Embed Size (px)
Citation preview

INFORMATION BULLETIN
UNDERSTANDING DETECTION CAPABILITY: LoB, LoD AND LoQ IN THE CLINICAL LABORATORY.
OVERVIEW
HISTORY
Detection capability is a broad term that relates to measurement accuracy. It has not always been necessary to
assess the accuracy and precision in measuring clinical analytes. Historically, the clinical decision points for most
analytes have been at concentrations much higher than the analytical limitations of the methods. For example,
the lower performance limits of methods for sodium and glucose have always been far below concentrations
where clinical decisions are made. Detection capability becomes exceedingly important for analytes such as
troponin, PSA and hCG, where most individuals have very low concentrations and the detection limits of the
methods are near the clinical decision limits. It is important to characterize low-end method performance for
analytes such as these in order for manufacturers to be assured that quality test methods have been developed
and for laboratories to trust that accurate patient results will be reported. This bulletin provides an overview of
detection capability with specific examples of statistical methods to assess accuracy and precision. The statistics
described are meant to be established by the manufacturer during development of the assay, however, laboratories
may wish to verify these in their institution as they prepare to implement a new test method. Additionally, these
statistics have application in the verification of analytical measuring range for immunoassays.
Analytical sensitivity was used to describe the upper limit of test results expected when testing a sample
containing no analyte. This statistic has no practical application alone, since there is no analyte in the sample.
It is difficult to apply this statistic to anything other than the measuring system. Analytical sensitivity is described
as a concentration limit, above which the user expects the observed signal to be the result of analyte presence,
in other words, the level at which the measured value is reflective of true analyte and not simply noise in the
system. Analytical sensitivity is typically calculated by repeatedly testing a blank (sample containing no analyte)
and determining a limit either 2 or 3 standard deviations (SD) above the observed mean. The number of replicates,
whether 2 or 3 SD is appropriate, and other details have not been established by consensus.
Functional sensitivity is a term that refers to measurement accuracy with a stated imprecision. The concept was
first applied to thyroid-stimulating hormone (TSH) assay, where “generations” of TSH assays were defined by the
concentration of TSH that could be measured at ≤20% coefficient of variance (CV). A CV of ≤10% is considered the
desired level of imprecision for other analytes, such as troponin. While functional sensitivity is related in a more
practical sense to clinical sample testing, there is no consensus protocol for establishing this statistic.
LAB FORWARD
DEFINITIONS
The preferred terms used to describe the detection capability of a measurement system are Limit of Blank (LoB), Limit of Detection (LoD) and Limit of
Quantitation (LoQ).
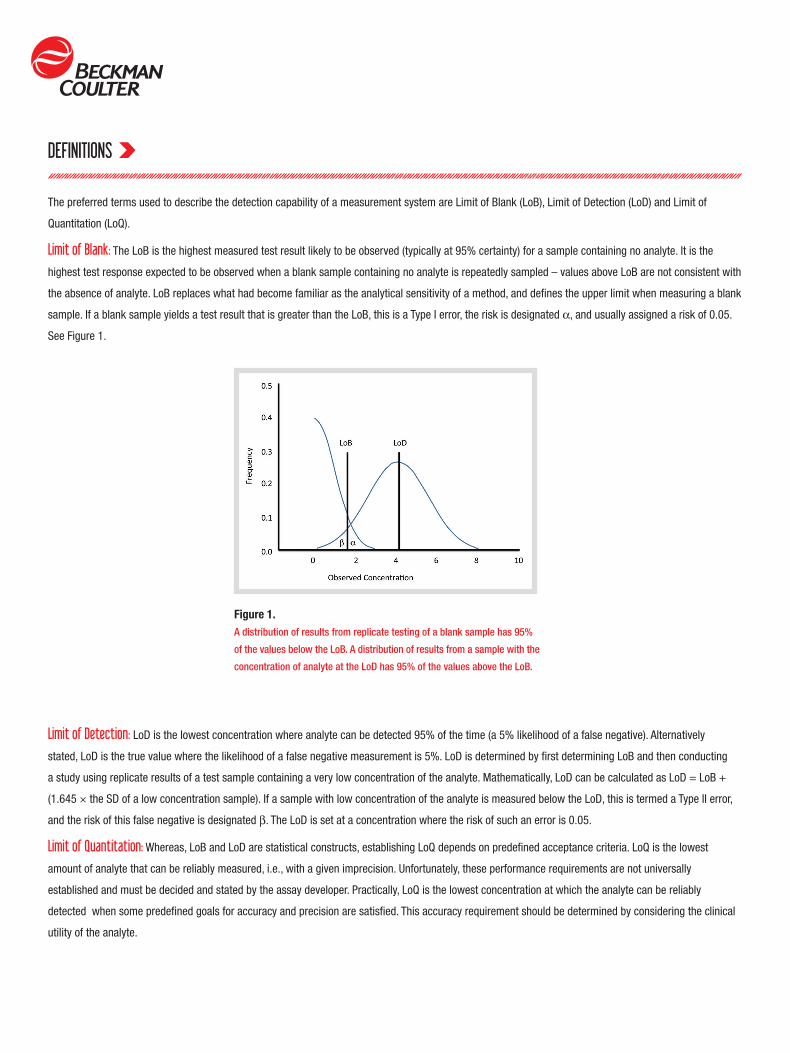
Limit of Blank: The LoB is the highest measured test result likely to be observed (typically at 95% certainty) for a sample containing no analyte. It is the
highest test response expected to be observed when a blank sample containing no analyte is repeatedly sampled – values above LoB are not consistent with
the absence of analyte. LoB replaces what had become familiar as the analytical sensitivity of a method, and defines the upper limit when measuring a blank
sample. If a blank sample yields a test result that is greater than the LoB, this is a Type I error, the risk is designated α, and usually assigned a risk of 0.05.
See Figure 1.
Limit of Detection: LoD is the lowest concentration where analyte can be detected 95% of the time (a 5% likelihood of a false negative). Alternatively
stated, LoD is the true value where the likelihood of a false negative measurement is 5%. LoD is determined by first determining LoB and then conducting
a study using replicate results of a test sample containing a very low concentration of the analyte. Mathematically, LoD can be calculated as LoD = LoB +
(1.645 × the SD of a low concentration sample). If a sample with low concentration of the analyte is measured below the LoD, this is termed a Type II error,
and the risk of this false negative is designated β. The LoD is set at a concentration where the risk of such an error is 0.05.
Limit of Quantitation: Whereas, LoB and LoD are statistical constructs, establishing LoQ depends on predefined acceptance criteria. LoQ is the lowest
amount of analyte that can be reliably measured, i.e., with a given imprecision. Unfortunately, these performance requirements are not universally
established and must be decided and stated by the assay developer. Practically, LoQ is the lowest concentration at which the analyte can be reliably
detected when some predefined goals for accuracy and precision are satisfied. This accuracy requirement should be determined by considering the clinical
utility of the analyte.
Figure 1.A distribution of results from replicate testing of a blank sample has 95%
of the values below the LoB. A distribution of results from a sample with the
concentration of analyte at the LoD has 95% of the values above the LoB.

SAMPLES TO CALCULATE LoB, LoD AND LoQ
PROTOCOLS TO CALCULATE LoB AND LoD
Blank Samples: Blank samples (containing no analyte) are used to establish the LoB. Ideally, the blank samples used should be natural, e.g., native serum,
and not an artificial matrix such as buffer. For therapeutic drugs, serum from an individual not medicated with the drug can be used. For endogenous
analytes, samples can be stripped, enzyme-treated, adsorbed with charcoal, antibody-precipitated, etc., to produce zero-analyte samples. The exact
minimum number of required blank replicate test results can be determined statistically, although four to five samples with 60 total replicates is usually
considered adequate.
Low-level Samples: Low-level samples are used to establish the LoD. The samples should be natural (e.g., native serum) as artificial or spiked samples may
behave differently in the measurement system. The number of samples and the concentrations depend on the protocol used (see below). Developers usually
have an initial estimate of the LoD. Sample concentrations should bracket this value.
Classical: The traditional approach for establishing LoB and LoD is based on the CLSI document EP17-A2. LoB is determined by repeatedly measuring the
blank sample using multiple reagent lots over multiple days. More rigorous testing could use multiple analyzers, with multiple calibrations, etc. The developer
must decide between parametric and nonparametric techniques, depending on the distribution of test results. If the test results have a normal distribution,
the mean and SD are determined and LoB can be calculated as:
LoB = Meanblank + cpSDblank
Because the SDblank is a biased estimate and not the true population SD, a correcting multiplier (cp) is used.
where cp is a multiplier to correct the SD estimate, B is the number of blank test results, and K is the number of blank samples. If the data is not normally
distributed, a nonparametric method should be used. A simple rank ordering method can be used to sort the data, then the ranked position of the 95th
percentile can be calculated:
Rank position = 0.5 + B(0.95)
For example, with 80 test results, the calculation is:
Rank position = 0.5 + 80(0.95) = 76.5
Because the resulting rank is not a whole integer, the results corresponding to the 76th and 77th ranked numbers are averaged to derive the LoD.
This protocol assumes the variability of the test method is nearly constant at concentrations near the LoD. At least four low level samples are needed for LoD
determination. These samples should be near the estimated LoD, typically one to five times the estimated LoB. The exact concentrations of the samples are
not important, because it is the SD that is used to calculate the LoD. LoD is derived from the calculated LoB and an expression of the determined SD of the
low level sample results.
LoD = LoB + cpSDlow level
where cp is the correcting multiplier, L is the total number of low level sample results, and J is the number of low level samples used.
PROTOCOLS TO CALCULATE LoB AND LoD
PROTOCOLS TO CALCULATE LoQ
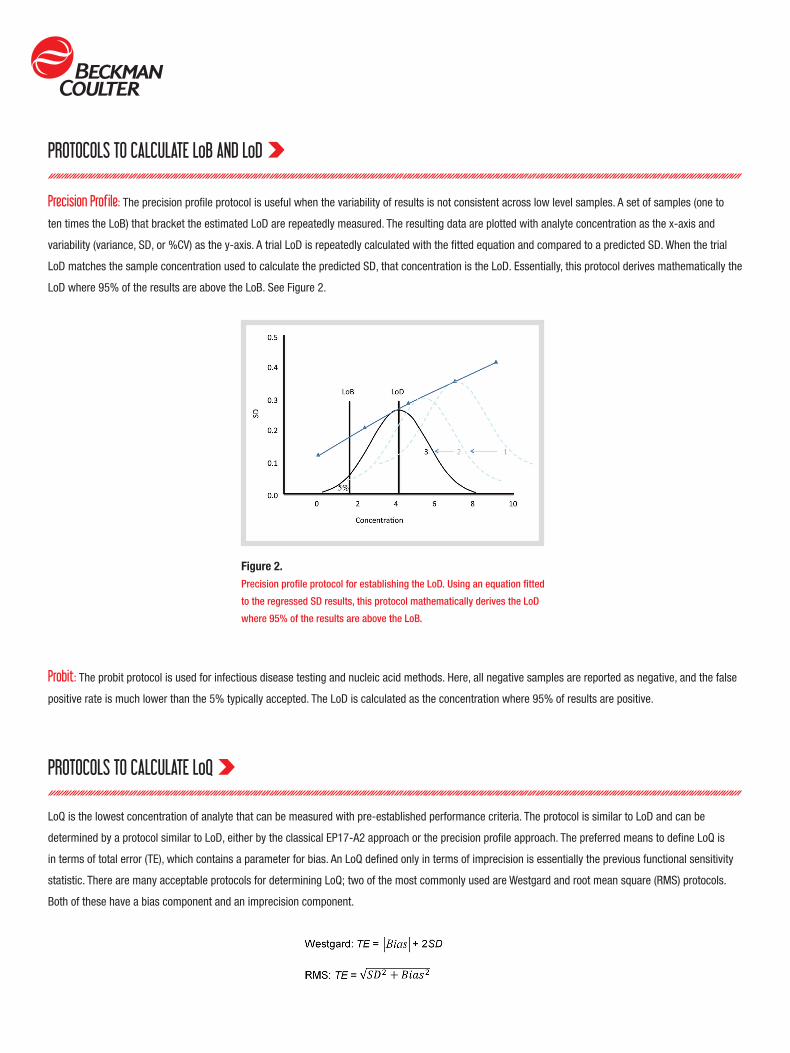
Precision Profile: The precision profile protocol is useful when the variability of results is not consistent across low level samples. A set of samples (one to
ten times the LoB) that bracket the estimated LoD are repeatedly measured. The resulting data are plotted with analyte concentration as the x-axis and
variability (variance, SD, or %CV) as the y-axis. A trial LoD is repeatedly calculated with the fitted equation and compared to a predicted SD. When the trial
LoD matches the sample concentration used to calculate the predicted SD, that concentration is the LoD. Essentially, this protocol derives mathematically the
LoD where 95% of the results are above the LoB. See Figure 2.
LoQ is the lowest concentration of analyte that can be measured with pre-established performance criteria. The protocol is similar to LoD and can be
determined by a protocol similar to LoD, either by the classical EP17-A2 approach or the precision profile approach. The preferred means to define LoQ is
in terms of total error (TE), which contains a parameter for bias. An LoQ defined only in terms of imprecision is essentially the previous functional sensitivity
statistic. There are many acceptable protocols for determining LoQ; two of the most commonly used are Westgard and root mean square (RMS) protocols.
Both of these have a bias component and an imprecision component.
Probit: The probit protocol is used for infectious disease testing and nucleic acid methods. Here, all negative samples are reported as negative, and the false
positive rate is much lower than the 5% typically accepted. The LoD is calculated as the concentration where 95% of results are positive.
Figure 2.Precision profile protocol for establishing the LoD. Using an equation fitted
to the regressed SD results, this protocol mathematically derives the LoD
where 95% of the results are above the LoB.

PROTOCOLS TO CALCULATE LoQ
There are several acceptable ways to estimate bias. Assigned standards are rarely available at the concentration of LoQ. Diluted or spiked samples are
acceptable, as long as the samples are commutable to actual patient samples. The design of the protocol can vary widely, but typically has replicates
sampled over multiple days using more than one analyzer, and more than one reagent lot. Multiple calibrations, etc., can be added to increase variability for
a more robust study design.
Similar to the LoD determination, a precision profile approach is typically used for LoQ. The main difference is the selection of low level samples, which must
have known concentrations. After all data has been collected, the LoQ is determined by the following steps.
1. Calculate the mean (x) and SD for each sample for all replicates within each reagent lot.
2. Caluclate the bias: Bias = x - R
where R is the known (assigned) value for the sample.
3. Calculate the TE for each sample.
4. Plot the known values for each sample on the x-axis and the calculated TEs on the y-axis.
5. Fit an equation to the resulting line (or simply use the plot) and determine the analyte concentration that corresponds to the pre-determined TE value.
This concentration is the LoQ. See Figure 3.
Figure 3.Precision profile (variant) protocol for establishing the LoQ. The LoQ is the sample
concentration that corresponds to the pre-determined Total Error.

LoB, LoD, LoQ AND LABORATORY REPORTING
SUMMARY
The laboratory must decide how to translate the LoB, LoD and LoQ into their patient reports. At a minimum, the report might provide:
A more elaborate reporting scheme might be:
(in these examples, the actual values for LoD and LoQ are stated)
LoB, LoD and LoQ are statistics that allow manufacturers to assess low level test performance of assays they develop. Additionally, laboratory directors can
compare test method performance to other manufacturers, as well as gain guidance for reporting test results. For further details on establishing and verifying
detection capability, consult CLSI document EP17-A2, published in June 2012.
RESULT REPORT< LoD “not detected”
LoB < result < LoQ “analyte detected”
≥ LoQ report the result
RESULT REPORT< LoD “not detected; result < LoD”
LoD ≤ result < LoQ report the result, “interpret result with caution due to higher assay imprecision”
≥ LoQ report the result
Beckman Coulter and the stylized logo are trademarks of Beckman Coulter, Inc. and are registered with the USPTO. For Beckman Coulter’s worldwide office locations and phone numbers, please visit www.beckmancoulter.com/contact IB-18039A B2013-14094 www.beckmancoulter.com © 2013 Beckman Coulter, Inc.





























