Embed Size (px)
Citation preview

71.
T R I G L Y C E R I D E A N D F A T T Y A C I D A N A L Y S I S B Y GAS CHROMATOGRAPHY
A . K U K S I S
ABSTRACT
Extension of gas chromatographic methods t o the analysis of high molecular weight compounds has permitted the study of natura l l i p i d m i x - tu res without a p r i o r transformation in to simple f a t t y ac id e s t e r s . It is now possible t o separate and measure i n a s ingle chromatogram the amounts of cholesterol , diglycerides, s t e r y l e s t e r s and t r ig lycer ides present i n a t o t a l l i p i d ex t rac t . has required a perfect ion i n instrument design and i n working procedures not o rd ina r i ly demanded of equipment used i n isothermal s t e ro id and f a t t y ac id analyses. the advanced apparatus design and optimized operating parameters have yielded improved resolut ion and b e t t e r quant i f ica t ion . When combined w i t h thin- layer chromatographic methods f o r the separation of l i p i d c lasses and f o r a resolut ion of saturated and unsaturated homologues, the modified gas chromatographic techniques have permitted a nearly complete f rac t iona t ion and quant i f ica t ion of a l l components of na tura l l i p i d mixtures.
The resolut ion of s t e r y l e s t e r s and t r ig lycer ides
When applied t o determinations of f a t t y acids, however,
The paper describes some of the advances made i n the design of the gas chromatographs and i n the preparation of high qua l i t y C O ~ I ~ S re- sponsible f o r these achievements. The discussion of operating parameters is l imi ted t o the consideration of programmed temperature separation of mixtures of t o t a l neut ra l l i p i d s , t r ig lycer ides and simple f a t t y acid e s t e r s .
INTRODUCTION
The invention of gas chromatographic techniques f o r the separa- t i o n and quant i ta t ive estimation of f a t t y acids (1) completely revolution- ized the f i e l d of l i p i d chemistry and f o r the first time provided an indi- cat ion of the t r u e complexity of na tura l fats and o i l s . This development l e d t o the eventual introduction of the polyester l i q u i d phase6 (2) which permitted the ana lys i s of many complex mixtures of f a t t y acids w i t h re la - t i v e ease and reproducibi l i ty . ings of thermally unstable polymers as s ta t ionary phases, a r e bes t su i ted f o r the study of the c16 and c18 f a t t y acids which make up the bulk of the common animal f a t s and vegetable o i l s . They a r e unsui table f o r the sepa- ra t ion of natura l f a t t y ac id e s t e r s such as glycerides and s t e r y l e s t e r s .
These methods, which u t i l i z e heavy coat-
Successful gas chromatography of i n t a c t na tura l l i p i d e s t e r s (3) had t o await the discovery of the t h i n - f i l m techniques (4) used i n s t e ro id separations, and temperature programming. achieved (5), however, have a l so required modifications i n the bas ic design
The separations present ly

72.
of the gas chromatographs, the perfect ion of the dual hydrogen flame ioniza- t i o n detector , and the development of methods of dual column operation. The success of the s tudies w i t h the high molecular weight materials and the sub- sequent refinements o f t h e general conditions of gas chromatography have yielded b e t t e r separations and higher recoveries a l so w i t h the lower mole- cu la r weight compounds including the simple alkyl e s t e r s of f a t t y acids .
This paper discusses some of these developments and indicates how the improved methods of gas chromatographic analysis can be e f f ec t ive ly combined with o ther chromtographic sys t em and chemical procedures of invest igat ion t o y i e ld an e s sen t i a l ly complete account of the composition of most mixtures of na tura l t r ig lycer ides .
GAS CRROMATOGRAPHY O F TRIGLYCERIDES
The procedures described here a r e chief ly those developed over Short, thin-fi lm the l as t few years i n the author 's laborator ies (3,s).
columns prepared by coating diatomaceous earth supports with s i l i cone polymers have been used f o r t he resolut ion of na tura l t r ig lycer ide mixtures s ince 1961. have taken place i n instrumentation and column technology. is a b r i e f account of the present s t a t e of the work together with a com- mentary on i t s more c r i t i c a l features.
The method has been gradually improved as new developments The following
1. Instrumentation
Effective gas chromatography of na tura l glycerides requires the Although the basic pr inciples optimum i n apparatus design and operation.
of good design have been wel l known f o r some time, there has been con- siderable reluctance on the p a r t of the manufacturers t o incorporate a l l the fea tures in to any one instrument. Testing of gas chromatographic apparatuses f o r t r ig lycer ide separation i n our laboratory has shown t h a t only one of the four leading manufacturers examined had produced an in- strument l i n e which could be used f o r glyceride separation without modi- f i c a t i o n . Although the unsat isfactory instruments were i n several re- spects superior t o the sa t i s f ac to ry instrument, they contained one o r more design f a u l t s which rendered them unsuitable f o r these separations. more adequate instrument makes a r e ident i f ied i n the legends t o the f igures . (6) t o be capable of the separation of na tura l glycerides without modifi- cat ion.
The
Only one o ther l i n e of gas chromatographs has been reported
a ) Temperatures
Triglyceride analyses require temperature pro gramrned ope rat ion Linear temperature programmers with propor-
The most
w i t h temperatures t o 35OoC. t i o n a l heat input are of the most general application, but ce r t a in non- l i n e a r programs may be b e t t e r su i ted f o r spec i f ic resolutions. usefu l temperature programs f o r metal columns have been those providing regular increases of 2-SoC per minute i n the 200-325% range of oven tem- perature . Glass and s i l iconized metal columns have permitted programs of up t o 2OoC per minute without any noticeable lack of equi l ibra t ion of the solutes .

73.
The column i n l e t heater should provide uniform heating of the in- l e t end of the column and the in j ec to r compartment and should be capable of maintaining i t s temperature s e t t i n g without superheating despi te the r i s e i n the oven temperature. For spec ia l appl icat ions there may be a need f o r a programmed temperature operation of the in j ec to r heater a l so . Isothermal s e t t i ngs of 280-325'C, however, have proved adequate f o r most appl icat ions.
The de tec tor compartment should be maintained a t a constant in-
Although temperature var ia t ions of dependent temperature, usua l ly the maximum working temperature. of 340-350°C has proved sa t i s fac tory . a f e w degrees may take place without any noticeable e f f e c t upon the resolu- t i o n o r recovery of the compounds, the retent ion times do change. For t h i s reason, high qua l i t y work involving the measurements of physical constants on the separated components requires r i g i d control of a l l temperatures.
A s e t t i n g
b) Injectors
The in j ec to r compartment should be joined d i r e c t l y t o the oven without any cold junctions t h a t require spec ia l i n l e t l i n e hea ters . The design should permit an on-column in jec t ion w i t h a conventional 10 yl Hamilton syringe equipped w i t h a 2 i n . needle. The in j ec to r compartment should be of a small volume capable of accepting the in j ec to r end of the column i n such a manner (Figure 1) as t o allow the c a r r i e r gas t o sweep around the end of the column and insure a rapid and complete entry of a l l sample in to the column. The c a r r i e r gas should be preheated by passing through a s t a in l e s s steel capi l la ry wrapped around the in j ec to r block, and should en te r the in j ec to r b a r r e l j u s t above the Swagelok f i t t i n g accepting the column. sharper peaks, bet ter resolution and higher recovery of the injected mate- r ia l . Furthermore, on-column in jec t ion allows a sa t i s f ac to ry admiss ion o f the solute t o the column a t temperatures of up t o 7OoC below those required f o r f lash evaporation (325-350OC). The apparatus should be capable of dual, compensating column operation to minimize the e f f ec t s of l i q u i d phase bleed- ing a t elevated temperatures. This requires t h a t both columns be equipped w i t h on-column in jec tors which are maintained a t constant and iden t i ca l temperatures throughout the operation.
c) Detectors
It has been shown (5) t h a t on-column in jec t ion r e su l t s i n much
The de tec tor should be located d i r e c t l y a t the end o f the column without any extra spacing which requires special detector l i n e heaters and complicates the operation of the chromatograph. A dual, compensating flame de tec tor is recommended. Those of the non-metallic construction give l e s s noise and a r e f r e e of competitive ion col lect ion. The flame skould be de- signed f o r operation a t high flow ra t e s and elevated temperatures. A t t he high column temperatures the argon ionizat ion de tec tor becomes eas i ly con- taminated with the bleed of the l i qu id phase and is l e s s s a t i s f ac to ry . The hydrogen flame detector , however, may a l so show unusual e f f ec t s which may be traced t o s i l i cone deposits on the flame j e t s o r the co l lec tor assembly. The presence of these deposits is demonstrated i n the l o s s of s igna l below a ce r t a in l e v e l of s ens i t i v i ty . The base l i n e appears extremely quie t and s t ab le as the background noise, column bleed and the minor components are not recorded. This l o s s i n s e n s i t i v i t y cannot be recovered by lowering the attenuation, and the instrument l i n e a r i t y is poor. S i l i c a and any carbon deposits can be readi ly remved i n the u l t rasonic bath.

74.
The extremely wide range of the hydrogen flame detector , which can be obtained w i t h careful adjustment of hydrogen and a i r flows under normal operation, permits the use of a wide range of sample concentrations and a quant i ta t ive estimation of both major and minor components in the same run. tained i n the range 0-45 pg and f o r t r i s t e a r i n i n the range 0-20 pg. l a t t e r compound has shown a d i f f e ren t slope i n the 20-45,ug range. under the usual operating conditions a full scale t r i s t e a r i n peak represents only 10-2Opg of t r ig lycer ide , t h i s non-linearity occurs only i n overloaded columns and does not present a problem i n analysis .
It has been shown ( 7 ) that a l i n e a r response f o r t r i l a u r i n may be ob- The
Since
d) Flow rates
High flow ra t e s (150-300 ml/min of nitrogen o r helium) of c a r r i e r gas obtained by means of low head pressures a re necessary if the tri- glycerides a r e t o be vo la t i l i zed and brought o f f the column as symmetrical peaks a t the lowest possible operating temperatures. The hydrogen flame detector i s r e l a t ive ly insens i t ive to small changes i n the flow r a t e of the c a r r i e r gas. Great f luc tua t ions i n the c a r r i e r flow, however, should be absolutely avoided as the concentration of the sample i n the de tec tor w i l l change w i t h the f l o w rate, which i n turn w i l l change the apparent detector response. Constant flow ra t e s of c a r r i e r gas may be maintained by means of d i f f e r e n t i a l f l o w controls which automatically ad jus t the flows t o com- pensate f o r the changes i n column resis tance during the temperature pro- gramming. Constant flows f a c i l i t a t e the operation of dual column systems and help t o maintain equal base widths of the peaks during l i n e a r temper- a tu re increase.
2 . Column Technology
Gas chromatography of t r ig lycer ides requires columns of especial ly high qua l i ty . Usually, however, those s i lan iz ing and support coating techniques which produce adequate packings f o r the resolution of s t e r o l s w i l l a l so serve t o prepare sa t i s f ac to ry columns f o r t r ig lycer ide work. optimum performance, ce r t a in fea tures of t he physical makeup of the column and the mode of operation a re important and deserve consideration. Specific columns and operating conditions a r e described i n the legends t o the f igures used t o i l l u s t r a t e various appl icat ions.
For
Glass, s t a in l e s s s t e e l , and aluminum tubes have been used w i t h equal success a s the column mater ia l . The g lass columns, however, provide b e t t e r peak resolut ion and allow f a s t e r programing r a t e s than s t e e l columns, which require more time f o r equi l ibra t ion due t o grea te r surface adsorption. Si lanizing the i n t e r i o r of the s t e e l tubes g rea t ly reduces the adsorption and gives columns,the performance of which is indistinguishable from t h a t of g lass columns. The dimensions of the columns employed have ranged from 6 in . t o 8 f t . i n length and 1/16-1/4 i n . i n diameter. of 18-24 i n . i n length and 1/8 i n . O.D. provide su f f i c i en t number of theo- r e t i c a l p l a t e s t o completely resolve most mixtures of na tura l t r ig lycer ides , without impaired recoveries. The s t e e l columns with 1/8 in . O.D. and 1/16 in . I.D. give the bes t resolution, but because of heavier w a l l require more time f o r heat equi l ibra t ion and permit only low ra t e s of programming. The
Columns

75.
highest resolut ions a r e obtained on thin-wall 1/16 in . O.D. s t e e l columns which y i e ld about 1000 theore t ica l p l a t e s pe r foo t of column length. The decreased flow rates, however, may reduce the recoveries of the higher molecular weight mater ia ls . The grea t disadvantage of g lass columns i s the lack of adequate means of maintaining a sa t i s f ac to ry s e a l between the g lass and the metal in le t and o u t l e t por t s during a l t e rna te heating and cooling cycles.
b ) Packing6
O f the l a rge va r i e ty of chromatographic supports t r i e d , the s i lan ized diatomaceous ear th preparations have yielded high qua l i ty columns most frequently. Mesh s i zes of 60-80 and 100-120 have given maximum column eff ic iency even w i t h shor t column lengths . high temperature s t a b i l i t y and low polar i ty , s a t i s f ac to ry t r ig lyce r ide separations have been real ized only with the s i l i cone l i qu id phases. (General E lec t r i c s i l i cone gum), W-1 (Dow Corning f luoroalkyl s i l i cone gum) and JXR (Applied Science polysiloxane polymer) have a l l given s imi la r separations. d i r e c t appl icat ion of the desired thickness of coating of high temperature s t a b i l i t y without extensive conditioning and s t r ipp ing of the v o l a t i l e mate- r i a l . Other s i l i cone polymers of high thermal s t a b i l i t y and of po ten t i a l appl icat ion i n t r ig lycer ide separations have been introduced under the t rade names of OV-1 and OV-17 (Applied Science Laboratories). s i l i cone polymers and have been claimed t o give some 25-30%less r i s e i n the base l i n e than SE-30 when programed t o 34OoC. Polysulfone (Applied Science Laboratories) is another polymer of high temperature poten t ia l with some separating cha rac t e r i s t i c s similar t o those of W-1. It is a s o l i d up t o approximately 24OoC and i ts thermal s t a b i l i t y has been ra ted above t h a t of JXR. Experimental techniques su i tab le f o r the preparation of the support, s i lan iz ing , and the appl icat ion of the coatings have been described by Homing e t a 1 (4). r e l a t ive ly low cost from re l i ab le manufacturers o f gas chromatographic supplies.
Because of the requirement of
SE-30
The extremely low bleed of the JXR polymer makes possible the
These a r e phenyl-
However, excel lent column packing6 can be obtained a t a --
c) F i l l i n g and conditioning of columns
The requirement o f maximum eff ic iency demands t h a t the shor t columns be uniformly packed and t h a t the packing re ta ins i t s s t ruc ture dur- ing the i n i t i a l conditioning and through the heating and cooling cycles of normal operation. a r e packed with the help of the suction of a water pump, but column loading under pos i t ive pressure (15-25 p s i ) of i n e r t gas may a l so be used. f i l l i n g the o u t l e t end of a su i tab ly shaped tube is closed with a l i g h t plug of s i l i conized g lass wool, extending a few mm from the end of the tube in to the column i n t e r i o r . If mechanical v ibra tors a r e used, care should be taken not t o f r ac tu re the support pa r t i c l e s and t o expose adsorptive s i t e s . The column i s f i rmly f i l l e d t o about 35 mm from the i n l e t end, and a small s i l i conized g lass wool plug i s pushed down the tube t o r e s t against the packing. under c losely similar conditions. very high flow ra t e s but because of loose packing require longer column lengths t o obtain column ef f ic ienc ies comparable t o those avai lable i n t i g h t l y packed shor t tubes.
Usually the narrow diameter (1/16-1/8 i n . ) s t e e l co i l s
Before
For a dual column instrument, two columns a re packed simultaneously Columns packed by gravi ty s e t t l i n g give

76.
Before use the columns a r e conditioned by heating t o maximum oper- a t ing temperatures and keeping there u n t i l the low molecular weight residues have been polymerized o r bled of f . To avoid contamination, t he o u t l e t end of the de tec tor i s l e f t f r e e and the c a r r i e r gas ( s l i gh t ly below i t s optimum flow rate) is vented without passing through the detector . Depending on the l i qu id phase, the time required f o r conditioning may vary from a f e w hours t o several days. The first f e w runs seldom give en t i r e ly sa t i s f ac to ry re- coveries and b r i e f periods of addi t ional conditioning are required.
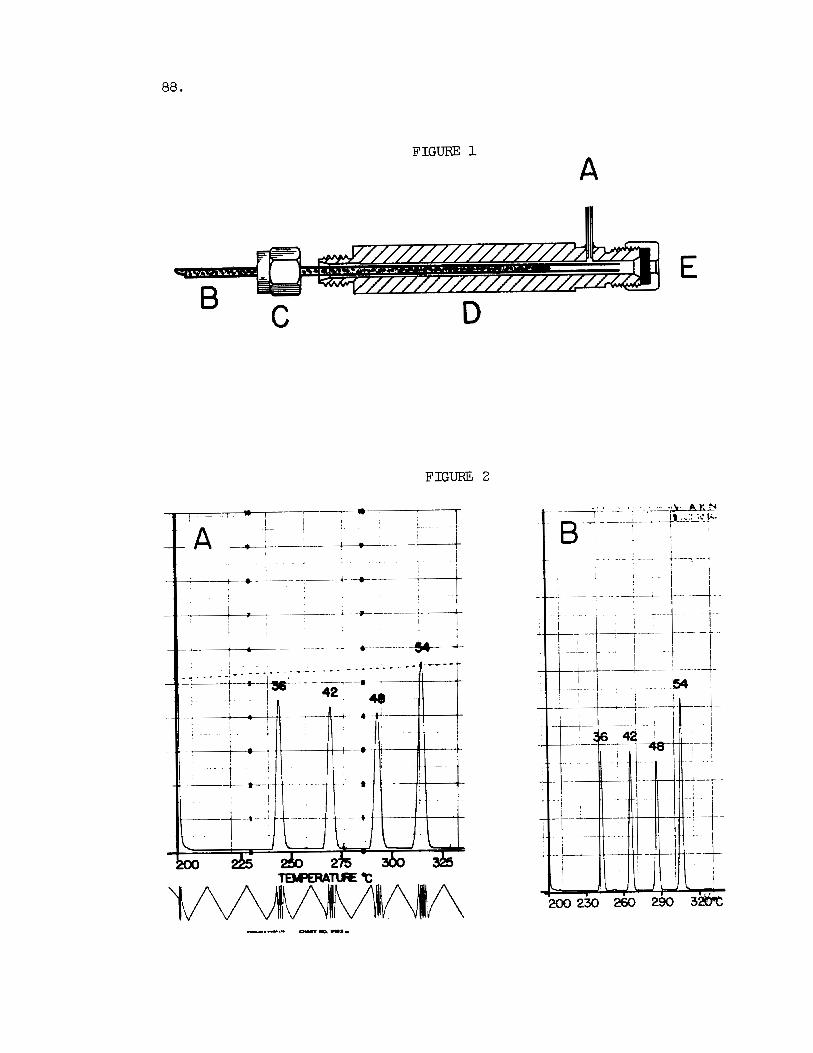
The conditioned column is tes ted f o r i t s a b i l i t y t o resolve a standard mixture of t r i l a u r i n , t r imyr is t in , t r ipa lmi t in and t r i s t e a r i n . Under properly adjusted working conditions, the four t r ig lycer ides should be eluted a s symmetrical peaks w i t h the approximate retent ion temperatures given i n Figure 2 . The base widths of the peaks should be narrow and the peak spacing su f f i c i en t ly wide t o accommodate two even carbon number tri- glycerides d i f f e r ing by two carbons between any two adjacent simple glyc- e r ides . Besides assessing resolution, the i n i t i a l runs w i t h the standard glycerides of known concentration a l so serve t o determine the extent of the ac tua l as well as the proportional recovery of the d i f f e ren t glycerides. With the common flame ionizat ion detectors , sample s i zes range from 1-50 jig o f t o t a l glyceride. The usual peak seen i n separations of t h i s kind is 0.5-2 pg f o r a s ing le component. The proportional recovery of t he standard glycerides m y be appraised from a comparison of the peak areas , which should r e f l e c t nearly correct weight proportions.
The l i m i t of s e n s i t i v i t y i s about 0 . 0 l p g .
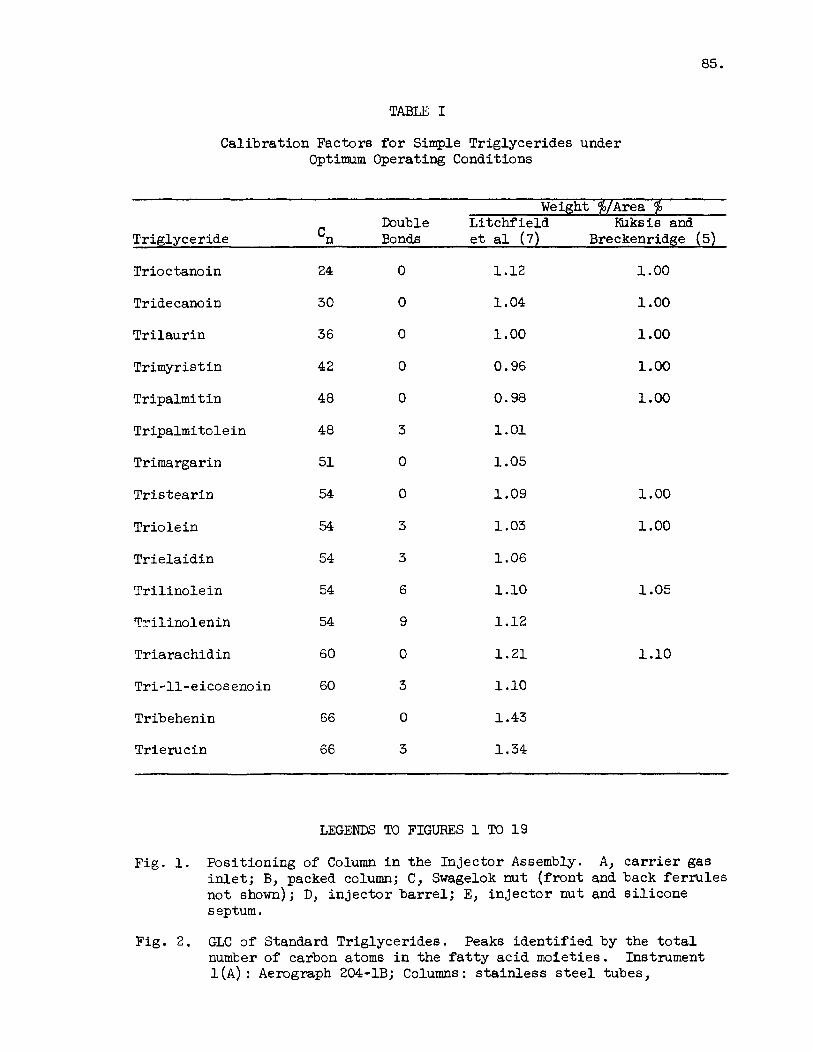
-act recoveries of the various glycerides necessary f o r quantita- t i v e work may be determined by careful ca l ibra t ion of the system wi th accurately measured amounts of su i tab le standards. The correction f ac to r s derived f r o m such ca l ibra t ions (Table I) may be used f o r work w i t h na tura l glyceride mixtures t o which they a r e applied on the bas i s of molecular weight. Because of gradual column de ter iora t ion and detector fouling it is necessary t o recheck the cal ibrat ions per iodical ly . For work w i t h erucic acid r ich o i l s and marine o i l s , the columns and the apparatus are t e s t ed w i t h glyceride standards containing t r ia rachid in and t r i e ruc in . Columns t h a t do not show responses i n the indicated range should be recon- di t ioned o r discarded. Since the performance o f the column is determined using the complete system, it should be kept i n mind t h a t fac tors o ther than the column may be respcnsible f o r poor recovery and resolut ion.
3 . Separation of Natural Glycerides
Work w i t h standards has shown t h a t the method separates glycerides according t o the molecular weight o r carbon number, although some resolut ion due t o differences i n molecular shape may also be demonstrated. Ordinarily, saturated glycerides a r e not resolved from t h e i r unsaturated homologues. For t h i s reason the most e f fec t ive separations are obtained wi th those na tura l glyceride mixtures which contain a la rge var ie ty of f a t t y acid chain lengths . Coconut o i l i s made up of t r ig lycer ides containing c6 t o C18 f a t t y ac ids . f o r new columns. S imi la r e lu t ion pa t te rns can be obtained f o r palm kernel
It is very easy t o resolve (3) and may be used as a t r i a l mixture
oil, and the o i l s of the Lauraceae, Lythraceae and Ulmaceae p lan t famil ies , a l l of which are r i c h i n l a u r i c acid (8).

77.
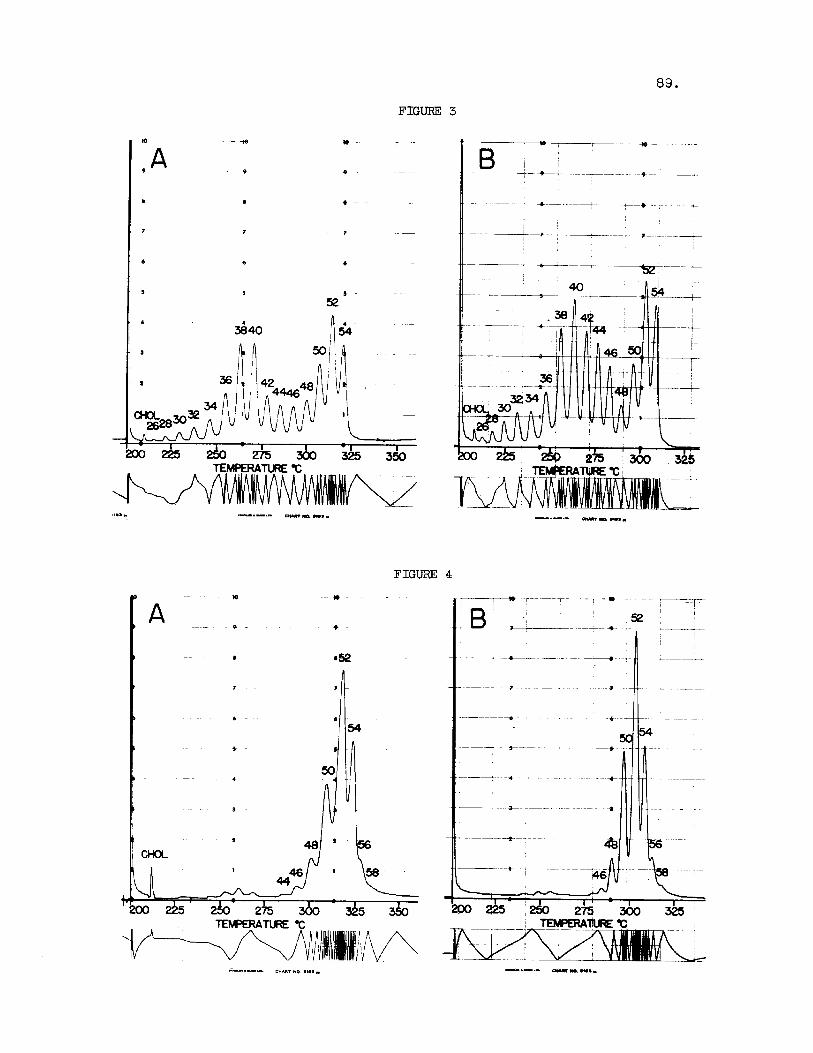
The bovine milk fa t , although somewhat more complex, contains a comparable range of t r ig lycer ides . The e lu t ion pat terns recorded f o r t r ig lycer ide mixtures of t h i s type have been shown by the author on several occasions (9,lO). and goat milk. Because of the presence of small amounts of odd carbon number and branched chain f a t t y acids i n the milk f a t s , the re turn t o base l i n e be- tween adjacent peaks i s not as complete as f o r coconut o i l . The two-hump e lu t ion pa t te rns a r e very cha rac t e r i s t i c and resemble those observed f o r cow's milk. Figure 4 shows the e lu t ion sequences noted f o r dog and human milks. Cue t o a v i r t u a l absence of bu tyr ic acid from these f a t s , the f i rs t hump i n the glyceride e lu t ion pa t te rn does not appear. In a l l cases the milk fats contain a much wider range of glycerides than the body f a t s of the corresponding species.
Figure 3 shows the e lu t ion pa t te rns recorded f o r sheep
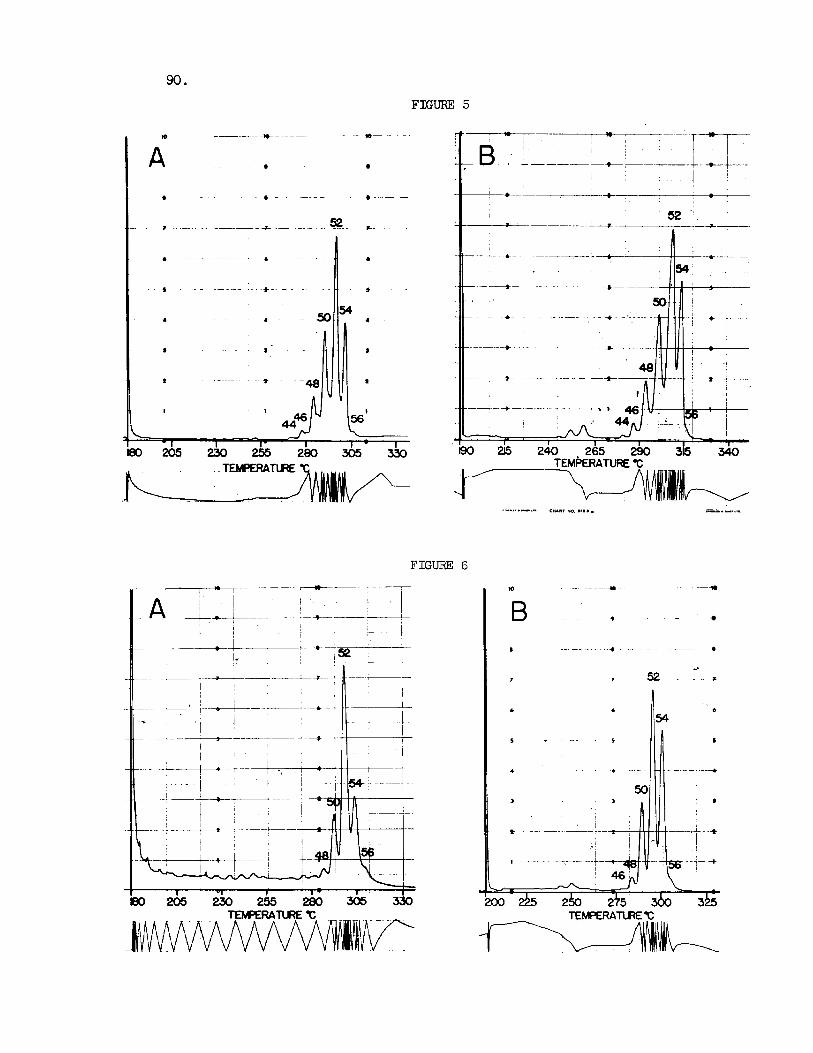
Figure 5 shows the e lu t ion pat terns recorded f o r the t r ig lycer ides of sheep sue t and beef tallow. peaks ind ica te t h a t most of the f a t is made up of t r ig lycer ides containing two c16 and one c$8 acid, and one c16 and two c18 ac ids . pa t te rns a r e obtained f o r t he t r ig lycer ide mixtures of o ther t i s sues of these animals.
The la rge proportions of the C50 and C 5 2
Comparable e lu t ion
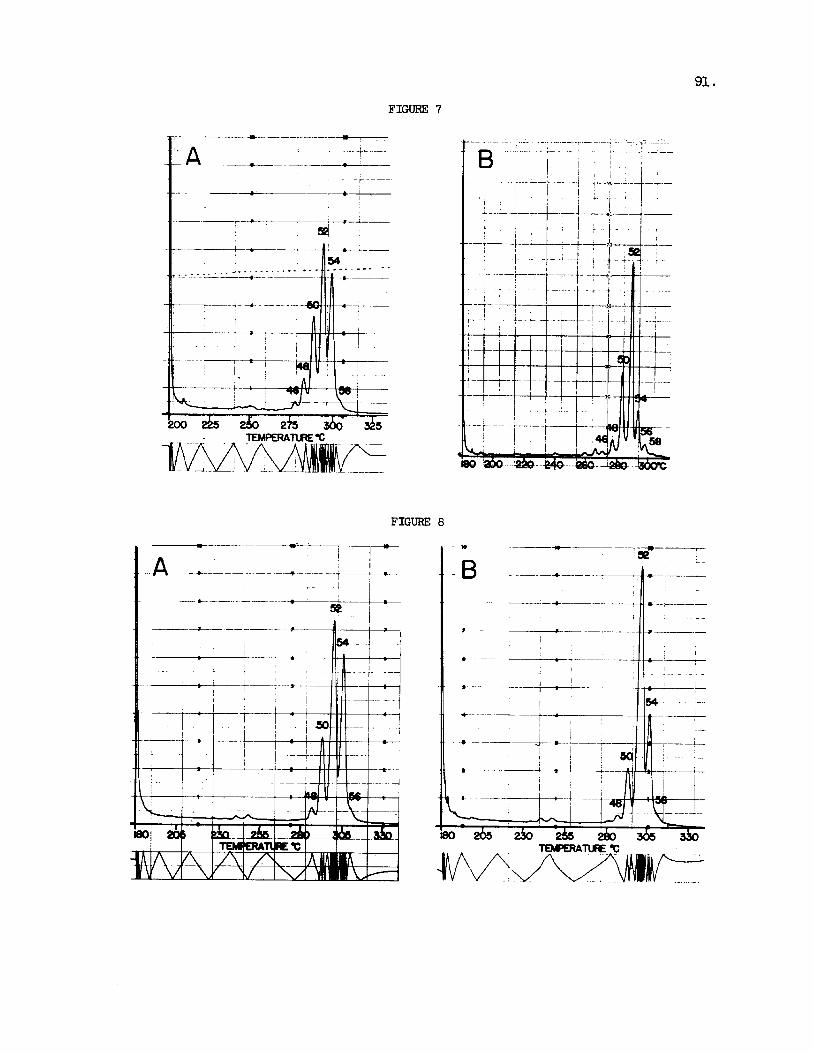
Figure 6 gives the e lu t ion pat terns recorded f o r the t r ig lycer ides from chicken and goose f a t . Again the t r ig lycer ides o f C 5 0 and C52 carbons predominate and there i s a supe r f i c i a l s imi l a r i t y t o the sheep and beef f a t t r ig lycer ides as well as t o the glycerides of the dog pancreas and rat l i v e r shown i n Figure 7. More extensive comparisons have shown t h a t the differences i n the glyceride e lu t ion pa t te rns a re frequently g rea t e r between d i f f e ren t t i s sues i n a s ingle animal than between the same t i s sues o f d i f - f e r e n t species. Thus the glyceride mixtures of plasma, l i v e r , kidney and hear t show considerable s i m i l a r i t i e s among some species, ye t the glycerides of t he kidney and the l i v e r of the same animal may d i f f e r widely. l a t t e r observation i s i l l u s t r a t e d with the elut ion pat terns obtained f o r the kidney and l i v e r of t he same goose shown i n Figure 8. s i m i l a r i t i e s o r differences may frequently be very pronounced, the e lu t ion pa t te rns could conceivably change with d i e t and even vary from one indi- vidual t o the other , and a r e unl ikely t o provide any well defined character- i s t i c s .
The
Although these
The separations obtained with a number of the common vegetable o i l s were shown i n one of the o r ig ina l publications from our laboratory ( 9 ) . The predominance of the C 1 8 f a t t y acids i n these o i l s r e su l t s i n a major peak f o r the C 5 4 glycerides with much smaller amounts contributed by the
:?go foun8'in these o i l s . A s shown by calculations of recovery (3) and by chromatography a f t e r hydrogenation, the areas of the eluted peaks a r e cor rec t even f o r the most highly unsaturated o i l s . i den t i ca l e lu t ion pa t te rns f o r raw and hydrogenated samples o f l inseed oil.
and C glycerides which account f o r the l e s s e r anounts of the c16 acids
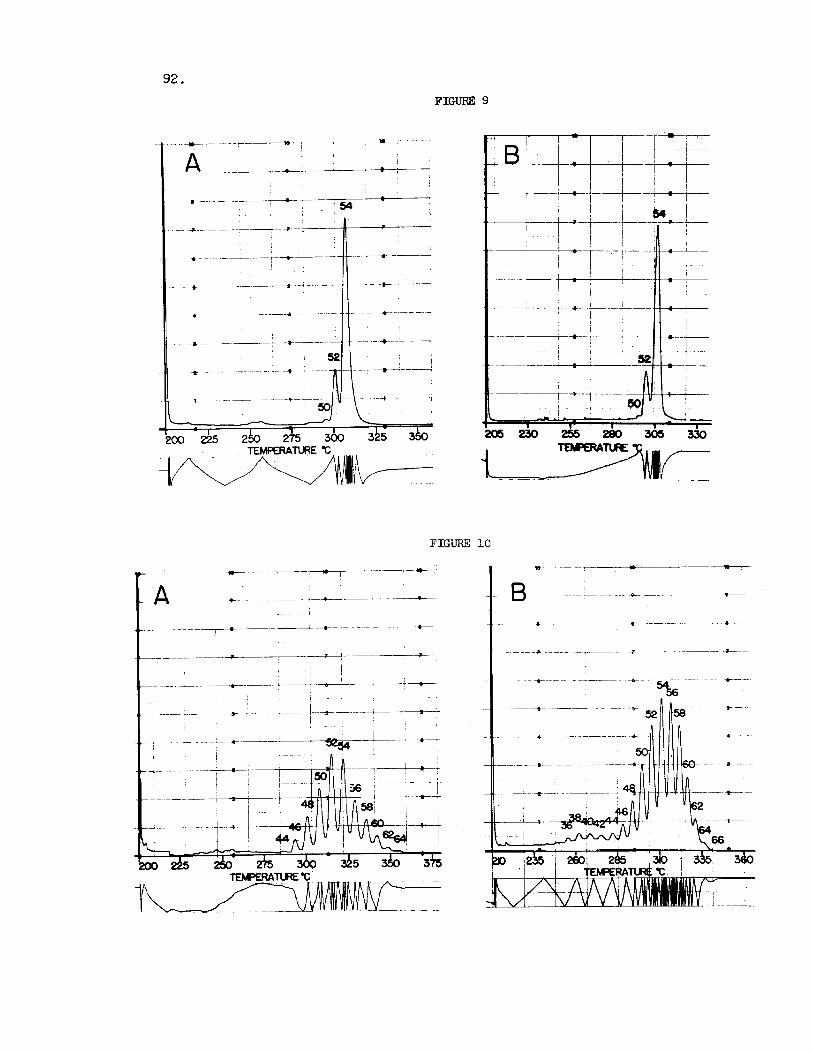
Thus Figure 9 shows
Figure 10 i l l u s t r a t e s the resolution achieved f o r menhaden and herr ing o i l s . acids and contain t r ig lycer ides with carbon numbers of up t o C 6 6 . Because of the progressively decreasing retent ion times observed f o r the higher members of the homologous se r i e s under l i n e a r temperature programming, the longer chain glycerides show Considerable overlap on the shor t columns.
These o i l s a r e r ich i n unsaturated C18, Czo, and C z 2 f a t t y

70.
Somewhat longer columns (24 in.) and narrower column diameters (1/16 i n . ) give b e t t e r peak spacing. The e lu t ion pa t te rn i s not mater ia l ly improved by a p r i o r hydrogenation of the o i l . obtained f o r o the r marine o i l s and erucic ac id r i ch p l an t o i l s (11).
Comparable e lu t ion pa t te rns have been
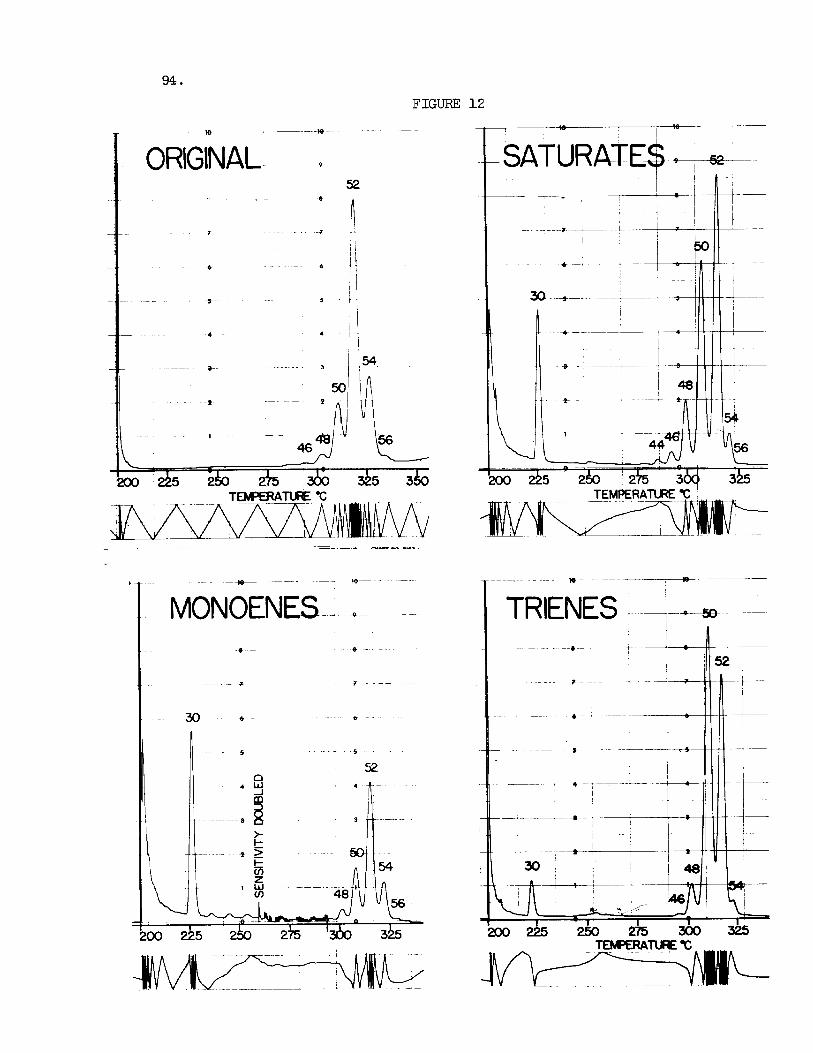
The comparisons of the t r ig lycer ide e lu t ion pa t te rns can be g rea t ly upgraded by a preliminary separation o f the t r ig lycer ide mixture on the basis of unsaturation on s i l i c a g e l p l a t e s impregnated with s i l v e r n i t r a t e (12). A s a r e s u l t of it, it is possible t o compare the d i s t r ibu - t i o n s among the saturated, and the mono-, di- , and t r i -unsaturated glyc- e r ides of d i f f e r e n t t i s sues and animal o r p l an t species . t r a t e s the separations obtained on s i l v e r n i t r a t e p l a t e s f o r a sample of l a r d . there has a l so been a resolut ion of ce r t a in pos i t iona l isomers, which may be separately recovered and analyzed i n the gas chromatograph. Figure 1 2 shows the e lu t ion pa t te rns recorded f o r the d i f f e ren t bands and the o r i g i - na l f a t . The quant i ta t ive proportions of the bands may be determined f r o m the peak area of tr idecanoin which can be conveniently added t o the t r i g lyc - e r ide mixtures recovered from the p l a t e .
Figure 11 i l l u s -
In addi t ion t o the segregation based on the number of double bonds,
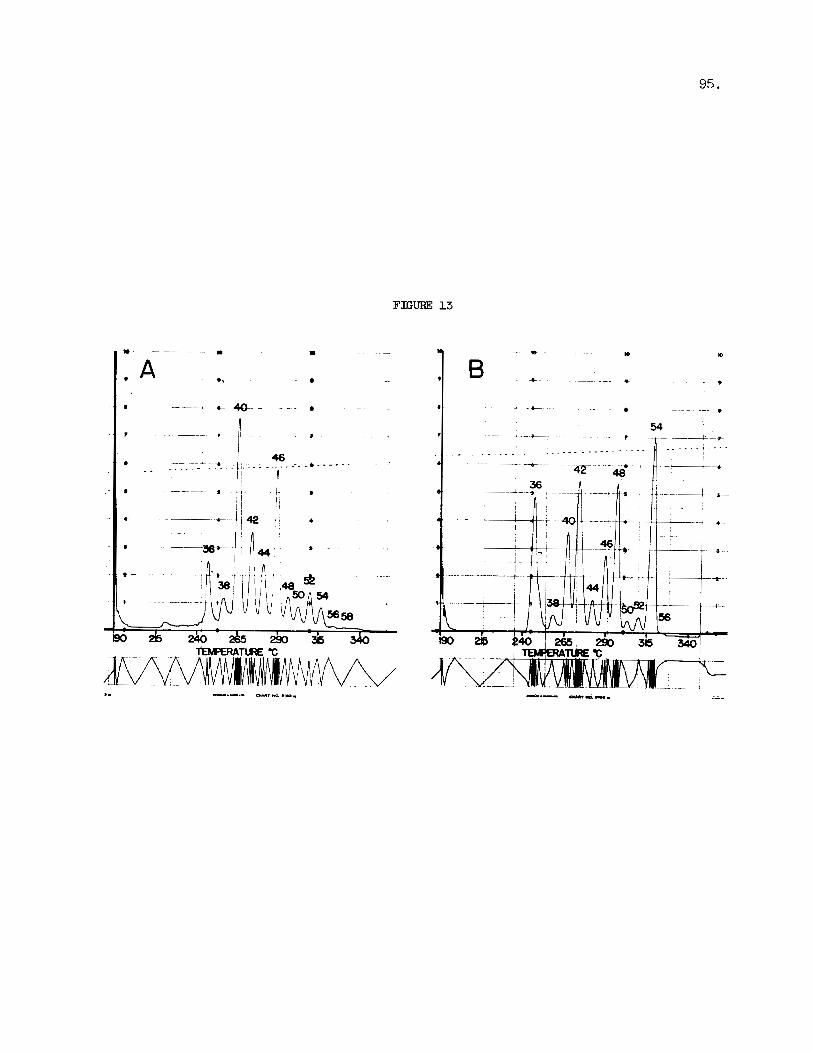
The composition of the d i - and t r i -unsaturated glycerides can be fu r the r analyzed by subjecting a port ion of the mater ia l recovered from the p l a t e t o a permanganate-periodate oxidation p r i o r t o gas chromatography. This oxidation quant i ta t ive ly converts the unsaturated triglycerid.es in to saturated glycerides containing aze l a i c ac id residues (13) . Following diazomethylation the oxidized glycerides behave i n the gas chromatograph as t r ig lycer ides containing one, two, o r three l a u r i c ac id residues. Figure 13 shows the e lu t ion pa t te rn recorded f o r the oxidation products of whole lard. the standard glycerides, the overlap between t r i a z e l a i n and t r i l a u r i n is not complete, but it is close enough t o permit e f fec t ive ana lys i s . f i r t h e r - more, the overlap with the corresponding even carbon number glycerides in- creases as the number of aze la ic acid residues per molecule decreases.
A s it can be seen from the p a r t of the p r i n t superimposed upon
This method of analysis i s equally well su i ted f o r the determina- t i o n of the t r i g lyce r ide composition of vegetable o i l s , and f i s h o i l s . Because of high unsaturation, the f i s h o i l t r ig lycer ides cannot be ade- quately resolved by the common techniques of s i l v e r n i t r a t e thin- layer chromatography, as glycerides of more than 5 double bonds remain a t the or ig in . This d i f f i c u l t y , however, is l i k e l y t o be overcome by modifica- t i ons i n the g e l and the developing solvents .
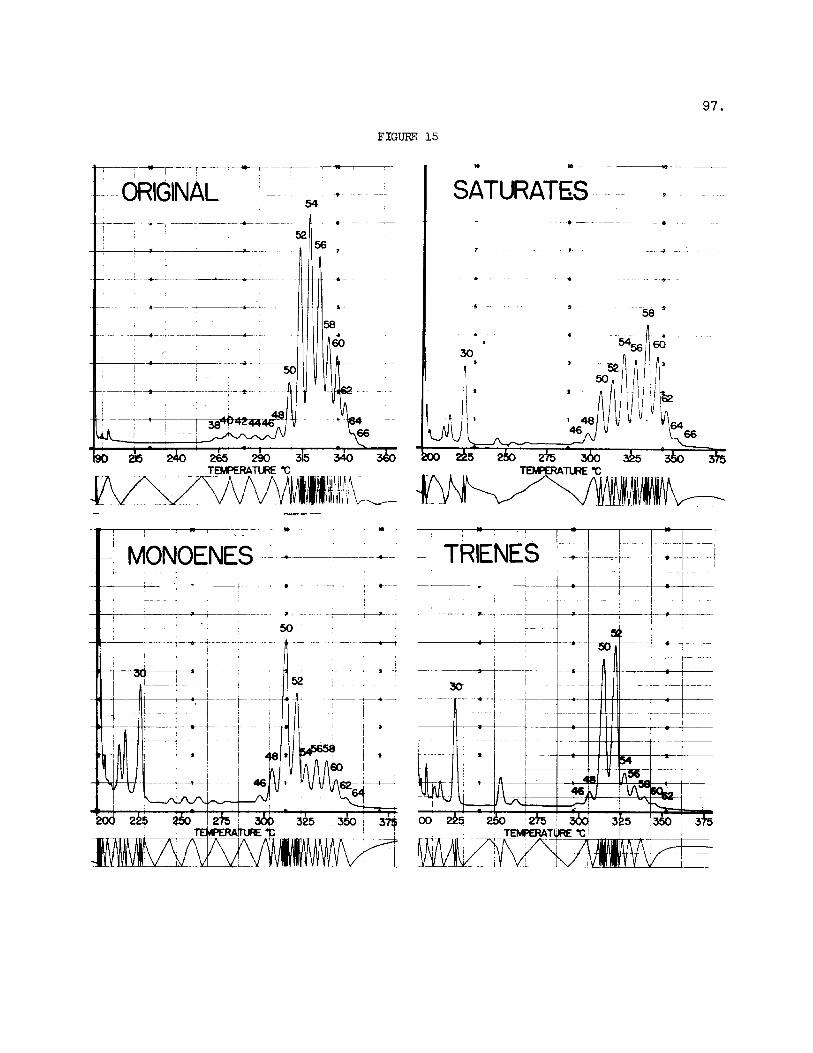
The appl icat ion of these methods t o the analysis of fats of in - d u s t r i a l importance is i l l u s t r a t e d with a sample of margarine prepared by p a r t i a l hydrogenation of f i s h o i l s . Figure 14 shows the resolut ion ob- tained on the s i l v e r n i t r a t e p l a t e . The combination p r i n t given i n Figure 15 shows the glyceride composition of the various bands recovered from the p l a t e . t r ig lycer ide bands can be e f f ec t ive ly estimated by means of tr idecanoin used as in t e rna l standard.
A s noted e a r l i e r , the proportions of the mater ia l i n the various
These gas chromatographic systems a r e equally e f f ec t ive f o r the resolut ion of t o t a l neut ra l and i n ce r t a in cases t o t a l l i p i d mixtures. chromatograms of t o t a l neut ra l l i p i d ex t rac ts of plasma and lymph show an
The

79.
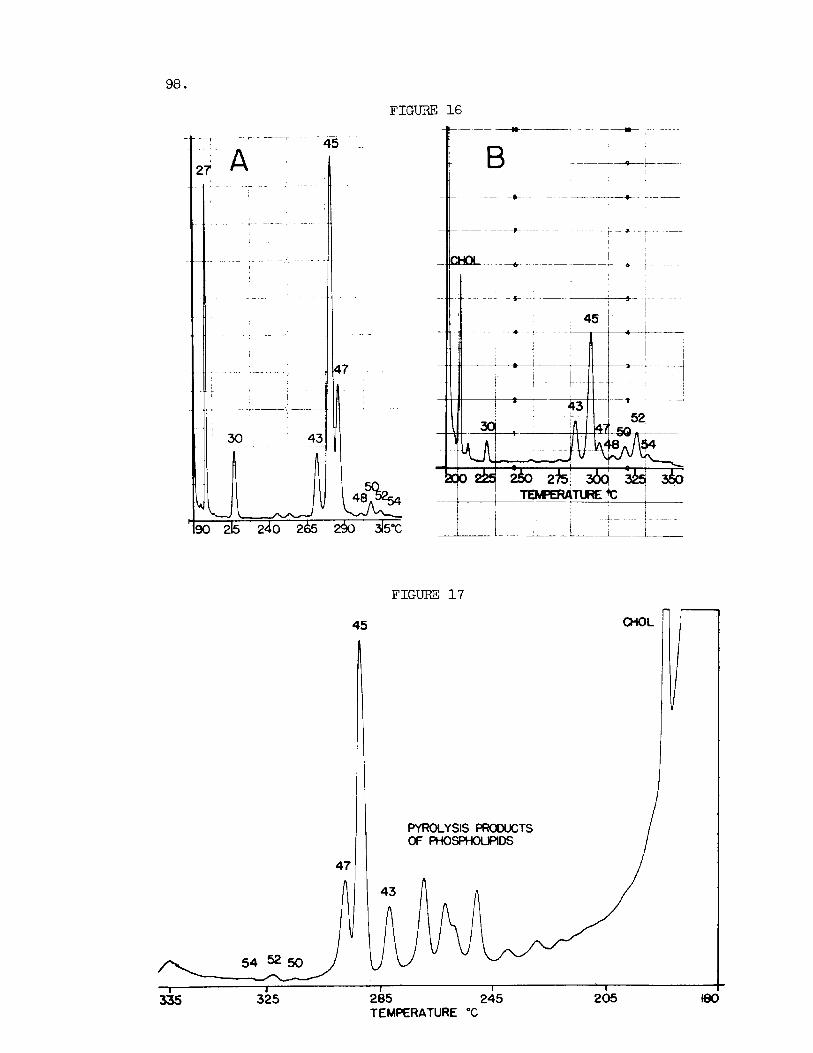
essen t i a l ly complete resolution of a l l the l i p i d components present (14) . There is no s ign i f i can t overlap between the s t e r y l e s t e r s and the t r i g lyc - er ides . It has s ince been demonstrated t h a t the t o t a l l i p i d ex t rac ts of these body f l u i d s can a l so be analyzed i n the gas chromatograph and good estimates f o r cholesterol , t o t a l phospholipid, cholesteryl e s t e r s and t r ig lycer ides obtained i n a s ingle run. Fig. 16 shows the neutral plasma l i p i d s of man and dog. Under the conditions of gas chromatography the phospholipids a r e pyrolyzed t o the corresponding propene d i o l d i e s t e r s i n about 60% yield. with retent ion times comparable t o those of diglycerides, and emerge well ahead of the steryl e s t e r s . chromatography f o r the determination of the l i p i d composition of plasma and o ther body f l u i d s without p r i o r extract ion. The l i p i d e s t e r s appar- en t ly completely sublime from t h e i r l ipoprotein complexes a t the in j ec to r temperatures and appear as symmetrical peaks i n the e f f luent . Figure 1 7 shows the e lu t ion pa t t e rn recorded f o r 3 pl of f a s t ing dog plasma.
These d i e s t e r s a r e e luted from the t r ig lycer ide column
Eventually it may be possible t o use gas
GAS CHROMATOGRAPHY OF FATTY ACIDS
Conventional gas chromatographic analyses of f a t t y acids a re per- formed on the methyl e s t e r s using heavy coatings of l i q u i d phases. the isothermal conditions generally employed, these methods a re bes t su i t ed f o r the determination of the common C 1 2 to C 1 8 saturated and unsaturated f a t t y acids . mixtures containing both shor t and long chain homologues. Posi t ional and s t e r i c isomers of the common unsaturated f a t t y acids a r e not resolved and there is d i f f i c u l t y i n the quant i ta t ive estimation of peaks of long re- ten t ion times. These problems may not be serious when dealing with ex- t r a c t s of t o t a l l i p i d s i n which the unusual acids make up negl igible pro- port ions. t a n t when attempting t o reconstruct the ove ra l l composition of a l i p i d m i x - t u r e from the fa t ty ac id determination of completely resolved l i p i d classes and individual e s t e r groups. Attempts t o solve t h i s problem have been made by seeking new l iqu id phases that would be capable of resolving a l l f a t t y acids , and by incorporating in to the present gas chromatographic systems those design and operation features which have proved successful i n the separations of o ther compounds. Other improvements in the methods of de te r - mining f a t t y acids have been sought through short cuts i n the preparation of f a t t y acid e s t e r s . appears t o be of l imited p r a c t i c a l app l i cab i l i t y and w i l l not be discussed.
Under
They a r e not fully sa t i s f ac to ry f o r the scanning of acid
Accurate estimates of a l l f a t t y acids become extremely impor-
The determination of f r e e f a t t y acids present ly
1. Column Technology and Instrumentation
Since s t ab le l i qu id phases of po la r i ty high enough f o r the resolu- t i o n of f a t t y acids have been slow i n forthcoming, the major improvements i n the gas chromatography of fa t ty acids have been made by designing in- struments t h a t permit work with v o l a t i l e phases. In these e f f o r t s the dual column systems have proved most s a t i s f ac to ry and have allowed temperature programed operations o f up t o 5OoC above the normal working l i m i t o f the common l i q u i d phases. With increasing i n t e r e s t in programed temperature separations there has been a gradual switch towards the use o f columns with thinner coatings of s ta t ionary phase.

80 .
a ) Liquid phases
Polar l i qu id phases i n the form of various polyesters have been most useful f o r f a t t y acid separations. l i q u i d phase have been applied extensively to the separation and iden t i f i - cat ion of f a t t y acids i n the form of meth 1 es te r s (1). These phases a r e l imited to temperatures not exceeding 220 C . proached a s ign i f i can t amount of the l i q u i d phase v o l a t i l i z e r s and bleeds in to the detector . the polyesters and thereby r a i se t h e i r m a x i m u m usefu l temperature l i m i t . By introducing various terminating agents and by a judicious choice of the e s t e r i f i ca t ion ca t a lys t it has been possible t o increase the upper l i m i t of the working range t o about 250-270°C. creasing the bleed r a t e of the polyester columns i n the working range has been the copolymerization of ethylene glycol, succinic acid and dimethyl- siloxane monomers (EGSS-X, Applied Science Laboratories). The mixed s i l i - cone-polyester polymers a re l e s s polar than the simple polyesters but a r e s t i l l capable of maintaining su f f i c i en t s e l e c t i v i t y f o r the unsaturated f a t t y acids t o give complete separations. The u t i l i z a t i o n of 1% coatings of t h i s phase permits temperature programming with s ingle columns i n the range of 120-225°C, without s ign i f i can t baseline elevation (15). The capec- i t y of such columns i s su f f i c i en t f o r work with flame ionizat ion detectors . A s a 10% coating t h i s s ta t ionary phase is idea l ly su i ted f o r t he isothermal separation of methyl e s t e r s i n the C14 to C24 range, a l l of which can be eluted within 30 minutes. The shor te r chain f a t t y acids can be e f fec t ive ly determined a t selected lower isothermal temperatures. Another po lar e s t e r packing t o which spec i f i c a t t en t ion has been ca l led i s the tetracyano- ethylated pentaery thr i to l (TCEPE) . Its upper thermal l i m i t is only 15OoC, but when used a s a 3% ccjating on Aeropak 30 i n a 10 f t . x 1/16 i n . O.D. column (Wilkens Instrument and Research) it has been claimed t o permit complete resolut ion t o a l l t he common f a t t y acid e s t e r s within 35 minutes a t 105OC.
Columns containing 10-25% (w/w)
B A s the upper l i m i t is ap-
Several techniques have been developed t o s t a b i l i z e
The most useful method of de-
Non-polar l i qu id phases have generally higher thermal s t a b i l i t i e s than the polyesters , but have the i n a b i l i t y t o separate the c18 and higher unsaturated f a t t y acids which contain 2 o r more double bonds. Because of t h e i r g rea te r temperature s t a b i l i t y , non-polar l i q u i d phases a re excel lent choice for separating saturated f a t t y acids w i t h chain length l a rge r than 22 carbons. Columns packed with 10-17$ Apiezon L grease have been widely used (1). Separations o f the c18 homologues a re possible, however, a l so with the non-polar l i qu id phases when packings of about 3% l i q u i d phase a r e used in combinatior! with 1/16 in . O.D. x 20 f t . columns (Wilkens In- strument and Research) which generate over 20,000 theore t ica l p l a t e s (calculated f o r methyl s t e a r a t e ) . polymers (SE-30, JXR) give the lowest bleed r a t e s and a re pa r t i cu la r ly well sui ted f o r temperature programming and quant i ta t ive work which does not require a resolut ion beyond the molecular weights. A s t ab le mater ia l recommended f o r preparative sca le gas chromatography of f a t t y acid e s t e r s because of low bleed r a t e (up t o 25OoC) i s the beta-cyclodextrin ace t a t e . This phase, however, w i l l not resolve methyl s t ea ra t e and methyl o lea te .
O f the non-polar phases the s i l i cone
b) Columns and supports
Although the f a t t y acid e s t e r s a r e r e l a t ive ly non-polar and highly v o l a t i l e , the gas chromatographic analysis of t h e i r mixtures can be

81.
considerably improved by adopting the techniques developed f o r dealing with more polar and much l e s s v o l a t i l e compounds. Thus s i lan iz ing the i n t e r i o r of metal C O ~ U m n f o r the use of g lass columns improves peak shape and reso- l u t ion . Signif icant improvements i n column eff ic iency a r e real ized by using s i l iconized supports of narrow mesh s izes . Gas Chrom Q (Applied Science Laboratories), Aeropak (Wilkens Instrument and Research), and the high performance Chromosorbs (Johns Yanville) a r e a l l w e l l su i t ed f o r pre- paring high qua l i ty columns. Micropak (Wilkens Instrument and Research) columns (1/16 in . O.D.) of 10 t o 20 f t . length give vas t ly improved resolutions with most l i q u i d phases because of the grea te r number of t heo re t i ca l p l a t e s provided per f o o t of column length.
Narrow bore 1/8 i n . 0 .D. columns and the
c) Instrumentation
Advanced equipment design and b e t t e r methods of operation have f a c i l i t a t e d the r ea l i za t ion of good f a t t y ac id separations even with poor packings and badly packed columns. placement of the f lash vaporization chamber with an on-column i n l e t . change has allowed the appl icat ion of the e s t e r mixtures t o the column i n a narrow zone giving much sharper peaks and b e t t e r resolution, and has per- mitted work a t considerably lower temperatures.
O f major s ignif icance has been the re- This
The replacement of the thermal conductivity c e l l and argon ioniza- t i o n detectors with flame ionizat ion u n i t s has resul ted i n a more s a t i s - factory in te rpre ta t ion of the detector response and a more rapid quantita- t i o n of the eluted components. Flame ionizat ion detect ion i n combination with dual column operation has been most e f f ec t ive i n temperature pro- gramming of columns containing thermally unstable l i qu id phases. Other improvements i n instrumentation of the type described f o r t r i g lyce r ide separations a r e valuable a l so i n work with f a t t y acid e s t e r s bu t have not been repeated here.
2. Method of Chromatography
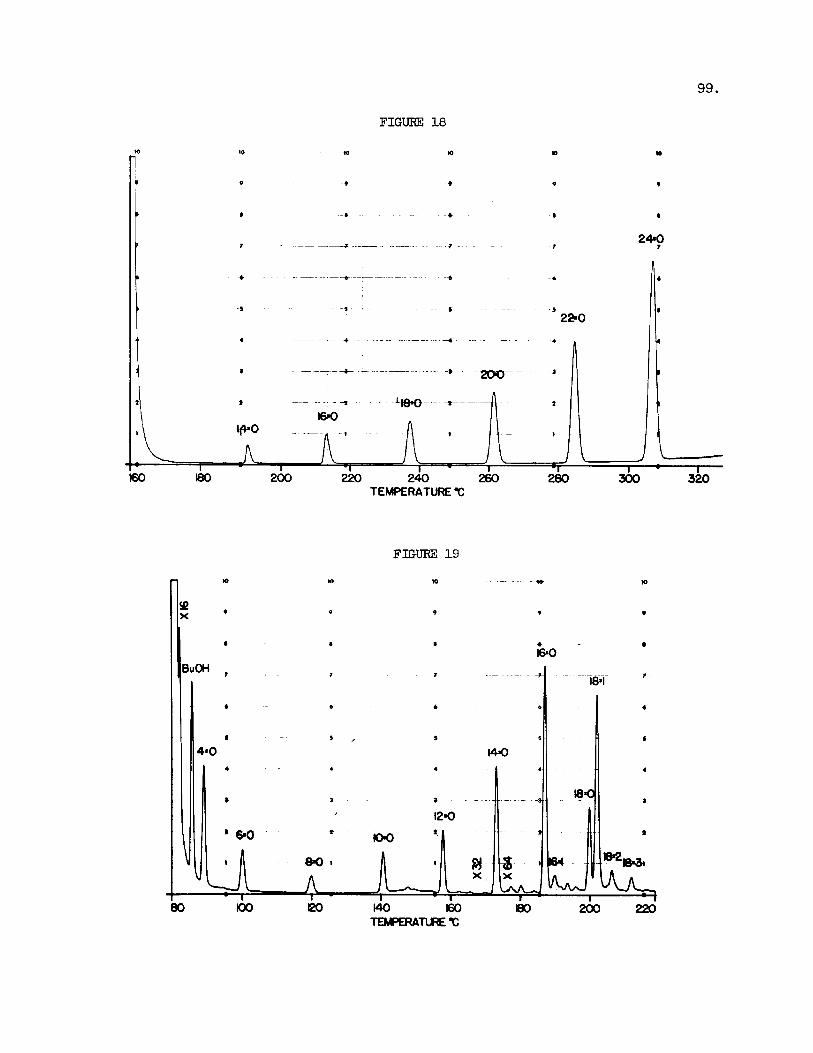
Wide ranges of f a t t y ac id methyl e s t e r s may be scanned on s u i t - able columns by the use of temperature programming. Excellent separations of the methyl o r bu ty l e s t e r s of fa t ty acids on the bas i s of molecular weight a r e obtained on 5% SE-30 columns (Figure 18) . This column provides the most effect ive means present ly avai lable f o r the quant i f icat ion of t he t o t a l f a t t y acids recovered from a pa r t i cu la r l i p i d preparation using an in t e rna l standard (methyl heptadecanoate) . The e lu t ion can be completed within 30 minutes using a temperature range of 160 t o 3OO0C and se lec t ing temperature increments and chart speeds t o s u i t the e s t e r mixture a t hand. Complete separations of a l l f a t t y acids can be made on 1% EGSS-X l i q u i d phase i n the temperature range 125OC t o 215OC using a 1 2 f t . column (15). Good batches of t h i s packing y i e ld excel lent separations o f not only the monoenes and dienes but a l so of t r ienes and te t raenes of various chain lengths. These packings, however, a r e qui te var iable and frequently batches a r e found that give no resolut ion of anything. Highly e f f i c i e n t packings prepared w i t h t h i s phase a l so allow the resolut ion of c16 and c18 dimethyl ace t a l s from the c16 and c18 f a t t y ac id methyl e s t e r s . the SE-30 and the EGSS-X packings a r e idea l f o r instrumental area integra- t i on even with s ing le column instruments.
Both

82.
Temperature programmed runs w i t h the diethylene glycolsuccinic acid polyester l i qu id phases (DEGS) may be obtained i n dual column, dual flame detector instruments. Good resolut ion of the methyl e s t e r s of milk f a t t r ig lycer ides , f o r example, has been achieved in some 20 minutes i n the temperature range of 100-235°C, when programmed a t rates of 4 t o G°C/min. A run w i t h the methyl esters of menhaden o i l may be completed i n about 30 minutes giving complete separations of a l l the saturated and unsaturated f a t t y acids including the C z Z Z 5 and C22:6 e s t e r s . Although the e f f ic iency of the polyester columns may be somewhat increased by decreasing the thick- ness of the coating t o about 6$ and thus a l so minimizing the column bleed, the advantages gained are o f f s e t by the shortened l i f e t i m e of t he column. A loading of about 10% l i qu id phase appears to provide a s a t i s f ac to ry com- promise. Baseline s t a b i l i t y i n the compensated analysis i s very good u n t i l the end of the run when a tendency t o d r i f t below sca le develops. This can be avoided by l eve l l i ng the program of f a t about 2 l O o C and completing the run under isothermal conditions. Certain preparations of ethylene glycol adipate have been shown (2) t o possess a considerably higher thermal sta- b i l i t y than the diethylene glycol succinate polymer, as have the s t ab i l i zed polyesters. The advantages tha t such e s t e r s may o f f e r i n programmed t e m - perature analyses of f a t t y acids , however, have remained uncertain.
The separations obtained by temperature programming the Apiezon l iqu id phases a r e comparable to , and o f f e r no advantage over, those noted f o r s i l i cone gum columns. qua l i t a t ive i n nature and has been r e s t r i c t ed t o isothermal separations of the shor t and medium chain length f a t t y acid e s t e r s . the separation of pos i t iona l and geometric isomers of unsaturated f a t t y acids and some branched chain ac ids . phases have been most sa t i s f ac to ry f o r these s tud ies .
The work with capi l la ry columns has been la rge ly
It is applicable t o
Apiezon L and the polyester l i q u i d
3. Ident i f ica t ion of Fa t ty Acid Esters
Relative retent ion times a r e conveniently used f o r t en ta t ive iden- t i f i c a t i o n of f a t t y acid e s t e r s because considerable data of t h i s type has been published. For a systematic characterization of the e s t e r s the gas chromatographic data may be expressed in terms of the equivalent chain lengths (ECL) o r the rrodified equivalent chain length (MECL). The ECL i s obtained by p lo t t i ng the logaritki;!i of re tent ion times of normal saturated monocarboxylic acid e s t e r s against the number of carbon atoms i n the corre- sponding acids . rather than the saturated es te rs , and u t i l i z e s re tent ion times r e l a t ive t o methyl s t ea ra t e ra ther than absolute values. The chain lengths of o ther compounds may be measured by comparing the retent ion times of the standards t o that of the unknown. The use of two o r more columns is recommended.
The MECL is based upon a p l o t with the monounsaturated
Although these gas chromatographic techniques a r e qui te helpful f o r determination of the t en ta t ive iden t i ty of unknown f a t t y acid e s t e r s , o ther methods should be used t o ver i fy t h i s information. Oxidation with permanganate-periodate, reductive ozonolysis and mass spectrometry have a l l been demonstrated t o provide much of the required information about the s t ruc ture of unsaturated and saturated f a t t y acid e s t e r s . The considera- t i o n of these techniques i s beyond the scope of the present review.

83.
4. Preparation of Fa t ty Acid Esters
The f a t t y acid e s t e r s necessary f o r the gas chromatographic analy- sis may be conveniently obtained by transmethylation of the l i p i d s o r by a methylation of the f a t t y acids recovered a f t e r saponification. Both tech- niques have been extensively employed and a var ie ty o f reagents u t i l i z e d (16) . Special advantages have been claimed f o r 5% perchloric acid solut ion i n methanol a s a rapid (10 min.) methylating agent f o r free f a t t y acid6 a t l o w temperatures (55OC). Because of the small quant i t ies of l i p i d usua l ly involved i n work w i t h b io logica l materials, the preparations of the methyl e s t e r s a r e bes t conducted by means of transmethylation. extract ion of the acids inevi tably r e su l t s i n losses due t o foaming and numerous t r ans fe r s .
Saponification and
Useful i n quant i ta t ive work combining thin- layer and gas chroma- tographic systems is the finding t h a t complete t r anses t e r i f i ca t ions of a l l l i p i d s may be obtained i n the presence of the s i l i c a g e l scrapings from thin- layer p l a t e s , including those containing s i l v e r n i t r a t e . Refluxing o r heating i n sealed tubes with 10% (w/v) su l fu r i c acid i n dry methanol f o r 1 - 2 hours a t 8OoC i s su f f i c i en t t o convert a l l l i p i d s to the methyl e s t e r s except sphingomyelin which requires about 1 6 hours. The time f o r the t r anses t e r i f i ca t ion of the sphingomyelin may be shortened by increasing the reaction temperature t o l l O ° C . hydroquinone pro tec ts the l i p i d s from peroxidation. The use of methanolic su l fu r i c ac id f o r the procedure i s preferred i n view of the reports claim- ing contamination from the use of methanolic HLC a s a methylating agent i n the ultramicroanalyses. Nevertheless, transmethylations with methanolic HCL as w e l l as BF3-methanol have been found highly sa t i s f ac to ry i n a number of labora tor ies . The methanolic su l fu r i c acid procedure has been adopted a s a t en ta t ive method by the American O i l Chemists' Society f o r the pre- parat ion of the methyl e s t e r s of Cg-Cz4 saturated and unsaturated f a t t y acids ( 1 7 ) .
Nitrogen atmosphere and a c rys t a l of
The quant i ta t ive analysis of shor t chain f a t t y acids o r mixtures
These e s t e r s a r e somewhat less v o l a t i l e than the methyl of shor t and long chain f a t t y acids a re improved by a preparation of the buty l e s t e r s . e s t e r s and permit more sa t i s f ac to ry concentration during the evaporation of organic solvents. The buty l e s t e r s can be prepared by subs t i tu t ing butanol f o r methanol i n the c o m m es t e r i f i ca t ion mixtures catalyzed by mineral ac ids . e s t e r s of f a t t y acids derived from mi lk f a t t r ig lycer ides on diethylene glycol succinate. The f a t t y acid e s t e r s should be analyzed as soon as pre- pared. a t low temperature f o r 24 hours. For longer storage they should be sealed i n - a g lass ampule under vacuum and placed i n a f reezer .
Figure 1 9 shows a programmed temperature separation of the buty l
They may be kept i n an atmosphere of nitrogen i n a screw cap v i a l
CONCLUSION
The described techniques permit the accumulation of a f a n t a s t i c d e t a i l of information regarding the composition of any f a t . Such e f f o r t s , however, may not be wisely invested unless the source of the f a t i s ade- quately defined and reproducibly sampled. An analysis of a t o t a l l i p i d ex t rac t of a whole organ i s of l i t t l e biochemical i n t e r e s t . t i s sue and c e l l u l a r f rac t iona t ion must precede any l i p i d extract ion.
An ef fec t ive There

84.
may be a fu r the r need f o r the i so l a t ion of individual l ipoproteins o r l ipoprotein groups, if the design and purpose of the diverse mixtures o f na tura l l i p i d e s t e r s i s t o be determined.
REFERENCES
1. A. T, James, i n D. Glick (Editor), Methods of Biochemical Analysis, Vol. 8, Interscience Publishers, New York, 1960, p . l .
2. W. R . Supina, H. A. Szymanski (Editor), Biomedical Applications of Gas Chromatography, Plenum Press, New York, 1964, p .271.
3. A. Kuksis, J. Am. O i l Chemists' SOC. - 42, 269 (1965).
4. E. C, Horning, W. J. A. VandenHeuvel, and B. G. Creech, i n D. Glick (Editor), Methods of Biochemical Analysis, Vol. 11, Interscience Publishers, New York, 1963, p.69.
5. A. Kuksis and W. C . Breckenridge, J. Lipid Res. (1966), In press .
6. M. L. Yates, H. L. Conrad and J. S. Forrester , Bul le t in AB-509, Micro Tek Instruments, Inc. , Baton Rouge, Louisiana.
7. C . Li tchf ie ld , R. D. Harlow and R. Reiser, J. Am. O i l Chemists? SOC. 42, - 849 (1965).
8. C . Li tchf ie ld , E. Miller, R. D. Harlow and R . Reiser, J. Am. O i l Chemists' SOC. - 43, 102A (1966).
9. A . m k s i s and M. J. McCarthy, Can. J. Biochem. Physiol. 40, 679 (1962).
10.
11. R. D. Harlow, C . L i tchf ie ld and R . Reiser, Lipids (1966), In press .
A. Kuksis and M. J. McCarthy, J. Am. O i l Chemists' SOC. 41, - 17 (1964).
12. F. B. Padley, i n M. Lederer (Editor), Chromatographic Reviews, Vol. 8, Elsevier Publishing Co., Amsterdam, 1966, In press.
13.
14.
C . G. Youngs, J. Am. O i l Chemists' SOC. - 38, 62 (1961).
A. Kuksis, Can. J. Biochem. - 42, 419 (1964).
15. E. C. Horning and W. J. A. VandenHeuvel, J. Am. O i l Chemists' Soc. 41, - 707 (1964).
16. A. Kuksis, i n M. Lederer (Edi tor) , Chromatographic Reviews, Vol. 8, Elsevier Publishing Co., Amsterdam, 1966, I n press .
1 7 . Report o f t he Instrumental Techniques Committee AOCS 1964-65, J. Am. O i l Chemists' SOC. 43, - 1OA (1966).
85.
TABLE I
Calibration Factors f o r Simple Triglycerides under Optimum Operating Conditions
Weight $/Area F Double Li tchf ie ld m k s i s and
Triglyceride 'n Bonds e t a 1 (7) Breckenridge (5)
Trio c tano i n 24 0 1 . 1 2 1.00
Tride can0 i n 30 0 1.04 1 .oo
Tri laur in 36 0 1.00 1 .oo Trimyristin 42 0 0.96 1.00
Tripalmitin 48 0 0.98 1.00
Tripalmitolein 48 3 1.01
Trimargarin 51 0 1.05
Tr i s tear in 54 0 1.09
Triolein 54 3 1.03
Tr ie la id in 54 3 1.06
Tr i l ino le i n 54 6 1.10
Tri l inolenin 54 9 1.12
Triarachidin 60 0 1 . 2 1
Tri-11-eicos enoin 60 3 1.10
Tribehenin 66 0 1.43
Trierucin 66 3 1.34
1.00
1 .oo
1.05
1.10
LEGENDS TO FIGURES 1 TO 19
Fig. 1. Posit ioning of Column i n the In jec tor Assembly. A, c a r r i e r gas i n l e t ; B, packed column; C, Swagelok nut ( f ront and back ferrules not shown); D, i n j ec to r bar re l ; E, i n j ec to r nut and s i l i cone s ep tum .
Fig. 2 . GLC o f Standard Triglycerides. Peaks iden t i f i ed by the t o t a l number of carbon atoms i n the f a t t y ac id moieties. Instrument 1 (A) : Aerograph 204-1B; Columns : s t a in l e s s s t e e l tubes,

86.
20 in . x 1/8 in . O.D., packed with 3% (w/w) JXR on Gas Chrom Q (100-120 mesh) and assembled as shown i n Fig. 1; Nitrogen flow r a t e : 120 ml/min. a t room temperature; Injector , 28OoC; Detector, 34OoC; Sample: 1 y1 of 1% (w/v) solut ion in chloroform. Attenua- t i o n 1 x 16. Temperature program as shown. Chart speed, 6 min/in.
Instrument 2(B) : Beckman GC-4 with modified on-column i n l e t and spec ia l heater; Columns: s t a in l e s s s t e e l tubes, 20 i n . x 1/8 in . O.D., packed with 3$ (w/w) JXR on Gas Chrom Q (100-120 mesh); Nitrogen flow r a t e : 120 ml/min. a t room temperature; Special in- jec tor , 28OoC; I n l e t l i n e , 30OoC; Detector l i n e , 325OC; Detector, 34OoC; Samp e : 1 pl of 1% (w/v) solut ion i n chloroform. Attenua- t i o n 5 x 10 . Temperature program as shown. Chart speed, 0.2 in/min.
3
Fig. 3. GLC of Triglycerides of Sheep (A) and Goat (B) Milk. Experi- mental conditions a s given f o r Instrument 1 i n Fig. 2. Peak i d e n t i t i e s as i n Fig. 2.
Fig. 4 . GLC of Triglycerides o f dog (A) and Human (B) Milk. Experi- mental conditions and peak i d e n t i t i e s as i n Fig. 3 .
Fig. 5. GLC of Triglycerides of Sheep Suet (A) and Beef Tallow (B) . Experimental conditions and peak i d e n t i t i e s a s i n Fig. 3 .
Fig , 6 . GLC of Triglycerides of Chicken (A) and Goose (B) fa t . Experi- mental conditions and peak iden t i t i e s as i n Fig. 3 .
Fig. 7. GLC of Triglycerides of Dog Pancreas (A) and R a t Liver (B) . Experimental conditions and peak iden t i t i e s as i n Fig. 2 .
Fig. 8. GLC of Triglycerides of Goose Kidney (A) and Liver (B) . mental conditions and peak i d e n t i t i e s as i n Fig. 3.
Experi-
Fig. 9. GLC of Triglycerides of Raw (A) and Eiydrogenated (B) Linseed O i l . Experimental conditions and peak i d e n t i t i e s a s i n Fig. 3.
Fig. 10. GLC of Triglycerides o f Menhaden (A) and Herring (B) O i l s . In- j ec tor , 325OC; Nitrogen flow ra t e : 200 ml/min. a t mom temgera- tu re . Other experimental conditions and peak i d e n t i t i e s as i n Fig. 3.
Fig. 11. Separation of Lard by TLC w i t h Plates Coated with S i l i c a Gel G Impregnated with S i lve r Nitrate and Developed with 0.7% Methanol i n Chloroform. tri, t e t r a , and polyunsaturated glyceride c l a s s .
Usually two peaks a r e seen f o r each mno, d i ,
Fig. 1 2 . GLC of Triglycerides of Lard Before and After S i lve r Ni t ra te TLC. Experimental conditions and peak i d e n t i t i e s a s i n Fig. 3.
Fig. 13. GLC of Oxidation Products of Whole Lard. A, oxidation products only; B, oxidation products plus standard t r ig lycer ide mixture. Experimental conditions as in Fig. 3. Peak i d e n t i t i e s a s ex- plained i n the t e x t .

87.
Fig. 14. Separation of the Triglycerides of a Fish O i l Margarine by TLC with P la tes Coated with S i l i c a Gel G Impregnated with S i lve r Nitrate and Developed with 0.7% Methanol i n Chloroform. i d e n t i t i e s as i n Fig. 11.
Band
Fig. 15. GLC of Triglycerides of the Fish O i l Margarine Before and After S i lve r Nitrate TLC. as given in Fig. 10.
Experimental conditions and peak i d e n t i t i e s
Fig. 16. GLC of neut ra l Plasma Lipids of Dog (A) and Man (B) . Experimen- t a l conditions as i n Fig. 3. Peak i d e n t i t i e s a s explained i n the t e x t .
Fig. 1 7 . GLC of Total Plasma Lipids of Dog. Peak i d e n t i t i e s as i n Fig. 16. Instrument 3: Aerograph Hy-Fi 600 D; Column: s t a i n l e s s s t e e l tube, 18 in . x 1/8 i n . O.D., packed with 3% (w/w) JXR on Gas Chrom Q (100-120 mesh) and assembled as shown i n Fig. 1. Nitrogen flow r a t e : 300 ml/min. a t room temperature; In jec tor , Z90°C; Detector, equal t o column temperature; Sample: 3 9 of whole dog plasma. Attenuation 1 x 16 . Temperature program as shown. C h a r t speed, 1 x 3 in/min.
Fig. 18. GLC of Fa t ty Acid Methyl Esters . C1+ t o C24 saturated f a t t y ac ids . Instrument 3 : Aerograph Hy-Fi 600 D. Column: s t a i n l e s s s t e e l tube, 5 f t . x 1/8 in. O.D., packed with 5% (w/w) SE-30 on s i lan ized Chromosorb W (60-80 mesh) and assembled a s shown i n Fig. 1. Injector , 25OoC; Detector, temperature equal t o column tempera- ture; Sample: N I H standard mixture F, 1 p l of a 1% (w/v) solu- t i o n i n chloroform. Attenuation 1 x 16. Temperature program as shown.
Nitrogen flow ra t e : 60 ml/min. a t room temperature;
Fig. 19. GLC of Fa t ty Acid Butyl Esters . sa tura ted acids . Chromatograph, Model 402 with modified i n j e c t o r i n l e t . Colums: glass U tubes, 4 f t . x 1/4 i n . O.D., 1/8 i n . I.D., packed with 20$ (w/w) diethylene glycol succinate (DEGS) on Gas Chrom P (80-100 mesh) ; Injector , 23OoC; Hydrogen Flame detector , 23OoC; Sample: f a t t y acids of bovine mi lk f a t t r ig lycer ides , 1 p1 of a 1% (w/v) so lu t ion i n chloroform. Attenuation 10 x 32. Tempera- t u re program as shown.
C4 t o C2o saturated and un- Instrument 4: F & M High Efficiency Gas
88.
FIGURE 1
A
FIGURE 2
1 I
89.
FIGURE: 3
m
,A b
7
6
i e
9
. ..
6 4 -
5 5 5 .-
52 b
36 40
3-- - --- 1- A .+--
40 I- -
FIGURE 4
i
A
- , A5
90.
FIGURF: 5
.- .
A
, . _. .. +.-
4 4 - - 501P
*-
5
I JJ k' ' 44% . -
I- I I - I 0 2 0 5 230 w 2 8 0 3 0 5 330
TEMWtATLRE
R-
-w-- 501
TEMPERATURE c
FIGURFl 6
4
91.
FIGURE 7
FIGURE 8
92. FIGURE 9
t -It ---- --- FIGURE 10
e-
- x) - , I -It-

93.
FIGURE ll
SATURATES MONOENES
DIENES
TRIENES
TETRAENES
POLYENES
94.
F I G W 1 .2
7- = I '"
CSATURAJE$---
te- - - - 't I MONOENES + -
a - 0 -
~
I I TRIENES --------- I---
95.
F I G m 13
* A - *s
4 .~
m
?
- 6
S
4
8
Ie m
t
a
7
- - - .
- e
io
- c

96.
FIGURE 14
SATURATES MONOENES
DIENES
TRIENES
TETRAENES
POLYENES
97.
F I G W 15
SATURATES P . - . .
5 58
5 -
TEMPERATURE C
i 1 1- I - - 1 - --
- TRIENES '--+-+r,
I
98.
FIGUIIF: 1 6
45 - r -- - ,
r7 ' A . . .- - - ,
30 43
&?
- * i ' I
7-- , r- ------- - - 1
. t B I
47 n
FIGUIiE 1 7
45
PYROLYSIS PROOUCTS OF PHOSPHOIJPIDS
WOL
i
I
I
1 1 I I I 335 325 285 245 205 la
TEMPERATURE 'C
99.
F I G W 18
+ . . - b
S
4
200 220 240 280 320 TEMPERATURE C
FIGURE 19
H) P -
0 9
m
I I .~
b 6
5 , 5
4 4
3 a
129
le
9
? -
6
5
4
a
¶

100.
DR. CARPENTER: I would l i k e now t o c a l l on D r . Donald J. Lisk who is head of the Pest ic ide Residue Laboratory here a t Cornell University. has been doing a grea t deal of work i n this area and of course h i s pes t ic ide and residue s tudies a r e o f v i t a l i n t e r e s t t o us in our la rge animal re- search a t the present time. D r . Lisk.
He
# # # # # # # # # # # # #





























