Embed Size (px)
Citation preview

Vol. 14, No. 1MOLECULAR AND CELLULAR BIOLOGY, Jan. 1994, p. 427-4360270-7306/94/$04.00+0Copyright X 1994, American Society for Microbiology
Target Cell Death Triggered by Cytotoxic T Lymphocytes: aTarget Cell Mutant Distinguishes Passive Pore Formation
and Active Cell Suicide MechanismsDAVID S. UCKER,* JON D. WILSON, AND LEILA D. HEBSHI
Division ofImmunology, Medical Biology Institute, La Jolla, California 92037
Received 24 August 1993/Accepted 11 October 1993
The role of the target cell in its own death mediated by cytotoxic T lymphocytes (CTL) has beencontroversial. The ability of the pore-forming granule components of CTL to induce target cell death directlyhas been taken to suggest an essentially passive role for the target. This view of CTL-mediated killing ascribesto the target the single role of providing an antigenic stimulus to the CTL; this signal results in the vectoraldegranulation and secretion of pore-forming elements onto the target. On the other hand, by a number ofcriteria, target cell death triggered by CTL appears fundamentally different from death resulting frommembrane damage and osmotic lysis. CTL-triggered target cell death involves primary internal lesions of thetarget cell that reflect a physiological cell death process. Orderly nuclear disintegration, including laminphosphorylation and solubilization, chromatin condensation, and genome digestion, are among the earliestevents, preceding the loss of plasma membrane integrity. We have tested directly the involvement of the targetcell in its own death by examining whether we could isolate mutants of target cells that have retained the abilityto be recognized by and provide an antigenic stimulus to CTL while having lost the capacity to respond bydying. Here, we describe one such mutant, BW87. We have used this CTL-resistant mutant to analyze themechanisms of CTL-triggered target cell death under a variety of conditions. The identification of a mutabletarget cell element essential for the cell death response to CTL provides genetic evidence that target cell deathreflects an active cell suicide process similar to other physiological cell deaths.
The mechanism by which cytotoxic T lymphocytes (CTL)trigger the death of their target cells has remained contro-versial. Although target cell death is not cell autonomousand does not require target cell macromolecular synthesis(48, 59; but see references 25 and 73), the metabolic activityof the targeted cell appears to be required (30), and in manymorphological and biochemical respects, the cell death pro-cess is indistinguishable from cell autonomous physiologicalcell deaths (64). Indeed, the concept of internal disintegra-tion, with prelytic nuclear events, including genome diges-tion, preceding the loss of plasma membrane integrity,comes from studies of CTL-induced target cell death (48).We recently described intracellular structural changes, in-cluding mitotic-like phosphorylation and solubilization ofnuclear lamins, as among the earliest events detectable in theCTL-triggered death of target cells (67).The nearly ubiquitous presence in effector cells of pore-
forming cytolytic components sequestered in secretory gran-ules (5, 6, 12, 27, 28, 33) has been taken to suggest that targetcells die when they trigger the exocytosis of these lyticgranules from CTL (18). The granule exocytosis model ofCTL action (54) proposes that the target, in providing anantigenic stimulus to the CTL, triggers a degranulationresponse from the CTL, resulting in the vectoral depositionof pore-forming elements onto the target. Perforin, thepore-forming protein of effector cell granules (also termedcytolysin), can kill nucleated target cells in a calcium-dependent manner, but the cell lysis that results is not aphysiological cell death (9, 10, 14, 67).
* Corresponding author. Present address (as of 1 January 1994):Department of Microbiology and Immunology, E829, University ofIllinois College of Medicine, 835 South Wolcott M/C 790, Chicago,IL 60612. Phone: (312) 996-6648. Fax: (312) 996-6415.
Together with perforin, cytolytic granules contain a vari-ety of molecules that may be involved in cell death, includinga family of serine proteases (for example, granzyme A andgranzyme B/fragmentin 2 [31, 36, 42, 52, 53]) and an RNA-binding protein (TIA-1 [61]). These granule components,under certain circumstances and in purified form, have beenshown to induce genome digestion or even physiological celldeath (15, 52, 53, 61), giving rise to the proposal that targetcell death actually results from the action of these non-pore-forming components following their transfer from the effec-tor cell to the target cell (via a polyperforin pore).The validity of these models remains unresolved. Genetic
manipulations to date have yielded only equivocal results.Antisense ablation of expression of perforin (1) exertedpartial loss of cytolytic effector function under conditions ofredirected lysis (see below); the effect on antigen-specificCTL activity was not tested. Similarly, ablation of granzymeA expression did not correlate consistently with loss ofcytolytic activity among the antisense transfectant clonesand populations examined (60). The expression of perforinfollowing gene transfer into exocytic but noncytotoxic cellsconferred potent lytic activity against erythrocytes but onlyweak cytolytic activity against nucleated target cells (54).The weak cytolytic activity against nucleated targets gener-ated by the expression of both perforin and granzyme A wasassociated additionally with genome digestion (55); whetherthis DNA breakdown occurred prelytically and representsan authentic physiological cell death has not been deter-mined.A more fundamental reservation regarding the role of
cytolytic granule components in CTL activity concerns thelack of correlation of expression of these presumptive lyticelements with the ability of effector cells to trigger target celldeath. Among CTL populations with roughly comparable
427
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.

428 UCKER ET AL.
effector cell activities, levels of expression of perforin andgranzymes vary widely. While some long-term in vitro CTLclones display substantial levels of perforin and granzymeactivity, primary CTL express low levels of perforin andgranzyme mRNA, and some CTL generated in vivo haveundetectable levels of expression (5, 6, 17, 19). On the otherhand, abundant expression of granule components has beenobserved consistently in natural killer and lymphokine-activated killer cells (19, 45, 52, 53, 56, 73). It may be thatcytolytic granule molecules are more relevant to the actionof these effector populations than to the action of antigen-specific CTL. The lack of correlation between cytolyticgranule activity and CTL-triggered target cell death is evi-denced strikingly with calcium-independent target cells.CTL can trigger the death of these targets equally in thepresence of calcium, associated with typical CTL degranu-lation, and in the absence of calcium, when CTL degranu-lation and granzyme release are undetectable (39, 62).Whether this reflects the absolute absence of degranulationor only the limits of the sensitivity of the assay is not easilyresolved.We have explored the mechanism of CTL-triggered target
cell death by characterizing genetically the involvement ofthe target cell in its own death. Our approach is motivated bythe hypothesis that mutable elements of the target cell whichreveal response elements necessary for the cell death pro-cess induced by CTL can be identified. Recently, we re-ported one target cell mutant that fails to digest its genomeduring cell death, allowing us to identify the enzyme respon-sible for genome digestion as DNase I and to conclude thatgenome digestion is a dispensable consequence of CTL-triggered target cell death (67). Here we describe anothersomatic cell mutant that is recognized by CTL and triggersCTL degranulation but that fails to die in response. We haveused this mutant to probe the mechanisms by which targetcells are triggered to die by CTL under a variety of circum-stances. The behavior of the CTL-resistant mutant demon-strates that normal, antigen-specific CTL-triggered targetcell death is a physiological cell death process, involving theactive participation of the target cell in its own death.
MATERIALS AND METHODS
Cell culture. All cells were grown in RPMI 1640 medium(Whittaker Bioproducts, Walkersville, Md.) supplementedwith glutamine (2 mM), 2-mercaptoethanol (50 ,M), andheat-inactivated fetal bovine serum (10% [vol/vol]; TissueCulture Biologicals, Tulare, Calif.). The cell lines usedincluded the AKR-derived (H-2) thymoma BW5147 (seebelow), the C57BL/6-derived (H-2b) thymoma EL4, and theDBA/2-derived (H-2") mastocytoma P815.CTL effector populations were harvested on day 5 from
primary mixed lymphocyte cultures (MLC): 7 x 106 spleencell responders and 3 x 106 irradiated (3,000 R) allogeneicspleen cell stimulators per 2 ml of culture. CTL clonesNancy (specific for H-2Dk [66]), Jennifer (specific for H-2q[67]), and CTL 3 (a heteroclitic clone specific for H-2Kk [49])were stimulated on a 7-day cycle with appropriate targetsand interleukin-2 (IL-2) (2.5 x 105 CTL and 5 x 106irradiated spleen cells from AKR, DBA/1, and BALB/cmice, respectively, per 2 ml of culture containing 10 U ofIL-2 per ml). CTL clone 36 (CTL 36), specific for the peptidecorresponding to residues 365 to 380 of the A/PR/8/34influenza virus nucleoprotein (NP) sequence presented byH-2Db, was similarly maintained with peptide-pulsedC57BL/6 spleen cells as described previously (21).
Mutant isolation. BW1083, a freshly subcloned glucocor-ticoid-resistant derivative of the AKR-derived (H-2!), thy-midine kinase-negative thymoma BW5147, was treatedwith H-2k_specific CTL (from a C57BL/6 anti-AKR MLC).From two rounds of treatment (coculture of 107 nonmuta-genized target cells with 5 x 107 CTL for 3 days, followed byrecovery and outgrowth of surviving target cells for 1 week),a CTL-resistant population was obtained. The CTL-resistantBW87 clone was derived by plating this population atlimiting dilution. In all of the experiments reported here, weused a subclone of the BW87 mutant, BW87.G7. The dou-bling time of the BW87 mutant (approximately 16 h) isindistinguishable from that of its parent.
Somatic cell fusion. IL-2-independent, hypoxanthine-amin-opterin-thymidine-resistant hybrids from the fusion of theC57BL/6 (H-2b)-derived Mlsa-reactive, IL-2-dependenthelper T-cell clone L2 (13) (kindly provided by Andy Glase-brook) with BW87 or BW1083 were prepared as describedpreviously (35). Hybrids were tested for Mlsa reactivity bythe production IL-2 in response to irradiated CBA/J (Mlsa)spleen cells (106 per microwell) (65). Positive cells weresubcloned at limiting dilution.
Transfection. Cells were transfected by a modification ofthe spheroplast fusion technique of Sandri-Goldin et al. (51).Bacterial spheroplasts were prepared essentially as de-scribed elsewhere (51). T cells (107) were washed once inserum-free medium, collected by centrifugation (5 min at 500x g, 25°C), and resuspended by gentle pipetting in thespheroplast suspension (approximately 2 x 109 spheroplastsin 5 ml). The mixture was transferred to a 60-mm-diameterdish and centrifuged at 1,500 x g for 7 min. After gentleaspiration of the supernatant, 1.5 ml of polyethylene glycolsolution (70% [wt/vol] PEG-4000 [Roth] and 15% [vol/vol]dimethyl sulfoxide in phosphate-buffered saline [PBS]) wasadded to the film of cells and spheroplasts left on the surfaceof the plate. The dish was spun at 750 x g for 60 s. Cells werecollected by gently pipetting prewarmed serum-free mediumacross the plate. Cells were resuspended in growth mediumcontaining kanamycin (100 ,ug/ml) and plated in 96-welldishes at 2 x 104 cells per well. After 24 h, G418-containingmedium (1.2 mg/ml) was added.
Cytotoxicity assays. Cells were labeled with 5'-[125I]iodo-2'-deoxyuridine ([125I]IUdR; 5 Ci/mg; Amersham, ArlingtonHeights, Ill.) and/or sodium [51Cr]chromate (400 to 1,200mCi/mg; New England Nuclear, Boston, Mass.) as de-scribed previously (50). Cells (5 x 106) were incubated in 0.5ml with 20 p,Ci of [12 I]IUdR and/or 100 pCi of [51Cr]chro-mate for 90 min at 37°C, washed, and incubated for another60 min at 37°C without label. For redirected lysis, cells werederivatized with 3 mM trinitrobenzene sulfonate in PBS for15 min at 37°C after labeling as described previously (41).Cells were tested for susceptibility to CTL-triggered celldeath essentially as described previously (67). Reactions(200-,ul volume) were performed in V-bottom 96-well plates,included 104 labeled targets and effector CTL at the indi-cated effector-to-target cell (E:T) ratios, and were incubatedat 37°C for 4 h or as indicated. Plates were centrifuged (5 minat 500 x g, 25°C), and 100 pl of each supernatant wascollected for determination of 51Cr and/or Triton-indepen-dent [1"I]IUdR release. Then 100 pl of 0.4% Triton X-100 inPBS was added to the wells, and another 100 ,ul of eachsupernatant was collected after recentrifugation for determi-nation of Triton-dependent ['251]IUdR release. Specific 51Crrelease was calculated according to the following formula:[(experimental release - spontaneous release) x 100]/(max-imal release - spontaneous release). Spontaneous release
MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.

ACTIVE TARGET CELL INVOLVEMENT IN CTL-TRIGGERED DEATH 429
was generally less than 10%. Maximal release was deter-mined by lysing targets with 0.25% sodium dodecyl sulfate.Specific [1 I]IUdR release was calculated similarly; total[1 5IJIUdR release included both Triton-dependent and Tri-ton-independent release (49).
Lysis by cytolytic granules and ATP-mediated cell deathwere assessed similarly. Serial dilutions of ATP were incu-bated with 51Cr-labeled target cells in medium additionallybuffered as described previously (4). Cytolytic granulespurified from a rat large granular lymphocyte tumor (32)(graciously provided by Pierre Henkart) were diluted seriallyin calcium-free PBS and incubated with the labeled targetcells in complete medium for 1 h.BLT serine esterase release. N-benzyloxycarbonyl-L-ly-
sine thiobenzyl ester (BLT) serine esterase activity in super-natants collected after 3 h of incubation was determined bythe ability to cleave the chromogenic BLT substrate asdescribed previously (67). Briefly, 20 RI of each culturesupernatant was added to 180 RI of reaction buffer [2 x 10-4M BLT ester and 1.1 x 10-4 M 5,5'-dithio-bis-(2-nitroben-zoic acid) in PBS, pH 7.4], and the reaction mixtures wereincubated at 25°C for 60 min. Optical densities were mea-sured at 405 nm in an automated enzyme immunoassay platereader (EL309; Bio-Tek Instruments, Burlington, Vt.).
Cytofluorimetric analyses. Cells were stained for expres-sion of total H-2k class I molecules with M1.42 antibody (ratantibody specific for mouse major histocompatibility com-plex class I [58]) and for specific expression of H-2Db with28-14-8S antibody (mouse antibody specific for H-2Db [40]).Cells (106) were incubated with 100 p.l of culture supernatantof the primary antibody at 4°C for 45 min. The washed cellswere resuspended in 100 ,ul of buffer (Hanks' balanced saltsolution with 0.2% bovine serum albumin) containing 10 p,lof fluorescein isothiocyanate-conjugated secondary antibody(goat anti-mouse or mouse anti-rat immunoglobulin; FisherBiotech, Pittsburgh, Pa.) and incubated at 4°C for another 45min. Cells were analyzed cytofluorimetrically on a FACScananalyzer (Becton Dickinson, San Jose, Calif.).Conjugates of CTL and target cells were analyzed by a
method previously described (26). CTL effectors were la-beled with fluorescein diacetate, mixed with target cells at anE:T ratio of 2 (so as to favor the formation of targetsconjugated with a single effector; 4 x 105 labeled effectorsand 8 x 105 unlabeled targets in a 200-p,l reaction), andbriefly centrifuged (3 min at 500 x g, 25°C). Pelleted cellswere gently resuspended and diluted into 500 ,ul of buffer forcytofluorimetric analysis after approximately 10 min. Targetcells exhibited greater light scatter properties than did effec-tors. Conjugates exhibiting both CTL-like fluorescein label-ing and target cell-like high light scatter properties wereidentified.
Reagents and chemicals. ATP and kanamycin were pur-chased from Sigma Chemical Co. (St. Louis, Mo.). G418(G418 sulfate; Geneticin) was purchased from GIBCO(Grand Island, N.Y.). Concanavalin A (Sigma) was resus-pended and dialyzed in tissue culture medium without serumat 1 mg/ml. The synthetic glucocorticoid dexamethasone(Sigma) was prepared as a stock solution (10-' M) in 95%ethanol. Ionomycin and valinomycin (Calbiochem, La Jolla,Calif.) were resuspended in dimethyl sulfoxide at 1.0 mM.
RESULTS
Recovery of a CTL-resistant mutant. That a defect inDNase I expression in a target cell results in the failure ofthat cell to digest its genome without altering its ability to die
in response to CTL indicates that genome digestion is adispensable target cell function associated with CTL-trig-gered cell death (67). To identify target cell functions that areessential to the CTL-induced cell death process, we haveselected for target cell mutants that are resistant to CTL-triggered death while retaining the ability to be recognizedby CTL. BW87 is one such antigenic and resistant clone.BW87 was isolated from BW1083, a glucocorticoid-resis-
tant subclone of the AKR-derived (H-2k) thymoma BW5147(see Materials and Methods). BW87 was selected for resis-tance to alloantigen-specific CTL. As demonstrated in asimple 51Cr release assay (Fig. 1), BW1083, like its parentBW5147, expresses H-2k determinants (Fig. 2) and is sensi-tive to cell death triggered by H-2k_specific CTL effectors.Independently, activation of CTL specific for an irrelevantH-2 haplotype and agglutination with target cells by thelectin concanavalin A will induce the death of the CTL-sensitive cells (Fig. 1). In contrast, CTL can induce neitherantigen-specific nor lectin-dependent death of the CTL-resistant BW87 mutant (Fig. 1).How much less sensitive to CTL-triggered cell death is
BW87 than its parent? We have quantified relative CTLsusceptibility in terms of the number of effector cells pertarget (Effector dose) required to trigger 50% specific 5Crrelease (ED50). The data in Fig. 1, for primary MLC-derivedCTL effectors, yield an observed ED50 for BW1083 of 11 inthe absence and 2 in the presence of concanavalin A. Incontrast, the calculated ED50 for BW87 with the sameeffectors is greater than 1,000 with or without lectin. Datacompiled from 20 experiments with MLC-derived effectorsand long-term CTL clones indicate that BW87 cells are atleast 400 times less sensitive to CTL-triggered death than areBW1083 cells (Table 1).These 51Cr release experiments are consistent with other
measures of CTL-triggered death. The plating efficiency ofBW1083 cells is reduced to less than 1% of the control valuefollowing treatment with antigen-specific or lectin-dependentCTL (at an E:T ratio of 20); in contrast, BW87 cells retain80% of colony-forming ability following CTL treatment (datanot shown). The release of free fatty acids from target cellsis another indication of CTL-triggered death (46). WhileBW1083 cells release substantial levels of free fatty acidswhen subjected to antigen-specific or lectin-dependent CIL,no free fatty acid release is induced from BW87 cells (22).BW87 resistance is not due to a failure in recognition by
CTL. The failure of BW87 to die in response to CTL is notdue to an inability to be recognized by CTL. BW87 cellscontinue to express H-2k determinants, although at levelsreduced to about 10% of that of the parental cells (based onmean immunofluorescence staining intensities; Fig. 2). Thisreduced H-2k expression is sufficient, nonetheless, to allowBW87 cells to be recognized and form antigen-specificconjugates effectively with H-2k-specific CTL (Fig. 3).
Still, to obviate the possibility that the failure of BW87 todie in response to CTL is due to this quantitative or other,subtle alteration in the expression of endogenous H-2kmolecules, we introduced a distinct H-2 gene into BW87cells and tested whether expression of the new recognitionmolecule conferred sensitivity to CTL-triggered death.BW87 cells were transfected with an H-2D" construct drivenby a Moloney murine leukemia virus long terminal repeatpromoter and bearing a G418 resistance gene (3). Transfec-tant clones, selected for G418 resistance, were screened forfunctional expression of the transfected H-2 determinant byvirtue of the ability to present an H-2Db-restricted viralpeptide antigen to a CIL clone. Presentation was assessed
VOL. 14, 1994
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.
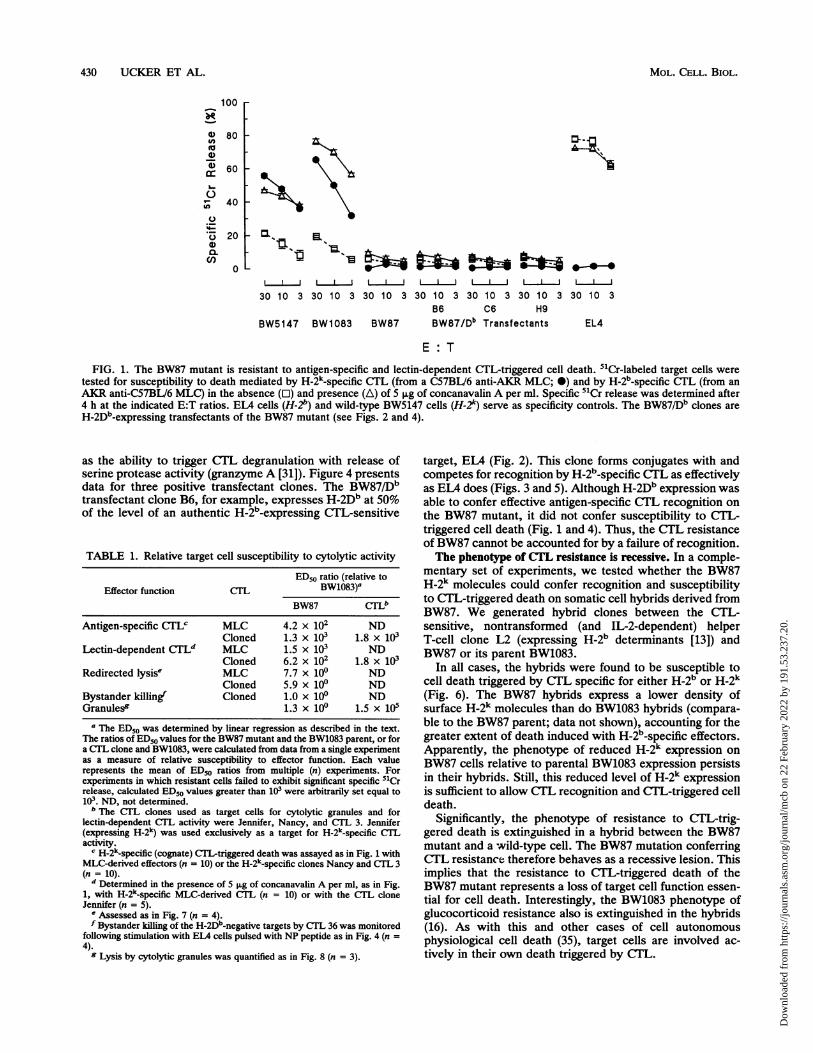
430 UCKER ET AL.
100 rH
(D 800_
a)0) 60
,,, 4040
U Zo 200)cLCO
0
30 10 3 30 10 3 30 10 3 30 10 3 30 10 3 30 10 3 30 10 3B6 C6 H9
BW5147 BW1083 BW87 BW87/Db Transf ectants EL4
E: T
FIG. 1. The BW87 mutant is resistant to antigen-specific and lectin-dependent CTL-triggered cell death. 51Cr-labeled target cells weretested for susceptibility to death mediated by H-2k-specific C7L (from a C57BL/6 anti-AKR MLC; 0) and by H-2b-specific CTL (from anAKR anti-C57BL/6 MLC) in the absence (O) and presence (A) of 5 jig of concanavalin A per ml. Specific 51Cr release was determined after4 h at the indicated E:T ratios. ELA cells (H-2b) and wild-type BW5147 cells (H-2k) serve as specificity controls. The BW87/Db clones areH-2Db_expressing transfectants of the BW87 mutant (see Figs. 2 and 4).
as the ability to trigger CTL degranulation with release ofserine protease activity (granzyme A [31]). Figure 4 presentsdata for three positive transfectant clones. The BW87/Dbtransfectant clone B6, for example, expresses H-2Db at 50%of the level of an authentic H-2b-expressing CTL-sensitive
TABLE 1. Relative target cell susceptibility to cytolytic activity
ED50 ratio (relative toEffector function CTL BW1083)-
BW87 CTLb
Antigen-specific CTLC MLC 4.2 x 102 NDCloned 1.3 x 103 1.8 x 103
Lectin-dependent CTLd MLC 1.5 x 103 NDCloned 6.2 x 102 1.8 x 103
Redirected lysise MLC 7.7 x 10° NDCloned 5.9 x 100 ND
Bystander killingf Cloned 1.0 x 100 NDGranulesO 1.3 x 100 1.5 x 102
a The ED50 was determined by linear regression as described in the text.The ratios of ED50 values for the BW87 mutant and the BW1083 parent, or fora CTL clone and BW1083, were calculated from data from a single experimentas a measure of relative susceptibility to effector function. Each valuerepresents the mean of ED50 ratios from multiple (n) experiments. Forexperiments in which resistant cells failed to exhibit significant specific 51Crrelease, calculated ED50 values greater than 103 were arbitrarily set equal to103. ND, not determined.b The CIL clones used as target cells for cytolytic granules and for
lectin-dependent CTL activity were Jennifer, Nancy, and CTL 3. Jennifer(expressing H-2k) was used exclusively as a target for H-2k-specific CTLactivity.
' H-2k-specific (cognate) CTL-triggered death was assayed as in Fig. 1 withMLC-derived effectors (n = 10) or the H-2k-specific clones Nancy and CTL 3(n = 10).d Determined in the presence of 5 ,g of concanavalin A per ml, as in Fig.
1, with H-2k_specific MLC-derived CTL (n = 10) or with the CTL cloneJennifer (n = 5).
' Assessed as in Fig. 7 (n = 4).f Bystander killing of the H-2Db-negative targets by CTL 36 was monitored
following stimulation with ELA cells pulsed with NP peptide as in Fig. 4 (n =4).g Lysis by cytolytic granules was quantified as in Fig. 8 (n = 3).
target, EIA (Fig. 2). This clone forms conjugates with andcompetes for recognition by H-2b-specific CTL as effectivelyas EL4 does (Figs. 3 and 5). Although H-2Db expression wasable to confer effective antigen-specific CTL recognition onthe BW87 mutant, it did not confer susceptibility to CTL-triggered cell death (Fig. 1 and 4). Thus, the CTL resistanceof BW87 cannot be accounted for by a failure of recognition.The phenotype of CTL resistance is recessive. In a comple-
mentary set of experiments, we tested whether the BW87H-2k molecules could confer recognition and susceptibilityto CTL-triggered death on somatic cell hybrids derived fromBW87. We generated hybrid clones between the CTL-sensitive, nontransformed (and IL-2-dependent) helperT-cell clone L2 (expressing H-2b determinants [13]) andBW87 or its parent BW1083.
In all cases, the hybrids were found to be susceptible tocell death triggered by CIL specific for either H-2b or H-2k(Fig. 6). The BW87 hybrids express a lower density ofsurface H-2k molecules than do BW1083 hybrids (compara-ble to the BW87 parent; data not shown), accounting for thegreater extent of death induced with H-2b-specific effectors.Apparently, the phenotype of reduced H-2k expression onBW87 cells relative to parental BW1083 expression persistsin their hybrids. Still, this reduced level of H-2k expressionis sufficient to allow CIL recognition and CTL-triggered celldeath.
Significantly, the phenotype of resistance to CTL-trig-gered death is extinguished in a hybrid between the BW87mutant and a wild-type cell. The BW87 mutation conferringCTL resistance therefore behaves as a recessive lesion. Thisimplies that the resistance to CTL-triggered death of theBW87 mutant represents a loss of target cell function essen-tial for cell death. Interestingly, the BW1083 phenotype ofglucocorticoid resistance also is extinguished in the hybrids(16). As with this and other cases of cell autonomousphysiological cell death (35), target cells are involved ac-tively in their own death triggered by CTL.
MOL. CELL. BIOL.
I
.,.a, EL'ala 'o
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.
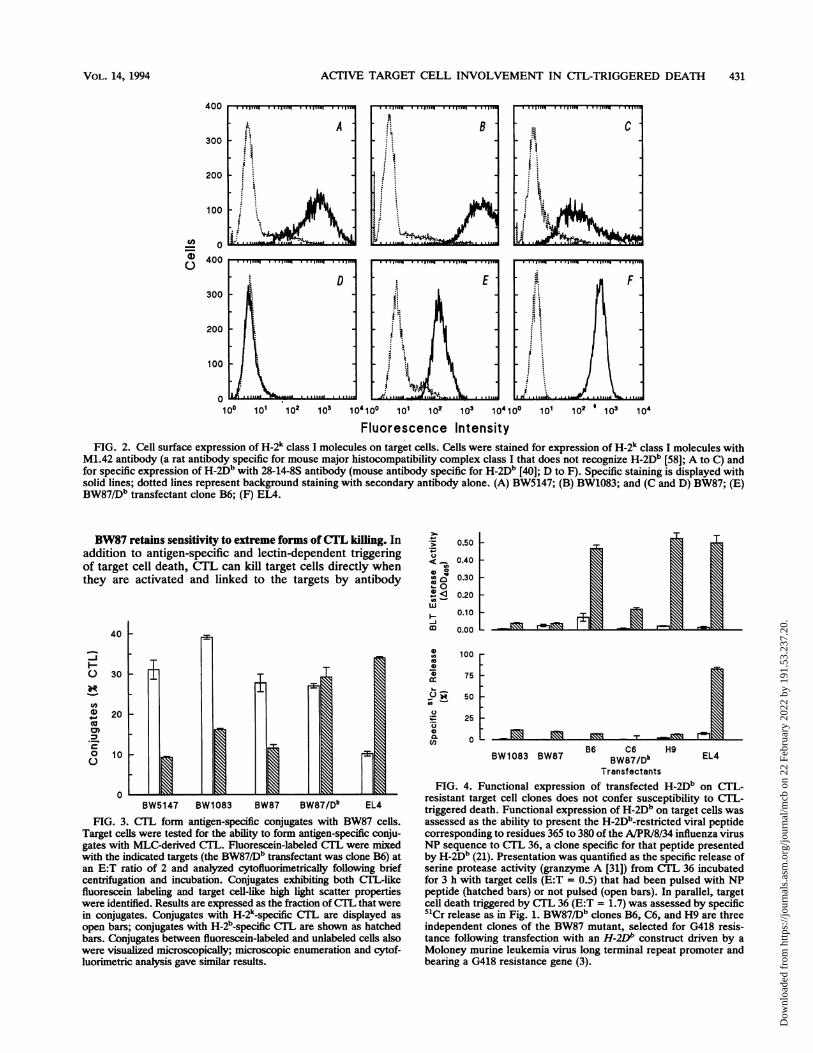
ACTIVE TARGET CELL INVOLVEMENT IN CTL-TRIGGERED DEATH 431
400 ..j.."..I.wl|WlBw glW
A300
200
100
'~ 0
o 400
300
200
100
0 .-
100 10 102 103 104100
Fluc1o0 102 103 104100 1o0 102 103 104
orescence IntensityFIG. 2. Cell surface expression of H-2k class I molecules on target cells. Cells were stained for expression of H-2k class I molecules with
M1.42 antibody (a rat antibody specific for mouse major histocompatibility complex class I that does not recognize H-2Db [58]; A to C) andfor specific expression of H-2Db with 28-14-8S antibody (mouse antibody specific for H-2Db [40]; D to F). Specific staining is displayed withsolid lines; dotted lines represent background staining with secondary antibody alone. (A) BW5147; (B) BW1083; and (C and D) BW87; (E)BW87/Db transfectant clone B6; (F) EL4.
BW87 retains sensitivity to extreme forms of CTL killing. Inaddition to antigen-specific and lectin-dependent triggeringof target cell death, CT7L can kill target cells directly whenthey are activated and linked to the targets by antibody
40
-j
4-
(a
0(0
0)0
0
3 0.50
<.... 0.40
t 0.30 -
L.0 000.00.10
30
20
10 BW1083 BW87
U, 1000
cc 75-
OR 50
25-
0Xen O
Em -M_ I
B6 C6 H9BW87/Db EL4
Transfectants
FIG. 3. CTarget cells i
gates withwith the indian E:T raticcentrifugatio:fluorescein 1
were identificin conjugateopen bars; obars. Conjugwere visualizluorimetric a
I1 I1g1I1I1g IN FIG. 4. Functional expression of transfected H-2Db on CTL-resistant target cell clones does not confer susceptibility to CTL-
BW5147 BW1083 BW87 BW87/Db EL4 triggered death. Functional expression of H-2Db on target cells was
TL form antigen-specific conjugates with BW87 cells. assessed as the ability to present the H-2Db1restricted viral peptidewere tested for the ability to form antigen-specific conju- corresponding to residues 365 to 380 of the A/PR/8/34 influenza virusILC-derived CTL. Fluorescein-labeled CTL were mixed NP sequence to CTL 36, a clone specific for that peptide presentedcated targets (the BW87/Db transfectant was clone B6) at by H-2Db (21). Presentation was quantified as the specific release of) of 2 and analyzed cytofluorimetrically following brief serine protease activity (granzyme A [31]) from CTL 36 incubatedIn and incubation. Conjugates exhibiting both CTL-like for 3 h with target cells (E:T = 0.5) that had been pulsed with NPabeling and target cell-like high light scatter properties peptide (hatched bars) or not pulsed (open bars). In parallel, targeted. Results are expressed as the fraction of CTL that were cell death triggered by CTL 36 (E:T = 1.7) was assessed by specifics. Conjugates with H-2k-specific CTL are displayed as 51Cr release as in Fig. 1. BW87/Db clones B6, C6, and H9 are threeonjugates with H-2b-specific CTL are shown as hatched independent clones of the BW87 mutant, selected for G418 resis-;ates between fluorescein-labeled and unlabeled cells also tance following transfection with an H-2Db construct driven by azed microscopically; microscopic enumeration and cytof- Moloney murine leukemia virus long terminal repeat promoter andmalysis gave similar results. bearing a G418 resistance gene (3).
.. ...q . I.... . . I....I . . I...
Ij
VOL. 14, 1994
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.
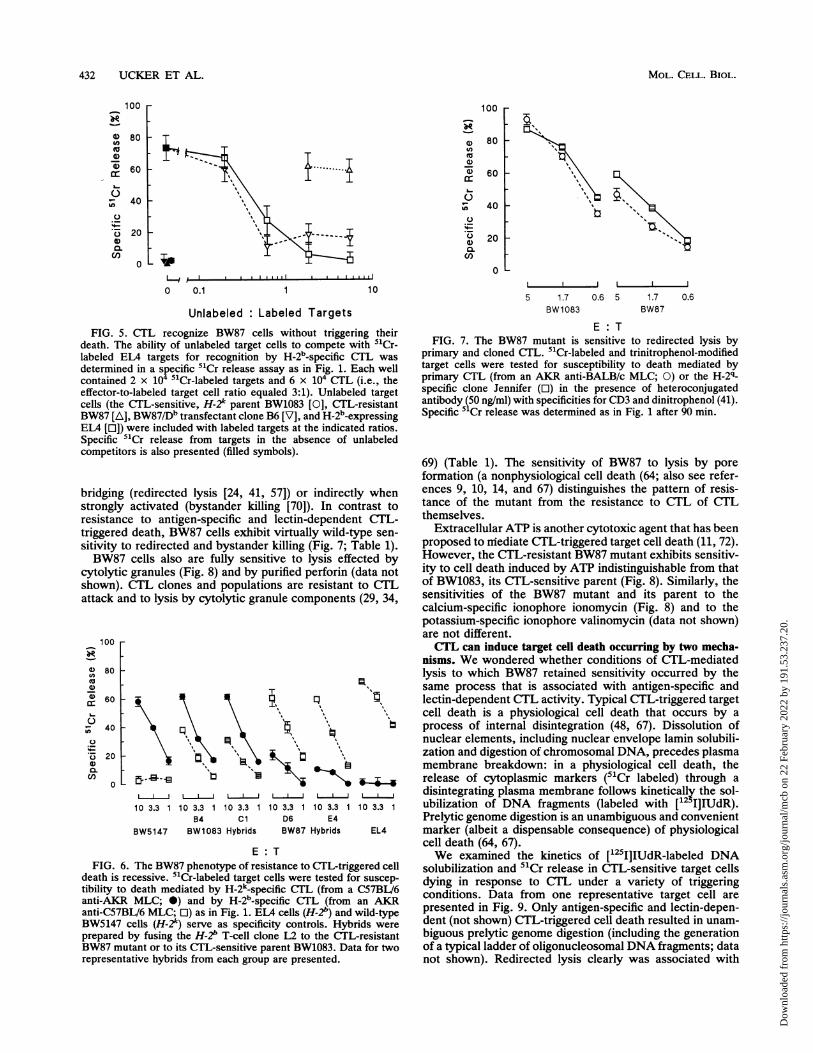
432 UCKER ET AL.
100
4) 800(D0 60
, 400
0 200
C,)0
100
K
0
(U)0
SIA
(a
._.)Ln
Lq0 0.1 10
Unlabeled : Labeled TargetsFIG. 5. CTL recognize BW87 cells without triggering their
death. The ability of unlabeled target cells to compete with 5"Cr-labeled ELA targets for recognition by H-2b_specific CTL wasdetermined in a specific 51Cr release assay as in Fig. 1. Each wellcontained 2 x 104 "Cr-labeled targets and 6 x 104 CTL (i.e., theeffector-to-labeled target cell ratio equaled 3:1). Unlabeled targetcells (the CTL-sensitive, H-2k parent BW1083 [0], CTL-resistantBW87 [A], BW87/Db transfectant clone B6 [V], and H-2b-expressingELA [E]) were included with labeled targets at the indicated ratios.Specific 51Cr release from targets in the absence of unlabeledcompetitors is also presented (filled symbols).
bridging (redirected lysis [24, 41, 57]) or indirectly whenstrongly activated (bystander killing [70]). In contrast toresistance to antigen-specific and lectin-dependent CTL-triggered death, BW87 cells exhibit virtually wild-type sen-sitivity to redirected and bystander killing (Fig. 7; Table 1).BW87 cells also are fully sensitive to lysis effected by
cytolytic granules (Fig. 8) and by purified perforin (data notshown). CTL clones and populations are resistant to CTLattack and to lysis by cytolytic granule components (29, 34,
10 3.3 1 10 3.3 1B4
I LL
10 3.3 1 10ci
BW5147 BW1083 Hybrids
'K:I
I
3.3 1 10 3.3 1 10 3.3 1D6 E4BW87 Hybrids EL4
E: TFIG. 6. The BW87 phenotype of resistance to CTL-triggered cell
death is recessive. 5"Cr-labeled target cells were tested for suscep-tibility to death mediated by H-2k_specific CTL (from a C57BV/6anti-AKR MLC; *) and by H-2bNspecific CTL (from an AKRanti-C57BL/6 MLC; El) as in Fig. 1. ETA cells (H-2") and wild-typeBW5147 cells (H-2k) serve as specificity controls. Hybrids were
prepared by fusing the H-2' T-cell clone L2 to the CTL-resistantBW87 mutant or to its CTL-sensitive parent BW1083. Data for tworepresentative hybrids from each group are presented.
80
60
40
20
O L
5 1.7 0.6 5 1.7 0.6BW1083 BW87
E: TFIG. 7. The BW87 mutant is sensitive to redirected lysis by
primary and cloned CTL. 51Cr-labeled and trinitrophenol-modifiedtarget cells were tested for susceptibility to death mediated byprimary CTL (from an AKR anti-BALB/c MLC; 0) or the H-2q_specific clone Jennifer (E) in the presence of heteroconjugatedantibody (50 ng/ml) with specificities for CD3 and dinitrophenol (41).Specific 5 Cr release was determined as in Fig. 1 after 90 min.
69) (Table 1). The sensitivity of BW87 to lysis by poreformation (a nonphysiological cell death (64; also see refer-ences 9, 10, 14, and 67) distinguishes the pattern of resis-tance of the mutant from the resistance to CTL of CTLthemselves.
Extracellular ATP is another cytotoxic agent that has beenproposed to niediate CTL-triggered target cell death (11, 72).However, the CTL-resistant BW87 mutant exhibits sensitiv-ity to cell death induced by ATP indistinguishable from thatof BW1083, its CTL-sensitive parent (Fig. 8). Similarly, thesensitivities of the BW87 mutant and its parent to thecalcium-specific ionophore ionomycin (Fig. 8) and to thepotassium-specific ionophore valinomycin (data not shown)are not different.CTL can induce target cell death occurring by two mecha-
nisms. We wondered whether conditions of CTL-mediatedlysis to which BW87 retained sensitivity occurred by thesame process that is associated with antigen-specific andlectin-dependent CTL activity. Typical CTL-triggered targetcell death is a physiological cell death that occurs by aprocess of internal disintegration (48, 67). Dissolution ofnuclear elements, including nuclear envelope lamin solubili-zation and digestion of chromosomal DNA, precedes plasmamembrane breakdown: in a physiological cell death, therelease of cytoplasmic markers (51Cr labeled) through adisintegrating plasma membrane follows kinetically the sol-ubilization of DNA fragments (labeled with [12 I]IUdR).Prelytic genome digestion is an unambiguous and convenientmarker (albeit a dispensable consequence) of physiologicalcell death (64, 67).We examined the kinetics of ['"I]IUdR-labeled DNA
solubilization and 51Cr release in CTL-sensitive target cellsdying in response to CTL under a variety of triggeringconditions. Data from one representative target cell arepresented in Fig. 9. Only antigen-specific and lectin-depen-dent (not shown) CTL-triggered cell death resulted in unam-biguous prelytic genome digestion (including the generationof a typical ladder of oligonucleosomal DNA fragments; datanot shown). Redirected lysis clearly was associated with
-100
° 80(U0)0 60
o40
.)U 20
CL)cn0
MOL. CELL. BIOL.
1
........ 11Ii
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.
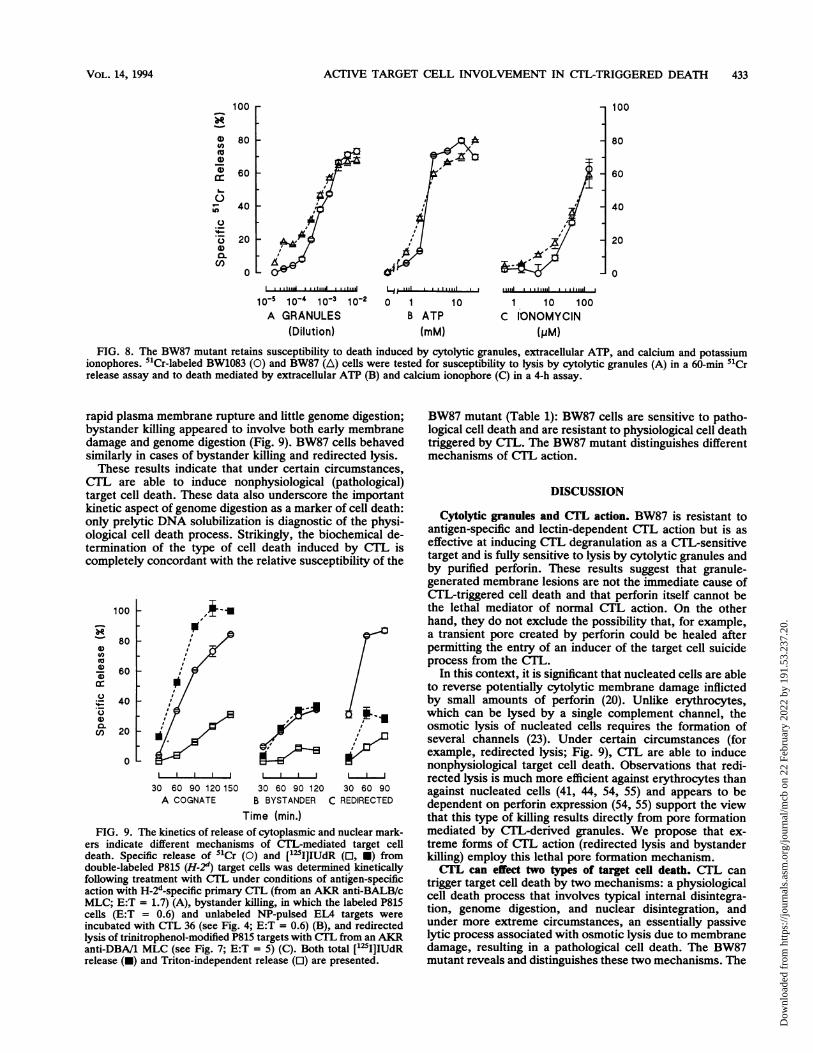
ACTIVE TARGET CELL INVOLVEMENT IN CTL-TRIGGERED DEATH 433
100 r
0
U)
ii0
._.)
n
80
60
40
20
0 K
100
80
60
40
20
0
10-5 10-4 10-3 10-2A GRANULES
(Dilution)
0 1 10B ATP
(mM)
, ~~~~~~~....I............ ....
1 10 100C IONOMYCIN
(PM)FIG. 8. The BW87 mutant retains susceptibility to death induced by cytolytic granules, extracellular ATP, and calcium and potassium
ionophores. 51Cr-labeled BW1083 (0) and BW87 (A) cells were tested for susceptibility to lysis by cytolytic granules (A) in a 60-min 51Crrelease assay and to death mediated by extracellular ATP (B) and calcium ionophore (C) in a 4-h assay.
rapid plasma membrane rupture and little genome digestion;bystander killing appeared to involve both early membranedamage and genome digestion (Fig. 9). BW87 cells behavedsimilarly in cases of bystander killing and redirected lysis.These results indicate that under certain circumstances,
CTL are able to induce nonphysiological (pathological)target cell death. These data also underscore the importantkinetic aspect of genome digestion as a marker of cell death:only prelytic DNA solubilization is diagnostic of the physi-ological cell death process. Strikingly, the biochemical de-termination of the type of cell death induced by CTL iscompletely concordant with the relative susceptibility of the
30 60 90 120150A COGNATE
30 60 90 120 30 60 90
B BYSTANDER C REDIRECTED
Time (min.)FIG. 9. The kinetics of release of cytoplasmic and nuclear mark-
ers indicate different mechanisms of CTL-mediated target celldeath. Specific release of 51Cr (0) and ['251IIUdR (O, *) fromdouble-labeled P815 (H-2") target cells was determined kineticallyfollowing treatment with CTL under conditions of antigen-specificaction with H-2d-specific primary CTL (from an AKR anti-BALB/cMLC; E:T = 1.7) (A), bystander killing, in which the labeled P815cells (E:T = 0.6) and unlabeled NP-pulsed ELA targets wereincubated with CTL 36 (see Fig. 4; E:T = 0.6) (B), and redirectedlysis of trinitrophenol-modified P815 targets with CTL from an AKRanti-DBA/1 MLC (see Fig. 7; E:T = 5) (C). Both total [125I]IUdRrelease (-) and Triton-independent release (0) are presented.
BW87 mutant (Table 1): BW87 cells are sensitive to patho-logical cell death and are resistant to physiological cell deathtriggered by CTL. The BW87 mutant distinguishes differentmechanisms of CTL action.
DISCUSSION
Cytolytic granules and CTL action. BW87 is resistant toantigen-specific and lectin-dependent CTL action but is aseffective at inducing CTL degranulation as a CTL-sensitivetarget and is fully sensitive to lysis by cytolytic granules andby purified perforin. These results suggest that granule-generated membrane lesions are not the immediate cause ofCTL-triggered cell death and that perforin itself cannot bethe lethal mediator of normal CTL action. On the otherhand, they do not exclude the possibility that, for example,a transient pore created by perforin could be healed afterpermitting the entry of an inducer of the target cell suicideprocess from the CTL.
In this context, it is significant that nucleated cells are ableto reverse potentially cytolytic membrane damage inflictedby small amounts of perforin (20). Unlike erythrocytes,which can be lysed by a single complement channel, theosmotic lysis of nucleated cells requires the formation ofseveral channels (23). Under certain circumstances (forexample, redirected lysis; Fig. 9), CTL are able to inducenonphysiological target cell death. Observations that redi-rected lysis is much more efficient against erythrocytes thanagainst nucleated cells (41, 44, 54, 55) and appears to bedependent on perforin expression (54, 55) support the viewthat this type of killing results directly from pore formationmediated by CrL-derived granules. We propose that ex-treme forms of CT7L action (redirected lysis and bystanderkilling) employ this lethal pore formation mechanism.CTL can effect two types of target cell death. CTL can
trigger target cell death by two mechanisms: a physiologicalcell death process that involves typical internal disintegra-tion, genome digestion, and nuclear disintegration, andunder more extreme circumstances, an essentially passivelytic process associated with osmotic lysis due to membranedamage, resulting in a pathological cell death. The BW87mutant reveals and distinguishes these two mechanisms. The
100
bA
0
00.
C')
80
60
40
20
0
VOL. 14, 1994
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.

434 UCKER ET AL.
ability of CTL to induce different types of target cell deathhas been suggested previously (38, 73) and is confirmed bydifferences in the patterns of release of nuclear and cytoplas-mic markers during target cell death. Normal CTL activity,triggered in an antigen-specific or lectin-dependent manner,employs the physiological process.
Target cells are involved actively in their death. That theBW87 phenotype of resistance to physiological cell deathtriggered by CTL is recessive indicates that the failure of thetarget cell to die results from the loss of a function or activitynecessary for the cell death response. This is not a trivialdeficit, for example, the inability of the target to be recog-nized or the failure of the mutant to trigger a degranulationresponse from the CTL. Instead, the defect of the mutantreveals an essential functional contribution that the targetcell makes normally to its own death. In this sense, evendeath triggered by a CTL effector is an induced cell suicideprocess.
CTL-triggered physiological target cell death is unusualamong physiological cell deaths in a number of respects.Most obviously, it is not a cell autonomous process. Therapidity of the process (within minutes; Fig. 9) reflects theabsence of a requirement for macromolecular synthesis (48,59). We hypothesize that the effector cell is able to providea molecule or signal introduced into the target (for example,via a polyperforin lesion) which obviates earlier steps of thephysiological cell death process that include de novo geneexpression (as well as rigorous cell cycle control and sensi-tivity to BCL-2 [64, 68]). Nonetheless, target cell metabolicactivity appears to be necessary for cell death (30), consis-tent with the notion that the target cell plays an active role inits own death.The results presented here for the BW87 mutant indepen-
dently demonstrate that the target cell actively contributesan essential function to the cell death process. The loss ofthat function prevents target cells from responding to CTLby dying. Because the CTL-triggered process can occur inthe absence of macromolecular synthesis, the function ab-sent in BW87 is not one that must be induced in susceptiblecells for cell death to occur; further, the absence of thisfunction in the BW87 mutant is not deleterious for cellsurvival. We conclude that an activity constitutively presentin the target cell is essential for CTL-triggered cell death.BW87 resistance and physiological cell death. Simplisti-
cally, the resistance to CTL action of the BW87 mutantcould result either from an inability of the mutant to receivean inductive signal from the CTL or from the mutant'sfailure to respond to that signal. BW87 is not pleiotropicallyresistant to the induction of physiological cell death. Forexample, the CTL-resistant mutant retains susceptibility toATP-triggered cell death (Fig. 8), to death resulting fromelevated intracellular calcium levels (Fig. 8), and to deathinduced by low-dose irradiation and by elevated intracellularcyclic AMP levels (16). Thus, the physiological cell deathresponse in BW87 cells appears generally to be intact. Thisimplies either that these cells are defective specifically in oneof the afferent signaling steps between the lethal hit deliveredby the CTL and distal elements of the cell death process orthat the CTL-triggered cell death process is geneticallydistinct from that of other physiological cell deaths.We have argued that all physiological cell deaths likely
share common components (64; also see reference 8). How-ever, this hypothesis, particularly relating to CTL-triggeredcell death, remains unproven. Previously, we reported thatanother target cell mutant, 4R.D, appeared to be resistant tocell death induced both by glucocorticoids and by CTL as a
result of a single genetic lesion, suggesting that a commonmutable element governs the ability of a cell to die (63).Subsequent work (7) has shown that the 4R.D mutant cannotbe considered generally resistant to CTL, however. There-fore, no genetic linkage among CTL-triggered death andother cell suicide processes yet has been established.
Mediators of CTL-triggered physiological cell death. Anumber of potential mediators of CTL-triggered target celldeath have been proposed, including extracellular ATP,granule-derived molecules, and elevated intracellular ionfluxes (2, 11, 15, 43, 52, 53, 61, 72). Our observation that theCTL resistance of BW87 does not confer resistance toATP-mediated cell death complements another recent so-matic cell genetic study of ATP sensitivity. Avery et al. (4)found that target cell mutants resistant to ATP-induceddeath were not resistant to CTL-triggered death. Together,these results demonstrate that the induction of cell death byCTL and that by ATP occur by genetically distinct mecha-nisms.While the role of calcium in cell death in general is
controversial, it is clear that elevated free calcium within thetarget cell (2, 43) is not sufficient to account for CTL-triggered target cell death (64). For example, the induction ofphysiological cell death in T cells by calcium ionophores isdependent on macromolecular synthesis (71). The undimin-ished sensitivity of the BW87 mutant to elevated intracellu-lar calcium levels caused by calcium ionophore furtherdemonstrates that a calcium flux likely is not involved inmediating CTL-triggered cell death. The potassium iono-phore valinomycin, in contrast, has been shown to mimicCTL-triggered cell death kinetically and with respect toindependence from macromolecular synthesis requirements(2, 37). The observation that CTL-resistant BW87 cellsexhibit unaltered sensitivity to cell death induced by valino-mycin argues, however, that cell deaths triggered by CTLand by presumptive intracellular potassium changes also aredistinct.
It also is worth noting that cells derived from the BW5147thymoma are not susceptible to CTL action in the absence ofcalcium. Consequently, the Fas antigen, which has beenproposed to serve in triggering target cell death undercalcium-independent conditions of CTL action (47), likely isnot involved here.The involvement of non-pore-forming granule components
such as granzyme A, granzyme B/fragmentin 2, and TIA-1(15, 52, 53, 61) have not been tested with the BW87 mutant.It may be revealing to examine whether the BW87 mutant issusceptible to these molecules when the molecules areintroduced with low doses of perforin. It also will beinteresting to explore the sensitivity of this mutant to otherforms of cell-mediated target cell death, including helperT-cell cytolysis (59). CTL-resistant target cell mutants suchas BW87 will permit the role of these and other potentialmediators to be tested rigorously.
ACKNOWLEDGMENTS
We thank Linda Burkly for the Moloney murine leukemia virus/H-2Db construct, Pierre Henkart for cytolytic granules, Andy Glase-brook for L2 cells, Matt Mescher and Kevin Kane for CTL 36 andNP peptide, John Russell for CTL 3, and David Segal for hetero-conjugated antibody. We also are grateful for the constructivecomments of C. Cowing, S.-T. Ju, A. Kleinfeld, D. Mosier, R.Riblet, and J. Russell.D.S.U. is a Scholar of the Leukemia Society of America. This
work was supported by grant GM38800 from the National Institutesof Health to D.S.U.
MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.

ACTIVE TARGET CELL INVOLVEMENT IN CTL-TRIGGERED DEATH 435
REFERENCES1. Acha-Orbea, H., L. Scarpellino, S. Hertig, M. Dupuis, and J.
Tschopp. 1990. Inhibition of lymphocyte mediated cytotoxicityby perforin antisense oligonucleotides. EMBO J. 9:3815-3819.
2. Allbritton, N. L., C. R. Verret, R. C. Wolley, and H. N. Eisen.1988. Calcium ion concentration and DNA fragmentation intarget cell destruction by murine cloned cytotoxic T lympho-cytes. J. Exp. Med. 167:514-527.
3. Allen, H., J. Fraser, D. Flyer, S. Calvin, and R. Flavell. 1986.,B2-Microglobulin is not required for cell surface expression ofthe murine class I histocompatibility antigen H-2Db or of atruncated H-2Db. Proc. Natl. Acad. Sci. USA 83:7447-7451.
4. Avery, R. K., K. J. Bleier, and M. S. PasternacL 1992. Differ-ences between ATP-mediated cytotoxicity and cell-mediatedcytotoxicity. J. Immunol. 149:1265-1270.
5. Berke, G., and D. Rosen. 1988. Highly lytic in vivo primedcytolytic T lymphocytes devoid of lytic granules and BLT-esterase activity acquire these constituents in the presence of Tcell growth factors upon blast transformation in vitro. J. Immu-nol. 141:1429-1436.
6. Dennert, G., C. G. Anderson, and G. Prochazka. 1987. Highactivity of Na-benzyloxycarbonyl-L-lysine thiobenzyl esterserine esterase and cytolytic perforin in cloned cell lines is notdemonstrable in in vivo-induced cytotoxic effector cells. Proc.Natl. Acad. Sci. USA 84:5004-5008.
7. Dennert, G., C. Landon, and M. Nowicki. 1988. Cell-mediatedand glucocorticoid-mediated target cell lysis do not appear toshare common pathways. J. Immunol. 141:785-791,2187.
8. Dowd, D. R., and R. L. Miesfeld. 1992. Evidence that glucocor-ticoid- and cyclic AMP-induced apoptotic pathways in lympho-cytes share distal events. Mol. Cell. Biol. 12:3600-3608.
9. Duke, R. C., P. M. Persechini, S. Chang, C.-C. Liu, J. J. Cohen,and J. D.-E. Young. 1989. Purified perforin induces targetcell lysis but not DNA fragmentation. J. Exp. Med. 170:1451-1456.
10. Duke, R. C., K. S. Sellins, and J. J. Cohen. 1988. Cytotoxiclymphocyte-derived lytic granules do not induce DNA fragmen-tation in target cells. J. Immunol. 141:2191-2194.
11. Filippini, A., R. E. Taffs, and M. V. Sitkovsky. 1990. Extracel-lular ATP in T-lymphocyte activation: possible role in effectorfunctions. Proc. Natl. Acad. Sci. USA 87:8267-8271;88:6899.
12. Garcia-Sanz, J. A., G. Plaetinck, F. Velotti, D. Masson, J.Tschopp, H. R. MacDonald, and M. Nabholz. 1987. Perforin ispresent only in normal activated Ly2+ T lymphocytes and not inL3T4' cells, but the serine protease granzyme A is made byboth subsets. EMBO J. 6:933-938.
13. Glasebrook, A. L., M. Sarmiento, M. R. Loken, D. P. Dialynas,J. Quintans, L. Eisenberg, C. T. Lutz, D. Wilde, and F. W. Fitch.1981. Murine T lymphocyte clones with distinct immunologicalfunctions. Immunol. Rev. 54:225-266.
14. Gromkowski, S. H., T. C. Brown, D. Masson, and J. Tschopp.1988. Lack of DNA degradation in target cells lysed by granulesderived from cytolytic T lymphocytes. J. Immunol. 141:774-778.
15. Hayes, M. P., G. A. Berrebi, and P. A. Henkart. 1989. Inductionof target cell DNA release by the cytotoxic T lymphocytegranule protease granzyme A. J. Exp. Med. 170:933-946.
16. Hebshi, L. D., and D. S. Ucker. Unpublished data.17. Helgason, C. D., J. A. Prendergast, G. Berke, and R. C.
Bleackley. 1992. Peritoneal exudate lymphocyte and mixedlymphocyte culture hybridomas are cytolytic in the absence ofcytotoxic cell proteinases and perforin. Eur. J. Immunol. 22:3187-3190.
18. Henkart, M. P., and P. A. Henkart. 1981. Lymphocyte mediatedcytolysis as a secretory phenomenon, p. 227-247. In W. R.Clark and P. Golstein (ed.), Mechanisms of cell-mediated cyto-toxicity. Plenum Press, New York.
19. Joag, S. V., C.-C. Liu, B. S. Kwon, W. R. Clark, and J. D.-E.Young. 1990. Expression of mRNAs for pore-forming proteinand two serine esterases in murine primary and cloned effectorlymphocytes. J. Cell. Biochem. 43:81-88.
20. Jones, J., M. B. Hallett, and B. P. Morgan. 1990. Reversible celldamage by T-cell perforins: calcium influx and propidium iodide
uptake into K562 cells in the absence of lysis. Biochem. J.267:303-307.
21. Kane, K. P., A. Vitiello, L. A. Sherman, and M. F. Mescher.1989. Cytolytic T-lymphocyte response to isolated class I H-2proteins and influenza peptides. Nature (London) 340:157-159.
22. Kleinfeld, A. M. Personal communication.23. Koski, C. L., L. E. Ramm, C. H. Hammer, M. M. Mayer, and
M. L. Shin. 1983. Cytolysis of nucleated cells by complement:cell death displays multi-hit characteristics. Proc. Natl. Acad.Sci. USA 80:3816-3820.
24. Kranz, D. M., S. Tonegawa, and H. N. Eisen. 1984. Attachmentof an anti-receptor antibody to non-target cells renders themsusceptible to lysis by a clone of cytotoxic T lymphocytes. Proc.Natl. Acad. Sci. USA 81:7922-7926.
25. Landon, C., M. Nowicki, S. Sugawara, and G. Dennert. 1990.Differential effects of protein synthesis inhibition on CTL andtargets in cell-mediated cytotoxicity. Cell. Immunol. 128:412-426.
26. Lebow, L. T., and B. Bonavida. 1990. Purification and charac-terization of cytolytic and noncytolytic human natural killer cellsubsets. Proc. Natl. Acad. Sci. USA 87:6063-6067.
27. Lu, C.-C., S. Rafii, A. Granelli-Piperano, J. A. Trapani, andJ. D.-E. Young. 1989. Perforin and serine esterase gene expres-sion in stimulated human T cells: kinetics, mitogen require-ments and effects of cyclosporin A. J. Exp. Med. 170:2105-2118.
28. Lu, P., J. A. Garcia-Sanz, M. G. Lichtenheld, and E. R. Podack.1992. Perforin expression in human peripheral blood mononu-clear cells: definition of an IL-2-independent pathway of per-forin induction in CD8+ T cells. J. Immunol. 148:3354-3360.
29. Luciani, M.-F., J.-F. Brunet, M. Suzan, F. Denizot, and P.Goistein. 1986. Self-sparing of long-term in vitro-cloned oruncloned cytotoxic T lymphocytes. J. Exp. Med. 164:962-967.
30. MacDonald, H. R., and C. J. Koch. 1977. Energy metabolismand T-cell-mediated cytolysis I. Synergism between inhibitorsof respiration and glycolysis. J. Exp. Med. 146:698-709.
31. Masson, D., and J. Tschopp. 1987. A family of serine esterasesin lytic granules of cytolytic T lymphocytes. Cell 49:679-685.
32. Millard, P. J., M. P. Henkart, C. W. Reynolds, and P. A.Henkart. 1984. Purification and properties of cytoplasmic gran-ules from cytotoxic rat LGL tumors. J. Immunol. 132:3197-3204.
33. Nagler-Anderson, C., M. Lichtenheld, H. N. Eisen, and E. R.Podack. 1989. Perforn mRNA in primary peritoneal exudatecytotoxic T lymphocytes. J. Immunol. 143:3440-3443.
34. Nagler-Anderson, C., C. R. Verret, A. A. Firmenich, M. Berne,and H. N. Eisen. 1988. Resistance of primary CD8+ cytotoxic Tlymphocytes to lysis by cytotoxic granules from cloned T celllines. J. Immunol. 141:3299-3305.
35. Nickas, G., J. Meyers, L. D. Hebshi, J. D. Ashwell, D. P. Gold,B. Sydora, and D. S. Ucker. 1992. Susceptibility to cell death isa dominant phenotype: triggering of activation-driven T-celldeath independent of the T-cell antigen receptor complex. Mol.Cell. Biol. 12:379-385.
36. Ojcius, D. M., L. M. Zheng, E. C. Sphicas, A. Zychlinsky, andJ. D.-E. Young. 1991. Subcellular localization of perforin andserine esterase in lymphokine-activated killer cells and cyto-toxic T cells by immunogold labeling. J. Immunol. 146:4427-4432.
37. Ojcius, D. M., A. Zychlinsky, L. M. Zheng, and J. D.-E. Young.1991. lonophore-induced apoptosis: role of DNA fragmentationand calcium fluxes. Exp. Cell Res. 197:43-49.
38. Ostergaard, H. L., and W. R. Clark. 1989. Evidence for multiplelytic pathways used by cytotoxic T lymphocytes. J. Immunol.143:2120-2126.
39. Ostergaard, H. L., K. P. Kane, M. F. Mescher, and W. R. Clark.1987. Cytotoxic T lymphocyte mediated lysis without release ofserine esterase. Nature (London) 330:71-72.
40. Ozato, K., T. H. Hansen, and D. H. Sachs. 1980. Monoclonalantibodies to mouse MHC antigens. II. Antibodies to the H-2Ldantigen, the products of a third polymorphic locus of the mousemajor histocompatibility complex. J. Immunol. 125:2473-2477.
41. Perez, P., R. W. Hoffman, S. Shaw, J. A. Bluestone, and D. M.
VOL. 14, 1994
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.

436 UCKER ET AL.
Segal. 1985. Specific targeting of cytotoxic T cells by anti-T3linked to anti-target cell antibody. Nature (London) 316:354-356.
42. Peters, P. J., J. Borst, V. Oorschot, M. Fukuda, 0. Krahenbuhl,J. Tschopp, J. W. Slot, and H. J. Geuze. 1991. Cytotoxic Tlymphocyte granules are secretory lysosomes, containing bothperforin and granzymes. J. Exp. Med. 173:1099-1109.
43. Poenie, M., R. Y. Tsien, and A.-M. Schmitt-Verhulst. 1987.Sequential activation and lethal hit measured by [Ca2"]i inindividual cytolytic T cells and targets. EMBO J. 6:2223-2232.
44. Redegeld, F. A., S. Chatterjee, N. A. Berger, and M. V.Sitkovsky. 1992. Poly-(ADP-ribose) polymerase partially con-tributes to target cell death triggered by cytolytic T lympho-cytes. J. Immunol. 149:3509-3516.
45. Reynolds, C. W., D. Reichardt, M. Henkart, P. Millard, and P.Henkart. 1987. Inhibition ofNK and ADCC activity by antibod-ies against purified cytoplasmic granules from rat LGL tumors.J. Leukocyte Biol. 42:642-652.
46. Richieri, G. V., and A. M. Kleinfeld. 1991. Free fatty acids areproduced in and secreted from target cells very early in cyto-toxic T lymphocyte-mediated killing. J. Immunol. 147:2809-2815.
47. Rouvier, E., M.-F. Luciani, and P. Golstein. 1993. Fas involve-ment in Ca2+-independent T cell-mediated cytotoxicity. J. Exp.Med. 177:195-200.
48. Russell, J. H. 1983. Internal disintegration model of cytotoxiclymphocyte-induced target damage. Immunol. Rev. 72:97-118.
49. Russell, J. H., and C. B. Dobos. 1983. Characterization of a"heteroclitic" cytotoxic lymphocyte clone: heterogeneity ofreceptors or signals? J. Immunol. 130:538-541.
50. Russell, J. H., V. R. Masakowski, and C. B. Dobos. 1980.Mechanisms of immune lysis. I. Physiological distinction be-tween target cell death mediated by cytotoxic T lymphocytesand antibody plus complement. J. Immunol. 124:1100-1105.
51. Sandri-Goldin, R. M., A. L. Goldin, M. Levine, and J. C.Glorioso. 1981. High-frequency transfer of cloned herpes sim-plex virus type I sequences to mammalian cells by protoplastfusion. Mol. Cell. Biol. 1:743-752.
52. Shi, L., C. M. Kam, J. C. Powers, R. Aebersold, and A. H.Greenberg. 1992. Purification of three cytotoxic lymphocytegranule serine proteases that induce apoptosis through distinctsubstrate and target cell interactions. J. Exp. Med. 176:1521-1529.
53. Shi, L., R. P. Kraut, R. Aebersold, and A. H. Greenberg. 1992.A natural killer cell granule protein that induces DNA fragmen-tation and apoptosis. J. Exp. Med. 175:553-566.
54. Shiver, J. W., and P. A. Henkart. 1991. A noncytotoxic mastcell tumor line exhibits potent IgE-dependent cytotoxicity aftertransfection with the cytolysin/perforin gene. Cell 64:1175-1181.
55. Shiver, J. W., L. Su, and P. A. Henkart. 1992. Cytotoxicity withtarget DNA breakdown by rat basophilic leukemia cells ex-pressing both cytolysin and granzyme A. Cell 71:315-322.
56. Smyth, M. J., Y. Norishisa, and J. R. Ortaldo. 1992. Multiplecytolytic mechanisms displayed by activated human peripheralblood T cell subsets. J. Immunol. 148:55-62.
57. Staerz, U. D., 0. Kanagawa, and M. J. Bevan. 1985. Hybridantibodies can target sites for attack by T cells. Nature (Lon-don) 314:628-633.
58. Stallcup, K. C., T. A. Springer, and M. F. Mescher. 1981.Characterization of an anti-H-2 monoclonal antibody and its usein large-scale antigen purification. J. Immunol. 127:923-930.
59. Strack, P., C. Martin, S. Saito, R. H. Dekuyff, and S.-T. Ju.1990. Metabolic inhibitors distinguish cytolytic activity of CD4and CD8 clones. Eur. J. Immunol. 20:179-184.
60. Talento, A., M. Nguyen, S. Law, J. K. Wu, M. Poe, J. T. Blake,M. Patel, T.-J. Wu, C. L. Manyak, M. Silberklang, G. Mark, M.Springer, N. H. Sigal, I. L. Weissman, R. C. Bleackley, E. R.Podack, M. L. Tykocinski, and G. C. Koo. 1992. Transfection ofmouse cytotoxic T lymphocyte with an antisense granzyme avector reduces lytic activity. J. Immunol. 149:4009-4015.
61. Tian, Q., M. Streuli, H. Saito, S. F. Schlossman, and P.Anderson. 1991. A polyadenylate binding protein localized tothe granules of cytolytic lymphocytes induces DNA fragmenta-tion in target cells. Cell 67:629-639.
62. Trenn, G., H. Takayama, and M. V. Sitkovsky. 1987. Exocytosisof cytolytic granules may not be required for target cell lysis bycytotoxic T-lymphocytes. Nature (London) 330:72-74.
63. Ucker, D. S. 1987. Cytotoxic T lymphocytes and glucocorticoidsactivate an endogenous suicide process in target cells. Nature(London) 327:62-64.
64. Ucker, D. S. 1991. Death by suicide: one way to go in mamma-lian cellular development? New Biol. 3:103-109.
65. Ucker, D. S., J. D. Ashwell, and G. Nickas. 1989. Activation-driven T cell death I. Requirements for de novo transcriptionand translation and association with genome fragmentation. J.Immunol. 143:3461-3469.
66. Ucker, D. S., J. Meyers, and P. S. Obermiller. 1992. Activation-driven T cell death. II. Quantitative differences alone distin-guish stimuli triggering nontransformed T cell proliferation ordeath. J. Immunol. 149:1583-1592.
67. Ucker, D. S., P. S. Obermiller, W. Eckhart, J. R. Apgar, N. A.Berger, and J. Meyers. 1992. Genome digestion is a dispensableconsequence of physiological cell death mediated by cytotoxicT lymphocytes. Mol. Cell. Biol. 12:3060-3069.
68. Vaux, D. L., H. L. Aguila, and I. L. Weissman. 1992. Bcl-2prevents death of factor-deprived cells but fails to preventapoptosis in targets of cell mediated killing. Int. Immunol.4:821-824.
69. Verret, C. R., A. A. Firmenich, D. M. Kranz, and H. N. Eisen.1987. Resistance of cytotoxic T lymphocytes to the lytic effectsof their toxic granules. J. Exp. Med. 166:1536-1546.
70. Vitiello, A., W. R. Heath, and L. A. Sherman. 1989. Self-presentation of peptide antigen by CTL. J. Immunol. 143:1512-1517.
71. Wyllie, A. H., R. G. Morris, A. L. Smith, and D. Dunlop. 1984.Chromatin cleavage in apoptosis: association with condensedchromatin morphology and dependence on macromolecularsynthesis. J. Pathol. 142:67-77.
72. Zanovello, P., V. Bronte, A. Rosato, P. Pizzo, and F. Di Virgilio.1990. Responses of mouse lymphocytes to extracellular ATP.II. Extracellular ATP causes cell type-dependent lysis and DNAfragmentation. J. Immunol. 145:1545-1550.
73. Zychlinsky, A., L. M. Zheng, C.-C. Liu, and J. D.-E. Young.1991. Cytolytic lymphocytes induce both apoptosis and necrosisin target cells. J. Immunol. 146:393-400.
MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 22
Feb
ruar
y 20
22 b
y 19
1.53
.237
.20.








![Tumour-infiltrating cytotoxic T lymphocytes in …...such as CD8+ and natural killer lymphocytes [17], indu-cing a cytotoxic cascade resulting in tumour cell death, while other TILs](https://img.pdfslide.net/doc/110x75/5f4838f3212d137c1c54d55d/tumour-infiltrating-cytotoxic-t-lymphocytes-in-such-as-cd8-and-natural-killer.jpg)




















