Embed Size (px)
Citation preview

The Adrenal Medulla and Extra-adrenal Paraganglia:Then and Now
Arthur S. Tischler & Karel Pacak & Graeme Eisenhofer
# Springer Science+Business Media New York 2013
Abstract The past 25 years have witnessed revolutionarychanges in the care of patients with pheochromocytomas andextra-adrenal paragangliomas. Germline mutations of at least13 genes are now associated with tumor development, agreater degree of hereditary susceptibility than for any otherhuman neoplasm. Somatic mutations, either of the same genesor of several additional ones with closely related functions, arealso increasingly recognized. Clinicians are now aware of thegenetic implicat ions of a pheochromocytoma orparaganglioma. All patients are therefore offered genetic test-ing and receive lifelong surveillance. Almost all of the mutat-ed genes have well-described correlations with clinical andbiochemical phenotypes. Tumors arising in patients with mu-tations of the SDHB gene have at least a 30 % chance ofmetastasizing and typically produce norepinephrine and/ordopamine. Assay of plasma-free metanephrines serves as ahighly sensitive and specific biochemical screen for the pres-ence of catecholamine-producing tumors, and the dopaminemetabolite methoxytyramine serves as a useful marker fordetecting minimally functional tumors or their metastases.New functional imaging techniques provide highly sensitivetumor localization. In addition to differential diagnosis,
pathologists play new roles in helping to identify hereditarydisease and guiding the sequence of genetic testing.
Keywords Pheochromocytoma . Paraganglioma . Genetics .
Biochemistry . Imaging . Pediatrics . Treatment
Introduction
A Clinical Vignette
A 41-year-old man presented in 1989 with a history of hyper-tension fluctuating markedly and increasing in severity overseveral years. He reported intermittent severe headaches butwas otherwise asymptomatic. He denied having palpitationsor abnormal sweating. Blood pressure measurements variedwidely through multiple visits to his physician, who eventu-ally considered the possibility of a pheochromocytoma. Bio-chemical testing of 24-h urine samples showed elevated nor-epinephrine, borderline elevation of dopamine, and normalepinephrine, with additional elevations of total metanephrinesand vanillylmandelic acid (VMA). CT imaging showed a 5-cm well-circumscribed abdominal mass near the aortic bifur-cation. The mass was resected uneventfully after pharmaco-logical blockade of alpha and beta adrenergic receptors, and adiagnosis of pheochromocytoma was rendered by the pathol-ogist. Biochemical testing up to 6 months postoperatively waswithin normal limits. However, after 3 years the patientreturned complaining of bone pain and was found to havemetastases histologically identical to the resected tumor in-volving bone, liver, and lung. Treatment was palliative and thepatient died after 1 month. Seven years later, one of his threechildren presented at age 16 with disseminated metastases of asimilar tumor. This suggested the possibility of a hereditarydisease, but the identity of the disease and required course ofaction were unknown.
A. S. Tischler (*)Department of Pathology, Tufts Medical Center, 800 WashingtonSt, Boston, MA 02111, USAe-mail: [email protected]
K. PacakProgram in Reproductive and Adult Endocrinology, EuniceKennedy Shriver National Institute of Child Health and HumanDevelopment, National Institutes of Health, 10 Center Drive,MSC-1583, Bethesda, MD 20892-1583, USAe-mail: [email protected]
G. EisenhoferInstitute of Clinical Chemistry and Laboratory Medicine andDepartment of Medicine III, University of Dresden, Fetscherstrasse74, 01307 Dresden, Germanye-mail: [email protected]
Endocr PatholDOI 10.1007/s12022-013-9286-3

The 25 years since 1989 have seen dramatic changes inevery aspect of this patient’s management. Those changesreflect interrelated advances in genetics, biochemistry, imag-ing, and pediatrics as well as in pathology. There would nowbe a different therapeutic outcome and different follow-up forfamily members, with extra considerations of biology andbehavior of these particular tumors. In addition, in accordancewith the current (2004) World Health Organization nomencla-ture, the term pheochromocytoma (PCC) is used only forintra-adrenal tumors. The diagnosis would therefore beextra-adrenal paraganglioma (PGL). The current diagnosticterminology serves to emphasize phenotypic and genotypicdifferences associated with adrenal versus extra-adrenal loca-tion. However, the somewhat awkward annotation “PCC/PGL” is often used in scientific literature in recognition ofthe fact that a PCC is an intra-adrenal PGL with many simi-larities to its extraadrenal counterparts.
Genetics
Rapid progress in genetics has created a new understanding ofPCC/PGL and in many ways can be viewed as having set thefoundation on which other advances were built. In 1989,pheochromocytomas and other PGL were essentially curiosi-ties. Although there was a recognized association with neuro-fibromatosis, von Hippel–Lindau disease and multiple endo-crine neoplasia syndromes, the actual responsible genes wereunknown. PCC and other PGL associated with the sympathet-ic nervous system appeared to be only distantly related to head
and neck PGL assoc i a t ed wi th the vagus andglossopharyngeal nerves, which were often called “glomustumors” or “chemodectomas.” All of the tumors were consid-ered to be almost always benign.
The NF1 , VHL , and RET genes were identified between1990 and 1994 [1], and by then pheochromocytoma (the termthen encompassing both adrenal and extra-adrenal sympathet-ic PGL) was firmly established as “the 10 % tumor”: 10 %extra-adrenal, 10 % hereditary, and 10 % malignant. The firstmajor breakthrough leading to the modern era came in 2000,with a paper by Baysal et al. showing that germline mutationsof the SDHD gene are responsible for a subset of hereditaryPGL in the head and neck, principally arising in the carotidbody [2]. SDHD is one of four separate genes encodingsubunits of the Krebs cycle enzyme succinate dehydrogenase(SDH). Attention therefore is rapidly focused on other SDHsubunits and soon implicated both SDHC [3] and SDHB [4].Importantly, SDHB mutations were associated with suscepti-bility to both head and neck and abdominal PGL [4]. Thehereditary component of the 10 % rule was definitivelyoverturned with a 2002 paper by Neumann and colleagues[5] showing that substantial numbers of patients with appar-ently sporadic PCC/PGL-harbored occult germline mutationsof the susceptibility genes known at that time. The search fornew genes subsequently accelerated, with the 10 % tumor onlybriefly becoming “the 10 gene tumor” [1]. At least 13 genes arenow known to drive the development of hereditary PCC/PGL,including the genes encoding all of the SDH subunits (collec-tively “SDHx”) and SDHAF2 , which encodes an associatedprotein (Tables 1 and 2). As of 2013, at least 33% of PCC/PGL,
Table 1 Tumor distributions in major familial paraganglioma syndromes
Gene (chromosome) Syndrome Paraganglioma distribution Other tumorsa
Adrenal Other sympathetic Head and neck
RET (10q11) MEN2 √√√ √– MTC and parathyroid adenoma (MEN2A only)
VHL (3p25–26) VHL √√ √√ √– RCC , hemangioblastoma, i-NET, pan-NET, and pituitaryadenoma [56]
NF1 (17q11) NF1 √ Neurofibroma , i-NET, GIST, and pituitary adenoma [57]
SDHD (11q23) PGL1 √ √ √√√ RCC and pituitary adenoma [9]
SDHC (1q21–23) PGL3 √– √– √√√ RCC, GIST, and pituitary adenoma [58]
SDHB (1p36) PGL4 √ √√√ √√ RCC, GIST, and pituitary adenoma
Exceptions to usual genotype correlations, including infrequently associated tumors and unusual tumor distributions, are likely to be recognized moreoften with widespread genetic testing. In some instances, causal relationships between the germline mutation and infrequently associated tumors are notfully established. Most hereditary PCC and PGL are attributable to the above genes. In addition to the syndromes named according to single involvedgenes, the “Carney–Stratakis dyad,” consisting of PGL and GIST, can be caused by mutations of SDHB , SDHC , or SDHD . A hereditary basis has notbeen identified for the Carney triad, which also includes pulmonary chondroma [59]. Note that the combination of sympathoadrenal PGL withparasympathetic PGL in the head and neck strongly suggests mutations of SDHB or SDHD. SDHD-mutated PGL in the head and neck are frequentlymultifocal but do not often metastasize
MTC medullary thyroid (C cell) carcinoma, i-NET intestinal neuroendocrine tumor, pan-NET pancreatic neuroendocrine tumor, RCC renal cellcarcinoma, GIST gastrointestinal stromal tumor. √– unusual tumor distributions described in only occasional case reportsa Frequently associated tumors are in bold font
Endocr Pathol

both in the abdomen and in the head and neck [6], are consid-ered to be hereditary, allowing for regional variations. This isthe highest percentage for any human tumor.
It is now known that any familial PCC/PGL aggregation ismore likely to involve a SDHx mutation than a mutation ofany other gene, including susceptibility genes recognized justa few years ago [7]. SDHx mutations are also the mostcommon cause of PCC or PGL in children [7]. Furthermore,syndromes associated with hereditary PCC/PGL now bothoverlap and include new components. For example, renal cellcarcinoma, previously associated with mutated VHL , is alsoassociated with SDHx mutations [8]. Gastrointestinal stromaltumor (GIST) is associated with several syndromes [7, 8], asare occasional GI and pancreatic neuroendocrine tumors andpituitary adenomas [9, 10] (Tables 1 and 2). The presence ofsyndromically associated lesions in a patient or family mem-ber is a clue to the presence of hereditary disease, but somehereditary PCC/PGL (TMEM127 , SDHA , andKIF1B) are notsyndromic, i.e., are not associated with other stigmata, whileothers (SDHD and SDHAF2 ) show a pattern of inheritanceconsistent withmaternal imprinting and becomemanifest onlyafter paternal transmission.
Hereditary PCC and PGL are characterized by markedgenotype–phenotype correlations that have profound implica-tions for diagnosis and management of affected patients.Several key points must now be remembered:
& Tumor location depends on the specific mutated gene& Biochemical phenotype depends on both the genotype and
tumor location (see Biochemistry)& The likelihood of metastasis depends on the geno-
type and correlates with both biochemical phenotype andtumor location.
Clinicians who treat patients with PCC or PGL currentlyemploy paradigms based on these points to choose the most
logical and cost-effective sequence of genetic testing, to aid inthe selection of imaging modalities and to stratify long-termrisk [11]. The last point is obviously paramount. While overallrisk of metastasis is still approximately 10–15 %, the risk islower for adrenal (∼10 %) than for extra-adrenal abdominaltumors (>30 %) and highest for tumors with SDHB mutations(∼30–50 %), which are most often intra-abdominal and extra-adrenal [11]. SDHB mutations are estimated to account for upto 30 % of all metastatic PCC and up to 50 % of all metastaticPGL [12].
Hereditary PCC or PGL with different genotypes exhibitdistinct profiles of gene expression and can be divided accord-ing to their profiles into two clusters. “Cluster 1” tumors arecharacterized by pseudohypoxic gene expression profiles,with increased expression of hypoxia-inducible transcriptionfactors and their target genes. This cluster contains all tumorscaused by mutations of genes encoding SDH subunits orVHL. Selective expression of subsets of these target genespartly separates the tumors with SDHx mutations from thosewith VHL mutations [13]. However, the extent of separationdepends on tumor location. PGLs with SDHD mutations inthe head and neck are more similar to VHL PCCs than toSDHD abdominal PGLs [14]. “Cluster 2” is more heteroge-neous than cluster 1, consisting of tumors caused by suscep-tibility genes associated with increased RAS signaling (RETand NF1 ), mitogenesis (MAX ), protein trafficking(TMEM127), and other functions. These findings suggest thatdivision of the two main clusters into subclusters will benecessary for better understanding of pathogenic mechanismsand improved treatment strategies [14].
Somatic mutations of hereditary susceptibility genes haveuntil recently been considered uncommon causes of trulysporadic PCC/PGL. In general that is still relatively the case,although estimated involvement of RET and VHL has some-what increased [15]. An exception isNF1 , which was recentlyfound to be mutated in as many as 40 % of PCC and
Table 2 Relatively uncommon mutated genes causing hereditary PCC/PGL
Gene Syndrome Paraganglioma distribution Other tumors
Adrenal Other sympathetic Head and neck
SDHAF2 [60] PGL2 √SDHA [61] √ √– Pituitary adenoma[10, 58]
TMEM127 [62] √ √–MAX [63] √ V
PHD2 (EGLN1) [64] √HIF2A(EPAS1) [65] √ Duodenal somatostatinoma[66]
KIF1B [67] √
HIF2A mutations identified thus far are most often somatic, although one patient with a germline mutation has been reported [65]. The finding ofidentical somatic mutations in PGLs and a duodenal somatostatinoma remains unexplained [66]. Germline mutations of both VHL and HIF2A areknown to cause polycythemia [65], which was also found in patients with somaticHIF2A mutations in their tumors. Mosaicism has been suggested as apossible explanation for both the polycythemia and the somatostatinoma [68]
Endocr Pathol

occasional abdominal PGL [16]. The precise phenotypic par-allels between hereditary versus somatic mutations of thesame genes remain to be determined. In addition, some spo-radic PCC or PGL harbor somatic mutations of signalingmolecules predicted to act downstream of mutated hereditarysusceptibility genes rather than in the hereditary susceptibilitygenes themselves [15]. Examples includeHIF2A [17, 18] andHRAS [19]. The HIF2A protein would be a secondary medi-ator of pseudo-hypoxic signaling in hereditary tumors withSDHx or VHL mutations because cluster 1 mutations lead tostabilization of HIF proteins. HIF2A mutations result in resis-tance to hydroxylations and thus comparable stabilization. Bycontrast, the HRAS protein might be secondarily activated intumors with either gain-of-function RET mutations or loss-of-function NF1 mutations (see “Biochemistry”).
Biochemistry
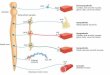
Several important concepts regarding the biochemical func-tions of PCC/PGL and biochemical testing of affected patientsdeveloped in concert with the revolutionary changes in genet-ics [20] (Fig. 1). First, it was realized that that at least 90 % ofcatecholamine catabolism occurs continuously within tumorcells, mostly through the action of catecholamine O-methyltransferase, and is independent of catecholamine secretion.Elevated circulating levels ofO-methylated metabolites there-fore point to the presence of pheochromocytoma or PGL with
greater sensitivity and specificity than elevated levels of theparent amines, which may result from increased sympatheticnervous system activity or come from other sources. Thisrealization occurred together with the development of im-proved analytical techniques for distinguishing free plasmametanephrines produced directly by tumor cells from sulfatedconjugates that circulate in plasma at much higher concentra-tions and potentially mask tumor sources. The sulfated conju-gates are produced in the intestine, where up to 50 % of thebody’s norepinephrine is synthesized, and they are slowlycleared by the kidneys. New analytical methods also facilitat-ed distinction between the O -methylated metabolites of epi-nephrine, norepinephrine and dopamine, i.e., metanephrine,normetanephrine, and methoxytyramine. Methods were alsodeveloped for analyzing fractionated urinary metanephrines.The best test for initial screening of patients is still debated[11]. Current assessments suggest that plasma freemetanephrines and urinary-fractionated metanephrines havecomparable sensitivity but the urine test is less specific [20].
A second important new concept is that catecholaminemetabolite profiles can point toward both a tumor’s locationand its genetic basis. Hereditary PCC/PGL can for the mostpart be assigned to either of the two clusters simply bywhether or not they produce significant amounts of epineph-rine, optimally determined by relative levels of plasma-freemetanephrine and normetanephrine [11]. As shown in Tables 1and 2, hereditary tumors in the adrenal are most likely to beassociated with cluster 2. Almost all of the body’s epinephrineis normally synthesized in the adrenal, and it is therefore not asurprise that epinephrine-producing tumors segregate to clus-ter 2. However, additional genotype information is provided bythe fact that PCC associated with von Hippel–Lindau diseaseare an exception and are usually noradrenergic, despite theirintra-adrenal location. The reason for this is not yet known.However, a PCC that only produces norepinephrine andnormetanephrine points strongly to VHL. Intra-adrenal tumorswith SDHx mutations are relatively rare compared with thosewith mutated VHL , but are also typically noradrenergic.
For many years, it has been known that the hormonalmanifestations of PCC/PGL are highly variable from patientto patient and dependent in part on biochemical phenotype[21]. For example, sweating, palpitations, and tachycardia aremore often associated with adrenergic than noradrenergictumors. In addition, some patients are asymptomatic or haveonly minimal, ambiguous symptoms. Although many tumor-and patient-dependent factors might influence the severity ofsymptoms, foremost are the amount and type of hormoneproduced. Head and neck PGL usually show little or nocatecholamine synthesis and therefore typically present asclinically silent and often biochemically undetectable masslesions. To a lesser extent, this is also true of abdominalPGL, particularly those with SDHB mutations [22]. It hasrecently been demonstrated that ∼70 % of tumors with SDHB
Fig. 1 Current concepts of catecholamine metabolism and production ofcirculating catecholamine metabolites. The contribution of the adrenalmedulla or a pheochromocytoma is shown in relation to other organs andtissues. Abbreviations: MAO monoamine oxidase, COMT catechol-O-methyltransferase, SULT sulfotransferase type 1A3, NE norepinephrine,E epinephrine, NMN normetanephrine, MN metanephrine, DHPG 3,4-dihydroxyphenylglycol, MHPG 3-methoxy-4-hydroxyphenylglycol,VMA vanillylmandelic acid, NMN-S normetanephrine sulfate, MN-Smetanephrine sulfate. Note that E and NE are O-methylated in the samecells in which they are synthesized, producing free MN and NMN thatdirectly enter the bloodstream
Endocr Pathol

or SDHD mutations, including these minimally functionalPGL, often produce significant quantities of dopamine andare detectable by testing for free plasma methoxytyramine[11]. Methoxytyramine levels therefore now serve as a sensi-tive new screen to test for the presence of previously unde-tectable primary tumors or metastases and, potentially, as ameans to prospectively stratify the risk of metastasis from aprimary tumor that has not metastasized. The potential role ofmethoxytyramine in prospective risk assessment applies toadrenal as well as extra-adrenal tumors [11]. Other circulatingbiomarkers have also been evaluated for this purpose, asrecently reviewed [11].
Imaging
The advanced anatomic imaging modalities of the1980s, i.e., CT scans and MRIs, have been supplement-ed by new functional imaging techniques. By definition,functional imaging exploits specific physiological prop-erties of tumor cells in order to increase sensitivity andspecificity. In addition, functional imaging can potential-ly guide subsequent therapy. Well-known examples ofthe latter include 131I-MIBG treatment of tumors imagedby 123I-MIBG, which is taken up by the norepinephrinetransporter in the cell membrane, or use of somatostatinanalogs to treat tumors localized by means of octreotidescans, which detect somatostatin receptors [23].
Many new tracers and new technologies are now avail-able. New technologies include single-photon emissioncomputed tomography (SPECT) and positron emissiontomography (PET). Further, hybrid machines can combinethese modalities with anatomic imaging (SPECT/CT, PET/CT, and PET/MRI) for even greater sensitivity [23, 24].PET or PET/CT together with new tracers for somatostatinreceptor imaging are rapidly supplanting conventional pla-nar 111In-octreotide somatostatin receptor scintigraphy(Octreoscan) or SPECT for more sensitive somatostatinreceptor imaging. The most utilized of the current gener-ation of PET tracers for somatostatin receptor imaging are68Ga-labeled somatostatin analog peptides bearing namesthat begin with “68Ga-DOTA-” (for Ga-68-(1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-). These in-clude 68Ga-DOTATOC (DOTA-D-Phe1-Tyr3-octreotide),68Ga-DOTATATE (DOTA-D-Phe1-Tyr3-octreotate), 68Ga-DOTANOC (DOTA-1-NaI3-octreotide) [23, 25] and relat-ed compounds. A new specific target for highly specificfunctional imaging of PCC/PGL is the vesicular mono-amine transporter (VMAT1) which, in contrast to the NEtransporter, is localized on the membrane of neurosecreto-ry granules and can be imaged by 18F-fluorodopaminePET [26].
The choice of imaging modality is influenced by the ana-tomic site of a tumor and its functionality, both of which are inlarge part determined by its genotype [26] [27]. The initiallocalization of PCC and extra-adrenal sympathetic PGL usu-ally is based on CT or MRI. MRI is sometimes preferred forPGL in the heart, mediastinum, and urinary bladder; while CTis preferred for tumors in the lung [28]. Subsequent functionalimaging serves to confirm that an anatomically identifiedlesion is in fact a PCC or PGL rather than an unrelatedincidentaloma. In addition, functional imaging displays thewhole body, and is therefore valuable for detecting multiple ormetastatic tumors, particularly in patients with hereditaryPCC/PGL [29]. Use of either 123I-MIBG SPECT or 18F-FDOPA PET is currently recommended for PCC and sympa-thetic PGL [27] (Fig. 2). As MIBG is taken up by the NEtransporter, 123I-MIBG SPECT has relatively low sensitivityfor minimally functional tumors, which often include PGLassociated with SDHB mutations. It is nonetheless importantfor tumors that cannot be removed surgically and requireradiotherapy with 131I-MIBG. It is now known that PCC/PGL with little or no catecholamine production often avidlytake up 18F-fluorodeoxyglucose (FDG) via glucose trans-porters on the cell membrane [26, 27]. Although FDG uptakeis not specific for PCC/PGL, FDG-PET can be very effectivefor detecting both primary and metastatic tumors with SDHBmutations [27]. Somatostatin receptor imaging can be alsoused for that purpose [30]. As 18F-FDG PET is nonspecific,somatostatin receptor imaging is increasingly favored. PET/MRI is expected to be especially useful for parasympatheticPGL in the head and neck and has been advocated even forbaseline imaging [31] and for sympathetic PGL in unusuallocations [24, 32].
Fig. 2 Coronal 18F-fluorodopa (left) and 18F-fluorodopamine (right)images of a female patient with extensive metastatic PGL. Overall, 18F-fluorodopa images showmore numerous lesions, predominantly in bones
Endocr Pathol

Pediatrics
Pediatric patients who present with an apparently sporadicPCC or PGL are especially likely to be manifesting a hered-itary disease. Fewer than 20 % of PCC or PGL occur inpatients younger than 18 years. Germline mutations of knownsusceptibility genes have been reported in almost 60 % ofpatients presenting before age 18 and 70% before age 10 [33].The youngest age of onset varies somewhat for different genesbut is 6 years or less for patients with SDHB mutations [33].PCC caused by a VHL mutation has been reported in oneinfant at age 2.75 years [34]. Genetic testing is thereforeadvisable for all children presenting with PCC or PGL andfor children of parents known to harbor mutations. Childrenwho test positive require lifelong screening for new or recur-rent tumors. The type of biochemical testing and type ofimaging are tailored to the tumor genotype [33], as for adultpatients. However, pediatric patients require especially strin-gent efforts to use imaging modalities such as MRI or ultra-sound to minimize radiation exposure.
Treatment
Complete surgical excision is the definitive treatment for anyindividual PCC or PGL. There have been important surgicaladvances since 1989, and patients have greatly benefittedfrom the fact that many surgeries are now performedlaparoscopically. In addition, patients can now sometimes beoffered cortex-sparing adrenal surgery in specialized treatmentcenters. This can especially benefit patients with MEN2 orVHL, who are at risk for bilateral adrenal tumors, by elimi-nating the need for lifelong corticosteroid maintenance [35].Robotic surgery with dye-enhancement has recently beenintroduced for this purpose [36].
Unfortunately, up to 30 % of PCC/PGL metastasize, andthere are still few treatment options after metastases occur.There is also no way to prevent the development of newtumors in patients with hereditary disease, and those tumors,particularly in patients with SDHB mutations, are the mostlikely to metastasize. Historically, chemotherapy or radiother-apy with 131I-MIBG or other agents have been employed,resulting in symptomatic improvement and transient diseasestabilization in some patients, but little or no improvement insurvival. Trials using various treatment approaches to targetthe pseudohypoxia-driven hallmarks of cancer may showsome promise in the near future [37] [12].
A probable deficiency of many treatment strategies is thatthey do not account for the fact that, in contrast to other typesof malignant tumor, metastatic PCC/PGLs usually grow veryslowly and most of the cells at any given time are not repli-cating. Treatments that target DNA replication or tumor an-giogenesis have therefore met with only limited success.
However, new types of targeted therapy are on the horizon.One potentially interesting new drug known as Gamitrinibspecifically targets the mitochondrial complex containingSDHB and associated chaperone proteins and is highly toxicto non-dividing human PCC cells in cell culture studies [38].Other potentially effective drugs target a variety of transcrip-tional mechanisms and signaling pathways that are not spe-cifically related to tumor growth [39].
An important aspect of modern treatment is that all patientspresenting with a PCC or PGL are offered genetic testing andgenetic counseling. The order of genes to be tested is based onclinical parameters including tumor distribution, biochemicalphenotype, associated lesions and family history, and is oftenfurther guided by pathology. Next-generation sequencing tech-nologies might soon alter this approach bymaking simultaneoustesting of all susceptibility genes more cost-effective [40].
Pathology
In 1989, the main function of pathologists in managing patientswith PCC/PCLwas to diagnose primary and metastatic tumors.In a few cases, the presence of adrenal medullary hyperplasiacould point pathologists and clinicians to the presence ofMEN2A or 2B. In 2013, making a correct diagnosis is stillparamount, but other roles are increasingly important.
PCC and PGL can usually be correctly diagnosed byroutine H&E histology. The tumors often exhibit a classichistologic pattern with prominent “Zellballen.” However,many architectural and cytological variations can be seen,as recently reviewed [41]. In difficult cases, immunostain-ing for chromogranin A usually suffices to establish aneuroendocrine phenotype and the absence of stainingfor keratins usually rules out other types of neuroendo-crine tumor. Staining for tyrosine hydroxylase (TH), whichis required for catecholamine synthesis, can provide furtherconfirmation, though it is not entirely specific to PCC orPGL. The use of immunostaining for TH can also provideuseful information because some abdominal PGL andmany head and neck PGL are TH negative and thereforebiochemically nonfunctional. Persistently increasedmetanephrines in a patient after removal of a TH-negative PGL suggest the presence of an additional tumor.
The most recent WHO terminology for PCC and PGLdefines malignancy by the development of metastases [42].There is currently no way for a pathologist to diagnose aprimary PCC or PGL as benign or malignant unless metastasesoccur, and all patients therefore require long-term follow up.However, pathologists have developed protocols for stratifyingthe risk of metastasis [43–45]. These protocols may soon helpto determine the type and duration of follow up required.
The pheochromocytoma of the adrenal scaled score (PASS)proposed by Thompson in 2002 [44], applied only to the
Endocr Pathol

adrenal. It assigns numerical weights to 12 histological pa-rameters including necrosis, vascular and capsular invasion,mitotic count, and extension into periadrenal fat. As originallyreported, tumors with a summated score of <4 did not metas-tasize. Some institutions now report good prognosticationwith the PASS system or modified versions of it [46], whileothers do not. The differences might be attributable partly todifferent genotypes of the patients studied and partly to poorconcordance between pathologists in assigning scores [47]. Asecond system described by Kimura in 2005 [45] applies toboth PCC and extra-adrenal PGL. It considers some the samehistological parameters as the PASS together with biochemi-cal phenotype and Ki-67 labeling to derive a three-tier riskstratification. A large multi-institutional study underway inJapan shows good prognostication with this system despitethe absence of genetic data, with scoring done by a singlepathologist. Various immunohistochemical markers have alsobeen proposed to correlate with metastatic potential, withmixed results as recently reviewed [11].
In addition to its traditional uses in differential diagnosis,functional characterization, and identification of biomarkers,immunohistochemistry plays an important new role in guidingthe genetic testing of patients with PCC and PGL. It was firstshown in 2005 that immunoreactivity for SDHB protein is lostin PCC or PGL with mutations of either the SDHB or SDHDgene [48]. Subsequently this loss was found to occur as aconsequence of mutations in any of the genes encoding sub-units of the SDH complex, i.e., SDHA , SDHB , SDHC SDHD ,or SDHAF2 . IHC for SDHB thereby serves as a pre-screen forall SDHx mutations[49] (Fig. 2). A complementary test isimmunostaining for SDHA, which is lost only with SDHAgene mutations [50]. These immunohistochemical geneticscreening tests initially developed for PCC were rapidly ap-plied to syndromically associated lesions and can now be usedto identify SDHB and SDHA mutations in both GISTs andrenal cell carcinomas [51].
A question raised early on concerning IHC screening forSDHx mutations is whether the benefits of the tests outweighthe potentially tragic consequences of missing a mutationbecause of high background staining seen in some tumorseven when the test is performed in an expert setting [52]. Inaddition, some tumors without a SDHB mutation show loss ofSDHB protein [52]. It is likely that both types of potentiallymisleading result are associated with mutations of VHL or ofgenes encoding hypoxia inducible transcription factors, be-cause VHL mutation can lead to downregulation of SDHBthrough a regulatory loop mediated by HIFα [53]. Immuno-blots suggest that this downregulation could be nearly com-plete or partial [54]. Optimal interpretation of the IHC test isaided by adhering to a guideline that SDHB immunoreactivityin tumor cells should be as intense as in neighboring endothe-lial cells and should be in a granular mitochondrial distributionin order for a tumor to be called positive for SDHB (negativefor mutation) (Fig. 3). As both IHC and molecular genetictesting will always entail some potential for error, both mustalways be interpreted in clinical context.
An important responsibility of pathologists is to help clini-cians interpret the significance of pathologic findings. This rolehas always existed. However, it assumes particular importancein the shifting complex landscape of PCC/PGL. To that end, asynoptic reporting template has been proposed that incorpo-rates an optional clinicopathologic correlation section Whenpossible, this encourages the pathologist to consider all aspectsof a case including tumor distribution history of other lesions,biochemical data, H&E histology, and immunohistochemistryin order to formulate an optimally informative report [55]. Thisis particularly important with respect to the possibility ofhereditary diseases, in which associated lesions can be moreimportant than PCC or PGL themselves. The data summarizedin Tables 1 and 2 are approximations as of September 2013.However, genetic data are changing almost daily. Textbooksare no longer a reliable way to remain up to date.
Fig. 3 Immunohistochemical staining for SDHB in PGLs with mutatedor wild-type SDHB genes. a Typical positive staining and wild-typegene; b typical negative staining and mutated gene; c unusually highbackground and mutated gene. Endothelial cells (E) serve as intrinsicpositive controls for this IHC test, showing coarsely granular cytoplasmic
staining corresponding to the mitochondrial localization of the SDHcomplex. Note that tumor cells (T) in (a), stain as intensely as endothelialcells and have the same granular staining pattern. In (c), the staining oftumor cells is lighter than that of endothelial cells and lymphocytes (L) inthe same field and is diffuse rather than granular
Endocr Pathol

The Standard of Care
A Clinical Vignette, 2014
A 41-year-old man presents with hypertension fluctuatingmarkedly in severity over the past year. He reports intermittentsevere headaches but is otherwise asymptomatic. He denieshaving palpitations or abnormal sweating. The physician con-siders the possibility of a pheochromocytoma despite theabsence of the classic symptom triad. Biochemical testing offree plasma metanephrines shows elevated normetanephrine,normal metanephrine and borderline elevation ofmethoxtyramine. CT imaging shows a 5-cm wellcircumscribed abdominal mass near the aortic bifurcation.The mass is resected uneventfully after pharmacologicalblockade and diagnosed as a PGL. IHC staining shows thetumor is negative for SDHB. The patient is offered genetictesting, which shows a germline mutation of the SDHB gene.Testing of family members shows the same mutation in thepatient’s 13-year-old son. Follow-up biochemical testingshows persistent normetanephrine elevation in the patientand functional imaging studies show a second smallparaspinal thoracic tumor. Elevated normetanephrine andmethoxytyramine levels are also found in his son, in whomtwo small abdominal tumors are discovered. All tumors aresuccessfully resected, and both affected individuals are toldthat they will require lifelong surveillance.
The precise details of this 2013 scenario will vary some-what depending on clinicians’ preferences and institutionalavailability of analytical and imaging techniques. The keypoints are the following:
& The possibility of PCC or PGL is considered early in thecourse of disease
& Assay of plasma free metanephrines serves as a highlysensitive biochemical screen for the presence ofcatecholamine-producing tumors
& Functional imaging techniques provide highly sensitivetumor localization
& Clinicians are aware of the genetic implications of a PCCor PGL and all patients are offered genetic testing
& All patients receive lifelong surveillance.
All of these points represent recent improvements in patientcare.
References
1. Gimenez-Roqueplo AP, Dahia PL, Robledo M: An update on thegenetics of paraganglioma, pheochromocytoma, and associated he-reditary syndromes. Horm Metab Res 44:328–33, 2012
2. Baysal BE, Ferrell RE, Willett-Brozick JE, et al.: Mutations inSDHD, a mitochondrial complex II gene, in hereditaryparaganglioma. Science 287:848–51, 2000
3. Niemann S, Muller U: Mutations in SDHC cause autosomal domi-nant paraganglioma, type 3. Nat Genet 26:268–70, 2000
4. Astuti D, Latif F, Dallol A, et al.: Gene mutations in the succinatedehydrogenase subunit SDHB cause susceptibility to familial pheo-chromocytoma and to familial paraganglioma. Am J Hum Genet 69:49–54, 2001
5. Neumann HP, Bausch B, McWhinney SR, et al.: Germ-linemutations in nonsyndromic pheochromocytoma. N Engl J Med346:1459–66., 2002
6. Boedeker CC, Hensen EF, NeumannHP, et al.: Genetics of hereditaryhead and neck paragangliomas. Head Neck 2013
7. Pasini B, Stratakis CA: SDH mutations in tumorigenesis andi n h e r i t e d e n d o c r i n e t umo u r s : l e s s o n f r om t h ephaeochromocytoma-paraganglioma syndromes. J Intern Med266:19–42, 2009
8. Malinoc A, Sullivan M, Wiech T, et al.: Biallelic inactivation of theSDHC gene in renal carcinoma associated with paraganglioma syn-drome type 3. Endocr Relat Cancer 19:283–90, 2012
9. Xekouki P, Stratakis CA: Succinate dehydrogenase (SDHx)mutations in pituitary tumors: could this be a new role formitochondrial complex II and/or Krebs cycle defects? EndocrRelat Cancer 19:C33–40, 2012
10. Beckers A: Means, motive, and opportunity: SDH mutations aresuspects in pituitary tumors. J Clin Endocrinol Metab 98:2274–6, 2013
11. Eisenhofer G, Tischler AS, de Krijger RR: Diagnostic tests andbiomarkers for pheochromocytoma and extra-adrenalparaganglioma: from routine laboratory methods to disease stratifi-cation. Endocr Pathol 23:4–14, 2012
12. Jimenez C, Rohren E, HabraMA, et al.: Current and future treatmentsfor malignant pheochromocytoma and sympathetic paraganglioma.Curr Oncol Rep 15:356–71, 2013
13. Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, et al.:Research resource: Transcriptional profiling reveals differentpseudohypoxic signatures in SDHB and VHL-related pheochromo-cytomas. Mol Endocrinol 24:2382–91, 2010
14. Shankavaram U, Fliedner SM, Elkahloun AG, et al.: Genotypeand tumor locus de te rmine expres s i on pro f i l e o fpseudohypoxic pheochromocytomas and paragangliomas.Neoplasia 15:435–47, 2013
15. Dahia PL: The genetic landscape of pheochromocytomas andparagangliomas: somatic mutations take center stage. J ClinEndocrinol Metab 98:2679–81, 2013
16. BurnichonN, Buffet A, Parfait B, et al.: Somatic NF1 inactivation is afrequent event in sporadic pheochromocytoma. Hum Mol Genet 21:5397–405, 2012
17. Toledo RA, Qin Y, Srikantan S, et al.: In vivo and in vitro oncogeniceffects of HIF2A mutations in pheochromocytomas andparagangliomas. Endocr Relat Cancer 20:349–59, 2013
18. Pacak K, Jochmanova I, Prodanov T, et al.: New syndrome ofparaganglioma and somatostatinoma associated with polycythemia.J Clin Oncol 31:1690–8, 2013
19. Crona J, Delgado Verdugo A, Maharjan R, et al.: SomaticMutations in H-RAS in Sporadic Pheochromocytoma andParaganglioma Identified by Exome Sequencing. J ClinEndocrinol Metab 2013
20. Eisenhofer G, Goldstein DS, Kopin IJ, Crout JR: Pheochromocytoma:rediscovery as a catecholamine-metabolizing tumor. Endocr Pathol 14:193–212, 2003
21. Manger WM: The protean manifestations of pheochromocytoma.Horm Metab Res 41:658–63, 2009
22. Timmers HJ, Pacak K, Huynh TT, et al.: Biochemically silent ab-dominal paragangliomas in patients with mutations in the succinate
Endocr Pathol

dehydrogenase subunit B gene. J Clin Endocrinol Metab 93:4826–32, 2008
23. Balogova S, Talbot JN,Nataf V, et al.: 18F-fluorodihydroxyphenylalaninevs other radiopharmaceuticals for imaging neuroendocrine tu-mours according to their type. Eur J Nucl Med Mol Imaging40:943–66, 2013
24. Hartung-Knemeyer V, Rosenbaum-Krumme S, Buchbender C, et al.:Malignant pheochromocytoma imaging with [124I]mIBG PET/MR.J Clin Endocrinol Metab 97:3833–4, 2012
25. Kabasakal L, Demirci E, Ocak M, et al.: Comparison of (6)(8)Ga-DOTATATE and (6)(8)Ga-DOTANOC PET/CT imaging in the samepatient group with neuroendocrine tumours. Eur J Nucl Med MolImaging 39:1271–7, 2012
26. Taieb D, Neumann H, Rubello D, Al-Nahhas A, Guillet B, Hindie E:Modern nuclear imaging for paragangliomas: beyond SPECT. J NuclMed 53:264–74, 2012
27. Timmers HJ, Taieb D, Pacak K: Current and future anatomical andfunctional imaging approaches to pheochromocytoma andparaganglioma. Horm Metab Res 44:367–72, 2012
28. Ilias I, Pacak K: Current approaches and recommended algorithm forthe diagnostic localization of pheochromocytoma. J Clin EndocrinolMetab 89:479–91, 2004
29. Taieb D, Timmers HJ, Hindie E, et al.: EANM 2012 guidelines forradionuclide imaging of phaeochromocytoma and paraganglioma.Eur J Nucl Med Mol Imaging 39:1977–95, 2012
30. Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, et al.:Imaging work-up for screening of paraganglioma and pheo-chromocytoma in SDHx mutation carriers: a multicenter pro-spective study from the PGL.EVA Investigators. J ClinEndocrinol Metab 98:E162-73, 2013
31. Sharma P, Thakar A, Suman KCS, et al.: 68Ga-DOTANOC PET/CTfor baseline evaluation of patients with head and neck paraganglioma.J Nucl Med 54:841–7, 2013
32. Mayerhoefer ME, Ba-Ssalamah A, Weber M, et al.: Gadoxetate-enhanced versus diffusion-weighted MRI for fused Ga-68-DOTANOC PET/MRI in patients with neuroendocrine tumours ofthe upper abdomen. Eur Radiol 23:1978–85, 2013
33. Waguespack SG, Rich T, Grubbs E, et al.: A current reviewof the etiology, diagnosis, and treatment of pediatric pheo-chromocytoma and paraganglioma. J Clin Endocrinol Metab95:2023–37, 2010
34. Sovinz P, Urban C, Uhrig S, et al.: Pheochromocytoma in a 2.75-year-old-girl with a germline von Hippel–Lindau mutation Q164R.Am J Med Genet A 152A:1752–5, 2010
35. Benhammou JN, Boris RS, Pacak K, Pinto PA, Linehan WM,Bratslavsky G: Functional and oncologic outcomes of partialadrenalectomy for pheochromocytoma in patients with vonHippel–Lindau syndrome after at least 5 years of followup. JUrol 184:1855–9, 2010
36. Manny TB, Pompeo AS, Hemal AK: Robotic partial adrenalectomyusing indocyanine green dye with near-infrared imaging: the initialclinical experience. Urology 82:738–42, 2013
37. Ayala-Ramirez M, Chougnet CN, Habra MA, et al.: Treatment withsunitinib for patients with progressive metastatic pheochromocyto-mas and sympathetic paragangliomas. J Clin Endocrinol Metab 97:4040–50, 2012
38. Chae YC, Angelin A, Lisanti S, et al.: Landscape of the mitochon-drial Hsp90 metabolome in tumours. Nat Commun 4:2139, 2013
39. Nolting S, Garcia E, Alusi G, et al.: Combined blockade of signallingp a t hway s s hows ma rk ed an t i - t umou r po t e n t i a l i nphaeochromocytoma cell lines. J Mol Endocrinol 49:79–96, 2012
40. Welander J, Garvin S, Bohnmark R, et al.: Germline SDHAmutationdetected by next-generation sequencing in a young index patient withlarge paraganglioma. J Clin Endocrinol Metab 98:E1379–80, 2013
41. Tischler AS: Pheochromocytoma and extra-adrenal paraganglioma:updates. Arch Pathol Lab Med 132:1272–84, 2008
42. DeLellis RA, Lloyd RV, Heitz PU, Eng C: Tumours of EndocrineOrgans. In World Health Organization Classification of Tumors.IARC Press, Lyon, 2004
43. Linnoila RI, Keiser HR, Steinberg SM, Lack EE: Histopathology ofbenign versus malignant sympathoadrenal paragangliomas: clinico-pathologic study of 120 cases including unusual histologic features.Hum Pathol 21:1168–80, 1990
44. Thompson LD: Pheochromocytoma of the Adrenal gland ScaledScore (PASS) to separate benign from malignant neoplasms: a clin-icopathologic and immunophenotypic study of 100 cases. Am J SurgPathol 26:551–66, 2002
45. Kimura N, Watanabe T, Noshiro T, Shizawa S, Miura Y: Histologicalgrading of adrenal and extra-adrenal pheochromocytomas and rela-tionship to prognosis: a clinicopathological analysis of 116 adrenalpheochromocytomas and 30 extra-adrenal sympatheticparagangliomas including 38 malignant tumors. Endocr Pathol 16:23–32, 2005
46. Strong VE, Kennedy T, Al-Ahmadie H, et al.: Prognostic indicatorsof malignancy in adrenal pheochromocytomas: clinical, histopatho-logic, and cell cycle/apoptosis gene expression analysis. Surgery 143:759–68, 2008
47. Wu D, Tischler AS, Lloyd RV, et al.: Observer variation in theapplication of the Pheochromocytoma of the Adrenal Gland ScaledScore. Am J Surg Pathol 33:599–608, 2009
48. Dahia PL, Ross KN, Wright ME, et al.: A HIF1alpha RegulatoryLoop Links Hypox ia and Mi tochondr ia l S igna l s inPheochromocytomas. PLoS Genet 1:e8, 2005
49. van Nederveen FH, Gaal J, Favier J, et al.: An immunohistochemicalprocedure to detect pat ients with paraganglioma andphaeochromocytoma with germline SDHB, SDHC, or SDHD genemutations: a retrospective and prospective analysis. Lancet Oncol 10:764–71, 2009
50. Korpershoek E, Favier J, Gaal J, et al.: SDHA immunohistochemistrydetects germline SDHA gene mutations in apparently sporadicparagangliomas and pheochromocytomas. J Clin Endocrinol Metab96:E1472–6, 2011
51. Gill AJ: Succinate dehydrogenase (SDH) and mitochondrial drivenneoplasia. Pathology 44:285–92, 2012
52. Erlic Z, Neumann HP: Diagnosing patients with hereditaryparaganglial tumours. Lancet Oncol 10:741, 2009
53. Dahia PL, Ross KN,Wright ME, et al.: A HIF1alpha regulatory looplinks hypoxia and mitochondrial signals in pheochromocytomas.PLoS Genet 1:72–80, 2005
54. Dahia PL: Transcription association of VHL and SDHmutations linkhypoxia and oxidoreductase signals in pheochromocytomas. Ann NYAcad Sci 1073:208–20, 2006
55. Mete O, Tischler AS, R dK, et al.: Protocol for the Examination ofSpecimens from Patients with Pheochromocytomas and Extra-adrenal Paragangliomas. Arch Pathol Lab Med 2013
56. Tudorancea A, Francois P, Trouillas J, et al.: Von Hippel–Lindaudisease and aggressive GH-PRL pituitary adenoma in a young boy.Ann Endocrinol (Paris) 73:37–42, 2012
57. Kurozumi K, Tabuchi A, Ono Y, et al.: [Pituitary adenoma associatedwith neurofibromatosis type 1: case report]. No Shinkei Geka 30:741–5, 2002
58. Dwight T, Mann K, Benn DE, et al.: Familial SDHA mutationassociated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab 98:E1103-8, 2013
59. Stratakis CA, Carney JA: The triad of paragangliomas, gastric stro-mal tumours and pulmonary chondromas (Carney triad), and thedyad of paragangliomas and gastric stromal sarcomas(Carney–Stratakis syndrome): molecular genetics and clinical impli-cations. J Intern Med 266:43–52, 2009
60. Bayley JP, Kunst HP, CasconA, et al.: SDHAF2mutations in familialand sporadic paraganglioma and phaeochromocytoma. Lancet Oncol11:366–72, 2010
Endocr Pathol

61. Burnichon N, Briere JJ, Libe R, et al.: SDHA is a tumor suppressorgene causing paraganglioma. Hum Mol Genet 19:3011–20, 2010
62. Qin Y, Yao L, King EE, et al.: Germline mutations inTMEM127 confer susceptibility to pheochromocytoma. Nat Genet42:229–33, 2010
63. Burnichon N, Cascon A, Schiavi F, et al.: MAX mutations causehereditary and sporadic pheochromocytoma and paraganglioma. ClinCancer Res 18:2828–37, 2012
64. Astuti D, Ricketts CJ, Chowdhury R, et al.: Mutation analysis of HIFprolyl hydroxylases (PHD/EGLN) in individuals with features ofphaeochromocytoma and renal cell carcinoma susceptibility.Endocr Relat Cancer 18:73–83, 2011
65. Lorenzo FR, Yang C, Ng Tang Fui M, et al.: A novel EPAS1/HIF2Agermline mutation in a congenital polycythemia with paraganglioma.J Mol Med (Berl) 91:507–12, 2013
66. Zhuang Z, Yang C, Lorenzo F, et al.: Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl JMed 367:922–30, 2012
67. Schlisio S, Kenchappa RS, Vredeveld LC, et al.: The kinesinKIF1Bbeta acts downstream from EglN3 to induce apoptosisand is a potential 1p36 tumor suppressor. Genes Dev 22:884–93, 2008
68. Maher ER: HIF2 and endocrine neoplasia: an evolving story. EndocrRelat Cancer 20:C5–7, 2013
Endocr Pathol