Embed Size (px)
Citation preview

Eur. J . Biochem. 62, 401 -410 (1976)
The Enzymic Addition of Poly(A) to the 3’-End of RNA using Bacteriophage MS 2 RNA as a Model System
Rene DEVOS, Elie GILLIS, and Walter FTERS
Ldboratorium voor Fysiologische Scheikunde and Ldboratorium voor Moleculaire Biologie, Rijksuniversiteit Gent
(Received July 14/0ctober 24, 1975)
ATP : RNA adenyltransferase, purified from Escherichia coli, was used to add a series of adenosine residues to the 3‘-end of MS2 RNA. Incubations of the order of a few minutes at 37 “C were sufficient for synthesis of a short poly(A) chain that did not appreciably alter the hydrodynamic or electrophoretic properties of MS2 RNA. The size of the poly(A) tails was estimated by gel electro- phoresis after prior hydrolysis of the primer RNA with pancreatic ribonuclease. These results were in good agreement with the values calculated on the basis of the relative amount of incorporated AMP. After the addition of a short poly(A) tail, approximately 50 ”/, of the treated material binds specifically to an oligo(dT)-cellulose column. The majority of the recovered poly(A)-containing RNA was still intact, as shown by analysis on polyacrylamide gel.
After incubations beyond 6 min, slowly sedimenting material, also showing reduced electro- phoretic mobility, was formed. Presumably this material corresponds to RNA chains to which long poly(A) tails are linked.
Enzymes catalyzing the addition of ribonucleotides from ribonucleoside triphosphates (most frequently ATP) to the 3‘-end of various RNA’s, have been shown to exist in a wide variety of systems [l]. The physiological role of most of these enzymes is obscure; however, the presence of poly(A) stretches at the 3’-end of most eukaryotic mRNA’s is very well documented. Although the enzymic activities isolated from different sources have often not been fully characterized, the reactions seem to proceed in the same way by sequential addition of monomer residues to the primer molecules, and/or synthesis of long poly(A) chains de now. These enzymes could prove very useful for synthesis in vitro of RNA-poly(A) molecules (mimics of eukaryotic mRNA’s), but have not been extensively exploited to date [2,3].
Recently, Sippel [4] has characterized an enzyme from Escherichia coli catalyzing a primer-dependent synthesis of poly(A). It can be distinguished from a variety of other bacterial enzymes synthesizing poly(A). This enzyme, designated as ATP : RNA adenyltrans-
This is paper XXX of a series; paper XXIX was by Fiers et al., Nature (Lond.) 256, 273 (1975).
Abbreviations. Poly(A), poly(adeny1ic acid) ; oligo(dT), oligo- (thymidylic acid); (rA)ss and (rA)w, poly(adeny1ic acid) with an average of 25 and 90 residues respectively; poly(U), poly(uridy1ic acid).
Enzymes. ATP : RNA adenyltransferase (EC 2.7.7.19); ribo- nuclease A (EC 3.1.4.22); polynucleotide phosphorylase (EC 2.7. 7.8).
ferase, was extensively purified. Characterization of the product, using tRNA as a primer, showed that short poly(A) chains were added to the 3’-end of the RNA. The majority of the incorporated AMP, however, was present as very long poly(A) chains. By shortening the time of incubation we were able to avoid the incorporation of AMP into these long poly(A) chains.
The purpose of this report is to show the pos- sibility of using ATP : RNA adenyltransferase, puri- fied according to Sippel, for adding poly(A) tails to a wide variety of natural RNA’s and more specifically to bacteriophage RNA’s, like MS2 RNA.
Recently, Gilvarg et af. [5] used a terminal ribo- adenylate transferase purified from calf thymus [6] to add a poly(A) tail to the 3‘-end of bacteriophage QB RNA.
MATERIALS AND METHODS
Materials
All labeled nucleotides were purchased from The Radiochemical Centre, Amersham. Unlabeled nucleo- tides and dithiothreitol were obtained from Sigma. For column chromatography, cellulose P11 (7.4 mequiv./g) from Whatman, Sephadex G-25 and G-1 00 from Pharmacia, and oligo(dT)-cellulose from Collaborative Research were used. Escherichia coli

402 Addition of Poly(A) to the 3'-End of MS2 RNA
B/r was kindly provided by Dr A. E. Sippel. 32P- labeled and 14C-labeled MS2 RNA, unlabeled MS2 RNA and Qfl RNA were prepared by dodecylsulphate/ phenol extraction of purified virus.
1 6 3 ribosomal RNA was obtained by treatment of a ribosome pellet from E. coli D11 with phenol and further purified by glycerol gradient centrifuga- tion. 5-S ribosomal RNA was purified by chromato- graphy on a Scphadex G-100 column. Encephalomyo- carditis (EMC) RNA was kindly supplied by Dr K. Marcker. E. coli tRNA was a product of Boehringer, while (rA)c and (rA), from Miles were used for calibration of gel electrophoresis. Pancreatic ribo- nuclease A was purchased from Worthington.
Enzyme Assuys
The incubation temperature for all reactions was 37 "C.
ATP : RNA Adenyltransferase. The standard assay measured the conversion of [3H]ATP into acid- insoluble material. The incubation mixtures (0.1 ml) contained 50 mM Tris-HC1 (pH 7.9), 1 mM dithio- threitol, 200mM NaC1, 10mM MgCI,, 2.5 mM MnCl,, 0.2 mM [3H]ATP (10 pCi/pmol), 5- 10 pg MS2 RNA or 15pg tRNA and enzyme (where possible, an amount corresponding to approximately 2 pg of purified ATP : RNA adenyltransferase). After incubations for appropriate times, 3 ml cold 5 % tri- chloroacetic acid + 0.01 M sodium pyrophosphate was added. The acid-insoluble material was collected on glass filters (Whatman, type GF/C), and the filters were washed three times with cold 5 % trichloroacetic acid and with ethanol. Radioactivity was counted in the Packdrd Tricarb scintillation spectrometer model 3380.
Polynucleotide Phosphorylase. The assay (0. I ml) contained 85 mM Tris-HC1 (pH 8.5), 15 mM MgCl,, 0.16 mM [14C]ADP (1 pCi/ymol), 15 pg tRNA and enzyme. About 0.6 pg of purified polynucleotide phosphorylase (Sephadex G-200 step [7]) was used as a reference. The radioactivity was counted as above.
Buffers
Buffer A contained 0.1 M NaCl, 5 mM Tris-HC1, pH 7.0, 1 mM MgCI,, 0.1 mM EDTA. Electrophoresis buffer contained 0.04 M Tris-acetate, pH 7.2, 0.02 M sodium acetate, 1 mM sodium EDTA.
Phenol/chloroform/isoamylalcohol (25/24/1, by vol.) [8], was equilibrated with acetate buffer (0.1 M NaC1, 10 mM sodium acetate pH 6.0, 1 mM EDTA).
Poiyacrylamide Gel Electrophoresis of R N A
Electrophoresis of high-molecular-weight RNA was carried out in 2.4% polyacrylamide gels in elec-
trophoresis buffer containing 0.2 sodium dodecyl sulphate [9], while electrophoresis of the poly(A) sequences was carried out in 10 % polyacrylamide gels in 0.05 M Tris-acetate, pH 7.0 buffer [lo, 111. The gels were 6 mm in diameter and 7 cm long; pre- electrophoresis was for 30 min at 5 mA/gel. Samples were applied in electrophoresis buffer containing 6 % sucrose and bromophenol blue as a marker. After electrophoresis, the gels were scanned in a Joyce Loebl Chromoscan, using a 260-nm interference filter. After freezing, the gels were cut into 1-mm slices. 0.35 ml50 mM Tris-HC1 pH 7.5,O. 1 % sodium dodecyl sulphate was added to each slice in a scintillation vial and the mixture was incubated overnight at 37 "C. Next, 3 ml Triton/toluene scintillation mixture was added (4 g PPO, 0.1 g POPOP, 333 ml Triton X-100, 667 ml toluene), and the radioactivity was measured.
Glycerol Gradient Centrifugation
After ethanol precipitation the polyadenylated product was dissolved in 0.1 ml 2 mM Tris-HC1, pH 7.5, 1 mM EDTA and loaded on a 10-30% glycerol gradient made up in buffer A. The gradients were centrifuged for 5 h at 40000 rev./min in a Spinco SW-41 rotor. The absorbance at 260 nm was monitored continuously using an Isco gradient frac- tionator, ultraviolet detector and recorder. The acid- precipitable radioactivity of each fraction was mea- sured.
Oligo (dT) -cellulose Chromatography
The RNA from the peak fractions of the glycerol gradients was precipitated with ethanol and dissolved in 10 pl water. 0.3 ml 0.5 M KC1, 10 mM Tris-HC1, pH 7.5 was added, and the RNA was loaded at room temperature onto an oligo(dT)-cellulose column (0.3 ml bed volume), packed in a pasteur pipette [12, 131. The column was then washed with five 0.3-ml portions loading buffer and eluted with seven 0.3-ml portions 10 mM Tris-HC1, pH 7.5. The acid-pre- cipitable radioactivity of each fraction was measured.
RESULTS
Characterization of the Purified Enzyme
ATP : RNA adenyltransferase of Escherichia coli B/r was purified according to Sippel [4], using phos- phocellulose chromatography in the presence of a high salt concentration as the last purification step. The AMP-incorporating activity was completely prim- er-dependent (Fig. 1). The activity shoulder at the lower side of the salt gradient was presumably due to nucleolytic activity, as was shown by incubation of the fractions with MS2 [32P]RNA and determina-

R. Devos, E. Gillis. and W. Fiers 403
70
60 '
20
10
0
12
10 .: 0
6 %
4
2
0
. 2 4 N zz2 - /\
0 10 20 30 40 Fraction number
Fig. 1. Purificuiion of A T P : RNA ad~,i?.r.li,.urisJerasr on a phospho- cellulo.se cohmn. ATP : RNA adenyltransferase was assayed on 10-pl aliquots as described in Materials and Methods ( b O ) , using ["HIATP at a specific activity of 10 pCi/pmol. To dctermine the primer-independent activity (A- -A), the specific activity of the ['HIATP was raised to 46pCi/pmol. Ribonuclease test (0-0): a 10 pl aliquot of each fraction was incubated for 1 h at 37 "C with 25 pg Escherichia coli rRNA, 3 pg MS2 [32P]RNA (30000 counts/min) in 50 p1 adenyltransferase assay mixture but without ATP. After addition of 0.2 ml 5 trichloroacetic acid + 0.01 M sodium pyrophosphate and centrifugation of the precip- itate. 0.1 rnl of the supernatant was used for determination of the acid-soluble radioactivity. A control with 10 p1 H,O resulted in 900 counts/min acid-soluble material (not subtracted). Polynucleo- tide phosphorylase tests (A-A) were carried out using 10 pl of each fraction and ['4C]ADP at a specific activity of 1 pCi/pmol. Thc NaCl gradient was determined by conductivity measurements (*---a)
tion of the acid-solubilized radioactivity. Polynucleo- tide phosphorylase activity was negligible as measured by AMP incorporation with [14C]ADP as a sub- strate. When UTP was used as a substrate at pH 10 a significant incorporation into acid-precipitable ma- terial was observed. It is not known whether this activity is due to a contaminating enzyme [14], or to the ATP : RNA adenyltransferase itself.
The enzyme responds to various RNA species as primers for addition of a poly(A) stretch at their 3'-end (Table 1). At the primer concentrations used the rate of the reaction for most types of RNA was nearly independent of the amount of RNA, except for tRNA.
Bacteriophage MS2 R N A as a Primer .for ATP: R N A Adenyltransferase
The NaCl concentration for optimal poly(A)- polymerizing activity, using MS2 RNA as a primer, was between 0.15 M and 0.20 M.
Table 1 . Stimulation of AMP incorporation by wrious R N A species Limited synthesis was carried out by incubation for 5 min at 37 'C with ATP : RNA adenyltransferase in the standard assay (0.1 ml total volume). The specific activity of the [3H]ATP was 50 pCi/pmol
Primer Amount of RNA Total AMP incor- poration
Kg pmol
MS2 RNA 2.70 2.4 10.5 9.5
QB RNA 3.75 2.8 12.5 9.6
tRNA 0.054 2.2 0.27 11
1 6 3 RNA 1.2 2.1 6.0 11
EMC RNA 5.44 2.7
pmol
1490 1150
1420 990
430 900
1450 1520
1580
MS2 RNA ( k g / O . l m l )
Fig.2. Ejfect of MS2 R N A concentration on the rate of A M P in- corporation by ATP : RNA adenyltransfrrase. Standard ATP : RNA adenyltransferase assay mixtures (0.1 ml) were used except for the variable concentrations of MS2 RNA. The incubation was for 5 min at 37 "C. ( a d ) AMP incorporation; ( x - x ) molar ratio of AMP incorporated to MS2 RNA present
The effect of increasing amounts of MS2 RNA on the rate of AMP incorporation, measured after 5-min incubation, closely resembles saturation (Fig. 2). At low primer levels (0.1 - 3 pg/O.l ml) calculation of the ratio of incorporated AMP versus molar amount of MS2 RNA present shows that the average length of the synthesized poly(A) chains decreases sharply with increasing amounts of primer.
After 15-min incubation at 37 "C at high MS2 RNA concentrations, 5 nmol (25% of the input) of [3H]ATP was incorporated in acid-insoluble material.

404
10
Addition of Poly(A) to the 3'-End of MS2 RNA
10 min
5. -
I 0
10
10 min
5. -
0 15min '2
) OO 1 2 3 4 5 Migration (cm)
Fig.3. Gel electrophoretic analysis of the enzymic product of M S 2 R N A and ATP: R N A adenyltransferase as afunction of the incubation time. ATP:RNA adenyltransferase was incubated in the standard system (0.1 ml) with 10 pg MS2 RNA for 0, 5, 10 and 15 min at 37 "C. The reaction was stopped with 2 pl 20 % dodecylsulphate and treated with 0.1 ml phenol/chloroform/isoamylalcohol mixture. After precipitation of the RNA with 0.1 vol. 2 M potassium acetate (pH 7.0) and 2 vol. ethanol, the RNA was dissolved in 25 p1 electrophoresis buffer and loaded on 2.4 "/, polyacrylamide gels. The arrows indicate the position of MS2 RNA
CHARACTERIZATION OF THE REACTION PRODUCTS USING MS2 RNA AS A PRIMER
Gel Filtration
After treatment with phenol and ethanol pre- cipitation the enzymic products of the reaction were isolated by gel filtration on a Sephadex G-25 or a G-100 column. The profiles showed that the [3H]AMP was incorporated into high-molecular-weight material, eluting in the exclusion volume.
Polyacrylamide Gel Electrophoresis
Electrophoresis in 2.4 % polyacrylamide gels of the products after 0, 5, 10 and 15 min of incubation showed that part of the PHIAMP label coincided with MS2 RNA (Fig.3), suggesting a covalent linkage between MS2 RNA and poly(A). If the incubations exceeded 5 min, however, a second more slowly migrating peak of radioactivity appeared. Presumably this material corresponds to MS2 RNA to which a long and rather rigid poly(A) strand is attached (further characterization of the product is given below).
Glycerol Gradient Centrifugation
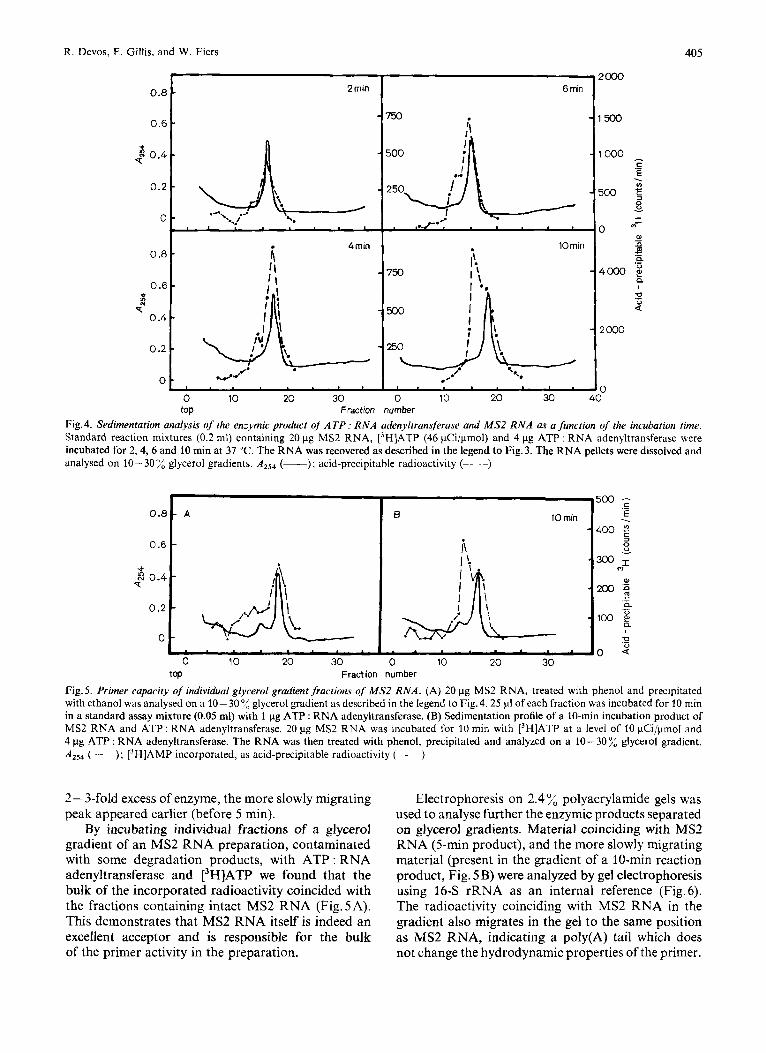
Zone centrifugation of the reaction products was used to show a physical attachment of the poly(A) tail to the 3'-end of MS2 RNA molecules. Standard assay mixtures containing [3H]ATP, ATP : RNA adenyltransferase and MS2 RNA were incubated for 2, 4, 6 and 10 min at 37 "C, and then analysed on 10- 30 % glycerol gradients (Fig. 4). The radioactivity profiles show a characteristic pattern, comparable to the results obtained by gel electrophoresis. After short incubation periods (0- 5 min) the incorporated label coincided with MS2 RNA. After longer incuba- tion periods, however, a second peak appeared which now sedimented more slowly in the gradients. The special structure of the poly(A) tail presumably affects the hydrodynamic properties of the product, resulting in a considerable drag once a critical number of adenosine residues has been reached. That the slower sedimentation is not due to degradation, but corre- sponds to a stiff, bulkier molecule, is shown by the fact that this component is identical to the material which has a smaller mobility in polyacrylamide gel than MS2 RNA (cf. below).
The overall rate of the reaction was directly pro- portional to the amount of enzyme added. Using a
R. Devos, E. Gillis. and W. Fiers
0.8
0.6
7 J 0 - 4 1
0 . 2
0 -
0.8
O a 6 : a TN 0.4
0.2
0 -
405
2min 6 min - - 750
\$, ~ -=)J - . i0J ,
- 1. -. .-',* .-. I -../ 4. , ,.I /-.-* ,
I \ 750 II
4min 10min - r;
.' ' ! '. I
- 250 - . dfl' ,/ %*..
0 10 2 0 30 0 10 20 30
Fig.4. Sedimentation unulysis of' the enzymic product of' ATP : RNA adenyltruns/&me und MS2 RNA a.? a function of the incubution time. Standard reaction mixtures (0.2 ml) containing 20 pg MS2 RNA, [3H]ATP (46 pCi/pmol) and 4 pg ATP : RNA adenyltransferase were incubated for 2,4, 6 and 10 min at 37 "C. The RNA was recovered as described in the legend to Fig. 3. The RNA pellets were dissolved and analysed on 10- 30 yd glycerol gradients. (-); acid-prccipitable radioactivity (-- --)
500 .- E . 10 min 0.8 - A B
-400 f 0 h u
\t - 3 0 0 m I
i - 100 g
0.6 -
(u
-200; 8 0.4 -
T w ._ a ._ 0 . 2 - -
0 - 0
o a i . . 0 10 20 30 0 10 20 30
top Fraction number Fig.5. Primer capacity of individual glvcerol gradient fractions of' MS2 RNA. (A) 20 pg MS2 RNA, treated with phenol and Precipitated with ethanol was analysed on a 10- 3004 glycerol gradient as described in the legend to Fig.4. 25 p1 of each fraction was incubated for 10 rnin in a standard assay mixture (0.05 ml) with 1 pg ATP : RNA adenyltransferase. (B) Sedimentation profile of a 10-min incubation product of MS2 RNA and ATP : RNA adenyltransferdse. 20 pg MS2 RNA was incubated for 10 rnin with [3H]ATP at a level of 10 pCi/pmol and 4 pg ATP : RNA adenyltransferase. The RNA was then treated with phenol, precipitated and analyzed on a 10-300,: glycerol gradient. A,,, (----): [3H]AMP incorporated, as acid-precipitable radioactivity (----)
2 - 3-fold excess of enzyme, the more slowly migrating peak appeared earlier (before 5 min).
By incubating individual fractions of a glycerol gradient of an MS2 RNA preparation, contaminated with some degradation products, with ATP : RNA adenyltransferase and [3H]ATP we found that the bulk of the incorporated radioactivity coincided with the fractions containing intact MS2 RNA (Fig. 5A). This demonstrates that MS2 RNA itself is indeed an excellent acceptor and is responsible for the bulk of the primer activity in the preparation.
Electrophoresis on 2.4 % polyacrylamide gels was used to analyse further the enzymic products separated on glycerol gradients. Material coinciding with MS2 RNA (5-min product), and the more slowly migrating material (present in the gradient of a 10-min reaction product, Fig. 5 B) were analyzed by gel electrophoresis using 16-S rRNA as an internal reference (Fig.6). The radioactivity coinciding with MS2 RNA in the gradient also migrates in the gel to the same position as MS2 RNA, indicating a poly(A) tail which does not change the hydrodynamic properties of the primer.

406
16-S rRNA S h i n A h
Addition of Poly(A) to the 3’-End of MS2 RNA
1 I I I I
1 2 3 4 5 Migratlon (crn)
Fig.6. Gel electrophoresis o j the 5-min and 10-min enrymic pr.ottucts of’ MS2 RNA and A T P : RNA adenyltransjerasr afrer purification on 10-30% glycerol grudienfs. Two reaction mixtures (0.4 ml) each containing 40 pg MS2 RNA, 8 pg ATP : RNA adenyltransferase and [3H]ATP (46 pCi/pnol) were incubated for respectively 5 and 10 min. The cnzymic products were analyzed on glycerol gradients. The material (A) coinciding with MS2 RNA and (B) sedimenting morc slowly than MS2 RNA, was precipitated in the presence of 16-S rRNA with 2 vol. of ethanol and dissolved in electrophoresis buffer. AZhO (O--O) (including marker 16-S RNA); [3H]AMP incorporated (0--0)
The more slowly sedimenting peak, however, pene- trates more slowly in the gel than MS2 RNA; this means that this material is not a degradation product. The concept of a critical length of poly(A), creating a rather drastic hydrodynamic change (slower sedi- mentation, slower mobility in gels), arises from the observation that, after more than 5 min of incubation, the second discrete radioactivity peak is found, rather than a gradual shift of the profile.
Estimation ojthe Length of the Poly(A) Chain
After incubations for 2, 4 and 6 min the average lengths of the poly(A) stretches attached to MS2 RNA were calculated on the basis of the acid-insoluble radioactivity relative to the AZ6,, units of MS2 RNA in the glycerol gradients, assuming that all MS2 RNA molecules were indeed used as a primer (Table 2).
A direct estimation of the size of the synthesized poly(A) tails after isolation of the enzymic product by glycerol gradient centrifugation was obtained by digestion with pancreatic ribonuclease in the presence
Table 2. E.stimnted length of the poly(A) stretches attached to MS2 RNA, calculated on lhe basis of precursor incorporution and assuming homogeneous primer ejJiciency The absorbance at 260 nm and acid-precipitable radioactivity of the MS2 RNA peak fractions after glycerol gradient centrifuga- tion of the 2, 4 and 6-min reaction products werc measured. The chain lengths (i.e. number of adenosine rcsidues added) are the calculated average of five experiments carried out as described in the legend to Fig.4. The amounts (pmol RNA) refer to a typical experi- ment
Incubation MS2 RNA Incorporated Chain time AMP length
+ _ n
min pmol pmol
2 5.0 4 4.6 6 4.6
100 26 k 4 220 51 k 6 375 84 9
of a high salt concentration [15- 171. After treatment with phenol, 4-S and 5-S RNA’s were added, and the mixtures were analyzed by electrophoresis on 10 %

R. Devos, E. Gillis. and W. Fiers 407
2 min 5s 4s
1 .o J J . Table 3. Direct measurement of the average length of the pol?( A ) chains The length estimates are based on a calibration curve, reported by Pinder and Gratzer [ l l ] and checked by means of commercial (rA)z and ( r A h preparations. RM (tRNA) is the ratio of the mobilities of the top of the radioactivity profile versus that of the top of the tRNA absorbance peak. The values are the averages of two different experiments, one of which is shown in Fig. 7
~
Incubation R, (tRNA) M , of poly(A) Chain time length
min
2 0.96 10 500 20 - 30 4 0.65 20 000 50 - 60 6 0.51 27 000 80 - 90
polyacrylamide gels. Broad bands for the isolated poly(A) stretches were found, representing a hetero- geneous distribution of sizes (Fig. 7).
The use of 4-S and 5-S RNA’s as references to determine the length of the poly(A) chain, does not take into account any special effect of the poly(A) structure on the electrophoretic mobility. Applica- tion of a calibration curve [ I l l , for determining molecular weight and mobility in 10 % polyacrylamide gels for poly(A) chains relative to tRNA, allowed an estimation of the size. The mean values of the poly(A) chains were calculated using the mobilities of the top of each radioactivity profile (Fig. 7). The results (Table 3) are in good agreement with the previous calculations. They show directly that the size of the added poly(A) tail is indeed a linear function of the incubation time with ATP : RNA adenyltransferase. It should be noted, however, that the calibration curve may not be exactly applicable to our electrophoresis conditions. Therefore, the curve was checked by the use of commercial @A)% and (rA)g preparations as standards. The 50 % range of monomer length of these fractions, based on sedimentation velocity, was re- spectively 25 - 51 and 62- 108 (information provided by the manufacturer). The products moved indeed in the expected regions, but they were too hetero- geneous for exact calibration.
Migration (cm)
Fig. 7. Gel electrophoresis o f p ~ l y ( A ) frugments bolated from MS2 R N A after incubation with ATP : RNA adenyltransferase. Poly- adenylated MS2 RNA isolated from glycerol gradients, as de- scribed in the legend to Fig. 4, was precipitated with ethanol together with 40 pg MS2 RNA. The RNA was dissolved in 0.1 ml buffer A containing 0.25 M NaCl and digested with 2.5 pg pancreatic ribonuclease A for 30 min at 37 “C. The reaction was stopped by addition of 10 p1 10% sodium dodecylsuiphate and 0.1 ml phenol/ chloroform/isoamylalcohol mixture. The RNA was again pre- cipitated with ethanol together with 4-S and 5-S RNA, and then run on a 10 % polyacrylamide gel. The position of the absorbance markers is indicated by arrows. The results are summarized in Table 3
Purification of MS2 RNA-poly(A) by Chromatography on Oligo(dT) -cellulose
In order to separate the MS2 RNA-poly(A) from the non-poly(A)-containing MS2 RNA, we hybridized the incubation products to oligo(dT)-cellulose [12,13]. Fig. 8A represents the elution profile of the reaction product after 4-min incubation of MS2 [‘“C] RNA with ATP : RNA adenyltransferase in the presence of [3H]ATP. The RNA was first purified by zonal centrifugation and the MS2 RNA peak fractions were then applied to an oligo(dT)-cellulose column. In the

408 Addition of Poly(A) to the 3'-End of MS2 RNA
5 10
I C
I I I I I I
5 10 Fraction number
Fig.8. OIigo(dT)-cellulose chromatography of MY2 RNA-poly(AJ and MS2 RNA and primer cupaciiy of the non-retained material. (A) 23.5 pg MS2 [14C]RNA (1600 counts min-' pg-'), incubated for 4 min with 6 pg ATP : RNA adenyltransferase and with [3H]ATP (SO pCi/pmol), was purified on a glycerol gradient. The peak fractions were precipitated with ethanol, the pellet was quickly dissolved in 10 pI HLO, and 0.3 ml 0.5 M KCl, 10 mM Tris-HCI, pH 7.5, was added. The RNA was chromatographed on an oligo(dT)-cellulose column and 0.3-ml fractions were collected. SO pI of each fraction were taken for determination of the acid-precipitable radioactivity. Insert: 40 p1 of fractions 1-5 were used in a standard adenyltransferase assay mixture (without NaCI); the incubation was for 4 min at 37 "C. (B) 23.5 pg MS2 [I4C]RNA (1600 counts min-' pg-'), incubated for 4 min without ATP and with 6 pg ATP : RNA adenyltransferase was centrifuged and chromatographed over oligo(dT)-cellulose under the same conditions as described for (A). Insert: 20 pI 0.5 M KCI, 10 mM Tris-HCI, pH 7.5, were added to 20 p1 of fractions 1 - S , which were used in a standard adenyltransferase assay mixture as described for (A). (C) The material retained by an oligo(dT)-cellulose column similar to (A) was precipitated with ethanol, and rechromatographed on the same column. 0.1 ml of each fraction was used for determination of the acid-precipitable radioactivity. Hatched area: MS2 [I4C]RNA. Dotted area: [3H]AMP attached to the original MS2 RNA. Insert: [3H]AMP attached to the run-through fractions in a subsequent reaction
presence of 0.5 M KC1, approximately 50% of the MS2 [14C]RNA and more then 90% of the [3H]- poly(A) were retained on the column and could be eluted in the low-salt buffer. On the other hand MS2 Ei4C]RNA, similarly incubated without [3H]ATP and treated otherwise in the same way, failed to bind to the column in the presence of 0.5 M KCl (Fig8B). These results seem to indicate that 50% of the MS2 RNA molecules are not able to act as a primer for the enzyme, due to the lack of a free 3'-hydroxyl group or to some conformational hindrance. However, by testing the primer capacity of the oligo(dT)-cellulose effluents, it was shown that in both cases the RNA molecules in the run-through still acted a s a primer (inserts in Fig. 8 A and B), indicating that the amount of MS2 RNA not capable of being adenylated is very small. Some conformational rearrangement could of course have occurred between the first and the second enzymic reaction. The material, bound to the oligo(dT> cellulose column and eluted in low salt, was analyzed
by electrophoresis on a 2.4% polyacrylamide gel. The majority of the [3H]poly(A) coincided with the MS2 [I4C]RNA and about 25 was present as free poly(A) (Fig.9A). Since the original load of the oligo(dT)- cellulose column was coincident with MS2 RNA when purified by zonal centrifugation this free poly(A) fraction must be derived from some breakdown of polyadenylated MS2 RNA. However, the extent of breakage is very low and is not sufficient to explain the lack of retention of about 50% of the MS2 RNA after polyadenylation. The non-retained fraction was still physically intact on electrophoresis and showed that some adenylation had in fact occurred (Fig. 9 B). It seems likely, therefore, that under the conditions used, some products with very short poly(A) chains were not retained.
When the bound material was rechromatographed on the same column in high salt, 90% was retained by the oligo(dT)-cellulose, indicating that the chro- matographic separation was reliable (Fig. 8 C).

R. Devos, E. Gillis, and W. Fiers 409
2
... e ._ E . a c
= 8 -
m= 1
? ’ 0
C
1 A
2 3 4 5 6 2 3 4 5 6 0 1 1 Migration (crn)
B
h
2
,.. c .- E . v) - E = 8 - V
P 3 0
1
3
Fig.9. Polyacrylamide gel electrophoresis of’ the non-retained and the bound muteriul qf the oligo(dT)-cellulose chromatogruphy. The bound (A) and the non-retained (B) material of the oligo(dT)-cellulose chromatography were precipitated separately with 2 vol. of ethanol at - 20 “C. Each RNA pellet was dissolved in 15 p1 electrophoresis buffer and applied to a 2.4% polyacrylamide gel. (-0) 14C; ( O d ) 3H
Table 4 presents the amounts of retained and unbound material on oligo(dT)-cellulose of poly- adenylated MS2 [14C]RNA both as a function of the incubation time at a constant primer level (32 pg MS2 RNA/0.3 ml), and as a function of the RNA concentration for a specified time of incubation (4 min). It is shown that within the primer concentra- tion range of 5- 10 pg/O.l ml, the molar ratio of incorporated AMP versus MS2 RNA is only deter- mined by the incubation time and is almost independ- ent of RNA concentration. The amount of non- adenylated MS2 RNA is also not dependent on the RNA concentration and is probably an inherent property of the MS2 RNA preparation used as a primer.
It may be noted that the molar ratio of AMP incorporated per viral RNA molecule is somewhat higher in the fractions retained on oligo(dT)-cellulose than found upon direct isolation of the poly(A) segment (Table 3). This may be due in part to an underestimation of the isolated poly(A) segments (as mentioned above, exact calibration is difficult) and in part it may be due to a selection of molecules with longer poly(A) tails in the oligo(dT)-cellulose chromatographic procedure (this is certainly so in the 2-min sample).
DISCUSSION
Most eukaryotic mRNA’s contain a poly(A) segment at or near their 3’-terminus. Enzymes capable of adding a 3’-linked poly(A) segment to an RNA chain are found in different tissues and organisms. Nevertheless, it is difficult to precisely define the function in vivo of these widespread enzymes or to assess their role in mRNA maturation. Possibly some of these enzymes catalyze a reaction in vitro which is only distantly related to their function in vivo.
In order to study several biochemical properties of eukaryotic mRNA, it would be of interest to have available an mRNA of known nucleotide sequence and containing a poly(A) tail. For this purpose we have defined the conditions for synthesis of MS2 RNA- poly(A). The methods described in the present work, using ATP : RNA adenyltransferase from E. coli, enable the addition of a poly(A) tail to most natural RNA’s. The fact that the initiation of the poly(A) polymerizing activity of the enzyme on the primer is very fast, has been exploited to minimize residual ribonuclease activity, and to avoid de novo synthesis of rather long poly(A) chains. Although the usefulness of pancreatic ribonuclease to estimate the length of the poly(A) has its limitations, reasonable agreement was obtained between the calculated and the estimated

410 R. Devos, E. Gillis, and W. Fiers: Addition of Poly(A) to the 3'-End of MS2 RNA
Table 4. OIigo(dT)-cellulose chromatography of .WS2 [14C]RNA- (3H]po ly (A) as a.function uf the incubation time ( A ) and as a func- tion of the RNA concentration ( B ) (A) MS2 [14C]RNA (32 pg, 1200 counts min" pmol-') in 0.3 ml of the standard incubation mixture was polyadenylated for 2, 4 and 6 min at 37 "C. The product was purified on glycerol gradients and the MS2 RNA peak fractions were chromatographed on oligo(dT)- cellulose as described in the legend to Fig. 8. (B) 1 5 pg (2600 counts min-' pmol-I), 23.5 pg (1700 counts min-' pmol-') and 32 pg (1200 counts min-' pmol-I) MS2 [I4C]RNA was polyadenylated for 4 min at 37 "C in a 0.3-ml reaction mixture. After incubation, unlabeled MS2 RNA was added to a final total amount of 32 pg RNA. The enzymic products were purified by zonal centrifugation and then chromatographed on the same column. The total amount of recovered material was taken as 100 yi and represented more than 95 "/, of the load
(A) Incuba- Incorporated Non- Eluted in Incor- tion AMP/MS2 retained low salt porated time RNA in the AMP/MS2
glycerol RNA in the gradient cluted peak material fractions
min mol/mol yc:
2 49 72.0 4 96 54.0 6 134 52.0
-
~ -__ .
(B) Amount of RNA
pg/0.3 ml
15 65 50.5 23.5 68 51 .O 32 60 56.0
~~
28.0 46.0 48.0
~
mol/mol
137 184 233 ___.
49.5 49.0 44.0
117 130 115
value of the mean size of the poly(A) stretches attached to MS2 RNA.
It was shown by chromatography on oligo(dT)- cellulose columns that approximately 50 % of the treated MS2 RNA molecules are bound. This suggests that the conformation of the RNA molecules plays a role in allowing the adenyltransferase to bind to the 3'-end. However, the presence of free poly(A) chains in the material bound to oligo(dT)-cellulose also indicates that some breakage of the poly(A) tail from the high-molecular-weight RNA can occur. If neces- sary, this contaminating poly(A) can be eliminated by zonal centrifugation of the eluted polyadenylated RNA.
Using calf thymus terminal riboadenylate trans- ferase to add a poly(A) tail to the 3'-end of QP RNA, Gilvarg et al. [5] found that approximately 50 % of the treated Qfl RNA was retained on a poly(U)-Sephadex column. It is not known whether lack of complete
adenylation was due to conformational inaccessibility of the acceptor end of the primer. Using ATP : RNA adenyltransferase from E. coli to polyadenylate QP RNA, we found that more than 90% of the treated QP RNA could be hybridized to oligo(dT)-cellulose (unpublished results). In this sense, Q j RNA can be considered an even better primer than MS2 RNA for the E. coli adenyltransferase. The addition of poly(A) tails to other RNA's has already been useful in studies on the lifetime expectancy of eukaryotic mRNA in vivo [18] and many other possibilities for bio- chemical experimentation exist. Furthermore, on this basis new methods may be devised for nucleotide sequence determination of RNA's.
We are grateful to Dr A. Sippel and to Dr K. Marcker for kind gifts of matcrial. This research was supported by the Nationaal Fonds voor Wetenschappelijk Onderzoek of Belgium. One of us (R.D.) holds a fellowship from the I.W.O.N.L.
REFERENCES
1 .
2.
3.
4. 5.
6.
7.
8.
9.
10.
1 1 .
12.
13.
Payne, K. J. & Boezi, J. A. (1970) J . B id . Chem. 245, 1378-
Getz, M. J. , Birnie, G. D. & Paul, J. (1974) Biochemistry, 13,
Thrall, C . L., Park, W. D., Rashba, H. W., Stein, J. L., Mans, R. J. & Stein, G. S. (1974) Biochem. Biuphys. Res. C'ommun.
1387.
2235 - 2240.
61, 1443- 1449. Sippel, A. E. (1973) Eur. J. Biochem. 37, 31 -40. Gilvarg, C., Bollum, F. J. & Weissmann, C. (1975) Proc. Nut/
Acad. Sci. U.S.A. 72, 428-432. Tsiapolis, C. M., Dorson, J. W., De Sante, D. M. & Bollum,
F. J. (1973) Biochem. Biophys. Res. Commun. 50, 737- 743. Kimhi, Y. & Littauer, U. Z. (1968) J. Biol. Chem. 243, 231-
240. Perry, R. P., La Torre, J. , Kelley, D. E. & Greenberg, J . R.
(1972) Biochim. Biophys. Actu, 262, 220-226. Bishop, D. H. L., Claybrook, J. R. & Spiegelman, S. (1967)
J. Mol. B i d . 26, 373-387. Geroch, M. E., Richards, E. G. & Davies, G. A. (1968) B c r . .I .
Biochem. 6,325 - 330. Pinder, J. C. & Gratzer, W. B. (1974) Biochim. Biophys. Arm,
Aviv, H. & Leder, P. (1972) Proc.. Natl Acad. Sci. U.S.A. 69,
Soreq, H., Nudel, U., Salomon, R., Revel, M. & Littauer, U.
349,47 - 52.
1408- 1412.
Z. (1974) J . Mol. Biol. 88. 233-245. 14. Shiifer, R.,' Zillig, W. & Pricss, H. (1972) FEES Lett. 2.5,
15 . Darnell, J. E., Wall, R. & Tushinski, R. J . (1971) Proc.. Nut1 Acad. Sci. U.S.A. 68, 1321-1325.
16. Mansbridge, J. N., Crossley, J. A,, Lanyon, W. G. & William- son, R. (1974) Eur. J . Biochem. 44,261 -269.
17. Gorski, J . , Morrison, M. R., Merkel, C. G. & Lingrel, J. B. (1974) J . Mol. B i d . 86, 363-371.
18. Huez, G., Marbaix, G., Hubert, E., Cleuter, Y., Leclercq, M., Chantrenne, H., Devos, R., Soreq, H., Nudel, U. & Littauer, U.Z. (1975) Eur. J . Biochem. SY, 589-592.
87 - 90.
R. Devos and E. Gillis, Laboratorium voor Fysiologische Scheikunde, Fakulteit voor Geneeskunde, Rijksuniversiteit tc Gent, K. Lod. Ledeganckstraat 35, B-9000 Gent, Bclgium
W. Fiers, Laboratorium voor Moleculaire Biologie, Fakulteit der Wetenschappen, Rijksuniversiteit tc Gent, K. Lod. Ledeganckstraat 35, B-9000 Gent, Belgium





























