
Apoptosis (2006) 11:1945–1957DOI 10.1007/s10495-006-0090-0
N-acetyl-cysteine protects liver from apoptotic death in an animalmodel of fulminant hepatic failureB. San-Miguel · M. Alvarez · J. M. Culebras ·J. Gonzalez-Gallego · M. J. Tunon
Published online: 4 October 2006C© Springer Science + Business Media, LLC 2006
Abstract Background: This work was undertaken to in-vestigate whether treatment with N-acetyl-cysteine (NAC)prevents oxidative stress and inhibits the apoptotic pathwaysin an animal model of fulminant hepatic failure. Methods:Rabbits were experimentally infected with 2 × 104 hemag-glutination units of a rabbit hemorrhagic disease virus isolate.Results: The spontaneous mortality rate of infected animalswas 67% at 36 h post infection (pi) and 90% at 48 h pi.This percentage decreased significantly in animals receivingan i.p. injection of NAC (150 mg/kg body way/daily), for7 days prior to infection. From 36 h pi marked increaseswere detected in blood levels of transaminases, lactate de-hydrogenase, bilirubin and the oxidised/reduced glutathioneratio. All these effects were significantly prevented by NACtreatment. The Bax to Bcl-2 relative expression, the expres-sion of FasL, cytochrome c and PARP-1, and the activityof caspase 3 were significantly increased at 36 and 48 hpi in infected animals. These changes were markedly re-duced in animals treated with NAC, with the exception ofFasL. Conclusion: Our results suggest a potential hepatopro-tective role of NAC in fulminant hepatic failure, mediatedpartially through the modulation of the intrinsic pathway ofapoptosis.
Keywords Fulminant hepatic failure . N-acetyl-cysteine .
Oxidative stress
B. San-Miguel · J. Gonzalez-Gallego · M. J. Tunon (�)Department of Physiology, University of Leon, 24071 Leon, Spaine-mail: [email protected]
M. AlvarezDepartment of Animal Health, University of Leon, Leon, Spain
J. M. CulebrasHospital of Leon, Leon, Spain
Introduction
Apoptotic cell death of hepatocytes emerges as a funda-mental component of virtually all acute and chronic liverdiseases [1]. Fulminant hepatic failure (FHF) is character-ized by a severe hepatocellular dysfunction with a massivedeath of hepatocytes, in which apoptosis may play a rolein addition to necrosis [2]. Recent data indicate that serumcytochrome c is a possible new marker for acute liver fail-ure in humans and correlates to serum levels of AST andALT [3], and novel therapeutic approaches try to target theapoptotic cascade [4]. There are two overlapping signallingpathways leading to apoptosis, termed the intrinsic and ex-trinsic pathway. The extrinsic or death receptor-dependentpathway is initiated by death ligands like TNF or Fas-ligand,leading to cleavage of procaspase 8 to its active form, whichsubsequently activates downstream effectors such as cas-pase 3. The intrinsic pathway is characterized by mitochon-dria dysfunction, and various stimuli, particularly oxidativestress, can lead to damage of the mitochondrial inner mem-brane, resulting in mitochondrial permeability transition, cy-tochrome c release and activation of downstream effectorcaspases [5]. The Bcl-2 protein family plays a role in theregulation of the intrinsic pathway. Two key representativemembers of this family are the antiapoptotic Bcl-2 and thepro-apoptotic Bax [6]. The two apoptotic pathways are notmutually exclusive in hepatocytes, but are closely interre-lated, as the mitochondrial pathway is often required to am-plify the relatively weak death receptor-induced apoptoticsignal [1].
The involvement in, and activation of, the intrinsic path-way by reactive oxygen species leads to the assumption thatantioxidants might prevent fulminant hepatic failure by in-terfering with the deadly cascade. Different recent studieshave demonstrated that treatment with the thiol antioxidant
Springer

1946 Apoptosis (2006) 11:1945–1957
N-acetyl-cysteine (NAC) consistently inhibits the pro-grammed cell death induced by a variety of stimuli [7],blocking apoptosis induced by cytotoxic agents [8–10], andprotecting from apoptotic death in chronic renal failure [11],chronic obstructive pulmonary disease [12] or exercise inadrenalectomized animals [13].
The rabbit hemorrhagic disease virus (RHDV) is a mem-ber of the Caliciviridae family that causes in wild and domes-tic rabbits an acute highly fatal disease [14]. Hepatic damageplays a central pathogenic role, and the viral antigens aremainly detected in the liver [15–17]. The most consistentpathological findings are fulminate acute hepatitis and dis-seminated intravascular coagulation; neurological symptomsare present and the disease evolves rapidly to death within48 to 72 h after infection in about 90% of the cases [15, 16].We have recently shown by data on animal survival, clin-ical feature, histological data, changes in blood chemistryand intracranial pressure monitoring that the rabbit hemor-rhagic disease fulfils many of the requirements of an animalmodel of FHF [18], being useful to improve our insight intothe metabolic and physiological derangements of FHF andto facilitate the development of new therapeutic modalities[19]. We have also reported that in this model there is aloss in the oxidant/antioxidant balance, with a marked in-crease in the oxidised/reduced glutathione ratio and dimin-ished superoxide dismutase activity [20]. The present workwas undertaken to investigate whether the RHDV causes liverapoptosis and whether treatment with NAC prevents oxida-tive stress and inhibits the apoptotic pathways in this animalmodel of FHF.
Materials and methods
Virus and experimental model
Study protocols were reviewed and approved by the Univer-sity of Leon Animal Care Committee. Nine-week-old NewZealand White rabbits were kept in a climatized room at21◦C, with a 12 h light cycle. They were given a standarddry rabbit food and water ad libitum. For survival and DNAfragmentation studies, rabbits received an i.p. injection ofNAC (150 mg/kg body way/daily; Sigma, St Louis, MO,USA) or the same volume of vehicle (saline) for seven days(n = 12 each). The last day all the animals were injected in-tramuscularly with 2 × 104 hemagglutination units of RHDVisolate Ast/89 [16]. Animals in those groups were left to dyespontaneously. Blood and liver studies were carried out inbatches of control rabbits and of infected animals, treatedwith NAC or vehicle, that were killed at 12 h, 24 h, 36 h and48 h post infection (pi) (n = 5 each). Animals were killedwith sodium pentobarbital.
Haemagglutination test
Titration of the virus was made by a haemagglutination testaccording to the protocol for RHDV of the O.I.E [21]. Briefly,infected rabbit livers were homogenised in 10% (w/v) PBSand clarified by to consecutive low speed centrifugations(500 × g for 20 min and 6000 × g for 30 min). Serial twofolddilutions of clarified supernatant liver tissue homogenateswere incubated with equal volumes of 0.75% human group0 red blood cells suspension in PBS in sealed round bottommicrotitre plates at 4◦C for 2 h. Virus titre (haemagglutinating(HA) units) was expressed as the reciprocal of the highestdilution causing complete human group 0 red blood cellsagglutination.
Histology and immunohistochemistry
Tissue samples were recovered, fixed in 10% buffered forma-lin, and embedded in paraffin. Sections (4 µm) were stainedwith hematoxylin-eosin (HE) for morphologic examinationunder microscopy. Formalin fixed tissues were also used todetect the expression of activated caspase-3. Briefly, sam-ples were dewaxed and hydrated through graded ethanols,cooked in 25 mM citrate buffer, pH 6.0, in a pressure cookerfor 10 min, transferred into boiling deionized water and letto cool for 20 min. Tissue sections were then treated with3% hydrogen peroxide to inactivate endogenous peroxidaseactivity. The slides were incubated with rabbit polyclonal ac-tivated caspase-3 (Cell Signaling Technology, Danvers, MA)overnight at 4◦C, followed by incubation with biotinylatedsecond antibody (Biotinylated Anti-Rabbit IgG; Vector Lab-oratories, Burlingame, CA) for 1 h at room temperature.After 45 min of avidin-biotin amplification (ABC Standard;Vector Laboratories), samples were incubated with the sub-strate 0.1% 3′,3′-diaminobenzidine (DAB/Ni Substrate; Vec-tor Laboratories) at room temperature for 10 min. Patholog-ical findings were assessed by one of the authors blinded tothe group allocations. For transmission electron microscopyanalysis, livers were fixed with a modified Karnovsky fixa-tive [2% glutaraldehyde + 4% buffered formalin (0.1 mol/Lphosphate buffer)] for 2 h, followed by osmication (2% OsO4
for 2 h). Samples were then dehydrated and embedded inEpon resin. Ultrathin sections were stained with uranyl ac-etate and lead nitrate and observed under a JEM-1010 trans-mission electron microscope (JEOL, Tokyo, Japan).
Blood chemistry
Laboratory determinations included alanine transaminase(ALT), aspartate transaminase, (AST), and bilirubin. Anal-yses were done in the Hospital of Leon Clinical ChemistryLaboratory using standard techniques.
Springer

Apoptosis (2006) 11:1945–1957 1947
DNA fragmentation
DNA was extracted from liver tissue by the QIAquick GelExtraction kit (QIAGEN GmbH, Hilden, Germany). DNApellets were dissolved in 100 µl of TE buffer (10 mM Tris-HCl pH 7.4, 1 mM EDTA) and incubated with 40 µg/ml ofRNase at 37◦C for 1 h. 1 µg DNA samples were subjectedto electrophoresis on a 2% agarose gel containing ethidiumbromide and visualized under UV illumination.
Caspase-3 activity
Lysates were prepared by homogenizing liver tissue in0.25 mM sucrose, 1 mM EDTA, 10 mM Tris and a proteaseinhibitor cocktail (Roche Diagnostics GmbH, Mannheim,Germany). The lysates were then centrifuged at 14,000 × gfor 10 min at 4◦C, and supernatants (50 µg protein) wereincubated for 1 h at 37◦C in HEPES buffer containing100 µM concentrations of the specific fluorogenic substrateAc-DEVD-AMC. Cleavage of the caspase-3 substrate wasmonitored at excitation wavelength of 360 and emissionwavelength of 460 nm, respectively using a spectrofluorime-ter (Hitachi F-2000 fluorimeter, Hitachi LTD., Tokyo, Japan).Protein content of samples was determined [22] using bovineserum albumin as standard.
Oxidised and reduced glutathione
Oxidised (GSSG) and reduced (GSH) glutathione analysiswas performed fluorimetrically by the method of Hissin andHilf [23]. Briefly, 250 mg of liver tissue was homogenisedin 0.1 M sodium phosphate 5 mM EDTA buffer (pH 8.0)with 25% phosphoric acid at a proportion of 1:20. The mix-ture was centrifuged at 100,000 × g for 30 min at 4◦C, thesupernatant was collected and 500 µL were diluted with4.5 mL of buffer. Two spectrophotometry cuvettes per sam-ple were prepared with 1.8 mL phosphate-EDTA buffer,100 µL supernatant and 100 µL O-phthalaldehyde. Afterincubating for 15 min at 4◦C, a spectrofluorometric readingwas obtained at an excitation wavelenght of 350 nm and anemission wavelenght of 420 nm. To find the percentage ofglutathione corresponding to oxidised and reduced forms,500 µL of the sample supernatant was incubated with 20 µL4-vinylpyridine for 30 min; to this mixture 4.5 mL of 0.1N NaOH was added. A 100 µL portion of this mixture wasthen processed as described above to determine GSSG. GSHwas obtained by subtracting GSSG from total glutathione.
Western blot analysis
For Western blot analysis of Bcl-2, Bax, cytochrome c andFasL, liver tissue was homogenised in 10 mM Tris buffer (pH7.4) containing 100 mM NaCl and a protease inhibitor cock-
tail (Roche Diagnostics GmbH, Mannheim, Germany) andcentrifuged at 8000 × g for 15 min. For detection of cleav-age products of poly(ADP) ribose polymerase 1 (PARP-1),nuclear extracts were prepared from liver homogenates asdescribed previously [24]. Protein concentration of the cy-tosolic and nuclear liver fractions were determined [22] us-ing bovine serum albumin as standard. Protein extracts (50–100 µg) were separated by 10–15% sodium dodecyl sulphate(SDS)-polyacrylamide gel electrophoresis for 1.5 h at 100 Vand then blotted on polyvinyllidene difluoride (PVDF) mem-branes (Amersham Pharmacia, Buckinghamshire, UK). Themembranes were then blocked with 5% non-fat dry milkin Tris-buffered saline containing 0.05% Tween 20 (TBST)for 30 min at 37◦C and probed overnight at 4◦C with poly-clonal anti-Bcl-2, Bax, cytochrome-c, FasL and PARP (SantaCruz Biotechnology, Santa Cruz, CA, USA; 1:200–1:400 di-lution with PBST containing 3% non-fat dry milk). Equalloading of protein was demonstrated by probing the mem-branes with a rabbit anti-β-actin polyclonal antibody (Sigma,St Louis, MO, USA; 1:1000 dilution). After washing withTBST, the membranes were incubated for 1 h at room tem-perature in TBST containing 4000-fold diluted anti-swineIgG rabbit antibody conjugated with horseradish peroxidase(Dako, Glostrup, Denmark; 1:4000). Finally, the membraneswere developed with an enhanced chemiluminescence west-ern blotting determination kit (ECL-Plus, Amersham Phar-macia), and Hyperfilm ECL (Amersham Life Science,Buckinghamshire, UK) was exposed to them. Relative pro-tein levels were determined using a Fluor-S Multimager in-strument (BioRad). The optical density of tissue sampleswere normalized to a control sample in an arbitrary densito-metry unit.
Statistical analysis
Means and SEMs were calculated. Significant differencesbetween means were evaluated by analyses of variance andin the case of significance, a Newman-Keul’s test was alsoapplied. A difference was considered significant when p wasless than 0.05. Survival data were evaluated with Fisher’sexact test. Calculations were performed with SPSS + 13.0statistical software (Chicago, IL).
Results
Survival rate findings
For survival studies, rabbits received an i.p. injection of NACor the same volume of vehicle for seven days. The last dayall the animals were injected intramuscularly with 2 × 104
hemagglutination units of RHDV isolate and left to dye spon-taneously. The mortality rate of infected animals was 67%
Springer
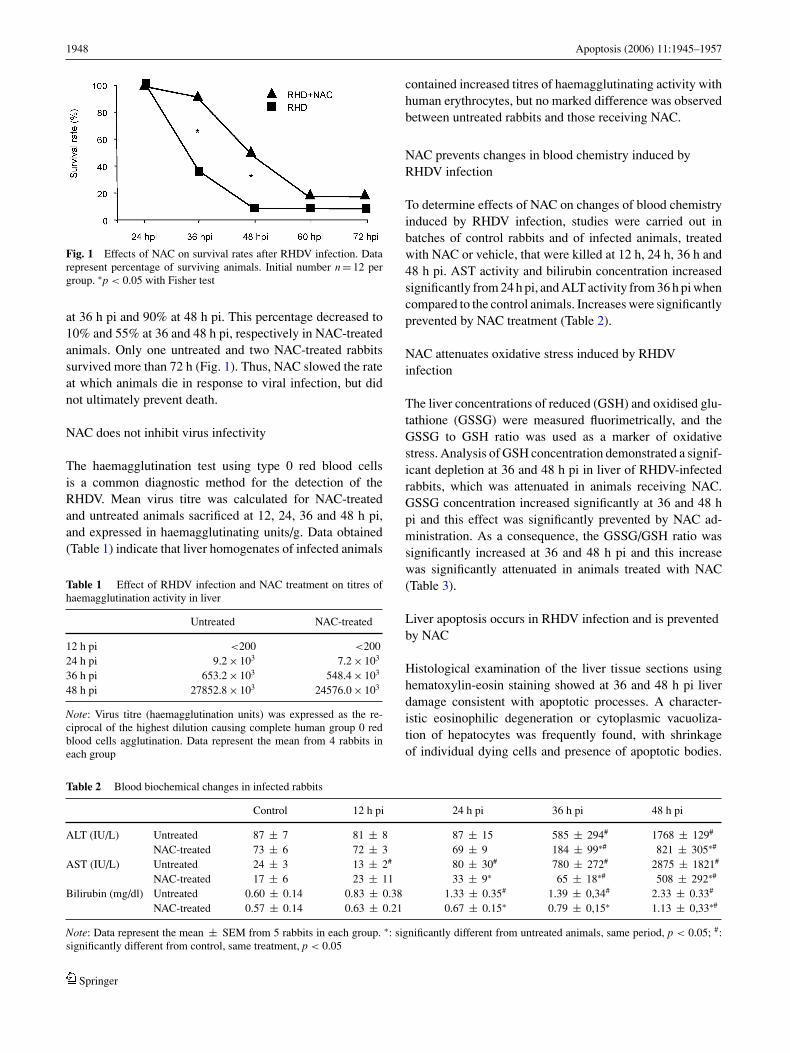
1948 Apoptosis (2006) 11:1945–1957
Fig. 1 Effects of NAC on survival rates after RHDV infection. Datarepresent percentage of surviving animals. Initial number n = 12 pergroup. ∗p < 0.05 with Fisher test
at 36 h pi and 90% at 48 h pi. This percentage decreased to10% and 55% at 36 and 48 h pi, respectively in NAC-treatedanimals. Only one untreated and two NAC-treated rabbitssurvived more than 72 h (Fig. 1). Thus, NAC slowed the rateat which animals die in response to viral infection, but didnot ultimately prevent death.
NAC does not inhibit virus infectivity
The haemagglutination test using type 0 red blood cellsis a common diagnostic method for the detection of theRHDV. Mean virus titre was calculated for NAC-treatedand untreated animals sacrificed at 12, 24, 36 and 48 h pi,and expressed in haemagglutinating units/g. Data obtained(Table 1) indicate that liver homogenates of infected animals
Table 1 Effect of RHDV infection and NAC treatment on titres ofhaemagglutination activity in liver
Untreated NAC-treated
12 h pi <200 <20024 h pi 9.2 × 103 7.2 × 103
36 h pi 653.2 × 103 548.4 × 103
48 h pi 27852.8 × 103 24576.0 × 103
Note: Virus titre (haemagglutination units) was expressed as the re-ciprocal of the highest dilution causing complete human group 0 redblood cells agglutination. Data represent the mean from 4 rabbits ineach group
contained increased titres of haemagglutinating activity withhuman erythrocytes, but no marked difference was observedbetween untreated rabbits and those receiving NAC.
NAC prevents changes in blood chemistry induced byRHDV infection
To determine effects of NAC on changes of blood chemistryinduced by RHDV infection, studies were carried out inbatches of control rabbits and of infected animals, treatedwith NAC or vehicle, that were killed at 12 h, 24 h, 36 h and48 h pi. AST activity and bilirubin concentration increasedsignificantly from 24 h pi, and ALT activity from 36 h pi whencompared to the control animals. Increases were significantlyprevented by NAC treatment (Table 2).
NAC attenuates oxidative stress induced by RHDVinfection
The liver concentrations of reduced (GSH) and oxidised glu-tathione (GSSG) were measured fluorimetrically, and theGSSG to GSH ratio was used as a marker of oxidativestress. Analysis of GSH concentration demonstrated a signif-icant depletion at 36 and 48 h pi in liver of RHDV-infectedrabbits, which was attenuated in animals receiving NAC.GSSG concentration increased significantly at 36 and 48 hpi and this effect was significantly prevented by NAC ad-ministration. As a consequence, the GSSG/GSH ratio wassignificantly increased at 36 and 48 h pi and this increasewas significantly attenuated in animals treated with NAC(Table 3).
Liver apoptosis occurs in RHDV infection and is preventedby NAC
Histological examination of the liver tissue sections usinghematoxylin-eosin staining showed at 36 and 48 h pi liverdamage consistent with apoptotic processes. A character-istic eosinophilic degeneration or cytoplasmic vacuoliza-tion of hepatocytes was frequently found, with shrinkageof individual dying cells and presence of apoptotic bodies.
Table 2 Blood biochemical changes in infected rabbits
Control 12 h pi 24 h pi 36 h pi 48 h pi
ALT (IU/L) Untreated 87 ± 7 81 ± 8 87 ± 15 585 ± 294# 1768 ± 129#
NAC-treated 73 ± 6 72 ± 3 69 ± 9 184 ± 99∗# 821 ± 305∗#
AST (IU/L) Untreated 24 ± 3 13 ± 2# 80 ± 30# 780 ± 272# 2875 ± 1821#
NAC-treated 17 ± 6 23 ± 11 33 ± 9∗ 65 ± 18∗# 508 ± 292∗#
Bilirubin (mg/dl) Untreated 0.60 ± 0.14 0.83 ± 0.38 1.33 ± 0.35# 1.39 ± 0,34# 2.33 ± 0.33#
NAC-treated 0.57 ± 0.14 0.63 ± 0.21 0.67 ± 0.15∗ 0.79 ± 0,15∗ 1.13 ± 0,33∗#
Note: Data represent the mean ± SEM from 5 rabbits in each group. ∗: significantly different from untreated animals, same period, p < 0.05; #:significantly different from control, same treatment, p < 0.05
Springer

Apoptosis (2006) 11:1945–1957 1949
Table 3 Effect of RHDV infection and NAC treatment on reduced (GSH) and oxidised (GSSG) glutathione concentrations in liver
Control 12 h pi 24 h pi 36 h pi 48 h pi
GSH (nmol/mg protein)Untreated 5.60 ± 0.88 4.71 ± 0.36 5.41 ± 1.01 4.08 ± 0.54# 3.09 ± 0.24#
NAC-treated 6.29 ± 0.92 5.19 ± 1.52 5.16 ± 0.47 4.45 ± 0.64# 3.66 ± 0.69#
GSSG (nmol/mg protein)Untreated 0.12 ± 0.01 0.13 ± 0.01 0.14 ± 0.03 0.25 ± 0.01# 0.26 ± 0.01#
NAC-treated 0.12 ± 0.01 0.13 ± 0.02 0.13 ± 0.01 0.17 ± 0.03∗# 0.18 ± 0.04∗#
GSSG/GSH × 100Untreated 2.19 ± 0.22 2.78 ± 0.18# 2.65 ± 0.23 6.18 ± 0.28# 8.36 ± 0.32#
NAC-treated 1.91 ± 0.11 2.48 ± 0.21# 2.48 ± 0.09∗# 3.79 ± 0.35∗# 4.92 ± 0.39∗#
Note: Data represent the mean ± SEM from 5 rabbits in each group. ∗: significantly different from untreated animals, same period, p < 0.05; #:significantly different from control, same treatment, p < 0.05
Fig. 2 Photograph of agarose gel containing electrophoretically sep-arated low-molecular-weight DNA fragments from controls (C andC-NAC groups), infected untreated (RHD) and infected NAC-treated(RHD-NAC) rabbits at 36 h p.i.
These elements were found dispersed throughout the hepaticlobule, or in confluent in foci of dead cells that distorted thenormal acinar arrangement in homogeneous hepatic lami-nae. In rabbits pretreated with NAC, the liver structure wasbetter preserved.
The presence of DNA ladders, characteristic of the apop-totic cleavage of nuclear DNA into small fragments betweennucleosomes, was studied in DNA samples extracted fromliver tissue, incubated with RNase and electrophoresed onagarose gel containing ethidium bromide. Laddering wasdemonstrated from 36 h pi in liver of infected rabbits. DNAfragmentation was not observed in animals that had beentreated with NAC (Fig. 2).
To further confirm that hepatocytes committed apoptosis,we performed transmission electron microscopy analysis.
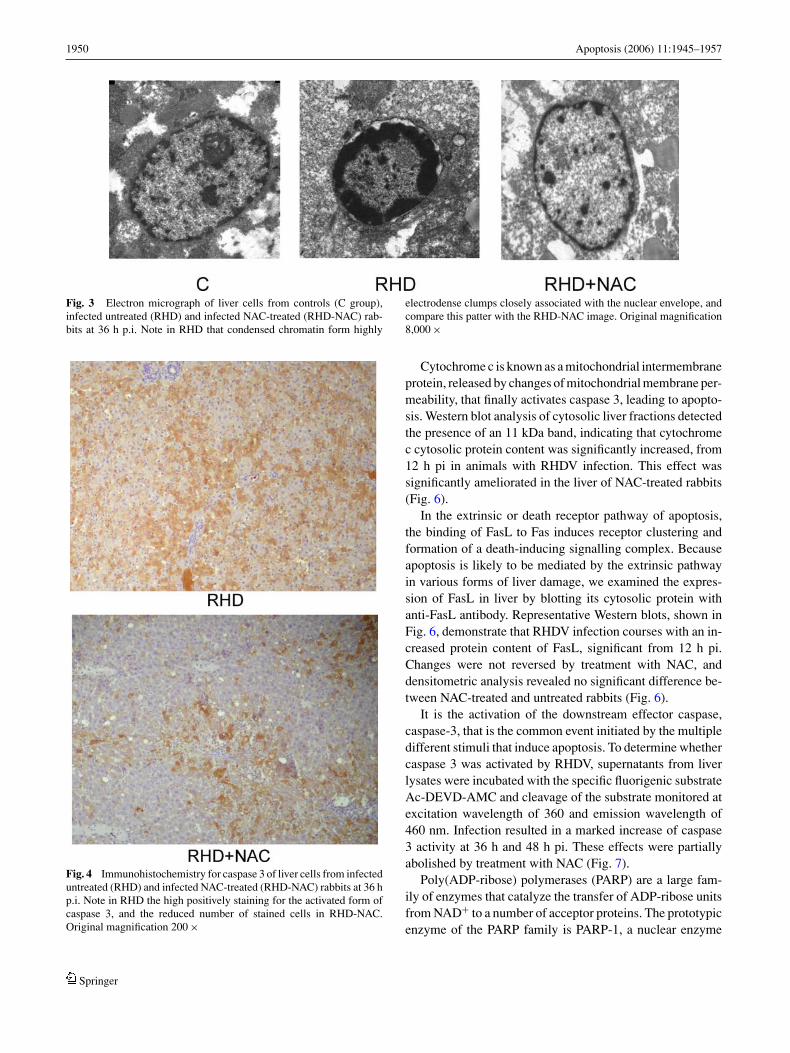
As shown in Fig. 3, nuclei of liver cells from RHDV-infectedanimals presented condensation of the chromatin forminghighly electrodense clumps closely associated with thenuclear envelope. The ultrastructure of the liver of NAC-treated animals was practically unaltered, and similar tocontrol animals.
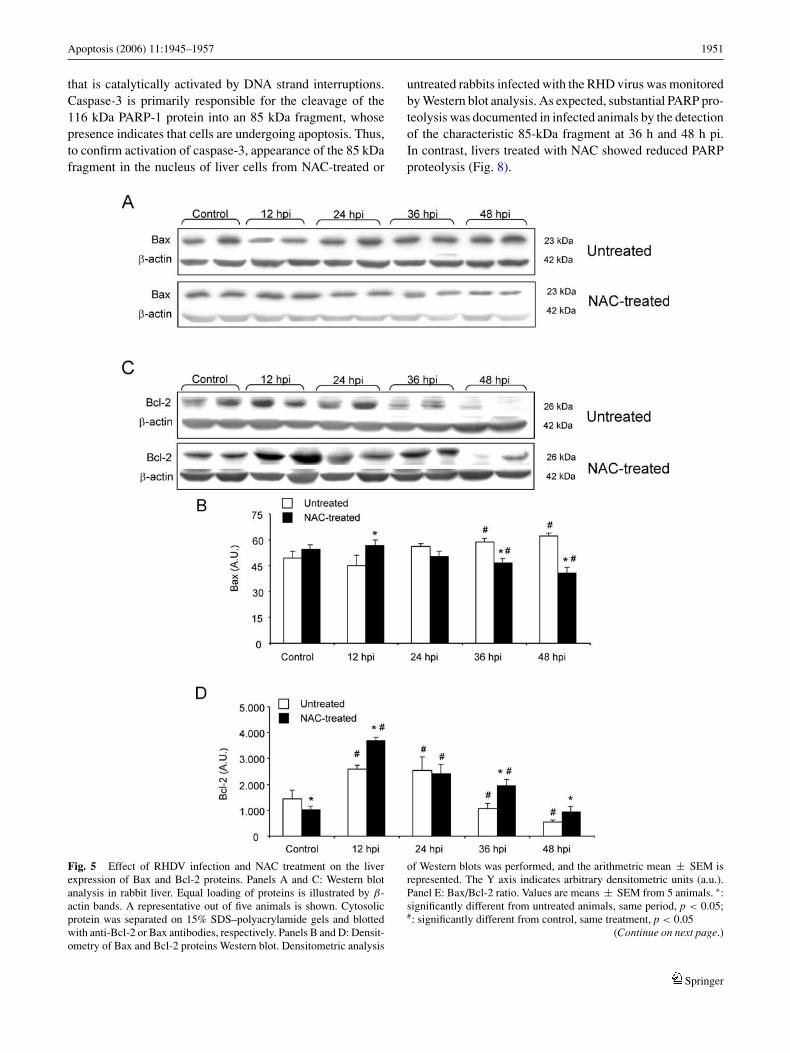
Immunochemistry for caspase-3 was performed to de-termine the number of cells undergoing apoptosis. In liversections from RHDV-infected rabbits numerous positively-stained hepatocytes were seen scattered throughout theparenchyma. However, in section from NAC-treated animals,staining was mainly observed in periportal hepatocytes, al-though a few individual cells in other areas of the hepatic lob-ule were also stained (Fig. 4). At 36 h pi a mean of 34.7 ± 6.6positively stained cells were detected in the RHDV-infectedgroup. In livers treated with NAC, a mean of 12.1 ± 3.2 pos-itively stained cells were detected (p < 0.01). There were nopositively stained cells in control animals.
NAC inhibits expression and activity of apoptotic markers
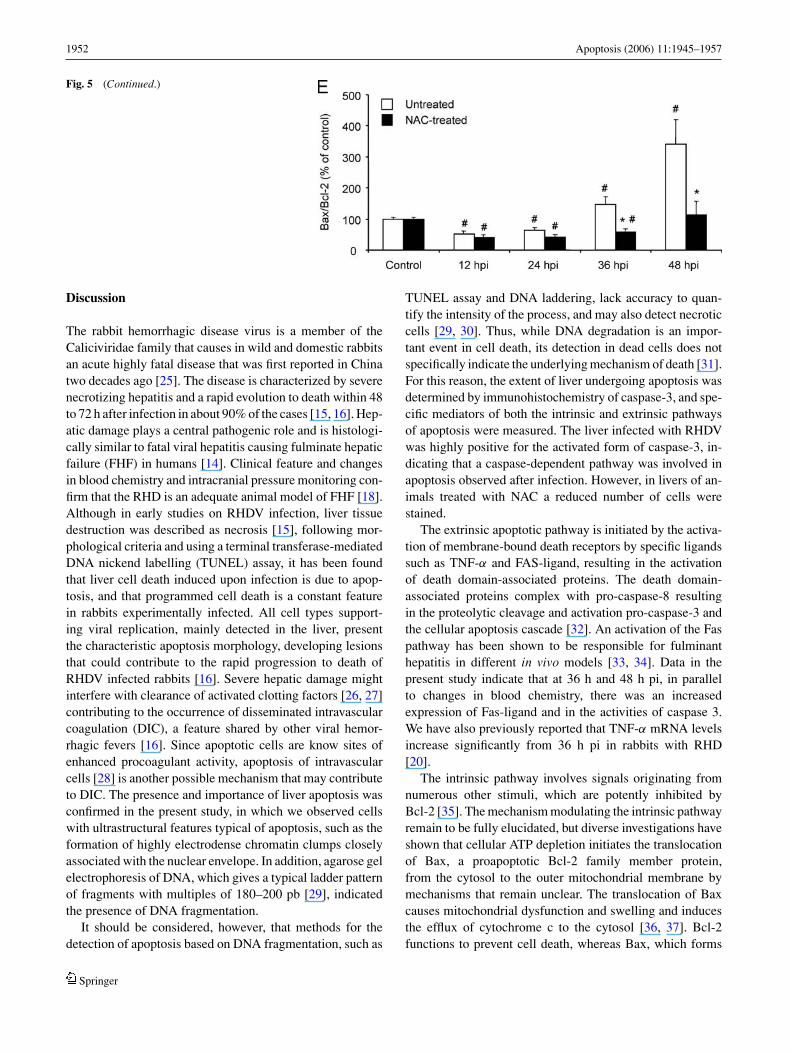
The Bcl-2 family proteins are important apoptotic regula-tory molecules. Members of this family can be divided intoan antiapoptotic family, which includes Bcl-2, and an apop-totic family, which includes Bax. The ratio of antiapoptoticBcl-2 to pro-apoptotic Bax contributes to the susceptibilityof a given cell to apoptosis. Thus, cytosolic protein contentfor both molecules was studied by Western blot. Cytoso-lic protein was separated on 15% SDS–polyacrylamide gelsand blotted with anti-Bcl-2 or Bax antibodies, respectively.Figure 5 shows representative Western blots of Bcl-2 andBax expressions in animals without and with NAC treatment.Densitometric analysis revealed that Bax expression was sig-nificantly increased and Bcl-2 expression significantly re-duced at 36 and 48 h pi in untreated rabbits. Treatment withNAC significantly prevented those changes, resulting in asignificantly lower Bax/Bcl-2 ratio when compared to un-treated rabbits (Fig. 5).
Springer
1950 Apoptosis (2006) 11:1945–1957
Fig. 3 Electron micrograph of liver cells from controls (C group),infected untreated (RHD) and infected NAC-treated (RHD-NAC) rab-bits at 36 h p.i. Note in RHD that condensed chromatin form highly
electrodense clumps closely associated with the nuclear envelope, andcompare this patter with the RHD-NAC image. Original magnification8,000 ×
Fig. 4 Immunohistochemistry for caspase 3 of liver cells from infecteduntreated (RHD) and infected NAC-treated (RHD-NAC) rabbits at 36 hp.i. Note in RHD the high positively staining for the activated form ofcaspase 3, and the reduced number of stained cells in RHD-NAC.Original magnification 200 ×
Cytochrome c is known as a mitochondrial intermembraneprotein, released by changes of mitochondrial membrane per-meability, that finally activates caspase 3, leading to apopto-sis. Western blot analysis of cytosolic liver fractions detectedthe presence of an 11 kDa band, indicating that cytochromec cytosolic protein content was significantly increased, from12 h pi in animals with RHDV infection. This effect wassignificantly ameliorated in the liver of NAC-treated rabbits(Fig. 6).
In the extrinsic or death receptor pathway of apoptosis,the binding of FasL to Fas induces receptor clustering andformation of a death-inducing signalling complex. Becauseapoptosis is likely to be mediated by the extrinsic pathwayin various forms of liver damage, we examined the expres-sion of FasL in liver by blotting its cytosolic protein withanti-FasL antibody. Representative Western blots, shown inFig. 6, demonstrate that RHDV infection courses with an in-creased protein content of FasL, significant from 12 h pi.Changes were not reversed by treatment with NAC, anddensitometric analysis revealed no significant difference be-tween NAC-treated and untreated rabbits (Fig. 6).
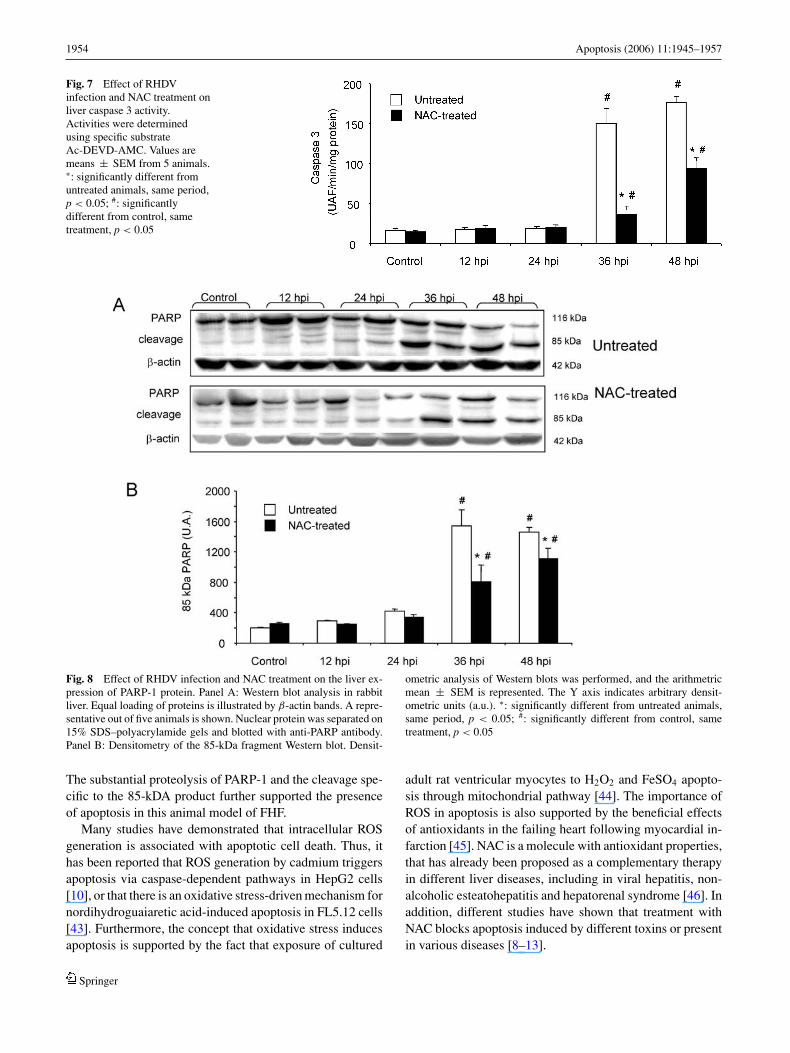
It is the activation of the downstream effector caspase,caspase-3, that is the common event initiated by the multipledifferent stimuli that induce apoptosis. To determine whethercaspase 3 was activated by RHDV, supernatants from liverlysates were incubated with the specific fluorigenic substrateAc-DEVD-AMC and cleavage of the substrate monitored atexcitation wavelength of 360 and emission wavelength of460 nm. Infection resulted in a marked increase of caspase3 activity at 36 h and 48 h pi. These effects were partiallyabolished by treatment with NAC (Fig. 7).
Poly(ADP-ribose) polymerases (PARP) are a large fam-ily of enzymes that catalyze the transfer of ADP-ribose unitsfrom NAD+ to a number of acceptor proteins. The prototypicenzyme of the PARP family is PARP-1, a nuclear enzyme
Springer
Apoptosis (2006) 11:1945–1957 1951
that is catalytically activated by DNA strand interruptions.Caspase-3 is primarily responsible for the cleavage of the116 kDa PARP-1 protein into an 85 kDa fragment, whosepresence indicates that cells are undergoing apoptosis. Thus,to confirm activation of caspase-3, appearance of the 85 kDafragment in the nucleus of liver cells from NAC-treated or
untreated rabbits infected with the RHD virus was monitoredby Western blot analysis. As expected, substantial PARP pro-teolysis was documented in infected animals by the detectionof the characteristic 85-kDa fragment at 36 h and 48 h pi.In contrast, livers treated with NAC showed reduced PARPproteolysis (Fig. 8).
Fig. 5 Effect of RHDV infection and NAC treatment on the liverexpression of Bax and Bcl-2 proteins. Panels A and C: Western blotanalysis in rabbit liver. Equal loading of proteins is illustrated by β-actin bands. A representative out of five animals is shown. Cytosolicprotein was separated on 15% SDS–polyacrylamide gels and blottedwith anti-Bcl-2 or Bax antibodies, respectively. Panels B and D: Densit-ometry of Bax and Bcl-2 proteins Western blot. Densitometric analysis
of Western blots was performed, and the arithmetric mean ± SEM isrepresented. The Y axis indicates arbitrary densitometric units (a.u.).Panel E: Bax/Bcl-2 ratio. Values are means ± SEM from 5 animals. ∗:significantly different from untreated animals, same period, p < 0.05;#: significantly different from control, same treatment, p < 0.05
(Continue on next page.)
Springer
1952 Apoptosis (2006) 11:1945–1957
Fig. 5 (Continued.)
Discussion
The rabbit hemorrhagic disease virus is a member of theCaliciviridae family that causes in wild and domestic rabbitsan acute highly fatal disease that was first reported in Chinatwo decades ago [25]. The disease is characterized by severenecrotizing hepatitis and a rapid evolution to death within 48to 72 h after infection in about 90% of the cases [15, 16]. Hep-atic damage plays a central pathogenic role and is histologi-cally similar to fatal viral hepatitis causing fulminate hepaticfailure (FHF) in humans [14]. Clinical feature and changesin blood chemistry and intracranial pressure monitoring con-firm that the RHD is an adequate animal model of FHF [18].Although in early studies on RHDV infection, liver tissuedestruction was described as necrosis [15], following mor-phological criteria and using a terminal transferase-mediatedDNA nickend labelling (TUNEL) assay, it has been foundthat liver cell death induced upon infection is due to apop-tosis, and that programmed cell death is a constant featurein rabbits experimentally infected. All cell types support-ing viral replication, mainly detected in the liver, presentthe characteristic apoptosis morphology, developing lesionsthat could contribute to the rapid progression to death ofRHDV infected rabbits [16]. Severe hepatic damage mightinterfere with clearance of activated clotting factors [26, 27]contributing to the occurrence of disseminated intravascularcoagulation (DIC), a feature shared by other viral hemor-rhagic fevers [16]. Since apoptotic cells are know sites ofenhanced procoagulant activity, apoptosis of intravascularcells [28] is another possible mechanism that may contributeto DIC. The presence and importance of liver apoptosis wasconfirmed in the present study, in which we observed cellswith ultrastructural features typical of apoptosis, such as theformation of highly electrodense chromatin clumps closelyassociated with the nuclear envelope. In addition, agarose gelelectrophoresis of DNA, which gives a typical ladder patternof fragments with multiples of 180–200 pb [29], indicatedthe presence of DNA fragmentation.
It should be considered, however, that methods for thedetection of apoptosis based on DNA fragmentation, such as
TUNEL assay and DNA laddering, lack accuracy to quan-tify the intensity of the process, and may also detect necroticcells [29, 30]. Thus, while DNA degradation is an impor-tant event in cell death, its detection in dead cells does notspecifically indicate the underlying mechanism of death [31].For this reason, the extent of liver undergoing apoptosis wasdetermined by immunohistochemistry of caspase-3, and spe-cific mediators of both the intrinsic and extrinsic pathwaysof apoptosis were measured. The liver infected with RHDVwas highly positive for the activated form of caspase-3, in-dicating that a caspase-dependent pathway was involved inapoptosis observed after infection. However, in livers of an-imals treated with NAC a reduced number of cells werestained.
The extrinsic apoptotic pathway is initiated by the activa-tion of membrane-bound death receptors by specific ligandssuch as TNF-α and FAS-ligand, resulting in the activationof death domain-associated proteins. The death domain-associated proteins complex with pro-caspase-8 resultingin the proteolytic cleavage and activation pro-caspase-3 andthe cellular apoptosis cascade [32]. An activation of the Faspathway has been shown to be responsible for fulminanthepatitis in different in vivo models [33, 34]. Data in thepresent study indicate that at 36 h and 48 h pi, in parallelto changes in blood chemistry, there was an increasedexpression of Fas-ligand and in the activities of caspase 3.We have also previously reported that TNF-α mRNA levelsincrease significantly from 36 h pi in rabbits with RHD[20].
The intrinsic pathway involves signals originating fromnumerous other stimuli, which are potently inhibited byBcl-2 [35]. The mechanism modulating the intrinsic pathwayremain to be fully elucidated, but diverse investigations haveshown that cellular ATP depletion initiates the translocationof Bax, a proapoptotic Bcl-2 family member protein,from the cytosol to the outer mitochondrial membrane bymechanisms that remain unclear. The translocation of Baxcauses mitochondrial dysfunction and swelling and inducesthe efflux of cytochrome c to the cytosol [36, 37]. Bcl-2functions to prevent cell death, whereas Bax, which forms
Springer
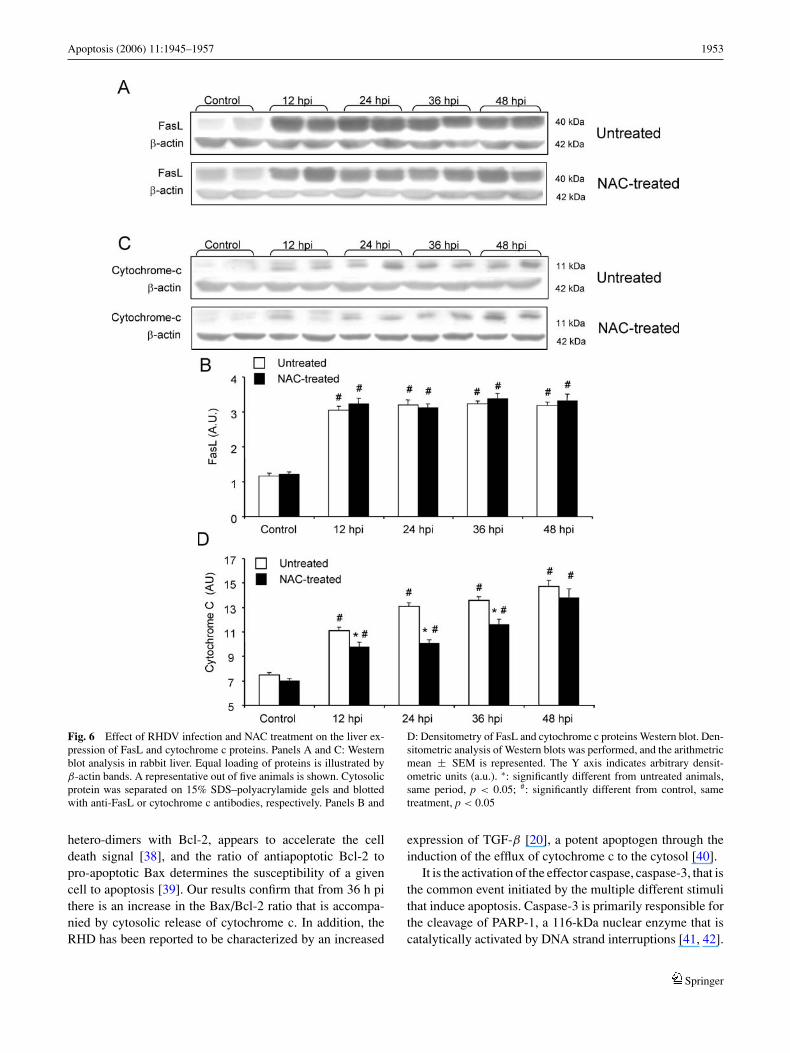
Apoptosis (2006) 11:1945–1957 1953
Fig. 6 Effect of RHDV infection and NAC treatment on the liver ex-pression of FasL and cytochrome c proteins. Panels A and C: Westernblot analysis in rabbit liver. Equal loading of proteins is illustrated byβ-actin bands. A representative out of five animals is shown. Cytosolicprotein was separated on 15% SDS–polyacrylamide gels and blottedwith anti-FasL or cytochrome c antibodies, respectively. Panels B and
D: Densitometry of FasL and cytochrome c proteins Western blot. Den-sitometric analysis of Western blots was performed, and the arithmetricmean ± SEM is represented. The Y axis indicates arbitrary densit-ometric units (a.u.). ∗: significantly different from untreated animals,same period, p < 0.05; #: significantly different from control, sametreatment, p < 0.05
hetero-dimers with Bcl-2, appears to accelerate the celldeath signal [38], and the ratio of antiapoptotic Bcl-2 topro-apoptotic Bax determines the susceptibility of a givencell to apoptosis [39]. Our results confirm that from 36 h pithere is an increase in the Bax/Bcl-2 ratio that is accompa-nied by cytosolic release of cytochrome c. In addition, theRHD has been reported to be characterized by an increased
expression of TGF-β [20], a potent apoptogen through theinduction of the efflux of cytochrome c to the cytosol [40].
It is the activation of the effector caspase, caspase-3, that isthe common event initiated by the multiple different stimulithat induce apoptosis. Caspase-3 is primarily responsible forthe cleavage of PARP-1, a 116-kDa nuclear enzyme that iscatalytically activated by DNA strand interruptions [41, 42].
Springer
1954 Apoptosis (2006) 11:1945–1957
Fig. 7 Effect of RHDVinfection and NAC treatment onliver caspase 3 activity.Activities were determinedusing specific substrateAc-DEVD-AMC. Values aremeans ± SEM from 5 animals.∗: significantly different fromuntreated animals, same period,p < 0.05; #: significantlydifferent from control, sametreatment, p < 0.05
Fig. 8 Effect of RHDV infection and NAC treatment on the liver ex-pression of PARP-1 protein. Panel A: Western blot analysis in rabbitliver. Equal loading of proteins is illustrated by β-actin bands. A repre-sentative out of five animals is shown. Nuclear protein was separated on15% SDS–polyacrylamide gels and blotted with anti-PARP antibody.Panel B: Densitometry of the 85-kDa fragment Western blot. Densit-
ometric analysis of Western blots was performed, and the arithmetricmean ± SEM is represented. The Y axis indicates arbitrary densit-ometric units (a.u.). ∗: significantly different from untreated animals,same period, p < 0.05; #: significantly different from control, sametreatment, p < 0.05
The substantial proteolysis of PARP-1 and the cleavage spe-cific to the 85-kDA product further supported the presenceof apoptosis in this animal model of FHF.
Many studies have demonstrated that intracellular ROSgeneration is associated with apoptotic cell death. Thus, ithas been reported that ROS generation by cadmium triggersapoptosis via caspase-dependent pathways in HepG2 cells[10], or that there is an oxidative stress-driven mechanism fornordihydroguaiaretic acid-induced apoptosis in FL5.12 cells[43]. Furthermore, the concept that oxidative stress inducesapoptosis is supported by the fact that exposure of cultured
adult rat ventricular myocytes to H2O2 and FeSO4 apopto-sis through mitochondrial pathway [44]. The importance ofROS in apoptosis is also supported by the beneficial effectsof antioxidants in the failing heart following myocardial in-farction [45]. NAC is a molecule with antioxidant properties,that has already been proposed as a complementary therapyin different liver diseases, including in viral hepatitis, non-alcoholic esteatohepatitis and hepatorenal syndrome [46]. Inaddition, different studies have shown that treatment withNAC blocks apoptosis induced by different toxins or presentin various diseases [8–13].
Springer

Apoptosis (2006) 11:1945–1957 1955
Changes in the oxidant/antioxidant balance constitute acharacteristic feature of the RHD, with significant decreasesin GSH liver concentration and SOD activity, and a markedincrease in the GSSG/GSH ratio present in infected animals[20]. Falls in GSH levels may be due to different reasons, in-cluding a lowered synthesis or increased degradation to gen-erate cysteine required in other potentially damaged tissues[47]. In any case, the scavenger NAC protected, as expected,infected rabbits against oxidative stress, because the GSSGconcentration and the GSSG/GSH ratio were significantlyreduced in treated animals. This was accompanied by an in-creased survival percentage at 36 and 48 h pi, together withan absence of DNA ladders, characteristic of the apoptoticcleavage of nuclear DNA, and of cells with ultrastructuralapoptotic features. When specific effects on mediator of theextrinsic and intrinsic pathways were investigated, it wasfound that NAC treatment resulted in a marked decrease inthe Bax/Bcl-2 ratio, cytochrome c release, caspase 3 activityand PARP-1 proteolysis. This clearly demonstrated a pro-tective role of NAC against apoptotic death and indicatedthat oxidative stress-driven mechanisms may be involved inmitochondrial disruption.
Although results by different authors indicate that ROSmay constitute a direct cause of mitochondrial dysfunctionand to perform certain functions in the early stages of apop-tosis [10, 48], it has also been reported that ROS are capableof inducing the expression of Fas and FasL mRNA duringapoptosis in intestinal cells, indicating that ROS might befunctioning upstream the mitochondria [49]. Previous stud-ies have also shown that antioxidants such as GSH, catalaseor NAC can effect inhibition of Fas-induced apoptosis in dif-ferent cell types [50]. However, we were unable to detect anychange in Fas-ligand expression in infected animals treatedwith NAC. This absence of effects on the death receptor de-pendent pathway, which resulted in values for both caspase3 activity and expression of the 85 kDa PARP-1 fragmentstill significantly elevated in comparison to control animals,would explain why NAC slows the rate at which animals dyein response to viral infection, but does not prevents death.Therefore, the fact that death is not ultimately prevented byNAC does not necessarily means that apoptosis is a minormechanism of cell death in this model. Experiments usingdifferent caspase inhibitors (separately or in combination)would help to clarify this point.
The haemagglutination test using human type 0 red bloodcells allows to calculate the number of infecting particlespresent in homogenates of different organs and is a techniquewidely adopted to quantify calicivirus inocula [51]. Usingthis technique high haemagglutinating titres of RHDV anti-gens have been previously demonstrated in liver of RHDV-infected rabbits [52, 53]. This was confirmed in the presentstudy, with higher titres reached at 48 h pi. Haemaggluti-nation activity was not clearly modified by NAC treatment,
which suggests that inhibition of virus infectivity does notcontribute to the protective effects induced by NAC.
It is important to consider that antioxidants not only pro-tect cells from oxidative and apoptotic damage, but also playa critical role in immunoregulation. Glutathione participatesin the early stages of lymphocyte activation necessary forhost defense, enhancing activation of cytotoxic T-cells, andits depletion inhibits mitogen-induced lymphocyte activa-tion and natural killer cell function [54]. Different studieshave shown that NAC and other antioxidants, by modulat-ing glutathione levels enhance T-cell function, increasingthe release of chemokines and decreasing susceptibility toinfection in HIV-infected patients [55, 56]. Because the in-nate immune system plays a crucial role in the liver and cellswhich express T-cell receptors might be involved in hepaticinjury in fulminant hepatic failure [57], contribution of animmunoregulatory role of NAC to its protective role in RHDcannot be ruled out.
In any case, our data support that, in the RHD model ofFHF, apoptotic cell death is induced via both the intrinsicand the extrinsic signalling pathways, and that the antiapop-totic action of NAC probably is related to the modulationof Bcl-2 and Bax genes. The protective effect provided bythe antioxidant NAC suggests that oxidative stress is a pri-mary pathway for apoptosis in this model of FHF. Becausefulminant hepatic failure is associated with a high risk oflethal results and orthotopic liver transplantation is not al-ways available in a timely fashion, the inhibition of oxidativestress and/or direct intervention on apoptotic pathways couldbe molecular targets for potential therapeutic use of antiox-idants, contributing to temporary support while awaiting aliver transplant.
Acknowledgments This work was partially supported by grants fromthe Fondo de Investigacion Sanitaria (PI/021121) and the Junta deCastilla y Leon, Spain.
References
1. Gavhami S, Hashemi M, Kadkhoda K, Alavian SM, Bay GH, LosM (2005) Apoptosis in liver diseases – detection and therapeuticapplications. Med Sci Monit 11:337–345
2. Pretet JL, Pelletier L, Bernard B, Coumes-Marquet S, KantelipB, Mouglin C (2003) Apoptosis participates to liver damage inHSV-induced fulminat hepatitis. Apoptosis 8:655–663
3. Sakaida I, Kimura T, Yamasaki T, Fukumoto Y, Watanabe K,Aoyama M et al (2005) Cytochrome c is a possible new markerfor fulminant hepatitis in humans. J Gastroenterol 40:179–185
4. Schuchmann M, Galle PR (2001) Apoptosis in liver disease. EurJ Gastroenterol Hepatol 13:785–790
5. Delhalle S, Duvoix A, Schnekenburger M, Morceau F, Dicato M,Diederich M (2003) An introduction to the molecular mechanismsof apoptosis. Ann NY Acad Sci 1010:1–8
6. Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, MatsudaH et al (1998) Bax interacts with the permeability transitionpore to induce permeability transition and cytochrome c release
Springer

1956 Apoptosis (2006) 11:1945–1957
in isolated mitochondria. Proc Natl Acad Sci USA 95:14681–14686
7. D’Agostini F, Izzotti A, Balansky RM, Bennicelli C, Flora SD(2005) Modulation of apoptosis by cancer chemopreventiveagents. Mutat Res 591:173–186
8. Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ(2005) Thiol reducing compounds prevent human amylin-evokedcytotoxicity. FEBS J 272:4949–4959
9. Wu YJ, Muldoon LL, Neuwelt EA (2005) The chemoprotectiveagent N-acetylcysteine blocks cisplatin-induced apoptosis throughcaspase signalling pathway. J Pharmacol Exp Ther 312:424–431
10. Oh SH, Lim SC (2006) A rapid and transient ROS gener-ation by cadmium triggers apoptosis via caspase-dependentpathway in HepG2 cells and this is inhibited through N-acetyl-cysteine-mediated catalase upregulation. Toxicol Appl Pharmacol212:212–223
11. Zachwieja J, Zaniew M, Bobkowski W, Stefaniak E, WarzywodaA, Ostalka-Novicka D et al (2005) Beneficial in vitro effectof N-acetyl-cysteine on oxidative stress and apoptosis. PediatrNephrol 20:725–731
12. Sadowska AM, Manuel-y-Keenoy B, De Backer BA (in press)Antioxidant and anti-inflammatory efficacy of NAC in thetreatment of COPD: discordant in vitro and in vivo dose-effects:a review. Pulm Pharmacol Ther. doi:10.1016/j.pupt.2005.12.007
13. Quadrilatero J, Hoffman-Goetz L (2005) N-acetyl-L-cysteineprotects intestinal lymphocytes from apoptotic death after acuteexercise in adrenalectomized mice. Am J Physiol 288:R1664–R1672
14. Mikami O, Park JH, Kimura T, Ochiai K, Itakura C (1999)Hepatic lesions in young rabbits experimentally infected withrabbit haemorrhagic disease virus. Res Vet Sci 66:237–242
15. Park JH, Lee Y, Itakura C (1995) Pathogenesis of acute necrotichepatitis in rabbit hemorrhagic disease. Lab Anim Sci 45:445-449
16. Alonso C, Oviedo JM, Martin-Alonso JM, Diaz E, Boga JA, Parra,F (1998) Programmed cell death in the pathogenesis of rabbithemorrhagic disease. Arch Virol 143:321–332
17. Ferreira PG, Costa-e-Silva A, Monteiro E, Oliveira MJ, AguasAP (2004) Transient decrease in blood heterophils and sustainedliver damage caused by calicivirus infection of young rabbits thatare naturally resistant to rabbit haemorrhagic disease. Res Vet Sci76:83–94
18. Tunon MJ, Sanchez-Campos S, Garcıa-Ferreras J, Alvarez M,Jorquera F, Gonzalez-Gallego J (2003) Rabbit hemorrhagic viraldisease: characterization of a new animal model of fulminant liverfailure. J Lab Clin Med 141:272–278
19. Belanguer M, Butterworth RF (2005) Acute liver failure: a criticalappraisal of available animal models. Metab Brain Dis 20:409–423
20. Sanchez-Campos S, Alvarez M, Culebras JM, Gonzalez-GallegoJ, Tunon MJ (2004) Pathogenic molecular mechanisms in ananimal model of fulminant hepatic failure: rabbit hemorrhagicviral disease. J Lab Clin Med 144:215–222
21. O.I.E. (2000) Rabbit haemorrhagic disease. In: Manual ofStandards for diagnostic tests and vaccines. World Organizationfor Animal Health, Paris, pp 762–776
22. Markwell MA, Haas SM, Bieber LL, Tolbert NE (1978) A modi-fication of the Lowry procedure to simplify protein determinationin membrane and lipoprotein samples. Anal Biochem 87:206–210
23. Hissin PJ, Hilf RA (1976) Fluorimetric method for determinationof oxidised and reduced glutathione in tissues. Anal Biochem74:214–26
24. Tunon MJ, Sanchez-Campos S, Gutierrez B, Culebras MJ,Gonzalez-Gallego J (2003) Effects of FK506 and rapamycin ongeneration of reactive oxygen species, nitric oxide productionand nuclear factor kappa B activation in rat hepatocytes. BiochemPharmacol 66:439–445
25. Liu SJ, Xue HP, Pu BQ, Quia NH (1984) A new viral disease inrabbits. Anim Husb Vet Med 16:253–255
26. Prieto JM, Fernandez F, Alvarez V, Espi A, Garcıa Marın JF,Alvarez M et al (2000) Immunohistochemical localisation ofrabbit haemorrhagic disease virus VP-60 antigen in early infectionof young and adult rabbits. Res Vet Sci 68:181–187
27. Ferreira PG, Costa e Silva A, Oliveira MJR, Monteiro E, CunhaEM, Aguas AP (2006) Severe leukopenia and liver biochemistrychanges in adult rabbits after calicivirus infection. Res Vet Sci80:218–225
28. Ramiro-Ibanez F, Martın-Alonso JM, Garcıa Palencia P, ParraF, Alonso C (1999) Macrophage tropism of rabbit hemorrhagicdisease virus is associated with vascular pathology. Virus Res60:21–28
29. Kumar D, Kirshenbaum L, Li T, Danelisen I, Singal P (1999)Apoptosis in isolated adult cardiomyocytes exposed to adriamycin.Ann NY Acad Sci 874:156–168
30. Ohno M, Takemura G, Ohno A, Misao R, Hayakawa Y,Minatoguchi S et al (1998) “Apoptotic” myocytes in infarct area inrabbit hearts may be oncotic myocytes with DNA fragmentation:analysis by immunogold electron microscopy combined with insitu nick end-labeling. Circulation 98:1422–1430
31. Fink SL, Cookson BT (2005) Apoptosis, pyroptosis and necrosis:mechanistic description of dead and dying eukaryotic cells. InfectImmun 73:1907–1916
32. Nagata S (1997) Apoptosis by death factor. Cell 88:355–36533. Galle PR, Hofmann WJ, Walczak H et al (1995) Involvement of
the CD95 (APO-1/Fas) receptor and ligand in liver damage. J ExpMed 182:1223–1230
34. Zhang H, Cook J, Nickel J, Yang P, Wang Z, Wang X, Curiel DT,Zhou T, Mountz JD et al (2000) Reduction of liver Fas expressionby an antisense oligonucleotide protects mice from fulminanthepatitis. Nat Biotechnol 18:862–867
35. Green DR, Reed JC (1998) Mitochondria and apoptosis. Science281:1309–1312
36. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD (1997)The release of cytochrome c from mitochondria: a primary site forBcl-2 regulation of apoptosis. Science 275:1132–1136
37. Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, MachoA et al (1996) Bcl-2 inhibits the mitochondrial release of anapoptogenic protease. J Exp Med 184:1331–1341
38. Oltvai ZN, Millima CL, Korsmeyer SJ (1993) Bcl-2 heterodimer-izes in vivo with a conserved homology bax, that acceleratesprogrammed cell death. Cell 74:609–619
39. Yang J, Korsmeyer SJ (1996) Molecular apoptosis: a discourse onthe BCL2 family and cell death. Blood 88:386–401
40. Cain K, Freathy C (2001) Liver toxicity and apoptosis: role ofthe TGF-β1, cytochrome c and the apoptosome. Toxicol Lett120:307–315
41. Mauriz JL, Gonzalez P, Jorquera F, Olcoz JL, Gonzalez-GallegoJ (2003) Caspase inhibition does not protect against liver damagein hemorrhagic shock. Shock 19:33–37
42. Soldani C, Scovassi AI (2002) Poly(ADP-ribose) polymerase-1 cleavage during apoptosis. An update. Apoptosis 7:321–328
43. Deshpande VS, Kehrer JP (2006) Oxidative stress-driven mecha-nisms of nordihydroguaiaretic acid-induced apoptosis in FL5.12cells. Toxicol Appl Pharmacol 214:230–236
44. Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, MatsuzakiM et al (2002) Direct activation of mitochondrial apoptosismachinery of c-Jun N-terminal kinase in adult cardiac myocytes.J Biol Chem 277:10244–10250
45. Oskarsson HJ, Coppey L, Weiss RM, Li WG (2000) Antioxidantsattenuate myocyte apoptosis in the remote non-infarcted my-ocardium following large myocardial infarction. Cardiovasc Res45:679–687
Springer

Apoptosis (2006) 11:1945–1957 1957
46. Majano PL, Medina J, Zuiba I, Sunyer L, Lara-Pezzi E,Maldonado-Rodrıguez A et al (2004) N-acetyl-cysteine modu-lates inducible nitric oxide synthase gene expression in humanhepatocytes. J Hepatol 40:632–637
47. Sanchez-Campos S, Lopez-Acebo R, Gonzalez P, CulebrasJM, Tunon MJ, Gonzalez-Gallego J (1998) Cholestasis andalterations of glutathione metabolism induced by FK506 in therat. Transplantation 68:84–88
48. Simon HU, Haj-Yehia A, Levi-Schaffer F (2005) Role of re-active oxygen species (ROS) in apoptosis induction. Apoptosis5:415–418
49. Aronis A, Melendez JA, Golan O, Shilo S, Dicter N, TiroshO (2003) Potentiation of Fas-mediated apoptosis by attenuatedproduction of mitochondria-derived reactive oxygen species. CellDeath Differ 10:335–344
50. Devadas S, Hinshaw JA, Zaritskaya L, Williams MS (2003) Fas-stimulated generation of reactive oxygen species or exogenousoxidative stress sensitize cells to Fas-mediated apoptosis. FreeRadic Biol Med 35:648–661
51. Chasey D, Lucas MH, Westcott DG, Sharp G, Kitching A, HughesSK (1995) Development of diagnostic approaches to the iden-tification of rabbit haemorrhagic disease. Vet Record 137:158–160
52. Granzow H, Weiland F, Strebelow HG, Liu CM, Schirrmeier H(1996) Rabbit hemorrhagic disease virus (RHDV): ultrastructureand biochemical studies of typical and core-like particles in liverhomogenates. Virus Res 41:163–172
53. Shien JH, Shieh HK, Lee LH (1998) Characterization of rabbithaemorrhagic disease virus field isolates in Taiwan. J Virol Meth27:27–33
54. Lioy J, Ho WH, Cutilli JR, Polin RA, Douglas SD (1993) Thiolsuppression of human immunodeficiency virus type 1 replicationin primary cord blood monocyte-derived macrophages in vitro. JClin Invest 91:495–498
55. Cavallini L, Alexandre A (2000) Oral N-acetylcysteine increasesthe production of anti-HIV chemokines in peripheral bloodmononuclear cells. Life Sci 67:147–154
56. Kalamasz D, Long SA, Taniguchi R, Buckner JH, Berenson RJ,Bonyhadi M (2004) Optimization of human T-cell expansionex vivo using magnetic beads conjugated with anti-CD3 andanti-CD28 antibodies. J Immunother 27:405–418
57. Migayawa R, Ichida T, Yamagiwa S, Miyaji C, Watanabe H,Sato Y et al (2005) Hepatic natural killer and natural killer Tcells markedly decreased in two cases of drug-induced fulminanthepatic failure rescued by living donor liver transplantation. JGastroenterol Hepatol 20:1126–1130
Springer
Recommended

















