavo em mortalidade entre mulheres (Ferlay, Shin et al., 2010).
Estes dados demonstram que este tumor é altamente fatal, sendo a sobrevida em 5
anos menor que 10%, principalmente devido ao diagnóstico tardio da doença
(Parkin, 2001). A sintomatologia aparece somente em estágios bem avançados, o
que resulta em um tratamento ineficaz (Allen, Richardson et al., 1997; Mclarty,
Deschamps et al., 1997). Existe uma melhora significativa no prognóstico quando os
tumores são ressecados em estágios iniciais, portanto, o diagnóstico precoce se faz
necessário.
No Brasil, o câncer de esôfago está em sexto lugar em incidência e é o quarto
mais fatal entre homens, enquanto em mulheres ocupa o décimo quinto lugar em
incidência e o décimo terceiro em mortalidade. Segundo dados do Ministério da
Saúde, 10.350 novos casos são esperados para o ano de 2012. As maiores taxas de
incidência estão na região sul e sudeste, sendo que no Rio Grande do Sul, São
Paulo e Rio de Janeiro as taxas brutas de incidência são de 18,01; 9,32 e 9,15/105
para homens e 6,60; 2,26 e 3,00/105 para mulheres, respectivamente (Inca, 2011).
Os tipos histológicos de câncer de esôfago mais prevalentes são o carcinoma
epidermóide (CEE) e adenocarcinoma (ADE) (Montesano, Hollstein et al., 1996). O
CEE aparece como um dos tipos de câncer mais frequentes no mundo, enquanto as
taxas de incidência de ADE têm crescido nos países desenvolvidos nos últimos anos
(Parkin, Pisani et al., 1999; Pickens e Orringer, 2003). No Brasil, o CEE representa
cerca de 90% do total dos casos de câncer de esôfago e acomete principalmente
pacientes do sexo masculino com idade entre 55 e 65 anos, onde os tumores se
localizam preferencialmente no terço médio do esôfago (Ribeiro Pinto, Teixeira
Rossini et al., 2003; Rossini, Rapozo et al., 2007).

13
A etiologia do CEE é multi-fatorial e apresenta agentes comuns em diversos
países e outros restritos a regiões específicas. Em países ocidentais, o CEE surge
tipicamente após muitos anos de abuso de álcool e tabaco. Estes fatores quando
combinados mostram um efeito multiplicativo, mesmo quando o consumo de álcool é
baixo (Castellsague, Munoz et al., 1999; Castellsague, Munoz et al., 2000; Rossini,
Rapozo et al., 2007). Porém, outros fatores contribuem para a gênese do CEE. No
sul do Brasil, Argentina e Uruguai, o hábito de consumir “chimarrão” muito quente e
em grandes volumes é um fator de risco importante para o desenvolvimento de CEE
(Victora, Munoz et al., 1987; Rolon, Castellsague et al., 1995; De Stefani, Deneo-
Pellegrini et al., 1999; Castellsague, Munoz et al., 2000). Em diversos países
orientais, a ingestão de bebidas quentes (normalmente na forma de chá) também
representa um fator de risco para CEE, normalmente associado a outros fatores,
como nitrosaminas presentes na dieta (Craddock, 1993; Islami, Malekshah et al.,
2009). Nos últimos anos, a infecção por HPV tem sido avaliada em CEE em diversas
populações e, em algumas delas, representa fator de risco para o desenvolvimento
deste tumor (Dreilich, Bergqvist et al., 2006; Shuyama, Castillo et al., 2007).
1.1. Alterações Genéticas em CEE
Apesar de ainda ser um câncer pouco estudado (dentre os 10 tipos de tumor
mais incidentes, o CEE é aquele com o menor número de publicações científicas em
periódicos indexados), algumas alterações genéticas já foram descritas em CEE.
Dentre elas, as mutações no gene TP53 são as mais comumente observadas. Este
é um gene supressor de tumor que regula a parada do ciclo celular, o reparo de DNA
e a apoptose, além de ser o gene mais frequentemente mutado em cânceres
humanos (Hollstein, Sidransky et al., 1991; Olivier, Eeles et al., 2002). Em CEE,
mutações em TP53 são consideradas um evento precoce e o perfil destas mutações
varia de acordo com a região geográfica, o que pode ter uma relação com fatores
etiológicos associados com a doença em cada região (Hainaut e Hollstein, 2000).
Um estudo do nosso laboratório mostrou uma prevalência de mutações de 34,5%
em amostras de CEE provenientes da região sudeste do Brasil (Rio de Janeiro e
São Paulo). Além disso, foi observada uma alta frequência de mutações em pares
de bases A:T, o que já foi descrito em outros países como a França e pode estar
relacionado ao consumo de etanol (Rossini, De Almeida Simao et al., 2010).
Outras alterações genéticas que parecem contribuir para o desenvolvimento de
CEE incluem polimorfismos em genes envolvidos no controle do ciclo celular e

14
enzimas que participam do metabolismo de xenobióticos. Por exemplo, mutações e
deleções no gene p21waf1/CIP1 são raras em neoplasias humanas, mas polimorfismos
têm sido correlacionados ao aparecimento de alguns tumores, como o CEE.
Especialmente, polimorfismos nos códons 31 e 149 do exon 2 podem desempenhar
um papel fundamental no processo de carcinogênese esofágica (Bahl, Arora et al.,
2000). Um outro estudo do nosso grupo mostrou que polimorfismos em genes que
codificam enzimas de fase II do processo de biotransformação modificam o risco
para desenvolvimento de CEE em brasileiros. Neste caso, o polimorfismo GSTP1
Ile105Val e o genótipo selvagem para GSTT1 foram apontados como fatores de
risco independentes (Rossini, Rapozo et al., 2007).
Além de mutações e polimorfismos, a superexpressão de alguns genes também
já foi descrita em CEE. A superexpressão de Ciclina D1, uma proteína importante
para o controle do ciclo celular pela via p16-pRb, já foi reportada em CEE e foi
correlacionada à amplificação gênica (Tsuruta, Sakamoto et al., 1993). Este perfil é
observado em 23-65% dos casos e já é evidente em displasias e tumores precoces
(Adelaide, Monges et al., 1995; Nakagawa, Zukerberg et al., 1995; Shamma, Doki et
al., 2000). Outro gene superexpresso em CEE é o EGFR (Hanawa, Suzuki et al.,
2006; Sunpaweravong, Suwiwat et al., 2009). Este gene codifica o receptor do fator
de crescimento epidérmico que, na presença de ligantes como o EGF, sofre homo
ou heterodimerização (com outros membros da mesma família) e tem sua atividade
quinase estimulada (Olayioye, Neve et al., 2000). Estes receptores regulam vias de
sinalização importantes, como proliferação celular e apoptose, e sua superexpressão
é normalmente associada à amplificação gênica (Hanawa, Suzuki et al., 2006;
Rodemann, Dittmann et al., 2007; Sunpaweravong, Suwiwat et al., 2009).
Por fim, a expressão de alguns genes é encontrada diminuída em CEE. Este é o
caso, por exemplo, do gene que codifica a proteína retinoblastoma (pRb). Esta
proteína forma um complexo com o fator de transcrição E2F, impedindo a
transcrição de genes envolvidos na progressão do ciclo celular (Ezhevsky, Nagahara
et al., 1997). Deleções e mutações neste gene são raras em CEE, ao contrário do
que é observado em outros tumores, enquanto a perda de heterozigosidade parece
ser um evento frequente (Xing, Yang et al., 1999). Outro importante regulador do
ciclo celular encontrado alterado em CEE é o gene p16INK4a. Sua expressão
encontra-se diminuída nestes tumores por diferentes mecanismos, como deleção
homozigótica e metilação da região promotora (Maesawa, Tamura et al., 1996;
Fujiwara, Noguchi et al., 2008).

15
1.2. Alterações Epigenéticas em CEE
As alterações epigenéticas também parecem ser fundamentais no
desenvolvimento de CEE. Observou-se, por exemplo, que diversos genes
supressores de tumor parecem ser silenciados por metilação da região promotora
neste tipo de tumor. Este é o caso do gene p16INK4a, cujo promotor foi encontrado
metilado em 40-88% dos tumores e em 37% das mucosas normais adjacentes,
sugerindo um papel importante deste gene em estágios precoces do processo de
carcinogênese esofágica (Xing, Yang et al., 1999; Wang, Sasco et al., 2008). Outro
gene frequentemente encontrado hipermetilado em CEE é o FHIT. Cerca de 14-84%
destes tumores apresentam este gene supressor de tumor silenciado, sendo que
este perfil parece estar relacionado à exposição ao tabaco (Tanaka, Shimada et al.,
1998; Kim, Park et al., 2009). Soma e colaboradores demonstraram que a nicotina
pode induzir a hipermetilação de FHIT por meio da superexpressão da DNMT3a
(Soma, Kaganoi et al., 2006). Além disso, modificações de histonas também
parecem contribuir para o silenciamento deste gene, sendo que a desacetilação da
histona H4 foi encontrada em cerca de 70% dos casos (Tzao, Sun et al., 2006).
O silenciamento por metilação da região promotora em CEE também afeta genes
envolvidos no reparo de DNA. Foi demonstrado que a enzima de reparo MGMT (O6-
metilguanina-DNA metiltransferase) tem sua expressão diminuída em CEE e isto foi
relacionado à hipermetilação do gene. A frequência desta metilação varia de 27 a
72% no tecido tumoral, é de cerca de 67% no epitélio displásico, enquanto no tecido
normal fica entre 11 e 29% (Fang, Jin et al., 2005; Wang, Sasco et al., 2008). Isto
mostra um aumento progressivo da metilação de acordo com a evolução da doença,
sugerindo que as alterações epigenéticas desempenham um papel importante não
só na iniciação do tumor, mas também na sua progressão.
Outros genes já foram encontrados hipermetilados em CEE, como RAR-β,
RASSF1A, hMLH1, CDX2, entre outros (Tzao, Hsu et al., 2005; Guo, Ren et al.,
2006; Wong, Tao et al., 2006; Guo, House et al., 2007). Outras alterações
epigenéticas também já foram encontradas em CEE. Hoseok e colaboradores
demonstraram, por exemplo, que a avaliação dos níveis globais de modificações de
histonas pode ser um fator prognóstico de sobrevida livre de doença em pacientes
com CEE (I, Ko et al., 2010). Além disso, algumas análises em larga escala do perfil
de expressão de miRNAs em CEE também foram feitas. Elas mostraram que estes
perfis podem predizer sobrevida e são capazes de diferenciar os subtipos
histológicos de câncer de esôfago, além de diferenciar os tecidos tumoral e normal

16
adjacente (Feber, Xi et al., 2008; Guo, Chen et al., 2008). Dessa forma, ainda existe
um longo caminho a ser trilhado até que a biologia deste tumor seja conhecida e isto
resulte em melhorias para os pacientes.
Além disso, a maioria dos estudos foi feita em populações orientais, ou seja, em
populações com um “background” genético muito diferente da população brasileira e
expostas a fatores etiológicos diferentes. Além disso, nestes estudos um ou poucos
genes foram selecionados de acordo com os dados da literatura e/ou com sua
importância em vias celulares. Dessa forma, nosso estudo pretende não só traçar
um perfil de metilação do DNA em CEE para a nossa população, como também
fazê-lo de uma maneira não tendenciosa.

17
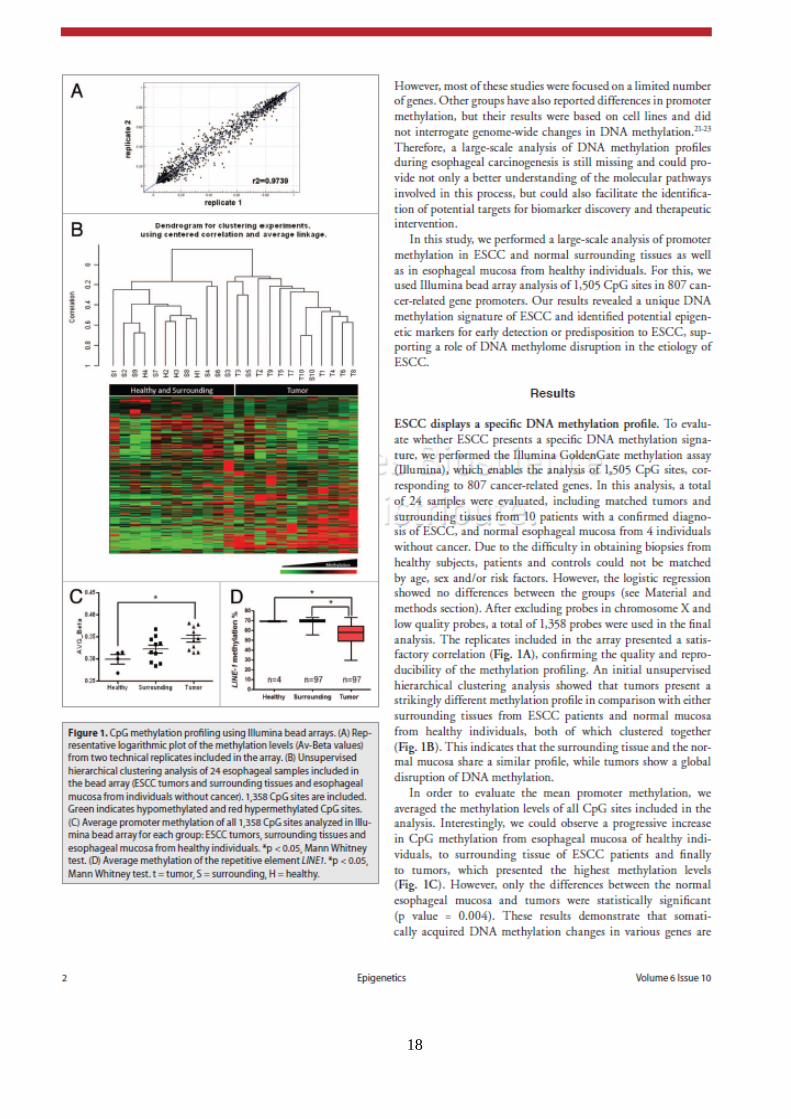
18
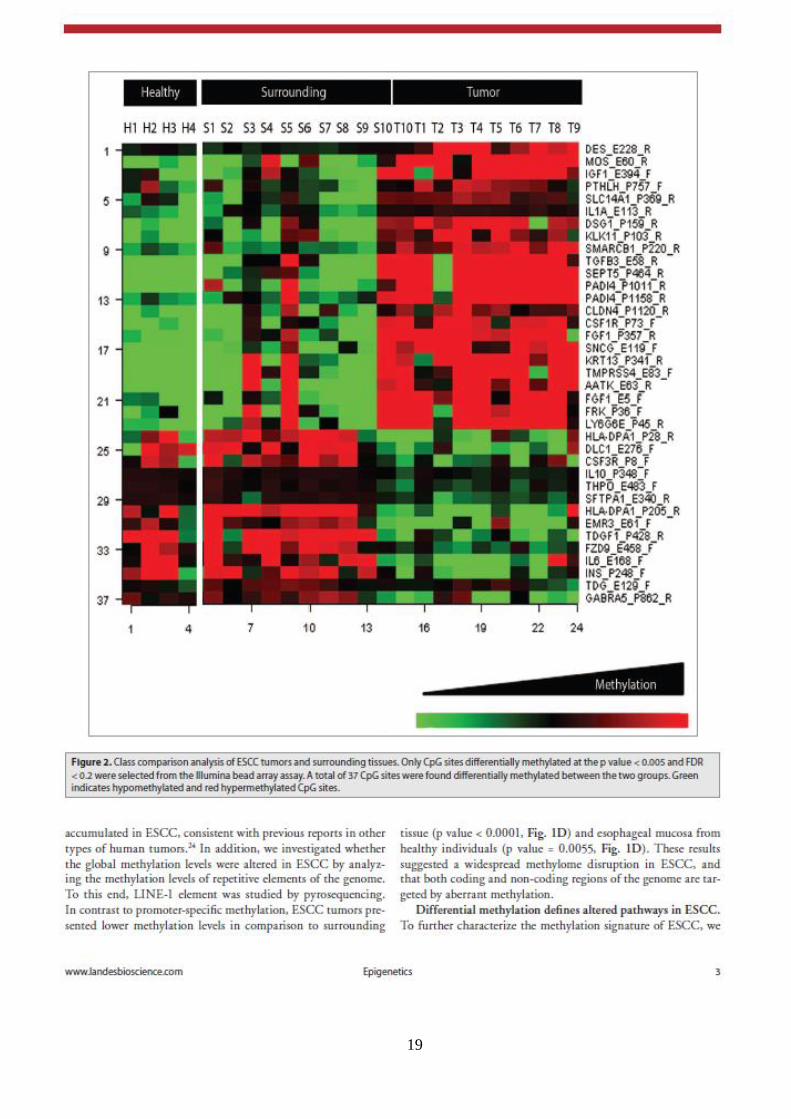
19
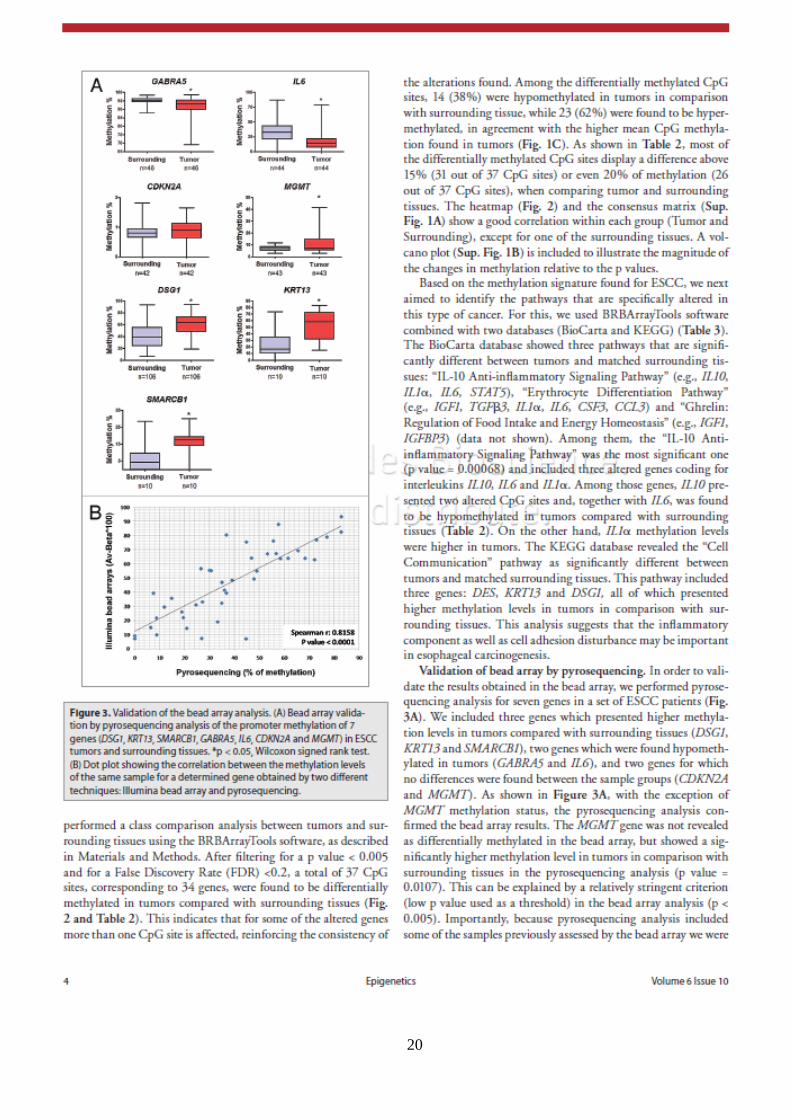
20
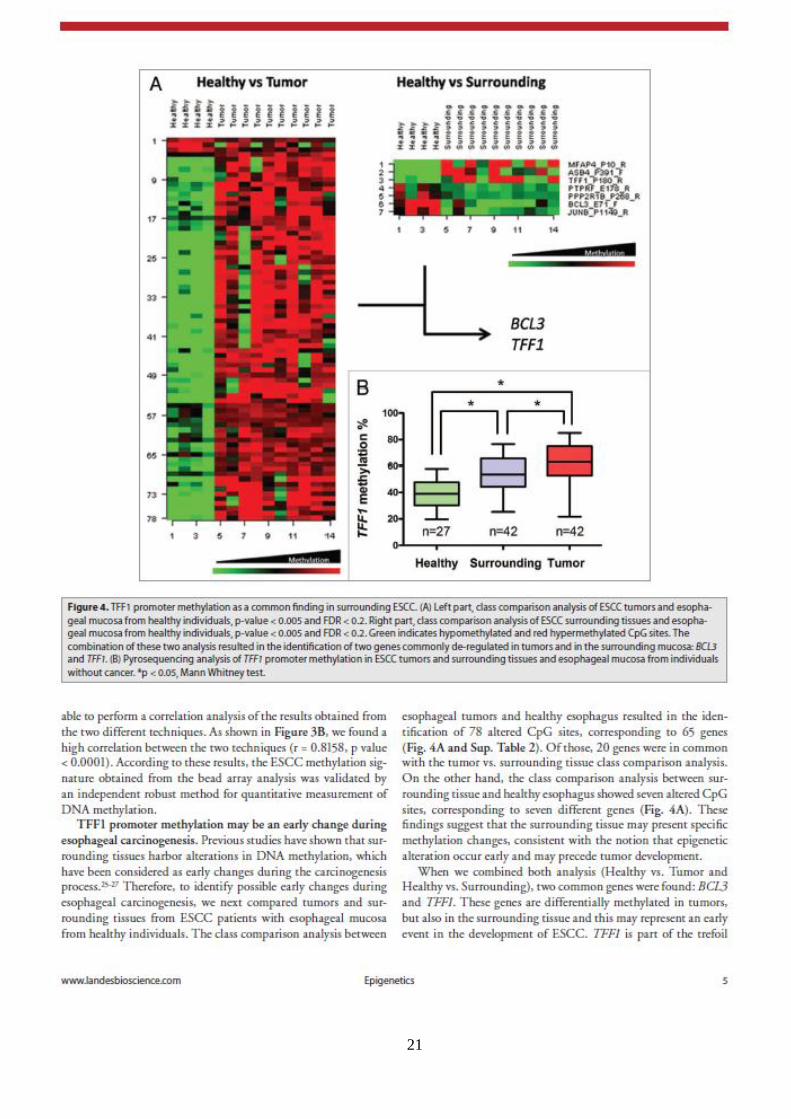
21

22

23
24
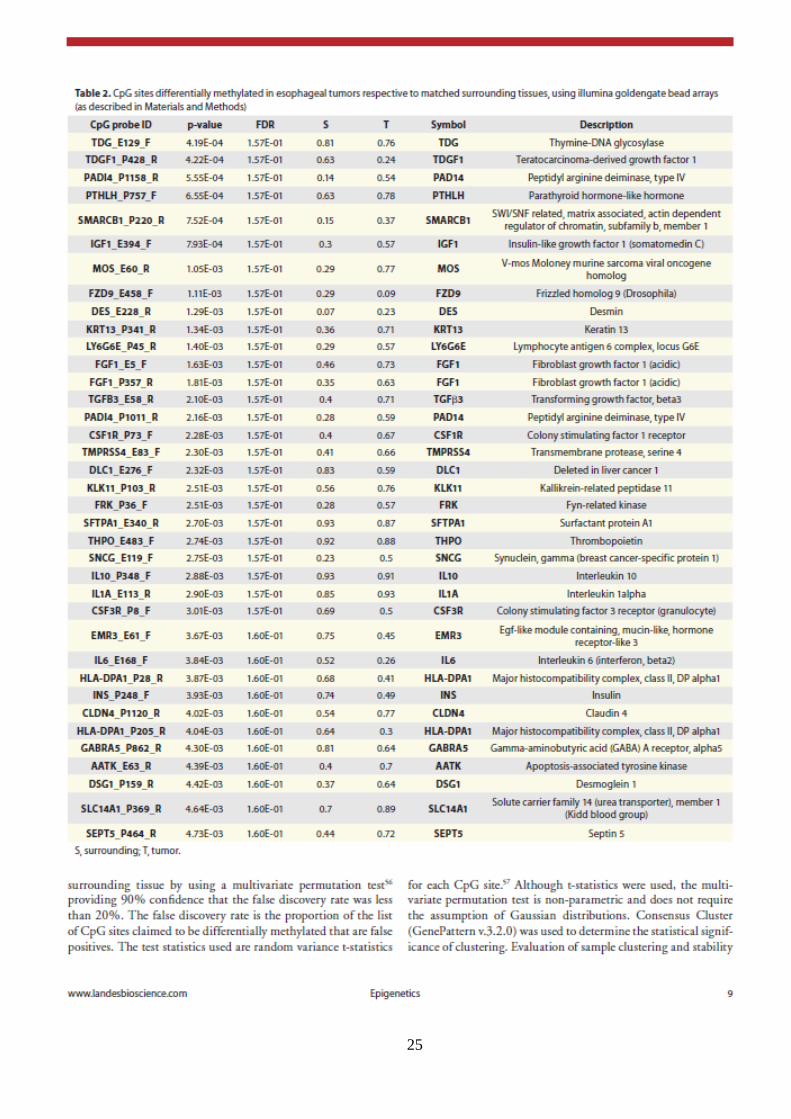
25
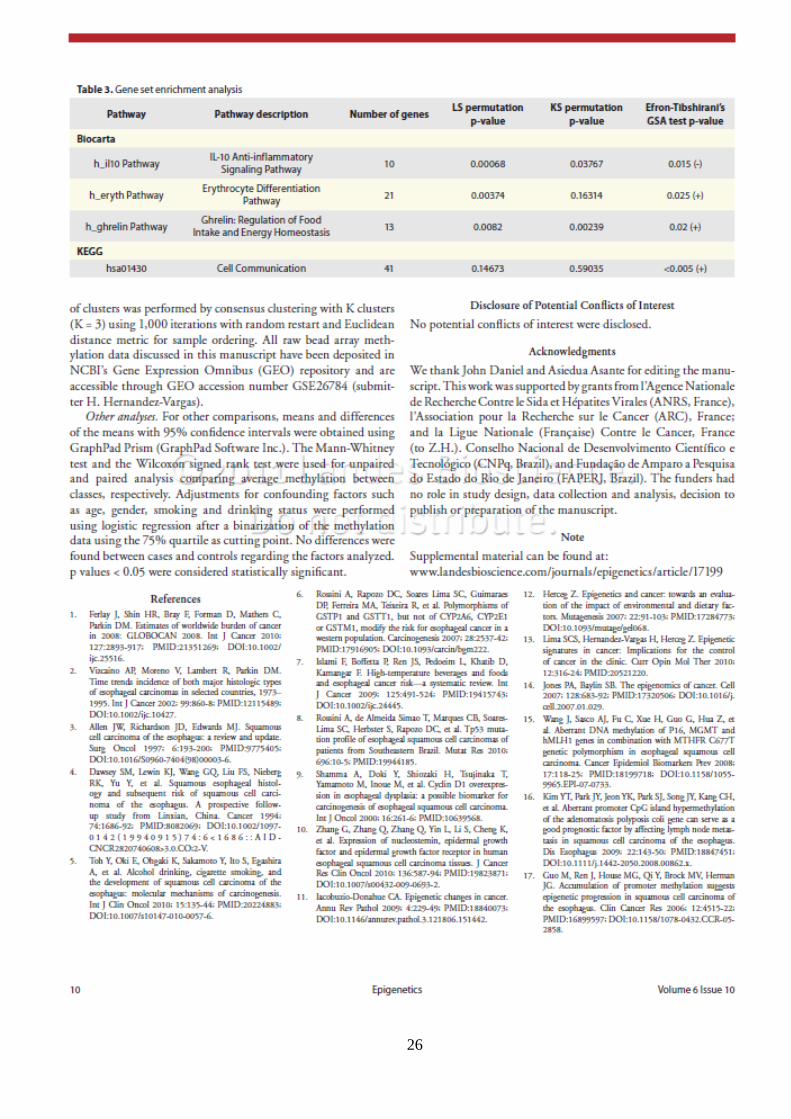
26

27
28
Identification of a methylome signature of esophageal squamous
cell carcinoma and potential early epigenetic biomarkers
Supplementary Data
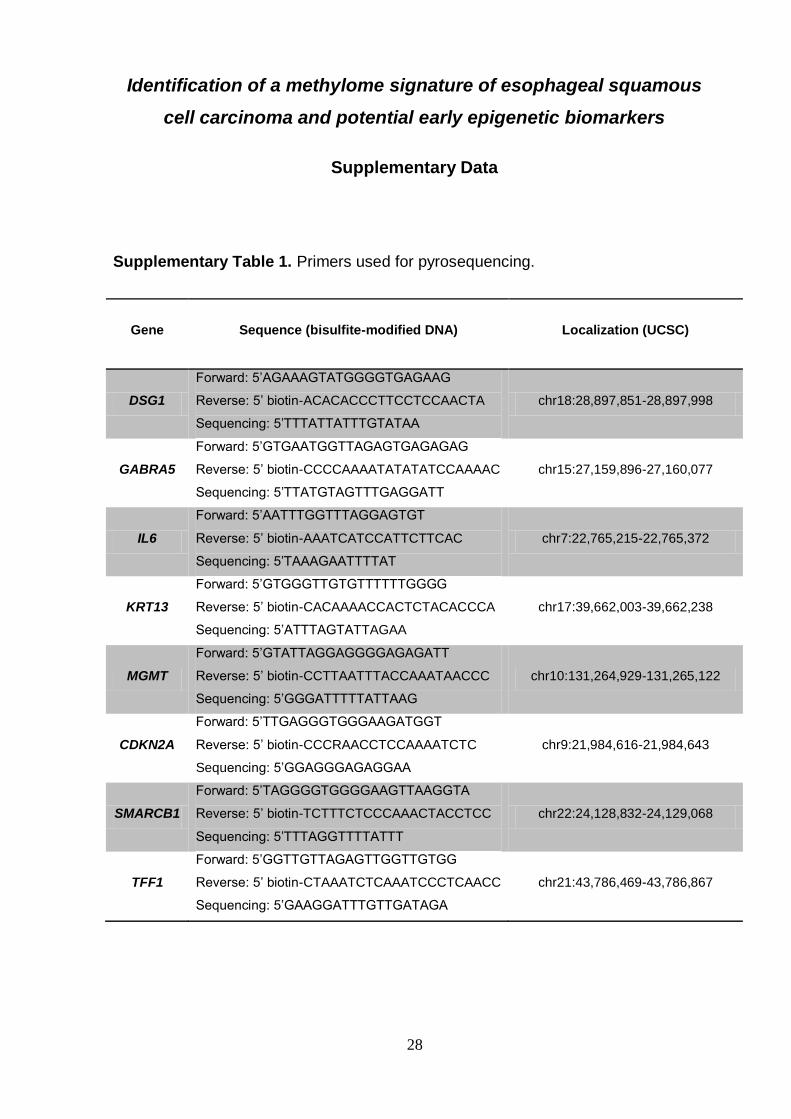
Supplementary Table 1. Primers used for pyrosequencing.
Gene
Sequence (bisulfite-modified DNA)
Localization (UCSC)
DSG1
Forward: 5’AGAAAGTATGGGGTGAGAAG
Reverse: 5’ biotin-ACACACCCTTCCTCCAACTA
Sequencing: 5’TTTATTATTTGTATAA
chr18:28,897,851-28,897,998
GABRA5
Forward: 5’GTGAATGGTTAGAGTGAGAGAG
Reverse: 5’ biotin-CCCCAAAATATATATCCAAAAC
Sequencing: 5’TTATGTAGTTTGAGGATT
chr15:27,159,896-27,160,077
IL6
Forward: 5’AATTTGGTTTAGGAGTGT
Reverse: 5’ biotin-AAATCATCCATTCTTCAC
Sequencing: 5’TAAAGAATTTTAT
chr7:22,765,215-22,765,372
KRT13
Forward: 5’GTGGGTTGTGTTTTTTGGGG
Reverse: 5’ biotin-CACAAAACCACTCTACACCCA
Sequencing: 5’ATTTAGTATTAGAA
chr17:39,662,003-39,662,238
MGMT
Forward: 5’GTATTAGGAGGGGAGAGATT
Reverse: 5’ biotin-CCTTAATTTACCAAATAACCC
Sequencing: 5’GGGATTTTTATTAAG
chr10:131,264,929-131,265,122
CDKN2A
Forward: 5’TTGAGGGTGGGAAGATGGT
Reverse: 5’ biotin-CCCRAACCTCCAAAATCTC
Sequencing: 5’GGAGGGAGAGGAA
chr9:21,984,616-21,984,643
SMARCB1
Forward: 5’TAGGGGTGGGGAAGTTAAGGTA
Reverse: 5’ biotin-TCTTTCTCCCAAACTACCTCC
Sequencing: 5’TTTAGGTTTTATTT
chr22:24,128,832-24,129,068
TFF1
Forward: 5’GGTTGTTAGAGTTGGTTGTGG
Reverse: 5’ biotin-CTAAATCTCAAATCCCTCAACC
Sequencing: 5’GAAGGATTTGTTGATAGA
chr21:43,786,469-43,786,867
29
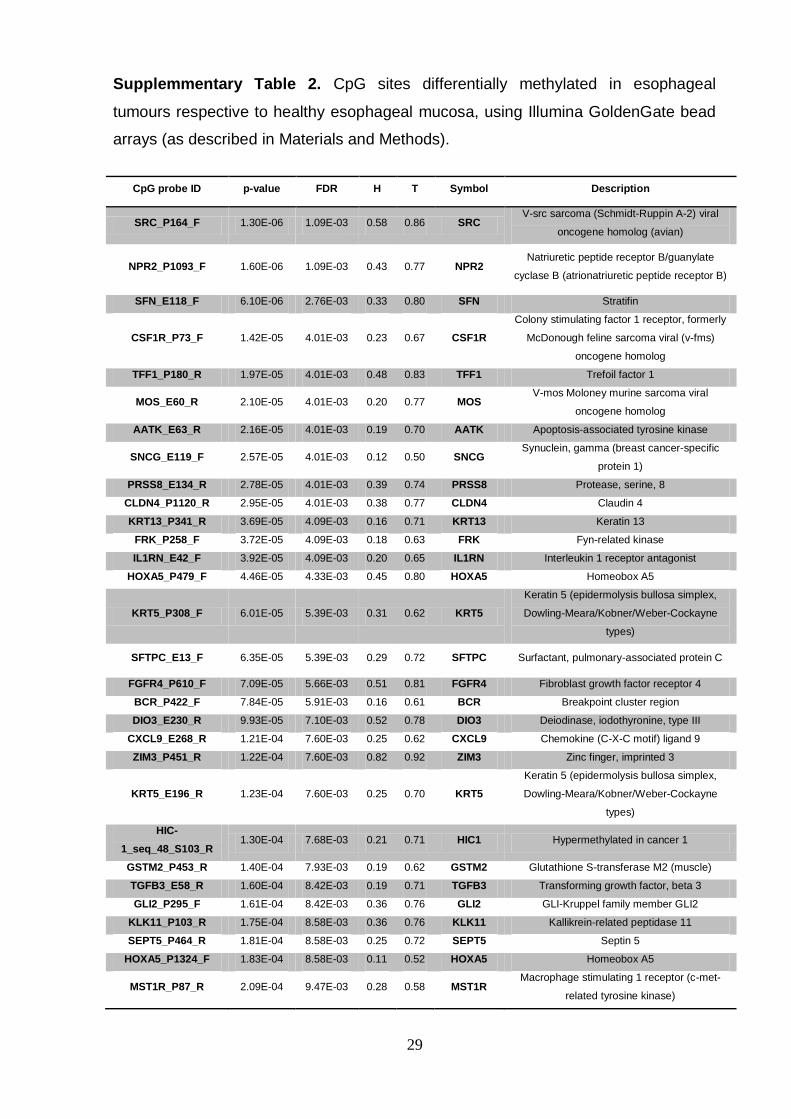
Supplemmentary Table 2. CpG sites differentially methylated in esophageal
tumours respective to healthy esophageal mucosa, using Illumina GoldenGate bead
arrays (as described in Materials and Methods).
CpG probe ID p-value FDR H T Symbol Description
SRC_P164_F 1.30E-06 1.09E-03 0.58 0.86 SRC V-src sarcoma (Schmidt-Ruppin A-2) viral
oncogene homolog (avian)
NPR2_P1093_F 1.60E-06 1.09E-03 0.43 0.77 NPR2 Natriuretic peptide receptor B/guanylate
cyclase B (atrionatriuretic peptide receptor B)
SFN_E118_F 6.10E-06 2.76E-03 0.33 0.80 SFN Stratifin
CSF1R_P73_F 1.42E-05 4.01E-03 0.23 0.67 CSF1R
Colony stimulating factor 1 receptor, formerly
McDonough feline sarcoma viral (v-fms)
oncogene homolog
TFF1_P180_R 1.97E-05 4.01E-03 0.48 0.83 TFF1 Trefoil factor 1
MOS_E60_R 2.10E-05 4.01E-03 0.20 0.77 MOS V-mos Moloney murine sarcoma viral
oncogene homolog
AATK_E63_R 2.16E-05 4.01E-03 0.19 0.70 AATK Apoptosis-associated tyrosine kinase
SNCG_E119_F 2.57E-05 4.01E-03 0.12 0.50 SNCG Synuclein, gamma (breast cancer-specific
protein 1)
PRSS8_E134_R 2.78E-05 4.01E-03 0.39 0.74 PRSS8 Protease, serine, 8
CLDN4_P1120_R 2.95E-05 4.01E-03 0.38 0.77 CLDN4 Claudin 4
KRT13_P341_R 3.69E-05 4.09E-03 0.16 0.71 KRT13 Keratin 13
FRK_P258_F 3.72E-05 4.09E-03 0.18 0.63 FRK Fyn-related kinase
IL1RN_E42_F 3.92E-05 4.09E-03 0.20 0.65 IL1RN Interleukin 1 receptor antagonist
HOXA5_P479_F 4.46E-05 4.33E-03 0.45 0.80 HOXA5 Homeobox A5
KRT5_P308_F 6.01E-05 5.39E-03 0.31 0.62 KRT5
Keratin 5 (epidermolysis bullosa simplex,
Dowling-Meara/Kobner/Weber-Cockayne
types)
SFTPC_E13_F 6.35E-05 5.39E-03 0.29 0.72 SFTPC Surfactant, pulmonary-associated protein C
FGFR4_P610_F 7.09E-05 5.66E-03 0.51 0.81 FGFR4 Fibroblast growth factor receptor 4
BCR_P422_F 7.84E-05 5.91E-03 0.16 0.61 BCR Breakpoint cluster region
DIO3_E230_R 9.93E-05 7.10E-03 0.52 0.78 DIO3 Deiodinase, iodothyronine, type III
CXCL9_E268_R 1.21E-04 7.60E-03 0.25 0.62 CXCL9 Chemokine (C-X-C motif) ligand 9
ZIM3_P451_R 1.22E-04 7.60E-03 0.82 0.92 ZIM3 Zinc finger, imprinted 3
KRT5_E196_R 1.23E-04 7.60E-03 0.25 0.70 KRT5
Keratin 5 (epidermolysis bullosa simplex,
Dowling-Meara/Kobner/Weber-Cockayne
types)
HIC-
1_seq_48_S103_R 1.30E-04 7.68E-03 0.21 0.71 HIC1 Hypermethylated in cancer 1
GSTM2_P453_R 1.40E-04 7.93E-03 0.19 0.62 GSTM2 Glutathione S-transferase M2 (muscle)
TGFB3_E58_R 1.60E-04 8.42E-03 0.19 0.71 TGFB3 Transforming growth factor, beta 3
GLI2_P295_F 1.61E-04 8.42E-03 0.36 0.76 GLI2 GLI-Kruppel family member GLI2
KLK11_P103_R 1.75E-04 8.58E-03 0.36 0.76 KLK11 Kallikrein-related peptidase 11
SEPT5_P464_R 1.81E-04 8.58E-03 0.25 0.72 SEPT5 Septin 5
HOXA5_P1324_F 1.83E-04 8.58E-03 0.11 0.52 HOXA5 Homeobox A5
MST1R_P87_R 2.09E-04 9.47E-03 0.28 0.58 MST1R Macrophage stimulating 1 receptor (c-met-
related tyrosine kinase)
30
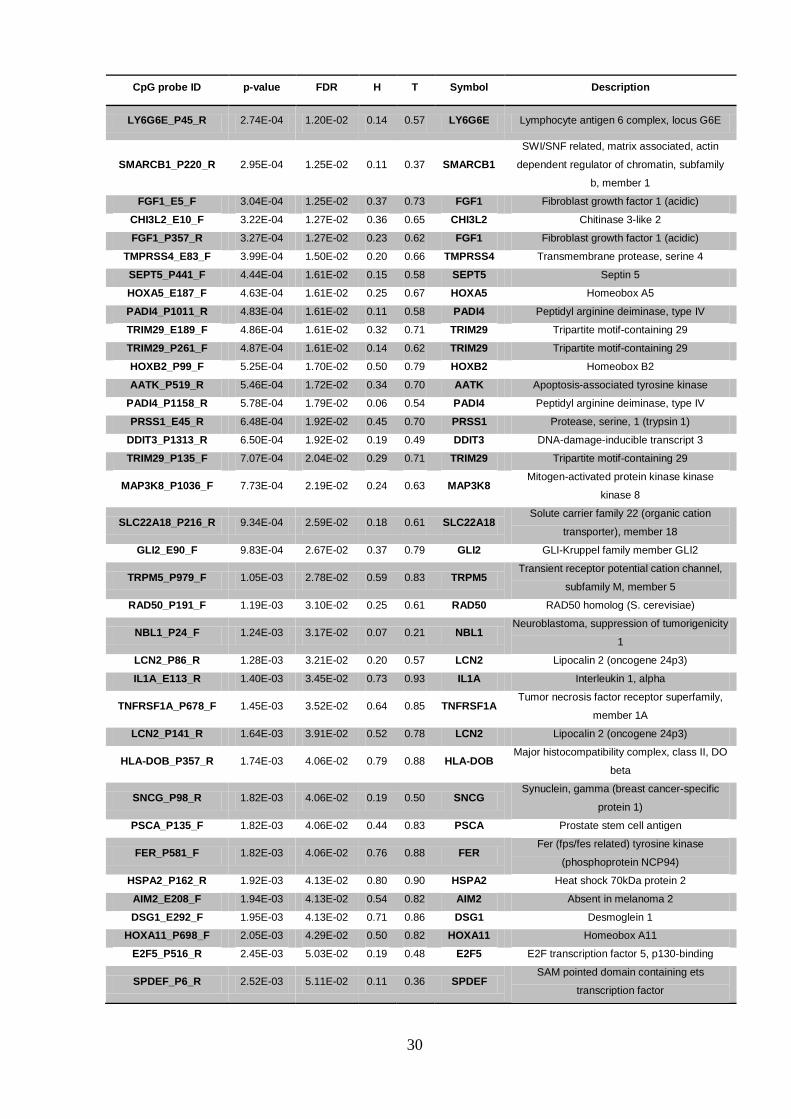
CpG probe ID p-value FDR H T Symbol Description
LY6G6E_P45_R 2.74E-04 1.20E-02 0.14 0.57 LY6G6E Lymphocyte antigen 6 complex, locus G6E
SMARCB1_P220_R 2.95E-04 1.25E-02 0.11 0.37 SMARCB1
SWI/SNF related, matrix associated, actin
dependent regulator of chromatin, subfamily
b, member 1
FGF1_E5_F 3.04E-04 1.25E-02 0.37 0.73 FGF1 Fibroblast growth factor 1 (acidic)
CHI3L2_E10_F 3.22E-04 1.27E-02 0.36 0.65 CHI3L2 Chitinase 3-like 2
FGF1_P357_R 3.27E-04 1.27E-02 0.23 0.62 FGF1 Fibroblast growth factor 1 (acidic)
TMPRSS4_E83_F 3.99E-04 1.50E-02 0.20 0.66 TMPRSS4 Transmembrane protease, serine 4
SEPT5_P441_F 4.44E-04 1.61E-02 0.15 0.58 SEPT5 Septin 5
HOXA5_E187_F 4.63E-04 1.61E-02 0.25 0.67 HOXA5 Homeobox A5
PADI4_P1011_R 4.83E-04 1.61E-02 0.11 0.58 PADI4 Peptidyl arginine deiminase, type IV
TRIM29_E189_F 4.86E-04 1.61E-02 0.32 0.71 TRIM29 Tripartite motif-containing 29
TRIM29_P261_F 4.87E-04 1.61E-02 0.14 0.62 TRIM29 Tripartite motif-containing 29
HOXB2_P99_F 5.25E-04 1.70E-02 0.50 0.79 HOXB2 Homeobox B2
AATK_P519_R 5.46E-04 1.72E-02 0.34 0.70 AATK Apoptosis-associated tyrosine kinase
PADI4_P1158_R 5.78E-04 1.79E-02 0.06 0.54 PADI4 Peptidyl arginine deiminase, type IV
PRSS1_E45_R 6.48E-04 1.92E-02 0.45 0.70 PRSS1 Protease, serine, 1 (trypsin 1)
DDIT3_P1313_R 6.50E-04 1.92E-02 0.19 0.49 DDIT3 DNA-damage-inducible transcript 3
TRIM29_P135_F 7.07E-04 2.04E-02 0.29 0.71 TRIM29 Tripartite motif-containing 29
MAP3K8_P1036_F 7.73E-04 2.19E-02 0.24 0.63 MAP3K8 Mitogen-activated protein kinase kinase
kinase 8
SLC22A18_P216_R 9.34E-04 2.59E-02 0.18 0.61 SLC22A18 Solute carrier family 22 (organic cation
transporter), member 18
GLI2_E90_F 9.83E-04 2.67E-02 0.37 0.79 GLI2 GLI-Kruppel family member GLI2
TRPM5_P979_F 1.05E-03 2.78E-02 0.59 0.83 TRPM5 Transient receptor potential cation channel,
subfamily M, member 5
RAD50_P191_F 1.19E-03 3.10E-02 0.25 0.61 RAD50 RAD50 homolog (S. cerevisiae)
NBL1_P24_F 1.24E-03 3.17E-02 0.07 0.21 NBL1 Neuroblastoma, suppression of tumorigenicity
1
LCN2_P86_R 1.28E-03 3.21E-02 0.20 0.57 LCN2 Lipocalin 2 (oncogene 24p3)
IL1A_E113_R 1.40E-03 3.45E-02 0.73 0.93 IL1A Interleukin 1, alpha
TNFRSF1A_P678_F 1.45E-03 3.52E-02 0.64 0.85 TNFRSF1A Tumor necrosis factor receptor superfamily,
member 1A
LCN2_P141_R 1.64E-03 3.91E-02 0.52 0.78 LCN2 Lipocalin 2 (oncogene 24p3)
HLA-DOB_P357_R 1.74E-03 4.06E-02 0.79 0.88 HLA-DOB Major histocompatibility complex, class II, DO
beta
SNCG_P98_R 1.82E-03 4.06E-02 0.19 0.50 SNCG Synuclein, gamma (breast cancer-specific
protein 1)
PSCA_P135_F 1.82E-03 4.06E-02 0.44 0.83 PSCA Prostate stem cell antigen
FER_P581_F 1.82E-03 4.06E-02 0.76 0.88 FER Fer (fps/fes related) tyrosine kinase
(phosphoprotein NCP94)
HSPA2_P162_R 1.92E-03 4.13E-02 0.80 0.90 HSPA2 Heat shock 70kDa protein 2
AIM2_E208_F 1.94E-03 4.13E-02 0.54 0.82 AIM2 Absent in melanoma 2
DSG1_E292_F 1.95E-03 4.13E-02 0.71 0.86 DSG1 Desmoglein 1
HOXA11_P698_F 2.05E-03 4.29E-02 0.50 0.82 HOXA11 Homeobox A11
E2F5_P516_R 2.45E-03 5.03E-02 0.19 0.48 E2F5 E2F transcription factor 5, p130-binding
SPDEF_P6_R 2.52E-03 5.11E-02 0.11 0.36 SPDEF SAM pointed domain containing ets
transcription factor
31
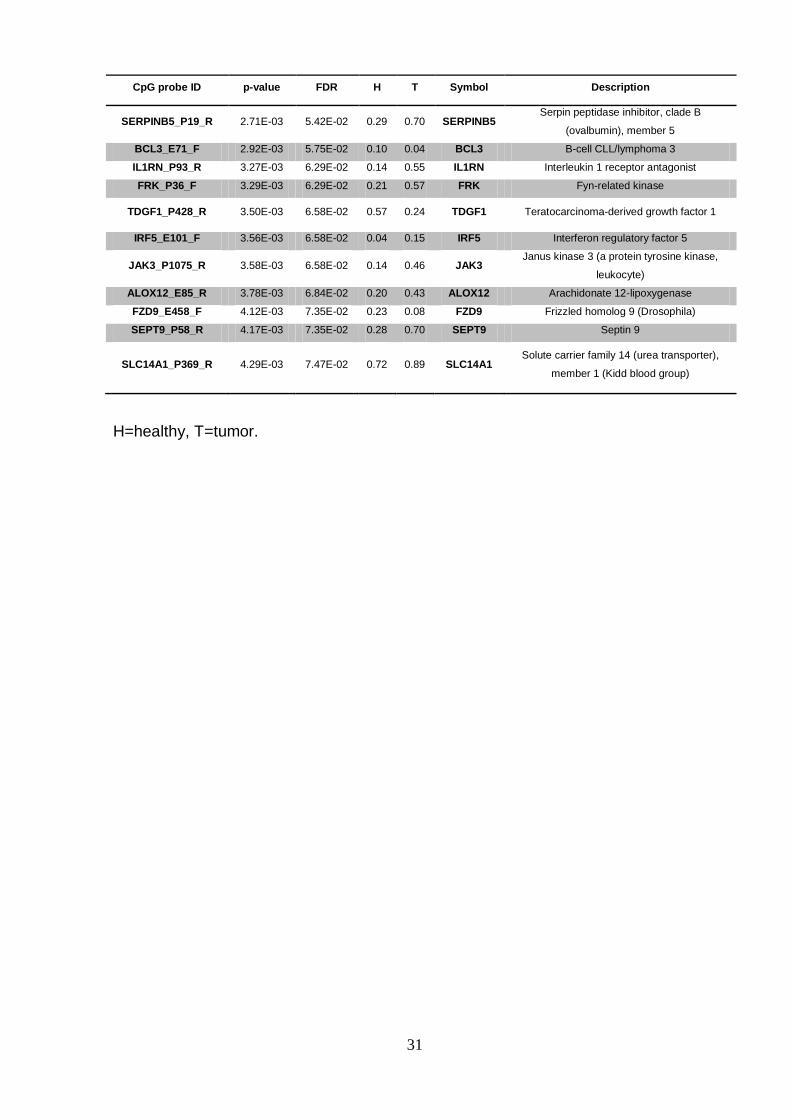
CpG probe ID p-value FDR H T Symbol Description
SERPINB5_P19_R 2.71E-03 5.42E-02 0.29 0.70 SERPINB5 Serpin peptidase inhibitor, clade B
(ovalbumin), member 5
BCL3_E71_F 2.92E-03 5.75E-02 0.10 0.04 BCL3 B-cell CLL/lymphoma 3
IL1RN_P93_R 3.27E-03 6.29E-02 0.14 0.55 IL1RN Interleukin 1 receptor antagonist
FRK_P36_F 3.29E-03 6.29E-02 0.21 0.57 FRK Fyn-related kinase
TDGF1_P428_R 3.50E-03 6.58E-02 0.57 0.24 TDGF1 Teratocarcinoma-derived growth factor 1
IRF5_E101_F 3.56E-03 6.58E-02 0.04 0.15 IRF5 Interferon regulatory factor 5
JAK3_P1075_R 3.58E-03 6.58E-02 0.14 0.46 JAK3 Janus kinase 3 (a protein tyrosine kinase,
leukocyte)
ALOX12_E85_R 3.78E-03 6.84E-02 0.20 0.43 ALOX12 Arachidonate 12-lipoxygenase
FZD9_E458_F 4.12E-03 7.35E-02 0.23 0.08 FZD9 Frizzled homolog 9 (Drosophila)
SEPT9_P58_R 4.17E-03 7.35E-02 0.28 0.70 SEPT9 Septin 9
SLC14A1_P369_R 4.29E-03 7.47E-02 0.72 0.89 SLC14A1 Solute carrier family 14 (urea transporter),
member 1 (Kidd blood group)
H=healthy, T=tumor.
32
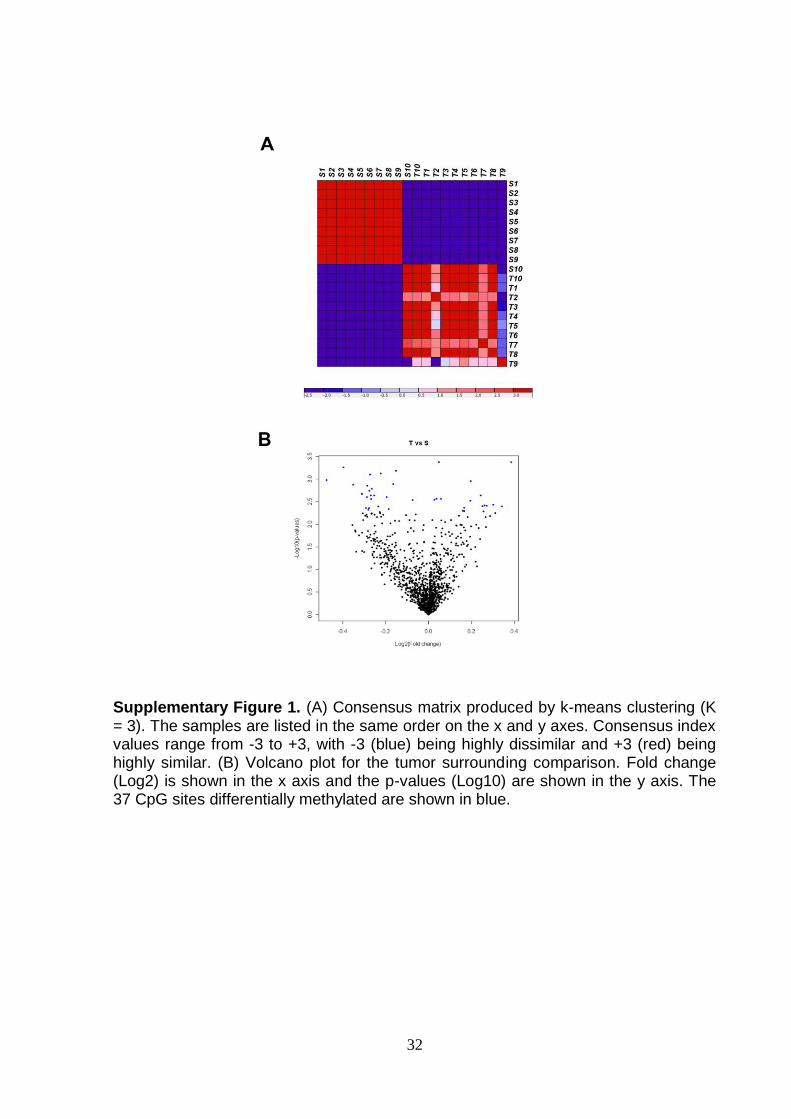
Supplementary Figure 1. (A) Consensus matrix produced by k-means clustering (K = 3). The samples are listed in the same order on the x and y axes. Consensus index values range from -3 to +3, with -3 (blue) being highly dissimilar and +3 (red) being highly similar. (B) Volcano plot for the tumor surrounding comparison. Fold change (Log2) is shown in the x axis and the p-values (Log10) are shown in the y axis. The 37 CpG sites differentially methylated are shown in blue.

33
Discussão
O CEE é um tumor extremamente fatal, principalmente em países em
desenvolvimento, e ainda muito pouco conhecido. Este trabalhou visou elucidar, pelo
menos em parte, a importância de alterações no perfil de metilação para este tipo de
tumor. De maneira bastante satisfatória, pudemos estabelecer um perfil tumor-
específico de alterações em metilação do DNA que revelou um enriquecimento de
determinadas vias celulares possivelmente fundamentais para o processo de
carcinogênese esofágica. Dentre elas, a perda de adesão celular e a indução do
processo inflamatório foram as mais evidentes. O epitélio esofágico, assim como
outros epitélios, apresenta uma organização bem específica na qual a hierarquia
celular é muito importante. A camada basal deste epitélio é formada por uma única
camada de células não diferenciadas com o potencial de formar todo o restante do
epitélio. A partir de divisões não simétricas de células desta camada, temos a
formação das camadas superiores deste tecido que sofrem um processo de
diferenciação celular conforme se aproximam do lúmen esofágico (Seery e Watt,
2000). Durante este processo, a modificação do perfil de expressão gênica é muito
importante, sendo que a substituição de determinados tipos de citoqueratina e a
alteração do tipo de adesão celular são as modificações mais bem descritas
(Bernerd e Asselineau, 1997; Del Bino, Vioux et al., 2004). Neste trabalho, de
maneira bastante interessante, encontramos alterações no perfil de metilação dos
promotores tanto de citoqueratinas como de moléculas de adesão, gerando o
enriquecimento da via de adesão celular. De uma maneira geral, as alterações
encontradas apontaram para uma perda de diferenciação celular, uma vez que tal
perfil se assemelhava àquele esperado para células basais do epitélio. A
hipermetilação de DSG1 encontrada nos tumores esofágicos, por exemplo, levaria a
uma menor expressão desta molécula desmosomal, o que já foi relacionado a uma
perda de diferenciação celular em pele. Getsios e colaboradores mostraram que a
DSG1 suprime a sinalização via EGFR, promovendo assim a diferenciação
epidérmica (Getsios, Simpson et al., 2009). Sendo assim, na camada basal do
epitélio a expressão de DSG1 não é encontrada, permitindo a ativação da via de
EGFR e, conforme a diferenciação ocorre, as células passam a expressar esta
molécula de adesão, inibindo esta via. Além disso, a hipermetilação de CK13, um
marcador de diferenciação epitelial, encontrada nos tumores esofágicos avaliados
neste estudo levaria ao silenciamento deste gene, perturbando assim a organização

34
do tecido (Ohta, Ogawa et al., 2010). De acordo com este panorama e com os
nossos dados, podemos sugerir que a mudança do perfil de expressão gênica
induzida por alterações na metilação do DNA em tumores esofágicos que resultaria
em uma perda de diferenciação celular pode ser fundamental para o processo de
carcinogênese esofágica.
A outra via apontada como alterada neste estudo, o componente inflamatório,
nos remete a um assunto bastante em voga atualmente que é o microambiente
tumoral (Hanahan e Weinberg, 2011). De uma maneira geral, é cada vez mais aceito
que células inflamatórias são importantes constituintes de tumores. Apesar de se
acreditar inicialmente que a presença destas células estaria correlacionada à
tentativa de erradicar células tumorais, sabe-se hoje que células inflamatórias podem
potencializar o processo de carcinogênese de diferentes maneiras. A liberação de
fatores de crescimento, quimiocinas e citocinas que amplificam a resposta
inflamatória é apenas uma das maneiras pelas quais a inflamação pode contribuir
para o desenvolvimento tumoral. Sendo assim, nosso estudo mostrou que alterações
do componente inflamatório podem ser fundamentais também para a carcinogênese
esofágica. Especificamente, a hipometilação de IL6 pode ser um fator importante
para este processo, uma vez que uma alta expressão desta citocina já foi
correlacionada a diversos tumores epiteliais (Schafer e Brugge, 2007). Tal
associação se deve principalmente à ativação de STAT3 que, por sua vez, regula
vias importantes para o processo de carcinogênese, como proliferação celular,
angiogênese e apoptose (Jarnicki, Putoczki et al., 2010). STAT3 bloqueia a
apoptose por ativar a expressão de genes anti-apoptóticos, como Bcl-2 e Bcl-XL, e
por induzir outras proteínas que suprimem indiretamente a apoptose, como a
chaperona Hsp70 (Bromberg, Wrzeszczynska et al., 1999; Stephanou, Brar et al.,
2000; Sikora e Grzesiuk, 2007). Além disso, STAT3 também bloqueia a via
extrínseca pela repressão transcripcional de FAS (Ivanov, Krasilnikov et al., 2002).
Já em relação à proliferação celular, STAT3 a promove principalmente por meio da
indução de ciclina B1, cdc2, c-myc e ciclina D1, assim como pela repressão de p21
(Bromberg, Wrzeszczynska et al., 1999; Masuda, Suzui et al., 2002; Bollrath, Phesse
et al., 2009). Por fim, STAT3 promove a angiogênese por induzir a transcrição de
VEGF e H1F1α, clássicos fatores de crescimento pró-angiogênicos (Niu, Wright et
al., 2002; Wei, Kuo et al., 2003) . Sendo assim, uma maior expressão de IL6 no
tecido esofágico levaria a uma maior ativação da via de STAT3 que poderia culminar
não só na promoção da sobrevivência celular, mas também em um maior potencial

35
angiogênico, eventos fundamentais para o crescimento tumoral. Um estudo recente
mostrou ainda que IL6 é capaz de alterar a metilação do DNA em células de
carcinoma oral, induzindo hipometilação global e hipermetilação de promotores
específicos, como CHFR, GATA5 e PAX6 (Gasche, Hoffmann et al., 2011). Sendo
assim, esta citocina pode ter um papel ainda mais abrangente no processo de
carcinogênese. Esta é a primeira vez que um papel de IL6 é sugerido no
desenvolvimento de CEE, porém o seu papel é mais claro para outros tumores,
conforme mencionado anteriormente. Em câncer de mama, por exemplo, a maior
expressão de IL6 é correlacionada ao estágio do tumor, a um maior número de sítios
metastáticos e a um mau prognóstico (Jiang, Yang et al., 2000; Salgado, Junius et
al., 2003). Baseado nisto, novas terapias que têm como alvo IL6 e STAT3 têm sido
propostas e algumas já têm mostrado resultados promissores no tratamento de
inflamações crônicas (Jarnicki, Putoczki et al., 2010). Dessa forma, a identificação de
alterações epigenéticas em IL6 feita neste estudo pode nos ajudar não só a
compreender melhor a biologia do CEE, como pode representar também um novo
alvo terapêutico para esta neoplasia, fatal na maioria dos casos.
Apesar de extremamente relevantes biologicamente, as alterações em
metilação do DNA em CEE identificadas neste estudo diferem daquelas previamente
reportadas. É evidente que o presente trabalho acessou tais alterações de uma
maneira muito mais abrangente, mas alguns genes classicamente apontados como
alterados em CEE por outros trabalhos não tiveram a mesma importância neste
estudo. Esse é o caso, por exemplo, dos genes p16INK4A e FHIT, o primeiro um
importante regulador do ciclo celular e o segundo envolvido no metabolismo de
purinas, ambos reportados como silenciados por hipermetilação do promotor em
uma série de neoplasias, incluindo CEE (Kuroki, Trapasso et al., 2003; Salam,
Hussain et al., 2009; Ling, Li et al., 2011). Neste caso, entretanto, a disparidade
encontrada pode ser explicada pelas populações estudadas, uma vez que tais
alterações foram identificadas em populações orientais e nosso estudo contou com
uma população ocidental. A principal diferença entre tais populações acerca do CEE
são os fatores de risco. Enquanto as populações orientais apresentam como
principais fatores etiológicos para CEE o consumo de bebidas quentes e a
contaminação de alimentos por nitrosaminas, no Brasil (assim como em outras
populações ocidentais) o tabagismo e o etilismo são apontados como mais
importantes. Além disso, a rigorosa análise utilizada neste estudo pode também ter
contribuído para a ausência de alguns genes entre aqueles diferencialmente

36
metilados. Este é o caso, por exemplo, do gene MGMT que não mostrou diferenças
entre os tumores e os tecidos normais adjacentes quando a plataforma
GoldenGate® foi utilizada, porém apresentou uma hipermetilação em tumores na
análise por pirosequenciamento, quando um valor de p mais permissivo foi utilizado.
Por fim, algumas disparidades em relação à literatura podem também ser explicadas
pela diferença de abordagem. Nosso estudo visou identificar diferenças do perfil de
metilação de DNA de tumores em comparação ao tecido normal adjacente, sem
qualquer tentativa de correlação com prognóstico. Isso poderia explicar, por
exemplo, porque o gene RASSF1A, previamente reportado como diferencialmente
metilado em CEE e associado à progressão do tumor, não foi identificado no nosso
estudo (Mao, Li et al., 2011). Na verdade, quando realizamos uma análise “piloto”
comparando tumores pouco diferenciados com tumores bem e moderadamente
diferenciados encontramos uma hipermetilação de RASSF1A no primeiro grupo
(dados não mostrados). Baseado nestes aspectos, podemos então compreender
melhor as disparidades do nosso estudo em relação à literatura.
Um outro aspecto bastante interessante deste trabalho foi a identificação de
potenciais alterações precoces em CEE. Uma vez que nosso trabalho contou com
amostras esofágicas de indivíduos sem câncer, pudemos comparar tal grupo com as
amostras de pacientes com CEE e identificar alterações na metilação do DNA que já
estivessem presentes no tecido adjacente de pacientes, considerado
patologicamente normal. A partir desta análise, dois genes foram identificados como
diferencialmente metilados e, para nossa surpresa, ambos pareciam ser de extrema
relevância biológica para o tecido esofágico. Um deles, o gene trefoil factor 1 (TFF1),
faz parte de uma família de peptídeos que desempenham um papel fundamental
para a restituição do epitélio gastrointestinal após uma injúria (Taupin e Podolsky,
2003). Estes fatores são pequenas proteínas resistentes a proteases que são
secretadas de maneira abundante na superfície de mucosas, normalmente em
conjunto com mucinas (Ribieras, Tomasetto et al., 1998; Taupin e Podolsky, 2003).
A regulação de TFF1 já foi bem estudada em câncer de mama e estômago e este
gene parece desempenhar um papel conflitante nestes dois tipos de tumor. Em
carcinoma de mama, foi demonstrado que a indução de TFF1 resulta em um
aumento da proliferação celular e sobrevivência, do crescimento independente de
ancoragem, da migração celular e invasão, assim como leva a um aumento do
tamanho de tumores em modelos de xenoenxerto (Amiry, Kong et al., 2009). Neste
tecido, o TFF1 é regulado principalmente por estradiol, uma vez que é um gene

37
regulado por estrogênio (Sun, Spencer et al., 2005). Já no estômago, o TFF1 tem
sido apontando como um gene supressor de tumor. Neste tecido, a regulação por
estrogênio parece não ser importante, uma vez que seu receptor normalmente não é
expresso, e este gene é encontrado silenciado por perda de heterozigose ou
hipermetilação do promotor em tumores gástricos (Carvalho, Kayademir et al., 2002).
Além disso, alterações em TFF1 parecem ser um evento precoce durante o
processo de carcinogênese gástrica, já sendo encontradas em lesões pré-
neoplásicas (Fujimoto, Yasui et al., 2000; Shi, Cai et al., 2006). Este panorama se
assemelha bastante àquele encontrado para nossas amostras, onde a
hipermetilação de TFF1 já é encontrada no tecido normal adjacente de pacientes
com CEE. Sendo assim, poderíamos propor que o TFF1 age como um gene
supressor de tumor para o esôfago assim como o faz para o estômago. Entretanto,
os dados encontrados na literatura até então são um pouco escassos. O único
trabalho que acessou a expressão de TFF1 em CEE até então mostrou apenas que
seus níveis são mais baixos neste tipo de tumor do que no adenocarcinoma de
esôfago (Mitas, Almeida et al., 2005). Dessa forma, estudos para comprovar a
importância de alterações em TFF1 para o CEE, assim como as suas consequências
funcionais são de fundamental importância.
O outro gene encontrado diferencialmente metilado já no tecido normal de
pacientes com CEE foi o BCL3. Este gene, ao contrário do que podemos imaginar,
não se assemelha em termos de sequência e função ao já bem caracterizado BCL2,
sendo que seus nomes são semelhantes apenas por terem sido descobertos em um
mesmo contexto, ou seja, alterados em leucemias/linfomas. Na verdade, o BCL3
ainda é um gene pouco conhecido, mas parece ter um papel fundamental da
regulação da transcrição mediada por NF-κB (Franzoso, Bours et al., 1993; Bauer,
Villunger et al., 2006). O gene BCL3 já foi encontrado superexpresso em diversos
tipos de tumor, como mama, linfoma, carcinoma de nasofaringe e carcinoma
hepatocelular, porém os mecanismos que levam a esta regulação ainda não são
claros (Cogswell, Guttridge et al., 2000; Thornburg, Pathmanathan et al., 2003;
Canoz, Rassidakis et al., 2004; O'neil, Buzkova et al., 2007). Em determinados tipos
de linfoma, as translocações incluindo o gene BCL3 são frequentes, mas para outros
tipos de tumor elas não são comuns (Szymanowska, Klapper et al., 2008). Sendo
assim, este trabalho propõe pela primeira vez uma perda de regulação epigenética
de BCL3 que poderia levar a uma alta expressão deste gene não só em CEE, mas
também em outros tumores.

38
De uma maneira geral, este trabalho identificou importantes alterações em
metilação do DNA em CEE assim como trouxe à tona possíveis alterações precoces
para este tipo de tumor. Para alguns dos genes identificados neste estudo, a
correlação inversa entre metilação do promotor e expressão gênica já é bem
estabelecida, como é o caso do TFF1 (Fujimoto, Yasui et al., 2000). Porém, para
outros esta correlação não foi acessada até então. Dessa forma, apesar de diversos
trabalhos afirmarem que esta correlação é real na maioria das vezes, seria
interessante confirmá-la caso a caso. Infelizmente, devido à falta de acesso a
amostras, não foi possível fazê-lo no presente estudo. Porém, tal investigação é de
nosso interesse e pretendemos realizá-la em um futuro próximo.

39
Capítulo II: Alterações epigenéticas no gene BCL3 e
suas consequências funcionais
Introdução
1. BCL3: um membro diferenciado da família IB
NF-B designa um grupo de fatores de transcrição definidos em parte por sua
habilidade de se ligar a uma região consenso no DNA de cerca de 10-11 pares de
base e inclui cinco membros: p65 (RelA), RelB, c-Rel, p50/p105 (NF-B1) e
p52/p100 (NF-B2) (Ohno, Takimoto et al., 1990; Massoumi, Chmielarska et al.,
2006). Estes fatores regulam uma vasta gama de processos fisiológicos e
patológicos, como inflamação, sistema imune, sobrevivência celular e proliferação e
eles se diferenciam em parte pelo estado em que são sintetizados (Ghosh e Karin,
2002). Enquanto os membros p65, RelB e c-Rel são sintetizados como proteínas
maduras associadas a proteínas inibitórias IB, os membros p50 e p52 são
sintetizados como grandes precursores denominados p105 e p100, respectivamente
(Massoumi, Chmielarska et al., 2006). Uma variedade de sinais pode liberar estes
fatores, normalmente retidos no citoplasma, e desencadear sua translocação para o
núcleo mediante a degradação dos inibidores IB ou pela clivagem de p105 e p100
(Massoumi, Chmielarska et al., 2006). Uma vez no núcleo, p65, RelB e c-Rel são
capazes não só de se ligar a regiões consenso no DNA por meio de seu domínio de
ligação ao DNA (domínio Rel), mas também de ativar a transcrição gênica via seu
domínio de transativação (Israel, 2000; Ghosh e Karin, 2002). Já p50 e p52
apresentam um domínio de ligação ao DNA, mas não possuem o domínio de
transativação, ou seja, eles são capazes de promover a transcrição gênica apenas
quando formam heterodímeros com outros membros da família (p65, RelB ou c-Rel)
ou quando recrutam seu co-ativador, o BCL3 (Fujita, Nolan et al., 1993; Watanabe,
Iwamura et al., 1997).
O gene BCL3 foi originalmente descrito em pacientes com leucemia linfocítica
crônica que apresentam a translocação t(14;19) (Asou, Takechi et al., 1993). Esta
translocação justapõe este gene ao locus da cadeia pesada de imunoglobulinas,
resultando em sua superexpressão (Ohno, Takimoto et al., 1990). Sabe-se hoje que

40
o BCL3 faz parte da família IB que compreende importantes reguladores dos
fatores de transcrição NF-B e é constituída por sete membros, sendo que todos
apresentam uma série de repetições “ankyrin” responsáveis pelas interações
proteína-proteína (Thornburg, Pathmanathan et al., 2003). O BCL3 se distingue dos
outros membros da família IB por se localizar no núcleo, não ser degradado
mediante ativação de NF-B e por possuir sete, no lugar de seis repetições “ankyrin”
(Ahmed e Milner, 2009).
O BCL3 pode tanto ativar quanto inibir a transcrição gênica. Carmody e
colaboradores demonstraram que BCL3 age como um regulador negativo da
sinalização mediada por receptores Toll-like em macrófagos murinos por meio do
bloqueio da ubiquitinização de p50 (Carmody, Ruan et al., 2007). Já a ativação da
transcrição dependente de BCL3 é um pouco controversa. Alguns trabalhos
mostraram que o BCL3 favorece a transativação mediada por NF-B por remover
homodímeros inibitórios p50 de sítios consenso B, possibilitando assim a ligação de
outros dímeros NF-B ativos (Franzoso, Bours et al., 1992; Bours, Franzoso et al.,
1993; Fujita, Nolan et al., 1993; Hayden e Ghosh, 2008). Além disso, BCL3 pode se
associar a dímeros p52, formando um complexo ternário capaz de ativar diretamente
a expressão gênica (Bours, Franzoso et al., 1993). Sendo assim, outros estudos são
necessários para melhor definir o modo de ação de BCL3 que parece ser
dependente do tipo celular, estímulo e associação a diferentes dímeros de NF-B.
2. BCL3, proliferação e sobrevivência celular
O gene BCL3 já foi encontrado superexpresso em alguns tipos de tumor,
como mama, linfoma, carcinoma de nasofaringe e carcinoma hepatocelular, porém
os mecanismos que levam a esta superexpressão, assim como as consequências
funcionais de tais alterações ainda não são claros (Cogswell, Guttridge et al., 2000;
Thornburg, Pathmanathan et al., 2003; Canoz, Rassidakis et al., 2004; O'neil,
Buzkova et al., 2007). Maldonado e colaboradores tentaram elucidar, ao menos em
parte, as vias reguladas por BCL3. Estes autores analisaram em larga escala as
alterações de expressão de mRNA após o silenciamento de BCL3 em uma linhagem
celular de colo de útero e, de maneira bastante interessante, identificaram uma
gama de genes que parecem ser direta ou indiretamente regulados por BCL3
(Maldonado, Espinosa et al., 2010). Ao todo, cento e dezoito genes tiveram a
expressão aumentada e sessenta e nove genes tiveram sua expressão diminuída

41
após o silenciamento de BCL3, sendo que a análise de enriquecimento de vias
mostrou uma maior representação das vias de crescimento celular, proliferação,
morte celular e regulação da expressão gênica. Além disso, neste mesmo trabalho
foi mostrado que STAT3, um importante oncogene em tumores humanos, era o
nódulo central destas vias.
Outros trabalhos demonstraram mais especificamente o papel de BCL3 na
proliferação celular. Gua e colaboradores mostraram que a expressão ectópica de
miR-25b, encontrado silenciado em câncer de ovário, induz parada do ciclo celular e
leva a uma redução da proliferação celular, sendo tais efeitos mediados pelo
silenciamento de BCL3 (Guan, Yao et al., 2011). Outro estudo mostrou ainda que o
complexo p52-BCL3 é capaz de induzir a expressão de Ciclina D1 em células
epiteliais humanas do brônquio por meio da ligação ao sítio IB de seu promotor
(Wang, Shi et al., 2010). Além disso, trabalhos com camundongos transgênicos que
superexpressam Bcl3 mostraram que tais animais apresentam esplenomegalia e um
acúmulo de células B maduras em nódulos linfáticos, na medula óssea e na
cavidade peritoneal (Ong, Hackbarth et al., 1998). Tal fenótipo foi diretamente
correlacionado a um aumento da proliferação celular e pode explicar, ao menos em
parte, o papel do Bcl3 em leucemias/linfomas.
Outra importante via que parece ser regulada por BCL3 é a vida da apoptose.
Kashatus e colaboradores mostraram que BCL3 pode ser induzido por danos ao
DNA e é necessário para a indução de HDM2 e supressão da atividade persistente
de p53, bloqueando assim a apoptose induzida por este importante fator de
transcrição (Kashatus, Cogswell et al., 2006). Além disso, foi demonstrado que BCL3
interage com a proteína CtBP1, promovendo sua estabilização por bloquear sua
ubiquitinização (Choi, Lee et al., 2010). Sabe-se que CtBP1 reprime uma série de
genes pró-apoptóticos, o que poderia explicar, ao menos em parte, o efeito anti-
apoptótico de BCL3. Além disso, tal evidência sugere que os efeitos de BCL3 vão
além daqueles mediados por NF-κB, dando início assim a uma busca por outros
parceiros desta proteína.
Apesar destas evidências acerca dos efeitos celulares e moleculares do
BCL3, seu papel real no processo de carcinogênese ainda não é bem definido. Não
se sabe ao certo qual das funções desempenhadas pelo BCL3, na proliferação
celular ou apoptose, seria mais importante durante este processo ou até mesmo se
ambas seriam desempenhadas. A realidade hoje é que este gene apresenta um
grande potencial para ser correlacionado a importantes vias celulares, mas suas

42
funções ainda são pouco conhecidas. Tal panorama é ainda mais obscuro quando o
assunto é sua regulação.
3. Estrutura e regulação de BCL3
O gene BCL3 encontra-se no braço longo do cromossomo 19 (Fig. 2.1A),
possui nove exons e três ilhotas CpG, sendo uma na região promotora e duas no
corpo do gene (Fig. 2.1B). Apesar de possuir ilhotas tão bem definidas, apenas um
trabalho sugeriu até então uma função de tais elementos na regulação da expressão
de BCL3. Nishikori e colaboradores mostraram que a expressão deste gene
encontra-se aumentada em linhagens celulares de linfoma de grandes células
anaplásticas em comparação a linhagens de doença de Hodgkin, sendo que este
aumento foi correlacionado à amplificação gênica em alguns casos e à
hipometilação da ilhota CpG 3 em outros casos (Nishikori, Maesako et al., 2003).
Entretanto, nenhuma correlação foi encontrada entre a expressão de BCL3 e o perfil
de metilação da ilhota CpG que se sobrepõe ao promotor do gene, uma vez que tal
região foi encontrada desmetilada em todos os casos testados.
Figura 2.1: Organização do gene BCL3 no genoma e suas potencias regiões de controle. (A) Localização cromossômica do gene BCL3. (B) Distribuição de exons do gene BCL3 e suas ilhotas CpG, representadas pelos retângulos verdes. (C) Representação esquemática do gene BCL3, ressaltando suas ilhotas CpG (estrelas verdes) e seu “enhancer”. Figura obtida a partir do site http://genome.ucsc.edu/, acessado em 10/12/2011.

43
A regulação de BCL3 vai além de sua transcrição. Já foi demonstrado que a
proteína BCL3 é constitutivamente fosforilada in vivo, sendo tal processo mediado
pela enzima GSK3 (Viatour, Merville et al., 2004). Uma vez fosforilada, a proteína é
então direcionada à degradação pelo proteasoma, o que previne seu acúmulo na
célula (Viatour, Merville et al., 2004). Acredita-se que tal mecanismo previna a
atividade oncogênica de BCL3 em situações normais, porém, uma vez que BCL3 é
superexpresso, sua fosforilação e consequente degradação podem não ser
suficientes para evitar tal potencial.
Em termos de sinais extracelulares, já foi demonstrado que a expressão de
BCL3 pode ser regulada por algumas citocinas. Rebollo e colaboradores mostraram
que IL4 é capaz de induzir a expressão do mRNA e protéica de Bcl3 em células T
murinas, sendo tal indução mediada por fatores de transcrição AP1 e/ou AP1-like,
que se ligam a regiões consenso no promotor do gene Bcl3 (Rebollo, Dumoutier et
al., 2000). Além disso, também já foi descrito que IL6 é capaz de induzir a expressão
de BCL3 em células de mieloma (Brocke-Heidrich, Kretzschmar et al., 2004). Neste
caso o mediador identificado foi STAT3, que se liga a uma região regulatória no
íntron 2 posteriormente caracterizada com um “enhancer” (Fig. 2.1C) (Brocke-
Heidrich, Ge et al., 2006). Por fim, a transcrição de BCL3 pode ser reprimida por seu
próprio produto gênico, sendo que tal feedback negativo se dá via os sítios de
ligação a NF-κB presentes no promotor e no enhancer do gene (Brocke-Heidrich, Ge
et al., 2006).
Dentre as alterações encontradas neste estudo a partir da análise em larga
escala, a hipometilação de BCL3 se destacou não só pela relevância biológica deste
gene, mas também por ter sido encontrada já no tecido normal adjacente de
pacientes. Além disso, relatos da literatura mostram que BCL3 é regulado por
inflamação, mais especificamente por IL6, o que torna este gene ainda mais
relevante para a carcinogênese esofágica. Nosso estudo mostrou que o componente
inflamatório é uma das principais vias alteradas em CEE e, dessa forma, sua
interação com as células epiteliais esofágicas poderia explicar as alterações
encontradas em BCL3. Entretanto, até então, estas são apenas especulações já que
não há qualquer outro relato na literatura da importância de BCL3 para CEE ou da
regulação deste gene pelo componente inflamatório no esôfago. Sendo assim,
decidimos concentrar os estudos funcionais na regulação de BCL3 em células
esofágicas, assim como em sua participação para a homeostase do tecido.
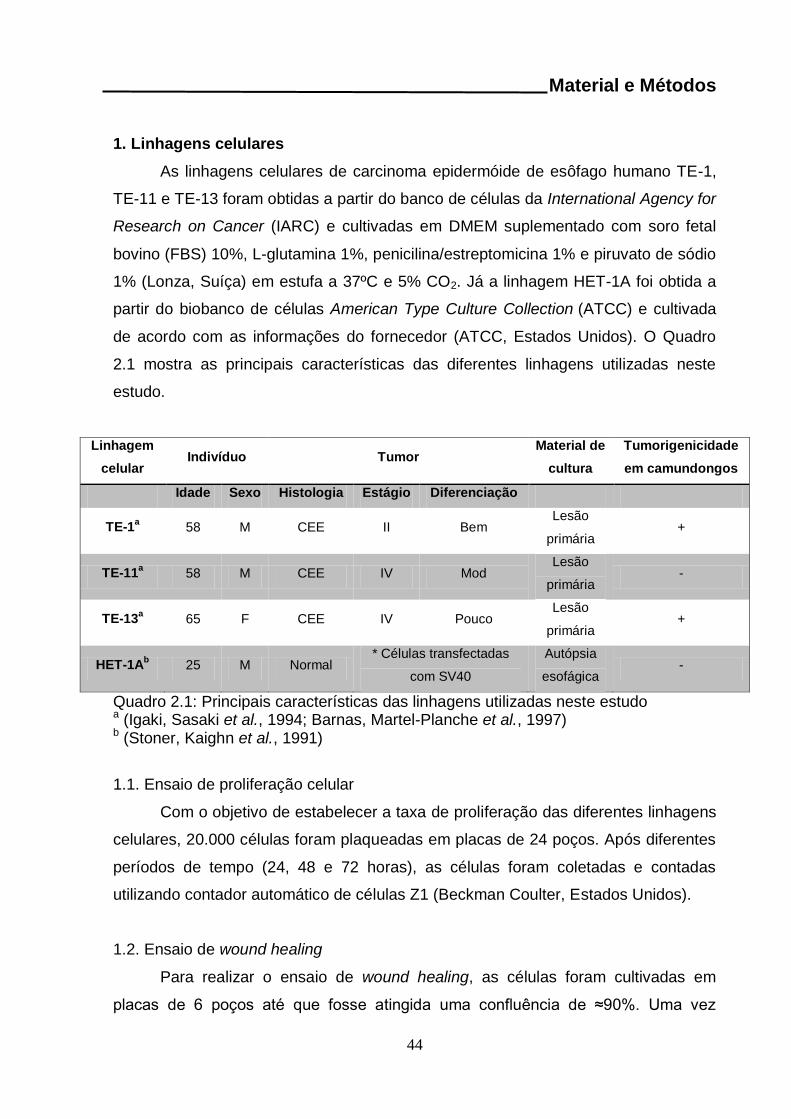
44
Material e Métodos
1. Linhagens celulares
As linhagens celulares de carcinoma epidermóide de esôfago humano TE-1,
TE-11 e TE-13 foram obtidas a partir do banco de células da International Agency for
Research on Cancer (IARC) e cultivadas em DMEM suplementado com soro fetal
bovino (FBS) 10%, L-glutamina 1%, penicilina/estreptomicina 1% e piruvato de sódio
1% (Lonza, Suíça) em estufa a 37ºC e 5% CO2. Já a linhagem HET-1A foi obtida a
partir do biobanco de células American Type Culture Collection (ATCC) e cultivada
de acordo com as informações do fornecedor (ATCC, Estados Unidos). O Quadro
2.1 mostra as principais características das diferentes linhagens utilizadas neste
estudo.
Linhagem
celular Indivíduo Tumor
Material de
cultura
Tumorigenicidade
em camundongos
Idade Sexo Histologia Estágio Diferenciação
TE-1a 58 M CEE II Bem
Lesão
primária +
TE-11a 58 M CEE IV Mod
Lesão
primária -
TE-13a 65 F CEE IV Pouco
Lesão
primária +
HET-1Ab 25 M Normal
* Células transfectadas
com SV40
Autópsia
esofágica -
Quadro 2.1: Principais características das linhagens utilizadas neste estudo
a (Igaki, Sasaki et al., 1994; Barnas, Martel-Planche et al., 1997) b (Stoner, Kaighn et al., 1991)
1.1. Ensaio de proliferação celular
Com o objetivo de estabelecer a taxa de proliferação das diferentes linhagens
celulares, 20.000 células foram plaqueadas em placas de 24 poços. Após diferentes
períodos de tempo (24, 48 e 72 horas), as células foram coletadas e contadas
utilizando contador automático de células Z1 (Beckman Coulter, Estados Unidos).
1.2. Ensaio de wound healing
Para realizar o ensaio de wound healing, as células foram cultivadas em
placas de 6 poços até que fosse atingida uma confluência de ≈90%. Uma vez

45
atingida tal confluência, os poços foram “riscados” utilizando uma ponteira de P200,
de modo a formar uma “fenda” na região central do poço. As placas foram então
acompanhadas e fotografadas após diferentes períodos de tempo para que fosse
avaliado o potencial de repopulação da “fenda” formada.
1.3. Tratamento com Interleucina 6 (IL6)
As linhagens celulares foram tratadas com IL6 (Peprotech, Estados Unidos)
em diferentes concentrações e por diferentes períodos de tempo pela adição direta
da citocina ao meio de cultura de modo a atingir a concentração desejada.
1.4. Extração de DNA a partir de linhagens celulares
As células foram coletadas e centrifugadas de modo a formar um pellet. A
este pellet foram adicionados 500 µL de tampão TAIL (50 mM Tris-HCl pH 8,0, 100
mM EDTA, 100mM NaCl, 1% SDS) e 25 µL de proteinase K 10 mg/mL e tal mistura
foi incubada overnight a 55ºC. Foram então adicionados 200 µL de NaCl 6M que
foram homogeneizados pela inversão dos tubos. Seguiu-se uma centrifugação de 10
minutos à velocidade máxima à temperatura ambiente. O sobrenadante foi
transferido para um novo tubo, ao qual foram adicionados 500 µL de isopropanol
(Merck, Alemanha). A solução foi homogeneizada pela inversão dos tubos e seguiu-
se uma centrifugação de 10 minutos à velocidade máxima à temperatura ambiente.
O sobrenadante foi então descartado e o pellet foi lavado com 500 µL de etanol
(Merck, Alemanha) 70%. Seguiu-se uma centrifugação de 10 minutos à velocidade
máxima à temperatura ambiente após a qual o etanol foi descartado. O pellet foi
então ressuspendido em tampão TE (10 mM Tris-HCl; 1mM EDTA- pH 7,5).
1.4.1. Quantificação do DNA
Após a extração, os DNAs foram quantificados através da absorbância
medida em 260 e 280 nm em espectrofotômetro (UV- 160, Shimadzu/Japão). A
razão de A260 e A280 deve ser igual ou superior a 1,75 para, então, calcular-se a
concentração do DNA (Sambrook e Russel, 2001). Uma unidade de absorbância
corresponde a uma concentração de 50 μg/mL de DNA.
1.4.2. Tratamento com Bissulfito de Sódio
Com o objetivo de transformar as citosinas não metiladas em uracila (as
citosinas metiladas permanecem inalteradas), 500 ng de DNA genômico foram
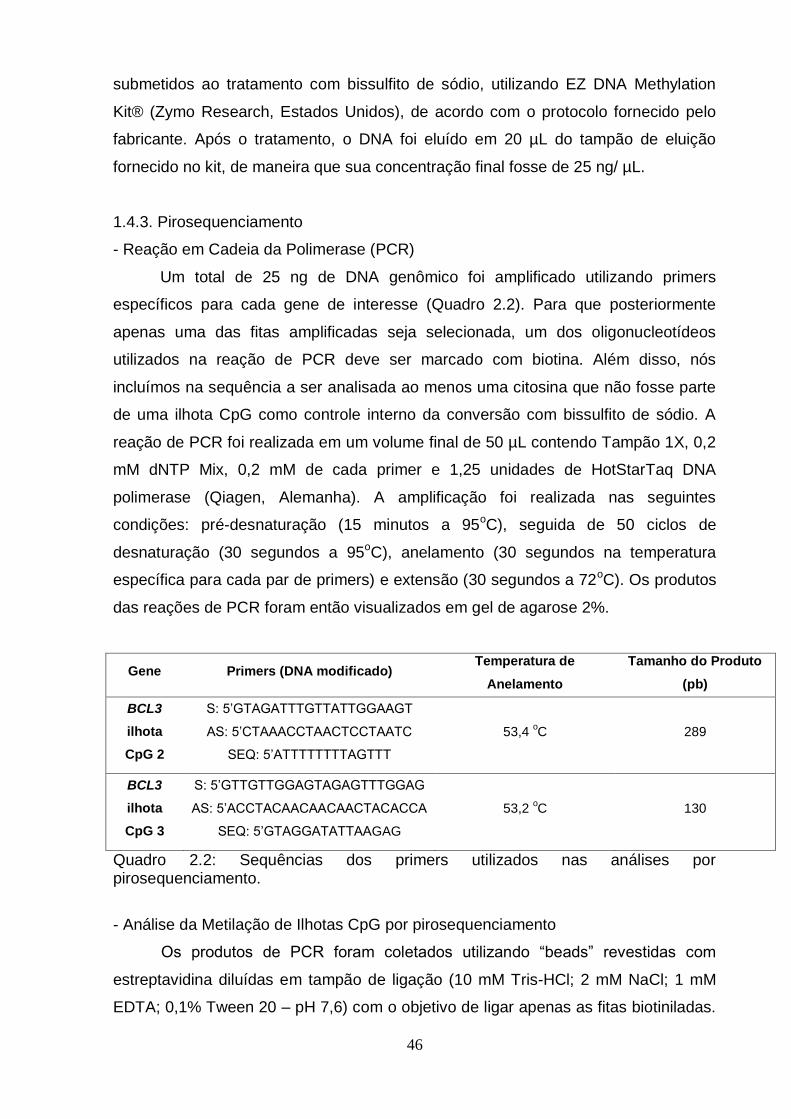
46
submetidos ao tratamento com bissulfito de sódio, utilizando EZ DNA Methylation
Kit® (Zymo Research, Estados Unidos), de acordo com o protocolo fornecido pelo
fabricante. Após o tratamento, o DNA foi eluído em 20 µL do tampão de eluição
fornecido no kit, de maneira que sua concentração final fosse de 25 ng/ µL.
1.4.3. Pirosequenciamento
- Reação em Cadeia da Polimerase (PCR)
Um total de 25 ng de DNA genômico foi amplificado utilizando primers
específicos para cada gene de interesse (Quadro 2.2). Para que posteriormente
apenas uma das fitas amplificadas seja selecionada, um dos oligonucleotídeos
utilizados na reação de PCR deve ser marcado com biotina. Além disso, nós
incluímos na sequência a ser analisada ao menos uma citosina que não fosse parte
de uma ilhota CpG como controle interno da conversão com bissulfito de sódio. A
reação de PCR foi realizada em um volume final de 50 µL contendo Tampão 1X, 0,2
mM dNTP Mix, 0,2 mM de cada primer e 1,25 unidades de HotStarTaq DNA
polimerase (Qiagen, Alemanha). A amplificação foi realizada nas seguintes
condições: pré-desnaturação (15 minutos a 95oC), seguida de 50 ciclos de
desnaturação (30 segundos a 95oC), anelamento (30 segundos na temperatura
específica para cada par de primers) e extensão (30 segundos a 72oC). Os produtos
das reações de PCR foram então visualizados em gel de agarose 2%.
Gene Primers (DNA modificado) Temperatura de
Anelamento
Tamanho do Produto
(pb)
BCL3
ilhota
CpG 2
S: 5’GTAGATTTGTTATTGGAAGT
AS: 5’CTAAACCTAACTCCTAATC
SEQ: 5’ATTTTTTTTAGTTT
53,4 oC 289
BCL3
ilhota
CpG 3
S: 5’GTTGTTGGAGTAGAGTTTGGAG
AS: 5’ACCTACAACAACAACTACACCA
SEQ: 5’GTAGGATATTAAGAG
53,2 oC 130
Quadro 2.2: Sequências dos primers utilizados nas análises por pirosequenciamento.
- Análise da Metilação de Ilhotas CpG por pirosequenciamento
Os produtos de PCR foram coletados utilizando “beads” revestidas com
estreptavidina diluídas em tampão de ligação (10 mM Tris-HCl; 2 mM NaCl; 1 mM
EDTA; 0,1% Tween 20 – pH 7,6) com o objetivo de ligar apenas as fitas biotiniladas.

47
Após 10 minutos de agitação, as “beads” foram aspiradas utilizando a “Vacuum Prep
Workstation”, lavadas com etanol 70%, seguida por desnaturação com NaOH e
lavagem (10 mM Tris-Acetato – pH 7,6). Após a adição de 40 µL da solução de
primer de sequenciamento 0,4 mM diluído em tampão de anelamento (20 mM Tris-
Acetato; 2 mM MgAc2 – pH 7,6), o MIX (“beads” e oligonucleotídeos) foi desnaturado
por 2 minutos a 80oC e então resfriado por 20 minutos para atingir a temperatura de
anelamento dos primers. Os volumes de dNTPs, enzimas e substrato foram
definidos de acordo com a sequência a ser injetada. A fita biotinilada foi então
sequenciada com o sistema PSQ TM96MA (Biotage, Suécia).
Esta técnica baseia-se no sequenciamento por síntese, onde temos a adição
e incorporação sequencial de nucleotídeos (Fig. 2.2). O nucleotídeo só é
incorporado pela DNA polimerase se for complementar à fita molde, sendo que essa
incorporação pode ser monitorada em tempo real (Dejeux, El Abdalaoui et al., 2009).
Inicialmente, um oligonucleotídeo desenhado especialmente para o sequenciamento
se anela à fita simples do produto gerado na reação de PCR, a qual serve como
molde para a síntese. São então adicionadas as enzimas DNA polimerase, ATP
sulfurilase, luciferase e apirase, assim como os substratos adenosina 5’ fosfosulfato
(APS) e luciferina. Em seguida, o primeiro desoxiribonucleotídeo trifosfato (dNTP) é
adicionado à reação. Caso ele seja complementar à fita molde, a DNA polimerase o
incorpora à fita que está sendo sintetizada. Cada incorporação é acompanhada da
liberação de pirofosfato (PPi) em uma quantidade equimolar à quantidade de
nucleotídeo incorporada. A enzima ATP sulfurilase converte então o PPi em ATP na
presença de APS. O ATP produzido é utilizado na conversão de luciferina em
oxiluciferina (reação catalisada pela enzima luciferase) que gera luz em uma
intensidade proporcional à quantidade de ATP. A luz produzida é captada e
visualizada como um pico no gráfico luz X tempo, sendo que a altura de cada pico é
proporcional à quantidade de nucleotídeos adicionados. Enquanto isso, a apirase
degrada continuamente os nucleotídeos não incorporados e o ATP. Quando esta
etapa é concluída, um novo nucleotídeo é adicionado (Dejeux, El Abdalaoui et al.,
2009).

48
Figura 2.2: Esquema geral da técnica de pirosequenciamento. (A) Anelamento do primer para sequenciamento à fita simples do produto gerado na reação de PCR e incorporação do desoxiribonucleotídeo trifosfato (dNTP) à fita que está sendo sintetizada com consequente liberação de pirofosfato (PPi). (B) A enzima sulfurilase converte adenosina 5’ fosfosulfato (APS) e PPi em ATP que é posteriormente utilizado na conversão de luciferina em oxiluciferina. Nesta etapa há a liberação de luz, que é captada e representada como um pico no gráfico luz X tempo. (C) Durante todo o experimento, a enzima apirase degrada os nucleotídeos não incorporados, assim como o ATP. Adaptado de: http://www.pyrosequencing.com/DynPage.aspx?id=7454.
O método de pirosequenciamento trata cada sítio CpG como um polimorfismo
C/T e gera um dado quantitativo da proporção relativa do alelo metilado versus o
alelo não metilado. Os níveis de metilação dos sítios CpG de interesse foram
avaliados convertendo os pirogramas em valores numéricos correspondentes às
alturas dos picos obtidos (Fig. 2.3) e calculando a média de todos os sítios CpG
analisados para um promotor específico (Vaissiere, Hung et al., 2009).
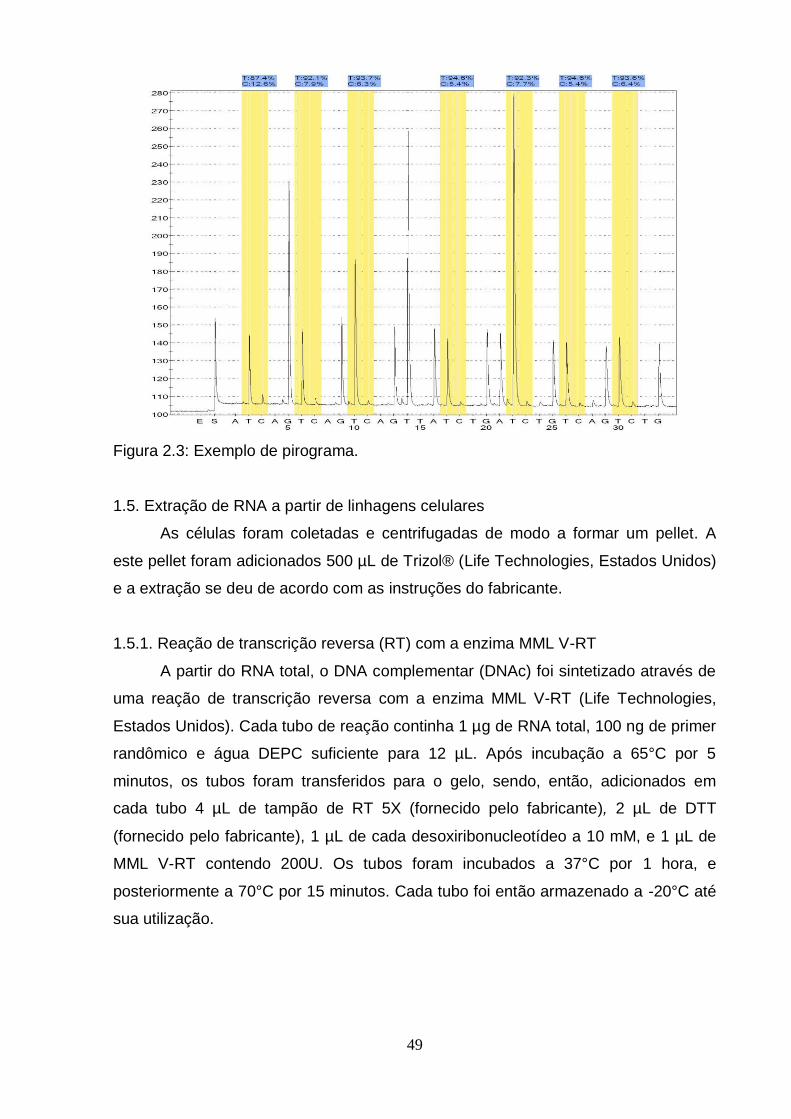
49
Figura 2.3: Exemplo de pirograma.
1.5. Extração de RNA a partir de linhagens celulares
As células foram coletadas e centrifugadas de modo a formar um pellet. A
este pellet foram adicionados 500 µL de Trizol® (Life Technologies, Estados Unidos)
e a extração se deu de acordo com as instruções do fabricante.
1.5.1. Reação de transcrição reversa (RT) com a enzima MML V-RT
A partir do RNA total, o DNA complementar (DNAc) foi sintetizado através de
uma reação de transcrição reversa com a enzima MML V-RT (Life Technologies,
Estados Unidos). Cada tubo de reação continha 1 µg de RNA total, 100 ng de primer
randômico e água DEPC suficiente para 12 µL. Após incubação a 65°C por 5
minutos, os tubos foram transferidos para o gelo, sendo, então, adicionados em
cada tubo 4 µL de tampão de RT 5X (fornecido pelo fabricante), 2 µL de DTT
(fornecido pelo fabricante), 1 µL de cada desoxiribonucleotídeo a 10 mM, e 1 µL de
MML V-RT contendo 200U. Os tubos foram incubados a 37°C por 1 hora, e
posteriormente a 70°C por 15 minutos. Cada tubo foi então armazenado a -20°C até
sua utilização.

50
1.5.2. Reação em cadeia da polimerase quantitativa (PCRq)
A amplificação de cada DNAc foi feita pelo sistema de detecção de seqüência
Mx3000P (Stratagene, Estados Unidos). A reação de PCRq foi realizada em placas
de 96 poços com 5 µL do reagente SYBR Green PCR Master Mix 2X (Applied
Biosystems, Estados Unidos), 10 pmoles de cada par de oligonucleotídeos (ver
Quadro 2.3), 2 µL do DNAc (diluído 10 X) a ser analisado e água deionizada estéril
para completar o volume final de 10 L. A reação de amplificação foi realizada nas
seguintes condições: pré-desnaturação (5 minutos a 95oC), seguida de 40 ciclos de
desnaturação (15 segundos a 95oC), anelamento e extensão (1 minuto a 60oC).
Todas as reações foram feitas adicionando a este programa a curva de dissociação.
Isto permitiu avaliar, caso houvesse, a formação de produtos inespecíficos e/ou
contaminação.
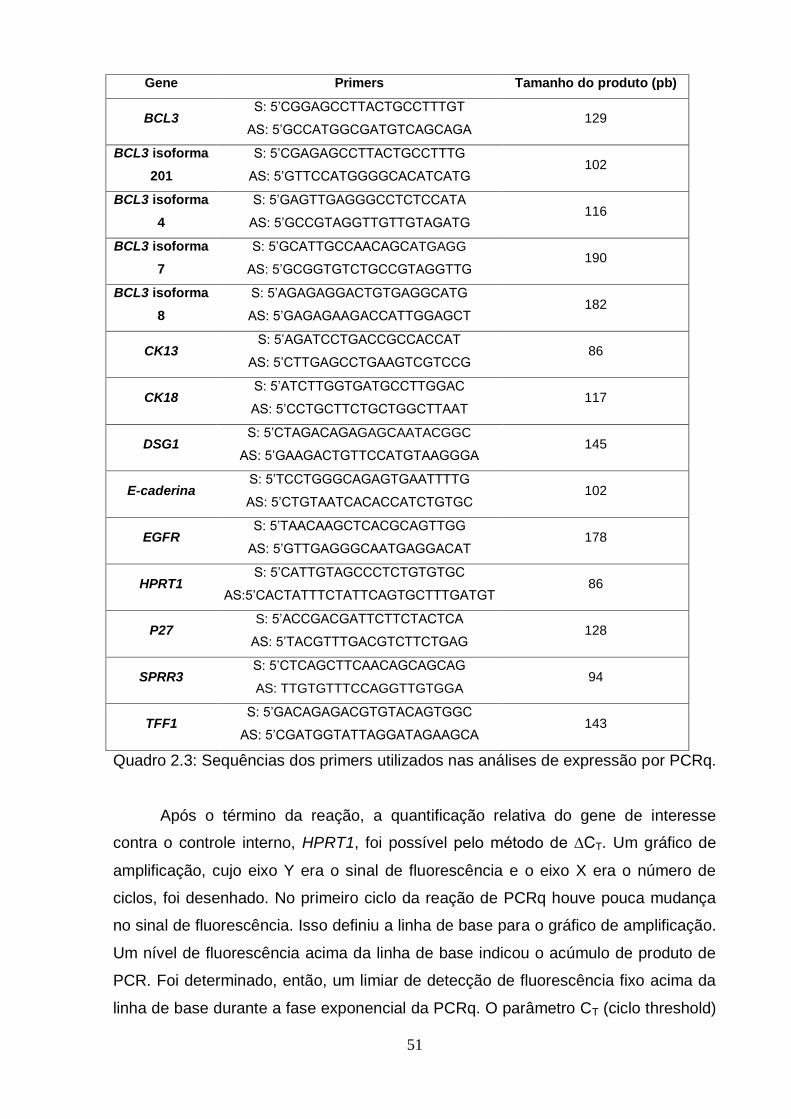
51
Gene Primers Tamanho do produto (pb)
BCL3 S: 5’CGGAGCCTTACTGCCTTTGT
AS: 5’GCCATGGCGATGTCAGCAGA 129
BCL3 isoforma
201
S: 5’CGAGAGCCTTACTGCCTTTG
AS: 5’GTTCCATGGGGCACATCATG 102
BCL3 isoforma
4
S: 5’GAGTTGAGGGCCTCTCCATA
AS: 5’GCCGTAGGTTGTTGTAGATG 116
BCL3 isoforma
7
S: 5’GCATTGCCAACAGCATGAGG
AS: 5’GCGGTGTCTGCCGTAGGTTG 190
BCL3 isoforma
8
S: 5’AGAGAGGACTGTGAGGCATG
AS: 5’GAGAGAAGACCATTGGAGCT 182
CK13 S: 5’AGATCCTGACCGCCACCAT
AS: 5’CTTGAGCCTGAAGTCGTCCG 86
CK18 S: 5’ATCTTGGTGATGCCTTGGAC
AS: 5’CCTGCTTCTGCTGGCTTAAT 117
DSG1 S: 5’CTAGACAGAGAGCAATACGGC
AS: 5’GAAGACTGTTCCATGTAAGGGA 145
E-caderina S: 5’TCCTGGGCAGAGTGAATTTTG
AS: 5’CTGTAATCACACCATCTGTGC 102
EGFR S: 5’TAACAAGCTCACGCAGTTGG
AS: 5’GTTGAGGGCAATGAGGACAT 178
HPRT1 S: 5’CATTGTAGCCCTCTGTGTGC
AS:5’CACTATTTCTATTCAGTGCTTTGATGT 86
P27 S: 5’ACCGACGATTCTTCTACTCA
AS: 5’TACGTTTGACGTCTTCTGAG 128
SPRR3 S: 5’CTCAGCTTCAACAGCAGCAG
AS: TTGTGTTTCCAGGTTGTGGA 94
TFF1 S: 5’GACAGAGACGTGTACAGTGGC
AS: 5’CGATGGTATTAGGATAGAAGCA 143
Quadro 2.3: Sequências dos primers utilizados nas análises de expressão por PCRq.
Após o término da reação, a quantificação relativa do gene de interesse
contra o controle interno, HPRT1, foi possível pelo método de ∆CT. Um gráfico de
amplificação, cujo eixo Y era o sinal de fluorescência e o eixo X era o número de
ciclos, foi desenhado. No primeiro ciclo da reação de PCRq houve pouca mudança
no sinal de fluorescência. Isso definiu a linha de base para o gráfico de amplificação.
Um nível de fluorescência acima da linha de base indicou o acúmulo de produto de
PCR. Foi determinado, então, um limiar de detecção de fluorescência fixo acima da
linha de base durante a fase exponencial da PCRq. O parâmetro CT (ciclo threshold)

52
foi definido como o número de ciclos, inteiro ou não, no qual a fluorescência
ultrapassou os limiares previamente fixados. A diferença (∆CT) entre as médias (de
três experimentos) do gene de interesse e do HPRT1 foi calculada pelo programa
Microsoft Excel e o valor de quantificação relativa foi expresso como 2-∆CT.
1.6. Imunofluorescência de células cultivadas em monocamada
Para que a imunomarcação fosse realizada, as células foram cultivadas em
placas de 24 poços sobre uma lamínula. Após os tratamentos de interesse, o meio
de cultura foi retirado e foram adicionados 500 µL de metanol:acetona (1:1) a cada
poço, para que as células fossem fixadas. As placas foram então mantidas a -20ºC
por 15 minutos. Após este período, foi adicionado 1 mL de PBS contendo BSA
(albumina sérica bovina) 3% e as placas foram mantidas por 1 hora à temperatura
ambiente. Seguiu-se uma lavagem com PBS (Lonza, Suíça) por 5 minutos sob
agitação leve, após a qual foi adicionado o anticorpo primário diluído em PBS. Após
incubação por 1 hora à temperatura ambiente, as células foram lavadas 3 vezes com
PBS por 5 minutos cada. Foi então adicionado o anticorpo secundário diluído em
PBS, que foi incubado por 1 hora à temperatura ambiente. Seguiram-se 3 lavagens
com PBS por 5 minutos cada. Após as lavagens, as lâminas foram montadas com
meio de montagem contendo DAPI (Vector Laboratories, Estados Unidos). Após a
secagem, as células foram visualizadas em microscópio de fluorescência Nikon
Eclipse Ti (Nikon, Japão).
1.7. Transfecção com plasmídeo e/ou esiRNA
As transfecções das diferentes linhagens celulares com o plasmídeo de
expressão para BCL3 (gentilmente doado pelo Dr Kunitada Shimotohno da Kyoto
University, Japão) e/ou com o esiRNA contra BCL3 (Life Technologies, Estados
Unidos) foram feitas utilizando Lipofectamina 2000 (Life Technologies, Estados
Unidos), de acordo com o protocolo do fabricante com algumas alterações.
Resumidamente, as células foram cultivadas em placas de 24 poços até que a
confluência atingisse cerca de 50%. Um dia antes da transfecção, o meio de cultura
foi substituído por 500 µL de meio completo sem antibióticos. No dia da transfecção,
1 µg de plasmídeo e/ou 1 µg de esiRNA foram diluídos em 50 µL de DMEM, assim
como 2 µL de Lipofectamina 2000 foram diluídos em 50 µL de DMEM. Ambas as
soluções foram incubadas por 5 minutos à temperatura ambiente e após este
período elas foram misturadas. A nova solução contendo Lipofectamina 2000 e

53
plasmídeo ou esiRNA foi então incubada por 20 minutos à temperatura ambiente.
Após esta incubação, o volume total da mistura foi adicionado ao poço da placa de
24 poços. O meio de cultura foi trocado após 24 horas. Acessamos então o
silenciamento gênico e a superexpressão de BCL3 por RT-PCRq e
imunofluorescência após 48 horas.
2. Cultivo de células em matrigel
Com o objetivo de estabelecer um modelo de diferenciação celular para
células esofágicas, cultivamos as células em matrigel (BD Biosciences, Estados
Unidos) seguindo o protocolo descrito por Debnath e colaboradores para células
mamárias, com algumas adaptações (Debnath, Muthuswamy et al., 2003). De
maneira resumida, placas de vidro de 8 poços (Nunc, Estados Unidos) foram
colocadas no fluxo laminar sobre uma placa de gelo. Em seguida, foram adicionados
100 µL de matrigel a cada poço e a matriz foi espalhada utilizando uma ponteira de
P200. A placa foi então incubada a 37ºC por 1 hora. Enquanto a matriz estava
solidificando, uma placa confluente de células foi tripsinizada e ressuspendida em
meio de cultura. Em seguida, as células foram contadas e diluídas de modo a atingir
uma concentração de 5.000 células/mL. Em paralelo, foi preparada uma solução de
matrigel 4% diluída em meio de cultura. A solução de células a 5.000 células/mL foi
então misturada com a solução de matrigel 4% em uma proporção 1:1 e 400 µL
dessa mistura foram adicionados a cada poço. As células foram cultivadas em estufa
a 37ºC e 5% CO2 por 14 dias, sendo que o meio de cultura foi trocado a cada 3-4
dias, sendo substituído por meio contendo 2% matrigel.
2.1. Extração de RNA de células cultivadas em matrigel
Para uma extração de RNA eficiente, foi necessário remover as células da
cama de matrigel através da diluição desta matriz. Para isso, utilizamos o protocolo
descrito por Lee e colaboradores com algumas alterações (Lee, Kenny et al., 2007).
De maneira resumida, o meio de cultura foi removido de cada poço e em seguida as
células foram lavadas 2 vezes com 400 µL de PBS gelado. Foram então adicionados
800 µL de PBS-EDTA (5 mM EDTA, 1 mM NaVO4, 1.5 mM NaF em PBS) e a matriz
foi “descolada” da superfície da placa com o auxílio de uma ponteira de P200. A
placa foi então submetida a agitação leve por 30 minutos a 4ºC. Em seguida, a
solução foi transferida para um tubo tipo Falcon de 15 mL e o poço foi lavado com
400 µL de PBS-EDTA, que também foram transferidos para o tubo tipo Falcon. O

54
tubo foi submetido a agitação leve por 30 minutos a 4 ºC e em seguida verificamos
se a matriz estava completamente dissolvida. Caso não estivesse, o tubo era
mantido por mais tempo sob agitação e/ou era adicionado mais PBS-EDTA. Os
tubos foram então centrifugados por 5 minutos a ≈ 115 g de modo a formar um
pellet. A este pellet foram adicionados 800 µL de Trizol® e a extração se deu de
acordo com as instruções do fabricante. As análises que se seguiram, como RT e
PCRq foram feitas como descritas anteriormente nesta seção.
2.2. Imunofluorescência de células cultivadas em matrigel
A imunofluorescência de células cultivadas em matrigel foi feita diretamente
na placa onde essas células foram cultivadas de acordo com Debnath e
colaboradores, com algumas alterações (Debnath, Muthuswamy et al., 2003).
Resumidamente, o meio de cultura foi retirado, foram adicionados 400 µL de
paraformaldeído 4% e as placas foram mantidas à temperatura ambiente por 45
minutos. Em seguida, o paraformaldeído foi retirado com bastante cuidado, uma vez
que a matrigel pode se dissolver após essa etapa (dependendo do lote) e é
fundamental que as células não sejam aspiradas. Após a retirada completa do
paraformaldeído, as células foram deixadas por 5 minutos à temperatura ambiente
para que “secassem”. Seguiu-se então uma etapa de permeabilização com 400 µL
de PBS contendo 0.5% Triton X-100 por 15 minutos à temperatura ambiente. Após
esta etapa, foi feito o bloqueio com 400 µL de tampão IF (PBS contendo 10% FBS e
0,05% Tween-20) por 1 hora à temperatura ambiente. Após este período, foram
adicionados 100 µL de anticorpo primário diluído em PBS contendo 0,05% Tween-20
e seguiu-se uma incubação à temperatura ambiente overnight. No dia seguinte,
foram realizadas 3 lavagens sob agitação leve com PBS contendo 0,05% Tween-20
à temperatura ambiente por 10 minutos cada. Seguiu-se a incubação com 100 µL de
anticorpo secundário diluído em PBS contendo 0,05% Tween-20 por 1 hora à
temperatura ambiente. Após esta incubação, foram realizadas 3 lavagens sob
agitação leve com PBS contendo 0,05% Tween-20 à temperatura ambiente por 10
minutos cada. Para corar o núcleo, foi feita uma incubação com TOPRO-3 5 µM
diluído em PBS por 15 minutos à temperatura ambiente. Seguiu-se uma lavagem
com PBS por 5 minutos. Após a lavagem, foi removida a grade de plástico, foi
adicionado meio de montagem e foi colocada uma lamínula sobre a lâmina. Após a
secagem da lâmina, esta foi visualizada em microscópio Nikon Eclipse Ti (Nikon,
Japão) e/ou microscópio confocal 200M (Zeiss, Estados Unidos).

55
2.3. Inclusão das células cultivadas em matrigel em parafina
Para incluir as células cultivadas em matrigel em parafina, o meio de cultura
foi retirado, as células foram lavadas com PBS 2 vezes e, em seguida, foram fixadas
com paraformaldeído 4% por 45 minutos. Após a fixação, a matriz diluiu restando
apenas células na placa que foram retiradas com o auxílio de um bisturi. Estas
células foram então colocadas sobre uma cama de agarose de baixo ponto de fusão
e foram cobertas em seguida também com agarose. Os “sanduíches” de agarose
foram processados segundo técnicas histológicas convencionais, e incluídos
separadamente em parafina (Paraplast, Sigma). De cada bloco foram obtidos
inicialmente 6 cortes histológicos em micrótomo com espessura de 3 a 5 µm, que
foram corados por Hematoxilina & Eosina (HE) e analisados ao microscópio óptico.
Novos cortes foram obtidos para a realização da técnica de imunofluorescência.
3. Análises estatísticas
As análises estatísticas deste trabalho foram feitas com o auxílio do programa
GraphPad Prism (GraphPad Software Inc.). Primeiramente, os grupos analisados
foram testados quanto a normalidade. Caso a distribuição dos dados fosse normal, o
teste T de Student (pareado ou não, dependendo do caso) era utilizado. Já quando
os dados não apresentavam uma distribuição normal, no caso de amostras
pareadas, utilizamos o teste de Wilcoxon; no caso de amostras não pareadas,
utilizamos o teste de Mann-Whitney e no caso de mais de dois grupos, utilizamos o
teste de Kruskal-Wallis com pós-teste de comparação múltipla de Dunn. Em todos os
casos, as diferenças foram consideradas estatisticamente significativas quando
p<0,05.

56
Resultados e Discussão
1. Regulação da expressão de BCL3 por IL6
Com o objetivo de verificar se BCL3 é induzido por IL6 em células esofágicas,
duas linhagens celulares de esôfago foram tratadas com diferentes doses de IL6
(10, 20 e 100 ng/mL) por diferentes períodos de tempo (30 minutos, 24 e 72 horas).
A Fig. 2.4 mostra que a expressão do mRNA de BCL3 é induzida em ambas as
linhagens, com algumas diferenças em relação à dose e ao tempo de tratamento. No
caso da linhagem TE-11 (Fig. 2.4A e B), que é uma linhagem obtida a partir de um
CEE, as doses de 10 ng/mL e 100 ng/mL foram capazes de induzir de maneira
estatisticamente significativa a expressão de BCL3 após 24 horas, e após 30
minutos e 24 horas, respectivamente (Fig. 2.4A). O tratamento com a dose de 100
ng/mL por 30 minutos foi então repetido (Fig. 2.4B), confirmando a indução de BCL3
por IL6 em TE-11, e estas condições foram escolhidas para os experimentos que se
seguiram com esta linhagem. Já a linhagem HET-1A apresentou uma indução
significativa de BCL3 por IL6 em praticamente todas as doses e tempos de
tratamento testados, com exceção da dose de 10 ng/mL por 24 horas (Fig. 2.4C). O
tratamento com a dose de 10 ng/mL por 30 minutos foi então repetido (Fig. 2.4D),
confirmando a indução de BCL3 por IL6 em HET-1A, e estas condições foram
escolhidas para os experimentos que se seguiram com esta linhagem. A linhagem
HET-1A foi obtida a partir de uma autópsia de tecido esofágico saudável, o que pode
explicar a indução mais eficiente neste caso. De acordo com os nossos resultados
obtidos no capítulo anterior, a hipometilação de IL6 e sua provável superexpressão
são alterações frequentes durante a carcinogênese esofágica e, portanto, já
estariam presentes em linhagens derivadas de CEE. Sendo assim, a linhagem TE-
11 já teria uma ativação da via de sinalização desencadeada por IL6 e, portanto, o
tratamento com a citocina exógena não desencadearia os mesmos efeitos que em
uma célula não tumoral.
57
Figura 2.4: Expressão do mRNA de BCL3 em diferentes linhagens esofágicas após o tratamento com IL6 em diferentes doses (10, 20 e 100 ng/mL) por diferentes períodos de tempo (30 minutos, 24 e 72 horas). (A) TE-11. (B) TE-11 tratada com IL6 100 ng/mL por 30 minutos. (C) HET-1A. (D) HET-1A tratada com IL6 10 ng/mL por 30 minutos. Teste T não-pareado *p<0,05.
2. Avaliação do perfil de metilação do gene BCL3 em amostras humanas e em
linhagens celulares após o tratamento com IL6
Com o objetivo de melhor compreender a regulação do gene BCL3,
investigamos mais a fundo suas potenciais regiões de controle. A ilhota CpG que se
sobrepõe ao primeiro exon do gene BCL3 foi a ilhota acessada pela plataforma
GoldenGate® e corresponde à região promotora do gene, regulando portanto a sua
expressão. Infelizmente, por motivos técnicos, não foi possível desenhar um ensaio
de pirosequenciamento para avaliar o perfil de metilação desta região em um
número maior de amostras ou em linhagens celulares. A região promotora do gene
BCL3 é muito rica em sítios CpG, além de se tratar de uma região repetitiva, o que
dificultou o desenho e o anelamento de primers, impossibilitando a análise.
Já a ilhota CpG 2 está presente no corpo do gene, é a menos densa das
ilhotas encontradas em BCL3 e está posicionada bastante próxima ao enhancer do
gene. O perfil de metilação desta ilhota foi avaliado por pirosequenciamento e não

58
foram encontradas diferenças estatisticamente significantes entre os grupos de
amostras humanas (tecido esofágico de indivíduos saudáveis e tecido normal
adjacente e tumoral de pacientes com CEE) considerando-se tanto a metilação
média dos 5 sítios CpG avaliados (Fig. 2.5A) quanto cada sítio separadamente (Fig.
2.5C). Além disso, esta análise evidenciou a presença de outliers para todos os
sítios analisados, porém sem qualquer associação com a identidade da amostra
(Fig. 2.5B). Esta mesma região foi avaliada em linhagens celulares (TE-11 e HET-
1A) tratadas com IL6, contudo todos os sítios CpG apresentaram 0% de metilação
em todas as condições testadas. Tais resultados se mostraram surpreendentes, uma
vez que os corpos dos genes normalmente encontram-se hipermetilados,
principalmente se o gene estiver ativo transcripcionalmente (Ball, Li et al., 2009).
Porém, os baixos níveis de metilação encontrados na ilhota CpG 2 podem ser
explicados por sua proximidade ao enhancer do gene. Esta é uma região bastante
susceptível à ligação de fatores de transcrição e, como descrito em diversos
trabalhos, a metilação normalmente impede a ligação de tais fatores (Deaton e Bird,
2011). Sendo assim, seria importante que essa região fosse mantida desmetilada
para permitir a indução da expressão de BCL3.

59
Figura 2.5: Perfil de metilação da ilhota CpG 2 do gene BCL3 em amostras esofágicas de indivíduos saudáveis e pacientes com CEE (tecido normal, adjacente e tumor). (A) Metilação média dos 5 sítios CpG analisados por grupo de amostras. (B) Distribuição dos níveis de metilação para cada sítio avaliado. (C) Perfil de metilação de cada grupo por sitio CpG. Teste de Mann-Whitney.
Por fim, a ilhota CpG 3, que também está presente no corpo do gene, se
mostra a região com a maior densidade de sítios CpG do gene BCL3. O perfil de
metilação desta região foi acessado por pirosequenciamento e foram analisados ao
todo 5 sítios CpG. A partir da análise da média desses 5 sítios, não foram
encontradas diferenças estatisticamente significantes entre os diferentes grupos de
amostras humanas (Fig. 2.6A). Porém, ao avaliar a distribuição dos níveis de
metilação para cada sítio analisado, percebemos que o sítio CpG 5 apresentava
níveis consideravelmente mais baixos em comparação aos sítios vizinhos (Fig.
2.6B). Sendo assim, avaliamos cada sítio separadamente e, de maneira bastante
interessante, percebemos que no sítio CpG 5 tanto o tecido normal adjacente quanto
o tecido tumoral de pacientes com CEE apresentavam níveis de metilação mais altos
em comparação ao tecido esofágico de indivíduos saudáveis (Fig. 2.6C). Este perfil
é exatamente o oposto daquele encontrado para a região promotora dos mesmos
grupos de amostra, onde o tecido normal adjacente e tumoral de pacientes
apresentam uma hipometilação em comparação ao tecido saudável.

60
Figura 2.6: Perfil de metilação da ilhota CpG 3 do gene BCL3 em amostras esofágicas de indivíduos saudáveis e pacientes com CEE (tecido normal, adjacente e tumor). (A) Metilação média dos 5 sítios CpG analisados por grupo de amostras. (B) Distribuição dos níveis de metilação para cada sítio avaliado. (C) Perfil de metilação de cada grupo por sitio CpG. Teste de Mann-Whitney *p<0,05.
As mesmas análises foram então feitas para linhagens celulares tratadas com
IL6. De uma maneira geral, o perfil de metilação das linhagens HET-1A e TE-11 se
apresentou bastante semelhante àquele das amostras humanas. Ou seja, a média
de metilação dos 5 sítios analisados foi alta, sendo que não foram encontradas
diferenças estatisticamente significantes quando comparamos as células tratadas
com IL6 com seus respectivos controles (Fig. 2.7A). Entretanto, quando a
distribuição de metilação de cada sítio foi analisada, mais uma vez o sítio CpG 5 se
destacou por apresentar níveis de metilação inferiores aos dos outros sítios (Fig.
2.7B). A análise de cada sítio individualmente nos mostrou então que para o sítio
CpG 5, ambas as linhagens celulares tratadas com IL6 apresentaram um aumento
dos níveis de metilação em comparação aos seus respectivos controles (Fig. 2.7C).
Além disso, a linhagem TE-11, proveniente de um CEE, apresentou níveis mais
elevados de metilação neste mesmo sítio em comparação à linhagem HET-1A,
proveniente de um tecido esofágico saudável.

61
Figura 2.7: Perfil de metilação da ilhota CpG 3 do gene BCL3 em linhagens celulares esofágicas HET-1A e TE-11 tratadas com IL6. (A) Metilação média dos 5 sítios CpG analisados por grupo. (B) Distribuição dos níveis de metilação para cada sítio avaliado agrupando as amostras tratadas e não tratadas com IL6. (C) Perfil de metilação de cada grupo por sitio CpG. Teste de Mann-Whitney *p<0,05.
Os resultados encontrados para a ilhota CpG 3 vão de acordo com a literatura
no que diz respeito aos altos níveis de metilação encontrados no corpo de genes
transcripcionalmente ativos (Ball, Li et al., 2009). Além disso, após o tratamento com
IL6, que conforme mostrado anteriormente levou a indução de expressão do mRNA
de BCL3, tais níveis se mostraram ainda mais elevados para o sítio CpG 5. De
maneira bastante interessante, tal aumento foi semelhante àquele observado em
amostras de pacientes com CEE em comparação ao tecido esofágico de indivíduos
saudáveis, sugerindo que a citocina IL6 possa mimetizar in vitro o que acontece in
vivo durante o processo de carcinogênese esofágica. O significado de tais
diferenças de metilação no corpo do gene BCL3 ainda não é claro, mas trabalhos da
literatura sugerem que uma região com tal densidade de sítios CpG como a ilhota
CpG 3 possa representar um promotor alternativo (Maunakea, Nagarajan et al.,
2010). Sendo assim, decidimos avaliar o possível papel da ilhota CpG 3 na
regulação de transcritos alternativos.

62
3. Avaliação da expressão de transcritos alternativos do gene BCL3 em
linhagens celulares esofágicas tratadas com IL6
O gene BCL3 apresenta um total de 9 transcritos alternativos além do
transcrito “clássico” (até então analisado neste trabalho), sendo que apenas um
deles codifica uma proteína (Fig. 2.8). Decidimos então avaliar a expressão destes
transcritos alternativos em linhagens celulares esofágicas tratadas com IL6 para
investigar se a ilhota CpG 3 poderia regular a expressão de algum deles. Foram
desenhados então primers para cada transcrito, sendo que para apenas 4 deles foi
possível fazê-lo de modo a obter uma amplificação específica, uma vez que para os
outros primers havia sempre um anelamento com mais de um transcrito, não
permitindo a amplificação seletiva. Dessa forma, foi possível apenas a análise da
expressão dos transcritos BCL3-201, BCL3-004, BCL3-007 e BCL3-008.
Figura 2.8: Transcritos do gene BCL3. O transcrito BCL3-001 representa o transcrito “clássico”, enquanto os outros seriam os transcritos alternativos. Os transcritos alternativos circundados em vermelho são aqueles analisados neste estudo. Obtido a partir do site http://www.ncbi.nlm.nih.gov/mapview/, data de acesso 30/11/2011.
Após o tratamento com IL6, a expressão de todos os transcritos analisados foi
induzida de maneira significativa em ambas as linhagens testadas, com exceção do
transcrito BCL3-007 na linhagem TE-11 (Fig. 2.9). Isto sugere que o perfil de
metilação da ilhota CpG 3 não regula diretamente a expressão de tais transcritos,
uma vez que foi observado um aumento da metilação desta região após o
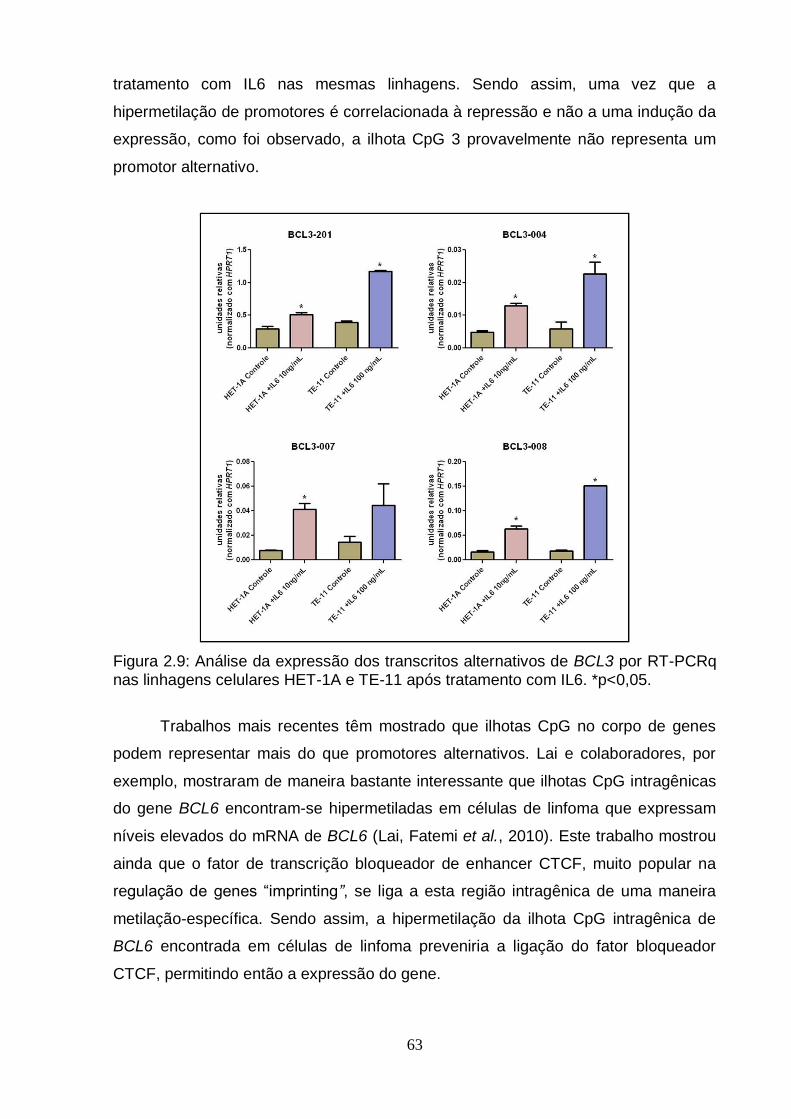
63
tratamento com IL6 nas mesmas linhagens. Sendo assim, uma vez que a
hipermetilação de promotores é correlacionada à repressão e não a uma indução da
expressão, como foi observado, a ilhota CpG 3 provavelmente não representa um
promotor alternativo.
Figura 2.9: Análise da expressão dos transcritos alternativos de BCL3 por RT-PCRq nas linhagens celulares HET-1A e TE-11 após tratamento com IL6. *p<0,05.
Trabalhos mais recentes têm mostrado que ilhotas CpG no corpo de genes
podem representar mais do que promotores alternativos. Lai e colaboradores, por
exemplo, mostraram de maneira bastante interessante que ilhotas CpG intragênicas
do gene BCL6 encontram-se hipermetiladas em células de linfoma que expressam
níveis elevados do mRNA de BCL6 (Lai, Fatemi et al., 2010). Este trabalho mostrou
ainda que o fator de transcrição bloqueador de enhancer CTCF, muito popular na
regulação de genes “imprinting”, se liga a esta região intragênica de uma maneira
metilação-específica. Sendo assim, a hipermetilação da ilhota CpG intragênica de
BCL6 encontrada em células de linfoma preveniria a ligação do fator bloqueador
CTCF, permitindo então a expressão do gene.

64
Outro trabalho publicado recentemente evidenciou ainda a versatilidade de
CTCF, ligando este fator diretamente à regulação epigenética e a doenças. Ohlsson
e colaboradores mostraram que CTCF pode funcionar não apenas como repressor
da transcrição, mas também como ativador e insulador (Ohlsson, Renkawitz et al.,
2001). Dessa forma, este fator, anteriormente associado apenas às regiões de
controle por “imprinting”, pode ajudar a elucidar o papel de alterações em metilação
do DNA no desenvolvimento de doenças, incluindo o câncer.
Tais evidências nos direcionam a especular que o mesmo tipo de regulação
de BCL6 possa ocorrer em BCL3. De acordo com os nossos achados, a ilhota CpG
3 poderia representar um sítio de ligação para CTCF que se encontra hipermetilado,
bloqueando assim a ligação deste fator, em situações em que o gene BCL3 é
expresso. Sendo assim, uma vez que esta expressão é induzida, teríamos um
aumento concomitante dos níveis de metilação. Entretanto, estudos posteriores
devem ser feitos para comprovar tal hipótese.
4. Estabelecimento do modelo de diferenciação celular em matrigel utilizando
células esofágicas.
Alguns modelos que visam o estabelecimento da organização e polaridade
epiteliais in vitro já foram estabelecidos. Tais modelos utilizaram como substrato
membranas basais reconstituídas ou colágeno e linhagens celulares animais, por
exemplo: “Madin-Darby canine kidney” – MDCK, células primárias de pâncreas de
rato e células glândula mamária normal murina - NMuMG (Hall, Farson et al., 1982;
Montesano, Mouron et al., 1983; Barcellos-Hoff, Aggeler et al., 1989; Montesano,
Schaller et al., 1991). O modelo de diferenciação epitelial de células mamária
humanas em matrigel foi primeiramente descrito por Petersen e colaboradores e foi
descrito como um modelo capaz de diferenciar células normais de células malignas
(Petersen, Ronnov-Jessen et al., 1992). Neste trabalho os autores mostraram que
células mamárias normais formam estruturas semelhantes a ácinos, enquanto
células tumorais falham em fazê-lo. Dessa forma, este estudo mostrou que o cultivo
de células em matrigel não só mimetiza a organização e polaridade epiteliais in vitro,
como também é capaz de prever a identidade tumoral das células. De acordo com
este panorama, visamos o estabelecimento de um modelo semelhante utilizando
células esofágicas de modo que pudéssemos melhor compreender o papel do gene
BCL3 na organização epitelial esofágica normal, assim como no processo de
tumorigênese.
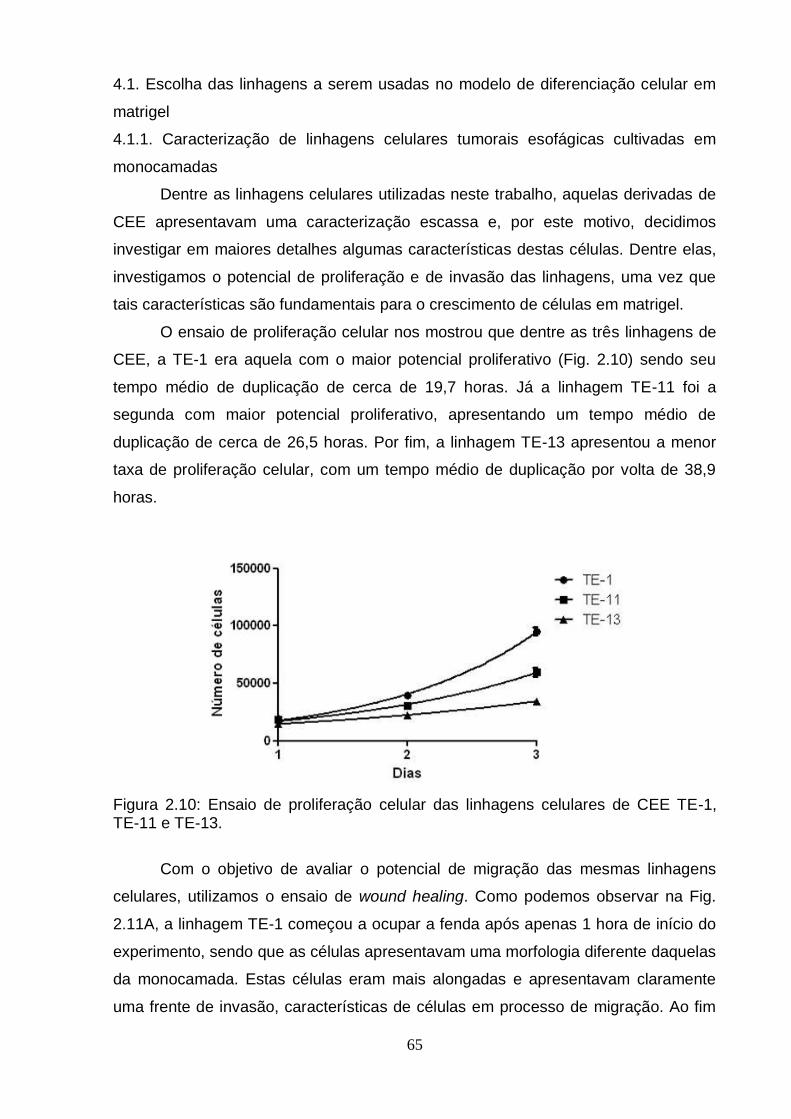
65
4.1. Escolha das linhagens a serem usadas no modelo de diferenciação celular em
matrigel
4.1.1. Caracterização de linhagens celulares tumorais esofágicas cultivadas em
monocamadas
Dentre as linhagens celulares utilizadas neste trabalho, aquelas derivadas de
CEE apresentavam uma caracterização escassa e, por este motivo, decidimos
investigar em maiores detalhes algumas características destas células. Dentre elas,
investigamos o potencial de proliferação e de invasão das linhagens, uma vez que
tais características são fundamentais para o crescimento de células em matrigel.
O ensaio de proliferação celular nos mostrou que dentre as três linhagens de
CEE, a TE-1 era aquela com o maior potencial proliferativo (Fig. 2.10) sendo seu
tempo médio de duplicação de cerca de 19,7 horas. Já a linhagem TE-11 foi a
segunda com maior potencial proliferativo, apresentando um tempo médio de
duplicação de cerca de 26,5 horas. Por fim, a linhagem TE-13 apresentou a menor
taxa de proliferação celular, com um tempo médio de duplicação por volta de 38,9
horas.
Figura 2.10: Ensaio de proliferação celular das linhagens celulares de CEE TE-1, TE-11 e TE-13.
Com o objetivo de avaliar o potencial de migração das mesmas linhagens
celulares, utilizamos o ensaio de wound healing. Como podemos observar na Fig.
2.11A, a linhagem TE-1 começou a ocupar a fenda após apenas 1 hora de início do
experimento, sendo que as células apresentavam uma morfologia diferente daquelas
da monocamada. Estas células eram mais alongadas e apresentavam claramente
uma frente de invasão, características de células em processo de migração. Ao fim

66
de 24 horas, a fenda já estava praticamente preenchida. Já a linhagem TE-11 não
apresentou o mesmo perfil (Fig. 2.11B). Neste caso, as células não pareciam migrar,
uma vez que não encontramos células com o perfil clássico deste processo. Porém,
ao fim de 24 horas o preenchimento da fenda também era praticamente completo,
aparentemente em consequência da proliferação das células que se encontravam na
borda da fenda. Por fim, a linhagem TE-13 apresentou um perfil completamente
diferente da outras linhagens, sendo que mesmo ao fim de 24 horas não
observamos qualquer mudança em termos de largura da fenda (Fig. 2.11C).
Figura 2.11: Ensaio de wound healing utilizando diferentes linhagens celulares. (A) TE-1. (B) TE-11. (C) TE-13. Fotografias obtidas em microscópio invertido, aumento de 4X.
O potencial de preenchimento da fenda no ensaio de wound healing parece
estar pelo menos parcialmente associado ao potencial de proliferação celular. A
linhagem TE-13, como mostramos antes, apresenta uma baixa taxa de proliferação
celular e, de acordo com este experimento, não possui um alto potencial migratório.
Já a TE-1 e a TE-11 apresentam taxas de proliferação mais altas, o que poderia
estar contribuindo para o preenchimento da fenda. Porém, no caso da TE-1, este
não parece ser o único fator, uma vez que já observamos a presença de células na
fenda apenas 1 hora após o início do experimento. Sendo assim, podemos esperar
comportamentos bastante diferentes das três linhagens no modelo de diferenciação
celular em matrigel.

67
4.1.2. Estabelecimento do modelo de diferenciação celular em matrigel.
De maneira resumida, o modelo de diferenciação celular em matrigel visa a
formação de esferas a partir de células únicas que se tornam ocas ao fim de 14 dias
como resultado de um processo de apoptose e de um processo de polarização
celular (Fig. 2.12). Tais processos levam à formação de estruturas semelhantes a
ácinos mamários, circundados por uma única camada de células organizadas e
polarizadas, mimetizando assim o processo de diferenciação epitelial.
Figura 2.12: Esquema do modelo de diferenciação celular em matrigel. Adaptado de Debnath et al., 2003.
Com o objetivo de estabelecer o modelo de diferenciação celular em matrigel
para células esofágicas, utilizamos inicialmente as três linhagens celulares derivadas
de CEE previamente caracterizadas neste estudo. A linhagem TE-1 apresentou um
perfil bastante semelhante àquele reportado para a linhagem celular maligna de
mama MCF10CA1a (Imbalzano, Tatarkova et al., 2009). Ou seja, não só as esferas
falharam em formar o lúmen, como cresceram de forma desordenada, formando
estruturas que nada se assemelhavam às esferas diferenciadas (Fig. 2.13).
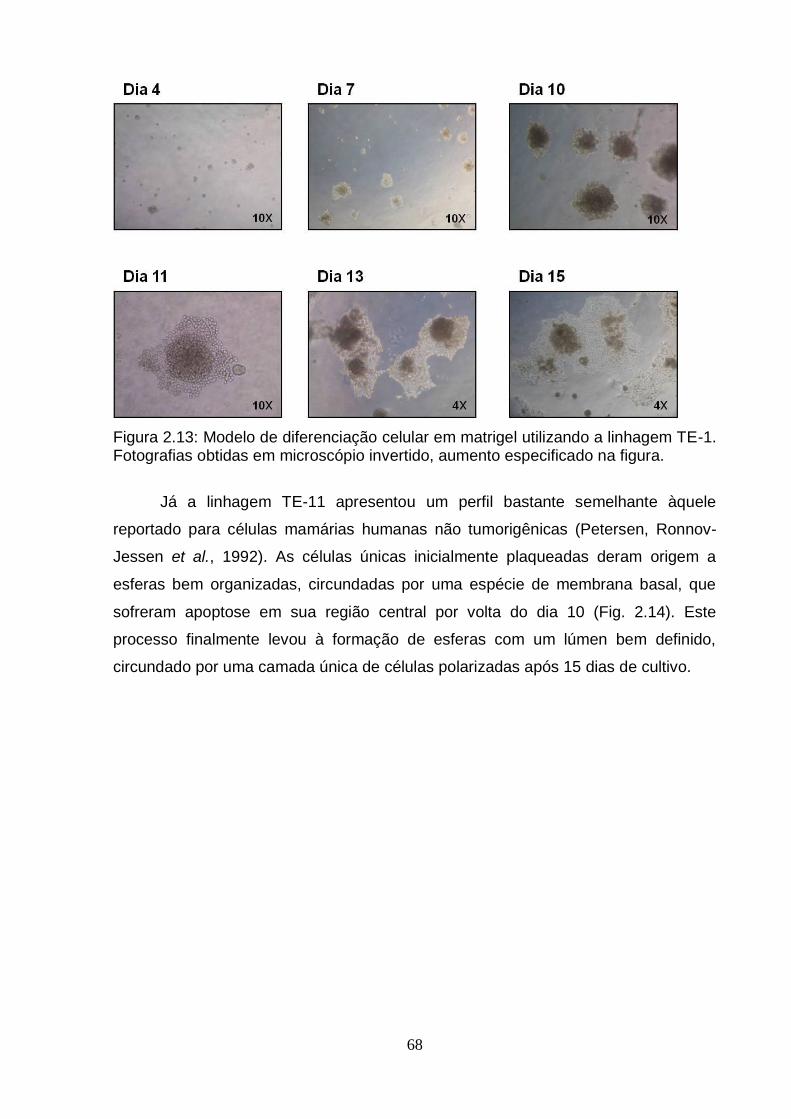
68
Figura 2.13: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-1. Fotografias obtidas em microscópio invertido, aumento especificado na figura.
Já a linhagem TE-11 apresentou um perfil bastante semelhante àquele
reportado para células mamárias humanas não tumorigênicas (Petersen, Ronnov-
Jessen et al., 1992). As células únicas inicialmente plaqueadas deram origem a
esferas bem organizadas, circundadas por uma espécie de membrana basal, que
sofreram apoptose em sua região central por volta do dia 10 (Fig. 2.14). Este
processo finalmente levou à formação de esferas com um lúmen bem definido,
circundado por uma camada única de células polarizadas após 15 dias de cultivo.
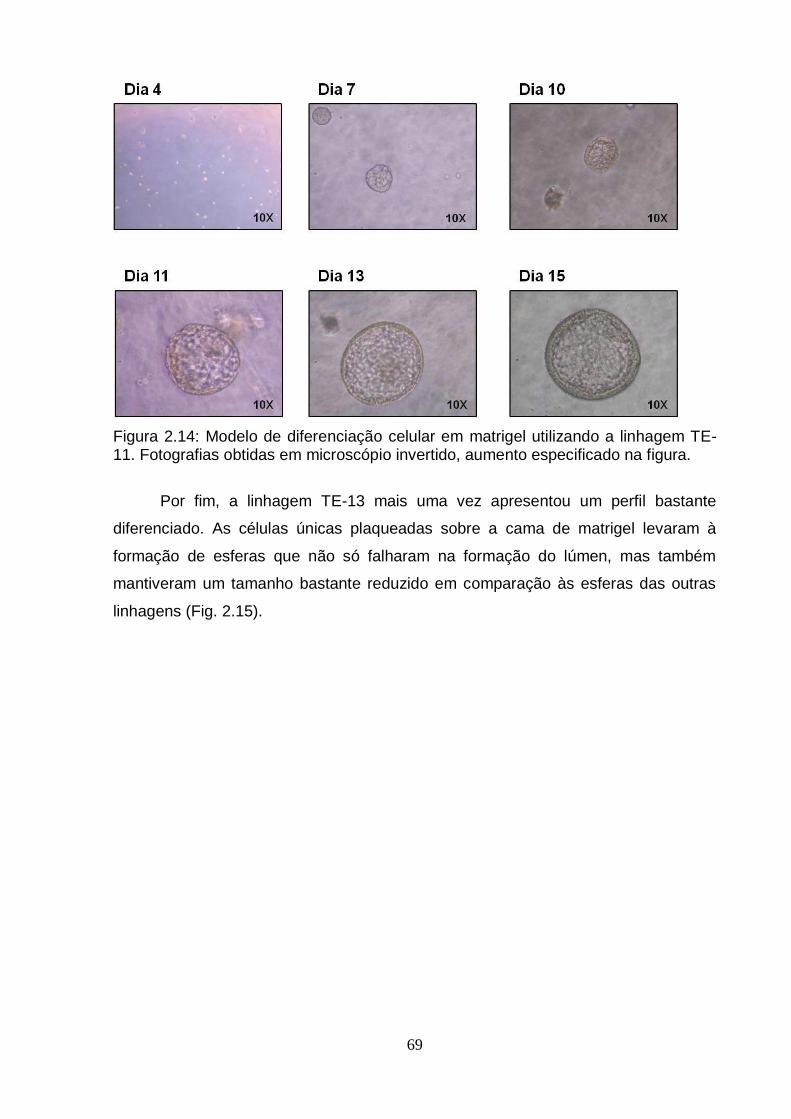
69
Figura 2.14: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-11. Fotografias obtidas em microscópio invertido, aumento especificado na figura.
Por fim, a linhagem TE-13 mais uma vez apresentou um perfil bastante
diferenciado. As células únicas plaqueadas sobre a cama de matrigel levaram à
formação de esferas que não só falharam na formação do lúmen, mas também
mantiveram um tamanho bastante reduzido em comparação às esferas das outras
linhagens (Fig. 2.15).
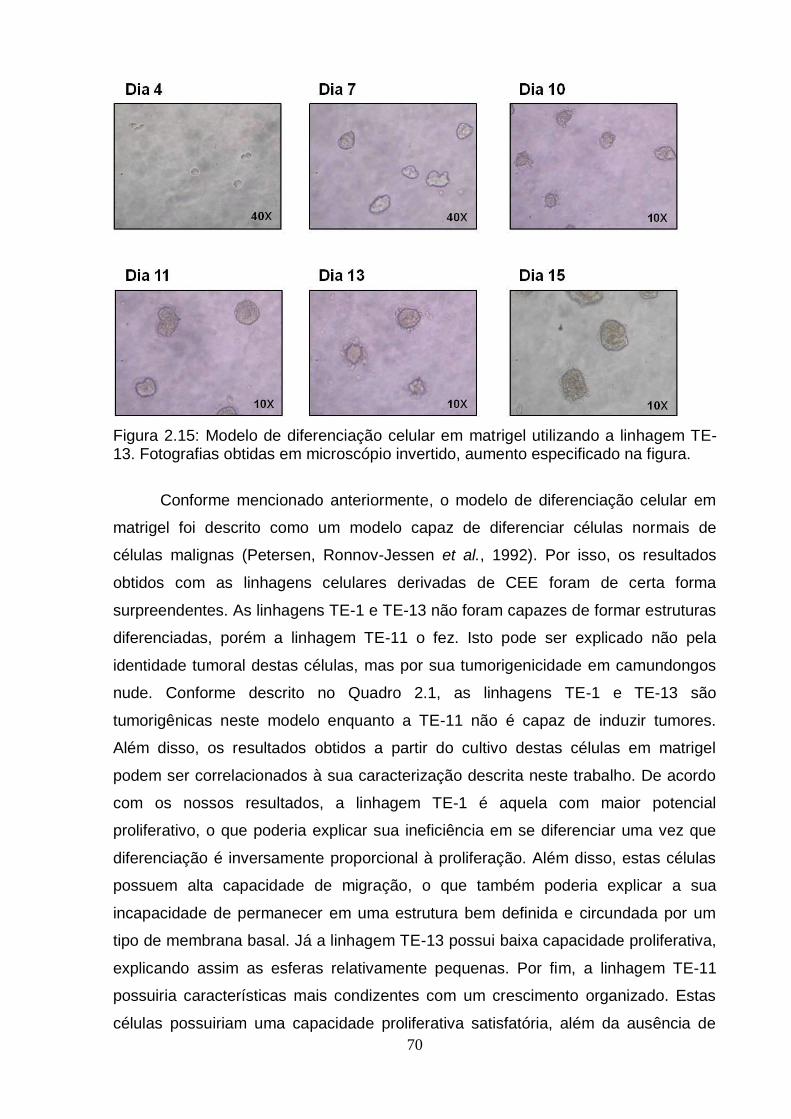
70
Figura 2.15: Modelo de diferenciação celular em matrigel utilizando a linhagem TE-13. Fotografias obtidas em microscópio invertido, aumento especificado na figura.
Conforme mencionado anteriormente, o modelo de diferenciação celular em
matrigel foi descrito como um modelo capaz de diferenciar células normais de
células malignas (Petersen, Ronnov-Jessen et al., 1992). Por isso, os resultados
obtidos com as linhagens celulares derivadas de CEE foram de certa forma
surpreendentes. As linhagens TE-1 e TE-13 não foram capazes de formar estruturas
diferenciadas, porém a linhagem TE-11 o fez. Isto pode ser explicado não pela
identidade tumoral destas células, mas por sua tumorigenicidade em camundongos
nude. Conforme descrito no Quadro 2.1, as linhagens TE-1 e TE-13 são
tumorigênicas neste modelo enquanto a TE-11 não é capaz de induzir tumores.
Além disso, os resultados obtidos a partir do cultivo destas células em matrigel
podem ser correlacionados à sua caracterização descrita neste trabalho. De acordo
com os nossos resultados, a linhagem TE-1 é aquela com maior potencial
proliferativo, o que poderia explicar sua ineficiência em se diferenciar uma vez que
diferenciação é inversamente proporcional à proliferação. Além disso, estas células
possuem alta capacidade de migração, o que também poderia explicar a sua
incapacidade de permanecer em uma estrutura bem definida e circundada por um
tipo de membrana basal. Já a linhagem TE-13 possui baixa capacidade proliferativa,
explicando assim as esferas relativamente pequenas. Por fim, a linhagem TE-11
possuiria características mais condizentes com um crescimento organizado. Estas
células possuiriam uma capacidade proliferativa satisfatória, além da ausência de

71
um potencial migracional, o que contribuiria para um crescimento mais organizado,
atingindo a diferenciação celular. Mediante a isto, a linhagem TE-11 foi escolhida
para a realização dos experimentos posteriores utilizando o modelo de diferenciação
celular em matrigel.
5. Caracterização do modelo de diferenciação celular em matrigel utilizando a
linhagem TE-11
5.1. Avaliação da presença de caspase 3 clivada por imunohistoquímica
De acordo com o modelo proposto por Debnath e colaboradores, durante o
processo de diferenciação celular em matrigel as esferas formadas sofrem apoptose
por volta do dia nove de modo a originar o lúmen. Sendo assim, decidimos avaliar no
nosso modelo se o mesmo processo ocorria por meio da análise por
imunofluorescência da presença de caspase 3 clivada. Na Fig. 2.16, podemos
observar diferentes exemplos da ocorrência deste evento. No dia nove de cultivo, as
esferas sofrem apoptose em sua região central, onde teremos a formação do lúmen
posteriormente. Sendo assim, podemos afirmar que o modelo de diferenciação de
células esofágicas em matrigel é semelhante àquele descrito para células mamárias.
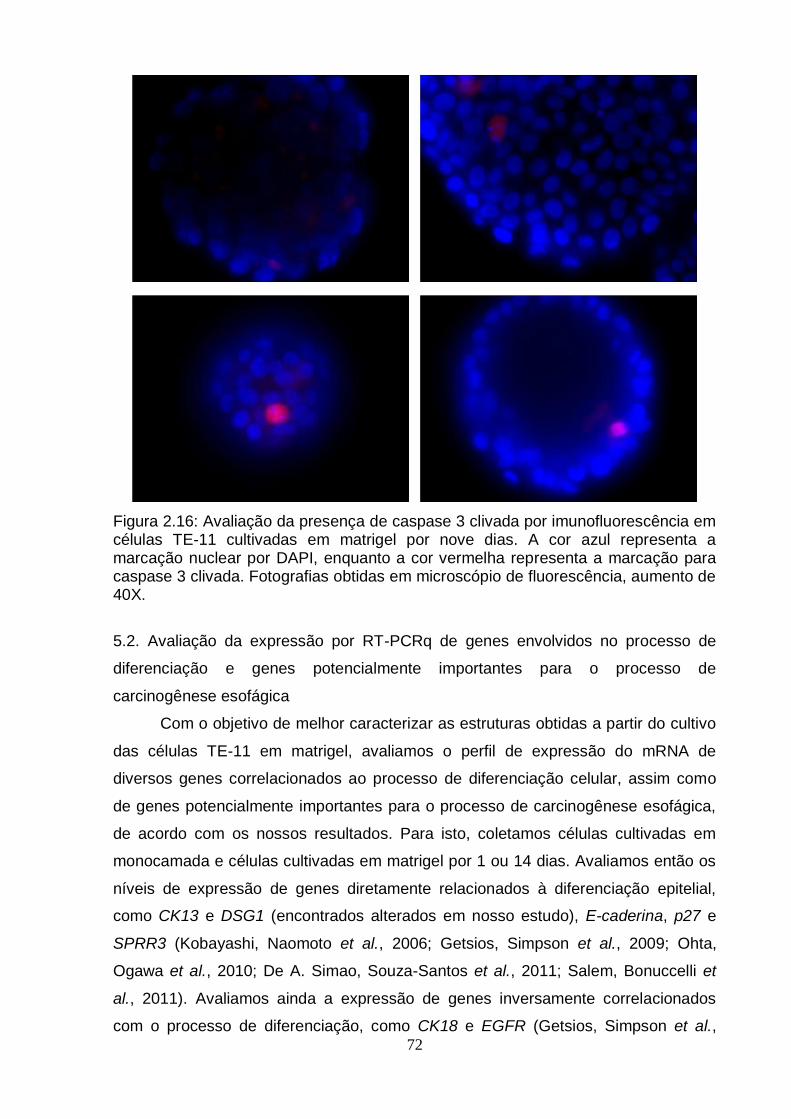
72
Figura 2.16: Avaliação da presença de caspase 3 clivada por imunofluorescência em células TE-11 cultivadas em matrigel por nove dias. A cor azul representa a marcação nuclear por DAPI, enquanto a cor vermelha representa a marcação para caspase 3 clivada. Fotografias obtidas em microscópio de fluorescência, aumento de 40X.
5.2. Avaliação da expressão por RT-PCRq de genes envolvidos no processo de
diferenciação e genes potencialmente importantes para o processo de
carcinogênese esofágica
Com o objetivo de melhor caracterizar as estruturas obtidas a partir do cultivo
das células TE-11 em matrigel, avaliamos o perfil de expressão do mRNA de
diversos genes correlacionados ao processo de diferenciação celular, assim como
de genes potencialmente importantes para o processo de carcinogênese esofágica,
de acordo com os nossos resultados. Para isto, coletamos células cultivadas em
monocamada e células cultivadas em matrigel por 1 ou 14 dias. Avaliamos então os
níveis de expressão de genes diretamente relacionados à diferenciação epitelial,
como CK13 e DSG1 (encontrados alterados em nosso estudo), E-caderina, p27 e
SPRR3 (Kobayashi, Naomoto et al., 2006; Getsios, Simpson et al., 2009; Ohta,
Ogawa et al., 2010; De A. Simao, Souza-Santos et al., 2011; Salem, Bonuccelli et
al., 2011). Avaliamos ainda a expressão de genes inversamente correlacionados
com o processo de diferenciação, como CK18 e EGFR (Getsios, Simpson et al.,

73
2009; Makino, Yamasaki et al., 2009) e de genes encontrados diferencialmente
metilados em nosso estudo, como TFF1 (gene importante para proteção da mucosa
– (Ribieras, Tomasetto et al., 1998)) e BCL3 (gene anti-apoptótico – (Choi, Lee et al.,
2010)). Para nossa surpresa, todos os genes analisados apresentaram uma
diminuição de expressão após o cultivo em matrigel por um dia em comparação ao
cultivo em monocamada (Fig. 2.17). Além disso, praticamente nenhum dos genes
avaliados apresentou qualquer variação de expressão após treze dias de cultivo em
matrigel (Fig. 2.17). Isto pode ser explicado pelo fato deste modelo não mimetizar a
diferenciação epitelial estratificada clássica, uma vez que temos a presença de
apenas uma camada de células polarizadas e, portanto, consideradas diferenciadas.
O epitélio esofágico humano é formado por múltiplas camadas de células, sendo que
a diferenciação ocorre de acordo com a aproximação destas células ao lúmen
(Squier e Kremer, 2001). Sendo assim, o modelo estabelecido neste trabalho não
reflete o mesmo processo de diferenciação esofágico in vivo e não deve ser utilizado
com o objetivo de reproduzir tal processo.
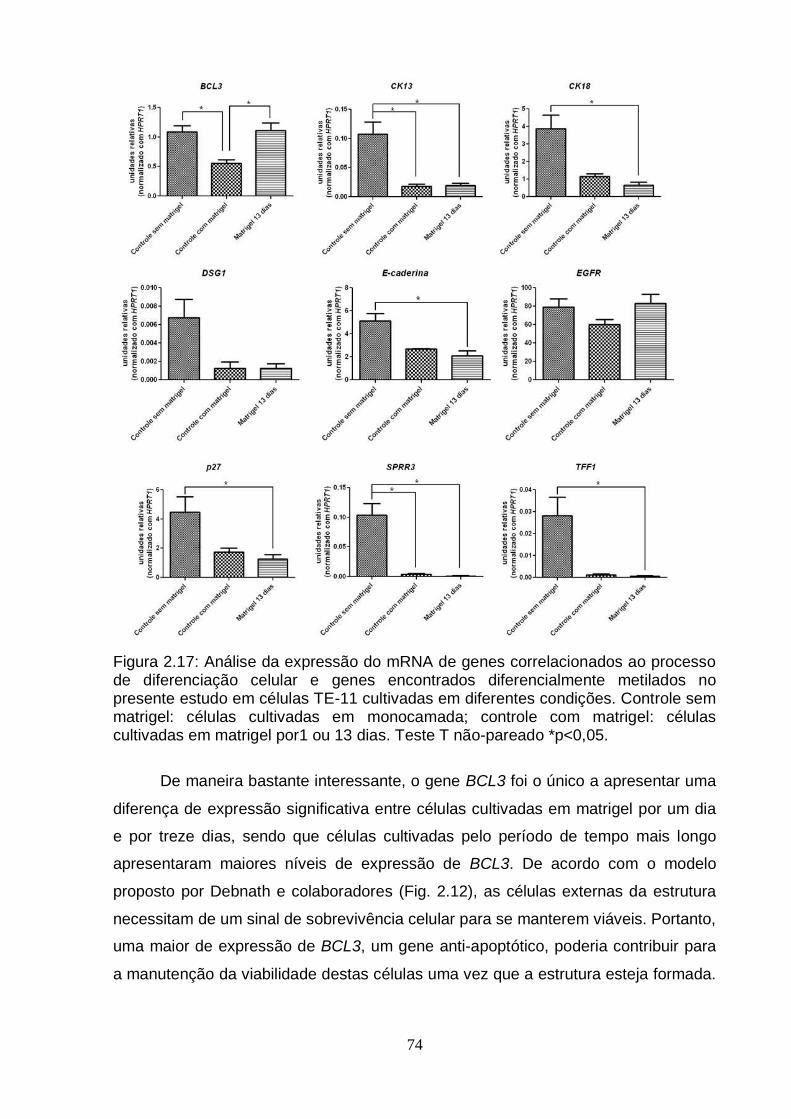
74
Figura 2.17: Análise da expressão do mRNA de genes correlacionados ao processo de diferenciação celular e genes encontrados diferencialmente metilados no presente estudo em células TE-11 cultivadas em diferentes condições. Controle sem matrigel: células cultivadas em monocamada; controle com matrigel: células cultivadas em matrigel por1 ou 13 dias. Teste T não-pareado *p<0,05.
De maneira bastante interessante, o gene BCL3 foi o único a apresentar uma
diferença de expressão significativa entre células cultivadas em matrigel por um dia
e por treze dias, sendo que células cultivadas pelo período de tempo mais longo
apresentaram maiores níveis de expressão de BCL3. De acordo com o modelo
proposto por Debnath e colaboradores (Fig. 2.12), as células externas da estrutura
necessitam de um sinal de sobrevivência celular para se manterem viáveis. Portanto,
uma maior de expressão de BCL3, um gene anti-apoptótico, poderia contribuir para
a manutenção da viabilidade destas células uma vez que a estrutura esteja formada.

75
Para estudar em maiores detalhes esta correlação entre a expressão de BCL3,
decidimos então modular tal expressão por diferentes mecanismos.
6. Efeito da IL6 sobre a diferenciação celular em matrigel
Conforme demonstrado anteriormente neste estudo, o tratamento de
linhagens celulares esofágicas com IL6 é capaz de induzir a expressão do mRNA de
BCL3. Sendo assim, com o objetivo de investigar a influência de BCL3 sobre a
diferenciação celular em matrigel, tratamos as células cultivadas em matrigel com
esta interleucina. Para facilitar a interpretação dos resultados, classificamos as
esferas observadas como estruturas verdadeiras, ou seja, aquelas esferas com o
lúmen completamente formado e circundado por uma única camada de células
polarizadas e não-estruturas ou esferas que falharam parcial ou completamente em
formar o lúmen (Fig. 2.18).
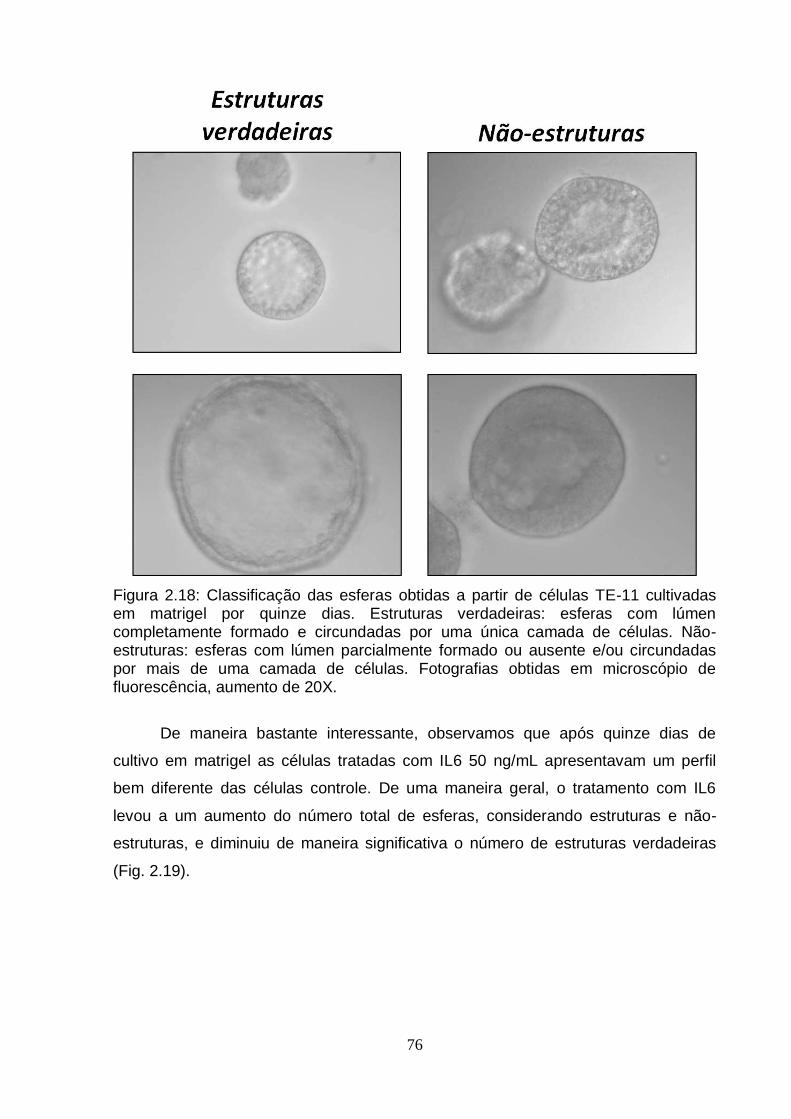
76
Figura 2.18: Classificação das esferas obtidas a partir de células TE-11 cultivadas em matrigel por quinze dias. Estruturas verdadeiras: esferas com lúmen completamente formado e circundadas por uma única camada de células. Não-estruturas: esferas com lúmen parcialmente formado ou ausente e/ou circundadas por mais de uma camada de células. Fotografias obtidas em microscópio de fluorescência, aumento de 20X.
De maneira bastante interessante, observamos que após quinze dias de
cultivo em matrigel as células tratadas com IL6 50 ng/mL apresentavam um perfil
bem diferente das células controle. De uma maneira geral, o tratamento com IL6
levou a um aumento do número total de esferas, considerando estruturas e não-
estruturas, e diminuiu de maneira significativa o número de estruturas verdadeiras
(Fig. 2.19).
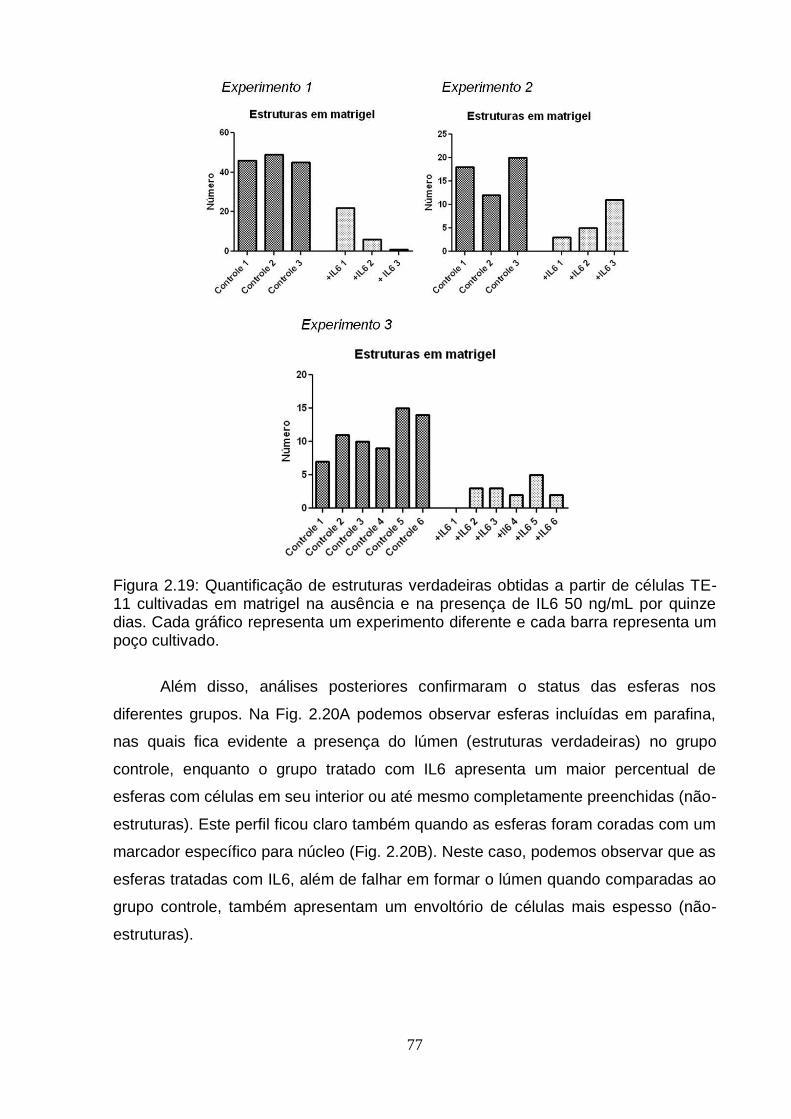
77
Figura 2.19: Quantificação de estruturas verdadeiras obtidas a partir de células TE-11 cultivadas em matrigel na ausência e na presença de IL6 50 ng/mL por quinze dias. Cada gráfico representa um experimento diferente e cada barra representa um poço cultivado.
Além disso, análises posteriores confirmaram o status das esferas nos
diferentes grupos. Na Fig. 2.20A podemos observar esferas incluídas em parafina,
nas quais fica evidente a presença do lúmen (estruturas verdadeiras) no grupo
controle, enquanto o grupo tratado com IL6 apresenta um maior percentual de
esferas com células em seu interior ou até mesmo completamente preenchidas (não-
estruturas). Este perfil ficou claro também quando as esferas foram coradas com um
marcador específico para núcleo (Fig. 2.20B). Neste caso, podemos observar que as
esferas tratadas com IL6, além de falhar em formar o lúmen quando comparadas ao
grupo controle, também apresentam um envoltório de células mais espesso (não-
estruturas).
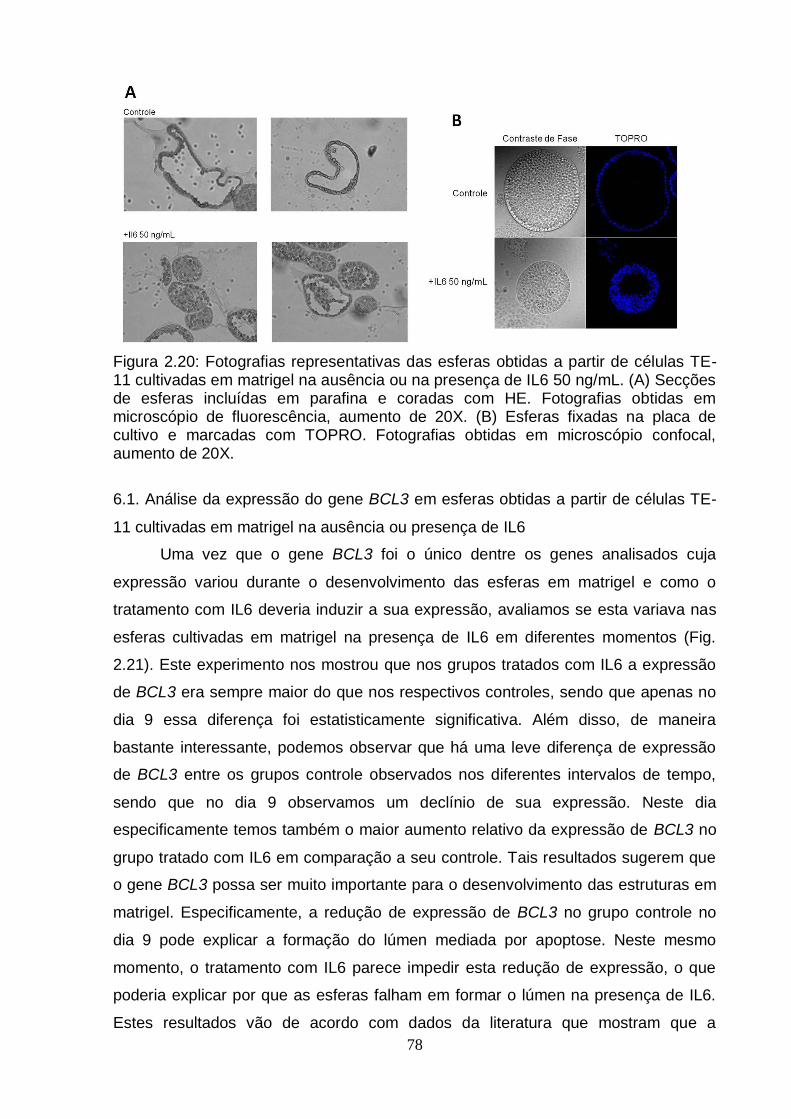
78
Figura 2.20: Fotografias representativas das esferas obtidas a partir de células TE-11 cultivadas em matrigel na ausência ou na presença de IL6 50 ng/mL. (A) Secções de esferas incluídas em parafina e coradas com HE. Fotografias obtidas em microscópio de fluorescência, aumento de 20X. (B) Esferas fixadas na placa de cultivo e marcadas com TOPRO. Fotografias obtidas em microscópio confocal, aumento de 20X.
6.1. Análise da expressão do gene BCL3 em esferas obtidas a partir de células TE-
11 cultivadas em matrigel na ausência ou presença de IL6
Uma vez que o gene BCL3 foi o único dentre os genes analisados cuja
expressão variou durante o desenvolvimento das esferas em matrigel e como o
tratamento com IL6 deveria induzir a sua expressão, avaliamos se esta variava nas
esferas cultivadas em matrigel na presença de IL6 em diferentes momentos (Fig.
2.21). Este experimento nos mostrou que nos grupos tratados com IL6 a expressão
de BCL3 era sempre maior do que nos respectivos controles, sendo que apenas no
dia 9 essa diferença foi estatisticamente significativa. Além disso, de maneira
bastante interessante, podemos observar que há uma leve diferença de expressão
de BCL3 entre os grupos controle observados nos diferentes intervalos de tempo,
sendo que no dia 9 observamos um declínio de sua expressão. Neste dia
especificamente temos também o maior aumento relativo da expressão de BCL3 no
grupo tratado com IL6 em comparação a seu controle. Tais resultados sugerem que
o gene BCL3 possa ser muito importante para o desenvolvimento das estruturas em
matrigel. Especificamente, a redução de expressão de BCL3 no grupo controle no
dia 9 pode explicar a formação do lúmen mediada por apoptose. Neste mesmo
momento, o tratamento com IL6 parece impedir esta redução de expressão, o que
poderia explicar por que as esferas falham em formar o lúmen na presença de IL6.
Estes resultados vão de acordo com dados da literatura que mostram que a
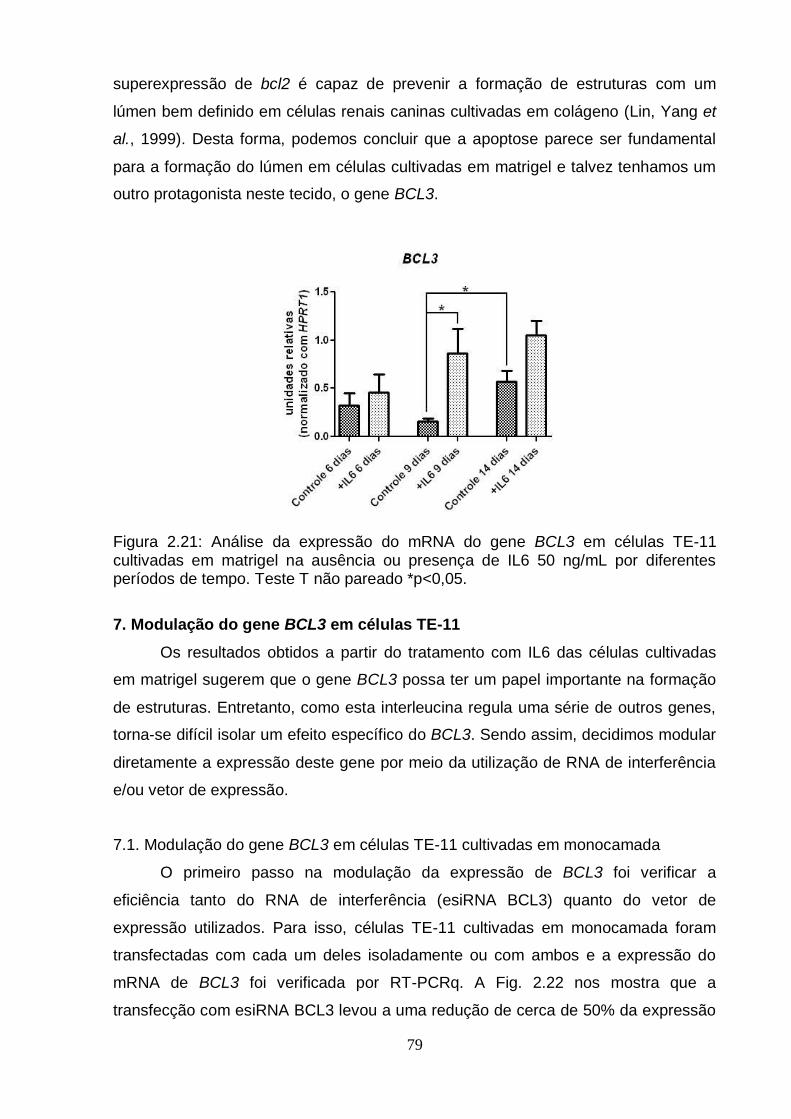
79
superexpressão de bcl2 é capaz de prevenir a formação de estruturas com um
lúmen bem definido em células renais caninas cultivadas em colágeno (Lin, Yang et
al., 1999). Desta forma, podemos concluir que a apoptose parece ser fundamental
para a formação do lúmen em células cultivadas em matrigel e talvez tenhamos um
outro protagonista neste tecido, o gene BCL3.
Figura 2.21: Análise da expressão do mRNA do gene BCL3 em células TE-11 cultivadas em matrigel na ausência ou presença de IL6 50 ng/mL por diferentes períodos de tempo. Teste T não pareado *p<0,05.
7. Modulação do gene BCL3 em células TE-11
Os resultados obtidos a partir do tratamento com IL6 das células cultivadas
em matrigel sugerem que o gene BCL3 possa ter um papel importante na formação
de estruturas. Entretanto, como esta interleucina regula uma série de outros genes,
torna-se difícil isolar um efeito específico do BCL3. Sendo assim, decidimos modular
diretamente a expressão deste gene por meio da utilização de RNA de interferência
e/ou vetor de expressão.
7.1. Modulação do gene BCL3 em células TE-11 cultivadas em monocamada
O primeiro passo na modulação da expressão de BCL3 foi verificar a
eficiência tanto do RNA de interferência (esiRNA BCL3) quanto do vetor de
expressão utilizados. Para isso, células TE-11 cultivadas em monocamada foram
transfectadas com cada um deles isoladamente ou com ambos e a expressão do
mRNA de BCL3 foi verificada por RT-PCRq. A Fig. 2.22 nos mostra que a
transfecção com esiRNA BCL3 levou a uma redução de cerca de 50% da expressão
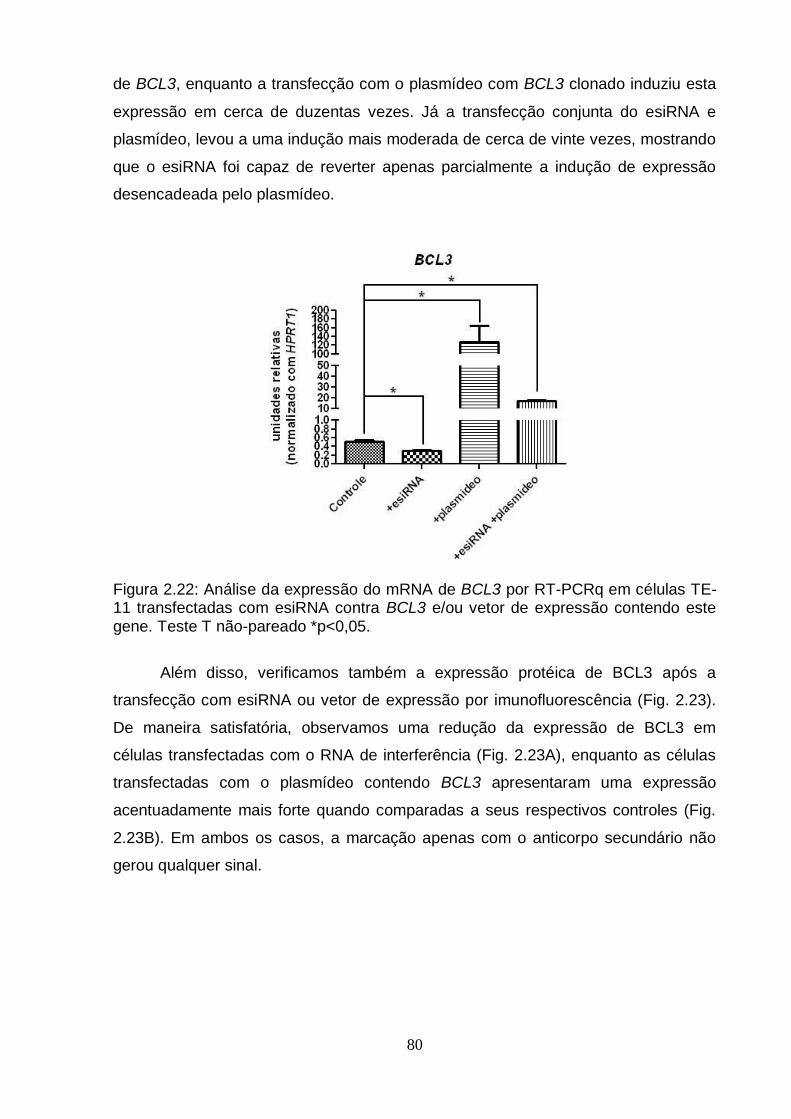
80
de BCL3, enquanto a transfecção com o plasmídeo com BCL3 clonado induziu esta
expressão em cerca de duzentas vezes. Já a transfecção conjunta do esiRNA e
plasmídeo, levou a uma indução mais moderada de cerca de vinte vezes, mostrando
que o esiRNA foi capaz de reverter apenas parcialmente a indução de expressão
desencadeada pelo plasmídeo.
Figura 2.22: Análise da expressão do mRNA de BCL3 por RT-PCRq em células TE-11 transfectadas com esiRNA contra BCL3 e/ou vetor de expressão contendo este gene. Teste T não-pareado *p<0,05.
Além disso, verificamos também a expressão protéica de BCL3 após a
transfecção com esiRNA ou vetor de expressão por imunofluorescência (Fig. 2.23).
De maneira satisfatória, observamos uma redução da expressão de BCL3 em
células transfectadas com o RNA de interferência (Fig. 2.23A), enquanto as células
transfectadas com o plasmídeo contendo BCL3 apresentaram uma expressão
acentuadamente mais forte quando comparadas a seus respectivos controles (Fig.
2.23B). Em ambos os casos, a marcação apenas com o anticorpo secundário não
gerou qualquer sinal.
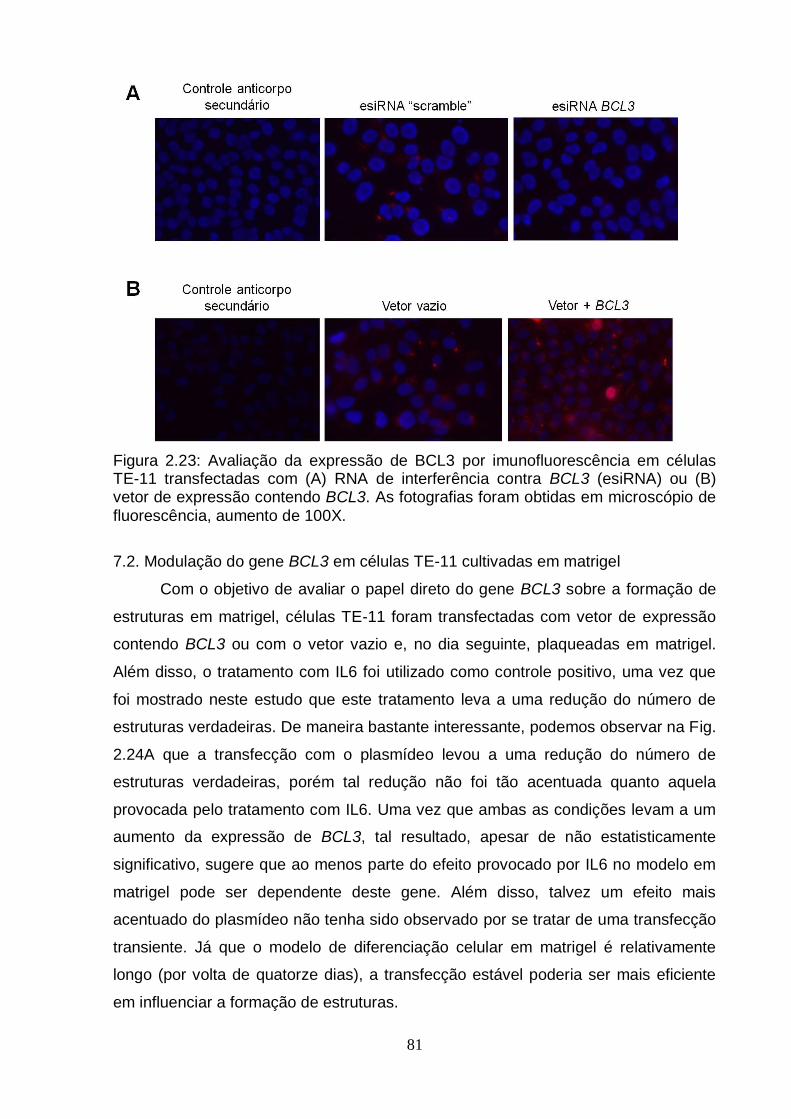
81
Figura 2.23: Avaliação da expressão de BCL3 por imunofluorescência em células TE-11 transfectadas com (A) RNA de interferência contra BCL3 (esiRNA) ou (B) vetor de expressão contendo BCL3. As fotografias foram obtidas em microscópio de fluorescência, aumento de 100X.
7.2. Modulação do gene BCL3 em células TE-11 cultivadas em matrigel
Com o objetivo de avaliar o papel direto do gene BCL3 sobre a formação de
estruturas em matrigel, células TE-11 foram transfectadas com vetor de expressão
contendo BCL3 ou com o vetor vazio e, no dia seguinte, plaqueadas em matrigel.
Além disso, o tratamento com IL6 foi utilizado como controle positivo, uma vez que
foi mostrado neste estudo que este tratamento leva a uma redução do número de
estruturas verdadeiras. De maneira bastante interessante, podemos observar na Fig.
2.24A que a transfecção com o plasmídeo levou a uma redução do número de
estruturas verdadeiras, porém tal redução não foi tão acentuada quanto aquela
provocada pelo tratamento com IL6. Uma vez que ambas as condições levam a um
aumento da expressão de BCL3, tal resultado, apesar de não estatisticamente
significativo, sugere que ao menos parte do efeito provocado por IL6 no modelo em
matrigel pode ser dependente deste gene. Além disso, talvez um efeito mais
acentuado do plasmídeo não tenha sido observado por se tratar de uma transfecção
transiente. Já que o modelo de diferenciação celular em matrigel é relativamente
longo (por volta de quatorze dias), a transfecção estável poderia ser mais eficiente
em influenciar a formação de estruturas.
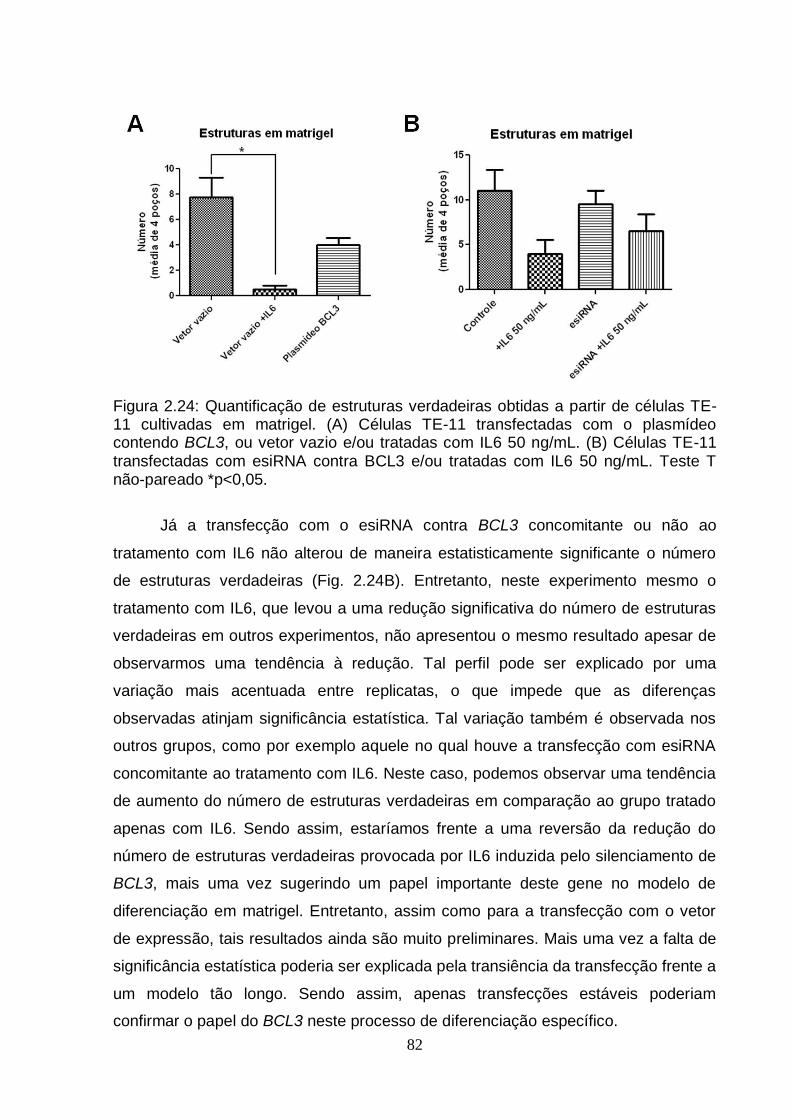
82
Figura 2.24: Quantificação de estruturas verdadeiras obtidas a partir de células TE-11 cultivadas em matrigel. (A) Células TE-11 transfectadas com o plasmídeo contendo BCL3, ou vetor vazio e/ou tratadas com IL6 50 ng/mL. (B) Células TE-11 transfectadas com esiRNA contra BCL3 e/ou tratadas com IL6 50 ng/mL. Teste T não-pareado *p<0,05.
Já a transfecção com o esiRNA contra BCL3 concomitante ou não ao
tratamento com IL6 não alterou de maneira estatisticamente significante o número
de estruturas verdadeiras (Fig. 2.24B). Entretanto, neste experimento mesmo o
tratamento com IL6, que levou a uma redução significativa do número de estruturas
verdadeiras em outros experimentos, não apresentou o mesmo resultado apesar de
observarmos uma tendência à redução. Tal perfil pode ser explicado por uma
variação mais acentuada entre replicatas, o que impede que as diferenças
observadas atinjam significância estatística. Tal variação também é observada nos
outros grupos, como por exemplo aquele no qual houve a transfecção com esiRNA
concomitante ao tratamento com IL6. Neste caso, podemos observar uma tendência
de aumento do número de estruturas verdadeiras em comparação ao grupo tratado
apenas com IL6. Sendo assim, estaríamos frente a uma reversão da redução do
número de estruturas verdadeiras provocada por IL6 induzida pelo silenciamento de
BCL3, mais uma vez sugerindo um papel importante deste gene no modelo de
diferenciação em matrigel. Entretanto, assim como para a transfecção com o vetor
de expressão, tais resultados ainda são muito preliminares. Mais uma vez a falta de
significância estatística poderia ser explicada pela transiência da transfecção frente a
um modelo tão longo. Sendo assim, apenas transfecções estáveis poderiam
confirmar o papel do BCL3 neste processo de diferenciação específico.

83
Discussão Geral
Por muito tempo o câncer foi visto como uma doença genética, porém com o
avanço científico esta visão vem mudando consideravelmente. Sabe-se hoje que os
tumores surgem como resultado de uma interação dos genes com o ambiente e tal
interação induz tanto alterações genéticas quanto epigenéticas. Porém, apesar
deste conhecimento em linhas gerais, a identificação de tais alterações ainda está
no início, especialmente no caso de determinados tipos de câncer. Este é o caso do
foco do nosso estudo, o CEE. Este é um tumor extremamente fatal normalmente
diagnosticado em estágios tardios principalmente devido à ausência de sintomas até
o fechamento parcial ou total da luz do esôfago. Sendo assim, torna-se essencial
encontrar meios de diagnosticar o CEE precocemente e, para isso, é necessário
compreender mais a fundo a biologia deste tumor.
Conforme mencionado anteriormente, algumas alterações genéticas já foram
descritas para CEE, enquanto os estudos que investigaram a presença de
alterações epigenéticas ainda são escassos. Devido a este panorama e à cada vez
mais recorrente identificação de alterações em metilação do DNA como alterações
precoces em outros tipos de câncer, decidimos investigar de maneira abrangente e
não tendenciosa a presença de tais alterações em CEE. Para isso, contamos com
uma plataforma que interroga o perfil de metilação do promotor de mais de 800
genes e, de maneira bastante satisfatória, identificamos um perfil de metilação
tumor-específico e alterações presentes já na mucosa normal adjacente de
pacientes, o que poderia representar alterações precoces. Este trabalho foi pioneiro
não só pelo ineditismo dos resultados, mas também pela gama de possibilidades de
outros estudos que abriu.
A identificação de potencias via alteradas em CEE reforçou uma suspeita já
levantada pelo nosso e por outros grupos. O processo inflamatório já foi apontado
como fundamental para diversos tipos de tumor, como para o câncer coloretal e até
mesmo para o adenocarcinoma de esôfago. Porém, seu papel no CEE ainda não é
claro. Acredita-se hoje que virtualmente todas as lesões neoplásicas contenham
células imunes, cuja presença varia entre infiltrados sutis a inflamações acentuadas
(Pages, Galon et al., 2010). Porém, tal participação foi vista inicialmente como uma
tentativa do sistema imune de erradicar as células tumorais. Resultados do nosso
grupo mostram, entretanto, que o infiltrado inflamatório parece agir como um
promotor da tumorigênese em CEE (Rapozo, 2009). Neste estudo, foi mostrado que

84
camundongos submetidos a uma dose sub-carcinogênica de um carcinógeno
químico (N-nitroso-dietilamina) desenvolviam tumores quando submetidos a uma
injúria esofágica que induzia um processo inflamatório crônico. O mesmo resultado
não foi observado em camundongos tratados apenas com o carcinógeno, sugerindo
um papel fundamental da inflamação durante a carcinogênese esofágica.
Sendo assim, nosso estudo não só corrobora resultados anteriores do nosso
grupo, como também traz possíveis explicações para tais correlações.
Primeiramente, a hipometilação de IL6 encontrada em tumores sugere esta citocina
como um possível efetor da promoção tumoral. Diversos trabalhos já mostraram uma
associação de IL6 com aumento de proliferação e sobrevivência celular, porém
diversos alvos moleculares desta citocina já foram descritos e a relevância de cada
um não é clara (Hirano, Ishihara et al., 2000). É possível que mais de um alvo seja
importante para o processo de carcinogênese, assim como é possível que tal
importância dependa do tipo de tumor. Para CEE, nosso resultados sugerem que
BCL3 seja um alvo importante. Comprovamos aqui que a expressão deste gene é
induzida por IL6 em células esofágicas, assim como já foi demonstrado para outros
tipos celulares, e que tal indução pode resultar em, pelo menos, um bloqueio da
apoptose. Tal correlação parece clara no modelo de diferenciação celular em
matrigel desenvolvido em nosso estudo. Neste modelo, o controle rigoroso de
proliferação e morte celular é fundamental para o desenvolvimento correto das
estruturas de células diferenciadas (Debnath, Muthuswamy et al., 2003). Nossos
resultados mostram que o tratamento com IL6 é capaz de perturbar este
desenvolvimento e isto parece ser mediado, ao menos em parte, pela indução da
expressão de BCL3. Sendo assim, estabelecemos um link entre um mediador
inflamatório e um processo específico de diferenciação celular in vitro que sugere um
mecanismo pelo qual a inflamação poderia promover a carcinogênese esofágica.
Além disso, nossos resultados também sugerem um efeito direto de IL6 sobre
o perfil de metilação do gene BCL3. Um trabalho recente corrobora este resultado,
uma vez que foi demonstrado em linhagens celulares de carcinoma epidermóide oral
que a inflamação crônica mediada por IL6 é capaz de induzir alterações em
metilação do DNA (Gasche, Hoffmann et al., 2011). Neste estudo, foi observada uma
indução da hipometilação de LINE1, assim como uma indução da hipermetilação de
promotores específicos, como os de CHFR, GATA5 e PAX6. Sendo assim, nossos
resultados não só sugerem possíveis consequências funcionais da inflamação
crônica mediada por IL6, como também possíveis mecanismos.

85
Foi mostrado ainda que IL6 é capaz de regular a atividade de DNMT1 e a
expressão de genes supressores de tumor por modular micro-RNAs (miR-148a e
miR-152) em colangiosarcoma (Braconi, Huang et al., 2010). Sendo assim, os
efeitos epigenéticos de IL6 podem ir além da metilação do DNA e isto deve ser
investigado também em CEE em um futuro próximo.
A importância das alterações encontradas em BCL3 vai além do seu papel de
possível mediador de algumas das consequências funcionais induzidas por IL6.
Outro fator importante foi a identificação de tais alterações já na mucosa normal
adjacente de pacientes com CEE em comparação à mucosa esofágica de indivíduos
saudáveis. A partir desta mesma análise, o gene TFF1 também foi identificado,
sugerindo um papel de ambos na formação do campo de cancerização no epitélio
esofágico. O paradigma amplamente aceito de tumorigênese tem início com
mutações em genes-chave para o crescimento celular, resultando em um
crescimento imediato da lesão. A progressão ocorreria então conforme as células
iniciadas adquirissem mutações adicionais. Entretanto, o processo de carcinogênese
pode começar bem antes do aparecimento das primeiras lesões detectáveis
clinicamente e até mesmo bem antes do aparecimento das chamadas lesões pré-
neoplásicas. Neste contexto, o campo de cancerização, ou seja, a substituição de
uma população de células normais por um clone de células histologicamente não
displásicas e pró-tumorigênicas tem sido correlacionado ao desenvolvimento de
diversos tipos de tumor (Graham, Mcdonald et al., 2011). Além disso, uma
possibilidade bastante aceita ultimamente é a de que tais células pró-tumorigênicas
não necessariamente carreguem mutações, mas sim alterações epigenéticas.
Alterações em metilação do DNA já foram encontradas na mucosa normal adjacente
de pacientes com diferentes tipos de tumor. Este é o caso, por exemplo, do
hepatocarcinoma (Kondo, Kanai et al., 2000), câncer coloretal (Issa, Ahuja et al.,
2001), adenocarcinoma de esôfago (Eads, Lord et al., 2000), câncer de pulmão
(Guo, House et al., 2004) e gástrico (Ushijima, 2007).
No caso do CEE, apenas um trabalho acessou até então a presença de um
campo de cancerização epigenético. Lee e colaboradores mostraram que os níveis
de metilação de quatro marcadores (HOXA9, NEFH, UCHL1 e MT1M) aumentam
progressivamente da mucosa esofágica de indivíduos saudáveis não expostos a
fatores de risco para aqueles que sofreram tal exposição, passando pela mucosa
normal adjacente de pacientes com CEE, sendo que os maiores níveis de metilação
são detectados nos tumores dos mesmo pacientes (Lee, Wang et al., 2011). Além

86
disso, neste mesmo trabalho foi mostrado que a exposição cumulativa ao álcool
aumenta a metilação do promotor do HOXA9, enquanto fumantes apresentam
maiores níveis de metilação de todos os marcadores avaliados. Sendo assim, nosso
trabalho vem sugerir novos marcadores (BCL3 e TFF1) que devem ser avaliados
com maior cuidado não só em relação aos fatores de risco para CEE, como também
em lesões pré-neoplásicas, se possível.
Uma outra potencial utilidade para os marcadores precoces identificados
neste trabalho é na detecção precoce de segundos tumores primários de esôfago
(STPE) em pacientes com câncer de cabeça e pescoço. No caso de pacientes com
tumores orais ou de orofaringe, a incidência de segundos tumores primários é
relativamente alta, entre 17 e 30%, incluindo tumores sincrônicos e metacrônicos
(Braakhuis, Tabor et al., 2002). Dentre estes, os STPE estão entre os mais
frequentes e normalmente apresentam um prognóstico ruim. Sendo assim,
marcadores que possam permitir uma detecção precoce destes tumores seriam de
grande valia.
Nosso trabalho identificou ainda uma série de alterações em outros genes
que podem ser importantes para a carcinogênese esofágica, como aqueles
envolvidos em adesão celular, que também merecem ser investigados mais a fundo.
Entretanto, não houve tempo hábil para que tais análises mais detalhadas fossem
feitas. Talvez tenhamos levantado mais dúvidas do que solucionado, mas este
trabalho foi o primeiro passo para melhor compreender quais e como alterações
epigenéticas podem contribuir para a carcinogênese esofágica.

87
Conclusões
Com base em nossos resultados, podemos concluir que:
- O CEE apresenta um perfil único de metilação de promotores quando
comparado ao tecido normal adjacente;
- A via de adesão celular e o componente inflamatório parecem ser importantes
para o desenvolvimento de CEE;
- O CEE apresenta uma hipometilação global;
- A hipermetilação do promotor de TFF1 e a hipometilação do promotor de
BCL3 parecem ser alterações precoces em CEE;
- A expressão de BCL3 é regulada por IL6 em células esofágicas e esta
regulação provavelmente envolve mecanismos epigenéticos;
- O tratamento com IL6 é capaz de perturbar o processo de diferenciação in
vitro utilizando o modelo em matrigel e isto parece ser dependente, ao menos
em parte, da regulação da expressão de BCL3.

88
Perspectivas
Nossos resultados mostraram a hipermetilação de TFF1 e a hipometilação de
BCL3 como potencias alterações precoces em CEE. Neste contexto, pretendemos
ainda avaliar:
- Expressão (gênica e protéica) e metilação de TFF1 e BCL3 (Ilhotas CpG 1 e
3) em CEE, tecido normal adjacente e mucosa esofágica de indivíduos
saudáveis de modo a correlacionar expressão e metilação, assim como
investigar se tais alterações são fatores prognósticos para CEE.
- Expressão (gênica e protéica) e metilação de TFF1 e BCL3 (Ilhotas CpG 1 e
3) em mucosa de indivíduos saudáveis divididos em quatro grupos: (1) não
fumam e não bebem, (2) não fumam e bebem, (3) fumam e não bebem, (4)
fumam e bebem. Estas análises poderão responder se a regulação dos genes
TFF1 e BCL3 em mucosa esofágica é dependente, ao menos em parte, dos
fatores etiológicos para CEE.
- Expressão (gênica e protéica) e metilação de TFF1 e BCL3 (Ilhotas CpG 1 e
3) em pacientes com câncer de cabeça e pescoço, de modo a avaliar se estes
genes também são importantes para estes tumores, assim como se eles
podem ser utilizados como biomarcadores para o desenvolvimento de
segundos tumores primários de esôfago
Nosso trabalho mostrou ainda que o gene BCL3 é induzido por IL6 em células
esofágicas, provavelmente por mecanismos epigenéticos, e que tal indução parece
perturbar o processo de diferenciação in vitro utilizando o modelo em matrigel. De
modo a investigar este fenômeno mais a fundo, pretendemos:
- Estabelecer linhagens celulares que superexpressam BCL3 constitutivamente
para avaliar se o processo de diferenciação in vitro é perturbado, assim como
no caso do tratamento com IL6;
- Avaliar se outros genes anti-apoptóticos, como BCL2, poderiam ser induzidos
por IL6 e colaborar para o bloqueio do processo de diferenciação in vitro;
- Investigar se a hipermetilação encontrada na ilhota CpG 3 do gene BCL3
poderia induzir a expressão do gene por impedir a ligação de CTCF;
- Avaliar se IL6 é capaz de alterar direta ou indiretamente a expressão de
mediadores epigenéticos, como as DNMTs, em células esofágicas.

89
Referências Bibliográficas
ADAMS, R. L. et al. Mouse DNA methylase: methylation of native DNA. Biochim
Biophys Acta, v. 561, n. 2, p. 345-57, Feb 27 1979.
ADELAIDE, J. et al. Oesophageal cancer and amplification of the human cyclin D
gene CCND1/PRAD1. Br J Cancer, v. 71, n. 1, p. 64-8, Jan 1995.
AHMED, S. U.; MILNER, J. Basal cancer cell survival involves JNK2 suppression of a
novel JNK1/c-Jun/Bcl-3 apoptotic network. PLoS One, v. 4, n. 10, p. e7305, 2009.
ALLEN, J. W.; RICHARDSON, J. D.; EDWARDS, M. J. Squamous cell carcinoma of
the esophagus: a review and update. Surg Oncol, v. 6, n. 4, p. 193-200, Dec 1997.
AMIRY, N. et al. Trefoil factor-1 (TFF1) enhances oncogenicity of mammary
carcinoma cells. Endocrinology, v. 150, n. 10, p. 4473-83, Oct 2009.
ASOU, H. et al. Japanese B cell chronic lymphocytic leukaemia: a cytogenetic and
molecular biological study. Br J Haematol, v. 85, n. 3, p. 492-7, Nov 1993.
BAHL, R. et al. Novel polymorphism in p21(waf1/cip1) cyclin dependent kinase
inhibitor gene: association with human esophageal cancer. Oncogene, v. 19, n. 3, p.
323-8, Jan 20 2000.
BALL, M. P. et al. Targeted and genome-scale strategies reveal gene-body
methylation signatures in human cells. Nat Biotechnol, v. 27, n. 4, p. 361-8, Apr
2009.
BARCELLOS-HOFF, M. H. et al. Functional differentiation and alveolar
morphogenesis of primary mammary cultures on reconstituted basement membrane.
Development, v. 105, n. 2, p. 223-35, Feb 1989.
BARNAS, C. et al. Inactivation of the p53 protein in cell lines derived from human
esophageal cancers. Int J Cancer, v. 71, n. 1, p. 79-87, Mar 28 1997.

90
BARTEL, D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, v.
116, n. 2, p. 281-97, Jan 23 2004.
BAUER, A. et al. The NF-kappaB regulator Bcl-3 and the BH3-only proteins Bim and
Puma control the death of activated T cells. Proc Natl Acad Sci U S A, v. 103, n. 29,
p. 10979-84, Jul 18 2006.
BERNERD, F.; ASSELINEAU, D. Successive alteration and recovery of epidermal
differentiation and morphogenesis after specific UVB-damages in skin reconstructed
in vitro. Dev Biol, v. 183, n. 2, p. 123-38, Mar 15 1997.
BERNSTEIN, B. E.; MEISSNER, A.; LANDER, E. S. The mammalian epigenome.
Cell, v. 128, n. 4, p. 669-81, Feb 23 2007.
BIRD, A. DNA methylation patterns and epigenetic memory. Genes Dev, v. 16, n. 1,
p. 6-21, Jan 1 2002.
BOLLRATH, J. et al. gp130-mediated Stat3 activation in enterocytes regulates cell
survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer
Cell, v. 15, n. 2, p. 91-102, Feb 3 2009.
BOURS, V. et al. The oncoprotein Bcl-3 directly transactivates through kappa B
motifs via association with DNA-binding p50B homodimers. Cell, v. 72, n. 5, p. 729-
39, Mar 12 1993.
BOVERI, T.; BOVERI, M. The Origin of Malignant Tumors. Williams and Wilkins,
Baltimore, 1929.
BRAAKHUIS, B. J. et al. Second primary tumors and field cancerization in oral and
oropharyngeal cancer: molecular techniques provide new insights and definitions.
Head Neck, v. 24, n. 2, p. 198-206, Feb 2002.
BRACONI, C.; HUANG, N.; PATEL, T. MicroRNA-dependent regulation of DNA
methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in
human malignant cholangiocytes. Hepatology, v. 51, n. 3, p. 881-90, Mar 2010.

91
BROCKE-HEIDRICH, K. et al. BCL3 is induced by IL-6 via Stat3 binding to intronic
enhancer HS4 and represses its own transcription. Oncogene, v. 25, n. 55, p. 7297-
304, Nov 23 2006.
BROCKE-HEIDRICH, K. et al. Interleukin-6-dependent gene expression profiles in
multiple myeloma INA-6 cells reveal a Bcl-2 family-independent survival pathway
closely associated with Stat3 activation. Blood, v. 103, n. 1, p. 242-51, Jan 1 2004.
BROMBERG, J. F. et al. Stat3 as an oncogene. Cell, v. 98, n. 3, p. 295-303, Aug 6
1999.
CALIN, G. A.; CROCE, C. M. MicroRNA signatures in human cancers. Nat Rev
Cancer, v. 6, n. 11, p. 857-66, Nov 2006.
CANOZ, O. et al. Immunohistochemical detection of BCL-3 in lymphoid neoplasms:
a survey of 353 cases. Mod Pathol, v. 17, n. 8, p. 911-7, Aug 2004.
CARMODY, R. J. et al. Negative regulation of toll-like receptor signaling by NF-
kappaB p50 ubiquitination blockade. Science, v. 317, n. 5838, p. 675-8, Aug 3 2007.
CARVALHO, R. et al. Loss of heterozygosity and promoter methylation, but not
mutation, may underlie loss of TFF1 in gastric carcinoma. Lab Invest, v. 82, n. 10, p.
1319-26, Oct 2002.
CASTELLSAGUE, X. et al. Influence of mate drinking, hot beverages and diet on
esophageal cancer risk in South America. Int J Cancer, v. 88, n. 4, p. 658-64, Nov
15 2000.
CASTELLSAGUE, X. et al. Independent and joint effects of tobacco smoking and
alcohol drinking on the risk of esophageal cancer in men and women. Int J Cancer,
v. 82, n. 5, p. 657-64, Aug 27 1999.
CHENDRIMADA, T. P. et al. TRBP recruits the Dicer complex to Ago2 for microRNA
processing and gene silencing. Nature, v. 436, n. 7051, p. 740-4, Aug 4 2005.

92
CHOI, H. J. et al. Bcl3-dependent stabilization of CtBP1 is crucial for the inhibition of
apoptosis and tumor progression in breast cancer. Biochem Biophys Res
Commun, v. 400, n. 3, p. 396-402, Sep 24 2010.
COGSWELL, P. C. et al. Selective activation of NF-kappa B subunits in human
breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene, v. 19,
n. 9, p. 1123-31, Feb 24 2000.
CRADDOCK, V. M. Cancer of the Esophagus. Approaches to the Etiology.
Cambridge University Press, Cambridge, UK, 1993.
DE A. SIMAO, T. et al. Quantitative evaluation of SPRR3 expression in esophageal
squamous cell carcinoma by qPCR and its potential use as a biomarker. Exp Mol
Pathol, v. 91, n. 2, p. 584-9, Oct 2011.
DE STEFANI, E. et al. Meat intake and risk of squamous cell esophageal cancer: a
case-control study in Uruguay. Int J Cancer, v. 82, n. 1, p. 33-7, Jul 2 1999.
DEATON, A. M.; BIRD, A. CpG islands and the regulation of transcription. Genes
Dev, v. 25, n. 10, p. 1010-22, May 15 2011.
DEBNATH, J.; MUTHUSWAMY, S. K.; BRUGGE, J. S. Morphogenesis and
oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional
basement membrane cultures. Methods, v. 30, n. 3, p. 256-68, Jul 2003.
DEJEUX, E. et al. Identification and quantification of differentially methylated loci by
the pyrosequencing technology. Methods Mol Biol, v. 507, p. 189-205, 2009.
DEL BINO, S. et al. Ultraviolet B induces hyperproliferation and modification of
epidermal differentiation in normal human skin grafted on to nude mice. Br J
Dermatol, v. 150, n. 4, p. 658-67, Apr 2004.
DREILICH, M. et al. High-risk human papilloma virus (HPV) and survival in patients
with esophageal carcinoma: a pilot study. BMC Cancer, v. 6, p. 94, 2006.

93
EADS, C. A. et al. Fields of aberrant CpG island hypermethylation in Barrett's
esophagus and associated adenocarcinoma. Cancer Res, v. 60, n. 18, p. 5021-6,
Sep 15 2000.
ELLIS, L.; ATADJA, P. W.; JOHNSTONE, R. W. Epigenetics in cancer: targeting
chromatin modifications. Mol Cancer Ther, v. 8, n. 6, p. 1409-20, Jun 2009.
ESTELLER, M. Cancer epigenomics: DNA methylomes and histone-modification
maps. Nat Rev Genet, v. 8, n. 4, p. 286-98, Apr 2007.
EZHEVSKY, S. A. et al. Hypo-phosphorylation of the retinoblastoma protein (pRb)
by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci U S A, v.
94, n. 20, p. 10699-704, Sep 30 1997.
FAHRNER, J. A. et al. Dependence of histone modifications and gene expression on
DNA hypermethylation in cancer. Cancer Res, v. 62, n. 24, p. 7213-8, Dec 15 2002.
FANG, M. Z. et al. Promoter hypermethylation and inactivation of O(6)-
methylguanine-DNA methyltransferase in esophageal squamous cell carcinomas and
its reactivation in cell lines. Int J Oncol, v. 26, n. 3, p. 615-22, Mar 2005.
FEBER, A. et al. MicroRNA expression profiles of esophageal cancer. J Thorac
Cardiovasc Surg, v. 135, n. 2, p. 255-60; discussion 260, Feb 2008.
FEINBERG, A. P.; OHLSSON, R.; HENIKOFF, S. The epigenetic progenitor origin of
human cancer. Nat Rev Genet, v. 7, n. 1, p. 21-33, Jan 2006.
FERLAY, J. et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN
2008. Int J Cancer, v. 127, n. 12, p. 2893-917, Dec 15 2010.
FRANZOSO, G. et al. The oncoprotein Bcl-3 can facilitate NF-kappa B-mediated
transactivation by removing inhibiting p50 homodimers from select kappa B sites.
EMBO J, v. 12, n. 10, p. 3893-901, Oct 1993.

94
FRANZOSO, G. et al. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-
kappa B-mediated inhibition. Nature, v. 359, n. 6393, p. 339-42, Sep 24 1992.
FUJIMOTO, J. et al. DNA hypermethylation at the pS2 promoter region is associated
with early stage of stomach carcinogenesis. Cancer Lett, v. 149, n. 1-2, p. 125-34,
Feb 28 2000.
FUJITA, T. et al. The candidate proto-oncogene bcl-3 encodes a transcriptional
coactivator that activates through NF-kappa B p50 homodimers. Genes Dev, v. 7, n.
7B, p. 1354-63, Jul 1993.
FUJIWARA, S. et al. Hypermethylation of p16 gene promoter correlates with loss of
p16 expression that results in poorer prognosis in esophageal squamous cell
carcinomas. Dis Esophagus, v. 21, n. 2, p. 125-31, 2008.
FUTREAL, P. A. et al. A census of human cancer genes. Nat Rev Cancer, v. 4, n. 3,
p. 177-83, Mar 2004.
GARCIA-RAMIREZ, M.; ROCCHINI, C.; AUSIO, J. Modulation of chromatin folding
by histone acetylation. J Biol Chem, v. 270, n. 30, p. 17923-8, Jul 28 1995.
GARZON, R.; CALIN, G. A.; CROCE, C. M. MicroRNAs in Cancer. Annu Rev Med,
v. 60, p. 167-79, 2009.
GASCHE, J. A. et al. Interleukin-6 promotes tumorigenesis by altering DNA
methylation in oral cancer cells. Int J Cancer, v. 129, n. 5, p. 1053-63, Sep 1 2011.
GETSIOS, S. et al. Desmoglein 1-dependent suppression of EGFR signaling
promotes epidermal differentiation and morphogenesis. J Cell Biol, v. 185, n. 7, p.
1243-58, Jun 29 2009.
GHOSH, S.; KARIN, M. Missing pieces in the NF-kappaB puzzle. Cell, v. 109 Suppl,
p. S81-96, Apr 2002.

95
GRAHAM, T. A.; MCDONALD, S. A.; WRIGHT, N. A. Field cancerization in the GI
tract. Future Oncol, v. 7, n. 8, p. 981-93, Aug 2011.
GUAN, Y. et al. MiR-125b targets BCL3 and suppresses ovarian cancer proliferation.
Int J Cancer, v. 128, n. 10, p. 2274-83, May 15 2011.
GUO, M. et al. Promoter hypermethylation of resected bronchial margins: a field
defect of changes? Clin Cancer Res, v. 10, n. 15, p. 5131-6, Aug 1 2004.
GUO, M. et al. Epigenetic silencing of CDX2 is a feature of squamous esophageal
cancer. Int J Cancer, v. 121, n. 6, p. 1219-26, Sep 15 2007.
GUO, M. et al. Accumulation of promoter methylation suggests epigenetic
progression in squamous cell carcinoma of the esophagus. Clin Cancer Res, v. 12,
n. 15, p. 4515-22, Aug 1 2006.
GUO, Y. et al. Distinctive microRNA profiles relating to patient survival in esophageal
squamous cell carcinoma. Cancer Res, v. 68, n. 1, p. 26-33, Jan 1 2008.
HAINAUT, P.; HOLLSTEIN, M. p53 and human cancer: the first ten thousand
mutations. Adv Cancer Res, v. 77, p. 81-137, 2000.
HALL, H. G.; FARSON, D. A.; BISSELL, M. J. Lumen formation by epithelial cell lines
in response to collagen overlay: a morphogenetic model in culture. Proc Natl Acad
Sci U S A, v. 79, n. 15, p. 4672-6, Aug 1982.
HANAHAN, D.; WEINBERG, R. A. The hallmarks of cancer. Cell, v. 100, n. 1, p. 57-
70, Jan 7 2000.
HANAHAN, D.; WEINBERG, R. A. Hallmarks of cancer: the next generation. Cell, v.
144, n. 5, p. 646-74, Mar 4 2011.
HANAWA, M. et al. EGFR protein overexpression and gene amplification in
squamous cell carcinomas of the esophagus. Int J Cancer, v. 118, n. 5, p. 1173-80,
Mar 1 2006.

96
HAYDEN, M. S.; GHOSH, S. Shared principles in NF-kappaB signaling. Cell, v. 132,
n. 3, p. 344-62, Feb 8 2008.
HERCEG, Z. Epigenetics and cancer: towards an evaluation of the impact of
environmental and dietary factors. Mutagenesis, v. 22, n. 2, p. 91-103, Mar 2007.
HINSHELWOOD, R. A.; CLARK, S. J. Breast cancer epigenetics: normal human
mammary epithelial cells as a model system. J Mol Med (Berl), v. 86, n. 12, p. 1315-
28, Dec 2008.
HIRANO, T.; ISHIHARA, K.; HIBI, M. Roles of STAT3 in mediating the cell growth,
differentiation and survival signals relayed through the IL-6 family of cytokine
receptors. Oncogene, v. 19, n. 21, p. 2548-56, May 15 2000.
HOLLSTEIN, M. et al. p53 mutations in human cancers. Science, v. 253, n. 5015, p.
49-53, Jul 5 1991.
HOLST, C. R. et al. Methylation of p16(INK4a) promoters occurs in vivo in
histologically normal human mammary epithelia. Cancer Res, v. 63, n. 7, p. 1596-
601, Apr 1 2003.
HUTVAGNER, G. et al. A cellular function for the RNA-interference enzyme Dicer in
the maturation of the let-7 small temporal RNA. Science, v. 293, n. 5531, p. 834-8,
Aug 3 2001.
I, H. et al. Association of global levels of histone modifications with recurrence-free
survival in stage IIB and III esophageal squamous cell carcinomas. Cancer
Epidemiol Biomarkers Prev, v. 19, n. 2, p. 566-73, Feb 2010.
IACOBUZIO-DONAHUE, C. A. Epigenetic changes in cancer. Annu Rev Pathol, v.
4, p. 229-49, 2009.

97
IGAKI, H. et al. Highly frequent homozygous deletion of the p16 gene in esophageal
cancer cell lines. Biochem Biophys Res Commun, v. 203, n. 2, p. 1090-5, Sep 15
1994.
IMBALZANO, K. M. et al. Increasingly transformed MCF-10A cells have a
progressively tumor-like phenotype in three-dimensional basement membrane
culture. Cancer Cell Int, v. 9, p. 7, 2009.
INCA. Incidência de Câncer no Brasil, Estimativa 2012. Instituto Nacional de
Câncer José Alencar Gomes da Silva, Ministério da Saúde, 2011.
ISLAMI, F. et al. Patterns of food and nutrient consumption in northern Iran, a high-
risk area for esophageal cancer. Nutr Cancer, v. 61, n. 4, p. 475-83, 2009.
ISRAEL, A. The IKK complex: an integrator of all signals that activate NF-kappaB?
Trends Cell Biol, v. 10, n. 4, p. 129-33, Apr 2000.
ISSA, J. P. et al. Accelerated age-related CpG island methylation in ulcerative colitis.
Cancer Res, v. 61, n. 9, p. 3573-7, May 1 2001.
IVANOV, V. N.; KRASILNIKOV, M.; RONAI, Z. Regulation of Fas expression by
STAT3 and c-Jun is mediated by phosphatidylinositol 3-kinase-AKT signaling. J Biol
Chem, v. 277, n. 7, p. 4932-44, Feb 15 2002.
JARNICKI, A.; PUTOCZKI, T.; ERNST, M. Stat3: linking inflammation to epithelial
cancer - more than a "gut" feeling? Cell Div, v. 5, p. 14, 2010.
JIANG, X. P. et al. Reduction in serum IL-6 after vacination of breast cancer patients
with tumour-associated antigens is related to estrogen receptor status. Cytokine, v.
12, n. 5, p. 458-65, May 2000.
JONES, P. A.; BAYLIN, S. B. The fundamental role of epigenetic events in cancer.
Nat Rev Genet, v. 3, n. 6, p. 415-28, Jun 2002.

98
KASHATUS, D.; COGSWELL, P.; BALDWIN, A. S. Expression of the Bcl-3 proto-
oncogene suppresses p53 activation. Genes Dev, v. 20, n. 2, p. 225-35, Jan 15
2006.
KIM, Y. T. et al. Aberrant promoter CpG island hypermethylation of the
adenomatosis polyposis coli gene can serve as a good prognostic factor by affecting
lymph node metastasis in squamous cell carcinoma of the esophagus. Dis
Esophagus, v. 22, n. 2, p. 143-50, 2009.
KOBAYASHI, M. et al. Heparanase regulates esophageal keratinocyte differentiation
through nuclear translocation and heparan sulfate cleavage. Differentiation, v. 74, n.
5, p. 235-43, Jun 2006.
KONDO, Y. et al. Genetic instability and aberrant DNA methylation in chronic
hepatitis and cirrhosis--A comprehensive study of loss of heterozygosity and
microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in
microdissected specimens from patients with hepatocellular carcinoma. Hepatology,
v. 32, n. 5, p. 970-9, Nov 2000.
KUROKI, T. et al. Allele loss and promoter hypermethylation of VHL, RAR-beta,
RASSF1A, and FHIT tumor suppressor genes on chromosome 3p in esophageal
squamous cell carcinoma. Cancer Res, v. 63, n. 13, p. 3724-8, Jul 1 2003.
LAFON-HUGHES, L. et al. Chromatin-remodelling mechanisms in cancer. Mutat
Res, v. 658, n. 3, p. 191-214, Mar-Apr 2008.
LAI, A. Y. et al. DNA methylation prevents CTCF-mediated silencing of the
oncogene BCL6 in B cell lymphomas. J Exp Med, v. 207, n. 9, p. 1939-50, Aug 30
2010.
LAIRD, P. W. The power and the promise of DNA methylation markers. Nat Rev
Cancer, v. 3, n. 4, p. 253-66, Apr 2003.
LEE, G. Y. et al. Three-dimensional culture models of normal and malignant breast
epithelial cells. Nat Methods, v. 4, n. 4, p. 359-65, Apr 2007.

99
LEE, Y. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature,
v. 425, n. 6956, p. 415-9, Sep 25 2003.
LEE, Y. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J, v.
23, n. 20, p. 4051-60, Oct 13 2004.
LEE, Y. C. et al. Revisit of field cancerization in squamous cell carcinoma of upper
aerodigestive tract: better risk assessment with epigenetic markers. Cancer Prev
Res (Phila), v. 4, n. 12, p. 1982-92, Dec 2011.
LENGAUER, C.; KINZLER, K. W.; VOGELSTEIN, B. Genetic instabilities in human
cancers. Nature, v. 396, n. 6712, p. 643-9, Dec 17 1998.
LIN, H. H. et al. Bcl-2 overexpression prevents apoptosis-induced Madin-Darby
canine kidney simple epithelial cyst formation. Kidney Int, v. 55, n. 1, p. 168-78, Jan
1999.
LING, Z. Q. et al. Hypermethylation-modulated down-regulation of CDH1 expression
contributes to the progression of esophageal cancer. Int J Mol Med, v. 27, n. 5, p.
625-35, May 2011.
LOIZOU, J. I. et al. Epigenetic information in chromatin: the code of entry for DNA
repair. Cell Cycle, v. 5, n. 7, p. 696-701, Apr 2006.
LOPEZ, J. et al. The context and potential of epigenetics in oncology. Br J Cancer,
v. 100, n. 4, p. 571-7, Feb 24 2009.
LU, J. et al. MicroRNA expression profiles classify human cancers. Nature, v. 435,
n. 7043, p. 834-8, Jun 9 2005.
MAESAWA, C. et al. Inactivation of the CDKN2 gene by homozygous deletion and
de novo methylation is associated with advanced stage esophageal squamous cell
carcinoma. Cancer Res, v. 56, n. 17, p. 3875-8, Sep 1 1996.

100
MAKINO, T. et al. Cytokeratins 18 and 8 are poor prognostic markers in patients with
squamous cell carcinoma of the oesophagus. Br J Cancer, v. 101, n. 8, p. 1298-306,
Oct 20 2009.
MALDONADO, V. et al. Gene regulation by BCL3 in a cervical cancer cell line. Folia
Biol (Praha), v. 56, n. 4, p. 183-93, 2010.
MAO, W. M. et al. Hypermethylation-modulated downregulation of RASSF1A
expression is associated with the progression of esophageal cancer. Arch Med Res,
v. 42, n. 3, p. 182-8, Apr 2011.
MASSOUMI, R. et al. Cyld inhibits tumor cell proliferation by blocking Bcl-3-
dependent NF-kappaB signaling. Cell, v. 125, n. 4, p. 665-77, May 19 2006.
MASUDA, M. et al. Constitutive activation of signal transducers and activators of
transcription 3 correlates with cyclin D1 overexpression and may provide a novel
prognostic marker in head and neck squamous cell carcinoma. Cancer Res, v. 62, n.
12, p. 3351-5, Jun 15 2002.
MAUNAKEA, A. K. et al. Conserved role of intragenic DNA methylation in regulating
alternative promoters. Nature, v. 466, n. 7303, p. 253-7, Jul 8 2010.
MCLARTY, A. J. et al. Esophageal resection for cancer of the esophagus: long-term
function and quality of life. Ann Thorac Surg, v. 63, n. 6, p. 1568-72, Jun 1997.
MITAS, M. et al. Accurate discrimination of Barrett's esophagus and esophageal
adenocarcinoma using a quantitative three-tiered algorithm and multimarker real-time
reverse transcription-PCR. Clin Cancer Res, v. 11, n. 6, p. 2205-14, Mar 15 2005.
MOLLOY, P. L.; WATT, F. DNA methylation and specific protein-DNA interactions.
Philos Trans R Soc Lond B Biol Sci, v. 326, n. 1235, p. 267-75, Jan 30 1990.
MOLNAR, V. et al. Changes in miRNA expression in solid tumors: an miRNA
profiling in melanomas. Semin Cancer Biol, v. 18, n. 2, p. 111-22, Apr 2008.

101
MONTESANO, R.; HOLLSTEIN, M.; HAINAUT, P. Genetic alterations in esophageal
cancer and their relevance to etiology and pathogenesis: a review. Int J Cancer, v.
69, n. 3, p. 225-35, Jun 21 1996.
MONTESANO, R. et al. Collagen matrix promotes reorganization of pancreatic
endocrine cell monolayers into islet-like organoids. J Cell Biol, v. 97, n. 3, p. 935-9,
Sep 1983.
MONTESANO, R.; SCHALLER, G.; ORCI, L. Induction of epithelial tubular
morphogenesis in vitro by fibroblast-derived soluble factors. Cell, v. 66, n. 4, p. 697-
711, Aug 23 1991.
NAKAGAWA, H. et al. Human cyclin D1 oncogene and esophageal squamous cell
carcinoma. Cancer, v. 76, n. 4, p. 541-9, Aug 15 1995.
NAN, X.; CROSS, S.; BIRD, A. Gene silencing by methyl-CpG-binding proteins.
Novartis Found Symp, v. 214, p. 6-16; discussion 16-21, 46-50, 1998.
NISHIKORI, M. et al. High-level expression of BCL3 differentiates t(2;5)(p23;q35)-
positive anaplastic large cell lymphoma from Hodgkin disease. Blood, v. 101, n. 7, p.
2789-96, Apr 1 2003.
NIU, G. et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor
angiogenesis. Oncogene, v. 21, n. 13, p. 2000-8, Mar 27 2002.
O'NEIL, B. H. et al. Expression of nuclear factor-kappaB family proteins in
hepatocellular carcinomas. Oncology, v. 72, n. 1-2, p. 97-104, 2007.
OHLSSON, R.; RENKAWITZ, R.; LOBANENKOV, V. CTCF is a uniquely versatile
transcription regulator linked to epigenetics and disease. Trends Genet, v. 17, n. 9,
p. 520-7, Sep 2001.
OHNO, H.; TAKIMOTO, G.; MCKEITHAN, T. W. The candidate proto-oncogene bcl-3
is related to genes implicated in cell lineage determination and cell cycle control.
Cell, v. 60, n. 6, p. 991-7, Mar 23 1990.

102
OHTA, K. et al. Histopathological evaluation including cytokeratin 13 and Ki-67 in the
border between Lugol-stained and -unstained areas. Oncol Rep, v. 24, n. 1, p. 9-14,
Jul 2010.
OLAYIOYE, M. A. et al. The ErbB signaling network: receptor heterodimerization in
development and cancer. EMBO J, v. 19, n. 13, p. 3159-67, Jul 3 2000.
OLIVIER, M. et al. The IARC TP53 database: new online mutation analysis and
recommendations to users. Hum Mutat, v. 19, n. 6, p. 607-14, Jun 2002.
ONG, S. T. et al. Lymphadenopathy, splenomegaly, and altered immunoglobulin
production in BCL3 transgenic mice. Oncogene, v. 16, n. 18, p. 2333-43, May 7
1998.
PAGES, F. et al. Immune infiltration in human tumors: a prognostic factor that should
not be ignored. Oncogene, v. 29, n. 8, p. 1093-102, Feb 25 2010.
PARKIN, D. M. Global cancer statistics in the year 2000. Lancet Oncol, v. 2, n. 9, p.
533-43, Sep 2001.
PARKIN, D. M.; PISANI, P.; FERLAY, J. Global cancer statistics. CA Cancer J Clin,
v. 49, n. 1, p. 33-64, 1, Jan-Feb 1999.
PETERSEN, O. W. et al. Interaction with basement membrane serves to rapidly
distinguish growth and differentiation pattern of normal and malignant human breast
epithelial cells. Proc Natl Acad Sci U S A, v. 89, n. 19, p. 9064-8, Oct 1 1992.
PICKENS, A.; ORRINGER, M. B. Geographical distribution and racial disparity in
esophageal cancer. Ann Thorac Surg, v. 76, n. 4, p. S1367-9, Oct 2003.
PITOT, H. C. Oncogenes and human neoplasia. Clin Lab Med, v. 6, n. 1, p. 167-79,
Mar 1986.

103
RAPOZO, D. C. Efeito da lesão térmica na carcinogênese esofágica. Doutorado,
Universidade do Estado do Rio de Janeiro, Instituto de Biologia Roberto
Alcântara Gomes, 2009.
REBOLLO, A. et al. Bcl-3 expression promotes cell survival following interleukin-4
deprivation and is controlled by AP1 and AP1-like transcription factors. Mol Cell Biol,
v. 20, n. 10, p. 3407-16, May 2000.
RIBEIRO PINTO, L. F. et al. Mechanisms of esophageal cancer development in
Brazilians. Mutat Res, v. 544, n. 2-3, p. 365-73, Nov 2003.
RIBIERAS, S.; TOMASETTO, C.; RIO, M. C. The pS2/TFF1 trefoil factor, from basic
research to clinical applications. Biochim Biophys Acta, v. 1378, n. 1, p. F61-77,
Aug 19 1998.
RODEMANN, H. P.; DITTMANN, K.; TOULANY, M. Radiation-induced EGFR-
signaling and control of DNA-damage repair. Int J Radiat Biol, v. 83, n. 11-12, p.
781-91, Nov-Dec 2007.
RODRIGUEZ, A. et al. Identification of mammalian microRNA host genes and
transcription units. Genome Res, v. 14, n. 10A, p. 1902-10, Oct 2004.
ROLON, P. A. et al. Hot and cold mate drinking and esophageal cancer in Paraguay.
Cancer Epidemiol Biomarkers Prev, v. 4, n. 6, p. 595-605, Sep 1995.
ROSSINI, A. et al. TP53 mutation profile of esophageal squamous cell carcinomas
of patients from Southeastern Brazil. Mutat Res, v. 696, n. 1, p. 10-5, Feb 2010.
ROSSINI, A. et al. Polymorphisms of GSTP1 and GSTT1, but not of CYP2A6,
CYP2E1 or GSTM1, modify the risk for esophageal cancer in a western population.
Carcinogenesis, v. 28, n. 12, p. 2537-42, Dec 2007.
SALAM, I. et al. Aberrant promoter methylation and reduced expression of p16 gene
in esophageal squamous cell carcinoma from Kashmir valley: a high-risk area. Mol
Cell Biochem, v. 332, n. 1-2, p. 51-8, Dec 2009.

104
SALEM, A. F. et al. Caveolin-1 promotes pancreatic cancer cell differentiation and
restores membranous E-cadherin via suppression of the epithelial-mesenchymal
transition. Cell Cycle, v. 10, n. 21, p. 3692-700, Nov 1 2011.
SALGADO, R. et al. Circulating interleukin-6 predicts survival in patients with
metastatic breast cancer. Int J Cancer, v. 103, n. 5, p. 642-6, Feb 20 2003.
SAMBROOK, J.; RUSSEL, D. W. T. Molecular Cloning—A Laboratory Manual.
Cold Spring Harbor Laboratory Press, New York, 2001.
SCHAFER, Z. T.; BRUGGE, J. S. IL-6 involvement in epithelial cancers. J Clin
Invest, v. 117, n. 12, p. 3660-3, Dec 2007.
SEERY, J. P.; WATT, F. M. Asymmetric stem-cell divisions define the architecture of
human oesophageal epithelium. Curr Biol, v. 10, n. 22, p. 1447-50, Nov 16 2000.
SHAMMA, A. et al. Cyclin D1 overexpression in esophageal dysplasia: a possible
biomarker for carcinogenesis of esophageal squamous cell carcinoma. Int J Oncol,
v. 16, n. 2, p. 261-6, Feb 2000.
SHI, S. Q.; CAI, J. T.; YANG, J. M. Expression of trefoil factors 1 and 2 in
precancerous condition and gastric cancer. World J Gastroenterol, v. 12, n. 19, p.
3119-22, May 21 2006.
SHUYAMA, K. et al. Human papillomavirus in high- and low-risk areas of
oesophageal squamous cell carcinoma in China. Br J Cancer, v. 96, n. 10, p. 1554-
9, May 21 2007.
SIKORA, A.; GRZESIUK, E. Heat shock response in gastrointestinal tract. J Physiol
Pharmacol, v. 58 Suppl 3, p. 43-62, Aug 2007.
SLACK, J. M. Conrad Hal Waddington: the last Renaissance biologist? Nat Rev
Genet, v. 3, n. 11, p. 889-95, Nov 2002.

105
SOMA, T. et al. Nicotine induces the fragile histidine triad methylation in human
esophageal squamous epithelial cells. Int J Cancer, v. 119, n. 5, p. 1023-7, Sep 1
2006.
SQUIER, C. A.; KREMER, M. J. Biology of oral mucosa and esophagus. J Natl
Cancer Inst Monogr, n. 29, p. 7-15, 2001.
STEPHANOU, A. et al. Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and
Bcl-x promoters. Cell Death Differ, v. 7, n. 3, p. 329-30, Mar 2000.
STONER, G. D. et al. Establishment and characterization of SV40 T-antigen
immortalized human esophageal epithelial cells. Cancer Res, v. 51, n. 1, p. 365-71,
Jan 1 1991.
SUN, J. M. et al. Estrogen regulation of trefoil factor 1 expression by estrogen
receptor alpha and Sp proteins. Exp Cell Res, v. 302, n. 1, p. 96-107, Jan 1 2005.
SUNPAWERAVONG, P. et al. Correlation of epidermal growth factor receptor
mutation, immunohistochemistry, and fluorescence in situ hybridization in esophageal
squamous cell carcinoma. J Med Assoc Thai, v. 92, n. 9, p. 1136-42, Sep 2009.
SZYMANOWSKA, N. et al. BCL2 and BCL3 are recurrent translocation partners of
the IGH locus. Cancer Genet Cytogenet, v. 186, n. 2, p. 110-4, Oct 15 2008.
TANAKA, H. et al. Methylation of the 5' CpG island of the FHIT gene is closely
associated with transcriptional inactivation in esophageal squamous cell carcinomas.
Cancer Res, v. 58, n. 15, p. 3429-34, Aug 1 1998.
TAUPIN, D.; PODOLSKY, D. K. Trefoil factors: initiators of mucosal healing. Nat Rev
Mol Cell Biol, v. 4, n. 9, p. 721-32, Sep 2003.
THORNBURG, N. J.; PATHMANATHAN, R.; RAAB-TRAUB, N. Activation of nuclear
factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma.
Cancer Res, v. 63, n. 23, p. 8293-301, Dec 1 2003.

106
TSURUTA, H. et al. Amplification and overexpression of EXP1 and EXP2/Cyclin D1
genes in human esophageal carcinomas. Biochem Biophys Res Commun, v. 196,
n. 3, p. 1529-36, Nov 15 1993.
TZAO, C. et al. Promoter methylation of the hMLH1 gene and protein expression of
human mutL homolog 1 and human mutS homolog 2 in resected esophageal
squamous cell carcinoma. J Thorac Cardiovasc Surg, v. 130, n. 5, p. 1371, Nov
2005.
TZAO, C. et al. Reduced acetylated histone H4 is associated with promoter
methylation of the fragile histidine triad gene in resected esophageal squamous cell
carcinoma. Ann Thorac Surg, v. 82, n. 2, p. 396-401; discussion 401, Aug 2006.
USHIJIMA, T. Epigenetic field for cancerization. J Biochem Mol Biol, v. 40, n. 2, p.
142-50, Mar 31 2007.
VAISSIERE, T. et al. Quantitative analysis of DNA methylation profiles in lung
cancer identifies aberrant DNA methylation of specific genes and its association with
gender and cancer risk factors. Cancer Res, v. 69, n. 1, p. 243-52, Jan 1 2009.
VIATOUR, P. et al. Protein phosphorylation as a key mechanism for the regulation of
BCL-3 activity. Cell Cycle, v. 3, n. 12, p. 1498-501, Dec 2004.
VICTORA, C. G. et al. Hot beverages and oesophageal cancer in southern Brazil: a
case-control study. Int J Cancer, v. 39, n. 6, p. 710-6, Jun 15 1987.
WANG, F. et al. p52-Bcl3 complex promotes cyclin D1 expression in BEAS-2B cells
in response to low concentration arsenite. Toxicology, v. 273, n. 1-3, p. 12-8, Jun 29
2010.
WANG, J. et al. Aberrant DNA methylation of P16, MGMT, and hMLH1 genes in
combination with MTHFR C677T genetic polymorphism in esophageal squamous cell
carcinoma. Cancer Epidemiol Biomarkers Prev, v. 17, n. 1, p. 118-25, Jan 2008.

107
WANG, Y. et al. Beyond the double helix: writing and reading the histone code.
Novartis Found Symp, v. 259, p. 3-17; discussion 17-21, 163-9, 2004.
WATANABE, N. et al. Regulation of NFKB1 proteins by the candidate oncoprotein
BCL-3: generation of NF-kappaB homodimers from the cytoplasmic pool of p50-p105
and nuclear translocation. EMBO J, v. 16, n. 12, p. 3609-20, Jun 16 1997.
WEI, L. H. et al. Interleukin-6 promotes cervical tumor growth by VEGF-dependent
angiogenesis via a STAT3 pathway. Oncogene, v. 22, n. 10, p. 1517-27, Mar 13
2003.
WONG, M. L. et al. Aberrant promoter hypermethylation and silencing of the critical
3p21 tumour suppressor gene, RASSF1A, in Chinese oesophageal squamous cell
carcinoma. Int J Oncol, v. 28, n. 3, p. 767-73, Mar 2006.
XING, E. P. et al. Loss of heterozygosity of the Rb gene correlates with pRb protein
expression and associates with p53 alteration in human esophageal cancer. Clin
Cancer Res, v. 5, n. 5, p. 1231-40, May 1999.
YI, R. et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short
hairpin RNAs. Genes Dev, v. 17, n. 24, p. 3011-6, Dec 15 2003.

108
Anexo I - The Effects of Diet on
Epigenetic Processes

109

110

111

112

113

114

115

116

117

118

119
Anexo II - Epigenetic Signatures in
Cancer: Implications for the Control
of Cancer in the Clinic

120

121

122

123
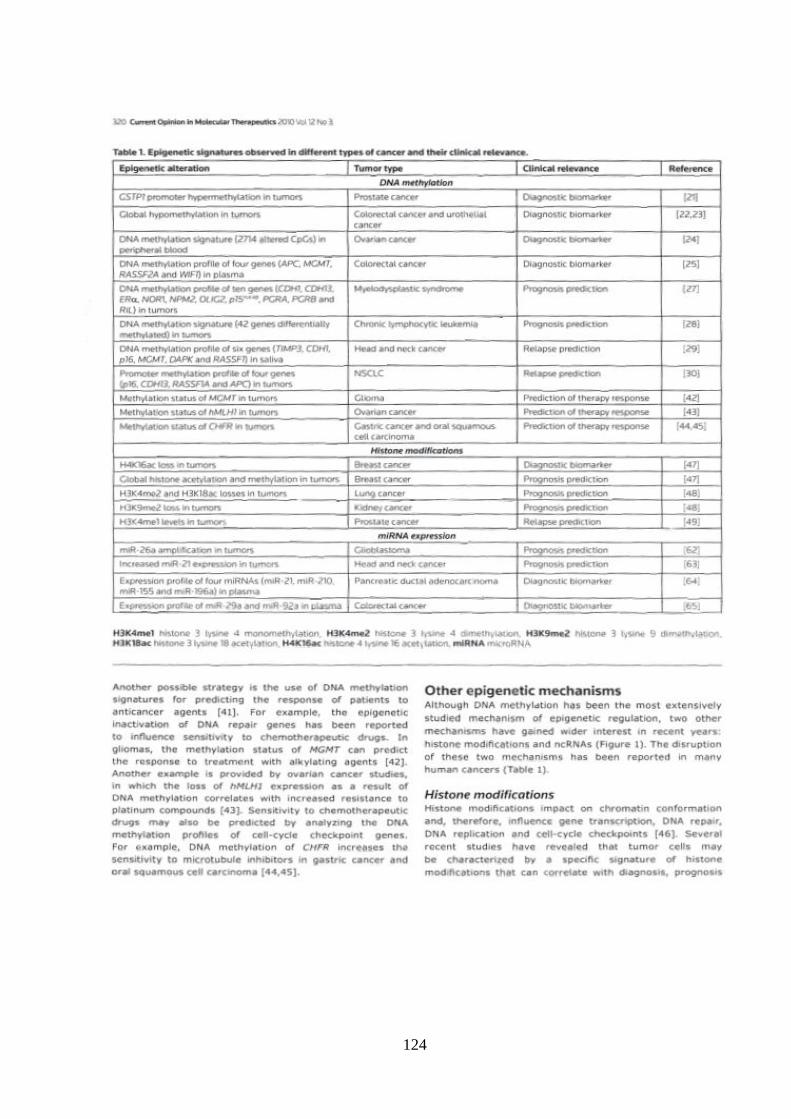
124

125

126

127

128

129
Anexo III - Aberrant DNA
methylation of cancer-associated
genes in gastric cancer in the
European Prospective Investigation
into Cancer and Nutrition (EPIC–
EURGAST)

130

131

132

133
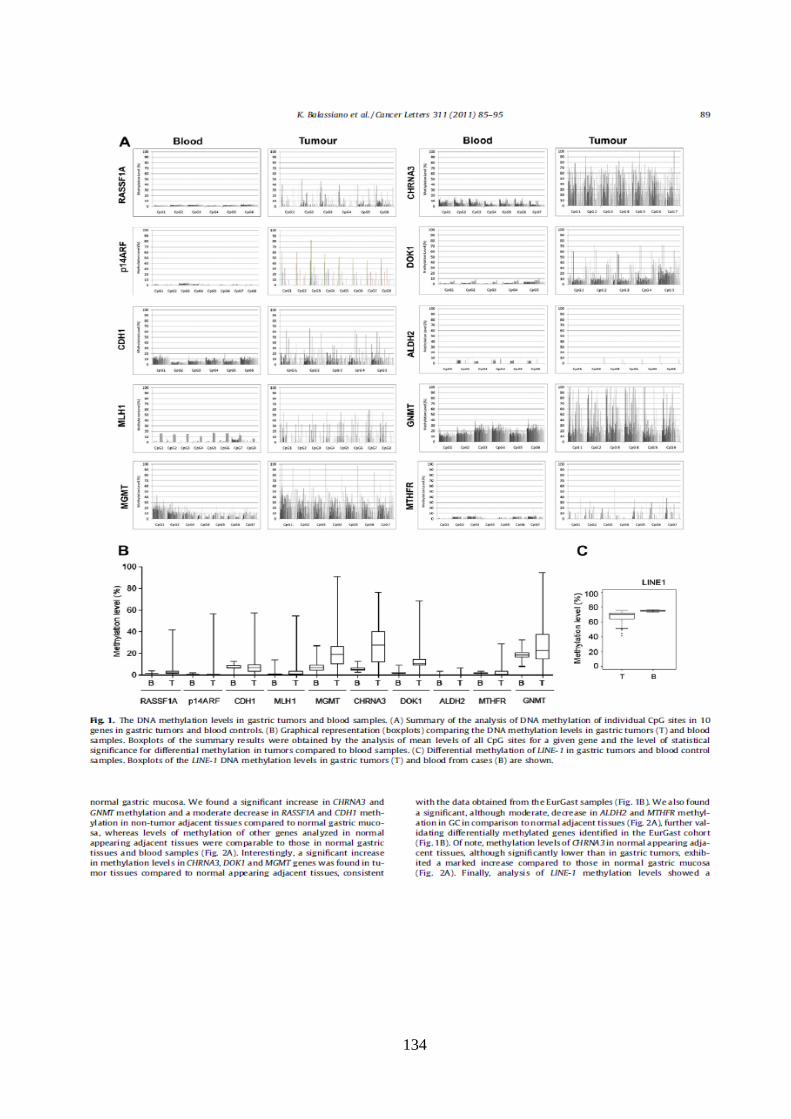
134

135

136

137

138

139

140

141
Anexo IV:
ARTIGO ACEITO PARA PUBLICAÇÃO NA REVISTA EPIGENETICS
DNA methylation changes associated with risk factors in
tumours of the upper aerodigestive tract
Samson Mani1†, Kasia Szymańska1†, Cyrille Cuenin1, David Zaridze2, Karen
Balassiano1, Sheila C.S. Lima1,9
, Elena Matos3, Alexander Daudt4, Sergio Koifman5,
Victor Wunsch Filho6, Ana M. B. Menezes7, Maria Paula Curado8, Gilles Ferro1,
Thomas Vaissière1, Bakary Sylla1, Massimo Tommasino1, Luis Felipe Ribeiro Pinto9,10
,
Paolo Boffetta8,11, Pierre Hainaut1, Paul Brennan1, Zdenko Herceg1*
1International Agency for Research on Cancer (IARC), Lyon, France;
2Cancer Research Centre, Moscow, Russia;
3Institut of Oncology Angel H. Roffo, University of Buenos Aires, Argentina;
4Hospital de Clinics de Porto Alegre, Porto Alegre, Brazil;
5Escola Nacional de Saude Publica, Rio de Janeiro, Brazil;
6Universidade de Sao Paulo, Sao Paulo, Brazil;
7Universidade Federal de Pelotas, Pelotas, Brazil;
8The International Prevention Research Institute, Lyon, France;
9Divisão de Genética, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
10Departamento de Bioquímica, Universidade do Estado do Rio de Janeiro, Instituto
de Biologia Roberto Alcantara Gomes, Rio de Janeiro, Brazil
11Present address: The Tisch Cancer Institute and Institute for Translational
Epidemiology; Mount Sinai School of Medicine, New York, NY, USA.
†These authors made equal contribution to the work

142
Running title: DNA methylation changes in UADT cancers
*Corresponding author: Zdenko Herceg, PhD; Epigenetics Group, International
Agency for Research on Cancer (IARC), 150 cours Albert Thomas, Lyon, 69008,
France
Tel.: +33 4 72 73 83 98; Fax: +33 4 72 73 83 22; E-mail: [email protected]
Key words: DNA methylation, upper aerodigestive tract, cancer, risk factors,
biomarkers
ABSTRACT
Cancers of the upper aerodigestive tract (UADT) are common forms of malignancy
associated with tobacco and alcohol exposures, although human papillomavirus and
nutritional deficiency are also important risk factors. While somatically acquired DNA
methylation changes have been associated with UADT cancer, what triggers these
events and precise epigenetic targets are poorly understood. In this study, we
applied quantitative profiling of DNA methylation states in a panel of cancer-
associated genes to a case-control study of UADT cancers. Our analyses revealed a
high frequency of aberrant hypermethylation of several genes, including MYOD1,
CHRNA3, and MTHFR in UADT tumours, whereas CDKN2A was moderately
hypermethylated. Among differentially methylated genes, we identified a new gene
(the nicotinic acetycholine receptor gene CHRN3) as target of aberrant
hypermethylation in UADT cancer, suggesting that epigenetic deregulation of
nicotinic acetycholine receptors in non-neuronal tissues may promote the
development of UADT cancer. Importantly, we found that sex and age is strongly
associated with the methylation states, whereas tobacco smoking and alcohol intake
may also influence the methylation levels in specific genes. This study identifies
aberrant DNA methylation patterns in UADT cancer and suggests a potential
mechanism by which environmental factors may deregulate key cellular genes
involved in tumour suppression and contribute to UADT cancer.