Embed Size (px)
Citation preview

Vol. 77 p-FLUOROPHENYLALANINE IN EXOPENICILLINASE 135
REFERENCES
Boyd, W. C. (1956). Fundamentals of Immunology, 2nd ed.New York: Interscience Publishers Inc.
Charlwood, P. A. & Gordon, A. H. (1958). Biochem. J. 70,433.
Cohen, G. N., Halvorson, H. 0. & Spiegelman, S. (1959). InMicro8omal Partices and Protein Synthe8is, p. 100. Ed.by Roberts, R. B. New York: Pergamon Press Ltd.
Cohen, G. N. & Munier, R. (1959). Biochim. biophy8. Acta,31, 347.
Cowie, D., Cohen, G. N., Bolton, E. T. & de Robichon-Szulmajster, H. (1959). Biochim. biophys. Acta, 34, 39.
Grabar, P. (1959). Meta. biochem. Anal. 7, 1.Grabar, P. & Williams, C. A., jun. (1955). Biochim. biophys.
Acta, 17, 67.Halvorson, H. 0. & Spiegelman, S. (1952). J. Bact. 64, 207.Kogut, M., Pollock, M. R. & Tridgell, E. J. (1956).
Biochem. J. 62, 391.Kruh, J. & Rosa, J. (1959). Biochim. biophys. Acta, 34,561.Kunkel, H. G. (1954). Meth. biochem. Anal. 1, 141.
Lowry, 0. H., Rosebrough, N. J., Farr, A. L. & Randall,R. S. (1951). J. biol. Chem. 193, 265.
McQuillen, K. & Roberts, R. B. (1954). J. biol. Chem. 207,81.
Mandelstam, J. & Rogers, H. J. (1959). Biochem. J. 72,654.
Mitchell, P. D. (1949). J. gen. Microbiol. 4, 399.Munier, R. & Cohen, G. N. (1956). Biochim. biophy8. Acta,
21, 592.Munier, R. & Cohen, G. N. (1959). Biochim. biophy8. Acta,
31, 378.Newton, G. G. F. & Abraham, E. P. (1954). Biochem. J.
58, 103.Pollock, M. R. & Kramer, M. (1958). Biochem. J. 70,
665.Richmond, M. H. (1959). Biochem. J. 73, 155.Richmond, M. H. (1960). Biochem. J. 77, 112.Trudinger, P. A. & Cohen, G. N. (1956). Biochem. J. 62,
488.Urba, R. C. (1959). Biochem. J. 71, 513.Vaughan, M. & Steinberg, D. (1958). Fed. Proc. 17, 328.
Biochem. J. (1960) 77, 135
Argininosuccinic Aciduria: Identification and Reactions of the AbnormalMetabolite in a Newly Described Form of Mental Disease,
with Some Preliminary Metabolic Studies
BY R. G. WESTALLMedical Unit, University CoUege Hospital Medical School, London, W.C. 1
(Received 22 February 1960)
Allan, Cusworth, Dent & Wilson (1958) havereported a new form of severe mental diseasewhich they believe to be due to an inbom error ofmetabolism. The authors described a family withfour children in which two of the sibs were normaland the other two were seriously mentally retarded.Paper-chromatographic examination of the urineof the family showed that the two affected childrenwere excreting large amounts of a ninhydrin-reacting substance which was absent in the urineof the rest of the family. Moreover, further testson the affected children showed a relatively highconcentration of the abnormal metabolite in thecerebrospinal fluid and a somewhat lower concen-tration in the plasma. The unknown substancecould not be identified as any of the substancesusually found in normal urine, nor with any of thecompounds found only in trace amounts (Westall,1955). Hence at the time when the full report ofthe case was published the particular metabolitewas unidentified but the authors were of theopinion that it was probably a peptide of a typeresistant to acid hydrolysis.
Further work on this substance (reportedbriefly by Westall, 1958) has now established thatit is argininosuccinic acid. This acid was firstpostulated on theoretical grounds to be an inter-mediate in the pathway of synthesis of argininefrom citrulline and aspartic acid in the presence ofliver enzymes (Ratner & Pappas, 1949). It waslater synthesized, isolated and characterized froma similar system (Ratner, Petrack & Rochovansky,1953). These authors demonstrated that the re-action takes place in two stages. In the first,argininosuccinic acid is formed from citrulline andaspartic acid under the action of a 'condensingenzyme' present in mammalian liver and kidney;this requires adenosine triphosphate. The secondstage requires the presence of a 'splitting enzyme',which catalyses the breakdown of argininosuccinicacid to form arginine and fumaric acid. They wereable to prepare argininosuccinic acid from both ofthese systems since the splitting-enzyme system isreversible. The natural accumulation of arginino-succinic acid in mammalian tissues or body fluidshas not been demonstrated before and it may be

R. G. WESTALL
assumed that under normal conditions it occursonly transiently. However, it has been found inpea-meal extracts (Davison & Elliott, 1952) and amutant strain of Neurospora cra8sa has beendescribed which accumulates argininosuccinic acidin the mycelium (Fincham & Boylen, 1955).In the present paper details are given of the
formal identification of argininosuccinic acidobtained for the first time from human material.
RESULTS
Chemical 8tudie8I8olation of argininosuccini acidfrom the patient'8
urine. Several attempts were made to isolate theunknown substance by using a system of coupledion-exchange-resin columns (Westall, 1952), butthe results were extremely confusing and it becameobvious that the substance being studied wasbeing degraded during fractionation. However, itwas possible to isolate a white crystalline substancewhich moved to a different position on paperchromatography from that taken up by the originalunknown substance. This newly isolated substancewas stable to acid hydrolysis but gave aspartic acidand ornithine after alkaline hydrolysis. This infor-mation provided the clue and led to the recognitionthat the original substance was probably arginino-succinic acid (ASA) and that the isolated crystallinesubstance was one of the cyclic forms described byRatner et at. (1953).As soon as it was supposed that the unknown
substance was ASA a simple method for its isola-tion was immediately available. Ratner et al.(1953), in their studies on the biosynthesis of ASA,successfully isolated it from tissue homogenates asthe barium salt, which is soluble in water butvirtually insoluble in aqueous 75% ethanol. Thismethod works equally well for isolation of ASAfrom urine, and 160 g. of the crude barium salt wasobtained from 251. of urine from one of theaffected sibs (K.R.). As Ratner et al. (1953)pointed out, ASA does not in practice take up theexpected 3 equiv. of barium but has a variablecontent of between 2 and 2-5 equiv. of bariumwhen precipitated in the presence of excess ofBa(OH)2 at about pH 10-11. ASA is convenientlystored as the barium salt, which seems to be stableif kept dry. The production of the free basepresents some difficulty since it forms cycliccompounds spontaneously in aqueous solution.The anhydride formation is favoured by hightemperatures and acidic conditions. Hence if anaqueous solution of ASA remains at room tempera-ture for a short time considerable cyclization takesplace, since the isoelectric point of the free ampho-lyte is about pH 3-4. The time required to removebarium quantitatively as BaSO4 ahd to produce a
freeze-dried powder leads to the formation of about10% of anhydride. However, removal of bariumby exchange with NH+4 ion on a cation-exchange-resin column at 20 and freeze-drying of the effluentproduced a much purer sample of ASA. Thismethod of producing the free base is an apparentcontradiction of the statement made earlier thatASA is unstable on ion-exchange resins. This can beexplained by the fact that the conditions werecarefully selected to avoid cyclization. Thetemperature was kept at 20, the amount of resinused was only slightly more than that required toremove barium and exchange with NH4+ ion. ThusASA did not exist as the free acid until NH3 wasremoved by volatilization during the freeze-drying.Subsequent analysis gave no evidence for ammo-nium salt formation. The passage of the solutionthrough the resin column was completed in4-5 min. Unfortunately, in spite of the limitedanhydride formation it has still not been possible tosecure crystalline ASA. Therefore the example ofRatner et al. (1953) has been followed and theanhydride forms, which crystallize readily fromaqueous ethanol, have been used for elementaryanalyses.
Behaviour of ar,qininosuccinic acid on filter-paperchromatogram,8. Fig. 1 (a) represents the standardtwo-way paper chromatogram most commonlyused for screening urines to detect amino aciduriasof various types (Dent, 1951). Aspartic acid,glutamic acid and glycine were put in as markerssince the lutidine-water solvent runs off the leadingedge of the paper and absolute Rp values do notapply. When the purified barium salt of ASA,obtained by repeated precipitation of the crudematerial, was run in this system a single spot wasobtained in the position indicated. A sample ofDr Ratner's barium salt of ASA moved to thesame position. When, however, free ASA, re-generated from the barium salt, was dissolved inwater and allowed to stand at room temperaturebefore being examined by paper chromatographythree well-defined spots were obtained. Onecorresponded with the original position ofASA andthe others took up the positions marked as B and C.Ratner et al. (1953) were aware that ASA changedinto a modified form, on standing in aqueoussolution, which they postulated to be a cyclicanhydride. They isolated and analysed this sub-stance. When a sample of this anhydride (this andthe sample ofbarium argininosuccinate were kindlysupplied by Dr J. B. Jepson from gift samples fromDr S. Ratner) was run in the same chromatographicsystem it moved to the position marked C. Thus itwould appear that there are two modified, probablyboth cyclic, forms of ASA. It is understandablethat Ratner et al. (1953) were not aware of thisbecause they used one-way paper chromatograms
136 1960
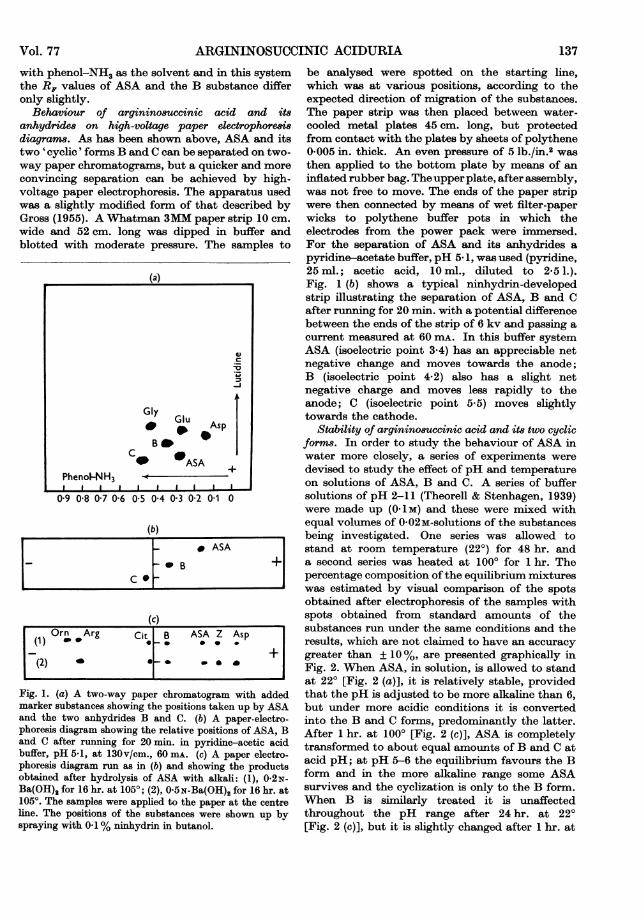
ARGININOSUCCINIC ACIDURIA
with phenol-NH3 as the solvent and in this systemthe R, values of ASA and the B substance differonly slightly.
Behaviour of argininosuccinic acid and itsanhydrides on high-voltage paper electrophoresisdiagrans. As has been shown above, ASA and itstwo 'cyclic' forms B and C can be separated on two-way paper chromatograms, but a quicker and moreconvincing separation can be achieved by high-voltage paper electrophoresis. The apparatus usedwas a slightly modified form of that described byGross (1955). A Whatman 3MM paper strip 10 cm.wide and 52 cm. long was dipped in buffer andblotted with moderate pressure. The samples to
(a)
Phenol-NH3I I I I
.)
IGlyGlu
Asp
B *
C0 ASA
I I I I I
09 0-8 0-7 0-6 05 04 03 02 0-1 0
(b)
t*ASA
C *r
(c)Orn Arg Cit B ASA Z Asp
(2) do
Fig. 1. (a) A two-way paper chromatogram with addedmarker substances showing the positions taken up by ASAand the two anhydrides B and C. (b) A paper-electro-phoresis diagram showing the relative positions of ASA, Band C after running for 20 min-. in pyridine-acetic acidbuffer, pH 5-1, at 130v/cm., 60 m&. (c) A paper electro-phoresis diagram run as in (b) and showing the productsobtained after hydrolysis of ASA with alkali: (1), 0-2N-Ba(OH)2 for 16 hr. at 1050; (2), 0 5N-Ba(OH)2 for 16 hr. at1050. The samples were applied to the paper at the centreline. The positions of the substances were shown up byspraying with 0.1% ninhydrin in butanol.
be analysed were spotted on the starting line,which was at various positions, according to theexpected direction of migration of the substances.The paper strip was then placed between water-cooled metal plates 45 cm. long, but protectedfrom contact with the plates by sheets of polythene0 005 in. thick. An even pressure of 5 lb./in.2 wasthen applied to the bottom plate by means of aninflated rubber bag. Theupper plate, after assembly,was not free to move. The ends of the paper stripwere then connected by means of wet filter-paperwicks to polythene buffer pots in which theelectrodes from the power pack were immersed.For the separation of ASA and its anhydrides apyridine-acetate buffer, pH 5. 1, was used (pyridine,25 ml.; acetic acid, 10 ml., diluted to 2-5 1.).Fig. 1 (b) shows a typical ninhydrin-developedstrip illustrating the separation of ASA, B and Cafter running for 20 min. with a potential differencebetween the ends of the strip of 6 kv and passing acurrent measured at 60 mA. In this buffer systemASA (isoelectric point 3.4) has an appreciable netnegative change and moves towards the anode;B (isoelectric point 4.2) also has a slight netnegative charge and moves less rapidly to theanode; C (isoelectric point 5-5) moves slightlytowards the cathode.
Stability of argininosuccinic acid and its two cyclicforms. In order to study the behaviour of ASA inwater more closely, a series of experiments weredevised to study the effect of pH and temperatureon solutions of ASA, B and C. A series of buffersolutions of pH 2-11 (Theorell & Stenhagen, 1939)were made up (0 1M) and these were mixed withequal volumes of 0-02M-solutions of the substancesbeing investigated. One series was allowed tostand at room temperature (22°) for 48 hr. anda second series was heated at 100° for 1 hr. Thepercentage composition of the equilibrium mixtureswas estimated by visual comparison of the spotsobtained after electrophoresis of the samples withspots obtained from standard amounts of thesubstances run under the same conditions and theresults, which are not claimed to have an accuracygreater than ± 10%, are presented graphically inFig. 2. When ASA, in solution, is allowed to standat 220 [Fig. 2 (a)], it is relatively stable, providedthat the pH is adjusted to be more alkaline than 6,but under more acidic conditions it is convertedinto the B and C forms, predominantly the latter.After 1 hr. at 1000 [Fig. 2 (c)], ASA is completelytransformed to about equal amounts of B and C atacid pH; at pH 5-6 the equilibrium favours the Bform and in the more alkaline range some ASAsurvives and the cycization is only to the B form.When B is similarly treated it is unaffectedthroughout the pH range after 24 hr. at 220[Fig. 2 (c)], but it is slightly changed after 1 hr. at
Vol. 77 137

R. G. WESTALL
1000 [Fig. 2 (d)]; about 25% is changed to the Cform at pH 2 and about 10% ofASA is regeneratedat pH 11. The C substance is stable at pH 2-5 at220 but is more readily converted back into ASAthan B when the conditions become more alkaline[Fig. 2 (e)]. At 100°, C is still stable at pH 2-3 butat pH 5 the ring is opened rapidly with the forma-tion of ASA, which presumably partly recyclizes toform B. It is curious that when C is heated at 1000at pH 5-7 there is a loss (30-40%) of total nin-hydrin colour. Since the ninhydrin reaction is dueto the amino group of the ornithine moiety itwould seem that there may be some formation ofintermediary substances involving some modifica-tion of the amino group. To sulmmarize, ASA ismore stable at alkaline pH provided that thetemperature is not raised. The optimum conditionsfor forming the B anhydride is to heat ASA atpH 6; C, on the other hand, is best prepared byacidifying ASA and standing at room temperature.
Treatment of arginino8uccinwo acid and it8 an-hydrides with 8trong acid. When ASA is refluxed for24 hr. with 5-5N-HC1 it is rapidly converted intothe B and C forms and there is some further degra-dation (not exceeding 10%) leading to the forma-tion of arginine, ornithine and aspartic acid. Similarexperiments with B and C individually showedthe same relative stability and the same productsof degradation but there was a slight tendency forC to be more stable than B under these conditions.
Degradation 8tudie8 on argininouccminic acid with
alkali. As Ratner et al. (1953) have observed, ASAbreaks down completely to ornithine and asparticacid after refluxing with 3N-Ba(OH)2 for 24 hr.They also stated that this was true also for theiranhydride (C) although the liberation of NH8 wasslower and the final yield of aspartic acid was lowerthan for ASA. Repeat experiments under theseconditions, where the products of hydrolysis havebeen examined by paper electrophoresis, have con-firnned that ASA and C yield ornithine and asparticacid only. Further, B also gave the same degrada-tion products. Presumably B and C are first con-verted into the open ASA forms and furtherdegradation proceeds from there. Reference toFig. 2 (d) shows that B is more resistant to ringopening at alkaline pH than C [Fig. 2 (f)]. Indiscussing the route of degradation of ASA withalkali, Ratner et al. (1953) considered it might beby a simple multiple cleavage to give ornithine,aspartic acid, C02 and NH3, but they stated thatwith milder conditions there might be somecitrulline formed first which would later breakdown to yield ornithine. With alkali treatment oftheir anhydride (C) they had evidence for theformation of ureido acids and postulated break-down by a route through ornithine and 5-(acetic-acid) hydantoin, the latter being further degradedto ureidosuccinic acid and finally to aspartic acid,C02 and NH3.
Degradative studies of ASA with varyingstrengths of alkali have shown that the series of
100
RO
600
0 40
0-0220
100 (b) (d)
80 /--- I ~ ~ ~ ~
50 60 ;kF .
60~40A
20
2 3 4 5 6 7 8 9 1011 2 3 4 5 6 7 8 9 1011 2 3 4 5 6 7 8 9 1011
pH pH pHFig. 2. Graphs showing the effect of temperature and pH on 0-01 m-solutions of ASA (0), B (A) and C (0)(a) ASA stood at 220 for 48 hr. (b) ASA heated at 1000 for 1 hr. (c) B stood at 220 for 48 hr. (d) B heated at1000 for I hr. (e) C stood at 220 for 48 hr. (f) C heated at 1000 for 1 hr.
138 1960

ARGININOSUCCINIC ACIDURIA
events that leads to the ultimate production ofornithine and aspartic acid is complex. Hydrolysiswith 0-2N-Ba(OH)2 for 16 hr. at 1050 in a sealedtube gives a number of products: arginine, orni-thine, citrulline, an unknown substance 'Z' and atrace of aspartic acid together with unchangedASA and B. When 0-5N-Ba(OH)2 was used for thesame hydrolysis time no arginine was found and theamount ofornithine and aspartic acid was increased.Fig. 1 (c) illustrates the position taken up by thesesubstances on the high-voltage electrophoresisdiagram. The B and C forms, if treated similarly,give basically the same results but no trace of theZ substance was found. The occurrence of this un-known substance was difficult to explain until itwas discovered that the sample of partially purifiedbarium argininosuccinate used in the hydrolysisstudies contained, as an impurity, another un-known substance 'Y'. Y behaves as an ampholyte,since it is retained on both anion- and cation-exchange resins and can be recovered withoutdegrading. It does not give a colour with ninhydrinnor, in fact, with any of the routine spray reagentsused in the chromatographic or electrophoreticstudies. It was found by chance, since its presenceon a particular electrophoresis diagram caused adistortion in the shape of the spot given by a knownsubstance in a neighbouring position on the paperstrip. It has now been found that its presence onpaper electrophoresis diagrams can be shown byspraying with the alkaline nitroprusside-ferri-cyanide reagent (Kirby-Berry, Sutton, Cain &Berry, 1951). It gives a red spot which fadeswithin 15 min. but it can still be seen some hourslater since it gives a greenish fluorescence whenviewed in u.v. light. Y has now been isolated butnot fully characterized. However, it is convertedinto Z by mild alkaline hydrolysis, which explainsthe occurrence of Z in the hydrolysis studies; bothY and Z yield only ornithine and aspartic acid when
Aspartic acid
zz CASA
B
Citrulline HCI
Ornithine
2 4 6 8 1012 14 1618 20 22 24 26 28 30 32 34Fraction no.
Fig. 3. Amino acid composition of the fractions obtainedafter displacement with 01 N-HCl from a column ofDowex-2 ion-exchange resin. The mixture applied to thecolumn was a partial hydrolysate ofASA with 0-5N-NaOH.
fully hydrolysed with 2N-NaOH for 24 hr. Henceit would seem that Y must have some closestructural relationship with ASA.
I8olation of aub8tance8 yielded by mild alkalitreatment of arginino8uccinic acid. In order toisolate some of the degradation products of ASAwith alkali, and also the contaminating unknownsubstance Z, an hydrolysis of 10 g. of the crudebarium salt was undertaken (as described in theExperimental section). The flow diagram ofseparation and the amino acid composition of thefractions is shown in Fig. 3. The early fractionscontain ornithine only, followed by two fractionsmixed with citrulline and then pure citrulline andso on. Z was easily isolated since it has a lowsolubility in water and, in fact, crystallized out asa fine white powder in the fraction tubes.
Quantitative determination of arginino8uccinicacid. The method of Moore & Stein (1954) for thequantitative estimation of amino acids in bodyfluids with a column of the cation-exchange resinDowex-50 provides a useful means of estimatingASA. It was used by Allan et al. (1958) for measur-ing the relative concentration of their unknownmetabolite (now known to be ASA) in the bloodplasma, cerebrospinal fluid and urine of theirpatients. Their more detailed analyses are nowavailable (Cusworth & Dent, 1960). Further workon the chemistry of ASA has now led to a probableexplanation of the behaviour of ASA on such acolumn. In the body fluids of these patients,within the range of physiological pH values, ASAexists predominantly in the open-chain form.However, in the early stages of the passage of thesamples down the Moore & Stein column, the pH ofthe ambient solution is at 3-4 and is even more acid(2.0) than this when the sample is applied to thecolumn. Under these conditions ASA forms anequilibrium mixture with its two anhydrides(called for convenience B and C) in the approximateratio of 2:3:5. Since these three substances havedifferent isoelectric points (ASA 3 4, B 4-2, C 5-5),ASA, being more acidic, moves ahead down thecolumn and this leads to a new equilibrium withfurther production of more B and C. Hence thequantity of free ASA is soon decreased to minimalamounts because, under the conditions of thecolumn procedure, there is no regeneration of ASAfrom the separated anhydrides. This all takes placequite rapidly since fairly sharp twin peaks are seenon the colunm flow diagram, which can be shown tocorrespond with the cyclic forms B and C. Theyoccur near the position taken up by leucine andisoleucine and in the urine samples from thepatient under study the B and C peaks are so largethat the smaller leucine and isoleucine peaks werefrequently overlapped. Paper-chromatographicanalysis of the fractions from the column showed
Vol. 77 139

R. G. WESTALL
that the first of the two peaks was due to the Bform and the second showed the C form with asmall amount of free ASA. The occurrence of thesmall quantity ofthe open-chain form was probablyan artifact and it was probably regenerated from Cduring the desalting of the fractions on snall ion-exchange columns before the paper-chromato-graphic analysis. The explanation of the behaviourof ASA on the Moore & Stein column outlinedabove was further corroborated by carrying outtest runs on the columns with isolated ASA (freebase) and the B and C anhydrides. The ninhydrincolour values of B and C are 94 and 100% re-spectively of that given by leucine. A typical flowsheet of the Moore & Stein analysis of the urineof a child with argininosuccinic aciduria is shown inFig. 4.
Clinical metabolic studie8Effects of dietary change on the excretion of
arginino8uccinic acid. Since ASA is an intermediatein the urea cycle it might be expected that variationin the quantities of the substances made availableto the cycle could cause variation in the excretionof ASA.
The patient was an 8-year-old male child [previouslydescribed as case 2 of Allan et al. (1958)] admitted to theMetabolic Ward of University College Hospital, London,under the care of Professor C. E. Dent. The child was fedwith a mixed constant diet of his own choice containing30 g. of protein/day. The dietary supplements were fed inaddition to the basic diet and were given in an orange-juicedrink, one-quarter of which was consumed at each of fourmeals during the day. Urine samples (24 hr.) were collectedthroughout the study. L-Arginine was added at 2 g./dayover a period of 3 days and the urine collected on the thirdday was selected for analysis. L-Aspartic acid, (5.0 g.) L-ornithine (4.0 g.) and L-citrulkine (4.0 g.) were fed for oneday only but as the last two substances are not found inthe normal diet in appreciable quantities, a small test dosewas given on the previous day in case of any ill-effects. Thechild was put back on the basic diet for several daysbetween each experimental feed. As a final trial the proteinlevel of the basic diet was dropped to 10 g./day over aperiod of 5 days, other dietary constituents being kept asconstant as possible. The urine sample collected on thefifth day was selected for analysis.
The child remained physically well and activethroughout the period of study and there appearedto be no appreciable change in his mental state. Theresults are shown in Table 1. ASA was determined
Glutamine4-
asparagineSerine
ThreonineAspartic acid
200-1u
Glutamic acid
A/1
Glycine LeucineIsoleucine
Alanine Methionine
Valine
/\-I A ;300 400
Cno
j4-Influent (0*2M-sodium acetate buffer, pH 3-1) ÷I*- Gradient elution-
Ammonia Ornithine
03 LysineTyrosine Histidine
0 ~~~Phenylalanine)5Q 0-2 Arginine
1 -Methyihistidine
0.1
0
600 700 lil 800 900 1000I ~ ~~~~~750
From 0-2m, pH 3-1 to 2-0m, pH 5B1Fig. 4. A Moore & Stein column 2 flow diagram of the amino acid analysis of 2 ml. of urine (from total 24 hr.
urine volume of 770 ml.) from patient K.R. Peaks B and C represent the anhydride derivatives of ASA.
Urea
H03>: 0.3 -
e .OOE0 (l
zEfflen(m)
50 1000in
B C
500
1960140
I. -1

Table 1. Urinary excretion of arginino8uccinic acid and urea of a patient with arginino8uccinic aciduria
Diet/day30 g. of protein30 g. of protein + 2 g. of arginine30 g. of protein + 5 g. of aspartic acid30 g. of protein + 4 g. of ornithine30 g. of protein + 4 g. of citrulline10 g. of protein
Total Nin diet
(g.)4*85.45-35-65.71-6
* Mean of 3 days.
ASA(g./24 hr.)
3.1*2-82-63-84-22.7t
Urea(g./24 hr.)
7-1*6-45.47-05-24.5t
t Mean of 5 days.
(as above) by the Moore & Stein method andurea by the standard hypobromite method. Nofirm conclusions can be drawn from a trial of thisnature where the tests were made on one child onlyand were of short duration. However, from theresults obtained it would appear that the output ofASA in the urine was little affected by the amountof protein in the diet as on the low protein intakehe was still excreting 2-7 g./day, although he musthave been in negative N balance as the urine N was
greater than dietary N. In view of this, one wouldnot expect additional arginine to increase the out-put of ASA and this in fact was the case. On theother hand, ornithine and citrulline, which are notfound in appreciable quantities in normal diets, andwhich in the Krebs-Henseleit urea cycle modifiedby Ratner & Pappas (1949) are nearer precursors ofASA, increased the output of ASA quite appreci-ably.
DISCUSSION
In view of the chemical evidence that has beenpresented, coupled with the paper-chromatographicand paper-electrophoretic comparisons with auth-entic samples, there seems little doubt that theunknown substance excreted by the patient studiedis argininosuccinic acid (ASA). Its complicatedbehaviour in solution is, however, worthy of furthercomment. In discussing this, Ratner et al. (1953)postulated that of the several forms of cycizationthat are possible the two forms shown opposite werethe most likely (B, C).
Ratner and her colleagues concluded that theywere dealing with only one of these two forms and,since it is difficult, analytically, to differentiatebetween them, they could not definitely decidewhich form they had. However, since their an-
hydride gave a Jaff6 reaction by analogy withcreatinine, and since, with an isoelectric point atpH 5 5, it would seem that the stronger of the twocarboxyl groups of the succinic acid moiety is in-volved in the cyclization, it is probable that theiranhydride was the five-membered-ring form.Comparative tests have shown that substance C isidentical with Ratner's anhydride and it has beenassumed provisionally in the absence of absolute
proof that C has the five-membered-ring structure.With the same reservations substance B has beenallocated the six-membered-ring structure. It hasan isoelectric point at pH 4-2, which might beexpected if the weaker carboxyl group of thesuccinic acid moiety (i.e. the y-carboxyl of asparticacid) was involved in the cyclization. It also failsto give a Jaffe reaction or any colour with thecreatinine reagent of Van Pilsum, Martin, Kito &
Hess (1956), but it will give a weak blue colourwith the alkaline nitroprusside-ferricyanide re-
agent (Kirby-Berry et al. 1951).
H H C02H
CH%2-N--C-N-H2CH2 NH CH2
qH2 OO2H
CH .NH2
CO2H
(Argininosuccinic acid)
H C02H
CH2-N--CN--CH2
CH2 HN OH2
OH2 C_O
CHNH2
CO2H
(B)H
H N C=0
0H2-N--O=N-CH,
OH2 OH2
CH2 C02HOH*NH2
CO2H(C)
ASA N +urea N
(g./24 hr.)3.93.53-04*03-22*6
ASA Nas % oftotal N1413-51516-522-518-0
Vol. 77 ARGININOSUCCINIC ACIDURIA 141

R. G. WESTALL
Both cyclic forms, apart from some conversion ofB into C, are almost completely stable to acidhydrolysis. On the other hand, in alkaline condi-tions the rings are operned, more slowly with B thanwith C, to regenerate ASA, which is then susceptibleto further breakdown. The degradation is complexand presumably is related to the resonating effectof the double bond, which causes a variation in thesite that is the weakest link in the chain. Arginineand citrulline have both been detected after mildalkaline hydrolyses but the formation of arginineby hydrolysis would also yield malic acid, whichultimately would affect the final yield of asparticacid. Ratner et al. (1953), however, found thatASA, after strong alkaline hydrolysis, gaveornithine and aspartic acid in close to theoreticalyield. To explain this it must either be postulatedthat the mechanism of breakdown is differentunder strong alkaline conditions or that the cleav-age to form arginine is not hydrolytic and thatdirect cleavage forms fumaric acid, which laterreacts with free NH3 to form aspartic acid. Theformation of aspartic acid from fumaric acid andammonium carbonate heated in aqueous solution ina closed tube can readily be demonstrated.With regard to the biological significance of
these curious substances derived from ASA, it maywell be that they are only chemical artifacts whichhave no place in the metabolism of ASA. Ratneret al. (1953) found that the anhydride form C wasinert in various enzyme systems. Although B hasso far not been tested, it is potentially the more
interesting substance since, as Ratner (1954) hasobserved, it can be regarded as a substituted di-hydro-orotic acid, a substance which has beenshown to be on the pathway to pyrimidine syn-thesis (Wright et al. 1951).
In addition to the chemical problems, believed toconcern cyclization of ASA as mentioned above,there still remains the problem of the unidentifiedsubstances designated Y and Z. It is now estab-lished that Z is not a chemical degradation productof ASA, as was thought earlier, but is derived fromY, which was present in the crude barium arginino-succinate preparation. The possibility still re-
mained that Y might be an artifact formed byprocedures employed for the isolation of ASA.However, it was detected on electrophoresisdiagrams (by the use of the alkaline nitroprusside-ferricyanide reagent) of fresh urine specimens fromthe patient excreting large amounts of ASA. Itwas not present in a number of normal urines,including those from the unaffected members ofthe patient's own family. Hence it would seem
reasonable to assume that Y is a naturally occurringmetabolite that, like ASA itself, is normally an
intracellular substance which is only rarelyexcreted in the urine. With this assumption and
with the chemical evidence that Y must have astructure somewhat similar to that of ASA (sinceit gives the same final degradation products,namely, aspartic acid and ornithine), it can bespeculated that Y may be another intermediate inthe series of reactions which make up the Krebs-Henseleit urea cycle. On the other hand, Y mayrepresent an attempt by the patient to conjugatesome of the excess of ASA formed in the tissues andto produce a derivative which is less harmful andbetter tolerated. It is hoped that when both Y andZ are fully characterized it will be possible toexplain their function.
In considering the pathogenesis of the clinicalsyndrome in the two affected children we are insome difficulty because of inadequate data. At themoment no other similar patients have beendescribed and enough family data is thereforelacking. Nevertheless, it seems probable that thedisease is hereditary and probably due to someenzyme defect, similar to those already found inother metabolic diseases, e.g. phenylketonuria,galactosaemia, etc. On considering the urea cycle,Ratner et al. (1953) have shown that an enzyme(argininosuccinase) occurs in the liver and catalysesthe breakdown of ASA with the formation ofarginine and fumaric acid. One might expecttherefore that the genetic defect might be a de-ficiency in this enzyme. If our current ideas arecorrect, this should lead to an inability to formurea whereas Allan et al. (1958) showed that in thedisease the ability to form urea seemed unimpaired.Hence it is difficult to believe that the splittingenzyme is absent in the liver, which is regarded asthe seat of urea production.The second unusual feature is that the concentra-
tion of ASA is nearly three times as high in thecerebrospinal fluid as in the systemic blood plasma(Allan et al. 1958). This was a very striking findingsince most of the amino acids found in cerebro-spinal fluid are present in much higher concentra-tion in plasma. This makes it most unlikely that thecerebrospinal fluid derives its ASA from theplasma, and in any case ASA has poor permeabilityproperties (Walker & Myers, 1953) and couldhardly be expected to penetrate the blood-brainbarrier. Thus at present there is no better sugges-tion than that already discussed by Allen et al.(1958) in which they speculate that the unknownmetabolite (ASA) may be formed in the brain, orin some other structure closely associated with thecerebrospinal fluid, and leaks from there to theblood, from which it is rapidly cleared by thekidney. This suggestion is not as bizarre as it mightseem. For years it has been assumed that urea isformed only in the liver and that its presence inthe cerebrospinal fluid was due to infiltration fromthe blood plasma. Recent work by Sporn, Dingman,
142 1960

ARGININOSUCCINIC ACIDURIA
Defalco & Davies (1959) has shown that rat brain iscapable of urea synthesis in vivo and Walker(1958) has shown that argininosuccinase (splittingenzyme), previously found in mammals only in theliver and kidney (Ratner, 1954), is also present invarious other organs of the dog, including the brain.The feeding experiments were undertaken to see
how far the excretion ofASA could be influenced bythe diet. In this way it was hoped that some indica-tion might be given which would lead to a rationaltherapy which would lessen the synthesis of ASAand lower its concentration in the body fluids. If,for instance, arginine had increased the excretion ofASA, then it would be reasonable to attempt tolower the intake of arginine. However, the onlycompounds found to increase ASA output wereornithine and citrulline, substances not found assuch in appreciable quantities in the normal diet.At the moment therefore it is difficult to see whatdietary therapy can be usefully tried.The increased output of ASA after taking
additional ornithine and citrulline serves as addi-tional evidence for its identification, and for itsclose relationship. metabolically with these com-pounds. The effect of a higher protein diet was nottried since the child seemed disinclined to eat moreprotein than was given in the 30 g./day regimen.
EXPERIMENTAL
Isolation of barium argininosuccinate. Urine (25 1.) fromcase 2 (Allan et al. 1958) was decreased in volume to 41. ina rotating evaporator (Van Heyningen, 1949). A hotsaturated solution containing 350 g. of Ba(OH)2,8H20 wasadded with stirring and the mixture was allowed to standovernight. The precipitate was removed by filtration and tothe filtrate (5-4 1.) 3 vol. of ethanol and 50 g. of solidBa(OH)2,8H20 were added. After standing for 4 hr. theprecipitate was separated by centrifuging, transferred to alarge Buchner funnel and washed with 75% ethanol.After a final washing with 95% ethanol the solid cake wasbroken up and allowed to dry at room temperature. Thepowder was finally dried in a desiccator over H2SO4. Yieldof crude barium argininosuccinate was 160 g. Two-waypaper-chromatographic analysis for amino acids withphenol-NH3 and lutidine-water (2.2: 1, v/v) as solvents and25 leg. of the sample showed a large spot, due to ASA, andweak spots, due to traces of aspartic acid and glutamicacid. The crude barium salt (60 g.) was dissolved in 200 ml.of water and an insoluble residue was discarded. Ethanol(600 ml.) was added to the clear solution and the reprecipi-tated barium salt was filtered and dried. The yield was 47 g.Paper-chromatographic analysis showed ASA as the onlyninhydrin-reacting constituent.
Preparation offree argininosuccinic acid (1). The purifiedbarium salt of ASA (5 g.) was dissolved in 25 ml. of waterand the small insoluble residue was discarded. The clearsolution was chilled (40) and N-H2S04 (19 ml.), also cooled,was added to bring the pH of the mixture to 3-4. At theexact end point no precipitate was obtained when eithermore acid or weak Ba(OH)2 solution was added to a centri-
fuged portion of the mixture. The BaSO4 was removed bycentrifuging and the clear solution was freeze-dried. Afluffy pale-cream-coloured powder was obtained. Yield was2*4 g. (Found: N, 18-9. Theory: N, 19.3 %).
Preparation offree argininosuccinic acid (2). A glass tube15 cm. long and 2-4 cm. in diameter constricted to capillarybore at the lower end and plugged with glass wool wasfilled with Dowex-50 cation-exchange resin (4% cross-linked and 100-200 mesh; 7-5 g. dry wt.). The resin waspacked as a slurry after pretreatment with 2N-HC1 andwashing with water by decantation. Aq. N-NH3 soln. wasrun down the column until the effluent was stronglyalkaline. The column was then washed with water to re-move the excess of NH3 and kept at 40 until required. Thepurified barium salt of ASA (5 g.) was dissolved in 40 ml. ofwater and a small insoluble residue discarded. The solutionwas chilled and then passed through the prepared resincolumn. The effluent containing the soluble ammoniumsalt of ASA was collected and freeze-dried. Final dryingwas carried out in a Fischer drier at 800. Yield was 2-1 g.(Found: N, 19-1. Theory: N, 19-3%).
Preparation of argininosuccinic acid anhydride (sub-stance C). Free ASA (1.5 g.) was dissolved in 10 ml. ofwater and was allowed to stand at room temperature for2 days. Ethanol (10 ml.) was added and as soon as crystalsappeared a further 20 ml. of ethanol was added gradually.After standing at 40 overnight the crop of crystals wasfiltered off and dried. Yield 0 7 g. [Found: C, 44-1; H, 6-10;N, 20-4 (Dumas). C1oH16805N4 requires C, 44.1; H, 5-92; N,20-6%].
Isolation of substances derived from partial degrada-tion of crude argininosuccinic acid with alkaliThe crude barium salt of ASA (10 g.) was dissoved in
100 ml. of water and 2N-H2S04 was added cautiously untilthe mixture was adjusted to pH 3-4. BaSO4 was removedby centrifuging and the supernatant was mixed with anequal volume of N-NaOH and refluxed for 4 hr. Thehydrolysate was cooled and diluted to 11. with water andthen run through a resin-column system (Westall, 1952),consisting of two columns coupled one beneath the other,filled with the anion-exchange resin Dowex-2 (8% cross-linked and 100-200 mesh). The upper column was madefrom a filtration tube 1-7 cm. in diameter and having aresin bed 12 cm. long (containing 10 g. of dry resin). Thesecond column had a tube 1-2 cm. in diameter with a resinbed 8 cm. high (3.5 g. of dry resin). The tubes were packedwith a slurry of resin in water and then coupled togetherwith capillary glass tubing. The resin was converted intothe OH form by passing 2N-NaOH down the columnsfollowed by washing with water until the pH of theeffluent was between 7 and 8. After the diluted hydrolysatewas passed down the columns a further 100 ml. ofwater wasrun on and this was followed by the displacing agent 01N-HCI. As soon as a positive ninhydrin reaction was obtainedon testing the effluent, fractions (8-10 ml.) were collecteduntil the effluent became strongly acid. Samples (51.u.)from the fractions were spotted on paper strips and thestrips were analysed by electrophoresis. The amino acidcomposition of the fractions is illustrated in Fig. 3.
Isolation of ornithine hydrochloride. Fractions 1-11 weremixed, adjusted to pH 6 with N-HCI and evaporated atreduced pressure to 5 ml. Crystals were obtained after thegradual addition of ethanol to give a final volume of 20 ml.
Vol. 77 143

144 R. G. WESTALL 1960The crystals were collected and recrystallized from waterand ethanol. Yield was 0-82 g. (Found: C, 35*8; H, 7*1;N, 16-7. C5H180N2C1 requires C, 35-6; H, 7-7; N, 16.6%).
Isolation of citrulline- Fractions 13-16 were combinedand evaporated under reduced pressure to 2 ml. Crystalswere obtained after the addition of ethanol. Yield afterrecrystallization was 0-21 g. (Found: C, 41-6; H, 7 7; N,23-8. CH1303N3 requires C, 41-1; H, 7-5; N, 24 0%).
Isolation of substance B. Fractions 17-19 were mixed andevaporated under reduced pressure to 2 ml. A crystallineproduct was obtained by the addition of ethanol. Yieldwas 0-14 g. This material gave a lower figure for its Ncontent than was expected and it was ultimately discoveredthat it was contaminated with an unknown, non-ninhydrin-reacting ampholyte which had a lower N content. A puresample of B was obtained later by repeated recrystalliza-tion, from aqueous ethanol, of a sample obtained from alarger-scale fractionation carried out in a similar manner.Yield was 1-7 g. [Found: C, 43-9; H, 6-1; N, 20-8 (Dumas).CloH1605N4 requires C, 44-1; H, 5-9; N, 20-6%].
Isolaton of substance Z. Substance Z formed a whitedeposit shortly after the fractions were collected. Thiswhite powder was filtered off from fractions 22-26 and thefractions were decreased in volume to 10 ml. and a furthercrop was obtained. This powder was very slightly solublein water and was washed well with cold water beforedrying. Yield was 0-32 g. (Found: C, 44-61; H, 5-46; N,15-3%).
Isolation of aspartic acid. Small amounts of aspartic acidcrystallized spontaneously in fraction tubes 28-32. Thismaterial was filtered off and a second crop was obtained bydecreasing the volume of the mother liquor to 5 ml. Bothcrops were combined and recrystallized from hot water.Yield was 0-21 g. (Found: C, 36-37; H, 5-33; N, 10-70.C4H704N requires C, 36-1; H, 5-3; N, 10.52%).
SUMMARY
1. The unknown metabolite previously de-scribed as the predominant amino acid in the urineof two mentally defective sibs has been isolated andhas proved to be argininosuccinic acid.
2. The behaviour of argininosuccinic acid inaqueous solution has been studied. It apparentlyforms an equilibrium mixture with two othermodified forms which are probably cyclizedanhydrides.
3. The complicated behaviour ofargininosuccinicacid on resin columns can be explained on the basisof its known reactivity. It is, nevertheless,possible to assay it by the method of Moore &Stein (1954). Each patient with argininosuccinicaciduria excreted about 3 g. of argininosuccinicacid per 24 hr. in his urine.
4. Argininosuccinic acid and its two anhydrideforms yield ornithine and aspartic acid after long
hydrolysis with strong alkali, but with weakeralkali for a shorter time a number of intermediatesare forned which include arginine and citrulline.
5. An unknown substance has been found in theurine of one patient with argininosuccinic aciduriawhich after treatment with strong alkali breaksdown to yield aspartic acid and ornithine.
6. The gross excretion of argininosuccinic acidseemed little affected by lowering the proteincontent of the diet over a short period or by addingarginine, but it was increased by feeding ornithineand citrulline.
The author would like to thank Professor C. E. Dent forhis interest and encouragement throughout this work. Hewould also like to acknowledge gratefully the help ofMiss E. Davies, who carried out the Moore & Stein analyses,Dr F. V. Flynn, who did the urea analyses and also the co-operation afforded to him by the Dietitian (Mrs B. Hartland)and the Sister (Miss M. Wilmot) and nurses ofthe MetabolicWard, University College Hospital, London.
REFERENCES
Allan, J. D., Cusworth, D. C., Dent, C. E. & Wilson, V. K.(1958). Lancet, i, 182.
Cusworth, D. C. & Dent, C. E. (1960). Biochem. J. 74, 550.Davison, D. C. & Elliott, W. H. (1952). Nature, Lond., 169,
313.Dent, C. E. (1951). Recent Advance8 in Clinical Pathology,2nd ed., p. 238. London: J. and A. Churchill Ltd.
Fincham, J. R. S. & Boylen, J. B. (1955). Biochem. J. 61,xxi.
Grows, D. (1955). Nature, Lond., 176, 72.Kirby-Berry, H., Sutton, H. E., Cain, L. & Berry, J. S.
(1951). Texa8 Univ. Publ. No. 5509, 22.Moore, S. & Stein, W. H. (1954). J. biol. Chem. 211, 893.Ratner, S. (1954). Advanc. Enzymol. 15, 319, 357.Ratner, S. & Pappas, A. (1949). J. biol. Chem. 179, 1183.Ratner, S., Petrack, B. & Rochovansky, 0. (1953). J. biol.Chem. 204, 95.
Sporn, M. B., Dingman, W., Defalco, A. & Davies, R. K.(1959). Nature, Lond., 183, 1520.
Theorell, T. & Stenhagen, E. (1939). Biochem. Z. 299, 417.Van Heyningen, W. E. (1949). Brit. J. exp. Path. 30, 302.Van Pilsnm, J. F., Martin, R. P., Kito, E. & Hess, J.
(1956). J. biol. Chem. 222, 225.Walker, J. B. (1958). Proc. Soc. exp. Biol., N. Y., 98, 7.Walker, J. B. & Myers, J. (1953). J. biol. Chem. 203, 143.Westall, R. G. (1952). Biochem. J. 52, 638.Westall, R. G. (1955). Biochem. J. 60, 247.Westall, R. G. (1958). Ab8tr. Comm. 4th int. Congr.
Biochem., Vienna, no. 13-34, p. 168.Wright, L. D., Miller, C. S., Skeggs, H. R., Huff, J. W.,Weed, L. L. & Wilson, D. W. (1951). J. Amer. chem. Soc.73, 1898.





























