Embed Size (px)
Citation preview

XXXVII Reunión Anual de la Asociación
Andaluza de Hematología y HemoterapiaMarbella, 18 y 19 de mayo de 2017
Con el Aval de:

Edita:
Asociación Andaluza de Hematología y Hemoterapia
Coordina:
Triana Congresos
Maquetación:
Punto DIP Triana
ISBN:
978-84-697-3132-1
Dep. Legal:
SE-1013-2017

3MARBELLA - 18 Y 19 DE MAYO DE 2017
Índice
CARTA DE BIENVENIDA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
COMITÉS. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
PROGRAMA CIENTÍFICO . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
PONENCIAS
Enfoque biológico del tratamiento de la Leucemia Linfática Crónica. Dr. Francesc Bosch, Dra. Anna Varela, Dr. Pau Abrisqueta . . . 27 Linfoma folicular: novedades terapéuticas en primera línea y en recaída/refractariedad. Dr. Antonio Salar. . . . . . . . . . . . . . . . . . . 31 Tratamiento adaptado a la respuesta metabólica evaluada por FDG/PET en linfoma de Hodgkin clásico. Dr. Antonio Rueda
Domínguez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33 Marco regulatorio de los Medicamentos Biológicos. Julio Sanchez Fierro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35 Implicaciones pronósticas y terapéuticas de las alteraciones genético-moleculares en LAM: recomendaciones en 2017.
Dres. Josefina Serrano, C. Martínez-Losada, J. Serrano-López y Joaquín Sánchez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36 Citogenética y estado mutacional en síndromes mielodisplásicos. Francesc Solé . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44 Papel actual del trasplante hematopoyético en el tratamiento de pacientes adultos con leucemias mieloblásticas agudas y
síndromes mielodisplásicos. Ildefonso Espigado . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48 Transfusión masiva en Obstetricia. Magdalena Carmona González . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52 Hemovigilancia y calidad transfusional. Miguel Ángel Alvarez Rivas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56 Tratamiento de la trombocitemia esencial y de la policitemia vera. Juan Carlos Hernández Boluda . . . . . . . . . . . . . . . . . . . . . . . . . . 60 Alteraciones moleculares en las neoplasias mieloproliferativas crónicas Filadelfia negativas. Beatriz Bellosillo . . . . . . . . . . . . . . . 62 Supervivencia libre de tratamiento en la leucemia mieloide crónica: ¿en qué pacientes es posible la curación? Antonio Jiménez
Velasco. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63 ¿Es necesario seguir haciendo estudios de trombofilia hoy día? Dr. Javier Rodríguez Martorell. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66 Anticoagulantes orales directos como tratamiento de primera línea en FA no valvular. Alejandro Pérez Cabeza . . . . . . . . . . . . . . . 69 Anticoagulantes orales directos como tratamiento de primera línea en FA no valvular (FANV). Punto de vista del Hematólogo.
Rosa María Campos Álvarez. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70 ¿Hacia nuevos paradigmas terapéuticos en la primera línea de tratamiento del Mieloma Múltiple? (Nuevos fármacos, nuevas
combinaciones de fármacos, la incorporación del mantenimiento y la revisión de la escala de edad). Dr. Joan Bladé Creixenti . . 74
CASOS CLÍNICOS CITOLÓGICOS
Varón adulto con fiebre, cuadro poliadenopático y bicitopenia. Mª Paz Garrastazul Sánchez, Irene Millán Ortega, Raquel de la Varga Martínez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
Neoplasia Mieloproliferativa Crónica (NMPc) con reordenamiento de PDGFRß. M. Gómez Rosa, R. Morales Camacho, I. Simón Pilo, S. Burillo Sanz, E. Ríos Herranz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
Adolescente de 13 años con poliadenopatías y bicitopenia. M.D. Serrano Chacón, R.M. Morales-Camacho, C. Prats-Martín, R. López Irizo, T. Caballero-Velázquez, O. Pérez López, E Carrillo, M.T. Vargas, R. Bernal Ruiz, J.J. Borrero . . . . . . . . . . . . . . . . . . . . . . . . 92
Leucemia eosinofílica maligna. Irene Vico Herrera, María del Carmen Martínez Losada, Víctor Arqueros Martínez, Joaquín Sánchez García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
Hombre de 50 años VIH, con fiebre y pancitopenia progresiva. M. Reinoso, C. Prats-Martín, R.M. Morales-Camacho, J.J. Borrero, N. Al Kadi, O. Pérez, E. Carrillo, J.M. de Blas, M.T. Vargas, T. Caballero-Velázquez, R. Bernal, J.A. Pérez-Simón. . . . . . . . . . . . . . . . . . . . 103

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA4
COMUNICACIONES ORALES
Hematología Clínica
Relación entre el perfil inmune de la médula ósea con variables clínico/biológicas en pacientes con patología de células plasmáticas. E. García Torres, C. Martínez Losada, M.A. Álvarez Rivas, J. Serrano-López, J. Serrano, J. Sánchez-García . . . . . . . . . . . . 111
Manifestaciones dermatológicas en trombocitemia esencial. Evolución y seguimiento a largo plazo: Experiencia en un hospital comarcal. M.M. Herráez-Albendea, S.R. Verdesoto-Cozzarelli, R. Cruz Conde-de Boom . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
Evaluación pronóstica en pacientes con síndrome mielodisplásico de bajo riesgo. Parámetros relacionados con la probabilidad de progresión leucémica. J.F. Falantes, J.F. Márquez-Malaver, C. Calderón Cabrera, I. Espigado, J.A. Pérez-Simón . . . . . . . . . . . . . . . 115
Factores de riesgo y tratamiento de segunda línea en la Enfermedad Injerto Contra Huésped aguda (EICHa). A. I. Álvarez Sánchez, M. Yébenes Ramírez, E. García Torres, C. Martín Calvo, R. Rojas Contreras, C. Herrera Arroyo. . . . . . . . . . . . . . . . . . . . . . . . . . 118
Leucemias agudas de linaje ambiguo, experiencia en un único centro. B. Pedrote Amador, J. Falantes González, I. Montero Cuadrado, M.L. Martino Galiana, J.A. Perez Simón, J. González Campos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
Hemostasia y Trombosis
Déficit hereditario de antitrombina: experiencia de nuestro centro. M. Ángeles Domínguez-Muñoz, M.A. Blum-Domínguez, Reyes Jiménez-Bárcenas, Ramiro Nuñez-Vázquez, Rosario Pérez Garrido, F.J. Rodríguez-Martorell . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
Hemorragias graves en pacientes anticoagulados con anticoagulantes de acción directa. S. Ordóñez Vahi, J.A. Raposo Puglia, B. Díez del Corral, R. Campos Álvarez, A. Estella García, S. Garzón López . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
Morfología y Biología Celular
Importancia de la secuenciación masiva para la detección de mutaciones en TP53. en la leucemia linfática crónica. E. Carrillo Cruz, M. Siuto Alcántara, F. De la Cruz Vicente, M. Ruiz Mercado, J.R. García Lozano, J.A. Pérez Simón . . . . . . . . . . . . . . . . . . . . . . . . . . 133
COMUNICACIONES PÓSTERS
Gestión y Automatización en Hematología
Consulta virtual telefónica como herramienta de mejora de la calidad asistencial: experiencia en nuestro centro. M. Anguita Arance, J.D. Tallon Pérez, B. Soler Gabarrón, M.C. Avellaneda Molina . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
Agilización de Procesos Asistenciales: Plan Piloto de Inclusión de Interconsultas en Hospital de Día de Hematología (HDH). M.J. Llamas Poyato, D. Moreno Garrido, I. García Díez. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
Estudio retrospectivo de los linfomas T diagnosticados en nuestro centro en los últimos 17 años. E. Morente Constantín, P.A. González Sierra, A. Núñez García, L. Moratalla López, E. López Fernández, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
Implantación de algoritmos diagnósticos de anemias en la gerencia de área integrada de Puertollano. Silvia del Rocio Verdesoto Cozzarelli, María Mar Herráez Albendea, María del Pilar García Fernández, María Carmen Lorenzo Lozano . . . . . . . . . . . . . . . 141
Papel del hematólogo de guardia: análisis de la demanda asistencial. J.A. Raposo Puglia, S. Ordóñez Vahí, M.V. Verdugo Cabeza de Vaca, C. Blázquez Goñi, A. Salamanca Cuenca, S. Garzón López . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 143
Hematología Clínica
Implicación de la mutación de la calreticulina en la presentación clínica y repercusión pronostica en la trombocitemia esencial. Y. Moatassim de la Torre, E. Clavero Sánchez, L. Entrena Ureña, A. Alba Sosa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 149
Tratamiento con radiofrecuencia del plasmocitoma óseo solitario. Experiencia en nuestro centro. Soraya Lorente de Uña, E. Morales Muñoz, M. González Bernal, J. Gutiérrez de Guzmán . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 150
Trasplante alogénico de progenitores hematopoyéticos en síndromes mielodisplásicos. I. Vico Herrera, A.I. Álvarez Sánchez, C. Martín Calvo, E. García Torres, F.J. Casaño Sánchez, J. Sánchez García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 152
Inmunoterapia en linfoma de zona gris quimiorresistente. C. Díaz Aizpún, M. Revelles Peñas, M. Espeso de Haro . . . . . . . . . . . . . . 153 Rituximab en monoterapia puede ser una opción óptima de tratamiento para pacientes con linfoma MALT primario de
pulmón. A.I. Álvarez Sánchez, L. Burgos Vigara, C. Martínez Losada, J. Sánchez García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154 Inmunomodulación con lenalidomida en linfoma no Hodgkin B folicular refractario. A propósito de un caso. J. Díez Pastor,
M.M. Alcalá Peña, M. Espeso de Haro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156

5MARBELLA - 18 Y 19 DE MAYO DE 2017
Hemodiálisis como tratamiento del fracaso renal en el Mieloma Múltiple con exceso de Cadenas Ligeras. N. Domínguez Velasco, R. Duro Millán, K. Kestler González, B. Herruzo Delgado, J. Montero Benítez, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . 158
Transplante cardiaco y de Médula ósea en paciente con Amiloidosis AL e Insuficiencia cardiaca: a propósito de un caso. N. Domínguez Velasco, R. Duro Millán, C. Muñoz García, B. Herruzo Delgado, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . . . . . . 159
Revisión de la implantación del protocolo de transfusión masiva por hemorragia masiva en nuestro centro hospitalario. M. Molina, V. Calama, E. García, E. Martínez, M. Gómez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 160
Leucemia mieloide aguda en primera remisión completa: estudio descriptivo y análisis de supervivencia de un único centro. M. Molina, V. Calama, M. Gómez, C. Couto, I. Simón, E. Ríos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
Análisis epidemiológico y serológico del virus de la hepatitis B y C en un grupo de síndromes linfoproliferativos. Resultados de un centro. V. Calama. Ruiz-Mateos, M.E. Molina Rodríguez, M. Gómez Rosa, J.C. López MartÍn, I. Simon Pilo, E. Ríos Herranz . . . . . . 163
Síndrome de Sweet histiocitoide asociado a trasplante autólogo de progenitores hematopoyéteicos. A propósito de un caso. I. Sánchez Bazán, C. Díaz Aizpún, S. Simonsen, C. Bethencourt Mateos, A.I. Heineger Mazo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
Análisis de la correlación de la afectación de médula ósea al diagnóstico en linfomas indolentes mediante PET y BMO. V. Calama Ruiz-Mateos, M.E. Molina Rodríguez, M. Gómez Rosa, I. Simon Pilo, C. Couto Caro, E. Ríos Herranz . . . . . . . . . . . . . . . . . . . 166
Trasplante alogénico de progenitores hematopoyéticos en aplasia medular severa. A.I. Álvarez Sánchez, M. Yebenes Ramírez, E. Gracia Torres, C. Martín Calvo, J. Serrano López, V. Arqueros Martínez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
Linfocitosis monoclonal B tipo LLC asociada a linfoma no Hodgkin T. Myriam Revelles Peñas, Carola Díaz Aizpún, Manuel Espeso de Haro . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
Era de los inhibidores de la tirosin cinasa (ITK)en Leucemia Mieloide Crónica (LMC), manejo y experiencia en nuestro centro. M.J. Llamas Poyato, D. Moreno Garrido, I. García Díez. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
Tratamiento con Ibrutinib en la leucemia linfática crónica en recaída o refractaria. Revisión de casos en nuestro centro. J. Díez Pastor, O. Benítez Hidalgo, M. Ortiz Pareja, A.I. Heiniger Mazo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
Seguridad y eficacia de los esquemas basados en pomalidomida en pacientes con mieloma múltiple en recaída o refractario: serie de casos de un centro. A.M. Alba Sosa, Y. Moatassim de la Torre, E. Clavero Sánchez. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
Eficacia de Nivolumab en enfermedad de Hodgkin clásica refractaria. O. Benítez Hidalgo, D.E. Barrios Decoud, M. Espeso de Haro 174 Mucormicosis cutánea: a propósito de un caso. S. Ordóñez Vahi, A. Salamanca Cuenca, V. Rubio Sánchez, S. Garzón López . . . . . . . 175 Tumores sólidos concomitantes con neoplasias hematológicas: caso a caso. M.J. Llamas Poyato, D. Moreno Garrido, I. García Díez 177 Bacteriemias en pacientes hematológicos hospitalizados. S. Ordóñez Vahi, V. Verdugo Cabeza de Vaca, H.J. Campo Palacio,
L. Domínguez Acosta, V. Rubio Sánchez, S. Garzón López . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 178 Perfil de seguridad de Ibrutinib en leucemia linfática crónica, linfoma del manto y macroglobulinemia de Waldentröm en recaída o
refractario. Experiencia de un centro. M. Ruiz Mercado, F. De la Cruz Vicente, M. Solé Rodríguez, E. Carrillo Cruz, J.A. Pérez- Simón . . . 181 Papel de los inhibidores de Tirosin Kinasa de Bruton en la excepcional leucemia linfática crónica con infiltración
leptomeníngea. M. Ruiz Mercado, E. Carrillo Cruz, M. Solé Rodríguez, F. De la Cruz Vicente, J.A. Pérez- Simón . . . . . . . . . . . . . . . . . . . 183 5-Azacitidina como nueva indicación de tratamiento en pacientes con leucemia mieloide aguda (LMA): a propósito de un caso.
María Eugenia Molina Rodríguez, Virgilio Calama, María Vahí, Carmen Couto, Juan Carlos Martin, Eduardo Ríos . . . . . . . . . . . . . . . . 184 Histiocitosis sistémica refractaria. Tratamiento con Macob-B como puente a trasplante alogénico. A propósito de un caso.
I. Sánchez Bazán, O. Benítez Hidalgo, M. Espeso de Haro, A.I. Heineger Mazo. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185 Lupus eritematoso sistémico incompleto en niña de 10 años con diagnóstico de trombocitopenia inmune primaria.
A propósito de un caso. M.A. Ruiz Cobo, K. Gómez Correcha, R. Cabra, R. Zapata Bautista, J.N. Rodríguez Rodríguez . . . . . . . . . . . . . 186 Histiciocitosis de células de Langerhans. Revisión de la experiencia del Hospital Regional Universitario de Málaga. A. González
Fernandez, F. Cabrera Ruiz, M. Espeso de Haro. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 187 Infecciones bacterianas en el paciente hematológico hospitalizado. C. Muñoz García, B. Herruzo Delgado, N. Domínguez Velasco,
J. Gálvez Acebal, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 189 Segundo trasplante autólogo de progenitores hematopoyéticos en pacientes con mieloma múltiple en recaída. Experiencia en
un hospital de 2º nivel. M.A. Ruiz Cobo, A.J. Palma Vallellano, R. Zapata Bautista, B. Díaz Roldán, J.N. Rodríguez Rodríguez . . . . . . 190 Tratamiento del linfoma anaplásico de células grandes B asociado a prótesis mamaria. M. Ruiz Mercado, M. Solé Rodríguez,
N. Alkadi Fernández, E. Carrillo Cruz, F. De la Cruz Vicente, J.A. Pérez Simón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 192 Manejo diagnóstico y terapéutico del linfoma primario del sistema nervioso central. M. Solé Rodríguez, M. Ruiz Mercado,
F. De La Cruz Vicente, E. Carrillo Cruz, J.J. Borrero Martín, J.A. Pérez Simón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 193 Necrosis de médula ósea en paciente con neoplasia hematológica. B. Díaz Roldán, A. Palma Vallellano, R. Zapata Bautista,
M.A. Ruíz Cobos, J.A. Quesada P., M.V. Moreno Romero. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 194 Ruxolitinib en pacientes con hemodialisis: a propósito de un caso de mielofibrosis e insuficiencia renal terminal. M. Ángeles
Domínguez-Muñoz, Patricia Jiménez Guerrero, Begoña Pedrote Amador, José González Campos, Isabel Montero Cuadrado . . . . . . . 195 Tratamiento de linfoma difuso de células grandes B con afectación del sistema nervioso central con esquema de quimioterapia
R-IDARAM. María Solé Rodríguez, N. Rodríguez Torre, M. Ruiz Mercado, F. De La Cruz Vicente, I. Montero Cuadrado, J.A. Pérez Simón 196

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA6
Episodios de neutropenia febril durante un semestre hospitalario: análisis epidemiológico. J.A. Raposo Puglia, S. Ordóñez Vahi, L. Domínguez Acosta, A. Salamanca Cuenca, M.V. Verdugo Cabeza de Vaca, V. Rubio Sánchez . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
Aplasia medular grave con respuesta a agonista de los receptores de la Trombopoyetina: una nueva oportunidad. M.M. Herráez-Albendea, S.R. Verdesoto-Cozzarelli . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
La leucemia de linfocitos grandes granulares y células NK, una entidad muy poco frecuente. E. Morente Constantín, A.B. Rivera Ginés, M.P. Garrido Collado, A. Núñez García, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 202
Síndrome de Schnitzler: la gammapatía monoclonal dentro de una entidad poco común y de difícil diagnóstico. E. Morente Constantín, B. Mesa Simón, M.P. Garrido Collado, R. Ríos Tamayo, A.B. Rivera Ginés, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . 203
Utilidad del F18-FDG-PET/TC y/o biopsia ósea para el estadiaje de los linfoma B difusos de células grandes. M. Yébenes Ramírez, I. Vico Herrera, C. Martínez Losada, C. Chic Acevedo, J. Sánchez García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
El síndrome hemofagocítico: revisión de los casos diagnosticados en nuestro centro en el último año. J. Badiola González, E. Morente Constantín, B. Mesa Simón, P.A. González Sierra, E. López Fernández, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
Factores de riesgo asociados a la mortalidad en los pacientes con hemopatías que requieren ingreso en la unidad de cuidados intensivos. E. Morente Constantín, A.B. Rivera Ginés, P.A. González Sierra, F. Manzano Manzano, E. López Fernández, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
Evaluación de la EMR mediante citometría de flujo pre y post auto trasplante en miloma múltiple. M. Yébenes Ramírez, M.A. Álvarez Rivas, C. Martínez Losada, J. Sánchez García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 210
Tuberculosis (TBC) diseminada en una paciente con leucemia linfocítica crónica, a propósito de un caso. V. Calama Ruiz-Mateos, M.E. Molina Rodríguez, I. Simón Pilo, M. Vahí Sánchez de Medina, M. Gómez Rosa, E. Ríos Herranz . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
Sarcoma granulocítico: la importancia del diagnóstico y tratamiento precoces en una entidad agresiva. E. Morente Constantín, A.J. Cruz Díaz, P.A. González Sierra, A. Romero Aguilar, L. Moratalla López, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
Coexistencia de dos hemopatías: leucemia mieloide crónica y gammapatía monoclonal de significado incierto en un mismo paciente. ¿Cuál es el camino a seguir? E. Morente Constantin, J.M. Puerta Puerta, P. García Martín, B. Mesa Simón, A. Núñez García, M. Jurado Chacón. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
Análisis de la mortalidad en la Unidad de Cuidados Intensivos en pacientes hematológicos. N. Domínguez Velasco, C. Muñoz García, J.R. Jiménez del Valle, K. Kestler González, B. Herruzo Delgado, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 217
Estudio descriptivo de los linfomas MALT diagnosticados en nuestro centro en los últimos 15 años. E. Morente Constantín, A. Núñez García, P. Romero García, P.A. González Sierra, E. López Fernández, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 219
Citopenia hemtológica en enfermedad autoinmune sistémica: a propósito de un caso. J. Morán Sánchez, F.J. Capote García, A. Santisteban Espejo, M.C. Fernández Valle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
Linfoma Difuso de Células Grandes B coexistente con otros linfomas. F.J. Cabrera Ruiz, A. González Fernández, M. Espeso de Haro 221 El CRTS de Granada-Almería organiza la primera donación masiva de plasma a nivel nacional. E. Morente Constantín, A. López
Berrio, P. Romero García, M.A. García Ruíz, M.A. Hernández Vidaña, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 222 Estudio descriptivo de las características clínico/biológicas y supervivencia en pacientes con síndrome mielodisplásico de bajo
riesgo con variables pronósticas no desfavorables. Werner González Molina, José Francisco Falantes González, Ildefonso Espigado Tocino, Cristina Calderón Cabrera, Francisco José Márquez Malaver, José Antonio Pérez Simón . . . . . . . . . . . . . . . . . . . . . . . 223
La experiencia de nuestro centro en pacientes diagnosticados de mastocitosis. E. Morente Constantín, A. Núñez García, F. Hernández Mohedo, P. Navarro Álvarez, R. Ríos Tamayo, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 225
Casos de hemofilia adquirida vistos en nuestro hospital durante los últimos 12 años. D. Jiménez Fernández, E. Morente Constantín, L. Entrena Ureña, M.J. Gutiérrez Pimentel, M.A. García Ruiz, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 226
Leucemia aguda postrasplante renal: revisión de dos casos. N. Alkadi Fernández, M. Ruiz Mercado, M.I. Montero, T. Caballero, J. González Campos, J.A. Pérez-Simón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 227
Evaluación de la Trombocitopenia Inmune Primaria (TIP) en la infancia. Experiencia en nuestro centro. R. Zapata Bautista, S. Ramírez García, Mª. A. Ruíz Cobo, I. Vazquez-Pastor Jiménez, B. Díaz Roldán, J.N. Rodríguez Rodríguez . . . . . . . . . . . . . . . . . . . . . . . . . . 228
Evaluación de la experiencia como centro trasplantador en pacientes con Síndromes linfoproliferativos en nuestro centro. R. Zapata Bautista, A. Palma Vallellano, I. Vázquez-Pastor Jiménez, E. Gil Espárraga, K. Gómez Korrecha, J.F. Domínguez . . . . . . . . . 229
P53 como factor pronóstico individual en síndromes mielodisplásicos. Experiencia en Hospital Universitario Nuestra Señora de Valme. María Eugenia Molina Rodríguez, Virgilio Calama, María Vahí, Isabel Simón, Elvira Martínez, Eduardo Ríos. . . . . . . . . . . . . . 230
Evaluación de la experiencia como centro trasplantador en pacientes con Mieloma Múltiple y otras discrasias de células plasmáticas. R. Zapata Bautista, A. Palma Vallellano, E. Gil Espárraga, J.N. Rodríguez Rodríguez, I. Vázquez-Pastor Jiménez, K. Gómez Korrecha . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 231
Evaluación de los factores de riesgo de recaída en SNC en los pacientes con Linfoma B difuso de células grandes. R. Zapata Bautista, E. Gil Espárraga, M.ªA. Ruíz Cobo, B. Díaz Roldán, K. Gómez Korrecha, A. Palma Vallellano . . . . . . . . . . . . . . . . . . . . . . . . . . 232
Linfoma no Hodgkin con afectación cerebral. Experiencia en nuestro hospital. V. Melero Cruz, G. García Fernández, F. Almagro Torres, J.A. López López, M.S. Durán Nieto . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 233

7MARBELLA - 18 Y 19 DE MAYO DE 2017
Linfomas de células B de alto grado doble Hit o doble Protein tratados con R-Daepoch. Experiencia de un centro. P. Jiménez Guerrero, M.A. Domínguez Muñoz, M. Ruiz Mercado, E. Carrillo Cruz, F. De la Cruz Vicente, I. Montero Cuadrado . . . . . . . . . . . . . . . . . 234
Tratamiento con Pomalidomida-Dexametasona de pacientes con Mieloma Múltiple en recaída. J. Capote García, M.E. Rodríguez Mateos, M.C. Fernández Valle, M.J. Huertas Fernández, G. Blanco Sánchez, F.J. Capote Huelva . . . . . . . . . . . . . . . . . . 235
Paciente con linfoma anaplásico primario cutáneo en Recidiva. Tratamiento con Brentuximab Vedotina. J. Capote García, F.J.Capote Huelva . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 236
Valor pronóstico de la enfermedad mínima residual inmunológica en pacientes adultos con leucemia aguda sometidos a trasplante alogénico de precursores hematopoyéticos en primera remisión completa. C. Martínez Losada, E. García-Torres, J. Serrano, S. Tabares, J. Sánchez-García . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 237
Diagnóstico sincrónico de procesos neoplásicos en hematología. Julia Morán Sánchez, F.J. Capote García, A. Santisteban Espejo, M.C. Fernández Valle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 238
Trasplante secuencial en LMA primariamente refractarias. M.D. Madrigal Toscano, R. Saldaña Moreno, A. Salamanca Cuenca, L. Domínguez Acosta, V. Rubio Sánchez, S. Garzón López . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 239
Infecciones fúngicas invasoras (IFI) por Mucor en pacientes oncohematológicos y receptores de trasplantes de progenitores hematopoyéticos. J.A. Rojas Martínez, M. Solé Rodríguez, M. Aguilar Guisado, I. Montero Cuadrado, M.L. Martino Galiano, J.A. Pérez-Simón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 240
Tabaquismo en afectados de trastorno hematológico (cuidadores, familiares y pacientes). A. García Nieto, F.J. Capote Huelva . . . . 242 Paciente con Macroglobulinemia de Waldeström (MW) y amiloidosis renal AL. M.A. Suito Alcántara, E. Carrillo Cruz, J. Martín
Sánchez, M.L. Martino Galiana . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243 Anemia hemolítica severa como debut de un caso de macroglobulinemia de Waldenstrom. B. Herruzo, M.C. Muñoz García,
I. Fernández Román, J. Montero Benítez, K.E. Kestler Domínguez, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 244 Trasplante hematopoyético autólogo bajo régimen domiciliario. Un nuevo modelo de atención eficaz y seguro. P. González
Sierra, S. De Linares Fernández, E. Fernández López, L. Moratalla López, Z. Mesa Morales, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . 245
Hemoterapia e Inmunohematología
Análisis según criterios clínico/analíticos de la actividad trasfusional en un hospital comarcal durante el año 2016. M.E. Clavero Sánchez, Y. Moatassim De la Torre, A.M. Alba Sosa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
Análisis de las necesidades transfusionales en el trasplante autólogo movilizado con Plerixafor + G-CSF VS. G-CSF. J.A. Raposo Puglia, M.V. Verdugo Cabeza de Vaca, A. Salamanca Cuenca, S. Ordóñez Vahí, M.A. Correa Alonso . . . . . . . . . . . . . . . . . . . . . . . . . . . . 250
Trasplante autólogo en pacientes movilizados con G-CSF + Plerixafor versus G-CSF: análisis del prendimiento de neutrófilos y plaquetas en nuestra serie. J.A. Raposo Puglia, A. Salamanca Cuenca, M.V. Verdugo Cabeza de Vaca, S. Ordóñez Vahí, H.J. Campo Palacio, M.A. Correa Alonso . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 251
Sistema de hemovigilancia en un hospital de nivel 2. Análisis de reacciones adversas, errores asociados y casi incidentes relacionados con el acto transfusional. V. Calama Ruiz-Mateos, M.E. Molina Rodríguez, M. Gómez Rosa, E. Martínez Estéfano, M. Vahí Sánchez de Medina, E. Ríos Herranz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252
Resultados de un sistema integral corporativo de seguridad transfusional electrónica completa multicentro. M.A. García Ruiz, F.J. Pérez Zenni, E. Morente Constantín, F. Hernández Mohedo, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 253
Evaluación de los resultados de movilización de progenitores hematopoyéticos en el trasplante autólogo: experiencia en nuestro centro. R. Zapata Bautista, A. Palma Vallellano, Mª.A. Ruíz Cobo, A. Lara Bohórquez, M.A. Castilla Reyes, M. Bendala. . . . . 254
¿Podrá ser la transfusión de hematíes un factor de riesgo para enterocolitis necrotizante en recién nacidos de bajo peso al nacer? K. Kestler, A. Marin Cassinello, I. Fernández Román, I. Tallón Ruiz, A. Rodríguez Fernández, J. Montero Benítez. . . . . . . . . . . . 255
Hemostasia y Trombosis
Perfil clínico y eventos en pacientes anticoagulados con apixaban. Estudio en vida real. A.I. Pérez Cabeza, A. Valle Alberca, M.E. Moreno Beltrán, J.A. González Correa, R. Bravo Marqués, P.A. Chinchurreta Capote. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 259
Persistencia al tratamiento con apixaban en pacientes con fibrilación auricular. no valvular. A.I. Pérez Cabeza, A. Valle Alberca, M.E. Moreno Beltrán, J.A. González Correa, R. Bravo Marqués, P.A. Chinchurreta Capote. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 260
Indicación de tratamiento con apixaban en fibrilación auricular: Adherencia a ficha técnica y al informe de posicionamiento terapéutico de la AEMPS. A.I. Pérez Cabeza, A. Valle Alberca, M.E. Moreno Beltrán, J.A. González Correa, R. Bravo Marqués, P.A. Cinchurreta Capote . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 261
Prevención de eventos tromboembólicos en pacientes con fibrilación auricular tras la creación de una consulta monográfica. A.I. Pérez Cabeza, R. Bravo Marqués, M.E. Moreno Beltrán, A. Valle Alberca, F. Ruiz Mateas, P.A. Chinchurreta Capote . . . . . . . . . . . . 262

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA8
Papel de la anticoagulación oral en la prevención de eventos cardiovasculares en pacientes con fibrilación auricular atendidos de forma ambulatoria. A.I. Pérez Cabeza, M.E. Zambrano Medina, M.E. Moreno Beltrán, F. Mesa Prado, S. López Tejero, P.A. Chinchurreta Capote . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263
Discordancias en la posología de los anticoagulantes orales directos al emplear fórmulas de filtrado glomerular estimado en vez de la ecuación de Cockcroft Gault. A.I. Pérez Cabeza, R. Bravo Marqués, M.E. Moreno Beltrán, A. Alberca Valle, F. Mesa Prado, P.A. Chinchurreta Capote . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 264
Trombosis cardíaca en paciente con enfermedad de Von Willebrand tipo I severa. I. Sánchez Bazán, M.E. Mingot Castellano. . . . . 265 Romiplostin en trombopenia inmune: experiencia de un Hospital Comarcal. M.C. Galán Fernández, M.O. García Montero,
A.I. Gallardo Morillo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 266 Análisis de la incidencia de hiperhomocisteinemia y eventos trombóticos en una serie de casos obtenida de la consulta de
Hematología de un hospital comarcal. M.E. Clavero Sánchez, Y. Moatassim De la Torre, I. García Cabrera, A.M. Alba Sosa . . . . . . . . 267 Efecto del calcitriol y paracalcitol en la expresión del factor tisular y PAR-2 en las células de músculo liso vasculares.
J.M. Marténez Moreno, C. Herencia, I. Vico, Y. Almadén, F. Velasco, A.I. Álvarez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 268 Terapia puente con bemiparina sódica a dosis terapéuticas: experiencia en nuestro centro. M.A. García Ruiz, E. Morente
Constantín, P. Romero García, M. Gómez Morales, M.D. Fernández Jiménez, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 269 Síndrome de hemofilia adquirida: estudio a contrarreloj. I. Vico (1), A.I. Álvarez (1), M. Yébenes (1), F. Velasco (1),
M. Fernández (1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 270 La eosinofilia como síndrome paraneoplásico favorecedor de episodios trombóticos. E. Morente Constantín, D. Fernández
Jiménez, L. Castillo Portellano, M.A. García Ruíz, M.J. Gutiérrez Pimentel, M. Jurado Chacón . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 271 Inhibidores adquiridos contra factores de la coagulación: experiencia de nuestro centro. M.A. Blum-Domínguez,
M.A. Domínguez-Muñoz, R.J. Núñez-Vázquez, R. Jiménez-Bárcenas, R. Pérez-Garrido, F.J. Rodríguez-Martorell . . . . . . . . . . . . . . . . . 272 Disfibrinogenemias. Experiencia en Nuestro Centro. G. García Fernández, V. Melero Cruz, F. Almagro Torres, J.A. López López,
M.M. Nieto Hernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 273 Sinoviortesis radioisotópica en pacientes hemofílicos pediátricos y adolescentes. M.A. Blum-Domínguez, L. Pérez-Ortega,
R. Jiménez-Bárcenas, R.J. Núñez-Vázquez, F.J. Rodríguez-Martorell, R. Pérez-Garrido . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 275 Mujer con Linfoma B de Células Grandes y tos persistente con disfonía: A propósito de un caso de Síndrome de Ortner.
K. Kestler, G. Rodríguez García, A. García Guerrero, M. García Díez, N. Domínguez Velasco, B. Herruzo Delgado . . . . . . . . . . . . . . . . . . 276
Morfología y Biología Celular
Patrones de recaída fenotípica y genotípica en leucemias agudas mieloblásticas bajo riesgo citogenético/molecular. C. Martínez-Losada, J. Serrano-López, J. Serrano, J. Sánchez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 281
Necrosis de médula ósea secundaria a hemopatía malgina: A propósito de un caso. B. Herruzo Delgado, M. Manzanares Pérez, F.R. Acosta Maestre, N. Domínguez Velasco, J. Montero Benítez, A. Rodríguez Fernández . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 282
Linfoma de Hodgkin anal primario: una entidad infrecuente. M.J. Llamas Poyato, D. Moreno Garrido, I. García Díez . . . . . . . . . . . . 284 Síndrome hemofagocítico como complicación en un paciente con cáncer de recto: la importancia de la morfología de médula
ósea en el diagnóstico de una entidad agresiva. E. Morente Constantín, J. Badiola González, R. Ríos Tamayo, M.P. Garrido Collado, P.A. González Sierra, M. Jurado Chacón. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
Síndrome hemofagocítico secundario a leishmaniasis visceral. F.J. Cabrera Ruiz, M. Revelles Peñas, A. Campos Garrigues . . . . . . . 287 Estudio descriptivo de la Linfocitosis B monoclonal CD 5 positivo en un hospital de tercer nivel. P. Jiménez Guerrero, O. Pérez
López, M. Ruíz Mercado, C. Prats Martín, E. Carrillo Cruz, T. Caballero Velázquez . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289 Análisis genómico y funcional de las MO-MSC y de los exosomas derivados de las mismas en pacientes diabéticos con isquemia
arterial periférica. Implicaciones en terapia celular. Vanesa Martín, María Muñoz, Gustavo Díez, Rosario Jiménez, Sonia Nogueras, Inmaculada Herrera . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 290
Serie Roja
Haptoglobina, evaluación en el laboratorio de Eritropatología. R. López Rodríguez, M.P. Garrastazul Sánchez, M.J. Hernández Alfaro, C. De Cos Höhr . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 293
Identificación de una mutación en el gen HBA2 como causa de hemoglobina variante (hemoglobina yuda). A propósito de un caso. Silvia Del Rocio Verdesoto Cozzarelli, María Mar Herráez-Albendea, María Castillo Jarilla-Fernández, Manuel Martínez-Fernández, Esther González-Real . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 297
Anemia hemolítica autoinmune por anticuerpos calientes de tipo IGM y complemento: a propósito de un caso. J.A. Raposo Puglia, A. Atienza Saborido, A. Salamanca Cuenca, M.A. Correa Alonso, S. Ordóñez Vahí, M.D. Madrigal Toscano . . . . . . . . . . . . . . . . 300

9MARBELLA - 18 Y 19 DE MAYO DE 2017
Bienvenida
Queridos compañeros:
Deseamos que esta edición de la Reunión Anual de la Asociación Andaluza de Hematología y Hemoterapia
que se celebra en Marbella, sirva una vez más para actualizar nuestros conocimientos de las enfermedades
hematológicas, así como para compartir inquietudes ligadas a nuestro trabajo, con el objetivo de mejorar
la atención que los hematólogos prestamos diariamente a nuestros pacientes.
Para la unidad de Hematología y Hemoterapia del Hospital Costa del Sol ha sido motivo de satisfacción
haber participado en la organización de este congreso y agradecemos de todo corazón el apoyo y trabajo
de los miembros del comité organizador, del comité científico y del comité evaluador de comunicaciones,
así como de todos los ponentes y moderadores.
Pero, sobre todo, esperamos disfrutar con vuestra asistencia.
Un fuerte abrazo,
María Ángeles Medina Pérez
Presidenta Comité Organizador
XXXVII Reunión Anual AAHH


11MARBELLA - 18 Y 19 DE MAYO DE 2017
Excmo. Sr. D. José Bernal Gutiérrez
Alcalde de Marbella
Ilmo. Sr. D. Aquilino Alonso Miranda
Consejero de Salud de la Junta de Andalucía
Dr. D. Torcuato Romero López
Gerente del Hospital Costa del Sol
Dr. D. Francisco Martos Pérez
Director Médico del Hospital Costa del Sol
Dr. D. Juan José Sánchez Luque
Presidente del Colegio de Médicos de Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA12
Dra. Dña. M.ª Luz Martino Galiano
Dra. Dña. Margarita Jiménez Jambrina
Dr. D. Manuel Espeso de Haro
Dr. D. Antonio Fernández Jurado
Almería: Dr. D. Manuel Suárez Solís
Cádiz: Dr. D. Antonio Paz Coll
Córdoba: Dr. D. José Ramón Molina Hurtado
Granada: Dr. D. José Manuel Puerta Puerta
Huelva: Dra. Dña. Karoll Gómez Corecha
Jaén: Dra. Dña. Francisca Almagro Torres
Málaga: Dra. Dña. Carmen Galán Fernández
Sevilla: Dr. D. José Falantes González

13MARBELLA - 18 Y 19 DE MAYO DE 2017
Dra. Dña. Ángeles Medina Pérez
Dra. Dña. M.ª Isabel Mata Vázquez
Dra. Dña. Esperanza Moreno Beltrán
Dra. Dña. María Casanova Espinosa
Dr. D. Antonio Fernández Jurado
Dra. Dña. Gema Fornés Torres
Dra. Dña. Ana Isabel Heiniger Mazo
Dra. Dña. M.ª Luz Martino Galiana
Dr. D. Eduardo Ríos Herranz
Dr. D. José Joaquín Ruiz Arredondo

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA14
Dra. Dña. María Casanova Espinosa
Dr. D. Miguel Ángel Álvarez Rivas
Dr. D. Ricardo Bernal Ruiz
Dr. D. Francisco Javier Capote Huelva
Dr. D. Carlos Clavero Farre
Dr. D. Ildefonso Espigado Tocino
Dr. D. José Falantes González
Dra. Dña. Gema Fornés Torres
Dr. D. Antonio Jiménez Velasco
Dra. Dña. M.ª Isabel Mata Vázquez
Dra. Dña. Ángeles Medina Pérez
Dra. Dña. Eva Mingot Castellano
Dra. Dña. Esperanza Moreno Beltrán
Dr. D. Eduardo Ríos Herranz
Dr. D. Francisco Javier Rodríguez Martorell
Dr. D. Joaquín Sánchez García
Dra. Dña. Josefina Serrano López

15MARBELLA - 18 Y 19 DE MAYO DE 2017
Dr. D. Manuel Barrios García
Dra. Dña. Rosa Campos Álvarez
Dra. Dña. Isabel Caparrós Miranda
Dra. Dña. M.ª Ángeles Correa Alonso
Dra. Dña. M.ª Carmen Couto Caro
Dr. D. Manuel Espeso de Haro
Dr. D. José Falantes González
Dra. Dña. Soledad Durán Nieto
Dra. Dña. Margarita Fernández de la Mata
Dra. Dña. Gracia García Gemar
Dra. Dña. Ricarda García Sánchez
Dra. Dña. Lourdes Hermosín Ramos
Dra. Dña. Francisca Hernández Mohedo
Dr. D. Juan Antonio López López
Dra. Dña. Carmen Martín Calvo
Dr. D. Eusebio Martín Chacón
Dra. Dña. M.ª Eva Mingot Castellano
Dra. Dña. Rosario Morales Camacho
Dra. Dña. Marian Cuesta Casas
Dr. D. José Manuel Puerta Puerta
Dra. Dña. Mari Paz Queipo de Llano
Dra. Dña. M.ª José Ramírez Sánchez
Dr. D. Eduardo Ríos Herranz
Dr. D. Rafael Rios Tamayo
Dra. Dña. Alicia Rodríguez Fernández
Dr. D. Guillermo Rodríguez García
Dr. D. Juan Nicolás Rodríguez Rodríguez
Dra. Dña. Concepción Ruiz Nuño
Dr. D. Joaquín Sánchez García
Dr. D. Francisco Sánchez Gordo

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA16
Programa Científico
JUEVES 18 DE MAYO
Entrega documentación
Asamblea general de la AAHH
Pausa-Café
Dr. Francesc Bosch Albareda Serv. Hematología. Hospital de la Vall d’Hebron, Barcelona
Dr. Antonio Salar Silvestre Serv. Hematología. Hospital del Mar. Parc de Salut Mar. Barcelona
Dr. Antonio Rueda Domínguez Unidad de Onco- Hematología. Hospital Costa del Sol, Marbella
Moderadores: Dr. Eduardo Rios Herranz Hospital Universitario Virgen de Valme, Sevilla Dra. Ángeles Medina Pérez Hospital Costa del Sol, Marbella
Patrocinado por Janssen
D. Julio Sánchez FierroLorenzo Abogados
Patrocinado por Roche Farma
Acto inaugural
Cóctel de bienvenida

17MARBELLA - 18 Y 19 DE MAYO DE 2017
VIERNES 19 DE MAYO
Dra. Josefina Serrano López Serv. Hematología. Hospital Universitario Reina Sofía, Córdoba
Dr. Francesc Solé Ristol Institut Josep Carreras (IJC). Director Científico Campus ICO-Germans Trias i Pujol, Barcelona
Dr. Ildefonso Espigado Tocino Serv. Hematología. Hospital Universitario Virgen del Rocío, Sevilla
Moderadores: Dr. José Falantes González Hospital Universitario Virgen del Rocío, Sevilla Dr. Joaquín Sánchez García Hospital Universitario Reina Sofía, Córdoba
Dra. Magdalena Carmona González Serv. Hematología. Hospital Universitario Virgen del Rocío, Sevilla
Dr. Miguel Ángel Álvarez Rivas Serv. Hematología. Hospital Universitario Reina Sofía, Córdoba
Moderadores: Dra. Gema Fornés Torres Centro de Transfusión Sanguínea, Córdoba
Pausa-Café
Visita y comentario de pósters
Dr. Juan Carlos Hernández Boluda Serv. de Hematología. Hospital Clínico Universitario, Valencia

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA18
Dra. Beatriz Bellosillo Paricio Serv. de Patología y Área de Biología Molecular. Hospital del Mar, Barcelona
Dr. Antonio Jiménez Velasco Serv. Hematología. Hospital Regional, Málaga
Moderadoras: Dra. Isabel Mata Vázquez Hospital Costa del Sol, Marbella Dra. Pilar López Garrido Complejo Hospitalario Universitario, Granada
Dr. Francisco Javier Rodríguez Martorell
Serv. Hematología. Hospital Universitario Virgen del Rocío, Sevilla
Punto de vista del Cardiólogo: Dr. Alejandro Pérez Cabeza Serv. de Cardiología. Hospital Costa del Sol, Marbella
Punto de vista del Hematólogo: Dra. Rosa María Campos Álvarez
Serv. de Hematología. Hospital de Jerez Moderadoras:
Dra. Esperanza Moreno Beltrán Hospital Costa del Sol, Marbella Dra. Eva Mingot Castellano Hospital Regional Universitario, Málaga
Moderador: Dr. Antonio Fernández Jurado. AAHH
Dr. Antonio Fernández Jurado Consejero delegado de la AAHH
Dr. Miguel Canales AlbendeaHospital La Paz. Madrid

19MARBELLA - 18 Y 19 DE MAYO DE 2017
Dr. Antonio Salar Silvestre Hospital del Mar. Barcelona
Dra. Beatriz Pérez Sanz Roche Farma
Patrocinado por Roche Farma
Ponentes: Dra. Ana Marín Niebla
Hospital Vall d’Hebrón. BarcelonaDr. Norberto López Navarro Hospital Virgen de la Victoria. Málaga
Dr. Norberto López Navarro
Dra. Ana Marín Niebla
Dr. Norberto López Navarro
Dra. Ana Marín Niebla Patrocinado por Takeda
Experto: Dr. Antonio Jiménez Velasco Hospital Regional Carlos Haya. Málaga
Patrocinado por Incyte
Moderador: Dr. José Joaquín Ruiz Arredondo Hospital Universitario Virgen de la Victoria Agenda:
Dra. Carolina Moreno Atanasio Hospital Santa Creu i Sant Pau. Barcelona
Dra. Paula Rodríguez Otero Clínica Universidad de Navarra. Pamplona
Patrocinado por Janssen

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA20
Moderador: Dr. Francisco Javier Rodríguez Martorell Hospital Virgen del Rocío. Sevilla
Expertos: Dra. María Eva Mingot Castellano
Hospital Regional Universitario. MálagaDra. Raquel Garrido Ruiz Hospital Puerto Real. Cádiz
Dra. Garrido Ruiz
Dra. Mingot Castellano
Dra. Mingot Castellano
Patrocinado por Leo
Experto: Dr. José Ángel Hernández Rivas Hospital Infanta Leonor. Madrid
Patrocinado por Gilead
Pausa-Café
Visita y comentario de pósters
Dr. Joan Bladé Creixenti Servicio Hematología. Hospital Clinic, Barcelona
Dr. Juan José Lahuerta Palacios Serv. Hematología. Hospital 12 de octubre, Madrid

21MARBELLA - 18 Y 19 DE MAYO DE 2017
Moderadores: Dra. María Casanova Espinosa Hospital Costa del Sol, Marbella Dr. Sebastian Garzón López Hospital de Jerez de la Frontera, Cádiz Patrocinado por Celgene y Janssen
Moderadores: Dr. Ricardo Bernal Ruiz Hospital Universitario Virgen del Rocío, Sevilla Dra. Macarena Ortiz Pareja Hospital Regional Universiario, Málaga
Mª Paz Garrastazul Sánchez, Irene Millán Ortega, Raquel de la Varga Martínez Hospital Universitario Puerta del Mar de Cádiz
M. Gómez Rosa, R. Morales Camacho1, I. Simón Pilo, S. Burillo Sanz1, E. Ríos Herranz
H.U. Ntra. Sra. De Valme y 1H.U. V. del Rocío, Sevilla
M.D. Serrano Chacón, R.M. Morales-Camacho, C. Prats-Martín, R. López Irizo, T. Caballero-Velázquez, O. Pérez López, E. Carrillo, M.T. Vargas, R. Bernal Ruiz, J.J. Borrero Hospital Universitario Virgen del Rocío, Sevilla
Irene Vico Herrera, María del Carmen Martínez Losada, Víctor Arqueros Martínez, Joaquín Sánchez García
Hospital Universitario Reina Sofía, Córdoba
M. Reinoso, C. Prats-Martín, R.M. Morales-Camacho, J.J. Borrero, N. Al Kadi, O. Pérez, E. Carrillo, J.M. de Blas, M.T. Vargas, T. Caballero-Velázquez, R. Bernal, J.A. Pérez-Simón
Hospital Universitario Virgen del Rocío (HUVR). Sevilla

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA22
Moderadores: Dr. Antonio Paz Coll Hospital Puerto Real, Cádiz
Dr. Manuel Jurado Chacón Complejo Hospitalario Universitario, Granada
E. García Torres, C. Martínez Losada, M.A. Álvarez Rivas, J. Serrano-López, J. Serrano, J. Sánchez-García Hospital Universitario Reina Sofía
M. Ángeles Domínguez-Muñoz, M.A. Blum-Domínguez, Reyes Jiménez-Bárcenas, Ramiro Nuñez-Vázquez, Rosario Pérez Garrido, F.J. Rodríguez-Martorell Hospital Universitario Virgen del Rocío
M.M. Herráez-Albendea, S.R. Verdesoto-Cozzarelli, R. Cruz Conde-de Boom Hospital Santa Bárbara. Puertollano
E. Carrillo Cruz, M. Siuto Alcántara, F. De la Cruz Vicente, M. Ruiz Mercado, J.R. García Lozano, J.A. Pérez Simón
Hospital Universitario Virgen del Rocío
J.F. Falantes, J.F. Márquez-Malaver, C. Calderón Cabrera, I. Espigado, J.A. Pérez-Simón Hospital Universitario Virgen del Rocío
A. I. Álvarez Sánchez, M. Yébenes Ramírez, E. García Torres, C. Martín Calvo, R. Rojas Contreras, C. Herrera Arroyo
Hospital Universitario Reina Sofía.IMIBIC. Universidad de Córdoba

23MARBELLA - 18 Y 19 DE MAYO DE 2017
S. Ordóñez Vahi, J.A. Raposo Puglia, B. Díez del Corral, R. Campos Álvarez, A. Estella García, S. Garzón López
Hospital de Jerez de la Frontera
B. Pedrote Amador, J. Falantes González, I. Montero Cuadrado, M.L. Martino Galiana, J.A. Perez Simón, J. González Campos
Hospital Universitario Virgen del Rocío


PONENCIAS


PONENCIAS 27
INTRODUCCIÓN
En los últimos 5 años se ha producido notables avances en el diagnós-tico molecular y el tratamiento de la leucemia linfática crónica (LLC). Una de las claves ha sido el acercamiento producido entre los conoci-mientos biológicos y las posibilidades de tratamiento, de tal forma que en el momento actual podemos usar tratamientos destinados a modi-fi car aspectos clave en la biología del tumor. Ello ha permitido disponer de terapias mucho más efi caces y que, además, se pueden administrar a un abanico más amplio de enfermos con LLC, una enfermedad ca-racterizada por afectar a una población de edad avanzada. El futuro va encaminado al uso de combinaciones de fármacos no genotóxicos que modifi carán diversos mecanismos de linfomagénesis a la vez, lo que probablemente nos acercará a la curación de la enfermedad.
1. TRATAMIENTOS GENOTÓXICOS:LA QUIMIO-INMUNOTERAPIA
Durante más de 30 años el tratamiento más importante de la LLC era el clorambucil. Con él se conseguían respuestas parciales y un discreto control de la enfermedad. Posteriormente se combinaron quimiote-rápicos alrededor del análogo de purinas fl udarabina. A principios de este siglo la quimioterapia se combinó con anticuerpos monoclonales frente a antígenos linfocitarios, en especial anti-CD20, lo que ha pasa-do a denominarse quimioinmunoterapia.
La combinación FCR se considera el tratamiento estándar de los pa-cientes con LLC que pueden tolerar o recibir fl udarabina. Este trata-miento consigue una tasa de respuestas globales superiores al 90% con porcentaje de respuestas completas (RC) entre el 40 y 70% depen-diendo de los estudios. De forma interesante, en más de la mitad de dichas RC no se detecta enfermedad mínima residual (EMR), lo que se asocia a una prolongada duración de la respuesta y supervivencia.
Enfoque biológico del tratamiento de la Leucemia Linfática CrónicaDr. Francesc Bosch, Dra. Anna Varela, Dr. Pau Abrisqueta
Servicio de Hematología, Hospital Universitario Vall d’Hebron, Barcelona
Para los pacientes que no podrán tolerar el tratamiento con FCR, se han diseñado diferentes combinaciones de anticuerpos monoclonales anti-CD20 y clorambucil. Estas combinaciones permiten el abordaje terapéutico con quimio-inmunoterapia incluso en pacientes con di-ferentes comorbilidades que años atrás eran tratados únicamente de forma paliativa. Una de las combinaciones más desarrolladas es la de clorambucil combinada con el anticuerpo monoclonal obinutuzumab (GA101). Este nuevo anti-CD20 presenta diferentes mecanismos de acción y parece ser más activo que rituximab. Administrado conjun-tamente con clorambucil produce un 20% de RC, con un 38% de ca-sos ERM negativos. De forma interesante, los pacientes tratados con obinu-clorambucil presentan un tiempo hasta el siguiente tratamiento superior a 4 años, lo que hace esta combinación atractiva para un gru-po de enfermos de edad avanzada y comorbilidades.
2. TRATAMIENTOS DIRIGIDOS A DIANAS BIOLÓGICAS
En la LLC, al igual que en otros síndromes linfoproliferativos B, exis-ten dos vías biológicas que contibuyen al desarrollo y perpetuación del proceso tumoral. En primer lugar, una inhibición de la apoptosis derivada de una expresión constitutiva y muy elevada de la proteína anti-apoptótica BCL2. La otra vía fundamental es la del receptor de células B (BCR). Las señales mediadas a través de la activación del BCR, en general activado por auto-antígenos, inducen proliferación, maduración del linfocito de la LLC y su interacción con las células del microambiente. El gran avance producido en los últimos 5 años ha sido el desarrollo de inhibidores de estas dos vías mediante moléculas de pequeño tamaño, administración oral y que permiten una aproxima-ción diferente terapéutica completamente diferente a la quimioterapia convencional.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA28
2.1. Inhibidores de la vía del BCR
Los inhibidores de la vía del BCR inducen un bloqueo de la interacción del linfocito con las células del microambiente y estromales, lo que modifica el tráfico de dichos linfocitos desde y hacía los órganos linfoi-des secundarios (ganglio linfático y médula ósea). Las consecuencias clínicas en la LLC de dicha inhibición son comunes: los ganglios linfá-ticos tumorales se reducen, la médula ósea queda menos infiltrada y
a cambio se produce un aumento de los linfocitos en sangre periférica en la mayoría de casos.
Otra de las propiedades comunes a estos fármacos es que su mecanis-mo de acción es independiente de la acción de la proteína TP53. Dicho gen se encuentra mutado o delecionado (del17p) hasta en un 40% de casos de LLC refractaria a los tratamientos quimioterápicos, por lo que la presencia de dicha alteración está asociada a una falta de respuesta

PONENCIAS 29
al tratamiento convencional y a un pronóstico infausto. Los inhibidores del BCR (al igual que os de BCL2) permiten un abordaje terapéutico en dichas poblaciones de pacientes.
Los fármacos más desarrollados son ibrutinib, inhibidor selectivo y co-valente de la tirosin-quinasa de Bruton (BTK), e idelalisib, un inhibidor de la isoforma delta de la kinasa PI3K.
En primera línea, Ibrutinib consigue unas altísimas tasas de respuesta global y una supervivencia libre de progresión (SLP) del 90% a los 30 meses. Su actividad es independiente del estado mutacional de las inmunoglobulinas y de la presencia de alteraciones de TP53. Como tratamiento de rescate también es un fármaco muy activo tanto en pa-cientes de bajo riesgo (SLP del 80% a los 3 años) como en aquellos con alteraciones de TP53 (SLP mediana de 28 meses). Por su lado, idelalisib combinado con rituximab presenta una excelente tasa de respuestas completas con una mediana de tiempo hasta la progresión superior a los dos años. De nuevo, se trata de una combinación activa frente a casos que presentan anomalías de TP53. Idelalisib se asocia a una mayor tasa de infecciones oportunistas y a fenómenos autoinmunes, en especial cuando se administra en primera línea.
Ibrutinib está indicado en el tratamiento de primera línea de pacientes con o sin comorbilidades, así como en el tratamiento de rescate de los pacientes con LLC o linfoma linfocítico. Idelalisib combinado con rituximab está indicado en el tratamiento de rescate, mientras que no se recomienda en primera línea por los efectos adversos comentados anteriormente.
Estos fármacos, a pesar de no considerarse quimioterapias, no están exentos de cierta toxicidad. Así, ibrutinib se asocia a un mayor riesgo de sangrado, fibrilación auricular e hipertensión arterial. Por su parte, idelalisib produce neumonitis,hepatitis y colitis de origen autoinmune así como una mayor reactivación de P. Jirovecii y citomegalovirus. El uso y contraindicaciones de estas drogas están perfectamente detallados en sus correspondientes fichas técnicas.
2.2. Inhibidores de BCL2
La LLC se caracteriza por una elevada expresión de la proteína BCL2 lo que le confiere una resistencia constitutiva a la muerte celular pro-gramada o apoptosis. La proteína BCL2 se caracteriza por poseer 4 dominios BH3y forma junto a MCL1 BCL-XL y otras las denominadas proteínas anti-apoptóticas. Por todo ello, una diana de enorme interés en esta enfermedad es el bloqueo del mecanismo de acción de BCL2.
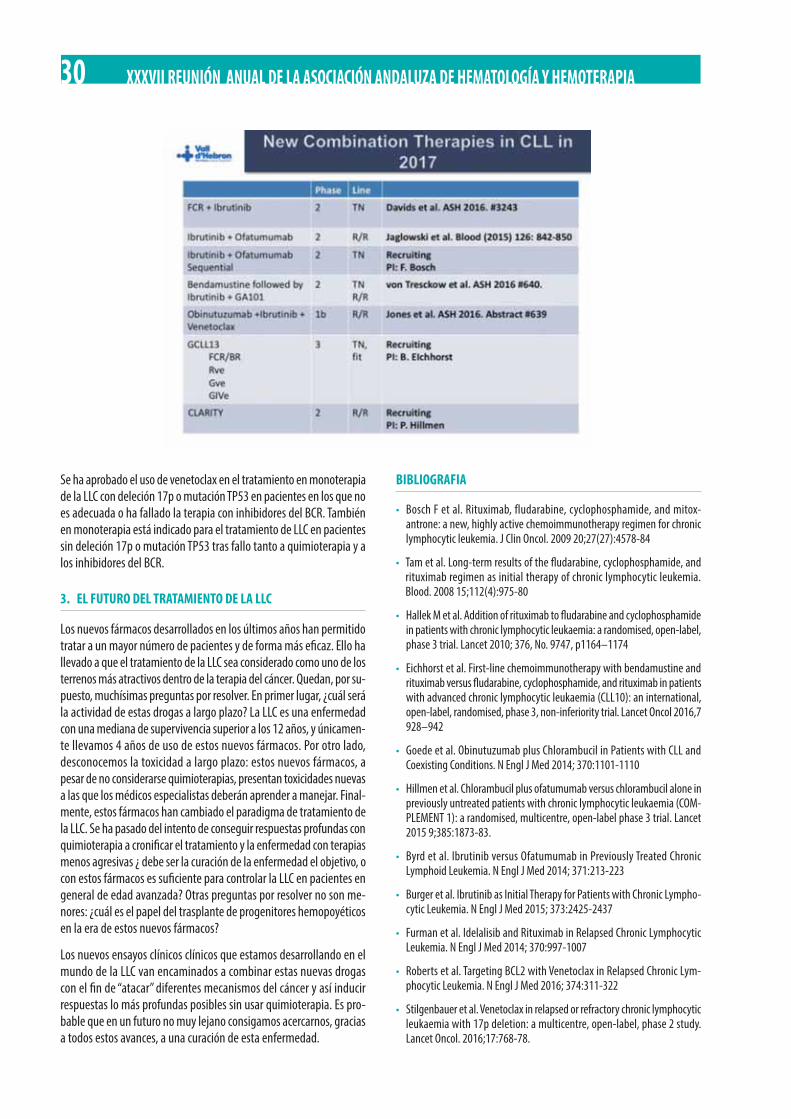
Venetoclax es una pequeña molécula de absorción oral que se une al sitio de unión de las proteínas proapoptóticas (BAX, BAD). Venetoclax es capaz de inducir muerte celular tumoral en cultivos tumorales, lí-neas celulares y en ratones transgénicos.
En el momento actual existen más de 400 pacientes que han sido tra-tados con venetoclax dentro de ensayos clínicos. Esta droga es muy activa toda vez que induce una rápida reducción de la carga tumoral. Ello se refleja en el riesgo aumentado de desarrollar un síndrome de lisis tumoral (SLT). Como consecuencia, los pacientes que reciben ve-netoclax deben, en primer lugar, estratificarse en el riesgo de SLT y tomar las medidas adecuadas en función del mismo. Posteriormente deben recibir venetoclax en dosis crecientes semanales, desde 20 mg/día hasta 400 mg/día. Con la clasificación del SLT y el escalado de dosis el riesgo de SLT es prácticamente inexistente.
Venetoclax se ha demostrado muy activo desde el punto de vista clí-nico. En pacientes muy tratados, la tasa de respuestas globales es del 80% con un 20% de RC. De forma interesante, dichas respuestas son profundas de tal suerte que se han descrito casos con ERM negativa.En esta población de pacientes muy tratados la mediana de duración de la respuesta es de alrededor de 25 meses. Al igual que las otras drogas, venetoclax es activo en pacientes con anomalías de TP53. Muy interesante es la combinación de venetoclax con rituximab, capaz de conseguir en ensayos clínicos hasta un 50% de RC y un 20% de ERM negativa. Su enorme actividad ha llevado a la designación de veneto-clax como breaktrough therapy designation en el tratamiento de la LLC.
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA30
Se ha aprobado el uso de venetoclax en el tratamiento en monoterapia de la LLC con deleción 17p o mutación TP53 en pacientes en los que no es adecuada o ha fallado la terapia con inhibidores del BCR. También en monoterapia está indicado para el tratamiento de LLC en pacientes sin deleción 17p o mutación TP53 tras fallo tanto a quimioterapia y a los inhibidores del BCR.
3. EL FUTURO DEL TRATAMIENTO DE LA LLC
Los nuevos fármacos desarrollados en los últimos años han permitido tratar a un mayor número de pacientes y de forma más eficaz. Ello ha llevado a que el tratamiento de la LLC sea considerado como uno de los terrenos más atractivos dentro de la terapia del cáncer. Quedan, por su-puesto, muchísimas preguntas por resolver. En primer lugar, ¿cuál será la actividad de estas drogas a largo plazo? La LLC es una enfermedad con una mediana de supervivencia superior a los 12 años, y únicamen-te llevamos 4 años de uso de estos nuevos fármacos. Por otro lado, desconocemos la toxicidad a largo plazo: estos nuevos fármacos, a pesar de no considerarse quimioterapias, presentan toxicidades nuevas a las que los médicos especialistas deberán aprender a manejar. Final-mente, estos fármacos han cambiado el paradigma de tratamiento de la LLC. Se ha pasado del intento de conseguir respuestas profundas con quimioterapia a cronificar el tratamiento y la enfermedad con terapias menos agresivas ¿ debe ser la curación de la enfermedad el objetivo, o con estos fármacos es suficiente para controlar la LLC en pacientes en general de edad avanzada? Otras preguntas por resolver no son me-nores: ¿cuál es el papel del trasplante de progenitores hemopoyéticos en la era de estos nuevos fármacos?
Los nuevos ensayos clínicos clínicos que estamos desarrollando en el mundo de la LLC van encaminados a combinar estas nuevas drogas con el fin de “atacar” diferentes mecanismos del cáncer y así inducir respuestas lo más profundas posibles sin usar quimioterapia. Es pro-bable que en un futuro no muy lejano consigamos acercarnos, gracias a todos estos avances, a una curación de esta enfermedad.
BIBLIOGRAFIA
Bosch F et al. Rituximab, fludarabine, cyclophosphamide, and mitox-antrone: a new, highly active chemoimmunotherapy regimen for chronic lymphocytic leukemia. J Clin Oncol. 2009 20;27(27):4578-84
Tam et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008 15;112(4):975-80
Hallek M et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010; 376, No. 9747, p1164–1174
Eichhorst et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 2016,7 928–942
Goede et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N Engl J Med 2014; 370:1101-1110
Hillmen et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COM-PLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 2015 9;385:1873-83.
Byrd et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med 2014; 371:213-223
Burger et al. Ibrutinib as Initial Therapy for Patients with Chronic Lympho-cytic Leukemia. N Engl J Med 2015; 373:2425-2437
Furman et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 2014; 370:997-1007
Roberts et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lym-phocytic Leukemia. N Engl J Med 2016; 374:311-322
Stilgenbauer et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17:768-78.

PONENCIAS 31
El linfoma folicular (LF) es el subtipo más frecuente de linfoma no Hodgkin indolente en nuestro medio. Aparece en individuos de edad media o avanzada y tiene una distribución según el sexo casi igual. En el momento del diagnóstico, la mayoría de pacientes con LF presenta enfermedad diseminada, incluyendo afectación de la médula ósea en más de un 40%, aunque muy frecuentemente están asintomáticos. La historia natural de los pacientes con LF se caracteriza por presentar un curso clínico indolente con una alta tasa de respuestas globales con cualquiera de los tratamientos y una supervivencia relativamente pro-longada. Después de la recaída inicial, tanto la tasa de respuesta como la supervivencia libre de progresión (SLP) después del tratamiento subsiguiente disminuyen de forma constante. La transformación a un subtipo histológico agresivo da como resultado un comportamiento clínico más agresivo.
El tratamiento convencional para el LF avanzado era la quimioterapia con agentes alquilantes, antibióticos antraciclínicos, análogos de las purinas, entre otros, bien fueran solos o en combinación. La introduc-ción del rituximab, anticuerpo monoclonal antiCD20, ha revolucionado el tratamiento de los linfomas, incluyendo al LF. De hecho, rituximab demostró actividad en LF recaído y/o refractario, y ésta fue su primera indicación autorizada por la FDA y la EMA.
En la primera línea del tratamiento del LF, cuatro grupos cooperati-vos diseñaron sendos estudios de fase 3 aleatorizados para evaluar la actividad de rituximab en combinación con varios esquemas de quimioterapia (CHOP, CVP, entre otros) y todos ellos demostraron beneficio en términos de SLP y supervivencia global (SG) a favor de los grupos de pacientes que recibían rituximab. Recientemente, dos estudios han mostrado la no inferioridad de la combinación rituximab-bendamustina frente a rituximab-CHOP en la primera línea del LF. Tras alcanzar respuesta con el tratamiento de inducción, se pueden iniciar estrategias terapéuticas para prolongar la respuesta, entre las que destaca el mantenimiento con rituximab. El estudio PRIMA evaluó en pacientes en respuesta tras inmunoquimioterapia el mantenimiento con rituximab durante 2 años frente a observación y demostró una mejoría significativa en la SLP, aunque por el momento no se han ob-servado diferencias en cuanto a la SG. En la actualidad, en pacientes tratados en primera línea con inmunoquimioterapia con rituximab, el tratamiento post-inducción con trasplante autólogo no ha demostrado beneficio en pacientes respondedores (RC o RP) y debiera limitarse a ensayos clínicos exclusivamente. Con la generalización del uso de rituximab, se ha desarrollado una nueva formulación que permite su administración subcutánea en tan solo 5-7 minutos, y se ha confirma-do su actividad y seguridad tanto en el contexto de inducción como en el mantenimiento del LF. También recientemente, se han presentado resultados preliminares de eficacia y seguridad de varios biosimilares de rituximab para administración endovenosa.
Recientemente, se ha evaluado la actividad de un nuevo anticuerpo anti-CD20 de tipo 2 denominado obinutuzumab en el tratamiento de primera línea del LF. En un estudio aleatorizado de fase 3 se ha demostrado que obinutuzumab más quimioterapia (CHOP, CVP o bendamustina) mejora la SLP respecto al tratamiento con rituximab y quimioterapia (HR 0,66 (IC95% 0,51-0,85), p=0,0012), ambos tra-tamientos seguidos de su mantenimiento correspondiente durante 2 años. Además, se aumenta el tiempo hasta el siguiente tratamiento en el grupo de pacientes con obinutuzumab y quimioterapia (HR 0,68 (IC95% 0,51-0,91), p=0,0094), y también aumenta el número de pa-cientes en los que no se detecta enfermedad residual mínima a favor de los casos que recibieron tratamiento con obinutuzumab. Reciente-mente se están incorporando a la primera línea de tratamiento del LF nuevos fármacos que no se consideran citostáticos, principalmente en combinación con rituximab. Así, la adición de lenalidomida a rituxi-mab en la inducción obtiene una mayor SLP, duración de la respuesta y tiempo al siguiente tratamiento con respecto a la administración de rituximab en monoterapia, y todo ello con un perfil de seguridad aceptable. Otros agentes están en estudio en estas combinaciones denominadas “chemo-free” (sin quimioterapia).
Tal y como ocurre en primera línea, no existe un consenso sobre cuál ha de ser el tratamiento de los pacientes con LF en la recaída. Las posibilidades terapéuticas van desde la abstención terapéutica hasta tratamientos intensivos, incluyendo estrategias de trasplante autólogo y alogénico. Es importante considerar si la recaída/progresión cumple criterios de refractariedad a la quimioterapia, al rituximab o a ambos, si bien las definiciones empleadas por los diferentes grupos para estas situaciones son múltiples y no hay una definición de consenso.
En la última década se está produciendo una auténtica revolución en el tratamiento de los síndromes linfoproliferativos, motivada princi-palmente por el gran avance en los conocimientos biológicos de los mismos, así como por las nuevas técnicas de bioingeniería en el de-sarrollo de fármacos. En el momento actual, hay múltiples fármacos en vía de experimentación preclínica y clínica que interfieren en las múltiples vías de señalización desreguladas en los linfomas y también que interaccionan con el microambiente. Un número cada vez mayor de fármacos se está evaluando ya sea solo o en combinaciones, inclu-yendo nuevos agentes citostáticos (palbociclib). Agentes inhibidores del proteasoma como bortezomib o carfilzomib están en investigación en el LF. Otros agentes más específicos incluyen anticuerpos monoclo-nales frente al CD20 (obinutuzumab, TRU-015) o frente otros antígenos (CD19, CD79a, CD22), bien sean desnudos o conjugados con fármacos o toxinas. Agentes que inhiben diversas vías celulares activadas por el receptor de células B, incluyendo la tirosina quinasa de bazo (SYK) y la tirosina quinasa de Bruton (BTK), y otras vías intracelulares tales como la diana mamífera de rapamicina (mTOR), PI3K y apoptosis. También, fármacos que se dirigen al microambiente tumoral, especialmente el agente inmunomodulador lenalidomida, o bien los nuevos inhibidores
Linfoma folicular: novedades terapéuticas en primera línea y en recaída/refractariedadDr. Antonio Salar
Servicio de Hematología. Hospital del Mar, Barcelona

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA32
del checkpoint están evaluándose en LF recaído o refractario. En la tabla 1 se enumeran los agentes en fase de investigación más avan-zada. El desarrollo de combinaciones de estos agentes debe basarse en la lógica científica con estudios correlativos para mejorar nuestra
comprensión de los mecanismos de acción y la resistencia de los fár-macos y la biología del tumor para mejorar aún más el resultado de los pacientes con LF.
TABLA 1. Nuevos agentes en el tratamiento del linfoma folicular
Diana Tipo Agentes
Proteasona Inhibidor proteasoma Bortezomib, Carfilzomib, NPI 00-52
Ciclo celular Inhibidor CDK 4 y 6 Palbociclib
CD19 MAb Medi-551, SAR3419
CD19 MAb biespecífico Blinatumomab
CD20 MAb Obinutuzumab, Ofatumumab, TRU-015
CD22 MAb Epratuzumab, Inotuzumab ozogamicin
CD79a MAb conjugado MMAE Polatuzumab
Desconocida IMID Lenalidomida
SYK Inhibidor de SYK Fostamatinib
Aurora Kinasa Inhibidor Aurora Kinasa A Alisertib
mTOR Inhibidor mTOR Temsirolimus, Everolimus
PI3K Inhibidor PI3K Idelalisib, Copanlisib, Duvelisib, TGR-1202,
INCB040093
BTK Inhibidor BTK Ibrutinib, Acalabrutinib, BGB-3111
BCL2 Antiapoptóticos Venetoclax
HDAC Inhibidor HDAC Vorinostat, Panobinostat, Belinostat,
Romidepsina
PD1/PD-L1 Inhibidor checkpoint Nivolumab, Pembrolizumab, Pidilizumab
CTLA-4 Inhibidor checkpoint Ipilimumab
MAb: anticuerpo monoclonal; IMID: Derivado inmunomodulador; HDAC: inhibidor deacetilasa de histonas.

PONENCIAS 33
El tratamiento adaptado a la respuesta metabólica en el linfoma de Hodgkin ha surgido como necesidad de reducir las toxicidades tardías graves asociadas ala radioterapia (RT) y la quimioterapia (QT). Esta estrategia permitiría reducir la intensidad del tratamiento en pacientes seleccionados por la obtención de una excelente respuesta precoz a la terapia primaria, y facilitaría la intensificación del tratamiento en los que obtienen una respuesta más pobre. El resultado de un PET precoz realizado tras dos ciclos de ABVD (adriamicina, bleomicina, vinblastine y dacarbacina) ha demostrado ser un fuerte predictor de la supervivencia libre de progresión (SLP) con un alto valor predictivo negativo (VPN) y un moderado valor predictivo positivo (VPP)1.
El concepto de tratamiento adaptado a la respuesta evaluada medi-ante PET interino se ha evaluado en varios ensayos clínicos para paci-entes con estadios iniciales y avanzados. Straus y cols2 realizaron un PET interino (PETi) después del segundo ciclo de ABVD en pacientes con estadios iniciales sin enfermedad bulky; aquellos en los que se observó negatividad en el PETi fueron tratados con dos ABVD adicio-nales y no recibieron RT. En los casos con PETi positivo la QT se cambió a eBEACOPP (bleomicina, etoposido, doxorubicina, ciclofosfamida, vincristina, prednisona, and procarbazina) por dos ciclos seguido de RT sobre campos afectados (IFRT). La SLP a 3 años fue del 92% y del 66% para las cohortes con PETi negativo y positivo, respectivamente. Recientemente se han publicado los resultados del ensayo RAPID3, en el que 602 pacientes con estadios IA y IIA fueron aleatorizados a recibir 3 ciclos de ABVD y fueron evaluados con PETi; aquellos con PETi nega-tivo no recibieron más tratamiento mientras que los pacientes con PETi + recibieron un cuarto ciclo de ABVD seguido de IFRT. El grupo control recibió 3 ciclos de ABVD y IFRT (no se evaluó la respuesta interina). La SLP a 3 años fue del 95% en el grupo control comparada con el 91% en el grupo PETi negative que no recibió RT. Aunque no se alcanzó el objetivo de no inferioridad de la estrategia de tratamiento adaptada al PETi, este estudio demostró que un significativo número de pacientes puede evitar la irradiación y alcanzar una excelente SLP. En el ensayo European Organisation for Research and Treatment of Cancer (EORTC)/Lymphoma Study Association (LYSA)/Fondazione Italiana Linfomi (FIL) HD104 se incluyeron 1950 pacientes con estadios iniciales favorables o desfavorables. La SLP a 5 años para los pacientes tratados exclusi-vamente con quimioterapia ABVD tras un PETi negativo fue del 87% (favorables) y del 89% (desfavorables); aunque la omisión de la RT en estos pacientes disminuyó significativamente la SLP, la supervivencia global no se afectó y cerca del 90% de los casos evitaron la RT. En am-bos estudios (RAPID y HD10) queda pendiente evaluar la supervivencia a largo plazo en relación a posibles efectos tardíos graves.
También se ha evaluado el papel del tratamiento adaptado a la respu-esta interina evaluada por PET en pacientes con enfermedad avanzada. En el ensayo RAHTL5 (Response-Adapted Therapy in Hodgkin Lymphoma
trial) 1214 pacientes recibieron 2 ciclos de ABVD seguido de evaluación de la respuesta mediante PETi. El 83.7% que obtuvo un PETi negativo fue aleatorizado a 4 ciclos más de ABVD o a 4 ciclos de AVD (omitiendo la bleomicina), sin administrar RT en ninguno de los dos grupos. La SLP fue del 85.7% y del 84.4% (supervivencia global 97.2% y 97.6%), respectivamente. La toxicidad fue inferior en el grupo tratado con AVD. En otros estudios, eBEACOPP fue utilizado como tratamiento inicial con intención de “desescalar” en los pacientes con PETi negativo. El grupo LYSA alatorizó a los pacientes con PETi negativo tras 2 ciclos de eBEACOPP a continuar con 4 ciclos más del mismo esquema o a 4 ciclos de ABVD. La SLP a dos años fue similar (94% vs 92%) y la toxicidad muy inferior en el grupo tratado con ABVD6.
Otros estudios han evaluado la intensificación del tratamiento en los pacientes con un PETi positivo. Press y cols han comunicado reciente-mente los resultados de un ensayo fase II (US Intergroup study S0816)7. Los pacientes fueron tratados con 2 ciclos de ABVD y sometidos a PETi; aquellos con PETi negativo (82%) recibieron 4 ciclos más de ABVD y los que presentaron PETi positivo (18%) continuaron con 6 ciclos de eBEACOPP. La SLP a 2 años para el grupo PETi + fue de un prometedor 64%. En el ensayo RATHL5 los pacientes con PETi positivo (16% del total) fueron tratados con 4 ciclos de eBEACOPP o con 6 ciclos de BEA-COPP-14 (a criterio del investigador), obteniendo una SLP a 3 años del 68% (supervivencia global, 87%) en este grupo de mal pronóstico. En pacientes con estadios iniciales, el ensayo EORTC/LYSA/HD104 aleator-izó a los pacientes con PETi + a continuar con 4 ABVD+IFRT o a recibir 4 ciclos de eBEACOPP+IFRT, resultando este último regimen en una SLP a 5 años estadísticamente superior (77% vs 91%).
Recientemente, Zinzani y cols han comunicado los resultados del en-sayo HD08018. En el que 103 pacientes con PETi + fueron tratados con quimioterapia de segunda línea (ifosfamida, gemcitabina, vinorelbina) seguido de transplante autologo de progenitores hematopoyéticos, alcanzando una SLP del 76% a 2 años (aunque el 20% de los pacien-tes no consiguió finalizar toda la terapia de rescate). Estos resultados podrían apoyar la realización de un estudio aleatorizado que evaluara el papel del transplante autologo en estos pacientes.
En conclusión, los datos agregados de los estudios comentados apoyarían la reducción de la intensidad del tratamiento en los pa-cientes con PETi negativo (aunque ningún ensayo ha alcanzado el objetivo de no inferioridad en los estadios iniciales, casi el 90% de los pacientes pueden evitar irradiarse) y la “escalada” del tratamiento en los pacientes con PETi + (especialmente en los estadios avanzados). Desafortunadamente, eBEACOPP es un esquema poco atractivo por su excesiva toxicidad a corto y largo plazo. Por ello, deben investigarse estrategias alternativas que incluyan los nuevos fármacos aprobados para el tratamiento del linfoma de Hodgkin recurrente o refractario (brentuximab vedotin, nivolumab, pembrolizumab).
Tratamiento adaptado a la respuesta metabólica evaluada por FDG/PET en linfoma de Hodgkin clásicoDr. Antonio Rueda Domínguez
Área de Oncohematología. Hospital Costa del Sol. Marbella

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA34
REFERENCIAS
1. Zinzani PL, Rigacci L, Stefoni V, et al. Early interim 18F-FDG PET in Hodg-kin’s lymphoma: evaluation on 304 patients. Eur J Nucl Med Mol Imaging 2012; 39: 4-12.
2. Straus DJ, Pitcher B, Kostakoglu L, et al. Initial results of US intergroup trial of response-adapted chemotherapy or chemotherapy/radiation therapy based on PET for non-bulky stage I and II Hodgkin lymphoma (CALGB/Alliance 50604). Blood 2015; 126: abstr 578.
3. Radford J, Illidge T, Counsell N, et al. Results of a trial of PET-directed therapy for early-stage Hodgkin’s lymphoma. N Engl J Med 2015; 372: 1598-607.
4. André MP, Girinsky T, Federico M, et al. Early Positron Emission Tomography Response-Adapted Treatment in Stage I and II Hodgkin Lymphoma: Final Results of the Randomized EORTC/LYSA/FIL H10 Trial. J Clin Oncol 2017; 14: JCO2016686394. doi: 10.1200/JCO.2016.68.6394.
5. Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med 2016; 374: 2419-29.
6. Casasnovas O, Brice P, Bouabdallah R, et al. Randomized phase III study comparing an early PET driven treatment de-escalation to a not PET-monitored strategy in patients with advanced stages Hodgkin lymphoma: interim analysis of the AHL2011 LYSA study. Blood 2015; 126: abstr 577.
7. Press OW, Li H, Schoder H, et al. US Intergroup trial of response-adapted therapy for stage III to IV Hodgkin lymphoma using early interim fluorode-oxyglucose-positron emission tomography imaging: Southwest Oncology Group S0816. J Clin Oncol 2016; 34: 2020-7.
8. Zinzani PL, Broccoli A, Gioia DM, et al. Interim positron emission tomog-raphy response-adapted therapy in advanced-stage Hodgkin lymphoma: final results of the phase II part of the HD0801 study. J Clin Oncol 2016; 34: 1375-85.

PONENCIAS 35
Son notorias la creciente participación de los medicamentos biologicos en el gasto farmacéutico, su impacto real sobre las cuentas públicas, las expectativas que generan para los pacientes con enfermedades graves o mortales y la importancia que estos tratamientos tienen para mejorar los resultados en salud.
Por ello, los medicamentos biologicos exigen gran atención por parte de los clinicos y no sólo desde la perspectiva asistencial, sino también desde su enfoque regulatorio.
Las especiales características de los biologicos, originales o similares, demandan una normativa específica, como así reconoce la Ley de Ga-rantias y Uso Racional de los Medicamentos y Productos Sanitarios.
Los puntos críticos a examinar son la autorización, la prescripcion por marca, la farmacovigilancia, la sustitución y la intercambiabilidad. También son cuestionees sensibles los condicionamientos al ejercicio de la función prescriptora y el desarrollo de la relacion medico-paci-ente, así como las eventuales responsabilidades que puedan derivarse en caso de inobservancia de la legislación aplicable.
Marco regulatorio de los Medicamentos BiológicosJulio Sanchez Fierro

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA36
La Leucemia Aguda Mieloblástica (LAM), es una enfermedad muy he-terogénea originada en la Stem Cell Hematopoyética (HSC), y caracte-rizada por un bloqueo en la diferenciación y una proliferación clonal incontrolada de precursores inmaduros neoplásicos, que sustituyen a la hematopoyesis medular normal.
La incidencia de LAM es de aproximadamente 3-5 casos/100.000 ha-bitantes/año, aumentando con la edad, lo cual sitúa la mediana de edad en 70 años, por lo que en su mayor parte podría considerarse una enfermedad de pacientes adultos mayores.
El pronóstico de la enfermedad es variable dependiendo de las caracte-rísticas clínicas del paciente (edad, comorbilidades, estado funcional), así como de las características biológicas de la leucemia, incluyendo las alteraciones genéticas y moleculares. Durante las 3 últimas décadas, de forma paralela al desarrollo de las modernas técnicas genómicas y de secuenciación masiva, se han producido importantes avances en el descubrimiento de numerosas lesiones genético-moleculares subyacentes y su implicación en la patogénesis de la LAM. Ello ha com-portado diversos cambios tanto en la clasificación de la LAM, como en el abordaje diagnóstico y terapéutico.
NOVEDADES EN CLASIFICACIÓN WHO 2016
Algunas de las alteraciones genéticas descritas se correlacionan con entidades clínico-patológicas diferenciales, lo cual ha quedado refleja-do en la actualización de la clasificación WHO 2008/2016 de neoplasias mieloides, que integra el diagnóstico citomorfológico, inmunofeno-típico y genético-molecular (Tabla 1)1,2. En dicha actualización se recogen algunos cambios en las categorías ya existentes, entre los que destacan la inclusión de 2 nuevas entidades consideradas provi-sionales: LAM con expresión BCR-ABL1 y LAM con RUNX1mut. Por su parte, LAM con NPM1mut y LAM con CEBPA bialélica mutada pasan a ser entidades definitivas, e independientemente de la presencia de displasia multilínea, y de la presencia de alteraciones cromosómicas asociadas.
Se mantienen los criterios de diagnóstico de LAM con cambios rela-cionados con mielodisplasia (presencia de displasia multilínea, he-mopatía mieloide previa, alteraciones citogenéticas relacionadas), aunque con algunas modificaciones en dichas alteraciones genéticas relacionadas (se ha excluido del9q..) Ver descripción en Tabla 1.
Aparece como nueva categoría la de Neoplasias Mieloides con predis-posición línea-germinal (Neoplasias Familiares Mieloides) que inclu-yen tanto LAM y SMD (Tabla 2). El reconocimiento de estos casos es de gran relevancia clínica, ya que además de cuidados clínicos y estudios biológicos especiales, dichos pacientes y familias van a requerir consejo genético especializado.
PANORAMA MOLECULAR.-
El descubrimiento de las numerosas lesiones genéticas y moleculares subyacentes e implicadas en la patogénesis de la LAM ha llevado a con-siderar hoy día la LAM como una enfermedad compleja3, multifactorial y dinámica, caracterizada por la adquisición de múltiples mutaciones somáticas iniciadoras (driver), donde coexisten clones competitivos entre si y que pueden evolucionar en el tiempo.
Empleando las tecnologías actuales, se ha descrito al menos una mu-tación somática en >95% de las LAM, con aproximadamente doce alteraciones genómicas por muestra leucémica, incluyendo una media de 3 mutaciones driver. Sin embargo, el perfil es notablemente hetero-géneo y sólo unas pocas alteraciones (FLT3, NPM1 Y DNMT3A) estarán presentes en más de un cuarto de las LAM. Dichas mutaciones gené-ticas han sido clasificadas hasta en 9 categorías funcionales4,5 (Tabla 3): 1) Fusiones entre genes codificantes de factores de transcripción, 2) NPM1; 3) Genes supresores de tumores; 4) Genes relacionados con la metilación del DNA; 5) Genes en vías Señalización y Receptores ti-rosin-kinasas; 6) Genes modificadores de la cromatina; 7) Mutaciones en genes de factores transcripción mieloides; 8) Genes del complejo cohesina; y 9) Genes del complejo spliceosoma.
Sin embargo, recientemente6 se han añadido otras 3 nuevas categorías heterogéneas con un fenotipo clínico y pronóstico diferenciado: a) LAM con mutaciones en la cromatina y en genes reguladores RNA-splicing; b) LAM con TP53mut y/o aneuplodías cromosómicas; c) y provisonal-mente LAM con IDH2R172 mut. (Figura 1).
Los estudios de evolución clonal en pacientes y en modelos murinos indican que las mutaciones en genes implicados en la regulación de la modificación DNA y cromatina (DNMT3A, TET2 y ASXL1) están a menudo presentes en progenitores considerados como preleucémi-cos y que ocurren precozmente en el desarrollo de la leucemogénesis. Tales mutaciones pueden persistir después del tratamiento y llevar a la expansión clonal durante la remisión y ocasionar la recidiva de la enfermedad.
Por tanto, actualmente parece razonable incluir entre las pruebas diag-nósticas (además del cariotipo convencional y/o FISH para detección de reordenamientos habituales7) los siguientes estudios mutacionales, como se recoge entre las recomendaciones del ELN 20178 y en la Plata-forma Diagnóstica Pethema-2017: a) NPM1, CEBPA y RUNX1, porque definen entidades diferenciales; b) mutaciones de FLT3 (DTI y su Ratio alélico, D835 e I836, no sólo por el pronóstico, sino porque pueden beneficiarse de Inhibidores T-K; c) TP53 y ASXL1 por conllevar un muy mal pronóstico con las terapias convencionales e incluso tras Alo-TPH.
Implicaciones pronósticas y terapéuticas de las alteraciones genético-moleculares en LAM: recomendaciones en 2017Dres. Josefina Serrano, C. Martínez-Losada, J. Serrano-López y Joaquín Sánchez
Servicio Hematología. Hospital Universitario Reina Sofía. IMIBIC. Córdoba

PONENCIAS 37
TABLA 1. Clasificación OMS 2016 para leucemias mieloides agudas
I. Leucemias mieloides agudas con alteraciones genéticas recurrentes.
RUNX1-RUNX1T1
LAM con NPM1 mutada
LAM con CEBPA bialélica mutada
Entidad provisional: LAM con BCR-ABL1
LAM con RUNX1 mutada
II. Leucemia mieloide aguda con cambios relacionados con mielodisplasia*.
III. Leucemias mieloides agudas relacionadas con tratamientos previos.
IV. Leucemias mieloides agudas no incluidas en otras categorías.
V. Sarcoma granulocítico
VI. Proliferación mieloide relacionada con S. Down
VII. Neoplasia células dendríticas blástica plasmocitoide
VIII. Leucemia agudas de línea ambigua
(*): Deben mostrar: 1) historia de SMD o SMD/SMP; 2) alteraciones citogenéticas relacionadas con SMD (3q-, -5, 5q-, -7, 7q-, -13, 13q,
11q-, 12p-, t(11;16); t(3;21); t(1;3), t(5;7), t(2;11), t(5;12), t(5;10); t(3;5) y cariotipos complejos; 3) displasia en ≥ 50% células en
al menos 2 líneas mieloides.
FACTORES PRONÓSTICOS:
La caracterización molecular no sólo permite una subclasificación más detallada, sino que contribuye a una mejor definición del pronóstico en un mayor número de pacientes proporcionando las bases para un tratamiento más individualizado y dirigido hacia dichas dianas moleculares.
A lo largo de los años se han dedicado muchos esfuerzos a tratar de de-finir factores pronósticos9 en una enfermedad tan heterogénea como
la LAM, con objeto de adaptar el tratamiento al riesgo y curabilidad de la enfermedad en un intento de minimizar la toxicidad.
El manejo actual8 de pacientes afectos de LAM está determinado por una serie de parámetros pre-tratamiento que incluyen:
a) Factores relacionados con el paciente: fundamentalmente la edad, asociada de manera independiente a peor pronóstico; comorbilida-des clínicas específicas y estado general del paciente que modulan la tolerancia a la quimioterapia, así como presencia de alteraciones
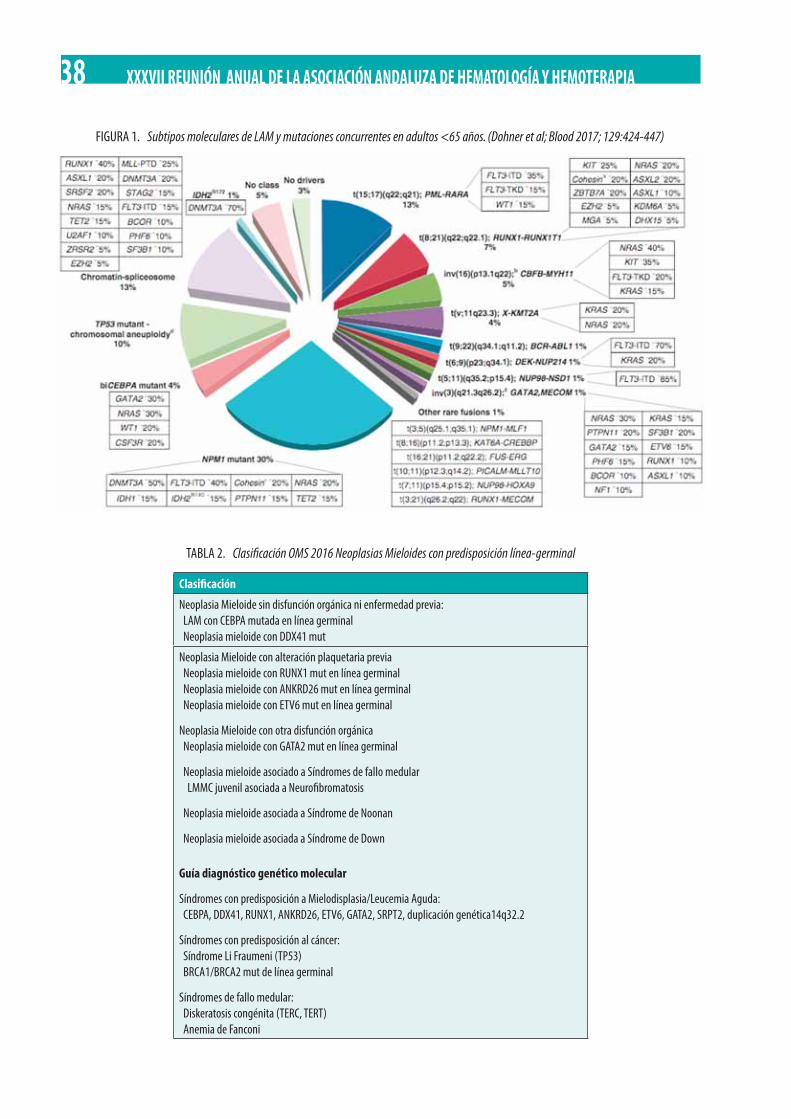
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA38
TABLA 2. Clasificación OMS 2016 Neoplasias Mieloides con predisposición línea-germinal
Clasificación
Neoplasia Mieloide sin disfunción orgánica ni enfermedad previa:
LAM con CEBPA mutada en línea germinal
Neoplasia mieloide con DDX41 mut
Neoplasia Mieloide con alteración plaquetaria previa
Neoplasia mieloide con RUNX1 mut en línea germinal
Neoplasia mieloide con ANKRD26 mut en línea germinal
Neoplasia mieloide con ETV6 mut en línea germinal
Neoplasia Mieloide con otra disfunción orgánica
Neoplasia mieloide con GATA2 mut en línea germinal
Neoplasia mieloide asociado a Síndromes de fallo medular
LMMC juvenil asociada a Neurofibromatosis
Neoplasia mieloide asociada a Síndrome de Noonan
Neoplasia mieloide asociada a Síndrome de Down
Guía diagnóstico genético molecular
Síndromes con predisposición a Mielodisplasia/Leucemia Aguda:
CEBPA, DDX41, RUNX1, ANKRD26, ETV6, GATA2, SRPT2, duplicación genética14q32.2
Síndromes con predisposición al cáncer:
Síndrome Li Fraumeni (TP53)
BRCA1/BRCA2 mut de línea germinal
Síndromes de fallo medular:
Diskeratosis congénita (TERC, TERT)
Anemia de Fanconi
FIGURA 1. Subtipos moleculares de LAM y mutaciones concurrentes en adultos <65 años. (Dohner et al; Blood 2017; 129:424-447)

PONENCIAS 39
TABLA 3. Mutaciones recurrentes en LAM clasificadas por grupos funcionales
Grupo funcional Mutación Frecuencia
Vías Señalización/Kinasas FLT3, KRAS, NRAS, KIT, PTPN11 y NF1 >60%
Modificadores Epigenéticos:
- Metilación DNA
- Modificadores Cromatina
DNMT3A, IDH1, IDH2, TET2, ASXL1, EZH2 y MLL/
KMT2A>50%
Nucleofosmina NPM1 >30%
Factores Transcripción CEBPA, RUNX1 y GATA2 20-25%
Supresores Tumores TP53 5-15%
Complejo Spliceosoma SRSF2, U2AF1, SF3B1 y ZRSR2 10%
Complejo Cohesina RAD21, STAG1, STAG2, SMC1A y SMC3
genéticas relacionadas con LAM de pacientes mayores que com-portan mayor resistencia al tratamiento, (asociadas SMD/SMPc ó exposición a citotóxicos previos).
b) Factores relacionados con la leucemia: las alteraciones genético-moleculares detectables en el clon leucémico parecen influenciar notablemente la respuesta ó resistencia al tratamiento. La cla-sificación pronóstica ha sufrido algunas modificaciones, según quedan reflejadas en las recomendaciones del panel de expertos de la ELN 2017, respecto a 2010 (Tabla 4). Aquí se recoge que tanto las mutaciones en RUNX1 y en ASLX1 son más frecuentes en pacientes mayores y comportan un peor pronóstico, al igual que las mutaciones en TP53 que se asocia frecuentemente a cariotipo complejo, monosómico ó con aneuploidías cromosómicas.
Sin embargo, el impacto pronóstico de ciertas mutaciones depende de la presencia/ausencia de otras, como es el caso de la interacción entre NPM1 y FLT3-DTI. Presentan un pronóstico favorable sólo los pacientes con NMP1mut en ausencia de FLT3-DTI ó con Ratio alélico bajo (Ratiolow), mientras que se catalogan de riesgo adverso aquellos con FLT3-DTI Ratiohigh con NPM1wt. Incluso en recientes estudios5 se han identificado firmas genéticas que parecen comportar neoplasia mieloides de muy alto riesgo (t-LAM, SMD/SMPc) donde se suman mutaciones en RNA spliceosoma, en cromatina ó en genes reguladores de la transcripción (SRSF2, ASXL1, RUNX1, …).
También, en LAM-CBF se han asociado mutaciones cooperantes, que bien podrían empeorar el pronóstico (KIT con alta expresión) y otras cuyo significado es aún más incierto, como la suma de mutaciones en genes señalizadores (NRAS, KRAS, NF1) frecuentes tanto en t(8;21) como en inv(16) (Figura 1).
Mientras que otros marcadores genéticos carecen de significado pro-nóstico conocido hoy en día, pero su importancia radica en la posibili-dad de emplear terapias diana, como con IDH1, IDH2, y KMT2A (MLL).
Factores post-tratamiento: La monitorización de EMR, medida tanto por CMF como por técnicas moleculares, supone una herramien-ta de gran utilidad en el periodo post-remisión, ayudando a la toma de decisiones terapéuticas tanto pre como post-TPH. La profundidad de la respuesta proporciona una valiosa información que debe ser realizada en laboratorios experimentados.
TATRAMIENTO ACTUAL:
A lo largo de los años se han dedicado muchos esfuerzos a tratar de definir factores pronósticos en una enfermedad tan heterogénea como la LAM, con objeto de adaptar el tratamiento al riesgo y curabilidad de la enfermedad en un intento de minimizar la toxicidad.
Sin embargo, durante las últimas 4 décadas el tratamiento de la LAM ha sufrido pocos cambios. Mientras que 2/3 de los adultos jóvenes alcanzan la RC, menos de la mitad de los mayores lo consiguen y sólo el 10% se curarán. Ello es consecuencia de una peor tolerancia al tra-tamiento estándar y superior tasa de resistencias, de citogenéticas adversas y de LAM secundarias en estos pacientes mayores. Sin em-bargo, la edad por sí misma no debe ser la razón para no administrar tratamiento intensivo.
La implementación en los criterios de valoración empleados para con-siderar un paciente como candidato a tratamiento de quimioterapia intensiva constituye una herramienta fundamental sobre todo en los pacientes mayores (>65-70 años). Debido en parte a la mejora de los cuidados de soporte y del estado general de salud en los pacientes ma-yores, diversos estudios han mostrado un beneficio en pacientes con-siderados “fit” del tratamiento intensivo ó activo, frente al tratamiento de soporte/paliativo en términos de calidad de vida y supervivencia. Para la consideración “unfit” no existen criterios firmes, recomendán-dose valorar sólo el mal estado general del paciente, comorbilidades significativas y presencia de alteraciones genético-moleculares ad-

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA40
versas, ya que en estos casos el beneficio del tratamiento no supera al riesgo.
A) Tratamiento Intensivo de inducción a la Remisión
La quimioterapia de inducción estándar incluye un agente ciclo espe-cífico, citarabina 100-200 mg/m2, administrada en infusión continua 7 días, combinada con un antibiótico antracíclico no ciclo-celular es-pecífico (daunorubicina/Idarubicina) durante 3 días. Con este régimen la tasa de RC oscila entre el 60% y el 80% y de SG 40-60%, según los grupos de riesgo. La decisión de tratar debe ser inmediata, ya que un retraso de más de 5 días en el inicio del tratamiento parece compro-meter el pronóstico en pacientes <60 años.
Se han intentado diversas estrategias para mejorar dichas tasas de RC, que incluyen el empleo de altas dosis de antracíclicos ó de citarabina, adición de un tercer agente citotóxico como el etopósido, fludarabina ó topotecan; uso de factores de crecimiento (citoquinas), como “pri-mado” para reclutar células leucémicas en ciclo celular, lo cual las vol-vería más sensibles a la quimioterapia citotóxica. Sin embargo, para la mayoría de los pacientes, ninguna de estas estrategias ha demostrado
beneficios respecto al régimen estándar 3+7, con dosis de al menos 60mg/m2 de Dauno y 12 mg/m2 Ida.
Papel de nuevos fármacos:
a) Inhibidores FLT3. Son fundamentalmente inh. tirosin-cinasas ó serin-treonin-cinasas. FLT3 pertenece a la familia de receptores tirosin-cinasa clase III y juega un papel importante en la prolife-ración y diferenciación hematopoyética, expresándose tanto en progenitores hematopoyéticos normales como en los blastos de LAM. Debido al impacto negativo de dichas mutaciones, se han ensayado estudios clínicos con diversos inhibidores de primera generación: PKC-412 (Midostaurin), CEP-701 (Lestaurtinib), MLN-518 (Tandutinib), SU11248 (Sunitinib), Bay 43-9006 (Sorafenib), los cuales inactivan también otras tirosin-cinasas (c-Kit, VEGF, PDGFR, ABL, etc), produciendo un bloqueo en la proliferación e induciendo apoptosis. Los ensayos en fase I-II con estos fármacos, administrados vía oral y en monoterapia, reportan sólo respues-tas transitorias con disminución de blastos en médula ósea y/o sangre periférica. Lestaurnitib en combinación con quimioterapia también falló en demostrar una mejora en la SG. Sin embargo,.
TABLA 4. Clasificación pronóstica genético/molecular en LAM según ELN 2017
Pronóstico Alteración Genética Alter. Molecular
FAVORABLE t(15;17)(q22;q12-21) PML-RARA
t(8;21)(q22;q22) RUNX1-RUNX1T1
inv(16)(p13q22)/(t(16;16)(p13;q22)
CBFB-MYH11
NPM1+/FLT3-DTI (-) o low
CEBPA bialélica
INTERMEDIOCariotipo Normal
NPM1-/FLT3- o low
NPM1mut/FLT3-DTI high
Citogenéticas no incluidos en grupos favorable y adverso
t(9;11)(p22;q23) MLLT3-KMT2A
ADVERSO - Cariotipo complejo (≥3 alt. Crom, no CBF)
- Mutaciónes balanceadas:
inv(3)(q21;q26) t(3;3)(q21;q26)
t(6;9)(p23;q34)
t(v;11)(;q23)
t(9;22)(q34.1;q11.2)
- 5; del(5q); -7; -17/abn(17p)
- Cariotipo Monosómico
FLT3-DTI high/NPM1wt
GATA2, MECOM (EVI1)
DEK-NUP214
KMT2A reorden
BCR-ABL1
RUNX1 mut
ASXL1 mut
TP53 mut

PONENCIAS 41
recientemente se han publicado los resultados del estudio ran-domizado fase III multicéntrico internacional (RATIFY) en el que participan entre otros, los grupos españoles PETHEMA Y CETLAM, comparando quimioterapia intensiva con/sin Midostaurin. Dicha asociación aumenta las tasas de RC y de SG en pacientes <60 años con LAM con FLT3 mutado, en primera línea.. Por tanto, en Febrero 2016 la FDA aprobó en fármaco en esta indicación y se abrió un programa global de uso compasivo, siendo recomendable su uso actualmente.
También se han comunicaron resultados prometedores con otros inhibidores de segunda generación más selectivos: Quizartinib (AC220), Crenolanib, Gilteritinib (ASP2215), los cuales han mos-trado actividad en ensayos fase 1-2 en pacientes con LAM mutFLT3 rec/ref tanto en monoterapia como en combinación con quimio-terapia. Actualmente están en marcha diversos Fase III de dichos fármacos en combinación con quimioterapia en pacientes de novo.
b) Gentuzumab Ozogamicina (GO) es un anticuerpo monoclonal humanizado anti-CD33, unido a un potente citotóxico, la caliquea-micina. Aunque inicialmente varios estudios no consiguieron de-mostrar ventajas en SG del uso de GO asociado a quimioterapia, si sugerían que estaba asociado a una mayor RFS en pacientes de riesgo favorable (CBF-LAM) y en algunos de riesgo intermedio. Por tanto, hoy en día sólo está disponible en ensayos clínicos y en uso compasivo en USA.
c) CPX-351. Asociación sinérgica con una ratio 5:1 molar de citara-bina/daunorubicina en liposomas encapsulados. Los estudios en fase 2 sugerían beneficio en primera línea de pacientes con LAMt y secundaria. Y en un posterior fase 3 en pacientes mayores con LAM alto riesgo, el CPX-351 mostró > tasa de respuestas (CR/CRi 47% vs 33%) y de SG, independientemente del Alo-TPH.
B) Estrategias post-remisión
El 90% de los pacientes en RC recaerían en el plazo de 4-6 meses si el tratamiento no continuara tras la inducción. Todos los tipos de qui-mioterapia post-remisión (consolidación, intensificación) prolongan la duración de la remisión, pero durante las últimas décadas se ha demostrado que las mejores tasas de SLE y posible curación se obtienen con una terapia post-remisión corta con 2-3 ciclos de máxima intensidad que contengan uno o dos ciclos de ARA-C a altas dosis +/- Auto-TPH, y en los casos de pronóstico intermedio-adverso un trasplante Alogénico de progenitores hematopoyéticos (Alo-TPH) como terapia final. Con dicha estrategia, de quimioterapia de conso-lidación intensiva, actualmente sabemos que podemos conseguir más del doble de supervivencia pero aumentando la morbi-mortalidad, habiéndose obtenido beneficio, por tanto, sólo en los adultos jóvenes, no así en los mayores.
Sin embargo, el objetivo en los últimos años comprende fundamental-mente la adaptación del tratamiento a los distintos grupos de riesgo citogenético/molecular y de respuesta al tratamiento (Tabla 5). Así, hoy día nadie duda que el TPH alogénico es el único procedimiento po-tencialmente curativo en LAM primariamente refractaria, al igual que en los casos de recidiva, así como en los casos de riesgo desfavorable.
Por el contrario, carece de indicación en pacientes en primera remisión completa con LAM de bajo riesgo, mientras que persiste la polémica en pacientes con citogenética de riesgo intermedio. Así, generalmente se recomienda el TPH alogénico en pacientes cuya tasa de recaída esperable sea superior al 35%.
En cualquier tipo de donante (familiar o no emparentado), puede reducirse la intensidad de la dosis del acondicionamiento (el llamado mini-alotrasplante o no mieloablativo), con la intención de aprovechar el efecto injerto contra leucemia (ICL), reduciendo la toxicidad del pro-cedimiento en pacientes de edad avanzada o mala situación clínica. El empleo de índices de comorbilidad y de riesgo de forma conjunta proporcionan una guía de gran utilidad en la toma de decisiones de forma individualizada.
C) Pacientes no candidatos a tratamiento intensivo:
En pacientes mayores (probablemente ≥65-70) con ECOG>2, comorbi-lidades clínicas significativas y/o genética desfavorable y considerados no candidatos a terapia intensiva, se debería emplear un tratamiento alternativo que oscila entre el cuidado de soporte, de baja intensidad y la inclusión en ensayos clínicos con drogas investigacionales (Tabla 6).
Destacamos el empleo de Terapias Epigenéticas, por encontrarse en fases más avanzadas de desarrollo clínico:
– Hipometilantes: tanto Decitabina como Azacitidina han mostrado superioridad en términos de SG frente al tratamiento estándar, en estudios fase III, siendo esta última especialmente ventajosa en pacientes con citogenética adversa y alteraciones relacionadas con mielodisplasia. Se requieren hasta 6 ciclos para observar la máxima respuesta, y parecen alterar la historia natural de la enfermedad sin alcanzar remisión completa. Además existen ya hipometilantes de 2ª generación como Guadecitabina (SGI-110), en fase 3 de desarrollo.
– Inhibidores IDH1-IDH2, se han mostrado eficaces en estudios preco-ces con respuestas prometedoras y están actualmente en desarrollo clínico tanto en monoterapia como en asociación con hipometilantes y con Venetoclax (inh Bcl-2), entre otras.
BIBLIOGRAFIA
1. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revission of the World Health Organization (WHO) classification of myeloid neoplasms anda cute leucemia: rationale and important changes. Blood 2009; 114 (5):937-951.
2. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision tothe WHO clas-sification of myeloid neoplasms and and acute leukemia. Blood 2016; 127 (20): 2391-2405.
3. Torres A, García-Castellanos JM, Serrano López J, y col. Leucemia Agudas. Medicine 10ª Serie 2008; 10:1390-401.
4. Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment out-come in cytogenetically normal acute myeloid leucemia. N Engl J Med. 2008;358:1909-1918.
5. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374:2209-2221.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA42
TABLA 5. Resultados comparativos y toxicidad asociada al procedimiento tras Trasplante Autólogo y Alogénico de Progenitores Hematopoyéticos en LAM
QT/Auto-TPH Alo-TPH/Fam Alo-TPH/DNE Alo-Intensidad Reducida
1ªRC
C. Favorable
t(15;17)
CBF
NPM1/CEBPA
No indicación
SLE 60-80%
MRT 4-8%
No indicación
No indicación
No indicación
No indicación
No indicación
No indicación
C. Intermedio SLE 42-55%
MRT 4-6%
SLE 48-62%
MRT 16-20%
C. Desfavorable SLE 18-25%
MRT 4-8%
SLE 34-45%
MRT 18-20%
SLE 30-40% (5años)
MRT 30%
SLE 50%
(2 años)
Rescate
2ªRC SLE 30%
SLE 60-80%, en t(15;17)
SLE 40% Pediátrica 40%
Adultos SLE 30%
MRT 30%
SLE 40-50%
(2 años)
Recaída No indicación SLE 20-30% Pediátrica 20%
Adultos SLE 10%
(5 años)
SLE 10-30%
(2 años)
Fracaso Inducción No indicación SLE 30-40% (3 años)
20% LAM 2aria
SLE 20-30% SLE 15-30% (1 año)
6. DiNardo CD and Cortes JE. Mutation in AML: prognostic and therapeutic implications. Hematology AM Soc Hematol Educ Program. 2016: 348-355
7. Döhner H, Estey EH, Amadori S, et al. Diagnosis and manegement of acute myeloid leucemia in adults: recommendations from an internac-ional expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115:453-474.
8. Döhner H, Estey EH, Grimwade D, et al. Diagnosis and manegement of AML in adults: 2017 ELN recommendations from an internacional expert panel. Blood 2017; 129:424-447.
9. Grimwade D and Hills RK, Moorman AV et al. Refinement of cytogenetic classification in AML: determination of prognostic significace of rare recu-rring chromosomal abnormalities amongst 5635 younger adults treated in the UK MRC trials. Haematologica/Haematology J. 2009; 94:217.

PONENCIAS 43
TABLA 6. Nuevas terapias en desarrollo clínico en LAM
Mecanismo
A) Inh. Tirosin-Kinasas . Inh FLT3 (Midostaurin, AC220, Gilteritinib, Crenolanib)
- Inh KIT
- Inhib. PI3K/AKT/m-TOR
- Inhib. Aurora-cinasa, CDK4/6, MPS1
B) Moduladores Epigenéticos - Inh. DNA-Metiltransferasa (Azacitidina/Decitabina y Guadecitabina
- Inhibidores Histona-Deacetilasa
- Inh. IDH1 e IDH2
- Inh. BET-bromodominio, DOT1L
C) Inductores Apoptosis/Inh mitocondriales - Inh. Bcl-2, Bcl-xL y Mcl-1
D) Terapias dirigidas al microambiente - Antiangiogénicos
- Antagonistas CXCR4 y CXCL12
E) Citotóxicos:
- CPX-351
- Vosaroxin
- Análogos Nucleósidos
F) Inmunoterapia:
- Anticuerpos Monoclonales anti-CD33, Cd44, CD47, CD123, CLEC12A
- Inmunoconjugados (GO, SGN33A)
- BITEs y DARTs
- CAR T cells
- Inh. Checkpoint inmunes (PD-1/PD-L1, CTLA-4)
- Anticuerpos Anti-KIR
- Vacunación Células Dendríticas

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA44
Los síndromes mielodisplásicos (SMD) comprenden un conjunto de in-suficiencias medulares crónicas relativamente frecuentes en la práctica diaria. En general se manifiestan en personas mayores de 50 años de edad, suelen expresarse en forma de mono, bi o pancitopenia con alte-raciones morfológicas y dishemopoyéticas que en un porcentaje variable (20-30%) evolucionan a una leucemia aguda (LA).
Los SMD son un grupo heterogéneo de neoplasias hematopoyéticas clonales caracterizadas por una diferenciación y maduración celular anómala. Cursan, generalmente, con pancitopenia, médula normo o hipercelular y con una hematopoyesis intensamente alterada a nivel morfológico y/o funcional. Presentan un curso clínico variable, con una supervivencia mediana inferior a 2 años que está en estrecha relación con el subgrupo de SMD y, frecuentemente, evoluciona a leucemia aguda (Greenberg y cols., 1997).
Los SMD se deben a una alteración clonal de las células hematopo-yéticas pluripotentes, según se ha podido estudiar con técnicas de biología molecular. Actualmente, se acepta que los SMD se originan como consecuencia de la acumulación de sucesivas lesiones genómicas de las células germinales hematopoyéticas.
El diagnóstico es, esencialmente, morfológico y se basa en la presencia de rasgos displásicos en sangre y medula ósea. Los SMD inciden princi-palmente en la población adulta, por encima de los 50 años de edad.
Los estudios citogenéticos proporcionan una importante ayuda pronóstica. En la mitad de los pacientes se observan alteraciones ci-togenéticas, y pueden alcanzar el 80% en los SMD secundarios. La mitad de las anomalías corresponden a hiperdiploidías y el resto son hipodiploidías y cariotipos complejos. La detección de translocaciones equilibradas es muy poco frecuente, pero es muy importante para en-contrar nuevos genes implicados en esta enfermedad (Tabla 1 y 2).
Las principales alteraciones citogenéticas son: deleción 5q, monosomía 7 o deleción 7q, trisomía 8, deleción 20q, deleción 11q, deleción 12p,… Además, las principales anomalías citogenéticas presentan una importante correlación citológica.
La detección de alteraciones citogenéticas es un hallazgo importante para el diagnóstico final y pronóstico de la patología, sin embargo, sólo un 50% de los casos presentan alteraciones cromosómicas, de forma que el 50% restante (que presentan un cariotipo normal) requieren de técnicas diagnósticas adicionales como la hibridación in situ fluo-rescente (FISH), para poder determinar la posible presencia de altera-ciones cromosómicas. La aplicación de técnicas adicionales en aquellos casos que presentaban un cariotipo complejo, normal o sin divisiones ha demostrado que se puede detectar hasta en un 15% de los casos alteraciones crípticas, no detectadas por citogenética convencional.
Una de las alteraciones citogenéticas más frecuentes en los SMD de novo es la deleción intersticial del brazo largo del cromosoma 5, su-
Citogenética y estado mutacional en síndromes mielodisplásicosFrancesc Solé
Institut de Recerca contra la Leucèmia Josep Carreras (IJC). Campus ICO-GTIP (Badalona). Barcelona
TABLA 1. Principales anomalías citogenéticas
Más frecuentes: Otras alteraciones:
del(5q) t(1;3)(p36;q21)
del(7q) y –7 t(1;7)(p11;p11)
+8 t(2;11)(p21;q23)
i(17)(q10) del(9)(q13q22)
del(20)(q11q13) 11q-, del(11q)
-Y +11
12p-, del(12p)
+13
13q-
-18/18q-
-20
+21
TABLA 2.Principales anomalías citogenéticas con utilidad para diagnóstico
diferencial
Alteraciones citogenéticas sugestivas de SMD en casos con citopenia
persistente pero en ausencia de rasgos morfológicos definitivos de SMD:
-5/5q- t(11;16)(q23;p13)
-7/7q- t(3;21)(q26;q22)
i(17)(q10), del(17p) t(1;3)(p36;q21)
-13, del(13q) t(2;11)(p21;q23)
del(12p) inv(3)(q21q26)*
del(9q) t(6;9)(p23;q34)*
idic(X)(q13)
Complejo con alguna de las alteraciones anteriores
* Dichas alteraciones en la nueva versión de la WHO se consideraran leucemias agudas
a pesar de no presentar el paciente un 20% de blastos.
poniendo un 10-15% de los SMD (Solé y cols., 2005; Haase y cols., 2007; Mallo y cols., 2011; Schanz y cols., 2012). La deleción de 5q puede presentarse como alteración aislada o asociada a alteraciones citogenéticas. El 5q- como única alteración junto con rasgos carac-terísticos en médula ósea constituye una nueva entidad, reconocida

PONENCIAS 45
por la OMS, dentro de los SMD, como “SMD con del(5q) aislada“. El término “Síndrome 5q-“ se utiliza para designar un subgrupo de casos con anemia macrocítica, plaquetas normales o elevadas e hipoplasia eritroide en médula ósea.
Actualmente, hay un nuevo fármaco en el mercado, la lenalidomida, dirigido a aquellas personas con SMD y que como alteración citogené-tica presentan un 5q-. Ensayos clínicos han demostrado que el fármaco es eficaz en SMD con 5q- como única alteración y acompañada de otras anomalías citogenéticas. La lenalidomida ha demostrado ser muy ac-tiva en pacientes con SMD portadores de la deleción 5q, dando una independencia transfusional y una respuesta citogenética en el 70% de los pacientes tratados.
En la tabla 3 se detalla un resumen de los cambios más importantes que incorporará la WHO referente a los SMD.
CITOGENÉTICA CONVENCIONAL E HIBRIDACIÓN IN SITU FLUO-RESCENTE (FISH)
El 50% de los pacientes diagnosticados de SMD presentan alteraciones citogenéticas. La determinación de estas anomalías se lleva a cabo, inicialmente, mediante la técnica de citogenética convencional que permite la visualización de todos los cromosomas. En ocasiones, el resultado es el de un cariotipo normal, en dichos casos se procede a la aplicación de la técnica de FISH para detectar alteraciones citoge-néticas específicas en núcleos en interfase (Haferlach y cols., 2008; Mallo y cols., 2008). La Tabla 1 muestra las principales alteraciones citogenéticas de los SMD.
Microarrays genómicos
Los microarrays genómicos se utilizan para el estudio de alteracio-nes en el número de copias de ADN o para el estudio del genotipo mediante cambios en un único nucleótido (SNP, Single Nucleotide
MDS with excess blasts
* MDS-SLD with pancytopenia or 1% circulating blasts (x2) should be considered MDS-U; MDS-RS and MDS-MLD with ring sideroblasts include either >=15% ring-sideroblasts or any ring-sideroblasts and SF3B1 mutation; MDS with isolated del(5q) allows one additional but non-high-risk chromosomal abnormality.
TABLA 3.Novedades de la nueva WHO referente a los SMD
Polymorphism). De microarrays genómicos se distinguen dos tipos: microarrays de hibridación genómica comparada (HGC) y de SNP. Los microarrays de SNP permiten detectar, además de cambios en el número de copias, pérdidas de heterocigosidad. La técnica de arrays de CGH/SNP tiene como ventaja su elevada resolución, eficaz en la detección de cambios genéticos pequeños que nos permitirá localizar genes candidatos específicos en los SMD. Además, permite obtener importantes resultados en un único estudio. En la literatura podemos encontrar diferentes estudios que aplican la técnica de microarrays genómicos en los SMD (Gondek y cols., 2008; Thiel y cols., 2011; Tiu y cols., 2011) y han mostrado que la aplicación de técnicas con una mayor resolución permiten la detección de alteraciones adicionales a las ya detectas por citogenética convencional o en aquellos casos sin alteraciones citogenéticas, la detección de alteraciones crípticas.
Este conocimiento contribuirá no solo a un mejor tratamiento de los pacientes, pudiéndoles ofrecer una terapia que se ajuste a las caracte-rísticas de su enfermedad sino que también permitirá un mejor diag-nóstico de los pacientes estudiados atendiendo a sus características genéticas.
Recientes estudios sobre con CGH array (o SNP arrays) han demostrado que el 87% de los pacientes con SMD presentan alteraciones citogenéticas o que el 60% de los pacientes con cariotipo normal presentan ganancias o pérdidas de material genético.
Si bien los arrays de expresión son una herramienta de investigación, la CGH array o SNP arrays formaran pronto parte de las técnicas que deberán incluir los laboratorios de citogenética para utilizarlas en el diagnóstico de las neoplasias hematológicas. Las principales ventajas de estas me-todologías son que no requieren células en división, permiten detectar ganancias y pérdidas de material genético (estas son las principales alte-raciones en los pacientes con SMD), permiten detectar cambios genéticos con una mayor resolución (0.5 Mb frente las 10 Mb de la citogenética convencional) y además la técnica de SNP arrays permite detectar UPD, cambios genéticos que pueden aportar mucha información sobre la etio-logía de los SMD. Sin embargo, también tienen ciertas limitaciones, entre las que hay que destacar: si la proporción de células tumorales es inferior al 30% no se detectaran alteraciones genéticas del clon patológico, no detectan translocaciones equilibradas (poco frecuentes en los SMD, 1% y muy frecuentes en otras patologías como las leucemias agudas o linfo-mas), y coste económico algo más elevado.
Los estudios preliminares utilizando dicha metodología abalan su utilidad. En el año 2007, Gondek y cols. presentaron un estudio sobre pacientes con SMD en los que comparaban los hallazgos citogenéticos detectados por citogenética convencional (cariotipo) frente SNP arrays. Observaron que mediante SNP arrays el 80% de los pacientes presenta-ban alteraciones citogenéticas frente el 50% de la citogenética conven-cional, y además en el 68% de los pacientes con citogenética normal, con SNP arrays se detectaban alteraciones citogenéticas. De cinco pacientes que no presentaron mitosis por citogenética, cuatro (80%) presentaron alteraciones citogenéticas por arrays, Además en el 33% de los pacientes se observaron LOH o UPD. Un reciente trabajo de Arenillas y colaborado-res sobre una serie de 62 pacientes sin resultado citogenético demuestra mediante la aplicación de los SNP arrays que el 40% de los pacientes
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA46
presentan alteraciones citogenéticas y que resultado detectado con di-cha anomalía permite estratificar los pacientes según el IPSS e IPSS-R (Arenillas y cols., 2013).
Estos resultados y su relativo coste económico hacen plantear la necesidad de aplicar la CGH/SNP array en aquellos casos que no sean informativos (ausencia de metafases o cariotipo normal) mediante citogenética con-vencional.
Por el momento, aun existen pocas series para establecer el valor pro-nóstico de las alteraciones citogenéticas detectadas por CGH/SNP arrays, y la nueva propuesta de revisión del IPSS se basará únicamente en los cambios detectados por citogenética convencional.
SECUENCIACIÓN, NEXT GENERATION SEQUENCING (NGS)
La secuenciación del genoma humano gracias a los secuenciadores de nueva generación está permitiendo detectar nuevos marcadores ge-néticos asociados a patologías. La aplicación de la secuenciación a una patología como los SMD, que solo presentan alteraciones citogenéticas en el 50% de los pacientes con las técnicas convencionales permitirá demostrar nuevos genes relacionados con el origen de dicha enferme-dad. La técnica de secuenciación hasta el momento se ha aplicado en pocos pacientes afectos de otras neoplasias y su uso en SMD es muy limitado (Graubert y cols., 2009, Bejar y cols., 2011; Papaemmanuil y cols., 2011; Yoshida y cols., 2011; Haferlach y cols., 2014). Hasta el momento se han realizado estudios aplicando la metodología de la NGS que permite estudiar una selección de genes más implicados en los SMD. Los estudios preliminares han permitido detectar alteracio-nes moleculares en aproximadamente el 70-80% de los pacientes. Además se ha demostrado que dichas alteraciones conllevan valor pronóstico (Tabla 4). En el seno del grupo internacional de estudio de los SMD, IWG-PM (MDS Foundation) se está realizando un proyecto multicéntrico para definir el patrón molecular de una amplia serie de pacientes con SMD y determinar su valor pronóstico, dicho score se llamará IPSS-M.
CONSIDERACIONES FINALES
– El estudio del cariotipo (citogenética convencional) se debe realizar siempre ante la sospecha diagnóstica de SMD.
– El resultado citogenético es el parámetro con mayor peso en el pro-nóstico de los SMD (IPSS-R, Greenberg y cols., 2012).
– En pacientes en los que el estudio citogenético falla se recomiendo el estudio por CGH o SNP arrays ya que sus resultados sirven para aplicar el IPSS-R.
– Se recomienda el panel de los principales genes mutados en SMD mediante NGS para definir el pronóstico de los pacientes.
– Está en curso la preparación del IPSS basado en resultados mole-culares, IPSS-M.
TABLA 4.Resumen de las principales anomalías moleculares y su valor pronóstico
GEN INCIDENCIA (%) PRONÓSTICO
TET2 20 BUENO
ASXL1 14.4 BUENO
RUNX1 8.7 MALO
TP53 7.5 MALO
EZH2 6.4 MALO
NRAS 3.6 MALO
JAK2 3 INTERMEDIO
ETV6 2.7 MALO
CBL 2.3 MALO
IDH2 2.1 MALO
NPM1 1.8 INTERMEDIO
IDH1 1.4 BUENO
KRAS 0.9 INTERMEDIO
GNAS 0.7 DESCONOCIDO
PTPN11 0.7 DESCONOCIDO
BRAF 0.5 DESCONOCIDO
PTEN 0.2 DESCONOCIDO
CDKN2A 0.2 DESCONOCIDO
Abreviaciones:
TET2, ten eleven translocation-2; ASXL1, additional sex-comb-like-1; RUNX1, trans-
criptional core binding factor family name; TP53, Protein P53; EZH2, enhancer of zes-
te homolog 2; KRAS, v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; NRAS,
neuroblastoma RAS viral (v-ras) oncogene homolog IDH2, isocitrate dehidrogenase
gene 2; NPM1, nucleophosmin gene; IDH1, isocitrate dehidrogenase gene 1; CBL, casitas
B-cell lymphoma; JAK2, janus kinase 2; ETV6, ets variant; GNAS, GNAS complex locus;
PTPN11, protein tyrosine phosphatase, non-receptor type 11; BRAF, v-raf murine sar-
coma viral oncogene homolog B1; PTEN, phosphatase and tensin homolog; CDKN2A,
Cyclin-dependent kinase
BIBLIOGRAFÍA
Arenillas L, Mallo M, Ramos F, Guinta K, Barragán E, Lumbreras E, Lar-ráyoz MJ, De Paz R, Tormo M, Abáigar M, Pedro C, Cervera J, Such E, José Calasanz M, Díez-Campelo M, Sanz GF, Hernández JM, Luño E, Saumell S, Maciejewski J, Florensa L, Solé F (2.013): Single nucleotide polymorphism array karyotyping, a diagnostic and prognostic tool in primary myelod-ysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes and Cancer. Oct 7. doi: 10.1002/gcc.22112. [Epub ahead of print]. PMID 24123380.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, Ebert BL (2011): Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364:2496-2506. PMID 22869879.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sul-tan C. The FrenchAmericanBritish Cooperative Group (1982): Proposals for the classification of the myelodysplastic syndromes. Br J Haematol 51:189199. PMID:6952920.
Brunning RD, Orazi A, Germing U, Le Beau MM, Porwit A, Baumann I (2008) Myelodysplastic Syndromes. In: Swerdlow SH, Campo E, Lee Harris N, Jaffe

PONENCIAS 47
ES, Pileri SA, Stein H, Thiele J, Vardiman JW (eds): WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC, Lyon: pp 87-107.
Gondek LP, Tiu R, O’Keefe CL, Sekeres MA, Theil KS, Maciejewski JP (2008): Chromosomal lesions and uniparental disomy detected by SNP ar-rays in MDS, MDS/MPD, and MDS-derived AML. Blood 111: 1534-1542. PMID:17954704.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, Ben-nett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, Malcovati L, Cazzola M, Cermak J, Fonatsch C, Le Beau MM, Slovak ML, Krieger O, Luebbert M, Maciejewski J, Magalhaes SM, Miyazaki Y, Pfeil-stöcker M, Sekeres M, Sperr WR, Stauder R, Tauro S, Valent P, Vallespi T, van de Loosdrecht AA, Germing U, Haase D (2.012): Revised International Prognostic Scoring System (IPSS-R) for Myelodysplastic Syndromes. Blood 120 (20): 2454-2465. FI. 9.898. PMID: 22740453. PMID:22740453.
Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, Kundgen A, Lübbert M, Kunzmann R, Giagounidis AA, Aul C, Trümper L, Krieger O, Stauder R, Müller TH, Wimazal F, Valent P, Fonatsch C, Steidl C (2007): New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 pa-tients. Blood. 110: 4385-4395. PMID: 17726160.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnitt-ger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W, Ogawa S: Land-scape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014 Feb;28(2):241-7. doi: 10.1038/leu.2013.336. Epub 2013 Nov 13. PMID:24220272.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, Godfrey AL, Rapado I, Cvejic A, Rance R, McGee C, Ellis P, Mudie LJ, Stephens PJ, McLaren S, Massie CE, Tarpey PS, Varela I, Nik-Zainal S, Davies HR, Shlien A, Jones D, Raine K, Hinton J, Butler AP, Teague JW, Baxter EJ, Score J, Galli
A, Della Porta MG, Travaglino E, Groves M, Tauro S, Munshi NC, Anderson KC, El-Naggar A, Fischer A, Mustonen V, Warren AJ, Cross NC, Green AR, Futreal PA, Stratton MR, Campbell PJ; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. (2011): Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365:1384-1395 . PMID: 21995386.
Schanz J, Tuechler H, Solé F, Mallo M, Luño E, Cervera J, Granada I, Hilde-brandt B, Slovak Ml, Ohyashiki K, Steidl C, Fonatsch C, Pfeilstoecker M, Noesslinger T, Valent P, Giagounidis A, Aul A, Luebbert M, Stauder R, Krieger O, Garcia-Manero G, Faderl S, Pierce S, Le Beau Mm, Bennett J, Greenberg P, Germing U, Haase D (2012): A new, comprehensive cytogenetic scor-ing system for primary myelodysplastic syndromes and oligoblastic AML leukemia following MDS derived from an international database merge. Journal Clinical Oncology: 30(8):820-829. PMID 22331955.
Solé F, Luño E, Sanzo C, Espinet B, Sanz GF, Cervera J, Calasanz MJ, Cigudosa JC, Millà F, Ribera JM, Bureo E, Marquez ML, Arranz E, Florensa L. (2005): Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haema-tologica 90:1168-1178. PMID: 16154839.
Tiu RV, Gondek LP, O’Keefe CL, Elson P, Huh J, Mohamedali A, Kulasekara-raj A, Advani AS, Paquette R, List AF, Sekeres MA, McDevitt MA, Mufti GJ, Maciejewski JP (2011): Prognostic impact of SNP arrays karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood 117: 4552-4560. PMID:21285439.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawa-hata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S (2011): Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478:64-69. PMID: 21909114.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA48
LEUCEMIAS MIELOBLÁSTICAS AGUDAS.
La leucemia aguda mieloblástica es la indicación más frecuente de transplante alogénico con un crecimiento anual mundial del 10%. La utilización de donantes familiares haploidénticos, no emparentados y de sangre de cordón umbilical permite disponer de donante para la mayor parte de pacientes. Los acondicionamientos de intensidad redu-cida permiten el alotrasplante en edades muy avanzadas. Sin embargo sólo una minoría de pacientes con LMA recibe un trasplante alogénico, sea por edad avanzada, comorbilidades, toxicidad de la terapia previa, no alcanzar remisión, recaída precoz o refractariedad de la leucemia.
Indicaciones. En la evaluación del riesgo-beneficio para decidir hacer o no un alotrasplante se deben contraponer, por un lado, el riesgo asociado al trasplante de mortalidad no relacionada con recaída y de morbilidad, y por otro, la reducción del riesgo de recaída en base al riego citogenético y molecular de la leucemia, factores del paciente, del donante y del propio procedimiento de trasplante. Las LMA con genética de riesgo favorable no tienen indicación a priori de alo-trasplante en primera remisión completa. En general el alotrasplante se recomienda cuando la incidencia de recaída sin el mismo se espera superior a 35-40%. Cuanto mayor es el riesgo de recaída esperado, mayor es el riesgo de mortalidad no relacionada con la recaída que puede ser aceptable. Se admite que en el grupo de genética adversa el trasplante debe realizarse en cuanto se consiga la remisión completa. El trasplante alogénico es la única opción curativa para pacientes con enfermedad primariamente refractaria.
La monitorización secuencial de la enfermedad mínima residual (EMR) mediante PCR cualitativa en tiempo real (qPCR-RT) o citometría de flujo (CMF) proporciona una guía para la toma de decisiones. Los pacientes con EMR persistente o de nueva aparición pueden recibir quimioterapia de rescate previamente al alotrasplante o proceder directamente al alotrasplante en función de la probabilidad de éxito de la quimioterapia de rescate. Aunque el alotrasplante produce en general resultados superiores a la quimioterapia, no anula el efecto negativo de la genética desfavorable o de la positividad de la EMR pretrasplante. Los pacientes sin citogenética de pronóstico adverso ni EMR positiva pueden en primera remisión completa recibir quimiote-rapia solamente o un trasplante autólogo.
Acondicionamiento mieloablativo vs. de intensidad reducida
Potencialmente los acondicionamientos de intensidad reducida pue-den beneficiar del efecto curativo graft-versus-leukemia a pacientes de edad avanzada o jóvenes con comorbilidades significativas. Actual-mente los regímenes mielablativos se recomiendan generalmente para pacientes jóvenes sin comorbilidades y los de intensidad reducida para mayores o jóvenes con comorbilidades significativas. El régimen
mieloablativo busulfán/ciclofosfamida se prefiere al ciclofosfamida/irradiación total corporal.
Comorbilidades y escores de riesgo.
El índice de comorbilidad para el alotrasplante (HCT-CI) es una herra-mienta validada que traduce las comorbilidades de un paciente a una puntuación única que predice la probabilidad de mortalidad no debida a recaída de un determinado régimen mieloablativo o de intensidad reducida1. El Disease Risk Index2 se basa en el estadio de la enfer-medad y citogenética y predice la probabilidad de recurrencia de la enfermedad tras regímenes mieloablativos o de intensidad reducida, independientemente de la edad, la intensidad del acondicionamiento, el origen del injerto y el tipo de donante.
Nuevas modalidades de alotrasplante
La depleción T parcial o completa y la administración de ciclofosfamida postrasplante puede reducir el riesgo de enfermedad injerto contra huésped aguda y crónica, aunque la prevención de la recaída postras-plante continúa siendo un importante reto con estas estrategias. Están en investigación nuevos regímenes de acondionamiento con nuevos fármacos o anticuerpos monoclonales radiomarcados y el tratamiento en el postrasplante precoz con inhibidores de tirosinkinasa o agentes hipometilantes. También están en investigación terapias encaminadas a aumentar el efecto injerto contra leucemia tales como el enriqueci-miento con células natural killer, la inmunoterapia adoptiva y el uso de células T de ingeniería genética dirigidas contra antígenos específicos de células de la LMA.
SÍNDROMES MIELODISPLÁSICOS
A pesar de la progresiva comprensión de la patogénesis molecular de los síndromes mielodisplásicos (SMD), los agentes terapéuticos dispo-nibles en la actualidad consiguen prolongar la vida pero no consiguen la curación. Por este motivo el alotrasplante hematopoyético está sien-do utilizado de manera creciente como opción terapéutica curativa en pacientes seleccionados3 con SMD. La introducción de regímenes de acondicionamiento de intensidad reducida (RIC) ha ampliado la indicación de alotransplante a pacientes de edad avanzada, con co-morbilidades o mala situación física. El uso creciente de donantes no emparentados o familiares haploidénticos contribuye a su frecuente uso actual.
La decisión de proponer o no el alotrasplante hematopoyético a un paciente con síndrome mielodisplásico se debe basar en la evaluación del riesgo de progresión y muerte debido a su enfermedad según el Prognostic Scoring System (IPSS-R)4 y la presencia de comorbilidades
Papel actual del trasplante hematopoyético en el tratamiento de pacientes adultos con leucemias mieloblásticas agudas y síndromes mielodisplásicosIldefonso Espigado
HU Virgen del Rocío / Instituto de investigación biomédica de Sevilla / Universidad de Sevilla

PONENCIAS 49
evaluadas mediante el HSC Comorbidity Index (HCT-CI)5,6. Los pacientes candidatos son aquellos que tienen una buena situación física, eva-luada con el índice de comorbilidad, y un síndrome mielodisplásico de alto riesgo con el IPSS-R. También son candidatos los pacientes de bajo riesgo en el IPSS-R con citogenética de mal pronóstico, citopenias profundas y alta carga transfusional. Los pacientes con una puntuación de riesgo en el alotrasplante muy alta porque tienen edad avanzada, puntuación alta en el índice HCT-CI, citogenética y/o alteraciones mo-leculares de muy mal pronóstico y una puntuación alta en el índice IPSS-R tienen muy pocas opciones de curación con un alotrasplante por lo que serían candidatos a tratamientos de investigación. No hay evidencia científica basada en ensayos clínicos aleatorizados que per-mita hacer recomendaciones sobre la intensidad óptima del régimen de acondicionamiento. En general, los regímenes de intensidad reduci-da se deben considerar para aquellos pacientes con contraindicaciones para acondicionamientos de alta intensidad. La decisión del momento óptimo para realizar el transplante requiere la evaluación cuidadosa de la situación del paciente, de las estrategias de tratamientos disponi-bles y de la disponibilidad de donante. En pacientes de alto riesgo de recidiva tras el alotrasplante se recomienda la estrategia profiláctica de infusión de linfocitos del donante (ILD). Si se produce la recidiva más allá de seis meses después del alotrasplante se puede plantear la infusión de linfocitos del donante (ILD) o un segundo trasplante, en pacientes con buena situación física.
Se pueden hacer algunas recomendaciones específicas en función de aspectos concretos del paciente o de la enfermedad.
Características del paciente
El performance status, la fragilidad y las comorbilidades tienen un papel importante en los resultados del trasplante. La edad, por sí mis-ma, lo tiene menos. Un estudio reciente ha mostrado que pacientes de alto riesgo entre 60 y 70 años se benefician del alotrasplante con acondicionamientos de intensidad reducida7.
Factores de la enfermedad
A efectos de la toma de decisión para trasplante, la clasificación del IPSS-R se puede simplificar en tres grupos: bajo riesgo (muy bajo y bajo riesgo del IPSS-R), riesgo intermedio y alto riesgo (alto y muy alto riesgo del IPSS-R).
Mieloblastos en médula ósea: en pacientes de riesgo intermedio con mieloblastos entre 5% y 10% no hay consenso en si deben ser conside-rados para trasplante al diagnóstico o cuando desarrollen factores de riesgo adicionales. Sí hay consenso en realizar el alotrasplante precoz si el número de blastos en médula ósea es superior al 10%1.
La citometría de flujo puede ser usada, junto con las valoraciones de riesgo general, para la evaluación del riesgo, la elección del tratamien-to y la monitorización de la actividad de la enfermedad.
Las características citogenéticas tienen significado pronóstico también tras el alotrasplante. La categoría de muy mal pronóstico predice peor supervivencia tras el alotrasplante, al igual que la presencia de cario-tipos complejos, cariotipos monosómicos o ambos. La nueva clasifi-
Algoritmo terapéutico para pacientes adultos con SMD y muy bajo riesgo o riesgo intermedio evaluado con el IPSS-R (tomado de T de Witte et al, Blood 2017)

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA50
cación citogenética del IPSS-R ha cambiado el impacto pronóstico de la del(7q) aislada que ha pasado a considerarse de riesgo citogenético intermedio, lo que supone que el paciente podría cualificar como de bajo riesgo. No hay consenso sobre si en esta situación debería propo-nerse alotrasplante inmediatamente tras el diagnóstico1.
Características moleculares. Los SMD asociados a mutaciones de SF3B1 constituyen una entidad con pronóstico favorable. Las mutaciones en DNMT3A, TET2, IDH1 e IDH2 están asociadas a displasia multilínea y SRSF2, RUNX1, U2AF1, ASXL1 y TP53 se asocian a mal pronóstico. Es-pecialmente la asociación de cariotipos complejos con mutaciones de TP53 presentan mal pronóstico tras alotrasplante. Estos pacientes de mal pronóstico serían candidatos a ensayos clínicos con estrategias postrasplante para prevenir la recidiva.
Las citopenias y la dependencia transfusional están incluidas en el IPSS-R. Las citopenias sintomáticas y severas que son refractarias a factores de crecimiento o que requieren soporte transfusional intenso pueden ser indicaciones independientes para alotrasplante, aunque requieren un tiempo de observación antes de tomar la decisión. El IPSS-R ha cambiado de grupo de riesgo aproximadamente al 65% de los pa-cientes y predice mejor que el IPSS la supervivencia tras el trasplante8.
Otros subtipos de SMD
Leucemia mielomonocítica crónica (LMMC). Para establecer las indica-ciones de alotrasplante se puede usar el Sistema de puntuación es-
pecífico para LMMC (CMML-specific scoring system, CPSS) o el propio IPSS-R para pacientes con LMMC de tipo displásico. El factor pronóstico más importante que predice un resultado favorable es conseguir el mejor estado de remision pretrasplante posible.
SMD relacionados con la terapéutica. Tienen peor pronóstico que los SMD de novo, pero, en general sus indicaciones para alotrasplantes son las mismas.
SMD con myelofibrosis. Son pacientes que suelen asociar pancitopenia y en general deben ser considerados para alotrasplante antes del de-sarrollo de mielofibrosis severa.
SMD hipoplásicos. El IPSS-R predice la supervivencia de forma ade-cuada. Una minoría puede responder a tratamiento inmunosupresor. El momento del trasplante depende de la severidad de las citopenias, intensidad de las transfusiones y probabilidad de respuesta a la in-munosupresión.
SMD originados de mutaciones heredadas de línea germinal. Su diag-nóstico debe considerarse en pacientes jóvenes (<40-50 años) con his-toria familiar sugestiva (disqueratosis congénita, Anemia de Fanconi, Síndroem de Shwachman-Diamond, Anemia de Blackfan-Diamomd y mutaciones de GATA2). Suelen ser candidatos a alotrasplante en estadíos precoces. Se debe tener mucha atención a la especialidad sensibilidad al acondionamneto de algunos de estos síndromes (ane-mia de Fanconi) y la selección de donante familiar que pudiera portar la mutación implicada.
Algoritmo terapéutico para pacientes adultos con SMD y pobre pronóstico evaluado con el IPSS-R(tomado de T de Witte et al, Blood 2017)

PONENCIAS 51
CONCLUSIONES
El alotrasplante hematopoyético está siendo usado de manera cre-ciente para el tratamiento de la leucemia mieloblásticaaguda y los síndromes mielodisplásicos. Las innovaciones recientes en las técnicas de trasplante y de seguimiento de la enfermedad mínima residual pueden mejorar los resultados. Por otra parte, el desarrollo de nuevos fármacos y estrategias de tratamiento podrían cambiar el papel del alotrasplante en el futuro.
BIBLIOGRAFÍA
1. Sorror ML, Storb RF, Sandmaier BM, et al. Comorbity-age index: a clinical measure of biologic age before allogeneic hematopoietic cell transplanta-tion. J Clin Oncol. 2014;32(29):3249-3256.
2. Armand P, Kim HT, Logan BR, etal. Validation and refinement of the Disease Risk Index for allogeneic stem cell transplantation. Blood 2014; 123(23):3664-3671.
3. de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recomendations from an interna-tional expert panel. Blood 2017; 129: 1753-1762.
4. Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for Myelodisplastic Syndromes. Blood 2012;120(12):2454-2465.
5. Sorror ML, Maris MB, Storb R, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood 106:2912-2919, 2005.
6. Sorror ML, Sandmaier BM, Storer BE, et al. Comorbidity and disease status-based risk strattification of outcomes amomg patients with acute myeloid leukemia or myelodysplasia receiving allogeneic hematopoietic cell transplantatio. J Clin Oncol 25: 4246-4254, 2007.
7. Koreth J, Pidala J, Peres WS et al. Role of reduced-intensity conditioning allogeneic hematopoietic stem-cell transplantation in older patients with de novo myelodyslastic síndromes: an international collaborative decisión analysis. J Clin Oncol, 2013;31(21):2662-2670.
8. Della Porta MG, Alessandrino EP, Bacigalupo A et al. Predictive factors for the outcome of allogeneic transplantation in patients with MDS stratified according to the revised IPSS-R. Blood 2014;123(15):2333-2342.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA52
INTRODUCIÓN
La hemorragia postparto severa es una de las complicaciones obsté-tricas más temidas y es la primera causa de mortalidad materna en el mundo.
Aunque su incidencia es mayor en los países no desarrollados, estudios epidemiológicos recientes indican que las tasas de hemorragia pos-tparto (HPP) severa están aumentando en los países desarrollados, siendo en la actualidad de 6,7/1000 partos.
Constituye una entidad diferenciada dentro de las hemorragias masi-vas y como tal requiere un protocolo de manejo especializado.
Se ha demostrado, que la mayoría de las muertes maternas por he-morragia, se producen en las primeras cuatro horas tras su detección, existiendo una hora de oro, que va a determinar las posibilidades de supervivencia sin secuelas de las pacientes Es por tanto una emer-gencia que requiere un tratamiento agresivo inmediato y estrategias de prevención
Cualquier parturienta es susceptible de desarrollarla, y por tanto, cada hospital debe estar preparado para afrontar esta situación
Algunas pacientes con HPP severa requieren trasfusión masiva, aunque afortunadamente esta es poco frecuente, si se actúa precozmente, y este debe ser el objetivo que nos debemos plantear .
Para ello es fundamental la comunicación y el trabajo en equipo, así como la elaboración de protocolos de actuación, basados en la mejor evidencia disponible , que deben ser difundidos e implantados.
Definición de Hemorragia Obstétrica Severa
Tradicionalmente, se ha considerado HPP anormal a la pérdida san-guínea superior a 500 ml tras parto vaginal y a 1000 ml tras cesárea
Las definiciones más modernas de sangrado anormal, prestan mayor atención a los síntomas de la hemorragia y puede ser mejor definida, como el sangrado excesivo que provoca síntomas (debilidad, mareo, síncope) y/o signos de hipovolemia (taquicardia hipotensión, oliguria). Es importante recordar, que la mayoría de estos cambios únicamente ocurrirán cuando la paciente haya perdido una cantidad importante de sangre (a partir de 1000-1.500 ml), aunque en pacientes con escasa reserva funcional puede ocurrir con perdidas menores
Así, la definición más práctica de HPP severa , es la de un sangrado activo y continuado, que amenace la estabilidad hemodinámica de la paciente.
Algunos autores valoran la gravedad de la HPP, en función del número de unidades de hematíes transfundidos durante las primeras horas tras el parto, considerando como HPP grave, aquella que recibe mas de 4
unidades de hematíes en una hora y transfusión masiva, la transfusión de más de 8 -10 unidades de hematíes en 24 horas.
Etiologia de la Hemorragia Obstétrica
Las causas de la hemorragia postparto se pueden simplificar en la regla de las “4T” según el defecto detectado. Fue propuesta por la Sociedad Canadiense de Obstetricia y Ginecología para recordar las causas más frecuentes de HPP: Tono (atonía uterina responsable del 50-60% de las HPP), Tejido (retención de productos placentarios 20-30% de las HPP ), Trauma (laceraciones cervicales y/o vaginales responsable del 10% de las mismas ) y Trombina (trastornos de coagulación preexistentes )
La atonía uterina, además de ser la principal causa de HPP, también es responsable de las hemorragias más severas, siendo la principal causa de HPP severa.
Prevención de la Hemorragia Obstétrica
La mayoría de las HPP se producen sin factores de riesgo conocido
La valoración de los factores de riesgo antenatales, predice únicamente el 40% de los casos de HPP, siendo la placenta previa y la placenta ácrata, los factores de riesgo identificados más importantes de las hemorragias graves.
El manejo activo de la 3ª fase del parto, ha demostrado disminuir la incidencia de HPP, así como la anemia postparto, y la necesidad de transfusión
Diagnostico de la Hemorragia Obstétrica
El diagnostico se basa en la observación clínica rutinaria, el control de las constantes vitales y el examen físico durante las curas del postparto o postoperatorio
El California Maternity Care Collaborative (CMQCC), ha propuesto una serie de valores de corte para disparar la alarma: frecuencia cardiaca (FC) ≥ 110 spm, presión arterial (PA) ≤ 85/45 mmHg y saturación de oxigeno < 95%
Este nuevo enfoque obedece principalmente a tres “problemas” a la hora de realizar el diagnostico de la hemorragia anormal:
1. La estimación visual siempre es inexacta
Se ha demostrado que infraestima el sangrado entre un 30 y un 50%
2. Las modificaciones fisiológicas de la paciente puérpera, provocan que la mayoría de mujeres sanas no muestren signos o síntomas de inestabilidad hemodinámica, hasta que experimenten una he-morragia que alcance aproximadamente 1.200 ml.
Transfusión masiva en Obstetricia Magdalena Carmona Gonzalez
UGC de Hematología y Hemoterapia. Hospital Virgen del Rocío

PONENCIAS 53
3. La posibilidad de hemorragias ocultas : en el desprendimiento prematuro de placenta o en la hemorragia por atonía uterina, el útero puede retener gran cantidad de sangre , y en los hematomas disecantes del canal del parto, se producen hemorragias exangui-nantes sin evidencia de sangrado
Debe favorecerse el pesado de los materiales empapados en sangre (compresas tocológicas, quirúrgicas y empapadores) y de las bolsas colectoras del paritorío, pues aumenta la precisión en la cuantificación de la hemorragia.
Cambios fisiólogicos en la coagulación de la gestante
El embarazo esta asociado con un estado de hipercoagulabilidad y disminución de la capacidad fibrinolítica.
Durante el segundo y, especialmente, el tercer trimestre de la gesta-ción aumenta la síntesis y la actividad de varios factores de la coagu-lación que producen un estado de hipercoagulabilidad. disminuyendo así la posibilidad de complicaciones hemorrágicas relacionadas con el parto.
Aumenta el fibrinógeno. ( las gestantes a término tendrán niveles de fibrinógeno en el límite alto de la normalidad o claramente superiores ) y los factores.VII, VIII, IX,X, XII y factor von Willebrand,.Disminuye la actividad anticoagulante endógena caracterizada por aumento del cofactor II de la heparina , alfa 1 antitripsina , actividad de la proteína S y resistencia a la proteína C activada.
En cuanto al contaje plaquetario, la trombopenia gestacional es un fenómeno frecuente aunque no suele tener significación clínica. La cifra de plaquetas suele mantenerse superior a 80 x 109/L.
Coagulopatia de la hemorragia obstetrica
Al igual que en otros casos de hemorragias masivas, la coagulopatía, se puede desarrollar precozmente en la HPP severa, y constituye un factor pronóstico independiente de mortalidad
Los mecanismos de esta coagulopatía son actualmente bien conocidos y esto ha hecho cambiar las guías de manejo de esta entidad a nivel internacional
En primer lugar, existiría una pérdida relacionada con la hemorragia de factores de coagulación, fundamentalmente fibrinógeno y plaquetas, junto con un rápido consumo de los mismos, por una activación exa-gerada de la cascada de la coagulación (especialmente frecuente, pero no exclusiva, del desprendimiento de placenta, la embolia de líquido amniótico y la preeclampsia),
En segundo lugar, existe una coagulopatía dilucional por la adminis-tración de liquidos intravenosos.
Por otro lado, la hiperfibrinoíisis, presente de forma predominante en la hemorragia obstétrica, hace que se deshagan los incipientes coágulos de fibrina formados, y produce un consumo y depleción de factores de coagulación, fundamentalmente fibrinógeno. Además. los productos de degradación del fibrinógeno originados, pueden empeo-rar la función plaquetaria.
Por último, el trastorno metabólico derivado de la hipoperfusión ti-sular y la hipotermia, junto con la anemia , acidosis y los trastornos electrolíticos, derivados de la politransfusión, contribuyen a alterar el complejo equilibrio hemostático de estas pacientes.
El fibrinógeno es el primer factor en caer a niveles críticos, de tal ma-nera que es el parámetro más sensible para indicar el compromiso hemostático durante la HPP severa.
Monitorización de la Coagulopatia
El diagnostico de la coagulopatia es fundamental para orientar el tra-tamiento de esta pacientes y predecir el riesgo de sangrado. pero lo mas importante es que los resultados estén disponibles en el menor tiempo posible, y se repitan mientras persista la hemorragia, cada 30-60 minutos.
A pesar de todo lo publicado en los últimos años, aún no existe una clara evidencia, sobre cual es el mejor método de laboratorio para monitorizar la coagulopatía de las HPP severas.
Las técnicas convencionales de coagulación no se pueden realizar en un tiempo útil para orientar el tratamiento de estas pacientes y ade-más se correlacionan mal, con coagulopatías relevantes, y con el riesgo hemorrágico. No hay evidencia de alta calidad que sostenga que un alargamiento del TP y TPTA sea útil para el diagnostico de la coagu-lopatía hemorrágica, y además en muchas pacientes con HPP, estos parámetros son normales, pero tanto el nivel de fibrinógeno como el recuento plaquetario, han demostrado importante correlación con la severidad de la hemorragia y su determinación debe realizarse con urgencia.
Las técnicas viscoelásticas (Tromboelastografía o Tromboelastometria (TEG/ROTEM), han cobrado especial interés en los últimos años, siendo recomendadas por distintas sociedades científicas, como complemento a las técnicas de coagulación convencionales, para orientar el diagnos-tico y el tratamiento de la coagulopatía , en la HPP severa .
Se ha encontrado una fuerte correlación entre las técnicas convencio-nales de coagulación y las variables del ROTEM en la HPP, y los resulta-dos del fibrinógeno por FIBTEM se correlacionan también muy bien con el nivel de fibrinógeno por método estandard de laboratorio (Claus).
La ventaja más importante de las técnicas viscoelásticas, es la rapidez de respuesta, de manera que los resultados pueden estar disponibles en 10 minutos, y también que permiten un diagnostico rápido de la fibrinólisis.
Sin embargo estas técnicas son costosas, no están disponibles en todos los laboratorios, requieren especial preparación para la interpretación de los resultados y aún faltan estudios randomizados que demuestren su superioridad frente a los estudios convencionales de coagulación
Algunos grupos proponen crear un perfil hemostático de urgencia que incluya TP, Fibrinogeno y plaquetas, y tratar de obtener los resultados en 15 minutos, Posteriormente y en función de cada caso se puede completar el estudio de la coagulopatía.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA54
Los centros que dispongan de TEG/ROTEM, deben incorporar dichas técnicas en los protocolos de HPP severa, sin prescindir de las técnicas convencionales de coagulación y esto puede permitir una más rápida corrección de la coagulopatia.
TRATAMIENTO de la HPP severa
Las estrategias terapéuticas para el tratamiento de la HPP están bien estandarizadas y son de sobra conocidas (obstétricas, quirúrgicas y radiológicas ), jugando un papel fundamental en la evolución de las pacientes, pero el tratamiento médico , sobretodo la terapia trans-fusional y la estrategia prohemostática es también esencial y en los últimos años ha sufrido importantes cambios.
El objetivo del tratamiento hemostático es detener lo más precozmen-te posible la pérdida de sangre y reducir los requerimientos transfu-sionales, porque como es sabido el grado de transfusión aumenta la morbilidad y mortalidad de las pacientes
El concepto de resucitación hemostática, también se aplica a la HPP severa y engloba varias estrategias de reanimación:
1. Evitar la administración de coloides, asi como de grandes cantidades de cristaloides que provocan hemodilución y pue-den empeoran la coagulopatía
2. Transfusión precoz de componentes sanguíneos. según una ratio determinada de Hematíes, Plasma y Plaquetas
En la HPP hay pocos estudios sobre el impacto de la transfusión, según una ratio elevada, en la morbilidad y mortalidad, pero por similitud con la hemorragia traumática, se aconseja una ratio de al menos 2:1:1 (Hematies/Plasma/Plaquetas ) en las pacientes con HPP, que requieran transfusión masiva,
Pero realmente, la única recomendación potente sobre la transfusión en la HPP, es que las pacientes reciban hematíes tan pronto como sea posible, en el caso de HPP severa ( 5 minutos ) y esto se consigue me-jor aplicando, también en esta entidad, los protocolos de transfusión masiva y activandolos rápidamente.
Más que una ratio transfusional determinada, el precoz tratamiento de la coagulopatia, determina la morbimortalidad materna
Pero no debemos olvidar que la transfusión de sangre por desgracia, tiene efectos adversos importantes
Para disminuir la exposición a la transfusión y controlar la hemorra-gia, los agentes prohemostático ( ácido tranexámico y concentrado de fibrinógeno) , son cada vez más usados en la HPP severa, y su precoz administración forma parte de la resucitación hemostática
3. Administración de Acido Tranexámico y Fibrinógeno
3.1. Acido Tranexámico
Limitar la hiperfibrinolísis, se ha sugerido como el primer paso, en un algoritmo de tratamiento de la coagulopatía de la HPP severa.
El ácido tranexámico (TA) es un fármaco antifibrinolítico ampliamente usado en medicina, sobre todo, en el ámbito quirúrgico para reducir la pérdida de sangre y la necesidad de transfusión.
Al igual que en el paciente traumático, existe bastante evidencia médica, en torno a la reducción de la pérdida sanguínea, y la dismi-nución de los requerimientos transfusionales en obstetricia. La simi-litud fisiopatológica con la hemorragia del politraumatizado justifica su empleo con el objetivo también de aumentar la supervivencia en la HPP masiva.
Aunque está demostrada la efectividad del tranexámico en dicha en-tidad , aun no hay datos de ensayos randomizados, pero dado que se trata de un fármaco económico, fácil de administrar y con un bajo índice de efectos colaterales , muchas sociedades científicas recomien-dan su uso.
La mas reciente guía de la OMS, aconseja administrar TA para el tra-tamiento de la HPP, si la oxitocina u otros uterotónicos no controlan la hemorragia, o si la hemorragia puede ser en parte por trauma ( 1 g en 5 minutes, repetido en 30 a 60 minutos si es necesario).
El ensayo WOMAN ( The World Maternal Antifibrinolytic ) ha incluido miles de pacientes de todo el mundo y tiene como objetivo determinar el efecto de la precoz administración de Tranexámico en la mortalidad e índice de histerectomías, así como la incidencia de complicaciones mayores en mujeres con HPP .
Los resultados de dicho ensayo deben publicarse este año, y esto nos permitirá establecer conclusiones definitivas sobre el uso del TA en la HPP severa.
3.2. FIBRINOGENO
El fibrinógeno juega un papel crítico en mantener una adecuada he-mostasia, y es fundamental para una efectiva formación del coagulo.
Es el primer factor que se pierde en la HPP masiva y además se ha demostrado que el nivel de fibrinógeno está independientemente asociado con la severidad de la hemorragia, de tal manera que por cada gr /l que desciende, aumenta 2,6 veces el riesgo de HPP severa. Un nivel de fibrinógeno de < 2gr/l en el momento del comienzo de la hemorragia tiene un valor predictivo positivo del 100% para evolución a hemorragia severa.. La corrección de la hipofibrinogenemia es, por tanto, una medida de importancia vital.
La transfusión de Plasma no es óptima para corregir déficit de fibrinó-geno porque para aumentar 1 g/L el nivel de fibrinógeno se necesitan unos 20 ml/kg, de Plasma con el riesgo que esto supone (sobrecarga circulatoria, excesivo,tiempo de administración, posibilidad de TRALI)
Por el contrario por cada gramo de concentrado de fibrinógeno ad-ministrado se aumenta en 0.35-0.40 g/L aproximadamente, su nivel plasmático, y además su infusión es rápida y en muy poco volumen.
La administración de fibrinógeno debe ser precoz y guiada por los resultados de laboratorio que demuestren hipofibrinogenemia, Se su-giere mantener los niveles de fibrinógenos superiores a 1.5-2 g/L para asegurar una adecuada coagulación en este escenario.

PONENCIAS 55
El problema es que a veces, el retraso en los resultados de laboratorio, hace que se tenga que utilizar de forma empírica inicialmente, para evitar que la hemorragia se haga incontrolable.
Aún no hay un nivel de evidencia alto en la literatura, para establecer de forma firme, que el tratamiento con fibrinógeno sea un tratamiento curativo en las HPP, pero lo trabajos que hay en marcha. posiblemente lo acaben demostrando..
CONCLUSIONES
La hemorragia obstétrica severa, requiere un diagnostico precoz, un control inmediato de la hemorragia, una rápida estabilización de la paciente y una precoz participación multiprofesional y multidisciplinar.
Aunque no haya evidencias concluyentes, sobre la resucitación hemos-tática óptima, el uso de las técnicas viscoelásticas, la terapia transfu-sional, asi como la administración de ácido tranexámico y fibrinógeno son pilares claves del tratamiento..
BIBLIOGRAFIA:
1. Anne-Sophie Ducloy-Bouthors, MD,* Sophie Susen, MD, PhD,†‡ Cynthia A. Wong, MD Medical Advances in the Treatment of Postpartum Hemor-rhage Anesth Analg 2014;119:1140–7)
2. A.J. Butwick1 and L.T. Goodnough2 Transfusion and coagulation man-agement in major obstetric hemorrhage Curr Opin Anaesthesiol. 2015 June ; 28(3): 275–284
3. C. Solomon1*, R. E. Collis2 and P. W. Collins3 Haemostatic monitoring during postpartum haemorrhage and implications for management Brit-
ish Journal of Anaesthesia 109 (6): 851–63 (2012)P¨ar I. Johansson,1,2 Jakob Stensballe,1,3
4. KIM EKELUND1, GABRIELE HANKE1, JAKOB STENSBALLE2,3, Hemostatic resuscitation in postpartum hemorrhage – a supplement to surgery Acta Obstetricia Et Gynecologica Scandinavica · February 2015
5. Laura Green,1 Marian Knight,2 Frances Seeney,3 The haematological features and transfusion management ofwomen who required massive transfusion for major obstetric haemorrhage in the UK: a population based study British Journal of Haematology, 2016, 172, 616–624
6. Marie Pierre Bonnet , Dan Benhamou2 Management of postpartum haemorrhage 27 JUN 2016
7. Mousa HA, Blum J, Abou El Senoun G, Shakur H, Alfirevic Z Treatment for primary postpartum haemorrhage (Review) Cochrane Database of Systematic Reviews 2014,
8. M.C. Gutierrez,a L.T. Goodnough,b M. Druzin,c A.J. Butwicka Postpartum hemorrhage treated with a massive transfusion protocol at a tertiary obstetric center: a retrospective study International Journal of Obstetric Anesthesia (2012) 21, 230–235
9. Rezan Abdul-Kadir,1 Claire McLintock,2 Anne-Sophie Ducloy an cols Evalu-ation and management of postpartum hemorrhage:consensus from an international expert panel TRANSFUSION 2014;54:1756-1768
10. Roberto Oliveri,1 Charles E. Wade,2 Sisse R. Ostrowski,1 and John B. Holcomb. How I treat patients with massive hemorrhage (Blood. 2014;124(20):3052-3058)
11. Ruth Shaylor, BMBS, BMedSci,* Carolyn F. Weiniger, MB, ChB,* Naola Austin, MD,† National and International Guidelines for Patient Blood Management in Obstetrics: A Qualitative Review Anesthesia & Anal-gesia · August 2016.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA56
La transfusión sanguínea es un procedimiento médico que permite reponer un componente sanguíneo, que ha sido obtenido previamente de un donante, bien directamente o tras un proceso de fraccionamien-to, a un paciente que lo precisa. La transfusión es un procedimiento muy seguro y que conlleva importantes beneficios al receptor posibi-litando actuaciones terapéuticas que serían imposibles sin su disponi-bilidad, aunque por las características biológicas de la sangre no está exenta de riesgos para el receptor.
Hemovigilancia (HV) consiste en un sistema para la detección, registro y análisis de la información relativa a los efectos adversos e inespera-dos de la transfusión sanguínea. La implantación de sistemas de he-movigilancia ha permitido en primer lugar optimizar el esfuerzo de los servicios de transfusión, asegurando una recogida de la información de los efectos adversos de forma completa, rigurosa y objetiva; en segun-do lugar, analizar las causas responsables de algunas complicaciones de la transfusión e introducir medidas correctoras o preventivas; en tercer lugar, conocer las necesidades y prioridades reales de la trans-fusión sanguínea; y finalmente aumentar la calidad y la seguridad transfusional, poniendo los recursos humanos, técnicos y económicos allí donde la cadena transfusión se ha mostrado más frágil.
1. ANTECEDENTES SISTEMA NACIONAL DE HEMOVIGILANCIA:
Tras las experiencias previas del sistema francés e ingles (SHOT) de hemovigilancia implantados en el año 1994 y 1996 respectivamente, se creó en el año 1998 bajo los auspicios de Mº Sanidad y Consumo un grupo de trabajo en Hemovigilancia, constituido por 8 especialis-tas en medicina transfusional a los que se le encomendó: diseñar un programa de hemovigilancia adaptado a la estructura territorial del estado español y a sus características político administrativas; elaborar los documentos de trabajo necesarios lo que incluia un manual de hemovigilancia y los formularios específicos de recogida de reacciones y efectos adversos; y promover el desarrollo de la hemovigilancia en las 17 comunidades autónomas españolas, dado que las competencias sanitarias estaban ya transferidas1,2,3.
Dicho sistema quedo establecido en un modelo simple con notificación voluntaria por parte de los centros y servicios de transfusión, con unas fichas de notificación aceptadas por todas las comunidades lo que aseguraba una homogeneidad en los criterios de clasificación de las diferentes reacciones o efectos adversos a la transfusión.
En febrero de 2002 el Ministerio de Sanidad y Consumo organizo una “Jornada sobre Hemovigilancia” a fin de promover la implantación de programas de HV en todas las comunidades autónomas. Ese mismo años el SAS creo una comisión asesora sobre HV, de cuya actividad surgió la resolución SC 683/03 de 11 de Agosto 2003 del Sistema An-daluz de Salud por la que se constituía y se ponía en funcionamiento
el sistema de HV en los distintos centros del SAS4. Asi mismo en el año 2003 se crea el sistema estatal para la seguridad transfusional5.
Finamente se trasponen dos directivas del consejo de Europa
Directiva 2002/98/EC (27 Enero 2003) en el Real Decreto 1088/2005 20 de septiembre sobre los requisitos tecnicos y condiciones mini-mas de la hemodonación y de los centros y servicios de transfusión6
Directiva 2002/61/EC (30 Septiembre 2005) Orden SCO/322/2007 de 9 Febrero de 2007 sobre trazabilidad y notificacion de reacciones y efectos adversos graves de la sangre7.
En el año 2004 se presenta el primer informe de HV estatal en el que se recogian 769 notificaciones recibidas en 6 Comunidades Autono-mas sobre un total aproximadamente de 1.160.000 componentes transfundidos. En el años 2015 se ha publicado el último informe de hemovigilancia que reporta una tasa de 20.4 notificaciones por 10.000 componentes transfundidos sobre un total de aproximada-mente 1.900.000 productos transfundidos en todo el pais3.
1a) Efectos adversos de la transfusión
Se define un efecto adversos a la transfusión como cualquier suceso indeseable e inesperado antes, durante o después de la transfusión que podría estar relacionado con la administración de la misma. Puede ser el resultado de un error o un incidente, y puede o no, producir una reacción adversa en el paciente.
Asi se define reacción adversa como una respuesta indeseable en el paciente asociada temporalmente con la administración del com-ponente. Puede ser el resultado de un incidente o de la interacción entre el receptor y la sangre, un producto biologicamente activo.
Se considera un incidente cuando el paciente es transfundido con un componente que no cumple con los requerimientos previstos o que estaba destinado a otro paciente. Incluye los errores transfusio-nales y las desviaciones en los procedimientos o protocolos. Puede producir o no, una reaccion adversa en el paciente.
Un casi incidente Es un error o una desviación en los procedimientos o en los protocolos que se detecta antes de iniciar la transfusión, y que podria haber conducido a una transfusión errónea o a una reacción adversa en el paciente.
2. ÁMBITO DE LA HEMOVIGILANCIA EN ESPAÑA
Abarca toda la cadena transfusional, lo que incluye desde la selección de los donantes, a la extracción de la sangre y las complicaciones a la donación, a el procesamiento y análisis de los componentes san-guíneos y, finalmente, hasta la transfusión y los efectos adversos e inesperados en el receptor. La notificación de los diferentes incidentes
Hemovigilancia y calidad transfusionalMiguel Ángel Alvarez Rivas
FEA Hematologia y Hemoterapia. Hospital Universitario Reina Sofia. Cordoba
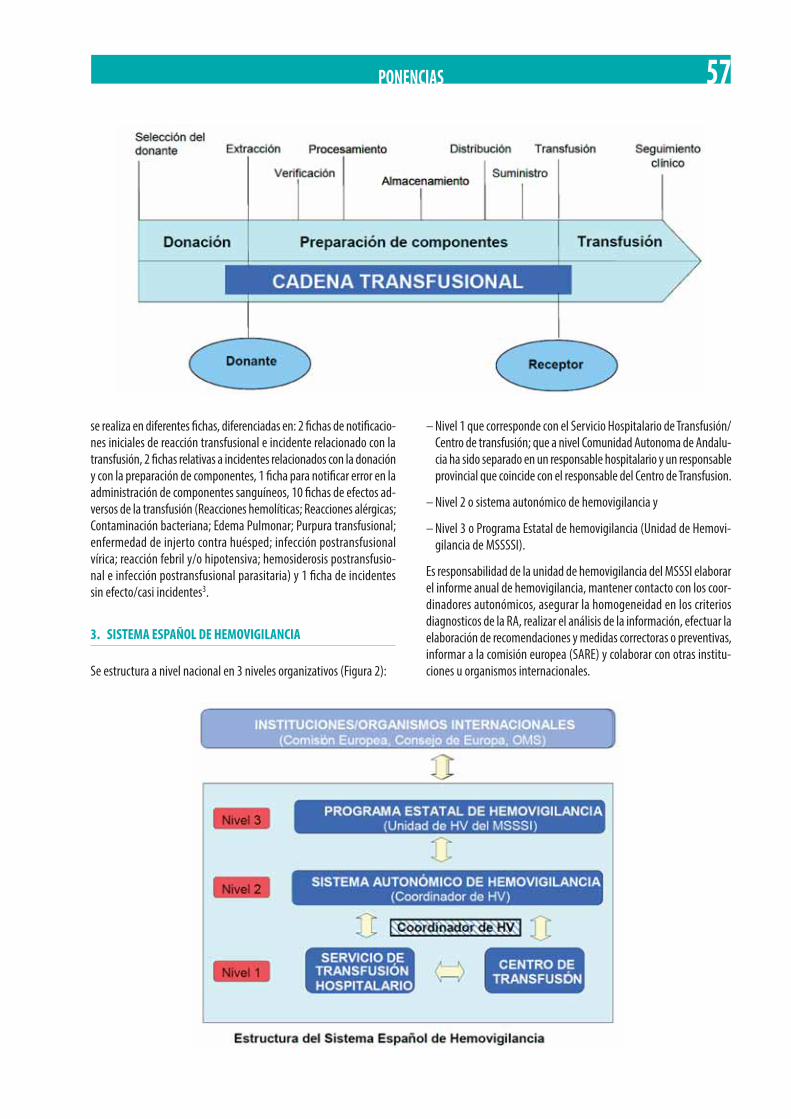
PONENCIAS 57
se realiza en diferentes fichas, diferenciadas en: 2 fichas de notificacio-nes iniciales de reacción transfusional e incidente relacionado con la transfusión, 2 fichas relativas a incidentes relacionados con la donación y con la preparación de componentes, 1 ficha para notificar error en la administración de componentes sanguíneos, 10 fichas de efectos ad-versos de la transfusión (Reacciones hemolíticas; Reacciones alérgicas; Contaminación bacteriana; Edema Pulmonar; Purpura transfusional; enfermedad de injerto contra huésped; infección postransfusional vírica; reacción febril y/o hipotensiva; hemosiderosis postransfusio-nal e infección postransfusional parasitaria) y 1 ficha de incidentes sin efecto/casi incidentes3.
3. SISTEMA ESPAÑOL DE HEMOVIGILANCIA
Se estructura a nivel nacional en 3 niveles organizativos (Figura 2):
– Nivel 1 que corresponde con el Servicio Hospitalario de Transfusión/Centro de transfusión; que a nivel Comunidad Autonoma de Andalu-cia ha sido separado en un responsable hospitalario y un responsable provincial que coincide con el responsable del Centro de Transfusion.
– Nivel 2 o sistema autonómico de hemovigilancia y
– Nivel 3 o Programa Estatal de hemovigilancia (Unidad de Hemovi-gilancia de MSSSSI).
Es responsabilidad de la unidad de hemovigilancia del MSSSI elaborar el informe anual de hemovigilancia, mantener contacto con los coor-dinadores autonómicos, asegurar la homogeneidad en los criterios diagnosticos de la RA, realizar el análisis de la información, efectuar la elaboración de recomendaciones y medidas correctoras o preventivas, informar a la comisión europea (SARE) y colaborar con otras institu-ciones u organismos internacionales.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA58
4 DE LA HEMOVIGILANCIA A LA CALIDAD Y SEGURIDAD TRANSFUSIONAL
4.1 Errores en la administración de componentes sanguíneos y “Casi incidentes”: En el sistema de HV se ha incluido la notificación de am-bas eventualidades, mediante la introducción de fichas especificas para cada uno de ellos. Esto ha permitido su registro y un posterior análisis individualizado de las diversas circunstancias responsables en cada caso, y que pueden haber desencadenado o no una reacción adversa en el receptor:
– Componente sanguíneo incorrectamente transfundido porque no cumple los requisitos establecidos o adecuados para el paciente o bien ha sido administrado a otro paciente independientemente de que sea ABO compatible, se haya transfundido solo una pequeña cantidad del mismo y que no haya determinado una reacción ad-versa.
– Incorrecta identificación de la muestra que se utiliza para la reali-zación de grupo sanguíneo, escrutinio de anticuerpos irregulares o en la realización de las pruebas cruzadas. Deberian notificarse independientemente que se haya detectado en los estudios pre-transfusionales habituales, o en segunda determinación si es el caso, independientemente de que no haya determinado una transfusión incorrecta o de que la muestra no fuera destinada a una transfusión inmediata. Tener en cuenta que dicho error no supone que se vaya a administrar una transfusión ABO incompatible.
– Distribucion de componentes sanguíneos inapropiados o inseguros, que incluiría aquellos componentes que en el momento de su envio no cumplia los requisitos para una transfusion adecuada.
Las incorporación de estas notificaciones ha permitido en la mayoría de los servicios de transfusión, a través de un proceso activo y continuo, la toma de medidas correctoras y preventivas que han determinado progresivamente en una mejora en la seguridad del proceso transfu-sional. Como ejemplo de estas medidas, se puede resaltar la incor-poración de los sistemas de identificación transfusional (pulseras, y utilización de sistema de código de barras en múltiples procedimientos del proceso de transfusion…), la incorporación de sistemas electró-nicos junto con check-list a la hora de extracción de las muestras y en el momento de inicio de la transfusión, o la realización de un segundo grupo a cabecera del enfermo previo al inicio de la transfusion. En este proceso de mejora ha tenido un efecto positivo la incorporación a los servicios de transfusión de diferentes sistemas de gestión de la calidad acreditados a través de diferentes agencias u organismos externos8. Su incorporación ha permitido detectar errores o desviacio-nes a los procedimientos y en el proceso de transfusión e incorporar medidas correctoras o preventivas a los mismos. Dichos errores pueden producirse en los diferentes pasos del proceso de transfusión:
– Durante la decisión clínica de transfusión.
– Durante la obtención de la muestra y la cumplimentación de la so-licitud de transfusión.
– En la realización de las pruebas pretransfusionales en el laboratorio.
– Entrega del componente solicitado al área clínica.
– Durante la administración del componente sanguíneo.
– En el proceso de vigilancia de la transfusión.
Asimismo, la implementación del uso de indicadores de transfu-sión permite monitorizar y evaluar la calidad del proceso hemoterapia asi como el cumplimiento de las diferentes guias clínicas. Los Indica-dores pueden ser internos o externos o bien indicadores de estructura, proceso o resultados. Su objetivo es cuantificar, evaluar tendencias y detectar desviaciones de los procesos/procedimientos durante las diferentes fases del proceso de transfusión.
4.2 Reduccion de reacciones transfusionales impredecibles: Los siste-mas de HV a través del análisis de los efectos adversos de la transfusión ha permitido y continúan permitiendo una reducción en determinadas complicaciones a la administración de componentes sanguíneos, y en la consiguiente mejora en la calidad y seguridad de los mismos.
Infecciones bacterianas: La contaminación bacteriana de productos sanguíneos es infrecuente pero comporta un resultado fatal en el re-ceptor cuando tiene lugar. Desde 2007, dos fallecimientos en España se ha producido por este motivo. En la mayoría de los casos se encuen-tran implicados concentrados de plaquetas contaminados por flora de origen cutáneo (3 casos de contaminación reportados en informe de 2015)8. Diversas estrategias han intentado disminuir su incidencia mediante el desvio de los primeros ml de sangre extraida o mediante la incorporación a los procedimientos de extracción de mejoras en el proceso de limpieza y desinfección del sitio de punción. Otras estrate-gias, ha sido reducir la caducidad de los concentrados de plaquetas a 4 dias, o bien mediante el uso de técnicas de cribaje de contaminación bacteriana que además pueden permitir un incremento de los días de almacenamiento y un consiguiente mejor aprovechamiento de este componente sanguíneo9.
Lesion pulmonar aguda secundaria a transfusión (TRALI): Las revisio-nes de los registros de incidentes tranfusionales mostraron una clara relación de esta complicación con los productos que contenían plasma (PFC y CP). Ese riesgo era 7-8 veces mayor en relación a los concen-trados de hematies. Se ha detectado una inequívoca relación con do-nantes de sexo femenino, lo que ha determinado recomendaciones del uso de donantes masculinos como fuente exclusiva de plasma, asi como la posible eliminación de donantes mujeres en los pooles de plaquetas o como uso de donantes de aféresis salvo que se realicen estudios previos para descartar la existencia de anticuerpos antiHLA o antiHNA10,11.
Purpura postransfusional y EICH secundaria a transfusión (EICH-t): Aun-que la introducción de la leucorredución universal fue motivada para prevenir determinados riesgos infecciosos, los datos actuales de que se disponen muestran que tras su incorporación se ha producido una reducción en la alosensibilizacion HLA y HPA lo que ha condicionado una reduccion en el número de casos reportados de purpura postrans-fusional, a la par que ha disminuido la refractariedad plaquetar. Por otro lado, pese a la notificación de casi incidentes motivados por la transfusión de productos sanguíneos no irradiados en pacientes con

PONENCIAS 59
una clara indicación para los mismos, no se ha detectado ningún caso de EICH-t en pacientes que han recibido productos leucodepleciona-dos12.
4.3 Uso óptimo de componentes sanguíneos. Consiste en asegurar un uso seguro de los componentes sanguineos sin reacciones adversas ni errores en su administración, con eficacia clínica de forma que cumple con su objetivo de beneficio al paciente al que se administra y con eficiencia realizándose en el momento que el paciente lo precisa evitando transfusiones innecesarias.
Edema Pulmonar Cardiogenico (TACO): Es una de las principales causas de mortalidad relacionadas con la transfusión, cuya incidencia (apro-ximadamente 1% de pacientes transfundidos) no ha descendido a lo largo de los años sino que ha ido en aumento, lo que sugiere que ha sido infrareportada a lo largo de estos años8,12. El análisis de los datos de HV muestran que ocurre fundamentalmente en la población anciana (60% con mas de 70 años), aunque también se encuentran en riesgo las mujeres embarazadas y los neonatos. La sobretransfusión es un factor evitable en su desarrollo. Las reglas de cálculo de incremento de valores de hemoglobina por número de concentrados de hematíes administrados no suelen tener en cuenta el peso de los pacientes. Por ello es importante su consideración, asi transfusiones de 4 mL/kg peso receptor determinaran un aumento de 1 gr/dL. Así mismo, hay que tener en consideración a la hora de prevenir su desarrollo la existencia de factores médicos adicionales, como la insuficiencia cardiaca o renal que acrecientan su riesgo. El desarrollo de iniciativas formativas en los diferentes centros hospitalarios y así como de guías clínicas podrían minimizar su riesgo en la práctica transfusional diaria13,14.
Transfusiones innecesarias, demoradas o infrautilizadas: las transfusio-nes no siempre son administradas apropiadamente. Errores analíticos o una insuficiente evaluación clínica de los pacientes puede motivar una transfusión innecesaria. Ejemplos son la tendencia a la sobretrans-fusion en pacientes con sangrado digestivo, que se puede reducir con una monitorización mas estrecha de los niveles de hemoglobina, o la trasfusión repetida de concentrados de plaquetas en pacientes con refractariedad inmune a las mismas. Asi mismo situaciones de apa-rente emergencia puede conllevar a un uso inapropiado de sangre Rh negativa – conlleva el despilfarro de un recurso escaso con un incremento del riesgo de alosensibilización-. Además las trasfusio-nes demoradas pueden suponer un riesgo para la seguridad de los pacientes. Un ejemplo puede ser un retraso en la toma de decisión para iniciar un procedimiento de transfusión masiva, o situaciones de baja disponibilidad de hemoderivados,…
Alosensibilizacion en pacientes con anemia hemolítica congénita (dre-panocitosis): Los análisis de los registros de reacciones hemolíticas han mostrado una sobrerrepresentación de pacientes con dreapanocitosis, que podría reducirse con un adecuado fenotipaje antes de la prime-ra transfusión o mediante un genotipaje de los antígenos de grupo sanguíneo12. Ademas mucho de estos pacientes acuden a diferentes centros que hace necesario crear mecanismos de acceso a la informa-ción de posibles aloanticuerpos previamente detectados.
5. RETOS DE FUTURO
Se hace necesario continuar incorporando medidas que puedan au-mentar la calidad y la seguridad en la transfusión sanguínea mediante sistemas que aseguren una adecuada formación y capacitación del personal que transfunde y prescribe, la utilización de check-list en la cabecera del paciente, tanto en la extracción de muestras como en la administración del componente sanguíneo, la utilización de guias y procedimientos que estén consensuados, la realización de auditorias al proceso de transfusión tanto al procedimiento como a las indicaciones. En este aspecto la necesidad de incorporar la figura de enfermero/a de hemovigilancia es un punto clave en las mejoras futuras.
BIBLIOGRAFIA
1. https://www.shotuk.org/
2. http://ansm.sante.fr/
3. http://www.msssi.gob.es/profesionales/saludPublica/medicinaTrans-fusional/home.htm
4. Resolucion de constitución y puesta en funcionamiento del sistema de hemovigilancia en los distintos centros del Sistema Andaluz de Salud 683/03 de 11 de Agosto de 2003.
5. Real Decreto 62/2003 de 17 de Enero, por el que se crea el sistema Estatal para la seguridad transfusional
6. Real Decreto 1088/2005 20 de septiembre sobre los requisitos tecnicos y condiciones minimas de la hemodonación y de los centros y servicios de transfusión
7. Orden SCO/322/2007 de 9 Febrero de 2007 sobre trazabilidad y notifica-cion de reacciones y efectos adversos graves de la sangre
8. Real Decreto 1343/2007, de 11 Octubre. Por el que se establecen normas y especificaciones relativas al sistema de calidad de los centros y servicios de transfusión.
9. Informe Estatal de Hemovigilancia 2015. Ministerio de Sanidad, Servicios Sociales e Igualdad. Diciembre 2015
10. Wagner B, Grabein B. “Sterility Testing of Blood Components and Advanced Therapy Medicinal Products” (Munich, April 29, 2010) Or-ganized by the DGTI Section “Safety in Hemotherapy” - Meeting Re-port. Transfusion Medicine and Hemotherapy. 2011;38(5):334-336. doi:10.1159/000331397.
11. Toy P, Gajic O, Bacchetti P, et al. Transfusion related acute lung injury: incidence and risk -factors. Blood. 2012, 119: 1757-67.
12. Eder AF, Herron RM, Strupp A, Dy A, White J, Notari EP, Dodd RY, Ben-jamin RJ. Effective reduction of transfusion-related acute lung injury with male-predominant plasma strategy in the American Red Cross (2006–2008) Transfusion. 2010;50:1732–42
13. PHB Bolton-Maggs (Ed) D Poles et al. on behalf of the Serious Hazards of Transfusion (SHOT) Steering Group. The 2015 Annual SHOT Report (2016).
14. Murphy EL, Kwaan N, Looney MR, et al. Risk Factors and Outcomes in Transfusion-associated Circulatory Overload. The American Journal of Medicine. 2013;126(4):357.e29-357.e38.
15. Roubinian, N. H., Hendrickson, J. E., Triulzi, D. J., et al. (2017), Incidence and clinical characteristics of transfusion-associated circulatory overload using an active surveillance algorithm. Vox Sang 2017; 112: 56–63.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA60
El tratamiento de la trombocitemia esencial (TE) y la policitemia vera (PV) va dirigido a prevenir la complicaciones trombohemorrágicas, que son la principal causa de morbimortalidad de estos pacientes1-3. Así, la edad superior a 60 años y el antecedente de trombosis o hemorragia mayor definen al grupo de alto riesgo en el que estaría indicado el tra-tamiento citorreductor. Los factores generales de riesgo cardiovascular (HTA, hipercolesterolemia, tabaquismo, diabetes), el perfil mutacional (JAK2 o CALR) y la leucocitosis se asocian al riesgo de trombosis, por lo que deben tenerse en cuenta de cara a individualizar el tratamiento1. En este sentido, se han publicado dos índices pronósticos para estimar de forma más precisa el riesgo trombótico en la TE: el IPSET-trombosis4 y el IPSET-trombosis revisado5.
La recomendación general para el manejo de los pacientes con PV es mantener el hematocrito (Hto) por debajo del 45%, en base a los resultados del estudio prospectivo aleatorizado CYTO-PV6. Asimismo, es práctica habitual el uso del ácido acetilsalicílico (AAS) a bajas dosis en todos los pacientes con TE y PV, independientemente del grupo de riesgo, en base a un ensayo clínico aleatorizado que demostró el bene-ficio de este fármaco en la PV7. Sin embargo, en estudios retrospectivos donde se comparó el empleo de AAS frente a la abstención terapéutica en pacientes con TE de bajo riesgo se observó que el beneficio del AAS se circunscribe a los pacientes con mutación del gen JAK2 (reducción del riesgo de trombosis venosa) o con factores de riesgo cardiovascular (reducción del riesgo de trombosis arterial)8. De hecho, el uso de AAS en pacientes de bajo riesgo con mutación de CALR no se asoció a una tasa menor de complicaciones trombóticas y, en cambio, sí a una ma-yor incidencia de sangrado9. Por otro lado, los pacientes con trombosis venosa se manejan con anticoagulación oral, de duración indefinida en la mayoría de casos10,11.
En la actualidad, la hidroxiurea (HU) es el agente citorreductor de elec-ción1, si bien está en curso un ensayo clínico internacional donde se compara su eficacia con la del interferón pegilado en pacientes con TE y PV de alto riesgo (NCT01259856). La seguridad a largo plazo de la HU se ha visto refrendada por los datos de un estudio sueco que incluyó más de 10.000 pacientes con neoplasias mieloproliferativas donde no se observó relación entre la exposición a HU y el riesgo de evolución a leucemia12. El tratamiento citorreductor disminuye la incidencia de trombosis, pero no está claro cuál es el mejor parámetro de respuesta que permite anticipar el pronóstico a largo plazo. Así, los dos estudios aleatorizados que establecieron a la HU como fármaco de elección de la TE de alto riesgo utilizaron distintos dinteles de cifra de plaquetas como objetivo del tratamiento citorreductor: <600 x 109/L en el es-tudio de Cortelazzo y cols. (HU vs placebo)13 y <400 x 109/L en el de Harrison y cols. (HU+AAS vs anagrelida+AAS)14. De cara a poder com-parar de una forma más uniforme la eficacia de los nuevos fármacos se han elaborado criterios estandarizados de respuesta y resistencia/intolerancia al tratamiento citorreductor de la TE y la PV15-17. En la práctica clínica, alrededor de una cuarta parte de los pacientes con TE y PV de alto riesgo cumple durante su evolución alguno de los criterios
de intolerancia o resistencia al tratamiento con HU18-20. Mientras la intolerancia a la HU carece de impacto pronóstico, el desarrollo de resistencia se asocia a una peor supervivencia. Por otro lado, datos recientes del Registro Español de Policitemia Vera sugieren que los pacientes con PV tratados con HU que requieren tres o más fleboto-mías al año para mantener el Hto < 45% tienen un mayor riesgo de trombosis21. El tratamiento de los pacientes resistentes o intolerantes a HU debe individualizarse, evitando en lo posible el empleo de agentes leucemógenos. Alternativas terapéuticas para estos pacientes son el busulfán22, el interferón pegilado23, la anagrelida24 y el ruxolitinib25.
BIBLIOGRAFIA
1. Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761-770.
2. Finazzi G. How to manage essential thrombocythemia. Leukemia. 2012;26(5):875-882.
3. Passamonti F. How I treat polycythemia vera. Blood. 2012;120(2):275-284.
4. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):5128-5133; quiz 5252.
5. Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5:e369.
6. Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22-33.
7. Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114-124.
8. Alvarez-Larran A, Cervantes F, Pereira A, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood. 2010;116(8):1205-1210; quiz 1387.
9. Alvarez-Larran A, Pereira A, Guglielmelli P, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica. 2016;101(8):926-931.
10. Hernandez-Boluda JC, Arellano-Rodrigo E, Cervantes F, et al. Oral anticoagulation to prevent thrombosis recurrence in polycythemia vera and essential thrombocythemia. Ann Hematol. 2015;94(6):911-918.
11. De Stefano V, Ruggeri M, Cervantes F, et al. High rate of recurrent venous thromboembolism in patients with myeloproliferative neoplasms and effect of prophylaxis with vitamin K antagonists. Leukemia. 2016;30(10):2032-2038.
12. Bjorkholm M, Derolf AR, Hultcrantz M, et al. Treatment-related risk factors for transformation to acute myeloid leukemia and
Tratamiento de la trombocitemia esencial y de la policitemia veraJuan Carlos Hernández Boluda
Servicio de Hematología, Hospital Clínico Universitario, INCLIVA, Valencia

PONENCIAS 61
myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410-2415.
13. Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332(17):1132-1136.
14. Harrison CN, Campbell PJ, Buck G, et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353(1):33-45.
15. Barosi G, Mesa R, Finazzi G, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778-4781.
16. Barosi G, Besses C, Birgegard G, et al. A unified definition of clinical resistance/intolerance to hydroxyurea in essential thrombocythemia: results of a consensus process by an international working group. Leukemia. 2007;21(2):277-280.
17. Barosi G, Birgegard G, Finazzi G, et al. A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol. 2010;148(6):961-963.
18. Hernandez-Boluda JC, Alvarez-Larran A, Gomez M, et al. Clinical evaluation of the European LeukaemiaNet criteria for clinicohaematological response and resistance/intolerance to hydroxycarbamide in essential thrombocythaemia. Br J Haematol. 2011;152(1):81-88.
19. Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363-1369.
20. Alvarez-Larran A, Kerguelen A, Hernandez-Boluda JC, et al. Frequency and prognostic value of resistance/intolerance to hydroxycarbamide in 890 patients with polycythaemia vera. Br J Haematol. 2016;172(5):786-793.
21. Alvarez-Larran A, Perez-Encinas M, Ferrer-Marin F, et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica. 2016.
22. Alvarez-Larran A, Martinez-Aviles L, Hernandez-Boluda JC, et al. Busulfan in patients with polycythemia vera or essential thrombocythemia refractory or intolerant to hydroxyurea. Ann Hematol. 2014;93(12):2037-2043.
23. Masarova L, Patel KP, Newberry KJ, et al. Pegylated interferon alfa-2a in patients with essential thrombocythaemia or polycythaemia vera: a post-hoc, median 83 month follow-up of an open-label, phase 2 trial. Lancet Haematol. 2017.
24. Hernandez-Boluda JC, Pereira A, Cervantes F, et al. Clinical evaluation of the European LeukemiaNet response criteria in patients with essential thrombocythemia treated with anagrelide. Ann Hematol. 2013;92(6):771-775.
25. Vannucchi AM. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(17):1670-1671.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA62
Las neoplasias mieloproliferativas Ph negativas (NMs Ph-neg.) comprenden, según la reciente clasificación de la OMS, las siguientes entidades: policitemia vera (PV), trombocitemia esencial (TE), mielofibrosis primaria (MFP), leucemia neutrofílica crónica (LNC), leucemia eosinofílica crónica y las neoplasias mieloproliferativas inclasificables. Todas ellas se caracterizan desde el punto de vista molecular, porque no presentan la translocación t(9;22) que origina el gen de fusión BCR-ABL1.
Las neoplasias mieloproliferativas Ph negativas clásicas, que comprenden la TE, la PV y la MFP, se caracterizan por la presencia de la mutación p.V617F que afecta al gen JAK2. La mutación JAK2V617F se detecta en el 90-95% de pacientes con PV, 60% de pacientes con TE y 60% de pacientes con MFP. En casos de PV y eritrocitosis idiopática negativas para JAK2V617F, se han descrito mutaciones en el exón 12 de JAK2. Por otra parte, se han descrito mutaciones en el gen MPL (que codifica el receptor de la trombopoyetina), en el 8% de MFP y en el 5% de TE, y mutaciones en el gen CALR en el 50-70% de los pacientes con TE y MFP que no presentan mutaciones ni en el gen JAK2 ni en el gen MPL.
La mutaciones en los genes JAK2, MPL y CALR, se consideran mutaciones driver de la PV, TE y MFP y forman parte de los actuales criterios diagnósticos de estas entidades.
Se han descrito mutaciones en un pequeño porcentaje de pacientes con NMs Ph-neg. en difentes genes que se consideran mutaciones non-driver. Por su función se clasifican en genes implicados en señalizacion intraceular: LNK, CBL; genes implicados en regulación epigenética:TET2, ASXL1, IDH1/IDH2, IKZF1, EZH2, DNMT3A; y genes implicados en el procesamiento del ARNmensajero (o splicing): SF3B1, SRSF2, U2AF1. Las mutaciones en estos genes se detectan principalmente en pacientes con MFP, y en el caso de los genes ASXL1, SRSF2, EZH2 e IDH1/2 podrían tener relevancia pronóstica.
Por último, la leucemia neutrofilica cronica se caracteriza por la presencia de mutaciones en el gen CSF3R que codifica para el receptor del factor estimulante de colonias de granulocitos. Las mutaciones del gen CSF3R pueden ser mutaciones que afectan al dominio transmembrana, de las cuales la mas frecuente es la mutación p.T618I, o bien mutaciones truncantes en el extremo carboxi-terminal. Las mutaciones en este gen también se pueden detectar en la neutropenia congénita severa y en la leucemia mieloide crónica atípica.
CONCLUSIONES
El diagnóstico de las NMs Ph-neg. se ha basado históricamente en criterios clínicos, biológicos e histológicos. La descripción de mutaciones en los genes JAK2, MPL, CALR y CSF3R, ha supuesto un cambio en la aproximación diagnóstica de estos pacientes.
Alteraciones moleculares en las neoplasias mieloproliferativas crónicas Filadelfia negativasBeatriz Bellosillo
Servicio de Anatomía Patológica, Hospital del Mar, Barcelona

PONENCIAS 63
INTRODUCCIÓN
El tratamiento de la leucemia mieloide crónica (LMC) con inhibido-res de la tirosín cinasa (ITC) ABL1 ha mejorado significativamente el pronóstico de los pacientes. Siendo actualmente la esperanza de vida para los que reciben tratamiento en primera fase crónica muy similar al de la población general de la misma edad1.
Hasta hace pocos años, el único tratamiento curativo en la LMC era el trasplante alogénico de precursores hematopoyéticos (alo-TPH), sin embargo en algunos de los pacientes tratados con imatinib y en una proporción aun mayor de los tratados con dasatinib o nilotinib, es posible alcanzar respuestas moleculares completas (RMC) o profundas (RMP) (BCR-ABL ratio ≤0,0032%) de forma mantenida. En muchos de ellos es posible discontinuar el tratamiento con ITC sin tener que rein-troducirlo posteriormente, es lo que conocemos como supervivencia libre de tratamiento (SLT)2.
ESTUDIOS CLÍNICOS DE DISCONTINUACIÓN DE IMATINIB
El primer estudio clínico de discontinuación de imatinib fue llevado a cabo por el grupo francés de LMC3. En él se interrumpió el trata-miento en 100 pacientes en RMP de al menos 2 años de duración y con una duración mínima de tratamiento con imatinib de 3 años. A los 24 meses de seguimiento, el 41% de los pacientes mantenían la RMP y a los 60 meses el 38% no ha tenido que reiniciar imatinib4. Tras este primer estudio, se han puesto en marcha otros cuyos resultados se resumen en la tabla 1. Uno de los más importantes por el número de pacientes y el diseño del mismo es el estudio europeo Euro-SKI (ClinicalTrials.gov numbers: NCT01596114)5. En el cual, un total de
448 pacientes tratados con imatinib y que presentaban al menos una RM 4.0 (BCR-ABL ≤0,01%) durante 1 año y al menos 3 años de tra-tamiento se les interrumpió imatinib. A los 6 meses de suspenderlo, en aquellos pacientes con un duración total de tratamiento superior a 5.8 años la probabilidad de mantener la respuesta molecular ma-yor (BCR-ABL ≤0,01%) fue del 65.5% frente al 42.6% en los tratados menos de 5.8 años.
ESTUDIOS DE DISCONTINUACIÓN CON DASATINIB O NILOTINIB.
Tanto dasatinib como nilotinib inducen una mayor proporción de respuestas moleculares profundas y en un tiempo menor cuando se comparan con imatinib, incrementándose de esta manera el número de pacientes con LMC que serán posibles candidatos a discontinuar el tratamiento. En el ensayo clínico ENESTnd con 5 años de seguimiento, la incidencia acumulada de RM 4.5 fue de el 54% de los pacientes tratados con nilotinib 300mg/12h frente al 31% de los que recibieron imatinib. En el DASISION, la incidencia acumulada de RM 4.5 a los 5 años fue del 42% y del 33% para dasatinib e imatinib respectiva-mente6,7.
La experiencia clínica en la discontinuación de ITC de segunda gene-ración es más limitada que con imatinib y en muchos de los estudios en marcha los pacientes previamente habían iniciado tratamiento con imatinib, recibiendo dasatinib o nilotinib por intolerancia o resistencia.
En la tabla 2 se resumen los datos actualizados de los principales es-tudios de discontinuación con Dasatinib o Nilotinib.
Supervivencia libre de tratamiento en la leucemia mieloide crónica: ¿en qué pacientes es posible la curación?Antonio Jiménez Velasco
UGC de Hematología y Hemoterapia. Hospital Regional Universitario de Málaga
TABLA 1. Principales estudios de discontinuación con imatinib2
STIM1 STIM2 ALLG-CML8 A-STIM
N 100 200 40 80
Tratamiento previo INF luego imatinib ≥3 años Imatinib ≥3 años Imatinib ≥3 años Imatinib ≥3 años
Respuesta requerida RMC
≥2 años
RMC
≥2 años
RMC
≥2 años
RMC
≥2 años
Definición de recaída Pérdida de la RMM o
incremento de BCR-ABL
≥1-log
Pérdida de la RMM o
incremento de BCR-ABL
≥1-log
Pérdida de la RMM o pérdida
confirmada de RM 4.5
Pérdida de la RMM
SLT%
mediana/tiempo
39%
(55 meses)
46%
(2 años)
45%
(42 meses)
64%
(23 meses)
Abreviaturas. IFN: interferón, RMC: respuesta molecular completa, RMM: respuesta molecular mayor (BCR-ABL ≤0,1%), RM 4.5: respuesta molecular 4.5 (BCR-ABL ≤0,0032%),
SLT: supervivencia libre de tratamiento.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA64
¿EN QUE PACIENTES PODEMOS PLANTEAR LA DISCONTINUACIÓN DEL TRATAMIENTO CON ITC?
Siguiendo las recomendaciones de la European LeukemiaNet, ésta sólo deberá realizarse en el contexto de ensayos clínicos (EECC) con-trolados o bajo ciertas circunstancias excepcionales, siendo requisito indispensable realizar una correcta monitorización molecular de los pacientes en un laboratorio con procedimientos estandarizados para el seguimiento del reordenamiento BCR-ABL8,9.
Recientemente Hughes et al. basándose en las evidencias de los tra-bajos publicados, han establecido una serie de requisitos clínicos y moleculares, que nos pueden ayudar a seleccionar mejor a los pacien-tes dependiendo del grupo en el que se encuadren10. En la tabla 3 se especifican estos criterios y grupos.
Aún existiendo evidencias que soportan que perfil de paciente tiene más posibilidades de éxito en la discontinuación, todavía hay muchas cuestiones sin resolver y cuyas respuestas sólo se podrán contestar mediante la realización de EECC.
FACTORES INMUNOLÓGICOS IMPLICADOS EN EL ÉXITO DE LA DISCONTINUACIÓN Y LA SUPERVIVENCIA LIBRE DE TRATAMIENTO CON ITC
A pesar de tener niveles similares de respuesta molecular profunda y ser la duración del tratamiento iguales, el éxito en la discontinuación varía entre pacientes. Sabemos que la célula madre leucémica es in-sensible a la acción de los ITC, pero a pesar de esto hay pacientes que tras discontinuar el ITC permanecen en respuesta molecular completa.
TABLA 2. Principales estudios de discontinuación con ITC de segunda generación (dasatinib o nilotinib)
DADI15 Stop
2G-ITC16range; 12-65ENEST freedom17 ENESTop18 DASfree19
N 63 60 190 125 30
ITC Dasatinib Nilotinib o dasatinib Nilotinib Nilotinib Dasatinib
RM antes de
discontinuar
RM 4.0 ≥ 1 año RM 4.5 ≥ 2 años RM 4.5 ≥ 1 año RM 4.5 ≥ 1 año RM 4.5 ≥ 1 año
Criterio para reiniciar
ITC
Pérdida de RM 4.0 Pérdida de RMM Pérdida de RMM Pérdida de RM 4.0 Pérdida de RMM
Mediana de
tratamiento con ITC
82 meses 76 meses 44 meses 88 meses NR
SLT al año 48% 63% 52% 58% 63%
Abreviaturas. RM: respuesta molecular, RMC: respuesta molecular completa, RMM: respuesta molecular mayor (BCR-ABL ≤0,1%), RM 4.0: respuesta molecular 4.0 (BCR-ABL
≤0,01%), RM 4.5: respuesta molecular 4.5 (BCR-ABL ≤0,0032%) , SLT: supervivencia libre de tratamiento.
TABLA 3. Criterios para la selección de pacientes que podrían ser candidatos a discontinuar el tratamiento con ITC (adaptado de Hughes et al.10)
Criterio Grupo A Grupo B Grupo C
Discontinuación Si * No
Sokal score No ser de alto riesgo Alto riesgo -
Reordenamiento BCR-ABL Típico (e13a2 o e14a2) Atípico pero cuantificable No cuantificable
Evolución de la LMC Fase Crónica Resistente previa o con mutaciones Fase acelerada o blástica previa
Respuesta al primer ITC Óptima Alarma Fallo
Duración total del tratamiento >8 años 3-8 años <3 años
Profundidad de la RM obtenida RM 4.5 RM 4.0 No RM 4.0
Duración de la RMP en un laboratorio
estandarizado
>2 años 1-2 años <1 año
Grupo A: Los pacientes que reúnen todos los criterios serían candidados para discontinuar tratamiento.
*Grupo B: Sólo se debería considerar la discontinuación bajo determinadas circunstancias como la presencia de una toxicidad clínicamente significativa o en casos de planificar
el embarazo.
Grupo C: La interrupción del tratamiento no es recomendada.
Abreviaturas. RM: respuesta molecular, RMP: respuesta molecular profunda, RM 4.0: respuesta molecular 4.0 (BCR-ABL ≤0,01%), RM 4.5: respuesta molecular 4.5 (BCR-ABL
≤0,0032%).

PONENCIAS 65
Es decir, deben existir otros factores biológicos propios del individuo que hagan que posible la curación de la LMC.
En este sentido, sabemos que el sistema inmune juega un papel funda-mental en la curación de la LMC. Hasta la llegada de los ITC a principios del siglo XX, el alo-TPH era el tratamiento de elección en la LMC debido a la capacidad curativa del efecto injerto contra la LMC, del mismo modo aquellos pacientes en recaída tras un alo-TPH son muy sensibles a la infusión de leucocitos del donante11,12.
Recientemente, en los EECC de discontinuación Euro-SKI y TIGER se ha estudiado el papel que desempeña el sistema inmune de los pa-cientes en el éxito de la discontinuación. Así, en aquellos que antes de interrumpir el tratamiento con ITC tenían una menor expresión de CD86 (ligando de la molécula inhibidora del linfocito T, CTLA-4) en sus células dendríticas, las probabilidades de permanecer libres de tratamiento se incrementan hasta el 70% frente al 30% en el otro subgrupo13. De igual manera, la supervivencia libre de tratamiento fue significativamente mayor en los pacientes que presentan en sus células NK un fenotipo activado frente a los que no, con una supervivencia libre de recaída molecular (pérdida de la RMM) a los 6 meses del 71% frente al 52% respectivamente14.
BIBLIOGRAFÍA
1. Hochhaus A, Larson RA, Guilhot F, et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017;376(10):917–927.
free remission in chronic myeloid leukemia. Leukemia. 2016;30(8):1638–1647.
3. Mahon FX, Rea D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete mo-lecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035.
4. Etienne G, Rea D, Guilhot J, et al. Long-Term Follow-up of the French 1 Stop Imatinib Study (STIM1) in Chronic Myeloid Leukemia Patients. J. Clin. Oncol. 2017;35(3):298–305.
5. Francois-xavier Mahon, Johan Richter, Joelle Guilhot, et al. Cessation of Tyrosine Kinase Inhibitors Treatment in Chronic Myeloid Leukemia Patients with Deep Molecular Response: Results of the Euro-Ski Trial [abstract]. Blood. 2016;128:632.
6. Hochhaus a, Saglio G, Hughes TP, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–1054.
7. Cortes JE, Saglio G, Kantarjian HM, et al. Final 5-year study results of DA-SISION: The dasatinib versus imatinib study in treatment-Naive chronic myeloid leukemia patients trial. J. Clin. Oncol. 2016;34(20):2333–2340.
8. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recom-mendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884.
9. Cross NCP, White HE, Colomer D, et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia. 2015;29(5):999–1003.
10. Hughes TP, Ross DM. Moving treatment-free remission into mainstream clinical practice in CML. Blood. 2016;128(1):17–23.
11. Ilander M, Hekim C, Mustjoki S. Immunology and immunotherapy of chronic myeloid leukemia. Curr. Hematol. Malig. Rep. 2014;9(1):17–23.
12. Radujkovic A, Guglielmi C, Bergantini S, et al. Donor Lymphocyte Infu-sions for Chronic Myeloid Leukemia Relapsing after Allogeneic Stem Cell Transplantation: May We Predict Graft-versus-Leukemia Without Graft-versus-Host Disease? Biol. Blood Marrow Transplant. 2015;21(7):1230–1236.
13. Schütz C, Inselmann S, Sausslele S, et al. Expression of the CTLA-4 li-gand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of dis-ease recurrence after treatment discontinuation in CML. Leukemia. 2017;31(4):829–836.
14. Ilander M, Olsson-Strömberg U, Schlums H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinua-tion in chronic myeloid leukemia. Leukemia. 2016;Dec 16(Epub ahead of print):PMID: 27890936.
15. Imagawa J, Tanaka H, Okada M, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol. 2015;2(12):e528–e535.
16. Rea D, Nicolini FE, Tulliez M, et al. Discontinuation of dasatinib or nilotinib in CML-interim analysis of the STOP2G-TKI study. Blood. 2016;129(7):846–855.
17. Hochhaus A, Masszi T, Giles FJ, et al. Treatment-free remission following frontline nilotinib in patients with chronic myeloid leukemia in chronic phase: results from the ENESTfreedom study. Leukemia. 2017;Mar 17(Epub ahead of print):PMID: 28218239.
18. Timothy P. Hughes, Carla Maria Boquimpani, Naoto Takahashi, et al. Treatment-free remission in patients with chronic myeloid leukemia in chronic phase according to reasons for switching from imatinib to nilo-tinib: subgroup analysis from ENESTop [abstract]. Blood. 2016;128:792.
19. Neil P. Shah, Ronald Paquette, Martin C. Müller, Susanne Saussele, Val-entin Garcìa-Gutiérrez, Antonio Jiménez-Velasco, et al. Treatment-free remission (TFR) in patients with chronic phase chronic myeloid leukemia (CML-CP) and in stable deep molecular response (DMR) to dasatinib — the DASfree Study [abstract]. Blood. 2016;128:1895.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA66
La trombofilia ha sido definida como “aquella situación de alteración o desequilibrio del sistema hemostático, congénita o adquirida, que condiciona una tendencia trombótica superior a la de sujetos norma-les, en ciertas situaciones de riesgo o incluso de manera espontánea” (1). Aunque, en ocasiones son situaciones adquiridas secundarias a otras patologías (neoplasias, colagenosis, enfermedad inflamatoria intestinal, síndrome nefrótico, etc), la predisposición hereditaria a la misma, o trombofilia congénita, ha sido objeto de una importante investigación básica y clínica desde el descubrimiento del déficit de antitrombina en 1965 (2), especialmente intensificada tras la descrip-ción de una tasa estimada de heredabilidad de la trombosis del 60% en el año 2000 por el trabajo colaborativo del grupo de hospital Sant Pau y la SFBR de San Antonio (3). Además, el cambio de concepción de la predisposición hereditaria a la enfermedad tromboembólica hacia una “enfermedad compleja” en la que interaccionan múltiples factores genéticos y ambientales (4) ha conducido a la búsqueda del mayor nú-mero posible de factores genéticos predisponentes con la finalidad de conseguir la mejor estimación del riesgo posible (5, 6). Sin embargo, una predisposición genética no es igual que una enfermedad, y no to-dos los individuos con trombofilia sufrirán un evento tromboembólico venoso (ETEV) durante su vida (7, 8) y los estudios biológicos de trom-bofilia son complejos y costosos. Como consecuencia, el diagnóstico de una situación de trombofilia subyacente es complicado en la práctica clínica y sólo suele realizarse después de que un paciente ya ha sufrido un ETEV, ya que el screening sistemático de trombofilia no se considera justificado por no ser coste-efectivo (9). Además, las indicaciones de realización de un estudio de trombofilia en la práctica clínica habitual
son todavía controvertidas y objeto de debate (10, 11) e, incluso, algu-nas sociedades científicas de Hematología han promovido campañas para que se dejen de realizar en algunas situaciones clínicas concretas (12, 13), al considerar que la detección de una trombofilia subyacente tiene escasa repercusión sobre el riesgo de recurrencia de un ETEV y que la duración del tratamiento anticoagulante debe determinarse por una serie de parámetros en los que no se incluye la trombofilia (14). Por esta razón algunas instituciones han recomendado analizar la rentabilidad de los estudios con la finalidad de implantar guías lo-cales de estudio de trombofilia que permitan aumentar su eficiencia y disminuir costes e inconvenientes para los pacientes (15).
Partiendo de la base de que no está justificado el estudio indiscrimi-nado de trombofilia y que se debe reservar para aquellos pacientes en que pueda cambiar el manejo preventivo o terapéutico (16, 17), se plantea la revisión de en qué pacientes sí se considera coste-efectiva la realización del estudio de trombofilia (porque pueda influir en el riesgo de recurrencia y, por tanto, en la duración del tratamiento an-ticoagulante, o bien por las implicaciones preventivas que pueda tener sobre los familiares en primer grado portadores asintomáticos), de qué pruebas debe constar dicho estudio y cuál es el momento adecuado de realizarlo. Aunque los pacientes candidatos ya habían sido pro-puestos por la Organización Mundial de la Salud (OMS) y la Sociedad Internacional de Trombosis y Hemostasia (ISTH) hace más de 20 años (18, 19), el tema ha seguido siendo objeto de intensa controversia existiendo revisiones recientes que definen la población a estudio (20) que, paradójicamente, coincide bastante con la ya definida por aquellos organismos (Tabla-I)
¿Es necesario seguir haciendo estudios de trombofilia hoy día?Dr. Javier Rodríguez Martorell
UGC de Hematología y Hemoterapia HH. UU. Virgen del Rocío, Sevilla
Paciente con ETEV y edad menor de 50 años.
TABLA I. Indicaciones propuestas para la realización de un estudio de trombofilia. Tomada de Colucci (20), traducida y ligeramente modificada

PONENCIAS 67
Respecto a las pruebas a realizar hay consenso general (16, 17, 21, 22) en que sólo debe incluir alteraciones epidemiológicamente fre-cuentes y relevantes para la toma de decisiones clínicas (Tabla II). El estudio se debe realizar fuera del momento agudo de la trombosis (para evitar falsos déficit de anticoagulantes naturales por consumo); es conveniente anticoagular durante 3 meses y, después, suspender o cambiar a HBPM durante 4 semanas, antes de extraer las muestras (la heparina, las antivitaminas-K y los anticoagulantes orales directos inducen diferentes artefactos en los resultados del estudio de trom-bofilia (Tabla-III). En mujeres no se debe hacer durante la gestación ni durante el uso de anticonceptivos hormonales combinados (ya que los estrógenos inducen alteraciones en la proteína-S y el test de resistencia a proteína-C activada que pueden originar falsos positivos).
Todos estos aspectos serán revisados y analizados en la presentación, así cómo las nuevas metodologías genómicas disponibles, con el objetivo de intentar definir el perfil de pacientes en los que estaría justificado, a día de hoy, realizar un estudio de trombofilia, las pruebas que debería incluir y el momento más oportuno de realizarlo.
BIBLIOGRAFIA
1. Schaffer AI. The hypercoagulable states. Ann Intern Med 1985: 102: 814-828.
2. Egeberg O. Inherited antithrombin III deficiency causing thrombophilia. Thromb Diath Haemorrh 1965; 13: 516-530.
3. Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM, et al. Genetic susceptibility to thrombosis and its relationship to physiological
risk factors: the GAIT study. Genetic Analysis of Idiophatic Thrombophilia. Am J Hum Genet 2000; 67: 1452-1459.
4. Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet 1999; 353: 1167-1173.
5. de Haan HG, Bezemer ID, Doggen CJM, Le Cessie S, Reitsma PH, Arellano AR, et al. Multiple SNP testing improves risk prediction of first venous thrombosis. Blood 2012; 120: 656-663.
6. Soria JM, Morange PE, Vila J, Souto JC, Moyano M, Trégouët DA, et al. Multilocus genetic risk scores for venous thromboembolism risk assess-ment. J Am Heart Assoc 2014; doi: 10.1161/JAHA.114.001060.
7. Simioni P, Sanson BJ, Prandoni P, Tormene D, Friederich PW, Girolami B, et al. Incidence of venous thromboembolism in families with inherited thrombophilia.Thromb Haemost 1999; 81:198-202.
8. Vossen CY, Conard J, Fontcuberta J, Makris M, Van der Meer FJ, Pabinger I, et al. Risk of a first venous thrombotic event in carriers of a familial thrombophilic defect. The European Prospective Cohort on Thrombo-philia (EPCOT). J Thromb Haemost 2005; 3: 459-464.
9. De Stefano V, Rossi E. Testing for inherited thrombophilia and conse-quences for antithrombotic prophylaxis in patients with venous throm-boembolism and their relatives. A review of the Guidelines from Scien-tific Societies and Working Groups. Thromb Haemost 2013; 110: 697-705.
10. Franchini M. The utility of thrombophilia testing. Clin Chem Lab Med 2014; 52: 495-497.
11. Favaloro EJ. The futility of thrombophilia testing. Clin Chem Lab Med 2014; 52: 499-503.
12. Hicks LK, Bering H, Carson KR, Kleinerman J, Kukreti V, Ma A, et al. The ASH Choosing Wisely® campaign: five hematologic tests and treatments to question. Blood 2013; 122: 3879-3883.
TABLA II. Pruebas recomendadas actualmente en el estudio biológico de trombofilia

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA68
13. Hillis CM, Schimmer AD, Couban S, Crowther MA. The Canadian Choosing Wisely campaign: the Canadian Hematology Society’s top five tests and treatments. Ann Hematol 2015; 94: 541-545.
14. Kearon C, Akl EA. Duration of anticoagulant therapy for deep vein throm-bosis and pulmonary embolism. Blood 2014; 123: 1794-1801.
15. Shen Y-M, Tsai J, Taiwo E, Gavva C, Yates SG, Patel V, et al. Analysis of thrombophilia test ordering practices at an academic center: a proposal for appropriate testing to reduce harm and cost. PLoS ONE 2016; 11(5): e0155326. doi:10.1371/journal.pone.0155326.
16. Middeldorp S. Evidence-based approach to thrombophilia testing. J Thromb Thrombolysis 2011; 31: 275-281.
17. Moll S. Thrombophilia: clinical–practical aspects. J Thromb Thrombolysis 2015; 39: 367-378.
18. Lane DA, Mannucci PM, Bauer KA, Bertina RM, Bochkov NP, Boulyjen-kov V, et al. Inherited thrombophilia: Part 1. Thromb Haemost 1996; 76: 651-662.
19. Lane DA, Mannucci PM, Bauer KA, Bertina RM, Bochkov NP, Boulyjen-kov V, et al. Inherited thrombophilia: Part 2. Thromb Haemost 1996; 76: 824-834.
20. Colucci G, Tsakir is DA. Thrombophil ia Screening: Univer-sal, Selected, or Neither? Clin Appl Thromb Hemost 2017; DOI: 10.1177/1076029616683803. Disponible en: journals.sagepub.com/home/cat.
21. Baglin T, Gray E, Greaves M, Hunt BJ, Keeling D, Machin S, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149: 209-220.
22. NICE Clinical Guideline 144: Venous thromboembolic diseases: diagnosis, management and thrombophilia testing; 2012; Updated Nov-2015.
TABLA III.Artefactos posibles si el esudio de trombofilia se realiza en el momento agudo de la trombosis o mientras el paciente está recibiendo anticoagulantes
Interpretación del EBT

PONENCIAS 69
Se repasarán los principales resultados de los ensayos pivotales con anticoagulantes orales directos (ACOD) en FA no valvular, los datos de vida real (estudios postautorización), las recomendaciones de las principales guías de práctica clínica así como del informe de po-sicionamiento terapéutico de la AEMPS. Por último se perfilarán los
futuros retos relacionados con los ACOD (lagunas de conocimiento, seguimiento adecuado de pacientes, aumento potencial de pacientes que se beneficiarán de la anticoagulación oral, el problema de la infra-dosificación, manejo de hemorragias, monitorización, etc).
Anticoagulantes orales directos como tratamiento de primera línea en FA no valvularAlejandro Pérez Cabeza
Hospital Costa del Sol, Marbella

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA70
INTRODUCCIÓN
La fibrilación auricular (FA) es la arritmia cardiaca más frecuente, en Europa y EEUU 1 de cada 4 adultos sufrirá FA. Además se prevé que el número de pacientes con FA aumente significativamente en los últi-mos años, en la Unión Europea en 2.030 la población de pacientes con FA será de 14-17 millones.1,2 Esto se puede atribuir a un mejor detec-ción de la FA silente, al envejecimiento de la población y al aumento de condiciones que favorecen el desarrollo FA.3
Esta arritmia sigue siendo una de las más importantes causas de acci-dente cerebrovascular (ACV)4, insuficiencia cardiaca (IC), muerte súbita y morbilidad cardiovascular (CV)5 en todo el mundo y en los últimos años se ha generado mucha información médica, lo que ha llevado a la Sociedad Europea de Cardiología a actualizar su guía de práctica clínica (GPC) sobre el manejo de FA y en el año 2016 se publica su segunda edición.
En esta GPC se propone un abordaje integral y estructurado para la atención del paciente con FA, para ello se requiere la cooperación de médicos de atención primaria, cardiólogos, cirujanos cardiacos, neu-rólogos, hematólogos, enfermeros y de los pacientes, para abordar cambios en el estilo de vida, el tratamiento de las enfermedades CV subyacentes y el tratamiento específico de la FA.
TRATAMIENTO ANTITROMBÓTICO EN FANV.
El tratamiento específico de la FA incluye la terapia antitrombótica, ya que ésta reduce considerablemente la incidencia de ACV y la morta-lidad de los pacientes.6,7
Otras intervenciones, como el control del ritmo y de la frecuencia cardiaca, mejoran los síntomas relacionados con la FA y pueden con-servar la función cardiaca, pero no se ha demostrado que reduzcan la morbimortalidad a largo plazo.8
El beneficio clínico neto de la anticoagulación oral es prácticamen-te universal con la excepción de pacientes con un riesgo muy bajo de ACV, por lo que debe indicarse en la mayoría de los pacientes con FA. Además, este tratamiento debe ser instaurado según la GPC de forma temprana y no requiere sistemáticamente la valoración de un especialista.
Hasta hace unos años, los únicos anticoagulantes orales disponibles eran los antivitamina K (AVK), y, a pesar de la evidencia disponible, era frecuente su infrautilización o su interrupción prematura. Las complicaciones hemorrágicas, tanto graves como leves, el alto riesgo «percibido» de sangrado y los esfuerzos necesarios para monitorizar y ajustar la dosis de AVK por su estrecho margen terapéutico eran las razones más comunes para ello.
Si los AVK se indican , no se interrumpen y se administran con un adecuado tiempo en rango terapéutico (TRT 65-70%) son muy eficaces para la prevención de ACV en pacientes con FA.
En 2006 el National Institute for Health and Clinical Excellence (NICE) estimó que le 46% de los pacientes con indicación de tratamiento con warfarina no la recibían y que muchos pacientes no conseguían un rango terapéutico optimo. 9
La intensidad de la anticoagulación, medida por el INR, se correlacio-na con los resultados clínicos. En un metaanálisis de 45 estudios que incluye 71.065 individuos que recibían AVK, la mitad de los eventos tromboembólicos sucedieron con INR por debajo de rango terapéutico y el 44% de las complicaciones hemorrágicas con INR por encima de rango terapéutico.10
En la GPC los AVK siguen teniendo una recomendación IA siempre y cuando el TRT sea adecuado. Recientemente se ha demostrado la uti-lidad de una nueva escala, la SAMe-TT2R2, capaz de identificar a los pacientes que no alcanzarán una calidad de anticoagulación óptima.11 Dicha escala se ha validado en la población española.12 Su aplicación incrementaría el porcentaje de pacientes con acceso a estos fármacos en los que no se prevé una anticoagulación estable, que supera el 40% de los pacientes en las diferentes series, y evitaría además los 6 meses de transición con AVK, según la normativa de nuestro país13.
Actualmente disponemos de 4 anticoagulantes orales directos (ACOD), dabigatrán, rivaroxabán, apixabán y edoxabán, que han sido autori-zados en la indicación de prevención del ictus y embolia sistémica en pacientes con FANV y que han demostrado ventajas sobre los AVK en esta indicación.14 En la GPC se establece la clara preferencia de iniciar tratamiento con los ACOD sobre los AVK en pacientes con indicación de anticoagulación con recomendación IA y también se recomienda el cambio a un ACOD si no se consigue un adecuado control del INR con AVK a pesar de una buena adherencia y si el paciente lo prefiere, recomendación IIbA. (en 2012 IB).15
En España no es posible, de forma estricta, seguir estas recomendacio-nes, ya que existen restricciones de uso, que se reflejan en el informe de posicionamiento terapéutico del Ministerio de Sanidad, Servicios Sociales e Igualdad (IPT)16 y en la necesidad de visado, estando solo financiado el uso de ACOD en FANV en unas determinadas situaciones clínicas y relacionadas con el control del INR. Por tanto, desde el punto de vista del hematólogo considero que debemos esforzarnos en man-tener a los pacientes con un buen control terapéutico confirmando cumplimiento, investigando posibles interacciones medicamentosas, valorando autocontrol, usando nuevos test para la monitorización (tiempo protrombina Fiix), debemos identificar rápidamente a los pacientes que no van a alcanzar una calidad de anticoagulación óptima
Anticoagulantes orales directos como tratamiento de primera línea en FA no valvular (FANV). Punto de vista del HematólogoRosa María Campos Álvarez
Servicio Hematología. Hospital de Jerez. Cádiz

PONENCIAS 71
y debemos cambiar de AVK a ACOD a todos los pacientes que no tengan contraindicación para recibirlos y que a pesar de todos los esfuerzos no consigan un buen control.
Existe dos situaciones clínicas no recogidas en el actual IPT16, y que a nuestro entender deberían tener financiación, el uso de ACOD para pacientes que presentan una complicación hemorrágica utilizando AVK, o la posibilidad de indicarlos como primera opción en pacientes con alto riesgo hemorrágico (HAS-BLED ≥ 3) situaciones en las cuales se ha demostrado una beneficio neto mayor con ACOD que con AVK.17-
19 De igual forma, tampoco se recoge en el actual IPT16 la indicación de uso de ACOD en pacientes con necesidad de cardioversión, cuando se ha demostrado que son igual de efectivos y seguros que los AVK en prevenir la aparición de embolias y además, se facilitaría la pro-gramación.20,21 En este sentido, la Sociedad Española de Trombosis y Hemostasia (SETH) en colaboración con otras sociedades científicas, entre ellas la de Cardiología, realizaron una propuesta de modificación del IPT sobre el uso y recomendaciones de ACOD en FANV,13 en la ac-tualización del IPT (necesario para incorporar Edoxaban), publicada el 21 de noviembre de 2016, de todas las cuestiones propuestas solo la inclusión de la escala CHA2DS2-vasc para valoración del riesgo trom-bótico en lugar de la CHADS2 se ha tenido en consideración.
Desde el punto de vista hematológico consideramos importante co-nocer las propiedades farmacológicas de los anticoagulantes orales actualmente disponibles, las ventajas y desventajas de la necesidad o no de monitorización, los resultados en cuanto a eficacia y seguridad demostrados en los estudios pivotales, esto nos ayudara a decidir que anticoagulante es de elección dependiendo del perfil del paciente. También debemos participar en la realización de protocolos de manejo de las complicaciones hemorrágicas asociadas al uso de anticoagu-lantes y de protocolos de actuación ante procedimientos invasivos o cruentos.
PROPIEDADES FARMACOLÓGICAS DE LOS ANTICOAGULANTES ORALES INDICADOS EN FANV. VENTAJAS E INCONVENIENTES.
Las características farmacológicas de los AVK se asocian a ventajas y a limitaciones prácticas. No se eliminan vía renal, por tanto, serán la opción mas conveniente en pacientes con insuficiencia renal grave. Por su efecto anticoagulante más prolongado el olvido o pérdida de una dosis, en principio, no conllevara un riesgo muy elevado de aparición de un evento trombótico inmediato. La necesidad de monitorización del INR, por su estrecho margen terapéutico, fomenta el contacto regu-lar del paciente con el personal sanitario, lo cual puede ser beneficioso desde punto de vista clínico, aunque no nos asegura una mayor adhe-rencia, pero para algunos pacientes puede ser un inconveniente, con impacto en su calidad de vida, además de conllevar costes adicionales (visitas frecuentes al centro de salud/hospital, pérdidas de horas labo-rables, sobrecarga personal enfermería ya que muchos pacientes, por su edad y movilidad reducida, precisan controles en domicilio…) Por su inicio de acción lento, si es preciso una anticoagulación inmediata, será necesario asociar antitrombóticos de rápida acción como las he-parinas y por su efecto anticoagulante más prolongado en ocasiones es necesario demorar procedimientos cruentos o cirugías no urgentes.
Las características farmacológicas de los ACOD ofrece ventajas prácticas sobre los AVK22 . Tienen un inicio de acción rápido (0.5-4 horas) y una VM corta por lo que rápidamente desaparece el efecto anticoagulante. Se eliminan por vía renal, aunque de forma diferente cada uno de ellos, por lo que el deterioro de la función renal puede conllevar acumulación y riesgo asociado de sangrado. No precisan de monitorización continua de su efecto farmacológico, dado que presentan unas características farmacocinéticas y farmacodinámicas similares entre individuos, po-seen una ventana terapéutica amplia y tienen menos interacciones alimentarias y farmacológicas.
Al igual que sucede con otros fármacos, el efecto anticoagulante de un ACOD se supone que es directamente proporcional a su concentración en plasma medida por la técnica de referencia que es la cromatografía líquida con espectrometría de masas, aunque el uso de esta técnica se limita al campo de la investigación.23
No se conoce el rango terapéutico de los ACOD, es decir, el rango anti-coagulante adecuado, lo que si se conoce son los niveles plasmáticos pico-valle y como podemos observar en la siguiente tabla son muy variables.24
Concentraciones plasmáticas (mediana) de los ACOD aprobados para FANV
Fármaco DosisTº para
pico
Valle (percentil
5th-95th)
Pico (percentil
5th-95th)
Dabigatran150 mg
c/12h1-2h
90ng/ml (31-
225)
184 ng/ml
(64-443)
Rivaroxaban 20 mg c/24h 2-4h 26 ng/ml(6-87)270 ng/ml
(189-419)
Apixaban 5 mg c/12h 3-4h103 ng/ml
(41-230)
171 ng/ml
(91-321)
Edoxaban 60 mg c/24h 1-2h22 ng/ml
(10-40)*
170 ng/ml
(120-250)*
* rango intercuartiles
Por no disponer del rango terapéutico se usa el término “rango en terapia” que incluye los niveles plasmáticos desde el percentil 5th del valle hasta el percentil 95th del pico (p.ej. para dabigatran el rango en terapia sería 31-443 ng/ml). La mayoría de los pacientes tendrán niveles en “rango en terapia” durante todo el tiempo de tratamiento. 25
En determinadas situaciones la medida del efecto anticoagulante o de los niveles plasmáticos del medicamento puede ser útil porque sospechemos que el paciente tiene niveles por debajo del “rango en terapia”: falta de eficacia, sospecha de no cumplimiento terapéu-tico, malabsorción gastrointestinal u obesidad o por encima del di-cho rango: sobredosis, hemorragia, edades avanzadas, insuficiencia hepato-renal o peso muy bajo. Sería también interesante disponer de esta información previo a una cirugía o procedimientos cruentos no demorables y si sospecha de interacciones medicamentosas de fármacos no evaluados.
Los ACOD interfieren con las pruebas básicas de coagulación y estas interferencias podemos usarlas para medición en situaciones espe-ciales. Disponemos de test de coagulación no específicos (TTPa, TP,

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA72
TT) que nos pueden informar de la presencia o ausencia del fármacos y test específicos (Tiempo de trombina, tiempo de ecarina, ensayos anti-FXa) que nos ofrecen información cuantitativa sobre niveles del fármaco. Los factores a tener en cuenta para la interpretación de los resultados de laboratorio son el método y reactivo usado, el fármaco en cuestión, cada uno altera los tiempos de una forma y el tiempo transcurrido desde la ingesta.26
ELECCIÓN ENTRE LOS DISTINTOS ACOD. PERFILES DE PACIENTES
La disponibilidad en la actualidad de 4 ACOD aprobados en FANV nos permite individualizar el tratamiento para cada paciente, en ausen-cia de comparaciones directas, la elección del anticoagulante estará probablemente influenciada por su eficacia en prevención del ictus isquémico, el riesgo de sangrado (intracraneal y GI), riesgo de infarto agudo de miocardio y beneficio sobre la mortalidad, además, debemos tener en consideración sus características farmacológicas y la adminis-tración cada 12 o 24 horas.27
COMPLICACIONES HEMORRÁGICAS Y MANEJO PERI-OPERATORIO DE LOS PACIENTES EN TRATAMIENTO CON ACOD. INDICACIÓN DEL USO DE ANTÍDOTOS EN SITUACIONES DE URGENCIA.
La complicación más grave del tratamiento con fármacos anticoagu-lantes en la práctica habitual es la aparición de hemorragia aguda, bien causada por un exceso de actividad de los fármacos, o bien de otro origen y que se ve agravada por el efecto inhibidor de la hemostasia de los anticoagulantes.
Analizando globalmente el riesgo de hemorragias mayores de los ACOD en los cuatro estudios pivotales, se observa una tendencia a la reducción de los mismas que no llega a ser significativa (p=0,06) del 14% (RR 0,86; IC 0,73-1,00).
La frecuencia con que se producen las hemorragias graves es difícil de estimar. En los ensayos clínicos se han descrito incidencias variables que oscilan entre 2 a 5.6 por 100 pacientes /año.28
De forma conjunta se observa en los ensayos una reducción de mas del 50% de las HIC y no diferencias en cuanto a las hemorragias mayores gastrointestinales.29
En la practica clínica real, donde los pacientes están menos contro-lados, la incidencia puede variar de 3.2 a 9 por 100 pacientes/año,
posiblemente condicionadas por las comorbilidades asociadas que presentan los pacientes.30,31
El manejo de las hemorragias graves asociadas a ACOD consiste en me-didas locales de control de sangrado, terapia de soporte, transfusión de hemoderivados, carbón activado, diálisis, agentes procoagulantes y antídotos.
Es recomendable disponer de protocolos de actuación en los hospitales y de un stock del único antídoto aprobado, Idarucizumab.
Con respecto al manejo peri-operatorio, el factor clave a tener en cuen-ta es la actividad anticoagulante existente que estará relacionada con el tiempo transcurrido desde la última toma, la función renal y las posibles interacciones con otros medicamentos que reciba el paciente. Es útil dividir el manejo entre procedimientos quirúrgicos o invasivos que se pueden realizar de forma electiva y procedimientos urgentes, en los que la situación clínica puede obligar a realizarlos independien-temente de la actividad del ACOD.
También es recomendable disponer de protocolos de manejo de estas situaciones. Estos protocolos deben ser desarrollados por un equipo multidisciplinar, consensuados por distintas especialidades en las Co-misiones de tratamiento antitrombótico con el objetivo de disminuir la variabilidad en las actuaciones y ofrecer a nuestros pacientes una asistencia de calidad.

PONENCIAS 73
BIBLIOGRAFÍA
1. Krijthe BP, Kunst A, Benjamin EJ et al. Projections on the number of indi-viduals with atrial fibrillation in the European Union, from 2000 to 2060. Eur Heart J. 2013;34:2746–51.
2. Colilla S, Crow A, Petkun W et al. Estimates of current and future inci-dence and prevalence of atrial fibrillation in the U.S. adult population. Am J Cardiol. 2013;112:1142–7.
3. Schnabel RB, Yin X, Gona P et al. 50 year trends in atrial fibrillation preva-lence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. 2015;386:154–62.
4. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983–8.
5. Stewart S, Hart CL, Hole DJ et al A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/ Paisley study. Am J Med. 2002;113:359–64
6. Hart RG, Pearce LA, Aguilar MI. Meta-analysis: antithrombotic therapy to prevent stroke in patients who have nonvalvular atrial fibrillation. Ann Intern Med. 2007;146:857–67.
7. Ruff CT, Giugliano RP, Braunwald E et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fi-brillation: a meta-analysis of randomised trials. Lancet. 2014;383:955–62.
8. Al-Khatib SM, Allen LaPointe NM, Chatterjee R et al. Rate and rhythm control therapies in patients with atrial fibrillation: a systematic review. Ann Intern Med. 2014;160:760–73.
9. National Institute for Health and Clinical Excellence (NICE). Clinical Guideline 36. National Cost Impact Report to accompany ‘Atrial Fibrilla-tion: the Management of Atrial Fibrillation’. London: NICE; 2006.
10. Oake N, Fergusson DA, Faster AJ et al Frequency of adverse events in patients with poor anticoagulation: a meta-analysis. CMAJ 2017; 176:1589-1594
11. Apostolakis S, Sullivan RM, Olshansky B, et al. Factors affecting qual-ity of anticoagulation control among patients with atrial fibrillation on warfarin: the SAMe-TT2R2 score. Chest. 2013;144:1555-1563.
12. Roldán V, Cancio S, Gálvez J, et al. The SAMe-TT2R2 score predicts poor anticoagulation control in af patients: a prospective ‘real-world’ inception cohort study. Am J Med. 2015;128:1237-1243.
13. Roldán I, Marín F. On the way to a better use of anticoagulants in non-valvular atrial fibrillation. Proposed amendment to the therapeutic positioning report UT/ V4/23122013. Rev Esp Cardiol. 2016;69:551-553.
14. Sterne JAC, Bodalia PN, Bryden PA,et al. Oral anticoagulants for primary prevention, treatment and secondary prevention of venous thromboem-bolic disease, and for prevention of stroke in atrial fibrillation: systematic review, network meta-analysis and cost-effectiveness analysis. Health Technol Assess 2017;21(9)
15. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the man-agement of atrial fibrillation developed in collaboration with EACTS: The Task Force for the management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC, endorsed by the European Stroke Organisation (ESO). Eur Heart J. 2016. Disponible en: https://doi.org/10.1093/eurheartj/ehw
16. Informe de posicionamiento terapéutico UT-ACOD/v5/21112016 Crite-rios y recomendaciones generales para el uso de los anticoagulantes orales directos (ACOD) en la prevención del ictus y la embolia sistémica
en pacientes con fibrilación auricular no valvular Fecha de publicación: 21 de noviembre de 2016
17. Ruff CT, Giugliano RP, Braunwald E et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fi-brillation: a meta-analysis of randomized trials. Lancet. 2014;383:955–62.
18. Friberg L, Rosenqvist M, Lip GY. Net clinical benefit of warfarin in patients with atrial fibrillation: a report from the Swedish atrial fibrillation cohort study. Circulation. 2012;125:2298–307.
19. Michalski F, Tittl L, Werth S et al . Selection, management, and outcome of vitamin K antagonist-treated patients with atrial fibrillation not switched to novel oral anticoagulants. Results from the Dresden NOAC registry. Thromb Haemost. 2015 Nov;114(5):1076-84. doi: 10.1160/TH15-02-0116.
20. Cappato R, Ezekowitz MD, Klein AL et al. Rivaroxaban vs. vitamin K antago-nists for cardioversion in atrial fibrillation. Eur Heart J. 2014;35:3346–55.
21. Santas E, Méndez J, Martínez-Brotons A, Núñez et al. Experiencia en la práctica clínica diaria con la cardioversión ambulatoria de fibrilación auricular en tratamiento con nuevos anticoagulantes orales. Rev Esp Cardiol. 2014;67:960-1.
22. Hellwig T, Gulseth M. Pharmacokinetic and pharmacodynamic drug in- teractions with new oral anticoagulants: what do they mean for patients with atrial fibrillation? Ann Pharmacother. 2013;47:1478–87.
23. Schmitz EM, Boonen K, van den Heuvel DJ, van Dongen JL, Sche- llings MW, Emmen JM, et al. Determination of dabigatran, rivaroxaban and apixaban by ultra-performance liquid chromatography - tandem mass spectrometry (UPLC-MS/MS) and coagulation assays for therapy monitor-ing of novel direct oral anticoagulants. Thromb Haemost 2014;12:1636-44.
24. Lippi G, Favaloro EJ. Recent guidelines and recommendations for labo-ratory assessment of the direct oral anticoagulants (DOACs): is there consensus? Clin Chem Lab Med 2015;53:185-89.
25. Samuelson BT and Cuker A. Measurement and reversal of the direct oral
anticoagulants Blood Reviews 2017; 31: 77–84.
26. Samuelson BT, Cuker A, Siegal DM et al. Laboratory Assessment of the
Anticoagulant Activity of Direct Oral Anticoagulants A Systematic Review CHEST 2017; 151(1):127-138
27. Schaefer JK, McBaneRD and Wysokinski WD. How to choose appropriate direct oral anticoagulant for patient with nonvalvular atrial fibrillation Ann Hematol 2016; 95:437–449
28. S Werth et al. Bleeding Risk, Management and Outcome in Patients Receiving Non-VKA Oral Anticoagulants (NOACs) Am J Cardiovasc Drugs 2015; 15:235-242.
29. Chai-Adisaksopha C, Crowther M, Isayama T et al. The impact of bleeding complications in patients receiving target-specific oral anticoagulants: a systematic review and meta-analysis. Blood. 2014; 124:2450-8.
30. Graham DJ, Reichman ME, Wernecke M et al. Cardiovascular, bleeding, and mortality risks in elderly Medicare patients treated with dabigatran or warfarin for nonvalvular atrial fibrillation. Circulation 2015; 131:157-64
31. Westendorf JB, Förster K, Pannach S et al. Rates, management, and outcome of rivaroxaban bleeding in daily care: results from the Dresden NOAC registry. Blood 2014;124:955-962
32. Doherty J, Ty J. Gluckman TJ, Hucker WJ et al. 2017 ACC Expert Consensus Decision Pathway for Periprocedural Management of Anticoagulation in Patients With Nonvalvular Atrial Fibrillation A Report of the American College of Cardiology Clinical Expert Consensus Document Task Force. Journal of the American College of Cardiology. 2017 DOI: 10.1016/j.jacc.2016.11.024 Article in Press.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA74
En el tratamiento de los pacientes con mieloma múltiple (MM) debe-mos distinguir aquellos que se pueden beneficiar de un tratamiento intensivo que incluya la práctica de un autotrasplante (TASPE) de los que, por razones de edad y/o co-morbilidad, se considera que no son candidatos a terapias intensivas. En los pacientes en los que no se contempla la práctica de TASPE, el tratamiento hasta hace una dé-cada ha consistido en la asociación de melfalán/prednisona (MP) o en regímenes basados en dexametasona. Con estos tratamientos se conseguía una tasa de remisiones completas inferiores al 5% y medianas de supervivencia que no superaban los 3 años. La dis-ponibilidad de nuevos fármacos, a saber, talidomida, bortezomib y lenalidomida ha cambiado el paradigma terapéutico en el MM. Así, la asociación de talidomida a MP (MPT) da lugar a una supervivencia libre de progresión(SLP) y global (SG) más prolongadas que MP. Sin embargo, la toxicidad neurológica de la talidomida, junto a los mejores resultados obtenidos con otros regímenes, ha hecho que el MPT ya no sea el tratamiento de elección. La adición de bortezomib a MP (MPV) produce una tasa de RC del 30% y una SLP y SG mucho más prolon-gados que el MP. El problema de la neurotoxicidad del bortezomib ha mejorado con la posología subcutánea y la administración semanal. Asimismo, la SLP se ha incrementado de forma significativa con un tratamiento de extensión, más allá de la pura “inducción” con 9 ciclos de MPV administrados en el estudio VISTA. Con la administración de lenalidomida/dexametasona de forma continua se ha obtenido mejo-res resultados que con MPT o con 18 ciclos de lenalidomida/dexame-tasona (estudio FIRST). De otro lado, el grupo americano SWOG acaba de publicar mejores resultados con VRD (bortezomib, lenalidomida y dexametasona) frente a lenalidomida/dexametasona. A fecha de hoy los regímenes indicados para pacientes no candidatos a TASPE serían MPV y lenalidomida/dexametasona administrada de forma continua. A la luz de los resultados publicados, parece imponerse un tratamiento de extensión más allá de la inducción. El VRD también
puede ser una buena opción. La elección de estos regímenes se basará fundamentalmente en la edad y co.morbilidad de cada paciente a fin de que la tolerancia sea buena. Con toda probabilidad los anticuerpos monoclonales (AcMo), en particular el daratumumab se incorporarán en el tratamiento inicial en un futuro no lejano. Con respecto a los pacientes candidatos a intensificación con TASPE el objetivo es con-seguir la máxima disminución de la masa tumoral con la inducción al objeto de alcanzar el máximo de RCs tras la intensificación con las altas dosis de melfalán siendo el objetivo final la obtención de SLP muy pro-longadas y que la mayor proporción posible de pacientes estén libres de progresión más allá de los 10 años tras el trasplante (“operational cure”). El tratamiento de inducción actualmente aprobado en dicha situación es la triple asociación VTD (bortezomib, talidomida y dexa-metasona) con tasa de RC pre-TASPE del 35% y post-TASPE cercanas al 50% así como una SLP de casi 5 años. Existen varios ECs en los que se está investigando la eficacia de VRD y también de KRD (carfilzomib, lenalidomida, dexametasona). El objetivo sería el utilizar el mejor IMid y el mejor inhibidor del proteasoma. Como intensificación, la dosis de melflán de 200 mg/m2 sigue siendo el estándar. Los resultados ob-tenidos con la administración de 2 a 4 ciclos de consolidación (mismo régimen que en la inducción) son muy prometedores. Finalmente, la Agencia Europea del Medicamento acaba de aprobar el tratamiento de mantenimiento post-TASPE con lenalidomida. Por tanto, en pacientes con MM candidatos a TASPE, la secuencia sería: inducción (inhibidor proteasoma, IMiD, dexametasona, intensificación con MEL-200, con-solidación con 2 a 4 ciclos como la inducción y mantenimiento basado en lenalidomida. En suma, sería lo que podríamos llamar “European Total Therapy”. Al igual que se ha mencionado para los pacientes no candidatos a TASPE, es altamente probable que en un futuro próximo se incorpore un AcMo en el tratamiento de inducción/consolidación y tal vez en el mantenimiento. Todo dependerá de los resultados de los ECs actualmente en curso.
¿Hacia nuevos paradigmas terapéuticos en la primera línea de tratamiento del Mieloma Múltiple? (Nuevos fármacos, nuevas combinaciones de fármacos, la incorporación del mantenimiento y la revisión de la escala de edad)Dr. Joan Bladé Creixenti
Unidad de Amiloidosis y Mieloma. Servicio de Hematología. Hospital Clínic. IDIBAPS. Barcelona

CASOS CLÍNICOS CITOLÓGICOS


CASOS CLÍNICOS CITOLÓGICOS 77
MOTIVO DE CONSULTA
Varón de 67 años que ingresó en el Servicio de Medicina Interna de nuestro hospital por deterioro general de 2 meses de evolución, con astenia, anorexia, pérdida de 4 kg de peso e inestabilidad en la marcha, con la adición de fiebre y sudoración profusa en la última semana.
ANTECEDENTES PERSONALES
Era originario de Guatemala y residía en nuestro país desde hacía 11 años. No refería antecedentes patológicos de interés.
EXPLORACIÓN FÍSICA
Temperatura 38 ºC. Aceptable estado general, bien perfundido e hidratado. En la palpación de territorios ganglionares, sólo se halló una adenopatía de 1.5 cm en la fosa supraclavicular derecha. La aus-cultación cardiopulmonar, la palpación abdominal y la exploración neurológica fueron normales.
PRUEBAS COMPLEMENTARIAS
Hemograma: cifra normal de leucocitos: 4.4 x 109/L (2.770 neutrófilos x 109/L, 690 linfocitos x 109/L, 870 monocitos x 109/L. Hay una modera-da bicitopenia, con Hb: 119 g/L y VCM: 82.9 fL, y plaquetas: 80 x109/L. Frotis de sangre periférica (Figura 1): destacaba la presencia de un 4% de linfocitos atípicos, cuya morfología recordaba a la de las células de Downey de la mononucleosis infecciosa, siento de tamaño mediano a grande, de forma irregular con una silueta que parecía querer ro-dear a los hematíes circundantes, un núcleo redondeado o irregular de cromatina laxa ocasionalmente con un nucleolo, y un citoplasma amplio y basófilo, sin vacuolas, donde a veces se apreciaba una fina
Varón adulto con fiebre, cuadro poliadenopático y bicitopeniaMª Paz Garrastazul Sánchez*, Irene Millán Ortega**, Raquel de la Varga Martínez***
Hospital Universitario Puerta del Mar de Cádiz, UGC de Hematología y Hemoterapia*, Servicio de Anatomía patológica** Servicio de Inmunología***
Extensión de sangre periférica, tinción de May-Grünwald-Giemsa (MGG) x 1000 aumen-
tos. Linfocito de aspecto estimulado de gran tamaño que presenta un núcleo irregular de
cromatina laxa y un citoplasma basófilo.
FIGURA 1
granulación. La citometría de flujo demostró que correspondían a lin-focitos T CD3+CD8+ activados, de gran tamaño y complejidad, con inmunofenotipo CD3+ CD5+ CD2+ CD7+ CD8+ CD4- CD56- CD57+ HLA DR++ CD25-.
Coagulación: Ligero descenso de la actividad de protrombina, del 53% (N: 70-100), con TPTA y fibrinógeno normales. Bioquímica: moderada elevación de creatinina de 1.5 mg/dL (N: 0.7-1.2), urea: 85 mg/dL (0.7-1.2), GOT: 74 U/L (N <37), triglicéridos: 365 mg/dL (N: 50-165), LDH: 1507 U/L (N: 135-225) y PCR: 178 mg/L (N < 5), siendo el resto normal. El estudio de anemia mostró una vitamina B12 y un ácido fólico normales, con elevación marcada de la ferritina, de 1812 μg/L. La hap-toglobina fue normal y el Coombs directo negativo. El proteinograma presentaba un trazado inflamatorio intenso y un descenso de la albú-mina: 2,32 mg/dL (N: 4,32-4,76). Estudios microbiológicos: se realizó la batería habitual de cultivos, serología y PCR para el estudio de fiebre de origen desconocido, siendo todo el estudio negativo. La serología para el virus de Epstein-Barr (VEB) fue compatible con infección pa-sada, con IgG EBNA positivo. En el aspirado medular, que se realizó posteriormente, se cuantificaron 5.6 copias de DNA de VEB x 107 /mL.
Pruebas de imagen: La Rx de tórax fue normal y la eco abdominal mostró una esplenomegalia de 15,5 cm. En la TAC de cuello, tórax y abdomen (figura 2) se observaron adenopatías múltiples y de pequeño tamaño, entre 12 Y 20 mm, en cadenas yugulares, fosas supraclavicula-res, mediastino y peritoneo. Había también un ligero derrame pleural bilateral, con atelectasia del lóbulo inferior derecho pulmonar, y una ligera esplenomegalia con lesiones nodulares hipodensas múltiples. La TAC craneal y la RMN de cabeza y cuello fueron normales.
Estudio de médula ósea: el aspirado medular (Tabla 1) fue realizado el día +8 desde la admisión del enfermo. Era ligeramente hipoce-
FIGURA 2
TAC abdominal. Se observa un discreto derrame pulmonar izquierdo con atelectasia de
la base pulmonar, así como una leve esplenomegalia con varias imágenes nodulares.
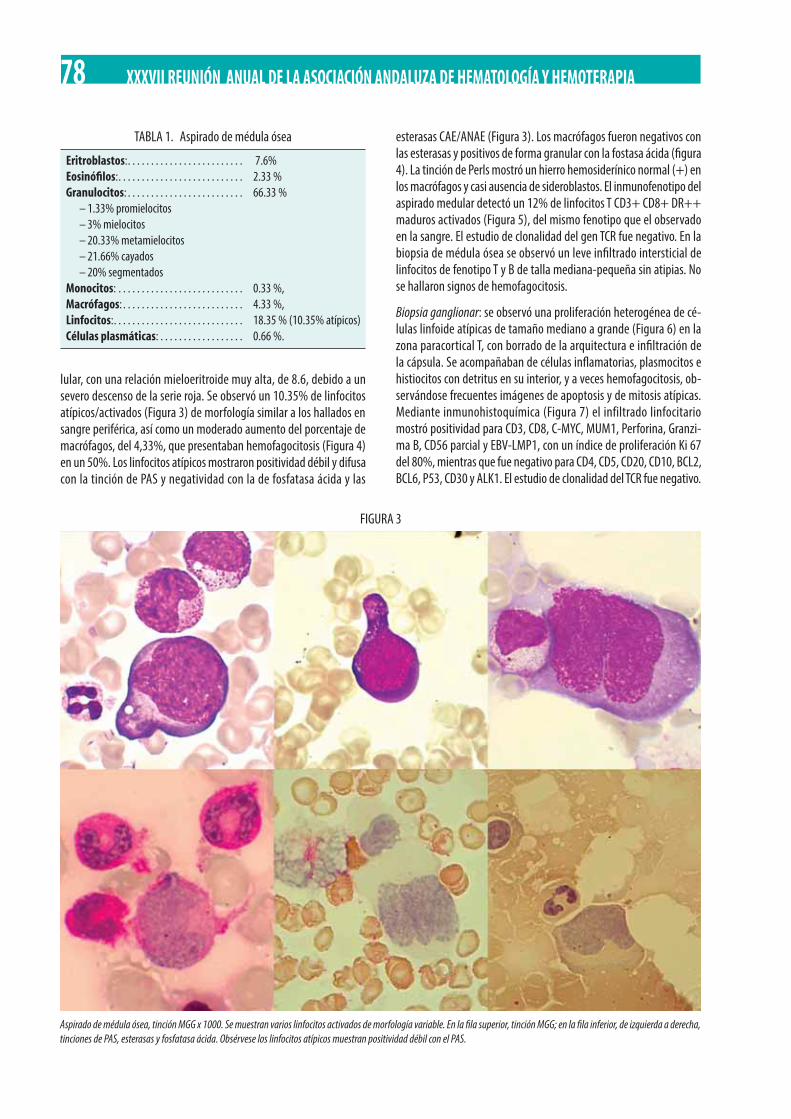
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA78
lular, con una relación mieloeritroide muy alta, de 8.6, debido a un severo descenso de la serie roja. Se observó un 10.35% de linfocitos atípicos/activados (Figura 3) de morfología similar a los hallados en sangre periférica, así como un moderado aumento del porcentaje de macrófagos, del 4,33%, que presentaban hemofagocitosis (Figura 4) en un 50%. Los linfocitos atípicos mostraron positividad débil y difusa con la tinción de PAS y negatividad con la de fosfatasa ácida y las
TABLA 1. Aspirado de médula ósea
Eritroblastos:. . . . . . . . . . . . . . . . . . . . . . . . . 7.6%
Eosinófilos:. . . . . . . . . . . . . . . . . . . . . . . . . . . 2.33 %
Granulocitos: . . . . . . . . . . . . . . . . . . . . . . . . . 66.33 %
– 1.33% promielocitos
– 3% mielocitos
– 20.33% metamielocitos
– 21.66% cayados
– 20% segmentados
Monocitos: . . . . . . . . . . . . . . . . . . . . . . . . . . . 0.33 %,
Macrófagos: . . . . . . . . . . . . . . . . . . . . . . . . . . 4.33 %,
Linfocitos:. . . . . . . . . . . . . . . . . . . . . . . . . . . . 18.35 % (10.35% atípicos)
Células plasmáticas: . . . . . . . . . . . . . . . . . . 0.66 %.
FIGURA 3
Aspirado de médula ósea, tinción MGG x 1000. Se muestran varios linfocitos activados de morfología variable. En la fila superior, tinción MGG; en la fila inferior, de izquierda a derecha,
tinciones de PAS, esterasas y fosfatasa ácida. Obsérvese los linfocitos atípicos muestran positividad débil con el PAS.
esterasas CAE/ANAE (Figura 3). Los macrófagos fueron negativos con las esterasas y positivos de forma granular con la fostasa ácida (figura 4). La tinción de Perls mostró un hierro hemosiderínico normal (+) en los macrófagos y casi ausencia de sideroblastos. El inmunofenotipo del aspirado medular detectó un 12% de linfocitos T CD3+ CD8+ DR++ maduros activados (Figura 5), del mismo fenotipo que el observado en la sangre. El estudio de clonalidad del gen TCR fue negativo. En la biopsia de médula ósea se observó un leve infiltrado intersticial de linfocitos de fenotipo T y B de talla mediana-pequeña sin atipias. No se hallaron signos de hemofagocitosis.
Biopsia ganglionar: se observó una proliferación heterogénea de cé-lulas linfoide atípicas de tamaño mediano a grande (Figura 6) en la zona paracortical T, con borrado de la arquitectura e infiltración de la cápsula. Se acompañaban de células inflamatorias, plasmocitos e histiocitos con detritus en su interior, y a veces hemofagocitosis, ob-servándose frecuentes imágenes de apoptosis y de mitosis atípicas. Mediante inmunohistoquímica (Figura 7) el infiltrado linfocitario mostró positividad para CD3, CD8, C-MYC, MUM1, Perforina, Granzi-ma B, CD56 parcial y EBV-LMP1, con un índice de proliferación Ki 67 del 80%, mientras que fue negativo para CD4, CD5, CD20, CD10, BCL2, BCL6, P53, CD30 y ALK1. El estudio de clonalidad del TCR fue negativo.

CASOS CLÍNICOS CITOLÓGICOS 79
FIGURA 4
Aspirado de médula ósea x1000. Macrófagos con signos de hemofagocitosis. En la fila superior tinción de MGG. En la fila inferior, de izquierda a derecha: tinciones de Perls, fosfatasa
ácida y esterasas. Obsérvese que muestran positividad con el Perls y la fosfatasa ácida.
FIGURA 5
Inmunofenotipo de médula ósea mediante citometría de flujo. En azul se muestran los linfocitos T CD4+, en verde linfocitos T CD8+ y en rojo linfocitos T CD8+ activados. A) Plot FSC
vs. SSC de la población de linfocitos T CD3+. B) Distribución de linfocitos T CD3+ mediante plot CD3 vs. SSC. C) Distribución de las subpoblaciones de linfocitos T mediante plot CD8 vs.
SSC. D) Plot que muestra que la población de linfocitos T de mayor tamaño y complejidad (rojo) están activados y expresan DR de alta intensidad.
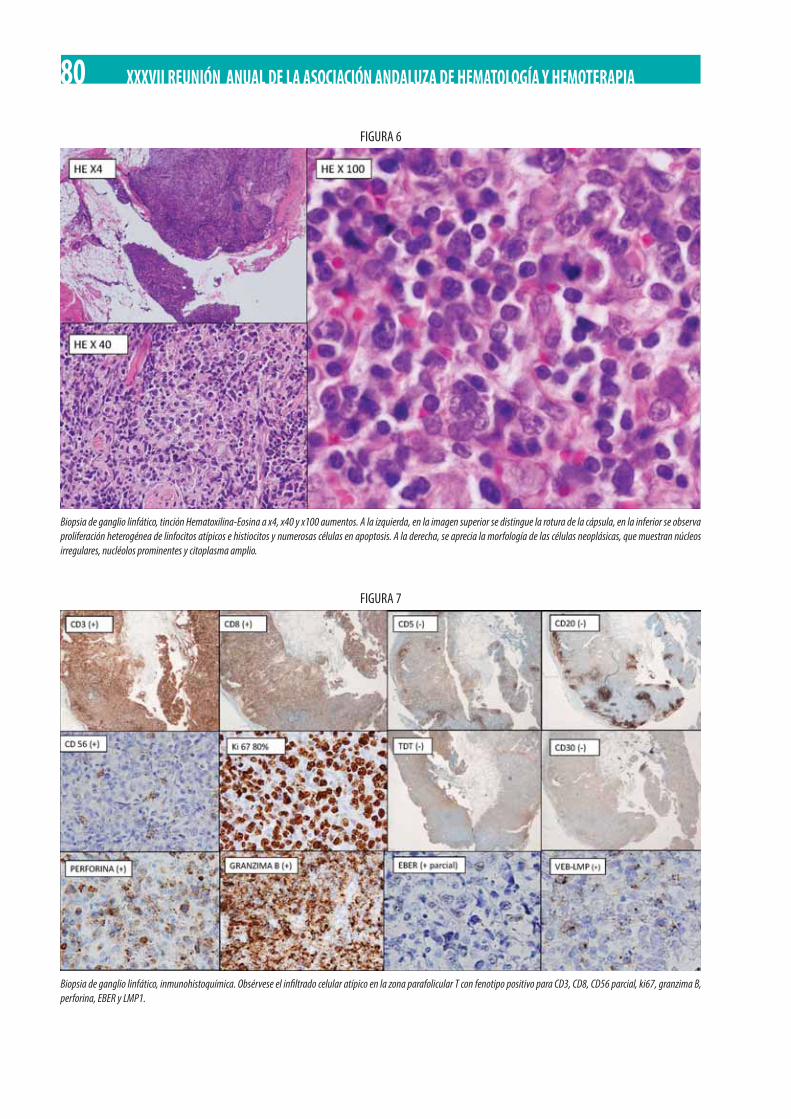
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA80
FIGURA 6
FIGURA 7
FIGURA 7
Biopsia de ganglio linfático, tinción Hematoxilina-Eosina a x4, x40 y x100 aumentos. A la izquierda, en la imagen superior se distingue la rotura de la cápsula, en la inferior se observa
proliferación heterogénea de linfocitos atípicos e histiocitos y numerosas células en apoptosis. A la derecha, se aprecia la morfología de las células neoplásicas, que muestran núcleos
irregulares, nucléolos prominentes y citoplasma amplio.
Biopsia de ganglio linfático, inmunohistoquímica. Obsérvese el infiltrado celular atípico en la zona parafolicular T con fenotipo positivo para CD3, CD8, CD56 parcial, ki67, granzima B,
perforina, EBER y LMP1.
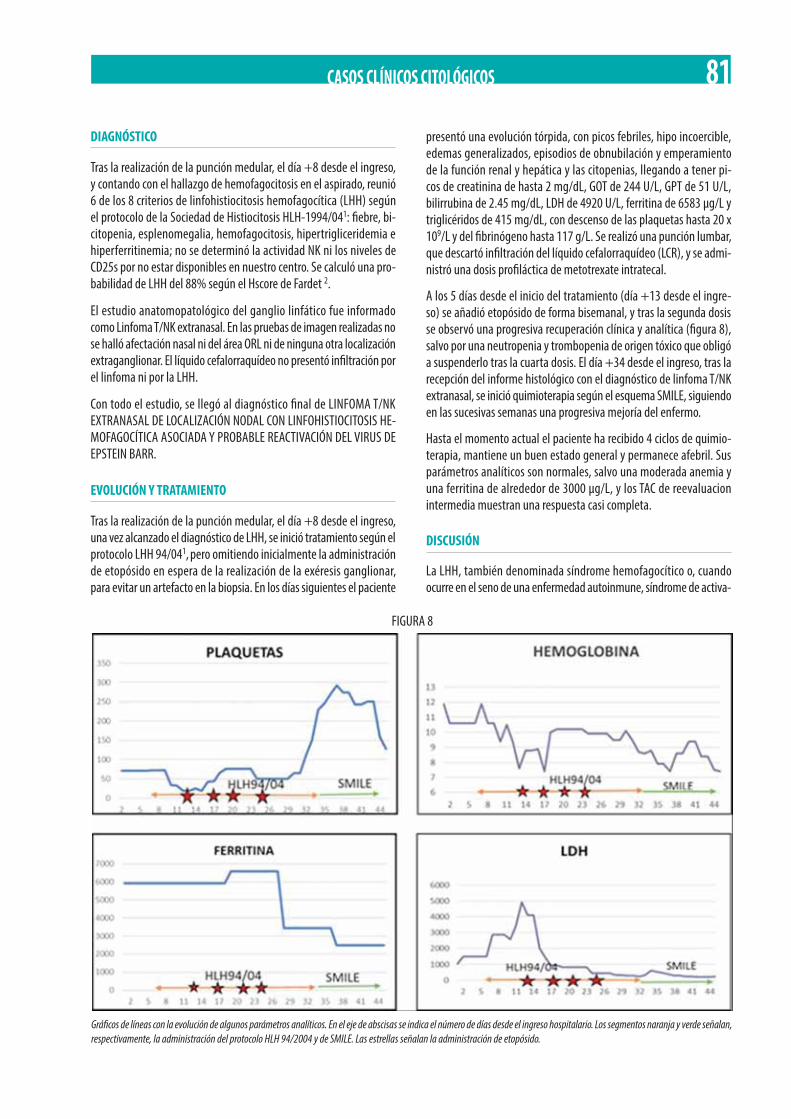
CASOS CLÍNICOS CITOLÓGICOS 81
DIAGNÓSTICO
Tras la realización de la punción medular, el día +8 desde el ingreso, y contando con el hallazgo de hemofagocitosis en el aspirado, reunió 6 de los 8 criterios de linfohistiocitosis hemofagocítica (LHH) según el protocolo de la Sociedad de Histiocitosis HLH-1994/041: fiebre, bi-citopenia, esplenomegalia, hemofagocitosis, hipertrigliceridemia e hiperferritinemia; no se determinó la actividad NK ni los niveles de CD25s por no estar disponibles en nuestro centro. Se calculó una pro-babilidad de LHH del 88% según el Hscore de Fardet 2.
El estudio anatomopatológico del ganglio linfático fue informado como Linfoma T/NK extranasal. En las pruebas de imagen realizadas no se halló afectación nasal ni del área ORL ni de ninguna otra localización extraganglionar. El líquido cefalorraquídeo no presentó infiltración por el linfoma ni por la LHH.
Con todo el estudio, se llegó al diagnóstico final de LINFOMA T/NK EXTRANASAL DE LOCALIZACIÓN NODAL CON LINFOHISTIOCITOSIS HE-MOFAGOCÍTICA ASOCIADA Y PROBABLE REACTIVACIÓN DEL VIRUS DE EPSTEIN BARR.
EVOLUCIÓN Y TRATAMIENTO
Tras la realización de la punción medular, el día +8 desde el ingreso, una vez alcanzado el diagnóstico de LHH, se inició tratamiento según el protocolo LHH 94/041, pero omitiendo inicialmente la administración de etopósido en espera de la realización de la exéresis ganglionar, para evitar un artefacto en la biopsia. En los días siguientes el paciente
presentó una evolución tórpida, con picos febriles, hipo incoercible, edemas generalizados, episodios de obnubilación y emperamiento de la función renal y hepática y las citopenias, llegando a tener pi-cos de creatinina de hasta 2 mg/dL, GOT de 244 U/L, GPT de 51 U/L, bilirrubina de 2.45 mg/dL, LDH de 4920 U/L, ferritina de 6583 μg/L y triglicéridos de 415 mg/dL, con descenso de las plaquetas hasta 20 x 109/L y del fibrinógeno hasta 117 g/L. Se realizó una punción lumbar, que descartó infiltración del líquido cefalorraquídeo (LCR), y se admi-nistró una dosis profiláctica de metotrexate intratecal.
A los 5 días desde el inicio del tratamiento (día +13 desde el ingre-so) se añadió etopósido de forma bisemanal, y tras la segunda dosis se observó una progresiva recuperación clínica y analítica (figura 8), salvo por una neutropenia y trombopenia de origen tóxico que obligó a suspenderlo tras la cuarta dosis. El día +34 desde el ingreso, tras la recepción del informe histológico con el diagnóstico de linfoma T/NK extranasal, se inició quimioterapia según el esquema SMILE, siguiendo en las sucesivas semanas una progresiva mejoría del enfermo.
Hasta el momento actual el paciente ha recibido 4 ciclos de quimio-terapia, mantiene un buen estado general y permanece afebril. Sus parámetros analíticos son normales, salvo una moderada anemia y una ferritina de alrededor de 3000 μg/L, y los TAC de reevaluacion intermedia muestran una respuesta casi completa.
DISCUSIÓN
La LHH, también denominada síndrome hemofagocítico o, cuando ocurre en el seno de una enfermedad autoinmune, síndrome de activa-
FIGURA 8
Gráficos de líneas con la evolución de algunos parámetros analíticos. En el eje de abscisas se indica el número de días desde el ingreso hospitalario. Los segmentos naranja y verde señalan,
respectivamente, la administración del protocolo HLH 94/2004 y de SMILE. Las estrellas señalan la administración de etopósido.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA82
ción macrofágica, es una rara enfermedad en la que un desencadenan-te infeccioso, con frecuencia el virus de Epstein-Barr (VEB), asociado a diversas condiciones predisponentes hereditarias o adquiridas, da lugar a una respuesta inmune prolongada, descontrolada e inefectiva a través de la excesiva activación de linfocitos T CD8+ y de células pre-sentadoras de antígenos, que proliferan desordenadamente infiltran-do médula ósea y otros tejidos y liberando una tormenta de citocinas proinflamatorias que dan lugar a una disfunción multisistémica4. La clave fundamental del síndrome es la existencia de un defecto en la función citotóxica de las células NK y los linfocitos T citotóxicos que impide el autocontrol del proceso inflamatorio mediante la elimina-ción de las células T activadas e histiocitos, lo que contribuye a una progresiva estimulación del sistema inmune.
Existen formas familiares de LHH y formas adquiridas. Las primeras son más frecuentes en el primer año de vida, se transmiten con ca-rácter AR y se deben a diversas mutaciones de proteínas implicadas en la función citotóxica. En los adultos, por el contrario, predominan las formas adquiridas, que se asocian mayoritariamente a neoplasias hematológicas, generalmente linfomas, aunque también se han des-crito en leucemias, mieloma y otras enfermedades malignas, así como en procesos autoinmunes, inmunodeficiencias o infecciones. Suele existir además un agente infeccioso que actúa como desencadenante del síndrome, destacando por su frecuencia el VEB. Es una enfermedad muy poco frecuente, algunas publicaciones refieren una incidencia de 0.9% de LHH asociados a neoplasias en adultos 5.
La Histiocyte Society propuso en 1994 una serie de criterios para el diagnóstico de LHH en niños que posteriormente fueron revisados en 20041 (Tabla 2), y que consistían en el hallazgo de un test molecu-lar consistente con LHH o bien en la reunión de al menos cinco de otros ocho criterios. Recientemente se ha publicado un HScore que permite calcular la probabilidad de diagnóstico de SHF en adultos2
y que consta de nueve variables (Tabla 3). Además de haber reunido un número suficiente de criterios según ambos protocolos, nuestro paciente presentó otras alteraciones que han sido descritas en casos
TABLA 2. Criterios diagnósticos de la HLH 94/2004
Debe estar presente uno de los dos criterios para establecer el diagnóstico:
I. Alteración molecular de HLH familiar (mutaciones PRF1, SH2K1A/SAP,
UNC13D, STX11, MUNC18-2, RAB27a)
II. Presencia de al menos 5 de 8 criterios:
1. Fiebre ≥ 38.5ºC
2. Esplenomegalia
3. Citopenias (afectando al menos dos líneas en sangre periférica):
Hemoglobina < 90 g/dL, Plaquetas < 100 x 109/L, Neutrófilos <
1 x 109/L
4. Hipertrigliceridemia (≥265 mg/dL) y/o hipofibrinogenemia (<150
mg/dL).
5. Hemofagocitosis en médula ósea, bazo, ganglio linfático o hígado
6. Baja o ausente actividad de células NK
7. Ferritina ≥ 500 mcg/L
8. CD25 soluble elevado ≥ 2400 U/mL
de LHH, como edemas, hiponatremia, hipoalbuminemia y elevación de LDH y PCR. Aunque tuvo clínica neurológica, como episodios de obnubilación e inestabilidad en la marcha, el líquido cefalorraquídeo no mostró infiltración celular.
Diversos estudios señalan a la hiperferritinemia como la prueba diag-nóstica de mayor sensibilidad y especificidad para el diagnóstico de LHH, con valores del 90% y 96% respectivamente para niveles supe-riores a 10.000μg/L6,7. En nuestro paciente, la ferritina, aunque muy elevada, no llegó a alcanzar esta cifra.
Histológicamente, el SHF presenta una proliferación de linfocitos, células NK e histiocitos en distintos órganos, como la médula ósea, los ganglios linfáticos, el bazo o el hígado, y, frecuentemente, se ob-serva hemofagocitosis. Diversas publicaciones8, han demostrado que los linfocitos expandidos tienen un inmunofenotipo T citotóxico CD8 + con fuerte expresión de DR y frecuente pérdida de CD5 o de otros marcadores T, como el CD3 o CD7, y pueden presentar clonalidad del TCR. En el caso que presentamos, la expresión de CD5 de los linfocitos atípicos era normal y no presentaban clonalidad del TCR.
Aunque el hallazgo de hemofagocitosis constituye uno de los criterios diagnósticos de la LHH, algunos autores9,10 han cuestionado la indi-cación de la punción medular dentro del proceso diagnóstico, ya que el hallazgo de hemofagocitosis no es un criterio obligado de LHH, y además tiene una baja sensibilidad y especificidad, entorno al 85% y 60%9 respectivamente. Esta baja sensibilidad se debe en parte a que no existe consenso en decidir qué cifra mínima de macrófagos con hemofagocitosis se requiere para considerarla significativa, con cifras que varían entre 0.05% y 3% según los distintos autores. No obstante, y a pesar de sus limitaciones, la mayoría de revisiones sobre LHH reco-miendan realizar estudio medular dentro del proceso diagnóstico4,5,11, ya que no sólo tiene utilidad para demostrar hemofagocitosis, sino que además permite investigar la presencia de neoplasias asociadas, como leucemias agudas o linfomas con infiltración medular.
El tratamiento de la LHH debe instaurarse de forma precoz, ya que una demora del mismo afecta negativamente a la supervivencia. Se basa en una combinación de inmunosupresores y agentes citotóxicos; el protocolo LHH 1994/041 consta de una fase de inducción de 8 semanas con dexametasona, etopósido y, en caso de afectación neurológica, metotrexate intratecal. Los casos familiares o los que presentan enfer-
TABLA 3. HS score (Fardet, 2014)
1. Inmunodepresión conocida
2. Temperatura elevada
3. Organomegalia
4. Hiperferritinemia
5. Citopenias
6. Hemofagocitosis
7. Hipertrigliceridemia
8. Hipofibrinogenemia
9. Elevación de GOT/GPT

CASOS CLÍNICOS CITOLÓGICOS 83
medad persistente o en recaída se siguen de una fase de continuación con dexametasona, etopósido y ciclosporina más una consolidación con alotrasplante medular. La mortalidad de la LHH es del 50-75% siendo aún mayor en los SHF asociados a malignidad5.
Nuestro paciente fue diagnosticado de un linfoma NK/T extranasal; la clasificación de la OMS de las neoplasias linfoides13 incluye este tipo de linfoma dentro de la categoría denominada linfoma extranodal NK/T nasal, que comprende tanto el tipo nasal como el extranasal, siendo ambos de localización predominantemente extranodal. Su prevalen-cia es mayor en ciertas zonas geográficas14, como los países asiáticos, América Central y Sudamérica, y es más frecuente en varones adultos.
El linfoma extranodal NK/T nasal es muy agresivo. A diferencia del tipo nasal15, que suele presentarse en estadios localizados, el extranasal se manifiesta generalmente de forma sistémica, con afectación de múltiples lugares extraganglionares como piel, tejidos blandos, tracto gastrointestinal, testículos, etc, pudiendo infiltrar de forma secundaria la médula ósea, la sangre periférica o los ganglios linfáticos. Aunque raros, han sido publicados algunos casos de linfomas NK/T con afec-tación primaria nodal en ausencia de afectación extranodal16, como en el caso de nuestro paciente. El linfoma NK/T nasal se asocia a LHH hasta en el 3% de los casos13.
Histológicamente, el linfoma extranodal NK/T, nasal o extranasal, se caracteriza por una gran destrucción, con necrosis prominente y ten-dencia a la invasión vascular. El infiltrado linfoide neoplásico puede ser muy polimorfo, con células linfoide de tamaño y morfología muy variable, que suelen acompañarse de células inflamatorias e histiocitos con detritus en su interior, dando lugar a la típica imagen en cielo estrellado14. Son también frecuentes las imágenes de apoptosis y de mitosis anómalas, y ocasionalmente se observan signos de hemofago-citosis. La mayoría de los casos presentan fenotipo NK, con expresión de CD56 y CD3 citoplasmático pero ausencia del CD3 de superficie y de reordenamiento de TCR; sin embargo algunos casos tienen un fenotipo de células T, y todos expresan proteínas citotóxicas (granzima B, TIA 1 y perforina). Este linfoma presenta una fuerte asociación el VEB, al que se le atribuye un probable papel patogénico, de modo que para su diagnóstico es obligada la demostración de partículas del virus en las células neoplásicas, mediante FISH (EBER) o inmunohistoquímica (LMP1)14. Diversos estudios han demostrado una correlación entre el número de copias de DNA del virus en sangre y la carga tumoral y el pronóstico, de forma que niveles superiores a 6.1 x 107/mL se han asociado significativamente a una inferior supervivencia libre de enfermedad17.
En cuanto a la genética del linfoma extranodal NK/T nasal, el antígeno del receptor de células T y los genes de inmunoglobulinas son de con-figuración centrogerminal en la mayoría de los casos. En una pequeña proporción de casos, 38% de los nasales y 27% de los extranasales15, el TCR muestra reordenamiento clonal, presumiblemente correspon-diente a los casos que derivan de linfocitos T citotóxicos. En nuestro paciente el estudio de clonalidad del ganglio fue negativo.
El pronóstico del linfoma NK-T históricamente ha sido pobre, con una supervivencia de 30-40%; sin embargo parece haber mejorado en los últimos años17 gracias al uso de quimioterapia más intensiva, como
el protocolo SMILE del NK Cell Tumor Study Group o el esquema Aspa-MetDex de los grupos GELA y GOELAMS.
PARA RECORDAR
El diagnóstico de la LHH supone un reto para el médico. Es funda-mental tener un alto grado de sospecha y dirigir la investigación diagnóstica con rapidez para instaurar precozmente el tratamiento. Dicha investigación debe ir dirigida no sólo hacia la confirmación del propio síndrome, sino también a la identificación de la patología subyacente y de la infección desencadenante.
La punción de médula ósea sigue teniendo un papel importante en el diagnóstico de la LHH, pues no sólo sirve para detectar la hemo-fagocitosis, sino que también contribuye al proceso de investigación etiológica del síndrome mediante la detección de leucemias o linfo-mas subyacentes que cursen con infiltración medular.
El hallazgo en la médula ósea de una proliferación de macrófagos con hemofagocitosis es frecuente en la LHH y constituye uno de sus criterios diagnósticos, aunque no es obligado, y tiene una baja sensibilidad y especificidad. Es importante que aprendamos a re-conocer la población expandida de linfocitos reactivos CD8+ que la suele acompañar.
La mayoría de casos de LHH en adultos se relacionan con linfomas. Muchos de ellos, como el linfoma T/NK nasal, están asociados al VEB, siendo importante para su diagnóstico la demostración de las partículas del virus en las células tumorales.
BIBLIOGRAFÍA
1. Henter J, Samuelsson-Horne A, Arico M, et al; Histocyte Society JL et al. HLH-2004: diagnostic and therapeutic guidelines for hematophagocytic lymphohistiocytosis. 2007;48(2): 124-131.
2. Fardet L, Galicier L, Lambotte O, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheum 2014;66(9):2613–20.
3. Alexandra Filipovich. Hemophagocytic lymphohistiocytosis (HLH) and re-lated sisorders. Hematology Am Soc Hematol Educ Program. 2009:127-31.
4. Hayden A, Park S, Giustini D, Lee AY, Chen Ly. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: A systematic scoping review. Blood Rev 2016; 30(6):411-420.
5. Lim SH, Park S, Jang JH, et al. Clinical significance of bone marrow he-mophagocytosis in adult patients with malignancy and non-malignan-cy-induced hemophagocytic lymphohistiocytosis. Ann Hematol. 2016 Jan;95(2):325-35.
6. Kai Lehmberg, Stephan Ehl. Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. British Journal of Haematology, 2013, 160, 275-287.
7. Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Pathol. 2014 Jan;141(1):62-71.
8. Chad M McCall, Shiyama Mudali, Robert J Arceci et al. Flow Cytometric findings in Hemophagocytic Lymphohistiocytosis. Am J clin Pathol, 2012; 137 (5): 786-794.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA84
9. Goel S, Polski JM, Imran H. Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci. 2012 Winter;42(1):21-5.
10. Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood, 2015; 7;125(19):2908-14.
11. Flavia G. N. Rosado MD, Annette S. Kim MD. Hemophagocytic Lymphohis-tiocytosis, an Update on Diagnosis and Pathogenesis. American Journal Clinical Pathology, 2013;139: 713-727.
12. Steven H. Swerdlow, Elias Campo, Stefano A. Pileri et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood, 2016; 127: 2375-2390.
13. Steven H Swerdlow, Elías Campo, Nancy Lee Harris et al. WHO classifica-tion of Tumours of Haematopoietic and Lymphoid Tissues. 4th edition. 2008.
14. Wing-yan Au, Dennis D. Weisenburger, Tanin Intragumtornchai et al. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood, 2009,113 (17): 3921-37.
15. Kagami, Yoshitoyo M.D.; Suzuki, Ritsuro M.D.; Taji, Hirohumi M.D. et al. Nodal cytotoxic Lymphoma Spectrum: A Clinicopathologic Study of 66 Patients. American Journal of Surgical Patholoy, 1999, 23 (10): 1184-1200.
16. Ritsuro Suzuki. Pathogenesis and treatment of extranodal natural killer/t-cell lymphoma. Seminars in Hematology, 2014; 51 (1): 42-51.
17. Wing-Yan Au, Annie Pang, Carolyn Choy et al. Quantification of circulating Epstein-Barr virus (EBV) DNA in the diagnosis and monitoring of natural killer cell and EBV-positive lymphomas in immunocompetent patients. Blood, 2004, 104: 243-249.

CASOS CLÍNICOS CITOLÓGICOS 85
MOTIVO DE CONSULTA
Varón de 56 años que consulta en Urgencias por dolor abdominal de varias semanas de evolución y síndrome constitucional desde hace 10 meses.
HISTORIA CLÍNICA
Como antecedentes personales de interés: HTA bien controlada des-de hace 10 años, con cardiopatía hipertensiva y fracción de eyección conservada. Estudiado recientemente en Dermatología por dermatitis granulomatosa. En la exploración física, destaca polo de bazo palpable y lesiones eritematosas sobre elevadas en placas de 1 cm, en cuello, escote y abdomen. Edemas en miembros inferiores.
PRUEBAS ANALÍTICAS
Hemograma: leucocitos 31.4 x109/L, Hb 95 g/L, plaquetas 268 x109/L. Fórmula manual: segmentados 31%, mielocitos 11%, metamielocitos 10%, cayados 28%, linfocitos 10%, monocitos 9%, eosinófilos 1%. Bioquímica: iones, perfil hepático y renal normal. LDH normal. Protei-nograma e inmunoglobulinas normales. β2-microglobulina 5.4mg/L, Vitamina B12 1700 pg/mL. Serología: hepática, virus herpes y VIH ne-gativa. Brucella, Leishmania, Coxiella y Sífilis negativas. Metabolismo del hierro: ferritina 203 μg/L, saturación 19.8%, capacidad de fijación 559 μg/dL, hierro sérico 31.6 μg/dL
ESTUDIOS COMPLEMENTARIOS Y PRUEBAS DE IMAGEN
Ecografía de abdomen: esplenomegalia de 16 cm. Tomografía axial computarizada (TAC): hepatoesplenomegalia. Mínimo derrame pleural bilateral. Líquido libre en gotiera paracólica derecha y pelvis. Adeno-patías mesentéricas no significativas (Figura 1)
ESTUDIOS CITOLÓGICOS E HISTOLÓGICOS
Frotis de sangre periférica: mielemia y desviación izquierda; 2 eri-troblastos por cada 100 células nucleadas; no se observan blastos. Anisopoiquilocitosis con presencia de frecuentes dacriocitos. Datos de displasia en serie granulocítica (hipogranulación) y ocasionales megatrombocitos (Figuras 2 a 4).
Aspirado médula ósea: mínima cantidad de grasa, material medular hipercelular (++) a expensas de hiperplasia granulocítica, sin displa-sia significativa. Se observó una población eosinófila aumentada, sin presentar características patológicas relevantes, tan sólo ocasional vacuolización citoplasmática. Megacariocitos sin alteraciones citomor-fológicas de interés. Mielograma: serie roja 5%, serie granulocítica 84 %, eosinófilos 9.5%, monocitos 0.5%, linfocitos 0.5%, blastos 0.5% (Figuras 5 a 10).
Biopsia médula ósea: médula ósea hipercelular, con proliferación gra-nulocítica compatible con neoplasia mieloproliferativa crónica. Fibrosis reticulínica, MF-1 de la Organización Mundial de la Salud (OMS) y evi-dencia de frecuentes mastocitos (Figuras 11 a 13).
CITOGENÉTICA Y BIOLOGÍA MOLECULAR
Citogenética convencional: en las 20 metafases analizadas (bandas G) se observa una translocación recíproca entre los cromosomas 5 y 10. Cariotipo: 46,XY,t(5;10)(q33;q21)[20]. No se detectaron otras anoma-lías (Figura 14).
Hibridación fluorescente in situ (FISH):
Reordenamientos bcr/abl (t(9;22)(q34;q11), FGFR1 (8p11), PDGFRα (FIP1L1-PDGFRα): negativos.
Reordenamiento PDGFRβ (5q32-q33): con la aplicación de esta son-da locus específica (LSI PDGFRβ (5q32-q33) Break Apart), se confir-ma en interfase (Figura 15) la presencia de un reordenamiento de PDGFRβ, en el 95% de las células.
WCP 5 y WCP10* se observa un reordenamiento de PDGFRβ. Se confirma con sondas de pintado cromosómico un reordenamiento recíproco entre los cromosomas 5 y 10 (Figura 16).
Biología Molecular: reordenamiento CCDC6 exón 7/PDGFR exón 11 (Figura 17).
DIAGNÓSTICO
Neoplasia mieloproliferativa crónica con eosinofilia asociada a reorde-namiento PDGFR (5q33) t(5;10).
EVOLUCIÓN
Tras confirmación molecular del diagnóstico el paciente inicia trata-miento con imatinib a dosis convencionales de 400 mg/día. Presenta muy buena evolución clínica inicial, con desaparición de edemas y mejoría de lesiones cutáneas (Figura 18). Desarrolla citopenias pro-gresivas que hacen necesaria reducción de dosis hasta estabilización de cifras hematimétricas. Actualmente, pendiente de reevaluación en médula, con hemograma normal, asintomático y excelente adherencia al tratamiento con imatinib 100 mg a días alternos.
DISCUSIÓN
Catalogada por la Organización Mundial de la Salud (OMS-2016) den-tro de las neoplasias mieloides y linfoides con eosinofilia y anomalías en PDGFRα, PDGFRβ, FGFR1 o con PCM1-JAK2 (como entidad provi-sional), la NMPc con reordenamiento PDGFRβ (5q33), es una entidad infrecuente. Este reordenamiento condiciona un gen de fusión con
Neoplasia Mieloproliferativa Crónica (NMPc) con reordenamiento de PDGFRßM. Gómez Rosa, R. Morales Camacho (1), I. Simón Pilo, S. Burillo Sanz (1), E. Ríos Herranz
H.U. Ntra. Sra. De Valme y H.U. V. del Rocío, Sevilla (1)

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA86
FIGURA 1
FIGURA 3
FIGURA 2
TAC. Hepatoesplenomegalia.
Sangre periférica (MGG x100). a. Dacriocitos. c. Forma monocitoide. d. Granulocito displásico.
Sangre periférica (MGG x400).
CASOS CLÍNICOS CITOLÓGICOS 87
FIGURA 5 FIGURA 6
FIGURA 4
Sangre periférica (MGG x1000). b. Megatrombocito. c. Eosinófilo con granulación gruesa. d. Granulocito displásico.
Aspirado de médula ósea (MGG x100). Hipercelularidad. Aspirado de médula ósea (MGG x200). Polimorfismo.
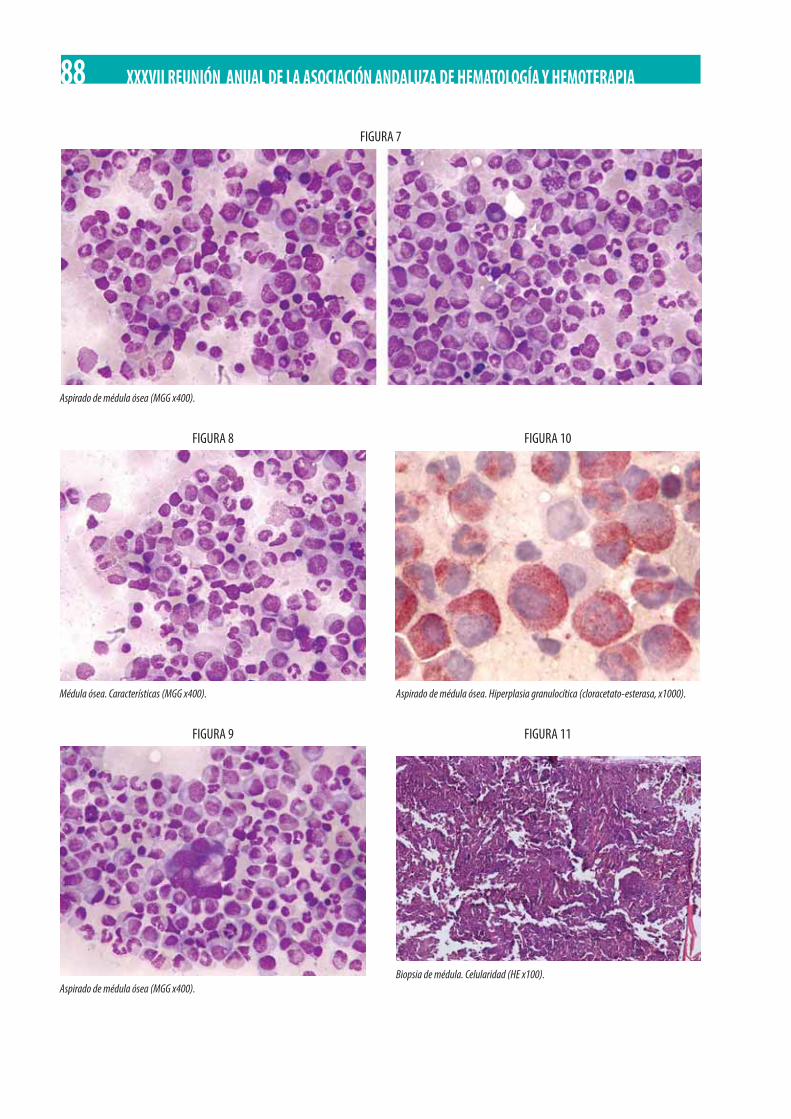
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA88
FIGURA 7
FIGURA 8 FIGURA 10
FIGURA 9 FIGURA 11
Aspirado de médula ósea (MGG x400).
Médula ósea. Características (MGG x400). Aspirado de médula ósea. Hiperplasia granulocítica (cloracetato-esterasa, x1000).
Aspirado de médula ósea (MGG x400).Biopsia de médula. Celularidad (HE x100).
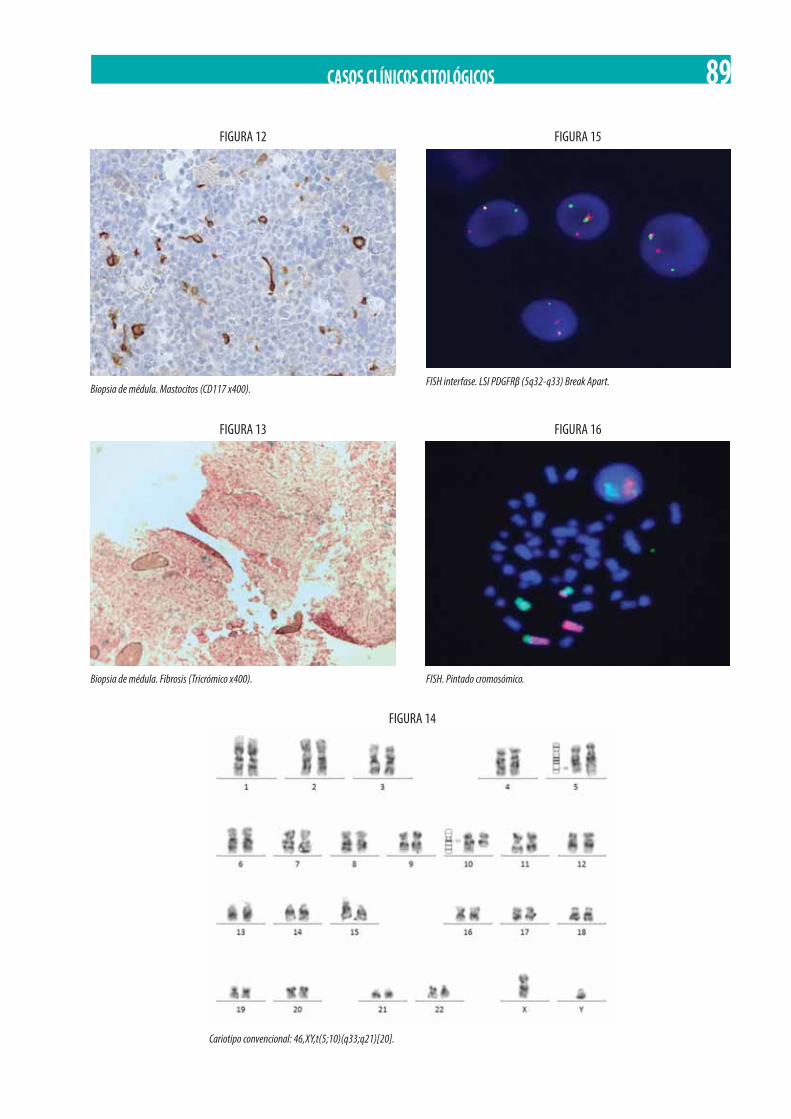
CASOS CLÍNICOS CITOLÓGICOS 89
FIGURA 12 FIGURA 15
FIGURA 13 FIGURA 16
FIGURA 14
Biopsia de médula. Mastocitos (CD117 x400).FISH interfase. LSI PDGFRβ (5q32-q33) Break Apart.
Biopsia de médula. Fibrosis (Tricrómico x400). FISH. Pintado cromosómico.
Cariotipo convencional: 46,XY,t(5;10)(q33;q21)[20].
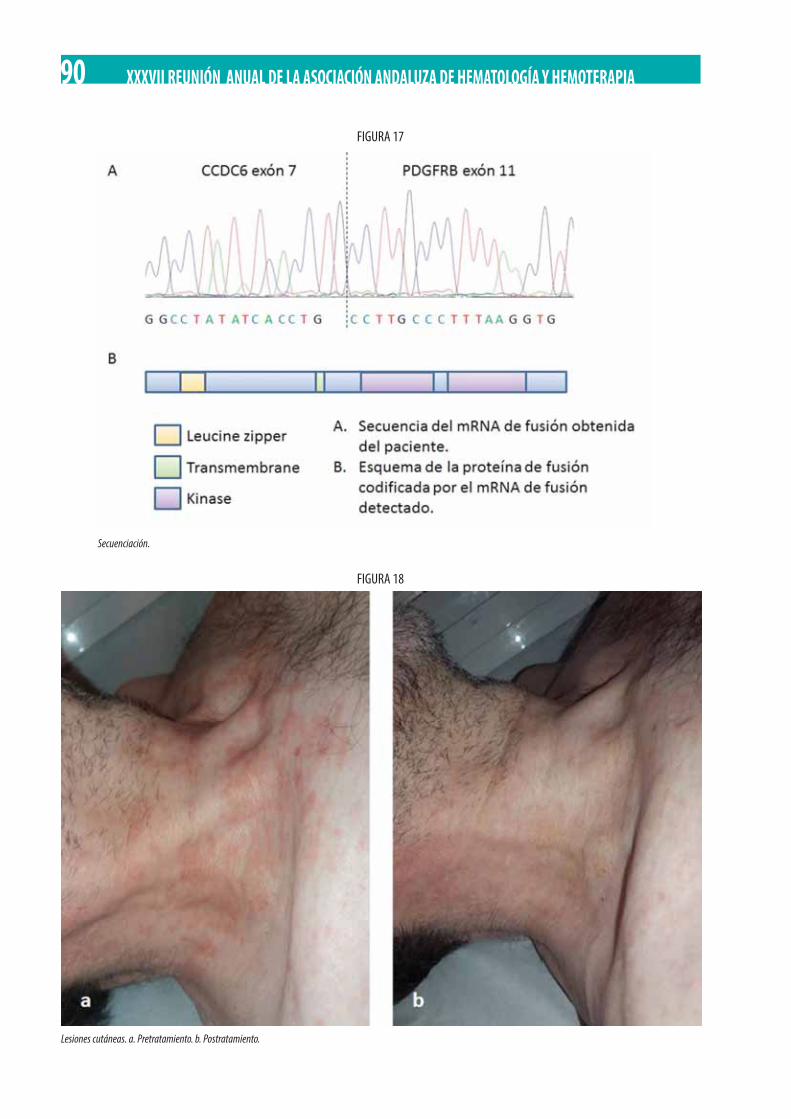
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA90
FIGURA 17
FIGURA 18
Secuenciación.
Lesiones cutáneas. a. Pretratamiento. b. Postratamiento.

CASOS CLÍNICOS CITOLÓGICOS 91
actividad tirosina kinasa y, consecuentemente, una señal mieloproli-ferativa continuada. La traslocación más frecuentemente encontrada es t(5;12), con la formación del gen de fusión ETV6-PDGFRβ. Presenta un pico de incidencia en la edad media de la vida y buena respuesta a tratamiento con inhibidores de tirosina kinasa, aunque sólo existen pequeñas series de casos. Sólo aparecen 5 casos publicados en la lite-ratura reciente de NMPc con reordenamiento PDGFRβ (5q33) asociadas a t(5;10), como el que presentamos. Todos ellos eran varones de edad media, con frecuente leucocitosis superior a 50 x109/L al diagnóstico -a diferencia de nuestro caso-, y esplenomegalia.
CONCLUSIÓN/A RECORDAR
Se trata de una entidad muy poco frecuente, que requiere un alto grado de sospecha para orientar el diagnóstico.
La eosinofilia hace sospechar el diagnóstico pero en muchos ca-sos –como el que se describe- está ausente, o sólo se expresa en médula ósea.
En nuestro caso fue clave la aportación de la Citogenética y Biología Molecular al diagnóstico, lo que pone de relevancia el papel cada vez más protagonista de estas técnicas en el diagnóstico hematológico.
A pesar de los pocos casos descritos con esta translocación, este reordenamiento presenta buena respuesta a inhibidores de tirosina kinasa. No obstante, existen escasas series de casos que permitan extraer conclusiones pronósticas a día de hoy.
BIBLIOGRAFÍA
1. Daniel A. Arber, Attilio Orazi, Robert Hasserjian, Jurgen Thiele, Michael J. Borowitz, Michelle M. Le Beau, Clara D. Bloomfield, Mario Cazzola, and James W. Vardiman. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, (Blood. 2016; 127(20):2391-2405)
2. Anastasiadou, E., Schwaller, J., Sternberg, D. W., Cain, D., Grieco, M., Siena, S., Mecucci, C., and Gilliland, D. G. H4(10S170), a gene frequently rear-ranged in papillary thyroid carcinoma, is fused to the platelet-derived b receptor (PDGF b R) in atypical chronic myeloid leukemia with a t(5;10)(q33;q22). Blood, 94 (Suppl. 1): 51a, 1999
3. Salvatore Siena, Gabriella Sammarelli, Maria Grazia Grimoldi, Roberta Schiavo, Andrea Nozza, Massimo Roncalli, Cristina Mecucci, Armando San-toro, Carmelo Carlo-Stella. New reciprocal translocation t(5;10)(q33;q22) associated with atypical chronic myeloid leukemia. Haematologica 1999; 84:369-372
4. Shashikant Kulkarni, Carol Heath, Sally Parker, Andrew Chase, Sameena Iqbal, Christopher F. Pocock, Jaspal Kaeda, Kate Cwynarski, John M. Gold-man, and Nicholas C. P. Cross. Fusion of H4/D10S170 to the Platelet-derived Growth Factor Receptor b in BCR-ABL-negative Myeloproliferative Disorders with a t(5;10)(q33;q21) CANCER RESEARCH 60, 3592–3598, July 1, 2000
5. Valeria AS De Melo, Alistair G Reid. A case of myeloproliferative disorder with t(5;10)(q33;q21.2) Atlas Genet Cytogenet Oncol Haematol. 2007;11(2)
6. Andrei M, Bandarchuck A, AbdelMalek C, Kundra A, Gotlieb V, Wang JC. PDGFRβ-Rearranged Myeloid Neoplasm with Marked Eosinophilia in a 37-Year-Old Man; And a Literature Review. Am J Case Report 2017 Feb 17

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA92
MOTIVO DE CONSULTA
Cefalea y desviación de la lengua.
HISTORIA CLÍNICA
Adolescente de 13 años sin antecedentes personales de interés, que consultó en otro centro por cefalea occipital de un mes de evolución y desviación de la lengua a la izquierda; tras realizar una tomografía axial computarizada(TAC) craneal se diagnosticó de mastoiditis e inició antibioterapia. A la semana siguiente acudió nuevamente por astenia y dolor retroesternal siendo remitida a nuestro hospital ante el hallazgo de anemia, trombopenia y poliadenopatías.
EXPLORACIÓN FÍSICA
Palidez, múltiples adenopatías de mediano tamaño y localización cervical, supraclavicular y axilar y dismetría mamaria con nódulos palpables diseminados, no dolorosos. No hepatoesplenomegalia.
PRUEBAS ANALÍTICAS
Hemograma: leucocitos 7.9 x109/L, neutrófilos 3.4 x109/L, hemoglo-bina 84 g/L, plaquetas 58 x109/L. Bioquímica:calcio 12,2 mg/dL, LDH 1.118 UI/L, ácido úrico 10,7 mg/dL. Coagulación normal. Marcadores tumorales: Enolasa Neuronal Específica 63.1 ng/mL (normal hasta 15 ng/mL). Líquido cefalorraquídeo sin hallazgos. Serología viral: IgG de Varicella-Zoster y Epstein Barr positivos, resto negativo (incluidos vi-rus hepatotropos, Parvovirus B19, Toxoplasma, VIH, Citomegalovirus y Herpes Virus).
ESTUDIOSCOMPLEMENTARIOS Y PRUEBASDE IMAGEN
Radiografía de tórax con ensanchamiento mediastínico y adenopatías hiliares bilaterales. Tomografía axial computarizada (TAC) toraco-ab-dominal con afectación adenopática, ósea (lesiones líticas encolumna vertebral, esternón, palas ilíacas, pubis y sacro) y pulmonar; nódulos mamarios bilaterales; implantes peritoneales y lesiones suprarrenales [Fig. 1]. Ecografía de mama: múltiples nódulos difusos de ecoge-nicidad heterogénea sugestivos de malignidad. La tomografía por emisión de positrones (PET-TAC) presentó extensa afectación linfática, pulmonar, pleural y ósea; implantes peritoneales, en suprarrenal iz-quierda y en partes blandas de ambas mamas, órbita y pierna izquier-da (flexor largo de los dedos). Resonancia nuclear magnética (RNM): imagen que sugiere tumor primario afectando al músculo flexor largo de los dedos izquierdo; metástasis óseas en metáfisis proximales de tibias y peronés.
ESTUDIOS CITOLÓGICOS, CITOMETRÍA E HISTOLOGÍA
Sangre periférica: leucocitos 7.9x109/L (mielocitos/metamielocitos 2%, cayados 14%, segmentados 35%, linfocitos 43%, monocitos 3%, basó-filos 1%, blastos 2%), eritroblastos 5% y plaquetas 58 x109/L[Fig. 2].
Aspirado de médula ósea (AMO): abundante celularidad monomórfica y frecuentes macrófagos con restos celulares en su interior. Las series eritroide y megacariocítica ausentes, neutrófilos segmentados 7%, macrófagos 4%, linfocitos 3% y células indiferenciadas de aspecto maligno 86% [Fig. 3], pleomórficas, con relación núcleo/citoplasma elevada, núcleos de cromatina laxa y nucléolos mal definidos; ocasio-nal binuclearidad; citoplasma con basofilia media y habitualmente vacuolado [Fig. 4]; a veces se observaba un material intracitoplas-mático amorfo y rosáceo [Fig.5]. Mostraban una elevada actividad mitóticay aunque solían verse individualizadas, también tendían a constituir agrupaciones o nidos, semejantes a metástasis [Fig. 6]. Destacaba su actividad fagocítica, encontrándose eritrofagocitosis e incluso fenómenos de canibalismo [Fig. 7]. Eran PAS positivas, con un patrón granular grosero y en mazacotes que podía coincidir o no con las vacuolas previamente descritas [Fig. 8], y fueron negativas para α-naftil-acetatoesterasa ( -NAE), cloracetatoesterasa (CAE), peroxi-dasa y sudan B negro.
El estudio de citometría de flujo (CMF) del AMO descartó infiltración por linfoma no Hodgkin B y leucemia aguda.
La biopsia de un nódulo mamariomostró una neoplasia altamente celular compuesta por células primitivas y uniformes con núcleo re-dondo monomorfo. Se estructuraban en pequeños nidos con áreas discohesivas periféricas separadas por septos fibrovasculares. Mos-traban diferenciación rabdomioblástica variable y frecuente presencia de células multinucleadas gigantes. Inmunohistoquímica: positividad para miogenina y desmina y negatividad CD34, CD45 y panCK [Fig. 9].
CITOGENÉTICA YBIOLOGÍA MOLECULAR
El estudio citogenético del AMO mostró un cariotipo 46,XX,t(2;13)(q35;q14)[2]/ 92,idemx2[5]/46,XX[1]. Se detectó una clona princi-pal con la traslocación t(2;13) y otra clona en torno a la tetraploidía que probablemente procediera de una duplicación de la línea principal [Fig. 10].
La hibridación fluorescente in situ (FISH) de la biopsia mamaria confir-mó un reordenamiento del gen FOXO1 (13q14) en todas las células tumorales evaluadas mediante la sonda «Vysis LSI FOXO1 (13q14) Dual Color, Break Apart» [Fig. 11].
Adolescente de 13 años con poliadenopatías y bicitopeniaM.D. Serrano Chacón, R.M. Morales-Camacho, C. Prats-Martín, R. López Irizo*, T. Caballero-Velázquez, O. Pérez López, E Carrillo, M.T. Vargas,
R. Bernal Ruiz, J.J. Borrero*
Unidad de Gestión Clínica (UGC) de Hematología y Hemoterapia, (*) UGC de Anatomía Patológica, Hospital Universitario Virgen del Rocío, Sevilla

CASOS CLÍNICOS CITOLÓGICOS 93
DIAGNÓSTICO
Rabdomiosarcoma Alveolar metastásico.
EVOLUCIÓN
A su ingreso se inició tratamiento del síndrome de lisis tumoral con hiperhidratación, alopurinol y rasburicasa más soporte hemodinámico y transfusión de hemoderivados. Se instauró tratamiento poliquimio-terápico (esquema “IVADo”) para sarcomas metastásicos (según pro-tocolo EpSSGRMS 2005) con ifosfamida, vincristina, actinomicina D y doxorrubicina. Se practicó exéresisdel músculo flexor del primer dedo del miembro inferior izquierdo junto con radioterapia local y una mas-tectomía simple bilateral. Inicialmente alcanzó la remisión completa, con recaída a los 10 meses del diagnóstico. Actualmente se mantiene en tratamiento paliativo.
DISCUSIÓN
En España la incidencia de neoplasias malignas en la infancia es de aproximadamente 13 casos por 100.000 habitantes y año, siendo el 60% tumores sólidos y el 40% linfomas y leucemias. Los rabdomio-
sarconas constituyen el 7% de los tumores sólidos infantiles, siendo el tumor de tejidos blandos más prevalente en niños y adolescentes1. Deriva de células mesenquimales relacionadas con la miogénesis de músculo esquelético, localizándose a menudo en cabeza y cuello, tracto genitourinario, extremidades y tronco. Se reconocen cuatro tipos histológicos: embrionario, alveolar, pleomórfico y esclerosante. El subtipo alveolar prevalece en aquéllos pacientes con características clínicas menos favorables al diagnóstico; hasta un 60% presentan la traslocación t(2;13)(q35;q14) que condiciona muy mal pronóstico. En esta traslocación se fusionan los oncogenes PAX3 y FOXO1 gene-rando una proteína quimérica (P3F) que provoca un bloqueo en la maduración de los mioblastos y un crecimiento descontrolado de los mismos2,12.
La infiltración de médula ósea varía entre el 2 y el 23 % dependiendo del subtipo y estadio de la enfermedad3. Las características citológicas descritas en la bibliografía corresponden a células de tamaño grande, pleomórficas, concitoplasma basófilo y vacuolado, fenómenos de eri-trofagocitosis y canibalismo, presencia de nidos y PAS positividad4,5.
La edad de la paciente, la concurrencia de un cuadro poliadenopático, citopenias y un aspirado medular con presencia de escasos nidos ce-
FIGURA 1 FIGURA 3
FIGURA 2
TAC toracoabdominal. Múltiples nódulos mamarios dispersos, bilaterales.
AMO (MGG, x1000). Abundantes células indiferenciadas de apariencia blástica.
Sangre periférica. May-Grünwald Giemsa (MGG), x1000.Células de aspecto indiferenciado (a y b); eritroblasto (c).

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA94
FIGURA 4
FIGURA 5
AMO (MGG, x1000). Pleomorfismo, a veces con grandes células blásticas binucleadas (a y b). A menudo se aprecia vacuolización citoplásmica (c, d y e).
AMO (MGG, x1000). Los citoplasmas contienen ocasionalmente un material amorfo de
color rosáceo.
lulares y de numerosas células indiferenciadas bien individualizadas plantea morfológicamente el diagnóstico diferencial con una leuce-mia aguda6. Concretamente la observación de vacuolas en las células sarcomatosas obliga a descartar que se trate de una infiltración por células de un linfoma de Burkitt. También agrupaciones celulares se han descrito como imágenes de pseudometástasis (pseudo-rosetas) o nidos de eclosión blástica en el aspirado medular de algunas leucemias agudas (p. ej. leucemia megacarioblástica7). Además, la infiltración mamaria, aunque inusual, también puede observarse ocasionalmente en diversos linfomas8.
Por tanto el principal diagnóstico diferencial morfológico se plantea con las células del linfoma/leucemia de Burkitt, que de forma distintiva presentan una actividad mitótica mayor, ausencia de canibalismo, nu-cléolos prominentes eintensa basofilia citoplasmática; habitualmente son negativas para la reacción del PAS y no suelen conformar figuras de pseudometástasis. Destacamos además la observación ocasional de un material rosáceo intracitoplasmático en las células malignas del caso presente, que debe corresponder a inclusiones rabdoides (actina y miosina principalmente) y que no se encuentra en células de origen hematopoyético.
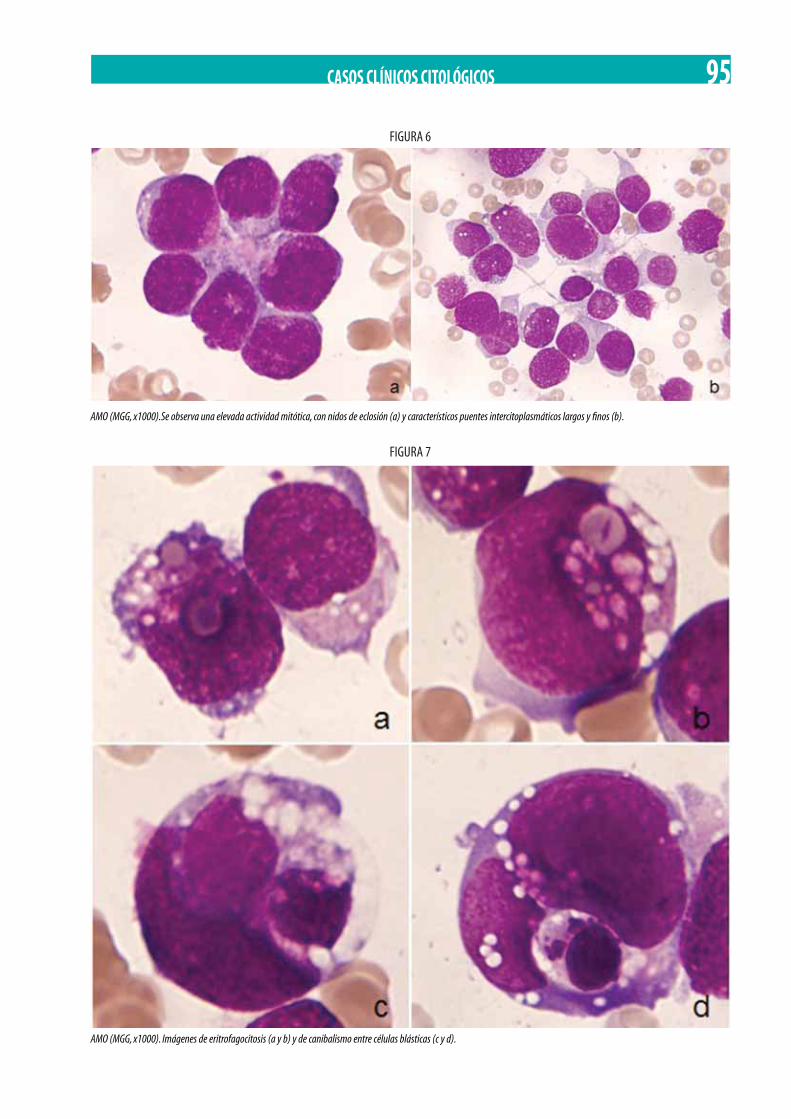
CASOS CLÍNICOS CITOLÓGICOS 95
FIGURA 6
FIGURA 7
AMO (MGG, x1000).Se observa una elevada actividad mitótica, con nidos de eclosión (a) y característicos puentes intercitoplasmáticos largos y finos (b).
AMO (MGG, x1000). Imágenes de eritrofagocitosis (a y b) y de canibalismo entre células blásticas (c y d).
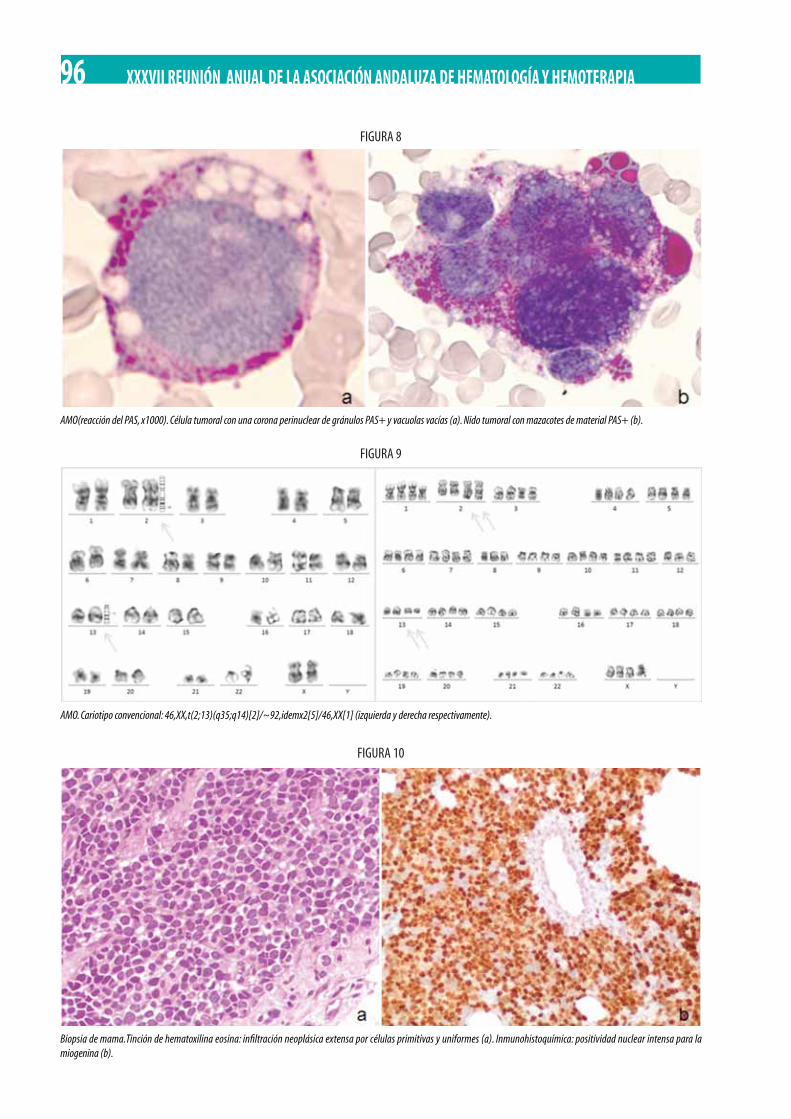
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA96
FIGURA 8
FIGURA 9
FIGURA 10
AMO(reacción del PAS, x1000). Célula tumoral con una corona perinuclear de gránulos PAS+ y vacuolas vacías (a). Nido tumoral con mazacotes de material PAS+ (b).
AMO. Cariotipo convencional: 46,XX,t(2;13)(q35;q14)[2]/~92,idemx2[5]/46,XX[1] (izquierda y derecha respectivamente).
Biopsia de mama.Tinción de hematoxilina eosina: infiltración neoplásica extensa por células primitivas y uniformes (a). Inmunohistoquímica: positividad nuclear intensa para la
miogenina (b).

CASOS CLÍNICOS CITOLÓGICOS 97
El diagnóstico definitivo precisa de la CMF para descartar que las cé-lulas malignas sean de origen hematológico; de estudios histológicos e inmunohistoquímicos para filiar el origen musculoesquelético de la neoplasia y de pruebas genéticas y moleculares para precisar una translocación típica del rabdomiosarcoma alveolar.
A RECORDAR
Las infiltraciones medulares de tumores sólidos en la infancia pueden asemejarse morfológicamente a una leucemia aguda, pudiendo orien-tar el diagnóstico un examen citológico exhaustivo.
El diagnóstico preciso exige el concurso de estudios citométricos, genéticos y moleculares que se pueden realizar en el aspirado de la médula ósea.
La realización de una biopsia de médula ósea es una opción útil en el estudio de pancitopenias severas en la infancia, siendo una prueba resolutiva para discriminar entre las infiltraciones por tumores sólidos o hemopatías malignas9,10,11.
BIBLIOGRAFÍA
1. Sierrasesumaga L.Tumores sólidos más frecuentes en la infancia. An Pediatr Contin. 2004;2:153-62.
2. Sun X, Guo W, Shen JK, Mankin HJ, Hornicek FJ, Duan Z. Rhabdo-myosarcoma: Advances in Molecular and Cellular Biology. Sarcoma. 2015;2015:232010.
3. Weiss AR, Lyden ER, Anderson JR, et al. Histologic and Clinical Charac-teristics Can Guide Staging Evaluations for Children and Adolescents With Rhabdomyosarcoma: A Report From the Children’s Oncology Group Soft Tissue Sarcoma Committee. Journal of Clinical Oncology. 2013;31(26):3226-3232.
4. Bien E, Maciejka-Kapuscinska L, Niedzwiecki M, et al. Childhood rhabdo-myosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clinical & Experimental Metastasis. 2010;27(6):399-407.
5. Stall JN, Bailey NG. Metastatic alveolar rhabdomyosarcoma to the bone marrow mimicking acute leukemia. Blood. 2012;120(18):3632.
6. Ortuño FJ, López-Poveda MJ. Rhabdomyosarcoma presenting with leukemic phase and massive bone marrow infiltration. Blood. 2016;127(26):3458.
7. Paredes-Aguilera R, Romero-Guzman L, Lopez-Santiago N, Trejo R. Biology, clinical, and hematologic features of acute megakaryoblastic leukemia in children. Am. J. Hematol. 2003;73:71–80.
8. Raj SD, Raj KM, Krishnamurthy S, Brahmaroutu A, Whitman GJ. Breast metastasis in an adult woman with alveolar rhabdomyosarcoma of the ethmoid sinus. Radiology Case Reports. 2014;9(3):858.
9. Karagiannis P, Guth N, Thoennissen GB, et al. Alveolar rhabdomyosar-coma confined to the bone marrow with no identifiable primary tumour using FDG-PET/CT. Clinical Sarcoma Research. 2015;5:24.
10. Kern JB, Hii A, Kruse MJ, et al. A Leukemic Presentation of Alveolar Rhab-domyosarcoma in a 52-Year-Old Woman Without an Identifiable Primary Tumor. International journal of surgical pathology. 2015;23(1):75-77.
11. Balogh P, Bánusz R, Csóka M, Váradi Z, Varga E, Sápi Z. Primary alveolar rhabdomyosarcoma of the bone: two cases and review of the litera-ture. Diagnostic Pathology. 2016;11:99.
12. Hu Q, Yuan Y, Wang C. Structural and Functional Studies of FKHR-PAX3, a Reciprocal Fusion Gene of the t(2;13) Chromosomal Translocation in Alveolar Rhabdomyosarcoma. Deb S, ed. PLoS ONE. 2013;8(6):e68065.
FIGURA 11
Biopsia de mama. FISH.Reordenamientodelgen FOXO1 en todas las células tumorales
(sonda «Vysis LSI FOXO1 (13q14) Dual Color, Break Apart»).

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA98
MOTIVO DE CONSULTA
Estudio de síndrome eosinofílico.
SINTOMATOLOGÍA
Varón de 54 años, sin antecedentes de interés, que acude a nuestro centro derivado de su hospital de referencia para estudio de síndrome eosinofílico. Refiere prurito generalizado desde hace 2 meses que se ha intensificado en los últimos días junto con aparición de lesiones eritematosas y tumefacción en extremidades. No otra sintomatología acompañante.
EXPLORACIÓN FÍSICA
A la exploración, el enfermo se encuentra hemodinámicamente esta-ble, con aceptable estado general, aunque clínicamente muy afectado por la gran intensidad del prurito. A nivel abdominal, esplenomegalia de unos 15 cm. Destaca miembro superior derecho edematizado con lesiones eritematosas, así como otras nodulares en miembros infe-riores y lesiones de rascado generalizadas. Se palpan adenopatías inguinales de forma bilateral de aproximadamente 1 centímetro de diámetro. El resto de la exploración es anodina.
PRUEBAS ANALÍTICAS
Hemograma: Hemoglobina 108 g/L; hematocrito 31%; VCM 94,3fL; HCM 32,9 pg; RDW 22,9%; plaquetas 209 ×109/L; leucocitos 66,98 ×109/L; neutrófilos 16,39 × 109/L (24,5%); linfocitos 3,8 ×109/L (5,7%); monocitos 1,7 ×109/L (2,5%); eosinófilos 43,6 ×109/L (65,2%); basófilos 0,77 ×109/L (1,2%). Estudio de anemia: hierro 115 mg/dL; ferritina 637,6 ng/mL; transferrina 157 mg/dL; vitamina B12 515 pg/mL; folato 2,8 nmol/L. β-2-microglobulina 2.756,6 mg/L. Se-rologías: negativas para hepatitis B, hepatitis C, HIV1 y 2, CMV, rubeola y VEB. Serología de áscaris, anisakis, leishmania, equinococo y brucella
Leucemia eosinofílica malignaIrene Vico Herrera, María del Carmen Martínez Losada, Víctor Arqueros Martínez, Joaquín Sánchez García
Servicio de Hematología y Hemoterapia. Hospital Universitario Reina Sofía, Córdoba
Torácico-abdominal, con corte a nivel esplénico: esplenomegalia de 17 cms de eje mayor.
Sangre periférica(MGG 100×): intensa eosinofilia con formas nucleares anómalas e hipogranulación.
FIGURA TAC
FIGURA 1
negativas. Anticuerpos IgG e IgM para toxoplasma positivos. Estudio de parásitos en heces, negativo. Dosificación de IgE 4,52 mg/dL (normal).
ESTUDIOS COMPLEMENTARIOS
Radiografía de tórax: sin hallazgos patológicos. Tomografía axial com-putarizada (TAC) toraco-abdominal: esplenomegalia de 17 cm sin otros hallazgos destacables (Figura TAC).
Frotis de sangre periférica: 78% de eosinófilos de aspecto displásico con núcleos hiposegmentados, vacuolización y aumento de granulación, 11% de neutrófilos, 6% de linfocitos, 2% monocitos, 2% basófilos, 1% de células de aspecto blástico (Figuras 1, 2, 3 y 4).
Aspirado de médula ósea: Extendido hipercelular con megacariocitos tromboformadores proporcionales. Hiperplasia mieloide conservando los diferentes estadios madurativos, sin alteraciones morfológicas, ob-jetivándose un aumento de la serie eosinofílica (43% sobre la pobla-ción mieloide total). Presencia de un 7% de blastos mieloides. Serie eritroide conservada sin presencia de displasia. No otros hallazgos. (Figura 5).
Citoquímica: Los eosinófilos presentaban positividad para la mielope-roxidasa, mientras que algunos granulocitos neutrófilos fueron ne-gativos. La cloracetatoesterasa resultó negativa para los eosinófilos maduros (Figura 6).
ESTUDIOS DE BIOLOGÍA MOLECULAR Y CITOGENÉTICA
Biología molecular: reordenamientos BCR-ABL (p190 y p210) y FIP1L1-PDGFRA negativos (Figura 7), mutaciones JAK2 (V617F y Exón 12), MPL y CALR negativas.
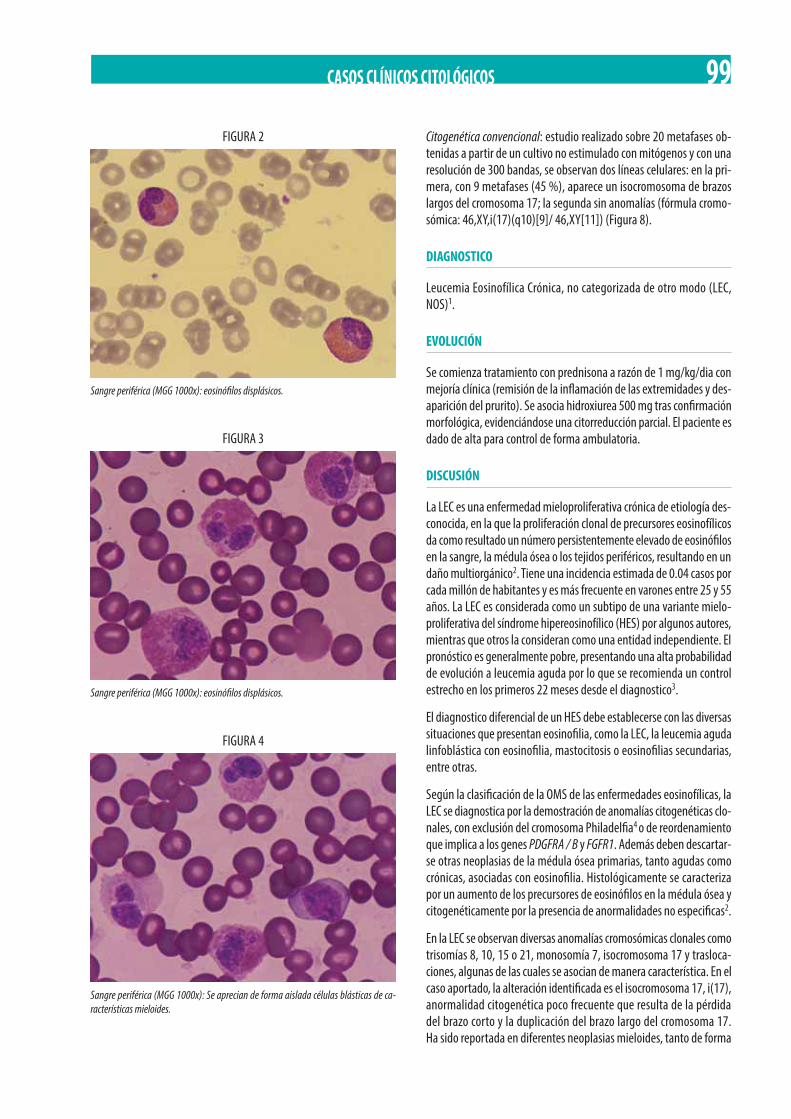
CASOS CLÍNICOS CITOLÓGICOS 99
Citogenética convencional: estudio realizado sobre 20 metafases ob-tenidas a partir de un cultivo no estimulado con mitógenos y con una resolución de 300 bandas, se observan dos líneas celulares: en la pri-mera, con 9 metafases (45 %), aparece un isocromosoma de brazos largos del cromosoma 17; la segunda sin anomalías (fórmula cromo-sómica: 46,XY,i(17)(q10)[9]/ 46,XY[11]) (Figura 8).
DIAGNOSTICO
Leucemia Eosinofílica Crónica, no categorizada de otro modo (LEC, NOS)1.
EVOLUCIÓN
Se comienza tratamiento con prednisona a razón de 1 mg/kg/dia con mejoría clínica (remisión de la inflamación de las extremidades y des-aparición del prurito). Se asocia hidroxiurea 500 mg tras confirmación morfológica, evidenciándose una citorreducción parcial. El paciente es dado de alta para control de forma ambulatoria.
DISCUSIÓN
La LEC es una enfermedad mieloproliferativa crónica de etiología des-conocida, en la que la proliferación clonal de precursores eosinofílicos da como resultado un número persistentemente elevado de eosinófilos en la sangre, la médula ósea o los tejidos periféricos, resultando en un daño multiorgánico2. Tiene una incidencia estimada de 0.04 casos por cada millón de habitantes y es más frecuente en varones entre 25 y 55 años. La LEC es considerada como un subtipo de una variante mielo-proliferativa del síndrome hipereosinofílico (HES) por algunos autores, mientras que otros la consideran como una entidad independiente. El pronóstico es generalmente pobre, presentando una alta probabilidad de evolución a leucemia aguda por lo que se recomienda un control estrecho en los primeros 22 meses desde el diagnostico3.
El diagnostico diferencial de un HES debe establecerse con las diversas situaciones que presentan eosinofilia, como la LEC, la leucemia aguda linfoblástica con eosinofilia, mastocitosis o eosinofilias secundarias, entre otras.
Según la clasificación de la OMS de las enfermedades eosinofílicas, la LEC se diagnostica por la demostración de anomalías citogenéticas clo-nales, con exclusión del cromosoma Philadelfia4 o de reordenamiento que implica a los genes PDGFRA / B y FGFR1. Además deben descartar-se otras neoplasias de la médula ósea primarias, tanto agudas como crónicas, asociadas con eosinofilia. Histológicamente se caracteriza por un aumento de los precursores de eosinófilos en la médula ósea y citogenéticamente por la presencia de anormalidades no especificas2.
En la LEC se observan diversas anomalías cromosómicas clonales como trisomías 8, 10, 15 o 21, monosomía 7, isocromosoma 17 y trasloca-ciones, algunas de las cuales se asocian de manera característica. En el caso aportado, la alteración identificada es el isocromosoma 17, i(17), anormalidad citogenética poco frecuente que resulta de la pérdida del brazo corto y la duplicación del brazo largo del cromosoma 17. Ha sido reportada en diferentes neoplasias mieloides, tanto de forma
FIGURA 2
Sangre periférica (MGG 1000x): eosinófilos displásicos.
FIGURA 3
Sangre periférica (MGG 1000x): eosinófilos displásicos.
FIGURA 4
Sangre periférica (MGG 1000x): Se aprecian de forma aislada células blásticas de ca-racterísticas mieloides.
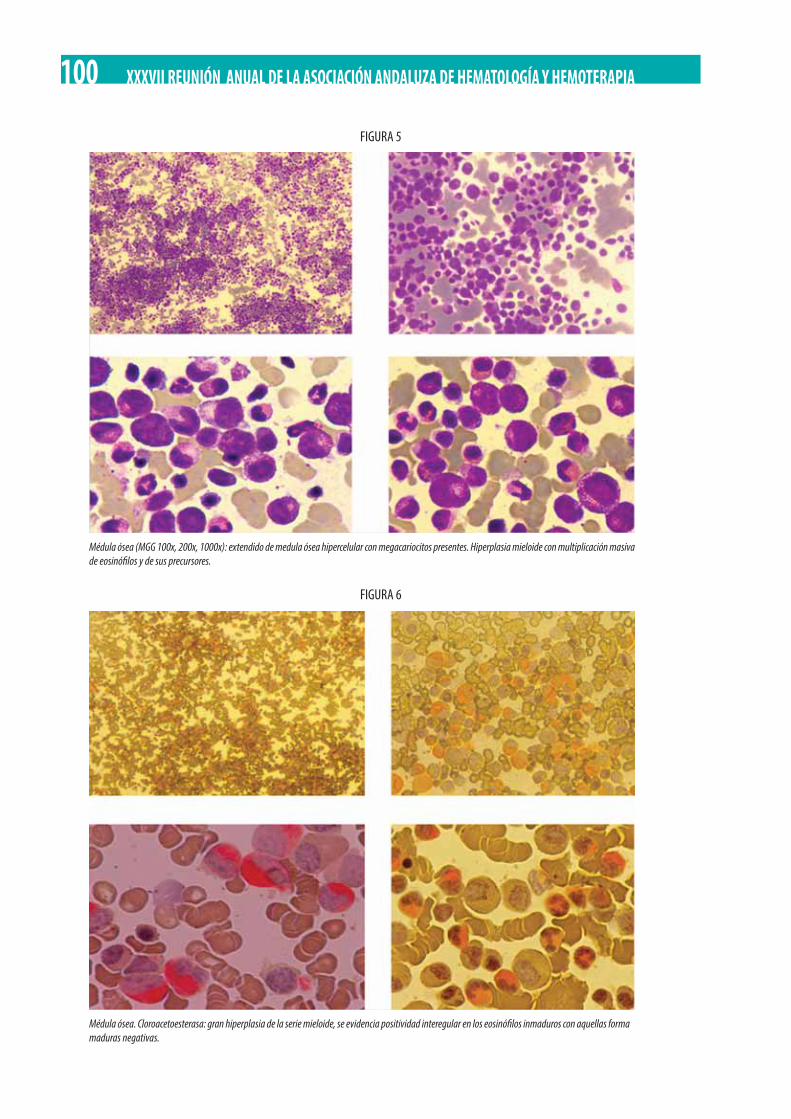
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA100
FIGURA 5
FIGURA 6
Médula ósea (MGG 100x, 200x, 1000x): extendido de medula ósea hipercelular con megacariocitos presentes. Hiperplasia mieloide con multiplicación masiva de eosinófilos y de sus precursores.
Médula ósea. Cloroacetoesterasa: gran hiperplasia de la serie mieloide, se evidencia positividad interegular en los eosinófilos inmaduros con aquellas forma maduras negativas.
CASOS CLÍNICOS CITOLÓGICOS 101
primaria como secundaria. Sin embargo, es la aberración secundaria que más frecuentemente se encuentra en la fase acelerada o crisis blástica de la Leucemia Mieloide Crónica, lo que le confiere a esta alte-ración un mal pronóstico. Por lo general el i(17) se suele hallar dentro de un cariotipo complejo, pero también se encuentra como anomalía única. Se estima que el i(17) como única anomalía define una entidad clínico-patológica distintiva con alto riesgo de progresión leucémica y mal pronóstico, con una deficiente respuesta a la quimioterapia y supervivencia corta tras la transformación leucémica.4,5 Los pacientes con i(17q) comparten varias características clínicas y hematológicas, tales como sexo masculino, edad avanzada, y esplenomegalia y/o hepatomegalia. El aspirado de médula ósea se caracteriza por hiper-celularidad, prominente basofilia y/o eosinofilia, disminución de la eri-tropoyesis y megacariocitos dismórficos6. La enfermedad suele seguir un curso crónico, aunque en la mayoría de ocasiones suele progresar y puede conducir a la muerte debido a las alteraciones orgánicas, ha-bitualmente por insuficiencia cardíaca, o como resultado de la trans-
FIGURA 7
Cariotipo de neoplasias hematológicas en M.O con técnica de bandas G: Isocromosoma de brazos largos del cromosoma 17.
FIGURA 8
Figura 8. Análisis de reordenamiento FIP1L1-PDGFRA en sangre por PCR: no se detecta.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA102
formación en leucemia aguda. No existe un tratamiento de elección, empleándose esteroides sistémicos, hidroxiurea e inhibidores de la tirosina quinasa (imatinib, nilotinib y sorafenib).7,8 En aquellos enfer-mos con un número de eosinófilos inferior a 5000/μl y que no tienen afectación orgánica no se requiere de una citorreducción rápida. En caso de fracaso del tratamiento se emplean citostáticos (hidroxiurea), con o sin IFN-α, seguido porvincristina o etopósido. Recientemente se contemplan opciones terapéuticas con mepolizumab o alemtuzumab. El único tratamiento con posibilidad de curación es el alotrasplante de progenitores hematopoyéticos.
A RECORDAR
La Leucemia Eosinofílica Crónica es una enfermedad muy poco fre-cuente, no bien definida, cuyo diagnóstico se establece en casos de síndrome hipereosinofílico idiopático en los que se demuestran anomalías citogenéticas clonales de mecanismo leucémico desco-nocido, que incluyen isocromosoma 17q, monosomía 7, trisomia 8 y del(20)(q11q12).3,4
Es imprescindible excluir anormalidades en BCR/ABL, PDGFRA, PD-GFRB y FGFR1.9
Puede presentarse con manifestaciones cutáneas de forma predo-minante, en lugar de manifestaciones sistémicas10.
BIBLIOGRAFÍA
1. Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syn-dromes. J Allergy Clin Immunol. 2012; 130(3):607-12.
2. Andreas Reiterand Jason Gotlib. Myeloid neoplasms with eosinophilia. Blood journal. 2017; 129: 704-14.
3. Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern-Med. 1968;68(6): 1220-9.
4. Citogenetic and molecular genetic aspects of eosinophilic leukaemias. British journal of haematology. 2003; 122: 173-9.
5. Eggendorfer M, Haferlach C, Zenger M. The landscape of myeloid neo-plasms with isochromosome 17q discloses a specific mutation profile and is characterized by an accumulation of pronosticalle adverse mo-lecular markers. Leukemia. 2016; 30: 1624-7.
6. Solid F, Torrabadella M, Granada T, et al. Isochromosome 17 as sole anomaly: a distinct myelodysplastic syndrome entity. Leukemia research. 1993; 17(8): 717-20.
7. Klion AD, Bochner BS, Gleich GJ, et al Approaches to the treatment of hypereosinophilic syndromes: a workshop summary reports. J Allergy Clin Immunol. 2006; 117:1292–302.
8. Ross DM, Altamura HK. Delayed diagnosis leading to accelerated-phase chronic eosinophilic leukemia due to a cytogenetically cryptic, imatinib-responsive TNIP1-PDFGRB fusion gene. Leukemia. 2016;30(6):1402-5.
9. Klion AD. Eosinophilic Myeloproliferative Disorders. Hematology Am Soc Hematol Educ Program. 2011; 2011: 257–63.
10. Vidyadharan S, Joseph B, Sukumaran PN. Chronic Eosinophilic Leukemia Presenting Predominantly with Cutaneous Manifestations. Indian J Der-matol. 2016 ; 61(4): 437–9.

CASOS CLÍNICOS CITOLÓGICOS 103
MOTIVO DE CONSULTA
Fiebre, diarrea y pérdida de peso.
HISTORIA CLÍNICA
Hombre de 50 años con antecedentes de apendicectomía, herniorrafia inguinal y colecistectomía. En Mayo de 2016 consultó por pérdida de 20 kg de peso, fiebre y diarrea de diez meses de evolución, sin hepato-esplenomegalia, y se diagnosticó de enfermedad VIH y sarcoma de Kaposi cutáneo. En los meses posteriores aparecieron adenopatías cer-vicales de gran tamaño, axilares e inguinales pequeñas, y se realizó en Octubre una biopsia ganglionar cervical con diagnóstico de Linfoma de Hodgkin (LH) clásico tipo Esclerosis Nodular (EN). Se inició trata-miento antirretroviral. Durante el estudio de extensión del linfoma se advirtió el desarrollo de una pancitopenia y se practicó un estudio de médula ósea.
EXPLORACIÓN FÍSICA
Regular estado general, consciente y ligeramente desorientado. Eupneico. Temperatura: 37.8ºC. Leucoplasia oral. Adenopatías late-rocervicales bilaterales de hasta 3 cms, axilares e inguinales de 1 cm. Hepatomegalia no dolorosa de 5 cms; no esplenomegalia. Lesiones violáceas sobreelevadas en cara, tronco y miembros superiores.
PRUEBAS ANALÍTICAS
Hemograma: leucocitos 1,11 x109/L (neutrófilos 0,4 x109/L), Hb 73 g/L (VCM 84 fL), plaquetas 51 x109/L. TP, TTPA y fibrinógeno normales. Bioquímica: hiperferritinemia de 8.589 μg/L (15-150), transferrina 159 mg/dL (200-330), albúmina 2,8 g/dL (3,5-5,3), calcio sérico 8.28 mg/dL, (8.5-10.5), PCR 120 g/L, β2-microglobulina 5,5 mg/L (1.5-2.5), triglicéridos 154 mg/dL (70-170), GOT 29 U/L (10-37), GPT 17 UI/L (10-40), GGT 92 UI/L (10-50), fosfatasa alcalina 568 U/L (40-130), LDH 195 U/L (135-225), VSG 58 mm/h. IgG 2765 mg/dL (700-1600), IgA 887 mg/dL (70-400), IgM 328 mg/dL (40-230). Marcadores tumora-les negativos. CD25 soluble 14.17 ng/mL (0-7.5). Serologías: Virus de Epstein Barr (VEB) IgG ELISA +, VEB IgM ELISA +, PCR-RT VEB 4.663 copias/mL.; VHA IgG +, HBs Ag +, anti Hbs IgG -, anti HBc IgG +, anti HBc IgM -, HBe Ag +, anti HBe IgG - (infección crónica por VHB). Citomegalovirus IgG +, PCR CMV 342 copias/mL; VHC negativo.
ESTUDIOS COMPLEMENTARIOS Y PRUEBAS DE IMAGEN
Tomografía axial computarizada (TAC): hallazgos altamente sugestivos de síndrome linfoproliferativo con adenopatías patológicas en cadenas cervicales, mediastino y retroperitoneo superior. Esplenomegalia de
Hombre de 50 años VIH, con fiebre y pancitopenia progresivaM. Reinoso (1), C. Prats-Martín (1), R.M. Morales-Camacho (1), J.J. Borrero (2), N. Al Kadi (1), O. Pérez (1), E. Carrillo (1), J.M. de Blas (1),
M.T. Vargas (1), T. Caballero-Velázquez (1), R. Bernal (1), J.A. Pérez-Simón (1)
(1) UGC de Hematología y Hemoterapia, (2) UGC Anatomía Patológica. Hospital Universitario Virgen del Rocío (HUVR). Sevilla
15 cms. PET-TAC: afectación linfática supra e infradiafragmática, de médula ósea, bazo e hígado.
ESTUDIOS CITOLÓGICOS E HISTOLÓGICOS
Biopsia de ganglio cervical: LH clásico tipo EN. Estudio inmunohisto-químico: CD45, CD20-, CD3-, CD15+, CD30+, VEB-. PCR-RT en ganglio EBV+.
Aspirado de médula ósea (MO) de celularidad moderadamente aumen-tada, serie eritroide 30.5% con leves alteraciones madurativas, granu-lopoyesis 55,5% bien representada con discretos signos de dismadu-rez, eosinófilos 2%, linfocitos 5.5%, monocitos 5%, macrófagos 1%, y plasmocitos 0.5%, en general grandes y con signos de reactividad. Serie megacariocítica conservada, con diversas dismorfias inespecífi-cas. Los macrófagos presentaban a menudo signos de activación, con fagosomas y emisión de largos seudópodos (Fig. 1); en un 30% se observaba hemofagocitosis de granulocitos (Fig. 2), eritroblastos, he-matíes, o/y plaquetas (Fig. 3). Resaltaba la presencia, inferior al 0.5%, de células grandes con un acentuado pleomorfismo, algunas gigantes (Fig. 4); de núcleo único o bien binucleadas (Fig. 5), o polilobuladas, con la cromatina densa, a veces reticulada (Fig. 6), siendo caracterís-tica la presencia de nucléolos basófilos, únicos o múltiples; citoplasma amplio con basofilia moderada y vacuolización variable, contorno en general irregular, observándose en algunas mamelones y refuerzo de la basofilia periférica. Estos hallazgos fueron altamente sugestivos de infiltración medular por LH. Cabe destacar que excepcionalmente se observaban macrófagos fagocitando estas células grandes (Fig. 7).
Biopsia de MO: se objetivó una celularidad media superior al 90%, en-tremezclándose una hematopoyesis de aspecto reactivo/hiperplásico
FIGURA 1
Aspirado de medula ósea (MGG x1000). Macrófago activado con una gran vacuola fagosómica.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA104
FIGURA 2
Aspirado de medula ósea (MGG x1000). Macrófagos fagocitando un número variable de granulocitos neutrófilos.
FIGURA 3 FIGURA 4
Aspirado de medula ósea (MGG x1000). Macrófago que ha internalizado simultánea-mente un eritroblasto, un granulocito y varias plaquetas.
Aspirado de medula ósea (MGG x1000). Una gran célula linfomatosa con un solo núcleo, y nucléolos mal definidos. Su citoplasma es basófilo, de bordes irregulares, y se aprecia una fina vacuolización.
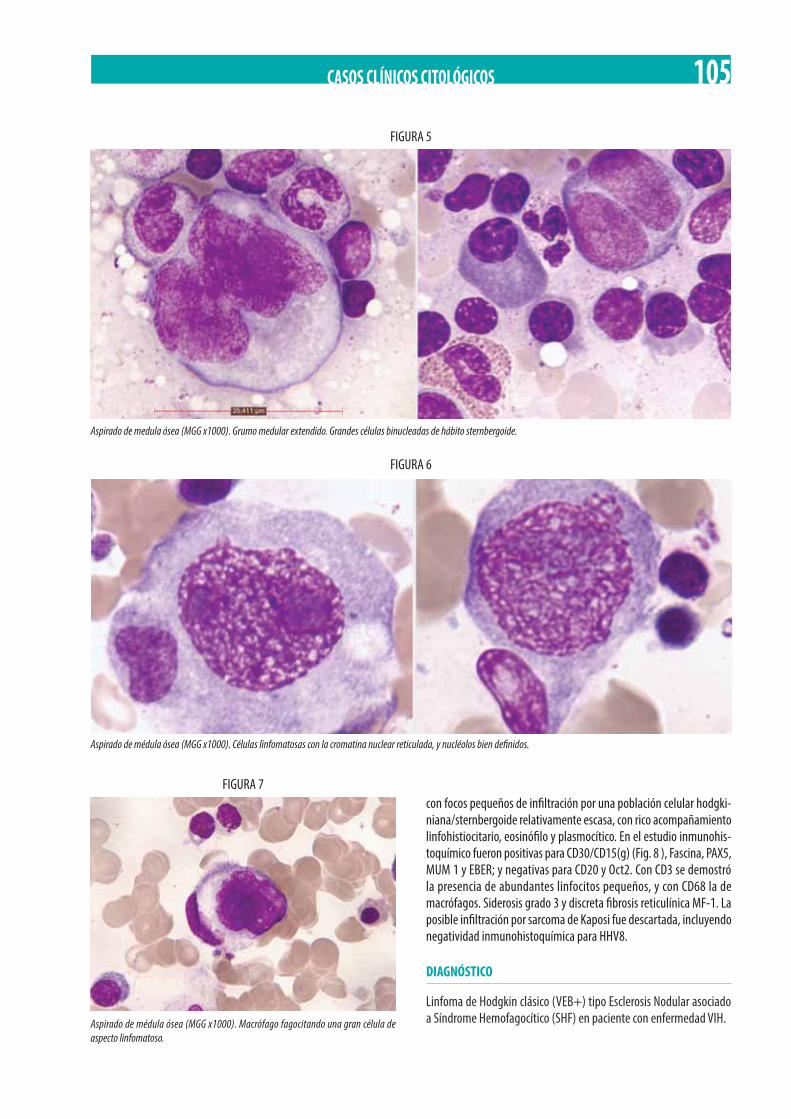
CASOS CLÍNICOS CITOLÓGICOS 105
FIGURA 5
FIGURA 6
Aspirado de medula ósea (MGG x1000). Grumo medular extendido. Grandes células binucleadas de hábito sternbergoide.
Aspirado de médula ósea (MGG x1000). Células linfomatosas con la cromatina nuclear reticulada, y nucléolos bien definidos.
FIGURA 7
Aspirado de médula ósea (MGG x1000). Macrófago fagocitando una gran célula de aspecto linfomatoso.
con focos pequeños de infiltración por una población celular hodgki-niana/sternbergoide relativamente escasa, con rico acompañamiento linfohistiocitario, eosinófilo y plasmocítico. En el estudio inmunohis-toquímico fueron positivas para CD30/CD15(g) (Fig. 8 ), Fascina, PAX5, MUM 1 y EBER; y negativas para CD20 y Oct2. Con CD3 se demostró la presencia de abundantes linfocitos pequeños, y con CD68 la de macrófagos. Siderosis grado 3 y discreta fibrosis reticulínica MF-1. La posible infiltración por sarcoma de Kaposi fue descartada, incluyendo negatividad inmunohistoquímica para HHV8.
DIAGNÓSTICO
Linfoma de Hodgkin clásico (VEB+) tipo Esclerosis Nodular asociado a Síndrome Hemofagocítico (SHF) en paciente con enfermedad VIH.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA106
EVOLUCIÓN
Tras el diagnóstico inició tratamiento con esquema ABVD (adriami-cina, bleomicina, vincristina y dexametasona). Tras el tercer ciclo, sin incidencias, no se palpan adenopatías, y se encuentra pendiente de TC intermedio de evaluación.
DISCUSIÓN
En el LH se demuestra histológicamente infiltración de la MO en el 4-20% de los casos¹,². En cambio, en el aspirado de MO la observación de células de tipo Reed-Sternberg y mononucleadas de Hodgkin es un hallazgo inusual (< 1%)³. En estos casos las células linfomatosas raramente se encuentran en porcentaje elevado4.
En el caso presentado identificamos células altamente sospechosas del LH en el aspirado de MO, destacando su pleomorfismo, los grandes núcleos con la cromatina densa y reticulada, presencia de nucléolos grandes (a menudo múltiples y mal delimitados), y marcada basofilia citoplasmática con numerosas vacuolas pequeñas. La biopsia de MO confirmó el diagnóstico de LH clásico tipo EN asociado a VEB, con fo-cos pequeños de infiltración compuestos por población hodgkiniana/sternbergoide con su habitual acompañamiento linfohistiocitario, eosinófilo y plasmocítico.
La observación adicional de signos de activación macrofágica con he-mofagocitosis relevante de diversas células, plantea la posibilidad de un SHF asociado al linfoma. Esta entidad clínica puede tener origen genético o ser reactiva a múltiples procesos, tales como infecciones, neoplasias o patologías autoinmunes5. Los criterios diagnósticos más usados son los establecidos en 2004 por la International Histiocyte Society6 (clínicos, analíticos e histológicos) (Tabla 1); son ocho, de los cuales son necesarios al menos cinco para establecer el diagnóstico. El paciente presenta fiebre, esplenomegalia, pancitopenia, hemofa-gocitosis, hiperferritinemia y CD25s elevado.
Recientemente se han publicado nuevos criterios de SHF basados en la puntuación de nueve variables, tres clínicas, cinco analíticas y una citológica7 (Tabla 2); el valor de corte considerado es 169, con una sensibilidad del 93% y una especificidad del 86%, permitiendo una precisión diagnóstica en el 90% de los pacientes. Aplicando dichos cri-terios a este caso la probabilidad de presentar un SHF alcanza un 92%.
Las neoplasias linfoides son una de las principales causas de SHF. En general se trata de Linfomas no Hodgkin, especialmente Linfoma Di-fuso de Células Grandes B y Linfomas de células T y NK 8,9. Han sido publicados casos aislados asociados a LH8,¹0,¹¹. En una única serie re-trospectiva de 34 pacientes9 se establece una fuerte correlación entre SHF, LH e infección por VEB hasta en un 94% de los sujetos, que llega al 100% en los VIH positivo (8/34 casos en la serie). Esta asociación resulta más común en el sexo masculino (76,4%) siendo los tipos his-tológicos predominantes celularidad mixta (45%) y depleción linfocí-tica (40%), en los que la infección concomitante por VEB y VIH resulta más habitual. Solo un 15% de los sujetos estaban diagnosticados de LH tipo EN. Con respecto a esta serie el caso aportado de asociación de LH y SHF muestra como rasgos más comunes que se trata de un hombre y es VEB+, y como hechos menos habituales la enfermedad VIH y el tipo histológico EN.
A RECORDAR
La observación de células de un Linfoma de Hodgkin en un aspirado medular es inusual, y su conservación es mejor en las extensiones que en las improntas de tejido, permitiendo reconocer mejor su diversidad morfológica.
El hallazgo simultáneo en la médula ósea de un Linfoma de Hodg-kin y de un Síndrome Hemofagocítico es excepcional, y debe hacer sospechar infección por virus de Epstein-Barr, especialmente en pacientes VIH.
FIGURA 8
Biopsia de médula ósea. A la izquierda hematoxilina-eosina, en el centro se observa una célula de tipo Reed-Sternberg. A la derecha inmunohistoquímica CD30.

CASOS CLÍNICOS CITOLÓGICOS 107
TABLA 1. Criterios diagnósticos para Síndrome Hemofagocítico establecidos por la International Histiocyte Society (2004)
1. Historia familiar o defecto genético conocido.
2. Presentar al menos cinco de los siguientes criterios:
BIBLIOGRAFÍA
1. Nadeem M, Naqi N, Hussain I, Khattak J, Ahmed, Khan B. Frequency of bone marrow involvement in Hodgkin´s Lymphoma on first presenta-tion. Journal of the College of Physicians and Surgeons Pakistan. 2009; 19 (12): 768-771.
2. Sudalaimuthu M, Basu D. Clinicopathological Features of Bone Marrow Infiltration in Hodgkin Lymphoma. Should bone marrow staging be done only in high risk patients. Turkish J of Pathol. 2013; 1: 1-5.
3. Howell SJ, Grey M, Chang J, Godfrey R et al. The value of bone marrow ex-amination in the staging of Hodgkin´s Lymphoma: A review of 955 cases seen in a regional cancer centre. Br J Haematol. 2002; 119: 408-411.
4. Lee SG, Paik SY, Sohn HJ, Kim SY, Kong SY. Numerous Reed-Sternberg cells in bone marrow aspirate from a patient with the syncytial variant of nodular sclerosis classical Hodgkin lymphoma. Int J Hematol. 2015; 101: 107-108.
5. Gupta S, Weitzman S. Primary and secondary hemophagocytic lympho-histiocytosis: clinical features, pathogenesis and therapy Expert Rev. Clin. Immunol. 2010; 6(1): 137–154.
6. Henter JI, Horne A, Aricó M, Egeler RM, FIlipovich AH, Imashuku E, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48: 124-131.
7. Farde L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 2014; 66: 2613-2620.
8. Morita Y, Kenzaka T, Yoshimoto H, Ohno N. Hodgkin´s lymphoma pre-ceded by haemophagocytic lymphohistiocytosis. BMJ Case Rep 2013.
9. Fanny Menard F, Besson C, Rince P, Lambotte O, Lazure T, Canioni D, et al. Hodgkin Lymphoma–Associated Hemophagocytic Syndrome: A Dis-order Strongly Correlated with Epstein-Barr Virus. Clin Infect Dis. 2008; 47: 531-534.
10. Mustafa Ali M, Ruano AL and Carraway HE. Hemophagocytic Lympho-histiocytosis in a patient with Hodgkin lymphoma and concurrent EBV, CMV, and Candida Infections. J Investig Med High Impact Case Rep. 2017 1; 5(1).
11. Domínguez-Muñoz MA, Morales-Camacho R, Prats-Martín C, Ávila R, Var-gas MT, Burillo S, Bernal R, Borrero JJ. Unusual co-occurrence of Hodgkin lymphoma and hemophagocytic lymphohistiocytosis in a bone marrow aspirate. Ann Hematol. 2016 May; 95(6):1019-21.
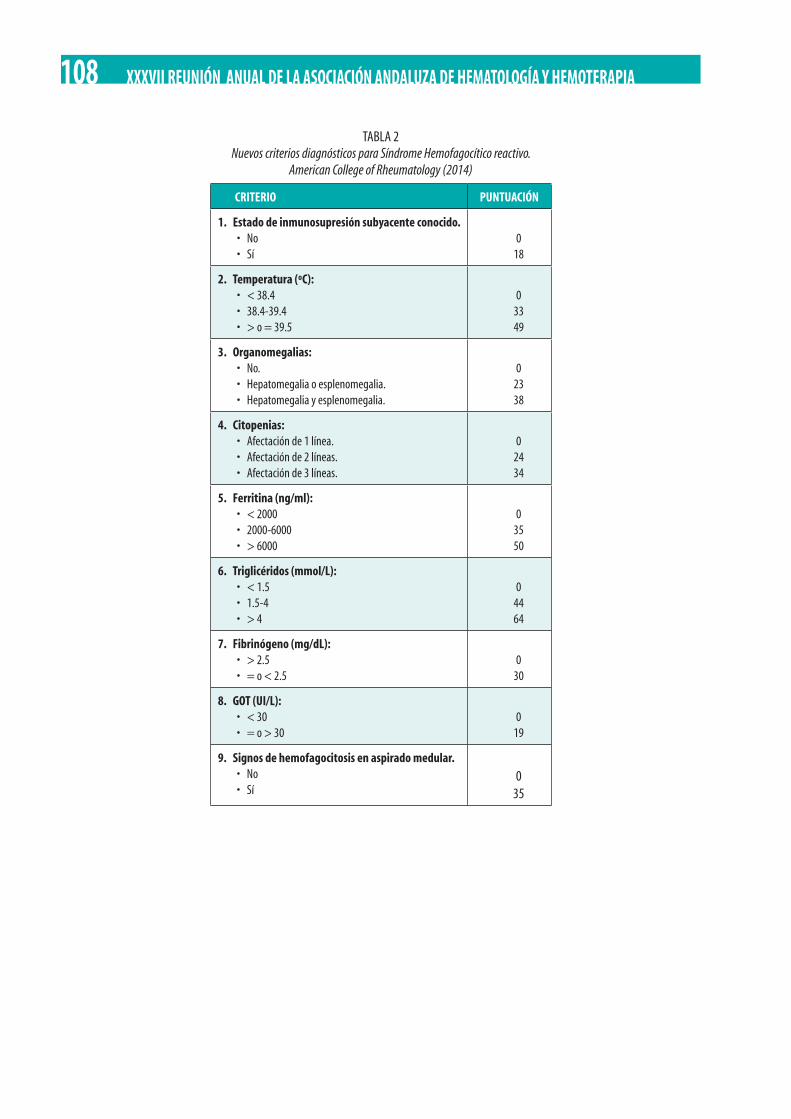
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA108
TABLA 2Nuevos criterios diagnósticos para Síndrome Hemofagocítico reactivo.
American College of Rheumatology (2014)
CRITERIO PUNTUACIÓN
1. Estado de inmunosupresión subyacente conocido.
18
2. Temperatura (ºC):
3. Organomegalias:
4. Citopenias:
5. Ferritina (ng/ml):
6. Triglicéridos (mmol/L):
7. Fibrinógeno (mg/dL):
8. GOT (UI/L):
9. Signos de hemofagocitosis en aspirado medular.

COMUNICACIONES ORALES
Hematología Clínica


COMUNICACIONES ORALES: Hematología Clínica 111
INTRODUCCIÓN/FUNDAMENTO:
Se ha descrito recientemente que el análisis del componente inmune tras el tratamiento del mieloma múltiple (MM) podría estar relacionado con las características clínicas y respuesta al tratamiento. Sin embargo, se desconoce el perfil inmune en la médula ósea al diagnóstico, así como su relación con variables clínico/biológicas e importancia pronóstica
OBJETIVO:
Analizar el componente inmune en pacientes con patología de células plasmáticas y su relación con las variables clínicas/laboratorio
PACIENTES, MATERIAL Y MÉTODO:
Estudio transversal de 60 pacientes consecutivos con patología de células plasmáticas diagnosticados en nuestro centro desde Junio 2015-Septiembre 2016. Se clasificaron en cuatro grupos: Gammapatía monoclonal de significado incierto (GMSI), MM quiescente, MM sintomático y MM tratados (MMt). Se determinaron las siguientes poblaciones celulares en médula ósea: linfocitos T (LT) (CD27-/CD27+), B (LB) (CD27-/CD27+), NK y células plasmáticas (CP) (normales CPN y patológicas CPP) mediante citometría de flujo multiparamétrica en FACSCanto II (Becton.Dickinson) y se correlacionó con las variables clínicas/laboratorio. El análisis de los datos fenotípicos se realizó mediante Software CellQuest, FACsDiva e Infinicyt (Becton-Dickinson).
RESULTADOS Y CONCLUSIONES:
Distribución de las poblaciones celulares: LT, LB, NK y células plasmáticas (CP) en la médula ósea. Tabla 1. Mayor cantidad de linfocitos B principalmente linfocitos B CD27+ y células NK en pacientes con GMSI que en el resto de los grupos (p= 0.084, p= 0.024 respectivamente).
Relación de poblaciones linfoides y células NK con variables clínicas-laboratorio:
* Mayor cantidad de LT CD27- se correlacionó con variables clínicas de peor pronóstico: mayor grado de anemia (p = 0,03), mayor afectación ósea, proteinuria, trombopenia y componente monoclonal ≥ 3 g/dl. Por el contrario, una mayor cantidad de linfocitos T y linfocitos B CD27+ se asoció con una menor frecuencia de lesiones líticas (p = 0,03). Mayor porcentaje de NK se asoció también con la presencia de afectación ósea, trombopenia, hipercalcemia y CM ≥ 3 g/dl.
* De forma notable, los pacientes que fallecieron en el primer trimestre tras el diagnóstico presentaban cifras más elevadas de linfocitos TCD27-, mientras que los pacientes que sobrevivieron presentaban cifras más elevadas de linfocitos B (p=0.07), especialmente LB CD27+ (p= 0.03).
* En el grupo de pacientes con MM tratados y EMR+ tenían un mayor número de LT CD27 (p = 0,08) que los pacientes con EMR-. El alcanzar una RP también presentaban mayor número de LT CD27- (p = 0,026) que los pacientes con RC o MBRP
* Los pacientes sin afectación lítica presentaban mayor cantidad de células plasmáticas normales mientras que los pacientes con insuficiencia renal presentaron mayor cantidad de CP, principalmente patológicas.
* No se encontró asociación estadística entre las diferentes poblaciones analizadas y los niveles de LDH, B2 microglobulina, albúmina y creatinina.
CONCLUSIONES
Nuestros resultados reflejan la relevancia del sistema inmune como factor pronóstico en la supervivencia global. Los pacientes con GMSI y MM que alcanzaron remisión completa estricta presentaban un mejor perfil inmunológico, caracterizado por un aumento de LB CD27+, NK y CP normales. El estudio de biomarcadores inmunológicos podría proporcionar un nuevo enfoque para la comprensión de los mecanismos de disfunción inmune en MM, así como para establecer nuevos scores pronósticos y objetivos terapéuticos.
Relación entre el perfil inmune de la médula ósea con variables clínico/biológicas en pacientes con patología de células plasmáticasE. García Torres (1), C. Martínez Losada (2), M.A. Álvarez Rivas (3), J. Serrano-López (4), J. Serrano (5), J. Sánchez-García (3)
(1) Hospital Universitario Reina Sofía. Instituto Maimónides Investigación Biomédica, (2) Hospital Universitario Reina Sofía. Instituto Maimónides Investigación
Biomédica, (3) Hospital Universitario Reina Sofía. Instituto Maimónides Investigación Biomédica, (4) Instituto Maimónides Investigación Biomédica,
(5) Hospital Universitario Reina Sofía. Instituto Maimónides Investigación Biomédica
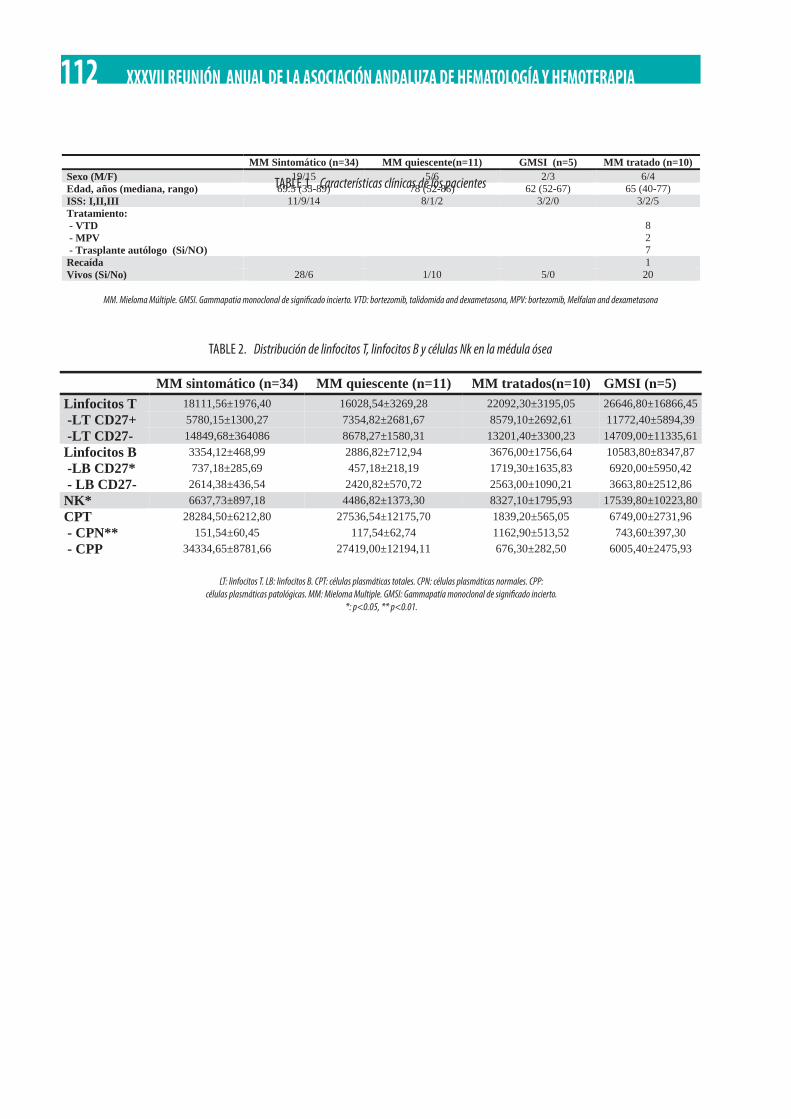
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA112
MM Sintomático (n=34) MM quiescente(n=11) GMSI (n=5) MM tratado (n=10) Sexo (M/F) 19/15 5/6 2/3 6/4 Edad, años (mediana, rango) 69.5 (35-89) 78 (52-86) 62 (52-67) 65 (40-77) ISS: I,II,III 11/9/14 8/1/2 3/2/0 3/2/5 Tratamiento: - VTD 8 - MPV 2 - Trasplante autólogo (Si/NO) 7 Recaída 1 Vivos (Si/No) 28/6 1/10 5/0 20
TABLE 1. Características clínicas de los pacientes
MM. Mieloma Múltiple. GMSI. Gammapatia monoclonal de significado incierto. VTD: bortezomib, talidomida and dexametasona, MPV: bortezomib, Melfalan and dexametasona
TABLE 2. Distribución de linfocitos T, linfocitos B y células Nk en la médula ósea
LT: linfocitos T. LB: linfocitos B. CPT: células plasmáticas totales. CPN: células plasmáticas normales. CPP:
células plasmáticas patológicas. MM: Mieloma Multiple. GMSI: Gammapatía monoclonal de significado incierto.
*: p<0.05, ** p<0.01.
MM sintomático (n=34) MM quiescente (n=11) MM tratados(n=10) GMSI (n=5)
Linfocitos T 18111,56±1976,40 16028,54±3269,28 22092,30±3195,05 26646,80±16866,45
-LT CD27+ 5780,15±1300,27 7354,82±2681,67 8579,10±2692,61 11772,40±5894,39
-LT CD27- 14849,68±364086 8678,27±1580,31 13201,40±3300,23 14709,00±11335,61
Linfocitos B 3354,12±468,99 2886,82±712,94 3676,00±1756,64 10583,80±8347,87
-LB CD27* 737,18±285,69 457,18±218,19 1719,30±1635,83 6920,00±5950,42
- LB CD27- 2614,38±436,54 2420,82±570,72 2563,00±1090,21 3663,80±2512,86
NK* 6637,73±897,18 4486,82±1373,30 8327,10±1795,93 17539,80±10223,80
CPT 28284,50±6212,80 27536,54±12175,70 1839,20±565,05 6749,00±2731,96
- CPN** 151,54±60,45 117,54±62,74 1162,90±513,52 743,60±397,30
- CPP 34334,65±8781,66 27419,00±12194,11 676,30±282,50 6005,40±2475,93

COMUNICACIONES ORALES: Hematología Clínica 113
INTRODUCCIÓN/FUNDAMENTO:
La trombocitemia esencial es una neoplasia mieloproliferativa crónica filadelfia negativa caracterizada por trombocitosis persistente, hiperplasia megacariocítica en médula ósea y un riesgo incrementado de complicaciones trombo-hemorrágicas. Las manifestaciones dermatológicas propias de la enfermedad son prurito, eritromelalgia, livedo reticularis, sangrados menores, acrocianosis y fenómeno de Raynaud. Otras manifestaciones desarrolladas en relación con los tratamientos recibidos son úlceras en mucosa oral y en extremidades inferiores, queratosis actínica, carcinoma espinocelular e hiperpigmentación de piel y uñas. La eficacia mostrada por diferentes fármacos utilizados en los últimos años, hidroxicarbamida, interferón-alfa (IFN-alfa) y anagrelida, ha condicionado la aparición de lesiones cutáneas inducidas por dichos agentes.
OBJETIVO:
Conocer la frecuencia y describir el tipo de eventos dermatológicos desarrollados, secundarios al tratamiento recibido, en los pacientes diagnosticados de trombocitemia esencial, en seguimiento en la consulta de Hematología de nuestro hospital.
PACIENTES, MATERIAL Y MÉTODO:
Se realiza un estudio retrospectivo de las manifestaciones cutáneas desarrolladas en pacientes diagnosticados de trombocitemia esencial en un hospital comarcal en los últimos 10 años. El diagnóstico de estas manifestaciones se realizó basándose en la clínica, anatomía patológica y exploraciones complementerias. Se recogen los tratamientos recibidos y la periodicidad con la que los pacientes habían consultado a un especialista en dermatología.
RESULTADOS:
Se incluyeron un total de 51 pacientes afectos de trombocitemia esencial, 24 (47,50%) eran varones y 27 (52,94 %) mujeres. La edad media fue de 66 años, con un rango entre 31 y los 87 años. Treinta y cuatro pacientes (66,66%) desarrollaron afectación cutánea secundaria al tratamiento recibido: 15 pacientes (44,11%) desarrollaron úlceras orales, 12 pacientes (35,29%) hiperpigmentación ungueal, 4 pacientes (11,76 %) úlceras maleolares, 2 pacientes (5,88%) queratosis actínica y 1 paciente (2,94%) carcinoma epidermoide (Figura1). El tiempo medio de evolución de las lesiones cutáneas fue de 28 meses. Previamente al diagnóstico de las lesiones dermatológicas ningún paciente había sido valorado por un dermatólogo. Los tratamientos empleados fueron: hidroxicarbamida 43 pacientes (84,31%), anagrelida 6 pacientes (11,66%). Dos pacientes (3,92%) no iniciaron tratamiento por no cumplir criterios. Ningún paciente de los incluídos recibió tratamiento con IFN-alfa ni con otra terapia. Ningún paciente en tratamiento con anagrelida desarrolló lesiones dérmicas.
CONCLUSIONES:
El desarrollo de manifestaciones cutáneas inducidas por diferentes agentes terapéuticos utilizados en trombocitemia esencial puede ser un efecto adverso cada vez más frecuente. El tipo de efecto adverso oscila desde manifestaciones benignas hasta otras manifestaciones como son neoplasias.
Identificar y tratar correctamente estas lesiones puede ayudar al paciente a optimizar la adherencia al tratamiento y mejorar el pronóstico de su enfermedad de base.
Se requiere un manejo multidisciplinar que nos permita realizar un diagnóstico precoz y seguimiento adecuado.
Manifestaciones dermatológicas en trombocitemia esencial. Evolución y seguimiento a largo plazo: Experiencia en un hospital comarcalM.M. Herráez-Albendea (1), S.R. Verdesoto-Cozzarelli (2), R. Cruz Conde-de Boom (2)
(1) Hospital Santa Bárbara, (2) Hospital Santa Bárbara. Puertollano

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA114
FIGURA 1
A Y B: Hiperpigmentación de uñas de manos y pies. C: Lesiones de queratosis actínica en cuero cabelludo. D: Úlcera maleolar en extremidad inferior

COMUNICACIONES ORALES: Hematología Clínica 115
INTRODUCCIÓN/FUNDAMENTO:
Los síndromes mielodisplásicos de bajo riesgo (SMD-BR) se caracterizan por una gran heterogeneidad clínica a pesar del desarrollo de modelos pronósticos específicamente dirigidos a este grupo de pacientes (pts)1. La evaluación de la severidad de las citopenias, la dependencia transfusional (DT) y el eventual riesgo de progresión a leucemia mieloblástica aguda (LMA) son los principales factores a considerar en la estratificación pronóstica. Dado que la mayoría de los pts categorizados como riesgo muy bajo/bajo/intermedio según el IPSS-R2 fallecieron sin evidencia de progresión a LMA, la identificación de los parámetros relacionados a la probabilidad de progresión leucémica es clave en la estratificación pronóstica de los pts con SMD-BR.
OBJETIVO:
Análisis de los parámetros relacionados a la probabilidad de progresión leucémica en una cohorte unicéntrica de pts con SMD-BR
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo de los pts con SMD-BR (definidos como riesgo bajo/intermedio-1 por el IPSS y categoría citogenética favorable: bueno/intermedio-1 según IPSS3 y muy bueno/bueno/intermedio según el IPSS-R). Los pts que recibieron agentes hipometilantes o trasplante fueron excluídos del análisis. La probabilidad de progresión leucémica se evaluó por el método de riesgos competidores [cmprsk package for R 2.14.0 software. R Development Core Team (2011); http://www.R-project.org/] y se calculó desde la fecha de diagnóstico del SMD hasta la fecha de progresión leucémica considerando el éxitus sin evidencia de evolución a LMA como evento competidor.
RESULTADOS
Un total de 437 pts fueron analizados. La mediana de edad fue 74 años (rango: 41-93). La anemia refractaria y la citopenia refractaria con displasia multilínea fueron los subtipos más frecuentes. El resto de las características basales se describen en la tabla 1. Tras una mediana de seguimiento de 103 meses (IC 95%: 81-124), 324 pts (74%) habían fallecido. La mediana de supervivencia global de la serie fue 39 meses (IC 95%: 33-45) y 77 pts (16.7%) progresaron a LMA. Considerando el éxitus sin evidencia de evolución a LMA como evento competidor, los siguientes parámetros tuvieron influencia en la probabilidad de progresión leucémica (Figura 1):Cariotipo diploide, -Y ó del5q vs. intermedio (p<0.001), trombopenia <50x10/L (p<0.001) y blastos 5-9% (p=0.002). En el análisis multivariante para identificar los parámetros predictivos para la progresión leucémica (subdistribution hazard regression model; SHR), los anteriores mantuvieron la significación estadística (Tabla 2).
CONCLUSIONES
En el presente estudio, parámetros clásicamente relacionados con pronóstico adverso debido a su impacto negativos obre la SG (DT), no se correlacionaron con la probabilidad de progresión leucémica. Trombopenia (<50x109/L), blastos (5-9%) y el cariotipo intermedio (vs. diploide/5q) se confirmaron como parámetros predictivos. La del5q y por tanto, el tratamiento con lenalidomida, no mostró impacto en la probabilidad de progresión. Los resultados obtenidos pueden facilitar la estratificación pronóstica de los pts con SMD-BR.
REFERENCIAS
1. Garcia-Manero G, et al. Leukemia. 2008.
2. Greenberg PL, et al. Blood. 2012.
3. Greenberg PL, et al. Blood 1989.
Evaluación pronóstica en pacientes con síndrome mielodisplásico de bajo riesgo. Parámetros relacionados con la probabilidad de progresión leucémicaJ.F. Falantes (1), J.F. Márquez-Malaver (2), C. Calderón Cabrera (2), I. Espigado (2), J.A. Pérez-Simón (2)
(1) Hospital Universitario Virgen del Rocío, (2) Hospital Universitario Virgen del Rocío

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA116
FIGURA 1. Probabilidad de progresión leucémica

COMUNICACIONES ORALES: Hematología Clínica 117
Parameter N (%)
Edad, años (mediana; rango) 74 (41-93)
Género
Masculino
Femenino
255 (58.1%)
183 (41.9%)
Subtipo FAB
AR
ARSA
LMMC
AREB
213 (48.7%)
131 (30%)
53 (12.1%)
40 (9.2%)
Subtipo WHO
5q
AR
ARSA
CRDM
AREB-1
AREB-2
SMD-U
LMMC
CRDU
ARSA-t
21 (4.9%)
81 (18.5%)
93 (21.2%)
142 (32.6%)
38 (8.7%)
1 (0.2%)
4 (0.9%)
53 (12.1%)
2 (0.45%)
2 (0.45%)
MDA score
Bajo
Intermedio
Alto
122 (25.1%)
206 (53.4%)
110 (21.5%)
IPSS-R
Muy bajo
Bajo
Intermedio
Alto
Muy alto
122 (27.7%)
123 (28%)
90 (20.7%)
90 (20.7%)
12 (2.9%)
Blastos MO %, mediana (rango)
<4%
5-9%
1 (0-9)
396 (90.5)
42 (9.5)
Hemoglobina (g/dL; mediana,
rango)
9.5 (3.5-14.5)
Plaquetas (x109/L; mediana,
rango)
< 50x109/L
50-150x109/L
>150x10e9/L
143 (7-1426)
78 (17.9%)
140 (32%)
219 (50.1%)
RAN (x109/L)
<0.5x109/L
>0.5x109/L
47 (10.8%)
390 (89.2%)
Dependencia transfusional
Si 198 (46%)
FAB: French-American-British classification, AR: Anemia refractaria, ARSA: AR con sideroblastos
en anillo, LMMC: Leucemia mielomonocítica crónica, AREB: AR con exceso de blastos, SMD-U:
SMD no clasificable, CRDU: Citopenia refractaria con displasia unilínea, ARSA-t: ARSA con
trombocitosis, MDA score: MD Anderson score para pacientes con SMD-BR, IPSS-R: International
Prognostic Scoring System-Revised, MO: Médula ósea, RAN: Recuento absoluto de neutrófilos
TABLA 1.
Características clínico/demográficas de los pacientes (N=437)
TABLA 2.
Progresión a LMA. Cox proportional hazard model
Covariable SHR 95% CI P value
Blastos MO (2-5%)1 1.13 0.4-2.7 0.78
Blastos MO (5-9%)1 2 1.1-3.8 0.01
Cariotipo (del 5q)2 0.9 0.2-2.6 0.9
Cariotipo (intermedio)2 2.2 1.2-4.2 0.01
Plaquetas (50-150x109/L)3 0.44 0.2-0.8 0.02
Plaquetas (>150x109/L)3 0.32 0.1-0.6 0.001
Dependencia transfusional 1.22 0.6-2.1 0.48
MO: Médula ósea1Referencia: Blastos (0-2%)2Referencia: Cariotipo normal 3Referencia: Plaquetas <50x109/L
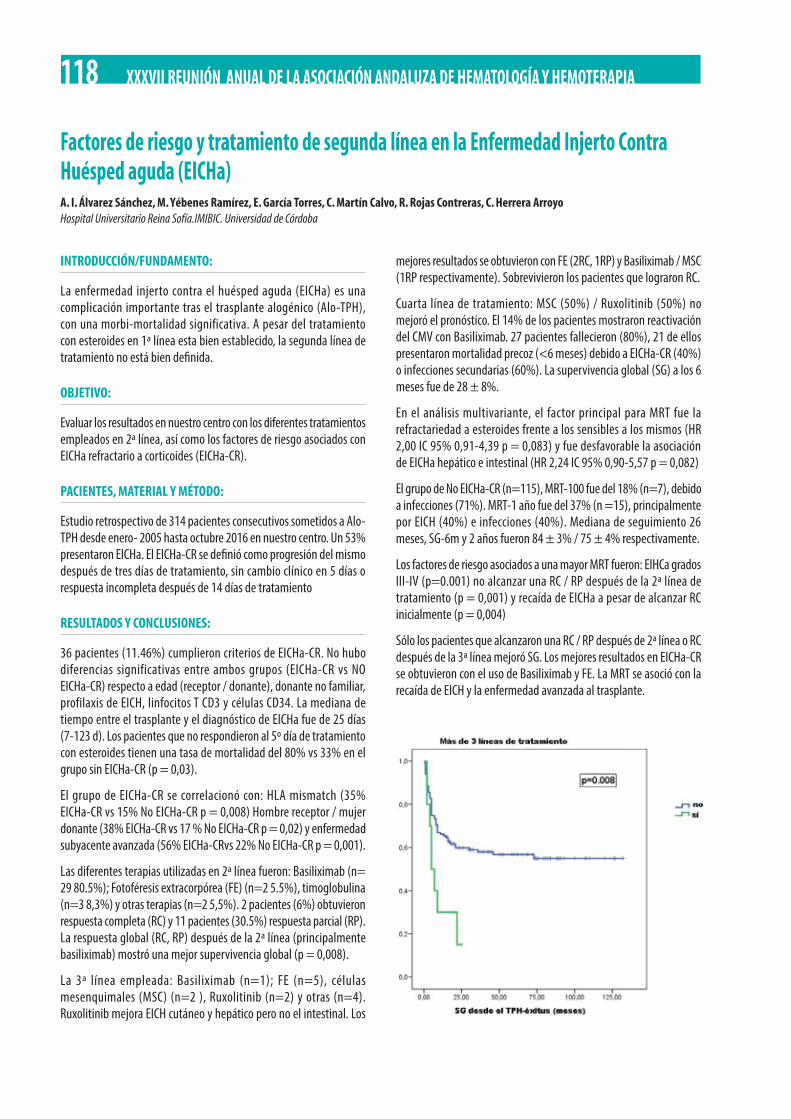
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA118
INTRODUCCIÓN/FUNDAMENTO:
La enfermedad injerto contra el huésped aguda (EICHa) es una complicación importante tras el trasplante alogénico (Alo-TPH), con una morbi-mortalidad significativa. A pesar del tratamiento con esteroides en 1ª línea esta bien establecido, la segunda línea de tratamiento no está bien definida.
OBJETIVO:
Evaluar los resultados en nuestro centro con los diferentes tratamientos empleados en 2ª línea, así como los factores de riesgo asociados con EICHa refractario a corticoides (EICHa-CR).
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de 314 pacientes consecutivos sometidos a Alo-TPH desde enero- 2005 hasta octubre 2016 en nuestro centro. Un 53% presentaron EICHa. El EICHa-CR se definió como progresión del mismo después de tres días de tratamiento, sin cambio clínico en 5 días o respuesta incompleta después de 14 días de tratamiento
RESULTADOS Y CONCLUSIONES:
36 pacientes (11.46%) cumplieron criterios de EICHa-CR. No hubo diferencias significativas entre ambos grupos (EICHa-CR vs NO EICHa-CR) respecto a edad (receptor / donante), donante no familiar, profilaxis de EICH, linfocitos T CD3 y células CD34. La mediana de tiempo entre el trasplante y el diagnóstico de EICHa fue de 25 días (7-123 d). Los pacientes que no respondieron al 5º día de tratamiento con esteroides tienen una tasa de mortalidad del 80% vs 33% en el grupo sin EICHa-CR (p = 0,03).
El grupo de EICHa-CR se correlacionó con: HLA mismatch (35% EICHa-CR vs 15% No EICHa-CR p = 0,008) Hombre receptor / mujer donante (38% EICHa-CR vs 17 % No EICHa-CR p = 0,02) y enfermedad subyacente avanzada (56% EICHa-CRvs 22% No EICHa-CR p = 0,001).
Las diferentes terapias utilizadas en 2ª línea fueron: Basiliximab (n= 29 80.5%); Fotoféresis extracorpórea (FE) (n=2 5.5%), timoglobulina (n=3 8,3%) y otras terapias (n=2 5,5%). 2 pacientes (6%) obtuvieron respuesta completa (RC) y 11 pacientes (30.5%) respuesta parcial (RP). La respuesta global (RC, RP) después de la 2ª línea (principalmente basiliximab) mostró una mejor supervivencia global (p = 0,008).
La 3ª línea empleada: Basiliximab (n=1); FE (n=5), células mesenquimales (MSC) (n=2 ), Ruxolitinib (n=2) y otras (n=4). Ruxolitinib mejora EICH cutáneo y hepático pero no el intestinal. Los
mejores resultados se obtuvieron con FE (2RC, 1RP) y Basiliximab / MSC (1RP respectivamente). Sobrevivieron los pacientes que lograron RC.
Cuarta línea de tratamiento: MSC (50%) / Ruxolitinib (50%) no mejoró el pronóstico. El 14% de los pacientes mostraron reactivación del CMV con Basiliximab. 27 pacientes fallecieron (80%), 21 de ellos presentaron mortalidad precoz (<6 meses) debido a EICHa-CR (40%) o infecciones secundarias (60%). La supervivencia global (SG) a los 6 meses fue de 28 ± 8%.
En el análisis multivariante, el factor principal para MRT fue la refractariedad a esteroides frente a los sensibles a los mismos (HR 2,00 IC 95% 0,91-4,39 p = 0,083) y fue desfavorable la asociación de EICHa hepático e intestinal (HR 2,24 IC 95% 0,90-5,57 p = 0,082)
El grupo de No EICHa-CR (n=115), MRT-100 fue del 18% (n=7), debido a infecciones (71%). MRT-1 año fue del 37% (n =15), principalmente por EICH (40%) e infecciones (40%). Mediana de seguimiento 26 meses, SG-6m y 2 años fueron 84 ± 3% / 75 ± 4% respectivamente.
Los factores de riesgo asociados a una mayor MRT fueron: EIHCa grados III-IV (p=0.001) no alcanzar una RC / RP después de la 2ª línea de tratamiento (p = 0,001) y recaída de EICHa a pesar de alcanzar RC inicialmente (p = 0,004)
Sólo los pacientes que alcanzaron una RC / RP después de 2ª línea o RC después de la 3ª línea mejoró SG. Los mejores resultados en EICHa-CR se obtuvieron con el uso de Basiliximab y FE. La MRT se asoció con la recaída de EICH y la enfermedad avanzada al trasplante.
Factores de riesgo y tratamiento de segunda línea en la Enfermedad Injerto Contra Huésped aguda (EICHa)A. I. Álvarez Sánchez, M. Yébenes Ramírez, E. García Torres, C. Martín Calvo, R. Rojas Contreras, C. Herrera Arroyo
Hospital Universitario Reina Sofía.IMIBIC. Universidad de Córdoba
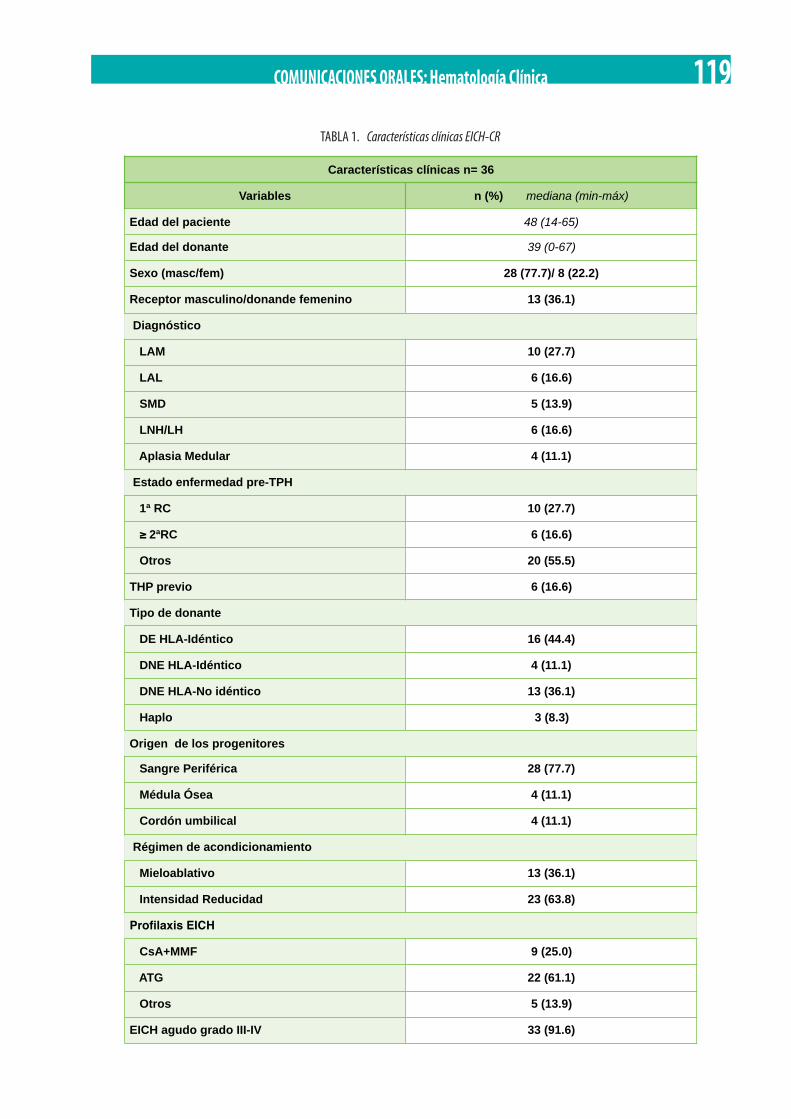
COMUNICACIONES ORALES: Hematología Clínica 119
Características clínicas n= 36
Variables n (%) mediana (min-máx)
Edad del paciente 48 (14-65)
Edad del donante 39 (0-67)
Sexo (masc/fem) 28 (77.7)/ 8 (22.2)
Receptor masculino/donande femenino 13 (36.1)
Diagnóstico
LAM 10 (27.7)
LAL 6 (16.6)
SMD 5 (13.9)
LNH/LH 6 (16.6)
Aplasia Medular 4 (11.1)
Estado enfermedad pre-TPH
1ª RC 10 (27.7)
2ªRC 6 (16.6)
Otros 20 (55.5)
THP previo 6 (16.6)
Tipo de donante
DE HLA-Idéntico 16 (44.4)
DNE HLA-Idéntico 4 (11.1)
DNE HLA-No idéntico 13 (36.1)
Haplo 3 (8.3)
Origen de los progenitores
Sangre Periférica 28 (77.7)
Médula Ósea 4 (11.1)
Cordón umbilical 4 (11.1)
Régimen de acondicionamiento
Mieloablativo 13 (36.1)
Intensidad Reducidad 23 (63.8)
CsA+MMF 9 (25.0)
ATG 22 (61.1)
Otros 5 (13.9)
EICH agudo grado III-IV 33 (91.6)
TABLA 1. Características clínicas EICH-CR

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA120
INTRODUCCIÓN/FUNDAMENTO:
Las leucemias agudas de linaje ambiguo son aquellas que no muestran clara evidencia de diferenciación hacia un determinado linaje. Suponen menos del 4% de las leucemias agudas totales. Se dividen en leucemias agudas indiferenciadas (AUL) sin antígenos específicos de linaje y leucemias agudas de fenotipo mixto (MPAL) con para más de una línea.
OBJETIVO:
Analizar las características clínicas de presentación, el tratamiento recibido y la evolución de los pacientes diagnosticados en nuestro centro de leucemia aguda de linaje ambiguo.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de todos los pacientes diagnosticados de leucemia aguda en nuestro centro entre enero de 2008 y marzo de 2017. Identificación de los pacientes con leucemia de linaje ambiguo según los criterios WHO 2008.
RESULTADOS Y CONCLUSIONES:
Un total de 359 pacientes fueron diagnosticados en nuestro centro de leucemia aguda, de ellos 15 (4.2%) cumplían criterios de linaje ambiguo. Ocho de ellos fueron varones y 7 mujeres, con una mediana de edad de 29 años (rango 1-86). Datos analíticos al diagnóstico reflejados en tabla1. Se dividieron en 3 subgrupos: Siete pacientes (47%) se identificaron como MPAL T/mieloide, 6 (40%) como MPAL B/mieloide y 2 (13%) como AUL. El 47% de los pacientes (7) presentaron
cariotipo adverso, definiéndose como tal aquellos con más de 3 de alteraciones citogenéticas, translocación (9;22) o reodenamiento MLL/11q23. Cuatro pacientes (27%) presentaban afectación extramedular al diagnóstico, 3 en SNC y 1 un líquido ascítico.
De los 15 pacientes, 7 (47%) recibieron esquemas quimioterápicos para leucemia aguda linfoblástica (LAL) (uno de ellos aún en tratamiento de inducción), 2 (13%) para leucemia mieloblástica aguda (LAM), 5 (33%) esquemas combinados y 1 (7%) tratamiento paliativo. Finalmente, 10 pacientes (69%) recibieron transplante alogénico de progenitores hematopoyéticos.
Un total de 9 pacientes (69%) consiguieron la remisión completa y de ellos 8 recayeron tras la primera línea de tratamiento. Con un seguimiento medio de 44 meses la mediana de supervivencia del grupo global es de 30 meses, con 7 pacientes vivos (47%) a los 2 años. Analizando por subgrupos diagnósticos, la mediana de supervivencia del subgrupo MPAL mieloide/T fue de 54 meses, MPAL mieloide/B 12 meses y AUL 11 meses. La mediana de supervivencia según el tratamiento recibido fue de 15 meses para los pacientes tratados con esquema de linfoblástica, 17 meses para los esquemas con mieloblástica y 13 para el tratamiento combinado.
CONCLUSIONES:
Las leucemias agudas de linaje ambiguo son una entidad clínica inusual, su presentación clínica es muy heterogénea y su tratamiento no está totalmente estandarizado.
En nuestra serie, aunque pequeña, presenta una alta proporción de pacientes con afectación extramedular y con cariotipo adverso.
El porcentaje de remisiones completas dentro de lo esperado pero con un muy alta porcentaje de recaídas.
Leucemias agudas de linaje ambiguo, experiencia en un único centroB. Pedrote Amador, J. Falantes González, I. Montero Cuadrado, M.L. Martino Galiana, J.A. Perez Simón, J. González Campos
Hospital Universitario Virgen del Rocío
TABLA 1. Datos analíticos al diagnóstico según subgrupo de leucemias de linaje ambiguo
Parámetro MPAL T/mieloide (N=7) MPAL B/mieloide (N=6) AUL (N=2)
Leucocitos (x10e9/L) 62,9 x10e9/L 66,4x10e9/L 189,3 x10e9/L
Hemoglobina (g/L) 107,83 g/L 78,8 g/L 83 g/L
Plaquetas (x10e9/L) 133 x10e9/L 60,8x10e9/L 45,5 x10e9/L
% blastos SP 43,7 % 60,4 % 84 %
% blastos MO 68,3 % 74,6 % 89%
Cariotipo adverso 42 % 50% -
Afectación extramedular 14% 50% -
SP: sangre periférica, MO: médula ósea

COMUNICACIONES ORALES: Hematología Clínica 121
Análisis de supervivencia


COMUNICACIONES ORALES
Hemostasia y Trombosis


COMUNICACIONES ORALES: Hemostasia y Trombosis 125
INTRODUCCIÓN/FUNDAMENTO:
El déficit de antitrombina (AT) es una trombofilia con elevado riesgo trombótico que se transmite de forma autosómica dominante (AD). Su deficiencia hereditaria se produce por la depresión total de su síntesis o por modificación de la molécula. El riesgo de ETEV (enfermedad tromboembólica venosa) aumenta 10 veces respecto a la población general. A los 25-35 años el 50% de los pacientes son sintomáticos. La trombosis se puede presentar de forma espontánea o en situaciones de riesgo. Es fundamental la profilaxis con heparina de bajo peso molecular (HBPM) y la administración de AT en determinados escenarios. Tras un evento trombótico la anticoagulación se mantiene habitualmente de forma indefinida
OBJETIVO:
Describir las características demográficas, clínicas y analíticas de nuestra cohorte con déficit de AT
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo y retrospectivo de 19 pacientes diagnosticados de déficit de AT hereditaria, excluyéndose aquellos con déficit adquirido, en un hospital de tercer nivel en el período de tiempo comprendido entre los años 2005-2016.
RESULTADOS Y CONCLUSIONES:
Analizamos un total de 19 pacientes diagnosticados de déficit de AT (12 mujeres y 7 hombres) con una mediana de edad al diagnóstico de 34 años (rango 11-60 años) y una media de nivel de AT de 58% (+/-9,7%). Un 52,6% (n=10) correspondió a casos sintomáticos; n=6 se objetivaron en mujeres tras presentar factores precipitantes para la trombosis (1 post operatorio, 1 gestación, 2 en relación con anticonceptivos orales, 1 paraneoplásica) y sólo
1 fue idiopática. Los otros casos (n=4) se presentaron en hombres sin causa desencadenante. Respecto al grupo sintomático el primer episodio trombótico objetivado ocurrió a la edad de 11 años (rango 11-58). El 60% (n=6) presentaban historia trombótica familiar y 40% (n=4) tenían familiares portadores de la trombofilia. Respecto a la localización de la trombosis hubo: 1 tromboembolismo pulmonar (TEP), 5 trombosis venosas profundas (TVP), 1 accidente cerebro vascular (ACVA) y 1 trombosis mesentérica; en 2 pacientes hubo coexistencia de TVP y TEP. Actualmente todos permanecen anticoagulados con AVK (n=7), ACODs (n=2) o HBPM (n=1); ver Tabla 1. Los casos asintomáticos (n=9) corresponden a diagnósticos realizados a raíz de estudio de portadores en parientes de primer grado, por el momento sin eventos trombóticos.
En nuestra serie también analizamos un total de 18 embarazos en 8 mujeres. Se documentaron 4 abortos y ninguna complicación vascular gestacional. En 4 de ellas se utilizó HBPM y aporte de AT periparto; 2 eran portadoras de otras trombofilias (F2-G20201A y MTHFR). En las que no se realizó profilaxis con HBPM, el diagnóstico de trombofilia fue a posteriori de la gestación y dos de éstas aún no han presentado eventos trombóticos (ver Tabla 2).
CONCLUSIONES
En nuestra serie el leve aumento de eventos trombóticos observada en pacientes de sexo femenino podría explicarse por la mayor frecuencia de exposición de las mujeres a factores de riesgo exclusivos de su género (gestación, anticonceptivos).
La administración de heparina y AT en el momento del parto mejora los resultados obstétricos y previene de la ETV materna.
Algunos de estos pacientes, a pesar de haber estado en situaciones de riesgo trombótico, no han desarrollado complicaciones lo que confirma el ETV como una enfermedad compleja con interacción de predisposición genética y factores ambientales.
Déficit hereditario de antitrombina: experiencia de nuestro centroM. Ángeles Domínguez-Muñoz (1), M.A. Blum-Domínguez (2), Reyes Jiménez-Bárcenas (2), Ramiro Nuñez-Vázquez (2),
Rosario Pérez Garrido (2), F.J. Rodríguez-Martorell (2)
(1) UGC de Hematología y Hemoterapia. Hospital Universitario Virgen del Rocío. Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla
(2) UGC de Hematología y Hemoterapia. Hospital Universitario Virgen del Rocío. Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA126
TABLA 1. Características generales pacientes con trombosis
Paciente SexoAntecedente
familiar trombosis
Historia
conocida de AT
familiar
Factor desencadenante %AT Trombosis Edad
1 Mujer Sí No Anticonceptivos orales 40% TEP 21
2 Mujer No Sí 48% No
3 Mujer Sí Sí 50% No
4 Mujer Sí No Paraneoplásico 60% TVP 58
5 Mujer No No Idiopático 60% ACV 39
6 Varón Sí Sí Idiopático 62% TVP+TEP 44
7 Mujer Sí Sí 45% No
8 Varón Sí Sí 57% No
9 Varón Sí Sí 49 % No
10 Varón Sí Sí 60 % No
11 Mujer Sí Sí 48% No
12 Mujer No No ACHO 78% TVP+TEP 24
13 Mujer No Sí 73% No
14 Mujer No No Gestación 59% TVP 32
15 Mujer Sí Sí Post iq 57% TVP 11
16 Mujer Sí Sí 70% No
17 Varón Sí No Idiopático 60% T.Mesentérica 45
18 Varón Sí Sí Idiopático 64% TVP 24
19 Varón Sí Sí Idiopático 62% TVP 50
TABLA 2. Características de las gestantes
PacienteAF de
trombosis
Hª familiar
déficit ATTrombosis Gestación
Diagnóstico
antes del
embarazo
Abortos HeparinaTto sustitutivo
con AT% AT
1 No Sí No G1P1 Sí 0 Sí Sí 48
2 Sí Sí No G3P2 No 1 No No 50
3 Sí No Sí G3P3 No 0 No No 60
4 No No Sí G3P1 No 2 No No 60
5 Sí Sí No G2P2 No 0 No No 45
6 Sí Sí No G1P1 Sí 0 Sí Si 48
7 No No Sí G3C1 Sí 1 Sí Si 70
8 Si Si No G2P2 Sí 0 Si SI 59

COMUNICACIONES ORALES: Hemostasia y Trombosis 127
INTRODUCCIÓN/FUNDAMENTO:
Más del 1% de la población española realiza tratamiento anticoa-gulante oral por diferentes motivos, siendo el más prevalente de ellos la fibrilación auricular crónica. A los anticoagulantes orales clásicos (warfarina, acenocumarol) se han unido en los últimos años los anticoagulantes de acción directa (ACOD) que ejercen inhibición selectiva de la trombina y del factor Xa.Por su cada vez mayor prevalencia justificada en estudios por datos de seguridad y eficacia es necesario conocer sus efectos secundarios, siendo la hemorragia el de mayor gravedad y repercusión
OBJETIVO:
Conocer la incidencia y características de las hemorragias grado III-IV de la OMS de pacientes en tratamiento con ACOD
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo descriptivo de los episodios hemorrágicos grado III-IV en pacientes anticoagulados con ACOD en el Hospital de Jerez de la Frontera de Enero a Diciembre de 2016.
Se recogieron factores demográficos, ACOD y dosis, motivo de anticoagulación, riesgos trombótico y hemorrágico –escalas CHA2DS2-VASC y HAS-BLED-,localización de la hemorragia, si fueron o no espontáneas,antiagregación concomitante,insuficiencia hepática o renal,niveles de hemogobina y plaquetas,necesidad de transfusión y número de concentrados transfundidos, uso de medidas reversoras y exitus
RESULTADOS Y CONCLUSIONES:
Actualmente en nuestro centro 1.065 personas realizan tratamiento con ACOD. En este tiempo se produjeron 13 hemorragias de estas características, lo que supone una incidencia deL 1.22%.
La media de edad fue de 76,5 años, con 69.2% varones frente a 30.8% mujeres.
Los pacientes realizaban tratamiento con: dabigatran 110 mg(N=5), rivaroxaban 15 mg(N=3), apixaban 5 mg(N=3), dabigatran 150 mg(N=1) y rivaroxaban 20 mg(N=1).
El 76.9% de los pacientes (N=10) presentaba CHA2DS2-VASC≥3; respecto al riesgo hemorrágico, un 38.4% (N=5) presentaba HAS-BLED≥3.
En el 76,9% de los casos(N=10) se produjo hemorragia digestiva (HD) mientras que en el restante 23,1% (N=3) la localización fue intracraneal (HIC; 1 con rivaroxaban, 1 con apixaban, 1 con dabigatran).La única hemorragia no espontánea se produjo en este último grupo (traumatismo craneoencefálico -TCE-), siendo las restantes 12 espontáneas.
El 15.3% (N=2) se encontraba también antiagregado (un paciente con AAS y otro con clopiogrel).
Casi la mitad de los pacientes (N= 6) presentaba un FG>50 ml/min (CDK-EPI). Únicamente un paciente presentaba FG<15 ml/min en el contexto de una insuficiencia renal crónica agudizada. Ningún paciente presentaba insuficiencia hepática
La media de cifra de Hb se situaba en 7.6 g/dl (máximo 16.6 g/dl, mínimo 4.5 g/dl). En todos los casos de HD se transfundieron concentrados de hematíes (CH);en la mitad de ellos se transfundieron 2 CH. Ningún paciente presentaba trombopenia(media 216 x 103 plq/μl).
Sólo en dos casos se realizó reversión con concentrado de complejo protrombínico –CCP- y en un caso (HIC tras TCE) se empleó el antídoto específico de dabigatran, Idarucizumab.
Ningún paciente falleció a causa de la hemorragia.
– Aunque las hemorragias graves con ACOD no son frecuentes en nuestra serie, se trata de una complicación potencialmente mortal, por lo que es fundamental un manejo rápido y adecuado del episodio aplicando las medidas específicas en función de cada caso (transfusión de concentrados, reversión)recomendándose que los centros dispongan de protocolos de actuación.
– Nuestra serie confirma la necesidad de realizar un seguimiento estrecho en estos pacientes, al menos en los primeros meses de tratamiento, para comprobar que factores de riesgo como la función renal o tratamientos concomitantes no afectan a la eficacia y seguridad del fármaco.
Hemorragias graves en pacientes anticoagulados con anticoagulantes de acción directaS. Ordóñez Vahi, J.A. Raposo Puglia, B. Díez del Corral, R. Campos Álvarez, A. Estella García, S. Garzón López
Hospital de Jerez de la Frontera

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA128
GRADO MANIFESTACIÓN
0
1
2
3
4
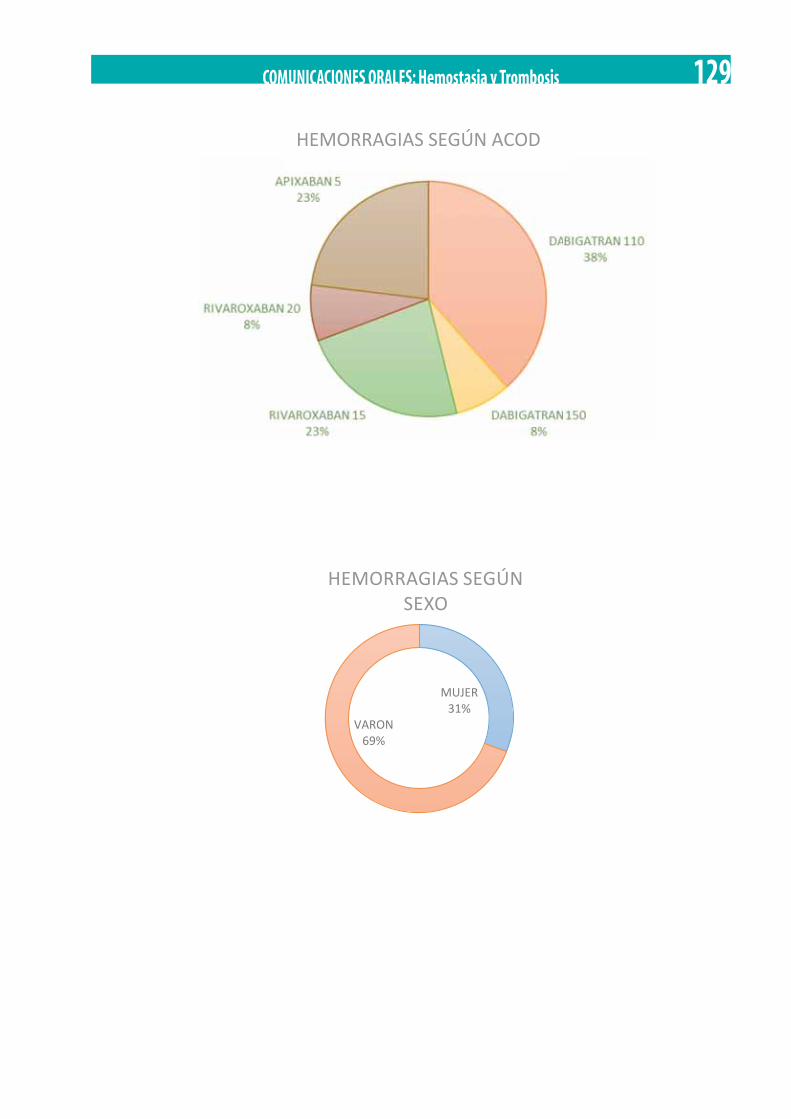
COMUNICACIONES ORALES: Hemostasia y Trombosis 129


COMUNICACIONES ORALES
Morfología y Biología Celular


COMUNICACIONES ORALES: Morfología y Biología Celular 133
INTRODUCCIÓN/FUNDAMENTO:
La deleción de 17p o la mutación de TP53 es uno de los factores pronósticos más importantes en la leucemia linfática crónica (LLC). Su presencia condiciona refractariedad a la inmunoquimioterapia y un pronóstico pobre. Se conoce que la mutación de TP53 puede estar presente de forma subclonal. En estos casos, la frecuencia del alelo mutado (VAF) es baja. El método estándar para detectar las mutaciones en TP53 es la secuenciación Sanger. Sin embargo, ésta técnica carece de sensibilidad para detectar mutaciones presentes en <10% de las células leucémicas. En los últimos años el uso de técnicas de secuenciación masiva y profunda han permitido la detección de mutaciones con VAF bajas.
OBJETIVO:
Describir los resultados de secuenciación masiva para la detección de mutaciones en TP53 en una serie de pacientes con leucemia linfática crónica y evaluar su importancia clínica.
PACIENTES, MATERIAL Y MÉTODO:
Se extrajo ADN de sangre periférica o medular mediante el equipo de extracción automática MagNa pure (Roche). El ADN fue cuantificado mediante Qubit® Fluorometer de Invitrogen™. La preparación de la librería, template y la carga del chip se realizó de forma automática con el Ion Chef System Ion Torrent. Se realizó secuenciación masiva con el equipo PGM (Ion Torrent). Se utilizó un panel de 24 amplicones que cubren las regiones codificantes del gen TP53 completo. Los resultados se analizaron usando el Ion Torrent Browser, coverage analysis plugin y el software Ion Reporter.
RESULTADOS Y CONCLUSIONES:
Se analizó la presencia de mutación en TP53 en 27 pacientes. Las características de los mismos se recogen en la tabla 1. El análisis se llevó a cabo en el momento del diagnóstico de la LLC en 6 pacientes y en el momento de la progresión en 21. Se observaron 7 mutaciones en 27 pacientes (25%). El 85% (6/7) se detectaron en pacientes en progresión con necesidad de tratamiento. La cobertura media fue de 1716, rango (1256-2000). La mediana de VAF fue de 5%, rango (3.5-64%). Los pacientes con mutaciones con VAF <5% (n=2) se analizaron por segunda vez reproduciéndose los mismos resultados.
Importancia de la secuenciación masiva para la detección de mutaciones en TP53 en la leucemia linfática crónicaE. Carrillo Cruz, M. Siuto Alcántara, F. De la Cruz Vicente, M. Ruiz Mercado, J.R. García Lozano, J.A. Pérez Simón
Hospital Universitario Virgen del Rocío
Todas las mutaciones se comprobaron en la base de datos COSMIC. Dos mutaciones con VAF elevado (43% y 67%) asociaron deleción de 17p documentado por FISH. En cambio, no se observó la deleción por FISH en casos con VAF más bajo (mediana 4,3, rango 3,1-7). En 4 pacientes que no presentaron la deleción por FISH, la demostración de la mutación de TP53 guió el tratamiento posterior.
CONCLUSIONES
Este método de secuenciación masiva tiene sensibilidad para detectar mutaciones de TP53 con VAF bajas.
La demostración de mutaciones en TP53 en pacientes con criterios de tratamiento y FISH negativa tiene implicaciones clínicas relevantes.
Características Pacientes: n (%)
Sexo
Hombre
Mujer
17 (63)
10 (37)
Edad al diagnóstico
Mediana, rango 61 (44-77)
Estadio Binet al diagnóstico
A
B
C
20 (74)
5 (18,5)
2 (7,5)
FISH al diagnóstico
Del13q
Trisomía 12
Del 11q
Del 17p
Negativa
9 (33,3)
2 (7,4)
7 (26)
2 (7,4)
7 (26)
Mutación TP53
Si
No
7 (26)
20 (74)
Momento del análisis de la mutación
Diagnóstico
Progresión
6 (22,2)
21 (77,7)
Tratamiento quimioterápico previo al análisis
mutacional (total 27)
Con mutacion TP53 (total 7)
Sin mutación TP53 (total 20)
10 (37)
3 (43)
7 (35)


COMUNICACIONES PÓSTERS
Gestión y Automatización en Hematología


COMUNICACIONES PÓSTERS: Gestión y Automatización en Hematología 137
INTRODUCCIÓN/FUNDAMENTO:
Las derivaciones a Hematología han experimentando un crecimiento exponencial en los últimos años. Se incrementan tiempos de demora, volumen de pacientes en consulta y, consecuentemente, se producen retrasos diagnóstico-terapéuticos. Es por ello imprescindible crear y potenciar planes de actuación que aumenten la eficiencia en la gestión de la demanda asistencial a Hematología.
Actualmente con la historia digital informatizada se garantiza el registro y confidencialidad de los informes elaborados siendo factible emitir diagnósticos y recomendaciones terapéuticas evitando la visita física del paciente.
OBJETIVO:
En nuestra Unidad se inicia en octubre´15 un proyecto de consulta no presencial (consulta virtual) cuyo principal objetivo es mejorar la calidad en la asistencia médica: reducir tiempos de espera, aumentar el tiempo dedicado a cada paciente y agilizar el proceso diagnóstico-terapéutico.
PACIENTES, MATERIAL Y MÉTODO:
Se genera una agenda de consulta semanal denominada Consulta Virtual [CV], utilizando la aplicación Estación Clínica [EC] del programa informático Diraya. Desde octubre/2015 se revisan, las derivaciones procedentes de especialidades hospitalarias y se seleccionan las susceptibles de ser citadas en CV.
La EC permite acceder a la historia clínica completa y elaborar un informe de consulta como respuesta a la derivación. Cualquier facultativo correctamente identificado tiene acceso al mismo. Se contacta telefónicamente con el paciente, que se identifica mediante un protocolo específico, se informa de la actitud a seguir y orientación diagnóstica enviándose por correo ordinario el informe.
RESULTADOS Y CONCLUSIONES:
Desde octubre/2015 hasta febrero/2017 han sido citados en CV 171 pacientes (el 30% del total de derivaciones de especialidades hospitalarias).
Han sido resueltas en CV (RCV) el 93.6%: el 54.4% (93) sin pruebas adicionales y el 39.2% (67) con estudio analítico ampliado previo a cita. Los pacientes que requieren valoración en consulta externa son citados en menos de 10 días y se consideran no resueltas en CV: 6.4%(11).
Los motivos de consulta (MC) valorados han sido: anemia 66 (38.6%), trombofilia 33 (19.3%), anticoagulantes 23 (13.4%), alteraciones de coagulación 13 (7.6%), leucocitosis 11 (6.4%), trombocitosis/trombopenia 9 (5.2%), otros (hiperferritinemia, hipergammaglobulinemia, leucopenia, poliglobulia) 16 (9.3%). De los principales MC han sido RCV: anemia 63 (95.5%), trombofilia 32 (97%), anticoagulantes 22 (95.6%), alteraciones de coagulación 9 (69.2%).
Las especialidades con mayor índice de derivación han sido: Medicina Interna 42 (24.6%), Urgencias 28 (16.4%), Digestivo 19 (11.1%) y Ginecología 17 (10%). De estas citas han sido RCV: MI 40 (95.2%), Urgencias 25 (89.3%), Digestivo 18 (94.7%), Ginecología 17 (100%).
CONCLUSIONES
La CV es una prometedora herramienta para mejorar la calidad asistencial por su efectividad, eficacia y eficiencia dado que: evita retrasos diagnóstico-terapéuticos, reduce lista de espera y sobrecarga asistencial, permitiendo la valoración rápida de un alto porcentaje de derivaciones.
Sería de gran interés fijar criterios de derivación a CV, trabajados con especialidades de alta tasa de derivación y sobre las patologías más demandadas, para conseguir citas directas en CV.
Dada nuestra buena experiencia y la excelente acogida de los pacientes creemos útil implantar un modelo mixto de CV+consulta externa.
El profesional y el paciente deben asumirla como una consulta médica, cumpliendo los principios básicos de relación médico-paciente, autonomía del paciente, secreto profesional y confidencialidad.
Consulta virtual telefónica como herramienta de mejora de la calidad asistencial: experiencia en nuestro centroM. Anguita Arance (1), J.D. Tallon Pérez (1), B. Soler Gabarrón (2), M.C. Avellaneda Molina (1)
(1) Hospital Universitario San Agustín. Área Sanitariar Norte de Jaén, (2) Atención Primaria. Área Sanitaria Norte de Jaén

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA138
INTRODUCCIÓN/FUNDAMENTO:
El Hospital de Día de Hematología (HDH) en el Hospital Infanta Margarita atiende pacientes con enfermedades hematológicas que requieren tratamiento activo o soporte transfusional de forma periódica, así como seguimiento de forma habitual.
El servicio que está abierto de 8 a 15 horas (L-J-V), cuenta con 2 enfermeros, un auxiliar y un médico. En este punto cabe destacar el papel del personal, que en su trato con los pacientes no solo llevan a cabo una labor terapéutica, sino que sirven de apoyo a pacientes y familiares, controlan síntomas y la evolución de los mismos y solucionan las dudas que van surgiendo a lo largo de su seguimiento.
Los casos que se atienden en esta Unidad proceden de la Consulta de Hematología (derivaciones desde AP y AE), desde donde tras valoración y estudio de cada caso se deriva a Hospital de Día para iniciar tratamiento.
OBJETIVO:
A lo largo de nuestra experiencia observamos que en esta Unidad se podría valorar a determinados pacientes derivados desde Urgencias (IC-Urg)/Atención Especializada (IC-AE)/atención Primaria (IC-AP) con preferencia si así se considera tras evaluación del hematólogo responsable.
Con este nuevo enfoque de Hospital de Día se pretende agilizar el proceso asistencial, ya que tanto las consultas como las IC en acto único suponen un avance en la calidad de la prestación sanitaria, facilitando la accesibilidad del ciudadano a los servicios sanitarios y recibiendo toda la atención necesaria en el menor tiempo posible.
Así mismo se consigue atender tanto a los pacientes ambulatorios como a las visitas no programadas, realizar exploraciones y tratamientos básicos con carácter inmediato y el seguimiento personalizado de los casos. Este hecho repercute directamente en la calidad de vida de los enfermos, con la reducción de los tiempos de espera.
PACIENTES, MATERIAL Y MÉTODO:
Ante lo comentado anteriormente , nos planteamos la posibilidad de incluir en nuestra agenda otras tareas como son: IC-AE, IC-Urg, valoración de pacientes post-hospitalización (Post-Hosp) e IC-AU (IC de acto único).
Este sistema se puso en marcha en Mayo de 2016, ya que a pesar de no tener agenda disponible para ello en los primeros cuatro meses de
Agilización de Procesos Asistenciales:Plan Piloto de Inclusión de Interconsultas en Hospital de Día de Hematología (HDH)M.J. Llamas Poyato, D. Moreno Garrido, I. García Díez
Hospital Infanta Margarita, AGS Sur de Córdoba
2016 ya se habían atendido en dicha Unidad 6 pacientes procedentes de AE.
El hematólogo responsable del HDH tras valorar la IC prioriza a aquellos pacientes de mayor gravedad o que se pueden resolver en consulta de acto único.
RESULTADOS Y CONCLUSIONES:
Desde la apertura de la nueva agenda en HDH (además de la labor asistencial habitual: tabla 1) en mayo de 2016 se han atendido las IC que aparecen en el gráfi co 1.
Las patologías atendidas en estas consultas han sido: linfomas y mielomas de nuevo diagnóstico favoreciendo el inicio precoz de tratamiento dado el tipo de patología, todos ellos procedentes de AE), mielofi brosis, anemias ferropénicas no responsivas a ferroterapia sustitutiva por vía oral (administrándose en el mismo i.v.), pacientes derivados desde hospitalización de nuestro hospital o del Hospital Universitario Reina Sofía, incluso valoración urgente de pacientes en tratamiento anticoagulante para retirada previa a intervención quirúrgica.
CONCLUSION: En defi nitiva, con este nuevo enfoque pretendemos mejorar la asistencia a los pacientes que acudan a nuestra consulta, siempre priorizando por tipo de patología, gravedad y benefi cio del acto médico; evitando citas y demoras innecesarias.
GRÁFICO 1.Nº y tipo de IC atendidas en plan piloto HDH
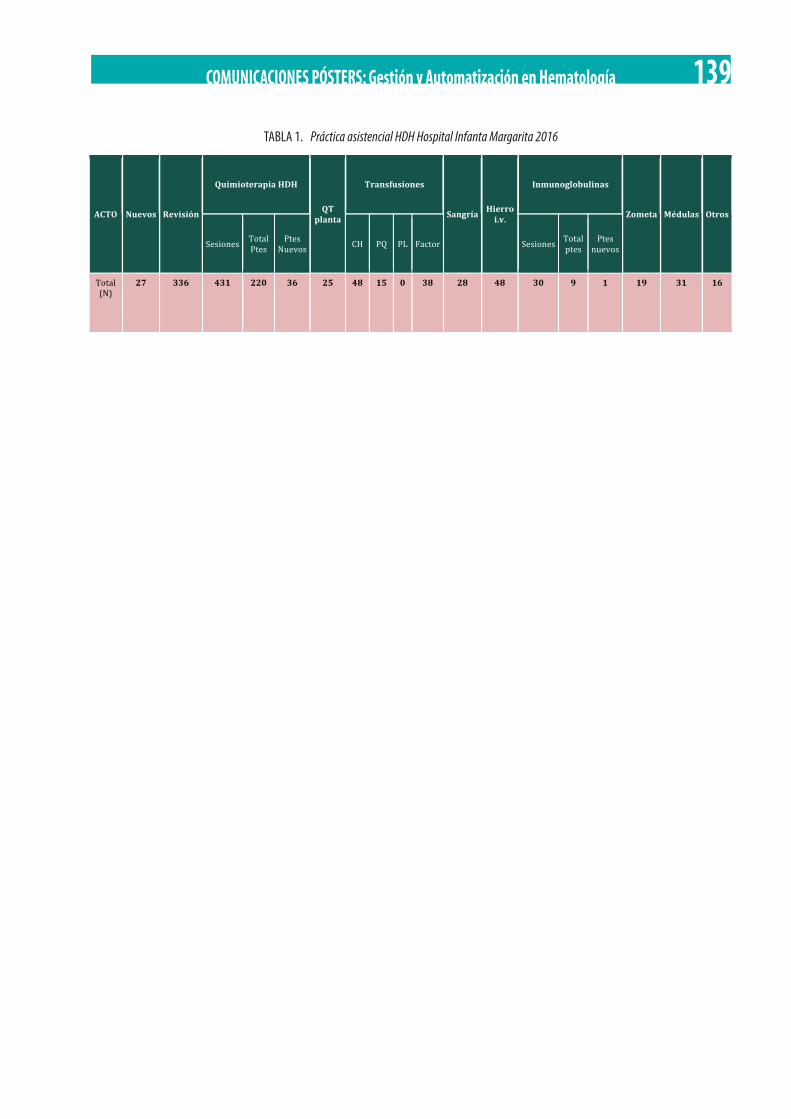
COMUNICACIONES PÓSTERS: Gestión y Automatización en Hematología 139
TABLA 1. Práctica asistencial HDH Hospital Infanta Margarita 2016

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA140
INTRODUCCIÓN/FUNDAMENTO:
Los linfomas T constituyen un % de los síndromes linfoproliferativos. La diversidad histológica de estos linfomas constituye la base que justifica el patrón heterogéneo de presentación con afectación extraganglionar con relativa frecuencia.
El perfil de agresividad de este subgrupo de linfomas y su escasa incidencia constituye el principal obstáculo para establecer la estrategia terapéutica adecuada en estos pacientes, según los resultados de ensayos aleatorizados.
OBJETIVO:
Estudio observacional retrospectivo con el fin de evaluar el tratamiento de los pacientes diagnosticados de neoplasias de estirpe T en nuestro centro desde 1.999 hasta mayo de 2.016, así como el estatus de la enfermedad en el último seguimiento, el estadio de Ann-Arbor y la edad al diagnóstico.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos 31 pacientes diagnosticados de neoplasias de estirpe T en nuestro centro entre 1.999 y mayo de 2.016. 14 pacientes fueron diagnosticados de linfoma T periférico NOS, 1 de linfoma T periférico en amígdala palatin a variante linfoepitelioide (linfoma de Lennert) y 1 de linfoma T periférico cutáneo primario de cuero cabelludo, 3 de linfoma angioinmunoblástico de células T, 4 de linfoma anaplásico T de células grandes tipo sistémico primario, 2 con linfoma T cutáneo, 5 con linfoma linfoblástico de células T (1 de ellos con infiltración del SNC) y 1 linfoma de células T asociado a enteropatía. El 53.13% de los
pacientes eran varones y el 46.87% eran mujeres. La media de edad al diagnóstico fue de 53.58 años (8-87 años).
El 9.68% de los pacientes se diagnosticaron en un estadio I de Ann-Arbor , el 9.68% se diagnosticaron en estadio II , el 38.71% se diagnosticaron en estadio III) y el 35.48% se diagnosticaron en estadio IV. Presentaron síntomas B un 67.1% al diagnóstico.
RESULTADOS Y CONCLUSIONES:
La mayoría de los pacientes (51.6%) recibió tratamiento según esquemas CHOP-like con una tasa de respuesta globales del 32.25% con alta tasa de recaída del 80%. El 16% recibió como primera línea tratamiento según esquema HyperCVAD con una tasa de respuestas globales del 20%. Otras opciones en primera línea fueron tratamiento paliativo (22.58%), PUVA (6.4%), cirugía (3.22%). Los esquemas de rescate empleados son ESHAP, bendamustina y gemcitabina. Unicamente fueron sometidos a Trasplante Hematopoyético 5 pacientes (16.12%), 3 alogénicos y 2 autólogos.
14 (45.1%)de los pacientes fueron éxitus y 15 permanecen vivos a fecha de hoy, perdiendo el seguimiento en 2 pacientes.
CONCLUSIONES
Dada la escasa incidencia de este tipo de linfoma no se ha establecido un tratamiento de referencia de primera línea. En nuestra serie existe gran diversidad de la opciones terapeuticas empleadas, si bien en la mayoría de los casos se ha utilizado esquemas CHOP like con baja tasa de respuesta y nbaja supervivencia libre d eprogresion.
Se requieren estudios randomizados con muestras amplias para definir el tratamiento de elección en estos pacientes.
Estudio retrospectivo de los linfomas T diagnosticados en nuestro centro en los últimos 17 añosE. Morente Constantín, P.A. González Sierra, A. Núñez García, L. Moratalla López, E. López Fernández, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

COMUNICACIONES PÓSTERS: Gestión y Automatización en Hematología 141
INTRODUCCIÓN/FUNDAMENTO:
Nos planteamos implantar un algoritmo diagnóstico como una herramienta necesaria para evitar peticiones mal dirigidas, redundantes e inadecuadas en pacientes con anemia. Se propone desde el Servicio de Análisis Clínicos del Hospital Santa Bárbara, la implementación de un algoritmo de anemias para mejorar la adecuación en los protocolos que establece el laboratorio para una mejora en la asistencia sanitaria. Se entiende por anemia la disminución de la masa eritrocitaria en sangre periférica con puntos de corte de VCM (según la OMS) que pueden verse en el algoritmo adjunto. La forma más lógica de evaluar una anemia es mediante una serie de parámetros analíticos y hematológicos que permita un correcto diagnóstico, usando esta aproximación con una extracción simple, única y dirigida.
OBJETIVO:
Implantar algoritmos diagnósticos en el estudio de anemias en la Gerencia de Área Integrada de Puertollano. Esto permitiría evaluar mediante una serie de pruebas bioquímicas y hematológicas encadenadas, el correcto enfoque de los diferentes tipos de anemia y contribuir así a la mejora asistencial para el usuario.
PACIENTES, MATERIAL Y MÉTODO:
La implantación del algoritmo se consensua desde el Servicio de Hematología y Análisis Clínicos y se comunicará a todos los médicos
peticionarios para una mejor realización del protocolo propuesto. Se realizará un seguimiento de la implantación de los algoritmos mediante la creación de unos indicadores en el plazo de un año. Además, este procedimiento se incluye en uno de los objetivos del sistema general de calidad de este laboratorio para 2017.
Se implantará una solicitud que se llamará “solicitud de estudio de anemias” en la que se hará constar historia clínica, cifras de Hb, VCM, tratamientos recibidos, y tubos requeridos para envío (EDTA y suero).
Los algoritmos se harán llegar a todos los médicos peticionarios para facilitar la correcta realización del protocolo (Imagen 1).
Los parámetros renal, hepático, tiroideo, índices hematimétricos, metabolismo férrico, Coombs directo, etc., quedan especificados en el algoritmo adjunto.
Se realizará un seguimiento mediante indicadores a lo largo de todo el año ya que forma parte de los objetivos de calidad.
RESULTADOS Y CONCLUSIONES:
Es preciso establecer el desarrollo de un circuito, así como la implicación y participación multidisciplinar de todo el personal responsable para poder obtener una mejor optimización de los recursos y aportar una gran mejora en la asistencia sanitaria, garantizando continuidad asistencial en todo el proceso.
Implantación de algoritmos diagnósticos de anemias en la gerencia de área integrada de PuertollanoSilvia del Rocio Verdesoto Cozzarelli (1), María Mar Herráez Albendea (2), María del Pilar García Fernández (2), María Carmen Lorenzo Lozano (2)
(1) Hospital Santa Bárbara- Gerencia Atención Integrada Puertollano, (2) Hospital Santa Bárbara-Gerencia Atención Integrada Puertollano
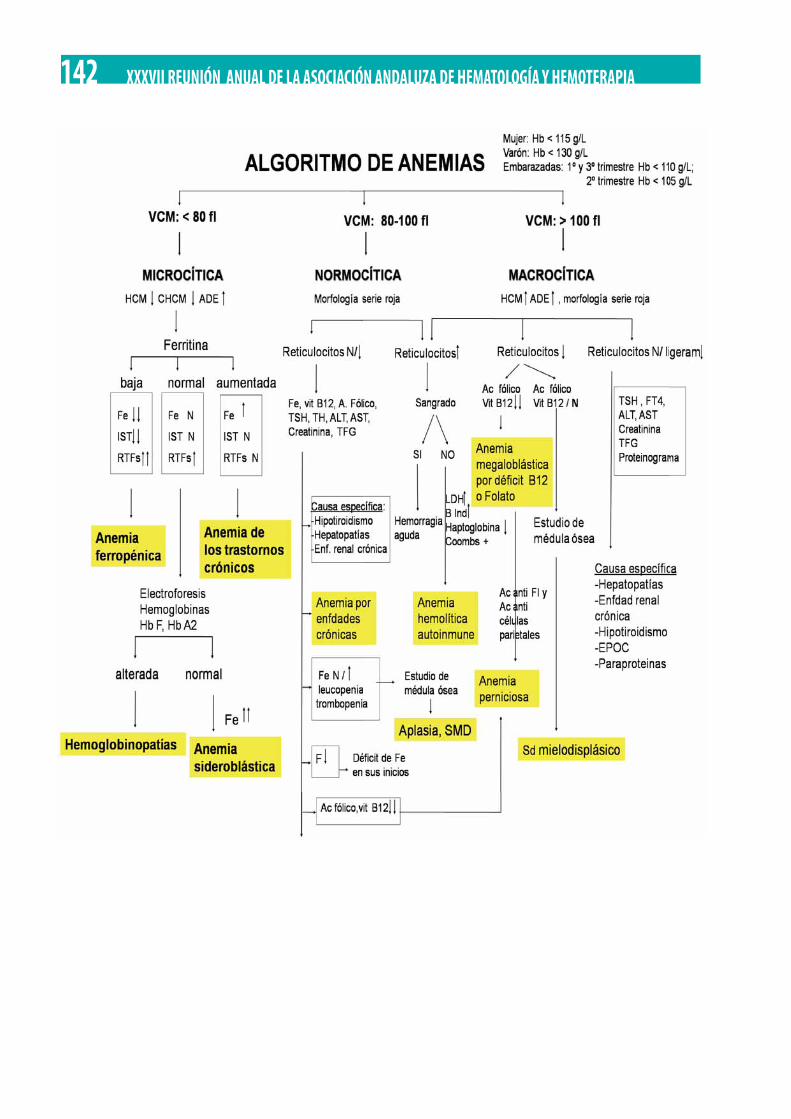
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA142

COMUNICACIONES PÓSTERS: Gestión y Automatización en Hematología 143
INTRODUCCIÓN/FUNDAMENTO:
Hematología y Hemoterapia es una especialidad médica diagnóstica con capacidad terapéutica que ejerce de apoyo al resto de servicios intra y extra hospitalarios.
Las consultas al hematólogo de guardia son frecuentes por parte de otras especialidades e incluso dentro de la propia unidad, y el nivel de dificultad abarca desde consultas sencillas a otras muy complejas en las que la celeridad en la respuesta puede condicionar la vida del paciente, más aún en aquellos centros que realizan trasplante alogénico.
OBJETIVO:
Analizar las características de las consultas al hematólogo de guardia, con presencia física, en el Hospital de Jerez de la Frontera, en el contexto de una Unidad con años de experiencia en trasplante, y con el fin de mejorar la formación, así como plantear la reestructuración del programa rotacional de los residentes en función de los datos obtenidos.
PACIENTES, MATERIAL Y MÉTODO:
Estudio prospectivo comprendido entre el 13/10/16 y el 14/3/17. Se registraron las consultas tanto telefónicas como presenciales al hematólogo durante 23 guardias que representaron todos los días de la semana en al menos 2 ocasiones.
Se empleó una plantilla en papel donde escribir de manera inmediata el asunto para proceder con posterioridad a su codificación informática. Se anotaron la fecha, la persona que realizaba el registro, el turno (Mañana, 8.15 a 15.00; Tarde, 15.01 a 20.00 y Noche, 20.01 a 8.14 horas), el servicio consultante, la sección, la categoría profesional y el tipo de demanda.
RESULTADOS Y CONCLUSIONES:
Se registraron un total de 489 consultas al hematólogo en las 23 guardias analizadas. Promedio 21,26 consultas/guardia, mediana: 20 [12-37]. Los promedios de demanda asistencial fueron 20,73 para el cómputo de lunes a jueves y 22,25 para viernes-sábados-domingos y festivos.
El porcentaje de consultas por turnos se muestra en el gráfico 1, procediendo de manera mayoritaria del propio servicio de Hematología, siendo la sección de planta de hospitalización la más demandante (144 consultas), seguido del Servicio de Urgencias generales (gráficas 2 y 3).
El motivo general de requerimientos y el perfil del consultante se detallan en las gráficas 4 y 5, destacando el peso de la hemostasia en su conjunto (123 consultas) suponiendo el 25,15%. Causas de valoración del paciente en hematología en gráfica 6.
CONCLUSIONES:
Se confirma la creencia generalizada de que la mayoría de consultas versan en torno a aspectos relacionados con la hemostasia
Vemos conveniente incrementar la oferta formativa interdisciplinaria en dicha materia
Podría ser necesario un reajuste en las rotaciones del residente para el afrontamiento adecuado de las consultas.
La demanda asistencial de la planta de hospitalización en hematología refleja la complejidad de los pacientes tratados.
El número de consultas pone de manifiesto la necesidad de guardias con presencia física.
Papel del hematólogo de guardia: análisis de la demanda asistencialJ.A. Raposo Puglia, S. Ordóñez Vahí, M.V. Verdugo Cabeza de Vaca, C. Blázquez Goñi, A. Salamanca Cuenca, S. Garzón López
Hospital Universitario Jerez de la Frontera

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA144
GRÁFICO 1. Demanda asistencial según el turno
GRÁFICO 2. Servicio consultante

COMUNICACIONES PÓSTERS: Gestión y Automatización en Hematología 145
GRÁFICO 3. Sección consultante del servicio de hematología
GRÁFICO 4. Motivo de consulta
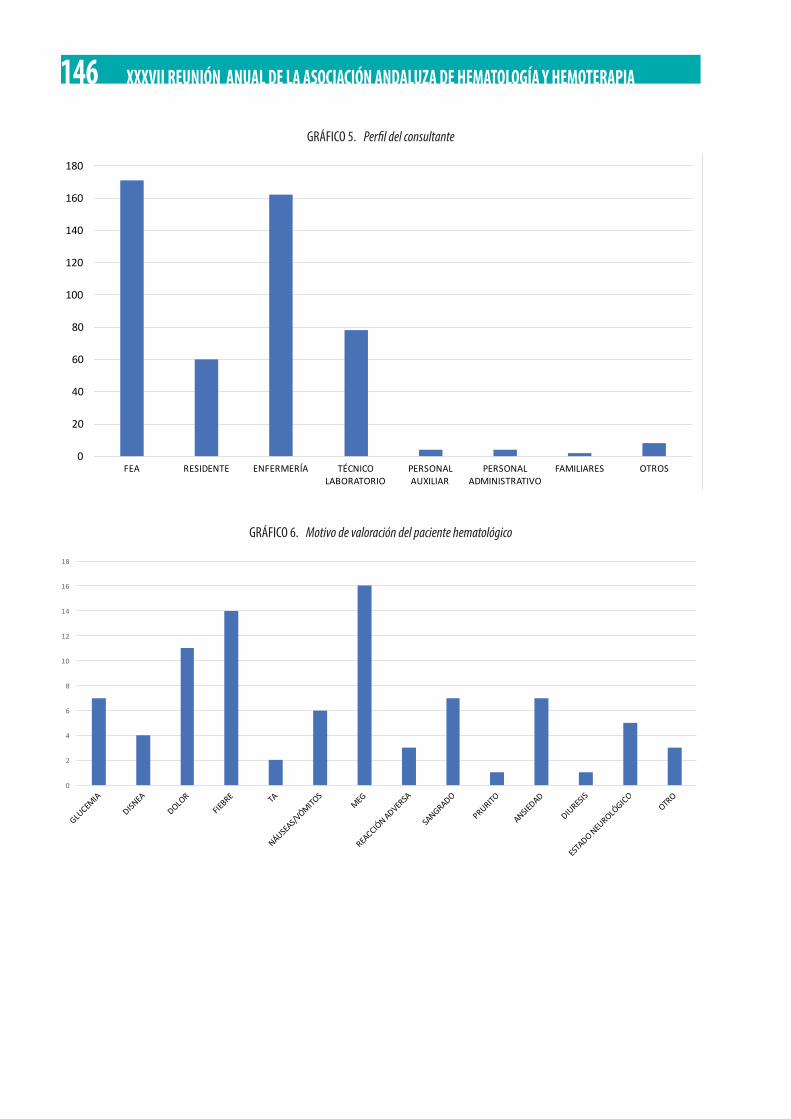
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA146
GRÁFICO 5. Perfi l del consultante
GRÁFICO 6. Motivo de valoración del paciente hematológico

COMUNICACIONES PÓSTERS
Hematología Clínica


COMUNICACIONES PÓSTERS: Hematología Clínica 149
INTRODUCCIÓN/FUNDAMENTO:
Las mutaciones del gen de la calreticulina (CALR) han sido recien-temente descritas en pacientes con trombocitemia esencial (TE) no portadores ni de las mutaciones JAK2 ni MPL.
OBJETIVO:
El objetivo de este estudio es analizar la relación entre mutaciones de CALR, los tipos 1 y 2, y las características clínico-analíticas de pacientes con TE, comprobando si la ratio mutacional de CALR muestra efecto sobre estas variables y comparar estas características con las de un grupo portador de la mutación JAK2V617F.
PACIENTES, MATERIAL Y MÉTODO:
Se ha realizado un estudio transversal con 34 pacientes diagnosticados de TE, en los que se ha detectado la mutación de CALR mediante PCR de análisis de fragmentos.
Posteriormente, se comparó este grupo con uno control de 31 pacien-tes diagnosticados de TE portadores de mutación JAK2V617F, detec-tada mediante PCR cuantitativa.
RESULTADOS Y CONCLUSIONES:
Tanto la mutación de tipo 1 como tipo 2 repetían la misma proporción a la descrita en la literatura, con un 50 % y 29,4 % respectivamente, encontrándose diferencias estadísticamente significativas (p<0,05) respecto a la cifra de plaquetas al diagnóstico, siendo superiores en la de tipo 2; sin poder demostrar diferencias estadísticamente significa-tivas en otras variables analizadas. En estas mutaciones de tipo 2 los valores de la ratio mutacional solían ser más homogéneos y superiores a 0,5, sugiriendo homocigosis. Se observaron diferencias en la forma de presentación entre los JAK2+ y CALR+, con cifras plaquetarias más elevadas y edades más temprana de aparición para CALR+ y mayor leucocitosis en JAK2+. La proporción de eventos trombohemorrági-cos fue superior en JAK2+, mientras que la incidencia de neoplasias secundarias fue mayor en CALR+.
El estudio de la mutación CALR y de sus subtipos tiene un interés diag-nóstico incuestionable. El perfil mutacional confiere un fenotipo par-ticular, un curso clínico y una evolución pronóstica singular. Además, la ratio mutacional podría ser de interés clínico.
Implicación de la mutación de la calreticulina en la presentación clínica y repercusión pronostica en la trombocitemia esencialY. Moatassim de la Torre (1), E. Clavero Sánchez (1), L. Entrena Ureña (2), A. Alba Sosa (1)
(1) Hospital Santa Ana , (2) Hospital Virgen de las Nieves Granada

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA150
INTRODUCCIÓN/FUNDAMENTO:
El plasmocitoma óseo solitario representa el 3-5% de la patología maligna de las células plasmáticas (CP). Se define como la lesión única ósea en la que se demuestra infiltración de CP clonales, siendo la localización más frecuente los cuerpos vertebrales. El estudio de médula ósea debe ser normal. La presencia de CP clonales en número <10% cambia el diagnóstico a “Plasmocitoma Solitario con afectación mínima de médula ósea”. La edad de presentación es más temprana que en el Mieloma Múltiple (MM), con una media entre 50-60 años. La Supervivencia Libre de Evento a los 10 años puede alcanzar al 40% de los casos, aunque prácticamente el 100% desarrollarán un MM a los 15 años del diagnóstico. Referente al tratamiento, establecidos como primera línea se encuentran, la radioterapia y la cirugía para estabi-lizar la zona afecta. Existen terapias minimamente invasivas como la ablación por radiofrecuencia, que ya se han utilizado en otro tipo de tumores con resultados exitosos.
OBJETIVO:
Evaluar la efectividad de la radiofrecuencia intraoperatoria, como al-ternativa a la radioterapia en el tratamiento.
PACIENTES, MATERIAL Y MÉTODO:
Tres pacientes fueron diagnosticados de plasmocitoma óseo solitario, entre 2013 y 2015, siendo tratados con radiofrecuencia intraoperatoria utilizando el sistema ‘Dfine Star’, instrumento quirúrgico compuesto por un electrodo bipolar de punta direccionable, que puede llegar a todos los puntos de la vértebra en que haya células tumorales, permi-tiendo la destrucción de las mismas.
RESULTADOS Y CONCLUSIONES:
1. Mujer de 54 años que consulta en Enero 2013 por lumbalgia y que en RM presenta imagen lítica en L3 con fractura aplastamiento junto a una Gammapatia Monoclonal IgG Lambda. Recibió inicial-mente tratamiento con radioterapia (5 sesiones). Ante persistencia
de dolor incoercible y tras estudio hematológico que descarta la existencia de CP clonales en médula ósea, en Octubre de 2013 se somete a cirugía con biopsia de la lesión que demostró Plasmoci-toma Óseo, ablación tumoral por radiofrecuencia, cementación y estabilización. Desde entonces la paciente se encuentra asintomá-tica y en seguimiento Hematológico sin presentar evolución a MM.
2. Varón de 59 años con lumbalgia desde hacía un año en quien se de-tecta en Agosto 2015 lesión en cuerpo vertebral D12. En el estudio de médula ósea no se observa infiltración por CP clonales. Se proce-de a intervención quirúrgica, junto con ablación por radiofrecuen-cia y cementación. La biopsia de la D12 demostró Plasmocitoma Óseo. Desde entonces en seguimiento hematológico presentando en Febrero 2017 inmunofijación positiva IgG Kappa, pendiente de estudio medular.
3. Mujer de 58 años con dorsalgia desde Marzo 2015. Tras detectar en RM lesión en T5, en Octubre 2015 se realiza biopsia ósea per-cutánea con el resultado de Plasmocitoma Óseo. El estudio hema-tológico mostró por Inmunofenotipo la existencia de un 0,24 % de CP clonales en médula ósea. En noviembre de 2015 se realiza intervención quirúrgica consistente en ablación por radiofrecuen-cia, descompresión y artrodesis posterior. En Noviembre 2016 se observa en estudio hematológico, progresión a MM por lo que recibe tratamiento con radioterapia y quimioterapia.
Conclusiones:
1. El uso de radiofrecuencia intraoperatoria podría ser una buena opción en el tratamiento de primera línea para el Plasmocitoma Solitario, ya que puede eliminar el tumor sólo en una sesión y en el mismo acto quirúrgico.
2. La radiofrecuencia no limita el uso futuro de la radioterapia si apa-rece un nuevo plasmocitoma, y disminuye el riesgo de toxicidad acumulada (como la mielitis transversa).
3. Se necesitan experiencias futuras para confirmar la seguridad y la rentabilidad de esta técnica.
Tratamiento con radiofrecuencia del plasmocitoma óseo solitario. Experiencia en nuestro centroSoraya Lorente de Uña, E. Morales Muñoz, M. González Bernal, J. Gutiérrez de Guzmán
Hospital Vitas Xanit Internacional, Málaga

COMUNICACIONES PÓSTERS: Hematología Clínica 151
Edad 55 59 58
Sexo Mujer Varón Mujer
Fecha diagnóstico Abril 2013 Agosto 2015 Octubre 2015
RM al diagnostico Lesión lítica L3 Lesión metastásica D12 Lesión lítica D4
Médula Ósea al diagnóstico 6% células plasmaticas no clonales
FISH negativo
Cariotipo 46XX
FISH negativo
Sin infiltración
Mínima afectación medular
Tratamientos - Radioterapia
- Radiofrecuencia y cirugía
-Radiofrecuencia y cirugía - Radiofrecuencia y cirugía
Nº Líneas de tratamiento 2 1 1
Estatus actual Respuesta completa Sospecha de progresión a MM Progresión a MM
Fecha recaida Febrero 2017 Noviembre 2016

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA152
INTRODUCCIÓN/FUNDAMENTO:
El Síndrome Mielodisplásico (SMD) es una enfermedad clonal carac-terizada por diferentes grados de citopenia, hematopoyesis ineficaz, médula displásica y riesgo de progresión a leucemia mieloide aguda (LAM). Son muchas las opciones de tratamiento aunque el trasplante alogenico de progenitores hematopoyéticos (Alo-TPH) de donante familiar o no emparentado se consideran el estándar de tratamiento, ofreciendo una potencial posibilidad de curación.
OBJETIVO:
Analizar los resultados del Alo-TPH como opción terapéutica en los pa-cientes diagnosticados con SMD en el Hospital Reina Sofia de Córdoba.
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo de 41 pacientes con SMD o LAM secundaria que recibieron un Alo-TPH en nuestro centro, desde Septiembre de 2001 a Octubre de 2015. Veintinueve de los pacientes eran hombres (70,7%). La mediana de edad en el momento en el que se realiza el trasplan-te fue de 55 años (14-69 años). Segun la clasificación de la OMS de 2008, el 58,5% fueron Anemia Refractaria con Exceso de Blastos tipo I y II (AREB I-II); 7.3% Anemia Refractaria con Sideroblastos en Anillo (ARSA); 12,2% de Anemia Refractaria / Citopenia (AR) y 22% LAM secundaria. La situación de la enfermedad antes del trasplante fue: 31,7% no habían recibido tratamiento quimioterapéutico previo, 22% en situación de Recaída / Progresión (R/P), 14,6% en respuesta com-pleta (RC), 21,9% en respuesta parcial (RP) y 9,7% en enfermedad estable (EE). Según el origen de los progenitores hematopoyéticos, en el 85,4% se realizaró con sangre periférica (SP). El 52% de los donantes fueron no emparentados y el 48% donantes familiares idénticos. La mayoría de ellos recibió acondicionamiento de intensidad reducida (68,3%).
RESULTADOS Y CONCLUSIONES:
La mediana de tiempo desde el diagnóstico al procedimiento fue de 11 meses (2,4-88,4 meses) con una mediana de seguimiento post-TPH de 15 meses (1-120 meses). Veintisiete (65,9%) pacientes alcanzaron RC post-TPH.
Complicaciones post trasplante más frecuentes: 26,8% Enfermedad Aguda Injerto contra Huésped (EICHa); 43,9% Enfermedad Crónica Injerto contra Huésped (EICHc) y 24% Enfermedad Veno-oclusiva Hepática (EVOH).
La Supervivencia Global (SG), Supervivencia Libre de Enfermedad (SLE) y la Probabilidad de Recaída (PR) a 5 años fue 40%, 45% y 6%, respectivamente. No se encontraron diferencias significativas en la SG, SLE y PR, según el tipo de Alo-TPH (relacionado vs. no relacionado), el origen de los progenitores o el tipo de acondicionamiento emplea-dos. Se observaron diferencias significativas en cuanto al estado de la enfermedad pre-TPH: pacientes con RC y los que no recibieron trata-miento previo al TPH presentaron una SG a 4 años de 57.9 +/- 11.3% vs 27.3 +/- 11% en el grupo de pacientes con EE, RP y enfermedad R/P (p = 0,050). Los pacientes que desarrollan EICHc tienen una SG mayor, 77,4 +/- 10% a los 5 años vs 22,7 +/- 8,9% de los que no lo desarrollaron (P <0,01). Estos resultados se confirman en el estudio multivariante (p = 0,002).El desarrollo de EICHc se considera factor protector en términos de supervivencia (5,2 veces el factor protector). Durante el seguimiento post-TPH, 23 pacientes (56,1%) fallecieron. Causas de exitus mas frecuentes: 21,9% procesos infecciosos; 7,3% EICHa; 9,8% EICHc; 2,4% EVOH y 2,4% fallo del injerto.
Conclusiones: El Alo-TPH en pacientes con SMD o LAM secundaria es una opción de tratamiento curativa, con una supervivencia aceptable. El esquema de acondicionamiento utilizado y el tipo de trasplante (familiar o no emparentado) no influyeron en los resultados. Obser-vamos una SG superior en aquellos pacientes en situación de RC previa al trasplante y en aquellos que desarrollan EICHc (5,2 veces factor de protección) en comparación con los que no lo desarrollan, siempre que dicha complicación no implique un difícil control terapéutico.
Trasplante alogénico de progenitores hematopoyéticos en síndromes mielodisplásicosI. Vico Herrera, A.I. Álvarez Sánchez, C. Martín Calvo, E. García Torres, F.J. Casaño Sánchez, J. Sánchez García
Hospital Universitario Reina Sofía de Cordoba

COMUNICACIONES PÓSTERS: Hematología Clínica 153
INTRODUCCIÓN/FUNDAMENTO:
La clasificación actual (OMS 2016) de las neoplasias linfoides es muy heterogénea, existiendo actualmente más de 100 entidades. Nos en-contramos ante un linfoma de células B inclasificable con característi-cas intermedias entre Linfoma Difuso de Células Grandes B (LDCGB) y Linfoma de Hodgkin clásico (LHc). Estos linfomas ‘híbridos’ presentan afectación mediastínica, aunque se ha descrito afectación ganglionar no mediastínica y en otros órganos; son más agresivos y presentan un peor pronóstico que LHc o LDCGB. Sin tratamiento específico definido actualmente.
OBJETIVO:
Presentamos el caso de Linfoma B inclasificable con características intermedias entre LDCGB y LHc refractario a 2 líneas de tratamiento quimioterápico, con remisión completa tras 3 ciclos de tratamiento inmunoterápico con antiCD30 (Brentuximab).
PACIENTES, MATERIAL Y MÉTODO:
Varón de 82 años al diagnóstico, sin antecedentes de interés, con síntomas B de 2 años de evolución, serologías virales negativas. Se realiza TC en diciembre de 2014 con adenopatías mínimas y se amplía estudio con PET-TC en Febrero de 2015 donde se objetiva captación hipermetabólica tumoral a nivel ganglionar supra e infradiafragmá-tica y heterogénea en bazo. En Junio de 2015 se biopsia adenopatía axilar izquierda donde se objetivan abundantes células grandes de hábito sternbergoide, y otras de morfología centroblástica e inmu-noblástica. Son positivas para CD30, marcadores B (CD20, CD79 alfa y PAX-5 intenso), BCL6, MUM1 y EBER, y CD15. Negatividad para CD3, CD45Ro (UCHL-1), CD10 y ALK. C-Myc positivo débil en menos del 40% de las células, y BCL2 focalmente positivo. Se distingue una celulari-dad acompañante constituida por eosinófilos, neutrófilos, histiocitos y linfocitos, principalmente T (CD3+). Índice proliferativo (KI-67) 55%. Médula ósea sin infiltración por linfoma. Se llega al diagnóstico de
Linfoma B inclasificable con características intermedias entre LDCGB NOS (C-Myc negativo por FISH) y LHc. Estadio IIIs-A. IPI 2.
El día 27/07/2015 inicia tratamiento con R-CHOP-Bleomicina a 66% de dosis. En PET-TC post-3º ciclo respuesta parcial >90% y persistencia de adenopatía hipermetabólica axilar izquierda. Tras 3 ciclos más se repite PET-TC (18/01/16) con resultado de aumento de tamaño y cap-tación de dicha adenopatía. Se realiza exéresis constatándose misma histología.
En ecografía abdominal de abril de 2016 se objetivan lesiones nodula-res en bazo e hígado, solicitándose PET-TC que confirma este hallazgo además de lesiones hipermetabólicas óseas, todo ello compatible con recaída de linfoma. El 23/05/16 inicia R-GPD a mitad de dosis por edad, del que recibe 6 ciclos, con buena evolución en control tras 3ºciclo. En PET-TC post 6º ciclo: aumento de la extensión de la afectación ósea con nuevas lesiones y reaparición de focos patológicos en hígado y bazo.
RESULTADOS Y CONCLUSIONES:
Se decide iniciar Brentuximab como 3ª línea de tratamiento el día 20/12/16. En PET-TC tras 3º ciclo el paciente se encuentra en remisión completa y sin efectos adversos, por lo que se prevén hasta 16 ciclos si la evolución lo permite.
Conclusiones
1. Brentuximab, fármaco aprobado para LHc CD30+ y Linfoma T Anaplásico de Células Grandes, ha superado la respuesta a la qui-mioterapia siendo eficaz y bien tolerado.
2. Brentuximab puede ser una opción en síndromes linfoproliferativos CD30+ cuyas líneas previas de tratamiento quimioterápico haya fracasado o en no candidatos a quimioterapia intensiva.
3. La evolución del caso nos dará más datos sobre duración de res-puesta y supervivencia.
Inmunoterapia en linfoma de zona gris quimiorresistenteC. Díaz Aizpún (1), M. Revelles Peñas (2), M. Espeso de Haro (1)
(1) Hospital Regional Universitario de Málaga, (2) Hospital Clínico Virgen de la Victoria

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA154
INTRODUCCIÓN/FUNDAMENTO:
El linfoma de la zona marginal extranodal (MALT) es la forma más co-mún de linfoma de la zona marginal. Su origen se produce fuera de los ganglios linfáticos y las localizaciones más frecuentes son estomago, intestino delgado, glándulas salivares, tiroides y pulmón.
El linfoma MALT con afectación pulmonar se ha relacionado con la ex-posición a tóxicos (tabaco), infecciones y enfermedades autoinmunes, produciendo una estimulación antigénica crónica del tejido pulmonar. La clínica de presentación suele ser inespecífica, presentandose como un hallazgo casual en una prueba de imagen torácica. El diagnostico se basa en el análisis histopatológico de una muestra por biopsia, donde se observa una infiltración difusa por células B clonales CD20, CD43, CD79a positivas con ausencia de expresión CD5 y CD103.
OBJETIVO:
Exponer el caso de un paciente diagnosticado de LINFOMA B DE LA ZONA MARGINAL EXTRANODAL PRIMARIO PULMONAR estadio II 2-E donde se realiza tratamiento con rituximab en monoterapia.
PACIENTES, MATERIAL Y MÉTODO:
Paciente varón de 82 años, fumador activo. Ingresa en Febrero´16 en Neumología para estudio por clínica de disnea de 4 meses de evolu-ción, dolor en hemitorax derecho, tos, expectoración blanquecina y esputos hemoptoicos.
Se realiza estudio analitico completo, estando todos los parámetros dentro de la normalidad. En gasometría arterial con oxigenoterapia (VMK 21%) presentaba insuficiencia respiratoria parcial y en la placa de tórax se observaba masa parahiliar con patrón micronodular aso-ciado y perdida de volumen en lóbulo inferior derecho (LID). En TC completo se observa masa de 35 mm de eje mayor en lóbulo superior derecho (LSD) rodeando bronquio principal derecho junto con ade-
nopatías paratraqueales, subcarinales e hiliares derechas de hasta 20 mm, sugestivo de carcinoma broncogénico (figura 1a). En biopsia por fibrobroncoscopia presenta infiltración por Linfoma B de bajo grado de la zona marginal con inmunofenotipo: CD20+, CD3-, CD43+, CD5-, CICLINA D1-, BCL6-, CD10- y CD23-.
El paciente es derivado a Hematología. En PET presenta captaciones hipermetabólicas en masa parahiliar derecha de LSD y actividad hi-permetabólica en región subcarinal y mediastino ipsi y contralateral (SUVmax 8).
RESULTADOS Y CONCLUSIONES:
Con diagnostico de LINFOMA B DE LA ZONA MARGINAL EXTRANODAL PRIMARIO PULMONAR estadio II 2-E (Ann-Arbor), inicia tratamiento con rituximab en monoterapia (4 ciclos). En reevaluación intratamien-to por fibrobroncoscopia, no se observan lesiones de malignidad y en TC se objetiva una disminución considerable de tamaño -6 mm- (figura 1b). Ante la buena respuesta, se administran otros 4 ciclos de Rituximab, con excelente tolerancia. En Mayo´16 el paciente finaliza tratamiento, asintomático, reevaluándose a través de PET, donde no se observa hipermetabolismo en pulmón derecho. Transcurridos 12 meses, el paciente esta estable desde el punto de vista hematológico.
El tratamiento del linfoma MALT pulmonar no está bien definido, basado en la resección quirúrgica y ocasionalmente la radioterapia sobre zona afecta, para los estadios localizados y quimioterapia o in-munoterapia para los estadios más avanzados o contraindicación de los anteriores. Los resultados a largo plazo son excelentes en la mayoría de los casos (SG del 70%-100% a 5 años). Como exponemos en nuestro caso, Rituximab es un buen fármaco para pacientes con diagnostico de Linfoma MALT que expresen CD20 y además ofrece como ventaja una baja tasa de toxicidad, asi como ofrecer una buena calidad de vida al paciente, al tratarse de una terapia que puede administrarse ambulatoria.
Rituximab en monoterapia puede ser una opción óptima de tratamiento para pacientes con linfoma MALT primario de pulmónA.I. Álvarez Sánchez, L. Burgos Vigara, C. Martínez Losada, J. Sánchez García
Hospital Universitario Reina Sofía de Cordoba
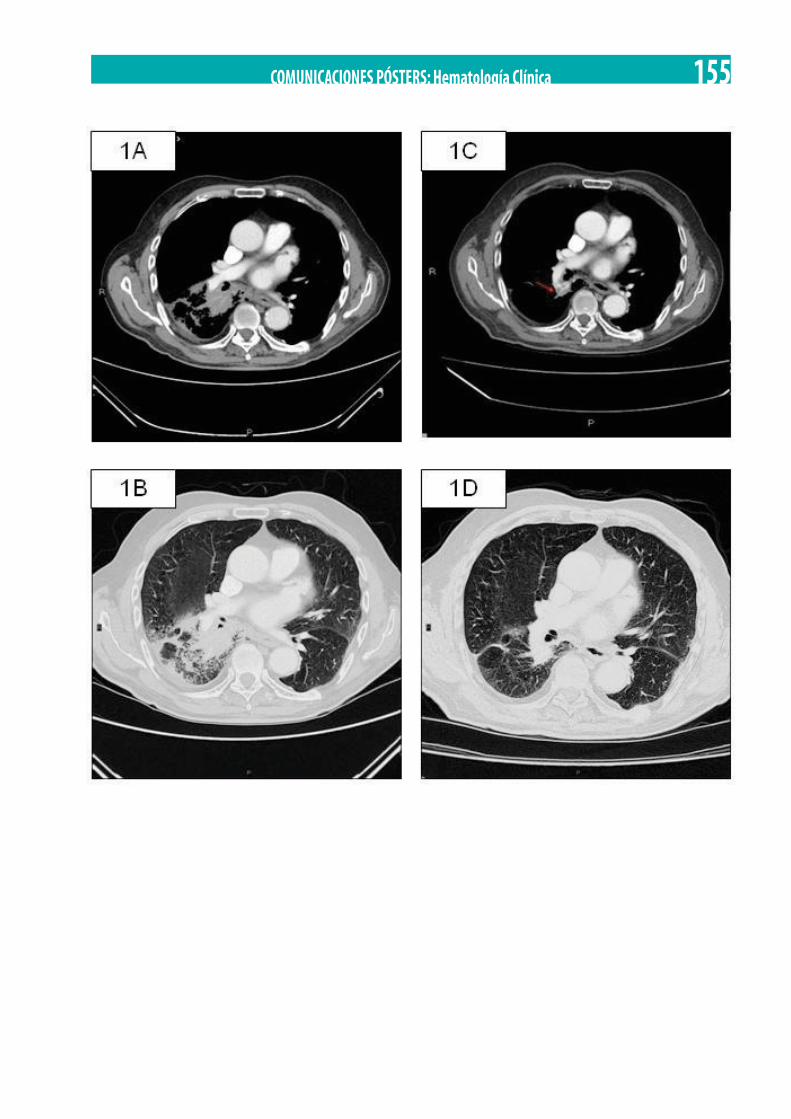
COMUNICACIONES PÓSTERS: Hematología Clínica 155

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA156
INTRODUCCIÓN/FUNDAMENTO:
La lenalidomida (Lena) tiene propiedades antineoplásicas, antiangio-génicas e inmunomoduladoras. Inhibe la proliferación de determina-das células hematopoyéticas, potencia la inmunidad celular, inhibe la angiogénesis y la producción de citocinas proinflamatorias por los monocitos.
Se propone un caso de linfoma no Hodgkin (LNH) B folicular quimio-rresistente en el que se muestra el papel de la lena como tratamiento de rescate.
OBJETIVO:
1. Mostrar el posible papel de la lena como agente terapéutico de rescate en una paciente con un linfoma quimiorresistente e im-posibilidad de recibir más quimioterapia intensiva por toxicidad.
2. Analizar la toxicidad derivada de este tratamiento y terapias pre-vias, especialmente toxicidad hematológica y segundas neoplasias.
PACIENTES, MATERIAL Y MÉTODO:
Paciente de 55 años. Diagnóstico de LNH B folicular grado IIIa REAL, estadio IV-A (afectación ganglionar supra e infradiafragmática); y extraganglionar en médula ósea (MO)), FLIPI 2 (diciembre de 2012). Hasta marzo de 2014 recibe los siguientes tratamientos:
1. R-CHOP x 6 ciclos, con progrsión de la enfermedad en PET-TC post 6º ciclo.
2. R-ESHAP x 3 ciclos, con persistencia de la enfermedad en PET-TC post 3º ciclo.
3. R-GPD x 3 ciclos, con progresión de la enfermedad.
4. R-MINE x 3 ciclos , con persistencia metabólica de la enfermedad (Figura 1).
Inicia tratamiento con Riruximab-lena (R-Lena) en marzo de 2014 (R días +1 y +15 y lena x 21 días consecutivos en ciclos de 28 días), recibiendo 4 ciclos. Evaluación: Respuesta parcial (RP). Consolidación con radioterapia (RT) local que finaliza en diciembre de 2014.
En biopsia de MO de junio de 2014, realizada por citopenias muy profundas y persistentes en paciente con múltiples líneas de quimio-terapia previa, se objetiva hipoplasia medular secundaria a terapia. 2 meses tras finalizar la RT de consolidación se constata progresión tumoral, por lo que recibe 6 ciclos de R-miniCHOEP, con progresión de la enfermedad a nivel supra e infradiafragmático y pulmonar tras finalizar.
Inmunomodulación con lenalidomida en linfoma no Hodgkin B folicular refractario. A propósito de un casoJ. Díez Pastor (1), M.M. Alcalá Peña (2), M. Espeso de Haro (2)
(1) Hospital Regional Universitario de Málaga , (2) Hospital Regional Universitario de Málaga
FIGURA 1
En mayo de 2016 inicia nuevamente R-Lena sin quimioterapia, con soporte hemoterápico intensivo concomitante.
RESULTADOS Y CONCLUSIONES:
Tras 5 ciclos de R-Lena, que la paciente recibe sin complicaciones, se realiza PET-TC que muestra respuesta metabólica parcial en todos los territorios ganglionares y a nivel pulmonar, con persistencia única-mente de adenopatías mediastínicas hipermetabólicas, siendo la me-jor respuesta obtenida en esta paciente a lo largo de su enfermedad (Figura 2).
Un mes más tarde, en revisión, se objetiva leucocitosis con aumento de LDH, por lo que se realiza estudio medular completo (morfología, inmunofenotipo, citogenética (Figura 3) y biología molecular), con diagnóstico de leucemia aguda de fenotipo mixto, B/mieloide (OMS 2016) con cariotipo complejo. Ingresa para tratamiento con FLAG-Ida, asintomática, pero tan sólo puede recibir la mitad del ciclo pues pre-senta infección por E. coli multisensible, con mala evolución clínica, desarrollo de insuficiencia respiratoria y renal, siendo finalmente éxitus por fallo multiorgánico.

COMUNICACIONES PÓSTERS: Hematología Clínica 157
CONCLUSIONES:
1. La lena como agente antineoplásico e inmunomodulador, puede tener un papel importante en el tratamiento de este tipo de LNH, posibilitando una terapia eficaz con relativa buena tolerancia y una toxicidad más limitada que la quimioterapia intensiva.
2. Existe riesgo de desarrollo de segundas neoplasias con el uso de lena, si bien en este caso no se puede determinar que sea la lena la exclusiva responsable del desarrollo de leucemia aguda, dadas las múltiples líneas de tratamiento quimioterápico previo.
FIGURA 2 FIGURA 3
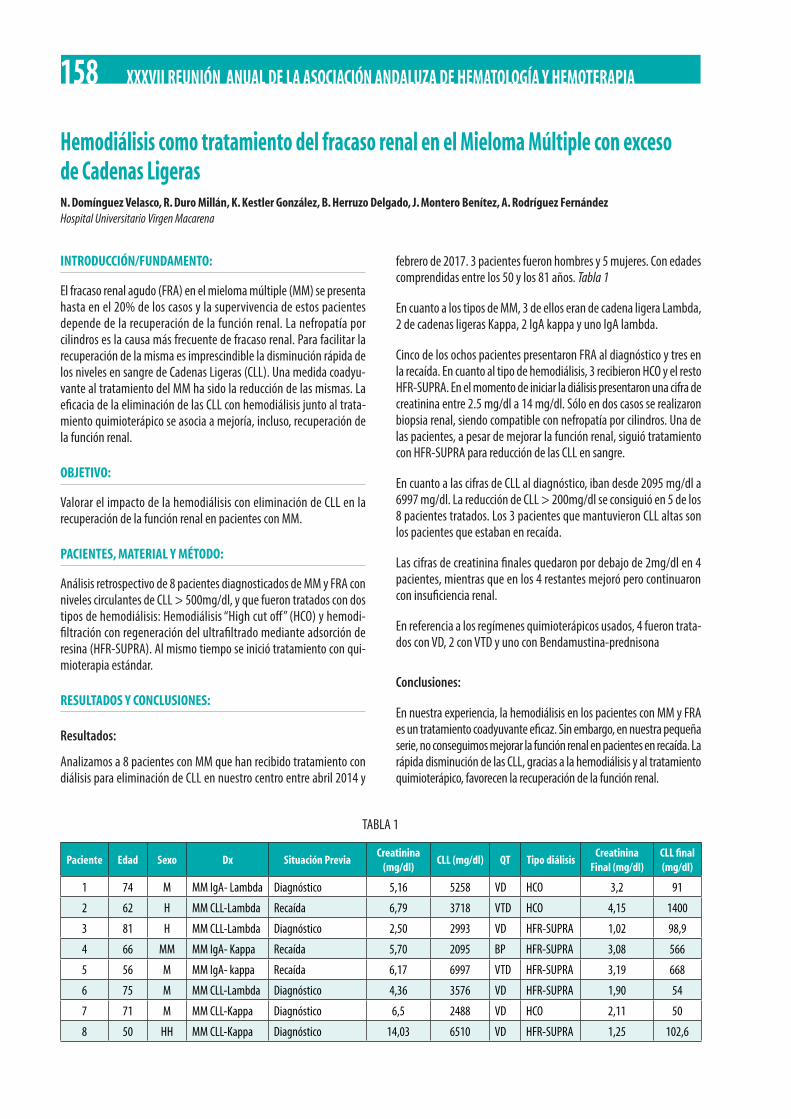
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA158
INTRODUCCIÓN/FUNDAMENTO:
El fracaso renal agudo (FRA) en el mieloma múltiple (MM) se presenta hasta en el 20% de los casos y la supervivencia de estos pacientes depende de la recuperación de la función renal. La nefropatía por cilindros es la causa más frecuente de fracaso renal. Para facilitar la recuperación de la misma es imprescindible la disminución rápida de los niveles en sangre de Cadenas Ligeras (CLL). Una medida coadyu-vante al tratamiento del MM ha sido la reducción de las mismas. La eficacia de la eliminación de las CLL con hemodiálisis junto al trata-miento quimioterápico se asocia a mejoría, incluso, recuperación de la función renal.
OBJETIVO:
Valorar el impacto de la hemodiálisis con eliminación de CLL en la recuperación de la función renal en pacientes con MM.
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo de 8 pacientes diagnosticados de MM y FRA con niveles circulantes de CLL > 500mg/dl, y que fueron tratados con dos tipos de hemodiálisis: Hemodiálisis “High cut off” (HCO) y hemodi-filtración con regeneración del ultrafiltrado mediante adsorción de resina (HFR-SUPRA). Al mismo tiempo se inició tratamiento con qui-mioterapia estándar.
RESULTADOS Y CONCLUSIONES:
Resultados:
Analizamos a 8 pacientes con MM que han recibido tratamiento con diálisis para eliminación de CLL en nuestro centro entre abril 2014 y
febrero de 2017. 3 pacientes fueron hombres y 5 mujeres. Con edades comprendidas entre los 50 y los 81 años. Tabla 1
En cuanto a los tipos de MM, 3 de ellos eran de cadena ligera Lambda, 2 de cadenas ligeras Kappa, 2 IgA kappa y uno IgA lambda.
Cinco de los ochos pacientes presentaron FRA al diagnóstico y tres en la recaída. En cuanto al tipo de hemodiálisis, 3 recibieron HCO y el resto HFR-SUPRA. En el momento de iniciar la diálisis presentaron una cifra de creatinina entre 2.5 mg/dl a 14 mg/dl. Sólo en dos casos se realizaron biopsia renal, siendo compatible con nefropatía por cilindros. Una de las pacientes, a pesar de mejorar la función renal, siguió tratamiento con HFR-SUPRA para reducción de las CLL en sangre.
En cuanto a las cifras de CLL al diagnóstico, iban desde 2095 mg/dl a 6997 mg/dl. La reducción de CLL > 200mg/dl se consiguió en 5 de los 8 pacientes tratados. Los 3 pacientes que mantuvieron CLL altas son los pacientes que estaban en recaída.
Las cifras de creatinina finales quedaron por debajo de 2mg/dl en 4 pacientes, mientras que en los 4 restantes mejoró pero continuaron con insuficiencia renal.
En referencia a los regímenes quimioterápicos usados, 4 fueron trata-dos con VD, 2 con VTD y uno con Bendamustina-prednisona
Conclusiones:
En nuestra experiencia, la hemodiálisis en los pacientes con MM y FRA es un tratamiento coadyuvante eficaz. Sin embargo, en nuestra pequeña serie, no conseguimos mejorar la función renal en pacientes en recaída. La rápida disminución de las CLL, gracias a la hemodiálisis y al tratamiento quimioterápico, favorecen la recuperación de la función renal.
Hemodiálisis como tratamiento del fracaso renal en el Mieloma Múltiple con exceso de Cadenas LigerasN. Domínguez Velasco, R. Duro Millán, K. Kestler González, B. Herruzo Delgado, J. Montero Benítez, A. Rodríguez Fernández
Hospital Universitario Virgen Macarena
TABLA 1
Paciente Edad Sexo Dx Situación PreviaCreatinina
(mg/dl)CLL (mg/dl) QT Tipo diálisis
Creatinina
Final (mg/dl)
CLL final
(mg/dl)
1 74 M MM IgA- Lambda Diagnóstico 5,16 5258 VD HCO 3,2 91
2 62 H MM CLL-Lambda Recaída 6,79 3718 VTD HCO 4,15 1400
3 81 H MM CLL-Lambda Diagnóstico 2,50 2993 VD HFR-SUPRA 1,02 98,9
4 66 MM MM IgA- Kappa Recaída 5,70 2095 BP HFR-SUPRA 3,08 566
5 56 M MM IgA- kappa Recaída 6,17 6997 VTD HFR-SUPRA 3,19 668
6 75 M MM CLL-Lambda Diagnóstico 4,36 3576 VD HFR-SUPRA 1,90 54
7 71 M MM CLL-Kappa Diagnóstico 6,5 2488 VD HCO 2,11 50
8 50 HH MM CLL-Kappa Diagnóstico 14,03 6510 VD HFR-SUPRA 1,25 102,6

COMUNICACIONES PÓSTERS: Hematología Clínica 159
INTRODUCCIÓN/FUNDAMENTO:
La amiloidosis AL es una enfermedad clonal de células plasmáticas en la que los fragmentos de cadena ligera de Ig monoclonales se de-positan en los tejidos, ocasionando de forma progresiva un fracaso lento y gradual del órgano afecto. El pronóstico de los pacientes con amiloidosis AL sistémica dependerá del número y severidad de órga-nos afectos. La amiloidosis cardiaca conlleva un pronóstico infausto.
OBJETIVO:
Presentamos un caso clínico de una paciente sometido a trasplante cardiaco (TxC) y TASPE como tratamiento de la amiloidosis AL con afectación cardiaca.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 41 años sin antecedenes personales de interés. En junio de 2015 ingresa en nuestro centro, a cargo de Medicina Interna en situación de anasarca y síndrome nefrótico, evidenciándose en eco-cardiografía una hipertrofia concéntrica de VI con función sistólica moderadamente deprimida, por lo que, ante síndrome nefrótico con afectación cardiaca, se plantea hipótesis de posible enfermedad por depósitos como principal sospecha clínica: amiloidosis.
Se realiza PAAF de grasa subcutánea con presencia de depósito ami-loide AL que se confirma posteriormente en biopsia de MO. En RMN cardiaca con realce tardio de gadolinio se objetiva miocardiopatía infiltrativa de tipo amiloidosis cardiaca (FEVI 40%). En julio, se inicia tratamiento con VD, recibiendo 3 ciclos, como complicación de una arritmia cardiaca relacionada con su miocardiopatía infiltrativa, sufrió trombosis en la aorta infrarrenal y en ambas arterias femorales, razón por la que se inicia segunda línea de quimioterapia esquema CyBorDex x 2 ciclos. A pesar del tratamiemto quimioterápico, la paciente muestra
empeoramiento clínico, y en nueva ecocardiografía se objetiva pro-gresión de la disfunción biventricular, estimándose FEVI del 20%, así como presencia de trombo en aurícula derecha.
Puesto que se trata de una paciente en situación de insuficiencia car-diaca avanzanda NYHA IV por amilodosis AL, con poca respuesta al tratamiento quimioterapico, debido al bajo número de ciclo recibidos, y dada la desaparición del síndrome nefrótico, se solicita valoración por la Unidad de Insuficiencia Cardiaca y Trasplante en su hospital de referencia, siendo aceptada y realizándose el mismo en noviembre de 2015 con buena funcion del injerto (FEVI ampliamente conservada, sin alteraciones segmentarias de la contractilidad)
En mayo de 2016, la paciente recibe TASPE, quedando en Respuesta Parcial tras el mismo.
En Enero de 2017, tras aumento de las cadenas ligeras en sangre y del Pro-BNP, además de nueva aparición de clínica de ICC, se decide iniciar de nuevo ciclo con CyBorDex, pendiente de nueva evolución de la respuesta alcanzada.
RESULTADOS Y CONCLUSIONES:
En la amiloidosis AL o primaria, el corazón se afecta en más de la mitad de los casos y condiciona un mal pronóstico, con una supervivencia menor de 1 año sin tratamiento. El tratamiento dirigido contra las células plasmáticas puede detener e incluso revertir los depósitos ya formados y mejorar la función de los órganos afectos, tanto con quimioterapia y/o TASPE. En casos seleccionados, y en centros con experiencia, la estrategia más aceptada en paciente con afectación cardiaca es realiza Trasplante Cardiaco (TxC) y después el tratamiento etiológico de la amiloidosis. Sin embargo, aun así, la supervivencia es muy pobre, menor de 5 años en la mayoría de los casos.
Transplante cardiaco y de Médula ósea en paciente con Amiloidosis AL e Insuficiencia cardiaca: a propósito de un casoN. Domínguez Velasco, R. Duro Millán, C. Muñoz García, B. Herruzo Delgado, A. Rodríguez Fernández
Hospital Universitario Virgen Macarena

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA160
INTRODUCCIÓN/FUNDAMENTO:
En situaciones de hemorragia masiva (HM), la necesidad de importan-tes cantidades de hemoderivados supone un gran consumo de recursos y la implicación de varias especialidades en múltiples escenarios. La elevada morbimortalidad en estos casos (30-69%), hace necesaria la realización de un protocolo de transfusión masiva (PTM), que en nuestro hospital se edita en diciembre de 2015, con el objetivo de es-tablecer un guion común en la práctica clínica en situaciones críticas y así conseguir un eficiente aporte de hemoderivados (cantidad-tiempo) utilizando componentes fijos de transfusión.
OBJETIVO:
Evaluar la calidad de la implantación del PTM en cuanto a su difusión, incidencias, unidades requeridas y almacenadas para ello y mortalidad en los pacientes incluidos en este protocolo.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo unicéntrico sobre 20 pacientes en nuestra área hospitalaria que han sufrido hemorragias masivas durante el año 2016. Los datos se recogieron de la estación clínica Diraya y e-Delphyn 8.0.5.0. Las variables incluidas fueron mortalidad , incidencias peri-transfusionales, componentes sanguíneos requeridos, especialidad destinataria y causas que justificaron la activación del protocolo.
RESULTADOS Y CONCLUSIONES:
Resultados: De un total de 20 casos de TM los grupos sanguíneos se distribuyen de la siguiente forma : 8 (A+), 7 (0+), 2 (B+), 2 AB+ y 1
(0-). La media de edad de estos pacientes fue de 57 años (32 – 81), 14 hombres (70%) y 6 mujeres (30%). Las causas más frecuentes de HM fueron : rotura de aneurisma aorta abdominal (4), digestivas (4), complicaciones en cirugía mayor abdominal (5), cirugía obstétrica (3), cirugía ortopédica (1) y complicaciones de pancreatitis graves (3). Fallecieron 5 pacientes (25%), 3 durante el procedimiento y 2 poste-riormente, por complicaciones secundarias a sus patologías tras una buena respuesta a la TM. Se enviaron 175 concentrados de hematíes (CH), 111 plasmas (PF) y 18 pool de plaquetas (PQ). En cuanto a los CH, 4 fueron (0-) reservadas para casos de extrema urgencia, 138 iso-grupo , y 33 isocompatibles. No se registraron incidencias a lo largo del procedimiento, inherentes a la actuación de los distintos especialistas implicados, a la cantidad y tiempo de envío de los hemoderivados, ni complicaciones en los pacientes por reacciones post-transfusionales.
Conclusiones: La inexistente notificación de incidencias durante el PTM, indica una implantación y difusión muy favorable, tanto en el personal del laboratorio como en los servicios hospitalarios implicados (mayoritariamente UCI, Cirugía y Anestesia). El stock de CH de nuestro laboratorio, es acordado con el Centro de Transfusión de referencia, en función de la proporción poblacional de grupo sanguíneo. Por ello, en tres de los pacientes con necesidades masivas de hemoderivados con grupos más minoritarios (B y AB), se transfunden concentrados isocompatibles, sin incidencias . La mortalidad global fue de un 25%, dato que constata el buen manejo de las situaciones críticas en estos pacientes tras la implantación del PTM. Sin embargo, en alguno de es-tos casos, no queda una clara constancia de la activación-desactivación del protocolo, aspecto que podría revisarse para su mejor funciona-miento y próximas evaluaciones.
Revisión de la implantación del protocolo de transfusión masiva por hemorragia masiva en nuestro centro hospitalarioM. Molina, V. Calama, E. García, E. Martínez, M. Gómez
Hospital Universitario Nuestra Señora de Valme

COMUNICACIONES PÓSTERS: Hematología Clínica 161
INTRODUCCIÓN/FUNDAMENTO:
Alcanzar remisión completa (RC) en la terapia de inducción es un factor pronóstico importante para decidir terapias posteriores en leucemia mieloide aguda (LMA). No alcanzarla es un un factor pronóstico ad-verso que obliga a plantear tratamiento con quimioterapia (QT) de reinducción seguido de trasplante alogénico de progenitores hema-topoyéticos (alo-TPH).
OBJETIVO:
Determinar el impacto en términos de supervivencia global (SG) en pacientes (pts) en primera RC tras uno o dos ciclos de inducción inde-pendientemente del esquema de tratamiento administrado y de la clasifi cación de riesgo ELN.
PACIENTES, MATERIAL Y MÉTODO:
Estudio restropectivo unicéntrico, sobre 83 pacientes diagnósticados entre 2010 y 2016 y clasifi cados según OMS/2016. Mediana de edad al diagnóstico 64 ± 18 años (17-91). Se recogieron datos demográfi cos ,
RC si o no y si necesitan uno o dos ciclos para alcanzarla, superviviencia libre de progresión (SLP) , éxitus y tiempo transcurrido hasta éxitus. Los esquemas de QT recibidos y las respuestas respectivas se describen en la tabla 1 ( excluyéndose las M3 (FAB) y t (15;17)).
RESULTADOS Y CONCLUSIONES:
Resultados: A fecha de análisis, han fallecido 58 pts (69,87 %). Glo-balmente, la mediana de SG fue de 7,2 meses (3,9- 10,5)) .Del total de 83 pts diagnosticados , fueron tratados 69 ( 83,13 %), la mayoría con los esquemas clásicos de inducción IDAC 3+7 ( n=31, 37,3% ) e IDAC 2+5 ( n=15, 18,1% ).) De los pts tratados excluyendo las promielocí-ticas ( n = 56) , solo alcanzan RC un 41,07 %, de los cuales el 73,91 % correspondió al esquema IDAC 3+7 y el 26.09% al IDAC 2+5 ( ( tabla 1 ). La mayoría de las RC se alcanzan tras el primer ciclo de inducción en ambos esquemas , con un total de 73,91% frente al 26,09 % que la logran con el segundo ciclo ( tabla 2) .Analizando la supervivencia ( SV ) a los 5 años en los que alcanzaron RC con un solo ciclo se obtiene un intervalo de 46,3 ± 14,2 %, frente al 44,4 ± 22,2 % que la obtuvieron tras dos ciclos (fi gura 1) .
Leucemia mieloide aguda en primera remisión completa: estudio descriptivo y análisis de supervivencia de un único centroM. Molina, V. Calama, M. Gómez, C. Couto, I. Simón, E. Ríos
Hospital Universitario Nuestra Señora de Valme
TABLA 1
TABLA 2
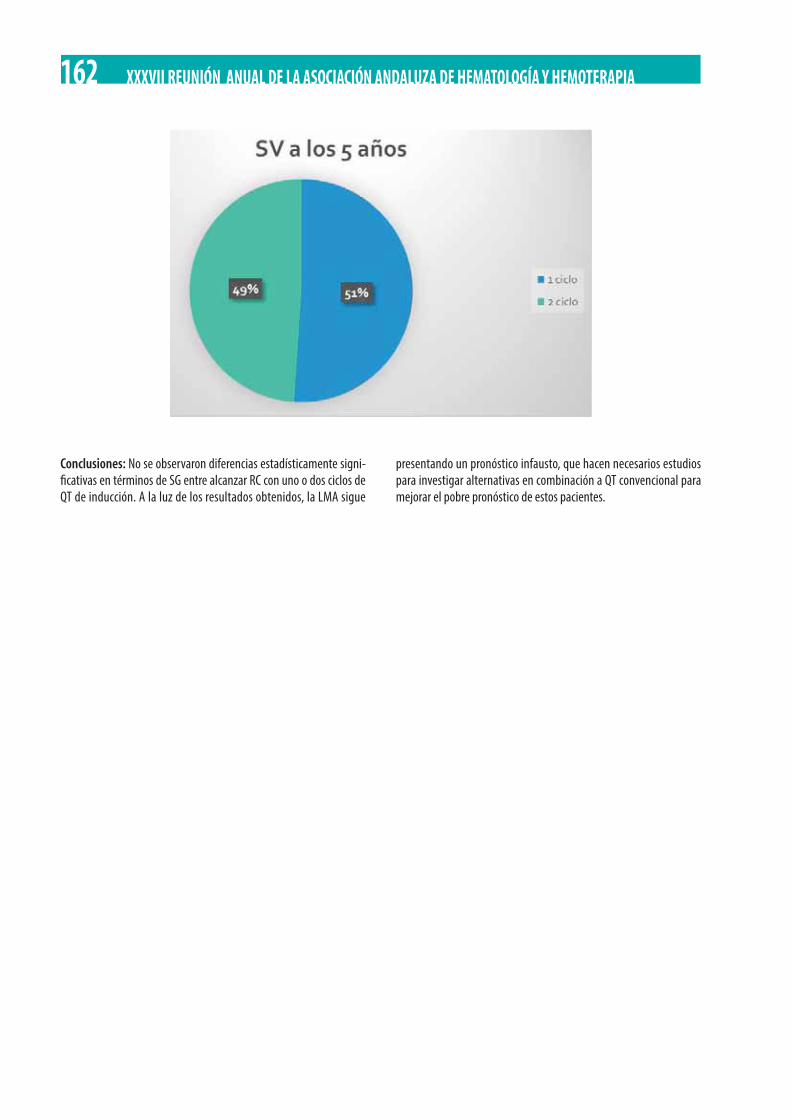
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA162
Conclusiones: No se observaron diferencias estadísticamente signi-ficativas en términos de SG entre alcanzar RC con uno o dos ciclos de QT de inducción. A la luz de los resultados obtenidos, la LMA sigue
presentando un pronóstico infausto, que hacen necesarios estudios para investigar alternativas en combinación a QT convencional para mejorar el pobre pronóstico de estos pacientes.

COMUNICACIONES PÓSTERS: Hematología Clínica 163
INTRODUCCIÓN/FUNDAMENTO:
Las infecciones víricas por VIH, VEB, VHH-8 o VLTH están asociadas a un incremento en el riesgo de desarrollar síndromes linfoproliferati-vos crónicos (SLPC). Esto se produce a través de mecanismos como integración directa al ADN, estimulación continua del sistema inmune o estados de inmunodeficiencias. Las infecciones crónicas por virus hepatotropos pueden estimular de forma permanente al sistema in-mune y a los linfocitos lo que puede conllevar a la aparición de un SLPC asociado.
OBJETIVO:
Determinar el estado serológico del virus de la hepatitis B y C (VHB–VHC) al diagnóstico y establecer una relación causal con el grupo de SLPC estudiado.
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo de la incidencia y estado serológico disponible al diagnóstico de un grupo de SLP [Linfoma folicular (LF), Linfoma de Hodgkin clásico (LH) y Linfoma difuso de células grandes B (LDCGB)] desde enero de 2008 a mayo de 2016.
Se estudiaron un total de 235 pacientes de los cuales 115 fueron muje-res (48,9%) y 120 varones (51,1%) con una edad media de 55,6 ± 17,5 (17-88). La distribución por diagnósticos fue de 102 casos (43.4%) LDCGB, 75 (31.9%) LCF y 58 (24.7%) LH clásico.
RESULTADOS Y CONCLUSIONES:
22 pacientes no tenían estudio serológico completo para VHB. De los 213 estudiados, 3 pacientes (1,4%) presentaron perfil VHB de infección crónica (HBsAg+ anti-HBc+ antiHBs-); 26 (12.2%) perfil de infección
pasada (HBsAg- anti-HBc+ antiHBs+); 25 (11.7%) perfil de vacunado (HBsAg- anti-HBc- antiHBs+) y 9 (4.2%) situación de post-infección aguda (HBsAg- anti-HBc+ antiHBs-)., 150 pacientes no tenían se-rología de contacto con virus B y 38 sí contacto pero no vacunal. La presencia de VHC es mucho menos prevalente (2.1%). La población vacunada VHB es más joven (ANOVA, p<0,01) que el resto. En cambio, la población HBC positiva es mayor (ANOVA, p=0,001) que la nega-tiva. No hay diferencias significativas para HbsAg y VHC (pero existe una muy baja prevalencia para afirmar conclusiones). No se evidenció correlación con el sexo (χ2 no significativa). No existe correlación con las patologías y la serología viral ni para VHB ni para VHC (salvo mayor presencia de pacientes anti-HBs positivos en LH).
Conclusiones:
1. No existen diferencias estadísticamente significativas entre la prevalencia de VHB y VHC (1.2% y 2.1% respectivamente) en po-blación con SLPC con respecto a la población general (0.27-1.69% en VHB y 1.6-2.6% en VHC).
2. No encontramos diferente patrón infeccioso asociado a los diferen-tes tipos de SLPC analizados por lo que no es posible afirmar un papel etiopatogénico de estos virus en el desarrollo de los mismos.
3. La tasa de vacunación VHB en los pacientes con SLPC estudiados es baja (11,7%) con excepción de pacientes jóvenes (lo que justifica el mayor patrón vacunal en LH).
4. El estado serológico más frecuente para el VHB es el de infección pasada.
5. La prevalencia de pacientes con riesgo de activación del VHB (an-tiHBc positivos) es del 17,4% lo que indica que aproximadamente 1 de cada 5 pacientes van a precisar profilaxis anti-VHB con la terapia.
Análisis epidemiológico y serológico del virus de la hepatitis B y C en un grupo de síndromes linfoproliferativos. Resultados de un centroV. Calama Ruiz-Mateos (1), M.E. Molina Rodríguez (2), M. Gómez Rosa (2), J.C. López Martin (2), I. Simon Pilo (2), E. Ríos Herranz (2)
(1) Hospital Universitario Nuestra Señora de Valme, (2) Hospital Universitario Nuestra Señora de Valme

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA164
INTRODUCCIÓN/FUNDAMENTO:
El síndrome de Sweet es una dermatosis caracterizada por sus llamati-vas manifestaciones clínicas y dermatopatológicas. Clínicamente cursa con la aparición brusca de fiebre y múltiples placas eritematoedema-tosas, sensibles o dolorosas, de distribución bilateral y asimétrica. His-tológicamente, lo más frecuente es encontrar un infiltrado neutrofílico dérmico. En raros casos, se observa una invasión por neutrófilos inma-duros que al microscopio óptico son indistinguibles de los histiocitos. La importancia de su diagnóstico radica en la gran repercusión sistémi-ca potencialmente grave, y su asociación con diversas enfermedades, especialmente neoplasias hematológicas y tumores malignos.
OBJETIVO:
Describir el caso de un síndrome de sweet histiocitoide en el contexto de autotrasplante de progenitores hematopoyéticos. -Remarcar la importancia del diagnóstico clínicopatológico de las manifestaciones dermatológicas durante el trasplante.
PACIENTES, MATERIAL Y MÉTODO:
Paciente mujer de 52 años, sin alergias medicamentosas conocidas, diagnosticada de linfoma no Hodgkin T CD8+ cutáneo. Tras tratamien-to con CHOPx8, ingresa en nuestro centro en 1ª RC para Trasplante Autólogo de progenitores hematopoyéticos, previa movilización con VP-16. En el dia +12, tras alcanzar implante, comienza con rash cutáneo pruriginoso en todo el cuerpo incluyendo cuero cabelludo, palmas y cara; con vesículas y fiebre de hasta 38ºC, sin repercusión hemodinámica. Desde un punto de vista clínico, se plantea el diag-nóstico diferencial entre etiología infecciosa (VVZ) y toxicodermia (en probable relación con estimulante de colonias granulocíticas o Pipera-zilina Tazobactam por correlación temporal). Se instaura tratamiento empírico con Aciclovir IV 10mg/Kg/8h. En el día +23, comienza con tos
Síndrome de Sweet histiocitoide asociado a trasplante autólogo de progenitores hematopoyéteicos. A propósito de un casoI. Sánchez Bazán, C. Díaz Aizpún, S. Simonsen, C. Bethencourt Mateos, A.I. Heineger Mazo
Hospital Regional de Málaga
e insuficiencia respiratoria, precisando oxigenoterapia para mantener saturaciones. En la Rx de tórax, se evidencia condensación retrocardía-ca en LII. Se descarta la etiología infecciosa por negatividad para la PCR de Adenovirus, HSV1, HSV2, Varicela Zoster, CMV y EB; tanto en el exudado de vesículas como en sangre. Los hemocultivos convenciona-les también fueron negativos. A favor de la toxicodermia secundaria a tratamiento con GCSF, la paciente había presentado, un ascenso brusco de las cifras leucocitarias; pasando de 300 granulocitos en el día +11 a 14.000 en el día +13. Realizamos biopsia cutánea, que revela un infiltrado histiocitario en dermis, con presencia de edema y leucoci-toclastia, sin signos de vasculitis; compatible con Sindrome de Sweet histiocitario. A la luz de los resultados, se suspende el tratamiento antiviral e inicia tratamiento con Metilprednisolona 1mg/kg/dia. Tras 21 días de tratamiento antimicrobiano y mejoría radiológica; se pro-cede a alta a domicilio, presentando mejoría de las lesiones cutáneas. La paciente sigue controles en consulta externa de Hematología con

COMUNICACIONES PÓSTERS: Hematología Clínica 165
buena evolución. Hasta la fecha no ha presentado recaída del cuadro cutáneo ni hematológico.
RESULTADOS Y CONCLUSIONES:
El síndrome de Sweet, debe ser opción diagnóstica ante la presen-cia de rash cutáneo y fiebre, sobre todo en el contexto de neoplasias
hematológicas. En ocasiones puede estar en relación con fármacos como el GCSF. El 70% de los casos son idiopáticos, por lo que no debe descartarse como posibilidad diagnóstica incluso en situación de RC. Para el diagnóstico es imprescindible realizar biopsia cutánea. El sín-drome de Sweet responde al tratamiento con corticoides sistémicos, aunque un 30% de los pacientes presentan recaídas tras su suspensión.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA166
INTRODUCCIÓN/FUNDAMENTO:
La evaluación de la infiltración de la médula ósea (MO) es un técni-ca crucial en la estadificación de linfomas. A pesar de que la biopsia de MO (BMO) es el referente para la evaluación de la infiltración, los criterios actuales de Lugano consideran que la BMO puede ser inne-cesaria en algunos casos. El PET/TC se ha convertido en una excelente herramienta para la estadificación y evaluación de la captación en MO en linfomas, especialmente en el difuso de células grandes y Hodgkin, no existiendo consenso en el resto de linfomas con menores tasas de proliferación celular (folicular, manto...).
OBJETIVO:
Identificar correlación en la afectación de MO al diagnóstico en linfo-mas indolentes mediante PET y BMO.
PACIENTES, MATERIAL Y MÉTODO:
Se estudiaron 44 pacientes (22 varones y 22 mujeres) con una edad media de 57 años (33-86) diagnosticados de linfoma folicular (n=36, 81.8%), linfoma de células del manto (n=6, 13.6%) y linfoma de la zona marginal (n=2, 4.5%).
RESULTADOS Y CONCLUSIONES:
En 30 de 36 (83.3%) pacientes con linfoma folicular el PET mostró infiltración medular; los 6 casos con linfoma del manto mostraron in-filtración de MO al PET y ninguno de los 2 casos con linfoma de la zona marginal fueron positivos al PET en MO (χ2, p=0.006)
En función de infiltración demostrada por BMO, 13 (36.1%) de los 36 pacientes con linfoma folicular mostraron infiltración en BMO, 4 (66.6%) de los pacientes con linfoma de células del manto y 1 (50%) de los 2 pacientes con linfoma de la zona marginal (χ2, p=0.35)
En 13 pacientes (29.5%) la afectación de MO fue identificada tanto por PET como por BMO; en 23 pacientes (52.3%) la afectación fue identi-ficada por PET pero no por BMO; 5 (11.4%) pacientes mostraron infil-
tración en BMO que no fue detectada por PET y finalmente 3 (6.8%) pacientes no mostraron afectación de MO ni por PET ni por BMO.
En conjunto, el PET identifica más del doble de pacientes afectos en MO que la BMO: 23 pacientes presentaron afectación al PET con BMO no infiltradas y un total de 5 pacientes presentaron infiltración en la BMO pero con PET negativo. El estudio de BMO respecto al PET presenta una sensibilidad de 36.1%, un VPP de 72.2% y un VPN de tan sólo 11.5%, lo que la hace poco útil para descartar infiltración medular respecto a la información aportada por el PET.
En los casos de pacientes con linfoma folicular, el estudio de BMO respecto al PET presenta una sensibilidad de 30%, un VPP de 69.2% y un VPN de tan sólo 8.7%.
En los pacientes con linfoma de células del manto, el estudio de BMO respecto al PET presenta una sensibilidad de 66,6%, con un VPP de 100% y un VPN nulo.
Por último, de los 2 pacientes con linfoma de la zona marginal los resultados indican mala sensibilidad del PET para la detección de infiltración de MO.
CONCLUSIONES:
La correlación de la afectación medular detectada por BMO y PET en linfomas histológicamente de baja tasa de proliferación celular es mala (índice exactitud = 0.36), especialmente en linfomas del manto en que la BMO puede no detectar enfermedad objetivable en PET y en linfomas de la zona marginal, que por el contrario el PET puede no detectar enfermedad evidenciable en la BMO.
El estudio por BMO en este tipo de linfomas es poco útil para des-cartar infiltración medular respecto a la información aportada por el PET.
En los linfomas foliculares, el PET supera claramente la detección de infiltración medular respecto a la BMO pero hasta un 12% de pacientes pueden tener enfermedad medular no detectable por PET lo que hace recomendable ambas pruebas para un estadiaje más exacto.
Análisis de la correlación de la afectación de médula ósea al diagnóstico en linfomas indolentes mediante PET y BMOV. Calama Ruiz-Mateos (1), M.E. Molina Rodríguez (2), M. Gómez Rosa (2), I. Simon Pilo (2), C. Couto Caro (2), E. Ríos Herranz (2)
(1) Hospital Universitario Nuestra Señora de Valme, (2) Hospital Universitario Nuestra Señora de Valme

COMUNICACIONES PÓSTERS: Hematología Clínica 167
INTRODUCCIÓN/FUNDAMENTO:
La Aplasia medular es una entidad clínica caracterizada por una dismi-nución (aplasia moderada) o desaparición (aplasia severa) del tejido hematopoyético de la medula ósea, produciendo una insuficiencia medular global. La Aplasia medular severa (AMS) es una enfermedad grave con una elevada tasa de mortalidad. El trasplante alogénico de progenitores hematopoyéticos (Alo-TPH) es la principal opción terapeutica, ya sea de donante familiar idéntico o de donante no emparentado.
OBJETIVO:
Evaluar las características y el pronóstico de los pacientes afectos de AMS sometidos a Alo-TPH en nuestro centro.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo que incluye 46 pacientes diagnosticados de AMS sometidos a Alo-TPH en nuestro centro entre Mayo´95-Mayo´15. Veinticinco pacientes eran varones (54%), con una mediana de edad al diagnóstico de 23 años (6-56 años). En el 87% de los casos la AMS era de origen primario. Las características de la serie se muestran en la Tabla 1. La serología para VIH, VHB y VHC fue negativa en todos los pacientes salvo en 2 (VHB positivo -infección aguda- y otro para VHC positivo -infección crónica-). La determinación del clon HPN fue positiva previa al TPH en 5 pacientes (10,9%). El 59 % de los casos (n=27) recibió Terapia Inmunosupresora (TIS) previa al Alo-TPH, con una mediana de tiempo del diagnóstico al inicio de la misma de 31 días (2-625 dias).
Trasplante alogénico de progenitores hematopoyéticos en aplasia medular severaA.I. Álvarez Sánchez, M. Yebenes Ramírez, E. Gracia Torres, C. Martín Calvo, J. Serrano López, V. Arqueros Martínez
Hospital Universitario Reina Sofía de Cordoba
TABLA 1. Características de los pacientes
Treinta pacientes (65%) recibieron un Alo-TPH de hermano idéntico (Alo-TPH Herm-ID) y 16 pacientes (35%) de donante no emparentado (Alo-TPH DNE). La fuente de progenitores fue medula ósea en 80% de los casos.
RESULTADOS Y CONCLUSIONES:
La mediana de tiempo desde el diagnóstico al Alo-TPH fue de 131 días (18-4.115 días).
Las complicaciones post-TPH se muestran en la Tabla 2.
Mediana de seguimiento post Alo-TPH de 72 meses (12-187 meses), con una supervivencia global (SG) de la serie a los 4 años es de 75 ± 6,8% (Figura 1C).
El estudio univariante de aquellos pacientes con edades comprendidas entre los 15 y 50 años (n=31) mostró los siguientes resultados:
1. Incidencia de enfermedad del injerto contra el huésped aguda (EICHa) moderada-severa significativamente mayor en el grupo Alo-TPH DNE respecto a Herm-ID (78% vs 23% (p=0,012).
2. Mayor SG en pacientes que recibieron un trasplante de médula ósea vs sangre periférica (SG a 4 años 84,±-6,5% vs 41,7±17,3%, p=0,009). Figura 1A.
3. Mayor SG en el grupo de pacientes que reciben un Alo-TPH Herm-ID vs DNE (SG a 4años de 96±4% vs 43± 12,6%, p<0,01). Figura 1B. En el estudio multivariante el Alo-TPH Herm-ID es 18 veces factor protector frente al TPH DNE, p=0,006.
TABLA 2.
Complicaciones Post-TPH según tipo de TPH (Herm.-ID vs DNE)
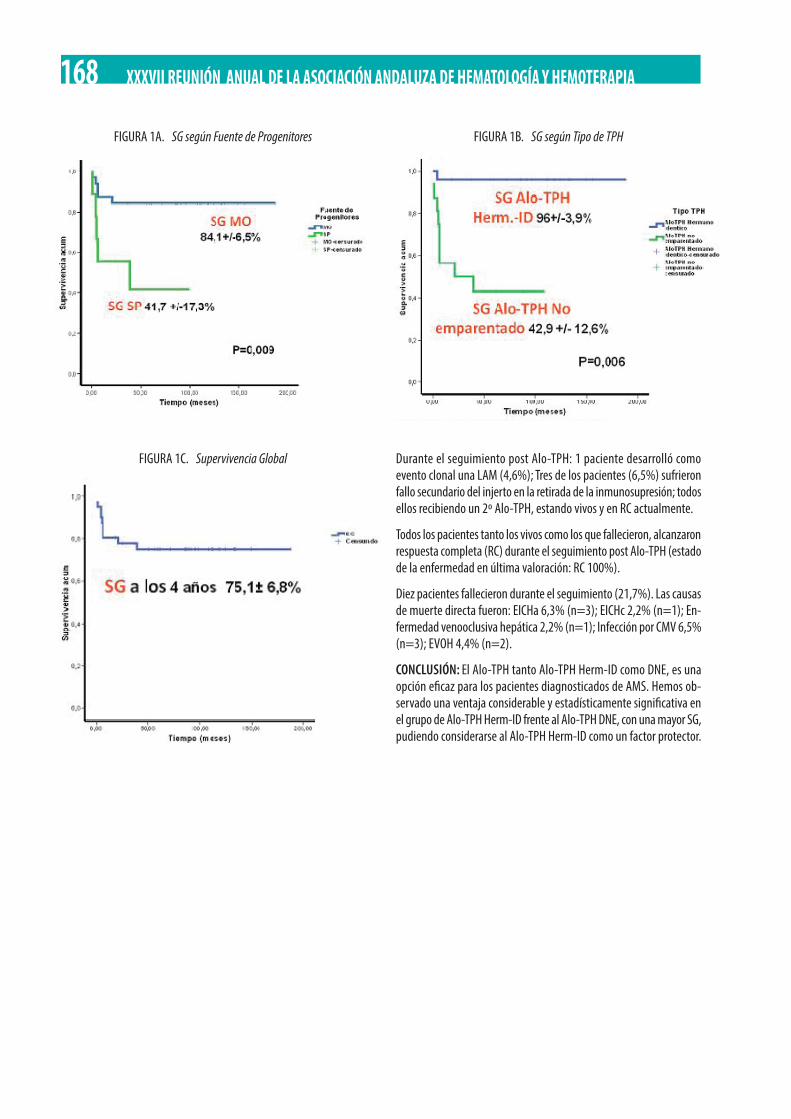
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA168
FIGURA 1A. SG según Fuente de Progenitores
FIGURA 1C. Supervivencia Global
FIGURA 1B. SG según Tipo de TPH
Durante el seguimiento post Alo-TPH: 1 paciente desarrolló como evento clonal una LAM (4,6%); Tres de los pacientes (6,5%) sufrieron fallo secundario del injerto en la retirada de la inmunosupresión; todos ellos recibiendo un 2º Alo-TPH, estando vivos y en RC actualmente.
Todos los pacientes tanto los vivos como los que fallecieron, alcanzaron respuesta completa (RC) durante el seguimiento post Alo-TPH (estado de la enfermedad en última valoración: RC 100%).
Diez pacientes fallecieron durante el seguimiento (21,7%). Las causas de muerte directa fueron: EICHa 6,3% (n=3); EICHc 2,2% (n=1); En-fermedad venooclusiva hepática 2,2% (n=1); Infección por CMV 6,5% (n=3); EVOH 4,4% (n=2).
CONCLUSIÓN: El Alo-TPH tanto Alo-TPH Herm-ID como DNE, es una opción eficaz para los pacientes diagnosticados de AMS. Hemos ob-servado una ventaja considerable y estadísticamente significativa en el grupo de Alo-TPH Herm-ID frente al Alo-TPH DNE, con una mayor SG, pudiendo considerarse al Alo-TPH Herm-ID como un factor protector.

COMUNICACIONES PÓSTERS: Hematología Clínica 169
INTRODUCCIÓN/FUNDAMENTO:
La Linfocitosis Monoclonal B tipo Leucemia Linfática Crónica (LMB LLC) puede ocurrir en un 3-5% de población adulta y hasta un 20% en > 60 años con contaje linfocitario normal. Hasta un 40% se transforma en Leucemia Linfática Crónica B (LLC-B), enfermedad de curso pro-longado e indolente con criterios específicos para iniciar tratamiento. El Linfoma No Hodgkin T (LNH T) conforma un grupo heterogéneo de neoplasias de células T de baja incidencia, < 15% de los linfomas, agresivo y mal pronóstico, cuya estrategia de tratamiento continúa sin consensuarse.
OBJETIVO:
Presentamos dos casos de pacientes en los que coexiste LNH T periféri-co NOS (sin especificar) con LLC-B y LMB LLC, uno de ellos con diagnos-tico simultáneo y otro con hallazgo de LLC-B posterior al diagnóstico de linfoma. Cada caso recibe una línea de tratamiento adaptado a edad y comorbilidad, con curso clínico opuesto.
PACIENTES, MATERIAL Y MÉTODO:
Caso 1: varón de 56 años, sin antecedentes de interés, que en Mayo de 2009 acude a Urgencias por tumoración supraclavicular izquierda. No síntomas B y serologías virales negativas. En TAC realizado se observan múltiples conglomerados adenopáticos submaxilares, supraclavicula-res y axilares bilaterales. Tras biopsia de adenopatía axilar izquierda es diagnosticado de LNH T periférico NOS estadío II-A. IPI 0. Biopsia de médula ósea con ausencia de infiltración por linfoma. Inicia tra-tamiento quimioterápico en Agosto de 2009 según esquema CHOP, del que completa 8 ciclos finalizando en Enero de 2010. Se realiza PET-TAC post 8º ciclo encontrándose en remisión completa (RC) por lo que se presenta para autotrasplante de progenitores hematopoyéti-
cos (autoTPH) como consolidación a la respuesta. En control previo a TPH se descubre linfocitosis con 8.4% células compatibles con LLC-B en inmunofenotipo de sangre periferica; 1% de células en biopsia de médula ósea. En Junio de 2010 se realiza autoTPH, quedando en RC del LNH T, no así de la LLC-B que persiste en progresión linfocitaria aunque actualmente sin criterios de iniciar tratamiento.
Caso 2: mujer de 77 años, sin antecedentes de interés, que en Mayo de 2012 acude a Urgencias por tumoración laterocervical izquierda y submandibular derecha. Presentaba sudoración vespertina y prurito generalizado, no otros síntomas B. Se realiza biopsia de adenopatía cervical en Abril de 2013 diagnosticándose de LNH T periférico NOS ALC+, CD3+, bcl6+, uchl1+, CD5+, CD4 +, CD8 +, CD20-, CD30 - , estadío III-A, IPI 3. En biopsia de médula ósea no se detecta infiltración por linfoma pero por inmunofenotipo se hallan células compatibles con LLC-B. Comienza tratamiento en Mayo de 2013 con CNOP (con antraciclinas atenuadas por > 70 años) del que recibe 6 ciclos con re-misión parcial en PET-TAC post 3º y 6º ciclo. Comienza tratamiento con GPD del que recibe 5 ciclos, alcanzando remisión completa en PET-TC de Junio de 2014. No candidata a autoTPH por edad. Ingresa en Sep-tiembre de 2014 por cuadro infeccioso con mala evolución falleciendo en Octubre de 2014 por fallo multiorgánico.
RESULTADOS Y CONCLUSIONES:
– Desconocemos el papel que representa LMB LLC / LLC-B en algunos casos (evolución a LNH T alto grado -Ritchter atípico-, reactiva a LNH T ó presentación paralela), se necesita más registro de casos para extraer conclusiones significativas.
– Independientemente de la clona LLC-B, debemos dirigir nuestro es-fuerzo terapéutico hacia la patología más agresiva como es el LNH T. CHOP seguido de autoTPH puede ser una opción eficaz.
Linfocitosis monoclonal B tipo LLC asociada a linfoma no Hodgkin TMyriam Revelles Peñas (1), Carola Díaz Aizpún (2), Manuel Espeso de Haro (2)
(1) Hospital Universitario Virgen de la Victoria , (2) Hospital Regional de Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA170
INTRODUCCIÓN/FUNDAMENTO:
LMC es una neoplasia que produce la expansión clonal de células di-ferenciadas de la línea mieloide, provocada por la producción incon-trolada de una tirosincinasa única BCR-ABL1 continuamente activa. Previamente a la introducción de imatinib, la esperanza de vida de un paciente diagnosticado de LMC a los 10 años era menor del 10%. Gracias a la introducción de los ITK se ha conseguido mejorar el pro-nóstico de estos pacientes, procuciéndose además el incremento de la prevalencia de la enfermedad (se duplica cada cinco años).
OBJETIVO:
Exponer la experiencia y manejo de los pacientes con LMC tratados con ITK en nuestro centro.
PACIENTES, MATERIAL Y MÉTODO:
Analizamos de forma retrospectiva los pacientes diagnosticados de LMC (N=10) y tratados con ITK en nuestro centro desde 2005 hasta la actualidad (todos ellos diagnosticados es fase crónica). El segui-miento de las respuestas se realizó según recomendaciones de la ELN. La mediana de edad fue de 60 años (24-83). Resto de características basales en tabla 1.
RESULTADOS Y CONCLUSIONES:
RESULTADOS:
De los 10 pacientes que han recibido tratamiento con ITK en nuestro centro por LMC 9 de ellos iniciaron tratamiento con Imatinib (ITK 1ª generación) y tan solo 1 de ellos (el más joven) lo hizo con Nilotinib (ITK 2ª generación).
Era de los inhibidores de la tirosin cinasa (ITK)en Leucemia Mieloide Crónica (LMC), manejo y experiencia en nuestro centroM.J. Llamas Poyato, D. Moreno Garrido, I. García Díez
Hospital Infanta Margarita, AGS Sur de Córdoba
pCARACTERÍSTICA N (%) Sexo femenino 3 (30) Mediana de Edad (años) 60 Tratamiento previo con Hydrea 6 (60%) Inicio tratamiento ITK 1ª generación 9 (90%) Síntomas sí 6 (60%) Factores de riesgo CV sí 4 (40%)
Índice Hasford Bajo 4 (40%) Intermedio 5 (50%) Alto 1 (10%)
Índice Sokal Bajo 3 (30%) Intermedio 6 (60%) Alto 1 (10%)
TABLA 1. Características basales de los pacientes
En cuanto a la respuesta obtenida tras el inicio del tratamiento, a los 3 meses 5 (50%) de ellos habían conseguido ratio BCR-ABL menor al 3% (en los otros 5 no se hizo cambio precoz).
En cuanto a reducción de dosis inicial por toxicidad 5 pacientes (50%) la requirieron (todos ellos en tratamiento con Imatinib), con posterior normalización de dosis, salvo en 2 de ellos que requirieron la suspen-sión definitiva por toxicidad cutánea (1 se cambió a Nilotinib y otro a Dasatinib). En la tabla 2 se muestra la toxicidad que presentaron nuestros pacientes con los ITK.
La suspensión del ITK de 1ª línea con cambio a un ITK de 2ª generación por falta de respuesta (pacientes con RCC sin RMM tras 18 meses de tratamiento) se dio en 2 pacientes (20%); los 2 se cambiaron a Nilo-tinib. El estudio mutacional para los pacientes sin RMM fue normal.
2 pacientes (> 80 años) con RMM reciben dosis reducidas de Imatinib (300 mg/24h) y el paciente joven que inició tratamiento con Nilo-tinib consiguió alcanzar RMM tras aumentar dosis de Nilotinib (400 mg/12h; paciente obeso).
Con una mediana de seguimiento de 34 meses (146-4), la SG y la SLP son del 100%.
Salvo los 2 pacientes de reciente diagnóstico (<1 año), todos se en-cuentran en RMM-RM5. 3 de nuestros pacientes presentan RMM4,5 o mejor desde hace más de 3 años, por lo que nos planteamos la posi-bilidad de discontinuar el tratamiento.
CONCLUSIONES:
Tras la introducción de los ITK se ha mejorado la esperanza de vida de los pacientes con LMC, siendo la tasa anual de fallecimientos menor del 5%, presentando además buena tolerancia a los mismos, como se demuestra en nuestra serie. Está aún por definir, mediante ensayos clínicos, qué enfermos son los más adecuados para la discontinua-ción, teniendo en cuenta el tratamiento previo y cuanto tiempo ha de mantenerse la RM profunda previamente a retirada para asegurarnos que se mantenga la RM profunda tras su suspensión.
TABLA 2. Eventos adversos

COMUNICACIONES PÓSTERS: Hematología Clínica 171
INTRODUCCIÓN/FUNDAMENTO:
La inmunoquimioterapia (IQ) es la opción estándar para los pacientes con leucemia linfática crónica (LLC) que necesitan tratamiento. Los pa-cientes con alteraciones citogenéticas de alto riesgo, p.ej. aberraciones de TP53 (deleción 17p13.1), presentan un elevado índice de recaída, resistencia a la quimioterapia e inferior supervivencia. La vía de P53 induce apoptosis ante un daño en el ADN celular, y su inactivación conduce a altas tasas de proliferación celular. En estos pacientes, las opciones de tratamiento son muy escasas. Ibrutinib es un inhibidor de la tirosín-kinasa de Bruton (BTK), disponible vía oral, que induce una inhibición de la señalización del receptor del linfocito B y de las interacciones de las células clonales con el microambiente.
OBJETIVO:
Mostrar la seguridad y la actividad de ibrutinib en los pacientes con LLC refractaria o en recaída tratados en nuestro centro entre enero de 2015 y enero de 2017.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo y retrospectivo. Se incluyen 9 pacientes, cuyas características antes del tratamiento se muestran en la tabla 1. Todos los pacientes tenían enfermedad activa que requería tratamiento, seis de ellos por insuficiencia medular progresiva (anemia o trombo-penia), dos por tiempo de duplicación linfocitaria ≤ 6 meses y uno por síntomas B. La delección 17p (FISH) la portaban el 67 %. Todos habían recibido IQ previa y uno se había realizado un alotrasplante de progenitores hematopoyéticos (aloTPH). Los pacientes recibieron 420 mg de ibrutinib al día vía oral.
RESULTADOS Y CONCLUSIONES:
Se evaluó la respuesta al tratamiento según los criterios del IWCLL, los efectos adversos según National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) y los eventos hemorrágicos.
La mediana de seguimiento fue de 9 meses (3-24). Todos los pacientes fueron evaluados a las 12 semanas de tratamiento, observando res-puesta parcial (RP) con linfocitosis. A las 24 semanas de tratamiento, se habían evaluado 7, todos manteniendo la RP con linfocitosis. De ellos, 2 pacientes que tenían Hb ≤ a 11 gr/dL y neutrófilos inferiores a 1500/mm3 previo ibrutinib habían alcanzado niveles normales de ambos. 5 tenían plaquetas ≤ a 100.000/mm3, que se normalizaron en tres casos. De 4 que presentaban esplenomegalia palpable, en 3 no se objetivaba y uno presentaba reducción al 50%. De 4 con adenopatías significativas, 3 no presentaban y uno (con masa Bulky) obtuvo una reducción del 50 %. Al año, se pudo evaluar a 3 pacientes, 2 perma-necían en RP con linfocitosis y un tercero en RP. 4 pacientes (44%) discontinuaron tratamiento, 2 por indicación de aloTPH, uno por trans-formación a LNH de alto grado y posterior fallecimiento y el último por cambio de tratamiento ante la necesidad de anticoagulación oral.
Durante los 3 primeros meses, 6 pacientes (66%) mostraron toxicidad: 33% diarrea grado 1-2, 33% rash cutáneo grado 1-2 y en un caso artralgias grado 1-2. Los eventos adversos se resolvieron espontánea-mente durante este período. Solamente un paciente precisó dismi-nución de dosis del fármaco durante un mes. Una paciente, afecta de enfermedad de von Willebrand, presentó gingivorragias y hematomas grado 2, que mejoraron con ácido tranexámico, y otro paciente pre-sentó hematuria grado 3 tras inicio de Dabigatrán.
CONCLUSIONES
1. Ibrutinib se muestra como una herramienta efectiva y con un buen perfil de seguridad para el tratamiento de la LLC, en especial para aquellos casos con citogenética de mal pronóstico.
2. Puede ser un excelente fármaco para utilizar como tratamiento puente en pacientes jóvenes con indicación de aloTPH.
Tratamiento con Ibrutinib en la leucemia linfática crónica en recaída o refractaria. Revisión de casos en nuestro centroJ. Díez Pastor (1), O. Benítez Hidalgo (2), M. Ortiz Pareja (2), A.I. Heiniger Mazo (2)
(1) Hospital Regional Universitario de Málaga , (2) Hospital Regional Universitario de Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA172
Mediana de edad, anos (rango) 68 (51-83)
Sexo, n (%)
- Masculino
-Femenino
4 (44%)
5 (56%)
ECOG, n (%)
- 0-1
- ≥2
8 (89%)
1 (11%)
Enfermedad Bulky, n (%)
< 10 cm
≥ 10 cm
8 (89%)
1 (11%)
Esplenomegalia, n (%)
-Si
-No
5 (56%)
45 (44%)
Estadio Binet, n (%)
-B
-C
3 (33%)
6 (67%)
FISH, n (%)
- Alto riesgo:
Del(17p)
Del(11q)
-Riesgo intermedio: Trisomía 12
6 (67%)
2 (22%)
1 (11%)
Media ß2 microglobulina, mg/L (rango) 5.01 (3.1-6.6)
Nivel de hemoglobina, g/dL, n (%)
- > 11
- ≤ 11
6 (67%)
3 (33%)
Recuento de neutrófilos, x 109/L, n (%)
> 1,5
≤ 1,5
7 (78%)
2 (22%)
Recuento de plaquetas, x 109/L, n (%)
> 100
≤ 100
3 (33%)
6 (67%)
Media de linfocitos, x 109/L (rango) 66.03 (0.6-156.2)
Mediana de terapias previas, n (rango) 2 (1-5)
Tipo de tratamiento previo, n (%)
- Quimioterapia
Análogos de las purinas
Alquilantes (incluida Bendamustina)
- Quimioinmunoterapia con anti-CD20
- Trasplante alogénico de PH de DNE
9 (100%)
6 (67%)
7 (78%)
8 (89%)
1 (11%)
TABLA 1. Características de los pacientes (n=9) previo al inicio de tratamiento con ibrutinib

COMUNICACIONES PÓSTERS: Hematología Clínica 173
INTRODUCCIÓN/FUNDAMENTO:
El mieloma múltiple (MM) se caracteriza por una proliferación clonal de células plasmáticas malignas en la médula ósea y lesiones osteo-líticas. La introducción de nuevos agentes, tales como talidomida, le-nalidomida, Y bortezomib, ha mejorado considerablemente las tasas de respuesta y la supervivencia, tanto en el momento del diagnóstico como en la recaída, sin embargo, el MM sigue siendo incurable, y la mayoría de los pacientes recaen y suelen convertirse en refractarios a las terapias disponibles. La pomalidomida, un análogo estrechamente relacionado de talidomida ha mostrado una actividad significativa en pacientes en recaída o refractarios a lenalidomida y / o bortezomib, aumentando las tasas de respuesta parcial y supervivencia libre de progresión.
OBJETIVO:
Ver la seguridad y eficacia de los esquemas basados en pomalidomina
PACIENTES, MATERIAL Y MÉTODO:
Se ha revisado de manera retrospectiva a 4 pacientes con mieloma múltiple que comenzaron tratamiento en el 2016 con regímenes basados en pomalidomida. 3 de los 4 pacientes pertenecen al géne-ro masculino y los 4 comprenden una edad entre los 43-77 años. El 75% tenía un componente monoclonal IgG kappa y el restante IgG lambda. De igual forma un 75% de los pacientes tenía un índice pro-nóstico internacional (ISS) estadio III y el 25% estadio II. Uno de los pacientes había recibido 6 líneas de tratamiento antes de comenzar el esquema terapéutico con pomalidomida, 2 habían recibido 4 líneas de tratamiento y uno 3 líneas de tratamiento. 2 de los 4 pacientes
había recibido un TPH. Al inicio del tratamiento solo uno presentaba insuficiencia renal (IR). El esquema de pomalidomida con ciclofosfami-da más dexametasona (PoCyDex) lo recibieron 2 pacientes y los otros dos recibieron pomalidomida más dexametasona (PoDex). La dosis de pomalidomida en los 4 pacientes fue de 4mg. Dos de los pacientes presentaron progresión de la enfermedad, uno progreso después del quinto ciclo de tratamiento con PoCyDex a un plasmocitoma cerebral y el otro progreso a una leucemia de células plasmática después del cuarto ciclo de PoCyDex. Los pacientes restantes habían recibido como línea de tratamiento PoDex, uno se encuentra con enfermedad estable después de 5 ciclos y el otro con una respuesta parcial después de 13 ciclos. Como toxicidad los 4 pacientes tuvieron a nivel hematológico, presentando los 4 neutropenia grado IV necesitando soporte con es-timulantes de colonias granulociticas sin llegar a hacer neutropenia febril. También los cuatro pacientes presentaron trombopenia llegando a necesitar soporte transfusional 2 de ellos.
RESULTADOS Y CONCLUSIONES:
La terapia combinada con pomalidomida más dexametasona con o sin ciclofosfamida, son unos esquemas de tratamiento emergentes importantes para su uso como terapia de rescate en pacientes con mieloma múltiple refractario y en recaída. Se ha observado que estos esquemas de tratamiento prolongan significativamente la supervi-vencia libre de progresión, la supervivencia global y el tiempo hasta la progresión de la enfermedad y mejora de las tasas de respuesta global en la población con intención de tratar. En los pacientes jóvenes se puede observa que el mieloma múltiple es más agresivo lo cual podemos ver en 2 de los 4 pacientes los cuales progresaron a pesar de tratamiento con pomalidomida. Estos esquemas ofrecen seguridad y una buena tolerabilidad incluso en pacientes mayores.
Seguridad y eficacia de los esquemas basados en pomalidomida en pacientes con mieloma múltiple en recaída o refractario: serie de casos de un centroA.M. Alba Sosa (1), Y. Moatassim de la Torre (2), E. Clavero Sánchez (2)
(1) Hospital Comarcal Santa Ana de Motril, (2) Hospital Comarcal Santa Ana de Motril

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA174
INTRODUCCIÓN/FUNDAMENTO:
La enfermedad de Hodkgin (EH) es un síndrome linfoproliferativo de origen B, representa el 10% de todos los linfomas y un 20-50% de casos se asocia al virus Epstein Bar (VEB). Distribución bimodal carac-terística: mujeres jóvenes (mediana 20 años), y hombre de edad más avanzada (mediana 65 años).
El tratamiento de elección suele basarse en quimioterapia tipo ABVD ± radioterapia local con porcentajes de remisión en torno a 70-80%. Inmunoterapia y fármacos dirigidos se reservan para casos que no responden a quimioterapia convencional.
OBJETIVO:
Presentamos el caso de una paciente con enfermedad de Hodgkin clásica tipo esclerosis nodular (EHc EN) refractaria. Vemos las terapias utilizadas y resultados tras la administración de Nivolumab.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 20 años sin antecedentes de interés que consulta por tumo-ración supraclavicular izquierda, pérdida de 6 kg de peso, sudoración nocturna y fiebre por lo que es ingresada para estudio. En TAC toraco-abdominal se objetiva mazacote adenopático mayor de 3 cm en espa-cio supraclavicular izquierdo. Adenopatías múltiples axilares izquierdas y paratraqueales derechas de tamaño significativo todas ellas, lesión tímica de 6 cm asi como mazacote adenopático en mediastino anterior.
La biopsia adenopática obtenida mediante mediastinoscopia (15/5/2014) es informada como EHc EN. Se completa estudio de extensión inicial mediante analítica donde destaca leucocitosis con linfocitosis sin alteración del resto de series, aumento de la VSG y se-rología VEB IgM+. Biopsia de médula ósea sin infiltración por linfoma, concluyéndose como EHc EN estadio II-B con 3 factores de riesgo (bulky mediastínico, VSG y ≥ 3 zonas ganglionares).
Se inicia tratamiento según esquema ABVD completando hasta 6 ciclos (último en Noviembre de 2014), con reevaluación posterior mediante PET-TAC en el que se objetiva progresión supradiafragmática. Siguiente línea con esquema ESHAP x 2 ciclos, pero aumento de tamaño del conglomerado adenopático laterocervical izquierdo a la exploración, continúa con esquema IGEV, administrando 3 ciclos del mismo. Se reevalúa mediante PET-TAC que es informado como enfermedad en progresión con persistencia del conglomerado adenopático laterocer-vical, supraclavicular, retropectoral izquierdo y adenopatías axilares ipsilaterales. Se plantea en este momento inicio de Brentuximab como 4ª línea de tratamiento, administrándose 6 ciclos del mismo, a pesar de lo cual la enfermedad progresa en el PET-TAC de reevaluación.
Tras dichos resultados se decide administración de dos ciclos con es-quema quimioterápico MiniBEAM pero la enfermedad continúa en progresión. Se inicia sexta línea de tratamiento con CMOPP por un ciclo con nueva progresión y posteriormente se comienza tratamiento con Nivolumab en Marzo de 2016, tras 8 ciclos se objetiva respuesta parcial de lesiones preexistentes y aparición de dudosos focos patoló-gicos en PET-TAC de reevaluación. Decidimos profundizar la respuesta y tras 24 ciclos con Nivolumab la paciente se encuentra por primera vez en respuesta metabólica completa, mediante PET-TAC a fecha de 13/03/2017.
RESULTADOS Y CONCLUSIONES:
1. El tratamiento abordado con “moléculas diana” tipo Nivolumab ha sido eficaz en caso de EH refractaria con buena tolerancia, sin toxicidad infecciosa y con anemia grado 1-2 de la OMS manejable.
2. Los resultados obtenidos y la buena tolerancia al fármaco hacen plantearnos el uso de Nivolumab tanto en monoterapia como en combinación en tratamiento de rescate (ensayos en desarrollo).
Eficacia de Nivolumab en enfermedad de Hodgkin clásica refractariaO. Benítez Hidalgo, D.E. Barrios Decoud, M. Espeso de Haro
Hospital Regional Universitario de Málaga

COMUNICACIONES PÓSTERS: Hematología Clínica 175
INTRODUCCIÓN/FUNDAMENTO:
La mucormicosis es una infección fúngica invasiva (IFI) poco frecuente -5% de IFI en hematológicos- causada por hongos de la familia Apo-physomyces elegans (orden de los mucorales).
Su ubicuidad y bajo potencial virulento los hace inocuos para gran parte de la población,siendo el paciente inmunodeprimido el principal candidato al desarrollo de la infección y potenciales complicaciones. Las neoplasias hematológicas son uno de los grupos de patologías más susceptibles,sólo por detrás de la Diabetes Mellitus.
Las afectaciones más frecuentes se producen en tracto sinusal, pulmones, sistema gastrointestinal, sistema nervioso central y piel(excepcional).
OBJETIVO:
Descripción de un caso de mucormicosis cutánea en paciente con neo-plasia hematológica.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos el caso de un paciente diagnosticado en 2003 de Leu-cemia Linfática Crónica B (LLC-B) con del17p,anemia hemolítica autoinmune(AHAI) e hipogammaglobulinemia severa (déficit IgA). Realizaba seguimiento en nuestras consultas habiendo recibido nume-rosos tratamientos a los que fue refractario (Fludarabina, Alemtuzu-mab, R-Clorambucil). Desde Marzo de 2016 realizaba tratamiento con corticoides diarios por nueva crisis hemolítica y trombopenia también posiblemente inmune. El último tratamiento instaurado–Ibrutinib- se suspendió el 20 de Abril(AHAI+cultivo de esputo con Pseudomonas Aeruginosa).
Ingresa en Mayo de 2016 por neutropenia febril en el contexto de infección pulmonar secundaria a hipogammaglobulinemia y corticoterapia,iniciando Meropenem al detectarse en hemocultivo Pseudomonas Aeruginosa.Realizaba desde una semana antes profi-laxis antifúngica(Fluconazol).En los primeros días de ingreso se añaden episodios de descompensación glucémica esteroidea.
Tras seis días ingresado manifiesta molestias en antebrazo izquierdo,donde días antes se había canalizado vía periférica.Ini-cialmente sólo presentaba un pequeño hematoma que se trató con analgesia y Mupirocina. A las 48 horas se advierte empeoramiento con gran hematoma violáceo y herida necrótica supurante en el centro, iniciándose Vancomicina y reducción de esteroides.
A los dos días avisan desde Microbiología por crecimiento de Mucor spp en exudado de la herida, comenzando Anfotericina B liposomal (7.5mg/kg/24h), y realizándose ese día desbridamiento en quirófano que se repite a las 48 horas.
Paralelamente se realizó despistaje de enfermedad micótica en otras localizaciones, descartándose afectación tanto pulmonar como sinusal, y cada 14/21 días recibió Ig IV.
RESULTADOS Y CONCLUSIONES:
Completó 27 días de tratamiento con Anfotericina B, realizándose du-rante ese tiempo curas periódicas con cultivos de exudados de la herida hasta el momento en que fueron repetidamente negativos, procedién-dose entonces a cobertura con autoinjerto de piel del mismo brazo.
Tras la retirada de la Anfotericina B se instauró profilaxis secundaria con Posaconazol -tres meses- tras los cuales se reinstauró Anfotericina B por infección por mucor rinosinusal, falleciendo en Noviembre de 2016.
Mucormicosis cutánea: a propósito de un casoS. Ordóñez Vahi, A. Salamanca Cuenca, V. Rubio Sánchez, S. Garzón López
Hospital de Jerez de la Frontera

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA176
– La mucormicosis cutánea es una entidad poco frecuente ante la que es necesario un alto índice de sospecha, siendo de vital importancia una rápida actuación diagnóstica (pruebas de imagen y toma de muestras aún en localizaciones no típicas y en patologías no con-sideradas de alto riesgo) y terapéutica (terapia antifúngica a dosis plenas, desbridamiento quirúrgico agresivo).
– En nuestro caso se cumplen varios de los factores de riesgo reco-nocidos para el desarrollo de IFI: DM, corticoterapia prolongada, neutropenia severa,inmunosupresión humoral,terapia previa con Alemtuzumab.

COMUNICACIONES PÓSTERS: Hematología Clínica 177
INTRODUCCIÓN/FUNDAMENTO:
Aunque es infrecuente, las enfermedades hematológicas pueden apa-recer de forma sincrónica con tumores sólidos.
OBJETIVO:
Describir 3 casos de enfermedades hematológicas concomitantes con tumores sólidos que aparecieron en el año 2015 en nuestro centro, así como su manejo práctico.
PACIENTES, MATERIAL Y MÉTODO:
Caso 1: mujer 47 años. Padre muerto por tumor cerebral. Hermana melliza diagnosticada de Ca de Mama. AP de Mieloma Múltiple IgA Kappa estadio III-A en Octubre de 2010; recibió tratamiento con es-quema VD y Auto-TPH en Marzo de 2011, sin presentar complicaciones durante y tras el mismo, alcanzando respuesta completa (RC). En Junio 2015 presenta datos clínicos y analíticos de recaída y en PET presenta progresión ósea de su enfermedad de base (mieloma múltiple) y re-fuerzo mamario subsidiario de completar estudio à Diagnóstico: Cáncer de mama ductal in situ.
Caso 2: hombre de 76 años, diagnosticado de LLC-B en 2014, en segui-miento en Consulta hasta agosto de 2015, donde presenta conglome-rado adenopático axilar izquierdo aislado de 6x7 cm à Diagnóstico de LBDCG (Sd.Richter). En esa misma fecha presenta molestias urinarias y en biopsia prostática se detecta adenomacarcinoma acinar poco diferenciado, valor patrón 8 (4,4) de Gleason.
Caso 3: hombre de 75 años, diagnosticado en junio de 2015 de linfoma linfocítico pequeño con ocupación de epigastrio y mesogastrio por
grandes masas adenopáticas en retroperitoneo perivascular superior la mayor de 17x16x10 cm y SUVmax 5.90 en PET. Durante el tratamiento quimioterápico se queja de diarrea explosiva, así como molestias tras la ingesta a pesar de presentar buena respuesta al tratamiento, por lo que se realiza interconsulta a Digestivo que lleva a cabo una colonos-copia donde se aprecia masa con oclusión casi completa de luz intes-tinal a nivel de recto con diagnóstico de adenocarcinoma intramucoso villiforme, de tipo adenomatoso no infiltrante.
RESULTADOS Y CONCLUSIONES:
Caso 1: La paciente recibió tratamiento con esquema VTD (x3 ciclos) y posteriormente se realizó mastectomía radical izquierda, sin requerir tratamiento QT, RT ni hormonal. Tras reevaluación encontrándose en RC se realizó 2º Auto-TPH en Abril de 2016, encontrándose actualmente en RC.
Caso 2: El paciente recibió tratamiento quimioterápico con esquema R-CHOP (x3 ciclos), mientras que se realizó bloqueo hormonal para Ca. Próstatico. Postriormente recibió RT para Ca.próstata y LBDCG pre-sentando buena tolerancia a RT; únicamente toxicidad genitourinaria grado 1. Actualmente se encuentra en RC.
Caso 3: Por nuestra parte el paciente finalizó tratamiento con esquema R-Benda. También se llevó a cabo la resección del adenocarcinoma colo-rectal. El paciente presenta actualmente reaparición de adeno-patías a nivel abdominal, pendiente de iniciar tratamiento de 2ª línea.
CONCLUSIONES: Aunque la incidencia de neoplasias sinérgicas es in-frecuente, hay que pensar en ellas antes datos clínicos discordantes con la enfermedad hematológica. El manejo de estos tumores con-comitantes debe ser interdisciplinar y priorizar según la gravedad de cada tumor.
Tumores sólidos concomitantes con neoplasias hematológicas: caso a casoM.J. Llamas Poyato, D. Moreno Garrido, I. García Díez
Hospital Infanta Margarita, AGS Sur de Córdoba

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA178
INTRODUCCIÓN/FUNDAMENTO:
La bacteriemia es una de las principales causas de morbimortalidad en el paciente hematológico. A la inmunosupresión intrínseca de estas patologías se unen la agresividad de los regímenes quimioterápicos,el aumento de incidencia de gérmenes multirresistentes o la creciente prevalencia de catéteres centrales(CVC).Es fundamental para el clínico conocer la etiología y factores relacionados con este tipo de complica-ción con el fin de aplicar el tratamiento más adecuado
OBJETIVO:
Conocer la incidencia,factores de riesgo y características de las bacte-riemias en nuestra planta de hospitalización
PACIENTES, MATERIAL Y MÉTODO:
Estudio observacional retrospectivo;se analizan los hemocultivos(HC) positivos en nuestra planta entre Enero-2014 y Diciembre-2015.Se registran factores demográficos,patología de base y estado actual,tratamiento esteroideo,neutropenia(en caso afirmativo, número de neutrófilos y si duración >7 días),germen detectado,idoneidad del tratamiento empírico previo al antibiograma y mortalidad relacionada con el episodio.
RESULTADOS Y CONCLUSIONES:
Durante este periodo se obtuvieron 101 HC positivos. La media de edad fue 40 años,con predominio de varones(57 frente a 44 mujeres).Un 28.7% de los episodios corresponden a pacientes con leucemia mieloblástica,seguida de la leucemia linfoblástica(24.7%),linfoma no Hodgkin(13.8%) y mieloma múltiple(8.9%).
Un 22.7% de la muestra se compone de pacientes en periodo peritrasplante;por detrás se encontraban pacientes en proceso de diagnóstico/recién diagnosticados y aquellos con enfermedad en progresión(15.8% cada grupo).El 25.7% estaba recibiendo en ese momento o había recibido en la semana previa corticoides.
Un 54.9% presentaba menos de 1000 neutrófilos en la extracción (me-dia de 20 neutrófilos en este grupo).Más de la mitad de éstos(57,1%) presentaba neutropenia de más de 7 días.
En el 17.8% de los casos pudo documentarse infección por CVC,siendo un 72,2% por gérmenes Gram+.
58 episodios fueron causados por microorganismos Gram+,destacando Staphylococcus epidermidis(N=33),Enterococcus (N=8) y Staphylo-coccus haemolyticus (N=6).Entre los Gram–(39 episodios) destacan E. Coli(N=13),Klebsiella Pneumoniae(N=10,tres productoras de carba-penemasas)y Pseudomonas Aeruginosa(N=3).Cuatro episodios fueron producidos por Candida Parapsilosis.
En el 66.3% de los episodios la antibioterapia empírica instaurada en el pico febril fue la adecuada de acuerdo con el antibiograma posterior En aquellos en que no fue adecuada,más de 1/3 eran infecciones por Staphylococcus epidermidis.En los otros 2/3 destacan 2casos de KPC y un 52% de pacientes no neutropénicos.
13 de los pacientes fallecieron(15.8% del total de episodios),11 de los cuales presentaban infección por Gram- (destacan 5 casos por Klebsiella).Sin embargo,sólo en 6 de los casos la mortalidad puede ser directamente atribuida al proceso infeccioso,siendo la progresión de la enfermedad y las complicaciones post-TPH las otras principales causas de exitus.
– En nuestra serie más de la mitad de las bacteriemias se producen en pacientes con leucemia aguda,de acuerdo con la literatura.El periodo peri-TPH constituye el momento de enfermedad de mayor riesgo para el desarrollo de estas infecciones.
– La neutropenia prolongada y los CVC son reconocidos factores de riesgo confirmados en nuestra serie.
– El mayor número de bacteriemias se produce por agentes Gram+,pero son las producidas por Gram- las de mayor tasa de mortalidad.
– La mortalidad ligada a sepsis por Gram- es frecuentemente un even-to final en pacientes con neoplasia en progresión o complicaciones severas post-TPH no controladas.
Bacteriemias en pacientes hematológicos hospitalizadosS. Ordóñez Vahi, V. Verdugo Cabeza de Vaca, H.J. Campo Palacio, L. Domínguez Acosta, V. Rubio Sánchez, S. Garzón López
Hospital de Jerez de la Frontera
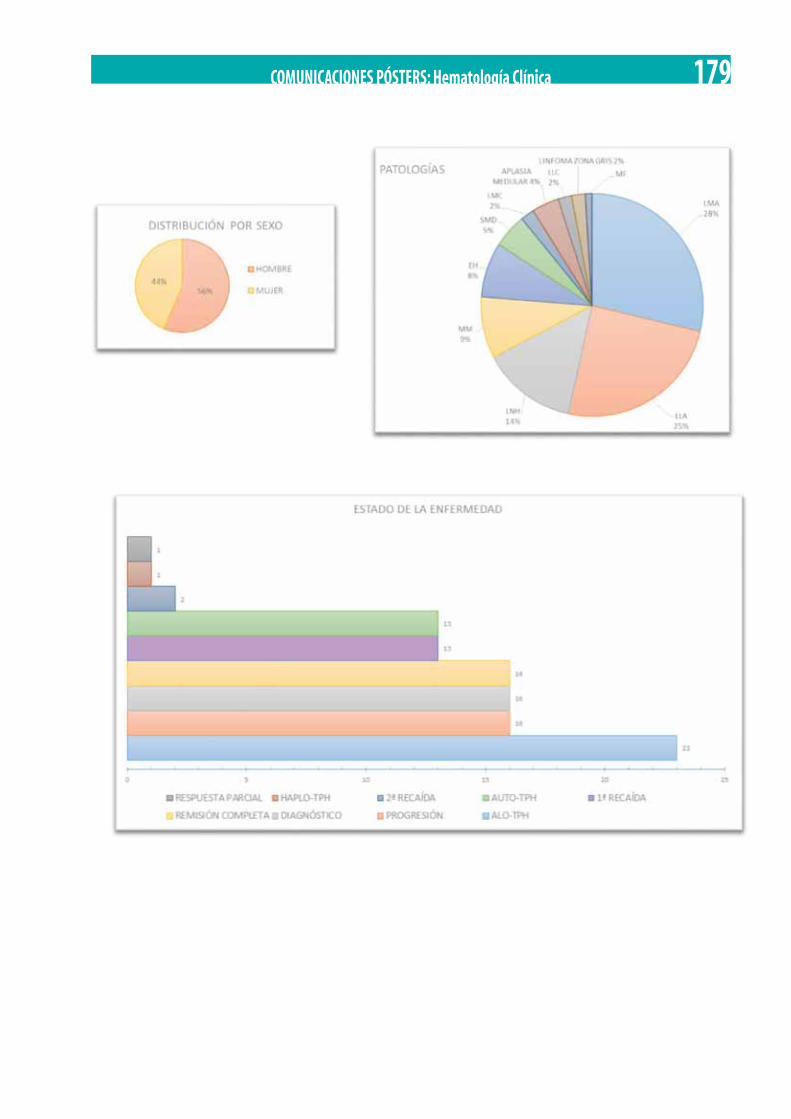
COMUNICACIONES PÓSTERS: Hematología Clínica 179
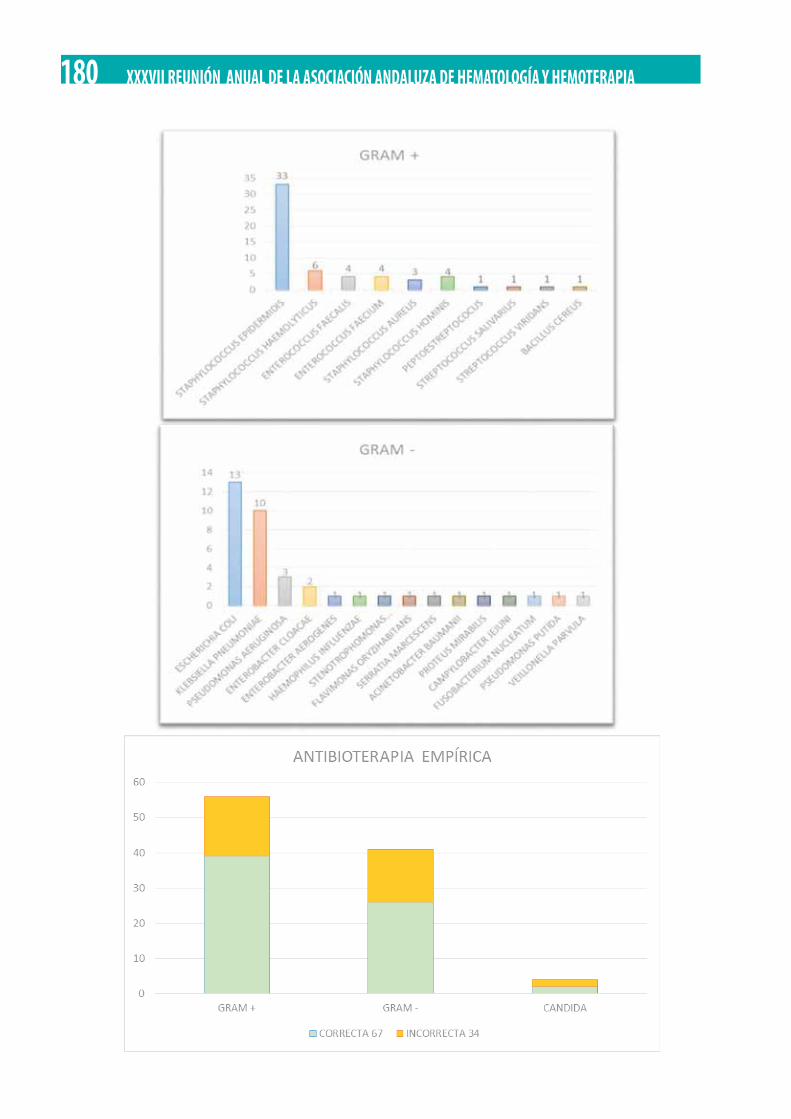
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA180
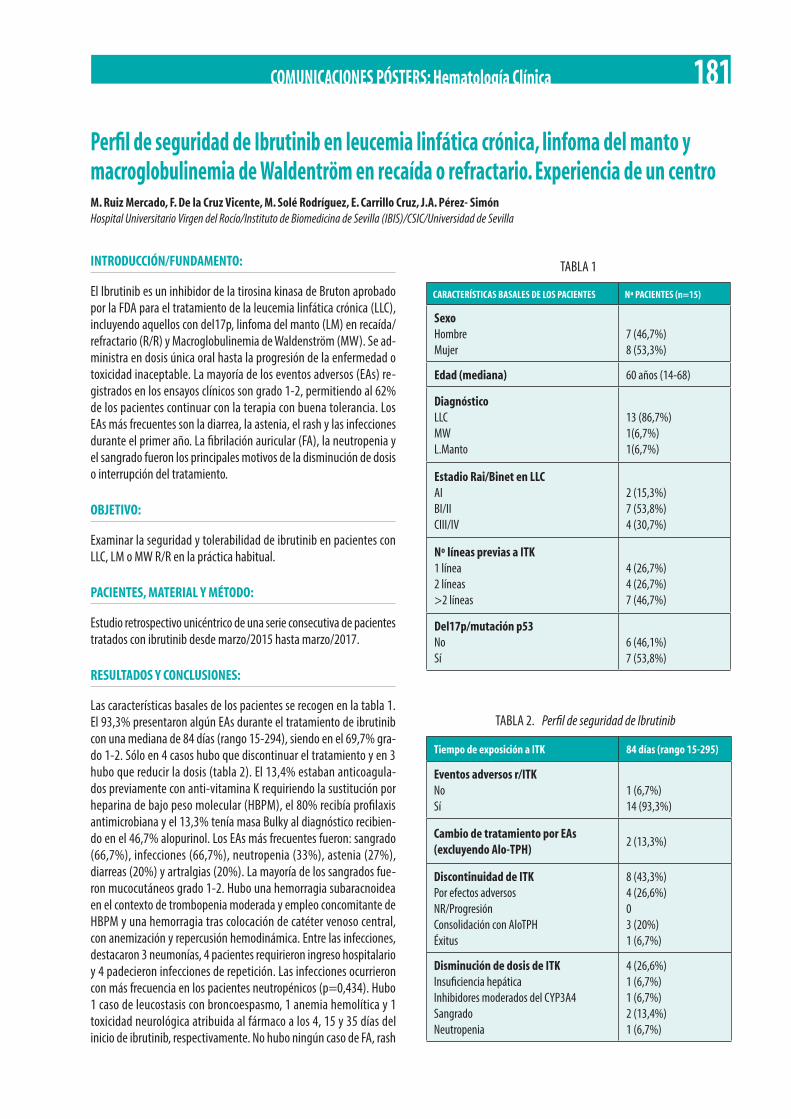
COMUNICACIONES PÓSTERS: Hematología Clínica 181
INTRODUCCIÓN/FUNDAMENTO:
El Ibrutinib es un inhibidor de la tirosina kinasa de Bruton aprobado por la FDA para el tratamiento de la leucemia linfática crónica (LLC), incluyendo aquellos con del17p, linfoma del manto (LM) en recaída/refractario (R/R) y Macroglobulinemia de Waldenström (MW). Se ad-ministra en dosis única oral hasta la progresión de la enfermedad o toxicidad inaceptable. La mayoría de los eventos adversos (EAs) re-gistrados en los ensayos clínicos son grado 1-2, permitiendo al 62% de los pacientes continuar con la terapia con buena tolerancia. Los EAs más frecuentes son la diarrea, la astenia, el rash y las infecciones durante el primer año. La fibrilación auricular (FA), la neutropenia y el sangrado fueron los principales motivos de la disminución de dosis o interrupción del tratamiento.
OBJETIVO:
Examinar la seguridad y tolerabilidad de ibrutinib en pacientes con LLC, LM o MW R/R en la práctica habitual.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo unicéntrico de una serie consecutiva de pacientes tratados con ibrutinib desde marzo/2015 hasta marzo/2017.
RESULTADOS Y CONCLUSIONES:
Las características basales de los pacientes se recogen en la tabla 1. El 93,3% presentaron algún EAs durante el tratamiento de ibrutinib con una mediana de 84 días (rango 15-294), siendo en el 69,7% gra-do 1-2. Sólo en 4 casos hubo que discontinuar el tratamiento y en 3 hubo que reducir la dosis (tabla 2). El 13,4% estaban anticoagula-dos previamente con anti-vitamina K requiriendo la sustitución por heparina de bajo peso molecular (HBPM), el 80% recibía profilaxis antimicrobiana y el 13,3% tenía masa Bulky al diagnóstico recibien-do en el 46,7% alopurinol. Los EAs más frecuentes fueron: sangrado (66,7%), infecciones (66,7%), neutropenia (33%), astenia (27%), diarreas (20%) y artralgias (20%). La mayoría de los sangrados fue-ron mucocutáneos grado 1-2. Hubo una hemorragia subaracnoidea en el contexto de trombopenia moderada y empleo concomitante de HBPM y una hemorragia tras colocación de catéter venoso central, con anemización y repercusión hemodinámica. Entre las infecciones, destacaron 3 neumonías, 4 pacientes requirieron ingreso hospitalario y 4 padecieron infecciones de repetición. Las infecciones ocurrieron con más frecuencia en los pacientes neutropénicos (p=0,434). Hubo 1 caso de leucostasis con broncoespasmo, 1 anemia hemolítica y 1 toxicidad neurológica atribuida al fármaco a los 4, 15 y 35 días del inicio de ibrutinib, respectivamente. No hubo ningún caso de FA, rash
Perfil de seguridad de Ibrutinib en leucemia linfática crónica, linfoma del manto y macroglobulinemia de Waldentröm en recaída o refractario. Experiencia de un centroM. Ruiz Mercado, F. De la Cruz Vicente, M. Solé Rodríguez, E. Carrillo Cruz, J.A. Pérez- Simón
Hospital Universitario Virgen del Rocío/Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla
TABLA 1
CARACTERÍSTICAS BASALES DE LOS PACIENTES Nª PACIENTES (n=15)
Sexo
Hombre
Mujer
7 (46,7%)
8 (53,3%)
Edad (mediana) 60 años (14-68)
Diagnóstico
LLC
MW
L.Manto
13 (86,7%)
1(6,7%)
1(6,7%)
Estadio Rai/Binet en LLC
AI
BI/II
CIII/IV
2 (15,3%)
7 (53,8%)
4 (30,7%)
Nº líneas previas a ITK
1 línea
2 líneas
>2 líneas
4 (26,7%)
4 (26,7%)
7 (46,7%)
Del17p/mutación p53
No
Sí
6 (46,1%)
7 (53,8%)
TABLA 2. Perfil de seguridad de Ibrutinib
Tiempo de exposición a ITK 84 días (rango 15-295)
Eventos adversos r/ITK
No
Sí
1 (6,7%)
14 (93,3%)
Cambio de tratamiento por EAs
(excluyendo Alo-TPH)2 (13,3%)
Discontinuidad de ITK
Por efectos adversos
NR/Progresión
Consolidación con AloTPH
Éxitus
8 (43,3%)
4 (26,6%)
0
3 (20%)
1 (6,7%)
Disminución de dosis de ITK
Insuficiencia hepática
Inhibidores moderados del CYP3A4
Sangrado
Neutropenia
4 (26,6%)
1 (6,7%)
1 (6,7%)
2 (13,4%)
1 (6,7%)

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA182
cutáneo o síndrome de lisis tumoral. En 8 de 12 pacientes con LLC se observó un aumento ≥ 50% de la cifra linfocitaria con respecto al valor basal al mes del inicio de ibrutinib. Todos los pacientes con LLC cuya respuesta fue evaluada a los 2 meses de tratamiento (7/13) y a los 6 meses (4/13), alcanzaron remisión parcial (RP). En 2 casos se realizó trasplante alogénico (Alo-TPH) a los 7 meses del inicio del tratamiento. El caso con MW, alcanzó muy buena RP pre-Alo-TPH a los 6 meses y el LM fue éxitus por progresión a las 2 semanas del inicio de ibrutinib.
CONCLUSIONES: Los eventos adversos más frecuentes durante el tra-tamiento con ibrutinib fueron las hemorragias y las infecciones, en su mayoría grado 1-2 y no requirieron la disminución de dosis ni la discontinuidad del fármaco, permitiendo su empleo a largo plazo en pacientes de edad avanzada con un perfil de toxicidad aceptable. En situación de recaída/refractariedad de LLC/MW, con múltiples líneas previas y citogenética adversa, ofreció alta tasa de respuesta y una terapia puente al Alo-TPH en pacientes seleccionados.

COMUNICACIONES PÓSTERS: Hematología Clínica 183
INTRODUCCIÓN/FUNDAMENTO:
La infiltración del sistema nervioso central (SNC) en la leucemia linfá-tica crónica (LLC) es una entidad rara, entorno al 0,8-2% y se presenta en más de la mitad de los casos en estadios precoces. La clínica neu-rológica es muy heterogénea e inespecífica. La citometría del líquido cefalorraquídeo (LCR) tiene un papel primordial en el diagnóstico mientras que las técnicas de neuroimagen son poco específicas. No hay un tratamiento estándar aunque la quimioinmunoterapia con-vencional que incluye fludarabina y, más recientemente, el ibrutinib, que atraviesa la barrera hemato-encefálica, ha demostrado resultados satisfactorios.
OBJETIVO:
Describir un caso de LLC con infiltración leptomeníngea en tratamiento con inhibidor de tirosin Kinasa de Bruton (BTK).
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 61 años con diagnóstico de LLC trisomía 12, ZAP70 22% en estadio A1 en 2004 que fue tratada con fludarabina-ciclofosfamida-mitoxantrone-rituximab por 5 ciclos alcanzando remisión completa con enfermedad mínima residual negativa. En mayo 2016 se detectó una linfocitosis de 31,2x10e9/L y 4 meses más tarde la paciente co-menzó con disminución de la agudeza visual del ojo derecho, amau-rosis fugax, cefalea de predominio frontal y terrores nocturnos. A la exploración física, no palpación de adenopatías ni organomegalias. La exploración neurológica era normal. Se realizó hemograma con leu-cocitos 41.42 x109/L, neutrófilos 2,1 x109/L linfocitos 30,6 x109/L, hemoglobina 125g/L y plaquetas 247x109/L y bioquímica con LDH normal. En el fondo de ojo: presencia de edema de papila bilateral con TAC de cráneo, sin hallazgos. La punción lumbar (PL) fue traumática
con LCR con contaminación periférica y estudios microbiológicos ne-gativos. Ante la alta sospecha de infiltración del SNC, se llevó a cabo resonancia magnética con contraste con evidencia de múltiples focos hiperintensos en la sustancia blanca subcortical y periventricular de ambos hemisferios, a nivel infratentorial en pedúnculo cerebeloso su-perior izquierdo y se repitió nueva PL que mostró un LCR claro con 26 cel/mm3 (85% células mononucleadas), glucosa 0,56 g/L, proteínas 0,32 g/L en ausencia de hematíes. En citología de LCR, se observaron linfocitos de aspecto patológico en un 71,6% y en la citometría del LCR un 43,2% de linfocitos B patológicos CD19+CD5+CD45+CD20-CD79+/-CD43+KAPPA+d-, confirmando la presencia de infiltración leptomeningea por su LLC. Se realizó estadiaje de la enfermedad con TAC de 4 áreas: ganglios cervicales en los límites superiores de la nor-malidad y agrupación de ganglios de pequeña entidad dispersos por raíz mesentérica y periaórticos, estableciendo un estadio A1. En el estudio medular con FISH se detectó la trisomía 12 en un 57%. Por ser recaída tardía, se decidió retratamiento con fludarabina-ciclofos-famida-rituximab recibiendo 1 ciclo en diciembre 2016 y cambian-do a ibrutinib al mes ante la identificación de una mutación TP53, p.Gly245Ser, c.733G>A, en el 7% del ADN analizado, y una mutación NOTCH1 c.7544_7545delICT. Desde el inicio del tratamiento, se apreció una marcada mejoría progresiva de la clínica neurológica, con reduc-ción paulatina del porcentaje de linfocitos monoclonales en el LCR, traduciendo respuesta a ibrutinib.
RESULTADOS Y CONCLUSIONES:
La infiltración sintomática del SNC por la LLC es muy infrecuente, se presenta usualmente en estadios precoces y requiere un alto nivel de sospecha ante pruebas de imagen inespecíficas. El ibrutinib resulta efectivo en el tratamiento de la afectación neurológica en LLC con mutación del p53, evitando la toxicidad de la quimioterapia sistémica.
Papel de los inhibidores de Tirosin Kinasa de Bruton en la excepcional leucemia linfática crónica con infiltración leptomeníngeaM. Ruiz Mercado, E. Carrillo Cruz, M. Solé Rodríguez, F. De la Cruz Vicente, J.A. Pérez- Simón
Hospital Universitario Virgen del Rocío/Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA184
INTRODUCCIÓN/FUNDAMENTO:
Los regímenes de tratamiento (tto) y los resultados tras inducción clá-sica en leucemia mieloide aguda (LMA) pueden diferir según la edad. El manejo de pacientes (ptes) mayores de 60 años es más complica-do al ser propensos a tener comorbilidades. Además, la enfermedad tiende a ser más resistente en subtipos de LMA desfavorables y las res-puestas son peores que en ptes más jóvenes que son tratados a dosis plenas.Por ello a raíz de diferentes estudios que han demostrado que el uso de otros fármacos alejados de los esquemas quimioterápicos de inducción clásicos aumentan la supervivencia global (SG), la tolerancia y la adhesión al tto (Drombret H. et all, Pleyer L. et all, etc..),es aprobada por la Organización Mundial de la Salud ( OMS ) la nueva indicación de 5-azacitidina ( 5- AZA) en ptes ancianos con comorbilidades por su menor toxicidad y buenos resultados en cuanto a respuesta, calidad de vida y SG.
OBJETIVO:
Impacto en cuanto a calidad de vida, necesidades transfusionales y respuesta tto en pte con LMA con cambios displásicos relacionados en tto con 5-AZA.
PACIENTES, MATERIAL Y MÉTODO:
Varón de 77 años, jubilado, independiente para actividades diarias, funciones superiores conservadas y vida activa. Como antecedentes presenta cólicos nefríticos de repetición de etiología úrica y escrófula paracervical derecha por M. tuberculosis (TBC) en diciembre/2015 en tto con tuberculostáticos con buena respuesta y seguimiento por Infec-cioso. En junio /2016 ingresa en planta de esta unidad por bicitopenia por hemoglobina ( hb) y plaquetas), pérdida de 9 kg en 6 meses y astenia leve-moderada.Por lo que se suspende tto de TBC.
Hemograma al ingreso: Hb 5,2 mg/dl, volumen corpuscular mé-dio (VCM) : 114 fl, , plaquetas 56x10e9/L , leucocitos 6080x10e9 8 neutrófilos 1450x10e9/L, monócitos 2550x10e9/L) . B12 y ferritina elevadas.
Bioquímica:LDH 379.
Mielograma: hipercelular +. Displasias en 3 líneas con 24 % de blastos con doble estirpe entre monoblásticos y mieloblásticos . Frecuentes bastones de Auer y aumento de monocitos aberrantes. Sideroblastos patológicos 44%. Mieloperoxidasas positivas 80%, ANAE 20%.
Cariotipo 46,XY (20).
Biología molecular no disponible.
Se diagnostica de LMA con cambios displásicos relacionados (riesgo no clasificado.Se inicia tto ambulatoriamente con 5- AZA esquema 5-2-2 a dosis plenas (75 mg/m2) el 25/julio/2016,permitiendo continuar tto con tuberculostáticos al no interferir en metabolización hepática.Se han administrado 8 ciclos completos con buena tolerancia, sin toxi-cidades importantes.
Antes del inicio del tto,fue transfundido por anemia sintomática 6 con-centrados de hematíes (CH). Se evalúan las necesidades de transfusión del pte cada 8 semanas siendo estos de 7 CH en las 8 primeras y de 4 CH en las últimas 8 semanas. En cuanto a revisiones, al inicio del tto fueron semanales para actualmente ser cada 15 días. Se evalúa respuesta al tto con aspirados de médula ósea tras tercer y sexto ciclo con persistencia de displasia multilínea, disminución de blastos a un 18 y 8 % respectivamente, citogenética sin crecimiento de metafases y enfermedad mínima residual positiva. No ha requerido ingresos hospitalarios y mantiene una buena calidad de vida.
RESULTADOS Y CONCLUSIONES:
Tras ocho meses desde el diagnóstico, se han disminuido las transfu-siones en un 27,27 % con respecto al periodo pre e inicio del tto, se espacian las revisiones y se han evitado ingresos con la disminución de costes derivados de estas causas y manteniendo una buena calidad de vida con respecto a su situación pre-hemopatía a pesar de no tener remisión completa.
5-Azacitidina como nueva indicación de tratamiento en pacientes con leucemia mieloide aguda (LMA): a propósito de un casoMaría Eugenia Molina Rodríguez, Virgilio Calama, María Vahí, Carmen Couto, Juan Carlos Martin, Eduardo Ríos
Hospital Universitario Nuestra Señora de Valme

COMUNICACIONES PÓSTERS: Hematología Clínica 185
INTRODUCCIÓN/FUNDAMENTO:
La histiocitosis de células de Langerhans (LCH) es una neoplasia mieloi-de poco frecuente caracterizada por lesiones óseas osteolíticas únicas o múltiples que en la anatomía patológica, muestran una infiltración por histiocitos. Estos histiocitos, junto con linfocitos, macrófagos y eosinófilos pueden infiltrar diversos órganos (especialmente piel, ganglios linfáticos, pulmones, timo, hígado, bazo, médula ósea o sistema nervioso central). Según la Histiocyte Society el tratamiento de 1ª línea está basado en regímenes con prednisona, vinblastina y citarabina; por el contrario, no existe un tratamiento estandarizado para refractariedad ó recaída.
OBJETIVO:
Poner de manifiesto la posibilidad de alcanzar remisión completa (RC) en estos pacientes, con esquemas agresivos tipo MACOP-B, y posterior Alotrasplante de progenitores hematopoyéticos (AloTPH).
PACIENTES, MATERIAL Y MÉTODO:
Presentamos el caso de una mujer de 27 años con LCH con afectación ósea craneal diagnosticada en 2005, a la edad de 15 años. Inicia pro-tocolo LCH-III de la SIOP con posterior mantenimiento con Vinblastina + Prednisona durante 1 año. Alcanza RC y es alta al cumplir 18 años. Seis años más tarde recae con adenopatías laterocervicales izquierdas, febrícula y síndrome constitucional. Inicia tratamiento con Vinblastina semanal + Prednisona, con aparición de nueva lesión parietal intra-
tratamiento, sin volver a alcanzar RC tras diversas líneas. Tratamientos recibidos: Cladribina x 4, CHOP x 6, autotrasplante de progenitores hematopoyéticos (AutoTPH) y ESHAP x 4. Por último, inicia tratamiento con MTX-AraC a altas dosis con profilaxis del SNC con TIT; durante el cual, se constata aparición de masa presacra, que obliga a suspender tratamiento por sospecha de absceso; pero con histología compatible con LCH tras biopsia. Tras resección de la misma, se trata con MACOP-B con doxorrubicina liposomal, recibiendo 9 semanas de dicho esque-ma, sin poder completarlo por disestesia palmoplantar 2ª a toxicidad por Bleomicina. Realiza el 03/02/2017 AloTPH de sangre periférica de donante familiar HLA idéntica (hermana) previo acondicionamiento TBF-2 y profilaxis de la EICR con Ciclofosfamida y Ciclosporina. En PET-TAC preTPH, únicamente persiste la captación inflamatoria perirrectal izquierda, con menor extensión e intensidad que en el estudio previo (Aprox. 7 mm SUVmax=2.6, previo: 4.3). Status preTPH: RP (> 3ª). Actualmente, la paciente se encuentra asintomática, a la espera de reevaluación de la enfermedad a los 3 meses del TPH.
RESULTADOS Y CONCLUSIONES:
1. En LCH el régimen MACOP-B ha sido demostrado eficaz en 1ª línea con tasas de respuesta de hasta 100% y RC del 73% (Derenzini et al. BMC Cancer -2015- 15:879).
2. Pacientes con LCH sistémica refractaria ó en recaída pueden benefi-ciarse e incluso alcanzar RC con esquemas agresivos tipo MACOP-B; y estos esquemas podrían estandarizarse para 2ª línea e incluso servir como “puente” al AloTPH.
Histiocitosis sistémica refractaria. Tratamiento con Macob-B como puente a trasplante alogénico. A propósito de un casoI. Sánchez Bazán, O. Benítez Hidalgo, M. Espeso de Haro, A.I. Heineger Mazo
Hospital Regional de Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA186
INTRODUCCIÓN/FUNDAMENTO:
La trombocitopenia inmune primaria (TIP), es la causa más frecuente de trombocitopenia en la infancia. Se caracteriza por disminución del numero de plaquetas, en ausencia de un trastorno sistémico asociado identificable y normalidad de la médula ósea, presentando una recu-peración espontánea del cuadro entre el 70-90% de los casos.
El lupus eritematoso sistémico (LES), es una enfermedad crónica, au-toinmune y multisistémica. El LES incompleto es aquel que se presenta en pacientes que no reúnen criterios de clasificación para LES, junto con Anticuerpos Antinucleares (ANA) positivos. A diferencia del LES, presenta menor número de manifestaciones renales y del sistema nervioso central.
Los factores de riesgo para desarrollar LES incluyen: inicio tardío, sexo femenino,TIP crónica y títulos elevados de ANA. Estos niños pueden presentar anticuerpos anti-Sm, anti-Lo, anti-Ro o anti-RNP positivos, por lo que deben tener un seguimiento cercano.
PACIENTES, MATERIAL Y MÉTODO:
Paciente mujer de 10 años, sin antecedentes personales de interés y con antecedente familiar de LES. Diagnosticada en Febrero de 2015 de TIP. Inició tratamiento con prednisona con normalización de la cifra de plaquetas. Al diagnóstico, se solicitó analítica con autoinmunidad donde destacaban: ANA positivos. Resto de anticuerpos negativos. Se derivó a consultas de pediatría para descartar enfermedad sistémica de base. Tras retirada del tratamiento, nuevo ingreso en Julio 2015 con buena evolución.
El abril de 2016, comienza cefalea y cervicalgia consultando en ur-gencias. Precisa varios ingresos en pediatría de forma intermitente realizándose RMN craneo-cervical que fué normal y presentando mejoría parcial del cuadro, por lo que es dada de alta. Tras persistir la sintomatología, se decide de nuevo ingreso para completar estudio. Durante el mismo, se realiza punción lumbar con presión elevada y
bioquímica normal, fondo de ojo con papiledema bilateral, electromio-grama y electroneurograma normales y cifra de plaquetas dentro de la normalidad. Ante los hallazgos y el empeoramiento clínico (cefalea, vómitos y debilidad muscular progresiva), se decide traslado a Hospital de referencia por hipertensión intracraneal (HIC).
Allí se realiza TAC de craneo con contraste, AngioTAC de troncos su-praaórticos y arteriografía, cuyo resultado fue: Trombosis venosa masiva de senos intracraneales. Infartos venosos con transformación hemorrágica.
Empeoramiento clínico a las 48 horas del ingreso en UCI-P, precisando sueroterapia intensiva y drogas vasoactivas para su estabilización. Se comienza tratamiento con heparina sódica en perfusión, bajo control estricto de la cifra de plaquetas. Ante la alta sospecha de LES, inician Rituximab, corticoides, inmunoglobulinas y plasmaféresis. Precisó dos intervenciónes quirúrgicas para colocación de un By-pass venoso cerebral y por trombosis el mismo.
A las 72 horas de la 2º intervención, empeoramiento del estado ge-neral, PIC elevadas y shock refractario al tratamiento, sufriendo final-mente parada cardiorrespiratoria y éxitus.
RESULTADOS Y CONCLUSIONES:
La prueba de ANA deben solicitarse siempre en niños con diagnós-tico de trombocitopenia.
ANA positivos se consideran de riesgo para el desarrollo posterior de LES, por lo que ante dicho hallazgo, deben solicitarse también anti-Sm, anti-Lo, anti-Ro y anti-RNP para completar el estudio de enfermedad sistémica.
Debe realizarse un seguimiento cuidadoso de los autoanticuerpos, para llegar a un diagnóstico y tratamiento temprano de enfermedad sistémica, evitando de esta forma daños importantes y mejorando el pronóstico.
Lupus eritematoso sistémico incompleto en niña de 10 años con diagnóstico de trombocitopenia inmune primaria. A propósito de un casoM.A. Ruiz Cobo, K. Gómez Correcha, R. Cabra, R. Zapata Bautista, J.N. Rodríguez Rodríguez
Hospital Juan Ramón Jimenez

COMUNICACIONES PÓSTERS: Hematología Clínica 187
INTRODUCCIÓN/FUNDAMENTO:
La Histiocitosis de células de Langerhans (LCH) es una proliferación clonal mieloide que expresa CD1a, langerina, proteína S100 y análisis ultraestructural con presencia de gránulos de Birbeck. Como sinónimos de esta enfermedad tenemos la histiocitosis X, el granuloma eosinofí-lico (si la lesión es solitaria), la enfermedad de Hand-Schüller-Cristian (si múltiples lesiones), enfermedad de Letterer-Siwe (si afectación visceral o diseminada).
La incidencia es aproximadamente de 5 casos por millón de pobla-ción y año, ocurriendo en la infancia. Existe una predilección por los hombres 3.7:1 y es más frecuente en blancos del norte de Europa y rara en negros.
OBJETIVO:
Exponer nuestra experiencia clínica sobre este tipo de patología y los resultados obtenidos con los tratamientos existentes hasta la fecha. Revisamos en nuestra serie el comportamiento de esta entidad a lo largo de 24 años.
PACIENTES, MATERIAL Y MÉTODO:
7 pacientes diagnosticados en nuestro centro entre 1993 y 2017. Se han analizado factores pronósticos, epidemiológicos y tratamientos administrados así como tasas de respuesta y supervivencias.
RESULTADOS Y CONCLUSIONES:
Resultados
La mediana de edad es de 45 años (14 – 68), discretamente más fre-cuente en hombres (57.1%; 4/3). Histología: Granuloma eosinofílico en 3 (42.8%), Histiocitosis sistémica en 3 (42.8%) e Histiocitosis si-nusal reactiva en 1 (14.2%). Tratamientos recibidos: Quimioterapia en 3 (Etopósido en 1 y protocolo LCH-III en 2)(42.8%), Cirugía en 1 (14.2%), Cirugía + radioterapia en 1 (14.2%), Radioterapia local clavicular en 1 (14.2%) y sin tratamiento por desaparición espontá-nea en 1 (14.2%). La mediana de seguimiento ha sido de 164 meses (36 - 282). La Supervivencia Libre de Enfermedad (SLE) ha sido de 64 meses (20 - 184). Todos los pacientes entran en remisión completa (RC) tras la primera línea, sólo 2 recaen (28.5%) pero se rescatan con tratamientos posteriores. Actualmente todos están vivos, 6 persisten en RC (85.7%) y 1 (14.3%) pendiente de evaluación tras Alotrasplante de progenitores hematopoyéticos (AloTPH).
Conclusiones
1. A pesar de ser una enfermedad rara observamos semejanzas en nuestra serie con respecto a literatura en cuanto a incidencia y sexo, no así en edad (por ver pacientes a partir de 15 años).
2. A pesar de ser una enfermedad rara presenta buena tasa de res-puesta global tras primera línea, buena RC y ningún fallecimiento ni por la enfermedad ni por el tratamiento.
3. Los distintos tratamientos administrados, desde observación hasta AloTPH, han sido consecuencia de la diversidad de afectación en estu-dio inicial de extensión y de la refractariedad en un caso.
Histiciocitosis de células de Langerhans. Revisión de la experiencia del Hospital Regional Universitario de MálagaA. González Fernandez (1), F. Cabrera Ruiz (1), M. Espeso de Haro (2)
(1) Hospital Virgen de la Victoria. , (2) Hospital Regional Universitario de Málaga
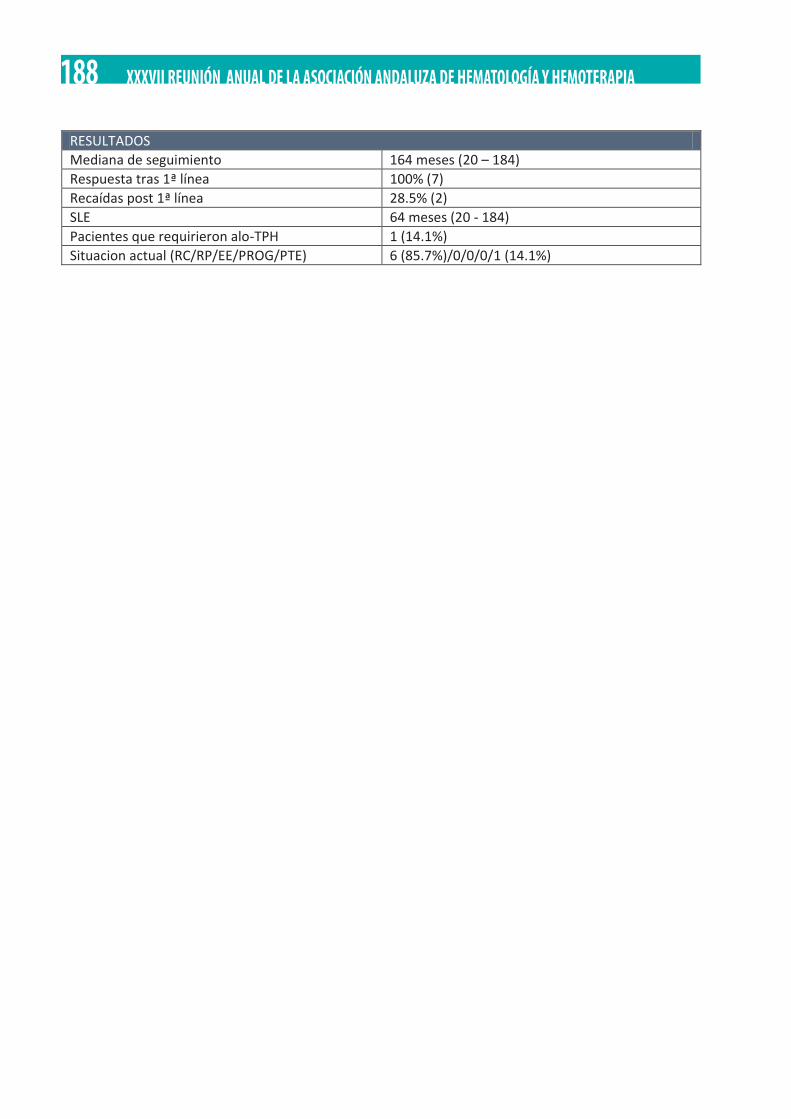
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA188
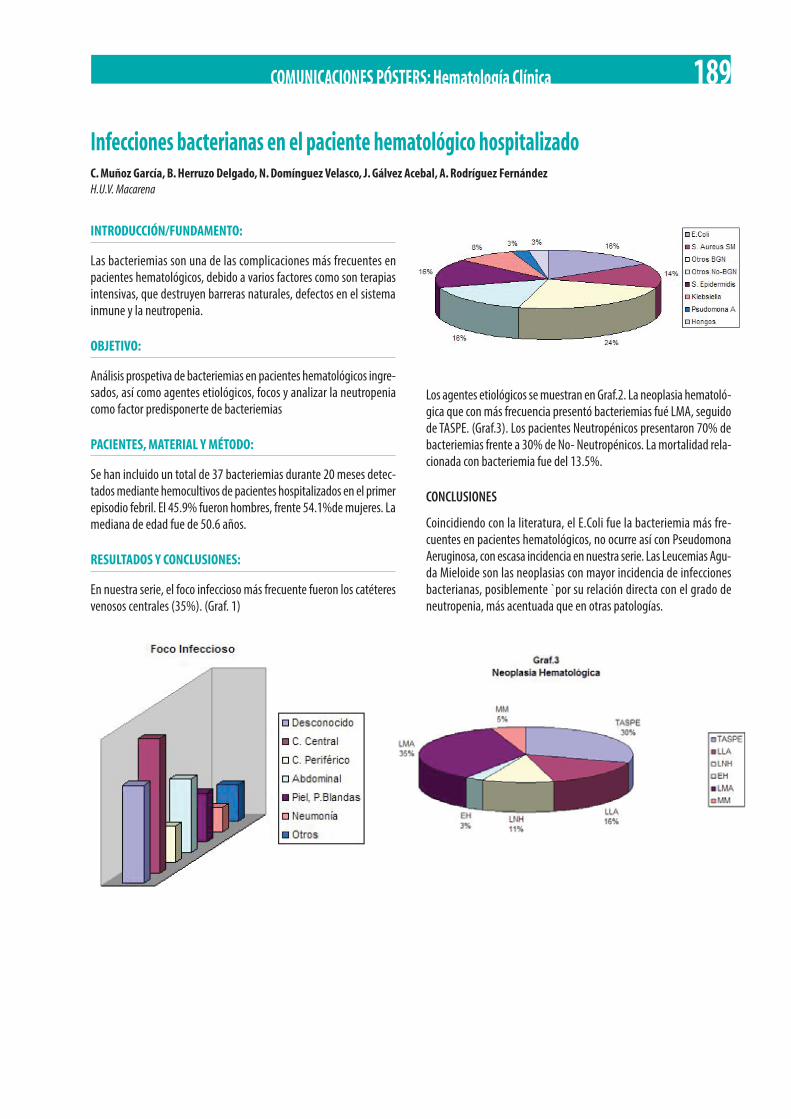
COMUNICACIONES PÓSTERS: Hematología Clínica 189
INTRODUCCIÓN/FUNDAMENTO:
Las bacteriemias son una de las complicaciones más frecuentes en pacientes hematológicos, debido a varios factores como son terapias intensivas, que destruyen barreras naturales, defectos en el sistema inmune y la neutropenia.
OBJETIVO:
Análisis prospetiva de bacteriemias en pacientes hematológicos ingre-sados, así como agentes etiológicos, focos y analizar la neutropenia como factor predisponerte de bacteriemias
PACIENTES, MATERIAL Y MÉTODO:
Se han incluido un total de 37 bacteriemias durante 20 meses detec-tados mediante hemocultivos de pacientes hospitalizados en el primer episodio febril. El 45.9% fueron hombres, frente 54.1%de mujeres. La mediana de edad fue de 50.6 años.
RESULTADOS Y CONCLUSIONES:
En nuestra serie, el foco infeccioso más frecuente fueron los catéteres venosos centrales (35%). (Graf. 1)
Infecciones bacterianas en el paciente hematológico hospitalizadoC. Muñoz García, B. Herruzo Delgado, N. Domínguez Velasco, J. Gálvez Acebal, A. Rodríguez Fernández
H.U.V. Macarena
Los agentes etiológicos se muestran en Graf.2. La neoplasia hematoló-gica que con más frecuencia presentó bacteriemias fué LMA, seguido de TASPE. (Graf.3). Los pacientes Neutropénicos presentaron 70% de bacteriemias frente a 30% de No- Neutropénicos. La mortalidad rela-cionada con bacteriemia fue del 13.5%.
CONCLUSIONES
Coincidiendo con la literatura, el E.Coli fue la bacteriemia más fre-cuentes en pacientes hematológicos, no ocurre así con Pseudomona Aeruginosa, con escasa incidencia en nuestra serie. Las Leucemias Agu-da Mieloide son las neoplasias con mayor incidencia de infecciones bacterianas, posiblemente `por su relación directa con el grado de neutropenia, más acentuada que en otras patologías.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA190
INTRODUCCIÓN/FUNDAMENTO:
El Mieloma Múltiple (MM) es una neoplasia caracterizada por infil-tración de células plasmáticas en médula ósea. Edad media 70 años.
Se han producido numerosos avances en el tratamiento del MM que han aumentado el tiempo libre de enfermedad, aunque continúa siendo una enfermedad incurable. Estas terapias seguidas de TASPE, aumentan la supervivencia de 5-36 meses en pacientes de alto riesgo.
La realización de un 2º TASPE en paciente en recaída es controvertido. Numerosos estudios han concluido estar indicado en pacientes me-nores de 65 años sin comorbilidades y sin complicaciones relevantes en el 1º TASPE, siendo la mortalidad tras en mismo menor de un 5% y alcanzando el 65% de los pacientes un tiempo libre de enfermedad entre 12- 32 meses. El factor pronóstico más importante, es el tiempo libre de enfermedad tras el 1º TASPE, demostrando eficacia cuando es mayor a 18 meses. Respecto a toxicidad y efectos secundarios com-parando 1º y 2º TASPE, no han encontrado diferencias significativas.
OBJETIVO:
Descripción de nuestra experiencia en pacientes sometidos a un se-gundo auto-TPH como forma de tratamiento del mieloma múltiple en recaída.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo retrospectivo de los pacientes con MM en recaída que recibieron un 2º TASPE en nuestro centro (Tabla 1).
Se registraron 4 pacientes con una media de edad de 58,5 años.
Respecto al tratamiento previo al 1º TASPE, todos de los casos recibie-ron esquema VAD, de los cuales 2 de 4 precisaron un nuevo esquema de quimioterapia intensiva para conseguir respuesta.
2 de 4 pacientes, consiguieron muy buena respuesta parcial, 1 de 4 una respuesta completa y 1 de 4 estabilidad de la enfermedad. Los pacien-
tes se sometieron a un TASPE sin complicaciones relevantes durante el mismo. Los 4 pacientes recibieron diferentes protocolos de tratamiento cuando se objetivó progresión de la enfermedad tras el primer TASPE: Dos líneas en 2 pacientes y tres líneas en otros 2. En todos ellos, la lenalidomida formó parte de alguna de dichas líneas de tratamiento.
Todos los pacientes tuvieron un tiempo libre de enfermedad entre los dos TASPE, mayor de 18 meses y no padecían comorbilidades impor-tantes (Karnosky 90%).
Se realizaron un 2º TASPE sin incidencias reseñables, recibiendo todos ellos acondicionamiento con Melfarán 200. Solamente 1 de los pa-cientes recibió tratamiento de consolidación y mantenimiento desde el postrasplante inmediato. Los 4 pacientes sufrieron progresión de la enfermedad a los 10 meses de media, comenzado de nuevo con tratamiento quimioterapico.
Respecto a la evolución, 3 de 4 pacientes fallecieron antes de los 25 meses de la realización del 2º TASPE y el 100% de ellos por una infec-ción respiratoria. En la actualidad, 1 de los pacientes, (tratamiento en el postrasplante inmediato), se encuentra en situación de enfermedad estable.
RESULTADOS Y CONCLUSIONES:
La toxicidad y efectos secundarios tras en 2º TASPE no fueron supe-riores a los del 1º TASPE, siendo los datos de mortalidad descritos en la literatura equiparables a los de un primer TASPE.
Solamente uno de los pacientes alcanzó un tiempo libre de enfer-medad tras el 2º TASPE mayor de 12 meses, si bien, la supervivencia tras es procedimiento en los fallecidos fue aproximadamente de dos años.
Cabe destacar que ninguno de los fallecidos recibió tratamiento in-mediato postrasplante, lo que demuestra la importancia de realizar un tratamiento completo con inducción, consolidación y manteni-miento en todos los casos.
Segundo trasplante autólogo de progenitores hematopoyéticos en pacientes con mieloma múltiple en recaída. Experiencia en un hospital de 2º nivelM.A. Ruiz Cobo, A.J. Palma Vallellano, R. Zapata Bautista, B. Díaz Roldán, J.N. Rodríguez Rodríguez
Hospital Juan Ramón Jiménez

COMUNICACIONES PÓSTERS: Hematología Clínica 191

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA192
INTRODUCCIÓN/FUNDAMENTO:
El linfoma anaplásico de células grandes asociado a prótesis mamaria (LACG-APM) es un linfoma T muy infrecuente (0.1-0.3 por 100,000 mujeres con implantes) que afecta a la cápsula (1/3 de los casos) o deriva de la formación de un seroma tardío periprotésico (2/3 de los casos). Uno de cada 8 presenta linfadenopatías. La mayoría tienen un pronóstico excelente únicamente con la retirada de la prótesis y capsulectomía. Sin embargo, la presencia de masas o adenopatías condiciona un curso más agresivo (SG 52.5% a 2 años) que justificaría el empleo de quimioterapia asociada a la cirugía protésica. La retirada del implante contralateral es considerado por algunos autores (4,6 % linfoma incidental en la mama sana).
OBJETIVO:
Describir dos casos de LACG asociado a prótesis mamaria diagnosti-cados en nuestro centro desde enero de 2007 hasta marzo de 2017.
PACIENTES, MATERIAL Y MÉTODO:
Caso 1: Mujer de 50 años con prótesis mamaria bilateral tras mastec-tomía por carcinoma ductal infiltrante multifocal grado II. A los 5 años de la cirugía, comenzó con dolor y endurecimiento progresivo de la mama izquierda, realizándose ecografía que reveló rotura protésica y ganglios axilares. La biopsia adenopática reveló un LACG-APM CD30+, ALK negativo. Para el estadiaje inicial, se realizó un PET con presencia de adenopatías hipermetabólicas supradiafragmáticas SUVmax 3,4 y actividad 18F-FDG en la cápsula fibrosada que rodea el implante ma-mario izquierdo (SUVMAX:2,8). La histología y la inmunohistoquímica
de la cápsula confirmó el diagnóstico de LACG-APM. Se trató con ritu-ximab, ciclofosfamida, vincristina, etopósido y prednisona (R-COEP) por 4 ciclos obteniendo respuesta completa en el PET intermedio con actividad 18F-FDG inflamatoria tras extirpación de la prótesis mamaria y capsulectomía (SUVmax 2,1). Se completaron 6 ciclos de tratamiento alcanzando remisión completa en la evaluación final, sin evidencia de recidiva a 2 años y 8 meses.
Caso 2: Mujer de 55 años con prótesis mamaria bilateral tras mastec-tomía con vaciamiento ganglionar por carcinoma ductal infiltrante en 2005. En junio de 2016, comenzó con progresivo aumento del tamaño de la mama izquierda, endurecimiento y dolor. Se realizó resonancia magnética mamaria que reveló gran colección líquida periprotésica izquierda de 8 x 3 cm compatible con seroma crónico. Se intervino para retirada de prótesis y capsulectomía con diagnóstico anatomo-patológico de LACG-APM CD30+ ALK negativo que afecta al líquido del seroma periprotésico e invade la cápsula. En PET realizado de estadiaje no se objetivó ningún depósito patológico del trazador en relación con su linfoma y la biopsia y citometría medular descartaron la infiltración. Se llevó a cabo en segunda instancia, retirada profiláctica de la pró-tesis mamaria contralateral y se optó por la abstinencia terapéutica, manteniendo respuesta completa.
RESULTADOS Y CONCLUSIONES:
El pronóstico del LACG-APM limitado al seroma y/o la cápsula peripro-tésica es excelente tras la retirada de la prótesis mamaria y la capsu-lectomía total, sin empleo de quimioterapia adicional. El LACG-APM con afectación adenopática requiere adicionalmente de quimioterapia sistémica con esquemas tipo CHOP para alcanzar respuestas duraderas.
Tratamiento del linfoma anaplásico de células grandes B asociado a prótesis mamariaM. Ruiz Mercado, M. Solé Rodríguez, N. Alkadi Fernández, E. Carrillo Cruz, F. De la Cruz Vicente, J.A. Pérez Simón
Hospital Universitario Virgen del Rocío/Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla

COMUNICACIONES PÓSTERS: Hematología Clínica 193
INTRODUCCIÓN/FUNDAMENTO:
El linfoma primario del sistema nervioso central (LPSNC) es una pato-logía infrecuente pero grave y de difícil diagnóstico por su localización. Algunos estudios sugieren que el tiempo de retraso entre el inicio de los síntomas y la confi rmación diagnóstica podría infl uir en el pronós-tico de estos pacientes y que la administración de corticoides podría interferir en la obtención e interpretación de las muestras.
OBJETIVO:
Evaluar el manejo diagnóstico y terapéutico de los LPSNC e identifi car posibles áreas de mejora y factores pronósticos.
PACIENTES, MATERIAL Y MÉTODO:
Los casos se identifi caron mediante una búsqueda retrospectiva en la base de anatomía patológica de nuestro centro de pacientes diag-nosticados de LPSNC entre los años 2005 y 2014. Posteriormente se han revisado las historias clínicas y se han excluido los pacientes con afectación sistémica y los secundarios a inmunosupresión.
RESULTADOS Y CONCLUSIONES:
Se identifi caron 7 pacientes con estos criterios cuyas características basales se resumen en la Tabla 1. En seis pacientes se realizó el diag-
nóstico mediante biopsia y en uno de ellos mediante resección com-pleta del tumor. El tiempo transcurrido de media entre la aparición de los primeros síntomas y la confi rmación diagnóstica ha sido de 78,2 días (rango 29-195). Sólo uno de los pacientes tuvo que realizarse una segunda biopsia para confi rmar el diagnóstico a pesar de que cuatro de ellos (57,1%) recibieron esteroides antes de la cirugía. Sólo un paciente tuvo una complicación postoperatoria y no fue infeccio-sa (neumoencéfalo). Salvo un paciente que se consideró paliativo al diagnóstico, todos recibieron quimioterapia de primera línea basada en metotrexato y citarabina. Tres de ellos progresaron durante el tra-tamiento, uno alcanzó remisión completa (RC) y dos respuesta parcial. El paciente que alcanzó RC se consolidó posteriormente con trasplante autólogo de progenitores hematopoyéticos y continúa vivo con un se-guimiento actual de 5 años y 3 meses. Este paciente es uno de los que se diagnosticó de forma más tardía desde el inicio de los síntomas (132 días) pero también era el más joven de la serie (19 años). Del resto de pacientes tres fallecieron como consecuencia del linfoma y en otros tres se perdió el seguimiento.
Conclusiones: El LPSNC en nuestro medio se diagnostica de forma tardía aunque no podemos establecer con nuestros datos si esto tiene o no impacto en la supervivencia de los pacientes. En nuestra serie el uso de esteroides de forma prequirúrgica no ha interferido en la interpretación de las muestras obtenidas por biopsia ni ha habido complicaciones infecciosas postoperatorias.
Manejo diagnóstico y terapéutico del linfoma primario del sistema nervioso centralM. Solé Rodríguez (1), M. Ruiz Mercado (2), F. De La Cruz Vicente (2), E. Carrillo Cruz (2), J.J. Borrero Martín (2), J.A. Pérez Simón (2)
Complejo hospitalario de Huelva-Hospital Juan Ramón Jiménez, (2) Hospital Universitario Virgen del Rocío, Sevilla
TABLA 1. Características basales de los pacientes

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA194
INTRODUCCIÓN/FUNDAMENTO:
La necrosis de médula ósea (NMO) consiste en la destrucción del estro-ma medular, incluyendo el parénquima hematopoyético. La etiología es diversa, aunque se observa un proceso neoplásico subyacente en el 90% de los casos, destacando entre los más frecuentes las neoplasias hematológicas (60%). La incidencia de la NMO en el aspirado oscila entre el 0.5% y el 33% según diferentes estudios. La fiebre y el dolor óseo son los síntomas más frecuentemente asociados a esta entidad, siendo la anemia y la trombocitopenia los hallazgos analíticos más comunes.
OBJETIVO:
Presentar un caso en el que la NMO se produce debido a una hemo-patía.
PACIENTES, MATERIAL Y MÉTODO:
Varón de 59 años. No antecedentes de interés. Consultó por edemas en miembros inferiores sin otros síntomas. No hallazgos en la exploración física, no adenopatías ni hepatoesplenomegalia palpable. Analítica: anemia normocítica (Hb 10g/dL, VCM 89.5fL), trombocitopenia (pla-quetas 47.000/μL) sin leucocitosis (leucocitos 6.730/μL con 4.210/μL neutrófilos, 1.850/μL linfocitos y 510/μL monocitos). Bioquímica, coagulación, test de Coombs directo, serología viral, cuantificación de inmunoglobulinas y marcadores tumorales sin alteraciones. Frotis de sangre periférica con 4% de eritroblastos y 1% de blastos de hábito mieloide. Inmunofenotipo de sangre periférica en el que se encuen-tran un 2% de blastos mieloides con expresión aberrante de CD71. Ecografía abdominal sin hallazgos. RNM de pelvis compatible con la presencia de un proceso proliferativo de la MO con reemplazamiento de la misma probablemente por infiltración celular. Aspirado de MO seco de celularidad global no valorable, con signos de dishemopoyesis.
Se observa una 4% de células blásticas de hábito mieloide. Estudio ci-togenético: normal. Biopsia de MO: El componente trabecular muestra un espacio medular de aspecto fibrinoide, quedando remanente una población hematopoyética con representación de las tres series y sin alteraciones displásicas ni infiltrativas. No se añade fibrosis reticulínica depósito de material amiloide, infiltrado histiocitario, granulomas ni patógenos. No hallazgos de leucemia mieloide aguda hipocelular ni neoplasia mieloproliferativa ni mielodisplasia. Diagnóstico: Necrosis de médula ósea de tipo fibrinoide. Mutación V617F en JAK2 por PCR en sangre periférica positiva. Reordenamiento BCR/ABL por PCR en sangre periférica no se detecta. En el FISH de sangre periférica no se detectan t(8;21), t(15;17), ni reordenamientos (11q23) o (16q22).
Juicio Clínico: Mielofibrosis Primaria (DIPSS-Plus de alto riesgo). El paciente ha presentado una evolución desfavorable siento necesario el soporte transfusional, por lo que está en tratamiento con Danazol y actualmente se encuentra pendiente de alotrasplante de progenitores hematopoyéticos.
RESULTADOS Y CONCLUSIONES:
Ante un paciente con necrosis de médula ósea sin una causa objetivada se debe realizar la búsqueda exhaustiva de una neoplasia. Infecciones, drepanocitosis e incluso algunos fármacos (ATRA, fludarabina, G-CSF) también se han escrito como potenciales causantes de este proceso.
La evolución tórpida con demandas transfusionales constituyen rasgos propios de procesos malignos con NMO, datos que se aprecian en el paciente descrito.
La NMO en pacientes con hemopatías se relaciona con un peor pro-nóstico y una respuesta pobre al tratamiento, aunque la supervivencia generalmente depende del pronóstico de la enfermedad subyacente.
Necrosis de médula ósea en paciente con neoplasia hematológicaB. Díaz Roldán, A. Palma Vallellano, R. Zapata Bautista, M.A. Ruíz Cobos, J.A. Quesada P., M.V. Moreno Romero
Hospital Juan Ramón Jimenez

COMUNICACIONES PÓSTERS: Hematología Clínica 195
INTRODUCCIÓN/FUNDAMENTO:
La mielofibrosis post Trombocitemia Esencial ocurre en un 5% de los pacientes siendo la mutación V617FJAK2 la mutación adquirida más prevalente. Una vez producida la transformación mielofibrótica, la ex-pectativa de vida ronda entre 5-10 años y la causas de muerte incluye la progresión a leucemia aguda (5-20%); sin embargo, la mayoría de las muertes son derivadas de las comorbilidades. Ruxolitinib (inhibidor selectivo de JAK1/JAK2), administrado por vía oral, ha demostrado reducir la esplenomegalia y mejorar la sintomatología de la enferme-dad, prolongando la supervivencia global de los pacientes. Es posible su utilización en pacientes con deterioro de la función renal, si bien, existe poca experiencia clínica comunicada al respecto.
OBJETIVO:
Presentamos el caso de un varón de 69 años con mielofibrosis post Trombocitemia Esencial en hemodiálisis y bajo tratamiento con Ruxo-litinib.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos el caso de un varón de 69 años con ERC G5A3 secundaria a enfermedad poliquística inicialmente bajo hemodiálisis y que recibe un trasplante renal de donante cadáver en el año 1999. En 2005 fue diagnosticado de Trombocitemia Esencial JAK2 V617F positivo inician-do tratamiento citorreductor con Hydrea más AAS. En 2014, tras cuadro
progresivo sintomático, datos de hipertensión portal con esplenome-galia de hasta 25 cm, presencia de dacriocitos, aumento de la LDH y anemización con requerimiento transfusional, se realiza aspirado y biopsia de médula ósea por sospecha de transformación mielofibrótica que se confirma. Inicialmente se mantiene el tratamiento con Hydrea con escasa respuesta, diagnosticándose simultáneamente a raíz de una pielonefritis de un oncocitoma renal. Se llevó a cabo nefrectomía izquierda con postoperatorio tórpido y descompensación de la hepa-topatía necesitándose paracentesis evacuadora y reanudación de la hemodiálisis. Dada la resistencia de su mielofibrosis al tratamiento con hidroxiurea, se decide iniciar tratamiento con ruxolitinib 15 mg los días de la diálisis con clara mejoría de la sintomatología constitucional y reducción del tamaño esplénico. Con el un seguimiento de un año des-de el inicio del fármaco, el paciente ha disminuido sus requerimientos transfusionales con lo que se ha incrementado actualmente la dosis a 20 mg, por el momento, con buena tolerancia .
RESULTADOS Y CONCLUSIONES:
La insuficiencia renal crónica avanzada en hemodiálisis no excluye a un paciente con mielofibrosis a ser candidato a tratamiento con ruxolitinib siempre que se ajuste correctamente la dosificación al procedimiento. En el caso presentado, se demostró como fármaco seguro y eficaz para evitar la progresión de la enfermedad, para el control de la sintoma-tología constitucional y, por consiguiente, para mejorar la calidad de vida y la supervivencia del paciente.
Ruxolitinib en pacientes con hemodialisis: a propósito de un caso de mielofibrosis e insuficiencia renal terminalM. Ángeles Domínguez-Muñoz, Patricia Jiménez Guerrero, Begoña Pedrote Amador, José González Campos, Isabel Montero Cuadrado
UGC de Hematología y Hemoterapia. Hospital Universitario Virgen del Rocío. Instituto de Biomedicina de Sevilla (IBIS)/CSIC/Universidad de Sevilla

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA196
INTRODUCCIÓN/FUNDAMENTO:
El pronóstico de los linfomas difusos de células grandes B (LDCG-B) ha mejorado en los últimos años gracias al tratamiento con inmu-noquimioterapia (Rituximab junto con quimioterapia convencional). La afectación del sistema nervioso central (SNC) es infrecuente pero conlleva un mal pronóstico especialmente en aquellos pacientes en los que la afectación se produce en la recaída con una superviven-cia media inferior a 6 meses. Debido a su baja frecuencia y a que los pacientes con afectación del SNC se excluyen habitualmente de los ensayos clínicos el tratamiento óptimo para este tipo de pacientes no está bien establecido. En primera línea la inmunoquimioterapia para el control de la afectación sistémica en combinación con fármacos que atraviesan la barrera hematoencefálica junto con terapia intratecal es el tratamiento más aceptado. En recaídas la consolidación con tras-plante autólogo es potencialmente curativa y también se ha usado la radioterapia aunque su toxicidad potencial limita su uso. Los esquemas IDARAM y más recientemente R-IDARAM (tabla 1) fueron diseñados para el tratamiento de este tipo de pacientes y han mostrado efi cacia especialmente en primera línea (remisiones completas (RC) 48-75% y supervivencia global a los dos años superior al 75%).
OBJETIVO:
Describir la experiencia de un único centro con el uso de R-IDARAM.
PACIENTES, MATERIAL Y MÉTODO:
Estudio restrospectivo mediante revisión de historias clínicas en nuestro centro de pacientes tratados con esquema R-IDARAM en los últimos dos años. Dos pacientes recibieron este esquema, uno de ellos en primera línea por tener afectación extensa de la base del cráneo al diagnóstico y el otro por recaída con afectación del SNC a menos de un año tras haber alcanzado remisión completa con esquema R-CHOP, tratamiento triple intratecal y metotrexato a dosis altas. Las caracterís-ticas basales de los pacientes se resumen en la Tabla 2. Los pacientes recibieron profi laxis antiinfecciosa con trimetoprim-sulfametoxazol y administración de factor estimulador de colonias granulocíticas desde el día +7 del ciclo hasta la recuperación de neutrófi los por encima de 1.5x10e9/L.
RESULTADOS Y CONCLUSIONES:
Los pacientes recibieron 4 y 5 ciclos de R-IDARAM respectivamente alcanzando ambos RC tanto de la afectación sistémica como de SNC confi rmada por pruebas de imagen y citometría de líquido cefalorra-quídeo tras el cuarto ciclo. La principal toxicidad ha sido infecciosa grados 3-4: la paciente en 1ª línea presentó 3 episodios infecciosos grado, uno de ellos grado 4 que requirió ingreso en UCI con retraso del siguiente ciclo de quimioterapia y el paciente en recaída presentó 4 episodios de neutropenia febril grado 3. La paciente que lo recibió en primera línea continúa viva a 9 meses del diagnóstico. El paciente
Tratamiento de linfoma difuso de células grandes B con afectación del sistema nervioso central con esquema de quimioterapia R-IDARAMMaría Solé Rodríguez (1), N. Rodríguez Torre (2), M. Ruiz Mercado (2), F. De La Cruz Vicente (2), I. Montero Cuadrado (2), J.A. Pérez Simón (2)
(1) Complejo hospitalario de Huelva-Hospital Juan Ramón Jiménez, (2) Hospital Universitario Virgen del Rocío, Sevilla
TABLA 1. Esquema de inmunoquimioterapia R-IDARAM / 21 días
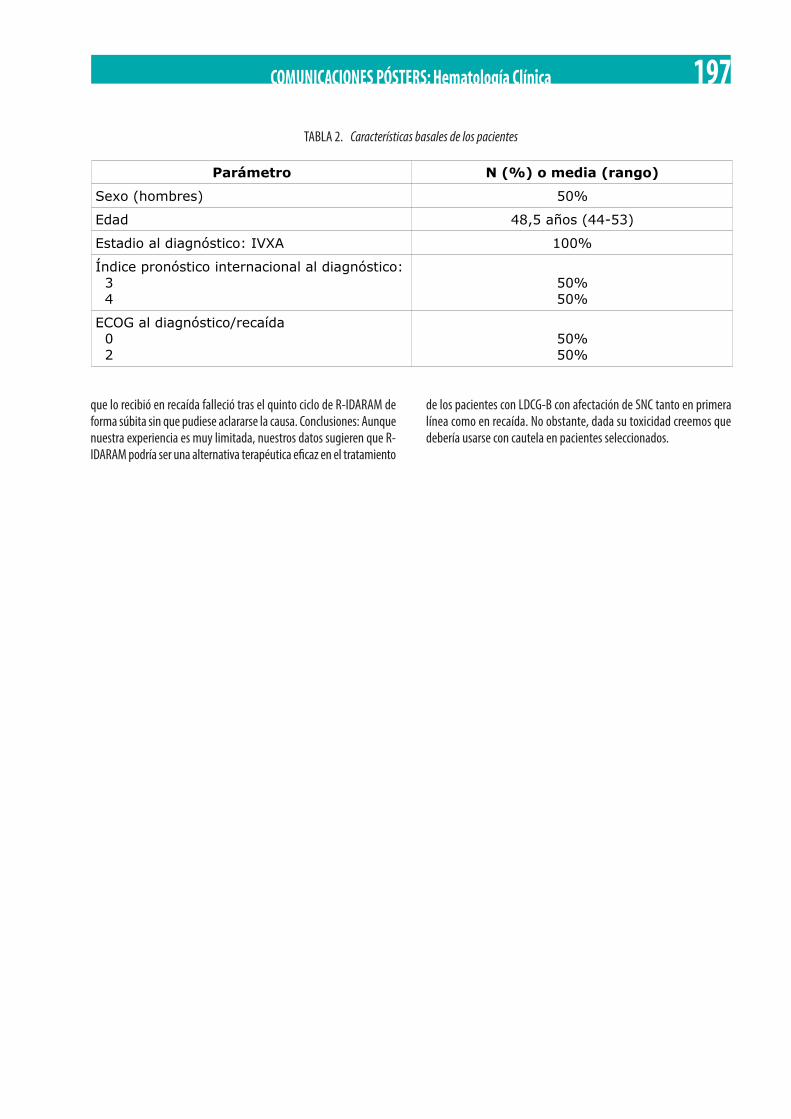
COMUNICACIONES PÓSTERS: Hematología Clínica 197
TABLA 2. Características basales de los pacientes
que lo recibió en recaída falleció tras el quinto ciclo de R-IDARAM de forma súbita sin que pudiese aclararse la causa. Conclusiones: Aunque nuestra experiencia es muy limitada, nuestros datos sugieren que R-IDARAM podría ser una alternativa terapéutica efi caz en el tratamiento
de los pacientes con LDCG-B con afectación de SNC tanto en primera línea como en recaída. No obstante, dada su toxicidad creemos que debería usarse con cautela en pacientes seleccionados.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA198
INTRODUCCIÓN/FUNDAMENTO:
Los episodios de neutropenia febril (NTF) son frecuentes en los pacien-tes hematológicos durante el tratamiento quimioterápico intensivo y cuando se someten a trasplante de progenitores hematopoyéticos (TPH).
No hay unanimidad de criterios, pero la Sociedad Americana de Enfer-medades Infecciosas (IDSA) define neutropenia como aquel recuento absoluto de neutrófilos (ANC) menor de 500 células/mL o menor de 1000 si se prevé el descenso en las próximas 48 horas. Se estima que la mitad de los pacientes con NTF tienen una infección establecida y una quinta parte de aquellos con ANC < 100 una bacteriemia.
Los principales factores de riesgo de infección son la duración de la neutropenia y el nadir de neutrófilos.
OBJETIVO:
Analizar la casuística del Servicio de Hematología del Hospital de Jerez de la Frontera.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo retrospectivo que recoge los NTF registrados en la planta de hospitalización en el semestre comprendido entre los meses de junio y noviembre de 2015. La muestra queda constituida por 49 casos. Se excluyeron a aquellos pacientes que presentaron fiebre antes de que se produjera la neutropenia.
RESULTADOS Y CONCLUSIONES:
En el periodo referido tuvimos un total de 227 ingresos de los cuales 19 fueron por NTF (8,37%). El total de NTF asciende a 49. La mediana de edad es de 57 años, con predominio de varones (30 frente a 19
mujeres). La Leucemia mieloide aguda (LMA), seguida del Linfoma No Hodgkin (LNH), constituye el grupo mayoritario de la muestra, supo-niendo en conjunto el 59.15 %. El motivo de atribución de la neutro-penia más representado es el tratamiento quimioterápico (Recuadro 1. Características de la muestra).
La duración promedio de la neutropenia es de 17 días (en este análisis se ha excluido a una paciente que nunca alcanzó recuperación por su enfermedad de base) y la cifra media de neutrófilos en el momento de aparición de la fiebre es 0.16 x 103 /mL. Estudiamos el tiempo transcurrido desde la constatación de la neutropenia hasta el NTF: mediana 3 días (P25= 0; P75= 9).
Mediante el estudio microbiológico dirigido se consiguió la docu-mentación en el 47% de los casos, observándose que las infecciones fúngicas ocurrieron en pacientes con LNH y Macroglobulinemia de Waldenström. Hubo dos casos con positividad del lavado nasofarín-geo para el Virus Sincitial Respiratorio (la muestra y el tipo de germen se detallan mediante gráficos).
CONCLUSIONES:
1. La proporción de ingresos por neutropenia febril fue pequeña.
2. El porcentaje de documentación microbiológica en nuestra serie concuerda con las estimaciones de infección establecida por las guías.
3. Observamos la tendencia al incremento de las infecciones por gérmenes gram negativos en detrimento de los gram positivos.
4. La mejora en el manejo de las vías venosas ha repercutido en el descenso de infecciones por S. epidermidis.
5. La ausencia de infección fúngica en los pacientes con LMA y TPH pone de manifiesto el beneficio de la profilaxis antifúngica.
Episodios de neutropenia febril durante un semestre hospitalario: análisis epidemiológicoJ.A. Raposo Puglia, S. Ordóñez Vahi, L. Domínguez Acosta, A. Salamanca Cuenca, M.V. Verdugo Cabeza de Vaca, V. Rubio Sánchez
Hospital Universitario Jerez de la Frontera

COMUNICACIONES PÓSTERS: Hematología Clínica 199
36,73%
22,45%
8,16% 8,16% 8,16%6,12%
4,08%2,04% 2,04% 2,04%
DISTRIBUCIÓN POR PATOLOGÍAS
LMA LNH LLA SMD LLC LH MM APLASIA MW LMC
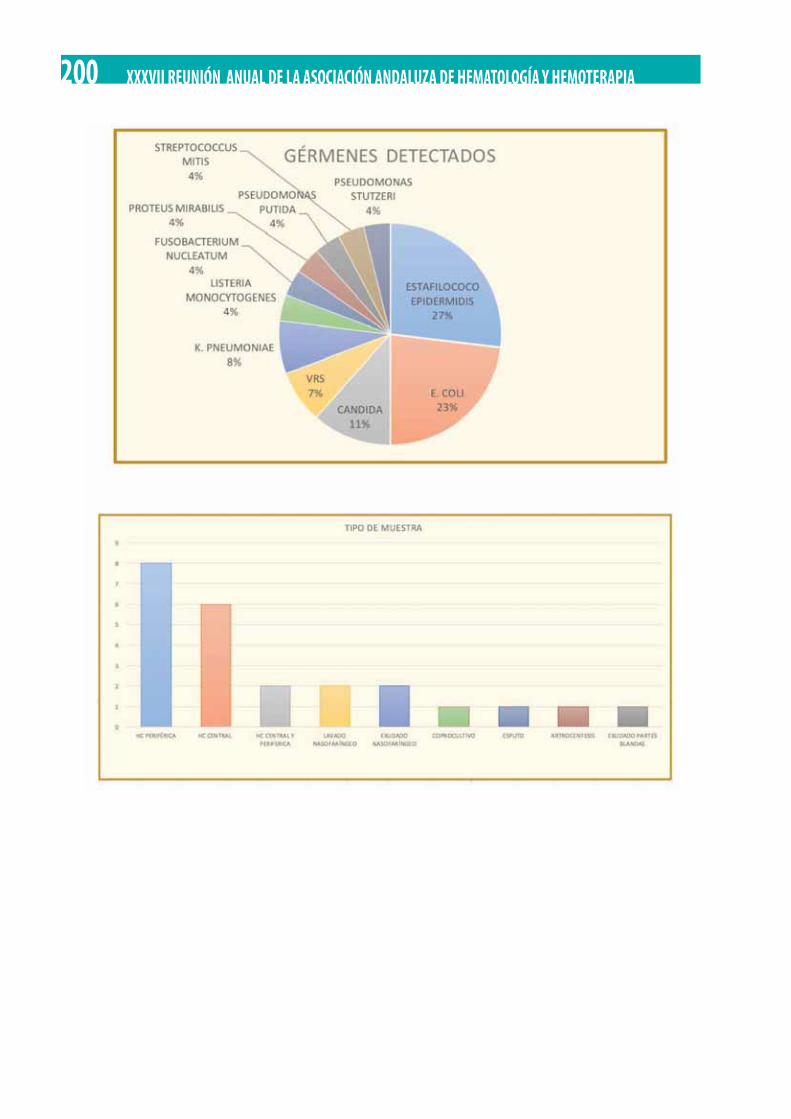
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA200

COMUNICACIONES PÓSTERS: Hematología Clínica 201
INTRODUCCIÓN/FUNDAMENTO:
La aplasia medular es una enfermedad caracterizada por pancitopenia periférica en el contexto de una médula ósea hipocelular, sustitución del tejido hematopoyético por grasa y ausencia de infiltración medular anormal. El desarrollo de nuevos agentes terapéuticos han permitido mejorar la supervivencia y calidad de vida de los pacientes.
OBJETIVO:
Paciente de 56 años, diagnosticado hace 33 años de anemia aplási-ca grave de etiología idiopática con buena respuesta a tratamiento inmunomodulador con globulina antitimocítica de origen equino, y soporte transfusional con obtención de una respuesta completa de los parámetros hemo-periféricos sanguíneos; presentando posterior reci-diva en forma de trombocitopenia y macrocitosis, motivo por el que se inició oximetolona, obteniendo respuesta hematológica satisfactoria, comportándose con efecto anabolizante-dependiente. El paciente no fue sometido a trasplante de progenitores hematopoyéticos al carecer de hermano histocompatible. El curso de la enfermedad ha sido cró-nico, con altibajos en las cifras de plaquetas y aparición de diferentes comorbilidades. Dieciséis años después, tras nueva biopsia medular se observó una hipoplasia megacariocítica con preservación cualitativa y cuantitativa de progenitores de la serie roja y blanca. Una citome-tría de flujo en sangre periférica mostró un clon HPN manifiesto en la serie granulocítica (34%), monocítica (38%) y eritroide (24%). Como efectos secundarios generados por oximetolona cabe destacar una dislipemia severa y un cuadro de rabdomiolisis crónica, motivos que llevaron a sustituir oximetolona por danazol, observándose descenso de la cifra de plaquetas (Plaquetas <30x109/L) obligando a introducir
oximetolona a una dosis mínima, recuperando la cifra de plaquetas en valores superiores a 50x109/L. El paciente fue objeto de seguimiento semestral con ecografía hepática en prevención de la aparición de ade-nomas o peliosis hepática. Un control manifestó imagen sugestiva de peliosis hepática. En 2005, ante la aparición de trombopenia se amplió el tratamiento con ciclosporina A. El paciente ha mantenido valores hematológicos estables desde el año 2012, motivo por el que, dadas las comorbilidades el tratamiento fue suspendido. Dada la naturaleza anabolizante-dependiente de su proceso y la presencia de un clon HPN en sangre periférica, se mantuvo seguimiento. Un año después, se ob-servó disminución progresiva del recuento plaquetario, manteniendo cifras superiores a 50x109/L). Hace 3 meses, el paciente desarrolló disminución de la cifra de plaquetas (plaquetas 33.000) precisando soporte transfusional ante la presencia de hematuria.
Ante la disminución del recuento plaquetario, dado los factores de riesgo cardiovascular, se planteó iniciar tratamiento con eltrombopag descartando la presencia de alteraciones citogenéticas. Eltrombopag fue iniciado a dosis de 25 mg cada 48 horas y según tolerancia se fue incrementando la dosis, siendo nuestro objetivo alcanzar cifras de plaquetas en un nivel seguro. Actualmente el paciente se encuentra con eltrombopag 75 mg cada 24 horas con buena tolerancia al mismo, no habiendo desarrollado hasta el momento ningún efecto adverso.
RESULTADOS Y CONCLUSIONES:
La aparición de eltrombopag permite desarrollar una mejor calidad de vida, una disminución de los efectos adversos a largo plazo por otras terapias, prolongando la supervivencia expresamente de aquellos pa-cientes no candidatos a trasplante de progenitores hematopoyéticos.
Aplasia medular grave con respuesta a agonista de los receptores de la Trombopoyetina: una nueva oportunidadM.M. Herráez-Albendea, S.R. Verdesoto-Cozzarelli
Hospital Santa Bárbara. Ciudad Real

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA202
INTRODUCCIÓN/FUNDAMENTO:
La leucemia de linfocitos grandes granulares (LLG) es una enferme-dad muy poco frecuente,asociada a una proliferación de clones de linfocitos atípicos de gran tamaño. Los linfocitos que más se afectan en Occidente son aquellos con fenotipo de linfocito T, dándose con muy poca frecuencia la afectación de los linfocitos NK. De hecho,en general,las neoplasias de células NK son entidades poco frecuentes y de difícil diagnóstico.
Atendiendo a esta subdivisión,las derivadas de los linfocitos T suelen tener un curso asintomático o bien dar simplemente la clínica derivada de la neutropenia acompañante,como las infecciones. Por lo tanto,el diagnóstico de este grupo de LLG suele realizarse tras observar una linfocitosis encontrada en una analítica que se realiza por otra causa o al encontrar en una exploración radiológica una esplenomegalia que puede formar parte de la enfermedad.
Por el contrario,aquellas derivadas de las células NK son más frecuentes en países orientales y son más agresivas, dando lugar frecuentemente a organomegalias y progresando con más frecuencia,por lo que es necesario instaurar tratamiento.
Se ha descrito su asociación con procesos autoinmunes,por ejemplo con la Artritis Reumatoide.
OBJETIVO:
Revisar una patología altamente infrecuente y de difícil diagnóstico a partir de un caso observado en nuestro servicio.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 81 años con antecedentes personales de hipertensión arterial,colelitiasis,fibrilación auricular,así como un ictus sufrido un año y medio antes a partir del cual mantiene inestabilidad secuelar.
Es derivada al servicio de Hematología desde las Urgencias (a las cuales acudió por dolor torácico) al serle detectada en la analítica realizada en este contexto una linfocitosis de 7.950 linfocitos/uL. También presentó
una anemia normocítica. La paciente no presentaba síntomas B ni cuadro constitucional.
Se decide solicitar una analítica completa con estudio de linfocitosis, inmunofenotipo de sangre periférica y perfil de anemias y también se citó en un mes para estudiar los resultados.
RESULTADOS Y CONCLUSIONES:
A la revisión, la paciente acudió refiriendo cansancio,mareos y dis-nea de esfuerzo,así como edematización ocasional de los miembros inferiores. A la exploración,no mostraba adenopatías ni hepatoesple-nomegalia.
En cuanto a la analítica solicitada,persistió la linfocitosis,así como el descenso de la hemoglobina. Además,se detecta una neutropenia leve no presente previamente. En el estudio de anemias no se encontró nada patológico salvo un ácido fólico por encima de los valores de normalidad. A nivel bioquímico presentó una elevación discreta de la urea, transaminasas y proteínas totales. La beta-2-Microglobulina estaba elevada.
En el informe del inmunofenotipo de sangre periférica llamó podero-samente la atención que la subpoblación mayoritaria eran las células NK (la mitad con fenotipo aberrante, CD56-, CD16+),por lo que se concluyó que se trataba de una LLG y células NK.
No se ha instaurado tratamiento,dadas las comorbilidades de la pa-ciente y la situación actual de estabilidad del cuadro.
Conclusiones:
Las LLG son muy poco frecuentes,pero sobre las que es necesario estar precavidos ya que pueden comprometer la morbimortalidad de nues-tros pacientes cuando también tienen fenotipo de células NK,puesto que confieren a esta entidad un comportamiento más agresivo. No obstante, se trata de patologías que pueden tener un curso crónico e indolente. Las indicaciones de tratamiento más frecuentes son las infecciones de repetición y,con menos frecuencia, la anemia,la esple-nomegalia o la aparición de síntomas B severos.
La leucemia de linfocitos grandes granulares y células NK, una entidad muy poco frecuenteE. Morente Constantín, A.B. Rivera Ginés, M.P. Garrido Collado, A. Núñez García, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

COMUNICACIONES PÓSTERS: Hematología Clínica 203
INTRODUCCIÓN/FUNDAMENTO:
El síndrome de Schnitzler es una entidad clínica poco frecuente descrita por primera vez en 1972. Comparte características con los denomina-dos síndromes autoinflamatorios y es más frecuente en varones entre los 40 y los 50 años.
Es una enfermedad de etiopatogenia desconocida en la cual se ha involucrado una alteración de la estabilidad de la Interleucina 1. Los síntomas que se pueden presentar son artralgias, fiebre intermitente y urticaria. También pueden aparecer adenopatías y un componente monoclonal IgM.
En la mayoría de los casos la enfermedad tiene un curso crónico y benigno, si bien los pacientes que la padecen presentan un riesgo de un 15% de evolucionar a un síndrome linfoproliferativo.
OBJETIVO:
Revisar una entidad poco frecuente en la práctica clínica y de difícil diagnóstico a través de un caso diagnosticado en nuestro servicio.
PACIENTES, MATERIAL Y MÉTODO:
Varón de 47 años con antecedentes personales de trastorno bipolar y vasculitis y con antecedentes familiares de madre afectada por ar-tritis reumatoide y padre fallecido posiblemente por un linfoma que es derivado al servicio de Hematología por sospecha de gammapatía IgM Kappa.
Clínicamente el paciente se encontraba asintomático salvo por un ligero prurito y la exploración no arrojó nada patológico. Se optó por solicitar un PET, una analítica completa y una PAMO y volver a revisar al paciente.
RESULTADOS Y CONCLUSIONES:
El PET informó de la presencia de múltiples focos hipermetabólicos en relación a adenopatías axilares, ilíaca externa derecha e inguinales, con aumento de captación difuso en médula ósea. La PAMO reflejó un 14% de linfocitos con presencia de linfoplasmocitos. En la analítica se evidenció una probable coagulopatía por déficit de factores que podría estar en relación a una infiltración hepática. Con la presunción diagnóstica de Gammapatía Monoclonal IgM Kappa (pobable Wal-denström) se decidió realizar una biopsia ganglionar.
La biopsia ganglionar mostró una hiperplasia folicular y plasmocitosis politípica, con policlonalidad en el estudio de genes IgH/K/L. Se con-cluyó que el paciente estaba afecto de una Gammapatía Monoclonal de Significado Incierto IgM Kappa en el contexto de un Síndrome de Schnitzler.
Conclusiones:
Se descartó, por tanto, la presencia de la una macroglobulinemia de Waldenstrom. No obstante, el Síndrome de Schnitzler presenta, como todas las Gammapatías Monoclonales de Significado Incierto, un ries-go de progresión a linfoma, mieloma IgM o macroglobulinemia de Waldeström. Se trata de una entidad muy poco diagnosticada, debido a su rareza y complejidad.
Síndrome de Schnitzler: la gammapatía monoclonal dentro de una entidad poco común y de difícil diagnósticoE. Morente Constantín, B. Mesa Simón, M.P. Garrido Collado, R. Ríos Tamayo, A.B. Rivera Ginés, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA204
INTRODUCCIÓN/FUNDAMENTO:
La valoración de la infiltración de la médula ósea (MO) al diagnóstico en el linfoma B difuso de células grandes (LBDCG) es primordial, en el estadiaje, pronóstico y tratamiento. La recomendación actual es la realización de la biopsia de médula ósea (BMO) al diagnóstico siendo un procedimiento invasivo, doloroso y con complicaciones. Aunque la utilidad actual del FDG-PET/TC es la reevaluación terapeútica, recientes publicaciones indican que podría sustituir a la BMO en el estadiaje
OBJETIVO:
El principal objetivo es observar si existe correlación entre los resul-tados de la BMO y el PET en la detección de afectación MO y como se-cundario determinar su impacto como factor de riesgo de mortalidad
PACIENTES, MATERIAL Y MÉTODO:
Seleccionamos a 138 pacientes diagnosticados de LBDCG, excluyendo afectación SNC primario, con BMO realizada en cresta íliaca al diag-nóstico en nuestro centro entre marzo-2005 y agosto-2016, de ellos 93 tenían realizado PET-TC (F18.FDG) al diagnóstico. Los resultados de ambas pruebas fueron analizadas por especialistas de Anatomía patológica y Medicina nuclear, en nuestro centro hospitalario.
Además de las variables sociodemográficas se recopilaron datos clí-nicos: estadiaje, síntomas B, índice pronóstico internacional (IPI) y tratamientos recogidos en la tabla 1.
RESULTADOS Y CONCLUSIONES:
Entre los 93 pacientes que tenían realizadas ambas pruebas al diag-nóstico, 73 PET no mostraron captación patológica en MO que se co-rrespondió en el 100% con el resultado de la BMO. Sin embargo los PET que presentaban infiltración ósea (n=20), sólo el 50% (n=10) de las BMO detectaron afectación ósea, captación era difusa en 5 casos,
3 con afectación múltiple incluyendo pelvis y en 2 casos la afectación estaba localizada únicamente en pelvis. De los 10 casos con PET posi-tivo y BMO negativa, las captaciones eran múltiples (n=6) sin afectar a pelvis o únicas (n=4). La tasa de concordancia entre el PET y la BMO fue del 89.25% (83 de 93) (gráfica 1)
Si consideramos la BMO como la prueba gold estándar, el PET muestra una sensibilidad del 50% y una especificidad del 100%. Si considera-mos cualquier afectación de MO ya sea por biopsia o PET, la sensibili-dad del PET sería del 100% (20 de 20) y la BMO del 50% (10 de 20) la especificidad para ambas sería del 100%.
La mediana de seguimiento fue de 45.5 (1-143) meses, con una super-vivencia global (SG) a los 5 años del 68.3 ± 4.1%, el 33.3% fallecieron, la principal causa fue la progresión linfomatosa (59.6%).
Los factores que influyeron en la SG en el estudio univariante por kaplan Meier, mostró significancia estadística: la presencia de sínto-mas B [no (82.5%) vs sí (47.2%) p=0.02], estadio IV [no (84.9) vs sí (61.1%) p=0.01], IPI ≥ 3 [no (86.1%) vs (60.5%) p=0.002], prímera recaída [no (81.5%) vs si (53.1%), p<0.001], cercano a la significación: afectación extranodal, edad>65 años y LDH ≥ 450 (tabla 2). Para la serie global, la afectación de MO detectada por PET y/o BMO mostró impacto significativo en la SG: [no (80.0%) vs sí (57.6%) p=0.02]
El estudio multivariante por regresión de Cox para determinar los fac-tores de mortalidad fueron: un IPI ≥ 3 (HR 1.92; IC 95% 1.03 a 3.56 p= 0.03) 1ª recaída linfomatosa (HR 3.64; IC 95% 2.03 a 6.53 p< 0.001) y padecer síntomas B (HR 2.14; IC 95% 1.15 a 4.02 p=0.01)
El PET muestra un alto valor predictivo negativo para la detectar infil-tración de la MO en el LBDCG, con PET negativo no es necesaria la BMO. Las discrepancias se deben a la afectación focal de la médula que no se localiza en la cresta ilíaca y no es detectada por la BMO. Por ello el PET tiene una mayor sensibilidad para la afectación focal, y permite sugerir que es una herramienta útil en la detección de infiltración de la MO, sustituyendo a la BMO.
Utilidad del F18-FDG-PET/TC y/o biopsia ósea para el estadiaje de los linfoma B difusos de células grandesM. Yébenes Ramírez, I. Vico Herrera, C. Martínez Losada, C. Chic Acevedo, J. Sánchez García
Hospital Universitario Reina Sofía. IMIBIC. Universidad de Córdoba

COMUNICACIONES PÓSTERS: Hematología Clínica 205
Auto-TPH
no 116 (84.1)
sí 22 (15.9)
1ª Recaída
no 106 (76.8)
sí 32 (23.2)
Causas éxitus (n=47)
progresión 28 (59.6)
infección 9 (19.1)
otras 10 (21.3)
Esquemas QT (1ªlinea)
R-CHOP 113 (81.8)RT 43 (34.4)
CHOP 12 (8.7)
COP 5 (3.7)RT 2 (20)
R-COP 5 (3.7)
Otros 3 (2.1)
Características clínicas n=
Variables n (%) mediana (min-máx)
Sexo (V/M) 74 (53.6)/ 64 (46.4)
Edad 61 (17-90)
LDH 450 37 (26.8)
Afectación extranodal 73 (52.9)
Síntomas B 31 (22.5)
Afectación médula ósea*
no 105 (76.1)
si 33 (23.9)
IPI 3 62 (44.9)
Estadio
I 16 (11.5)
II 35 (25.3)
III 27 (19.5)
IV 60 (43.6)
TABLA 1. Características clínicas. *Afectación médula ósea biopsia y/o PET

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA206
TABLA 2. Resultados de supervivencia estudio univariante; Kaplan-Meier. (n=138)
MO: médula ósea.
Variable 12 meses 24 meses p
Edad < 65 añosEdad > 65 años
86.5 ± 3.677.1 ± 6.1
75.9 ± 4.670.5 ± 6.6
0.07
Extranodal no sí
92.1 ± 3.476.5 ± 5.0
85.6 ± 4.564.7 ± 5.7
0.08
Sintomas B no sí
89.6 ± 3.064.1 ± 8.7
82.5 ± 3.747.2 ± 9.1
0.02
LDH > 450 no sí
87.9 ± 2.372.9 ± 7.3
79.4 ± 4.161.7 ± 8.1
0.05
Estadío IV no sí
93.3 ± 2.971.6 ± 5.8
84.9 ± 4.261.1 ± 6.4
0.01
Afectación MO no sí
88.3 ± 3.269.7 ± 8.0
80.0 ± 4.057.6 ± 8.6
0.02
IPI 3 no si
94.6 ± 2.670.8 ± 5.8
86.1 ± 4.160.5 ± 5.3
0,002
1ª Recaída no sí
85.6 ± 3.478.1 ± 7.3
81.5 ± 3.853.1 ± 8.8
< 0.001
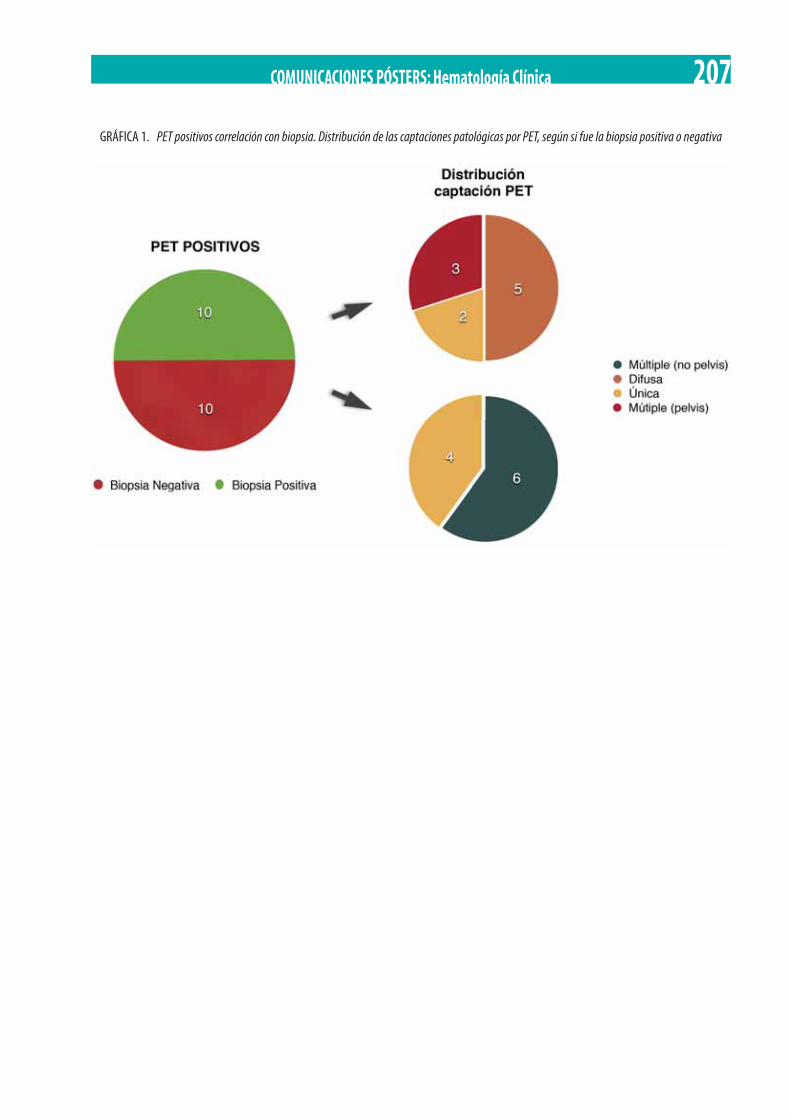
COMUNICACIONES PÓSTERS: Hematología Clínica 207
GRÁFICA 1. PET positivos correlación con biopsia. Distribución de las captaciones patológicas por PET, según si fue la biopsia positiva o negativa

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA208
INTRODUCCIÓN/FUNDAMENTO:
El síndrome hemofagocítico(SHF) es una entidad clínica agresiva y potencialmente mortal en la cual tiene lugar una activación in-mune excesiva. Puede ser un síndrome primario para el cual exista predisposición genética o aparecer de forma reactiva a procesos infecciosos,neoplásicos o autoinmunes.
El diagnosticar y así iniciar un tratamiento lo más precoz posible va a ser muy importante como determinante pronóstico de un síndrome que puede tener una mortalidad muy elevada. Sin embargo, a menudo se produce un retraso en el diagnóstico,debido a que se trata de una entidad muy poco frecuente y a que no existen datos que por sí solos confirmen el diagnóstico.
Para diagnosticarlo es necesario que se den al menos 5 situaciones de las siguientes: fiebre,esplenomegalia, citopenias,hipertrigliceridemia,hipofibrinogenemia, hemofagocitosis a nivel medular,ganglionar o esplénico, ferritina elevada, CD25 soluble > 2,400 U/mL,actividad de células NK nula o ausente.
OBJETIVO:
Para llevar a cabo una identificación precoz del SHF es necesario cono-cer las manifestaciones clínicas y de laboratorio con las que cursa. Por ello, se han recogido distintos datos de interés que suelen estar presen-tes en el cuadro clínico típico de la enfermedad. Se han seleccionado aquellos pacientes diagnosticados en el último año en nuestro centro.
PACIENTES, MATERIAL Y MÉTODO:
Se han analizado 4 pacientes con diagnóstico reciente(en el último año) de SHF,de los cuales se han recogido distintos datos demográficos,clínicos,analíticos y pronósticos.
RESULTADOS Y CONCLUSIONES:
En cuanto a los datos demográficos,3 de los pacientes eran hombres y 1 mujer;la edad media al diagnóstico fue de 52 años (rango de edad de los 45 a los 67).
La causa subyacente del SHF fue neoplásica en la mitad de los casos (un linfoma y una neoplasia de colon) e infecciosa en la otra mitad (por Leishmania en un caso y por CMV en el otro).
Es fundamental realizar un diagnóstico precoz de cara a mejorar la morbimortalidad de estos pacientes. En relación a esta circunstancia,el promedio de número de días transcurrido desde el inicio de los sínto-mas y el diagnóstico fue de 32.
Atendiendo a los criterios diagnósticos: los 4 pacientes presentaron fie-bre (las cifras oscilaron entre 38,8ºC y 40ºC), en al menos 3 de los 4 pa-cientes se objetivó la presencia de esplenomegalia (en uno de los casos no fue valorada), todos tuvieron pancitopenia (cifras de hemoglobina entre 5,6 y 7,8 g/dL;de leucocitos entre 1.160 y 2.390 por microlitro y de plaquetas entre 6.000 y 62.000 por microlitro). Todos presenta-ron una hiperferritinemia acusada (entre 5.528 y 14.046 ng/mL). En cuanto a la hipofibrinogenemia,la tenían 3 de los 4 casos(fibrinógeno entre 71 y 164 mg/dL). Se objetivó la presencia de hemofagocitosis en médula ósea en 3 de los 4 casos.
También se recogió si había hepatomegalia y adenopatías(presentes en la mitad de los casos),aumento de LDH y PCR (existente en todos los casos), de las transaminasas(elevadas en los 4 casos) y de la creatinina (elevada en 3 de los 4 casos).
En cuanto a la evolución,3 de los 4 casos necesitaron de traslado a la UCI. En cuanto al desenlace,1 de los 4 falleció.
Conclusiones:
Es necesario conocer los criterios diagnósticos que definen al SHF de cara a poder diagnosticarlo de forma precoz y, de esa manera,mejorar el pronóstico de una enfermedad que puede tener un desenlace fatal. Por tanto,sería conveniente incluirlo en el diagnóstico diferencial en todo paciente oncológico o infeccioso con pancitopenia y fiebre que presente hiperferritinemia y/o hipofibrinogenemia sin que existan razones justificadas para las mismas.
El síndrome hemofagocítico: revisión de los casos diagnosticados en nuestro centro en el último añoJ. Badiola González, E. Morente Constantín, B. Mesa Simón, P.A. González Sierra, E. López Fernández, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

COMUNICACIONES PÓSTERS: Hematología Clínica 209
INTRODUCCIÓN/FUNDAMENTO:
Los pacientes hematológicos a menudo requieren ingreso en la Unidad de Cuidados Intensivos (UCI), ya sea para el tratamiento de problemas derivados de su enfermedad o debido a complicaciones del tratamien-to que reciben.
OBJETIVO:
Analizar el pronóstico de los pacientes hematológicos que han reque-rido ingreso en la UCI, así como los factores que pueden modificar su pronóstico.
PACIENTES, MATERIAL Y MÉTODO:
Se ha elaborado una base de datos de 90 pacientes hematológicos que ingresaron en la UCI de nuestro hospital entre los años 2.010 y 2.015. Para cada uno se han recogido los siguientes datos: la fecha de ingreso en la UCI, el motivo del ingreso en la misma, la hemopatía de base padecida y si dicha hemopatía se encontraba activa en el momento del ingreso. También se registraron el sexo y la edad, si necesitaron o no ventilación mecánica invasiva, así como el número de días. Tam-bién se hizo constar si el paciente falleció durante el ingreso, si estaba trasplantado y en caso afirmativo el tipo de trasplante empleado, si presentaba Enfermedad Injerto Contra Huésped (EICH) y si estaba en situación de neutropenia.
RESULTADOS Y CONCLUSIONES:
La mortalidad global al ingreso fue del 67% (fallecieron 60 pacientes de los 90 con los que se contó). En total se registraron 62 varones (68% del total) y 28 mujeres (32%), siendo las cifras de mortalidad del 69% y del 64%, respectivamente. La edad media de los pacientes ingresados fue de 53 años, con un rango de edad de los 20 a 83.
Las patologías hematológicas de base que más se ingresaron fueron: Linfoma No Hodgkin, 32 pacientes (35,5%); Leucemia Mieloide Aguda,
19 (21,1%); Linfoma de Hodgkin, 9 (10%); Leucemia Linfoide Aguda y Mieloma Múltiple, con 6 (6,67%) cada uno; Síndrome Mielodisplá-sico y Leucemia Linfoide Crónica, con 4 (4,4%) cada uno; Leucemia Mieloide Crónica y Policitemia Vera, con 2 (2,2%) cada uno. También ingresaron en la UCI un caso de anemia aplásica, amiloidosis cardiaca y plasmocitoma intracraneal. Hay dos casos en los que se desconoce el diagnóstico.
Las causas más frecuentes que desencadenaron el ingreso son, por sí solas o asociadas, las siguientes: la más frecuente fue el fallo res-piratorio agudo (involucrado en 51 casos, el 56,67%), seguido de las infecciones en sus distintas modalidades (26, el 28,89%) y del fallo cardíaco (9 pacientes, 10%).
El hecho de requerir Ventilación Mecánica Invasiva (VMI) se asoció a una mortalidad del 71% (69 pacientes), cifra que se eleva al 74% si se necesitó por un periodo de 7 o más días.
De los 26 pacientes sometidos a un trasplante de progenitores hema-topoyéticos fallecieron 23, suponiendo un 88% de muertes (83% en los autólogos y 90% en los alogénicos).
De los 61 pacientes que tenían su hemopatía activa en el momento del ingreso fallecieron 37 (un 60%), cifra que alcanza el 93% (14 de 15) cuando lo que tenían activa era una EICH.
Por último, un total de 34 pacientes presentaron neutropenia en el ingreso en UCI, de los cuales fallecieron el 82% (28).
Conclusiones:
Los pacientes con enfermedades hematológicas tienen un mayor ries-go de padecer situaciones que requieran su ingreso en la UCI y, una vez en ella, suponen un tipo de paciente con un especial riesgo de desenlace fatal (mortalidad global del 67%).
Según nuestro estudio las situaciones que se vinculan a una mayor mortalidad son: tener una EICH activa (mortalidad del 93%), el haber sido receptor de un trasplante (88%), el haber presentado neutropenia (82%) y el requerir VMI (71%).
Factores de riesgo asociados a la mortalidad en los pacientes con hemopatías que requieren ingreso en la unidad de cuidados intensivosE. Morente Constantín, A.B. Rivera Ginés, P.A. González Sierra, F. Manzano Manzano, E. López Fernández, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA210
INTRODUCCIÓN/FUNDAMENTO:
La monitorización de la enfermedad mínima residual (EMR) mediante citometría de flujo multiparamétrica (CMF) es una herramienta útil para el seguimiento y valoración de la respuesta en neoplasias he-matológicas. En los últimos años, se ha demostrado su utilidad en pacientes con mieloma múltiple (MM) con el fin de poder subcatego-rizar mejor la calidad de la respuesta
OBJETIVO:
Describir nuestros resultados de EMR en pacientes sometidos a tras-plante autólogo
PACIENTES, MATERIAL Y MÉTODO:
Seleccionamos 15 pacientes diagnosticados de MM entre enero 2012 a febrero 2016, tratados con quimioterapia (QT) intensiva y consolidados con Auto-TPH los cuales tenían una determinación de EMR pre-TPH y post-TPH (3 meses) con un seguimiento ≥6 meses tras el Auto-TPH.
La mediana de edad al diagnóstico 60 años (47-69). Respecto al tra-tamiento, 2 pacientes recibieron QT según esquema VCD y 13 VTD (1 se complemento con RT). Mediana de ciclos de tratamiento 4. Todos se movilizaron con ciclofosfamida + G-CSF y recibieron acondiciona-miento con Melfalán-200. Las características clínicas de los pacientes se recogen en la tabla1.
Para el análisis inmunofenotípico se incubaron 50 μl de MO en EDTA durante 10 min a temperatura ambiente con los Anticuerpos Mono-clonales conjugados con 8 fluorocromos según panel modificado de EuroFlow: CD45, CD19, CD38, CD138, CD56, CD117, CD27, CD81. Tras la incubación, las muestras fueron lisadas con cloruro de amonio y resuspensión en solución salina con fosfato (PBS). La adquisición de al menos 500.000 eventos se realizó en Clitómetros de Flujo FACScalibur or FACSCanto II (Becton.Dickinson) El análisis de los datos fenotípicos se realizó mediante Software CellQuest, FACsDiva e Infinicyt
RESULTADOS Y CONCLUSIONES:
Tras el tratamiento de inducción recibido y según los criterios de res-puesta del Grupo Español Mieloma, la tasa de respuesta de la serie pre-trasplante es: 33.4% respuesta completa estricta (RCs), 20% res-puesta completa (RC), 20% muy buena respuesta parcial (MBRP) y 26.6% respuesta parcial (RP)
Se realizó estudio EMR por CMF previo al trasplante autólogo siendo en 8 pacientes negativa (5 RCs, 1 MBRP y 2RP) y en 7 positiva (3 RC, 2 MBRP y 2 RP).
En la evaluación post-TPH, 14 pacientes presentaban EMR negativa, entre ellos todos los que presentaban EMR negativa pre-TPH. Los 7 pacientes que presentaban EMR pre-TPH positiva, de ellos en el mo-mento actual 3 han recaído, otros 3 han mejorado su respuesta (2RCs y 1 RC), y uno continua en RC, no ha progresado, el 28,5% presentan inmunofijación en suero u orina positiva. (tabla 2)
De las 3 recaídas que ocurrieron a los 8, 12 y 24 meses postrasplante, los 3 pacientes presentaban una EMR negativa en la evaluación pos-trasplante. No obstante, uno de ellos presentaba una citología adversa (plasmoblástico), el cual falleció por progresión de la enfermedad.
La mediana de tiempo hasta la recaída: 12 meses (8-24 meses), con una mediana de seguimiento de 18 meses (6-29 meses), la SG y SLE a los 26 meses es 90.9 ± 8.7 y 45.5± 32.4.2 respectivamente. La SLE a los 12 meses en el grupo de EMR negativa es del 100% y con EMR positiva del 83.3%.
CONCLUSIONES: La detección de EMR por CMF es un método fácil, barato y rápido para monitorización del MM, que puede ser comple-mento a otros estudios para predecir que pacientes requieren un segui-miento más estrecho en consulta. La persistencia de EMR es siempre una característica de pronóstico adverso, incluso entre los pacientes que han alcanzado una RC. Una EMR negativa, presenta un mayor período de tiempo hasta la progresión que los pacientes que presenta una EMR positiva.
Evaluación de la EMR mediante citometría de flujo pre y post auto trasplante en miloma múltipleM. Yébenes Ramírez, M.A. Álvarez Rivas, C. Martínez Losada, J. Sánchez García
Hospital Universitario Reina Sofía.IMIBIC. Universidad de Córdoba

COMUNICACIONES PÓSTERS: Hematología Clínica 211
Características clínicas n=15
Variables n (%) mediana (min-máx)
Sexo (V/M) 11 (73.3)/4 (26.7)
Edad 60 (47-69)
Tipo de Mieloma
IgG kappa 4 (26.7)
IgG lambda 5 (33.3)
IgA lambda 2 (13.3)
Bence Jones kappa 2 (13.3)
Bence Jones lambda 1 (6.7)
No secretor 1 (6.7)
Estadio ISS
I 5 (33.3)
II 4 (26.7)
III 6 (40.0)
Lesiones óseas
No 3 (20.0)
Sí 12 (80.0)
Nivel de creatinina (g/dL) 0.93 (0.60-9.21)
Hemoglobina (g/dL) 11.6 (6.3-15.4)
Componente monoclonal 2.87 (0.00-7.26)
Plasmáticas M.O. (%) 25 (2-78)
Tratamiento
VTD 13 (86.66)
VCD 2 (13.34)
Nº ciclos
4 10 (66.7)
5 2 (13.3)
6 2 (13.3)
8 1 (6.7)

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA212
Negativa Positivo No disponible Recaída
*Mieloma no secretor.
EMR
actual
1
2
3
4
5
6
7
8
9 * *
10
11
12
13
14
15
TABLA 2. Comparación inmunofi jación, prueba de imagen y EMR, preTPH y post-TPH

COMUNICACIONES PÓSTERS: Hematología Clínica 213
INTRODUCCIÓN/FUNDAMENTO:
La leucemia linfocítica crónica (LLC) es el síndrome linfoproliferativo crónico (SLPC) de células B más frecuente en el mundo occidental. Las complicaciones más frecuentes son las infecciones, segundas neopla-sias y trastornos autoinmunes por defi ciencia de la inmunidad celular T, hipogammaglobulinemia y alteraciones en la inmunidad innata. Además, la toxicidad y efectos secundarios de los tratamientos (in-munomoduladores, anticuerpos monoclonales, agentes alquilantes, corticoides...) contribuyen a la disfunción global del sistema inmune. El riesgo relativo de padecer TBC en SLPC es de 2 a 40 veces superior al de la población general.
OBJETIVO:
Reporte un caso de TBC diseminada en una paciente con leucemia linfocítica crónica refractaria tras varias líneas de tratamiento.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 56 años sin antecedentes de interés, diagnosticada de LLC estadio B-I con del P53 en 2013. Hasta la fecha del evento que se co-munica, recibe 4 líneas de tratamiento (tabla 1) y como antecedentes familiares de interés, madre y dos tíos con tuberculosis pulmonar hace 10 y 12 años respectivamente.
Ingresa en Hematología procedente de Urgencias en Diciembre de 2016 por cuadro de astenia y fi ebre de 38ºC de 24 horas de evolución asociada a dorsalgia y síndrome emético. Analíticamente, destaca hiponatremia moderada (122 mEq/L) asintomática e hipokalemia (2,8 mEq/L) y cefalea occipital. Se iniciaron medidas de corrección de electrolitos y antibioterapia dirigida a foco urinario vs. digestivo.
A la exploración presentaba regular estado general, caquexia extre-ma (IMC 16 kg/m2), consciente y orientada aunque algo tendente al sueño, eupneica tolerando el decúbito, febril (38.2 ºC), HTA 190/110 mmHg. Tonos cardiaco rítmicos sin soplos, MVC sin ruidos sobreaña-
didos. Resto anodino. Se realizaron las siguientes pruebas comple-mentarias:
Analítica de ingreso: Na 122 mEq/L, K 2.8 mEq/L, osmolalidad 258, LDH 304 U/l. Hemograma sin citopenias
Rx tórax infi ltrado micronodular sugestivo de patrón miliar
Citología/Citometría LCR donde no se observan células patológicas. Linfocitosis de características reactivas
TC y RNM craneal sin hallazgos relevantes
Punción lumbar: aspecto claro; presión de salida normal; 565 leuco-citos (86% mononucleares), glucosa 56 mg/dl, proteínas totales 232 mg/dl, hematíes 0; ADA 17; cultivo y gram bacterias negativo; PCR virus herpes negativa; cultivo micobacterias se aíslaron M. tubercu-losis complex (resistencia a etambutol); PCR tuberculosis positiva; CMV negativo; Ag cryptococo negativo.
Evolución inicial tórpida con persistencia de la clínica neurológica, a pesar de inicio de tratamiento antituberculoso (isoniacida, rifampici-na, estreptomicina). Presenta dos episodios de convulsiones tónico-clónicas con Glasgow 3 tras varios días de fl uctuaciones en el nivel de conciencia siendo éxitus tras 23 días de ingreso.
RESULTADOS Y CONCLUSIONES:
1. La TBC meníngea es una complicación infrecuente, asociada a inmunosupresión, que debemos considerar en pacientes con LLC tratados con varias líneas de tratamiento.
2. El diagnóstico diferencial debe hacerse con otras infecciones opor-tunistas así como infi ltración por su enfermedad de base. Además puede ser interesante en estos paciente realizar pruebas diagnós-ticas orientadas a determinar el estado de infección latente.
3. El tratamiento debe instaurarse a la mayor rapidez posible y em-plea regímenes similares a los de la población general.
Tuberculosis (TBC) diseminada en una paciente con leucemia linfocítica crónica,a propósito de un casoV. Calama Ruiz-Mateos, M.E. Molina Rodríguez, I. Simón Pilo, M. Vahí Sánchez de Medina, M. Gómez Rosa, E. Ríos Herranz
Hospital Universitario Nuestra Señora de Valme
TABLA 1

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA214
INTRODUCCIÓN/FUNDAMENTO:
El sarcoma granulocítico (SG) es un tumor raro de localización extra-medular compuesto por células inmaduras de la serie granulocítica, originadas a partir de los mielobastos. El SG puede aparecer de novo o asociarse a una enfermedad hematológica (leucemia mieloide aguda, síndrome mielodisplásico o neoplasias mieloproliferativas crónicas), por lo tanto, al diagnosticar un sarcoma granulocítico hay que descar-tar la presencia de una enfermedad hematológica. El hecho de tener un SG confiere un muy mal pronóstico, con una supervivencia media a los 2 años del 6%.
OBJETIVO:
Realizar una revisión de una enfermedad rara y de difícil diagnósti-co a partir de dos casos vistos recientemente en nuestro servicio. En uno de los casos, la paciente ya tenía una enfermedad hematológica de base, en el segundo caso no había tenido lugar un proceso onco-hematológico.
PACIENTES, MATERIAL Y MÉTODO:
El primer caso corresponde a una mujer de 63 años sin antecedentes de interés que es derivada al servicio de Hematología desde su MAP por presentar una discreta leucocitosis y trombopenia, así como una monocitosis mantenida desde 2.010. Clínicamente se encontraba bien.
El segundo caso lo constituye un varón de 56 años sin más antecedente que el de hiperuricemia, derivado desde su MAP para estudio de una masa laterocervical izquierda dolorosa. Además, refiere en los días previos la aparición de fiebre con escalofríos y tiritona, así como una pérdida de 10-15 Kg en los últimos 6 meses.
RESULTADOS Y CONCLUSIONES:
En cuanto al primer caso, el inmunofenotipo mostró un 28% de mono-citos maduros (expresando la mitad CD56+). Con la sospecha de leuce-mia mielomonocítica crónica (LMMC) se llevó a cabo un seguimiento.
Dado que la cifra de plaquetas fue en descenso progresivamente, la siguiente conducta que se tomó fue la realización de una PAMO para estudio de cariotipo, FISH e inmunofenotipo de médula ósea.
Se llegó al diagnóstico de Leucemia Mielomonocítica Variante Mielo-proliferativa Tipo 1 con un IPSS de Bajo Riesgo y estudio JAK2/CALR/MPL negativos. Se inició tratamiento con Azacitidina.
Al cabo de un año, la paciente desarrolló una masa sangrante en la fosa nasal derecha, por lo que se indicó la realización de una biopsia, informada como un sarcoma mieloide. Se presentó el caso en sesión clínica para decidir la actitud a adoptar. Se cursó el ingreso y se solicitó un PET-TAC, así como se indicó inicio de tratamiento quimioterápico intensivo de LMA, necesario al contar con un SG asociado a la LMMC.
En lo que concierne al segundo caso, la biopsia de la adenopatía late-rocervical informó de la presencia de un sarcoma mieloide. Se realizó un PET-TAC que confirmó la aparición de focos hipermetabólicos a nivel laterocervical izquierdo. A partir de estos hallazgos, se realizó un estudio de extensión que reveló la presencia de una gammapatía policlonal, con cadenas Kappa libres en suero elevadas, así como el cociente Kappa/Lambda. También se encontró un aumento de la beta-2microglobulina. El frotis de sangre periférica no mostró alteraciones, pero en el inmunofenotipo de médula ósea se detectó un 30% de blastos. Se inició tratamiento según esquema de LMA.
Conclusiones:
El SG puede aparecer de forma secundaria a un proceso oncohemato-lógico (primera paciente), pero también puede antecederlo o acom-pañarlo (segundo paciente). Independientemente del momento, la conducta terapéutica ha de ser intensiva, ya que es un proceso que condiciona un mal pronóstico. Ante un paciente con LMA, la aparición de una masa, debe hacer pensar en un SG.
Sarcoma granulocítico: la importancia del diagnóstico y tratamiento precoces en una entidad agresivaE. Morente Constantín, A.J. Cruz Díaz, P.A. González Sierra, A. Romero Aguilar, L. Moratalla López, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

COMUNICACIONES PÓSTERS: Hematología Clínica 215
INTRODUCCIÓN/FUNDAMENTO:
La Leucemia Mieloide Crónica(LMC) se asocia en ocasiones a una Gam-mapatía Monoclonal(GM),siendo necesario el seguimiento estrecho de ambas entidades de cara al control clínico de estos pacientes.
OBJETIVO:
Revisar la coexistencia de una LMC y una GM a partir de la experiencia en nuestro centro de dos pacientes que sufren ambas enfermedades.
PACIENTES, MATERIAL Y MÉTODO:
El primer paciente es un varón de 67 años sin antecedentes de inte-rés que fue derivado a Hematología al detectarle una leucocitosis de 65.100 leucocitos/uL en una analítica realizada en Atención Primaria. En la primera valoración,el paciente no refería sintomatología de in-terés. En la exploración, no se hallaron adenopatías ni megalias y en la analítica se constataron una leucocitosis de 99.000/uL y un pico monoclonal IgM (1.050 mg/dL).
El segundo paciente es un varón de 62 años sin antecedentes perso-nales reseñables que fue diagnosticado en diciembre de 2009 de una LMC Phi+,detectándose también una GM IgA.
RESULTADOS Y CONCLUSIONES:
En lo que concierne al primer paciente,ante la sospecha de una LMC en el frotis de sangre periférica,se realizó PAMO+cariotipo,así como se solicitó una ecografía abdominal y un TAC toraco-abdominal. Se inició en ese momento tratamiento citorreductor con Hidroxicarba-mida junto a Alopurinol.
Efectivamente,la PAMO resultó compatible con una LMC,con un BCR/ABL positivo y un inmunofenotipo en el cual no se hallaron células plasmáticas. La terapia citorreductora fue resolutiva, al disminuirse la cifra de leucocitos a 36.900. Tanto el TAC como la ECO fueron nor-males desde el punto de vista hematológico. Quedó confirmado el diagnóstico de LMC Phi+ junto a una GM IgM y se inició tratamiento con Nilotinib,siguiéndose periódicamente en consulta.
Tras tres meses de tratamiento,el paciente presentó RHC+RCC+RM con un BCR/ABL de 0,014%,por lo que se mantuvo el tratamiento con Nilotinib. La GM persistió estabilizada.
En el segundo paciente, una vez diagnosticado de las dos patologías, se decidió implantar tratamiento con Imatinib (alcanzando al mes de tratamiento RHC+RCC y descenso del BCR/ABL) y llevar a cabo un seguimiento tanto de la LMC como de la GM.
Coexistencia de dos hemopatías: leucemia mieloide crónica y gammapatía monoclonal de significado incierto en un mismo paciente. ¿Cuál es el camino a seguir?E. Morente Constantin, J.M. Puerta Puerta, P. García Martín, B. Mesa Simón, A. Núñez García, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada
Células plasmáticas con un citoplasma característico de las células plasmáticas productoras de IgA (flameado)
Periódicamente, se han venido solicitando PAMOs con cariotipo e in-munofenotipo, así como también se ha repetido el BCR/ABL. Como parte del estudio,se han llevado a cabo varios PET-TAC sin que existie-ran focos hipermetabólicos.
En las sucesivas consultas, se ha constatado un aumento del nú-mero de células plasmáticas en médula ósea, presentando en el úl-timo aspirado una plasmocitosis del 9% (a expensas de elementos maduros),eosinofilia y monocitosis moderadas. En lo que afecta a la cuantificación de inmunoglobulinas, presenta un descenso de IgG e IgM y una IgA de 4.035 mg/dL(70-400). La ratio Kappa/Lambda está

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA216
elevada: 9,13(0,26-1,65). El paciente no presenta sintomatología CRAB propia del Múltiple Múltiple(MM).
En la actualidad, debido a la estabilidad de ambos cuadros,continúa con el tratamiento con Imatinib y acudiendo a las sucesivas citas de revisión.
Conclusiones:
Cuando coexisten una LMC y una GM es necesario llevar a cabo el tratamiento específico de la LMC,así como un seguimiento de
ambas entidades,de cara a actuar de forma precoz ante una po-sible progresión de la GM a MM. La situación de estos pacientes es comprometida,puesto que se plantea el gran problema de cómo tratarlos cuando la GM evolucione. Estos casos son muy poco frecuentes,existiendo muy poca información al respecto. Por tanto,sería recomendable la comunicación de este tipo de casos, con coexistencia de 2 hemopatías,para poder elaborar protocolos de ac-tuación.

COMUNICACIONES PÓSTERS: Hematología Clínica 217
INTRODUCCIÓN/FUNDAMENTO:
Los enfermos con neoplasias hematológicas presentan complicaciones graves durante su ingreso, tributarias de atención en una Unidad de Cuidados Intensivos (UCI). Por ello, conocer el desenlace de la estancia en la UCI y los factores predictores de mortalidad es de gran interés. Desde 2016, nuestro servicio ha implantado un equipo con intensivis-tas y hematólogos para el manejo conjunto de estos pacientes.
OBJETIVO:
El objetivo de nuestro estudio es conocer la mortalidad e identifi car el valor pronóstico de diferentes variables que presentan los pacientes hematológicos que requirieron ingreso en UCI
PACIENTES, MATERIAL Y MÉTODO:
Estudio Retrospectivo de 34 pacientes hematológicos ingresados en UCI, desde 2014 a marzo de 2017, analizando mortalidad con distintos factores que pueden estar relacionados con el pronóstico: Edad, QT intensiva, Neutropenia, Ventilación Mecánica (VM), y APACHE II (Acute Physiology and Chronic Health Evaluation II). Se ha empleado análisis estadístico mediante Ods Ratio y Test exacto de Fisher
RESULTADOS Y CONCLUSIONES:
Resultados:
Analizamos la mortalidad de 34 pacientes hematológicos que ingre-saron en UCI. El 64.7% de los pacientes eran hombre y 35.3% fueron mujeres. La mortalidad de los pacientes hematológicos fue del 55.9%.
Los diagnósticos hematológicos más frecuentes quedan recogidos en el gráfi co 1.
Con una mediana de edad, al ingreso, de 51 años y una media de de 49.3 años. Rango de 19-80 años.
Del total de ingresos, el 65% había recibido QT intensiva. El 56% estaban neutropénicos en el momento del ingreso y el 53% habían precisado VM. El índice APACHE II promedio fue de 19.6.
Analizamos la mortalidad con cada uno de estos factores pronósticos mediante de El Test Exacto de Fisher:
Relación entre mortalidad y edad: el 52.6% de los fallecido tenías >60 años, mientras que el 47.4% tenían ≤ de 60. Esta relación presenta un OR de 2.26. Con una p de 0.17.
Relación entre mortalidad y uso de QT intensiva: el 63.6% de los fallecidos la había recibido, mientras que el 41.7 restante no. Esta relación presenta una OR de 2.45. Con una p de 0.29.
Relación entre mortalidad y neutropenia: el 66.8% lo estaban en el momento del exitus y el 31.6% no. Esta relación presenta una OR de 3.2. Con una p 0.19.
Relación entre mortalidad y VM: el 77.8% de los paciente con VM terminan siendo exitus, mientras que el 26.3% de los fallecidos no habían recibido VM. Esta relación presenta una OR de 7. Con una p de 0.014.
Relación entre mortalidad y APACHE II: El 61.5% de los pacientes tenía un APACHE II>20, mientras que el 52.6% habían fallecido con un APACHE II ≤20. Esta relación presenta una OR de 2.01. Con una p de 0.73. Podemos observan la distribución en cuartiles de ambos grupos. Grafi co 2.
Análisis de la mortalidad en la Unidad de Cuidados Intensivos en pacientes hematológicosN. Domínguez Velasco (1), C. Muñoz García (1), J.R. Jiménez del Valle (2), K. Kestler González (1), B. Herruzo Delgado (1),
A. Rodríguez Fernández (1)
(1) UGC de Hematología de Hospital Universitario Virgen Macarena, (2) UGC de UCI de Hospital Universitario Virgen Macarena
LMA
LLA
LNH
LH
SMD
MM
Amiloidosis
PTT
GRÁFICO 1
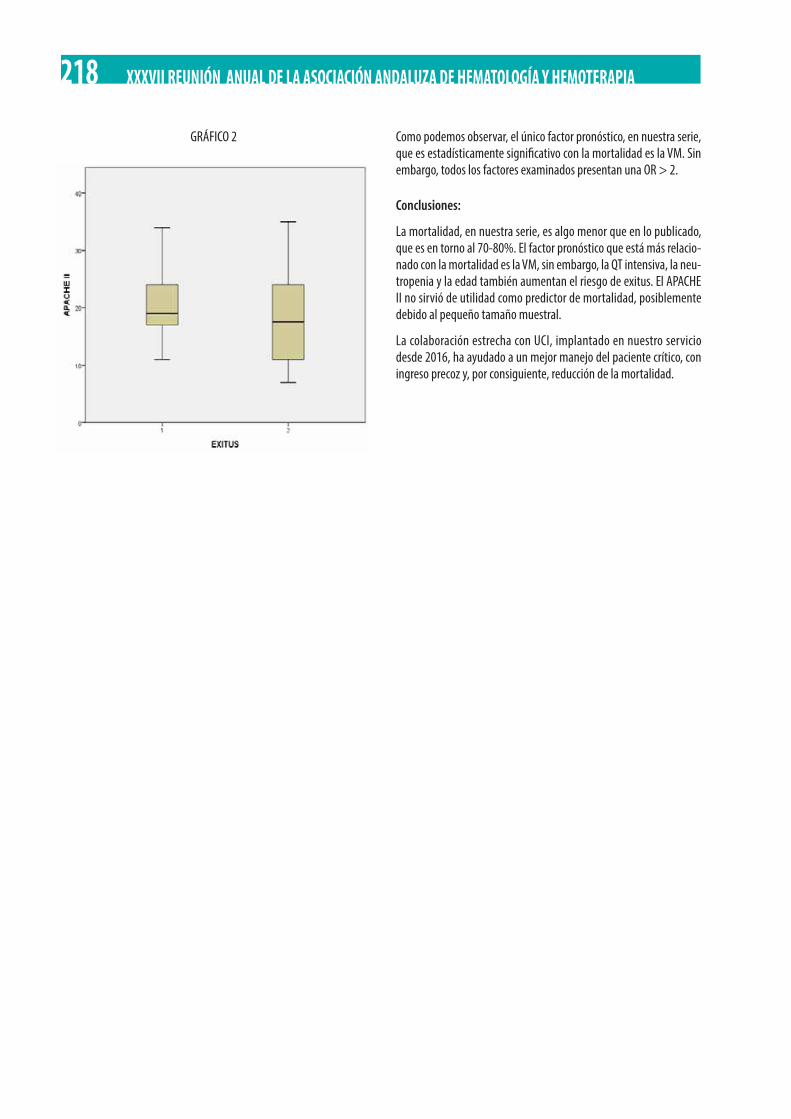
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA218
GRÁFICO 2 Como podemos observar, el único factor pronóstico, en nuestra serie, que es estadísticamente significativo con la mortalidad es la VM. Sin embargo, todos los factores examinados presentan una OR > 2.
Conclusiones:
La mortalidad, en nuestra serie, es algo menor que en lo publicado, que es en torno al 70-80%. El factor pronóstico que está más relacio-nado con la mortalidad es la VM, sin embargo, la QT intensiva, la neu-tropenia y la edad también aumentan el riesgo de exitus. El APACHE II no sirvió de utilidad como predictor de mortalidad, posiblemente debido al pequeño tamaño muestral.
La colaboración estrecha con UCI, implantado en nuestro servicio desde 2016, ha ayudado a un mejor manejo del paciente crítico, con ingreso precoz y, por consiguiente, reducción de la mortalidad.

COMUNICACIONES PÓSTERS: Hematología Clínica 219
INTRODUCCIÓN/FUNDAMENTO:
El linfoma MALT (7-10% de los linfomas no Hodgkin) está asociado a estimulación crónica por agentes infecciosos o enfermedades au-toinmunes.
Tradicionalmente en nuestra área hospitalaria los linfomas han sido tratados por las unidades de Oncología y Hematología.
OBJETIVO:
Estudio observacional retrospectivo de los pacientes diagnosticados de linfoma MALT en nuestro centro entre 2.001 y 2.015.
PACIENTES, MATERIAL Y MÉTODO:
En nuestra recogida de pacientes un 90% tenían más de 50 años de edad, con una mediana de edad de 65.5 años y una media de 63.4 años. La distribución por sexos distingue a 7 hombres y 3 mujeres. El 50% de los pacientes fueron éxitus, 2 de los cuales estaban en progresión de la enfermedad y los 3 restantes estaban en remisión completa; de éstos 3 pacientes, 2 fallecieron por causas ajenas a su hemopatía: uno por una neoplasia de pulmón y otro por un carcinoma epidermoide cuero cabelludo, desconociéndose la causa de la muerte del otro paciente.
RESULTADOS Y CONCLUSIONES:
El 80% de los pacientes consiguieron una remisión completa de su enfermedad, mientras que los 2 pacientes restantes progresaron y fallecieron (uno de ellos por shock séptico).
Un 30% estaba relacionado con Helicobacter pylori. Un 40% presenta-ban un estadío localizado frente a un 60% que presentaban un estadío más avanzado. Además, el 60% de los pacientes tenían síntomas B al diagnóstico y un paciente debutó con una masa bulky retroperitoneal. 8 pacientes fueron diagnosticados de linfoma MALT gástrico, uno de linfoma MALT intestinal y otro de linfoma MALT de glándula parótida.
Recibieron CHOP más tratamiento quirúrgico (gastrectomía) 2 pacien-tes, uno de los cuales también recibió radioterapia. Sólo recibieron CHOP 3 pacientes. Un paciente consig uió remisión de su linfoma MALT gástrico sólo con tratamiento erradicador de H. pylori. Un paciente recibió tratamiento erradicador más FC, 1 paciente recibió CVP. Un pa-ciente recayó tras CHOP y con MINE-ESHAP consiguió remisión com-pleta. Un paciente recayó tras realización de tratamiento erradicador y CHOP, con la aparición de un nódulo pulmonar, por lo que se optó por MINE más radioterapia local, consiguiéndose la remisión completa.
El tratamiento erradicador de H. pylori debería hacerse en todos los ca-sos de linfoma MALT gástrico, aunque éste sea negativo. En la revisión de nuestros pacientes, de un total de un 80% de afectación gástrica se realizó tratamiento erradicador en un 3.5%.
En la enfermedad diseminada no hay un tratamiento estándar: watch and wait, rituximab, CHOP, análogos de las purinas.
La mortalidad intra-tratamiento en nuestra serie fue del 20%. La mor-talidad tras el tratamiento (una vez alcanzada la remisión completa del linfoma MALT) debida a segundas neoplasias fue del 20%.
Estudio descriptivo de los linfomas MALT diagnosticados en nuestro centro en los últimos 15 añosE. Morente Constantín (1), A. Núñez García (1), P. Romero García (2), P.A. González Sierra (1), E. López Fernández (1), M. Jurado Chacón (1)
(1) Complejo Hospitalario Universitario de Granada, (2) Complejo Asistencial de Soria

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA220
INTRODUCCIÓN/FUNDAMENTO:
Las citopenias hematológicas representan una complicación relativa-mente frecuente dentro de las enfermedades autoinmunes sistémicas, por lo que estas deben siempre ser incluidas en el diagnóstico etioló-gico y abordaje de las mismas.
Por otra parte, el Síndrome de Evans, es una enfermedad atípica e infrecuente, autoinmune, de etiología idiopática o secundaria, que cursa con citopenias de aparición simultánea o secuencial, presen-tándose tanto en adultos como en niños, y representando un desafío diagnóstico y terapéutico para los hematólogos, siendo frecuente su asociación con el Lupus eritematoso sistémico (LES).
El LES es una enfermedad autoinmune crónica, de difícil diagnóstico, con afectación multiorgánica, que predomina en mujeres jóvenes, siendo su curso heterogéneo y de carácter intermitente (brotes), con un tratamiento amplio y variable, involucrando a una gran cantidad de especialistas en su proceso asistencial y manejo.
OBJETIVO:
Revisión del Síndrome de Evans asociado a LES, en relación a un caso diagnosticado en nuestro centro.
PACIENTES, MATERIAL Y MÉTODO:
Paciente con Anemia hemolítica autoinmune (AHAI) y posterior Púr-pura trombocitopénica inmune (PTI), diagnosticada de Síndrome de Evans con 12 años de edad. Recibió tratamiento corticoideo a dosis altas e Inmunoglobulinas, sin obtener respuesta mantenida. Se rea-lizó esplenectomía con recaída dos meses después por trombopenia de 2 x109/L. Como tratamiento de 2-3º línea recibió Ciclofosfamida y Bleomicina (con fibrosis pulmonar secundaria) obteniendo respuesta mantenida durante 5 años, aunque con recaida posterior, recibiendo tratamiento con Azatioprina y corticoides.
Dos años después acude a consultas refiriendo edemas perimaleola-res junto con lesiones cutáneas tipo livedo reticularis, sin objetivarse
deterioro de la función renal ni otra sintomatología añadida. Aparece síndrome nefrótico con proteinuria >300mg/dl en sistemático de orina y en analítica destacaba hiperlipidemia, hipoalbuminemia de 1,61 gr/dl, hiperfibrinogenemia, descenso de C3 y aumento de Ig M. ANA y ANCA y TSH normal. Radiografía de tórax con derrame pleural bilateral leve y ecografía abdominal sin hallazgos.
Se realizó biopsia renal izquierda con diagnóstico anatomopatológico de glomerulonefritis membranosa con glomerulonefritis proliferativa difusa asociada, siendo compatible con nefropatía lúpica.
Nefrología decidió retirar tratamiento con Azatioprina y sustituirlo por Micofenolato Mofetil (CellCept), reduciéndose la protenuria tras mes y medio de tratamiento, con normalización de niveles de albúmina, aunque manteniendo hipercolesterolemia, aumento de IgM y descen-so de C3. ANA y ANCA repetidamente negativos.
Apareció leucocitosis con expansión policlonal de linfocitos T grandes granulares en inmunofenotipo sp, asociado a positividad discreta Ig M al CMV.
Actualmente la paciente continúa en tratamiento con Micofenolato, y se mantiene estable clínica y analíticamente, aunque de forma es-porádica presenta crisis de anemia hemolítica, las cuales presentan buena respuesta a pulsos cortos de corticoides. El resto de las series permanecen normales.
RESULTADOS Y CONCLUSIONES:
– Las citopenias hematológicas son complicaciones relativamente frecuentes en enfermedades autoinmunitarias.
– Para conseguir un diagnóstico etiológico y tratamiento correctos, el abordaje de las citopenias hematológicas debe estar sistematizado y ser multidisciplinar.
– El seguimiento evolutivo de los pacientes a lo largo del tiempo, es de suma importancia para conseguir una correcta filiación y diagnóstico en todas las enfermedades atípicas e infrecuentes.
Citopenia hemtológica en enfermedad autoinmune sistémica: a propósito de un casoJ. Morán Sánchez, F.J. Capote García, A. Santisteban Espejo, M.C. Fernández Valle
Hospital Universitario Puerta del Mar

COMUNICACIONES PÓSTERS: Hematología Clínica 221
INTRODUCCIÓN/FUNDAMENTO:
La coexistencia de dos o más formas distintas de linfoma en un mismo paciente se denomina linfoma compuesto (LC). Es un hallazgo poco común, con pronóstico global del tipo histológico más agresivo. Lo más común es la presencia simultánea de linfoma folicular (LF) y difuso de células grandes (LDCGB).
OBJETIVO:
Describimos 3 casos de LC con LDCGB como nexo común, con diferentes líneas de tratamiento y mismo resultado, remisión completa (RC) en todos ellos.
PACIENTES, MATERIAL Y MÉTODO:
1. Varón de 64 años, en TAC se objetiva masa en cavum y afectación ganglionar supra e infradiafragmática. Biopsia de cavum con diagnostico de LDCGB centrogerminal. Debido a crecimiento de cavum con compromiso respiratorio inicia CHOP + Metotrexate + triple intratecal (TIT). La biopsia de medula ósea es diagnostica de Linfoma de Células del Manto clásico (LCMc). Se concluye en coexistencia de LDCGB centrogerminal ganglionar + LCMc medu-lar, estadio IV-A, IPI 5, MIPI 7.3. Por pronóstico más adverso, se decide continuar con esquema VR-CAP + TIT para LCMc de forma ambulatoria, completa 6 ciclos con PET-TAC post-3º y 6º ciclo en RC. Como tratamiento de consolidación se realiza autotrasplante de progenitores hematopoyéticos (autoTPH) el 28/10/16 previo acondicionamiento BEAM. Actualmente persiste en RC.
2. Varón de 67 años, exfumador y pluripatológico, diagnosticado de LF de bajo grado (II REAL) en biopsia axilar izquierda. En PET-TAC afectación ganglionar supra e infradiafragmática e infiltración muscular (psoas izquierdo) y ósea (ilíaco izquierdo). La médula
ósea está infiltrada por LDCGB sin especificar (NOS). Se concluye en LF (grado II REAL) ganglionar + LDCGB NOS medular, estadío IVe-A, IPI 3. Se trata con R-CHOP + Metotrexato + triple intratecal (TIT) x 6. PET-TAC post-3º y 6º ciclo en RC. Actualmente tratamiento con Rituximab bimensual x 2 años.
3. Varón de 82 años, pluripatológico, diagnosticado tras biopsia de adenopatía axilar izquierda de linfoma B inclasificable con carac-terísticas intermedias entre LDCGB NOS y Enfermedad de Hodgkin clásica (EHc). Se acompaña de síntomas B. En PET-TAC presenta afectación supra-infradiafragmática y bazo. Médula ósea sin in-filtración. Recibe R-CHOP + Bleomicina a 2/3 de dosis x 6, con remisión parcial (RP) tras 3er ciclo pero progresión axilar izquier-da tras el 6º. Se realiza exéresis del ganglio afectado con misma histología que al diagnóstico. En PET-TAC (12/05/2016) se confirma progresión a nivel hepático, esplénico y óseo. Inicia R-GPD a 1/2 de dosis x 6, alcanzando RP tras los 3 primeros. Sin embargo en la eva-luación post 6ºciclo con PET-TAC (25/11/16) presenta aumento de la extensión de la afectación ósea preexistente y nuevas lesiones, con reaparición de focos patológicos en hígado y bazo compatibles con malignidad. Ante progresión, se inicia nueva línea con Bren-tuximab, alcanzando RC tras 3 ciclos por PET-TAC (20/02/2017). Actualmente administrados 5 ciclos.
RESULTADOS Y CONCLUSIONES:
1. El diagnóstico de LC está aumentando a pesar de ser casos raros en literatura, quizás por disponer de técnicas de mayor precisión.
2. El LDCGB, tipo más frecuente de LNH, se ve implicado en la mayoría de casos, no pudiendo aseverar de forma concluyente presentación en paralelo ó transformación.
3. Ante la presencia de dos tipos histológicos diferentes, debe uti-lizarse el tratamiento correspondiente a la forma más agresiva.
Linfoma Difuso de Células Grandes B coexistente con otros linfomasF.J. Cabrera Ruiz (1), A. González Fernández (1), M. Espeso de Haro (2)
(1) Hospital Universitario Virgen de la Victoria, Málaga, (2) Hospital Regional Universitario, Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA222
INTRODUCCIÓN/FUNDAMENTO:
Los niveles de autosuficiencia en hemoderivados destinados a la transfusión sanguínea en Andalucía nos obligan a replantear el tipo de donación en los donantes altruistas de nuestra comunidad.
Somos autosuficientes y cubrimos las necesidades del fraccionamiento primario (hematíes,plaquetas,leucocitos), mientras que el resto de hemoderivados, como factor VIII, factor IX, albúmina,inmunoglobulinas intravenosas,antitrombina III y alfa-1-antitripsina siguen siendo un reto de la hemoterapia andaluza. Con este fin,la red transfusional de Andalucía se propone,mediante una campaña de plasmaféresis a nivel autonómico,aumentar la obtención de plasma para el fraccionamiento industrial.
OBJETIVO:
Poner de relieve la importancia de la donación de plasma.
PACIENTES, MATERIAL Y MÉTODO:
Las acciones para mejorar el nivel de autosuficiencia en los componentes plasmáticos comprenden el aumento de las donaciones, la optimización de la obtención del plasma en los procesos de separación de sangre total,la racionalización del uso de los hemoderivados,la optimización de las entregas rechazadas por la industria,el aumento de los rendimientos por parte de la industria y establecer los programas específicos de plasmaféresis. Para este último objetivo,se ha diseñado una campaña de plasmaféresis en la comunidad autónoma andaluza que tiene como objetivo aumentar el plasma disponible para fraccionamiento industrial hasta 75000 litros.
El diseño de la campaña comprende elegir de 5 a 10 colectas de más de 100 donantes que se repitan cuatro o más veces al año en cada provincia, con el objeto de obtener 30 plasmaféresis en cada una. Las
7800 plasmaféresis sumadas a los litros de plasma enviados en el año 2015 podrían completar el objetivo total.
El CRTS de Granada-Almería ha iniciado esta campaña organizado este año 2.016 una colecta extraordinaria, a lo largo de todo un día, exclusivamente de plasma, solicitando mediante carta la donación a donantes conocidos de plasma. Se emplearon 21 máquinas de plasmaféresis, solicitándose algunas de las mismas a otros centros de donación de España (Sevilla y Barcelona). Hubo un total de 109 donaciones,107 de donantes conocidos (62 hombres y 45 mujeres) y 2 de nuevos donantes (1 hombre y una mujer). El mayor número de donaciones entre los hombres se produjo en el grupo de edad comprendido entre los 41 y los 50 años,mientras que en las mujeres tuvo lugar en el grupo de edad de los 51 y los 60 años. En cuanto a las donaciones por grupo, la mayoría fueron del grupo A+, seguido de 0+, A-, B+, AB+, 0-, AB- y B- (éstos 2 últimos grupos con un solo donante cada uno),en este orden.
De cada 42 bolsas de plasma de +/- 200 mL se obtienen 21 viales de albúmina,1.3 viales de factor VIII,3 viales de IGIV,1 vial de A1P1 y 1 vial de FIX.
RESULTADOS Y CONCLUSIONES:
El aumento de la disponibilidad de componentes plasmáticos procedentes de plasma de donantes altruistas es un indicativo importante de mejora en la seguridad transfusional.
El fraccionamiento industrial del plasma procedente de donaciones altruistas y gestionado su fraccionamiento desde las instituciones públicas supone un mayor nivel de eficiencia económica para el siste ma sanitario público.
La experiencia con este tipo de campañas de promoción de la donación de plasma se objetiva como un método eficaz de aumentar la disponibilidad de plasma para su fraccionamiento.
El CRTS de Granada-Almería organiza la primera donación masiva de plasma a nivel nacionalE. Morente Constantín (1), A. López Berrio (1), P. Romero García (2), M.A. García Ruíz (1), M.A. Hernández Vidaña (1), M. Jurado Chacón (1)
(1) Complejo Hospitalario Universitario de Granada, (2) Complejo Asistencial de Soria

COMUNICACIONES PÓSTERS: Hematología Clínica 223
INTRODUCCIÓN/FUNDAMENTO:
A pesar de la mejoría en la estratificación de los pacientes con síndrome mielodisplásico (SMD) de bajo riesgo (Bajo/intermedio-1por IPSS1 y muy bueno/bueno/intermedio por IPSS-R2) a partir de scores predictivos específicamente desarrollados en estos subgrupos3,4, existe una gran heterogeneidad desde el punto de vista pronóstico, siendo la severidad de las citopenias y el cariotipo los principales factores de los que dependen esta variabilidad clínica. El subgrupo de pacientes con parámetros clínico/biológicos menos desfavorables está escasamente caracterizado.
OBJETIVO:
Descripción de la cohorte de pacientes con SMD-BR con parámetros clínico menos desfavorables, análisis de supervivencia de esta cohorte y comparación indirecta con la población de referencia sin diagnóstico de esta hemopatía.
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo unicéntrico sobre 90 pacientes con SMD-BR y características no desfavorables según los scores pronósticos actuales [citopenias no significativas (hemoglobina >10 g/dL; plaquetas >100x10e/L y recuento de neutrófilos >1x10e/L), cariotipo no
desfavorable y porcentaje de blastos <5%]. Periodo de estudio: 1990-2016. Variables analizadas: Mediana de supervivencia global (SG), causas de éxitus y su relación con el SMD así como comparación de la estimación de SG de este grupo de pacientes frente a la expectativa vital de la población de referencia sin diagnóstico de hemopatía (Fuente: Instituto Nacional de Estadística, año 2015. www.ine.es). Se evalúan como causa de éxitus relacionadas con el SMD5 las siguientes: progresión leucémica, infección, hemorragia o exacerbación comorbilidad tras progresión de citopenias.
RESULTADOS Y CONCLUSIONES:
La mediana de edad al diagnóstico fue 74 años (rango: 48-87). El subtipo más frecuente fue la anemia refractaria con sideroblastos en anillo. El resto de las características basales se describen en la tabla 1. En cuanto a la actitud terapéutica, la observación sin necesidad de tratamiento fue mayoritaria, siendo un reducido porcentaje de pacientes los que precisaron tratamiento con agentes eritropoyéticos/G-CSF durante el periodo de estudio. Únicamente el 7% de los pacientes experimentó progresión a leucemia aguda. La causa más frecuente de éxitus fue no relacionada con el SMD. Con una mediana de seguimiento de 5.2 años (IC 95%: 4.5-5.8 años), 30 pacientes habían fallecido, con una mediana de SG: 8.6 años (IC 95%: 5.5-11.7 años. Figura 1). Respecto a la población de referencia en nuestro medio a la
Estudio descriptivo de las características clínico/biológicas y supervivencia en pacientes con síndrome mielodisplásico de bajo riesgo con variables pronósticas no desfavorablesWerner González Molina, José Francisco Falantes González, Ildefonso Espigado Tocino, Cristina Calderón Cabrera,
Francisco José Márquez Malaver, José Antonio Pérez Simón
Hospital Universitario Virgen del Rocío
FIGURA 1
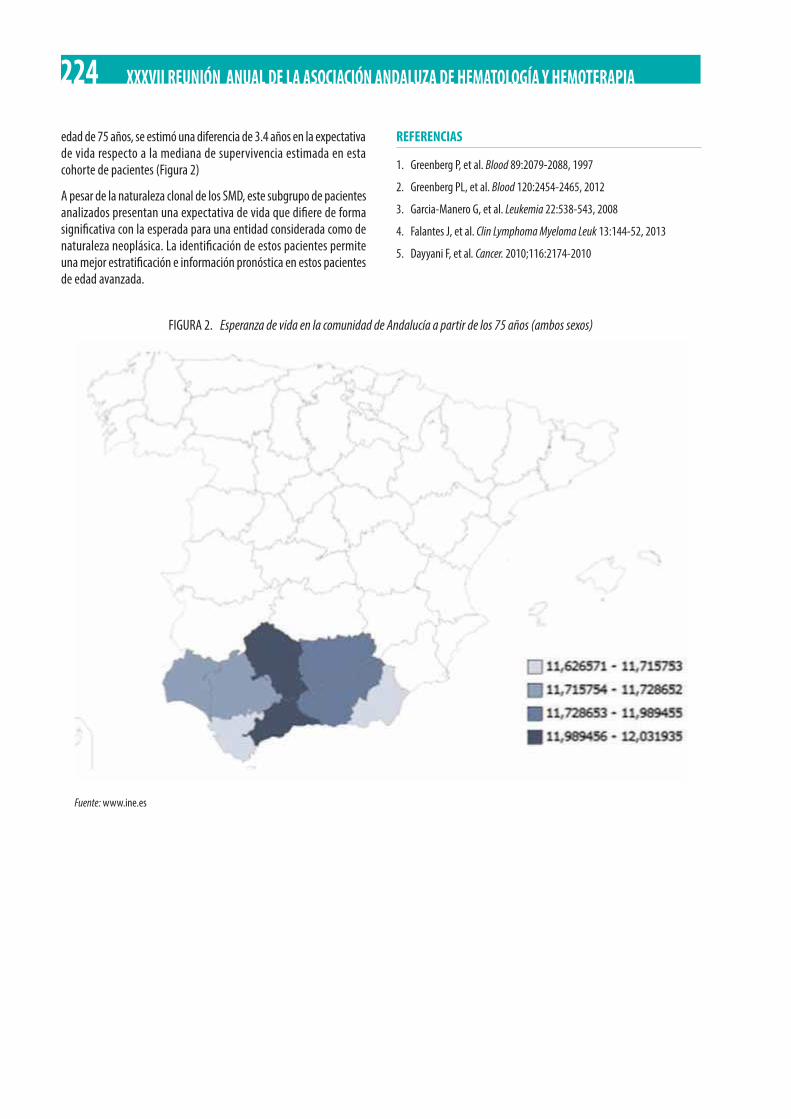
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA224
FIGURA 2. Esperanza de vida en la comunidad de Andalucía a partir de los 75 años (ambos sexos)
edad de 75 años, se estimó una diferencia de 3.4 años en la expectativa de vida respecto a la mediana de supervivencia estimada en esta cohorte de pacientes (Figura 2)
A pesar de la naturaleza clonal de los SMD, este subgrupo de pacientes analizados presentan una expectativa de vida que difiere de forma significativa con la esperada para una entidad considerada como de naturaleza neoplásica. La identificación de estos pacientes permite una mejor estratificación e información pronóstica en estos pacientes de edad avanzada.
REFERENCIAS
1. Greenberg P, et al. Blood 89:2079-2088, 1997
2. Greenberg PL, et al. Blood 120:2454-2465, 2012
3. Garcia-Manero G, et al. Leukemia 22:538-543, 2008
4. Falantes J, et al. Clin Lymphoma Myeloma Leuk 13:144-52, 2013
5. Dayyani F, et al. Cancer. 2010;116:2174-2010
Fuente: www.ine.es

COMUNICACIONES PÓSTERS: Hematología Clínica 225
INTRODUCCIÓN/FUNDAMENTO:
Bajo el término de mastocitosis se engloba un grupo de trastornos que se caracterizan por excesiva acumulación de mastocitos en los tejidos.Clasificada en cutánea o sistémica,abarcando también los tumores de mastocitos; además, existe una variante asociada a otras enfermedades hematológicas clonales con células no masto-citos. Sus síntomas pueden ser cutáneos(prurito, sofocos, ampollas , Darier),derivados de la liberación de mediadores de los mastocitos y consecuencia de la infiltración de órganos extracutáneos(médula ósea,tracto intestinal,hígado,tejido linfoide y bazo).
OBJETIVO:
Estudio observacional retrospectivo sobre la experiencia de nuestro centro en el manejo y evolución de los pacientes con mastocitosis.
PACIENTES, MATERIAL Y MÉTODO:
17 pacientes diagnosticados de mastocitosis desde 1.992 hasta 2.015. 12 varones y 5 mujeres, con una edad de tres a 70 años al diagnóstico.A nivel genético, 9 demostraron mutación del c-kit, destacando el sub-tipo D816V (presente en 8).
La forma más frecuente fue la sistémica,11 afectados. Se diagnostica-ron otras enfermedades hematológicas posteriormente:mielofibrosis, leucemia mieloide aguda y síndrome mielodisplásico.Órganos afec-tados en la forma extracutánea:hígado,médula,bazo y ganglios lin-fáticos.
El grado de infiltración de la médula ósea fluctuó de un 0,0001% a un 0,32%.10 pacientes tuvieron afectación dermatológica,destacando: urticaria(8) frente a las telangiectasias(3).
Analíticamente,12 pacientes presentaron histaminuria con valores en-tre los 65 y los 133.000 mcg/L.Se determinó la triptasa,siendo positiva en 12 con valores entre 3,81 y 189 mcg/L.
Cuatro pacientes presentaron sín tomas emocionales (ansiedad, depresión, insomnio) y u n caso importante deterioro del bienestar emocional y social, sin hallar relación con los niveles de triptasa, ni de infiltración medular.
Tratamiento: 10 pacientes recibieron tratamiento específico: prednisona,metotrexate,interferón y cromoglicato. Administrada quimioterapia citorreductora (hidroxiurea) a 4 pacientes.12 recibieron tratamiento sintomático (antihistamínicos,antileucotrienos,omeprazol,bifosfonatos).Se aconsejó evitar desencadenantes de la degranulación de mastocitos (calor,estrés emocional agudo,ejercicio extenuante,alcohol,picantes),con gran variación respecto a la tolerancia interindivual a estos desencadenantes.
2 pacientes fueron exitus: una fiebre sin foco en el paciente con mie-lofibrosis y una hemorragia cerebral masiva.
RESULTADOS Y CONCLUSIONES:
Se requiere tratamiento por un equipo multidisciplinar.
La fase emocional de la enfermedad es muy importante y no parece estar relacionada con el grado de infiltración por los mastocitos, pues incluso las formas con menor afectación mastocítica pueden presentar una calidad de vida muy deteriorada, pudiendo deberse a la liberación de células cebadas, lo que conllevaría el planteamiento de tratamiento específico de la enfermedad a pesar de tener controlada otra sintoma-tología (cutánea…)o con baja infiltración mastocítica.
La sintomatología cutánea se encuentra controlada,siendo la sintoma-tología emocional la más difícil de corregir.Considerable variación con respecto a la tolerancia de los desencadenantes específicos.
El diagnóstico y tratamiento precoz mejora su calidad de vida.Los antecedentes de la sintomatología en los pacientes con mastocitosis se remontan en casos años atrás del diagnóstico, con visitas a múl-tiples especialistas hasta alcanzar el diagnóstico. El peor pronóstico en nuestra serie de casos se dió en las mastocitosis que desarrollaron enfermedades hematológicas, a pesar del tratamiento de las mismas.
La experiencia de nuestro centro en pacientes diagnosticados de mastocitosisE. Morente Constantín, A. Núñez García, F. Hernández Mohedo, P. Navarro Álvarez, R. Ríos Tamayo, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA226
INTRODUCCIÓN/FUNDAMENTO:
La hemofilia adquirida (HA) es un trastorno autoinmune causado por autoanticuerpos contra epítopos activos del factor VIII, caracterizado por la aparición de sangrados. Pese a su infrecuencia, nos encontramos frente a una entidad con una gran importancia debido a la morbimor-talidad asociada y a las dificultades que puede llegar a presentar para su diagnóstico.
OBJETIVO:
Estudio observacional retrospectivo evaluando clínica, tratamiento y evolución de los pacientes diagnosticados de HA en nuestro centro entre 2.003 y 2.015.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos 6 pacientes con 7 episodios de hemofilia adquirida. El 66% de los pacientes son mujeres con una edad media de 53 años (29-84) y todos l os casos se diagnosticaron a raíz de sangrados: 6 de los episodios fueron sangrados cutáneos y musculares y uno de los ca-sos, que apareció en el postoperatorio de una cirugía de hernioplastia inguinal, fue un sangrado postquirúrgico excesivo.
Como antecedentes, 2 de los pacientes se encontraban en remisión de tumores de órgano sólido y una paciente había presentado un cuadro autoinmune con trombopenia y una determinación positiva de anti-cuerpos anticardiolipina (no confirmada posteriormente).
Dentro de los posibles desencadenantes tenemos 2 casos postparto y uno pocos meses tras un aborto tardío.
Todos los pacientes presentaban, al diagnóstico, niveles de factor VIII por debajo del 5% con una media de 1.4% (0%-4%) y una media de 17.31 UB (2.5-204.8).
En cuanto al tratamiento, 6 de los 7 episodios precisaron tratamiento hemostático con factor VIIa a dosis altas. El otro caso se trataba de
un sangrado cutáneo que se trató únicamente con antifibrinolíticos orale s.
Para la erradicación del inhibidor se admini straron esteroides asocia-dos a ciclofosfamida en 3 casos (43%), a rituximab en 2 casos (28.5%); se asoció ciclofosfamida y rituximab en una paciente y azatioprina en otra.
RESULTADOS Y CONCLUSIONES:
Los pacientes respondieron en 6 de los 7 episodios (85.7%) al trata-miento inmunosupresor con recuperación completa de la hemostasia y cese de la clínica de sangrado.
No obstante, de los 6 pacientes iniciales, 3 fueron éxitus (50%): 2 por complicaciones infecciosas del tratamiento inmunosupresor y una paciente por falta de respuesta.
La única paciente que no respondió se trató de una mujer que, tras un primer episodio tratado con corticoides y rituximab, presentó una recaída a los 3 años sin mejoría con tratamiento con ciclofosfamida, pero con un rápido deterioro físico, por el cual fallece.
CONCLUSIONES:
En nuestra experiencia, el factor VII recombinante demostró ser eficaz en el control de la clínica hemorrágica en todos los casos en los que s e usó. En cuanto al tratamiento inmunosupresor, nuestra muestra no permite analizar diferencias entre los distintos fármacos usados pero, con las opciones utilizadas, nos encontramos con tasas de respues-tas muy altas, si bien esto puede estar influido porque 3 de nuestros pacientes se trataban de hemofilias adquiridas relacionadas con la gestación, las cuales presentan un mejor pronóstico en todas las series.
Por otro lado, el hecho de que 2 de los pacientes fallecieran por com-plicaciones infecciosas nos hace pensar que, quizás, en una pobla-ción tan añosa, deberíamos ser muy cuidadosos con el tratamiento inmunosupresor, tratando de forma individualizada y planteando la administración de profilaxis antiinfecciosa.
Casos de hemofilia adquirida vistos en nuestro hospital durante los últimos 12 añosD. Jiménez Fernández, E. Morente Constantín, L. Entrena Ureña, M.J. Gutiérrez Pimentel, M.A. García Ruiz, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

COMUNICACIONES PÓSTERS: Hematología Clínica 227
INTRODUCCIÓN/FUNDAMENTO:
La supervivencia de los pacientes trasplantados renales ha mejorado en las últimas décadas, en parte como resultado de la introducción de nuevos fármacos inmunosupresores para prevenir el rechazo del aloinjerto. Sin embargo, la inmunosupresión prolongada en pacientes con trasplante de órgano sólido se relaciona con aumento de desarrollo de infecciones oportunistas y de un incremento de riesgo de neoplasias malignas, apoyando la oncogénesis causada por ciertos virus así como el deterioro de la vigilancia inmune. De hecho, la neoplasia maligna es la tercera causa más frecuente de muerte (después de eventos cardio-vasculares e infección) entre los receptores de trasplante renal. Aunque el desarrollo de la leucemia aguda es infrecuente en estos pacientes, está descrito que su incidencia aumenta cinco veces en los pacientes trasplantados renales.
OBJETIVO:
Presentamos dos casos de Leucemia Aguda (LA) postrasplante renal diagnosticados en un Hospital de Tercer nivel, durante el último año (2016-2017).
PACIENTES, MATERIAL Y MÉTODO:
CASO 1: Varón de 44 años, antecedentes de nefrectomía izquierda (año 2011 postraumatismo) y trasplante renal de donante vivo en Abril de 2012, incompatibilidad A, 2 B y 0 DR. Inmunosupresión (IS) recibida: Basiliximab, y posteriormente Prednisona, Tacrolimus y Mofetil Mi-cofenolato (MMF), con retirada esteroidea en 2013. En Septiembre
de 2016, se diagnostica de Leucemia Aguda Linfoblástica B común con cromosoma Filadelfia positiva (LAL Ph+), recibiendo tratamiento según Protocolo de PETHEMA LAL PH-08 (Inducción + Consolidación), e Imatinib 600 mg/día. Ha mantenido tratamiento con Prednisona y Tacrolimus. Evolución: remisión completa tras inducción con EMR negativa. Candidato a Trasplante alogénico de progenitores hemato-poyéticos (AloTPH) de donante no emparentado.
CASO 2: Varón de 42 años, antecedentes de enfermedad renal crónica estadio 5 secundaria a uropatía obstructiva, hemodiálisis en Abril de 2010 y trasplante renal de donante cadáver en Diciembre de 2011. IS recibida: Prednisona, Tacrolimus y MMF. En Abril 2012 se intenta retirada esteroidea por mal control de DM, frenándose por deterioro de función renal. En Marzo de 2017 se diagnostica de LA de linaje ambi-guo T/mieloide con cariotipo tetraploide. Actualmente en tratamiento con esquema de Inducción PETHEMA LAL-AR.
RESULTADOS Y CONCLUSIONES:
La LA postrasplante renal es una entidad muy infrecuente; en nuestra experiencia, la administración de quimioterapia intensiva concomitan-te al tratamiento inmunosupresor e incluso la utilización de imatinib no compromete el injerto renal y se muestra eficaz y segura. En los pacientes candidatos a AloTPH, debemos considerar también el HLA del donante renal para disminuir riesgo de rechazo del riñón trasplan-tado. Serían necesarios estudios prospectivos que permitan ampliar el conocimiento de los mecanismos carcinogénicos asociados con el trasplante de órganos y ayuden al diseño de estrategias terapéuticas de prevención de neoplasias en estos pacientes.
Leucemia aguda postrasplante renal: revisión de dos casosN. Alkadi Fernández (1), M. Ruiz Mercado (2), M.I. Montero (2), T. Caballero (2), J. González Campos (2), J.A. Pérez-Simón (2)
(1) Hospital Universitario Virgen del Rocío, (2) Hospital Universitario Virgen del Rocío

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA228
INTRODUCCIÓN/FUNDAMENTO:
La trombocitopenia inmune primaria (TIP) representa la trombocito-penia más frecuente en la infancia, presentándose en la mayoría de los casos, de forma aguda con resolución espontánea y sin identificarse un trastorno sistémico relacionado.
OBJETIVO:
Evaluar la TIP en la infancia diagnosticadas en nuestro centro desde hace 40 años. Hemos realizado un estudio retrospectivo que incluyó a 29 pacientes con TIP diagnosticados entre 0-15 años. Se recogieron antecedentes personales y familiares, manifestaciones hemorrágicas e infecciosas, datos analíticos (cifra de plaquetas, autoinmunidad y serología infecciosa) al diagnóstico, tratamientos y evolución clínica.
PACIENTES, MATERIAL Y MÉTODO:
Se analizaron 29 pacientes (52% varones y 48% mujeres), edad me-dia 6 años. Presentaban antecedentes familiares 3 pacientes, 2 con lupus eritematoso sistémico (LES) y 1 con síndrome antifosofolípido primario(SAF). Recuento de plaquetas inferior a 5x109 en 9 de ellos. Petequias y equimosis en 27 y sólo 2 sin manifestaciones. Los niveles de ANA fueron positivos en 3 casos (1 con niveles en el límite superior sin otros criterios sugestivos de enfermedad sistémica, un segundo caso con aPL positivo diagnosticado de LES y el tercero diagnosticado de posible LES que falleció por complicaciones de la misma), antiti-
roideos en 3 y aPL en 6 confirmándose este último en 1 sólo paciente. Serología positiva aguda para citomegalovirus en 2 casos, VHB en 4, VHS en 3 y Helicobacter pylori en 3. Tratamiento inicial: inmunoglo-bulinas con buena respuesta en 21 pacientes, corticoides con buena respuesta en 4 y esplenectomía en 8 con buena respuesta en 2 de ellos. 2 pacientes con los nuevos agentes agonistas de la trombopo-yetina con buena respuesta en ambos. Un paciente refractario a todo. Presentan una evolución crónica 5 pacientes, 8 persistente y 16 dados de alta actualmente.
RESULTADOS Y CONCLUSIONES:
La respuesta de nuestros pacientes al tratamiento es muy variable, con una mejor respuesta al tratamiento en primera línea con inmu-noglobulinas, con mayor refractariedad en pacientes que recibieron corticoides o esplenectomía como tratamiento inicial. En la actualidad 26 presentan cifras de plaquetas óptimas sin tratamiento, de ellos 16 dados de alta, 2 están en tratamiento de mantenimiento con esteroi-des y un paciente con agentes agonistas de la trombopoyetina.
Dos de los tres pacientes con ANA positivo que han desarrollado LES en la infancia, en los 3 casos las cifras de plaquetas al diagnóstico no descendieron por debajo de 5x109, precisando distintas líneas de tratamiento por refractariedad, siendo el tratamiento de elección es el de su enfermedad de base, de ahí la importancia de la realización al diagnóstico de un estudio completo de autoinmunidad para filiar trombocitopenias secundarias.
Evaluación de la Trombocitopenia Inmune Primaria (TIP) en la infancia. Experiencia en nuestro centroR. Zapata Bautista (1), S. Ramírez García (2), Mª. A. Ruíz Cobo (2), I. Vazquez-Pastor Jiménez (2), B. Díaz Roldan (2), J. N. Rodríguez Rodríguez (2)
(1) Hospital Juan Ramón Jiménez , (2) HJRJ

COMUNICACIONES PÓSTERS: Hematología Clínica 229
INTRODUCCIÓN/FUNDAMENTO:
La incidencia en España de Síndrome Linfoproliferativo (SLP) es de 30 a 70 nuevos casos cada año por millón de habitantes año. La indicación de autotrasplante de progenitores hematopoyético (TASPE) varía en función del SLP, siendo de primera línea, tras inducción previa con quimioterapia, en pacientes con Linfoma cerebral primario, Linfoma del Manto y LBDCG con afectación del SNC, y relegada a progresión de la enfermedad y/o recaídas en el resto de linfomas.
OBJETIVO:
Estudio retrospectivo de los pacientes con SLP en los que se ha reali-zado TASPE desde abril de 2011 a enero de 2017 que recoge las carac-terísticas evaluadas previamente y durante el procedimiento.
PACIENTES, MATERIAL Y MÉTODO:
Se realizaron 42 TASPE, 29 varones, 13 mujeres TASPE con una mediana de edad 46 años siendo 8 de ellos mayores de 60 años y de estos 4 ma-yores de 65. 81 % con linfoma no Hodgkin (LNH) y 19 % con linfomas de Hodgkin. De los LNH un 18 con linfoma B difuso de célula grande (LBDCG), 5 con linfoma folicular (LF), 4 L. Manto (LM), 2 linfomas T y 4 otros linfomas. La mediana de tiempo desde el diagnóstico de la enfermedad hasta el TASPE ha sido de 15.5 meses, recibiendo desde el diagnóstico diferentes líneas de tratamiento, trasplantándose en un 4,8% con una sola línea previa, un 69% con dos líneas previas, un 16,7% con tres líneas previas y un 9,5% con más de tres líneas. El estado alcanzado de los pacientes al TASPE fue en un 45% en respuesta
completa (RC) y un 53% en respuesta parcial (RP), un 2 % sin filiar. En cuanto a la evaluación del estado general del los pacientes presenta-ban comorbilidades asociadas un 10 %, con un Karnosfky mayor de 80 en todos los pacientes. Como régimen de acondicionamiento se utilizó BEAM en la totalidad de los pacientes salvo uno con linfoma cerebral primario que se utilizó BCNU/tiot Se infundieron progenitores hematopoyéticos de sangre periférica con una mediana de 6,2 × 10e6 CD34 +/kg y un volumen medio de DMSO de 21 mL, presentando un 35% toxicidad al DMSO de forma leve. No hubo fallo de implante. Los neutrófilos ≥ 0,5 × 10e9/L y plaquetas ≥ 20 × 10e9/L se recuperaron con una mediana de 11 días y 10 días respectivamente. Los pacien-tes fueron dados de alta con una mediana de ingreso de 25 días. Las complicaciones más frecuentes fueron digestivas (náuseas, vómitos y diarrea) en su mayoría de grado leve, mucositis en un 38% de los casos en grado III-IV y neutropenia febril en un 74 %, con aislamiento microbiológico en un 25,6% de los casos. Otras complicaciones de for-ma aislada fueron alteraciones hidroelectrolíticas, ansiedad, arritmias y dolor torácico. No ha habido muertes relacionadas con el TASPE ni antes de los 100 días postTASPE. Han fallecido 3 de los pacientes, por progresión de la enfermedad, estando 39 pacientes vivos con una me-diana de supervivencia de 26 meses tras el TASPE.
RESULTADOS Y CONCLUSIONES:
Nuestra experiencia como centro trasplantador realizando TASPE en pacientes con SLP nos ha permitido realizar este procedimiento en su mayoría como opción terapéutica de rescate con buenos resultados. Las complicaciones durante el mismo han sido bien toleradas por los pacientes con una buena evolución postTASPE.
Evaluación de la experiencia como centro trasplantador en pacientes con Síndromes linfoproliferativos en nuestro centroR. Zapata Bautista, A. Palma Vallellano, I. Vázquez-Pastor Jiménez, E. Gil Espárraga, K. Gómez Korrecha, J.F. Domínguez
Hospital Juan Ramón Jiménez
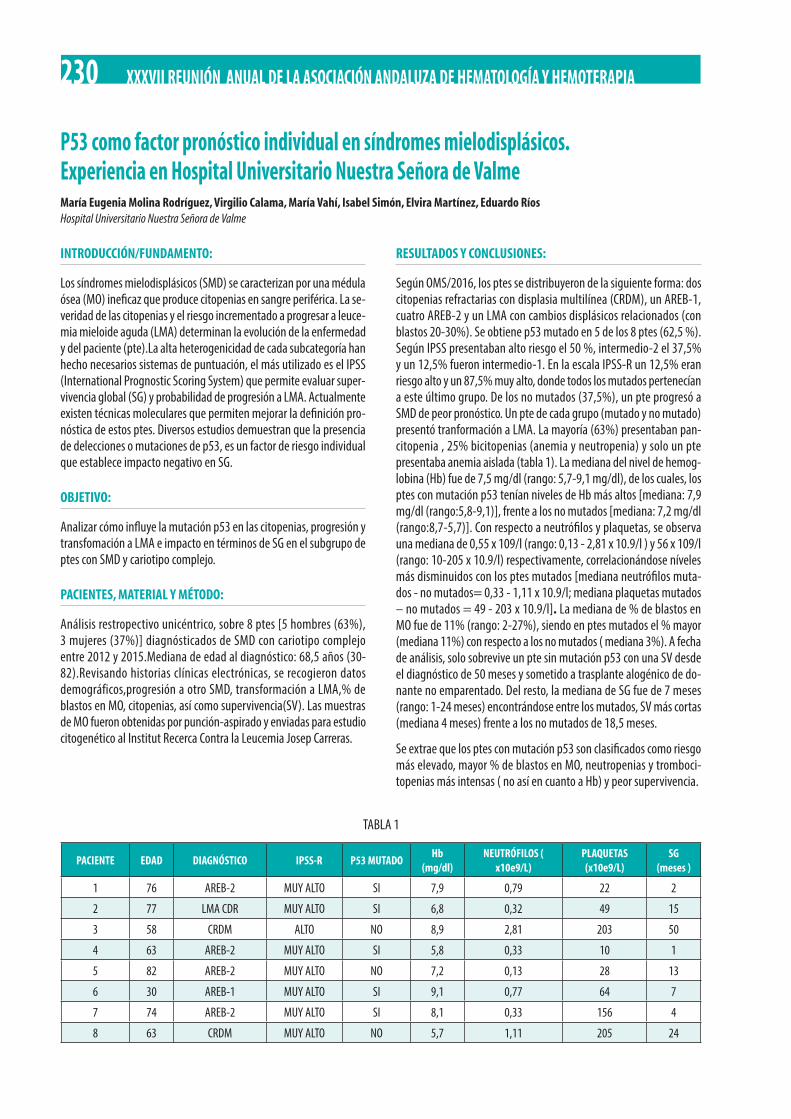
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA230
INTRODUCCIÓN/FUNDAMENTO:
Los síndromes mielodisplásicos (SMD) se caracterizan por una médula ósea (MO) ineficaz que produce citopenias en sangre periférica. La se-veridad de las citopenias y el riesgo incrementado a progresar a leuce-mia mieloide aguda (LMA) determinan la evolución de la enfermedad y del paciente (pte).La alta heterogenicidad de cada subcategoría han hecho necesarios sistemas de puntuación, el más utilizado es el IPSS (International Prognostic Scoring System) que permite evaluar super-vivencia global (SG) y probabilidad de progresión a LMA. Actualmente existen técnicas moleculares que permiten mejorar la definición pro-nóstica de estos ptes. Diversos estudios demuestran que la presencia de delecciones o mutaciones de p53, es un factor de riesgo individual que establece impacto negativo en SG.
OBJETIVO:
Analizar cómo influye la mutación p53 en las citopenias, progresión y transfomación a LMA e impacto en términos de SG en el subgrupo de ptes con SMD y cariotipo complejo.
PACIENTES, MATERIAL Y MÉTODO:
Análisis restropectivo unicéntrico, sobre 8 ptes [5 hombres (63%), 3 mujeres (37%)] diagnósticados de SMD con cariotipo complejo entre 2012 y 2015.Mediana de edad al diagnóstico: 68,5 años (30-82).Revisando historias clínicas electrónicas, se recogieron datos demográficos,progresión a otro SMD, transformación a LMA,% de blastos en MO, citopenias, así como supervivencia(SV). Las muestras de MO fueron obtenidas por punción-aspirado y enviadas para estudio citogenético al Institut Recerca Contra la Leucemia Josep Carreras.
RESULTADOS Y CONCLUSIONES:
Según OMS/2016, los ptes se distribuyeron de la siguiente forma: dos citopenias refractarias con displasia multilínea (CRDM), un AREB-1, cuatro AREB-2 y un LMA con cambios displásicos relacionados (con blastos 20-30%). Se obtiene p53 mutado en 5 de los 8 ptes (62,5 %). Según IPSS presentaban alto riesgo el 50 %, intermedio-2 el 37,5% y un 12,5% fueron intermedio-1. En la escala IPSS-R un 12,5% eran riesgo alto y un 87,5% muy alto, donde todos los mutados pertenecían a este último grupo. De los no mutados (37,5%), un pte progresó a SMD de peor pronóstico. Un pte de cada grupo (mutado y no mutado) presentó tranformación a LMA. La mayoría (63%) presentaban pan-citopenia , 25% bicitopenias (anemia y neutropenia) y solo un pte presentaba anemia aislada (tabla 1). La mediana del nivel de hemog-lobina (Hb) fue de 7,5 mg/dl (rango: 5,7-9,1 mg/dl), de los cuales, los ptes con mutación p53 tenían niveles de Hb más altos [mediana: 7,9 mg/dl (rango:5,8-9,1)], frente a los no mutados [mediana: 7,2 mg/dl (rango:8,7-5,7)]. Con respecto a neutrófilos y plaquetas, se observa una mediana de 0,55 x 109/l (rango: 0,13 - 2,81 x 10.9/l ) y 56 x 109/l (rango: 10-205 x 10.9/l) respectivamente, correlacionándose níveles más disminuidos con los ptes mutados [mediana neutrófilos muta-dos - no mutados= 0,33 - 1,11 x 10.9/l; mediana plaquetas mutados – no mutados = 49 - 203 x 10.9/l]. La mediana de % de blastos en MO fue de 11% (rango: 2-27%), siendo en ptes mutados el % mayor (mediana 11%) con respecto a los no mutados ( mediana 3%). A fecha de análisis, solo sobrevive un pte sin mutación p53 con una SV desde el diagnóstico de 50 meses y sometido a trasplante alogénico de do-nante no emparentado. Del resto, la mediana de SG fue de 7 meses (rango: 1-24 meses) encontrándose entre los mutados, SV más cortas (mediana 4 meses) frente a los no mutados de 18,5 meses.
Se extrae que los ptes con mutación p53 son clasificados como riesgo más elevado, mayor % de blastos en MO, neutropenias y tromboci-topenias más intensas ( no así en cuanto a Hb) y peor supervivencia.
P53 como factor pronóstico individual en síndromes mielodisplásicos. Experiencia en Hospital Universitario Nuestra Señora de ValmeMaría Eugenia Molina Rodríguez, Virgilio Calama, María Vahí, Isabel Simón, Elvira Martínez, Eduardo Ríos
Hospital Universitario Nuestra Señora de Valme
TABLA 1
PACIENTE EDAD DIAGNÓSTICO IPSS-R P53 MUTADOHb
(mg/dl)
NEUTRÓFILOS (
x10e9/L)
PLAQUETAS
(x10e9/L)
SG
(meses )
1 76 AREB-2 MUY ALTO SI 7,9 0,79 22 2
2 77 LMA CDR MUY ALTO SI 6,8 0,32 49 15
3 58 CRDM ALTO NO 8,9 2,81 203 50
4 63 AREB-2 MUY ALTO SI 5,8 0,33 10 1
5 82 AREB-2 MUY ALTO NO 7,2 0,13 28 13
6 30 AREB-1 MUY ALTO SI 9,1 0,77 64 7
7 74 AREB-2 MUY ALTO SI 8,1 0,33 156 4
8 63 CRDM MUY ALTO NO 5,7 1,11 205 24

COMUNICACIONES PÓSTERS: Hematología Clínica 231
INTRODUCCIÓN/FUNDAMENTO:
El MM sigue siendo una patología crónica incurable a pesar de la intro-ducción de nuevas estrategias de tratamiento y los nuevos fármacos. Dentro de las opciones terapéuticas la consolidación con autotras-plante de progenitores hematopoyéticos (TASPE) se ha convertido en primera línea de tratamiento tras inducción previa con quimioterapia.
OBJETIVO:
Estudio retrospectivo de los pacientes con MM en los que se ha reali-zado TASPE desde abril de 2011 a enero de 2017 que recoge las carac-terísticas evaluadas previamente y durante el procedimiento.
PACIENTES, MATERIAL Y MÉTODO:
Se realizaron 39 TASPE a 37 pacientes, 21 hombres y 16 mujeres, 35 con mieloma múltiple (MM), 1leucemia de células plasmáticas y 1 macroglobulinemia de Waldestrom (MW) con una mediana de edad 56 años siendo 15 de ellos mayores de 60 años y de estos 6 mayores de 65. La mediana de tiempo desde el diagnóstico de la enfermedad hasta el TASPE ha sido de 12,5 meses, recibiendo desde el diagnóstico diferentes líneas de tratamiento, trasplantándose en un 49% con una sola línea previa, un 25,6% con dos líneas previas, 12,8% con tres líneas previas y un 10,3% con más de tres líneas. El estado alcanzado de los pacientes al TASPE fue en un 23% en respuesta completa (RC), un 23 % en muy buen respuesta parcial (MBRP), un 36% en respuesta parcial (RP), un 8% con enfermedad en progresión (EP) y un 3% con enfermedad estable (EE); en un 7 % no disponemos del estado previo al TASPE. De los 39 TASPE 7 fueron realizados como segundo trasplante.
En cuanto a la evaluación del estado general del los pacientes presen-taban comorbilidades asociadas un 28 % , presentando un Karnosfky mayor de 80 en un 87 % de ellos. Como régimen de acondicionamiento se utilizó Melfalán 200mg en un 82%, 140mg en un 13% y BEAM en un 5%. Se infundieron progenitores hematopoyéticos de sangre periférica con una mediana de 3,8 × 10e6 CD34 + células/kg y un volumen de DMSO de mediana de 30 mL, con solo un 28% efectos secundarios, en su totalidad leve. No hubo fallo de implante. Los neutrófilos ≥ 0,5 × 10e9/L y plaquetas (≥ 20 × 10e9/L se recuperaron con una mediana de 12 días y 13 días respectivamente. Los pacientes fueron dados de alta con una mediana de ingreso de 20 días. Las complicaciones más frecuentes fueron digestivas (náuseas, vómitos y diarrea) en su ma-yoría de grado leve, mucositis en un 41% de los casos en grado III-IV y neutropenia febril que fue con aislamiento microbiológico solo en un 25,6% de los casos. Otras complicaciones de forma aislada fueron ototubaritis, epididimitis, y hepatoxicidad. No ha habido muertes relacionadas con el TASPE ni antes de los 100 días postTASPE. Han fallecido un 18% de los pacientes, por progresión de la enfermedad y por complicaciones no hematológicas solo en dos casos. La mediana de supervivencia tras el TASPE es de 28 meses.
RESULTADOS Y CONCLUSIONES:
En nuestra experiencia, la consolidación con TASPE se ha convertido en la opción terapéutica de primera línea en pacientes menores de 70 años, sobre todo si el enfermo presenta una adecuada respuesta al tratamiento de inducción previo al procedimiento. La aparición de complicaciones predecibles y controladas minimiza el riesgo de pre-sentar situaciones graves que pongan en riesgo la vida del paciente.
Evaluación de la experiencia como centro trasplantador en pacientes con Mieloma Múltiple y otras discrasias de células plasmáticasR. Zapata Bautista, A. Palma Vallellano, E. Gil Espárraga, J.N. Rodríguez Rodríguez, I. Vázquez-Pastor Jiménez, K. Gómez Korrecha
Hospital Juan Ramón Jiménez

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA232
INTRODUCCIÓN/FUNDAMENTO:
El riesgo de recaída del sistema nervioso central (SNC) de los pacientes con linfoma B difuso de células grandes (LBDCG) es del 5% y causante de mortalidad en la mayoría de los casos. Se pretende buscar factores de riesgo (LDH, afectación extraganglionar, etc) que puedan identificar los pacientes con mas riesgo de recaída y hacer una profilaxis de recaí-da en el SNC con terapia triple intratecal vs quimioterapia a altas dosis.
OBJETIVO:
Evaluamos retrospectivamente los casos de recaída en SNC de los LB-DCG durante el año 2016 en nuestro centro. Valorar los factores de riesgo que presentaban al diagnostico y si eran candidatos a profilaxis del SNC según las nuevas guías GELTAMO.
PACIENTES, MATERIAL Y MÉTODO:
Caso 1: Mujer de 75 años con recaída temprana de SNC. Paciente diag-nosticada en Julio de 2015 de Linfoma primario de SNC que alcanza remisión completa tras realizar tratamiento con meotrexato, rituximab y tamozolamida. Presenta al diagnóstico una LDH de 223 U/L , una sola afectación extranodal (SNC), IPI de 3. En Septiembre de 2016, comienza con cuadro de desorientación, se realiza TAC de cráneo que evidencia lesiones focales intraaxiales en lobulo temporal izquierdo y sustancia blanca subcortical frontal izquierda. La biopsia cerebral confirma diagnóstico compatible con de linfoma B inclasificable con características intermedias entre LBDCG y linfoma de Burkitt.
Caso 2: Mujer de 69 años con recaída tardía de SNC. Paciente diag-nosticada en 2012 de LBDCG que alcanza remisión completa tras 6 ciclos de RCHOP. Presenta al diagnóstico una LDH de 215 U/L una sola
afectación y extranodal (labio mayor vulvar) e IPI de 1. Al diagnóstico no presenta sintomatología neurológica no se realiza estudio de ex-tensión en SNC. En septiembre de 2016, presenta episodio sincopal por el que se realiza TAC de cráneo con presencia de lesiones ocupantes de espacio intraaxiales, se realiza biopsia cerebral con diagnóstico histológico de LBDCG.
Caso 3: Varón de años con recaída temprana leptomeníngea. Paciente diagnosticado en mayo de 2016 de LBDCG con masa mediastínica e infiltración de líquido pleural que alcanza respueta completa tras 6 ciclos de R-CHOP. Presenta al diagnóstico una LDH de 2688 U/L, afec-tación extranodal (pleural/pericárdico) e IPI de 2. En Agosto de 2016, presenta cuadro vertiginoso central con TAC de cráneo con infiltración metastásica leptomeníngea, con LCR con infiltración masiva por célu-las con IF y morfología compatibles con LBDCG.
Caso 4: Varón de años con recaída tardía de SNC. Diagnosticado en marzo de 2013 de LBDCG gastointestinal, renal y pancreático en respuesta completa tras 8 ciclos de R-CHOP. Presenta al diagnóstico una LDH de 876 U/L, varias afectaciones extranodales e IPI de 3. En Septiembre de 2016, presenta crisis convulsiva con TAC de cráneo con presencia de masa de la porción posterior de hueso temporal izquierdo con componente intracraneal y extracraneal y con confirmación histo-lógica tras biopsia de temporal de recaída de LBDCG.
RESULTADOS Y CONCLUSIONES:
Ninguno de nuestros pacientes recibió profilaxis del sistema nerviosos central a pesar de que los cuatro cumplían indicación según los nue-vos criterios. El 50% de los pacientes que presentaron recaída del SNC fallecieron. Consideramos imprescindible la necesidad de evaluar los factores de riesgo y la realización de profilaxis en aquellos pacientes que cumplan criterios.
Evaluación de los factores de riesgo de recaída en SNC en los pacientes con Linfoma B difuso de células grandesR. Zapata Bautista, E. Gil Espárraga, M.ªA. Ruíz Cobo, B. Díaz Roldán, K. Gómez Korrecha, A. Palma Vallellano
Hospital Juan Ramón Jiménez
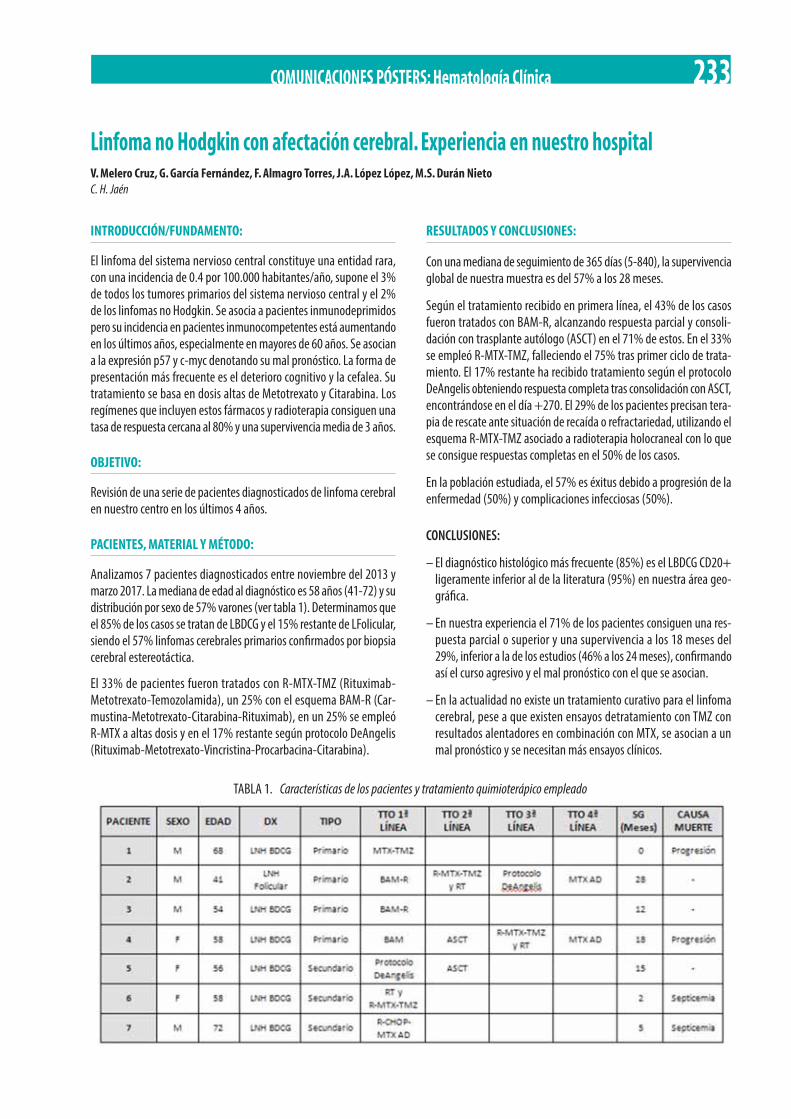
COMUNICACIONES PÓSTERS: Hematología Clínica 233
INTRODUCCIÓN/FUNDAMENTO:
El linfoma del sistema nervioso central constituye una entidad rara, con una incidencia de 0.4 por 100.000 habitantes/año, supone el 3% de todos los tumores primarios del sistema nervioso central y el 2% de los linfomas no Hodgkin. Se asocia a pacientes inmunodeprimidos pero su incidencia en pacientes inmunocompetentes está aumentando en los últimos años, especialmente en mayores de 60 años. Se asocian a la expresión p57 y c-myc denotando su mal pronóstico. La forma de presentación más frecuente es el deterioro cognitivo y la cefalea. Su tratamiento se basa en dosis altas de Metotrexato y Citarabina. Los regímenes que incluyen estos fármacos y radioterapia consiguen una tasa de respuesta cercana al 80% y una supervivencia media de 3 años.
OBJETIVO:
Revisión de una serie de pacientes diagnosticados de linfoma cerebral en nuestro centro en los últimos 4 años.
PACIENTES, MATERIAL Y MÉTODO:
Analizamos 7 pacientes diagnosticados entre noviembre del 2013 y marzo 2017. La mediana de edad al diagnóstico es 58 años (41-72) y su distribución por sexo de 57% varones (ver tabla 1). Determinamos que el 85% de los casos se tratan de LBDCG y el 15% restante de LFolicular, siendo el 57% linfomas cerebrales primarios confirmados por biopsia cerebral estereotáctica.
El 33% de pacientes fueron tratados con R-MTX-TMZ (Rituximab-Metotrexato-Temozolamida), un 25% con el esquema BAM-R (Car-mustina-Metotrexato-Citarabina-Rituximab), en un 25% se empleó R-MTX a altas dosis y en el 17% restante según protocolo DeAngelis (Rituximab-Metotrexato-Vincristina-Procarbacina-Citarabina).
RESULTADOS Y CONCLUSIONES:
Con una mediana de seguimiento de 365 días (5-840), la supervivencia global de nuestra muestra es del 57% a los 28 meses.
Según el tratamiento recibido en primera línea, el 43% de los casos fueron tratados con BAM-R, alcanzando respuesta parcial y consoli-dación con trasplante autólogo (ASCT) en el 71% de estos. En el 33% se empleó R-MTX-TMZ, falleciendo el 75% tras primer ciclo de trata-miento. El 17% restante ha recibido tratamiento según el protocolo DeAngelis obteniendo respuesta completa tras consolidación con ASCT, encontrándose en el día +270. El 29% de los pacientes precisan tera-pia de rescate ante situación de recaída o refractariedad, utilizando el esquema R-MTX-TMZ asociado a radioterapia holocraneal con lo que se consigue respuestas completas en el 50% de los casos.
En la población estudiada, el 57% es éxitus debido a progresión de la enfermedad (50%) y complicaciones infecciosas (50%).
CONCLUSIONES:
– El diagnóstico histológico más frecuente (85%) es el LBDCG CD20+ ligeramente inferior al de la literatura (95%) en nuestra área geo-gráfica.
– En nuestra experiencia el 71% de los pacientes consiguen una res-puesta parcial o superior y una supervivencia a los 18 meses del 29%, inferior a la de los estudios (46% a los 24 meses), confirmando así el curso agresivo y el mal pronóstico con el que se asocian.
– En la actualidad no existe un tratamiento curativo para el linfoma cerebral, pese a que existen ensayos detratamiento con TMZ con resultados alentadores en combinación con MTX, se asocian a un mal pronóstico y se necesitan más ensayos clínicos.
Linfoma no Hodgkin con afectación cerebral. Experiencia en nuestro hospitalV. Melero Cruz, G. García Fernández, F. Almagro Torres, J.A. López López, M.S. Durán Nieto
C. H. Jaén
TABLA 1. Características de los pacientes y tratamiento quimioterápico empleado

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA234
INTRODUCCIÓN/FUNDAMENTO:
Los linfomas de células B de alto grado con reordenamiento del gen c-myc, bcl2 y/o bcl6 presentan un pronóstico pobre con la quimioterapia estándar con una supervivencia libre de progresión (SLP) a los 2 años del 25%. Suponen del 7% al 10% de los linfoma B difuso de células grandes B (LDCG-B). Los doble protein sobreexpresan proteínas c-myc y bcl2 sin detectarse translocación. Su pronóstico es inferior a aquellos que no las sobreexpresan aunque superior al de los doble/triple hit.
Actualmente el régimen de inducción óptimo no está establecido. Diversos estudios retrospectivos han obtenido mejores resultados con R-daEPOCH vs R-hiperCVAD-MTX/AraC y R-CHOP, con respuestas completas cercanas al 70% y SLP a los 2 años del 67%.
OBJETIVO:
Describir las características clínico-biológicas de ocho pacientes diag-nosticados de linfoma B de alto grado doble hit ó doble protein tra-tados con R-daEPOCH y analizar los resultados obtenidos en cuanto a toxicidad y respuesta.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo observacional retrospectivo. Período: junio 2016 a marzo 2017. Se incluyeron todos los pacientes diagnosticado por biop-sia ganglionar de linfoma B doble/triple hit o doble protein tratados con R-daEPOCH. Se consideró linfoma double hit a aquel que reordenó c-myc con bcl-2ó bcl6, triple hit al que reordenó c-myc, bcl-2 y bcl-6 y double protein al que sobreexpresaba c-myc y bcl2 sin detectarse tras-locación. Para detectar reordenamientos se empleó FISH en interfase con sonda break apart sobre material parafinado y la sobreexpresión de proteínas se determinó mediante inmunohistoquímica. Análisis estadístico descriptivo se realizó con SPSS v.19.
RESULTADOS Y CONCLUSIONES:
Se diagnosticó 1 paciente de linfoma folicular (LF) de alto grado (3B) doble hit (c-myc+ bcl2+) y 7 pacientes como con LBDC-G no centro-germinal doble protein (en 4 de ellos no se pudo realizar FISH por material no adecuado o insuficiente). Cuatro eran mujeres (50%) y 4 varones (50%) La mediana de edad fue de 61 años (41-67 años). El 75% presentaban estadio IV al diagnóstico y el 25% estadio III. El 57% de los LDCG-B mostraron un IPI de alto riesgo, el 14,5% riesgo intemedio-alto y 28,5 % intermedio bajo riesgo. El LF presentaba FLIPI de riesgo intermedio. El 25% (2/8) presentaba infiltración de la médu-la ósea y un 12.5% (1/8) infiltración sistema nervioso central (SNC). Siete de ocho (87,5%) pacientes recibieron R-daEPOCH (rituximab, ciclofosfamida, prednisona, vincristina, etopósido, doxorrubicina) como tratamiento de primera línea (un caso lo recibió como segunda línea tras recaída tardía como LDCG-B con expresión doble protein) En el caso con infiltración SNC se añadió MTX a altas. En 42,8% (3/7) se realizó profilaxis del SNC con terapia intratecal. El 62.5% (5/8) presentó toxicidad grado III/IV, siendo la toxicidad más frecuente la neutrope-nia, generalmente de corta duración. De los pacientes evaluados con PET-TAC intermedio 4 pacientes (50%) alcanzaron respuesta completa y 2 respuesta parcial o progresión (25%), uno falleció por complicación infecciosa. La tasa de éxitus es del 12,5%.
En nuestro estudio la mayoría de los pacientes con linfomas B de alto grado tratados con R-da-EPOCH presentaban estadio avanzado al diagnóstico, edad inferior a 65 años y eran doble protein, aunque el porcentaje de doble hit pudiera ser mayor al reportado.En nuestra experiencia, la tasa de respuesta precoz con régimen R-da-EPOCH en primera línea fue similar a la tasa de respuesta descrita en la literatura, siendo la toxicidad aceptable.
Linfomas de células B de alto grado doble Hit o doble Protein tratados con R-Daepoch. Experiencia de un centroP. Jiménez Guerrero (1), M.A. Domínguez Muñoz (2), M. Ruiz Mercado (2), E. Carrillo Cruz (2), F. De la Cruz Vicente (2), I. Montero Cuadrado (2)
(1) Hospital Universitario Virgen del Rocío, (2) Hospital Universitario Virgen del Rocío

COMUNICACIONES PÓSTERS: Hematología Clínica 235
INTRODUCCIÓN/FUNDAMENTO:
Debido a las pocas alternativas disponibles en los pacientes refrac-tarios a lenalidomida y bortezomib, el uso de pomalidomida en este tipo de pacientes es relevante y significativo, habiendo demostrado un beneficio clínico tanto en SLP y SG (Informe de Posicionamiento Terapéutico de Pomalidomida)
Pomalidomida en combinación con dexametasona ha sido autoriza-do en el tratamiento de los pacientes adultos con mieloma múltiple resistente al tratamiento o recidivante que hayan recibido al menos dos tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el último tratamiento.
OBJETIVO:
Evaluar los resultados de eficacia del tratamiento con Pomalidomi-da-Dexametasona en pacientes con mieloma en recaída en nuestra Unidad.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de los pacientes con mieloma múltiple en recaída y que iniciaron tratamiento con pomalidomida y dexametasona en nuestra unidad desde septiembre de 2014 hasta diciembre de 2016.
TRATAMIENTO ADMINISTRADO
Pomalidomida: 4 mg los días 1 al 21 y Dexametasona: 40 mg los días 1, 8, 15 y 22, de cada ciclo de 28 días, hasta la progresión de la en-fermedad.
Toxicidad: se evaluó mediante NCI-CTC, versión 4.0
ANALISIS ESTADISTICO: Se utilizó IBM SPSS Statistics, versión 21
RESULTADOS Y CONCLUSIONES:
RESULTADOS:
Se han incluido 9 pacientes, 6 hombres y 3 mujeres, con una mediana de edad de 63 años (entre 51-70), que habían recibido entre 2-7 líneas de tratamiento previo. Todos habían recibido bortezomib y lenalido-mida y seis habían sido sometidos a TPH.
La mediana de ciclos administrados por paciente ha sido de 5 (entre 1-20). Seis pacientes han completado 3 o más ciclos.
La toxicidad ha sido fundamentalmente hematológica. TVP en un paciente
SEGUIMIENTO
Después de una mediana de seguimiento de 4.5 meses (2-20) desde el inicio del tratamiento la mediana de SLP y SG han sido de 7 y 14 meses, respectivamente.
CONCLUSIONES:
El tratamiento con pomalidomida y dexametasona se ha mostrado eficaz en nuestros pacientes, con resultados mejores en los que han completado 3 ó más ciclos.
Tratamiento con Pomalidomida-Dexametasona de pacientes con Mieloma Múltiple en recaídaJ. Capote García, M.E. Rodríguez Mateos, M.C. Fernández Valle, M.J. Huertas Fernández, G. Blanco Sánchez, F.J. Capote Huelva
Hospital Puerta del Mar

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA236
INTRODUCCIÓN/FUNDAMENTO:
El Linfoma Primario Anaplásico Cutáneo (Primary cutaneous anaplastic large cell lymphoma, pcALCL) forma parte del espectro de enferme-dades linfoproliferativas cutáneas CD30+. Clínicamente se presenta en forma de nódulos, usualmente menores de 5 cm, localizados en cualquier región corporal. Desde el punto de vista histológico expresan CD30 y son negativos para ALK. Aunque la tasa de supervivencia a los 5 años es superior al 85%, son muy frecuentes las recaídas. Para su tratamiento se recomiendan la escisión quirúrgica y la radioterapia.
Brentuximab Vedotina es un anticuerpo conjugado formado por un anticuerpo monoclonal dirigido contra CD30 (inmunoglobulina G1 [IgG1] quimérica recombinante, producida mediante tecnología de ADN recombinante en células de ovario de hámster chino) que se une de forma covalente al agente antimicrotúbulos monometil auristatina E (MMAE). Está autorizado su uso para el tratamiento del linfoma de Hodgkin en recaída o refractario y el linfoma anaplásico de células grandes (LACG) sistémico en recaída o refractario. Aunque son pocos los casos comunicados de su uso en el Linfoma Primario Anaplásico Cutáneo, los resultados son alentadores.
OBJETIVO:
Describir un caso de Linfoma Primario Anaplásico Cutáneo en recaída con buena respuesta al tratamiento con Brentuximab Vedotina.
PACIENTES, MATERIAL Y MÉTODO:
CASO CLINICO
Varón de 70 años, con antecedentes de insuficiencia renal crónica, diabetes y psoriasis, diagnosticado en diciembre de 2015 de Linfoma Primario Anaplásico Cutáneo tras biopsia de tumoración de 2 cms, de algo más de un mes de evolución, en surco nasogeniano izquierdo.
Biopsia tumoración: cilindro de piel de epidermis respetada que pre-senta en todo el espesor de la dermis y porción superior de la hipoder-mis una amplia y densa proliferación celular de morfología redonda y talla alta y media, de distribución difusa que desplaza los haces de colágeno y preserva la pared de los vasos sanguíneos, con positividad para ALC, CD3, CD4, CD5, CD30 y un índice de proliferación nuclear que alcanza prácticamente el 100% siendo la expresión negativa para ALK, CD7, TdT, CD8, CD20, CD56, S100, pancitoqueratina, CK20, Cromogra-nina y Sinaptofisina.
Se realizó en febrero de 2016 tratamiento quirúrgico y posterior radio-terapia por progresión al mes de la cirugía.
En octubre de 2016 se nos consultó nuevamente por progresión. En el estudio con PET-TAC se apreciaba un depósito patológico en el surco nasogeniano izquierdo de 10x9x14 mm y SUV de 6.2.
Una vez obtenida la correspondiente autorización, en enero de 2017 se comenzó la administración de Brentuximab Vedotina a dosis de 1,8 mg/kg, en perfusión intravenosa cada 3 semanas.
RESULTADOS Y CONCLUSIONES:
RESULTADOS:
Hasta el momento actual se han administrado 4 ciclos habiendo casi desaparecido la tumoración y sin que se hayan producido efectos ad-versos.
CONCLUSIONES:
En nuestro paciente el tratamiento con Brentuximab Vedotina se ha mostrado eficaz.
Hemos observado mejoría de las lesiones de psoriasis, aunque no po-demos establecer una relación causal por el momento.
Paciente con linfoma anaplásico primario cutáneo en Recidiva. Tratamiento con Brentuximab VedotinaJ. Capote García, F.J.Capote Huelva
Hospital Puerta del Mar

COMUNICACIONES PÓSTERS: Hematología Clínica 237
INTRODUCCIÓN/FUNDAMENTO:
El objetivo de los estudios de la enfermedad mínima residual (EMR) en pacientes con leucemia aguda es evaluar la carga leucémica residual y detectar la recaída temprana. En la leucemia aguda linfoblástica (LAL), se ha mostrado una fuerte correlación entre la EMR y la recaída de la enfermedad, mientras que en la leucemia aguda mieloblástica (LAM) esta relación es controvertida.
OBJETIVO:
Evaluar la EMR, tras la inducción y pre-trasplante, y determinar su valor pronóstico en la supervivencia global (SG) y la supervivencia libre de progresión (SLP) en pacientes con leucemia aguda
PACIENTES, MATERIAL Y MÉTODO:
Se incluyeron 110 pacientes adultos diagnosticados de leucemia aguda (34 LAL y 76 LAM) que recibieron tratamiento quimioterápico intensivo, según protocolo PETHEMA vigente y sometidos a trasplante alogenico (Alo-TPH) en 1ªremisión completa (RC) en nuestro centro (2005-2015). Se determinó de forma prospectiva la EMR tras RC mor-fológica post-inducción (EMRI) y pre-TPH (EMRpreTPH). La EMR se determinó por citometría de flujo multiparamétrica en FACSCanto II, hasta adquirir 10.000 eventos con el fin de detectar fenotipos abe-rrantes con una sensibilidad de 10-4, considerando EMR+ ≥ 0.01.
RESULTADOS Y CONCLUSIONES:
En la serie global con una mediana de seguimiento de 57 meses (8-135 meses), la SG a los 5 años fue 63.5%±4.9% (63±6.1% vs ±%63.3±8.6%, LAM vs LAL, p=NS).
Las variables que se asociaron con una menor SG fueron: EMRI+ (50.3±10.3% vs 76.4±6.6%, p=0.03), EMRpre-TPH+ (30.2±11.8% vs 81.3±5.8%, p<0.001), así como EMRI+/EMRpre-TPH+ (24.8±12.5% vs 76.3±7%, p<0.001). En el análisis multivariante los pacientes con
EMRpreTPH+ presentan 5 veces más riesgo de fallecer [HR 4.878 (IC 95% 2.118-11.236) p<0.001].
La SLE fue del 76±4.6% (73.3±5.8% vs 82±7.4%, LAM vs LAL, p=NS).La EMRI+ 62.4±11.8% vs 85±5.7%, (p=0.007), EMRpre-TPH 43.7±15% vs 89.1±4.7%, (p<0.001), EMRI+/ EMRpre-TPH 37.4±17% vs 85.9±5.9%, (p<0.001).En el análisis multivariable, los pacientes con EMRpreTPH+ presentan 6 veces más el riesgo de recaer [HR 5.973 (IC 95% 2.026-17.613) p<0.001].
No mostró significación estadística la edad, número de ciclos para alcanzarla RC, número de leucocitos ni grupo de riesgo citogenético/molecular tanto para la SG ni SLE.
Al evaluar el subgrupo LAM, la variable EMRpre-TPH se mantiene como mayor significación estadística en el análisis multivariable, tanto para la SG[HR 8.05 (IC 95% 2.119-30.611) p<0.001] como para la SLE[HR 10.605 (IC 95% 2.119-51.146) p=0.001]. Aunque los pacientes con alteraciones citogenéticas/moleculares de alto riesgo tienen con mayor frecuencia EMR+preTPH (58.8% vs 21.9%, alto vs int/bajo; p=0.01), no se mantiene como variable independiente en el análisis multivariable.
Asimismo, la SLE en pacientes con LAM fue mayor en aquellos con EMRpreTPH- (87.4±8.4% vs 28.2±19.3%, p<0.001), así como en LAL con EM RpreTPH-(84±8.5% vs 40±29.7%, p=0.05).
Otros factores post-TPH como tipo de acondicionamiento y de donante, profilaxis del EICH, origen de los progenitores y presencia de EICHa severo no mostró relación estadística con la recaída, mientras que EICHcrónico si (p<0.001).
Como conclusiones, podemos decir que la detección de EMR por CMF en pacientes adultos con leucemia aguda tanto LAM como LAL so-metidos a ALO-TPH en 1º RC contribuye a redefinir el pronóstico de la enfermedad y la calidad de la respuesta en el momento del TPH, constituyendo una herramienta indispensable en el manejo clínico y toma de decisiones de estos pacientes.
Valor pronóstico de la enfermedad mínima residual inmunológica en pacientes adultos con leucemia aguda sometidos a trasplante alogénico de precursores hematopoyéticos en primera remisión completaC. Martínez Losada, E. García-Torres, J. Serrano, S. Tabares, J. Sánchez-García
Hospital Universitario Reina Sofía

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA238
INTRODUCCIÓN/FUNDAMENTO:
Las neoplasias primarias múltiples son aquellas que se presentan en el mismo sujeto de forma simultánea o sucesiva. Su incidencia ha au-mentado en las últimas décadas, entre otros hechos por la mejora en el diagnóstico, siendo del 4-12% de los procesos oncológicos.
Se caracterizan porque cada una de las neoplasias tiene un patrón de malignidad definido, no es una metástasis de otra, con distinta histología y ninguna conexión entre ellas. Una puede ser un hallazgo en el curso del diagnóstico, estadificación o seguimiento de la otra. Si el diagnóstico es simultáneo se les llama sincrónicas.
OBJETIVO:
Revisión de dos casos de doble diagnóstico neoplásico en nuestro centro
PACIENTES, MATERIAL Y MÉTODO:
Caso 1: Paciente de 57 años, postmenopaúsica, con antecedentes fa-miliares de Ca mama, colon y endometrio, y AP sin interés salvo comor-bilidad moderada (obesidad), que fue diagnosticada en Julio de 2015 de Carcinoma ductal infiltrante mama derecha (posteriormente se vió que IIA triple negativo), a raíz de PAAF y biopsia de nódulo de 45 mm en cuadrante superoexterno de dicha mama, realizándose mastecto-mía derecha y linfadenectomía, mostrando el estudio anatomopatoló-gico invasión vascular y ganglionar axilar por la enfermedad, así como afectación ganglionar por linfoma linfocítico de células pequeñas /LLC, lo que se corroboró en el estudio de médula ósea de la paciente, que mostró infiltración intersticial de linfocitos de talla pequeña con inmunotinción positiva para CD20 y negativa para CD3. El estudio de extensión mostró pequeñas adenopatías cervicales, torácicas y ab-dominales, siendo la sangre periférica negativa para la enfermedad.
La paciente fue tratada con quimioterapia para su proceso neoplásico mamario seguido de radioterapia complementaria locoregional. Desde el punto de vista hematológico no ha cumplido criterios de inicio de
tratamiento, aunque sigue controles periódicos a través de nuestra consulta.
Caso 2: Paciente de 79 años, sin antecedentes familiares de interés y con AP de HTA, flutter auricular, histerectomía total por miomas y poliartralgias, que fue diagnosticada en Marzo 2016 tras biopsias de nódulos mamarios bilaterales, de Ca ductal infiltrante en mamas de-recha e izquierda, apareciendo en biopsia ganglionar del ganglio cen-tinela axilar izquierdo una afectación parcial por Linfoma de Hodgkin clásico celularidad mixta CD30 positivo, con alto índice proliferativo.
El estudio de extensión hematológico mostró adenopatías subcen-timétricas laterocervicales, axilares, inguinales e iliacas, así como pequeñas LOES esplénicas PET negativas. El PET sí fue positivo a nivel axilar izquierdo y mediastínico (sin adenopatías destacables a este nivel en TAC).
La paciente fue tratada con inhibidores de la aromatasa, al tratarse de 2 Ca ductales infiltrantes IA moderadamente diferenciados de bajo indice proliferativo, con receptores hormonales positivos, y con Radio-terapia bilateral, abarcando la axila izquierda, teniendo en cuenta la edad avanzada y la comorbilidad de la paciente, con buen resultado, siguiendo actualmente controles evolutivos en nuestra consulta ex-terna.
RESULTADOS Y CONCLUSIONES:
– En los últimos años existe un aumento progresivo en la incidencia y diagnóstico de las neoplasias primarias múltiples.
– Un correcto y exhaustivo estudio anatomopatológico impide ob-viar determinados procesos neoplásicos trascendentes que pudieran quedar “sepultados” por otros de mayor expresión o agresividad.
– La coexistencia de doble patología simultánea en los enfermos neoplásicos obliga a establecer prioridades en el abordaje terapéu-tico de los mismos.
Diagnóstico sincrónico de procesos neoplásicos en hematologíaJulia Morán Sánchez, F.J. Capote García, A. Santisteban Espejo, M.C. Fernández Valle
Hospital Universitario Puerta del Mar

COMUNICACIONES PÓSTERS: Hematología Clínica 239
INTRODUCCIÓN/FUNDAMENTO:
El pronóstico de los pacientes con Leucemia Mieloblástica Aguda (LMA) refractarios a la quimioterapia de inducción es sombrío. Se han desa-rrollado protocolos de tratamiento secuencial con quimioterapia de rescate y posterior trasplante alogénico cuyos resultados preliminares son favorables.
OBJETIVO:
Evaluar la tasa de respuesta, la SG y SLE de forma restrospectiva del tratamiento citorreductor con Clofarabina seguido de ALO-TPH secuen-cial en pacientes con LMA avanzada utilizado en nuestro centro en los últimos 5 años.
PACIENTES, MATERIAL Y MÉTODO:
Entre 2011-2016 un total de 6 pacientes (3 varones, 3 mujeres) con mediana de edad de 59 años (34-61) fueron tratados con Clofarabina 40 mg/m2 x 5 días seguido a los 7 días de ALO-TPH de intensidad reducida por fallo primario a la inducción. Todos los pacientes habían recibido al menos 2 ciclos de quimioterapia previa (3+7 IDA/ARA-C + FLAT/FLAGIDA/ICE) y persistían con >10 % blastos en M.O. 5 pacientes recibieron trasplante de un DE y 1 paciente de un hijo haploidéntico.
Después del TPH, 5 pacientes alcanzaron RC morfológica (tasa de res-puesta 83.3%) con un prendimiento adecuado (la media de días de recuperación de neutrófilos y plaquetas fueron +13 y +11 respecti-vamente). Dos de los pacientes permanecen vivos, tres recayeron a los 3,4 y 24 meses y uno fue refractaria al trasplante secuencial siendo exitus por persistencia de la enfermedad el dia +44. Ninguno murió por toxicidad relacionada con el procedimiento. La mayor toxicidad relacionada con la Clofarabina fue un rash cutánea reversible y tiempo muy limitado. La SG a los 2 años fue 44.4% y la SLE 50%. En todos los casos los inmunosupresores de retiraron de forma precoz (antes del día +100). No realizamos Infusiones de linfocitos del donante (ILD) profi-láctica, porque no objetivamos aumento de la EMR ni del quimerismo o porque desarrollaron EICR tras la retirada rápida de inmunosupre-sión. En total 3 de los 6 pacientes, presentaron datos de EICR crónica (2 de ellos son los que continúan vivos y en RC).
RESULTADOS Y CONCLUSIONES:
Basándonos en nuestros datos la citorreducción con clofarabina segui-do del ALO-TPH secuencial consigue alta tasa de respuestas completas, se asocia a pocas complicaciones, pero tiene gran riesgo de recaidas. Seria recomendable incluir en nuestra estrategia ILD preventivas a par-tir del dia +120. Como es ya conocido, la existencia de EICR reduce de forma importante la tasa de recaídas.
Trasplante secuencial en LMA primariamente refractariasM.D. Madrigal Toscano, R. Saldaña Moreno, A. Salamanca Cuenca, L. Domínguez Acosta, V. Rubio Sánchez, S. Garzón López
Hospital del SAS de Jerez de la Frontera
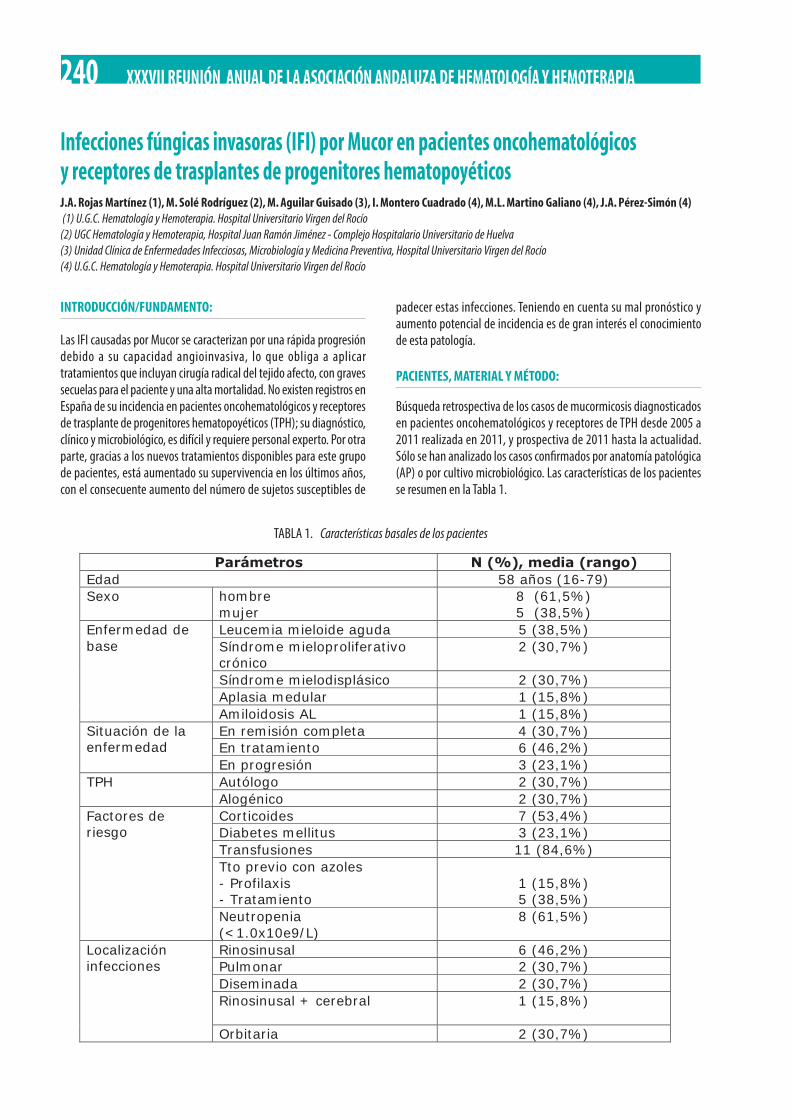
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA240
INTRODUCCIÓN/FUNDAMENTO:
Las IFI causadas por Mucor se caracterizan por una rápida progresión debido a su capacidad angioinvasiva, lo que obliga a aplicar tratamientos que incluyan cirugía radical del tejido afecto, con graves secuelas para el paciente y una alta mortalidad. No existen registros en España de su incidencia en pacientes oncohematológicos y receptores de trasplante de progenitores hematopoyéticos (TPH); su diagnóstico, clínico y microbiológico, es difícil y requiere personal experto. Por otra parte, gracias a los nuevos tratamientos disponibles para este grupo de pacientes, está aumentado su supervivencia en los últimos años, con el consecuente aumento del número de sujetos susceptibles de
padecer estas infecciones. Teniendo en cuenta su mal pronóstico y aumento potencial de incidencia es de gran interés el conocimiento de esta patología.
PACIENTES, MATERIAL Y MÉTODO:
Búsqueda retrospectiva de los casos de mucormicosis diagnosticados en pacientes oncohematológicos y receptores de TPH desde 2005 a 2011 realizada en 2011, y prospectiva de 2011 hasta la actualidad. Sólo se han analizado los casos confirmados por anatomía patológica (AP) o por cultivo microbiológico. Las características de los pacientes se resumen en la Tabla 1.
Infecciones fúngicas invasoras (IFI) por Mucor en pacientes oncohematológicos y receptores de trasplantes de progenitores hematopoyéticosJ.A. Rojas Martínez (1), M. Solé Rodríguez (2), M. Aguilar Guisado (3), I. Montero Cuadrado (4), M.L. Martino Galiano (4), J.A. Pérez-Simón (4)
(1) U.G.C. Hematología y Hemoterapia. Hospital Universitario Virgen del Rocío
(2) UGC Hematología y Hemoterapia, Hospital Juan Ramón Jiménez - Complejo Hospitalario Universitario de Huelva
(3) Unidad Clínica de Enfermedades Infecciosas, Microbiología y Medicina Preventiva, Hospital Universitario Virgen del Rocío
(4) U.G.C. Hematología y Hemoterapia. Hospital Universitario Virgen del Rocío
Edad 58 años (16-79) Sexo hombre
mujer 8 (61,5%) 5 (38,5%)
Enfermedad de base
Leucemia mieloide aguda 5 (38,5%) Síndrome mieloproliferativo crónico
2 (30,7%)
Síndrome mielodisplásico 2 (30,7%) Aplasia medular 1 (15,8%) Amiloidosis AL 1 (15,8%)
Situación de la enfermedad
En remisión completa 4 (30,7%) En tratamiento 6 (46,2%) En progresión 3 (23,1%)
TPH Autólogo 2 (30,7%) Alogénico 2 (30,7%)
Factores de riesgo
Corticoides 7 (53,4%) Diabetes mellitus 3 (23,1%) Transfusiones 11 (84,6%) Tto previo con azoles - Profilaxis - Tratamiento
1 (15,8%) 5 (38,5%)
Neutropenia (<1.0x10e9/L)
8 (61,5%)
Localización infecciones
Rinosinusal 6 (46,2%) Pulmonar 2 (30,7%) Diseminada 2 (30,7%) Rinosinusal + cerebral 1 (15,8%)
Orbitaria 2 (30,7%)
TABLA 1. Características basales de los pacientes

COMUNICACIONES PÓSTERS: Hematología Clínica 241
TABLA 2. Identificación de especies mediante cultivoRESULTADOS Y CONCLUSIONES:
Se han detectado 13 casos de mucormicosis identificados mediante diagnóstico anatomopatológico (11/13) y/o cultivo (9/13) (Tabla 2). En cuanto al tratamiento que recibieron los pacientes afectos, salvo dos que se trataron de forma paliativa y uno que recibió posaconazol en monoterapia, todos recibieron tratamiento antifúngico basado en anfotericina B liposomal (3 de ellos en combinación). En 7 de ellos se realizó cirugía radical. 10 pacientes fallecieron precozmente (media 15,3 días), y de los 4 supervivientes, uno presentó importantes secuelas que impidieron continuar con el tratamiento de su LA falleciendo por recaída a los 16 meses. 3 continúan vivos, a 42, 9 y 7 meses respectivamente. Estos cuatro pacientes con supervivencia mayor a los seis meses recibieron tratamiento antifúngico y cirugía radical en los primeros 3 días desde el inicio de los síntomas. Esta rápida intervención fue posible gracias a la sospecha clínica precoz, la toma apropiada y rápida de muestras, a la celeridad con que se interpretaron las mismas por un microbiólogo experto, y a la implicación en la toma de decisiones de todo el equipo médico-quirúrgico. En todos los pacientes que sobrevivieron se pudo continuar el tratamiento y la profilaxis secundaria con posaconazol por vía oral, sin evidencia de recidivas del mucor, aun en un paciente que han continuado recibiendo tratamiento quimioterápico y en otro que ha recibido quimioterapia y un TPH.
sp 4 (30,7%)
3 (23,1%)
spp 1 (15,8%)
1 (15,8%)
Mucor no especificado (diagnóstico por AP)
4 (30,7%)
Aunque el número de casos de nuestra serie es limitado, nuestros datos sugieren que el diagnóstico de sospecha basado en la presencia de factores de riesgo, la confirmación rápida del mismo y la instauración de un tratamiento precoz basado en cirugía radical y anfotericina pueden mejorar el pronóstico de estos enfermos. Dadas las graves secuelas que comporta la cirugía y la importancia de realizarla precozmente es de especial interés la coordinación y concienciación de todos los profesionales implicados para la toma eficaz y rápida de decisiones.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA242
INTRODUCCIÓN/FUNDAMENTO:
El consumo de tabaco es la principal causa prevenible de cáncer y de muertes por cáncer. Pero el consumo de cigarrillos no solo causa cáncer sino que también puede tener un impacto negativo en el tratamiento y la supervivencia de la persona con cáncer, al facilitar complicaciones infecciosas y segundas neoplasias, entre otras.
En cuanto a los cuidadores también se ha referido que en muchos casos aumentan el consumo de alcohol, fármacos y tabaco como consecuen-cia de la llamada sobrecarga del cuidador.
OBJETIVO:
Conocer datos sobre consumo de tabaco por pacientes con trastornos hematológicos y sus cuidadores y familiares.
PACIENTES, MATERIAL Y MÉTODO:
Han participado pacientes con trastornos hematológicos y sus cuida-dores y familiares.
Los sujetos respondieron a un cuestionario autoadministrado sobre consumo de tabaco. Se registraron datos sociodemográficos.
RESULTADOS Y CONCLUSIONES:
RESULTADOS:
Han participado 26 pacientes (linfoma: 15; mieloma: 6; leucemia: 5). Edad mediana: 62 años. . Sexo: 10 mujeres y 16 varones. Refieren no haber fumado nunca el 42.2%. Un 11.5% dicen seguir fumando a pesar de la enfermedad y sólo el 6.7% ha disminuido el consumo de cigarrillos por este motivo.
Han participado 129 cuidadores y familiares (linfoma: 49; mieloma: 22; leucemia: 58). Edad mediana: 51 años. . Sexo: 87 mujeres y 42 va-rones. Refieren no haber fumado nunca el 48.1%. Un 24% dicen seguir fumando a pesar de la enfermedad de su familiar y el 19.4% considera que ha aumentado el consumo de cigarrillos durante la enfermedad.
CONCLUSIONES:
Muchos pacientes siguen fumando durante la enfermedad a pesar del perjuicio adicional de este hábito durante la misma.
Un considerable número de cuidadores y familiares aumentan también el consumo en relación quizás con el estrés y la sobrecarga del cuidado.
Consideramos que sería útil la implementación de programas antita-baco para los afectados por enfermedades hematológicas.
Tabaquismo en afectados de trastorno hematológico (cuidadores, familiares y pacientes)A. García Nieto, F.J. Capote Huelva
Hospital Puerta del Mar

COMUNICACIONES PÓSTERS: Hematología Clínica 243
INTRODUCCIÓN/FUNDAMENTO:
La MW es un desorden proliferativo clonal B caracterizado por infil-tración medular por linfoma linfoplasmocítico asociado a paraprotei-nemia sérica IgM. La infiltración de órganos distintos a los ganglios linfáticos, hígado o bazo se da en <5% de los casos. De igual manera, la amiloidosis asociada a desórdenes de tipo IgM es una entidad de muy difícil diagnóstico pero que se está detectando cada vez con ma-yor frecuencia, en contraste con la característica afectación glomerular por oclusión de los capilares a causa de los grandes depósitos de IgM clonal descrita desde hace décadas en asociación con la MW en sus estadios más avanzados.
Presentamos un caso infrecuente de una paciente con asociación de amiloidosis renal AL y macroglobulinemia de Waldenström que inició estudio por una gammapatía IgG lambda.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 56 años en estudio por Medicina interna por equimosis pe-riorbitaria y pérdida de peso, que ante la sospecha de dermatomiositis inició corticoterapia con mejoría del cuadro. Un año después se derivó a consultas de Hematología para estudio de gammapatía IgG (compo-nente M 0.5 g/dl), presentando además hipoalbuminemia (1.4 g/dl), proteinuria, discreta elevación de IgM y una historia de brotes de lesio-nes equimóticas en miembros superiores de una semana de duración.
Ante la sospecha de amiloidosis, se realizaron biopsias de grasa abdo-minal y periorbitarias que fueron negativas. El proBNP, troponinas y el ecocardiograma fueron normales. El aspirado de médula ósea inicial mostró una población B monoclonal lambda con fenotipo poco espe-cifico y mínima presencia de células plasmáticas lambda positivas, y la biopsia de médula ósea tenía una escasa infiltración linfoplasmocítica lambda intersticial, con ausencia de depósito amiloide. La inmunofi-jación fue positiva tanto para suero como para orina con presencia de
cadenas lambda, siendo la primera de tipo IgG. El ratio k/l fue normal. Se realizó una TAC que no mostró adenopatías.
Los resultados del estudio medular mostraban hallazgos compatibles con un linfoma linfoplasmocítico. Se derivó a Nefrología para biopsia renal ante sospecha de amiloidosis con gammapatía IgG lambda (AL). Se evidenciaron depósitos mesangiales rojo congo positivos y en la inmunofluorescencia, depósitos periféricos de IgM, sin depósitos de cadenas ligeras kappa o lambda ni de IgG, IgA, C3 ni Cq1. Al existir discordancia entre la Ig de la gammapatía monoclonal y el depósito de IgM renal, se solicita revisión de la biopsia renal, encontrando en el material congelado un depósito lambda a nivel glomerular. Se solicitó además nueva inmunofijación sérica que resultó positiva para IgM lambda. En ADN extraído de la biopsia renal se confirmó la mutación MYD88 L256P, por lo que el diagnóstico final fue amiloidosis renal AL con depósito de cadenas ligeras lambda y afectación glomerular, asociada a MW.
Actualmente la paciente se encuentra bajo tratamiento con bortezo-mib, dexametasona y rituximab, pendiente de trasplante autólogo de progenitores hematopoyéticos.
RESULTADOS Y CONCLUSIONES:
Dentro de la patología renal asociada a la MW, la amiloidosis AL se ha convertido en una de las causas más frecuentes y está directamente relacionada con la producción clonal patológica de IgM y sus cadenas ligeras. Los pacientes con MW y nefropatía confirmada por biopsia tie-nen una menor supervivencia respecto a los pacientes sin daño renal, y es clínicamente relevante ya que su presencia es una indicación de tratamiento. La enfermedad renal asociada a MW es una complicación rara de una enfermedad poco frecuente, por lo que es importante la valoración de datos clínicos y analíticos durante el seguimiento de la gammapatía IgM que nos permitan detectar de manera precoz la necesidad de realizar pruebas diagnósticas más invasivas y por lo tanto de inicio de tratamiento oportuno.
Paciente con Macroglobulinemia de Waldeström (MW) y amiloidosis renal ALM.A. Suito Alcántara, E. Carrillo Cruz, J. Martín Sánchez, M.L. Martino Galiana
Hospital Universitario Virgen del Rocío

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA244
INTRODUCCIÓN/FUNDAMENTO:
La anemia hemolítica autoinmune (AHAI) es una entidad clínica en la que se produce una destrucción de hematíes por la acción de an-ticuerpos dirigidos contra antígenos eritrocitarios propios. Puede ser idiopática o secundaria a otras patologías, por lo que siempre debe descartarse una etiología desencadenante del cuadro, entre ellas a destacar infecciones víricas, fármacos, enfermedades autoinmunes y síndromes linfoproliferativos.
OBJETIVO:
Revisión de un caso clínico transcurrido en nuestro centro con Ma-croglobulinemia de Waldenström (MW) que debutó con una anemia hemolítica severa con hemoglobina (Hb) de 4 g/dL.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos un varón de 35 años con antecedentes personales de sensibilidad cutánea a la exposición al frío, refiriendo aparición de prurito y eritema en relación a la misma en tratamiento con 25 mg de atarax® diario durante los episodios.
Acude a nuestro centro por cuadro de astenia de 3 semanas de evo-lución, sumado a ictericia mucocutánea y orinas colúricas de 2 sema-nas. Febrícula el día anterior al ingreso. No adenopatías palpables. No pérdida de peso ni sintomatología constitucional. A su ingreso, Hb de 4.5 g/dL, buena cifra de reticulocitos y elevación de LDH y bilirrubina a expensas de bilirrubina indirecta. Se realiza test de Coombs directo que resulta positivo. En estudio de frotis de sangre periférica se constata anemia con anisopoiquilocitosis marcada y presencia de esquistocitos y policromasia. El estudio de orina resulta normal.
Establecida la anemia hemolítica de perfil autoinmune se realizan pruebas complementarias en busca de patologías asociadas. En el estudio de serologías destaca positividad para Ac anti-HBs y anti-HBc con antígeno de superficie negativo e IgG positiva para CMV y VEB. El estudio de M. Pneumoniae y otros virus respiratorios resulta negativo, así como para VIH y sífilis. Se realiza ecografía y TC de tórax, abdomen y pelvis que, salvo esplenomegalia de 13 cm sumado al hallazgo de bazo accesorio, no muestran adenopatías ni hallazgos destacables. Encontramos por otra parte un pico monoclonal IgM de 1.40 g/dL. El estudio inmuno-hematológico establece el proceso como anemia he-molítica autoinmune de tipo mixto. Se realiza panel de crioaglutininas que resulta positivo a título de 32.768.
De forma que, ante el hallazgo de pico monoclonal IgM, en el contexto de un cuadro de anemia hemolítica de perfil mixto, planteamos la
Anemia hemolítica severa como debut de un caso de macroglobulinemia de WaldenstromB. Herruzo, M.C. Muñoz García, I. Fernández Román, J. Montero Benítez, K.E. Kestler Domínguez, A. Rodríguez Fernández
Hospital Universitario Virgen Macarena
sospecha diagnóstica de MW, por lo que se decide realizar P/A y biopsia de médula ósea (MO).
RESULTADOS Y CONCLUSIONES:
En el estudio citomorfológico se observa un 20.6% de población linfo-citaria que la CMF identifica como población clonal B de inmunofeno-tipo inespecífico. El resultado de la biopsia informa de infiltración de médula ósea por proceso linfoproliferativo CD20 positivo compatible con linfoma linfoplasmocítico.
El paciente respondió a tratamiento con prednisona. La cifra de Hb experimentó un ascenso lento, más llamativo en cuanto insistimos en aplicar medidas para evitar la exposición al frío del paciente. Se mantuvo estable hemodinámicamente, asintomático y no presentó eventos hemorrágicos durante el ingreso.
Tras ser dado de alta ha realizado seguimiento en consultas externas de Hematología, con últimas cifras de Hb en torno a 12-13 g/dL y sin nuevas crisis hemolíticas. Por el momento no ha iniciado tratamiento y ya se le han retirado los corticoides.
Como conclusión, la AHAI es una entidad clínica que puede ser se-cundaria a múltiples patologías, entre las que cabe destacar los sín-dromes linfoproliferativos. En esta revisión, nos encontramos ante un paciente joven que presenta como debut de una MW un episodio de AHAI severa. Pese a la anodina clínica y la ausencia de adenopatías, el hallazgo de un pico monoclonal IgM nos hizo pensar pensar en la MW, que se confirmó posteriormente mediante estudio de MO. Actual-mente, el paciente se encuentra en seguimiento en consultas externas y asintomático, sin nuevas crisis recogidas tras finalizar tratamiento corticoideo.

COMUNICACIONES PÓSTERS: Hematología Clínica 245
INTRODUCCIÓN/FUNDAMENTO:
El Trasplante Autólogo de Progenitores Hematopoyéticos de Sangre Periférica (TASPE) es un procedimiento terapéutico empleado como terapia de consolidación en diferentes hemopatías agudas.
En la mayoría de los centros se realiza en el ámbito hospitalario con ingresos prolongados entre 3 y 4 semanas.
En los últimos años, en la mayoría de los centros hemos experimentado un aumento significativo del numero de procedimientos realizados por diferentes motivos (mayor incidencia de hemopatías, mayor edad de pacientes candidatos, aumento de la indicación). Además un in-greso hospitalario tan prolongado es experimentado por el paciente de modo traumático con una inadecuada conciliación con su entorno familiar y su esfera psico-emocional.
En nuestro centro hemos desarrollado un modelo de atención mix-to, con ingreso hospitalario durante las fases de acondicionamiento e infusión de Progenitores Hematopoyéticos, siendo dado de alta precoz el día +1. Desde este momento el paciente continúa con un modelo ambulatorio con un circuito de atención preferente. Para el seguimiento durante la estancia en el domicilio hemos desarrollado un app móvil (app THAD®) con diferentes utilidades (registro de constan-tes, adherencia al tratamiento, entrevista por videoconferencia) que nos permite un control durante las 24 horas del estado del paciente. De esta forma el paciente dispone de acceso a personal sanitario en todo momento.
OBJETIVO:
Analizar la eficacia y seguridad del Trasplante Hematopoyético bajo atención domiciliaria respecto al modelo convencional hospitalario.
Valorar la experiencia y la satisfacción del paciente sometido a TASPE domiciliario.
PACIENTES, MATERIAL Y MÉTODO:
Desde Junio de 2015 que iniciamos el programa a Febrero de 2017 he-mos realizado 13 TASPE bajo atención domiciliaria. Dado que han sido 11 MM y 2 linfomas, en el siguiente análisis únicamente incluiremos los pacientes con diagnóstico con MM por razones de homogeneidad de la muestra. En este periodo hemos realizado 32 TASPE en pacien-tes con MM, lo cual supone que la inclusión en el programa es del 34.3% (11/32). Las características de los pacientes están recogidas en la tabla 1.
Para evaluar la eficacia y seguridad del procedimiento hemos recogido el injerto leuco-plaquetar, el número de trasfusiones de hematíes y
Trasplante hematopoyético autólogo bajo régimen domiciliario. Un nuevo modelo de atención eficaz y seguroP. González Sierra (1), S. De Linares Fernández (2), E. Fernández López (2), L. Moratalla López (2), Z. Mesa Morales (2), M. Jurado Chacón (2)
(1) Hospital Virgen de las Nieves de Granada, (2) Hospital Virgen de las Nieves de Granada
TABLA 1. Características pacientes
Domi (n=11) Hospital (n=21) p <0.05
Sexo (Hombre) 81.8% 52.3% *
Edad (años) 57.1 (40-69) 59.8 (40-69)
Situación al TPH (RC) 63.6% 66.6%
Tipo de catéter
6 (54.5%)
5 (45.5%)
10 (47.6%)
11 (52.4%)
(x106/Kg)
3.01 3.14
Acondicionamiento
4 (36.3%)
4 (36.3%)
3 (27.4%)
9 (42.8%)
10 (47.6%)
2 (9.6%)
plaquetas, el empleo de Nutrición parenteral, los días de ingreso hospi-talario, la presencia de síndrome infeccioso, los días de fiebre, el número de antibióticos empleados y la mortalidad relacionada con el procedi-miento. Dichos datos se han comparado con los pacientes sometidos a TPH de modo hospitalario durante ese período de tiempo (n=21)
Para la valoración de la satisfacción del paciente se ha elaborado una encuesta con 8 items que se valoran de 0-10 y el paciente realiza al finalizar el procedimiento.
RESULTADOS Y CONCLUSIONES:
Resultados: El análisis estadístico comparando ambas muestras está recogido en la tabla 2. Los resultados de la encuesta de satisfacción está recogido en la tabla 3.
CONCLUSIONES
El ámbito bajo el que el desarrolle el procedimiento, tanto hospita-lario como domiciliario, no influye en la eficacia del mismo, presen-tando el mismo injerto leuco-plaquetar y las mismas necesidades trasfusionales.
En nuestra experiencia, el TASPE bajo régimen domiciliario es un procedimiento con menos complicaciones infecciosas, días con fie-bre, y empleo de antibióticos. Por lo que podemos concluir que es un procedimiento seguro.
La satisfacción percibida por los pacientes es alta, consiguiendo el objetivo de conciliación con el entorno psico-emocional del paciente.
XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA246
TABLA 2. Resultados
Domi (n=11) Hospital (n=21) P <0.05
PMN >500
(media/mediana días)
12.9/13 11.6/12
Plaq >20000
(media/mediana días)
14.2/15 12.4/12
Transfusión CH 0.36/0 0.61/0
Transfusión Plaquetas 1.91/2 1.85/2
Días procedimiento
(media/mediana días)
20/20 21.6/21 *
Días Ingreso
Hospitalario
(media/mediana días)
7.27/6 21.6/21 *
Fiebre (%) 2 (18.1%) 17 (80.9%) *
Días con fiebre
(media/mediana días)
0.45/0 3.76/3 *
Antibióticos empleados 0.45/0 2.71/2 *
MRT (30 días) 0% 0%
TABLA 3. Valoración de la satisfacción
2015 (n=4) 2016 (n=4) Mejoría
Seguridad 8.75 9.5
Atención Médica 9.25 9.75
Atención Enfermería 9.25 9.5
Atención Psicológica 9.25 9.5
Información 9.5 9.5 =
Organización 8.25 9.25
Respuesta 8.25 9.5
Disponibilidad 8.5 9.5
Satisfacción Global 8.75 9.5

COMUNICACIONES PÓSTERS
Hemoterapia e Inmunohematología


COMUNICACIONES PÓSTERS: Hemoterapia e Inmunohematología 249
INTRODUCCIÓN/FUNDAMENTO:
La finalidad de la transfusión de hematíes es la de aumentar la ca-pacidad de transporte de oxígeno a los tejidos. Están indicadas en el tratamiento de aquellas situaciones donde exista un déficit en esta capacidad, debido a anemia aguda o crónica, que causa un problema clínicamente importante y siempre que no haya una alternativa más inocua o no se pueda esperar a que haga efecto.
Los criterios transfusionales para concentrados de hematíes (CH) se definen claramente en diferentes guías, para nosotros la más cercana es la publicada por la SETS.
OBJETIVO:
Se plantea un estudio descriptivo de una serie de casos retrospectivo, en el que se recoge la actividad transfus ional del banco de sangre de un hospital comarcal realizada durante el año 2016.
PACIENTES, MATERIAL Y MÉTODO:
Se analizan los datos obtenidos con el software de analítica predictiva SPSS atendiendo a 3 variables: niveles de hemoglobina, plan de uso y unidades transfusionales.
RESULTADOS Y CONCLUSIONES:
Durante el año 2016 en el banco de sangre de nuestro hospital se recibieron 746 peticiones de pruebas cruzadas trasfundiendo un total de 1508 CH.
Dividimos las peticiones en 4 grupos según los valores de Hb, sobre estos datos realizamos un subanalisis para valorar por servicios de trasfusión y según el plan de uso de los hemoderivados.
El plan de uso se recoge en nuestra petición de trasfusión existiendo 4 opciones: cirugía programada (en la petición se detalla fecha y nunca
podrá ser superior a 72h tras la extracción), cruzar y reservar (se cruzan las bolsas y quedan reservadas en banco de sangre durante 72h), en el día y urgente para aquellas situaciones en las que la clínica del pacien te es urgente y precisa trasfusión con pruebas cruzad as en menos de 1h. A parte de estas situaciones está la trasfusión urgente sin pruebas cruzadas reservada para aquellas situaciones emergentes.
Las unidades transfusionales se reúnen en 4 grupos: unidades quirúr-gicas, médicas, servicio de urgencias y UCI y finalmente los pacientes oncohematológicos.
CONCLUSIONES:
– En términos generales las trasfusiones en nuestro centro se realizan atendiendo a los criterios transfusionales que se describen en la guía de la SETS. Esto lo confirma los datos: el 47% de las trasfusiones se realizan con hemoglobinas entre 7.1-8 gr/dl y el 92% con Hb<9gr/dl.
– Los pacientes oncohematológicos se trasfunden en su mayoría con hemoglobinas entre 7-9 gr/dl.
– La mayoría de las trasfusiones con un 83% de los casos se realizan en el día.
– La mayoría de las trasfusiones practicadas con Hb >9 gr/dl se rea-lizan en los diferentes servicios de cirugía, además at endiendo a los planes de uso de cirugía programada y cruzar y reservar. Al analizar con detenimiento estas peticiones la justificación que he-mos encontrado es que estas son entregadas en la consulta de pre anestesia para luego hacer la extracción de la muestra de sangre el día antes de la cirugía. En la petición queda anotado el valor de Hb del hemograma que se solicita para el estudio pre anestésico. Es durante o tras la cirugía cuando se ordena dicha trasfusión que obedece a criterios clínicos (sangrado quirúrgico, evolución clínica del paciente…) o criterios analíticos que no quedan reflejados en la petición del banco de sangre.
Análisis según criterios clínico/analíticos de la actividad trasfusional en un hospital comarcal durante el año 2016M.E. Clavero Sánchez, Y. Moatassim De la Torre, A.M. Alba Sosa
Hospital Básico Santa Ana Motril

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA250
INTRODUCCIÓN/FUNDAMENTO:
El trasplante autólogo de progenitores hematopoyéticos de sangre periférica (TASPE) forma parte con frecuencia del tratamiento de pa-cientes que padecen neoplasias hematológicas.
Plerixafor, antagonista del receptor de quimiocina CXCR4, en combi-nación con G-CSF, induce una mayor movilización en estos pacientes, que a menudo presentan factores de riesgo de fracaso.
Recientes publicaciones sugieren que su uso se asocia a un incremento de las necesidades transfusionales durante el TASPE.
OBJETIVO:
Analizar los requerimientos transfusionales en nuestra serie de pacien-tes autotrasplantados y movilizados con Plerixafor + G-CSF frente a aquellos que no necesitaron del uso del primero.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo que incluye a 52 pacientes movilizados y tras-plantados entre septiembre de 2009 y febrero de 2017. Selecciona-mos un grupo problema que precisó durante su movilización de la combinación G-CSF + Plerixafor (modalidad mayoritaria “just in time”, “pre-emptive” y en menor proporción “rescue”), y elegimos un grupo control de características similares que solo requirió de G-CSF (tabla 1).
Nuestro objetivo era conseguir 5x106/kg CD34+ para la realización de un trasplante y 10x106/kg CD34+ para dos, siendo el mínimo reque-rido 2x106/kg CD34+.
Se emplearon los separadores celulares COBE Spectra y Spectra Optia, y se procesaron al menos 2.5 volemias. Análisis estadístico mediante U de Mann-Whitney y Chi Cuadrado según el tipo de variable.
RESULTADOS Y CONCLUSIONES:
No encontramos diferencias estadísticamente significativas tanto en el consumo de hematíes como plaquetas, con mismo valor de mediana para cada tipo de hemoderivado en cada grupo. El análisis desglosado según el tipo de componente queda recogido en la tabla 2.
CONCLUSIONES:
Los pacientes que requirieron del empleo de Plerixafor tenían un perfil desfavorable estadísticamente significativo en cuanto al nú-mero de líneas de quimioterapia recibidas.
Plerixafor demostró ser un fármaco eficaz en la movilización y ob-tención de células CD34+ requeridas para al menos un trasplante en pacientes con factores de riesgo de fallo.
Análisis de las necesidades transfusionales en el trasplante autólogo movilizado con Plerixafor + G-CSF VS. G-CSFJ.A. Raposo Puglia, M.V. Verdugo Cabeza de Vaca, A. Salamanca Cuenca, S. Ordóñez Vahí, M.A. Correa Alonso
Hospital Universitario Jerez de la Frontera
TABLA 1. Caracerísticas
TABLA 2
Sólo se dio un fallo a Plerixafor en una paciente que quedó excluida de nuestro estudio.
No observamos diferencias estadísticamente significativas para el consumo de hemoderivados.
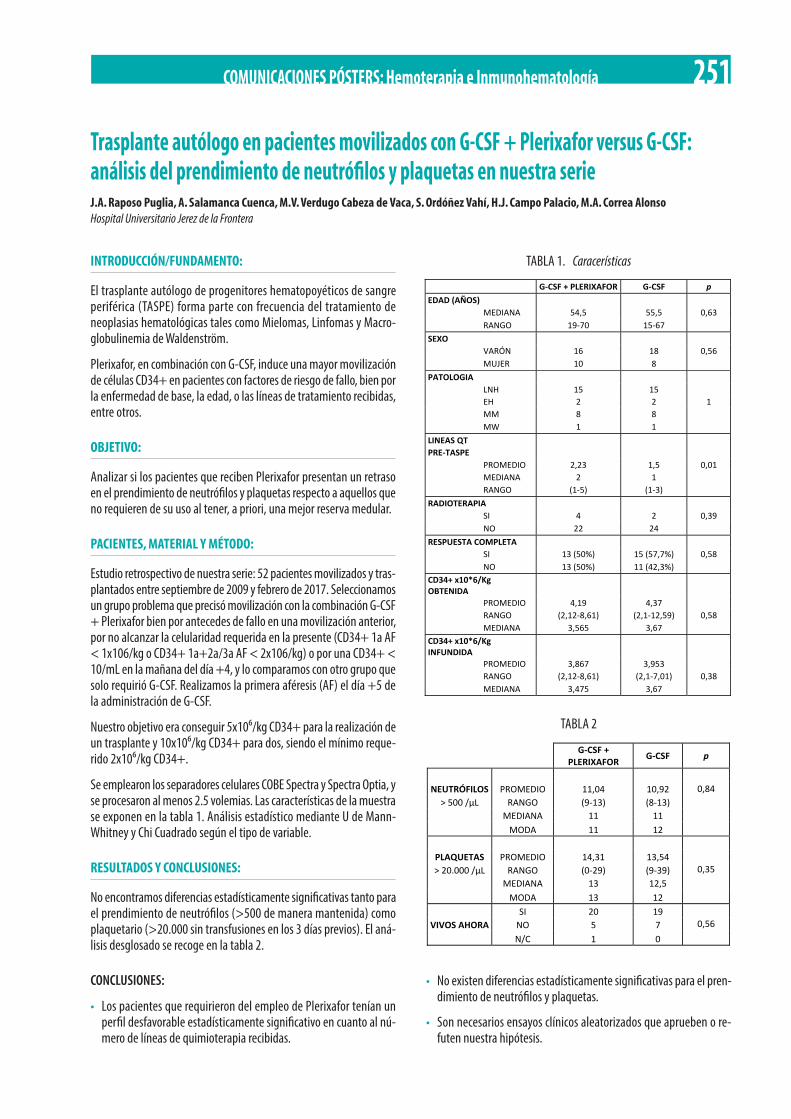
COMUNICACIONES PÓSTERS: Hemoterapia e Inmunohematología 251
TABLA 1. CaracerísticasINTRODUCCIÓN/FUNDAMENTO:
El trasplante autólogo de progenitores hematopoyéticos de sangre periférica (TASPE) forma parte con frecuencia del tratamiento de neoplasias hematológicas tales como Mielomas, Linfomas y Macro-globulinemia de Waldenström.
Plerixafor, en combinación con G-CSF, induce una mayor movilización de células CD34+ en pacientes con factores de riesgo de fallo, bien por la enfermedad de base, la edad, o las líneas de tratamiento recibidas, entre otros.
OBJETIVO:
Analizar si los pacientes que reciben Plerixafor presentan un retraso en el prendimiento de neutrófilos y plaquetas respecto a aquellos que no requieren de su uso al tener, a priori, una mejor reserva medular.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de nuestra serie: 52 pacientes movilizados y tras-plantados entre septiembre de 2009 y febrero de 2017. Seleccionamos un grupo problema que precisó movilización con la combinación G-CSF + Plerixafor bien por antecedes de fallo en una movilización anterior, por no alcanzar la celularidad requerida en la presente (CD34+ 1a AF < 1x106/kg o CD34+ 1a+2a/3a AF < 2x106/kg) o por una CD34+ < 10/mL en la mañana del día +4, y lo comparamos con otro grupo que solo requirió G-CSF. Realizamos la primera aféresis (AF) el día +5 de la administración de G-CSF.
Nuestro objetivo era conseguir 5x106/kg CD34+ para la realización de un trasplante y 10x106/kg CD34+ para dos, siendo el mínimo reque-rido 2x106/kg CD34+.
Se emplearon los separadores celulares COBE Spectra y Spectra Optia, y se procesaron al menos 2.5 volemias. Las características de la muestra se exponen en la tabla 1. Análisis estadístico mediante U de Mann-Whitney y Chi Cuadrado según el tipo de variable.
RESULTADOS Y CONCLUSIONES:
No encontramos diferencias estadísticamente significativas tanto para el prendimiento de neutrófilos (>500 de manera mantenida) como plaquetario (>20.000 sin transfusiones en los 3 días previos). El aná-lisis desglosado se recoge en la tabla 2.
CONCLUSIONES:
Los pacientes que requirieron del empleo de Plerixafor tenían un perfil desfavorable estadísticamente significativo en cuanto al nú-mero de líneas de quimioterapia recibidas.
Trasplante autólogo en pacientes movilizados con G-CSF + Plerixafor versus G-CSF: análisis del prendimiento de neutrófilos y plaquetas en nuestra serieJ.A. Raposo Puglia, A. Salamanca Cuenca, M.V. Verdugo Cabeza de Vaca, S. Ordóñez Vahí, H.J. Campo Palacio, M.A. Correa Alonso
Hospital Universitario Jerez de la Frontera
No existen diferencias estadísticamente significativas para el pren-dimiento de neutrófilos y plaquetas.
Son necesarios ensayos clínicos aleatorizados que aprueben o re-futen nuestra hipótesis.
TABLA 2

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA252
INTRODUCCIÓN/FUNDAMENTO:
El acto transfusional no está exento de riesgos, por ello mediante el Decreto 1088/2005 y posterior orden SCO/322/2007 es obligatorio que los Servicios de Transfusión cuenten con un Sistema de Hemovigilancia (SHV) que notifique a los organismos coordinadores correspondientes y permita la detección, registro, análisis y propuestas de mejora de los incidentes relacionados con la transfusión.
OBJETIVO:
Describir la incidencia y características de las reacciones adversas relacionadas con la transfusión (RAs), errores asociados (EAT) y casi incidentes (CI).
PACIENTES, MATERIAL Y MÉTODO:
Análisis retrospectivo de la incidencia y las características de las RAs, EAT y CI que han sido registrados en el SHV de nuestro centro desde Enero de 2011 a Diciembre de 2016. Durante dicho periodo se han transfundido un total de 52680 componentes de los cuales 44683 fue-ron concentrados de hematíes (CH), 3431 pooles de plaquetas (CP) y 4566 unidades de plasma fresco congelado (PFC). Presentamos un cierre transfusional en progresión en los últimos años con una media del 95%.
RESULTADOS Y CONCLUSIONES:
Se notificaron 132 RAs, de las cuales 118 (89.4%) se produjeron por CH, 12 (9.1%) por CP y 2 (1.5%) por PFC. 53 casos (40.2%) correspondieron a reacciones febriles y/o hipotensivas siendo la mayoría producidas por CH (92.4%). Le siguen en frecuencia las aloinmunizaciones con 45 casos (34.1%) registrados y producidos en su totalidad por CH. Otras menos frecuentes correspondieron a reacciones alérgicas/hipersen-sibilidad (16, 12.1%), edema pulmonar cardiogénico (14, 10.6%), 2 lesiones pulmonares agudas, 1 reacción hemolítica por incompatibi-lidad ABO y 1 refractariedad plaquetar. Destaca que 32 casos (24.3%) fueron registradas como graves (gravedad 2, 3 y 4); de ellos 3 fueron
mortales tratándose de 2 lesiones pulmonares agudas (LPART) y 1 edema pulmonar cardiogénico (EPC).
Se comunicaron 11 EAT, 8 (72.75%) por CH y 3 (27.25%) por PFC. Entre las causas de mayor gravedad destaca en 2 ocasiones la administración de CH distintos del paciente previsto, sólo 1 de ellos derivó en una reacción adversa por incompatibilidad de grupo y gravedad 3.
Además se notificaron 27 CI, de ellos 24 (88.8%) por CH, 2 (7.4%) por CP y 1 (3.8%) por PFC. De ellos cabe destacar que la mayoría (64%) se produjeron en el momento de la extracción, le sigue en frecuencia (22%) el error en la cabecera del paciente y en la prescripción del fa-cultativo (14%).
Conclusiones
El porcentaje de RAs fueron del 0.25%, EAT 0.02% y CI 0.05%.
Las RAs más frecuentes fueron las febriles/hipotensivas junto a las aloinmunizaciones, siendo en general la mayoría producidas por CH. Estos datos concuerdan con los registros autonómicos y nacionales disponibles.
Los CI muestran que la cadena transfusional permite en muchas ocasiones detectar errores antes de que derive en consecuencia por lo que es fundamental que quede registro y análisis de los mis-mos que nos permita en definitiva mejorar la seguridad y calidad transfusional. La mayor parte ocurrieron durante el momento de la extracción por errores en el etiquetado y/o identificación NST (número de seguridad transfusional).
Los EAT pueden prevenirse implementando medidas de seguridad adicionales como el uso de dispositivos electrónicos tipo PDA que faciliten la correcta identificación del paciente.
El análisis de los CI y EAT han servido para poner en marcha accio-nes correctoras no punitivas encaminadas a intentar prevenir en un futuro las situaciones de riesgo y así mismo promover activamente la conciencia profesional para el mantenimiento adecuado de la seguridad de la cadena transfusional.
Sistema de hemovigilancia en un hospital de nivel 2. Análisis de reacciones adversas, errores asociados y casi incidentes relacionados con el acto transfusionalV. Calama Ruiz-Mateos (1), M.E. Molina Rodríguez (2), M. Gómez Rosa (2), E. Martínez Estéfano (2), M. Vahí Sánchez de Medina (2),
E. Ríos Herranz (2)
(1) Hospital Universitario Nuestra Señora de Valme, (2) Hospital Universitario Nuestra Señora de Valme

COMUNICACIONES PÓSTERS: Hemoterapia e Inmunohematología 253
INTRODUCCIÓN/FUNDAMENTO:
La incorporación de la tecnología y los sistemas de información po-tencia la seguridad del proceso transfusional. Uno de los grandes retos de futuro, sino el mayor, es conseguir que la calidad y la seguridad de la transfusión sanguínea sean equiparables a la de los componentes sanguíneos que hoy en día se producen.
OBJETIVO:
Incorporación de un sistema integral y corporativo, multicentro de seguridad transfusional electrónico integrado dentro del sistema de gestión del servicio de transfusión para la trazabilidad completa del circuito de seguridad transfusional a nivel provincial, que gestione todos los procesos inherentes a la transfusión sanguínea en nuestra área de salud con base de datos única que almacene los datos centrali-zadamente, de manera que el paciente queda unívocamente definido en todos los centros.
PACIENTES, MATERIAL Y MÉTODO:
El facultativo realiza la Petición de transfusión electrónica desde la historia clínica Diraya que incluye el formulario de Consentimiento informado para transfusión del SAS. Además de los datos obligatorios incluye el plan de uso “Type and Screening” hasta 1 año para realizar en consulta de anestesia para cirugía programada sin fecha, que permite “Guardar” la petición y que se imprima al ingreso del paciente.
Identificación del paciente mediante una pulsera de identificación única que contiene un código de barras identificador (NUHSA), y la pulsera de seguridad transfusional con etiquetas preimpresas, (puesta en el momento de la extracción) para la muestra y petición.
El extractor de la muestra se identifica por un código personal (códi-go de barras de tarjetas personales o de forma manual). La muestra queda identificada con la etiqueta que se despega de la pulsera de
seguridad transfusional, quedando en este momento enlazado el paciente y la muestra.
Se incorpora una nueva aplicación para la lectura de ambas pulseras, la petición y la muestra a cabecera de paciente mediante estaciones clínicas móviles o PDA, vía WIFI con funcionamiento offline en zonas sin cobertura wifi. Se incrementa la seguridad con el módulo de Iden-tificación Positiva en la fase de extracción de muestra pretransfusional y en la de transfusión.
Información clínica: Desde Diraya se tiene acceso al historial trans-fusional del paciente, (unidades reservadas, transfusiones previas, incidentes).
Servicio transfusión: Reserva, prueba cruzada y entrega mediante un rígido control de seguridad transfusional. Limitación de validez de la muestra. Impresión de etiquetas. Funcionalidad especial para la extrema urgencia y peticiones especiales. Implementación albarán electrónico emitido por el CRTS para la introducción de los datos de los hemoderivados en el stock de forma electrónica.
Cierre transfusional y Hemovigilancia: Uso de formularios oficiales de España. Los datos del paciente y/o la unidad se transcriben automá-ticamente al formulario. El registro del incidente queda asociado al paciente y/o la unidad implicados.
RESULTADOS Y CONCLUSIONES:
Se ha realizado la implantación en varias fases. En un hospital co-marcal, y en la unidades de hospitalización de Hematología, de Re-animación, Hemodiálisis y UCI del hospital de referencia provincial. La automatización de cada eslabón del proceso reduce el riesgo de posibles errores. Se realizan peticiones correctas, se obliga a registrar el Consentimiento Informado, el cierre transfusional y los incidentes relacionados con la transfusión. Todo el proceso queda registrado en la Historia Clínica del paciente.
La implantación completa del sistema en todas las unidades clínicas de la provincia se completará en breve.
Resultados de un sistema integral corporativo de seguridad transfusional electrónica completa multicentroM.A. García Ruiz, F.J. Pérez Zenni, E. Morente Constantín, F. Hernández Mohedo, M. Jurado Chacón
Hospital universitario Virgen de las Nieves

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA254
INTRODUCCIÓN/FUNDAMENTO:
La movilización de progenitores hematopoyéticos (PH) para el tras-plante autólogo (TASPE) se ha beneficiado en los últimos años de di-ferentes estrategias que han optimizado la realización de la misma. El factor estimulante de colonias de granulocitos (G-CSF) sigue siendo el agente de movilización empleado ya sea de forma aislada o bien en combinación con quimioterapia (QTx) o Plerixafor. La utilización de QT ha aportado un beneficio teórico sobre GCSF aislado al eliminar la presencia de células residuales tumorales sin mejorar la supervivencia. Las nuevas moléculas movilizadoras interaccionan regulando la ho-meostasis de las CPH en el microambiente de la médula ósea liberando las células CD34 al torrente sanguíneo.
OBJETIVO:
Describir los resultados de la movilización de progenitores hemato-poyéticos para autotrasplante en nuestro centro desde 2013 a 2016.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de los episodios de movilización llevados a cabo. Se evalúa la estrategia empleada, la cantidad de CD34+/mcL pre-aféresis movilizados y los CD34+/Kg recolectados. Se consideraron movilizaciones satisfactorias aquellas con concentraciones preaféresis de CD34+/mcL mayores de 10 y más de 2x10e6 CD34+/Kg de peso recolectados, y fracasos de movilizaciones aquellas con concentracio-nes preaféresis de CD34+/mcL menores de 10 y/o menos de 2x10e6 CD34+/Kg de peso.
RESULTADOS Y CONCLUSIONES:
Desde Oct. 2013 se han realizado 80 sesiones de aféresis a 68 pacien-tes: 38 fueron síndromes linfoproliferativos (SLP), 28 gammapatías
monoclonales (GM) (27 mieloma múltiple, 1 Macroglobulinemia de Waldestrom) y 2 tumores germinales (TG). De los SLP 13 eran Linfoma B difuso de células grandes (LBDCG), 7 linfoma folicular, 8 linfoma de Hodgkin, 3 Linfoma del Manto (LM), 6 otros SLP y 1 Enfermedad de Castleman. De forma global los episodios de movilización se han realizado con G-CSF en un 25% de los casos, QTx asociada a G-CSF en un 64 % y Plerixafor asociada a G-CSF (+/- QTx) en un 11% de los casos. Los 28 pacientes con GM se han movilizado satisfactoriamen-te, 14 con GCSF, 9 con GCSF + ciclofosfamida y 5 plerixafor + GCSF (+/- Qtx), si bien en 7 casos hubo que recurrir a un segundo episodio de movilización por cifras preaféresis de CD34+ insuficientes. De los pacientes con SLP 31 realizaron una primera movilización satisfactoria, 30 con QTx asociada a G-CSF y uno con GCSF. 4 de los SLP tuvieron un fracaso de movilización previo, de los cuales se lograron movilizar 2 con plerixafor y otros 2 con citarabina a dosis intermedia tras fracaso previo a plerixafor. De los dos pacientes con TG sólo uno se movilizó satisfactoriamente precisando dos dosis de plerixafor. No se obtuvieron progenitores hematopoyéticos para trasplante en 4 pacientes: 3 por fallo de movilización que fueron 1 linfoma folicular multitratado, 1 TG y 1 Enf. de Castelman y 1 paciente con LBDCG por progresión de la enfermedad a pesar de una cifra de CD34+ preaféresis óptima. El porcentaje total de pacientes satisfactoriamente movilizados fue del 94 % con una mediana de 6,84 CD34+/Kg (2,81 – 21,9).
Se han descrito diversos factores de riesgo asociados a pobre movi-lización entre los que destacan la exposición previa a determinados agentes quimioterápicos y la infiltración medular por células tumora-les. Una adecuada evaluación previa de los pacientes es imprescindible para poner en práctica la estrategia óptima en base a dichos factores de riesgo, incluyendo el empleo anticipado de plerixafor en previsión de una mala movilización.
Evaluación de los resultados de movilización de progenitores hematopoyéticos en el trasplante autólogo: experiencia en nuestro centroR. Zapata Bautista, A. Palma Vallellano, Mª.A. Ruíz Cobo, A. Lara Bohórquez, M.A. Castilla Reyes, M. Bendala
Hospital Juan Ramón Jiménez

COMUNICACIONES PÓSTERS: Hemoterapia e Inmunohematología 255
INTRODUCCIÓN/FUNDAMENTO:
La Enterocolitis Necrotizante (ECN) es una patología pediátrica de etiología desconocida, con tratamiento controvertido y elevada mor-bimortalidad. Algunos estudios sugieren una posible relación entre ECN y la transfusión de concentrados de hematíes (CH) en prematuros, pudiendo ser por mecanismos inmunológicos o por una situación de isquemia/perfusión.
OBJETIVO:
Revisión de casos de prematuros de bajo peso al nacer (BPN) trans-fundidos y el desarrollo de ECN en ellos, valorando los factores de riesgo (FR) generales para ECN, así como riesgos relacionados con transfusión.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo, retrospectivo que analiza los prematuros de BPN transfundidos en la Unidad de Prematuros Hospital Virgen Macarena entre 2010-2014. (N = 122)
Se valoraran los principales FR para ECN como edad gestacional (EG) y peso al nacer, así como gravedad de enfermedad desarrollada.
Se realiza un sub-análisis de pacientes que desarrollan ECN y el tiempo desde la transfusión hasta el desarrollo de ECN, el volumen e indica-ción de la transfusión de CH y existencia de clínica abdominal previa.
RESULTADOS Y CONCLUSIONES:
Del total de recién nacidos estudiados la mitad presentó una EG en-tre 28 y 32 semanas. 37 (30%) fueron recién nacidos menores de 28
semanas y 23 (19%) entre 32 y 37 semanas. Sesenta (49%) fueron recién nacidos de muy BPN (1,001 a 1,500 gramos), 34 (28%) con extremadamente BPN (< 1,000 gramos) y 28 (23%) con BPN (1,501 y 2,500 gramos).
La principal indicación de transfusión fue exclusivamente analítica. A 96 pacientes (90%) se les indicó transfusión a razón de 15cc/Kg de peso. El 55% de ellos requirieron más de una transfusión, siendo el 37% de 3 o más transfusiones. La incidencia de ECN en la población estudiada fue del 18.9% (23 pacientes). De estos, el 77% presentaba menos de 1.500gr de peso al nacer y el 81% tenía EG de menos de 32 semanas. Según los Criterios de Estatificación de Bell, 12 casos fueron sospechosos (52%) y 5 casos fueron de enfermedad avanzada (21.7%). Un total de 14 pacientes (60%) presentaron clínica sugestiva de ECN previo a la transfusión, siendo estadísticamente significativo (p=0.000) para el desarrollo de ECN posterior. El resto de pacientes presentaron clínica dentro de las 48 horas siguientes a la transfusión. El 13% de los pacientes con ECN desarrollaron coagulación intravas-cular diseminada, el 100% de estos eran menores de 28 semanas de gestación y con peso al nacer menor a 1.000gr. El porcentaje de éxitus en paciente con ECN fue 21%.
En la literatura revisada, no hemos encontrado datos objetivos so-bre la incidencia de ECN en prematuros de BPN transfundidos. La asociación entre transfusión de CH y desarrollo de ECN es difícil de interpretar, considerando que se trata de una población (prematuros y BPN) con muchos FR, tanto para requerimiento de transfusión como para desarrollo de ECN. Especial mención merece la relación estadística encontrada entre el diagnostico de enterocolitis y clínica abdominal realizada previo a la transfusión, por ello no podemos considerar la transfusión como una causa definitiva de esta patología en la pobla-ción estudiada. Estos datos deberán analizarse posteriormente con es-tudios de casos y controles, para obtener resultados más concluyentes.
¿Podrá ser la transfusión de hematíes un factor de riesgo para enterocolitis necrotizante en recién nacidos de bajo peso al nacer?K. Kestler (1), A. Marin Cassinello (2), I. Fernández Román (3), I. Tallón Ruiz (3), A. Rodríguez Fernández (3), J. Montero Benítez (3)
(1) UGC Hematología. Hospital Universitario Virgen Macarena, (2) UCG Pediatría. Hospital Universitario Virgen Macarena
(3) UGC Hematología. Hospital Universitario Virgen Macarena


COMUNICACIONES PÓSTERS
Hemostasia y Trombosis


COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 259
INTRODUCCIÓN/FUNDAMENTO:
Los anticoagulantes orales directos son fármacos de primera elección en la fibrilación auricular (FA) no valvular. Sin embargo su entrada al mercado está limitada por distintos factores.
OBJETIVO:
Analizamos nuestra experiencia inicial con apixaban.
PACIENTES, MATERIAL Y MÉTODO:
Analizamos características basales y eventos de todos los pacientes con FA a los que se dispensó apixaban durante el primer trimestre de 2014, prescrito por cardiólogos o internistas del sistema sanitario público de nuestro distrito.
RESULTADOS Y CONCLUSIONES:
Incluidos 143 pacientes, con edad media 75±10años, 51% varones. El 95.1% tenían FA y el resto flutter auricular, siendo en 62,7% paroxísti-ca y encontrándose en ritmo sinusal el 50.4%. El riesgo tromboembó-lico fue elevado (CHA2DS2-VASc medio 3,8±1,76; CHADS2 2,28±1,38; HAS-BLED 1,46±0,76), con alta prevalencia de hipertensión (77%), diabetes (35,7%), enfermedad vascular (22,4%), dislipemia (42,7%), insuficiencia renal (21%, ClCr 78,1±28.02ml/min) y habiendo presen-tado ictus o embolismo previo un 21%. Cardiopatía de base presentaba un 69,2%, la mayoría hipertensiva (42,7%), con fracción de eyección preservada el 86,7%. El 30,1% consumía previamente AVK y la dosis de 5 mg/12h fue empleada en el 79%.
Tras un seguimiento medio de 33 meses se registraron 2 muertes de causa cardiovascular, un IAM no fatal y 4 eventos embólicos (en pa-cientes con CHA2DS2VASC medio de 4,5). Dos episodios de sangrado mayor (hemorragia subaracnoidea e ictus hemorrágico.
El perfil de riesgo cardiovascular fue alto en nuestros pacientes; sin embargo, la tasa de eventos fue baja, de acuerdo con resultados de estudios previos.
Perfil clínico y eventos en pacientes anticoagulados con apixaban. Estudio en vida realA.I. Pérez Cabeza (1), A. Valle Alberca (1), M.E. Moreno Beltrán (1), J.A. González Correa (2), R. Bravo Marqués (1), P.A. Chinchurreta Capote (1)
(1) Hospital Costa del Sol, (2) Facultad de Ciencias de la Salud

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA260
INTRODUCCIÓN/FUNDAMENTO:
La eficacia y seguridad de los anticoagulantes orales directos se ha demostrado en múltiples estudios. Conseguir una alta persistencia al tratamiento es vital para que esos resultados se reflejen en vida real.
OBJETIVO:
Conocer la persistencia al tratamiento con apixaban y los factores que influyen en la misma.
PACIENTES, MATERIAL Y MÉTODO:
Incluidos pacientes con FA no valvular a los que se dispensó apixaban de Enero-Marzo/2014, prescrito por un cardiólogo o internista del sistema sanitario público de nuestro distrito. Se realizan entrevistas telefónicas a los 12 y 24 meses.
RESULTADOS Y CONCLUSIONES:
Se incluyeron 143 pacientes de 75±10años, 51% varones, con un riesgo tromboembólico alto (CHA2DS2-VASc 3,8±1,76). La dosis em-pleada fue la recomendada por ficha técnica en el 95,8%, la mayoría con dosis altas (79%).
Continuaban con el tratamiento el 96.9% de los pacientes seguidos a un año (89,5%) y el 90,7% de los seguidos a 24 meses (75,5%). En un 6.1% se redujo la dosis durante el seguimiento. Se asoció la interrup-ción del tratamiento a: sexo masculino (17,3% vs 1,8%, p=0,007), enfermedad pulmonar (21,7% vs 5,9%; p=0,034), tipo de FA (per-sistente 15,6% vs paroxística 9,8% vs permanente 0%; p=0,045) y cifras mayores de HbA1c (6,7±1% vs 6.1±0,6%; p=0,032). La decisión de suspender el tratamiento fue del especialista en la mayoría de los casos (70%), siendo el motivo más frecuente los episodios de sangrado menor (22%). En el 60% no se sustituyó por ningún anticoagulante.
CONCLUSIONES: La persistencia del tratamiento con apixaban a dos años fue elevada, siendo su interrupción decisión del especialista en la mayoría de los casos.
Persistencia al tratamiento con apixaban en pacientes con fibrilación auricular no valvularA.I. Pérez Cabeza, A. Valle Alberca, M.E. Moreno Beltrán, J.A. González Correa, R. Bravo Marqués, P.A. Chinchurreta Capote
Hospital Costa del Sol

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 261
INTRODUCCIÓN/FUNDAMENTO:
Los anticoagulantes orales directos (ACOD) son fármacos de primera elección en la fibrilación auricular (FA) no valvular. Sin embargo, su acceso y uso sigue siendo restringido.
OBJETIVO:
Conocer el grado de adherencia a la ficha técnica de apixabán así como al informe de posicionamiento terapéutico (IPT) de la AEMPS.
PACIENTES, MATERIAL Y MÉTODO:
Incluimos todos los pacientes con FA no valvular a los que se dispensó apixaban durante el primer trimestre de 2014, prescrito por un car-diólogo o internista del sistema sanitario público de nuestro distrito. Analizamos si cumplen los criterios de prescripción y posología según
ficha técnica, así como las condiciones del informe de posicionamiento terapéutico (IPT) de la AEMPS.
RESULTADOS Y CONCLUSIONES:
Se incluyeron 143 pacientes, el 86% procedentes de cardiología. El 95,1% tenía diagnóstico de FA y el resto de flutter auricular. El CHA2DS2VASc era ≥2 en el 90,9% (medio 3,8±1,76). En el 95,1% de los casos se indicó apixaban de acuerdo con las recomendaciones de las guías europeas vigentes en ese momento. La indicación según ficha técnica también se seguía en la mayoría de los casos (93%). En cuanto a las recomendaciones de la AEMPS, no se presentaban las situaciones clínicas especiales requeridas en el 53.1% de nuestros pacientes, y en un 16.8% el riesgo según CHADS2 era menor al requerido (<2).
CONCLUSIONES: En nuestro distrito hay una adecuada adherencia a las guías de práctica clínica y a la ficha técnica de los ACOD; sin embargo en la mitad de los pacientes no se cumplen los criterios del IPT.
Indicación de tratamiento con apixaban en fibrilación auricular: Adherencia a ficha técnica y al informe de posicionamiento terapéutico de la AEMPSA.I. Pérez Cabeza (1), A. Valle Alberca (1), M.E. Moreno Beltrán (1), J.A. González Correa (2), R. Bravo Marqués (1), P.A. Cinchurreta Capote (1)
(1) Hospital Costa del Sol, (2) Facultad de Ciencias de la Salud

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA262
INTRODUCCIÓN/FUNDAMENTO:
La fibrilación auricular (FA) es una causa importante de eventos trom-boembólicos. Los anticoagulantes orales han demostrado reducir la incidencia de ictus y reducir la mortalidad. Es deseable un estudio y tratamiento precoz.
OBJETIVO:
Analizar los resultados iniciales tras la puesta en marcha de una con-sulta cardiológica monográfica de pacientes atendidos en Urgencias con FA sin anticoagulación oral previa.
PACIENTES, MATERIAL Y MÉTODO:
Estudio prospectivo de pacientes atendidos en una consulta monográ-fica de FA comparado con una consulta convencional de cardiología. Analizamos la influencia de esta nueva estructura en el tratamiento.
RESULTADOS Y CONCLUSIONES:
Se incluyen 457 pacientes, 51,2% varones, 69,3±14 años. 63,5% HTA, 23,4% DM, 10,5% IC/FEVI<40% y 6,9% Ictus/AIT. CHADS2 1,6±1,3, CHA2DS2VASc 2,8±1,8 y HASBLED 1,1±0,8; 44,4% FA paroxística, 49,7% persistente y 5,9% permanente. En consulta general cardioló-gica son valorados en 8 semanas (media), realizando ecocardiograma al 32,1% y se prescribe anticoagulantes al 62,2% (73,2% CHA2DS-2VASc>2), la mayoría con AVK. En consulta de FA son valorados en 12,1±8,9 días, disponiendo de ecocardiograma el 89,1% y se pres-criben anticoagulantes al 75,2% (44,4% ACOD, 28% AVK; 96,2% CHA2DS2VASc≥2). Los factores asociados con ACO fueron sexo feme-nino (p=0,029), edad (p<0,001), HTA (p=0,009), embolismo previo (p=0,036), descenso del filtrado glomerular (p<0,001) y presencia de FA persistente/permanente (p=0,006). Durante 55,4±19,3 semanas de seguimiento, la tasa de eventos tromboembólicos fue 0,7%.
Conclusiones: La creación de una consulta monográfica de FA se traduce en un valoración cardiológica más precoz y en una mayor prescripción de anticoagulantes orales, especialmente anticoagu-lantes directos, lo que redunda en una baja incidencia de eventos tromboembólicos.
Prevención de eventos tromboembólicos en pacientes con fibrilación auricular tras la creación de una consulta monográficaA.I. Pérez Cabeza, R. Bravo Marqués, M.E. Moreno Beltrán, A. Valle Alberca, F. Ruiz Mateas, P.A. Chinchurreta Capote
Hospital Costa del Sol

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 263
INTRODUCCIÓN/FUNDAMENTO:
La FA se asocia a una importante morbimortalidad. La anticoagulación oral (ACO) reduce el riesgo de ictus y mortalidad.
OBJETIVO:
Pretendemos conocer en nuestro medio si se reproducen estos bene-ficios clínicos.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de pacientes con FA atendidos de forma am-bulatoria en un centro hospitalario entre 2011 y 2012. Se analizan los eventos combinados (muerte, ACV, AIT, embolia periférica, SCA y sangrados mayores) hasta Enero 2017. La supervivencia libre de eventos en función de la prescripción de ACO se explora con las curvas de Kaplan Meier y se valora si existen diferencias significativas con el test de log Rank.
RESULTADOS Y CONCLUSIONES:
Se incluyen 390 pacientes con FA, edad media de 71 ± 11 años, 52,8% varones, con CHA2DS2VASc medio de 3,1 ± 1,6 y HAS-BLED 1,1 ± 0,7. El 69,7% (n=271) tenían prescritos anticoagulantes orales (249 AVK, 22 dabigatran). Tras un seguimiento medio de 46 ± 20 meses el 26,7% presentó algún evento combinado (72 muertes -34 cardiovas-culares- 34 eventos tromboembólicos, 8 SCA y 30 sangrados mayores). No hubo diferencias significativas en la supervivencia libre de even-tos en función de la prescripción de anticoagulantes orales (74,1% vs 71,4%, p=NS), ni en la reducción de éxitus; pero sí en la reducción de la incidencia de ictus (94,1% vs 84,9%, p=0,005).
Conclusiones: En nuestra muestra la ACO se relaciona con una menor incidencia de ictus, pero no con una reducción de la mortalidad total. Esto podría explicarse por la elevada proporción de exitus de causa no cardiovascular observada.
Papel de la anticoagulación oral en la prevención de eventos cardiovasculares en pacientes con fibrilación auricular atendidos de forma ambulatoriaA.I. Pérez Cabeza, M.E. Zambrano Medina, M.E. Moreno Beltrán, F. Mesa Prado, S. López Tejero, P.A. Chinchurreta Capote
Hospital Costa del Sol

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA264
INTRODUCCIÓN/FUNDAMENTO:
Los anticoagulantes orales directos (ACOD) precisan ajuste de dosis según el aclaramiento de creatinina estimado (eClCr) por la ecuación de Cockcroft Gault (CG). Existen discrepancias con las ecuaciones que estiman el filtrado glomerular (FGe).
OBJETIVO:
Analizamos cómo afectan a la posología recomendada para los ACOD el empleo de las ecuaciones CKD-EPI y MDRD-4 IDMS.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo, de pacientes con fibrilación auricular no valvu-lar atendidos en una consulta de cardiología entre noviembre 2012 y agosto 2014. Se reclasifican los pacientes según la posología recomen-dada para dabigatrán, rivaroxabán, apixabán y edoxabán, en función de la ecuación de FGe empleada. Se tienen en cuenta otros factores clínicos, según ficha técnica. Analizamos el porcentaje de discordancia.
RESULTADOS Y CONCLUSIONES:
Se estudian 454 pacientes, 53,5% hombres, con edad media de 68,7 ± 13,8. La media de las diferencias intraindividuales registradas res-pecto a la ecuación de GC fue de 3,9 ml/min/1,73m2 con MDRD-4 IDMS (IC95%: 1,4-6,4; p=0,003) y 11,3 ml/min/1,73m2 con CKD-EPI (IC95%: 8,9-13,7; p<0.001). Se observa un gradiente en la discor-dancia de la posología (Ej. Apixabán 1,1%, dabigatrán 3,5%, edoxa-ban 5,7%, rivaroxabán 8,4% con MDRD-4 IDMS). Las diferencias se limitaron a pacientes con eClCr < 60 ml/min. Fueron más manifiestas en ≥ 75 años en los cuales las ecuaciones de FGe sobreestiman la función renal.
Conclusiones: En pacientes con fibrilación auricular no valvular, es-pecialmente con insuficiencia renal y en ancianos, las ecuaciones de FGe tienden a sobreestimar la función renal respecto a CG y por ello a recomendar una sobredosificación de los ACOD.
Discordancias en la posología de los anticoagulantes orales directos al emplear fórmulas de filtrado glomerular estimado en vez de la ecuación de Cockcroft GaultA.I. Pérez Cabeza, R. Bravo Marqués, M.E. Moreno Beltrán, A. Alberca Valle, F. Mesa Prado, P.A. Chinchurreta Capote
Hospital Costa del Sol

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 265
INTRODUCCIÓN/FUNDAMENTO:
La enfermedad de Von Willebrand, es la coagulopatía hemorrágica congénita más frecuente. Dada su alta prevalencia, es habitual su aso-ciación con factores de riesgo cardiovascular que, en el caso de ocurrir eventos trombóticos/isquémicos; plantean un reto con respecto a su manejo. La frecuencia de eventos isquémicos en estos sujetos no ha sido establecida
OBJETIVO:
Presentamos el caso de un varón de 77 años con Enfermedad de von Willebran tipo I Severo (niveles basales de FVIIIc 38%, FvWRCo 5%, FvWAg 5%, mutación 1, c.3467C>T). Como antecedentes: HTA, disli-pemia, artritis reumatoide, cardiopatía isquémica con disfunción ven-tricular moderada e hipertensión pulmonar. Como clínica hemorrágica relevante sangrado digestivo con necesidad de ingreso y repercusión hemodinámica en dos ocasiones por angiodisplasia colónica y epistaxis de repetición.
PACIENTES, MATERIAL Y MÉTODO:
En Abril de 2016 es derivado a urgencias por su MAP para estudio de dolor torácico atípico de un mes de evolución con ECG patológico. Se objetiva mínima movilización enzimática e ingresa en el servicio de Cardiología. En situación de estabilidad hemodinámica y sin dolor torácico, se antiagrega con AAS 100mg/24h, a la espera de realización de pruebas diagnósticas. No se utilizó terapia hemostática sustitutiva.
Pruebas solicitadas:
– SPECT miocárdico: area de hipocaptación localizada en los segmen-tos anterodistal y apical, de carácter no reversible compatible con un
área de necrosis miocárdica. Hipocinesia generalizada; VTD estimado en 123 mL, FEVI 0.26 (Método de Germano-Cedars Sinai).
– ETT: Disfución sistólica isquémica de VI moderada con trombo apical. Esclerosis aórtica. Insuficiência aórtica leve. FEVI 53%.
– Hb 13,8 g/dL. Plaquetas 231 x10^9/L. Leu 7300 x10^9/L. Fórmula normal. TP 88%, ratio 1,1. TPTA 25 seg, ratio 1. Cr 0,9 mg/dL. FG 79 mL/min.
RESULTADOS Y CONCLUSIONES:
A la luz de los datos obtenidos, se suspende la antiagregación, admi-nistrando Bemiparina 5000 UI/24h sc, que se continúa al alta. El pa-ciente no presentó clínica hemorrágica durante su ingreso. Durante el mismo, en ningún momento se administró tratamiento hemostático.
Se realiza control a los 15 días, constatando hemoglobina estable, FvWAg 12%, FVIII 46%, FvWRCo 25% y DD 1500 , sin clínica hemo-rrágica ni síntomas cardiológicos, por lo que mantenemos con HBPM a mismas dosis. Se realiza seguimiento mensual. Tras primer mes se constata por ecografía la desaparición del trombo ventricular, suspen-diendo heparina que se sustituye por AAS 100mg/24h. El DDímero, permanece estable en torno a 2000. Ultimo control tras 6 meses del evento no datos de trombo. No clínica hemorrágica durante todo el seguimiento.
CONCLUSIONES:
– Las coagulopatías hemorrágicas, y por tanto la enfermedad de Von Willebrand, no protegen de eventos trombóticos o isquémicos.
– Es posible tratar los episodios trombóticos e isquémicos en pacien-tes con EvW con HBPM y antiagregación, con estrecho control de hemoglobina, ferritina y DD.
Trombosis cardíaca en paciente con enfermedad de Von Willebrand tipo I severaI. Sánchez Bazán, M.E. Mingot Castellano
Hospital Regional de Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA266
INTRODUCCIÓN/FUNDAMENTO:
Romiplostim es una proteína de fusión Fc-péptido (cuerpo peptídico) que señala y activa las rutas de transcripción intracelular a través del receptor de la TPO (también denominado cMpl) para aumentar la producción de plaquetas. Está indicado para pacientes adultos con Púrpura Trombocitopénica Inmune (Idiopática) (PTI) crónica que son refractarios a otros tratamientos como corticosteroides e inmunoglo-bulinas y en quienes la esplenectomía esta contraindicada.
OBJETIVO:
Analizar las características de los pacientes en tratamiento con ro-miplostin de nuestra área, valorando las dosis empleadas, los trata-mientos previos recibidos, las patologías asociadas, los posibles efectos secundarios así como la respuesta a romiplostin.
PACIENTES, MATERIAL Y MÉTODO:
Revisamos los pacientes en tratamiento con romiplostin desde febrero de 2013 hasta febrero de 2017, analizando las variables edad, sexo, patologías asociadas,cifra de plaquetas , tratamientos previos y dosis de romiplostin recibidas. Se incluyeron un total de 10 pacientes de los cuales 5 eran hombres y 5 mujeres. La mediana de edad fue de 67 años (rango 22-83).
De los pacientes analizados tres estaban diagnosticados de PTI en el contexto de leucemia linfática crónica B ( LLC-B)/linfoma linfocítico B (uno de ellos presentó la trombopenia transitoria y tan solo pre-cisó tratamiento con Romiplostin durante un mes, resolviéndose la trombopenia una vez recibido tratamiento especifico para su LLC.B (fludarabina, ciclofosfamida y rituximab)
Otros dos pacientes presentaban la PTI en relación a una macroglobu-linemia de Waldestrom.
Los otros 5 tenían diagnostico de PTI sin neoplasia hematológica aso-ciada.
Uno de ellos recibió romiplostin durante 6 meses y previo a esplenec-tomía (realizada sin incidencias ni necesidad de soporte)
Todos los pacientes habían recibido tratamientos previos que inclu-yeron uno o mas de los siguientes: corticoides, inmunoglobulina, azatioprina, danazol, rituximab y eltrombopag. Ningún paciente se había esplenectomizado previamente , aunque se realizó radioterapia esplénica en uno de ellos. Otro paciente recibió además tratamiento con esquema CDR (ciclofosfamida, dexametasona y rituximab) y otro bendamustina- rituximab e ibrutinib.
En todos nuestros pacientes se ha obtenido una respuesta plaquetar duradera, definida como >50x 109/L en al menos 6 ocasiones con-secutivas, salvo en dos de ellos de inicio reciente, que tan solo han recibido una y tres dosis respectivamente en el momento del estudio.
La mediana de dosis de inicio fue de 1mcg/kg. y la mediana de dosis de mantenimiento de 2 mcg/kg
La dosis mínima empleada fue de 0.7 mcg/kg y la máxima de 6 mcg/kg.
No se observaron efectos secundarios significativos
RESULTADOS Y CONCLUSIONES:
A pesar de tratarse de un numero pequeño de pacientes hemos podido observar una respuesta plaquetar duradera sin precisar tratamiento de rescate en prácticamente todos los pacientes del estudio.
Aunque se necesita un mayor número de pacientes con un plazo de seguimiento mayor, podemos afirmar que el empleo de Romiplostin parece efectivo y seguro a largo plazo.
Romiplostin en trombopenia inmune: experiencia de un hospital comarcalM.C. Galán Fernández, M.O. García Montero, A.I. Gallardo Morillo
Hospital de Antequera

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 267
INTRODUCCIÓN:
La trombosis venosa es una importante causa de morbilidad y mor-talidad en países occidentales, encontrándose asociada a factores genéticos y ambientales.
La homocisteína es un aminoácido producto intermediario del meta-bolismo de la metionina. Varios estudios epidemiológicos han demos-trado que el aumento de los niveles de homocisteína es un factor de riesgo independiente para ateroesclerosis y trombosis.
La hiperhomocisteinemia leve (16-24 mmol/l) o moderada (25-100 mmol/l) no es rara en la población general y está asociada a una va-riedad de factores genéticos o adquiridos que incluyen la edad, taba-quismo, insuficiencia renal, déficit de folato, vitamina B6 o B12 y defi-ciencias heterocigotas de enzimas que participan en su metabolismo. Entre ellas nos encontramos la mutación C677T o variante termolábil de la metil-tetrahidrofolato reductasa (MTHFR), siendo esta la causa genética más común de hiperhomocisteinemia leve-moderada.
Aunque la presencia del alelo T del polimorfismo C677T de la MTHFR se asocia con unos niveles más altos de homocisteína, tanto en la po-blación sana como en la población con enfermedad tromboembolica venosa (ETEV), no existe una asociación sólida entre este polimorfismo y la ETEV.
OBJETIVO:
Con el presente estudio se pretende describir la presencia y distribu-ción de las alteraciones en el gen C677T de la MTFHR tanto en su forma heterocigota como homocigota, hiperhomocisteinemia y clínica de ETEV en la consulta de hematología general de un hospital comarcal.
PACIENTES, MATERIAL Y MÉTODO:
Se realizó un estudio descriptivo no aleatorizado de serie de casos retrospectivo. Se obtiene la muestra de los pacientes que acudieron a la consulta de hematología de nuestro hospital comarcal entre diciem-bre de 2015 y diciembre de 2016. Se seleccionaron para el estudio los pacientes que presentaron la mutación C677T de la MTFHR, tanto en su forma heterocigota como homocigota. Se evaluó la presencia de hiperhomocisteinemia así como la presencia de algún evento clínico relacionado con la ETEV.
Se realiza el análisis de datos con el software de analítica predictiva SPSS. Sobre los datos obtenidos se estudia la posible asociación entre las variables niveles de homocisteína y la presencia de eventos trom-bóticos a través del test de Chi2 de independencia.
RESULTADOS Y CONCLUSIONES:
Las tasas de pacientes heterocigotos y homocigotos observada presen-ta similitud con las series de casos revisadas en la bibliografía.
Los pacientes con heterocigosis presentan mayor tasa de hiperhomo-cisteinemia los cual queda justificado por la etiología multicausal de la misma.
En nuestra muestra no encontramos asociación estadísticamente sig-nificativa entre hiperhomocisteinemia y ETEV.
A mayor edad la incidencia de eventos trombóticos alcanza el 100% de los pacientes, sin diferencia entre ambos sexos.
No se pueden obtener conclusiones poblacionales al tratarse de un estudio descriptivo sobre una muestra pequeña, sin controles pobla-cionales.
Análisis de la incidencia de hiperhomocisteinemia y eventos trombóticos en una serie de casos obtenida de la consulta de Hematología de un Hospital ComarcalM.E. Clavero Sánchez (1), Y. Moatassim De la Torre (1), I. García Cabrera (2), A.M. Alba Sosa (1)
(1) Hospital Santa Ana, (2) Hospital Virgen de las Nieves

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA268
INTRODUCCIÓN/FUNDAMENTO:
Estudios clínicos y epidemiológicos muestran una asociación entre el déficit de vitamina D y un aumento del riesgo de enfermedad car-diovascular.
OBJETIVO:
Debido a que el factor tisular (FT) en las células de musculo liso vascu-lares (VSMC) es un factor crítico en el desarrollo de trombosis arterial, hemos investigado si las moléculas de vitamina D, calcitriol y para-calcitol, pueden modular la expresión de FT inducido por citoquinas proinflamatorias como el TNF-a.
PACIENTES, MATERIAL Y MÉTODO:
Para ello se obtuvieron células del músculo liso aórtico humano, se cultivaron y analizaron mediante diferentes procediminetos.
RESULTADOS Y CONCLUSIONES:
Nuestros resultados muestran que, en comparación con controles, la incubación de dichas células con TNF-a aumenta la expresión de FT y
su actividad procoagulante. Todo ello a través de un mecanismo NK-kB dependiente, como se deduce de la translocación nuclear del p65-NF-kB y la directa interacción del NF-kB con la región promotora del FT. Este aumento en la señalización de FT se acompañó de un incremento de la expresión del receptor activado por proteasa tipo 2 (PAR-2) y una disminución, mediada por el miR-346, en la expresión del receptor de la vitamina D.
Cuando estos experimentos se realizaron en presencia de calcitriol o paracalcitol, comprobamos que dichas moléculas debilitaban la ex-presión y la actividad del FT inducida por el TNF- a (2.01 ± 0.24 y 1.32 ± 0.14 vs. 3.02 ± 0.39 pmol / mg proteína, R<0.05). Es más, estos resultados se acompañaron con una disminución en la señalización de NF-kB y una menor expresión de PAR-2. Todo ello, junto a unos niveles restablecidos del receptor de la vitamina D.
En su conjunto, estos resultados sugieren que la inflamación promueve un estado protrombótico, a través del aumento en la expresión del FT y PAR-2 en las VSMC. Sin embargo, tanto el calcitriol como el paracal-citol modulan dicho estado protrombótico. Un mecanismo que podría relacionarse con el efecto beneficioso de la vitamina D sobre el riesgo cardiovascular.
Efecto del calcitriol y paracalcitol en la expresión del factor tisular y PAR-2 en las células de músculo liso vascularesJ.M. Marténez Moreno (1), C. Herencia (2), I. Vico (3), Y. Almadén (2), F. Velasco (3), A.I. Álvarez (3)
(1) Instituto de Investigación Biomédica de Córdoba (IMIBIC), (2) Instituto de Investigación Biomédica de Córdoba (IMIBIC)
(3) Servicio de Hematología. Hospital Universitario “Reina Sofía”. Córdoba

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 269
INTRODUCCIÓN/FUNDAMENTO:
La terapia puente es la administración de un anticoagulante de acción rápida como la heparina de bajo peso molecular (HBPM) durante el período de cese del tratamiento anticoagulante oral. La decisión del establecimiento de la terapia puente se ha realizado de forma indi-vidualizada y teniendo en cuenta 3 factores: urgencia del proceso invasivo, riesgo hemorrágico del procedimiento y riesgo trombótico del paciente. Hay múltiples estudios que avalan a las HBPM como un tratamiento seguro y eficaz y más coste-efectivo que la heparina no fraccionada (HNF) en la profilaxis y tratamiento de la enfermedad tromboembólica venosa (ETEV), en trombosis venosa profunda (TVP) tromboembolismo pulmonar (TEP), considerándose fármacos de elec-ción en la prevención de ETEV. Existen varias HBPM comercializadas, con diferencias en peso molecular, relación antiXa-IIa y vida media. La bemiparina sódica es la que tiene una mayor relación anti Xa-IIa, (menor riesgo de sangrado), demostrando una baja incidencia de ETEV y de hemorragias en la práctica clínica real.
OBJETIVO:
Existen pocos datos publicados de la terapia puente a dosis terapéu-ticas en los pacientes tratados con anticoagulantes orales (AVK) y su manejo periquirúrgico. Se pretende evaluar la eficacia (recurrencia de trombosis) y seguridad del uso de bemiparina sódica a dosis anticoa-gulantes en la terapia puente y las posibles complicaciones trombó-ticas y/o hemorrágicas (hemorragias mayores y menores) derivadas de este uso.
PACIENTES, MATERIAL Y MÉTODO:
Hemos analizado 975 casos de terapia puente a dosis plenas en un año (hasta junio 2016), realizadas a 650 pacientes (315 hombres y 335 mujeres), entre 15-92 años, (media 69 años). Los motivos de an-ticoagulación fueron FA, prótesis mecánicas, TVP, TEP y trombosis en pacientes con trombofilia. En 70% de casos existían comorbilidades: insuficiencia cardíaca, EPOC, anemia, insuficiencia renal, hepatopatía y secuelas de ACV.
Terapia puente con bemiparina sódica a dosis terapéuticas: experiencia en nuestro centroM.A. García Ruiz (1), E. Morente Constantín (1), P. Romero García (2), M. Gómez Morales (1), M.D. Fernández Jiménez (1), M. Jurado Chacón (1)
(1) Hospital Universitario Virgen de las Nieves, (2) Complejo Asistencial de Soria
TABLA 1
La terapia puente ha consistido en suspender los AVK de 4 (acenocu-marol) a 6 días (warfarina) antes del procedimiento y sustitución por Bemiparina a dosis: <50 kg: 5.000 UI, 50–70 kg: 7.500 UI, 70–100 kg: 10.000 UI y >100 kg: 12.500 UI cada 24h, y una dosis profiláctica 3.500 UI, 12h preprocedimiento, y otra dosis a las 6-12h tras el mis-mo, en función del riesgo de sangrado de la intervención y del riesgo trombótico del paciente. (Tabla 1). La terapia puente se ha realizado en 225 casos de cirugía mayor (artroplastias, prótesis de cadera y rodilla, c. oftalmológica, sustituciones valvulares), 340 casos de cirugía menor (dermatológica, extracciones dentales complejas, implantes denta-rios), 295 casos de procedimientos invasivos, 50 casos por sangrados por AVK, 30 casos por ingresos con descompensación significativa del INR y 35 casos por estudio de trombofilia.
RESULTADOS Y CONCLUSIONES:
Como complicaciones del empleo de bemiparina sódica, se presenta-ron 40 casos de hematomas en zona de punción, 1 trombocitopenia (se confirmó que no fue inducida por heparina) y 2 casos de persistencia de metrorragia con anemización progresiva derivadas del procedi-miento quirúrgico. No hubo casos de hemorragia mayor ni de trom-bosis. La bemiparina administrada a dosis terapéuticas en período pe-rioperatorio, se asocia con una baja incidencia de recurrencia de ETEV y de sangrado. Las complicaciones presentadas en nuestra muestra han sido muy escasas y en pacientes con comorbilidades. En nuestro estudio, la bemiparina es segura y eficaz con mínimas complicaciones hemorrágicas. El tratamiento debe personalizarse en función de cada paciente y de factores relacionados con el procedimiento. Estudios posteriores confirmarán nuestros resultados.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA270
INTRODUCCIÓN/FUNDAMENTO:
Hemofilia adquirida (HA) es una patología rara aunque su impacto puede estar subestimado por la falta de conocimiento, la dificultad del diagnostico de laboratorio y la evolución fulminante. La incidencia de HA aumenta con la edad y la mitad de los pacientes presentan una condición clínica identificable subyacente asociada (malignidad, embarazo etc.); el resto son idiopáticos.
El desarrollo de autoanticuerpos circulantes inhiben algunas proteínas de la coagulación, más frecuentemente al factor VIII, producen un san-grado severo con una mortalidad entre un 9% y un 33%.
La presentación de este trastorno consiste en sangrados agudos típicos de patología de hemostasia primaria que pueden llegar a ser muy se-veros y que requieren un control hemostático muy temprano. El control con agentes bypaseadores es la prioridad en los sangrados agudos, sin embargo los pacientes mantienen un alto riesgo de sangrado mientras que persista el inhibidor por lo que el tratamiento con inmunosupre-sores debe iniciarse tan pronto como se tenga el diagnostico de HA.
OBJETIVO:
Describir cómo se llegó al diagnóstico de hemofilia adquirida siendo una patología poco frecuente.
PACIENTES, MATERIAL Y MÉTODO:
Varón de 92 años con antecedentes personales de HTA e insuficiencia renal crónica leve.
Ingresa en hospital de alta resolución por sospecha de infección res-piratoria con disnea y tos con expectoración. Dos días antes fue diag-nosticado de trombosis crónica parcialmente recanalizada, por lo que se encontraba en tratamiento con heparina ajustada a función renal y se le habían transfundido 2 unidades de concentrados de hematíes por Hb 7.4 gr/dl.
Se detecta reagudización de la anemia con Hb 5.8 gr/dl sin exteriori-zaciones hemorrágicas y en la exploración se palpa masa abdominal
por lo que se solicitó TAC de abdomen en el que se objetivó hematoma retroperitoneal. Debido a que el paciente presentó durante todo el ingreso un TTPa persistentemente elevado, se derivó a nuestro centro para estudio de forma ambulatoria y se suspendió heparina.
Días más tarde, consulta en nuestro servicio de urgencias por mele-nas con hipotensión y hematomas diseminados en abdomen, MMSS y MMII y hemorragia conjuntival. En analítica destacaba Hb de 8.1 gr/dl, plaquetas y bioquímica sin alteraciones. Coagulación con TTPa de 153.7 s, INR 1.07, TPact 90%. Trombina y reptilase normal.
Debido a la clínica hemorrágica y la alta sospecha de HA se realizan las siguientes determinaciones, TTPa 107 segundos (n 30”), mezcla paciente/control TTPa 50”. Dosificación de factores obteniéndose un Factor VIII:C < 1% con el resto de los factores normales junto con una dosificación de Unidades Bethesda en la que obtenemos un nivel de inhibidor de 40UB.
RESULTADOS Y CONCLUSIONES:
Tras la confirmación de HA iniciamos tratamiento con metilpredniso-lona a razón de 1 mg/kg/dia junto con Rituximab 375 mg/m2 dosis semanal por cuatro semanas.
El paciente durante los primeros días de ingreso mantiene episodios de sangrado digestivos con repercusión hemodinámica por lo que se administra FVIIa cediendo el sangrado tras la administración de dos dosis en 6 horas.
La evolución es muy favorable, siendo dado de alta sin clínica hemo-rrágica pendiente de nueva cuantificación de las unidades Bethesda de forma ambulatoria.
Ante un paciente de edad avanzada con clínica hemorrágica y TTPa alargado, sin antecedentes personales ni familiares de sangrado, hay que hacer despistaje HA. Es fundamental el diagnóstico de la misma teniendo en cuenta que su resolución depende de la ausencia total de inhibidor. A pesar de la gran mortalidad de esta patología, la edad avanzada de nuestro enfermo y el diagnostico tardío el paciente respondió adecuadamente sin llegar a diagnosticarse ningún factor condicionante.
Síndrome de hemofilia adquirida: estudio a contrarrelojI. Vico (1), A.I. Álvarez (1), M. Yébenes (1), F. Velasco (1), M. Fernández (1)
(1) Hospital Universitario Reina Sofía

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 271
INTRODUCCIÓN/FUNDAMENTO:
Los síndromes hipereosinofílicos son entidades en las cuales existe una sobreproducción de eosinófilos, asociando el daño de uno o más órganos provocado por la infiltración eosinofilica y la acción de los mediadores.
Una de las posibles causas de aparición de un síndrome hipereosi-nofílico es el desarrollo de un síndrome paraneoplásico. Se encuentra eosinofilia en aproximadamente un 1% de los pacientes con cáncer, alcanzando una cifra del 10% en el caso de los linfomas (en el caso de los tumores hematológicos, la eosinofilia podría estar estimulada por vía de las interleucinas). En el caso del cáncer de pulmón, en torno a un 3% de los pacientes presentarían esa alteración en el hemograma.
OBJETIVO:
Destacar los síndromes hipereosinofílicos como síndromes paraneoplá-sicos que pueden tener lugar en gran cantidad de tumores, siendo posible su asociación con la aparición de trombosis.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos el caso de una mujer de 48 años con antecedentes de hipercolesterolemia y de trastorno distímico, que padece un adeno-carcinoma de pulmón en estadio IV con metástasis cerebrales. Fue tratada con quimioterapia y radioterapia, a pesar de lo cual el tumor fue progresando, llegando a infiltrar el hígado. En el contexto de la enfermedad tumoral, la paciente desarrolló una eosinofilia progresiva como síndrome paraneoplásico.
Coincidiendo con la aparición de la eosinofilia, la paciente comenzó a desarrollar fenómenos trombóticos, en forma de trombosis veno-sas profundas de repetición en miembros inferiores. Por tal motivo se llevó a cabo un estudio de trombofilia completo, que resultó nor-mal. Se dedujo, por lo tanto, que la aparición de dichos fenómenos fue debido a la eosinofilia, además del riesgo de trombosis que lleva
asociado el cáncer per sé. Se inició en aquel momento tratamiento con Tinzaparina.
El desenlace de la paciente fue catastrófico, al sufrir un tromboem-bolismo en la arteria pulmonar principal izquierda, con extensión a arteria lobar inferior y a la porción proximal de las ramas segmentarias de la pirámide basal y língula. Dicho tromboembolismo pulmonar ma-sivo fue el desencadenante que propició el fallecimiento de la paciente.
RESULTADOS Y CONCLUSIONES:
Hay un número considerable de estudios que sugieren que tanto las arterias como las venas pueden dañarse en los síndromes hipereosi-nofílicos, si bien es cierto que los mecanismos son desconocidos. Ejem-plos de fenómenos oclusivos de la macrovasculatura son la oclusión de la arteria femoral, los tromboembolismos pulmonares o las trombosis de los senos intracraneales.
La trombosis de la microvasculatura y la subsecuente oclusión es de-bida a un daño endotelial favorecido por la infiltración del mismo por parte de los eosinófilos. Combinada con la anterior, influye la activa-ción de la cascada de la coagulación. Ejemplos de situaciones en las cuales las eosinofilias afectan a los vasos de pequeño calibre son el fallo renal agudo y el fenómeno de Raynaud, situación en la que podría llegar a producirse una necrosis digital.
Conclusiones:
Parece claro que los síndromes hipereosinofílicos pueden dar lugar a una situación protrombótica, al producir un daño endotelial y al tener un impacto a distintos niveles de la cascada de la coagulación. Sin embargo, los mecanismos precisos siguen siendo a día de hoy des-conocidos, motivo por el cual sería importante profundizar el estudio a este nivel.
Por lo tanto, se debe contemplar la posibilidad de que se desarrollen eventos trombóticos en aquellos pacientes oncológicos en los cuales ha tenido lugar la aparición de una eosinofilia como síndrome para-neoplásico.
La eosinofilia como síndrome paraneoplásico favorecedor de episodios trombóticosE. Morente Constantín, D. Fernández Jiménez, L. Castillo Portellano, M.A. García Ruíz, M.J. Gutiérrez Pimentel, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA272
INTRODUCCIÓN/FUNDAMENTO:
La hemofilia adquirida (HA) se define como una coagulopatía hemo-rrágica de origen inmune, sin antecedentes familiares o congénitos, caracterizada por la presencia de autoanticuerpos plasmáticos que inhiben la función procoagulante de alguno de los factores de la coa-gulación. Es muy infrecuente y de etiología desconocida, aunque el 50% de los casos se asocia a enfermedades autoinmunes, neoplasias, postparto o fármacos. Dado su elevada mortalidad, su diagnóstico y tratamiento precoces son de gran importancia para el pronóstico.
OBJETIVO:
Describir la presentación clínica y abordaje diagnóstico-terapéutico de casos de HA detectados en nuestro centro en un periodo de 9 años.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo retrospectivo de los casos de hemofilia adquirida atendidos en nuestro centro. Se incluyen 10 pacientes diagnosticados en el periodo de 2009-2017. Se analizan datos clínicos, analíticos y de tratamiento.
RESULTADOS Y CONCLUSIONES:
La tasa de incidencia estimada fué de 2 casos por millón de habitantes y año. Un 70% fueron varones, con una mediana de edad de 62 años (rango 27-79). Una paciente tenía antecedentes de leucemia promie-locítica en respuesta completa desde 1 año antes. Dos pacientes tenían enfermedades autoinmunes (tiroiditis, Raynaud) y una paciente se diagnosticó en el período postparto. El patrón hemorrágico al debut fue sobre todo mucocutáneo (80%), aunque en 3 pacientes coexistió con hematuria, en 1 con un hematoma muscular dorsal y en otro con hemorragia alveolar, muscular y de partes blandas complicado con
síndrome compartimental en miembro superior derecho. Un paciente debutó con sangrado en lecho quirúrgico (cavidad anoftálmica tras intervención). Un caso fué asintomático. La gravedad del sangrado fue considerada grave en el 50% de los pacientes a juicio del mé-dico responsable. La sospecha diagnóstica se basó en la clínica más alargamiento del TP o TTPA (o ambos) sin corrección del mismo tras incubación con plasma normal, por lo que se realizó dosificación de factores según la vía de coagulación implicada y estudio cuantitativo de inhibidores. Se excluyó la presencia de Ac. antifosfolípidos. Se en-contraron niveles disminuidos de factor VIII en 8 pacientes (media 2,38 U/dL), factor V en 1 paciente (0,5 U/dL), y de factor de Von Willebrand (5,5 U/dL) en 1 paciente. El test de Kasper fué positivo en todos los pacientes, y la cuantificación de inhibidores frente a los factores im-plicados ofreció una mediana de 43 unidades Bethesda (rango 1-208).
El tratamiento se basó en corticoides en monoterapia en 2 pacientes, y éstos más ciclofosfamida en 8 pacientes. El 80% precisó además so-porte hemostático con complejo protrombínico o FEIBA, precisando uno de los pacientes además FVIIa. En uno de los casos se administró F-VIII (paciente con auto-Ac vs factor de von Willebrand). Tres pacien-tes precisaron de soporte transfusional con concentrados de hematíes. La mediana de respuesta al tratamiento hemostático fue de 14,2 días (rango 3-40) y al inmunosupresor con negativización de inhibidores en plasma en promedio de 97 días. En ningún caso hemos observado recaída de la enfermedad, con un seguimiento de, al menos, 6 meses. Dos pacientes fueron éxitus por los eventos hemorrágicos.
CONCLUSIONES
La HA es una enfermedad muy infrecuente, con una mortalidad del 20% en nuestra población, muy similar a la descrita en la literatura.
El diagnóstico y tratamiento precoz y enérgico ayudan a mejorar el pronóstico de la enfermedad.
Inhibidores adquiridos contra factores de la coagulación: experiencia de nuestro centroM.A. Blum-Domínguez, M.A. Domínguez-Muñoz, R.J. Núñez-Vázquez, R. Jiménez-Bárcenas, R. Pérez-Garrido, F.J. Rodríguez-Martorell
Hospital Universitario Virgen del Rocio
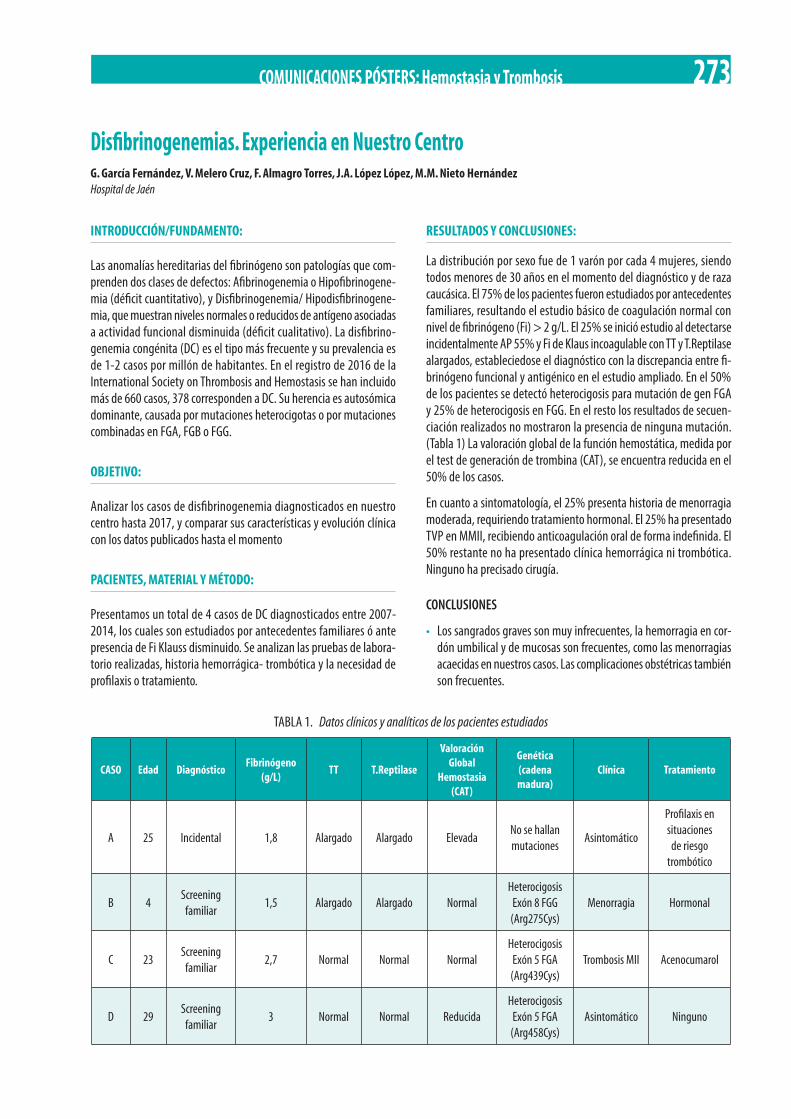
COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 273
INTRODUCCIÓN/FUNDAMENTO:
Las anomalías hereditarias del fibrinógeno son patologías que com-prenden dos clases de defectos: Afibrinogenemia o Hipofibrinogene-mia (déficit cuantitativo), y Disfibrinogenemia/ Hipodisfibrinogene-mia, que muestran niveles normales o reducidos de antígeno asociadas a actividad funcional disminuida (déficit cualitativo). La disfibrino-genemia congénita (DC) es el tipo más frecuente y su prevalencia es de 1-2 casos por millón de habitantes. En el registro de 2016 de la International Society on Thrombosis and Hemostasis se han incluido más de 660 casos, 378 corresponden a DC. Su herencia es autosómica dominante, causada por mutaciones heterocigotas o por mutaciones combinadas en FGA, FGB o FGG.
OBJETIVO:
Analizar los casos de disfibrinogenemia diagnosticados en nuestro centro hasta 2017, y comparar sus características y evolución clínica con los datos publicados hasta el momento
PACIENTES, MATERIAL Y MÉTODO:
Presentamos un total de 4 casos de DC diagnosticados entre 2007-2014, los cuales son estudiados por antecedentes familiares ó ante presencia de Fi Klauss disminuido. Se analizan las pruebas de labora-torio realizadas, historia hemorrágica- trombótica y la necesidad de profilaxis o tratamiento.
RESULTADOS Y CONCLUSIONES:
La distribución por sexo fue de 1 varón por cada 4 mujeres, siendo todos menores de 30 años en el momento del diagnóstico y de raza caucásica. El 75% de los pacientes fueron estudiados por antecedentes familiares, resultando el estudio básico de coagulación normal con nivel de fibrinógeno (Fi) > 2 g/L. El 25% se inició estudio al detectarse incidentalmente AP 55% y Fi de Klaus incoagulable con TT y T.Reptilase alargados, estableciedose el diagnóstico con la discrepancia entre fi-brinógeno funcional y antigénico en el estudio ampliado. En el 50% de los pacientes se detectó heterocigosis para mutación de gen FGA y 25% de heterocigosis en FGG. En el resto los resultados de secuen-ciación realizados no mostraron la presencia de ninguna mutación. (Tabla 1) La valoración global de la función hemostática, medida por el test de generación de trombina (CAT), se encuentra reducida en el 50% de los casos.
En cuanto a sintomatología, el 25% presenta historia de menorragia moderada, requiriendo tratamiento hormonal. El 25% ha presentado TVP en MMII, recibiendo anticoagulación oral de forma indefinida. El 50% restante no ha presentado clínica hemorrágica ni trombótica. Ninguno ha precisado cirugía.
CONCLUSIONES
Los sangrados graves son muy infrecuentes, la hemorragia en cor-dón umbilical y de mucosas son frecuentes, como las menorragias acaecidas en nuestros casos. Las complicaciones obstétricas también son frecuentes.
Disfibrinogenemias. Experiencia en Nuestro CentroG. García Fernández, V. Melero Cruz, F. Almagro Torres, J.A. López López, M.M. Nieto Hernández
Hospital de Jaén
TABLA 1. Datos clínicos y analíticos de los pacientes estudiados
CASO Edad DiagnósticoFibrinógeno
(g/L)TT T.Reptilase
Valoración
Global
Hemostasia
(CAT)
Genética
(cadena
madura)
Clínica Tratamiento
A 25 Incidental 1,8 Alargado Alargado ElevadaNo se hallan
mutacionesAsintomático
Profilaxis en
situaciones
de riesgo
trombótico
B 4Screening
familiar1,5 Alargado Alargado Normal
Heterocigosis
Exón 8 FGG
(Arg275Cys)
Menorragia Hormonal
C 23Screening
familiar2,7 Normal Normal Normal
Heterocigosis
Exón 5 FGA
(Arg439Cys)
Trombosis MII Acenocumarol
D 29Screening
familiar3 Normal Normal Reducida
Heterocigosis
Exón 5 FGA
(Arg458Cys)
Asintomático Ninguno

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA274
La predisposición a trombosis se relaciona con altos niveles de trom-bina y trastornos en la fibrinólisis; las más frecuentes son TVP en MMII e ICTUS, como en el 25% de nuestros pacientes.
Nuestra serie sigue una distribución similar a la descrita en la bi-bliografía en cuanto a presentación clínica: 26-30% hemorrágica; 20- 23% trombótica y 53- 60% asintomática (según los diferentes estudios)
El diagnóstico de laboratorio de CD es difícil y las pruebas más sen-sibles son la TT y la RT, aunque en algunos casos la mayoría de los parámetros de coagulación son normal y sólo el genotipo puede confirmar el diagnóstico.
Hasta la fecha se han descrito más de 100 mutaciones resultantes en CD. La mayoría de las mutaciones se identifican en el gen FGA, al igual que en nuestro caso.

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 275
INTRODUCCIÓN/FUNDAMENTO:
La hemofilia congénita es una enfermedad genética ligada al cro-mosoma X, que se caracteriza por sangrados espontáneos o tras traumatismos por déficit de factor VIII o IX de la coagulación. Su principal complicación es el hemartros, que de manera repetida ocasionan cambios articulares, principalmente una sinovitis crónica, que puede evolucionar a artropatía hemofílica (AH). El tratamiento inicial para evitar la progresión es el soporte hemostático profiláctico y fisioterapia. Sin embargo, ante una mala evolución la sinoviortesis radioisotópica (SR) puede tener un papel fundamental. La SR es un procedimiento intraarticular en el que, mediante la administración de un isótopo radiactivo, renio186 o itrio90 y posterior administración de corticoide intraarticular, se puede frenar la evolución de la sinovi-tis crónica a AH. Es mínimamente invasivo, de fácil realización y con escasos efectos secundarios.
OBJETIVO:
Describir los resultados obtenidos con la SR en los pacientes de edad pediátrica y adolescentes con sinovitis crónica / artropatía hemofílica en los últimos 12 años en nuestro hospital.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo retrospectivo de 23 pacientes sometidos a SR realizados en nuestro centro desde 2005. Analizamos características clínicas y analíticas, criterios para tratamiento y complicaciones asocia-das. Para la evaluación de la AH usamos la clasificación de Fernández-Palazzi.
RESULTADOS Y CONCLUSIONES:
Se han realizado 30 SR a 23 pacientes. Todos varones, con una mediana de edad de 14 años (rango 4-18). Del total, 17 fueron pacientes con
hemofilia A (12 graves y y 5 moderados), 1 con de enfermedad de Von Willebrand y 5 con hemofilia B (2 graves y 3 moderados). Dos de los pacientes con hemofilia A (HA) presentaban inhibidores contra FVIII de alto título. Respecto al tratamiento, todos los pacientes con HA grave y 2 moderada estaban bajo régimen profiláctico, y el resto de pacien-tes recibían tratamiento a demanda. Los pacientes con inhibidores se trataban con agentes by-pass. La mitad de los pacientes presentaban AH grado I, 40% grado II y 10% grado III. En cuanto a los hemartros previos, se obtuvo una mediana de 3 (rango 1-8).
La SR se realizó en la misma proporción en codos (11 casos) principal-mente en el derecho (8), que en rodillas, fundamentalmente ipsilateral (12). Asímismo, se realizaron 7 procedimientos en tobillo derecho y dos en el izquierdo. Se utilizó renio186 en 21 casos (promedio 2 MBq por procedimiento) e itrio90 (promedio de 4,5 MBq) en los nueve res-tantes. Los resultados obtenidos fueron: 15 excelentes (no hemartros, no sinovitis), 13 buenos (no hemartros y disminución de la sinovitis) y 2 respuestas parciaes (disminución del hemartros y persistencia de la sinovitis). En ningún caso hubo falta de respuesta. Se repitió el procedimiento en 7 pacientes que presentaron hemartros en los 6 meses siguientes. El procedimiento fue bien tolerado: Cuatro pacientes presentaron una reacción inflamatoria postratamiento que cedió con analgesicos y antiinflamatorios inhibidores de COX-2.
CONCLUSIONES
La sinoviortesis radioisotópica es un procedimiento eficaz para el control de sinovitis crónica en pacientes hemofílicos con hemartros de repetición, y evitar su progresión a artropatía hemofílica.
Presenta un índice de respuestas muy elevado, es mínimamente invasivo y no se asocia a complicaciones graves.
Sinoviortesis radioisotópica en pacientes hemofílicos pediátricos y adolescentes M.A. Blum-Domínguez (1), L. Pérez-Ortega (2), R. Jiménez-Bárcenas (3), R.J. Núñez-Vázquez (2), F.J. Rodríguez-Martorell (4), R. Pérez-Garrido (2)
(1) Hospital Universitario Virgen del Rocío, (2) Hospital Universitario Virgen del Rocio, (3) Hospital Universitario Virgen del RocioHospital Universitario Virgen del Rocio
(4) Hospital Universitario Virgen del Rocío

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA276
INTRODUCCIÓN/FUNDAMENTO:
La enfermedad tromboembólica es una complicación conocida de los procesos oncológicos, con repercusión en la evolución de la enferme-dad y aumentando morbimortalidad. Existe aumento del riesgo trom-bótico (RT) dependiendo de la patología de base, tratamiento recibido y el aumento de la supervivencia de los pacientes con cáncer. Se debe valorar individualmente el RT por si estuviera indicada profilaxis.
OBJETIVO:
Analizar un caso de trombosis con clínica inusual.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 45 años diagnosticada de Linfoma No Hodgkin B de Células Grandes que realiza tratamiento con R-CHOPx8 con remisión comple-ta. A los 2 meses de haber terminado el tratamiento, consulta por tos y disnea progresiva aumentando con esfuerzo. En radiografía de tórax (RxT) presenta cardiomegalia previamente inexistente. Se rea-liza ecocardiograma encontrando fracción de eyección (FEVI) 35%. Se sospecha tromboembolismo pulmonar (TEP), se realiza angio-TAC presentando un pequeño derrame pericárdico y cardiomegalia a ex-pensas de cámaras izquierdas, sin TEP. Se diagnostica de cardiomegalia por toxicidad de quimioterapia.
Al mes consulta de nuevo por afonía con tos seca desencadenada por toma de líquidos. A la exploración física sin hallazgos destacables. Se sospecha parálisis de cuerdas vocales, confirmándose por fibrolarin-goscopia: parálisis completa en aducción de cuerda vocal izquierda, con movilidad conservada de la derecha. Nueva RxT presenta aumento
Mujer con Linfoma B de Células Grandes y tos persistente con disfonía: A propósito de un caso de Síndrome de OrtnerK. Kestler (1), G. Rodríguez García (1), A. García Guerrero (2), M. García Díez (1), N. Domínguez Velasco (1), B. Herruzo Delgado (1)
(1) UGC Hematología. Hospital Universitario Virgen Macarena, (2) UGC Cardiología. Hospital Universitario Virgen Macarena
IMAGEN 1. Rx Tórax inicial: Cardiomegalia
IMAGEN 2. Rx Tórax control
de cardiomegalia. En TAC tórax hay aumento de derrame pericárdico y derrame pleural pequeño. En TAC cráneo se observan imágenes com-patibles con encefalomalacia secundaria a AVC previo. RMN de cráneo presenta lesiones en putamen relacionado a lesión isquémica antigua y pequeña lesión isquémica reciente (posible etiología embolica). En TAC cuello con deformidad glótica secundaria a parálisis de cuerda vocal izquierda, sin causa que lo justifique.
En ecocardiografía de control, presenta disfunción ventricular severa (FEVI <20%) con engrosamiento de pared de ventrículo izquierdo(VI), pudiendo corresponder a trombo mural y masa de 3cms en aurícula sugestiva de trombo. Se reinicia HBPM. En RMN presenta un trombo adherido en aurícula izquierda(AI) de 2 x 2.3cms. El análisis de las secuencias de realce tardío confirman existencia de trombos intraca-vitarios nodular en AI y laminar en VI. En pulmón izquierdo se aprecian lesiones sugestivas de infartos pulmonares.
RESULTADOS Y CONCLUSIONES:
Estamos ante una mujer con un LNHBCG que ha terminado recien-temente tratamiento quimioterápico sin profilaxis antitrombótica, todos factores aumentando el RT. Al completar estudio presenta mio-cardiopatía dilatada e insuficiencia cardíaca, asociado a un trombo secundario intraauricular-intraventricular, explicando los síntomas de la paciente dentro del Síndrome de Ortner o síndrome cardiovocal: la dilatación de la AI comprime el nervio recurrente izquierdo. También

COMUNICACIONES PÓSTERS: Hemostasia y Trombosis 277
IMAGEN 3. RMN cardíaca
IMAGEN 4. RMN cardíaca
IMAGEN 5. Rx Tórax control
presenta infartos y lesión isquémica de sistema nervioso central po-siblemente cardioembólica.
Se trata de una localización trombótica poco frecuente, con clínica inusual y difícil diagnóstico. Es importante recordar el aumento del RT de los pacientes oncológicos y los beneficios de la anticoagulación. Aunque en esta paciente la evolución fue favorable y sin secuelas per-manentes, no siempre es el caso. La mayoría de los eventos trombóti-cos se dan en paciente con tratamiento quimioterápico ambulatorio, ya que aumenta 6.5 veces el RT al compararlo con la población general. Aunque en pacientes oncohematológicos ingresados por razones mé-dicas se recomienda tromboprofilaxis, en pacientes ambulatorios no es una recomendación universal, pero sí se debe valorar necesidad de manera individual.
A los 24 meses del diagnóstico de trombosis presenta adecuada evolución de dilatación miocárdica, normalización de dimensiones de ventrículos y función sistólica, resolución absoluta de trombos. La disfonía tuvo mejoría progresiva en relación con normalización de dilatación auricular.


COMUNICACIONES PÓSTERS
Morfología y Biología Celular


COMUNICACIONES PÓSTERS: Morfología y Biología Celular 281
INTRODUCCIÓN/FUNDAMENTO:
A pesar de las altas tasas de curación con los tratamientos estándar, un porcentaje de pacientes con diagnóstico de LAM-bajo riesgo según clasificación de la EuropeanLeukemia Net (ELN),continuan recayendo.No está bien definido si la recaída es debida a una quimioresistencia intrínseca o a una evolución clonal con adquisición de nuevas muta-ciones drivers.
OBJETIVO:
Establecer mediante estudios fenotípicos y genotípicos la evolución clonal de pacientes diagnosticados de LAM- bajo riesgo citogenético/molecular que presentan recaída tras alcanzar remisión completa.
PACIENTES, MATERIAL Y MÉTODO:
Estudio retrospectivo de 26 pacientes con LAM-bajo-riesgo citogené-tico/molecular (no LAM-M3) (KN-NPM1mut/FLT3wt, N=16; t(8;21), N=4; inv(16), N=4; KN-CEBPAbi, N=2) según la clasificación ELN. Obtuvimos ADN procedente de muestras de médúla ósea en el mo-mento de recaída y diagnostico, y mediante secuenciacion directa Sanger [ABI 3130® GeneticAnalyzer (AppliedBiosystems Inc., Foster City, CA)], analizamos los genes NRAS, KRAS, DNMT3A, IDH1, IDH2 and TP53. Analizamos pérfil fenotípico en ambos momentos mediante FACSCanto II. Paralelamente, todas las mutaciones fueron estudiamos en las muestras del diagnóstico de 7 pacientes con LAM bajo riesgo citogenético-molecular que se encontraban aún en remisión completa
RESULTADOS Y CONCLUSIONES:
Patrones de recaída: En el análisis citométrico, hallamos dos patrones principales de recaída según la presencia o ausencia de marcadores aberrantes diagnostico/recaída: Patrón 1: 14 pacientes (53.8%) sin cambios. Patrón 2: 12 pacientes (46%) con cambios, afectando prin-
cipalmente a CD15 (58.3%), CD117, CD34 y CD56 (41.6%), CD7 y CD13 (33.3%), CD11b, CD4, CD33 y CD14 (25%). En el análisis molecular, se encontró mayor prevalencia de variaciones moleculares en los pacien-tes incluidos en el patrón 2 (36,3%), que afectaban a los genes NPM1, DNMT3A y FLT3 frente al patrón 1 (15,38%) para NPM1 y DNMT3a. No encontramos mutaciones en los genes TP53, N/KRAS. Un 75% de los pacientes que presentaban una mutación en NPM1 la mantenían también a la recaída, ocurriendo de la misma manera con DNMT3a (61.6%).
Impacto fenotípico/molecular en la supervivencia: Con una mediana de seguimiento tras la recaída de 35 meses y comparando el grupo de pacientes con cambios moleculares vs no cambios, encontramos que los pacientes con variaciones moleculares sobreviven menos (cambios: 20±17.9% vs no cambios 48.1±11.5%, p=,01), ocurriendo la muerte de forma más precoz (cambios: 38 meses vs no cambios: 94 meses, p=0.001).
Hallazgo de dos mutaciones nuevas y validación: Hallamos dos mu-taciones nuevas en el gen DNMT3a, no descritas en la literatura. Am-bas aparecieron en pacientes con cariotipo normal FLT3wt/NPM1mut: p.F902fs y p.D876Y. Ambas fueron validadas mediante pirosecuen-ciacion, no existiendo en muestras de individuos sanos ni en las del momento de la remisión completa de ambos pacientes y apareciendo en la recaída leucémica. Mediante análisis in silico comprobamos que producían una alteración en la proteína: una metilación pasiva, estan-do involucradas en la leucemogénesis.
CONCLUSIONES: i). Las mutaciones drivers son más frecuentes en pa-cientes con LAM de bajo riesgo que recaen que en aquellos en remi-sión completa.ii). Demostración de dos patrones de recaída: aquellos pacientes que mantienen la clona principal vs aquellos pacientes que recaen con una clona diferente.iii).Detección de dos nuevas mutacio-nes en gen DNMT3A: D876Y y p.F902fs, con afectación en la proteína y por tanto con probable repercusión en la aparición de la LAM.
Patrones de recaída fenotípica y genotípica en leucemias agudas mieloblásticas bajo riesgo citogenético/molecularC. Martínez-Losada (1), J. Serrano-López (2), J. Serrano (1), J. Sánchez (1)
(1) Hospital Universitario Reina Sofía, (2) IMIBIC

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA282
INTRODUCCIÓN/FUNDAMENTO:
La necrosis de médula ósea (NMO) es una entidad clínica infrecuente donde se produce una necrosis coagulativa que afecta de forma difusa al tejido hematopoyético. En un 90% de los casos se asocia a neopla-sias, siendo hasta el 60% hemopatías malignas. Presenta una rápida evolución y un mal pronóstico, con una clínica inespecífica. Realizar un diagnóstico precoz resulta complejo, lo que en la mayoría de casos sólo se consigue post mortem, lamentablemente.
OBJETIVO:
Revisión clínica de un caso acontecido en nuestro centro, el cual debutó con dolor dorsolumbar, plaquetopenia y elevación de LDH.
PACIENTES, MATERIAL Y MÉTODO:
Presentamos un varón de 47 años sin antecedentes personales médicos de interés, con ocupación profesional de guarda forestal (con historia de viajes a entornos rurales de varios países), que ingresa en M. Interna por dolor dorsolumbar intenso de 2 semanas de evolución junto con fiebre vespertina y equimosis múltiples en miembros y abdomen.
Ante la inicial sospecha de osteomielitis, inicia antibioterapia empírica. Se realiza batería de pruebas complementarias hallando un hemogra-ma normal a excepción de trombocitopenia grado III y una elevación de LDH y fosfatasa alcalina (FA) (7.743 U/L y 908 U/L respectivamente). En frotis de sangre periférica (FSP) se observa una reacción leucoeritro-blástica y trombocitopenia. Mediante gammagrafía actividad osteogé-nica incrementada en esqueleto axial y periférico que impresiona de carácter maligno. En TC-tórax, abdomen y pelvis destaca un derrame pleural bilateral. No otras imágenes sugestivas de neoplasia sólida y/o masas. Ante la sospecha de proceso neoplásico con posible afectación medular, se realiza punción/aspiración de médula ósea (MO) con toma de biopsia para estudio.
RESULTADOS Y CONCLUSIONES:
RESULTADOS:
En la citomorfología de MO se encuentra una pérdida de la arquitectura normal de MO, con celularidad hematopoyética escasa y un material amorfo eosinofílico de aspecto punteado que baña toda la extensión. Pequeños acúmulos celulares no hematológicos de 2-3 células difíciles de identificar, con núcleo picnótico, bordes citoplasmáticos irregulares y tamaño no superior a un hematíe. Este patrón celular es típico de las llamadas “ghost cells”. El informe de la biopsia da el diagnóstico de NMO grado III (afectación superior al 50%). Se realiza CMF con panel de cribado de agudas sin hallazgos concluyentes.
Necrosis de médula ósea secundaria a hemopatía malgina: A propósito de un casoB. Herruzo Delgado, M. Manzanares Pérez, F.R. Acosta Maestre, N. Domínguez Velasco, J. Montero Benítez, A. Rodríguez Fernández
Hospital Universitario Virgen Macarena
Tras 2 semanas de ingreso el paciente sufre un deterioro clínico rápido, falleciendo en el día +18 a consecuencia de una hemorragia cerebral masiva. Se solicita necropsia diagnóstica que informa finalmente de sarcoma mieloide/leucemia mieloide aguda ampliamente diseminada con afectación de médula ósea, compromiso ganglionar e infiltración multiorgánica (cerebro, corazón, hígado, bazo, costilla, testículos…).

COMUNICACIONES PÓSTERS: Morfología y Biología Celular 283
CONCLUSIONES:
La NMO es una entidad clínica infrecuente de rápida evolución, mal pronóstico y difícil diagnóstico, basado en el estudio citomorfológico de MO, requiriendo en ocasiones tomar múltiples biopsias. Las hemo-patías malignas siguen siendo la primera causa, seguidas de otras neo-plasias. Comporta una clínica inespecífica, donde la presencia de dolor óseo y/o fiebre, junto con citopenias y elevación de LDH y FA pueden orientarnos. En FSP puede observarse una reacción leucoeritroblástica.
Las opciones terapéuticas quedan limitadas en la mayoría de casos a tratamiento de soporte, puesto que la dificultad diagnóstica con-diciona el inicio del tratamiento y, lamentablemente, en la mayoría de los casos, el diagnóstico definitivo se obtiene tras el fallecimiento del paciente.

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA284
INTRODUCCIÓN/FUNDAMENTO:
El linfoma de Hodgkin anal primario es un tipo de linfoma raro, descrito principalmente en pacientes afectos de VIH o de enfermedad inflama-toria intestinal. Los linfomas de Hodgkin primarios gastrointestinales solo representan el 1-3% del total de casos y a nivel colorrectal solo 0,2%. La asociación a VEB positivo es muy frecuente.
OBJETIVO:
Presentamos el caso de una paciente de 67 años de edad, que consulta por tumoración anal en el Servicio de Urgencias, con diagnóstico de Linfoma de Hodgkin anal primario.
PACIENTES, MATERIAL Y MÉTODO:
Como antecedentes personales presentaba diabetes mellitus tipo 2 y Síndrome de Down, siendo VIH negativa y VEB positiva. Refería colitis de largo tiempo de evolución, con fisura, así como pérdida de peso de 15 Kg en 1 año, con pérdida de apetito. EDA normal. EDB previa: con orificio fistuloso en margen anal izquierdo. No tiene antecedentes de enfermedad inflamatoria intestinal ni afectación linfomatosa a otro nivel.
EA: Tras valoración por Cirugía, se realiza resección de dicha masa y realiza colostomía en flanco izquierdo, con diagnóstico anatomopa-tológico de Linfoma de Hodgkin anal primario.
En PRUEBAS COMPLEMENTRIAS, presentaba patrón de anemia ferro-pénica, con resto de hemograma normal. Serologías para VHB, VHC y VIH negativas. En TAC, no adenopatías ni masas a otro nivel, salvo tumoración rectal ya conocida. El diagnóstico anatomopatológico fue: fragmento de tejido perianal infiltrado por un proceso linfoprolife-rativo virus de Epstein-Barr positivo, con hallázgos histopatológicos compatibles con un linfoma de Hodgkin clásico subtipo celularidad mixta. Las células neoplásicas mostraban inmunotinción positiva para CD30, PAX 5, bcl2, bcl6, CD20, de manera focal para Oct-2, con detección masiva del Virus de Epstein-Barr (VEB) por medio de EBER y LMP1. Negativas para CD3, BOB.1, CD45, CD15 y ALK. Ecocardiograma: normal, con mínimo déficit de relajación de VI.
RESULTADOS Y CONCLUSIONES:
La paciente recibió, tras resección quirúrgica y colostomía, tratamiento con esquema AVBD del que ha recibido 3 ciclos sin incidencias, en-contrándose actualmente (4 meses fin de tratamiento) en Remisión Completa.
CONCLUSIONES: A pesar de que la presentación extranodal del Lin-foma de Hodgkin es una entidad rara, la posibilidad debe tenerse en cuenta, ya que la supervivencia depende de un tratamiento temprano y eficiente. Este subtipo de L.Hodgkin es más infrecuente aún en pa-cientes inmunocompetentes y sin enfermedad inflamatoria intestinal, como es el caso de nuestra paciente. El tratamiento habitual suele ser quirúrgico, con quimioterapia y/o radioterapia asociados.
Linfoma de Hodgkin anal primario: una entidad infrecuenteM.J. Llamas Poyato, D. Moreno Garrido, I. García Díez
Hospital Infanta Margarita, AGS Sur de Córdoba

COMUNICACIONES PÓSTERS: Morfología y Biología Celular 285
INTRODUCCIÓN/FUNDAMENTO:
El síndrome hemofagocítico o linfohistiocitosis hemofagocítica se pro-duce fruto de una activación patológica a nivel inmunológico. Existe, por tanto, una situación de inflamación desproporcionada en la cual pueden aparecer fiebre, citopenias, esplenomegalia, hipertrigliceride-mia e hipofibrinogenemia. También se caracteriza por la aparición de fenómenos de hemofagocitosis en médula ósea. Puede presentarse de forma primaria o por causa de tumores o infecciones.
Para poder diagnosticar un síndrome hemofagocítico se debe cumplir al menos uno de los siguientes supuestos:
Que haya un estudio molecular que así lo confirme (PRF1, UNC13D, Munc18-2, Rab27a, STX11, SH2D1A o BIRC4).
Que se den al menos 5 situaciones de las siguientes: fiebre, espleno-megalia, citopenias, hipertrigliceridemia, hipofibrinogenemia, hemo-fagocitosis a nivel medular, ganglionar o esplénico, ferritina elevada, CD25 soluble > 2.400 U/mL, actividad de células NK nula o ausente.
En cuanto al pronóstico de los pacientes que padecen un síndrome hemofagocítico, hay que señalar que éste ha mejorado en los últimos años, fruto del desarrollo e implantación en 1.994 de un protocolo internacional de tratamiento. Este tratamiento utiliza dexametasona, etopósido y metotrexato intratecal durante 8 semanas, reevaluando posteriormente cada caso.
OBJETIVO:
Realizar una revisión de una patología de difícil diagnóstico y cuyo pro-nóstico depende en gran medida de la sospecha y tratamiento precoz.
PACIENTES, MATERIAL Y MÉTODO:
Paciente de 57 años con antecedentes de hipertensión arterial y artritis gotosa que fue ingresado en la Unidad de Enfermedades Infecciosas debido a un shock séptico de origen urinario. Se resolvió el cuadro y fue dado de alta.
Posteriormente fue diagnosticado de un cáncer de recto con metástasis hepáticas, hecho que se descubrió en un nuevo ingreso. Durante dicho ingreso desarrolló un cuadro febril persistente y necesitó una reeva-luación por parte de Infecciosas. Como datos analíticos destacables, llamaba la atención una pancitopenia, junto con hipertrigliceridemia, hipofibrinogenemia e hiperferritinemia. Con la presunción diagnóstica de síndrome hemofagocítico y para aclarar el diagnóstico diferencial del cuadro se realizó una PAMO. En este contexto se decidió iniciar de forma precoz tratamiento empírico con Dexametasona.
RESULTADOS Y CONCLUSIONES:
El resultado del aspirado medular confirmó, junto a los datos clínicos y analíticos, que cumple criterios de síndrome hemofagocítico. Como el paciente tuvo una excelente respuesta al tratamiento corticoideo con
Síndrome hemofagocítico como complicación en un paciente con cáncer de recto: la importancia de la morfología de médula ósea en el diagnóstico de una entidad agresivaE. Morente Constantín, J. Badiola González, R. Ríos Tamayo, M.P. Garrido Collado, P.A. González Sierra, M. Jurado Chacón
Complejo Hospitalario Universitario de Granada
IMÁGENES DE LA MORFOLOGÍA DE MÉDULA ÓSEA
1. Imagen del estudio morfológico de médula ósea en la cual se observa un macrófago
cargado de detritus celulares, vacuolización citoplasmática y pigmento basófilo. 100x
2. Fenómeno de hemofagocitosis: macrófago de gran tamaño en cuyo citoplasma se
observan numerosos hematíes y varias plaquetas. 50x

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA286
Dexametasona se decidió mantenerlo, programando una reducción paulatina. Continuó el seguimiento de su cáncer de recto por parte de Oncología y se citó por Hematología para llevar a cabo una ree-valuación.
En las siguientes revisiones el paciente mostró un muy buen estado general, sin que aparecieran hallazgos patológicos en la exploración. La analítica se fue normalizando, por lo que se está procediendo a descender progresivamente la pauta de corticoides. Actualmente se encuentra pendiente de la próxima revisión.
Conclusiones:
El síndrome hemofagocítico es una enfermedad que asocia una mor-talidad elevada. Por lo tanto, es necesario conocerlo de cara a poder realizar un diagnóstico precoz y así iniciar de forma temprana el trata-miento del mismo. Los datos de la morfología de médula ósea son muy importantes para el diagnóstico, por lo que el papel del hematólogo es fundamental en esta patología tan heterogénea.
3. Digestión de un cayado, varias plaquetas y un hematíe por parte de un macrófago.
50x
4. Macrófago en cuyo citoplasma se encuentra atrapado un mielocito. 50x
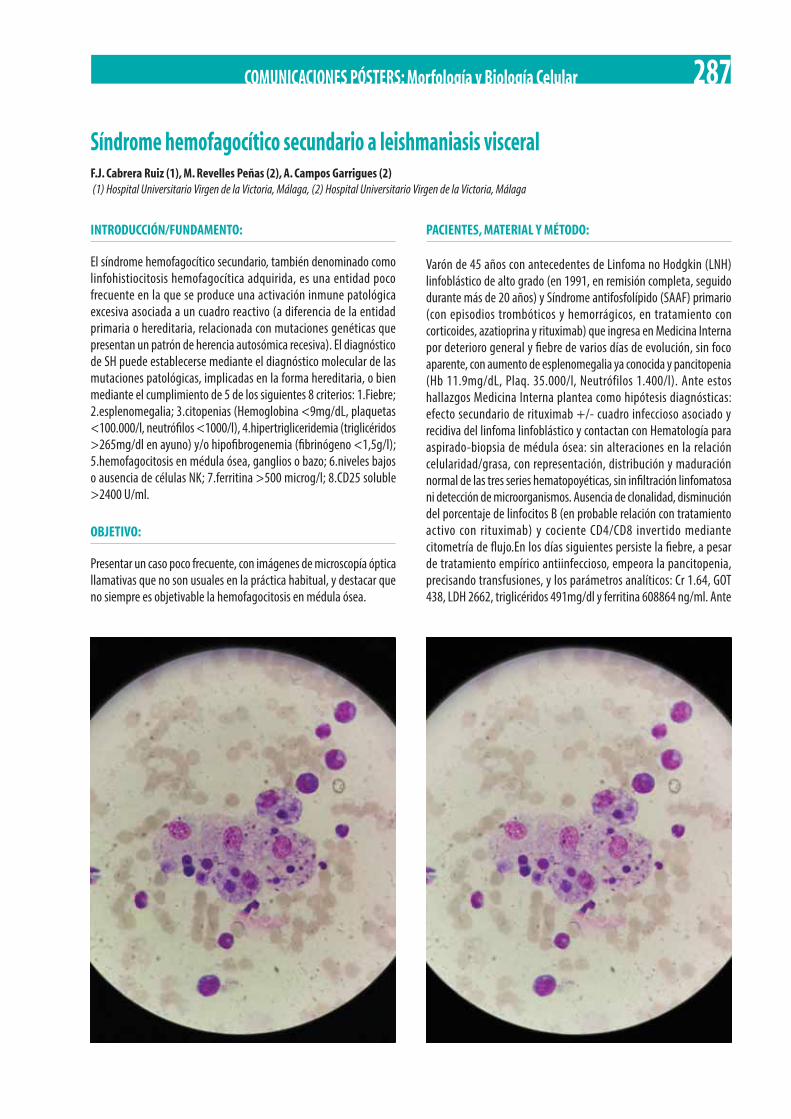
COMUNICACIONES PÓSTERS: Morfología y Biología Celular 287
INTRODUCCIÓN/FUNDAMENTO:
El síndrome hemofagocítico secundario, también denominado como linfohistiocitosis hemofagocítica adquirida, es una entidad poco frecuente en la que se produce una activación inmune patológica excesiva asociada a un cuadro reactivo (a diferencia de la entidad primaria o hereditaria, relacionada con mutaciones genéticas que presentan un patrón de herencia autosómica recesiva). El diagnóstico de SH puede establecerse mediante el diagnóstico molecular de las mutaciones patológicas, implicadas en la forma hereditaria, o bien mediante el cumplimiento de 5 de los siguientes 8 criterios: 1.Fiebre; 2.esplenomegalia; 3.citopenias (Hemoglobina <9mg/dL, plaquetas <100.000/l, neutrófilos <1000/l), 4.hipertrigliceridemia (triglicéridos >265mg/dl en ayuno) y/o hipofibrogenemia (fibrinógeno <1,5g/l); 5.hemofagocitosis en médula ósea, ganglios o bazo; 6.niveles bajos o ausencia de células NK; 7.ferritina >500 microg/l; 8.CD25 soluble >2400 U/ml.
OBJETIVO:
Presentar un caso poco frecuente, con imágenes de microscopía óptica llamativas que no son usuales en la práctica habitual, y destacar que no siempre es objetivable la hemofagocitosis en médula ósea.
PACIENTES, MATERIAL Y MÉTODO:
Varón de 45 años con antecedentes de Linfoma no Hodgkin (LNH) linfoblástico de alto grado (en 1991, en remisión completa, seguido durante más de 20 años) y Síndrome antifosfolípido (SAAF) primario (con episodios trombóticos y hemorrágicos, en tratamiento con corticoides, azatioprina y rituximab) que ingresa en Medicina Interna por deterioro general y fiebre de varios días de evolución, sin foco aparente, con aumento de esplenomegalia ya conocida y pancitopenia (Hb 11.9mg/dL, Plaq. 35.000/l, Neutrófilos 1.400/l). Ante estos hallazgos Medicina Interna plantea como hipótesis diagnósticas: efecto secundario de rituximab +/- cuadro infeccioso asociado y recidiva del linfoma linfoblástico y contactan con Hematología para aspirado-biopsia de médula ósea: sin alteraciones en la relación celularidad/grasa, con representación, distribución y maduración normal de las tres series hematopoyéticas, sin infiltración linfomatosa ni detección de microorganismos. Ausencia de clonalidad, disminución del porcentaje de linfocitos B (en probable relación con tratamiento activo con rituximab) y cociente CD4/CD8 invertido mediante citometría de flujo.En los días siguientes persiste la fiebre, a pesar de tratamiento empírico antiinfeccioso, empeora la pancitopenia, precisando transfusiones, y los parámetros analíticos: Cr 1.64, GOT 438, LDH 2662, triglicéridos 491mg/dl y ferritina 608864 ng/ml. Ante
Síndrome hemofagocítico secundario a leishmaniasis visceralF.J. Cabrera Ruiz (1), M. Revelles Peñas (2), A. Campos Garrigues (2)
(1) Hospital Universitario Virgen de la Victoria, Málaga, (2) Hospital Universitario Virgen de la Victoria, Málaga

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA288
el cumplimiento de cinco de los criterios diagnósticos establecidos por la International Histiocyte Society para el diagnóstico de linfohistiocitosis hemofagocítica, se establece esta entidad como causa del cuadro que sufre el paciente, y se propone la realización de un nuevo aspirado de médula ósea previo al inicio del tratamiento. En esta ocasión, se observan al microscopio óptico abundantes macrófagos de gran tamaño fagocitando células del sistema hematopoyético y presencia de parásitos compatibles con Leishmania, reforzándose el diagnóstico establecido y objetivándose el proceso etiológico asociado. Posteriormente, se recibieron resultados de complementarias enviadas a centros externos: PET-TAC con intensa captación metabólica a nivel hepatoesplénico y en médula ósea, y PCR de leishmania positiva.
RESULTADOS Y CONCLUSIONES:
La hemofagocitosis en médula ósea rara vez se presenta en los casos secundarios y puede no observarse hasta muy avanzada la enfermedad. Por lo tanto, en muchos casos, la repetición del aspirado puede ayudarnos a confirmar el diagnóstico de linfohistiocitosis hemofagocítica. Uno de los aspectos más importantes es su detección lo más precoz posible, por lo que hay que mantener un alto grado de sospecha.

COMUNICACIONES PÓSTERS: Morfología y Biología Celular 289
INTRODUCCIÓN/FUNDAMENTO:
La linfocitosis B monoclonal (LBM) es la detección en sangre periférica de linfocitos B clonales en número inferior a 5000/μL en ausencia de otros datos sugestivos de síndrome linfoproliferativo. Ésta presenta en la mayoría de los casos un inmunofenotipo típico de leucemia linfá-tica crónica (LLC), aunque también de LLC atípica o de no LLC (CD5-). Actualmente se subdividen en bajo y alto contaje en función de que la cifra de linfocitos B clonales sea inferior o igual/superior a 0.5 x 10e9/L respectivamente.
La LBM con fenotipo LLC presenta una incidencia de un 3.5% en sujetos mayor de 40 años. Se da con más frecuencia en hombre y su incidencia aumenta con la edad. Se considera una fase previa a LLC con un riesgo global de transformación anual del 1-2%/año, aunque en los de bajo contaje el riesgo es ínfimo por lo que no requiere seguimiento.
OBJETIVO:
Descripción de características epidemiológicas y clínicas de pacientes diagnosticados de LBM CD5+ en un hospital de tercer nivel.
PACIENTES, MATERIAL Y MÉTODO:
Estudio descriptivo y retrospectivo de una cohorte de pacientes diag-nosticados en los años 2015 y 2016 de LBM, mediante citometría de flujo (8 colores) en sangre periférica. Análisis mediante el paquete estadístico SPSS de variables cuantitativas y cualitativas.
RESULTADOS Y CONCLUSIONES:
En dicho período se han diagnosticado 34 pacientes de LBM , siendo mujeres el 53%(18/34). La mediana de edad al diagnóstico fue de 70 años (34-89). La mediana de leucocitos totales al diagnóstico fue de 11.38 x 109/L (4.3 – 23 x 109/L) y la de linfocitos B clonales de 3.8 x 109/L (0.09 - 4.92 x 109/L), de estos el 8,8% (3/34) son de bajo contaje (9/L ) y 91,2% (31/34) de alto contaje (≥ 0.5 x109/L). Se observó un predominio de restricción de cadenas Kappa débil (50%), frente a un 20.6% de lambda débil y un 29.4% ausencia de expresión de cadena en superficie. El 85.3% de los casos (29/34) presentó un fenotipo típico de LLC, mientras que el resto (5/34) lo presentó de LLC atípica por sobreexpresión de CD20.
Del total de casos, el 32.4% (11/34) progresaron a LLC-B, aunque ninguno ha requerido tratamiento por el momento(54,5% estadio A0, 45,5% estadío A1).De éstos, el CD38 fue negativo en todos los casos. La mediana de linfocitos B clonales al diagnóstico fue de 4.44 x 109/L (2.8 – 4.9 x 109/L), pertenecen todos al grupo de alto contaje y la mediana de tiempo hasta la progresión de 15 meses (2-18.5).
En nuestro estudio, en consonancia con lo descrito en la literatura, hemos observado que la gran mayoría de las LBM CD5+ presentan fenotipo típico LLC y se diagnostican en sujetos de edad avanzada. Ninguno de los pacientes con bajo contaje ha progresado mientras que aquellos con mayor número de linfocitos B clonales al diagnóstico parecen presentar mayor riesgo de progresar a LLC ya que en nuestro caso todos los pacientes que lo hicieron pertenecían al grupo de alto contaje.
Estudio descriptivo de la Linfocitosis B monoclonal CD 5 positivo en un hospital de tercer nivelP. Jiménez Guerrero (1), O. Pérez López (2), M. Ruíz Mercado (2), C. Prats Martín (2), E. Carrillo Cruz (2), T. Caballero Velázquez (2)
(1) Hospital Universitario Virgen del Rocío, (2) Hospital Universitario Virgen del Rocío

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA290
INTRODUCCIÓN/FUNDAMENTO:
La diabetes impide la correcta formación de nuevos vasos sanguíneos así como la cicatrización de las úlceras. Las células mensenquimales procedentes de médula ósea se están usando para favorecer la neo-vascularización, la curación de las úlceras y mejorar el flujo sanguíneo de las extremidades inferiores; aunque no se sabe con certeza si la diabetes menoscaba su capacidad funcional y terapeútica.
OBJETIVO:
El objetivo principal de nuestro estudio es demostrar si las MO-MSCs de pacientes diabéticos con isquemia arterial periférica y de individuos normales son diferentes en cuanto a morfología, diferenciación, fun-cionalidad proangiogénica in vitro y perfil de expresión génica
PACIENTES, MATERIAL Y MÉTODO:
1. ANÁLISIS MORFOLÓGICO realizamos cultivos de células mesenqui-males humanas (7 muestras de pacientes con diabetes tipo II y 4 de donantes sanos) procedentes de médula ósea de la fracción CMNS.Así mismo se reliazaron estudios con microscopía electrónica.
2. Se realizaron ESTUDIO DE DIFERENCIACIÓN hacia línea osteogénica y adipogénica en ambos grupos con MSCs procedentes de pase 2 sembradas en placa de diferenciación.
3. El ESTUDIO DE CAPACIDAD PROANGIOGÉNICA fue realizado en am-bos grupos utilizando medio EGM-2 durante 3 semanas. Al finalizar se realizó el recuento de vasos existentes en cada pocillo.
4. EL ESTUDIO DE PERFIL DE EXPRESIÓN GÉNICA fue realizado median-te microarrrays de expresión. Los resultados se validaron con PCR a tiempo real para 2 genes: MMP13 y SMOC 2 ambos involucrados en la formación de vasos sanguíneos; utilizando una serie de vali-dación de 26 pacientes.
5. AISLAMIENTO DE EXOSOMAS DE LAS CÉLULAS MSC Y ARRAY DE EX-PRESION DE MIRNA DEL CONTENIDO DE RNA DE DICHOS EXOSOMAS
RESULTADOS Y CONCLUSIONES:
1. IMPACTO DE LA DIABETES EN EL CULTIVO DE MSC: Los cultivos de cé-lulas presentaban una menor velocidad de crecimiento y apariencia envejecida. El 50% de las muestras no alcanzaban la confluencia.
2. La MISCROSCOPÍA ELECTRÓNICA mostraba diferencias entre los donante sanos y los pacientes diabéticos.
3. Por otra parte, LOS ESTUDIOS DE DIFERENCIACIÓN, mostraron dife-rencias entre los dos grupos: las MSCs de pacientes diabéticos en la mayor parte de las muestra no se diferenciaron a osteocitos; en el estudio de diferenciación a línea adipocitaria no encontramos diferencias.
Las MSCs de donantes sanos formaron más estructuras vasculares que las Paciente con DM. MSCs de pacientes diabéticos (153 es-tructuras vasculares vs 93 p=0.013).
4. EL perfil de expresión génica mostró un patrón de expresión glo-balmentedisminuido. Las MSCs de diabéticos se caracterizaron por una disminución en la expresión de MMP13 y SMOC2. Observamos un subgrupo de pacientes diabéticos con sobreexpresión de alguno de los dos genes estudiados, encontrando un aumento asímismo en la formación de estructuras vasculares en ese subgrupo.
5. Análisis del contenido de miRNA de los exosomas: Aparecieron un total de 7 genes (miRNA) significativos, 6 de ellos presentes en el control y ausentes en los pacientes, y uno de ellos presente en los pacientes y no en los controles.
Las MSC de pacientes diabéticos presentan diferencias morfológicas, funcionales y genéticas con respecto a MSC de donantes sanos.
Se ha demostrado un descenso en los niveles circulantes de miR 197 en pacientes con DM al igual que en los exosomas de MSC obtenidos de pacientes diabéticos en nuestro estudio.
El análisis de pérfil genético podria ayudarnos a discriminar pa-cientes que podría ser candidatos a terapias autólogas y cuales se beneficiarían de terapias alogenicas.
Análisis genómico y funcional de las MO-MSC y de los exosomas derivados de las mismas en pacientes diabéticos con isquemia arterial periférica. Implicaciones en terapia celularVanesa Martín, María Muñoz, Gustavo Díez, Rosario Jiménez, Sonia Nogueras, Inmaculada Herrera
HUR Sofía

COMUNICACIONES PÓSTERS
Serie Roja


COMUNICACIONES PÓSTERS: Serie Roja 293
INTRODUCCIÓN/FUNDAMENTO:
Entre las técnicas habituales que se realizan en el laboratorio de Eritro-patología se incluye la determinación de Haptoglobina, estudio fun-damental para el despistaje de Anemias hemolíticas intravasculares.
La Haptoglobina es una proteína sintetizada en el hígado. Las causas más frecuentes de niveles descendidos de esta proteína son: situacio-nes que impliquen afectación severa hepática, hemólisis intravascular, anemia megaloblástica, malnutrición, neonatos…
Nos proponemos evaluar los estudios realizados en nuestro Labora-torio.
OBJETIVO:
En el presente trabajo vamos a analizar las determinaciones de Hapto-globina realizadas en nuestro Laboratorio en el período de un año, eva-luando parámetros clínicos, analíticos y las conclusiones del estudio.
PACIENTES, MATERIAL Y MÉTODO:
En el último año hemos recibido 1279 muestras, realizando 261 estu-dios de Haptoglobina.
La determinación de Haptoglobina en nuestro Laboratorio puede ser solicitada directamente por el médico o formar parte del estudio de anemia, por sospecha de hemólisis.
Consideramos niveles bajos de Haptoglobina, los niveles menores de 0.5 mg/dl, habiendo encontrado 70 muestras con esta característica, que pasamos a analizar: motivo de estudio, edad y sexo de los pacien-tes, servicio peticionario, parámetros analíticos como LDH, Bilirrubina, reticulocitos, Coombs directo, presencia de esquistocitos y la conclusión del estudio.
RESULTADOS Y CONCLUSIONES:
Las muestras analizadas se corresponden con 58 pacientes (en 8 pa-cientes se han realizado varias determinaciones), 22 hombres y 36 mujeres.
Por servicios: 36 estudios proceden de Hematología; 9 de Digestivo; 7 de Medicina Interna: 6 de Urgencias; 3 de Neonatología; 2 de Cardio-logía y Dermatología; 1 de Anestesia, Nefrología y Medicina Intensiva.
Por edades: 24 pacientes tienen > 70 años, 19 con 50-70 años, 9 con 50-30 años y 5 niños (< 11 años).
El 61.5% (43) se corresponden con estudios de anemia. Los niveles de Hb son: entre 2,5 y 13 g/dl en 64 muestras, 43% (30) con Hb <9. y solo 6 casos con Hb > 13.
Del total de muestras, 46 presentan volumen corpuscular medio (VCM) normocitico y 22 con VCM > 100 fl.
Entre los índices sugestivos de hemólisis: Reticulocitos < 1% en 7 estudios, 1-3%: 13, > 3% en 45 casos; LDH >300 UI/l en 26 casos; niveles de Bilirrubina > 2 g/dl en 23 casos. En 10 estudios el frotis mostraba esquistocitos, en 5 esferocitos; Coombs directo positivo en 17 muestras.
En las conclusiones del estudio, 12 casos no fueron concluyentes y en los 46 restantes hemos encontrado: Hepatopatia (12 pacientes, 5 asociados a VHC), Anemia hemolítica autoinmune AHAI (12 casos, AHAI + Hepatopatía 3), hemólisis microangiopática en portadores pró-tesis valvulares metálicas (6), Esferocitosis (3), Neoplasia (3), Déficit de Vitamina B12 (2), 1 caso con Déficit Piruvato Kinasa, gestación, Hemofilia adquirida, Pénfigo, Crioaglutininas, infección viral, Sindrome hemolítico urémico y síndrome mielodisplásico.
Conclusiones
La determinación de Haptoglobina es un parámetro esencial en el Laboratorio de Eritropatología. En nuestra serie es un parámetro analizado en el 20% de las muestras.
Con los datos analizados las principales causas de descenso de la Haptoglobina son:– 12 pacientes (20.6%) no hemos obtenido datos concluyentes. – 40% (23) anemias hemolíticas (hemólisis autoinmune en 12;
microangiopática en 6, esferocitosis hereditaria en 3, Déficit de Piruvato Kinasa y Sindrome hemolítico urémico en 1)
– 20,6% (12) hepatopatías. – 19% (11) otras causas.
Haptoglobina, evaluación en el laboratorio de EritropatologíaR. López Rodríguez, M.P. Garrastazul Sánchez, M.J. Hernández Alfaro, C. De Cos Höhr
Hospital Universitario Puerta del Mar

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA294
GRÁFICOS ESTUDIOS DE HAPTOGLOBINA
Hombres
Mujeres
Estudios
Pacientes

COMUNICACIONES PÓSTERS: Serie Roja 295
Haptoglobina
0-0,1
0,1-0,3

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA296

COMUNICACIONES PÓSTERS: Serie Roja 297
INTRODUCCIÓN/FUNDAMENTO:
Existen mas de 900 variantes de hemoglobinas conocidas y dentro de estas aquellas variantes con afi nidad baja por el oxigeno con muy poca o nula sintomatología. La hemoglobina Yuda se describe por primera y única vez en 1992 en una mujer Japonesa con diabetes mellitus y hallazgo de pico anormal en HbA1c, que fi nalmente pone en eviden-cia estado mutacional en el gen HBA1: Ala sustituido por Asp en la posición alpha130 (H13).
Objetivo:
El objetivo del presente trabajo es describir un caso de una nueva mu-tación en estado heterocigoto del gen HBA2 responsable de la llamada Hemoglobina Yuda.
PACIENTES, MATERIAL Y MÉTODO:
Varon que a la edad de 13 años con anemia normocítica-normocrómica (Hb 10.9g/dL y VCM 83.60fl ) y solo astenia leve con el ejercicio (de-portista) como única sintomatología. No antecedentes familiares ni personales relevantes.
RESULTADOS Y CONCLUSIONES:
Se realizaron las siguientes pruebas complementarias: reticulocitos 3.3%, vitamina B12483pg/ml,acido fólico 9ng/ml, hierro 73ug/dL, Ferritina 47.9ng/ml, Transferrina 288mg/dL, LDH 334UI/L (normal), Test de Coombs negativo. Bilirrubinas total, directa e indirecta nor-males.Haptoglobina 80.1mg/dL. Eritropoyetina 2.42mU/ml. En la extensión de sangre periférica la morfologia eritrocitaria era normal. Test de HAM fue negativo, resistencia osmótica de los hematíes con: hemólisis inicial 6.5 por mil de ClNa (Test de Sacarosa (lisis eritrocitaria en presencia de sacarosa) 5% (sospechoso entre 5-10%). Electroforesis de Hemoglobinas (método sobre gel de
Agarosa) Hb A1 96.4%, Hb fetal 0.9%, HbS 0.%, Hb A2 2.7%. Es-tudio de enzima Glucosa 6 fosfato deshidrogenasa fue normal. En 2011 solicitan segunda opinión y es estudiado en Clínica Universitaria Navarra.En los estudios realizados se constató: Hemograma se objetivaba Hb 11.4g/dL, VCM 84fL, reticuloci-tos 3%, morfología de sangre periférica normal, Test de Ham negativo, resistencia globular osmótica normal, bilirrubinas total, directa, indirecta, metabolismo férrico, haptoglobina normal, hemosiderinuria negativo. Electroforesis capilar Hb A 97.6%, Hb A2 2.4%. (banda pequeña que emigra dentro de la hemoglobina A2). Estudio de cromatografía liquida de alta resolución (HPLC), no se eluyen cadenas de globinas anor-males ni hemoglobinas anormales. Hem-Piruvato quinasa 206mU/10e9eritrocitos. Se realiza estudio de médula ósea con hematopoyesis bien representada y sin alteraciones. Es-tudio molecular descarta la Delección de 3.7KB en el cluster de los genes Alfa. Se pierde en el seguimiento. Acude en 2016 a nuestras consultas por astenia acentuada con el ejercicio y control con hemograma. Se objetiva Hb 12.3g/dL, VCM 84f y restos de parámetros bioquímicos normales. Se solicita una nueva electroforesis capilar de hemoglobina, detectándose presencia de hemoglobina anómala de migración rápida no identifi cable (16.1%), HbA2 3%, HbA 79.7%, HbF <0.5%. (Imagen 1). Finalmente con estos hallazgos se solicita estu-dio molecular secuenciándose los genes de las globinas alfa y beta, detectándose la presencia en estado heterocigoto de la mutación HbA2:c.392C>A(p.Ala131Asp) que defi ne la hemog-lobina Yuda (Imagen 2)
Las nuevas técnicas de biología molecular permiten identifi car muta-ciones en las cadenas alfa o beta globina responsable de hemoglobinas variantes. Se describe un caso de una anemia sin causa no identifi cada, en la que el estudio de biología molecular muestra la existencia de una nueva mutación en el gen HBA2 que defi ne la hemoglobina Yuda, hasta ahora reportada en la bibliografía en un único caso.
Identifi cación de una mutación en el gen HBA2 como causa de hemoglobina variante (hemoglobina yuda). A propósito de un casoSilvia Del Rocio Verdesoto Cozzarelli (1), María Mar Herráez-Albendea (2), María Castillo Jarilla-Fernández (3), Manuel Martínez-Fernández (4),
Esther González-Real (5)
(1) Hospital Santa Bárbara-Gerencia Atención Integrada Puertollano, (2) Hospital Santa Bárbara-Gerencia Atención Integrada Puertollano
(3) Hospital Universitario General de Ciudad Real, (4) Laboratorios Reference. Barcelona, (5) Laboratorios Reference. Barcelona

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA298
IMAGEN 1

COMUNICACIONES PÓSTERS: Serie Roja 299
IMAGEN 2

XXXVII REUNIÓN ANUAL DE LA ASOCIACIÓN ANDALUZA DE HEMATOLOGÍA Y HEMOTERAPIA300
INTRODUCCIÓN/FUNDAMENTO:
La Anemia Hemolítica Autoinmune (AHAI) constituye todo un reto para el hematólogo cuando la inestabilidad hemodinámica provocada por la anemia hace necesaria la transfusión de hematíes y todas las uni-dades al realizar la prueba cruzada, resultan incompatibles.
Es importante tratar de identificar el Autoanticuerpo (Auto-Ac.) res-ponsable, tarea muchas veces imposible, pero más aún descartar la existencia de Alo-anticuerpos (Alo-Ac.) subyacentes que pudieran agravar más la hemólisis.
En los casos secundarios se puede asociar a procesos linfoproliferativos. Son más frecuentes por Ac. Calientes de tipo IgG y en raras ocasiones (1%) por IgA o IgM.
OBJETIVO:
Exponer la metodología de estudio de la AHAI a partir de un caso clínico real.
PACIENTES, MATERIAL Y MÉTODO:
Mujer de 65 años, con antecedentes personales de Hepatitis C erra-dicada, HTA no tratada, tiroidectomía hace 30 años en tratamiento sustitutivo y Herniorrafia umbilical. Trasladada desde otro hospital por diagnóstico de LNH, había sido transfundida con 8 concentrados de hematíes a raíz de hemorragia digestiva alta e iniciado tratamiento con corticoides por sospecha de AHAI.
La analítica inicial: Hb 7.8 gr/dl, VCM 131 fl. Reticulocitos 11.29%. LDH 1449 U/L. Bilirrubina indirecta 2.30 mg/dl.
Los estudios inmunohematológicos realizados fueron los siguientes:
GrupoABO/Rh(D): O positivo (control negativo). Fenotipo eritrocitario: C+, c+, E-, e+, K+. Control negativo (No se pudo ampliar más por los antecedentes transfusionales y el Test de Coombs Directo (TCD) positivo).
TCD con Antiglobulina Humana (AGH) poliespecífica: positivo +++, negativo con IgG e IgA, positivo con anti IgM ++ y con anti-C3c y anti C3d +++
El EAI fue positivo con los hematíes I, II y III (Autocontrol positivo).
Se procedió a realizar un panel de identificación con el suero proble-ma tanto en Liss-Coombs (L/C) como en enzimas con el resultado de panaglutinación.
Realizamos una elución ácida (Elukit) y posterior panel en L/C del eluido persistiendo la panaglutinación (panel sobrenadante negativo).
Titulación del eluido con hematíes diferenciales: R1R1: 2, R2R2: 1, rrJka+: 2 rrJkb+: 2
El panel del eluido a ½ también presentaba panaglutinación.
Titulación del suero con hematíes: R1R1 (16), R2R2 (16), rrJka+ (16), rrJkb+ (16) siendo negativo el panel del suero diluido a 1/16.
Para descartar la existencia de Alo-Ac. realizamos aloadsorciones di-ferenciales con hematíes R1R1 y rr con diferencias antigénicas para K, Fya, Fyb, Jka, Jkb, S, s.
El panel del suero aloadsorbido resultó negativo, lo que descartó la presencia de Alo-acs subyacentes.
La titulación del suero a 4ºC con hematíes O adulto, O de cordón y autocontrol resultó inferior a 1. El rango térmico del suero a menos de 37ºC. era negativo. Estos hallazgos descartan que el ac. de clase Ig M se trate de un ac. frío.
RESULTADOS Y CONCLUSIONES:
COMENTARIOS
En toda AHAI es fundamental descartar la presencia de Alo-acs. subyacentes y transfundir hematíes cuando sea necesario, con el fenotipo más parecido posible al del paciente.
Las AHAI por Acs. calientes de tipo IgM son poco frecuentes y se asocian a cuadros hemolíticos graves y de mal pronóstico.
En estos casos es obligado descartar la presencia de acs fríos.
La especificidad del Auto-ac a veces es imposible de determinar.
Anemia hemolítica autoinmune por anticuerpos calientes de tipo IGM y complemento: a propósito de un casoJ.A. Raposo Puglia (1), A. Atienza Saborido (2), A. Salamanca Cuenca (1), M.A. Correa Alonso (1), S. Ordóñez Vahí (1), M.D. Madrigal Toscano (1)
(1) Hospital Universitario Jerez de la Frontera, (2) Técnico Especialista de Laboratorio. Hospital Universitario Jerez de la Frontera

Secretaría Técnica:
Pagés del Corro, 80 - 1ª planta - 41010 SevillaT- 954 574240 F- 955 067479
[email protected] www.trianacongresos.com

COLABORADORES:





























