Embed Size (px)
Citation preview

B Y
D R . K I R U T H I K A . S
D R . L A T H A R A V I C H A N D R A N ,
S R I R A M A C H A N D R A M E D I C A L C O L L E G E .
UNUSUAL CAUSE FOR MACROCEPHALY

HISTORY
Ruthiresan 13 yr old male born out of non-consanguinous marriage
Large head
Chest deformity
Multiple skin lesions over trunk & extremities
No symptoms of raised ICP
No bony abnormalities / Fractures

PAST HISTORY
Parents noticed increase in the size of head since 9 months of age
But he was evaluated only at 4 yrs of age
Serial CT Scans done in various centres thereafter were normal
No significant medical illnesses

HISTORY
Antenatal H/O : No H/O fever with rash / Systemic Illnesses / radiation exposure / drug intake. AN USG scans - Normal
Natal H/O: FTNVD,b.wt 3kg. cried immediately
Postnatal H/O: uneventful
Developmental H/O: Early developmental milestones were delayed. Poor Scholastic performance
Immunization H/O: Immunized Up To Date. Optional vaccines not given.

GENERAL EXAMINATION
Afebrile, alert, no pallor / icterus/ cyanosis/ clubbing/ lymphadenopathy/ pedal edema
Vitals : RR 20/min
PR 102/min
BP 100/70 right UL sitting posture
CFT < 3 sec
PP well felt

ANTHROPOMETRY
ANTHRO.INDICES
CURRENT VALUE
EXPECTED VALUE
CENTILE INTERPRETATION
WT 30.5 40 < 3RD Undernutrition
HT 147 155 25TH Normal
HC 58 52 >97TH Macrocephaly
BMI 13 18 - 25 <3RD Undernutrition
SMR Stage 4 Postpubertal

GENERAL EXAMINATION
HEAD TO FOOT EXAMINATION
Macrocephaly
Pectus excavatum
Mutiple Café-au-lait spots >10 in no. >15mm size
Nipple & areola enlarged
Axillary freckling left side






SYSTEMIC EXAMINATION
RS –normal vesicular breath sounds present
CVS – S1 S2 present, no murmur
ABDOMEN –soft, no warmth, non tender
genitalia well developed, pubic hair thick ,plenty
• CNS –higher function normal
cranial nerves normal
motor /sensory/autonomic normal
spine ,cranium normal

INVESTIGATION
Blood routine – normal
Renal function test – normal
Thyroid function test – normal
Urine routine – normal
X ray wrist AP view for bone age – normal
USG abdomen – normal
Urine spot calcium, phosphorus - normal

FURTHER WORKUP
OPTHALAMIC EVALUATION : slit lamp examination revealed Lisch nodules in both eyes, fundus was normal, no evidence of optic glioma.
PSYCHOLOGICAL ASSESSMENT: his IQ was 55% with learning disability.
MRI BRAIN: showed Unidentified bright objects (UBOs) in the left frontal and hypothalamus.Size of ventricles were normal

MRI BRAIN
UBO

FINAL DIAGNOSIS –
NEUROFIBROMATOSIS - 1
CRITERIA FOR DIAGNOSIS :
Café-au-lait spots
Axillary or Inguinal freckling
Lisch nodules
Neurofibroma
Distinctive osseous lesion
Optic glioma
Ist degree relative with NF1
Our patient satisfied 3 out 7 criterias hence diagnosed as NEUROFIBROMA TYPE I

LITERATURE REVIEW

NEUROFIBROMA TYPE I
Also known as Von reckling hausen disease Autosomal Dominant Inheritance Neurocutaneous syndrome – defect in differentiation of
primitive ectoderm Consequence of abnormality of neural crest
differentiation and migration during early stages of embryogenesis
NF1 gene – 17q11.2 encodes all mRNA of 11 – 13 kb containing atleast 59 exons that produce a protein Neurofibromin.
More than 300 independent mutation reported. Majority of mutations in paternal germline

CLINICAL MANIFESTATION AND DIAGNOSTIC CRITERIA
1. Café-au-lait macules
6 or more over 5mm in greatest diameter in prepubertal individual
Over 15mm in postpubertal individuals
Hallmark of NF1 almost 100% in patients with predilection over trunk, extremities with sparing of face
2. Axillary or inguinal freckling
multiple hyperpigmented areas 2-3 mm in diameter
3. Lisch nodules
2 or more, hamartomas located within the iris.
Best identified by a slit lamp examination.>74% patients of NF1.

4. Neurofibroma
Small rubbery lesion with a slight purplish discoloration of the overlying skin
2 or more or one plexiform neurofibroma.
Neurofibroma typically involve skin but may also involve peripheral nerves, blood vessels within viscera.
characteristically appear during adolescence or pregnancy suggesting a hormonal influence.
Plexiform neurofibroma
Evident at birth and result from diffuse thickening of nerve trunks
frequently located in orbital or temporal region of face.
Produce overgrowth of extremity and a deformity of the corresponding bone.

5. Optic glioma • 15% patients,relatively benign tumour consisting of glial cells and a mucinous material.•Mostly asymptomatic. 20%have visual disturbance and precocious puberty.•MRI –a distinct focal mass originating from optic nerve or chiasma.
6. First degree relative with NF1 ,whose diagnosis was based on aformentioned criteria
7. DistinctiveOsseousLesion•Sphenoid Dysplasia causing pulsating exophthalmos. Cortical thinning of long bones.•Scoliosis most common orthopedic deformity

CAFÉ-AU-LAIT SPOTS
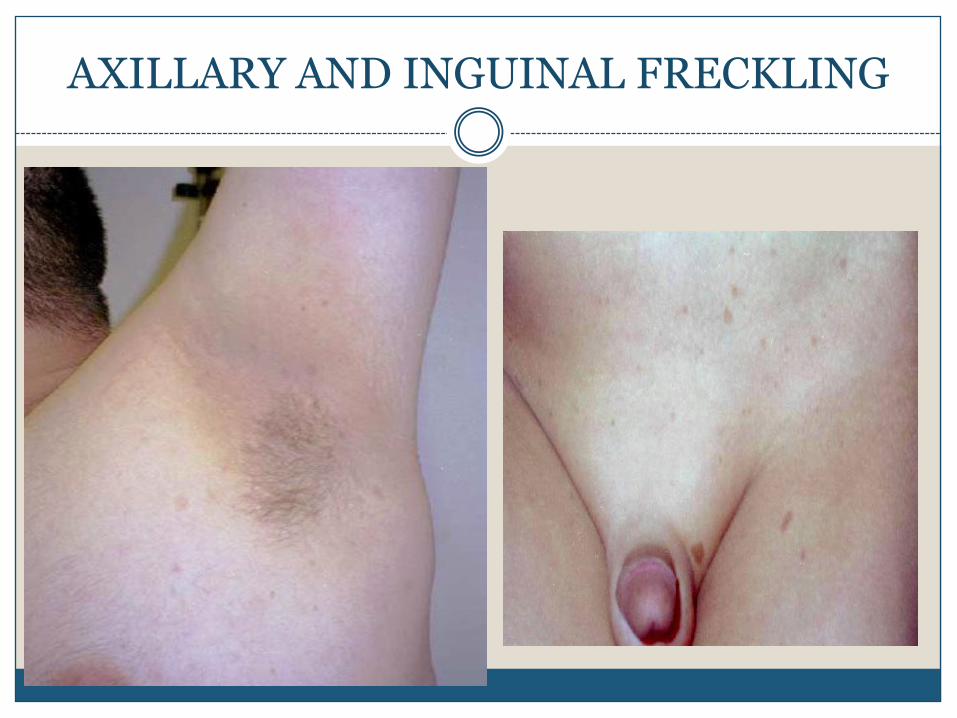
AXILLARY AND INGUINAL FRECKLING

LISCH NODULES

NEUROFIBROMA

OSSEOUS LESION

OPTIC GLIOMA
LEFT OPTIC N GLIOMA

COMPLICATIONS OF NF1
Cognitive abnormalities like learning disabilities are common and occur in 40 – 60% of NF1 patients.
Complex partial and generalised tonic clonic seizures are a frequent complications.
Hydrocephalus is a rare manifestation secondary to adueductal stenosis.Whereas macrocephaly with normal ventricles is a common finding.
MRI – hyperintense signals in optic track,brainstem,internal capsule, Cerebellum.These are ‘UNIDENTIFIED BRIGHT OBJECTS’(UBOs) that tend to disappear by 30yrs of age.
.

•Cerebral vessels may develop aneurysms or stenosis resulting in moyamoya disease.Neurologic sequelae of this vascular abnormality include transient ischemic attacks ,hemiparesis,cognitive defects.
•Precocious puberty become evident in the presence or absence of lesions in optic chiasma and hypothalamus
•Malignant neoplasms are also significant in NF1. Neurofibroma occasionally differentiates in to a neurofibrosarcoma or malignant schwannoma.
•Patient with NF1 at risk for hypertension,resulted from renal vascular stenosis or a pheochromocytoma.Incidence of pheochromacytoma, rhabdomyosarcoma, leukemia,wilms tumour is higher than in general population.
•Unusual association involving myeloid leukemia, juvenile xanthogranuloma and NF1.

UBOs
UBO

WORKUP
Molecular testing- in patient with a single clinical finding without positive family h/o
Linkage analysis in a patient with multiple affected family members.
Prenatal diagnosis via chorionic villus sampling when specific gene mutation is known
Preimplantation genetic diagnosis using in vitro fertilisation with selection of unaffected embryos for transfer.
24 hr urinary free catecholamine
EEG

Imaging studies
X ray long bones to visualise lytic lesions
MRI - UBOs are thought to represent areas of dysmyelination or focal areas of increased water content,not detected by CT Scan. Do not cause mass effect.
Gallium 67 scintigraphy – for patients with a large plexiform neurofibroma undergo malignant transformation
Myelography – to clarify the extent of a spinal cord tumor.

MANAGEMENT
MEDICAL CARE :
Detailed history,physical examination by a pediatrician.
Thorough annual ophthalmic examination by a pediatric opthalmologist until the age of 10.then routine visual assessment is recommended for early detection of optic glioma
Cutaneous examination at each visit to look for new lesions, or progression of prexisting ones.
Skeletal involvement should be determined.
Blood pressure checked at each visit and treated promptly if detected.

To be continued…..
Interval h/o should focus on subtle sensory or motor symptoms such as paresthesia, radiculopathy , weakness or muscle atrophy.
Chemotherapy to treat malignant peripheral nerve shealth tumors (MPNSTs) that are unresectable or metastatic.
Medications used -Farnesyl transferases with Lovastatin, Sorafenib,Rapamycin complex 1 inhibitor with Erlotinib,Carboplatin, Hyaluronan oligomers with Doxorubicin

MANAGEMENT
Surgical care : Neurofibroma that press on vital organs,obstruct
vision,grow rapidly deserve immediate attention.
Plastic surgery consultation is advisable for areas of great cosmetic concern like face.
Symptomatic peripheral nerve sheath tumors located along the nerve of brachial or pelvic plexus sometimes require surgical resection.
Resection of spinal cord tumors is difficult but often necessary to prevent progressive paraplegia or quadriplegia
Orthopedic intervention is indicated for rapidly progressive scoliosis and for some severe bony defects.

FROM ABTRACTS
Macrocephaly in NF1 patients is related to increased grey matter volume and white matter volume and corpus callosum enlargement
-Margariti PN, Blekas K, et al; Department of Radiology, University of Ioannina, Ioannina, Greece; Eur Radiol .2007;17(2):433-8
Delayed developmental apoptosis results in macrocephaly and delay in the development of appropriate neuronal connection in children with NF1.
-Moore BD, Slopis JM, Jackson EF,et al; Division of Paediatrcs, University of Texas Medical division, Anderson Cancer Centre,Houston. Neurology.2000;54(4): 914-20
The short stature was evident in adolescents girls of NF1 than NF1 boys but macrocephaly was evident in both sexes 31%.The 97th centile of the affected NF1 were above the 97th centile of healthy persons
-Rafia S,Garcia Pena JJ.et al; Serviciode neurologia pediatrica, Hospital Infantil Universitario Lapaz, Madrid, Spain; Rev Neurol.2004; 38(1):1009-12

REFERENCES
Nelson textbook of peditrics -18th
edition
Textbook of pediatric neurology –Fenichel
Neurofibromatosis center of New Jersey, Author Beth A Pletcha. Deparment of Pediatrics, University of medicine, New Jersey

THANK YOU





























