Embed Size (px)
Citation preview

1
Title: Dectin-1 is not required for controlling Candida albicans colonisation of the murine 1
gastrointestinal tract 2
Running title: Role of Dectin-1 in gastrointestinal candidiasis 3
Authors: Simon Vautier1, Rebecca A. Drummond1, Pierre Redelinghuys1, Graeme I. Murray2, 4
Donna M. MacCallum1§, Gordon D. Brown1§# 5
1Aberdeen Fungal Group, University of Aberdeen, Institute of Medical Sciences, Foresterhill, 6
Aberdeen, UK, AB25 2ZD. 7
2 Pathology, Division of Applied Medicine, University of Aberdeen, UK 8
9
#Corresponding author: 10
Professor Gordon D. Brown 11
Aberdeen Fungal Group, Section of Infection & Immunity, School of Medicine & Dentistry, 12
University of Aberdeen, Institute of Medical Sciences, Foresterhill, Aberdeen, UK, AB25 2ZD. 13
Email: [email protected] 14
Tel. +44 (0) 1224 437355 15
Fax. +44 (0) 1224 555766 16
17
§Equal contribution to senior authorship 18
Copyright © 2012, American Society for Microbiology. All Rights Reserved.Infect. Immun. doi:10.1128/IAI.00559-12 IAI Accepts, published online ahead of print on 17 September 2012
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

2
Abstract 19
Candida albicans is normally found as a commensal microbe, commonly colonising the 20
gastrointestinal tract in humans. However, this fungus can also cause mucosal and systemic 21
infections once immune function is compromised. Dectin-1 is an innate pattern recognition 22
receptor essential for the control of fungal infections in both mice and humans, however its role 23
in the control of C. albicans colonisation of the gastrointestinal tract has not been defined. Here, 24
we demonstrate that in mice Dectin-1 is essential for the control of gastrointestinal invasion 25
during systemic infection, with Dectin-1 deficiency associating with impaired fungal clearance 26
and dysregulated cytokine production. Surprisingly, however, following oral infection Dectin-1 27
was not required for the control of mucosal colonisation of the gastrointestinal tract, either in 28
terms of fungal burdens or cytokine response. Thus, in mice, Dectin-1 is essential for controlling 29
systemic infection with C. albicans, but appears redundant for the control of gastrointestinal 30
colonisation. 31
32
33
34
35
36
37
38
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

3
Introduction 39
Candida albicans is a member of the natural human commensal microbiota found in the 40
gastrointestinal and genitourinary tracts. However, C. albicans can cause both mucosal and 41
systemic infections, and is one of the most important nosocomial diseases with mortality rates of 42
around 40% for disseminated infections (29). Systemic C. albicans infections are commonly 43
thought to originate from the commensal mucosal microbiota disseminating to tissues following 44
damage to the mucosal barrier, alterations in the host immune system, and C. albicans 45
overgrowth following immunosuppressive drug and/or antibiotic treatments (15). It is important 46
to understand the host mechanisms involved in maintaining the natural microbiota and 47
controlling potential pathogens, such as C. albicans, if we are to learn how to prevent commensal 48
organisms becoming opportunistic infectious agents. 49
The innate immune system recognises C. albicans and other pathogens via pattern 50
recognition receptors (PRRs) which interact with ligands termed pathogen-associated molecular 51
patterns (PAMPs). Of the four major families of PRRs, the Toll-like receptor family (TLRs) are 52
the best studied, with TLR2 and TLR4, amongst others, being involved in fungal recognition (1). 53
However, anti-fungal immunity appears to be mediated primarily by members of the C-type 54
lectin receptor (CLR) family, including Dectin-1 and Dectin-2 (7). The β-glucan receptor, 55
Dectin-1, in particular, plays a non-redundant role in host defence against a number of medically 56
important fungal species, including C. albicans, Aspergillus fumigatus and Pneumocystis carinii 57
(24, 26, 27). Dectin-1 is primarily expressed on myeloid cells (neutrophils, macrophages and 58
dendritic cells) and is strongly expressed on immune cells in the lamina propria of the 59
gastrointestinal tract (21). In vitro experiments have shown that Dectin-1 mediates a number of 60
cellular functions, including phagocytosis, the respiratory burst and the production of 61
inflammatory mediators, including cytokines, chemokines and lipids (6). In vivo studies in mice 62
have demonstrated that Dectin-1 is vital for host defence in models of systemic candidiasis (26), 63
although this can be fungal strain-dependent (6, 24). In humans, Dectin-1 deficiency is 64
associated with a predisposition to mucocutaneous candidiasis, as well as increased 65
gastrointestinal (GI) colonisation in immune-compromised individuals, highlighting the 66
importance of this receptor in human mucosal defence (9, 20). 67
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

4
Our previous work suggested that Dectin-1 was required to prevent GI tract infection 68
with C. albicans during systemic infection in mice (26). In these studies, Dectin-1 deficiency was 69
associated with increased fungal burdens and gross morphological changes of the GI tract, 70
including enlargement of the stomach and discolouration of the small intestines (26). Here we 71
further investigated the role of Dectin-1 in controlling C. albicans GI infections in both systemic 72
and oral infections. 73
74
75
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

5
Materials and methods 76
Mice. Eight to twelve week old female C57BL/6 and Dectin-1-/- mice (26) backcrossed nine 77
times onto a C57BL/6 background were bred and maintained at the Medical Research Facility, 78
University of Aberdeen. Mice were separately housed in groups in individually ventilated cages, 79
unless otherwise indicated, and provided with food and water ad libitum. All experiments were 80
repeated at least two times with five or more mice per time point. All experimentation conformed 81
to the terms and conditions of United Kingdom Home Office licenses for research on animals 82
and the University of Aberdeen ethical review committee. 83
84
C. albicans strains, culture media, and growth conditions. C. albicans strain SC5314 (26), 85
AM2003-013 and AM2003-016 (19) were routinely grown and maintained on YPD agar (Sigma-86
Aldrich). For inoculum preparation a single colony was grown in Sabouraud broth (Oxoid) at 87
30oC for 24 h with shaking. Cells were washed twice in sterile phosphate-buffered saline (PBS) 88
and counted using a haemocytometer. Cell density was adjusted to the desired inoculum level 89
with PBS, and was confirmed by viable cell counts on agar plates. 90
91
Gastrointestinal model. The gastrointestinal model was performed essentially as described 92
previously (15) (See Fig. 4A). To reduce the commensal bacterial and endogenous fungal 93
microbiota, mice were provided with sterile water containing 2 mg/ml streptomycin (Invitrogen), 94
2000U/ml penicillin (Invitrogen) and 0.25 mg/ml fluconazole (Enzo) for three days and then 95
switched to water containing streptomycin and penicillin for a further 24 hr. Mice were then 96
provided with sterile water containing 1x107 CFU/ml C. albicans, 2 mg/ml streptomycin and 97
2000U/ml penicillin for five days. After C. albicans exposure, mice were maintained on sterile 98
water containing 2 mg/ml streptomycin, 2000U/ml penicillin and 0.2 mg/ml gentamicin 99
(Invitrogen). In some experiments, no antibiotics were used at any time point. To monitor 100
colonisation, stools were collected from individual mice on day 5 post-infection and every two 101
days thereafter. Stools were homogenised in 1 ml PBS, serially diluted and 25 µl of each dilution 102
plated on YPD agar containing 0.01 mg/ml vancomycin (Sigma) and 0.1 mg/ml gentamicin. 103
Plates were incubated overnight at 37oC under aerobic conditions and fungal content determined 104
by viable cell count and expressed as colony forming units (CFU) per g. Mice were sacrificed at 105
days 7, 14 and 21 post-exposure to C. albicans. Kidney, stomach, small intestine, caecum and 106
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

6
large intestine samples were harvested and washed 3 times with 1 ml sterile PBS to remove gut 107
contents. Tissue weights were determined, and samples were transferred into tubes containing 108
0.5 ml PBS, 0.05% (v/v) Triton X-100 and complete mini EDTA-free protease inhibitor cocktail 109
(as per the manufacturer’s instructions; Roche). The tissues were then homogenised, serially 110
diluted and plated on YPD as above. Cell debris was removed from the remaining tissue 111
homogenates by centrifugation at 13,000 rpm for 15 minutes at 40C and stored at -80oC for 112
subsequent cytokine analysis. 113
114
Systemic model. Mice were inoculated intravenously with 2x105 CFU C. albicans SC5314 in 115
100μl sterile PBS via the lateral tail vein. Mice were monitored daily and were sacrificed at day 3 116
post-infection or when judged to be moribund. Tissues were collected and processed for fungal 117
burden and cytokine analysis as described above. 118
119
Cytokine analysis. Cytokine levels were measured using the Bio-Plex ProTM Mouse 23-Plex kit 120
(Bio-Rad) and analysed on the Bio-Plex system using Bio-Plex ManagerTM software as per 121
manufacturer’s instructions. Stored tissue sample homogenate supernatants were thawed and 122
centrifuged for 15 minutes at 13,000 rpm at 4oC to remove debris. For each test, 50 µl of 123
undiluted sample was used and cytokine concentrations were normalised to sample protein 124
concentrations (BCA protein assay kit, Pierce). 125
126
Myeloperoxidase (MPO) activity. MPO activity in stored tissue supernatants were determined 127
using the Myeloperoxidase Activity Assay Kit (Abcam), as per manufacturer’s instructions. 128
129
Bile acid analysis. Small intestinal contents were centrifuged at 13,000 rpm. 20μl of the 130
supernatant was serially diluted and analysed for bile acid content using the Diazyme 131
colorimetric total bile acids assay kit as per manufacturer’s instructions. 132
133
Histology. Kidney, stomach, small intestine, caecum and large intestine samples were removed 134
from uninfected mice and infected mice, snap frozen in OCT (Sakura) and sectioned. Sections 135
were dehydrated with xylene and then rehydrated through a series of different ethanol solutions 136
and stained with hematoxylin and eosin (H & E) or periodic acid-Schiff (PAS) using 137
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

7
conventional staining methods. All individual segments of the alimentary tract were evaluated 138
for the presence and intensity of inflammation and for the presence of fungi. 139
140
Statistical analysis. The two tailed Student’s t-test was used to compare the two groups of mice. 141
The Mann-Whitney test was used to compare non-normally distributed data, as determined by 142
D'Agostino & Pearson omnibus normality test. Survival data was assessed by the log-rank test. 143
All statistical analyses were performed with GraphPad Prism software version 5.04. 144
145
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

8
Results 146
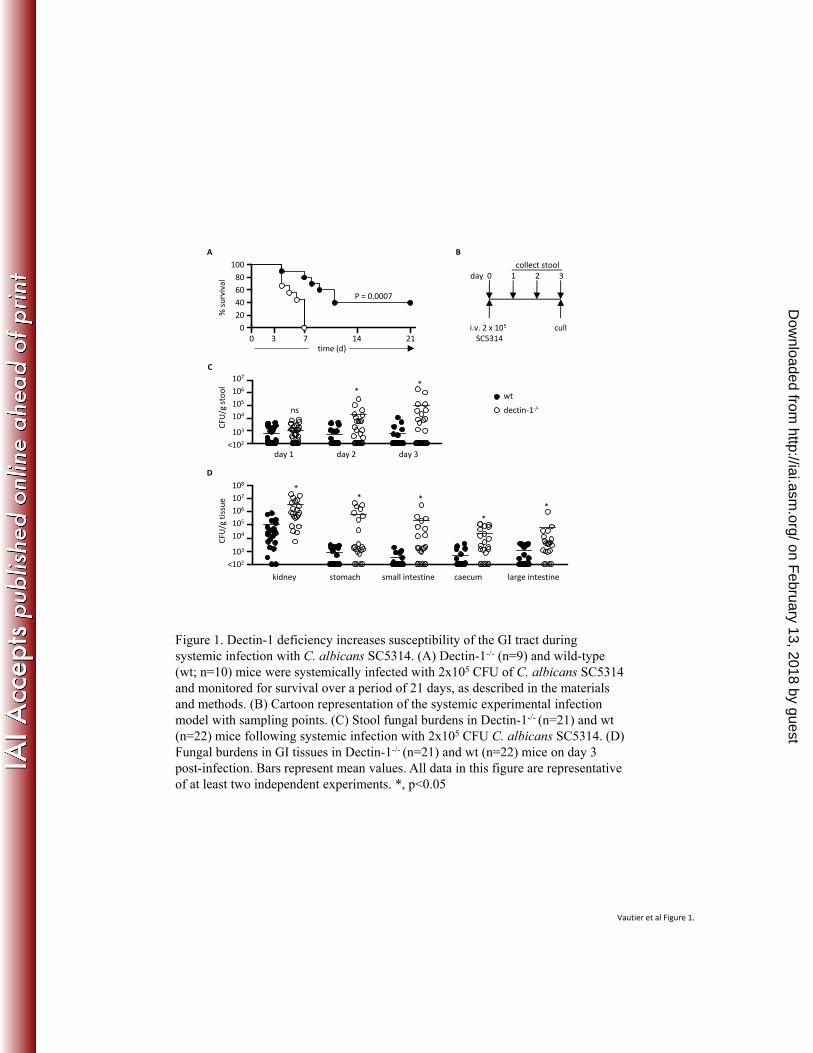
Dectin-1 deficient mice are more susceptible to systemic C. albicans infection. 147
We had previously observed that Dectin-1 deficiency correlated with GI tissue 148
colonisation during systemic infection with C. albicans (26). To further explore this 149
phenomenon, we systemically infected C57BL/6 wild-type and Dectin-1-deficient mice with 2 x 150
105 CFU C. albicans. This dose led to significantly enhanced mortality of the receptor-deficient 151
mice, as we had observed previously in the 129SvEv strain background (26) (Fig. 1A). We 152
examined stool fungal burdens over time (Fig. 1B & C) and assessed tissue fungal burdens at day 153
3 post infection (Fig. 1D); a time point chosen as it was prior to the onset of mortality in the 154
Dectin-1-/- mice. Within 2 days post-infection we observed significantly higher fungal burdens in 155
the stools of the Dectin-1-deficient animals which further increased by day 3. The identity of the 156
fungi in the stools was confirmed as C. albicans by 18s rDNA sequencing (data not shown). 157
Furthermore, stools plated from uninfected control animals showed no growth of fungal colonies 158
(data not shown). Interestingly, although wild-type mice also had C. albicans present in their 159
stools, these fungal burdens did not change during the course of the infection (Fig. 1C). 160
Similarly, on day 3, infected Dectin-1-deficient animals displayed significantly higher fungal 161
burdens in the kidney and GI tissues compared to wild type animals, similar to what we had 162
observed previously (26) (Fig. 1D). Thus Dectin-1 deficiency allows enhanced C. albicans 163
infection of the GI tract. 164
165
166
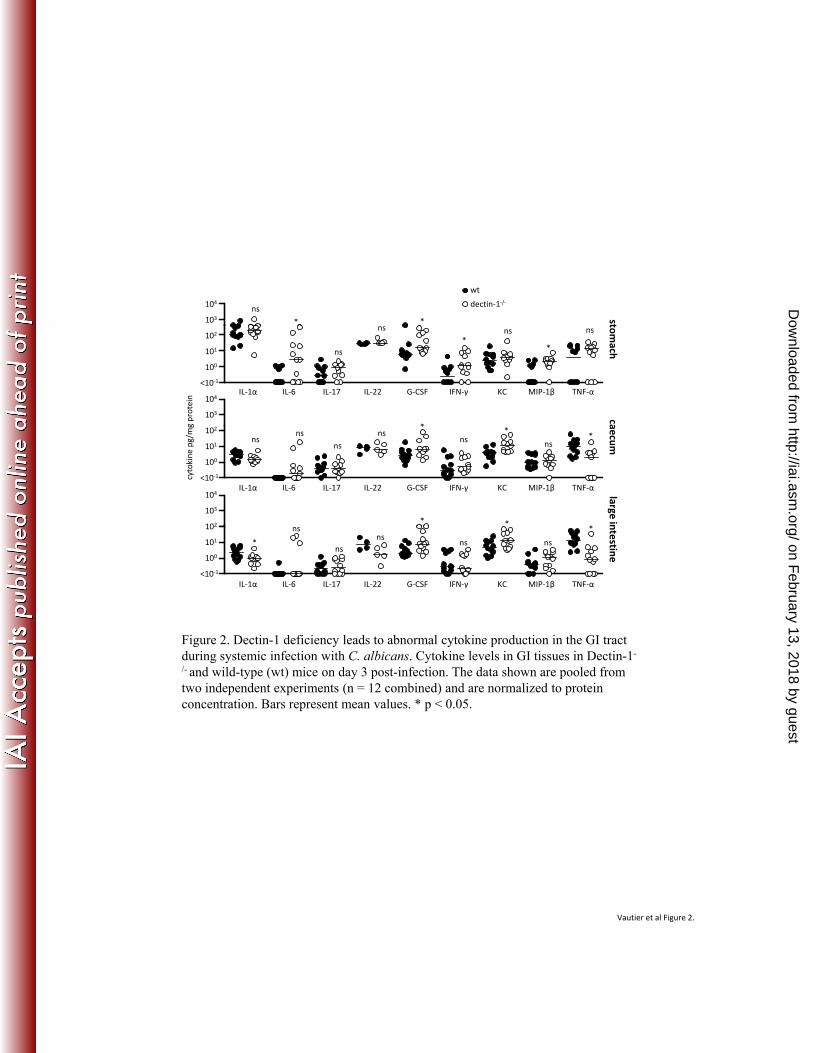
Dectin-1-deficient mice display dysregulated cytokine responses in the GI tract following 167
systemic infection. 168
To investigate the mechanism(s) underlying the increased susceptibility of Dectin-1-/- 169
mice to systemic GI infection, we measured cytokine levels in gastrointestinal tissues (Fig. 2). In 170
the stomachs of infected Dectin-1-/- mice we observed significantly increased levels of IL-6, G-171
CSF, IFN-γ and MIP-1β (CCL4) and, in the large intestine and caecum, increased levels of G-172
CSF and KC (CXCL1). Interestingly, in these latter two tissues we also observed significantly 173
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

9
reduced levels of TNF in the Dectin-1-/- mice, and significantly reduced IL-1α levels in the large 174
intestine. Thus, these data show that Dectin-1-deficiency results in dysfunctional cytokine 175
responses in the GI tissues during systemic infection with C. albicans, and that Dectin-1-176
mediated cytokine responses to this pathogen are tissue-specific. 177
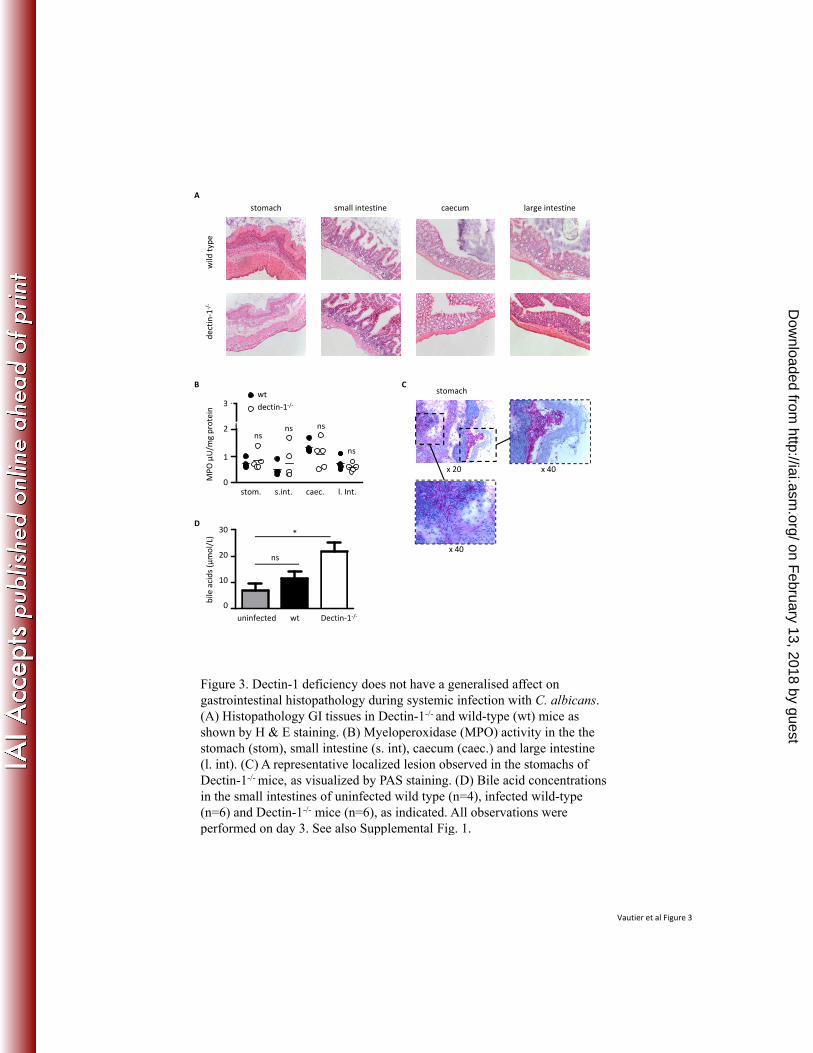
Enhanced fungal burdens and dysregulated cytokine responses do not correlate with 178
generalised GI tissue inflammation in the Dectin-1 deficient mice. 179
The increased fungal burdens and dysregulated cytokine responses in the Dectin-1-/- mice, 180
prompted us to examine the GI tissues for signs of pathology (Fig. 3). Surprisingly, we observed 181
no significant differences in inflammatory cell recruitment or pathology in any of the tissues 182
examined (Fig. 3A). Consistent with these observations, no difference in the levels of 183
myeloperoxidase (MPO) activity, a marker of neutrophil influx, were detected in these tissues 184
(Fig. 3B). PAS staining for fungi, however, did reveal localised invasive C. albicans lesions in 185
Dectin-1-/- mice which were not observed in the wild-type animals (Fig. 3C and data not shown). 186
We had previously observed that C. albicans infection of the GI tract resulted in gross-187
morphological changes, including enlarged stomachs and inflamed intestines (26). While we did 188
observe enlargement of the stomach upon infection, more extensive analysis revealed that this 189
phenotype was not limited to Dectin-1-/- mice and that it did not occur in all infected animals 190
(data not shown). However, the small intestines of Dectin-1-/- mice macroscopically appeared to 191
be reproducibly more inflamed than those of the wild type mice, which did not correlate with our 192
histological analysis, described above. On closer examination, the contents of the small intestines 193
in Dectin-1-/- mice appeared more yellow, producing an inflamed appearance (Supplemental Fig. 194
1). As dysregulated production of bile salts, which have a green/yellow appearance, has been 195
observed previously in other models of inflammation (5), we examined the level of bile acids in 196
the contents of the small intestine and observed that these were significantly increased in the 197
Dectin-1-/- mice (Fig. 3D). Thus, these results indicate that Dectin-1-deficiency results in an 198
enhanced, but localised, susceptibility to C. albicans infection in the GI tract. In addition Dectin-199
1-deficiency has broader effects, including dysregulated production of bile salts. 200
Dectin-1 deficiency does not alter susceptibility to C. albicans colonisation of the GI tract 201
during oral infection. 202
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

10
As Dectin-1-deficient mice were susceptible to GI tract colonisation during systemic 203
infections, we investigated the possibility that this CLR was also involved in controlling 204
colonisation of these tissues by C. albicans following oral infection. Mice are not normally 205
colonized by this fungal pathogen, so for these experiments, we made use of an established 206
model of GI colonisation (15), whereby mice are treated with antibiotics to displace the 207
endogenous microbiota and then orally-infected with C. albicans via the drinking water (Fig. 208
4A). C. albicans colonisation levels in the gastrointestinal tract were subsequently monitored by 209
examining fungal burdens in the stools, and in the tissues at specific time points (Fig. 4A). The 210
established level of GI colonisation (~107 CFU/g of stool) was unaffected by the dose of C. 211
albicans administered in the drinking water (Supplemental Fig. 2). 212
Initially we obtained extremely variable results with these experiments, where Dectin-1 213
deficiency had no effect or resulted in either significantly higher or lower GI fungal burdens 214
(Supplemental Figure 3 and data not shown). Notably, we also observed variation in results 215
between different cages of the same groups of animals (data not shown). As the microbial 216
composition of the GI tract is transferable between mice and influenced by colonisation with C. 217
albicans (8, 17), we performed all subsequent experiments with co-housed wild-type and 218
knockout animals. Under these conditions, we reproducibly found no difference in the 219
colonisation levels of the Dectin-1-/- mice, compared to wild-type animals, both in stool burdens 220
(Fig. 4B) and GI tissue fungal burdens at any of the time points examined (Fig. 4C). 221
Interestingly, however, in both wild-type and Dectin-1-/- mice, dissemination of C. albicans to 222
the kidneys was observed, although this had substantially decreased by the end of the 223
experiment, in both groups of animals (Fig. 4C). Importantly, none of the animals succumbed to 224
the infection during these experiments, even with increased infective doses (up to 108) or after 225
extended time periods (over 60 days). Similar results were also obtained in Dectin-1-deficient 226
mice in the 129SvEv background (data not shown) 227
Strains of C. albicans have different propensities in their ability to colonise the mucosa 228
and SC5314, in particular, is a poor coloniser of these tissues (18). Therefore, to confirm our 229
observations, we also tested the role of Dectin-1 using two additional clinical strains of C. 230
albicans (AM2003-013 and AM2003-016), which were originally isolated from infected mucosa 231
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

11
(19) . However, as we found with SC5314, Dectin-1 deficiency had no effect on GI colonisation 232
with either of these clinical strains (Supplemental Fig. 4). 233
We have recently shown that Dectin-1 deficiency can exacerbate colitis, and that this 234
phenotype was dependent on the composition of the microbiome (14). To exclude the possibility 235
that alterations in the microbiome following antibiotic treatment of mice were masking a 236
potential contribution of Dectin-1, we also examined GI colonisation in mice which did not 237
receive any antibiotics (Supplemental Fig. 5). In these experiments, C. albicans was rapidly 238
cleared from the GI tract, but, as we had observed in the antibiotic treated mice, there was no 239
effect of Dectin-1 deficiency. Interestingly, while C57BL/6 background mice rapidly cleared the 240
infection, 129/Sv retained a low, but persistent, level of infection throughout the course of the 241
experiment (Supplemental Fig. 5). 242
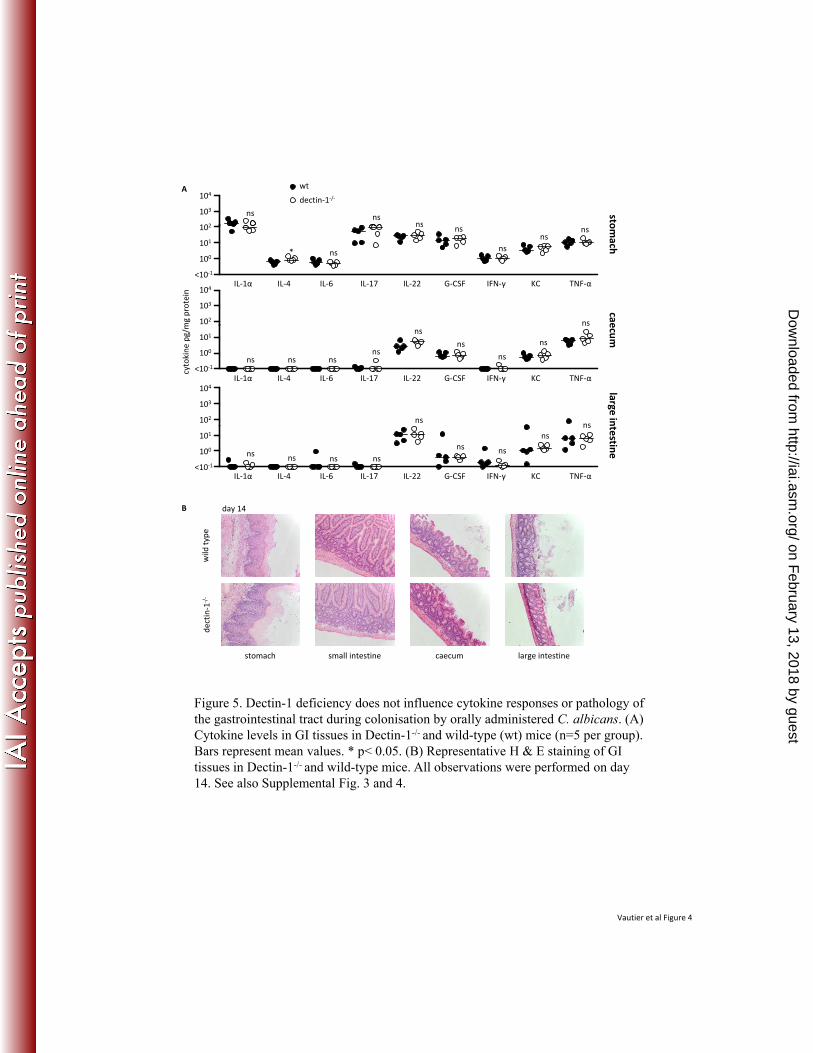
Dectin-1-deficiency does not affect cytokine responses or tissue pathology during C. 243
albicans GI colonisation following oral infection 244
Given our findings in the systemic model, we also examined cytokine responses and 245
histopathology in the gastrointestinal tissues from orally infected wild-type and Dectin-1-/- mice 246
(Fig. 5). Cytokine responses were examined at day 14 post-infection. Although we observed 247
increased levels of cytokines known to be important in controlling fungal infections, including 248
IL-17 and IL-22 (3), particularly in stomach tissue, there were no significant differences between 249
wild-type and Dectin-1-deficient animals (Fig. 5A). The one exception was IL-4, which was 250
slightly, but significantly, higher in the stomachs of Dectin-1-/- mice. Histological examination of 251
the GI tract also revealed no difference between the two groups, either at day 7, 14 or 21 post 252
infection (Fig. 5B and Supplemental Fig. 6). Localised fungal invasion of the stomach, with 253
resulting hyperplasia of the squamous epithelium was observed in both groups at days 7, 14 and 254
21 post-infection (Supplemental Fig. 7). Thus these data indicate that Dectin-1 is not involved in 255
controlling C. albicans colonisation of the GI tract following oral infection. 256
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

12
Discussion 257
C-type lectin receptors play a central role in host defence, mediating and directing both 258
innate and adaptive anti-fungal immunity (1, 28). Dectin-1, in particular, has an essential role in 259
the defence against a number of fungal pathogens, including C. albicans (26). Our previous work 260
characterising the role of this receptor had indicated that Dectin-1 was required for controlling 261
infection of the gastrointestinal tissues during systemic candidiasis (26). Here we confirmed 262
these findings, and demonstrated that loss of Dectin-1 led to dysregulated cytokine responses and 263
uncontrolled fungal growth in the GI tract at early time points post-infection. These results are 264
consistent with our previously established role for Dectin-1 in mediating protective innate 265
responses to C. albicans, including cytokine production and fungal uptake and killing (26). 266
Interestingly, whereas we had previously not observed any differences in cytokine levels in 267
infected kidneys at this early time point (day 3) (26), we could show that Dectin-1 was involved 268
in regulating cytokine responses in the GI tract, although this was varied and tissue specific. 269
Surprisingly, the Dectin-1-dependent defects were only observed in localised lesions and did not 270
correlate with generalised changes in tissue pathology or inflammation (including neutrophil 271
recruitment). However, we did reproducibly observe gross abnormalities of the small intestines 272
of Dectin-1-/- animals, which were not reflected in histopathological changes. Rather these 273
changes correlated with substantially increased production of bile acids, however the underlying 274
reasons for this require further exploration. 275
We also examined the role of Dectin-1 in gastrointestinal colonisation by C. albicans, 276
using an oral model of infection. Surprisingly, despite the essential requirement of this receptor 277
during systemic disease, we observed no differences in GI tract colonisation between wild-type 278
and Dectin-1 deficient animals. Notably, we only achieved reproducible results when the wild-279
type and Dectin-1 mice were housed together in the same cage. When the animals were caged 280
separately, we obtained variable results; in some experiments Dectin-1-deficiency conferred 281
susceptibility, whereas in others it conferred enhanced resistance. In fact, we even observed 282
differences between different cages of the same strain. Although not formally demonstrated here, 283
it is likely that this variation is due to differences in the microbiotia of the mice, which is known 284
to be transferrable between animals and to influence colonisation by C. albicans (8, 17). The lack 285
of an involvement of Dectin-1 in controlling GI colonisation under steady-state conditions may 286
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

13
reflect the expression of Dectin-1 in the lamina propria and not the epithelium (22). However, 287
Dectin-1 expression can be induced in epithelial tissues (25) and this receptor may play a role 288
under certain inflammatory conditions, such as we have recently shown during colitis (14). 289
Our results differ from two recent reports which have implicated Dectin-1 in the control 290
of C. albicans infections in the GI tract (2, 10). In the first study, mice with Dectin-1-deficient 291
macrophages (and neutrophils) presented with increased fungal burdens in the stomach and 292
caecum following oral infection with C. albicans (10). In the second report, Dectin-1 was found 293
to contribute to either resistance or susceptibility to infection, depending on mouse strain 294
background, which was related to the Dectin-1-isoform expressed in these animals (2, 11). 295
However, the infection models used were significantly different from our own; in both studies 296
mice were not pre-treated with antibiotics and only a single infective dose was administered (2, 297
10). However, we also found no role for Dectin-1 in mice which had not been treated with 298
antibiotics. The differences in susceptibility observed in these experiments may also reflect the 299
lack of co-housing between experimental groups and/or the different sources of these animals. 300
Indeed, mice from different locations can differ substantially in their microbiota (4). 301
While our results suggest that Dectin-1 is not involved in controlling the colonisation of 302
the GI tract, this receptor is required for controlling C. albicans infection at other mucosal sites. 303
In mouse models, Dectin-1 is necessary for controlling infection in both the oral and vaginal 304
mucosa (2, 13). Furthermore, in humans, individuals homozygous for a polymorphism which 305
renders them essentially Dectin-1 deficient have enhanced susceptibility to chronic 306
mucocutaneous candidiasis (CMC), which affects the skin, nail beds and oral and vaginal 307
mucosa (9). Importantly, these patients have defects in Th17 responses, which are critical for the 308
control of mucosal Candida infections (12). Consistent with these observations, we did not 309
observe any Dectin-1-dependent effects on the production of IL-17 or IL-22 in the GI tract, and it 310
is likely that other CLRs, such as Dectin-2, may play the major role in driving these responses in 311
these tissues (16, 23). However, preliminary analysis suggests that Dectin-2 is also not involved 312
in controlling GI-colonisation following oral infection (Supplemental Fig. 8). 313
In conclusion, our data demonstrate that the innate responses triggered by Dectin-1 plays 314
an essential role in controlling C. albicans infection of the GI tract during systemic infection. In 315
contrast, this receptor appears to have little, if any, role in controlling colonisation of the GI tract 316
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

14
following oral infection with this organism. Elucidating the mechanisms involved in this process 317
remains a priority if we are to fully understand how our immune system controls and responds to 318
colonisation by a pathogen which is a significant cause of human morbidity and mortality. 319
320
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

15
Acknowledgements 321
This study was supported by the University of Aberdeen and the Wellcome Trust. We thank S. 322
Vicky Tsoni for performing the 129SvEv systemic infections, Yoichiro Iwakura for the Dectin-2 323
mice, and Julie Taylor and the staff of our animal facility for the care and maintenance of our 324
animals. 325
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

16
References 326
1. Brown, G. D. 2011. Innate antifungal immunity: the key role of phagocytes. Annu Rev 327
Immunol 29:1-21. 328
2. Carvalho, A., G. Giovannini, A. De Luca, C. D'Angelo, A. Casagrande, R. G. 329
Iannitti, G. Ricci, C. Cunha, and L. Romani. 2012. Dectin-1 isoforms contribute to 330
distinct Th1/Th17 cell activation in mucosal candidiasis. Cell Mol Immunol 9:276-86. 331
3. Conti, H. R., and S. L. Gaffen. 2010. Host responses to Candida albicans: Th17 cells 332
and mucosal candidiasis. Microbes Infect 12:518-27. 333
4. Denning, T. L., B. A. Norris, O. Medina-Contreras, S. Manicassamy, D. Geem, R. 334
Madan, C. L. Karp, and B. Pulendran. 2011. Functional specializations of intestinal 335
dendritic cell and macrophage subsets that control Th17 and regulatory T cell responses 336
are dependent on the T cell/APC ratio, source of mouse strain, and regional localization. J 337
Immunol 187:733-47. 338
5. Dial, E. J., J. J. Romero, X. Villa, D. W. Mercer, and L. M. Lichtenberger. 2002. 339
Lipopolysaccharide-induced gastrointestinal injury in rats: role of surface hydrophobicity 340
and bile salts. Shock 17:77-80. 341
6. Drummond, R. A., and G. D. Brown. 2011. The role of Dectin-1 in the host defence 342
against fungal infections. Curr Opin Microbiol 14:392-399. 343
7. Drummond, R. A., S. Saijo, Y. Iwakura, and G. D. Brown. 2011. The role of 344
Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur J Immunol 41:276-81. 345
8. Elinav, E., T. Strowig, A. L. Kau, J. Henao-Mejia, C. A. Thaiss, C. J. Booth, D. R. 346
Peaper, J. Bertin, S. C. Eisenbarth, J. I. Gordon, and R. A. Flavell. 2011. NLRP6 347
inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745-57. 348
9. Ferwerda, B., G. Ferwerda, T. S. Plantinga, J. A. Willment, A. B. van Spriel, H. 349
Venselaar, C. C. Elbers, M. D. Johnson, A. Cambi, C. Huysamen, L. Jacobs, T. 350
Jansen, K. Verheijen, L. Masthoff, S. A. Morre, G. Vriend, D. L. Williams, J. R. 351
Perfect, L. A. Joosten, C. Wijmenga, J. W. van der Meer, G. J. Adema, B. J. 352
Kullberg, G. D. Brown, and M. G. Netea. 2009. Human dectin-1 deficiency and 353
mucocutaneous fungal infections. N Engl J Med 361:1760-7. 354
10. Gales, A., A. Conduche, J. Bernad, L. Lefevre, D. Olagnier, M. Beraud, G. Martin-355
Blondel, M. D. Linas, J. Auwerx, A. Coste, and B. Pipy. 2010. PPARgamma controls 356
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

17
dectin-1 expression required for host antifungal defense against Candida albicans. PLoS 357
Pathog 6:e1000714. 358
11. Heinsbroek, S. E., P. R. taylor, M. Rosas, J. A. Willment, D. L. Williams, S. Gordon, 359
and G. D. Brown. 2006. Expression of functionally different Dectin-1 isoforms by 360
murine macrophages. J Immunol 176:5513-5518. 361
12. Hernandez-Santos, N., and S. L. Gaffen. 2012. Th17 cells in immunity to Candida 362
albicans. Cell Host and Microbe 11:425-435. 363
13. Hise, A. G., J. Tomalka, S. Ganesan, K. Patel, B. A. Hall, G. D. Brown, and K. A. 364
Fitzgerald. 2009. An essential role for the NLRP3 inflammasome in host defense against 365
the human fungal pathogen Candida albicans. Cell Host Microbe 5:487-97. 366
14. Iliev, I. D., V. A. Funari, K. D. Taylor, Q. Nguyen, C. N. Reyes, S. P. Strom, J. 367
Brown, C. A. Becker, P. R. Fleshner, M. Dubinsky, J. I. Rotter, H. L. Wang, D. P. 368
McGovern, G. D. Brown, and D. M. Underhill. 2012. Interactions between commensal 369
fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336:1314-7. 370
15. Koh, A. Y., J. R. Kohler, K. T. Coggshall, N. Van Rooijen, and G. B. Pier. 2008. 371
Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS 372
Pathog 4:e35. 373
16. Leibundgut-Landmann, S., O. Gross, M. J. Robinson, F. Osorio, E. C. Slack, S. V. 374
Tsoni, E. Schweighoffer, V. Tybulewicz, G. D. Brown, J. Ruland, and C. Reis e 375
Sousa. 2007. Syk- and CARD9-dependent coupling of innate immunity to the induction 376
of T helper cells that produce interleukin 17. Nat Immunol 8:630-8. 377
17. Mason, K. L., J. R. Erb Downward, N. R. Falkowski, V. B. Young, J. Y. Kao, and G. 378
B. Huffnagle. 2012. Interplay between the gastric bacterial microbiota and Candida 379
albicans during postantibiotic recolonization and gastritis. Infect Immun 80:150-8. 380
18. Naglik, J. R., P. L. Fidel, Jr., and F. C. Odds. 2008. Animal models of mucosal 381
Candida infection. FEMS Microbiol Lett 283:129-39. 382
19. Odds, F. C., M. E. Bougnoux, D. J. Shaw, J. M. Bain, A. D. Davidson, D. Diogo, M. 383
D. Jacobsen, M. Lecomte, S. Y. Li, A. Tavanti, M. C. Maiden, N. A. Gow, and C. 384
d'Enfert. 2007. Molecular phylogenetics of Candida albicans. Eukaryot Cell 6:1041-52. 385
20. Plantinga, T. S., W. J. van der Velden, B. Ferwerda, A. B. van Spriel, G. Adema, T. 386
Feuth, J. P. Donnelly, G. D. Brown, B. J. Kullberg, N. M. Blijlevens, and M. G. 387
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

18
Netea. 2009. Early stop polymorphism in human DECTIN-1 is associated with increased 388
candida colonization in hematopoietic stem cell transplant recipients. Clin Infect Dis 389
49:724-32. 390
21. Reid, D. M., N. A. Gow, and G. D. Brown. 2009. Pattern recognition: recent insights 391
from Dectin-1. Curr Opin Immunol 21:30-7. 392
22. Reid, D. M., M. Montoya, P. R. Taylor, P. Borrow, S. Gordon, G. D. Brown, and S. 393
Y. Wong. 2004. Expression of the {beta}-glucan receptor, Dectin-1, on murine 394
leukocytes in situ correlates with its function in pathogen recognition and reveals 395
potential roles in leukocyte interactions. J Leukoc Biol 76:86-94. 396
23. Robinson, M. J., F. Osorio, M. Rosas, R. P. Freitas, E. Schweighoffer, O. Gross, J. S. 397
Verbeek, J. Ruland, V. Tybulewicz, G. D. Brown, L. F. Moita, P. R. Taylor, and C. 398
Reis e Sousa. 2009. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for 399
Th17 responses to fungal infection. J Exp Med 206:2037-51. 400
24. Saijo, S., N. Fujikado, T. Furuta, S. H. Chung, H. Kotaki, K. Seki, K. Sudo, S. 401
Akira, Y. Adachi, N. Ohno, T. Kinjo, K. Nakamura, K. Kawakami, and Y. Iwakura. 402
2007. Dectin-1 is required for host defense against Pneumocystis carinii but not against 403
Candida albicans. Nat Immunol 8:39-46. 404
25. Sun, W. K., X. Lu, X. Li, Q. Y. Sun, X. Su, Y. Song, H. M. Sun, and Y. Shi. 2012. 405
Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune 406
responses in human bronchial epithelial cells. Eur J Clin Microbiol Infect Dis. 407
26. Taylor, P. R., S. V. Tsoni, J. A. Willment, K. M. Dennehy, M. Rosas, H. Findon, K. 408
Haynes, C. Steele, M. Botto, S. Gordon, and G. D. Brown. 2007. Dectin-1 is required 409
for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31-8. 410
27. Werner, J. L., A. E. Metz, D. Horn, T. R. Schoeb, M. M. Hewitt, L. M. Schwiebert, 411
I. Faro-Trindade, G. D. Brown, and C. Steele. 2009. Requisite role for the dectin-1 412
beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol 413
182:4938-46. 414
28. Wuthrich, M., G. S. Deepe, Jr., and B. Klein. 2012. Adaptive immunity to fungi. Annu 415
Rev Immunol 30:115-48. 416
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

19
29. Zaoutis, T. E., J. Argon, J. Chu, J. A. Berlin, T. J. Walsh, and C. Feudtner. 2005. 417
The epidemiology and attributable outcomes of candidemia in adults and children 418
hospitalized in the United States: a propensity analysis. Clin Infect Dis 41:1232-9. 419
420
421
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

20
Figure Legends 422
Figure 1. Dectin-1 deficiency increases susceptibility of the GI tract during systemic infection 423
with C. albicans SC5314. (A) Dectin-1-/- (n=9) and wild-type (wt; n=10) mice were systemically 424
infected with 2x105 CFU of C. albicans SC5314 and monitored for survival over a period of 21 425
days, as described in the materials and methods. (B) Cartoon representation of the systemic 426
experimental infection model with sampling points. (C) Stool fungal burdens in Dectin-1-/- 427
(n=21) and wt (n=22) mice following systemic infection with 2x105 CFU C. albicans SC5314. 428
(D) Fungal burdens in GI tissues in Dectin-1-/- (n=21) and wt (n=22) mice on day 3 post-429
infection. Bars represent mean values. All data in this figure are representative of at least two 430
independent experiments. *, p<0.05 431
Figure 2. Dectin-1 deficiency leads to abnormal cytokine production in the GI tract during 432
systemic infection with C. albicans. Cytokine levels in GI tissues in Dectin-1-/- and wild-type 433
(wt) mice on day 3 post-infection. The data shown are pooled from two independent experiments 434
(n = 12 combined) and are normalized to protein concentration. Bars represent mean values. * p 435
< 0.05. 436
Figure 3. Dectin-1 deficiency does not have a generalised affect on gastrointestinal 437
histopathology during systemic infection with C. albicans. (A) Histopathology GI tissues in 438
Dectin-1-/- and wild-type (wt) mice as shown by H & E staining. (B) Myeloperoxidase (MPO) 439
activity in the the stomach (stom), small intestine (s. int), caecum (caec.) and large intestine (l. 440
int). (C) A representative localized lesion observed in the stomachs of Dectin-1-/- mice, as 441
visualized by PAS staining. (D) Bile acid concentrations in the small intestines of uninfected 442
wild type (n=4), infected wild-type (n=6) and Dectin-1-/- mice (n=6), as indicated. All 443
observations were performed on day 3. See also Supplemental Fig. 1. 444
Figure 4. Dectin-1 deficiency does not influence gastrointestinal tract colonisation by C. 445
albicans, following oral infection. (A) Cartoon representation of the experimental oral infection 446
model with sampling time points. (B) Stool fungal burdens in co-housed Dectin-1-/- (n=21) and 447
wild-type (wt; n=20) mice, following oral infection with C. albicans SC5314. (C) GI tissue 448
fungal burdens in Dectin-1-/- and wt mice on days 7, 14 and 21 (pooled data, n>16 per group). 449
Bars represent mean values. See also Supplemental Fig. 3. 450
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

21
Figure 5. Dectin-1 deficiency does not influence cytokine responses nor pathology of the 451
gastrointestinal tract during colonisation by orally administered C. albicans. (A) Cytokine levels 452
in GI tissues in Dectin-1-/- and wild-type (wt) mice (n=5 per group). Bars represent mean values. 453
* p< 0.05. (B) Representative H & E staining of GI tissues in Dectin-1-/- and wild-type mice. All 454
observations were performed on day 14. See also Supplemental Fig. 4 and 5. 455
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from
i 2 105
day 0 1 2 3
ll
collect stool
A B
% s
urvi
val
1008060
2040
0
P = 0.0007
C
CFU
/g s
tool
107
106
104
105
ns
** wt
dectin-1-/-
i.v. 2 x 105
SC5314cull0
0 3 7 14 21time (d)
/g ti
ssue
108
107
106
105
** *
**
D
C
<102
103
day 1 day 2 day 3
CFU
/
kidney stomach small intestine caecum large intestine
<102
104
103
Figure 1. Dectin-1 deficiency increases susceptibility of the GI tract during systemic infection with C. albicans SC5314. (A) Dectin-1-/- (n=9) and wild-type (wt; n=10) mice were systemically infected with 2x105 CFU of C. albicans SC5314 and monitored for survival over a period of 21 days, as described in the materials and methods. (B) Cartoon representation of the systemic experimental infection model with sampling points. (C) Stool fungal burdens in Dectin-1-/- (n=21) and wt p g p ( ) g ( )(n=22) mice following systemic infection with 2x105 CFU C. albicans SC5314. (D) Fungal burdens in GI tissues in Dectin-1-/- (n=21) and wt (n=22) mice on day 3 post-infection. Bars represent mean values. All data in this figure are representative of at least two independent experiments. *, p<0.05
Vautier et al Figure 1.
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from
wt
dectin-1-/-
103
104ns
* * stg
prot
ein
103
104
ca
*
<10-1
101
102
100
ns
ns*
ns ns
*
tomach
IL-1α IL-6 IL-17 IL-22 G-CSF IFN-γ KC MIP-1β TNF-α
cyto
kine
pg/
mg
102
103
104 large in* * *
<10-1
101
102
100
aecumnsns
nsns
* **ns ns
IL-1α IL-6 IL-17 IL-22 G-CSF IFN-γ KC MIP-1β TNF-α
<10-1
101
102
100
IL-1α IL-6 IL-17 IL-22 G-CSF IFN-γ KC MIP-1β TNF-α
ntestine
*
*ns
nsns
ns ns
Figure 2. Dectin-1 deficiency leads to abnormal cytokine production in the GI tract during systemic infection with C. albicans. Cytokine levels in GI tissues in Dectin-1-
/- and wild-type (wt) mice on day 3 post-infection. The data shown are pooled from two independent experiments (n = 12 combined) and are normalized to protein concentration. Bars represent mean values. * p < 0.05.
Vautier et al Figure 2.
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from
stomach small intestine caecum large intestineA
wild
type
tin-1
-/-
dect
ns2
3
otei
n
B Cstomachwt
dectin-1-/-
D
nsns ns
ns1
2
0stom. s.int. caec. l. Int.
MPO
μU
/mg
pr
x 20 x 40
bile
aci
ds (μ
mol
/L)
30
10
20
0
*
x 40 ns
uninfected wt Dectin-1-/-
Figure 3. Dectin-1 deficiency does not have a generalised affect on gastrointestinal histopathology during systemic infection with C. albicans. (A) Histopathology GI tissues in Dectin-1-/- and wild-type (wt) mice as shown by H & E staining. (B) Myeloperoxidase (MPO) activity in the thestomach (stom), small intestine (s. int), caecum (caec.) and large intestine (l. int). (C) A representative localized lesion observed in the stomachs of Dectin-1-/- mice, as visualized by PAS staining. (D) Bile acid concentrations in the small intestines of uninfected wild type (n=4), infected wild-type (n=6) and Dectin-1-/- mice (n=6), as indicated. All observations were performed on day 3 See also Supplemental Fig 1
Vautier et al Figure 3
performed on day 3. See also Supplemental Fig. 1.
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from

Asterile water +
streptomycin, penicillin,and fluconazole
sterile water +1 x 107 C. albicans SC5314
streptomycin and penicillin
tissue analysis
7 14 21
-4 0 time (d) 5 21
streptomycin and penicillinsterile water +
streptomycin, penicillin,and gentamicin
B
l
109
108 ns ns ns ns ns nsns ns ns
Ctime (d)
5 7 9 11 13 14 15 17 19 21
CFU
/g s
too 107
103
105
106
104
nsns ns ns
108108
107
106
<102
104
105
103
CFU
/g ti
ssue
nsns ns ns
day 7
d 14
ns
108
wt
dectin-1-/-
108
107 day 21
ns
nsns
nsns
day 14107
106
<102
104
105
103
CFU
/g ti
ssue
107
106
<102
104
105
103
kidney stomach s. intestine caecum l. intestine
CFU
/g ti
ssue
day 21
ns
nsns ns ns
Figure 4. Dectin-1 deficiency does not influence gastrointestinal tract g y gcolonisation by C. albicans, following oral infection. (A) Cartoon representation of the experimental oral infection model with sampling time points. (B) Stool fungal burdens in co-housed Dectin-1-/- (n=21) and wild-type (wt; n=20) mice, following oral infection with C. albicans SC5314. (C) GI tissue fungal burdens in Dectin-1-/- and wt mice on days 7, 14 and 21 (pooled data, n>16 per group). Bars represent mean values. See also
Vautier et al Figure 4
Supplemental Fig. 2
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from
wt
dectin-1-/-
102
103
104
ns nsns
stoA
102
103
104
<10-1
101
102
100
mg
prot
ein
* nsns
ns
ns
ns ns
ns
caeom
ach
IL-1α IL-4 IL-6 IL-17 IL-22 G-CSF IFN-γ KC TNF-α
<10-1
101
100
102
103
104
cyto
kine
pg/
m
ns
nsns
ns
nsnsns
ns ns
ns
large inecum
IL-1α IL-4 IL-6 IL-17 IL-22 G-CSF IFN-γ KC TNF-α
<10-1
101
100
IL-1α IL-4 IL-6 IL-17 IL-22 G-CSF IFN-γ KC TNF-α
nsns
nsns ns
nsns
ns
day 14
testine
B
wild
type
ectin
-1-/
-de
stomach small intestine caecum large intestine
Figure 5. Dectin-1 deficiency does not influence cytokine responses or pathology of the gastrointestinal tract during colonisation by orally administered C. albicans. (A) Cytokine levels in GI tissues in Dectin-1-/- and wild-type (wt) mice (n=5 per group). Bars represent mean values. * p< 0.05. (B) Representative H & E staining of GI tissues in Dectin-1-/- and wild-type mice. All observations were performed on day 14. See also Supplemental Fig. 3 and 4.
Vautier et al Figure 4
on February 13, 2018 by guest
http://iai.asm.org/
Dow
nloaded from





























