Embed Size (px)
Citation preview

1
Sumario ••••• Contents
ARTÍCULOS DE REVISIÓN
ESFEROCITOSIS HEREDITARIA: ASPECTOS CLÍNICOS,BIOQUÍMICOS Y MOLECULARES
Hereditary spherocytosis: Clinical, biochemical and molecularaspects
Mayelín Herrera García y Marianela Estrada del Cueto7
ASPECTOS CLÍNICOS Y EPIDEMIOLÓGICOS DE LA LEUCEMIAMIELOIDE AGUDA EN EL ANCIANO
Clinical and epidemiological aspects of acute myeloid leukemia inthe elderly
María Teresa Milanés Roldán, Rafael Losada Buchillón, Porfirio Hernández Ramírez, Olga M.Agramonte Llanes y Edelis Rosell Monzón
25
ARTÍCULOS ORIGINALES
INMUNOFENOTIPAJE Y SUPERVIVENCIA GLOBAL DE PACIENTESPEDIÁTRICOS CON LEUCEMIAS AGUDAS
Immunophenotyping and global survival of pediatric patients withacute leukemia
Vianed Marsán Suárez, Consuelo Macías Abraham, René Riviero Jiménez, Miriam Sánchez Segura,Beatriz Socarrás Ferrer, Anissa Gramatges Ortiz, María Teresa Milanés Roldán, Alejandro González
Otero y Porfirio Hernández Ramírez34

2
MECANISMOS DE ACCIÓN DE LA GAMMAGLOBULINA PARA USOENDOVENOSO
Mechanisms of action of gammaglobulins for intravenous useAmaury Noa Albelo, Boris Rodríguez Ramos y Arturo Vidal Tallet
41
EVALUACIÓN DE LA FUNCIÓN OPSONO FAGOCÍTICA DE LOSNEUTRÓFILOS EN PACIENTES INFECTADOS POR EL VIH
Evaluation of the opsonophagocytic function of neutrophils in HIV-infected patients
Randelys Molina Castro, Alejandro Álvarez García, Liliana Pérez Toledo, Lizet Sánchez Valdés,Yondel Torranzo Soto y Caridad Luzardo Suárez
48
POSIBLE PARTICIPACIÓN DEL SISTEMA COMPLEMENTO EN ELDESARROLLO DE MANIFESTACIONES AUTOINMUNES EN LA
LEUCEMIA LINFOCÍTICA CRÓNICAPossible role of the complement system in the development ofautoimmune manifestations in chronic lymphocytic leukemias
Rinaldo Villaescusa Blanco, Antonio Bencomo Hernández, Ada A. Arce Hernández, Julio C. MerlínLinares, Ana M. Guerreiro Hernández, Luz M. Morera Barrios y Porfirio Hernández Ramírez
55
ACTIVIDAD ANTICOMPLEMENTARIA EN PREPARACIONES DEINMUNOGLOBULINAS PARA USO ENDOVENOSO
Anti-complement activity in immunoglobulin preparations forintravenous use
Ada A. Arce Hernández, Rinaldo Villaescusa Blanco, Julio C. Merlín Linares, Ana M. GuerreiroHernández, Ed J. Nieuwahuys y Andrew J. Hannema
60
TÉCNICAS
COMPARACIÓN DEL ULTRAMICROMÉTODOINMUNOCITOQUÍMICO (UMICIQ) CON EL DE LA FOSFATASAALCALINA-ANTI FOSFOTASA ALCALINA (APAAP) PARA LA
CUANTIFICACIÓN DE SUBPOBLACIONES LINFOCITARIAS TComparison of the immunocytochemical ultramicromethod and thealkaline phosphatase – anti-alkaline phosphatase method for the
quantification of T lymphocyte subsetsBeatriz Socarrás Ferrer, Vianed Marsán Suárez, Miriam Sánchez Segura, Anissa Gramatges Ortiz,
Rinaldo Villaescusa Blanco y Consuelo Macías Abraham63

3
COMUNICACIONES BREVES
DETERMINACIÓN DE ANTICUERPOS ANTINUCLEARES EN LAANEMIA HEMOLÍTICA AUTOINMUNE Y LA PÚRPURA
TROMBOCITOPÉNICA AUTOINMUNEDetermination of antinuclear antibodies in autoimmune hemolytic
anemia and autoimmune thrombocytopenic purpuraAna M. Guerreiro Hernández, Rinaldo Villaescusa Blanco, Antonio Bencomo Hernández, Luz M.
Morera Barrios, Julio C. Merlín Linares, Ada A. Arce Hernández y Carlos García Guevara67
CARTAS AL DIRECTOR
CAPACIDADES FUNCIONALES EN ALUMNOS ATLETASPORTADORES DE HEMOGLOBINA S
Functional capacities in hemoglobin S-carrying athlete-studentsJuan J. González Pérez, Lay Hernández González y Ángel Aquino Pernas
70

4

5
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):7-24
Artículos de revisión
Instituto de hematología e Inmunología
ESFEROCITOSIS HEREDITARIA: ASPECTOS CLÍNICOS,BIOQUÍMICOS Y MOLECULARES
Lic. Mayelín Herrera García y Dra. Marianela Estrada del Cueto
RESUMEN
La esferocitosis hereditaria (EH) es una enfermedad caracterizada por anemiahemolítica de severidad variable, con presencia de esferocitos en sangre periféricay una respuesta clínica favorable a la esplenectomía. Con el desarrollo de nuevastécnicas se encontraron las primeras alteraciones bioquímicas de las proteínas dela membrana eritrocitaria, y posteriormente, se han podido precisar las alteracionesmoleculares mediante las técnicas del ADN recombinante. La EH es una enfermedadmuy heterogénea que se produce por un defecto intrínseco del glóbulo rojo, yexisten otras alteraciones secundarias a esta afección. La prueba más utilizadapara el diagnóstico de la EH es la fragilidad osmótica del glóbulo rojo. Se hademostrado que esta enfermedad es producida por defectos de las proteínas queintervienen en las interacciones verticales entre el esqueleto de la membrana y labicapa lipídica. El tratamiento de elección en la EH es la esplenectomía, ya quees el más efectivo en el control de la anemia, aunque la sobrevida de los glóbulosrojos permanece acortada y los esferocitos no desaparecen. Este proceder seindica en pacientes con anemia hemolítica severa o en individuos moderadamenteasintomáticos pero que presentan litiasis vesicular.
DeCS: ESFEROCITOSIS HEREDITARIA/diagnóstico; ESFEROCITOSISHEREDITARIA/ genética; ADN RECOMBINANTE; PROTEINAS DE LAMEMBRANA/química; PROTEINAS RECOMBINANTES/química;ESPLENECTOMIA; MEMBRANA ERITROCITICA/química.
ESTRUCTURADE LA MEMBRANAERITROCITARIA
La membrana del glóbulo rojo es laresponsable de las propiedades mecánicasy de la mayoría de las funciones fisiológicasde la célula.1
Está formada por una bicapa lipídicaplana, donde predominan en el 80 % losfosfolípidos y el colesterol y en menormedida los glicolípidos y aminofosfolípidos,distribuidos asimétricamente. De igualforma, se encuentran embebidas parcial ototalmente en ella las proteínas integralesde membrana, unidas fuertemente por

6
enlaces apolares. Su libre desplazamiento através de esta bicapa contribuye a mantenersu fluidez.2 Las proteínas periféricasinteractúan entre sí para formar una malla oenrejado que recubre la cara interior de ladoble capa de fosfolípidos y son lasresponsables de la estabilidad y las propie-dades viscoelásticas de la membrana.3 Entreestas proteínas se destacan la espectrina(Sp), la ankirina (banda 2.1, 2.2, 2.3 y 2.6), labanda 4.1, la banda 4.2, la banda 4.9, laaducina, la tropomiosina y la banda 7. Otrasproteínas periféricas se disponen hacia lacara exterior de la bicapa lipídica y ellas sonfundamentalmente antígenos de gruposanguíneo4 (fig.1).
La banda 3 representa el 25 % del totalde las proteínas integrales. Está constituidapor 2 dominios estructurales: el dominiocitoplasmático, que es el encargado de launión con las proteínas del esqueleto, y elsitio transmembranoso que mantiene elcontacto con el medio extra e intracelular,proporcionando los canales responsables deltransporte de iones bicarbonato (HCO
3−)
Banda 3 Glicoforina A
Ankirina4,2
4,9
p55
Actina Tropomiosina
Tropomodulina
β−espectrinaα−espectrina
Interacciones horizontales
Inte
racc
ione
s ve
rtica
les
Banda 3 Banda 3 Banda 3
Glicoforina C
Aducina
FIG. 1. Estructura de la membrana eritrocitaria.
y cloruros (CI−). Además, posee un sitio deglicosilación capaz de unir antígenos parael grupo sanguíneo I/i5 e intervieneactivamente en la eliminación de eritrocitosenvejecidos.6
Las glicoforinas son un grupo deproteínas integrales caracterizadas por suelevado contenido en ácido siálico. Lasglicoforinas A,B,C y D son las másimportantes y constituyen los sustratosantigénicos de los diferentes grupossanguíneos. La glicoforina C contribuye ala estabilidad de la membrana gracias a suinteracción con proteínas periféricas,además de participar en el intercambioiónico transmembranoso.7
La Sp es la proteína más abundante yademás la principal responsable delmantenimiento del enrejado proteico.8 Estácompuesta por 2 subunidades α y βenrolladas de forma antiparalela, las que seunen por sus extremos para formartetrámeros. Estas 2 subunidades estánconstituidas por secuencias repetitivas de106 aminoácidos, las que se enlazan paraformar una triple hélice9 (fig.2).

7
α−espectrina β−espectrina
β−espectrina α−espectrina
N
Sitio de auto-asociación
Sitio de nucleación
NH3
N C C
Unión a laankirina
Sitio de nucleación
N C
Uniónactina/4,1
Sitio deunión del
calcio
FIG.2. Estructura del tetrámero de espectrina (Sp). Se observa la triple hélice de las unidades repetitivas, los sitios de autoasociaciónde la Sp y los sitios de nucleación donde comienza la interacción lateral entre las cadenas α y β de la Sp.
La ankirina (ANK) está compuesta por3 subunidades estructurales correspon-dientes a 3 dominios funcionalmentediferentes: regulador, de unión a la membra-na y de unión a la Sp. Su contribución a laintegridad de la membrana es decisiva, yaque constituye un importante punto deanclaje a la doble capa lipídica a través de labanda 3.10
La actina es una proteína organizadaen forma de protofilamentos helicoidalesestabilizados por la interacción con la Sp, laproteína 4.1 y la tropomiosina.11
Otra de las proteínas que integran elcitoesqueleto es la proteína 4.1, cuyafunción fundamental es estabilizar la uniónespectrina-actina y contribuir a fijar elesqueleto a la bicapa lipídica.12
El mantenimiento de la forma yestabilidad de la membrana es tambiénresponsabilidad de otras proteínas. Entreellas está la proteína 4.2, que actúa comomodulador para estabilizar la interacciónankirina-banda 3.13 La banda 4.9, la aducinay la tropomiosina, protegen la estabilidadde la actina.14
Los estudios moleculares hanpermitido conocer la localización cromosó-mica de estas proteínas, así como la
secuencia nucleotídica de cada uno de losgenes (tabla 1).
Las interacciones proteicas determi-nantes de la integridad de la membranaeritrocitaria son de 2 tipos: verticales yhorizontales.2-4
Interacciones verticales→fijan elesqueleto a la doble capa lipídica.
− Unión entre la Sp y la banda 3,estabilizada por la ANK y modulada porla proteína 4.2.
− Unión entre la proteína 4.1 y la banda 3.
Interacciones horizontales→ sonresponsables de la estabilidad global delesqueleto.
− Unión entre moléculas de Sp para formardímeros y oligómeros de mayor pesomolecular.
− Unión entre la Sp, actina y la proteína4.1, estabilizada por la proteína 4.9.
Defectos en algunas de estas proteínaspueden dar lugar a trastornos clínicos enlos cuales está involucrada la estabilidaddel glóbulo rojo. Entre ellos los más

8
TABLA 1. Propiedades bioquímicas y moleculares de las proteínas de la membrana eritrocitaria
Símbolo PeriféricaBandas en gel Localización Tamaño del gen Número de Número de Peso molecular del (P) oPAGE-SDS Proteína cromosómica (Kb) exones aminoácidos (aprox/deducido) gen integral (l)
1 α espectrina 1q22-q25 80 52 2 429 240/281 SPTA1 P2 β espectrina 14q23-q24.2 >100 32 2 137 220/246 SPTB P2.1 Ankirina 8q11.2 ≅ 160 42 1 880 210/206 ANK1 P2.9 α aducina 4p16.3 - - - 103/81 - P
β aducina 2? - - - 97/80 - P3 AE1 17q12-q21 17 20 911 90-100/102 EPB3 l4.1 Proteína 4.1 1p33-p34.2 >250 23 588 88+78/66 EL1 P4.2 Palidina 15q15-q21 20 13 691 72/77 ELB42 P4.9 Dematina - - - - 48/52/43+46 - P
p55 Xq28 > 4 6 466 55/53 MPP1 P5 β actina 7pter-q22 - - - 43/42 ACTB P
Tropomodulina 9q22 - - - 43/41 TMOD P6 G3PD 12p13 - - - 35/36 G3PD P7 Estomatina 9q34.1 40 7 287 31/32 EPB72 I
Tropomiosina 1q31 - - - 27 +29/28 TPM3 P8 Proteína 8 - - - - 23/- - PPAS-1 Glicoforina A 4q31 - 7 131 36/14 GYPA IPAS-2 Glicoforina C 2q14-q21 14 4 128 32/14 GYPC IPAS-3 Glicoforina B 4q31 - 5 72 20/8 GYPB I
Glicoforina D 2q14-q21 14 4 106 23/11 GYPD I
frecuentes y mejor estudiados son laesferocitosis hereditaria (HS) y laeliptocitosis hereditaria (HE).4,7,8
ESFEROCITOSIS HEREDITARIA
ANTECEDENTES HISTÓRICOS
La esferocitosis hereditaria (EH) es unaenfermedad caracterizada por anemiahemolítica de severidad variable, conpresencia de esferocitos en sangre periféricay una respuesta clínica favorable a laesplenectomía.15
Fue descrita en 1871 por 2 médicosbelgas, Vanlair y Masius, cuando estudia-ron a una paciente joven que presentabadolor abdominal, esplenomegalia asociadacon íctero, vómitos, anemia y marcadaatrofia muscular. Los investigadoresnotaron que los glóbulos rojos tenían unaforma esférica y mucho más pequeña quelos normales, demostraron que el bazoestaba involucrado en el envejecimiento de
estas células y denominaron a esta enferme-dad “microcitemia”.16
Veinte años después fue redescubiertapor Wilson y Minkowsky , quienesregistraron 8 casos en 3 generacionesdiferentes de una misma familia.17
La mayor contribución al conocimientode esta enfermedad fue hecha por loshallazgos de Chauffard, el cual confirmó elaumento de la fragilidad osmótica de loseritrocitos, lo que explicaba la anemiahemolítica encontrada en estos casos.Observó también cómo se producía lacorrección de la hemólisis con laesplenectomía, y demostró la implicacióndel bazo en esta entidad.18
Posteriormente se encontró que losglóbulos rojos de pacientes con HSpresentaban una disminución de Na+
intracelular y una pérdida de lípidos de lamembrana, lo que explicaba la disminucióndel área superfical de la célula.19 Por estosestudios la entidad se conoce también comoenfermedad de Minkowsky-Chauffard.3-4
Después de la década de los 70, con eldesarrollo de nuevas técnicas, se

9
encontraron las primeras alteracionesbioquímicas de las proteínas de la membranaeritrocitaria y a partir de 1985, por medio delas técnicas del ADN recombinante, se hanpodido precisar las alteraciones molecularesen un número importante de casos.20
GENÉTICA Y PREVALENCIA
La EH es la anemia hemolítica másfrecuente en el mundo y se ha señalado unaprevalencia de 1:2000 en algunos paíseseuropeos.8 Sin embargo, estos datospueden no reflejar la frecuencia real de laenfermedad, ya que no se tienen en cuentalos portadores asintomáticos ni loshallazgos de una frecuencia del 1 % dedonantes de sangre con fragilidad osmóticaaumentada observada en algunos países.15
Aunque es más frecuente en individuos dela raza blanca, puede observarse ocasional-mente en otras razas o grupos étnicos.15,21
El 75 % de las familias afectadasmuestran un patrón autosómico dominante.El homocigótico de esta forma de herenciano ha sido identificado, lo que sugiere quesea incompatible con la vida.22 El 25 %restante corresponde a un patrónautosómico recesivo, nuevas mutacioneso pacientes con EH dominante conpenetrancia incompleta.2,23
FORMAS CLÍNICAS
La EH es una enfermedad muy hetero-génea desde el punto de vista clínico. Sepuede observar desde el portadorasintomático hasta pacientes que presentanuna anemia hemolítica crónica con grandesrequerimientos transfusionales.24,25
Dependiendo de la severidad delcuadro clínico, de las cifras de hemoglobina,los niveles de bilirrubina y el conteo dereticulocitos, esta enfermedad se clasifica
en 4 formas: portador asintomático, EHligera, EH típica y EH severa.26,27
Portador asintomático. En algunasfamilias se ha señalado un patrón deherencia autosómico recesivo. En estoscasos, los padres de un paciente afectadono presentan ninguna alteración. Enocasiones la afectación es muy leve, comoligero incremento de las cifras dereticulocitos, escasos esferocitos enperiferia o fragilidad osmótica incubadaalterada y puede no ser detectada por losexámenes de rutina. Debe tenerse en cuentatambién que pueden ocurrir nuevasmutaciones dentro de una familia aparentan-do un patrón de herencia autosómicorecesivo, por lo que siempre es importanteun estudio minucioso de todos losmiembros de la familia.3,4
EH ligera. Comprende entre el 20 y30 % de todos los pacientes con EHautosómica dominante, los que puedenpresentar una hemólisis ligera compen-sada.3,27 Los individuos son frecuentementeasintomáticos y algunos casos son difícilesde diagnosticar, ya que la anemia y laesplenomegalia son muy ligeras y enocasiones pueden estar ausentes. 28
Muchos de estos pacientes se diagnosticandurante estudios familiares o cuando en laetapa adulta aparece el íctero y laesplenomegalia. Episodios hemolíticospueden presentarse en el curso de algunosprocesos infecciosos como mononucleosis,parvovirus o citomegalovirus, así comodurante el embarazo, por esfuerzos físicosintensos o por sangramientos.27-30
EH típica. Entre el 50 y 60 % de lospacientes con EH autosómica dominantetienen esta forma clínica. Presentan unahemólisis compensada incompleta y unaanemia de ligera a moderada. El íctero escomún en niños, aunque se puede vertambién en los adultos y está asociado coninfecciones virales ligeras, debido a laestimulación reticuloendotelial y a unaumento de la hemólisis. Los requerimientos

10
transfusionales son esporádicos. Laesplenomegalia está presente en el 50 % delos niños y en el 75 % de los adultos.2,3,31,32
EH severa. Estos pacientes (5-10 %)evolucionan con una hemólisis severa ypresentan frecuentes requerimientostransfusionales. La mayoría de estos casostienen una forma autosómica recesiva de laenfermedad. Pueden presentar crisisaplásticas, retardo del crecimiento y de lamaduración sexual. La esplenectomía es eltratamiento de elección en esta formaclínica.33,34 Generalmente la enfermedaddebuta al nacimiento con ictericia yhemólisis y se requiere, en muchasocasiones, de exanguinotransfusión.30,32
ETIOLOGÍA
Es bien conocido que la EH se producepor un defecto intrínseco del glóbulo rojo yse han demostrado defectos moleculares dediferentes proteínas que conforman elesqueleto de la membrana eritrocitaria. Lapresencia de alteraciones del metabolismo,del transporte catiónico, de la fosforilaciónde las proteínas y de la composición de loslípidos de la membrana, son secundarias ala causa primaria de esta enfermedad.3,8,15
La lesión en la membrana está dada poruna pérdida del área de la célula, pero sedesconoce si esto se debe a una pérdidafísica (fragmentación ) o a una contracciónde la superficie de la membrana, y aunquela mayoría de las evidencias favorecen lafragmentación, no parece ser el únicomecanismo que explique este hallazgo. Seha demostrado que la fuerza requerida parafragmentar las células esferocíticas, así comosu elasticidad y deformabilidad, estándisminuidas, y que ésta es proporcional ala densidad de la espectrina en la membrana.Además, los glóbulos rojos esferocíticospierden membrana más rápidamente que losnormales cuando se deprime su metabolis-mo. La concentración de fosfolípidos y
colesterol es del 15 al 20 % menor de lonormal debido posiblemente a ladisminución de su superficie y se presumeque también se produce una pérdida de lasproteínas integrales de la membrana.2,4,25,31,35
FISIOPATOLOGÍA
El problema fundamental de la EH es laconsecuencia reológica de la disminuciónde la relación superficie/volumen. Lamembrana del glóbulo rojo es muy flexible,pero sólo puede incrementar su área un 3 %antes de romperse. Por consiguiente,mientras la célula se vuelve más esférica,es cada vez menos deformable. En el casode los hematíes esferocíticos, esta pobredeformabilidad es un obstáculo sólo parael bazo, ya que la mayoría de los esferocitossobreviven bien después de la esple-nectomía.4
- Eritrostasis:Ham, Castle y Dacie fueron los
primeros en señalar que los glóbulos rojosesferocíticos son particularmentevulnerables a la eritrostasis.
El glóbulo rojo sufre una serie decambios que conlleva a la autohemólisiscuando se incuba en ausencia de glucosa,proceso que se acelera en la HS, lo queconlleva a que los esferocitos se depletende ATP más rápidamente que lo normal. Amedida que los niveles de ATP disminuyen,falla la bomba de cationes y penetra agua ysodio en la célula. Cuando la célula alcanzamuy bajos niveles de ATP, el calciointracelular también aumenta, y da lugar aun fallo en la bomba de calcio, lo queproduce una salida del potasio intracelular.El mecanismo molecular de estos cambiosen la permeabilidad no es bien conocido,pero sus consecuencias están biendefinidas. A medida que el potasiodisminuye, el agua responde al cambio enosmolaridad y las células se encogen. Loshematíes esferocíticos no son capaces de

11
soportar estos cambios, ya que soninestables y se fragmentan excesivamentedurante la depleción metabólica, los lípidosde la membrana se pierden a una velocidadsuperior al doble de la normal y aunque nose sabe con exactitud, se plantea quetambién existe una pérdida proporcional deproteínas integrales de la membrana (almenos en la deficiencia primaria de banda 3se ha demostrado). Aunque la disminuciónde la superficie inicialmente se acompañade una deshidratación celular, la pérdida demembrana predomina, la célula excede suvolumen de hemólisis crítico (volumen/superficie > 100) y se produce laautohemólisis.1-4
- Dinámica del atrapamientoesplénico.
Uno de los aspectos no conocidosacerca de la fisiopatología de la EH es si loseventos que dan lugar al acondicionamientoy destrucción de los glóbulos rojosesferocíticos en el bazo son los mismos quedan lugar a un incremento en laesferoidicidad y autohemólisis durante laeritrostasis in vitro.
Es conocido que los hematíesesferocíticos son selectivamente retenidospor el bazo, y producen una pérdida de la
membrana, lo que promueve el atrapamientoesplénico y la destrucción eventual de lacélula. Estudios han demostrado que eltiempo de tránsito esplénico medio secorrelaciona inversamente con lasupervivencia de los glóbulos rojos en laHS. Parece ser que el atrapamiento esplénicoes promovido inicialmente por lainestabilidad del esqueleto de la membrana,pero los detalles de cómo el defectomolecular permite que éste se produzca, aúnno se han definido. Los mecanismos delcondicionamiento esplénico y de ladestrucción eritrocitaria son todavíadesconocidos. Estudios cinéticos sugierenque los glóbulos rojos son atrapadoscontinuamente dentro de los cordonesesplénicos durante el período que serequiere para inducir la forma esferoidalpasiva y la autohemólisis por depleciónmetabólica, aunque otra posibilidad puedeser la presencia de daños metabólicosrepetidos. Una especial susceptibilidad delos hematíes esferocíticos al medio ácidodel bazo y una intervención activa de losmacrófagos en el proceso de daño celulardurante la eritrostasis, pueden influirtambién, pero no hay evidencias directassobre estas 2 hipótesis (fig.3).2,4,15,23,31,35
? ? ?
Defecto primariodel esqueleto
de la membrana
Aumento de inestabilidad
de la membrana
Pérdida demembrana
Retenciónen el bazo
Disminución de lasuperficie con
relación al volumen“Esperocitos”
Disminución de ladeformabilidad
celular
Disminución de ATP
Daño inducidopor ácidos
Ataque por macrófagos
ERITROSTASIS* disminución de ATP* disminución de pH* aumento del contacto con macrófagos
HEMÓLISIS+? fagocitosis+? lisis osmótica
“CONDICIONAMIENTO”ESPLÉNICO
FIG. 3. Fisiopatología del “ condicionamiento” esplénico y destrucción de los glóbulos rojos en la esferocitosis hereditaria (EH).

12
DIAGNÓSTICO
Los hallazgos morfológicos encontra-dos en la EH son los mismos queencontramos en otros procesos hemolíti-cos: hiperplasia de precursores eritroidesen la médula ósea, cifra elevada dereticulocitos en sangre periférica, aumentode la bilirrubina no conjugada en el plasmay una elevada excreción de urobilinógeno.
La anemia generalmente es normo-cítica, normocrómica, con volumencorpuscular medio (VCM) normal osubnormal, aunque menor del esperado enotras condiciones con un grado similar dereticulocitosis.
La hemoglobina corpuscular media(HCM) es normal, pero la concentraciónhemoglobínica corpuscular media (CHCM)está aumentada en el 50 % de los pacientes,lo que refleja una deshidratación de unaparte de la población celular. No obstante,lo que caracteriza a la EH es la presencia deesferocitos en sangre periférica.2-4
Las diversas formas de la EH muestranun patrón morfológico diferente entre cadauna de estas. Los pacientes con EHautosómica dominante y algunos con laforma autosómica recesiva muestransolamente esferocitos en la lámina deperiferia, mientras que pacientes con unadeficiencia severa de β espectrina,presentan acantocitos y poiquilocitos,pudiendo en algunos casos, aparecer estosúltimos en un mayor porcentaje que losesferocitos. En los casos con unadeficiencia de banda 3 se ha observadotambién la presencia de pinceredcells.3,4,8,31,35
La prueba más utilizada para eldiagnóstico de la EH es la fragilidadosmótica del glóbulo rojo, la cual mide lahabilidad de los glóbulos rojos deincrementar su volumen cuando sonsometidos a soluciones hipotónicas de
cloruro de sodio (NaCl) de concentracionesvariables. Debido a que los esferocitostienen una relación superficie/volumendisminuida, tienen una capacidaddisminuida para aumentar su volumen y selisan a una concentración de sales máselevada que las células normales. Lasensibilidad de esta prueba puedeincrementarse con la preincubación de lascélulas. 31 Se han señalado otrasmodificaciones de la prueba de fragilidadosmótica. Entre estas estás la autohemólisis,la cual determina la hemólisis de los glóbulosrojos incubados sin glucosa en condicionesestériles, la prueba del glicerol y la pruebarosa,31 pero ninguna de ellas parece ser mássensible que la fragilidad osmóticaincubada. Se ha señalado que la prueba decriohemólisis hipertónica es 100 % sensiblepara el diagnóstico de la EH, pero estosresultados no han sido debidamenteconfirmados todavía.36 La introducción dela ectacitometría, que cuantifica ladeformabilidad de los eritrocitos por lamedición de la fuerza que induce laelongación de la célula, permitió eldesarrollo de la ectacitometría osmótica, lacual parece ser la prueba más sensible en laactualidad.37 Sin embargo, esta técnica soloestá disponible en un reducido grupo delaboratorios especializados. La fijación conglutaraldehído es útil para detectar lapresencia de alteraciones morfológicas:microcitos, acantocitos, pincered cells,poikilocitos, etcétera.37
El análisis de las proteínas de lamembrana eritrocitaria en electroforesis depoliacrilamida con SDS (PAGE-SDS) seutiliza para cuantificar las proteínas y/odetectar péptidos truncados con migraciónanormal, y el estudio molecular ha permitidoidentificar un número importante demutaciones en distintas proteínas de lamembrana eritrocitaria (fig.4).
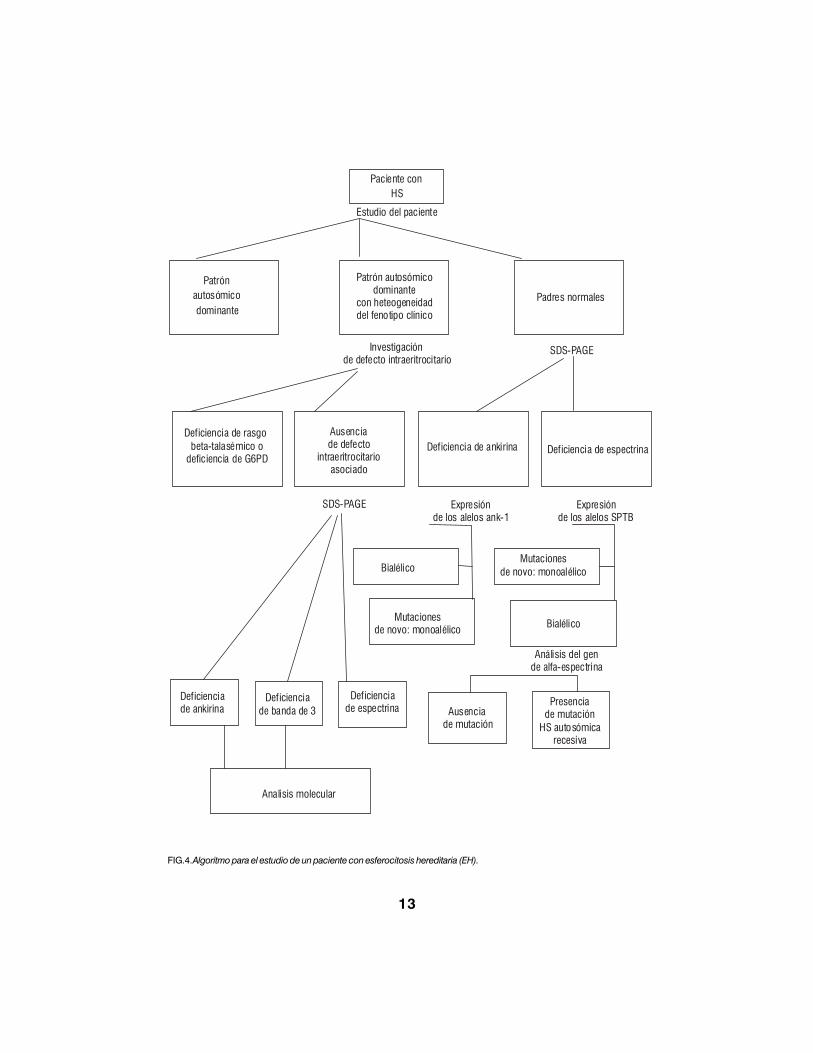
13
Paciente con HS
Estudio del paciente
Patrón autosómico dominante
Patrón autosómico dominante
con heteogeneidad del fenotipo clínico
Padres normales
Investigación de defecto intraeritrocitario
SDS-PAGE
Deficiencia de rasgo beta-talasémico o
deficiencia de G6PD
Ausencia de defecto
intraeritrocitario asociado
Deficiencia de ankirina Deficiencia de espectrina
SDS-PAGE Expresión de los alelos ank-1
Expresión de los alelos SPTB
BialélicoMutaciones
de novo: monoalélico
Mutaciones de novo: monoalélico Bialélico
Análisis del gen de alfa-espectrina
Deficiencia de ankirina
Deficiencia de banda de 3
Deficiencia de espectrina Ausencia
de mutación
Presencia de mutación
HS autosómica recesiva
Analisis molecular
FIG.4. Algoritmo para el estudio de un paciente con esferocitosis hereditaria (EH).

14
Es necesario señalar que para undiagnóstico preciso de la EH es importantesiempre realizar el estudio familiar.
DIAGNÓSTICO DIFERENCIAL
La forma típica de EH puede serdiagnosticada fácilmente, aunque debendescartarse otras causas de anemiahemolítica esferocítica tales como la anemiahemolítica autoinmune (prueba de Coombs),hemoglobinas inestables (electroforesis dehemoglobina, cuerpos de Heinz, etc.), laestomatosis hereditaria y el síndrome delRh nulo, entre otras. Durante el períodoneonatal, es difícil diferenciar la EH de laincompatibilidad ABO, en la que laesferocitosis es evidente. En estos casos,es imprescindible el estudio familiar, asícomo una reevaluación del niño entre los 4y 6 meses de nacido. No obstante, losesferocitos de la EH son distinguibles delas anemias hemolíticas autoinmunes por launiformidad de éstos, así como por elaumento de la CHCM.1-4,8
Se presentan también dificultadesdiagnósticas en los pacientes que debutancon una crisis aplástica. Al inicio, lanaturaleza de los síntomas sugiere laaparición de un proceso adquirido y laausencia de reticulocitos puede enmascararel diagnóstico de la anemia hemolítica.También se puede confundir el diagnósticocuando la EH se asocia con otrasenfermedades que elevan la relaciónvolumen/superficie, tales como ladeficiencia de hierro o el íctero obstructivo.La deficiencia de hierro normaliza lafragilidad anormal y la forma de losesferocitos, pero no mejora la sobrevida deéstos. En el íctero obstructivo desaparecela esferocitosis, debido a la acumulación decolesterol y fosfolípidos en la membranacelular. En individuos normales, este
proceso conlleva a la formación de targetcells, mientras que en la HS las célulasadoptan una forma discoidal y seincrementa la sobrevida de las células. Laβ-talasemia heterocigótica y algunasenzimopatías, también pueden interferir enel diagnóstico de la HS.15,23,31
Debido al curso asintomático de estaenfermedad en muchos pacientes, debedescartarse la presencia de una EH enaquellos casos con síntomas aislados talescomo esplenomegalia, íctero, litiasisvesicular en el adulto joven, anemias comoresultado de mononucleosis infecciosa uotras infecciones severas y durante elembarazo.1,8,15,35
Ocasionalmente se pueden observaresferocitos en pacientes con marcadaesplenomegalia (cirrosis y mielofibrosis) oen pacientes con anemias microangiopá-ticas, pero el diagnóstico diferencial deestas entidades no presenta grandesdificultades.15
El estudio familiar es muy importanteen el diagnóstico diferencial, sobre todopara poder precisar el carácter hereditariode la anemia, ya que las pruebas empleadaspara el diagnóstico de la HS pueden serpositivas en muchas de las patologíasmencionadas anteriormente.2-4,8
COMPLICACIONES
Las llamada crisis hemolíticas son muyfrecuentes en pacientes con EH. Cursandurante la aparición de una infección viraly la anemia en general es ligera, conreticulocitosis, ictericia y esplenomegalia.Cuando la hemólisis es severa, esrecomendable la administración detransfusiones de sangre.3,4
La crisis aplástica debida a la infecciónpor el parvovirus B19, es la complicaciónmás importante que pueden presentar estos

15
pacientes, la cual puede acompañarse defiebre, vómitos, dolor abdominal, cefalea,palidez y anemia severa.15,23,31
Las crisis megaloblásticas resultan deuna ingestión insuficiente de ácido fólico.Se observan frecuentemente durante elembarazo, donde los requerimientos deácido fólico son más elevados.4
La litiasis vesicular fue la primeracomplicación descrita en los pacientes conEH, la cual se ha observado en niñospequeños, aunque su aparición es másfrecuente en adolescentes y adultos. Entreel 55 y el 85 % de pacientes con EHpresentan litiasis, y la mitad de estos tienensíntomas de colelitiasis o de obstrucciónbiliar.2-15
Otras complicaciones, aunque muypoco frecuentes, han sido señaladas:hematopoyesis extramedular, gota, úlcerasen miembros inferiores y retraso en elcrecimiento y en el desarrollo sexual.26,28,31,35
TRATAMIENTO
La esplenectomía es el tratamiento deelección para los pacientes con EH, ya quees el más efectivo en el control de la anemia,aunque la sobrevida de los glóbulos rojospermanece acortada y los esferocitos nodesaparecen. Está indicada en pacientescon anemia hemolítica severa o enindividuos moderadamente sintomáticos,pero que presentan litiasis vesicular. En elresto de los casos con EH existediscrepancia en cuanto la recomendaciónde realizar la esplenectomía. Algunosinvestigadores sugieren realizarla a todopaciente con HS, aún en aquéllos con unaforma ligera o moderada de la enfermedad yen ausencia de litiasis vesicular, mientrasque otros plantean un manejo másconservador. La decisión de la esplenec-tomía en tales pacientes debe ser analizada
individualmente, evaluando los riesgos ybeneficios en cada caso. En los niños, esteproceder debe ser demorado lo más posibley no se recomienda su indicación antes delos 5 años de edad, ya que el riesgo deinfección es muy elevado.8,15,18,33
En la forma típica de la enfermedad,después de la esplenectomía, laesferocitosis persiste, pero el condiciona-miento de los microesferocitos desaparece,los niveles de reticulocitos se acercan a lanormalidad y aumenta el tiempo desobrevida de estos hematíes.8,15,31
Diferentes estudios han demostradoque el grado de respuesta a la esplenectomíaestá directamente relacionado con el gradode deficiencia de espectrina, sobre todo enla forma severa de la enfermedad(fundamentalmente la forma autosómicarecesiva), por lo que los pacientes mejoranel cuadro clínico y hematológico,desaparecen los requerimientos transfusio-nales, pero en muchos casos, se mantieneuna hemólisis de intensidad variable.26
La complicación más severa de estetratamiento es la sepsis posesplenectomía.Los datos reportados en la literatura indicanque esto ocurre en el 3,5 % de los pacientescon EH, y de éstos, en el 60 % es fatal.Teniendo en cuenta el riesgo de infección,estos pacientes se inmunizan con distintasvacunas: neumococo polivalente,Haemophilus influenzae y antimeningo-coco. Resultados satisfactorios se hanobtenido también con la terapia antibióticaprofiláctica con Fenoximetilpenicilinadurante 5 años después de la operación.Existen pacientes que no responden a laesplenectomía, lo cual puede deberse a: 1.La presencia de un bazo accesorio. 2. Eldesarrollo de esplenosis, la cual puedeocurrir varios años después de laesplenectomía. 3. La presencia de otra causade anemia hemolítica concomitante con laEH.4,15,31

16
Debido al riesgo inherente de lasinfecciones posesplenectomía, se hanempleado en los últimos años métodos másconservadores:
− La esplenectomía parcial. En pequeñosgrupos estudiados se ha demostrado suefectividad en la disminución de lahemólisis, mientras el bazo residualpreserva su función fagocítica.38,39
− Embolización esplénica parcial. Se haensayado con éxito en algunospacientes.40
DEFECTOS MOLECULARESDE LAS PROTEÍNASDE LA MEMBRANA
El análisis de las proteínas de lamembrana mediante electroforesis en gel depoliacrilamida (SDS-PAGE), ha sido unelemento muy importante para el estudiode las alteraciones de las proteínas delglóbulo rojo en la HS. Empleando un sistemade buffer continuo de Fairbanks, con ungradiente exponencial de poliacrilamida de3,5 al 17 %, se puede identificar la presenciade péptidos truncados o elongados, o unaconcentración anormal de las proteínas queconforman el citoesqueleto del glóbulorojo.3,8,41
Alteraciones de 4 proteínas de lamembrana eritrocitaria han sidoidentificadas en la EH y se ha demostradoque esta enfermedad se produce pordefectos de las proteínas que intervienenen las interacciones verticales entre elesqueleto de la membrana y la bicapalipídica,4,8,31,42-44 que son:
− Deficiencia de Sp.− Deficiencia combinada de Sp y ankirina.− Deficiencia de banda 3.− Deficiencia combinada de banda 3 y
proteína 4.2.− Defecto de proteína 4.2.
Diferentes estudios bioquímicos hanseñalado que del 30 al 45 % de los pacientestienen deficiencia combinada de Sp yankirina, el 30 % presenta deficiencia de Spaislada y el 20 % muestra deficiencia debanda 3. Sin embargo, se plantea que elporcentaje de individuos con deficiencia deankirina está subestimado por problemasde sensibilidad de la técnica, y ademásporque la reticulocitosis enmascara tambiénla deficiencia de esta proteína. Se hademostrado que cuando existe unadeficiencia de ankirina, la espectrina queestá en exceso es incapaz de ensamblarse ala membrana, debido a la pérdida de los sitiosde unión de la ankirina, y por este motivoes degradada. Esta es la razón por la quefrecuentemente se produce una deficienciade ambas proteínas; la deficiencia deespectrina es secundaria a una reducciónprimaria de ankirina. El defecto aislado deproteína 4.2 se ha encontrado en unreducido número de americanos yeuropeos, pero es más común en Asia,especialmente en Japón. Esta variedad defenotipos bioquímicos refleja la ampliaheterogeneidad de las alteracionesmoleculares que se producen en la EH.45-48
Al nivel molecular, las mutaciones quedan lugar a las alteraciones señaladasanteriormente se encuentran distribuidas alo largo de los genes afectados, y puedenser debidas a (tabla 2. fig.5):
1. Mutaciones que producen un corrimien-to de lectura debido a deleciones oinserciones de un número variable denucleótidos, lo que da lugar a unaalteración en el extremo C-terminal de laproteína.
2. Mutaciones sin sentido que resultan enuna terminación prematura de latranslación.
3. Mutaciones puntiformes debidas a lasustitución de una base.

17
Banda 3
N C
α−espectrina
β−espectrina
Ankirina
Unión actina/4,1
Unión membrana Unión espectrina
Zona reguladora
N
N
N
C
C
C
Unión del calcio
Citoplasma Transmembrana
Zona reguladora
FIG. 5. Esquema de los dominios de banda 3, ankirina, α y β espectrina con la ubicación de las mutaciones conocidas quecausan esferocitosis hereditaria (EH).
4. Mutaciones del sitio de empalme que danlugar a una anomalía en la transcripción.
5. Deleciones grandes del genoma.
Estos defectos moleculares puedenafectar la estabilidad de la transcripción, laestabilidad de la proteína mutada o lafunción específica de la proteína. Todasestas consecuencias conllevan a unadeficiencia de la proteína alterada. En el casode las mutaciones puntiformes, no seconoce el mecanismo por el cual se produceun defecto de la proteína. Sin embargo,algunas de estas mutaciones se encuentran
en residuos de aminoácidos altamenteconservados, que dan lugar a una proteínacon un daño funcional significativo. Esimportante señalar que, mientras laexpresión del gen de la α espectrina estálimitada a los eritrocitos, los genes de la βespectrina, ankirina, banda 3 y proteína 4.2se expresan en otras formas alternativas enotros tejidos, por lo que algunas mutacionesde estos genes se han reportado enasociación con manifestaciones extraeri-trocitarias.2-4,6,13,15,21-23,31,35,49,50
Los genes de la espectrina, ankirina ybanda 3 contienen o están flanqueados por
TABLA 2. Alteraciones moleculares encontradas en la esferocitosis hereditaria (EH)
Mutaciones que Mutaciones sin producen corrimiento sentido (codon de Mutaciones Mutaciones en el Deleciones del marco de lectura terminación) puntiformes sitio de empalme grandes delProteína Deleciones Inserciones genoma Total
Espectrinaα Sp 1 - 1 2 1 - 5β Sp 3 2 2 1 - 1 9Ankirina 7 2 2 7 - - 18Banda 3 6 4 2 12 - - 24Proteína 4.2 2 - 1 2 - - 5
Total 19 8 8 24 1 1 61

18
polimorfismos conocidos, que son útilespara estudios de ligamiento y consisten enmutaciones puntiformes, número variablede tandem repetidos (NVTR) o númerovariable de dinucleótidos repetidos (NVDR)(tabla 3).51,52
Deficiencia de espectrina:1. α espectrina: se sintetiza en exceso
(de 3 a 4 veces más que la incorporada a lamembrana), por lo que una produccióndeficiente de esta cadena en un alelo noproduce ninguna manifestación clínica. Eldefecto de los 2 alelos está relacionado conla forma recesiva de EH. La primera mutaciónreportada es una mutación puntiforme quese denominó α lla (969 GCT→GAT;Ala→Asp) y posteriormente se renombrócomo α espectrina Bughill (Sp αBH ). Estápresente en el 50 % de los individus con EHy deficiencia de espectrina, pero se sabeactualmente que no produce manifes-taciones clínicas, sino que se trata de unpolimorfismo silente (tabla 4).31,42,43
TABLA 3. Algunos polimorfismos silentes de la espectrina y la ankirina
Genes Sitio de restricción Características
α SpDominio α l Xba I (0,36/0,33 +0,03) Intron 2; tctaga/tccaga
Pvu II (1,4 +1,1/2,5) Intron 2; cagctg/cggctgMsp I (2,5/1,4 +2,1) Intron 2; ccag/ccgg
Dominio α II CGC/CAC;R701HATC/GTC;1809VACA/AGA; T853RGAT/GCT; D970A
Dominio α III AGA/ATA; R13331Dominio α V CTA/GTA; I 1857V
Ara I (8,40/5, 3+3,2) Intron 45; 6714 (-12 c→t)Intron 46; 6732 (-12t→a)
3´UT Secuencias repetidas GT y NVRTβ Sp
Hind III (17/4)Stu I(8/4,8 + 3,2)Taq I (3,2/2,7 + 0,8)
AnkirinaNco I(3,8/3,6)
3´UT NVRD
NVRT: número variable de repeticiones de tandem; NVRD: número variable de repeticiones de dinucleótidos.
2. β espectrina: se han encontrado variasmutaciones que afectan esta cadenapolipeptídica en individuos con la formadominante de la enfermedad. La primeramutación encontrada fue en la región N-terminal, que produce una unióndeficiente de la espectrina con la proteína4.1.4,8,15
Deficiencia de ankirina:Diferentes investigadores han
señalado que la deficiencia de ankirina estápresente en un número importante depacientes con HS. Puede ser debido atranslocaciones o a delaciones del brazocorto del cromosoma 8, aunque se hanencontrado también mutaciones puntifor-mes, sin sentido y en el sitio de empalme.Se ha demostrado que en la tercera parte delos pacientes con deficiencia combinada deankirina y espectrina, uno de los alelos deankirina tiene una expresión reducida. Estopuede ser debido a una reducción en latranscripción del gen o a una disminución

19
TABLA 4. Algunos polimorfismos silentes de la proteína banda 3
Nombre Cambio Exon Intron Consecuencia Dominio
Genas 89G→A 1 - ↓síntesis 3´UTPst I - - 3 - CP´DA38A´ 227A→C 4 - GAC→GCC; D38A CPMemphis I 280A→C - - AGG→GAG; K56E CPNapoli 411 ins T 5 - Corrimiento de lectura CPNachod(Hadreckralove II) 464(-3c→a) 5 - Deleción CPMondego 553C→T 6 _ CTT→TCT; P147S CPMemphis III 2675C→T 19 - CCG→CTG; P854L TM13
CP:citoplasmático; TM: transmembranoso.
de la estabilidad de los transcriptos. Tambiénse ha observado que las mutaciones denovo en uno de los alelos de la ankirina,que dan lugar a una disminución en suexpresión, se encuentran con ciertafrecuencia en los pacientes con HS cuyospadres son normales.44,53
Deficiencia de banda 3:La deficiencia de banda 3 en pacientes
con EH se ha demostrado mediante laelectroforesis en gel de poliacrilamida(PAGE-SDS) y se plantea que está presenteentre el 10 y el 20 % de los casos con laforma dominante. En ocasiones, puedeverse acompañada de una disminución dela proteína 4.2 como consecuencia demutaciones que afectan la porcióncitoplasmática de la banda 3. Muchospacientes tienen anemia ligera, esferoci-tosis y del 0,2 al 2,3 % de mushroom-shapedo pincered cells , lo cual no se haencontrado en otras formas de HS.8,54-56
Deficiencia de banda 4.2:Se ha demostrado la disminución
severa o ausencia de la proteína 4.2 en laforma recesiva de la EH. Los fenotiposdescritos en la literatura son muyheterogéneos: la morfología celular ha sidocaracterizada por la presencia de esferocitos,eliptocitos o esferoovalocitos. Se haencontrado fundamentalmente en Japón.57,58
La variabilidad del cuadro hematoló-gico, bioquímico y clínico de la EH dentrode una misma familia, ha hecho pensar enla presencia de otros factores intrínsecos omutaciones silentes que modulen laexpresión de los alelos patológicos.Recientemente se han presentado algunoshallazgos que pudieran explicar elcomportamiento tan heterogéneo de estaenfermedad.59,60
− Alelo Lyon y alelo Genas: el alelo Lyonpresenta un codon prematuro de parada(EH ligera) y el alelo Genas tiene unamutación puntiforme antes del codon deiniciación de la translación (silente). Lacombinación en trans de ambasmutaciones produce un agravamiento dela enfermedad.
− Alelo Coimbra y alelo Mondego: el aleloCoimbra presenta una mutaciónpuntiforme (EH ligera) y el alelo Mondegotiene 2 mutaciones puntiformes en eldominio citoplasmático (silente). Lacombinación en trans de ambasmutaciones produce un agravamiento dela enfermedad.
Futuras investigaciones permitiránprofundizar en la relación entre lasalteraciones moleculares y la expresiónclínica de la EH.

20
SUMMARY
Hereditary spherocytosis(HS) is a disease characterized by hemolytic anemia ofvariable severity, with spherocytes in peripheral blood and a clinical response tosplenectomy. The development of new techniques allowed finding out the firstbiochemical alterations in erythrocyte membrane proteins and later on, therecombinant DNA techniques made possible to detect molecular alternations.HS is a very heterogeneous disease caused by an intrinsic defect of red cells; thereare other secondary disorders to this affection. The most used test for diagnosingHS is the osmotic fragility of the red cell. It has been proved that this disease iscaused by defects in proteins participating in vertical interactions betweenmembrane skeleton and lipid bi-layer. The treatment of choice in HS issplenectomy, since this is the most effective method in the control of anemia,although red cell survival is still short and spherocytes do not disappear. Thisprocedure is recommended for patients with severe hemolytic anemia andmoderately asymptomatic individuals who have vesicular lithiasis.
Subject headings: SPHEROCYTOSIS, HEREDITARY/diagnosis; SPHEROCY-TOSIS, HEREDITARY/genetics; DNA RECOMBINANT; MEMBRANEPROTEINS/chemistry; RECOMBINANT PROTEINS/chemistry;SPLENECTOMY; ERYTHROCYTE MEMBRANE/chemistry.
REFERENCIAS BIBLIOGRÁFICAS
1. Berga L, Vives-Corrons JLl, Feliú E, Woessner S, Rozman C. Hemorreología. Bases teóricas y aplicacionesclínicas. Barcelona: Salvat, 1983.
2. Delaunay J. Genetic disorders in the red cell membrane. Crit Rev Oncol Hematol 1995;19:79-110. 3. Iolascon A, Miraglia del Giudice E, Perrotta S, Alloisio N, Morle L, Delaunay J. Hereditary spherocytosis:
from clinical to molecular defects. Haematologica 1998; 83:240-57. 4. Scriver SR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited disease.
Philadelphia: McGraw-Hill Company, CD-ROM, 1997. 5. Yu J, Steck TL. Isolation and characterization of band 3, the predominant polypeptide of the human
erythrocyte membrane. J Biol Chem 1975;250:9170-6. 6. Kay MMB, Folwers N, Goodman J. Alterations in membrane protein band 3 associated with acelerated
erythrocyte ageing. Proc Nall Acad Sci USA 1989;86:5834-8. 7. Marsh WL. Molecular defects associated with McLeod blood group phenotype. En. Salmon E, ed.
Blood groups and other red cells surface markers in health and disease 1982:17-82. 8. Tse WT, Lux SE. Red blood cell membrane disorders. Br J Haematol 1999;104:2-13. 9. Speicher DW, DeSilva TM, Speicher KD, Ursitti JA, Hembach P, Weglarz L. Location of human red
cell spectrin tetramer binding site and detection of a related “closed” hairpin loop dimer using proteolyticfootprinting. J Biol Chem 1993;268:4227-31.
10. Jordan C, Puschel B, Koob GR. Identification of a binding motif for ankirin on the a subunit of Na+ K+ATPase. J Biol Chem 1995;270:29971-4.
11. Fowler VM, Sussmann MA, Miller PG, Flucher BE, Daniels MP. Tropomodulin is associated with thefree (pointed) ends of the thin filaments in rat skeletal muscle. J Cell Biol 1993;120:411-4.
12. Mueller TJ, Jackson CW, Dockler ME, Morrison M. Membrane skeletal alterations during in vivomouse red cell ageing. Increase in the band 4.1a:4.1b ratio. J Clin Invest 1987;79:492-6.
13. Korsgren C, Lawler J, Lambert S, Speicher D, Cohen CM. Complete amino acid sequence and homologiesof human erythrocyte membrane protein band 4.2. Proc Natl Acad Sci USA 1990;87:613-5.
14. Horne WC, Leto TL, Marchesi VT. Differential phosphorylation of multiple sites in protein 4.1 andprotein 4.9 by phorbol ester-activated and cyclic AMP-dependent protein kinases. J Biol Chem1985;260:9073-7.

21
15. Beutler E, Lichtman MA, Coller BS, Kipps TJ. Williams Hematology. 5 ed. McGraw Hill, 1995.16. Vanlair CF, Masius JB. De la microcythemie. Bull R Acad Med Belg 1871;5:515.17. Wilson C. Some cases showing hereditary enlargement of the spleen. Trans Clin Soc 1890;23:162-4.18. Chauffard MA. Pathógene de l´ictere congenital de l´adulte. Semin Med 1907;27:25-7.19. Reed CF, Swisher SN. Erythrocyte lipid loss in hereditary espherocytosis. J Clin Invest
1966;45:777-81.20. Palek J, Jarolim P. Clinical expression and laboratory detection of red cells membrane protein mutations.
Semin Hematol 1993;30:249-51.21. Morton NE, MacKinney AA, Kosower N, Schilling RF, Gray MP. Genetics of spherocytosis. Am J Hum
Genet 1962;14:170-9.22. Gallagher PG, Forget BG, Lux SE. Disorders of the erythrocyte membrane. Nathan and Oski´s
Hematology of infancy and childhood. 5 ed. 1998; vol 1:544-664.23. Agre P, Orringer EP, Bennett V. Deficient red-cell spectrin in severe, recessively inherited spherocytosis.
N Engl J Med 1982;306:1155-9.24. Whitfield CF, Follweiler JB, Lopresti-Morrow L, Miller BA. Deficiency in the a-spectrin synthesis in
burst-forming units-erythroid in lethal hereditary spherocytosis. Blood 1991;78:3043-51.25. Iolascon A, Perrota S, Tavano R, Miraglia del Giudice E. Hereditary spherocytosis in newborn. Neonatal
Hematology and Inmunology 1997:197-202.26. Agre P, Asimos A, Casella JF, McMillan D. Inheritance pattern and clinical response to splenectomy
as a reflection of erythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med1986;315:1579-83.
27. Gehlback SH, Cooper BA. Hemolytic anemia in infectious mononucleosis due to inapparent congenitalspherocytosis. Scand J Haematol 1970;7:141-4.
28. Godal HC, Refsum HE. Haemolysis in athletes due to hereditary spherocytosis. Scand J Haematol1954;22:83-6.
29. Trucco Jl, Brown AK. Neonatal manifestations of hereditary spherocytosis. Am J Dis Child1967;113:263-7.
30. Burman D. Congenital spherocytosis in infancy. Arch Dis Child 1958;33:335-7.31. Hassoun H, Palek J. Hereditary spherocytosis: a review of the clinical and molecular aspects of the
disease. Blood Rev 1996;10:129-47.32. Shroter W, Kahsnitz E. Diagnosis of hereditary spherocytosis in newborn infants. J Pediatr
1983;103:460-3.33. Diamond LK. Indications for splenectomy in childhood. Results in fifthy-two operated cases. Am J
Surg 1938;39:400-4.34. Debre R, Lamy M, See G, Schrameck G. Congenital and familial hemolytic disease in children. Am J Dis
Child 1938;56:1189-92.35. Lux SE, Palek J. Disorder of a red cell membrane. En: Blood. Principles and practice of hematology
1995:1701-808.36. Streichman S, Gesheidt Y, Tatarsky l. Hypertonic cryohemolisis: a diagnostic test for hereditary
spherocytosis. Am J Hematol 1990;35:104-10.37. Clark MR, Mohandas N, Shohet SD. Osmotic gradient ektacytometry:comprehensive characterization
of red cell volume and surface maintenance. Blood 1983;61:899-903.38. Tchernia G, Bader-Meunier B, Berterottiere P, et al. Effectiveness of partial splenectomy in hereditary
spherocytosis. Curr Opin Hematol 1997;4:136-41.39. Pavón V, Estrada M, Fernández N, et al. Efectividad de la esplenectomía parcial en el tratamiento de
la esferocitosis hereditaria. Rev Invest Clin (en prensa).40. Israel DM, Hassal E, Culham GJA, Philips RR. Partial splenic embolization in children with hypersplenism.
J Pediat 1994;124:95-9.41. Fairbanks G, Steck TL, Wallach DFH. Electrophoresis on the major polypeptides of the human
erythrocyte membrane. Biochemistry 1971;10:2606-17.42. Agre P, Caella JF, Zinkham WH, McMillan C, Bennet V. Partial deficiency of erythrocyte spectrin in
hereditary spherocytosis. Nature 1985;314:380-3.43. Hanspal M, Yoon SH, Yu H, Hanspal JS, Lambert S, Palek J, et al. Molecular basis of spectrin and
anyirin defiencies in severe hereditary spherocytosis: evidence implicating primary defect of ankyrin.Blood 1991;77:165-73.

22
44. Eber SW, González JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant andrecessive hereditary spherocytosis. Nat Genet 1996;13:214-8.
45. Miraglia del Giudice E, lolascon A, Pinto L, Nobili B, Perrotta S. Erythrocyte membrane proteinalterations underlying clinical heterogeneity in hereditary spherocytosis. Br J Haematol1994;88:52-5.
46. Jarolim P, Murray JL, Rubin HL, Taylor M, Prchal JF, Ballas SK, et al. Characterization of 13 novelband 3 gene defects in hereditary spherocytosis with band 3 deficiency. Blood 1996;88:4366-74.
47. Eber SW, Armbrust R, Schöter W. Variable clinical severity of hereditary spherocytosis: relation toerythrocyte spectrin concentration, osmotic fragility and autohemolysis. J Pediatr 1990;117:409-16.
48. Lanciotti M, Perutelli P, Valetto A, Di Martino D, Mori PG. Ankirin deficiency is the most commondefect in dominant and non dominant hereditary spherocytosis. Haematologica 1997;82:460-2.
49. Petters LL, Birkenmeier CS, Bronson RT, et al. Purkinje cell degeneration associated with erythroidankirin deficiency in nb/nb mice. J Cell Biol 1991;114:1233-7.
50. McCann SR, Jacob HS. Spinal cord disease in hereditary spherocytosis: report of two cases withhypothesized common mechanism for neurologic and red cell abnormalities. Blood 1976;48:259-63.
51. Beeton L, Prchal JT, Coetzer TL. A Taql polymorphism in the human erytroid β spectrin gene. HumGenet 1995;95:365-6.
52. Gallagher PG, Romana M, Wong C, Forget BG. Genetic basis of the polymorphisms of the all domainof spectrin. Am J Hematol 1996;95:57-66.
53. Randon J, Miraglia del Giudice E, Bozon M, et al. Frequent de novo mutations of the ANK1 gen mimica recessive mode of transmisión in hereditary spherocytosis: three new ANK1 variants: ankyrins Bari,Napoli ll and Anzio. Br J Haematol 1997;96:500-6.
54. Jarolim P, Rubin HL, Liu SC, et al. Duplication of 10 nucleotides in erythroid band 3 (AE1) gene in akindred with hereditary spherocytosis and band 3 protein deficiency (band 3 PRAGUE). J Clin Invest1994;93:121-30.
55. Jarolim P, Rubin HL, Brabec V, Chrobak L, Zolotarev AS, Alper SL, et al. Mutations of conservedarginines in the membrane domain of erythroid band 3 lead to a decrease in membrane-associated band3 and to the phenotype of hereditary spherocytosis. Blood 1995;85:634-40.
56. Band 3 Campinas: a novel splicing mutation in the band 3 gene (AE1)associated with hereditaryspherocytosis. Hyperactivity of Na/Li countertransport and an abnormal renal bicarbonate handling.Blood 1997;90:2810-8.
57. Lima PRM, Gontijo JAR, López de Faria JB, Costa FF, Saad STO, Inoue T, et al. Even partial deficiencyof protein 4.2 is critical for integrity of skeletal network in situ and intramembrane particles in ahomozygous band 3 Fukuoka (G130R) with its impaired binding to protein 4.2. Blood 1996;88 (Supp1part 2):5b.
58. Cohen CM, Dotimas E, Korsgren C. Human erithrocyte membrane protein band 4.2 (Pallidin). SeminHematol 1993;30:119-37.
59. Allosio P, Texier A, Vallier ML, Ribeiro ML, Morle L, Bozon M, et al. Modulation of clinicalexpression and band 3 deficiency in hereditary spherocytosis. Blood 1997;90:414-20.
60. Allosio N, Maillet P, Carré G, Texier P, Vallier A, Baklouti F, et al. Hereditary spherocytosis with band3 deficiency. Association with a nonsense mutation of band 3 gene (allele Lyon), and aggravation by alow-expression allele occurring in trans (allele Genas). Blood 1996;88:1062-9.
Recibido: 5 de abril del 2001. Aprobado: 21 de diciembre del 2001.Lic. Mayelín Herrera García. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudadde La Habana, Cuba. Telef.(537)578268. Fax (537)338979.e-mail: [email protected]

23
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):25-33
Instituto de Hematología e Inmunología
ASPECTOS CLÍNICOS Y EPIDEMIOLÓGICOS DE LA LEUCEMIAMIELOIDE AGUDA EN EL ANCIANO
Dra. María Teresa Milanés Roldán, Dr. Rafael Losada Buchillón, Dr. Porfirio HernándezRamírez, Dra. Olga M. Agramonte Llanes y Dra. Edelis Rosell Monzón
RESUMEN
Se realizó una actualización sobre aspectos clínico-epidemiológicos de la leucemiamieloide aguda (LMA) en el anciano, donde se expusieron las característicasclínicas, morfológicas, citogenéticas y biológicas de la enfermedad. Se destacaque en los ancianos existen diferencias biológicas particulares con respecto alpaciente joven, las cuales le confieren un peor pronóstico al estar relacionadascon una respuesta pobre a la quimioterapia. Del mismo modo, se relacionaron losprincipales factores pronósticos que afectan el éxito del tratamiento de la LMAen el anciano, entre los que se observan la edad, el estado funcional al inicio de laenfermedad, las anormalidades cromosómicas y la expresión de la P- glicoproteína.Se destaca que tanto la incidencia como la mortalidad tienen una tendenciaascendente en el individuo viejo con respecto al joven, y que los resultadosgenerales del tratamiento de la LMA en estos pacientes, son cada vez menosalentadores, pues se observa un porcentaje de remisión completa y sobrevidalibre de enfermedad mucho menor que en pacientes jóvenes. Por tal motivo, sesugiere que en este tipo de pacientes debe realizarse una diferenciación terapéuticaencaminada a utilizar el esquema más adecuado para lograr mejores resultadosque los alcanzados hasta el momento.
DeCS: LEUCEMIA MIELOIDE/diagnóstico; LEUCEMIA MIELOIDE/epidemiología; LEUCEMIA MIELOIDE/ genética; ANCIANO.
La creencia popular de que la leucemiaaguda (LMA) es una enfermedad de niñosy adultos jóvenes contrasta con la realidad,pues la inmensa mayoría de los pacientescon LMA son ancianos. La edad media deincidencia es 64 años, y se ha visto queestá aumenta con la edad, la cual constituyeuno de los principales factores pronósticosadversos.1-4
En los últimos 20 años, la introducciónde estrategias de tratamientos más agresivosha mejorado marcadamente el pronósticoen pacientes jóvenes, con una tasa deremisión completa (RC) de más del 80 % ysobrevida libre de enfermedad (SLE) a los 5años superior al 40 %.5 Sin embargo, elpronóstico en pacientes ancianos tratadoscon quimioterapia (QMT) intensiva es aún

24
poco alentador, sin una mejoría significativaen la última década, donde se reportan tasasde RC de aproximadamente el 60 %, SLE alos 3 años menor del 10 % y sobrevidaglobal media de menos de 12 meses.6,7
La definición de ancianidad es difícilde determinar si se tiene en cuenta que laedad biológica puede diferir marcadamentede la cronológica. Sin embargo, se ha llegadoal consenso de que el paciente anciano esaquel que tiene alrededor de 65 años.
Cada vez es menos alentador elresultado general del tratamiento de la LMAen el anciano en relación con la poblacióngeneral. Diferencias intrínsecas en labiología de la enfermedad, factoresrelacionados con el paciente (por ejemplo:la tolerancia reducida a la quimioterapia yenfermedades concomitantes), anormalida-des citogenéticas desfavorables y laelevada actividad de genes resistenciamultidroga, pueden ayudar a explicar lasdiferencias en el éxito del tratamiento.7
EPIDEMIOLOGÍA
Aunque ningún grupo de edad esinmune al desarrollo de LMA, la mayoría delos pacientes con esta enfermedad sonancianos. Se señala un mayor número deenfermos a partir de la sexta década de lavida, debido probablemente al deterioro delsistema inmune, sobre todo de la inmunidadcelular, en particular la disminución de laactividad de las células citotóxicasnaturales (NK), las cuales tienen unaimportante función en la defensa delhuésped contra el cáncer, lo que se harelacionado con la mayor incidencia deneoplasias en edades geriátricas.8 En elhemisferio occidental, la incidencia de laLMA se incrementa con la edad, con unaincidencia estimada de 10/ 100 000, en lapoblación anciana.9 Estados Unidos en
1992 tuvo una incidencia en personas de 75años de 16/100 000, mientras que laincidencia total para todos los grupos deedades fue 2,6 /100 000. Datos recientes delnorte de Inglaterra indican que la incidenciaen ancianos es de 6/100 000, con unatendencia a desarrollarse más en hombresque en mujeres. Esta tasa es casi 6 veces lade la población general.9 La mortalidad tieneuna tendencia ascendente en el anciano conrespecto al joven. Este incremento de lamortalidad probablemente refleja elincremento en la incidencia de la LMA en elanciano, lo cual podría ser resultado demejores técnicas diagnósticas.6
Ciertos factores de riesgo han sidoestablecidos para el desarrollo de la LMA,como son: la exposición al benceno, laexposición a las radiaciones ionizantes ylas leucemias secundarias comoconsecuencia de QMT previa, por otraenfermedad maligna, cuyos esquemascontienen mostaza nitrogenada cisplatinoo etopóxido. Debido a que la LMAsecundaria es más frecuente en el ancianoy las enfermedades malignas a menudo sontratadas con estos medicamentos, lafrecuencia aumentada de LMA en al ancianopuede atribuirse en parte a estos riesgoiatrogénicos.9
CARACTERÍSTICAS CLÍNICAS
La LMA diagnosticada en pacientesancianos difiere de la diagnosticada enpacientes jóvenes no solamente en cuantoa características específicas de laenfermedad, sino también en característicasrelacionadas con la edad, como son:cambios fisiológicos en el funcionamientode los órganos, disminución en la habilidadde reacción al estrés, dependencia en lasactividades diarias, existencia de otrasenfermedades concomitantes, la necesidad

25
de tomar drogas para estas enfermedades yla reducida expectativa de vida.10
Desde el punto de vista clínico, en lospacientes ancianos existen 2 diferenciasesenciales: 1) alta incidencia de LMAdesarrollada a partir de un síndromemielodisplástico (SMD) y LMA trilineal sinevidencia de SMD previo; 2) pequeñaproporción (10 %) de pacientes con LMAhipocelular, una variedad vista casiexclusivamente en individuos mayores de50 años.11,12 En los pacientes ancianos conLMA, el estado funcional pobre al debut(según OMS aproximadamente 2) y laexistencia de enfermedades asociadas, esmucho más frecuente que en los jóvenes.13
El primero de estos 2 factores constituyeun conocido factor pronóstico adverso paramuchos cánceres y en particular para laLMA, lo que trae como consecuencia quese excluyan muchos pacientes ancianos delos ensayos clínicos. Exterman y otrosplantean que las patologías asociadas,aunque son comunes en los pacientesancianos con cáncer, no estáncorrelacionadas con el estado funcional.14
Esto sugiere que un gran número depacientes ancianos pudieran quedarexcluidos inapropiadamente de los ensayosclínicos del cáncer, y con esto se limitaría elconocimiento de nuevas modalidadesterapéuticas en la población de mayorriesgo.
Está claro que el resultado general delos pacientes ancianos con leucemia esmucho peor que para jóvenes similarmentetratados con la misma enfermedad. Mientrasun régimen de inducción agresivo tiene un70 % de probabilidad de RC en una personacon LMA menor de 60 años, el mismorégimen resultará en una tasa de RC de sóloel 45 al 50 % en aquellos pacientes mayoresde 60 años.15,16 Con quimioterapiamieloablativa o trasplante de médula ósea,el 45 % de los jóvenes adultos que logran
RC, experimentan un SLE prolongada.15,17
Sin embargo, sólo entre el 5 y el 15 % de lospacientes ancianos con LMA podrán lograruna SLE prolongada.18
Los adultos ancianos son muchosmenos capaces de resistir los rigores de laquimioterapia agresiva. La probabilidad demortalidad asociada con la quimioterapiade inducción es aproximadamente del 5 al15 % en pacientes menores de 60 años,comparados con aquellos mayores de 60años, en los que es muy elevada (25 %). Laprevalencia en un tercio de estos pacientesde enfermedades concomitantes como ladiabetes, la insuficiencia vascular y renal,hacen al paciente anciano menos tolerantea las infecciones que frecuentementeocurren durante la terapia de inducción,condicionadas por la immunosupresión queestá presente en estos pacientes. Elmetabolismo de las drogas quimioterapéu-ticas puede ser retardado debido a ladisminución de la función excretora renalrelacionada con la edad, lo cual conduce ala exposición a niveles mayores de la drogay mayor toxicidad.19,20 El efecto final de estasenfermedades concomitantes es unareducida habilidad del anciano a tolerar latoxicidad de los tratamientos intensivos.
Otra explicación para las altas tasas demortalidad en pacientes ancianos,relacionadas con el tratamiento, puede serla característica de resistencia intrínseca dela enfermedad, que permite un aclaramientomás lento de los blastos de la sangreperiférica y de la médula ósea, con elconsecuente retardo en la generación deelementos hematopoyéticos normales.21
CARACTERÍSTICASMORFOLÓGICAS
En relación con la morfología, la LMAen el anciano no muestra diferencia ninguna

26
comparada con los pacientes jóvenes.Varios estudios reportan las mismas tasasde subtipos FAB.22 Sin embargo, otrosplantean que la frecuencia del subtipo M3(leucemia promielocítica aguda) es muchomenor en la población anciana que en lapoblación joven, y puede estarcompletamente ausente en algunas series.7
La frecuencia de otros subtipos es similaren los 2 grupos. La LMA secundaria es máscomún en los ancianos que en los pacientesjóvenes.23
La mielodisplasia trilineal puede serreconocida morfológicamente en el 30 % delos pacientes ancianos con LMA primaria.3
Este hallazgo, enfatiza la posibilidad de queun SDM subclínico no diagnosticado podríaestar precediendo la aparición de la LMAen ancianos.12
Fialkow y otros encontraron que enlos pacientes ancianos al debut, loseritrocitos y plaquetas frecuentementederivan del clon leucémico; sin embargo,en pacientes jóvenes derivan de stem cellnormales. Tales hallazgos explican laelevada incidencia de remisión clonal en elanciano con persistencia del clon leucémicoy altas tasas de recaída.24
Estudios comparativos in vitro de laspropiedades del stem cell leucémico enpacientes mayores y menores de 60 años,reportan que no hay diferenciassignificativas en el crecimiento durante lafase-S del ciclo celular y la capacidad deautorrenovación del stem cell leucémico enambos grupos.6
CARACTERÍSTICASBIOLÓGICAS
Estudios recientes han permitido unmayor entendimiento de las diferenciasbiológicas inherentes de la LMA en elanciano comparado con la misma
enfermedad en el paciente joven. La edadrepresenta uno de los factores pronósticosadversos más importantes en la LMA.
En los pacientes ancianos con LMA lacélula maligna se origina de latransformación de un precursor másinmaduro (CD34+) que en los pacientesjóvenes, con una afectación más severa dela proliferación y la diferenciación. Losbastones de Auer son menos observadosen los pacientes ancianos y son máscomunes los SDM previos y los trastornosmieloproliferativos.25
La LMA que se origina de desórdenesprevios del stem cell de la médula ósea,probablemente sea más resistente a laquimioterapia debido a la alta exposiciónde genes resistentes a drogas como elMDR-1, los cuales le confieren un peorpronóstico en el anciano.26 La expresiónanormal de la proteína MSH2, implicada enla reparación de DNA desajustado y laprotección genómica, se observa con mayorprobabilidad en pacientes ancianos o en losque tienen tratamiento para la leucemia.27
Estas características biológicas intrínsecasde la enfermedad se relacionan con unarespuesta pobre a la quimioterapia.
ANORMALIDADESCITOGENÉTICAS
Una de las características pronósticasmás importantes para la LMA, es el statuscromosómico al diagnóstico.6
Rasgos cariotípicos anormales sonextremadamente comunes en la LMA. Laprevalencia de anormalidades citogenéti-cas, incluyendo la aneuploidía y lastranslocaciones, aparecen en el 50 % detodos los pacientes con LMA primaria, yalcanzan hasta el 87 % cuando se incluyenlos pacientes con LMA secundaria.3
Suficientes datos demuestran el efecto

27
adverso de las aberraciones cromosómicasen las tasas de RC y en la supervivencia.28
Fenaux y otros plantearon que el cariotipoes altamente predictivo del grado de RCdespués de QMT y también de la SLE.29
Grimwade y otros, en un estudiorealizado en 1 065 pacientes con LMA cuyaedad media era 66 años, consideraron queel cariotipo es el principal factor pronósticoen esta enfermedad, y no solo tiene valorpredictivo para las tasas de RC, sino tambiénpara los niveles de resistencia de laenfermedad y para el riesgo de recaída. Delmismo modo, plantearon que el patrón deanormalidades cromosómicas varía con laedad y encontraron tasas de RC superiores,menores niveles de enfermedad resistentey menor riesgo de recaída en aquellospacientes que presentaban anormalidadescromosómicas favorables: t(15,17), t(8,21)o inv.,16 las que predicen un buen resultadoal tratamiento con QMT intensiva y son másfrecuentes en pacientes jóvenes que enancianos.30 En contraste, alteracionescromosómicas de mal pronóstico, como sondel 5q-/monosomía 5 y del 7-/monosomía 7,frecuentemente observadas en lamielodisplasia o en la terapia relacionadacon la leucemia (particularmente si hanrecibido agentes alquilantes), son pocofrecuentes en los jóvenes, pero muycomunes en los pacientes ancianos (másdel 35 % de los casos).21,31 El mejor ejemplode incidencia de anormalidadescromosómicas dependientes de la edad conun pronóstico malo en la leucemia, es lat(9;22), la cual se observa en el 1 % de niñoscon leucemia linfoide aguda, pero solo enla tercera parte de los adultos con la mismaenfermedad.32
Teniendo en cuenta que el estadocitogenético es un factor pronósticoimportante y que el anciano está más amenudo afectado por estos cambios, estarájustificado un análisis citogenético
completo en pacientes ancianos con LMA,con el fin de obtener informaciónpronóstica.6
La región 5q31-33 del cromosoma 5contiene genes que codifican proteínasinvolucradas en la traducción y laregulación transcripcional de señales,especialmente el factor 1 regulador delinterferón y el de la respuesta 1 decrecimiento temprano. Esta región incluyegenes que codifican para el factorestimulador de crecimiento de coloniashematopoyéticas granulocitomacrófagas(GM-CSF) y las interleucinas IL-3, IL-4,IL-5 e IL-9, factores importantes en lasupervivencia y diferenciación de lascélulas hematopoyéticas.33 La región 7q22contiene genes involucrados en losmecanismos de reparación del DNA. Todoesto hace suponer que la pérdida de algunode estos genes puede predisponer a laleucemogénesis.33
FENOTIPO RESISTENCIAMULTIDROGA
Debido a que los regímenes detratamiento usuales han sido infructuososen el tratamiento de pacientes ancianos, enla última década se le ha dado una granatención a la resistencia multidroga (MDR)como un mecanismo fundamental del falloen el tratamiento de este grupo de pacientes.El gen MDR-1 de resistencia multidroga seencuentra en el brazo largo del cromosoma7. Está normalmente expresado en lascélulas parenquimatosas del hígado,páncreas, suprarrenales y riñón.6 Codificapara una P-glicoproteína (Pgp) que atraviesala membrana celular y funciona como unabomba de flujo ATP-dependiente.6
Se ha demostrado que en los ancianosexiste una alta tasa de expresión de Pgp, locual afecta el pronóstico en estos pacientes,

28
ya que está significativamente asociada conbajas tasas de RC y altas tasas de resistenciade la enfermedad.26
La Pgp está presente predominan-temente en las células blásticas de la LMACD34+, pero no en las células CD34-.34
Debido a que las células CD34+ tienenpotencial clonogénico, la resistenciamultidroga en estas células puede reducirla posibilidad de obtención de la remisión.
Estudios prospectivos recientes hananalizado la expresión de Pgp en pacientesancianos con LMA,26 lo que demuestra unaincidencia del 71 al 73 % de expresión dePgp en pacientes con LMA mayores de 55años, en contraste con solo el 37 % deincidencia en pacientes jóvenes.26,35
Otras proteínas de transporte no Pgp,son la proteína relacionada con laresistencia multidroga (MRP), la cualfunciona como una membrana, y la proteínarelacionada con el pulmón (LRP), la queparece funcionar bloqueando el transportede drogas del citoplasma al núcleo.36
Las proteínas no Pgp MRP y LRP,pueden también desempeñar una funciónimportante en la LMA. Aunque la expresiónMDR-1 parece ser similar en pacientes notratados o en recaída, la expresión de MRPse ha encontrado aumentada en la recaída.37
En un estudio prospectivo de LMA, lasobreexpresión de LRP fue fuertementecorrelacionada con la edad avanzada ycariotipo de mal pronóstico.37
La expresión de LRP puede afectar elpronóstico. En un estudio británicorealizado en un grupo de pacientes conLMA, se encontró que la expresión de LRPy la función de la Pgp están negativamentecorrelacionadas con la respuesta a la QMTy la supervivencia.38 En los pacientesmayores de 55 años, solo la LRP tuvo unsignificado predictor de la respuesta a laquimioterapia. Sin embargo, esto enfatiza laimportancia de considerar múltiples
características biológicas y mecanismoscomplejos de resistencia multidrogaresponsables del fallo del tratamiento en laLMA, particularmente en pacientesancianos.6
El estado citogenético y la expresiónde la Pgp están entre los factorespronósticos más importantes en la LMA,particularmente en el anciano, por la altafrecuencia con que se presentanalteraciones citogenéticas e incremento dela expresión de esta glicoproteína en estegrupo de edad.6
FACTORES PRONÓSTICOS
A pesar de los avances en elconocimiento de las característicasbiológicas y clínicas de la LMA en pacientesancianos, el pronóstico de estos pacientescontinúa siendo pobre. Estudios recienteshan demostrado tasas de supervivencia alos 5 años inferiores en pacientes mayoresde 55 años comparadas con las de pacientesmenores de 55 años. Para pacientes mayoresde 65 años las tasas estimadas desupervivencia a los 5 años fueron sólo del2,2 %.39 Debido al poco éxito del tratamientoen pacientes ancianos con LMA, ha sidomuy interesante estudiar los factoresresponsables de esa elevada tasa de falloen el tratamiento.6,40
Se han identificado varios factorespronósticos que afectan el éxito deltratamiento en pacientes ancianos conLMA:
1. Estado funcional pobre (según OMS ≥ 2). 2. Anormalidades citogenéticas. 3. Hepatoesplenomegalia. 4. Anemia. 5. Trombocitopenia. 6. Elevado conteo de blastos en sangre
periférica.

29
7. LMA secundaria. 8. Fenotipo resistencia multidroga MDR-1. 9. Sobreexpresión de LRP.10. Disgranulopoyesis.11. Enfermedades concomitantes.12. Presencia de infección al debut.13. Sexo masculino.
Varias características clínicas encontra-das en estudios de LMA en el ancianopueden afectar las tasas de RC y lasupervivencia.
En un modelo predictor de sobrevidaglobal desarrollado por Johnson y otros sedemostró que el estado funcional pobre, lahepatomegalia y el conteo de blastoselevados en sangre periférica, son factorespronósticos negativos, independientes dela supervivencia.41 Sin embargo, el conteoelevado de blastos (BL) en sangre periféricay el estado funcional pobre se ha visto queafectan negativamente. Este último, juntocon la edad avanzada (más de 60 años),fueron las variables clínicas másfuertemente asociadas con una peorsobrevida.6
Otras características de laboratorio,además del conteo de BL, se han identificado
como factores pronósticos. Los nivelesséricos de deshidrogenasa láctica (LDH)pueden ser importantes en los pacientesmayores de 60 años. Algunos estudios hanmostrado que cuando están elevados aldebut, se correlacionan de forma negativacon el logro de la RC, la SLE y la sobrevidaglobal.42 Otros, sin embargo, plantean queno existe correlación significativa entre losniveles séricos de LDH y la obtención deRC o la supervivencia mayor de 1año.22
Los cambios morfológicos pueden sermuy llamativos en pacientes ancianos conLMA. En diferentes estudios realizados seha observado que más del 30 % de estospresentan mielodisplasia trilineal, y más del60 % disgranulopoyesis.3 Del mismo modo,se encontró una correlación negativa entrela displasia hematopoyética y las tasas deRC y SLE, probablemente relacionada conla presencia de cariotipos adversos.43
Todo lo anteriormente señalado sugiereque en este tipo de paciente se debe realizaruna diferenciación terapéutica, con vistasa utilizar el esquema más adecuado paralograr mejores resultados que losalcanzados hasta el momento.
SUMMARY
An updating study of the clinical-epidemiological aspects of acute myeloidanemia in the elderly was conducted. It set forth the clinical, morphological,cytogenetic and biological characteristics of the disease. It was underlined thatthe elderly have specific biological differences in relation to the young patient,which give them a worse prognosis since such differences are related to a poorerresponse to chemotherapy. Similarly, the study listed the main prognostic factorsaffecting the success of AML treatment in the elderly, such as age, functionalstate at the onset of disease, chromosomal abnormalities and P-glycoproteinexpression. Both incidence and mortality are higher in older people than inyoung people and the general results of the AML treatment in these agedpatients are increasingly discouraging because it is observed that completeremission and disease-free survival rates in the elderly are much lower than inyoung patients. Therefore, a therapeutical differentiation aimed at using themost suitable scheme to obtain better results is suggested in this type of patients.
Subject headings: LEUKEMIA, MYELOID/diagnosis; LEUKEMIA, MYELOID/epidemiology; LEUKEMIA, MYELOID/genetics; AGED.

30
REFERENCIAS BIBLIOGRÁFICAS
1. Heath CW. Epidemiology and hereditary aspects of acute leukemia. En: Wiernik PH, Canellos GP,Dutcher JP, Kyle RA, eds. Neoplastic diseases of the blood, 3 ed. New York: Churchill Livingston,1996:177.
2. Cartwright RA, Staines A. Acute leukemias. Epidemiology of haematological diseases, Part 1. FlemingA, ed. Baillere Clin Haematol 1992;5:1-26.
3. Taylor PR, Reid MM, Stark AN, Bown N, Hamilton P. De novo acute myeloid leukemia in patientsover 55-years-old: A population-based study of incidence, treatment and outcome, Nother RegionHaematology Group. Leukemia 1995;9:231-5.
4. Wolfgang H, Wolfgan K, Shoch. Monagement of acute myeloid leukemia in elderly patients. J ClinOncol 1999;17:3569-76.
5. Mitus JA, Miller KB, Schenkein DP, Ryan HF, Parson SK, Wheller C, Antn JH. Improved survival forpatients whith acute myelogenous leukemia. J Clin Oncol 1995;13:560-8.
6. Jeffrey E Lancet, MD, Cheryl L, Willman MD, J M Bennett, MD. Acute myelogenous leukemia andaging. Hematol Oncol Clin N Am 2000;14:251-64.
7. Pinto A, Zagonel V, Ferrara F. Acute myeloid leukemia in the elderly: biology and therapeutic strategies.Rev Oncol Hematol 2001;39:275-87.
8. Romero Cabrera AJ. Inmunidad en el anciano. En: Temas de gerontogeriatría. Cienfuegos: Finlay;1990.p.49-53.
9. Groves F, Linet M, Devesa S. Epidemiology of Leukemia. En: Leukemia. 6a.ed. Henderson G, Lister M.Greaves H.eds. Philadelphia: Saunders;1996,p.152.
10. Cabrera M. Algunas afecciones hematológicas en el anciano En: Espinosa A, Romero AJ, eds. Cienfuegos:Finlay; 1990.p.175-84.
11. Nagai K, Kohno T, Chen XY. Diagnostic criteria for hypocellular acute leukemia: a clinical entitydistinct from overt acute leukemia and myelodysplastic syndrome. Leuk Res 1996;20:563-74.
12. Ferrara F, Annungiata M, Copia C, Magrin S, Mele G, Mirto S. Therapeutic options and treatmentresults for patients over 75 years of age whith acute myeloid leukemia. Haematologica 1998;83:126-31.
13. Repetto L, Venturino A, Vercelli M. Performance status and comorbidity in elderly cancer patientscompared with young patients with neoplasia and elderly patients without neoplastic conditions.Cancer 1998;82:760-3.
14. Exterman M, Overcsh J, Lyman GH. Comorbidity and functional status are independent in older cancerpatients. J Clin Oncol 1998;16:1582-5.
15. Mayer RJ, Davis RB, Schiffer CA. Intensive postremission chemotherapy in adults whith acute myeloidleukemia. N Engl J Med 1994;331:896-9.
16. Rees JK, Gray RG, Swirsky D, Hayhoe FG. Principal results of the medical research, council´s 8th acutemyeloid leukemia trial. Lancet 1986;2:1236-8.
17. Heil G, Hoelger D, Sang MA. A randomized double-blind placebo-controlled, phase II study of fitgostrimin remission induction and consolidation therapy for adults with the novo acute myeloid leukemia.Blood 1997;90:4710-6.
18. Stone RM, Berg DT, George SL, et al. Granulocyte-macrophage colony-stimulating factor after initialchemotherapy for elderly patients with primary acute myelogenous leukemia. N Engl J Med1995;332:1671-4.
19. Rubin EH, Andersen JW, Berg DT. Risk factor for high dose cytosine arabinoside neurotoxicity:analysis of a CALGB trial of post-remission cytosine arabinoside in patients with acute myeloidleukemia. J Clin Oncol 1992;10:948-50.
20. Smith GA, Damon LE, Rugo HS, Ries CA, Linker CA. High-dose cytarabine dose modification reducesthe incidence of neurotoxicity in patients with renal insuficiency. J Clin Oncol 1997;15:833-7.
21. Bloomfield CD, Lawence D, Byrd JC. Frequency of prolonged remission duration after high-dosecytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res1998,58:4173-7.
22. Latagliata R, Petti MC, Mandelli F. Acute myeloid leukemia in the elderly per aspera ad astra. LeukemiaRes 1999;23:603-13.
23. Baudard M, Marie JP, Cadio M, Viguie F, Zittoun R. Acute myelogenous leukemia in the elderly:restrospective study of 235 consecutive patients. Br J Haematol 1994;86:82-91.

31
24. Fialkow PJ, Singer JW, Raskind WH. Clinical remission in acute non lymphocytic leukemia. N Engl JMed 1997;317:468-72.
25. Green JP, Bach MR, Kinney MC. Acute myelogenous leukemia. En: Lee GR, ed. Wintrobe´s ClinicalHematology. 10ª. ed. Baltimore: William & Wilkins; 1998.p.2272-319.
26. Leith CP, Kopecky KJ, Godwin J. Acute myeloid leukemia in the elderly. Assessment of multidrugresistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responsesto standard chemotherapy. A Southwest Oncology Group Study. Blood 1997;89:3323.
27. Z hu Y-M, Das Gupta EP, Russel NH. Microsatellite inestability and P53 mutations are associated withabnormal expression of the MSH2 gene in adult acute leukemia. Blood 1999;94:733-6.
28. Lowenber B, Suciu S, Archimbaud E. Use of recombinat granulocyte-macrophage colony-stimulatingfactor during and after remission induction chemotherapy in patients aged 61 years and older withacute myeloid leukemia (LMA): Final report of AML-II, a phase III randomized study of the LeukemiaCooperative Group of the European Organisation for the Research and Treatment of Cancer (EORTC-LOG)and European The Dutch Belgian Hemato-Oncology Cooperative Group (HOVON). Blood1997;90:2952-63.
29. Fenaux P, Preudhome C, Lail JL. Cytogenetics and their prognostic value in the novo acute myeloidleukemia: A report on 283 cases. Br J Haematol 1989;73:61-4.
30. Grimwade D, Walker H, Harrison G, Oliver F. Value of hierarchical cytogenetic classification (AML):analysis of 1065 patients enterd into the United Kingdom Medical Research Council AML 11 trial.Bood 2001;98:1312-20.
31. Grimwade D, Walker H, Oliver F. The importance of diagnostic cytogenetics on outcome in AML:analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adultand Children´s leukemia Working Parties. Blood 1998;92:2322-8.
32. Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities inadult acute lymphoblastic leukemia. Blood 1998;91:3995-9.
33. Willian CL. Immuphenotyping and cytogenetics in older adults with acute myeloid leukemia: Significanceof expression of the multidrug resistance gene-1(MDR 1). Leukemia 1996;10S1:S33.
34. Uyl-de Groot CA, Lowenberg B, Vellenga E. Cost-effectiveness and quality-of-life assessment of GM-CSF as an adjunct to intensive remission induction chemotherapy in elderly patients with acutemyeloid leukemia. Br J Haematol 1998;100:629-4.
35. Willman CL. The prognostic significance of the expression and function of multidrug resistancetransporter proteins in acute myeloid leukemia: Studies of the Southwest Oncology Group LeukemiaResearch Program. Sem Hematol 1997;34:25-9.
36. List AF, Spier CS, Grogan TM. Overexpression of the mayor vault transporter protein lung-resistanceprotein predicts treatment outcome in acute myeloid leukemia. Leukemia 1996;10:937-9.
37. Schnaider E, Cowan KH, Bader H. Increased expression of the multidrug resistance-associated proteingene in relapsed acute leukemia . Blood 1995;85:186-91.
38. Borg AG, Burges R, Green LM. Overexpression of lung-resistance protein and increased P-glycoproteinfunction in acute myeloid leukemia cells predict a poor response to chemotherapy and reduced patientsurvival. Br J Haematol 1998;103:1083-7.
39. Ries LAG, Kosary CL, Hankey BF. SEER Cancer Statistics Review, 1993-1994. Bethes, MD, NationalCancer Institute. 1997, NIH Pub. No.97-2789.
40. Johnson PRE, Liu Yin JA. Prognostic factor in elderly patients with acute myeloid leukemia. LeukLymph 1994;6:51-6.
41. Johnson PRE, Ryder DJ, Liu Yin JA. Validaton of a model to predict survival in elderly patients withacute myeloid leukemia. Br J Haematol 1995;90:954-8.
42. Ferrara F, Mirto S. Serum LDH as a predictor of clinical outcome in acute myelogenous leukemia of theelderly. Br J Haematol 1996;92:627-31.
43. Gahn B, Haase D, Unterhalt M. De novo AML with dysplastic hematopoiesis. Cytogenetic andprognostic significance. Leukemia 1996;10:96-9.
Recibido: 14 de noviembre del 2001. Aprobado 30 de noviembre del 2001.Dra. María Teresa Milanés Roldán. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800,Ciudad de La Habana, Cuba. Teléf. (537)578268. Fax(537)442334. e-mail:[email protected]

32
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):34-40
Artículos originales
Instituto de Hematología e Inmunología
INMUNOFENOTIPAJE Y SUPERVIVENCIA GLOBALDE PACIENTES PEDIÁTRICOS CON LEUCEMIAS AGUDAS
Dra. Vianed Marsán Suárez, Dra. Consuelo Macías Abraham, Lic. René Rivero Jiménez,Dra. Miriam Sánchez Segura, Lic. Beatriz Socarrás Ferrer, Lic. Anissa GramatgesOrtiz, Dra. María Teresa Milanés Roldán, Dr. Alejandro González Otero y Dr. PorfirioHernández Ramírez
RESUMEN
El inmunofenotipaje celular (IFC) permite identificar la línea específica de origende las células leucémicas y su nivel de maduración. Se determinó la frecuencia delos distintos subtipos inmunológicos de leucemias agudas (LA) y su posiblerelación con la supervivencia global de los pacientes. Se estudiaron 117 niñoscon LA entre 1983 y 1999. El IFC se realizó mediante el ultramicrométodoinmunocitoquímico y el de fosfatasa alcalina-antifosfatasa alcalina. Del total deLA, 77(65,8 %) fueron linfoides agudas (LLA), 26 (22,2 %) mieloides agudas, 9(8 %) LA indiferenciadas y 5 (4 %) se clasificaron como LA híbridas. Del totalde LLA, 59 (76,6 %) fueron de fenotipo B y 18 (23,4 %) de fenotipo T. Seobservó una mayor sobrevida en los pacientes de linaje B en relación con los delinaje T y mieloide, con una diferencia muy significativa (p < 0,001). Lasupervivencia también se analizó entre las diferentes variedades de LA de linajeB y se observó una menor sobrevida de los pacientes con fenotipos más inmaduros.
DeCS: INMUNOFENOTIPIFICACION; LEUCEMIA MIELOIDE/diagnóstico;LEUCEMIA MIELOIDE/clasificación; LEUCEMIA MIELOIDE/inmunología;LEUCEMIA LINFOCITICA/diagnóstico; LEUCEMIA LINFOCITICA/clasificación; LEUCEMIA LINFOCITICA/inmunología; ANTICUERPOSMONOCLONALES/uso diagnóstico; TASA DE SUPERVIVENCIA; NIÑO.
Las leucemias agudas (LA) consti-tuyen un grupo heterogéneo de neoplasiascaracterizadas por una expansión clonal decélulas precursoras hematopoyéticastransformadas.1,2
En la actualidad, es posible identificarutilizando un panel de anticuerpos
monoclonales (AcMo), la línea específicade origen de las células leucémicas y sunivel de maduración. Estos estudios deinmunofenotipaje celular (IFC) sonesenciales para distinguir la leucemialinfoide aguda (LLA) de la leucemia mieloideaguda pobremente diferenciada (LMA-

33
M0), y clasificar la primera en subtiposinmunológicos.3,4
En nuestro estudio analizamos elinmunofenotipo de las células leucémicasen un grupo de niños con diagnóstico deLA para conocer la frecuencia de losdistintos subtipos inmunológicos y suposible relación con la supervivencia globalde los pacientes, así como comparar estosresultados con los publicados por otrosautores.
MÉTODOS
Se estudiaron 117 pacientes con LA,68 del sexo masculino y 49 del femenino,con una edad promedio de 7 años y unrango entre 5 meses y 15 años, diagnos-ticados en el Instituto de Hematología eInmunología en el período comprendidodesde julio de 1983 hasta diciembre de 1999.
El IFC de muestras procedentes demédula ósea o sangre periférica se llevó acabo mediante los métodos ultramicroinmu-nocitoquímico (UMICIQ)5,6 y de fosfatasaalcalina-antifosfatasa alcalina (APAAP).7
A continuación se relacionan losAcMo utilizados para el inmunofenotipajecelular:
1. Dirigidos contra antígenos expresadospor células B:− Anti-CD10(OKB-CALLA).− Anti-CD19; anti-CD20(B1).
• Institute of Cancer Research,Londres.
• Fundación Tettamanti, Italia.− Anti-CD22(Leu 14).− anti cadena µ.− Anti cadena κ.− anti cadena λ.
• Hospital Clínico de Barcelona.II. Dirigidos contra antígenos expresados
por células T:
− Anti-CD1(NA 134).− Anti-CD7(Leu 9).
• Institute of Cancer Research,Londres.
− Anti-CD2(OKT 11).− Anti-CD4(OKT 4).− Anti-CD8 (OKT 8).
• Filatov Institute of Immunology.− Anti-CD3(OKT 3).− Anti-CD5(Cris-1).
• Hospital Clínico de Barcelona.• Fundación Tettamanti, Italia.
III. Dirigidos contra antígenos expresadospor células mieloides:− Anti-CD 13(My7).− Anti-muramidasa.− Anti-CD41(943D).
• Hospital Clínico de Barcelona.• Fundación Tettamanti, Italia.
− Anti-CD33 (My 9).• Institute of Cancer Research.
− Anti-CD14.− Anti-CD15 (Leu M1).
• Filatov Institute of Immunology.• Fundación Tettamanti, Italia.
IV. Otros.− Anti-HLA-DR.− Anti-deoxi-nucleotidil transferasa
terminal (Tdt) .• Hospital Clínico de Barcelona.
De acuerdo con la expresión deantígenos celulares sobre las célulasblásticas, las LLA fueron clasificadas comode estirpe B (pro-B, común, pre-B y B) y deestirpe T (pro-T, pre-T, cortical y T) (tabla 1).
Se definieron como LA indiferenciadas(LAI), aquéllas que sólo expresaron losantígenos HLA-DR y Tdt, o al menos unode ellos.1
Se clasificaron como LLA-Mi+, aquellasLLA que expresaron hasta 2 antígenosmieloides8 y como LMA-Li+ aquellas LMAque expresaron hasta 2 antígenos linfoides.9
Las LA híbridas(LAH) se clasificaronsegún el criterio de Catovsky y otros.10,11

34
TABLA 1. Clasificación inmunológica de las leucemiaslinfoides agudas
Leucemias linfoblásticas inmunofenotipaje
Estirpe Bpro-B HLA-DR+/TdT+/CD19+común HLA-DR+/TdT+/CD19+/CD10+pre-B HLA-DR+/CD19+/CD10+/CD20+/Igc+B HLA-DR+/CD19+/CD10+/CD20+/IgS+Estirpe Tpro-T CD7+pre-T CD7+/CD5+cortical CD7+/CD5+/CD1+/CD3+/CD4+CD8+T CD7+/CD5+/CD3+/CD4+ o CD8 +
TdT: deoxinucleotidil transferasa terminal; Igc:inmunoglobulina intracitoplasmática; IgS: inmunoglobulinade superficie.
El cálculo de la supervivencia globalde las LA de estirpe B, T y mieloide serealizó por el método de Kaplan-Meier,12 yla comparación entre ellas por el método deIong-rang.13
RESULTADOS
Del total de pacientes estudiados, 77(65,8 %) fueron de linaje linfoide, 26 (22,2 %)de linaje mieloide, 9 (8 %) se clasificaroncomo LAI y 5 (4 %) como LAH. Del total deLLA, 59 (76,6 %) fueron de estirpe B y 18(23,4 %) de estirpe T. De las LLA de estirpeB, 48 (81,3 %) fueron común, 7 (11,9 %) pro-B, 3(5,1 %) pre-B y 1(1,7 %)B. De las LLAde estirpe T, 6 (33,4 %) presentaron fenotipode timocito común y se clasificaron comocorticales.
En 4 pacientes (22,2 %) se encontraronfenotipos pro-T, pre-T y T, respectivamente(tabla 2).
Del total de pacientes con LLA,13(16,9 %) fueron LLA-Mi+; de estas 9(69,2 %)con fenotipo B y 4 (30,8 %) con fenotipo T.Del total de LLA de linaje B Mi+,1 fueclasificada como pro-B y 8 de fenotipocomún (tabla 3).
Los antígenos mieloides expresados enlas LLA de estirpe B fueron CD 13, CD14,CD15, CD33 y glicoforina A.
TABLA 2. Frecuencia de distribución relativa de niños conleucemias agudas
Variedad de leucemia Frecuenciaaguda No. de casos (%)
LLA-Bpro-B 7(11,9)común 48(81,3)pre-B 3 (5,1)B 1(1,7)Total 59LLA-Tpro-T 4(22,2)pre-T 4(22,2)cortical 6(33,4)T 4(22,2)Total 18LMAM0 9(34,6)M3 1(3,8)M4 8(30,8)M5 7(27)M7 1(3,8)Total 26LAI 9(8)LAH 5(4)
Total 117
LLA: leucemia linfoide aguda; LMA: leucemia mieloideaguda; LAI: leucemia aguda indiferenciada; LAH: leucemiaaguda híbrida.
De las 4 LLA de estirpe T mi+,2 fueronpro-T y 2 pre-T. Los antígenos mieloidesexpresados fueron CD13, CD14, CD15 yCD33.
Del total de pacientes con LMA, 9(34,6 %) fueron clasificadas como M0, losantígenos CD13, CD33 y HLA-DR fueronpositivos en más del 75 % de los pacientesy la enzima TdT+ en el 25 % de éstos.
Sólo en 1 pacientes (3,8 %) se diag-nosticó la variedad M7, y se encontraronpositivos los antígenos CD13,CD33,HLA-DR y CD41.
Las células blásticas de los pacientesdiagnosticados con las variedades M0 yM7 no mostraron características mor-fológicas y citoquímicas típicas dediferenciación mieloide; no se encontraroncélulas cloronaftol positivas, lo que indicaque la actividad de la mieloperoxidasa fuenegativa.
35
TABLA 3. Fenotipos expresados en las leucemias linfoblásticasMi+ y en las leucemias mieloides agudas Li+
No. de casos Inmunofenotipaje
LLA-Mi+ 1 HLA-DR+/Tdt+/CD19+/CD13+
2 HLA-DR+/Tdt+/CD19+/CD10+/CD13+/CD33+
3 HLA-DR+/Tdt+/CD19+/CD10+/CD33+
4 y 5 HLA-DR+/Tdt+/CD19+/CD15+
6-8 HLA-DR+/Tdt+/CD19+/CD10+/CD14+
9 HLA-DR+/Tdt+/CD19+/D10+/glicoforina A+
10 CD7+/CD13+11 CD7+/CD13+/CD33+12 CD7+/CD5+/CD13+/
CD14+13 CD7+/CD5+/CD15+
LMA-Li+ 1 HLA-DR+/CD13+/CD33+/Tdt+
2 HLA-DR+/CD13+/CD33+/CD2+
3 HLA-DR+/CD13+CD33+/CD7+
4 HLA-DR+/CD13+/CD33+/CD10+
5 HLA-DR+/CD13+/CD33+/CD15+/CD2+
6 HLA-DR+/CD13+/CD33+/CD14+/CD5+
7 CD13+/CD33+/CD5+/HLA-DR-
8 HLA-DR+/CD13+/CD33+/CD15+/CD3+
LLA-Mi+: leucemia linfoide aguda con expresión de hasta 2antígenos mieloides; LMA-Li+: leucemia mieloide agudacon expresión de hasta 2 antígenos linfoides; Tdt:deoxinucleotidil transferasa terminal.
Se diagnosticaron 8 pacientes (30,8 %)con LMA-Li+. Los antígenos linfoidesencontrados en estos pacientes fueron CD2,CD3, CD5, CD7, CD10 y Tdt (tabla 3).
El análisis de la supervivencia globalde las LA de estirpe B, T y mieloide mostróuna mayor sobrevida en los pacientes delinaje B en relación con los de linaje T ymieloide, con una diferencia altamentesignificativa (p<0,001).
La sobrevida en los pacientes de linajeB al 1er., 2do., 4to., 5to. y 9no. años, fue de97 %, 88 %, 84 %,74 % y 58 %, respectiva-mente. Cuando se analizó la sobrevida en
los pacientes de linaje T al 1er., 2do., y 5to.años, esta fue de 82 %, 69 % y 46 %,respectivamente. Por su parte, los pacientescon LMA mostraron al 1er. y 2do. años dela enfermedad una sobrevida del 59 % y 49 %,respectivamente (fig. 1). La supervivenciatambién se analizó entre las diferentesvariedades de LA de linaje B, y se observóuna peor sobrevida de los pacientes confenotipos más inmaduros (fig. 2).
Maduraciónpre-B+B común pro-B
0 20 40 60 80 100 120Tiempo (meses)
1,0 0,9
0,8
0,7
0,6
0,5
0,4
0,3 0,2
0,1
0,0
FIG.1. Supervivencia global en niños con leucemias agudas.
TipoLMA LLA-T
0 20 40 60 80 100 120Tiempo (meses)
1,0 0,9
0,8
0,7
0,6
0,5
0,4
0,3
0,2
0,1
0,0
LLA-B
FIG. 2. Supervivencia global en niños con leucemiaslinfoides agudas B según estado de maduración.

36
DISCUSIÓN
Nuestros resultados demuestran quela distribución de los diferentes subtiposde LLA del niño en Cuba es similar a la quese observa en países como Inglaterra,Estados Unidos, Italia o Alemania. Estamisma distribución se ha descrito en otrospaíses como Taiwan, Australia, en niñosblancos de Sudáfrica, así como en niñosmexicanos y caucásicos chilenos.1,2,4,14,15
La mayoría de los pacientes con LLAde estirpe B se identificaron con el fenotipocomún (81,3 %), lo que confirma la conocidapreponderancia de este subtipo inmunoló-gico en la infancia. Esta variedad incide consimilar frecuencia en ambos sexos,preferentemente entre los 2 y 5 años deedad; la mayoría de los pacientes poseenbaja masa tumoral reflejada en el recuentoleucocitario y raramente poseen masamediastínica.1,2,4,16 La LLA común pudieraresultar de una respuesta anómala a unainfección común, probablemente viral, queocurre relativamente tardía en la infancia enlos países más desarrollados.14
La frecuencia de LLA pro-B en esteestudio es similar a informes de otrosinvestigadores. Esta variedad, antesdenominada nula, predomina en niñosmenores de 1 año, y probablementerepresenta el estadio más precoz de ladiferenciación de los precursores de célulaB.2,4,16,17
La incidencia de LLA pre-B essemejante a la informada por algunosgrupos iberoamericanos (del 4,5 al 11 %) ymenor que la señalada por gruposanglosajones (del 18 al 25 %).8,17 Si bien sedesconoce el motivo de esta diferencia, estopodría estar relacionado con factores gené-ticos y ambientales, como se ha planteadocon otros subtipos de leucemias.17
En nuestro estudio la frecuencia de laLLA-T (15,3 %), se comportó de forma
similar a la encontrada en paísesdesarrollados, y menor a la observada eninvestigaciones en India, Egipto, Áfricaecuatorial y en niños mapuches chilenos,en los cuales existe un incremento delfenotipo T,14,18,19 lo que pudiera explicarsepor la alta frecuencia de infeccionesprecoces en la infancia en los países menosdesarrollados, debido al hacinamiento y almenor nivel de protección con vacunas, loque estimula su inmunidad y aumenta laincidencia relativa de leucemia T.14
La mayoría de las LMA presentanfenotipos heterogéneos, en contraste conlas LLA, que generalmente muestranexpresión homogénea de sus marcadoresantigénicos. Virtualmente los antígenospan-mieloides CD13 y CD33 se expresanjuntos por los blastos mieloides; sinembargo, en una minoría de los casos, estosexpresan solo uno de estos antígenos.1
Existen algunos marcadores quemuestran una alta correlación con laclasificación FAB,20 como es el antígenomonocítico CD 14 en la M4 y en la M5, quepueden ser confirmadas con CD11c, CD36o ambos, y la expresión de glicoforina A enla M6. La variedad M3 se caracteriza por uninmunofenotipo más homogéneo, conexpresión de los antígenos CD13, CD33 ynegatividad del HLA-DR; por su parte, elCD15 puede encontrarse positivo entre el10 y el 25 % de los casos.1,8.9
El IFC es muy útil para el diagnósticode las variedades M0 y M7, ya que estosblastos no muestran características típicasde diferenciación mieloide, tantomorfológicas como citoquímicas, entre ellasla actividad de la mieloperoxidasa, por loque no es posible sobre estos datosdescartar el diagnóstico de una LLA.8,9
Para el diagnóstico de la variedad M0,las células leucémicas deben ser positivaspara uno de los antígenos pan-mieloidesCD13 o CD33 y ser negativas para antígenos

37
linfoides. La mieloperoxidasa es positivafrecuentemente por microscopia ultraestruc-tural.8
El diagnóstico de la variedad M7 debeser confirmado por la demostraciónultraestructural de peroxidasa plaquetariao por la detección de los antígenosplaquetarios CD41, CD42, CD61 y elrelacionado con el factor VIII.1,8
El IFC es útil además en la identificaciónde subgrupos de LA con pobrepronóstico.21 En nuestro estudio seencontró una mayor sobrevida en lospacientes con LLA de estirpe B en relacióncon los de estirpe T y mieloide, lo cual essimilar a lo encontrado por otros autores.4,14-16
La variedades de LLA pro-B, LLA-T(CD1-/CD3-) y LMA CD7+ se han iden-tificado como las de peor pronóstico.1,9,19
En nuestro estudio se comprobó que dentro
de las LLA de estirpe B, las pro-B mostraronuna menor sobrevida. La expresión demarcadores mieloides en la LLA del adultose ha asociado con un pobre pronóstico;sin embargo, en niños esta correlación noha sido demostrada,1,4,14,15,21 hecho que sepudiera explicar por la utilización dediferentes protocolos de tratamiento enestos 2 grupos etáreos.
En nuestro estudio, el IFC demostróser útil para el diagnóstico y la clasificaciónde la LLA, de las variedades M0 y M7 de laLMA, de la LAH, así como para laidentificación de grupos de peor pronóstico.Nuestros resultados demostraron que laincidencia de los distintos subtiposinmunológicos de LA y su relación con lasobrevida de los pacientes, es similar a laencontrada en la población caucásica enotras partes del mundo.
SUMMARY
Cell immunophenotyping (CIP) identifies the specific line of origin of leukemiccells and their maturing level. The frequency of the various immunologicalsubtypes of acute leukemias (AL) and their possible relationship with globalsurvival rate of patients were determined. One hundred and seventeen childrenwith AL were studied from 1983 to 1999. The CIP was conducted through theimmunocytochemical ultramicromethod and the alkaline phosphatase - anti-alkaline phosphatase method. Seventy seven (65,8%) of all AL were acutelymphoid leukemias (ALL), 26(22,2%) acute myeloid (AML), 9 (8%)undifferentiated AL, and 5 (4%) were hybrid AL. Fifty-nine (76,6%) ALLbelonged to phenotype B and 18 (23,4%) to phenotype T. A higher survival ratewas observed in phenotype B ALL patients than in phenotype T ALL andmyeloid anemia patients, with a very significant difference (p< 0,001). Survivalwas also analyzed among the various types of phenotype B acute leukemias; alower survival rate was observed in patients with more immature phenotypes.
Subject headings: IMMUNOPHENOTYPING; LEUKEMIA, MYELOID/diagnosis; LEUKEMIA, MYELOID/classification; LEUKEMIA MYELOID/immunology; LEUKEMIA, LYMPHOCYTIC/diagnosis; LEUKEMIA,LYMPHOCYTIC/classification; LEUKEMIA, LYMPHOCYTIC/immunology;MONOCLONAL ANTIBODIES/diagnostic use; SURVIVAL RATE; CHILD.

38
REFERENCIAS BIBLIOGRÁFICAS
1. Van Dongen JJM, Adraansen HJ. Immunobiology of Leukemia. In: Henderson ES, Lister TA, GreavesMF, eds. Leukemia. 6 ed. Philadelphia: WB Saunders; 1996.p.83-130.
2. Farhi DC, Rosenthal NS. Acute lymphopblastic leukemia. Clin Lab Med 2000;20(1):17-28. 3. Orfao A, Ciudad J, González M, López A, Abad M, Bouza P, et al. Flow cytometric in the diagnosis of
cancer. Scand J Clin Lab Invest 1995;55(Supp 222):141-52. 4. Malta A, Baez F, Floresa A, Ocampo E, Pacheco C, Biondi A, et al. Childhood acute lymphoblastic
leukemia. Int J Pediatr Hematol Oncol 1997;4:121-5. 5. Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Scand J Clin Lab Invest
1968;21(Supp 97):77-89. 6. Suárez L, Cruz C, Rivero RA. Ultramicrométodo inmunocitoquímico. Titulación de anticuerpos utilizados
para el inmunofenotipaje celular. Rev Cubana Hematol Inmunol Hemoter 1995;11(1):57-62. 7. Erber WN, Mason DY. Immunoalkalinephosphatase labelling of haematological samples. Technique
and Applications. Leuk Res 1985;9:829-30. 8. Kalidi HS, Chang KL, Medeiros LJ, Brynes RK, Slovak ML, Murata- Collins JL, et al. Acute lymphoblastic
leukemia. Survey of immunophenotype, French-American- British classification, frequency of myeloidantigen expression, and karyotypic abnormalities in 210 pediatric and adult cases. Am J Clin Pathol1999;111(4):467-76.
9. Reading C, Estey E, Huh Y, Claxton D, Sánchez G, Terstappen L, et al. Expression of unusualimmunophenotype combinations in acute myelogenous leukemia. Blood 1993;81(11):3083-90.
10. Catovsky D, Bucheri V, Matutes E, Dyer MJ, Shetty V, Yoshida N, et al. A classification of acuteleukaemia for the 1990´s. Ann Hematol 1991;62:16-21.
11. Bucheri V, Matutes E, Dyer MJS, Catovsky D. Lineage commitment in biphenotypic acute leukemia.Leukemia 1993;7:919-27.
12. Kaplan EL. Non parametric estimation for incomplete observations, J Am Stat Assoc 1958;53:457-81.13. Harrington D, Fleming TR. A class of rank test procedures for censored survival data. Biometrika
1992;69:133.14. Cabrera ME, Labra S, Ugarte S, Matutes E, Greaves MF. Inmunofenotipo, características clínicas y
laboratorio de la leucemia linfoblástica aguda en Chile. Rev Med Chile 1996;124:293-99.15. Paredes R, Romero L López N, Bravo A, Correa C, Joly E, et al. Inmunofenotipo de la leucemia aguda
linfoblástica en niños mexicanos. Sangre 1999;44(3):188-94.16. Melnick SJ. Acute lymphoblastic leukemia.Clin Lab Med 1999;19(1):169-86.17. Tiensiwakul P, Lertlum T, Nuchprayoon I, Seksam P. Immunophenotyping of acute lymphoblastic
leukemia in pediatric patients by three color flow cytometric analysis. Asian Pac J Allergy Immunol1999;17(1):17-21.
18. Burges R, Hansen-Hagge TE, Drexter HG, Gramatzki M. Heterogeneity of T-acute lymphoblasticleukemia (T-ALLL) cell lines: suggestion for classification by immunophenotype and T-cell receptorstudies. Leuk Res 1999;23:19-27.
19. Cascavilla N, Musto P, De Arena G, Ladogana S, Melillo L, Michele A, et al. Are “early” and “late” T-Acute lymphoblastic leukemias different diseases? A Single Center Study of 34 patients. Leuk Lymphoma1995;21:437-42.
20. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnic HR, et al. Proposal for theclassification of the acute leukaemias (FAB cooperative group). Br J Haematol 1976;33:451-8.
21. García JA, Monteserin MC, Delgado I, Benito L, Ona F. Aberrant immunophenotype detected by flowcytometric in acute lymphoblastic leukemia. Leuk Lymphoma 2000; 36(3-4):275-84.
Recibido: 11 de julio del 2001. Aprobado: 28 de septiembre del 2001.Dra. Vianed Marsán Suárez. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudadde La Habana, Cuba. Teléf. (537) 578262. Fax (537)33-8979. e-mail:[email protected]

39
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):41-7
Hospital Pediátrico Eliseo “Noel” Caamaño Matanzas
MECANISMOS DE ACCIÓN DE LA GAMMAGLOBULINAPARA USO ENDOVENOSO
Dr. Amaury Noa Albelo, Dr. Boris Rodríguez Ramos y Dr. Arturo Vidal Tallet
RESUMEN
Los preparados farmacéuticos de inmunoglobulinas para uso intravenosoconstituidos fundamentalmente por IgG, nos permiten la administración de éstasen dosis suprafisiológicas, lo que brinda la posibilidad de manipular la complejared de regulación a la que está sujeto el sistema inmune, y por lo tanto, variar elcurso de afecciones cuya patogenia se sustenta sobre la base de una disregulaciónde la respuesta inmuno reguladora.
DeCS: GAMMAGLOBULINAS/farmacología; INMUNOGLOBULINAS/farmacología; IGG.
La respuesta inmune está regulada poruna fina red de interacciones moleculares;la aberración de ésta se ha visto implicadaen la patogénesis de una serie de entidadesde etiología infecciosa, inflamatoria yautoinmune. Las inmunoglobulinas (IgS)son las moléculas efectoras de la respuestainmune humoral y desempeñan un papelprimordial en la colectividad de la red antesmencionada.
La elaboración de un preparadofarmacéutico de inmunoglobulinas para usointravenoso (IGIV), constituidasfundamentalmente por IgG, que es elcomponente mayoritario de la respuestainmune humoral, nos permite la administra-ción se estas en dosis suprafisiológicas, loque nos brinda la posibilidad de manipularesta compleja red de regulación, y por lo
tanto, variar el curso de afecciones cuyapatogenia se sustenta sobre la base de unadisregulación de la respuesta inmune.
En los últimos años, los avances enlos conocimientos moleculares de losmecanismos de regulación de la respuestainmune e inflamatoria nos han permitidodilucidar, al menos de forma parcial, losmecanismos mediante los cuales actúasobre este medicamento.
Las IgG están constituidas por unaporción Fc porción constante, responsablede las acciones biológicas de estas, comola fijación del complemento, la opsoniza-ción y la citotoxicidad mediada poranticuerpos (Ac), entre otras, y una porciónvariable o porción Fab, la cual tiene comoacción fundamental la interacción con elantígeno (Ag).

40
Los mecanismos de acción de las IGIV,si bien no dependen de forma absoluta deuna de estas porciones, se dividen para suestudio en:
I. Dependientes de la porción Fab:1. Regulación por redes idiotipo-antii-
diotipo.2. Inhibición de la síntesis de Ac por
la célula B.3. Bloqueo de superantígenos.
II. Dependientes de la porción Fc:1. Bloqueo funcional de receptores Fc.2. Inhibición del daño tisular mediado
por complemento.3. Regulación de redes citocinas-
anticitocinas.III. Otras:
1. Presencia de moléculas conactividad inmunomoduladora en lospreparados farmacéuticos de IGIV.
2. Potencialización de laremielinización.
I. Depedientes de la porción Fab:1. Regulación por redes idiotipo-
antiidiotipo.
El repertorio inmune es lo suficiente-mente amplio como para que losdeterminantes moleculares encargados deinteractuar con el Ag sean reconocidosentre sí de forma tal que un Ac 1, quereconoce a un Ag X, sea reconocido porun Ac 2; al Ac 1 se le denomina idiotipo y alAc2 antiidiotipo.1,2 De esta forma seestablece una red de reconocimiento einteracciones que determina un estado deequilibrio homeostático de la respuestainmune. Esta red de interacciones implicaademás a los receptores de reconocimientode la célula T y de la célula B. Del grado deconectividad de esta fina red dereconocimiento depende, en gran medida,la homeostasis del sistema inmune. El
equilibrio de esta conectividad depende deun continuo ajuste de 2 parámetros: elrepertorio activo (Ac producidos y célulasT activas) y la selección o deleción declonas al nivel de la médula ósea (células B)o al nivel de timo (células T). Estos 2parámetros actúan de forma dinámica einfluyen interactivamente, proporcionanajustes continuos a diferentes niveles deconectividad que mantienen el equilibrio,impiden que predomine de forma mantenidadeterminada dirección de respuesta, la cualpuede ser potencialmente dañina, por lotanto, se comporta como una red autónomay autorreactiva, que impide el comporta-miento autoagresivo de una clonadeterminada.
Las enfermedades autoinmunes sondefectos cuantitativos o cualitativos de laconectividad que se localizan más o menosen áreas específicas de la red y que estándirectamente relacionadas con la liberacióndel control de red en términos funcionaleso estructurales de determinadas clonasautoagresivas específicas. El mantenimien-to de la actividad autoagresiva quecaracteriza a estas afecciones está dadaporque el defecto en el repertorio periféricode respuesta no logra seleccionar unrepertorio adecuado al nivel central: médulaósea y timo.
La administración de altas dosis deIGIV, obtenidas de un número elevado deindividuos donde este proceso operaadecuadamente, en primer lugar compensade forma positiva los defectos deconectividad periférica, y en segundo lugar,intervendrían en la selección central delrepertorio de células T y de células B, loque pudiera definir a largo plazo que semantenga este equilibrio.
Existen modelos que demuestran quelas Igs en forma de dosis dependientesdeterminan la selección negativa de célulaspre-B ó B inmaduras al nivel de la médula

41
ósea. Esta depende directamente de laespecificidad antigénica de estas células,por lo tanto, ésta se relaciona directamentecon la reactividad del pool de Igs usadas.
La interacción de dichos Ac con susantiidiotipos en los complejos receptoresde membrana, inducen muerte celularprogramada (apoptosis); de manera similarocurre al nivel del timo con la célula T. Estoseventos experimentales justificarían loanteriormente expuesto, y ademásjustificaría tratar de definir el defectocuantitativo o cualitativo de conectividadpara seleccionar en ese sentidodeterminado repertorio de Igs, para controlarsin necesidad de usar dosis tan elevadas ydejar de actuar a ciegas, lo cual pudiera enocasiones ser ineficaz y en otras causardaño.
2. Inhibición de la síntesis de lgs por lacélula.
La interacción de las Igs mediante laporción Fab con la porción Fab de una Igde membrana del complejo receptor de Ag,del linfocito B, más la interacción de laporción Fc de la Ig en fase fluida con elreceptor Fc de la célula B, transduce señalesnegativas que inhiben de manera específicala progresión en la maduración a célulasplasmáticas productoras de Ac.3 Estainhibición evidentemente es selectiva paradeterminadas clonas, ya que elreconocimiento es a través de la parejaidiotipo-antiidiotipo. De esta manera, sepuede inhibir en periferia la producción dedeterminados tipos de autoanticuerpos conla administración de IGIV, en la cual se hanencontrado antiidiotipos contra un grannúmero de autoanticuerpos como:antiitiroglobulina, anti DNA y ANCA, porlo que dicho mecanismo resultaría útil enentidades en las cuales estosautoanticuerpos desempeñan un papel
patogénico, además la IGIV pudiera reducirla producción de autoanticuerpos que seencuentran presentes en los preparadosfarmacéuticos, con especificidad dirigidacontra la molécula CD5.4
3. Bloqueo de superantígenos.
Los Ags convencionales sonprocesados por células presentadoras deAg y expuestas en la membrana celular encombinación con moléculas MHC II o MHCI, según sea el caso. Este complejo esreconocido por al receptor de célula T porun sitio muy específico de reconocimientoantigénico. Este receptor es un complejomulticatenario, las cadenas alfa y beta sonlas principales responsables de estereconocimiento, mediado por sus porcionesvariables, de 30 a 500 segmentos génicos,conocidos como variables; son losresponsables de la codificación de estasporciones y la recombinación de estossegmentos definen la diversidad derepertorio de célula T, así como la diferenciaentre las clonas de células T de diferentesindividuos, aún entre gemelos homoci-góticos.
Dentro de las regiones variablesexisten zonas de menor viabilidad, y esta“monotonía” permite el mantenimiento dela estructura tridimensional de estasmoléculas. A estas zonas se unen lossuperantígenos, de forma tal que estimulana toda una familia de células T quecomparten secuencias específicas en estaregión de la cadena beta, a diferencia delos Ags nominales, que interactúan conregiones hipervariables de la porción dereconocimiento.
Además de lo anterior, los superantí-genos no requieren ser procesados por lascélulas presentadoras de Ag, sino que seunen directamente a regiones menospolimórficas del MHC, donde son

42
reconocidas por la célula T, activando a lamisma. De hecho, el crosslinking delreceptor de célula T (TCR) con el MHC IIpor el superantígeno, causa una extensablastogénesis de la célula T y de las célulaspresentadoras de antígenos (CPA), con elconsiguiente aumento de la síntesis decitocinas proinflamatorias (IL 12, IL 18, TNFalfa, IL 6, IL 1). La respuesta fisiológica alos superantígenos es similar a ladesencadenada por el lipopolisacárido(LPS).
Los superantígenos pueden causarexpansión de las células T autorrectivas ydesencadenar fenómenos autoinmunes. Seha descrito una gran cantidad de moléculascuya estructura determina esta actividadbiológica, entre las que se encuentran laexotoxina pirógena del estreptococo delgrupo A, enterotoxina del estafilococoáureo, toxina del síndrome del shock tóxico,entre otros.5,6 En la actualidad se conoce surelación con diversas entidades (escarlatina,síndrome de shock tóxico like, enfermedadde Kawasaki, intoxicación alimentaria porestafilococo), además sinergiza con el LPSy aumenta la letalidad del shock séptico. Enlos preparados farmacéuticos del IGIV seha demostrado la presencia de anticuerposcapaces de interactuar con éstos, queimpiden su interrelación con el sistemainmune y evitan la puesta en marcha de losmecanismos antes descritos.7 Hechosexperimentales en la práctica clínica así lodemuestran, y constituyen la terapia deelección en enfermedades en cuyapatogenia se invoca este mecanismo, comola enfermedad de Kawasaki.8,9
II. Dependientes de la porción Fc:1. Bloqueo funcional de receptores Fc.
Los receptores Fc son estructuras demembrana cuya función es interactuar conla porción Fc de las inmunoglobulinas. Este
fenómeno es la base del proceso llamadoopsonización por Ac, que además defacilitar la fagocitosis, la hace específica ydirigida según la especificidad del Ac, demanera que estructuras recubiertas de Ac,como pueden ser células, virus, bacterias,hongos, fragmentos de microorganismos,etcétera, contra las cuales se dirige larespuesta mediada por Ac, se hacensusceptibles de ser depuradas mediante elproceso de fagocitosis. Por otra parte, através de la interacción Fc-receptor de Fcse media otro proceso conocido comocitotoxicidad mediada por Ac, es decir, lacélula especializada (NK, por ejemplo), searma de Ac y le permite reconocerestructuras extrañas y destruirlas. Existenentidades en las cuales estos fenómenosson primordiales en su patogenia, como laPTI, la anemia hemolítica autoinmune(AHA), la neutropenia autoinmune, por loque al bloquearlas de forma competitiva,disminuye la depuración de estas célulasrecubiertas de Ac por las células del sistemamonocito-macrófago.
Evidentemente, la administración dedosis elevadas de IgG saturaría losreceptores Fc e impediría que células comolas plaquetas en la PTI,10,11 los hematíes enla AHA,12 y los neutrófilos en la neutropeniaautoinmune, sean eliminados. Este efectoes inmediato y transitorio, de manera que siel proceso es autolimitado como la PTI, selogra un aumento inmediato del recuentoplaquetario, hasta que otros mecanismosde este fármaco, al interactuar con losreceptores Fc, provocan un aumento delcatabolismo de Acs potencialmentepatógenos, dado por la saturación de losreceptores denominados FcRn. Estos seencuentran en el endotelio, y al unirse conlos Acs, los protegen de la degradación; lasaturación de estos por altasconcentraciones de inmunoglobulinas G,favorece el catabolismo de Acs endógeno.

43
2. Inhibición del daño tisular mediado porcomplemento.
Existe un número elevado de entidadescuya patogenia está asociada con lapresencia de inmunocomplejos (IC) y eldaño en éstas se relaciona en gran medidacon la capacidad de activar complemento,una vez depositado en el endotelio dedeterminados órganos y tejidos.
La administración de IGIV es capaz decambiar la relación estequiométrica de losIC, solubilizándolos o facilitando sufagocitosis por el sistema monocito-macrófago, lo cual impide que activen elcomplemento de manera no controlada conlos efectos deletéreos que traería comoconsecuencia.13-15
Por otra parte, las Igs suministradaspudieran formar dímeros, los cualesconsumirían C3b y C4b, lo cual limitaría porun lado la formación de la C3 convertasa dela vía alterna (mecanismo de ampliación deldaño), y por otro, la formación de la C5convertasa, que impide la generación delcomplejo de ataque a las membrana. Seconoce que estos efectos ofrecen bondadesterapéuticas en entidades como ladermatomiosistis, el lupus eritematososistémico (LES), algunas glomerulopatías yvasculitis.
3. Regulación de las redes citocinas-anticitocinas.
Las citocinas son glicoproteínas debajo peso molecular que actúan de formaautocrina, paracrina, yuxtacrina o endocrina.Media una serie de cambios metabólicosen determinadas células una vez queinteractúan con sus receptores demembrana. Su papel fundamental está dadoen situaciones de estrés y los cambiosmetabólicos celulares consisten enreajustes para eliminar la noxa en cuestióny reestablecer la homeostasia.
La aberración en los patrones desecreción de estos mediadores se ha vistoimplicada en un número cada vez mayor deentidades, muchas de las cuales producenriesgos para la vida, como son el shockséptico, el síndrome de shock tóxico, asícomo en afecciones de curso crónico(infecciones por microbacterias, atopia,etcétera.16,17
La IGIV es capaz de inhibir la secreciónde citocinas proinflamatorias (IL 1, IL 6, IL18, TNF alfa),18 de activar la secreción deotras con acción antiinflamatoria (IL 1ra, IL10),19 y a su vez incrementa la liberación dereceptores solubles de IL 1 beta, de INFalfa, de CD1120A (gp 55), CD12OB (gp 75),lo cual evidencia el carácter antiinflamatoriode la IGIV. Esta situación ha motivado elensayo de su uso en enfermedades queevolucionan con alteración de algunos deestos mediadores, como son los casos delshock séptico y la enfermedad de Kawasaki.
Se ha comprobado además que escapaz de reestablecer patrones aberrantesde secreción Th1/Th2, lo cual esfundamental en la recuperación deenfermedades infecciosas como la lepra yla tuberculosis, en las cuales un predominioTh 2 sería desastroso. Hoy se sabe que laaberración de estos patrones desempeñaun preponderante papel en la génesis de lainflamación atópica.
Se ha establecido la presencia de Acanticitocínicos en los preparados de IGIV,los que pudieran bloquear el efecto deéstas, lo cual nos orienta a pensar que lared descrita para la pareja idiotipo-antiidiotipo también incluirán Acs queinteraccionan con citocinas, que modularánsus acciones. Aún queda mucho porconocer en este campo.
III. Otras:
1. Presencia de moléculas con actividadinmunomoduladora en los preparadosde IGIV.

44
Este mecanismo de acción está dadopor la presencia en estos preparados demoléculas inmunológicamente activasdiferentes de las Igs, como son lassiguientes: MHCII solubles, INF gamma,CD8 y CD4 solubles,20 así como TGF beta,lo cual ejerce un efecto inmunomodulador,fundamentalmente inmunosupresor.21
La que se ha encontrado en mayoresconcentraciones y con mayor posibilidadde actividad terapéutica es la molécula deCD4, la cual, se ha sugerido, puede competircon las células T autorreactivo o lasmoléculas de MHCII de la célulapresentadora de antígenos, lo que puedeconducir a inmunosupresión inespecífica,que sería particularmente beneficioso enenfermedades autoinmunes.
2. Potencialización de la remielinización.
En modelos experimentales deencefalomielitis alérgica experimental yneuritis alérgica experimental, se hademostrado que la IGIV es capaz de producirremielinización, tanto al nivel central comoperiférico. Este mecanismo de acción noestá muy bien dilucidado molecularmente,aunque se conoce que la interacción de laIgG con receptores expresados al nivel dela célula de Schwann, es un eventoimportante. Este mecanismo pudieradesempeñar un papel de relieve en algunasenfermedades desmielinizantes humanascomo la esclerosis múltiple22 y algunasneuritis periféricas.22-24
SUMMARY
The pharmaceutical preparations of immunoglobulins for intravenous use, mainlymade up of IgG, allow us to administer them at supraphysiological dosage, whichprovides the possibility of handling the complex network of regulation governingthe immune system and thus changing the course of affection whose pathogenyis based on dysregulation of the regulating immune response.
Subject headings: GAMMAGLOBULINS/pharmacology; IMMUNOGLO-BULINS/pharmacology; IGG.
REFERENCIAS BIBLIOGRÁFICAS
1. Countinho A. The network theory: 21 years later. Scand Immunol 1995;42:3-8. 2. Dietrich G, Kaveri SV, Kazatchkine MD. Modulation of autoimmunity by idiotypic network. Clin
Immunol 1992;62:S73-S81. 3. Foster BJ, Duffy CM, Sharma AK. Systemic juvenile rheumatoide arthritis. Complicated by two
different renal lesions. Pediatric Nephrol 1998;12:113-6. 4. Dalakas MC. Mechanism of action of intravenous immunoglobin and therapeutic considerations in the
treatment of autoinmune neurologic disease. Neurology 1998; 51(Suppl05):S2-S8. 5. Kotzin BL, Leung DY, Kappler J, Meissner HC, Fuiton DR. Toxic shock syndrome toxin-secreted
staphylococcus aureaus in Kawasaki Syndrome. Lancet 1993:342:1385-88. 6. Abe J, Kotzin BL, Jujo K, Melish ME, Glode MP, Koshsaka T, et al. Selective expansion of T cells
expressing T- cell receptor variable regions V Beta 8 in Kawasaki disease. Procc Natl. Sci Academic.USA 1992;89:40,66-70.
7. Takei S, Arora YK, Walker SM. Intravenous immunoglobulin contain specific antibodies to activationT of cells by staphylococcal toxin superantigens. J Clin Invest 1993;91:602-7.

45
8. Kato H. Kawasaki disease. Proceedings of the 5th International Kawasaki disease Symphosium; 1995may 22-25; Fukuoka, Japan. Amsterdam:Elsevier;1995.
9. Fischer P, Uttenreuther MM, Naoe SH, et al. Kawasaki disease: Update undiagnosis, treatment, and stillcontroversial etiology. Pediatr Haematol Oncol 1996;13:487-501.
10. Farantino MD, Madden RM, Fennewald DL, et al. Treatment of acute immune thromboocytopeniapurpure with anti D immunoglobulin pooled immunoglobulin. J Pediatr 1999;134(1):21-6.
11. George JN, Woolf SH, Raskob GG, Wasser LM, Aledort PJ, Ballem PJ. Idiopatic thrombocytopeniapurpura: a practice guideline deveeloped by explicit methods for the American Society of Hematology.Blood 1996;88(1):3-40.
12. Domen RE. An overview of immune hemolytic anemias. Cleveland Clin J Med 1998;65:89-99.13. Basta M, Dalakas MC. High dose intravenous immunoglobulin exters its beneficial effect in patients
with dermatonyositin in blocking endomisial deposition of activated complement fragments. J ClinInvest 1994;94:1729-35.
14. Basta ME. Modulation of complement-mediated immune damage by intravenous immunoglobulin.Clin Ep Immunol 1996;196:7-12.
15. Rieben R, Roos A, Muizert Y, et al. Immunoglobulin M-enriched human intravenous immunoglobulinprevents complement activation in vitro and in vivo in a rat model of acute inflammation. Blood1999;93(3):9442-52.
16. Badolato R, Oppenheim JJ. Role of cytokines, acute-phase proteins and chemokines in the progressionof the rheumatoid arthritis. Semin Arthritis Rheumatism 1996;26(2):526-38.
17. Barret KE. Cytokines: sources, receptors and signalling. Bailliere´s Clinical Gastroenterology1996;10:1-5.
18. Abe Y, Horiuchi A, Miyake M, Kimura S. Anti-cytokine natural human immunoglobulin: one possiblemechanism of the clinical of intravenous immunoglobulin therapy. Immunol Rev 1994;139:5-19.
19. Anderson J, Skanse N, Sphir U, et al: Intravenous immunoglobulin affects cytokynes production in Tlymphocytes and monocytes-macrophages. Clin Exp Immunol 1996;104:10-21.
20. Blasezy R, Westhoff N, Grosse-Wilde H. Soluble CD4, CD8 and HLA molecules in commercialimmunoglobulin preparations. Lancet 1993;341:789-90.
21. Kekow J, Reinhol D, Pap T. Ansorge S. Intravenous Immunoglobulins and transforming growth factorb. Lancet 1998;351:184-5.
22. Archiron A, Gabbay U, Gilad, et al. Intravenous Immunoglobulin treatment in multiple sclerosis. Effecton relapses. Neurology 1998,50:398-402.
23. Van Engelson BGM, Miller DJ, Pavelkok D, et al. Promotion of remyelination by polyclonalimmunoglobulin in theiler´s virus-induced demyelination and in multiple sclerosis. J Neurol NeurosurgPsychatry 1994;57(Supply):65-8.
24. Gabriel CM, Gregson NA, Redford EJ, et al. Human inmunoglobulin ameliorales rat allergic neuritis.Brain 1997;120:1533-154.
Recibido: 28 de enero del 2001. Aprobado: 28 de junio del 2001.Dr. Amaury Noda Albelo.Hospital Pediátrico Eliseo “Noel”Caamaño. Matanzas.

46
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):48-54
Instituto de Medicina Tropical “Pedro Kourí”
EVALUACIÓN DE LA FUNCIÓN OPSONO FAGOCÍTICADE LOS NEUTRÓFILOS EN PACIENTES INFECTADOSPOR EL VIH
Lic. Randelys Molina Castro, Dr. Alejandro Álvarez García, Lic. Liliana Pérez Toledo,Lic. Lizet Sánchez Valdés, Téc. Yondel Torranzo Soto y Téc. Caridad Luzardo Suárez
RESUMEN
La disfunción de los neutrófilos podría contribuir a la susceptibilidad a infeccionesobservada en los pacientes infectados por el virus de inmunodeficiencia humana(VIH). Se realizó un estudio de corte transversal para evaluar la opsono fagocitosisde los neutrófilos en estos pacientes, en el que participaron 104 individuo: 25donantes de sangre (grupo control) y 79 VIH positivos (+). Estos últimos sedividieron en 3 grupos según su conteo global de linfocitos T CD4 + (CGLCD4):grupo 1 compuesto por 24 individuos con CGLCD4≥500 células/mm3; grupo 2por 26 pacientes con CGLCD4 de 200 a 499 células/mm3; y grupo 3 por 29 casoscon CGLCD4 < 200 células/mm.3 Se realizó la determinación de los valoresabsolutos de leucocitos y granulocitos y los valores relativos de linfocitos, lacuantificación de subpoblación linfoide T CD4 positiva (+) y la determinación delíndice opsono fagocítico. Se encontró una disminución significativa (p < 0,05) delconteo absoluto de leucocitos y granulocitos, así como de la función opsonofagocítica, en los grupos de pacientes VIH+ con respecto al grupo control. Seconstató una relación directa no lineal del conteo absoluto de linfocitos T CD4 +con el conteo global de leucocitos y de granulocitos y con la función opsonofagocítica.
DeCS: VIH/inmunología; NEUTROFILOS/inmunología; SINDROME DEINMUNODEFICIENCIA ADQUIRIDA/inmunología; RECUENTO DELINFOCITO CD4/análisis; FAGOCITOSIS; GRANULOCITOS/inmunología.
La fagocitosis mediada por polimorfo-nucleares (PMN) neutrófilos, constituyeuna de las principales defensas delorganismo hospedero en su lucha contralas infecciones producidas por bacterias yhongos. El proceso de fagocitosiscomprende varios pasos secuenciales:
quimiotaxis, adherencia de las partículasantigénicas a la superficie de los fagocitos,captación/ingestión (fagocitosis) y muerteintracelular mediada por mecanismosdependientes e independientes del oxígeno.Los fagocitos poseen receptores para elcomponente C3b del sistema de

47
complemento, así como para la regiónconstante de la molécula de inmuno-globulina. Esto permite que los microor-ganismos opsonizados puedan adherirse ala superficie de estas células, facilitando lafagocitosis.1
Una actividad fagocítica anormal puedeestar asociada con una gran variedad deenfermedades y puede estar ocasionada porun defecto de los neutrófilos, un problemafisiológico asociado con algún tipo deinmunoglobulina, con el sistema decomplemento, o ambos. La mayoría de losartículos revisados coincide en reconocerque se produce una alteración de la funciónde los neufrófilos en el curso de la infecciónpor el virus de inmunodeficiencia humana(VIH).
Dobmeyer y otros.2 comunicaron quelos granulocitos y monocitos de lospacientes asintomáticos infectados por elVIH mostraron una capacidad bacteriófagasignificativamente aumentada; sinembargo, los pacientes con síndrome deinmunodeficiencia adquirida (SIDA)mostraron una reducción significativa de lafagocitosis, lo cual se correlacionó con elnúmero de células T CD4 positivas (+).Además, Shalekoff y otros3 observaron unaumento significativo de la fagocitosis enlos granulocitos de individuos infectadospor el VIH-1; pero aquellos infectados porMycobacterium tuberculosis solamente, oen combinación con el VIH-1, mostraronuna capacidad significativamente disminui-da para fagocitar. Por otra parte, en untrabajo de Schaumann y otros,4 lafagocitosis de Escherichia coli y Sta-phylococcus aureus resultó significati-vamente disminuida en los PMN depacientes VIH + con bajos conteos decélulas T CD4+.
Otros autores5 estudiaron la opsonofagocitosis (OF) de Candida albicansempleando neutrófilos aislados de
individuos VIH + asintomáticos, pacientescon complejo relacionado con el SIDA,sometidos o no a tratamiento conzidovudina (AZT), así como pacientes conSIDA. Ellos observaron que esta actividaddisminuye progresivamente y secorrelaciona con la severidad de la infecciónpor VIH. En contraste, Wenisch y otros6
encontraron que los PMN de los pacientescon SIDA presentaban una actividadfagocítica aumentada contra Candida spp.
Según un estudio llevado a cabo porGruber y otros,7 las células viables deC.albicans tratadas con la gp160 o la gp41y suero, son fagocitadas por los PMN enuna menor cantidad que las C.albicanstratadas con la gp120 y suero, o suerosolamente. Esto permite concluir que lainteracción de la gp160 del VIH-1 conC.albicans puede aumentar lasusceptibilidad a este hongo en lospacientes VIH-1+.
Debido a las discrepancias encontra-das en la literatura revisada, y teniendo encuenta que en Cuba no se han reportadoestudios al respecto, nos propusimosevaluar la actividad opsono fagocítica delos neutrófilos frente a C.albicans enpacientes VIH +.
MÉTODOS
Población de estudio: participaron 104individuos: 25 donantes de sangrevoluntarios (grupo control) y 79 pacientesseropositivos, que asistieron para realizarseexámenes inmunológicos al laboratorioclínico del IPK y tenían edadescomprendidas entre 18 y 40 años. Lospacientes se dividieron en 3 gruposatendiendo a su conteo global de linfocitosT CD4 + (CGLCD4):
− Grupo 1: 24 individuos con CGLCD4≥500células/mm.3

48
− Grupo 2: 26 individuos con CGLCD4 de200 a 499 células/mm.3
− Grupo 3: 29 individuos con CGLCD4 <200células/mm.3
Colección y manejo de las muestras: acada individuo se le extrajeron 15 mL desangre periférica por punción venosa: 2 mLfueron colectados en tubos Vacutainer(Becton Dickinson Vacutainer Systems,Franklin Lakes, NJ) con EDTA K3 para ladeterminación de los valores absolutos deleucocitos y granulocitos, los valoresrelativos de linfocitos y para lacuantificación de subpoblación linfocitariaT CD4 +; 10 mL se colectaron en tubosVacutainer con EDTA K
3 para la
determinación del índice opsono fagocítico;del resto de la sangre se obtuvo suero paracompletar este estudio.
Valores absolutos de leucocitos ygranulocitos y valores relativos delinfocitos: se obtuvieron los valoresmediante un analizador hematológico de 18parámetros (COBAS MICROS; Roche).
Inmunofenotipaje por citometría deflujo: los valores relativos de los linfocitosT CD4 + se cuantificaron por citometría deflujo (citómetro de flujo FACScan; BectonDickinson) usando las técnicas estándarespara triple marcaje y los anticuerposmonoclonales anti CD3, anti CD45 y antiCD4 de la casa comercial BectonDickinson.8
Para determinar los valores absolutosde los linfocitos T CD4+ se tomaron losvalores absolutos de leucocitos y losrelativos de linfocitos.
Índice opsono fagocítico: la funciónopsono fagocítica se estudió según elmétodo descrito por Rivero y otros,9 con lamodificación de que los PMN provenían delpaciente y no de un donante sano de gruposanguíneo O. Se aislaron los PMN a partir
de sangre periférica y se preparó unasuspensión de estas células, así como otrade C.albicans a iguales concentraciones.Estos microorganismos se incubaron a 37 °Cen suero del paciente (opsonización). Seañadieron los PMN y se incubó a 37 °C(fagocitosis). Inmediatamente, a los 15 y alos 60 minutos, se contaron las C.albicansextracelulares en cámara de Neubahuer.Asumiendo que las C.albicans contadas enel tiempo cero constituyen el 100 %, lostotales contados en los tiempos 15 y 60representan, indirectamente, el porcentaje deOF a los 15 y 60 minutos, respectivamente.
Análisis estadístico de los resultados:los datos fueron almacenados comovariables. Se aplicó el test de Kolomogorov-Smirnov para el análisis de la normalidad deestas. Se realizó una comparación entre losgrupos de pacientes y entre estos y elcontrol, para lo cual se empleó el test deKruskal-Wallis. Se calculó el coeficiente decorrelación de Spearman para evaluar larelación entre todas las variables.
RESULTADOS
En nuestro estudio se determinaron losvalores absolutos de leucocitos y degranulocitos y el índice opsono fagocíticoa los 15 y 60 minutos (tabla 1).
Leucocitos: los conteos de leucocitosde los grupos 1,2 y 3 fueron signi-ficativamente más bajos que los del grupocontrol (p
1=0,0015; p
2=0,0003; p
3=0,0000).
No se encontraron diferencias significativasentre los valores de los distintos grupos depacientes VIH+ (tabla 1).
Granulocitos: el estudio de granu-locitos reveló disminuciones significativas enlos grupos 1,2 y 3 con respecto al grupo con-trol (p
1=0,0002; p
2=0,0000; p
3=0,0013). Entre
los grupos de pacientes VIH+ no se obser-varon diferencias significativas (tabla 1).
49
TABLA 1. Conteo global de leucocitos y de granulocitos e índice opsono fagocítico en los diferentes grupos de estudio (media± desviación estándar)
Grupo 1 Grupo 2 Grupo 3 Grupo controlDeterminación n= 24 n= 26 n = 29 n = 25
Leucocitos 6,6±1,8* 6,2±1,6* 5,8±1,7* 7,8±1,1(109/L) p1=0,0015 p2=0,0003 p3=0,0000Granulocitos 3,6±1,8* 3,2±1,2* 3,6±1,5* 4,8±0,8(109/L) p
1=0,0002 p
2=0,0000 p
3=0,0013
IOF 15 60,6 ±13,9* 60,4±11,8* 70,8±13,0* 42,6±9,4(%) p
1=0,0000 p
2=0,0000 p
3=0,0000
IOF 60 32,2±15,3* 35,2±15,4* 44,7±15,0* 19,4±4,8(%) p1=0,0004 p2=0,0000 p3=0,0000
* Diferencia significativa con respecto al grupo control.IOF 15: índice opsono fagocítico a los 15 minutos; IOF 60: índice opsono fagocítico a los 60 minutos.Grupo 1: 24 individuos con CGLCD4 ≥ 500 células/mm.3 Grupo 2: 26 individuos con CGLCD4 de 200-499 células/mm.3 Grupo3: 29 individuos con CGLCD4 < 200 células/mm.3 Grupo control: 25 donantes de sangre.
Actividad opsono fagocítica: seobservó una disminución significativa deesta función en los pacientes VIH+ conrespecto al grupo control (tiempo15:p
1=0,0000; p
2 =0,0000; p
3=0,0000; tiempo
60:p1=0,0004; p
2=0,0000; p
3=0,0000) (tabla 1).
Esta función resultó significativamentedisminuida en el grupo 3 respecto al resto delos pacientes VIH+ (tiempo 15: p
1-3= 0,0052;
p2-3
= 0,0051; tiempo 60:p1-3
=0,0032; p2-3
=0,0209).Relación entre variables: el estudio de
correlación entre las variables indicó queestas están relacionadas, aunque nolinealmente (-0,8 < coeficiente de correlaciónde Spearman > 0,8) (tabla 2).
El conteo absoluto de células T CD4 +se relacionó directamente con los conteos
TABLA 2. Coeficiente de correlación de Spearman entre las variables en los pacientes VIH+
Variables Leucocitos Granulocitos IOF15 IOF60 CGLCD4
R=0,796192 R =-0,467608 R=-0,533730 R=0,423939Leucocitos - p=0,000000 p=0,000001 p=0,000000 p=0,000011
R=-0,415867 R=-0,416040 p=0,269165Granulocitos - - p=0,000031 p=0,000030 p=0,008707
R=0,789374 R=-0,551048IOF15 - - - p=0,000000 p=0,000000
R=-0,590544IOF60 - - - - p=0,000000CGLCD4 - - - - -
IOF 15: índice opsono fagocítico a los 15 minutos; IOF 60: índice opsono fagocítico a los 60 minutos; CGLCD4: conteo globalde linfocitos T CD4+; NS: correlación no significativa; R: significativo cuando p < 0,05.
globales de leucocitos y de granulocitos ycon los valores de OF. Los conteos globalesde leucocitos y de granulocitos mostraronuna relación directa entre sí y con losvalores de OF. Dichos valores correspon-dientes al tiempo 15 y al 60 se relacionarondirectamente entre sí (tabla 2).
DISCUSIÓN
Una característica común de la mayorparte de las inflamaciones agudas inducidaspor procesos inmunológicos, es laacumulación de neutrófilos en los sitios dereacción en los que intervienen activamentepara mediar estos procesos. Estas células

50
desempeñan un papel importante comoprimera línea de defensa contra lasinfecciones en el hospedero; en sangre yespacios extravasculares, ejercen su efectoantimicrobiano a través de la interaccióncon anticuerpos, complemento y factoresquimiotácticos. Así, en la evaluación de lafunción de los neutrófilos no se puedeconsiderar la célula como una entidadindependiente, se debe tener en cuenta sudependencia de otros mecanismosinmunitarios, tanto celulares comohumorales, que participan como un sistemabien engranado. La alteraciones de estesistema pueden ser cuantitativas ycualitativas, lo cual predispone ainfecciones recurrentes en los pacientesinfectados por el VIH.10
La disminución de leucocitosobservada en los pacientes VIH+ (tabla 1)puede deberse a diversos factores queproducen una caída en el número delinfocitos o de neutrófilos. Entre las causasde linfopenia se reportan: el efecto citolíticodel virus sobre las células T CD4+,fenómenos autoinmunes, superantígenos yapoptosis.11-14
En la literatura revisada se reporta laexistencia de neutropenia en los pacientesVIH+. Entre las causas descritas seencuentran: la exposición a ciertas drogascomo AZT o ganciclovir y la infiltración dela médula ósea por células tumorales oagentes infecciosos.10 Estos factorespueden haber influido en la disminución delos granulocitos que se detecta en lospacientes VIH+ (tabla 1).
La disminución de la actividad opsonofagocítica podría deberse a una deficienciaen la capacidad opsonizante del suero delpaciente, a un defecto intrínseco de losneutrófilos, o a ambos factores.
En los individuos con infección porVIH-1 sintomática, se ha reportado laincapacidad de generar una respuesta
humoral de anticuerpos apropiada, lo cualpuede contribuir a las alteracionesfuncionales de los neutrófilos.6 A pesar deque existen altos niveles deinmunoglobulinas, la respuesta deanticuerpos a antígenos específicos estámarcadamente disminuida en los pacientesVIH+.10 Este factor pudo haber influidonegativamente en la opsonización de laC.albicans durante el ensayo de índiceopsono fagocítico y, por lo tanto, habercontribuido a la disminución de la OF (tabla1). La producción de una respuesta deanticuerpos inapropiada, posiblemente seagrave en los pacientes con conteos decélulas T CD4+ por debajo de 200 células/mm, 3 influyendo así en la marcadadisminución de la función opsono fagocíticaobservada en estos casos (tabla 1).
Se ha reportado, además, que ladisfunción de los neutrófilos puede ser unreflejo de la influencia de una serie deproductos liberados por agentesinfecciosos secundarios, o de citocinasproducidas en el curso de la respuestainmune del organismo contra ellos.15 Lafrecuencia y la severidad de infecciones esmayor en aquellos pacientes con conteosglobales de linfocitos T CD4+ inferiores a200 células/mm,3 lo que puede explicar lapronunciada disminución de la OFobservada en estos individuos (tabla 1).
La disminución de la OF encontradaen los pacientes infectados por el VIH (tabla1), también podría ser el resultado de lainteracción que durante el ensayo se pudoestablecer entre las proteínas del virus gp41o gp160 y la C.albicans (microorganismode prueba utilizado en nuestro estudio parala determinación del índice opsonofagocítico).7,16-18 Esta interacción podríaocurrir con mayor probabilidad en lospacientes con los conteos de linfocitos TCD4+ por debajo de 200 células/mm,3 debidoa que en estos casos, usualmente se reporta

51
una carga viral más elevada, queproporciona una mayor antigenemia.19
Las variables estudiadas en estetrabajo no se encontraron relacionadaslinealmente, lo que se interpreta comoevidencia de su carácter multifactorial.
Al disminuir el número de células TCD4+, aumenta la frecuencia de los diversosfactores anteriormente mencionados, quepueden contribuir al descenso del númerode leucocitos y de granulocitos, así como ala disfunción opsono fagocítica. Esto puedeexplicar la relación directa encontrada entreel conteo global de linfocitos T CD4+ y elde leucocitos y de granulocitos, así comocon los valores de OF (tabla 2).
Como era de esperar, dada la estrecharelación que existe entre los factores quepueden influir en la disminución deleucocitos, incluyendo los granulocitos yde la OF, se observó una relación directa
entre los conteos globales de dichas célulasy entre cada uno de estos y los valores deOF (tabla 2).
Los factores involucrados en ladisminución de la actividad opsonofagocítica inciden tanto en el incrementodel índice opsono fagocítico para el tiempode 15 minutos como para el de 60, lo que serefleja en la relación directa observada entreambos valores (tabla 2).
Finalmente, puede concluirse que eldeterioro paulatino de la OF detectado enlos pacientes VIH+, así como lasleucopenias y granulocitopenias, podríanprovocar una mayor susceptibilidad ainfecciones microbianas. Estas, aunquepueden presentarse desde el inicio de lainfección por VIH, son más numerosas yfrecuentes en las etapas más avanzadas dela enfermedad, en las que se acentúan dichasalteraciones.
SUMMARY
The dysfunction of neutrophils could contribute to susceptibility to infectionsobserved in HIV-infected patients. A cross-sectional study was conducted toevaluate the opsonophagocytosis of neutrophils in these patients. One hundredand four individuals participated in it: 25 blood donors (the control group) and79 HIV-positive patients who were distributed into 3 groups according to theglobal count of CD4 lymphocytes (GLCCD4): Group 1 composed of 24 individualswith GLCCD4³ 500 cell/mm3, Group 2 with 26 patients with GLCCD4 of 200-499 cell/mm3 and Group 3 composed by 29 patients with GLCCD4 < 200 cell/mm3 . Absolute values of leukocytes and granulocytes and relative values oflymphocytes were determined, CD4+ lymphocyte T subset was quantified andthe opsonophagocytic index was estimated. A significant reduction (p< 0,05) inabsolute count of leukocytes and granulocytes and in the opsonophagocyticfunction was found in HIV-infected patients compared with the control group.There was a non-linear direct relation of the absolute count of CD4+ Tlymphocytes with global count of leukocytes and granulocytes and theopsonophagocytic function.
Subject headings: VIH/immunology; NEUTROPHILS/immunology; ACQUIREDIMMUNODEFICIENCY SYNDROME/immunology; CD4 LYMPHOCYTECOUNT/analysis; PHAGOCYTOSIS; GRANULOCYTES/immunology.

52
REFERENCIAS BIBLIOGRÁFICAS
1. Roitt I, Brostoff J, Male D. Inmunología, 4ª ed. Madrid: Harcourt Brace; 1997:17.6-17.7. 2. Dobmeyer TS, Raffel B, Dobmeyer JM, Findhammer S, Klein SA, Kabelitz D, et al. Decreased function
of monocytes and granulocytes during HIV-1 infection correlates with CD4 cell counts. Eur J Med Res1995;1(1):9-15.
3. Shalekoff S, Tiemessen CT, Gray CM, Martin DJ. Depressed phagocytosis and oxidative burst inpolymorphonuclear leukocytes from individuals with pulmonar y tuberculosis with or without humanimmunodeficiency virus type 1 infection. Clin Diagn Lab Immunol 1998;5(1):41-4.
4. Schaumann R, Krosing J, Shah PM. Phagocytosis of Escherichia coli and Staphilococcus aureus byneutrophils of human immunodeficiency virus-infected patients. Eur J Med Res 1998;3(12):546-8.
5. Tachavanich K, Pattanapanyasat K, Sarasombath S, Suwannagool S, Suvattee V. Opsonophagocytosisand intracellular killing activity of neutrophils in patients with human immunodeficiency virus infection.Asian Pac J Allerge Immunol 1996;14(1):49-56.
6. Wenisch C, Parschalk B, Zedwitz-Liebenstein K, Graninger W, Rieger A. Dysregulation of thepolymorphonuclear leukocyte-Candida spp interaction in HIV-Positive patients. AIDS1996;10(9):983-7.
7. Gruber A, Lukasser-Vogl E, Borg-von Zepelin M, Dierich MP, Wurzner R. Human immunodeficiencyvirus type 1 gp 160 and gp 42 binding to Candida albicans selectively enhances candidal virulence invitro. J Infect Dis 1998;177(4):1057-63.
8. Nicholson JKA, Jones BM, Hubbard M. CD4 T-lymphocyte determinations on whole blood specimensusing a single-tube three-color assay. Cytometry 1993;14:685-9.
9. Rivero RA, Villaescusa R, González C, Palma LE, Ballester JM. Estandarización de una técnica paraevaluar la actividad opsonizante del suero humano fresco. Rev Cubana Hematol Inmunol Hemoter1985;1(2):200-8.
10. De Vita VT, Hellman S, Rosenberg SA. AIDS: etiology, diagnosis, treatment and prevention. 3ª ed.Philadelphia: J.B. Lippincott; 1992.p.62.
11. Voss TG, Fermin CD, Levy JA, Vigh S, Choi B, Garry RF. Alteration of intracellular potassium andsodium concentrations correlates with induction of cytopathic effects by human immunodeficiencyvirus. J Virol 1996;70:5447-54.
12. Silvestris F, Williams RC, Dammacco F. Autoreactivity in HIV-1 infections: the role of molecularmimicry. Clin Immunol Immunopathol 1995;75:197-205.
13. García S, Dadaglio G, Cilote V, Chenal H, Bondurand A, Gougeon ML. Evidence for an in vivosuperantigenic activity in human immunodeficiency virus-infected individuals. Blood1996;88:2151-61.
14. Gougeon ML, Lecoeur H, Dulioust A. Programmed cell death in peripheral lymphocytes from HIV-infected persons. J Immunol 1996;165:3509-20.
15. Wenisch C, Graninger W. Are extrinsic or intrinsic factor relevant for polymorphonuclear leukocytedysregulation septicemia? Clin Diagn Lab Immunol 1995;2:241-5.
16. Wurzner R, Gruber A, Stoiber H, Spruth M, Chen YH, Lukasser-Vogl E, et al. Human immunodeficiencyvirus type-1gp 41 binds to Candida albicans via complement C3-like regions. J Infect Dis1997;176(2):492-8.
17. Vartivarian SE. Virulence properties and non immune pathogenic mechanisms of fungi. Clin Infect Dis1992;14(supp 1):S30-6.
18. Wu T, Samaranayake LP, Cao BY, Wang J. In-vitro proteinase production by oral Candida albicansisolates from individual with and without HIV infection and its attenuation by antimycotic agents. JMed Microbiol 1996;44:311-6.
19. Richman DD, Whitley RJ, Hayden FG. Clinical Virology. New York: Churchill Livingstone; 1997.p.718.
Recibido: 22 de junio del 2001. Aprobado: 1 de octubre del 2001.Lic. Randelys Molina Castro. Instituto de Medicina Tropical “Pedro Kourí”. Dirección postal: P.0. Box:601, Marianao 13, Ciudad de La Habana, Cuba.Teléf. (537)22045. Fax (537)246051. e-mail:[email protected]

53
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):55-9
Instituto de Hematología e Inmunología
POSIBLE PARTICIPACIÓN DEL SISTEMA COMPLEMENTOEN EL DESARROLLO DE MANIFESTACIONES AUTOINMUNESEN LA LEUCEMIA LINFOCÍTICA CRÓNICA
Lic. Rinaldo Villaescusa Blanco, Lic. Antonio Bencomo Hernández, Lic. Ada A. ArceHernández, Lic. Julio C. Merlín Linares, Lic. Ana M. Guerreiro Hernández, Lic. Luz M.Morera Barrios y Dr. Porfirio Hernández Ramírez
RESUMEN
Se efectuó la medición de la actividad de la vía clásica, alternativa, factor B,factor D, así como la cuantificación de C1q, C3 y C4 del sistema complementoen 27 pacientes con leucemia linfocítica crónica de tipo B (LLC-B) CD5+estadificados en 2 grupos: 12 pacientes en fase poco avanzada de la enfermedad(estadios 0, I, II) y 15 en fase avanzada (estadios III, IV). Se demostró unadisminución de la actividad de la vía clásica y de la concentración de C1q y C4 en10 pacientes en fase avanzada, 7 de los cuales tenían asociada una anemiahemolítica autoinmune (AHAI); en el grupo en fase poco avanzada de laenfermedad no se demostraron deficiencias de complemento ni manifestacionesautoinmunes. Los datos obtenidos sugieren la posible participación del sistemacomplemento en el mantenimiento de la tolerancia inmunológica, cuya rupturapudiera provocar la síntesis de anticuerpos antieritrocitarios, y por lo tanto, laanemia hemolítica que se asocia en determinados pacientes en fase avanzada dela enfermedad.
DeCS: LEUCEMIA CRONICA DE CELULA B/inmunología; ENFERMEDADESAUTOINMUNES; COMPLEMENTO/inmunología; ANEMIA HEMOLITICAAUTOINMUNE/inmunología.
Con la leucemia linfocítica crónica(LLC) se asocian frecuentemente trastornosde la regulación inmune y fenómenos deautoinmunidad, y es la anemia hemolíticaautoinmune (AHAI) una de lasenfermedades que comúnmente sepresenta.1 La LLC-B se caracteriza por laacumulación de linfocitos B CD5+, loscuales se relacionan con la producción deautoanticuerpos naturales. Se ha planteado
también la participación de estas células enla patogénesis de la AHAI y se ha sugeridouna posible relación causal entre las célulasB CD5+ de ambas enfermedades.2 Lapresencia de autoanticuerpos contra loseritrocitos en algunos pacientes con LLC-B con AHAI no está esclarecida. Se haseñalado que los anticuerpos expresadospor las células B leucémicas CD5+ no seunen directamente con los eritrocitos, lo que

54
indica que estos no están involucrados ensu destrucción; se plantea que dichosanticuerpos antieritrocitarios sonproducidos por células B normalesremanentes.3
En diversas investigaciones se haobservado una asociación entre deficien-cias de los componentes iniciales de la víaclásica del complemento con enfermedadesautoinmunes, lo que sugiere ciertaparticipación del complemento en elmantenimiento de la tolerancia inmune.4,5 Entrabajos anteriores se han demostradodeficiencias del complemento en pacientescon LLC-B CD5+ en las fases másavanzadas de la enfermedad.6 En nuestrotrabajo nos propusimos estudiar la actividaddel sistema complemento y los niveles dealgunos de sus componentes en un grupode pacientes con LLC-B CD5+, en diferentesestadios, con AHAI asociada en algunosde los casos, con el objetivo de establecerel posible papel del sistema complementoen el desarrollo de enfermedadesautoinmunes en la LLC.
MÉTODOS
Se estudiaron 27 pacientes con eldiagnóstico de LLC-B CD5+: 16 hombres y11 mujeres, con una edad promedio de 60años (rango de 42 a 73 años). Los enfermosse estadificaron acorde con la clasificaciónde Rai.7 Doce pacientes se encontraban enfase poco avanzada de la enfermedad(estadios 0, I, II) y 15 en fase avanzada(estadios III, IV). Siete de los pacientes enfase avanzada de la enfermedad eranportadores de AHAI. En el momento delestudio ninguno de los pacientes teníainfección clínicamente manifiesta ni esta
pudo evidenciarse mediante cultivosmicrobiológicos.
La sangre se obtuvo por punciónvenosa y se conservó a -30 °C. Los sueroscontroles se obtuvieron y conservaron enlas mismas condiciones.
En todas las muestras estudiadas semidió la actividad de las vías clásica,alternativa, factor B, factor D y secuantificaron los componentes C1q, C3 yC4 del sistema complemento.
La actividad hemolítica de la vía clásicase determinó de acuerdo con el métododescrito por Mayer,8 y la vía alterna segúnlo propuesto por Platts-Mills y otros.9 Laactividad funcional del factor B y el factorD de la vía alterna se midieron por losmétodos descritos anteriormente.10,11
Los niveles de Clq, C3 y C4 secuantificaron por inmunodifusión radialsimple,12 para lo cual se emplearon placasde cuantificación elaboradas en nuestrolaboratorio. Para los valores de referencia,las pruebas anteriormente mencionadas serealizaron en 30 sujetos sanos.
RESULTADOS
Se demostró una disminución de laactividad del factor B, C3 y de la víaalternativa en 1 paciente (8,3 %), de los 12con LLC-B CD5+ en fase poco avanzada dela enfermedad (estadios 0, I, II), ningunoera portador de AHAI. En cambio, 10/15(66,6 %) pacientes en fase avanzada(estadios III, IV) presentaron fundamental-mente una disminución de los compo-nentes Clq, C4 y de la actividad de la víaclásica del complemento, 7 de los cualestenían asociada una AHAI (tablas 1,2).

55
TABLA 1. Niveles de complemento en pacientes con leucemia linfocítica crónica en fase poco avanzada de la enfermedad
Enfermo Estadio VC VA FB FD Clq C3 C4No. clínico (CH50) (%) (%) (%) (%) (g/L) (g/L) AHAI
1 0 27,3 91,3 82,8 92,2 89,1 1,71 0,33 Neg2 0 26,3 90,4 82,3 93,0 87,2 1,86 0,29 Neg3 0 26,5 88,5 80,4 87,3 90,3 1,90 0,31 Neg4 0 27,1 95,1 92,2 84,5 91,5 1,87 0,34 Neg5 0 25,4 96,4 93,3 89,0 92,3 2,01 0,40 Neg6 0 29,2 87,2 90,1 90,1 90,4 1,91 0,39 Neg7 0 27,3 89,2 88,8 89,3 87,2 1,89 0,42 Neg8 I 30,2 90,6 87,4 85,3 88,0 1,94 0,36 Neg9 I 29,3 94,0 91,1 92,2 94,6 2,02 0,30 Neg
10 I 28,4 99,0 96,3 97,1 92,2 1,79 0,42 Neg11 II 31,1 79,2 73,4 87,4 84,3 1,60 0,41 Neg12 II 29,0 92,0 94,2 88,0 87,6 1,86 0,37 Neg
Control 27,5 ± 98,0± 100± 95,0± 91,5± 2,02± 0,35±Normal 6,5 16,0 24,0 14,0 7,5 0,34 0,15
VC: vía clásica; VA: vía alternativa; FB:factor B; FD: factor D; AHAI: anemia hemolítica autoinmune.
TABLA 2. Niveles de complemento en pacientes con leucemia linfocítica crónica en fase avanzada de la enfermedad
Enfermo Estadio VC VA FB FD Clq C3 C4No. clínico (CH50) (%) (%) (%) (%) (g/L) (g/L) AHAI
1 III 27,1 901 79,5 89,2 86,2 1,74 0,34 Neg2 III 26,2 91,2 82,2 92,1 89,3 1,84 0,41 Neg3 III 28,3 88,7 81,4 88,8 91,3 1,81 0,32 Neg4 III 26,1 92,4 78,1 87,1 87,4 1,99 0,28 Neg5 III 23,3 80,1 77,0 93,4 92,2 1,53 0,39 Neg6 III 19,7 97,5 99,2 97,4 92,0 1,77 0,18 Neg7 III 18,6 99,2 96,4 101,1 79,4 1,96 0,17 Neg8 III 19,1 84,2 105,3 93,3 80,0 1,88 0,19 Pos9 III 17,3 87,5 98,6 95,2 79,4 1,93 0,16 Pos
10 III 17,8 92,3 94,1 84,7 77,7 2,02 0,17 Pos11 III 18,4 90,3 88,8 89,0 78,2 1,92 0,19 Pos12 IV 23,2 99,6 87,3 92,o 86,7 2,05 0,29 Neg13 IV 16,8 101,3 90,0 97,4 81,2 1,95 0,16 Pos14 IV 17,4 92,2 100,4 93,3 74,3 1,91 0,19 Pos15 IV 17,7 97,1 98,9 91,1 72,2 1,61 0,18 Pos
Control 27,5 ± 98,0± 100,0± 95,0± 91,5± 2,02± 0,35±Normal 6,5 16,0 24,0 14,0 7,5 0,34 0,15
VC: vía clásica; VA: vía alternativa; FB: factor B; FD: factor D; AHAI: anemia hemolítica autoinmune.
DISCUSIÓN
En la LLC-B se presentan trastornosde inmunorregulación con la pérdida de latolerancia a antígenos propios que semanifiestan en su asociación conenfermedades autoinmunes, entre las quese destaca la AHAI.1 Se ha señalado el papeldel sistema complemento en el manteni-
miento de la tolerancia inmune, ya quedeficiencias de los componentes inicialesde la vía clásica como el Clq, C2,C4 y enmenor medida el C3, se han relacionado conel desarrollo de enfermedades autoinmunescomo el lupus eritematoso sistémico.5
También es conocida la relación existenteentre enfermos con LLC-B y familiares deestos con primer grado de consanguinidad,

56
con desórdenes autoinmunes, cuyaocurrencia puede verse favorecida por lahipocomplementemia, lo que indica laposible existencia de factores genéticospredisponentes en esta enfermedad.13,14
En nuestro trabajo se obtuvieronvalores normales de complemento en lospacientes con LLC-B CD5+ en fase pocoavanzada de la enfermedad, donde enningún caso hubo asociación confenómenos autoinmunes. En cambio, en elgrupo de pacientes en fase avanzada, seobservaron niveles disminuidos de Clq, C4y de la actividad de la vía clásica en 10 delos 15 enfermos estudiados, 7 de los cualestenían asociado una AHAI. Estos datossugieren la posible participación del sistemacomplemento en el mantenimiento de la
tolerancia inmunológica, cuya rupturapudiera provocar la síntesis de anticuerposantieritrocitarios, y por lo tanto, la anemiahemolítica en determinados enfermos,fenómeno que pudiera estar genéticamentedeterminado, ya que en 3 pacientes seobtuvieron niveles disminuidos decomplemento sin enfermedad autoinmuneasociada, hasta el momento del estudio.Trabajos anteriores sugieren al complemen-to como un posible marcador temprano deldesarrollo de autoinmunidad en la LLC.15
Estudios futuros referentes aenfermedades malignas asociadas conmanifestaciones autoinmunes, nos permiti-rán profundizar en el posible papel delsistema complemento en el desarrollo yregulación de la respuesta inmune humoral.
SUMMARY
The measurement of the activity of the classical pathway, the alternative pathway,factor B, factor D, as well as the quantification of Clq, C3 and C4 of thecomplement system were made in 27 patients with type B chronic lymphocyticleukemia (CLL-B) CD5+, classified into 2 groups: 12 patients at little advancedstage of the disease (stages 0, I, II) and 15 patients at advanced stage (stages III,IV). It was proved that the activity of the classical pathway and the concentrationof Clq and C4 lowered in 10 patients at advanced stage, 7 of them also hadautoimmune hemolytic anemia. Neither complement deficiencies norautoimmune manifestations were shown in the group of patients at little advancedstage of the disease. Collected data indicates the possible involvement of thecomplement system in keeping the immune tolerance, the rupture of which maycause the synthesis of anti-erythrocyte antibodies and thus, the hemolytic anemiathat occurs in certain patients at advanced stage of the disease.
Subject headings: LEUKEMIA, B-CELL, CHRONIC/immunology;AUTOIMMUNE DISEASES; COMPLEMENT/immunology; ANEMIA,HEMOLYTIC, AUTOIMMUNE/immunology.
REFERENCIAS BIBLIOGRÁFICAS
1. Diehl LF, Ketchum LH. Autoimmune disease and chronic lymphocytic leukemia: autoimmune hemolyticanemia, pure red cell aplasia and autoimmune thrombocytopenia. Semin Oncol 1998;25 (1):80-97.
2. Youinou P, Pers JO, Jamin C, Lydyard PM. CD5-positive cells at the crossroads of malignancy andnonorgan-specific autoimmunity. Pathol Biol (Paris) 2000;48(6):574-6.

57
3. Efremov DG, Ivanovski M, Burrone OR. The pathologic significance of the immunoglobulins expressedby chronic lymphocytic leukemia B-cells in the development of autoimmune hemolytic anemia. LeukLymphoma 1998;28(3-4):285-93.
4. Carroll MC, Fischer MB. Complement and the immune response. Curr Opin Immunol 1997;9:64-9. 5. Nielsen CH, Fischer EM, Leslie RGQ. The role of complement in the acquired immune response.
Immunology 2000;100:4-12. 6. Villaescusa R, Borrego I, Merlín JC, Hernández P. Estudio seriado del sistema complemento e
inmunocomplejos circulantes en la leucemia linfoide crónica. Rev Cubana Hematol Inmunol Hemoter1989;5(1):37-47.
7. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternak BS. Clinical staging of chroniclymphocytic leukemia. Blood 1975;46(2):219-34.
8. Mayer MM. Complement and complement fixation. En: Kabat EA, Mayer MM (eds). ExperimentalImmunochemistry. Illinois: Thomas Publisher;1967.p.133-40.
9. Platts-Mills TAE, Ishizaka K. Activation of the alternative pathway of human complement by rabbitcells. J Immunol 1974;113:348-58.
10. Aguado MT, Celada A, Lambert PH. Medida de la función de la vía alternativa del sistema complemento:actividad hemolítica y solubilización de inmunocomplejos. Inmunología 1983;2:63-70.
11. Lesavre PH, Muller-Eberhard HJ. Mechanism of action of factor D of the alternative complementpathway. J Exp Med 1978:148:505-9.
12. Mancini G, Carbonara AD, Heremans JF. Single radial diffusion method for the immunologicalquantification of proteins. Peeters H (ed). Int Prot Biol Fluids. 11th Collogu Bruges. Oxford:Pergamon;1964;p.370-3.
13. Villaescusa R, Guerreiro AM, Merlín JC, Arce AA, García M, Hernández P. Hipocomplementemiafamiliar en la leucemia linfoide crónica. Rev Cubana Hematol Inmunol Hemoter 1998;14(2):101-6.
14. Yuille MR, Matutes E, Morossy A, Hildilth B, Catovsky D, Houlston RS. Familial chronic lymphocyticleukemia a survey and review of published studies. Br J Haematol 2000;109(4):794-9.
15. Lugassy G, Schlesinger M. The complement system in chronic lymphocytic leukemia: a possible rolein autoimmune manifestations. Leuk Lymphoma 1996;21(5-6):501-3.
Recibido: 20 de diciembre del 2001. Aprobado: 18 de enero del 2002.Lic. Rinaldo Villaescusa Blanco. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800,Ciudad de La Habana, Cuba. Teléf. (537) 578268, 544214. Fax(537)442334. e-mail:[email protected]

58
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):60-2
Instituto de Hematología e Inmunología
ACTIVIDAD ANTICOMPLEMENTARIA EN PREPARACIONESDE INMUNOGLOBULINAS PARA USO ENDOVENOSO
Lic. Ada A. Arce Hernández,1 Lic. Rinaldo Villaescusa Blanco,1 Lic. Julio C. MerlínLinares,1 Lic. Ana M. Guerreiro Hernández,1 Dr. Ed J. Nieuwahuys2 y Dr. Andrew J.Hannema2
RESUMEN
Se determinó la actividad anticomplementaria en muestras tratadas acidificandola solución ligeramente hasta pH 4,0, y sin tratar, de 10 lotes de preparados deinmunoglobulinas para uso endovenoso. Se demostró un aumento de la actividadanticomplementaria en 7/10 de las muestras sin tratar, lo que indica la presenciade agregados de inmunoglobulinas capaces de activar el sistema complemento.En las muestras acidificadas los niveles de actividad anticomplementaria semantuvieron dentro del rango permisible. Los resultados obtenidos indican laimportancia de la medición de la actividad anticomplementaria en los preparadosde inmunoglobulinas endovenosas para establecer la posible presencia de agregadoscuya administración puede causar serias reacciones sistémicas en el paciente.
DeCS: COMPOSICION DE MEDICAMENTOS/métodos; INMUNOGLO-BULINAS/farmacología; INMUNOGLOBULINAS/aislamiento & purificación;COMPLEMENTO/inmunología; IGG/análisis.
1 Instituto de Hematología e Inmunología. Cuba.2 Central Laboratory of Blood Transfusion. Holanda.
Es conocido que la IgG aisladapresenta marcadas propiedades anticomple-mentarias.1 Se ha observado que loscomponentes anticomplementarios en laspreparaciones de inmunoglobulinas sonagregados que se forman en el proceso deaislamiento o espontáneamente durante suconservación posterior, y limitan lasaplicaciones clínicas de éstas.2,3 Diversostratamientos se han empleado para prevenir
o controlar la formación de agregados enlas preparaciones de inmunoglobulinas,entre los que se destaca la disminución delpH, proteólisis y modificaciones químicas.4-6
Nuestro trabajo tiene el objetivo demedir la actividad anticomplementaria enmuestras de lotes de inmunoglobulinas parauso endovenoso, tratadas medianteacidificación ligera y sin tratar, con el fin deestablecer la presencia o no de agregados,capaces de activar el sistema complemento.

59
MÉTODOS
Se determinó la actividad anticom-plementaria en muestras tratadasacidificando la solución hasta 4,0 y muestrassin tratar, de 10 lotes de inmunoglobulinaspara uso endovenoso (CLB, Amsterdam).
La actividad anticomplementaria sedeterminó mediante una modificación delmétodo de Kabat y Mayer.7 Se mezclaron0,2 mL(10 mg de inmunoglobulina) de lapreparación de inmunoglobulina endo-venosa con 0,5 mL de suero de curiel quecontenía 20 unidades CH50 de actividadde complemento. Se incubó a 37 °C durante60 minutos. A continuación se añadieron0,5 mL de una suspensión de 5 × 108
eritrocitos de carnero × mL sensibili-zadoscon un antisuero contra eritrocitos decarnero preparado en conejos (hemolisina)y 1,5 mL de buffer veronal-salino, pH 7,4(BVS). Se incubó a 37 ° C durante 60minutos y se centrifugó 10 minutos a 1 500rpm. La actividad del complemento en lasmuestras se expresa en unidades CH50. Seincluye un control de complemento al quese añaden 0,2 mL de BVS en lugar de lapreparación de inmunoglobulinaendovenosa. La actividadanticomplementaria se calculó de la formasiguiente:
CH50 en el control-CH50 en la muestra × 100 CH50 en el control
RESULTADOS
Se demostró la presencia de agregadosanticomplementarios en 7/10 (70 %) de lasmuestras sin tratar, y se observó unadisminución de la actividad anticomplemen-taria en las muestras tratadas a pH 4,0(tabla ).
TABLA. Actividad anticomplementaria obtenida en lasmuestras tratadas a pH 4.0 y sin tratar de los lotes deinmunoglobulinas endovenosas
Inmunoglobulinas Actividadendovenosas anticomplementaria (%)(No.lote) Muestra Muestra sin tratar tratada
IEV 0202 32,52 12,34IEV 9427 11,07 8,73IEV 2005 14,94 10,12IEV 9505 24,05 9,92IEV 9508 39,76 11,55IEV 9009 24,06 13,06IEV 8502 16,82 7,09IEV 9506 51,07 16,41IEV 9511 42,54 13,02IEV 8906 25,95 10,28
Se considera un valor permisible hasta el 20 % de actividadanticomplementaria.
DISCUSIÓN
Las inmunoglobulinas endovenosasson productos biológicos y difieren muchocon respecto a los productos químicosfarmacéuticos. Es imprescindible garantizarun producto seguro y eficaz. La eficienciase logra si las inmunoglobulinas sonaisladas en su forma nativa y con unadistribución natural. La presencia deagregados activadores del complemento enlos preparados de inmunoglobulinas de usoendovenoso pueden causar severasreacciones colaterales, de ahí la importanciade la determinación en los mismos de laactividad anticomplementaria. En nuestrotrabajo se detectó un aumento de laactividad anticomplementaria en 7 de lasmuestras sin tratar de los 10 lotes deinmunoglobulinas endovenosas estudia-dos, lo que indica la elevada frecuencia deformación de agregados que ocurre duranteel proceso de elaboración del producto.
Existen diversos métodos que permitenobtener preparaciones de inmunoglobulinasendovenosas estables y tolerables, comoes el tratamiento con pepsina y lamodificación química de la molécula de

60
inmunoglobulina mediante agentesreductores o alquilantes, que eliminan, enambos casos, la posibilidad de activacióndel complemento,5 la precipitación selectivade agregados de inmunoglobulinas, conpolietilenglicol,1 cromatografía deadsorción6 o acidificando el preparado deinmunoglobulinas.4 En este último caso, seplantea que disminuyendo ligeramente elpH de la preparación a 4,0, se logran destruirlos agregados anticomplementarios sinafectar la estructura y función de losmonómeros de inmunoglobulinas. En losresultados obtenidos en nuestro estudio,
se observó una disminución de la actividadanticomplementaria hasta valorespermisibles en las muestras portadoras deagregados, al ser acidificadas a pH 4,0, loque indica la destrucción de estos.
Estos datos indican la importancia dela medición de la actividad anticomple-mentaria en los preparados deinmunoglobulinas endovenosas, tanto enel proceso de aislamiento como durante elalmacenamiento, para establecer la posiblepresencia de agregados cuya administra-ción puede causar serias reaccionessistémicas en el paciente.
SUMMARY
The anti-complement activity was determined in treated samples by slightly acidifyingthe solution up to a ph=4.0 and in untreated samples of 10 batches of immunoglobulinpreparations for intravenous use. A rise in the anti-complement activity of 7 out of10 untreated samples was shown, which indicated the presence of immunoglobulinaggregates capable of activating the complement system. The levels of anti-complement activity in the acidified samples remained within the allowable ranges.The results revealed the importance of measuring the anti-complement activity inintravenous immunoglobulin preparations so as to determine the possible presence ofaggregates that can cause serious systemic reactions if they are administered to apatient.
Subject headings: DRUG COMPOUNDING/methods; IMMUNOGLOBULINS/pharmacology; IMMUNOGLOBULINS/isolation & purification; COMPLEMENT/immunology; IGG/analysis.
REFERENCIAS BIBLIOGRÁFICAS
1. Miekka SI, Gozze I. Anticomplementary activity of human IgG.I. Mechanism of the artifactualincrease in anticomplementary activity of IgG during the assay. Vox Sang 1975;29(101):101-23.
2. Barandun S, Kistler P, Jeunet F, Isliker H. Intravenous administration of human gammaglobulin. VoxSang 1962;7:157-60.
3. Morell A, Schurch B, Ryser D, Hofer F, Skvaril F, Barandun S. In vivo behaviour of gamma globulinpreparations. Vox Sang 1980;38:272-5.
4. Kistler P, Stelger G. Human immunoglobulins for intravenous administration: preparation and properties.Prog Immunobiol Standard 1970;4:92-6.
5. Romer J, Morgenthaler JJ, Scherz R, Skvaril F. Characterization of various immunoglobulins preparationsfor intravenous use. I. Protein composition and antibody content. Vox Sang 1982;42:62-70.
6. Gronski P, Kanzy EJ, Ronneberger HJ, Geursen R, Seller FR. Quality criteria for i.v. immunoglobulins:importance of tests and product properties. Behring Inst Mitt 1986;80:16-20.
7. Mayer MM. Complement and complement fixation. En: Kabat EA, Mayer MM (eds). ExperimentalImmunochemistry. Illinois; Thomas Publisher,1964.p.211-25.
Recibido: 7 de noviembre del 2001. Aprobado: 20 de noviembre del 2001.Lic. Ada A. Arce Hernández. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800, Ciudadde La Habana, Cuba. e-mail: [email protected]

61
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):63-6
Técnicas
Instituto de Hematología e Inmunología
COMPARACIÓN DEL ULTRAMICROMÉTODOINMUNOCITOQUÍMICO (UMICIQ) CON EL DE LA FOSFATASAALCALINA-ANTI FOSFATASA ALCALINA (APAAP)PARA LA CUANTIFICACIÓN DE SUBPOBLACIONESLINFOCITARIAS T
Lic. Beatriz Socarrás Ferrer, Dra. Vianed Marsán Suárez, Dra. Miriam Sánchez Segura,Lic. Anissa Gramatges Ortiz, Lic. Rinaldo Villaescusa Blanco y Dra. Consuelo MacíasAbraham
RESUMEN
Se realizó un estudio comparativo entre el método inmunoenzimático que empleael complejo fofatasa alcalina-anti fofatasa alcalina (APAAP) y elultramicrométodo inmunocitoquímico (UMICIQ) utilizado para la detección demarcadores antigénicos y cuantificación de subpoblaciones de linfocitos T. Seestudiaron los antígenos celulares CD3, CD4 y CD8 en 30 individuos adultossupuestamente sanos. Al compararse los resultados por ambas técnicas, seencontraron diferencias estadísticamente significativas para una p < 0,05. Seconcluyó que aún cuando la eficacia diagnóstica de ambos métodos es similar, elAPAAP resultó más rápido, económico y menos laborioso que el UMICIQ, porlo que se recomienda como el procedimiento de preferencia para estos estudioscelulares.
DeCS: FOSFATASA ALCALINA; INMUNOHISTOQUIMICA/métodos;ANTIGENOS CD3/ análisis; ANTIGENOS CD4/análisis; ANTIGENOS CD8/análisis; SUBGRUPOS DE LINFOCITOS T/química; SUBGRUPOS DELINFOCITOS/inmunología; SUBGRUPOS DE LINFOCITOS/enzimología.
La integridad de la respuesta inmunecelular es importante en la defensa delorganismo frente a las infecciones, y dentrode estas respuestas, desempeñan un papelcentral los linfocitos humanos, que sepueden clasificar en dependencia de
determinados marcadores presentes en susuperficie celular.1-6
Tanto el diagnóstico de las hemopatíasmalignas como la determinación desubpoblaciones linfocitarias en pacientescon otras enfermedades, se puede realizar

62
por inmunofluorescencia directa eindirecta7,8 y por métodos inmunocito-químicos que utilizan anticuerposconjugados con enzimas como la peroxidasay la fofatasa alcalina. En este trabajo serealizó una comparación entre elultramicrométodo inmunocitoquímico(UMICIQ) previamente introducido ennuestro laboratorio, y el método de lafosfatasa alcalina-antifosfatasa alcalina(APAAP) para la cuantificación desubpoblaciones linfocitarias T en sangreperiférica humana.
MÉTODOS
Se tomaron muestras de sangreperiférica de 30 individuos supuestamentesanos, 16 hombres y 14 mujeres, queacudieron al Departamento de Hemoterapiadel Instituto de Hematología e Inmunología.El rango de edad osciló entre 23 y 46 años.
El estudio se realizó tanto por elUMICIQ como por la técnica APAAP.9-14
Los antígenos celulares fueron CD3, CD4 yCD8, determinados con anticuerposmonoclonales donados gentilmente por elHospital Clínico de Barcelona y el Institutode Inmunología Filatov de Rusia.
UMICIQ
Se extrajeron muestras de sangreperiférica heparinizada y se realizó elaislamiento de la células mononuclearessegún el método de Boyüm modificado.15
La concentración celular se ajustó a4x106 células/mL y se determinó la viabilidad(más del 95 %) según el método de exclusióncon tripán azul.
Se emplearon las células nodeshidratadas adheridas electrostática-
mente a las láminas porta objetos de cristalque contenían 21 pocillos tratados con poli-L- lisina fijados con vapores de for-maldehído.
Para detectar la reacción antígenoanticuerpo se emplearon anticuerposconjugados con peroxidasa, el peróxido dehidrógeno como sustrato y un agentecromógeno.9-12
APAAP
Se realizaron estudios de sangreproveniente de punción digital, en láminaporta objetos de cristal. El área del frotis aestudiar se delimitó con lápiz con punta dediamante. La láminas se fijaron con acetonapura. Posteriormente se distribuyó sobrecada lámina una fracción purificada deinmunoglobulina del antisuero de conejoanti-ratón (Linking) y luego se les añadió elcomplejo APAAP, que consistía encomplejos solubles de fosfatasa alcalina-anti fosfatasa (monoclonal de ratón).Además se utilizó como sustrato el naftol-AX-MX fosfato y un agente cromógeno.13,14
La lectura de ambos métodos se realizóen un microscopio óptico DIALUX 20EB.Se contaron al menos 100 células en cadalámina.
Se determinó la media aritmética y ladesviación estándar para los diferentesgrupos estudiados. Para la comparación seempleó la t de Student para muestraspareadas, para una p ≤ 0,05.16
RESULTADOS
No se encontraron diferenciasestadísticamente significativas entre ambosmétodos para ninguno de los marcadoresantigénicos estudiados (tabla).

63
El tiempo de procesamiento de lasmuestras por el método APAAP fue de 4 h,mientras que por el UMICIQ fue de 72 h.
DISCUSIÓN
Los métodos inmunocitoquímicos(APAAP y UMICIQ) se aplican con éxitoen el estudio de las inmunodeficiencias yen la caracterización inmunológica de lascélulas malignas en pacientes conleucemias, linfomas y otras enfermedadeshematológicas. 2,3,6,12,15 Estos métodospresentan algunas ventajas sobre los deinmunofluorescencia, como la posibilidadde visualizar las células con un microscopioóptico y además observar simultáneamentetanto la morfología celular como la reaccióninmunológica de sus antígenos con losanticuerpos específicos.
La técnica APAAP permite el inmuno-fenotipaje celular con las siguientes
TABLA. Resultados obtenidos en la comparación del métodoinmunoenzimático de la fosfatasa alcalina-anti fosfatasaalcalina (APAAP) con el ultramicrométodoinmunocitoquímico (UMICIQ).
Variables UMICIQ APAAP(%) n= 30 n = 30 X ± DE X ± DE P
CD3 61,60 ± 5,36 61,53 ± 4,58 NSCD4 44,13 ± 3,13 42,73 ± 3,43 NSCD8 24,13 ± 6,43 24,13 ± 5,69 NS
X: media aritmética; DE: desviación estándar; NS: nosignificativo, p ≤ 0,05; UMICIQ: ultramicrométodoinmunocitoquímico; APAAP: método fosfatasa alcalina-antifosfatasa alcalina.
ventajas: se realiza de extendidos de sangreperiférica o medular, por lo que no esnecesario el aislamiento de las célulasmononucleares y el uso del Ficoll, mientrasque en el UMICIQ las células mononu-cleares se obtienen a partir de un gradientede Ficoll y se adhieren por atracciónelectrostática de cargas, a láminasportaobjetos recubiertos con poli-L-lisina,y esto provoca un mayor tiempo deejecución y costo del sistema dediagnóstico.
El APAAP es más rápido, pues losresultados se obtienen a las 4 h de extraídaslas muestras, y el UMICIQ es un métodomás largo y laborioso. Por último, en elmétodo APAAP se pueden conservar lasláminas sin procesar, durante largosperíodos de tiempo, lo que permite estudiosretrospectivos; esto no es posible con elUMICIQ. Tiene la desventaja de quenecesita una lámina para cada anticuerpomonoclonal, mientras que con el UMICIQse pueden realizar 21 determinaciones encada lámina portaobjeto.
Por los resultados alcanzados,podemos concluir que el método APAAPes un procedimiento más rápido, menoslaborioso y tan eficaz como el UMICIQ,aplicable con confiabilidad para laclasificación inmunológica de lossíndromes linfo y mieloproliferativos, en laevaluación de poblaciones y subpo-blaciones celulares en nuestro laboratorio,por lo que pudiera hacerse extensivo a otroscentros del país.
SUMMARY
A comparative study was conducted on the immunoenzymatic method based onthe alkaline phosphatase – anti-alkaline phosphatase complex and theimmunocytochemical ultramicromethod used for detecting antigen markersand for the quantification of T-lymphocyte subsets Cell antigens CD3, CD4 and

64
CD8 from 30 apparently healthy adults were studied. When comparing theoutcome of both techniques, statistically significant differences were found,(p< 0,05). It was concluded that although the diagnostic efficiency of bothmethods are similar, the alkaline phosphatase – anti-alkaline phosphatase methodis quicker, more economical and less laborious than the immunocytochemicalultramicromethod, so it is recommended as a procedure of choice for these cellstudies.
Subject headings: ALKALINE PHOSPHATASE; IMMUNOHISTO-CHEMISTRY/methods; ANTIGENS, CD4/analysis; ANTIGENS, CD3/analysis;ANTIGENS, CD8/analysis; T-LYMPHOCYTE SUBSETS/chemistry; T-LYMPHOCYTE SUBSETS/immunology; T-LYMPHOCYTE SUBSETS/enzyme.
REFERENCIAS BIBLIOGRÁFICAS
1. Sánchez- Madrid F. Linfocitos T: diferenciación, subpoblaciones, activación. En: Larraga V, Fresno M,Enjuanes L. (eds). Inmunología. La Habana: Editorial Científico-Técnica, 1987:129-30 (EdiciónRevolucionaria).
2. Boffill M, Janossy G, Lee CA. Laboratory control values for CD4 and CD8 T lymphocytes. Implicationsfor HIV-1 diagnosis. Clin Exp Immunol 1992;88:242-52.
3. Rivero RA, Aranda RE, Cruz C, Lorigados LC, Hernández P. Subpoblaciones de linfocitos en la anemiadrepanocítica. Rev Cubana Hematol Inmunol Hemoter 1986;2(1):90-92.
4. Knapp W, Dorken B, Rieber P, Schidt RE, Stein H, von Borne AEG.CD Antigens-1989. Am J Pathol1989;135:420-1.
5. Schlossman SF. CD Antigens 1993. Immunol Today 1994;5:98-9. 6. Fournier AM, Sosenko JM. The relationship of total lymphocyte count to CD4 in patients infected
with human immunodeficiency virus. Am J Med Sc 1992;304:79-82. 7. Lorigados LC, Aranda RE, Rivero RA, Palma LE. Estandarización de la técnica de la inmunofluorescencia
indirecta para el estudio de las subpoblaciones linfocitarias con anticuerpos monoclonales. Rev CubanaHematol Inmunol Hemoter 1985;1(2);185-92.
8. Landay A, Gartland GL, Abo T, Cooper MO. Enumeration of human lymphocyte subpopulations byimmunofluorescence: a comparative study using automated flow microfluorometry and fluorescencemicroscopy. J Immunol Methods. 1983;58:337-47.
9. Rivero R , Bello M, Suárez L, Cruz C, Martínez M, Palma L. Introducción de un ultramicrométodoinmunocitoquímico para la cuantificación de subpoblaciones linfocitarias identificadas con anticuerposmonoclonales. Rev Cubana Hematol Inmunol Hemoter 1995;11(1):46-56.
10. Cruz C, Rivero R, Suárez L. Detección mejorada del antígeno CD2 por un ultramicrométodoinmunocitoquímico en células T no-deshidratadas unidas a láminas recubiertas con poli-L-lisina yfijadas con vapores de formaldehído. Rev Cubana Hematol Inmunol Hemoter 1995;11(1):71-72.
11. Cruz C, Pérez L, Rivero R, González C, Pérez J. Ultramicrométodo inmunocitoquímico (UMICIQ): suaplicación para cuantificar linfocitos T CD4 + en portadores del VIH y en pacientes con SIDA. RevCubana Hematol Inmunol Hemoter 1996;12(1):41-46.
12. Suárez L, Cruz C, Rivero R. Ultramicrométodo inmunocitoquímico. Titulación de anticuerpos utilizadospara el inmunofenotipaje celular. Rev Cubana Hematol Inmunol Hemoter 1995; 11 (1):57-62.
13. Cordell F, Falini B, Erber WN. Immunoenzymatic labelling of monoclonal antibodies using immunecomplex of alkaline phosphatase and monoclonal anti-alkaline phosphatase. J Histochem1984;3 2:219-29.
14. Houmand A, Abrahamsen B, Tinggaard Perdensen N. Revelance of Ki-67 expressión in the classificationof non-Hodking´s lymphomas: A morphometric and double-immunostaining study. Histopathology1992;20:13-20.
15. Boyüm A. Isolation of mononuclear cells and granulocytes from human blood. Scand J Clin Lab Invest1968;21 (suppl 97):77-89.
16. Freund JE. Estadística elemental moderna. 2da. ed. Londres: Universidad de Cambridge, 1960:85-89.
Recibido: 14 de noviembre del 2001. Aprobado: 24 de noviembre del 2001.Lic: Beatriz B. Socarrás Ferrer. Instituto de Hematología e Inmunología. Apartado 8070, CP 10800,Ciudad de La Habana, Cuba. e-mail:[email protected]

65
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):67-9
Comunicaciones breves
Instituto de Hematología e Inmunología
DETERMINACIÓN DE ANTICUERPOS ANTINUCLEARESEN LA ANEMIA HEMOLÍTICA AUTOINMUNE Y LA PÚRPURATROMBOCITOPÉNICA AUTOINMUNE
Lic. Ana M. Guerreiro Hernández, Lic. Rinaldo Villaescusa Blanco, Lic. Antonio BencomoHernández, Lic. Luz M. Morera Barrios, Lic. Julio C. Merlín Linares, Lic. Ada A. ArceHernández y Lic. Carlos García Guevara
RESUMEN
Se detectó la presencia de anticuerpos antinucleares mediante un método deinmunofluorescencia indirecta en uno de los 18 pacientes estudiados con anemiahemolítica autoinmune y en 3 de los 16 pacientes con púrpura trombocitopénicaautoinmune. Los resultados establecen la importancia de la detección de losanticuerpos antinucleares como elemento indicador de la existencia o desarrollode otra enfermedad autoinmune asociada.
DeCS: ANTICUERPOS ANTINUCLEARES/uso diagnóstico; ANEMIAHEMOLITICA AUTOINMUNE/inmunología; ANEMIA HEMOLITICAAUTOINMUNE/ diagnóstico; PURPURA TROMBOCITOPENICAIDIOPATICA/inmunología; PURPURA TROMBOCITOPENICA IDIOPATICA/diagnóstico; TECNICA DEL ANTICUERPO FLUORESCENTE INDIRECTA.
La anemia hemolítica autoinmune(AHAI) y la púrpura trombocitopénicaautoinmune (PTA) se caracterizan por lapresencia de anticuerpos dirigidos contraantígenos propios de los eritrocitos y lasplaquetas, respectivamente, y producenuna disminución de la sobrevida de estascélulas sanguíneas.1,2
La AHAI por anticuerpos calientesestá asociada con trastornos en lainmunorregulación; se ha observado comoentidad clínica secundaria en el 10 % de los
pacientes con lupus eritematoso sistémico(LES) y en el 15 % de las leucemiaslinfocíticas crónicas (LLC).3-5 En la PTA seha demostrado una frecuente asociacióncon el LES6 y ocasionalmente con otrosdesórdenes autoinmunes como la artritisreumatoidea, esclerodermia y la enfermedadmixta del tejido conectivo, así como condesórdenes linfoproliferativos.7
Se ha establecido la importancia de ladetección de los anticuerpos antinucleares(ANA) en el diagnóstico de diversas

66
enfermedades autoinmunes multisistémi-cas.8 En nuestro trabajo se detectó lapresencia de ANA, por un método deinmunofluorescencia indirecta,9 en unpaciente de los 18 estudiados con AHAI,en el se observó un patrón granular, y en 3de los 16 pacientes con PTA, 1 con patróngranular y 2 con homogéneo, provenientesdel Servicio de Inmunohematología delInstituto de Hematología e Inmunología.
La presencia de ANA en los 4 pacientesseñalados pudiera indicar la posibilidad deotra enfermedad autoinmune en desarrollo,o que la AHAI y la PTA diagnosticadas en
estos casos sean un trastorno secundarioa otra enfermedad subyacente de carácterautoinmune o linfoproliferativa. Lospatrones de ANA obtenidos, granular yhomogéneo, sugieren la posibilidad de unLES u otra enfermedad de tejido conectivoasociada.10
Los datos obtenidos indican laimportancia de incluir la detección de ANAen aquellas enfermedades autoinmunesrelacionadas con las células sanguíneas, yaque su presencia resulta un elementoindicador de la existencia o desarrollo deotra enfermedad autoinmune asociada.
SUMMARY
The presence of antinuclear antibodies was detected by an indirectimmunofluorescence method in one of the 18 studied patients with autoimmunehemolytic anemia and in 3 of the 16 patients with autoimmunethrombocytopenic purpura. The results showed the importance of the detectionof antinuclear antibodies as an indicating element of the existence or developmentof another associated autoimmune disease.
Subject headings: ANTIBODIES, ANTINUCLEAR/diagnostic use; ANEMIA,HEMOLYTIC AUTOIMMUNE/immunology, ANEMIA, HEMOLYTICAUTOIMMUNE/diagnosis; PURPURA, THROMBOCYTOPENICIDIOPATHIC/diagnosis; PURPURA, THROMBOCYTOPENIC IDIOPATHIC/immunology; INDIRECT FLUORESCENT ANTIBODY TECHNIQUE.
REFERENCIAS BIBLIOGRÁFICAS
1. Well JV. Enfermedades hematológicas. En: Stites DP, Abba I (eds). Inmunología Básica y Clínica. 7maed. México: Editorial El Manual Moderno, 1993:557-76.
2. MC Millan R. The pathogenesis of chronic immunethrombocytopenic purpura. Semin Hematol2000;37:5-9.
3. Williams RC, Kunkel HG, Capra JD. Antigenic specifications related to cold agglutinin activity ofgamma M globulins. Science 1968; 161:379-81.
4. Winfield JB, Mimura T, Fernsted PD. Antilymphocyte autoantibodies. En: Wallace DJ, Hahn BH (eds).Lupoid lupus erythematosus. Philadelphia: Lea and Febiger, 1993:254-59.
5. Bergasel DE. The chronic leukemias. A review of disease manifestations and the aims therapy. CanMed Assoc J 1967;96(25):1615-20.
6. Dubois EL. Lupus erythematosus: a review of the current status of discoid and systemic erythematosusand their variants. 2da. ed. California: Univ Southern Press, 1974:205-15.
7. Rose R, Mac RI. The autoimmune diseases. 3ra. ed. Illinois: Thomas Publisher, 1998:823-31. 8. Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell
biology. Adv Immunol 1989;44:93-151.

67
9. Fritzler MJ. Antinuclear antibodies in the investigation of rheumatic disease. Bull Rheum Dis1985;35(6):1-3.
10. Kenneth HF, Kenneth ES. Enfermedades reumáticas. En: Stites DP, Abba I (eds). Inmunología Básicay Clínica. 7ma. ed. México: Editorial El Manual Moderno, 1993:513-19.
Recibido: 15 de noviembre del 2001. Aprobado: 21 de noviembre del 2001Lic. Ana M. Guerreiro Hernández. Instituto de Hematología e Inmunología. Apartado 8070. CP 10800,Ciudad de La Habana, Cuba. e-mail:[email protected]

68
Rev Cubana Hematol Inmunol Hemoter 2002;18(1):70-1
Cartas al Director
CAPACIDADES FUNCIONALES EN ALUMNOS ATLETASPORTADORES DE HEMOGLOBINA S
DeCS: HEMOGLOBINA FALCIFORME/análisis; CAPACIDAD RESIDUAL FUNCIONAL;DEPORTES.
Al director:
Al estado de portador de hemoglobina S se le considera una condición biológica, quesólo en circunstancias especiales como muy baja tensión de oxígeno, acidosis ydeshidratación, tiene efectos adversos sobre la salud.1 Algunos autores asocian este estadocon un menor rendimiento deportivo, aunque las investigaciones realizadas sobre este aspectoofrecen resultados contradictorios.2,3
Se realizó una investigación con el objetivo de conocer las capacidades funcionales delos portadores de hemoglobina S, entre los alumnos matriculados en la Escuela de IniciaciónDeportiva de Sancti Spíritus, en el curso escolar 1999-2000.
A todos los alumnos se les registró la edad, el sexo, el deporte que practicaban y el colorde la piel. Además, se les realizó electroforesis de hemoglobina sobre acetato de celulosa yla prueba de solubilidad para la hemoglobina S.1
Con los resultados obtenidos se formaron 2 grupos de alumnos: un grupo compuestopor los 35 portadores de hemoglobina S identificados, y otro por 35 alumnos con hemoglobinaAA (grupo control). Para formar el grupo control se eligió al azar un alumno con hemoglobinaAA para cada uno, portador de hemoglobina S. La elección se realizó entre aquellos alumnosque coincidieron en el deporte que practicaban, edad, sexo y color de la piel.
A cada alumno de ambos grupos se le registraron los resultados obtenidos en lassiguientes pruebas: carrera de 60 metros planos para el sexo masculino y de 40 para elfemenino; carrera de 400 a 1 000 metros planos, según la edad y sexo del alumno; salto largoy salto alto sin impulso. Estas pruebas se realizaron según estaba previsto en el programadocente, y sus resultados se evaluaron en conjunto para cada alumno en bien, regular y mal,según la metodología vigente en la escuela (Plan de eficiencia física L.P.V. 2000. NormativasGenerales. Ciudad de La Habana: INDER,2000). Los resultados obtenidos se compararonmediante la prueba Chi cuadrado.
En la tabla se observa que los portadores de hemoglobina S obtuvieron menosevaluaciones de bien, con un total de 12 (34,3 %), en relación con los integrantes del grupocontrol (23, para el 65,7 %), para una X2/6,9 (p < 0,01).
69
TABLA. Resultados en las pruebas de eficiencia física, según el fenotipo de la hemoglobina de los alumnos-atletas. EIDE, SanctiSpíritus, 2000
Fenotipo Totalde la de Resultado en las pruebashemoglobina alumnos Bien Regular Mal No. % No. % No. %
AS 35 12* 34,3 15 42,9 8 22,9AA 35 23* 65,7 7 20,0 5 14,3
Total 70 35 50,0 22 31,4 13 18,6
* p >0,01.
Además, obtuvieron más evaluaciones de mal (8, para el 22,9 %), que los miembros delgrupo control (5, para el 14,2 %).
Los peores resultados obtenidos por los portadores de hemoglobina S en las pruebasrealizadas, se corresponde con el criterio de los autores que consideran que los atletasportadores de hemoglobina S, presentan resultados deportivos inferiores al de los atletascon hemoglobina AA.3 Estas diferencias pudieran estar relacionadas con las diferenciasobservadas entre los sistemas metabólicos energéticos de ambos grupos poblacionales.5
Sin embargo, es conveniente confirmar estos resultados en un número mayor de individuos,pues las diferencias observadas pudieran estar influenciadas por el pequeño tamaño de lasmuestras.
REFERENCIAS BIBLIOGRÁFICAS
1. Colombo B, Svarch E, Martínez G. Genética y clínica de las hemoglobinas humanas. La Habana: Ed.Pueblo y Educación;1993.
2. Bitanga E, Roullon JD. Influence of the sickle cell trait heterozygote on energy abilities. Pathol BiolParis 1998:46-52.
3. Bile A, Le Gallais D, Mercier J, Bogui P, Prefaut C. Sickle cell trait in Ivory Coast athetic throw andchampions, 1956-1995. Int J Sports Med 1998;19:215-19.
4. Bile A, Le Gallais D, Mercier B, Martínez P, Ahmaidi S, Prefaut C. Blood lactate concentrations duringincremental exercise in subjets with sickle cell trait. Med Sci Sports Exerc 1998;30:649-54.
Dr. Juan J. González PérezDra. Lay Hernández GonzálezLic. Ángel Aquino Pernas
Recibido: 21 de junio del 2001. Aprobado: 5 de septiembre del 2001.Dr. Juan J. González Pérez. Centro de Genética Médica. Frank País No. 352, entre B. Masó y B. Reeve,Sancti Spiritus.





















![Ch06 HIH [Street07]](https://img.pdfslide.net/doc/110x75/577cdf341a28ab9e78b0abb0/ch06-hih-street07.jpg)







