Embed Size (px)
Citation preview

GLÁNDULAS SUPRARRENALES
FISIOPATOLOGIA I
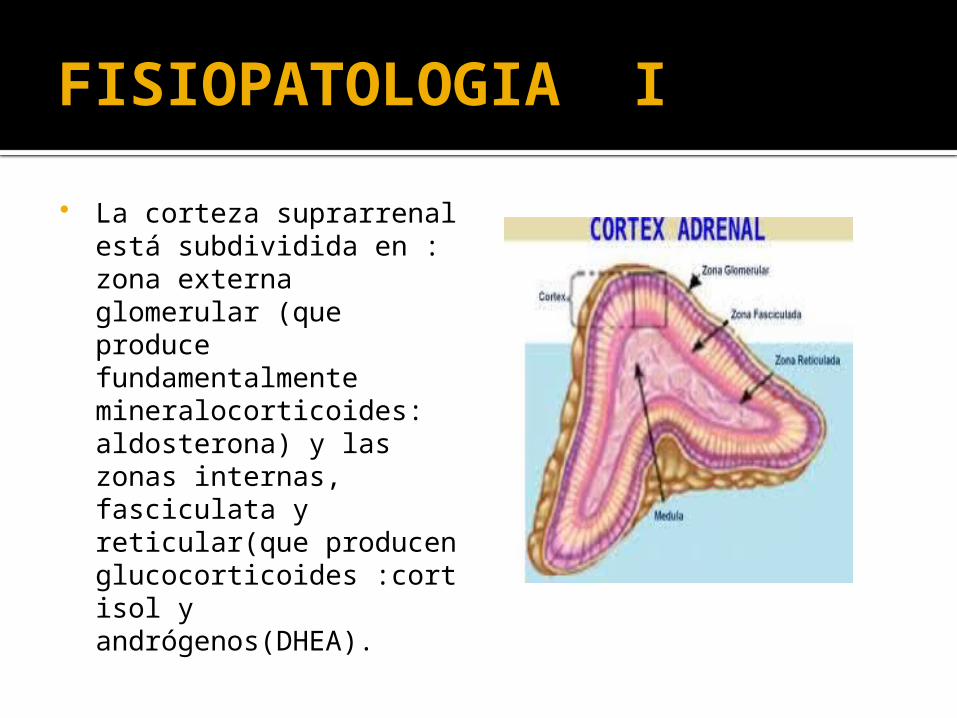
La corteza suprarrenal está subdividida en : zona externa glomerular (que produce fundamentalmente mineralocorticoides: aldosterona) y las zonas internas, fasciculata y reticular(que producen glucocorticoides :cortisol y andrógenos(DHEA).
SINDROME DE CUSHING
SIGNOS Y SINTOMAS I
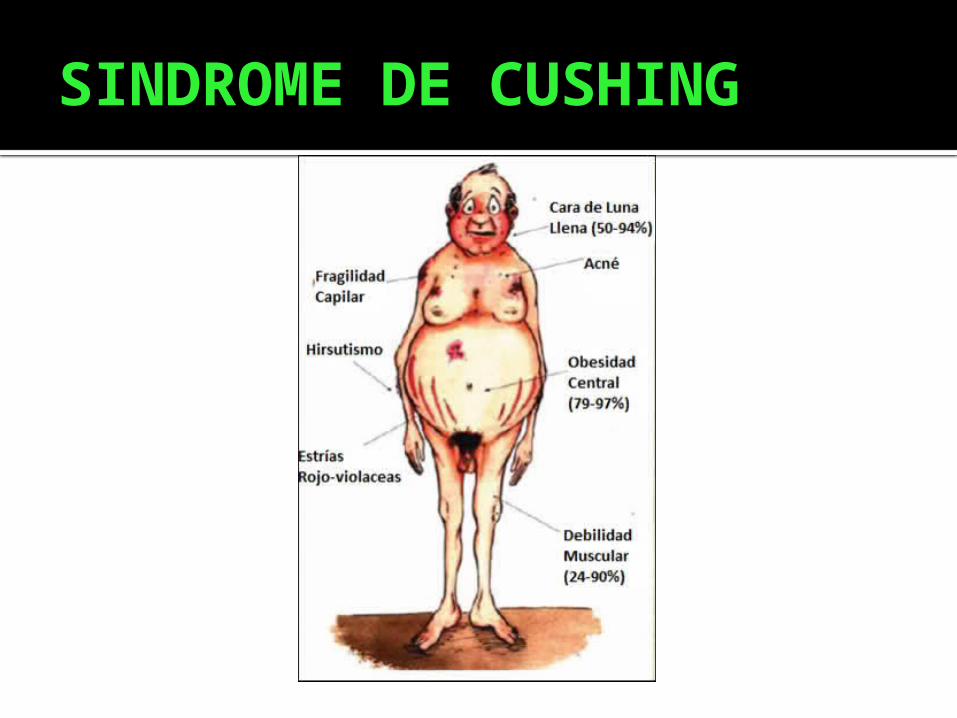
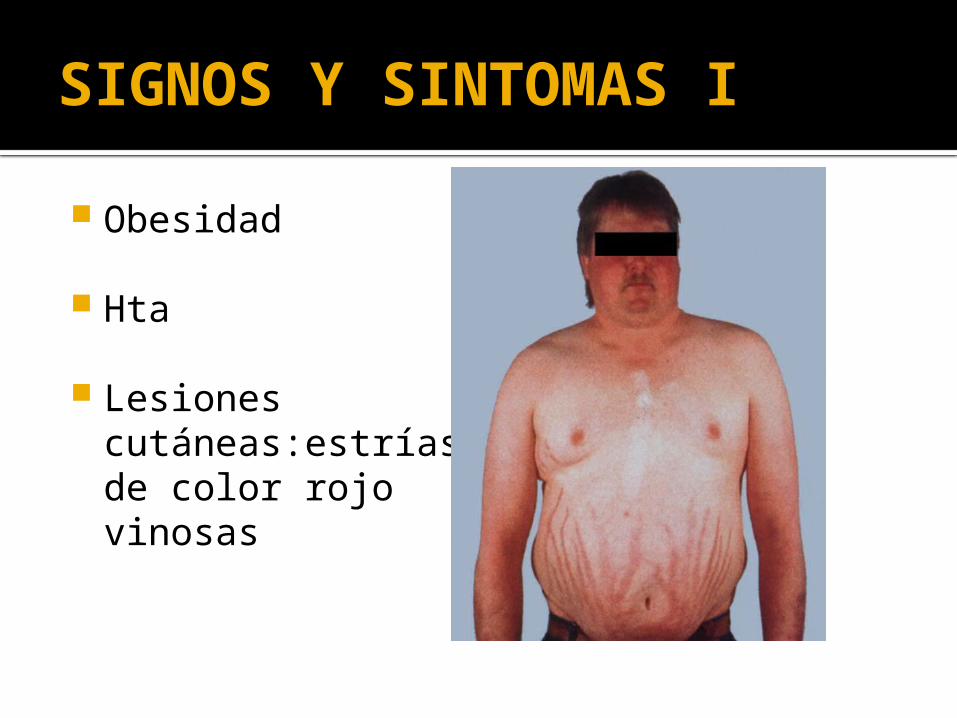
Obesidad
Hta
Lesiones cutáneas:estrías de color rojo vinosas

DISTRIBUCIÓN DEL TEJIDO ADIPOSO
En la cara-aspecto de luna llena
Region abdominal y la parte posterior del cuello-aspecto de joroba
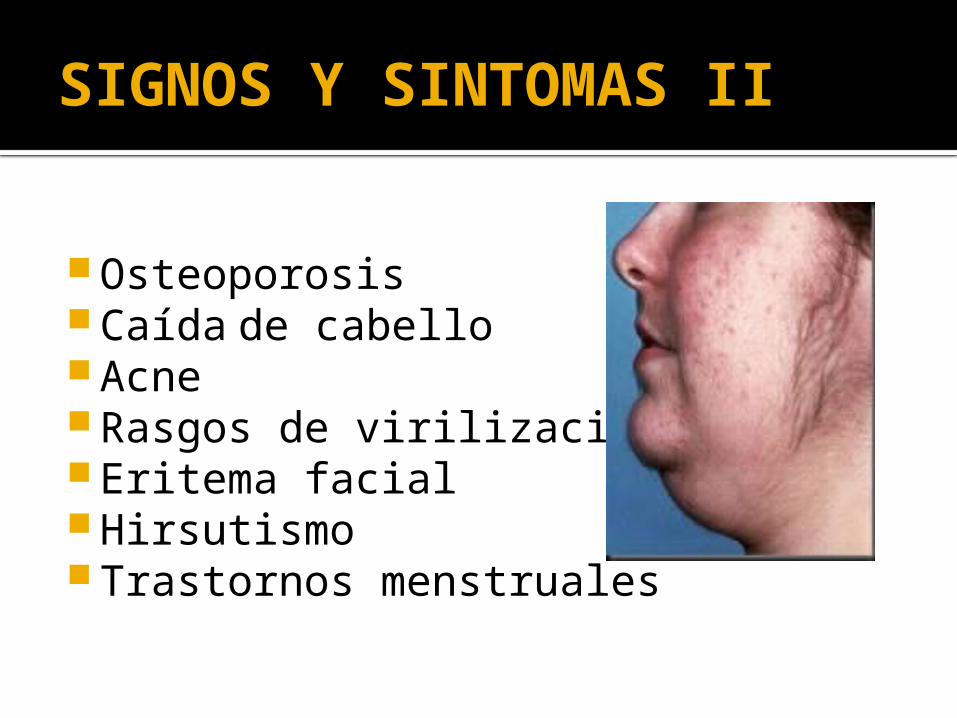
SIGNOS Y SINTOMAS II
Osteoporosis Caída de cabello Acne Rasgos de virilización Eritema facial Hirsutismo Trastornos menstruales

SIGNOS Y SINTOMAS III
Cambios neuropsiquiátricos y cognitivos con labilidad emocional, depresión,
irritabilidad, ansiedad , ataques de pánico, paranoia e insomnio
Euforicos o maniacos

Manifestaciones paraclinicas alcalosis
metabólica con hipopotasemia
hiperglucemia alteraciones
hematológicas alteraciones del
metabolismo lipídico
la supresión de las ejes tiroidea y gonádica
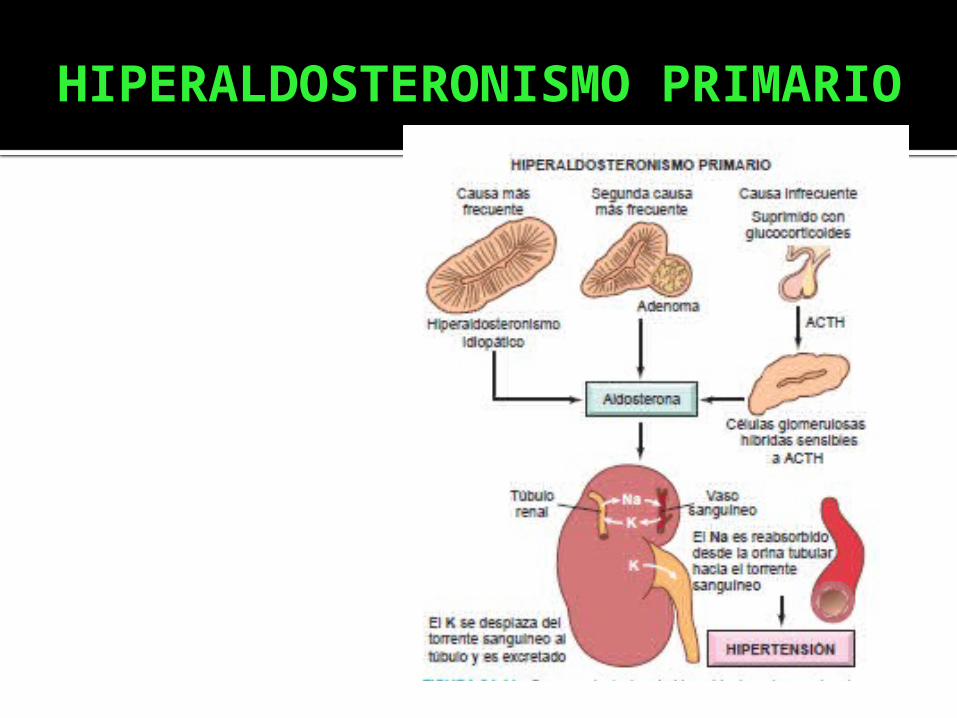
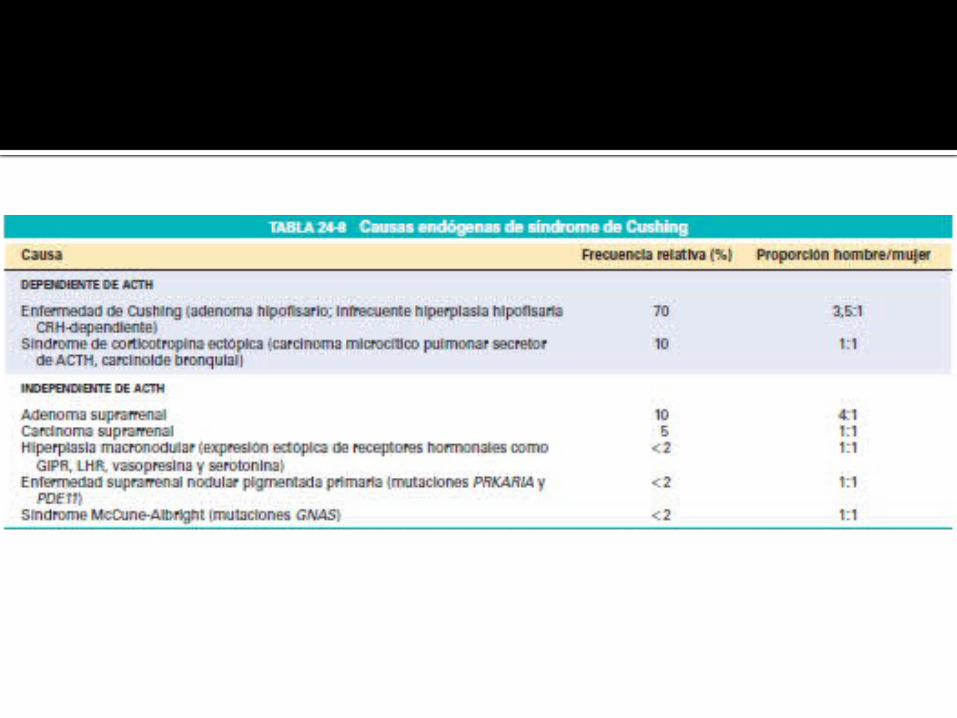
HIPERALDOSTERONISMO PRIMARIO
CONCEPTO
Esta caracterizado por: Hipertensión arterial (HTA): La
causa principal de hipertensión arterial secundaria.
Hipopotasemia. Disminución de la actividad de la
renina plasmática (ARP). Hipersecreción de la aldosterona.

ETIOLOGÍA ADENOMA PRODUCTOR DE LA
ALDOSTERONA O SÍNDROME DE CONN Son tumores benignos de la zona
glomerular de pequeño tamaño y bien encapsulados.
Suelen responder pobremente a la acción de la angiotensina II
Tiene una prevalencia del 4% de los pacientes con HAP.
CARCINOMA ADRENAL PRODUCTOR DE LA ALDOSTERONA Bastante infrecuente, se sospecha
ante la presencia de tumores adrenales mayores de 4cm.
Además de aldosterona suelen producir otras hormonas adrenales.
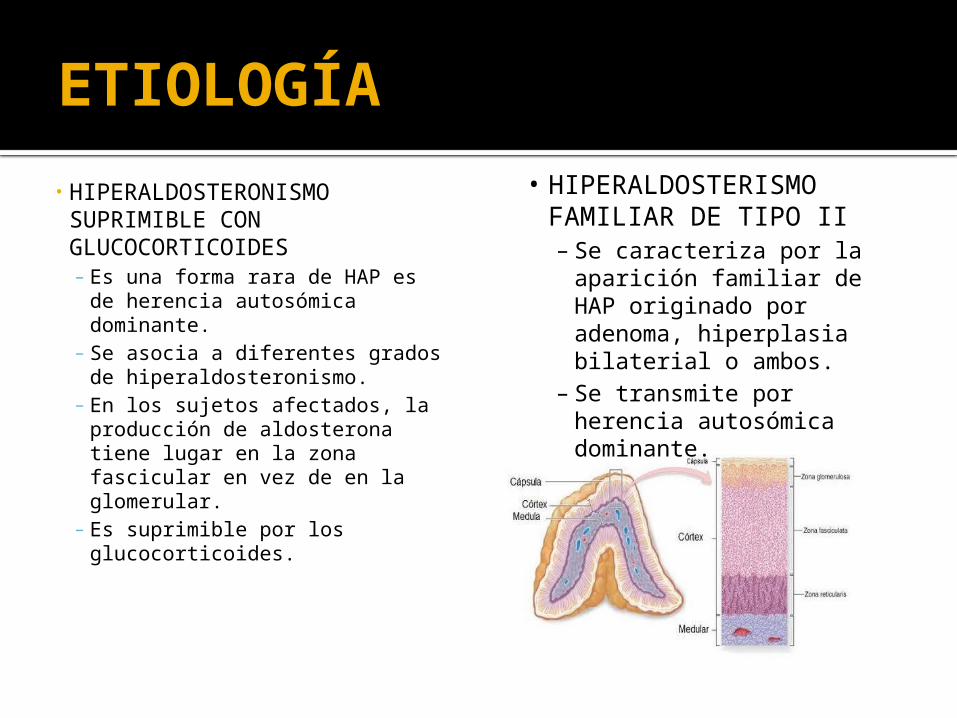
ETIOLOGÍA
• HIPERALDOSTERONISMO SUPRIMIBLE CON GLUCOCORTICOIDES – Es una forma rara de HAP es
de herencia autosómica dominante.
– Se asocia a diferentes grados de hiperaldosteronismo.
– En los sujetos afectados, la producción de aldosterona tiene lugar en la zona fascicular en vez de en la glomerular.
– Es suprimible por los glucocorticoides.
• HIPERALDOSTERISMO FAMILIAR DE TIPO II– Se caracteriza por la
aparición familiar de HAP originado por adenoma, hiperplasia bilaterial o ambos.
– Se transmite por herencia autosómica dominante.
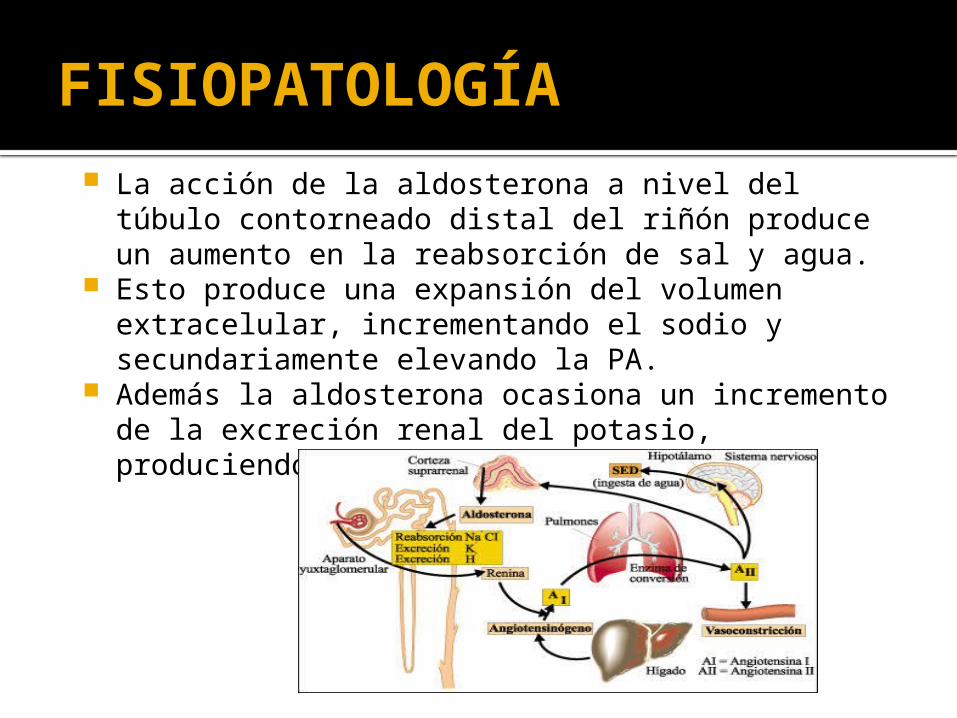
FISIOPATOLOGÍA La acción de la aldosterona a nivel del túbulo
contorneado distal del riñón produce un aumento en la reabsorción de sal y agua.
Esto produce una expansión del volumen extracelular, incrementando el sodio y secundariamente elevando la PA.
Además la aldosterona ocasiona un incremento de la excreción renal del potasio, produciendo una alcalosis metabólica.
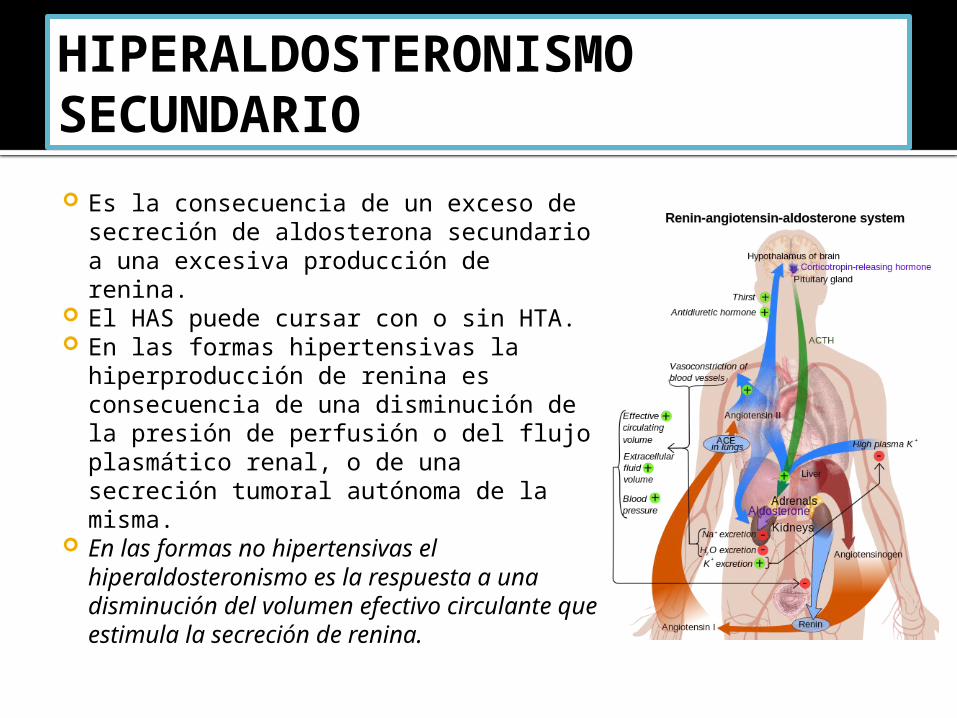
HIPERALDOSTERONISMO SECUNDARIO Es la consecuencia de un exceso de
secreción de aldosterona secundario a una excesiva producción de renina.
El HAS puede cursar con o sin HTA. En las formas hipertensivas la
hiperproducción de renina es consecuencia de una disminución de la presión de perfusión o del flujo plasmático renal, o de una secreción tumoral autónoma de la misma.
En las formas no hipertensivas el hiperaldosteronismo es la respuesta a una disminución del volumen efectivo circulante que estimula la secreción de renina.

FORMAS HIPERTENSIVAS
HIPERTENSIÓN RENOVASCULAREs la causa más común de HTA
dependiente de renina. Suele ser secundaria a
arterosclerosis o hiperplasia fibromuscular de las arterias renales.
En ambas situaciones se origina una disminución de la presión de perfusión en el riñón afectado con un estímulo de la liberación de renina y de la producción de angiotensina II.
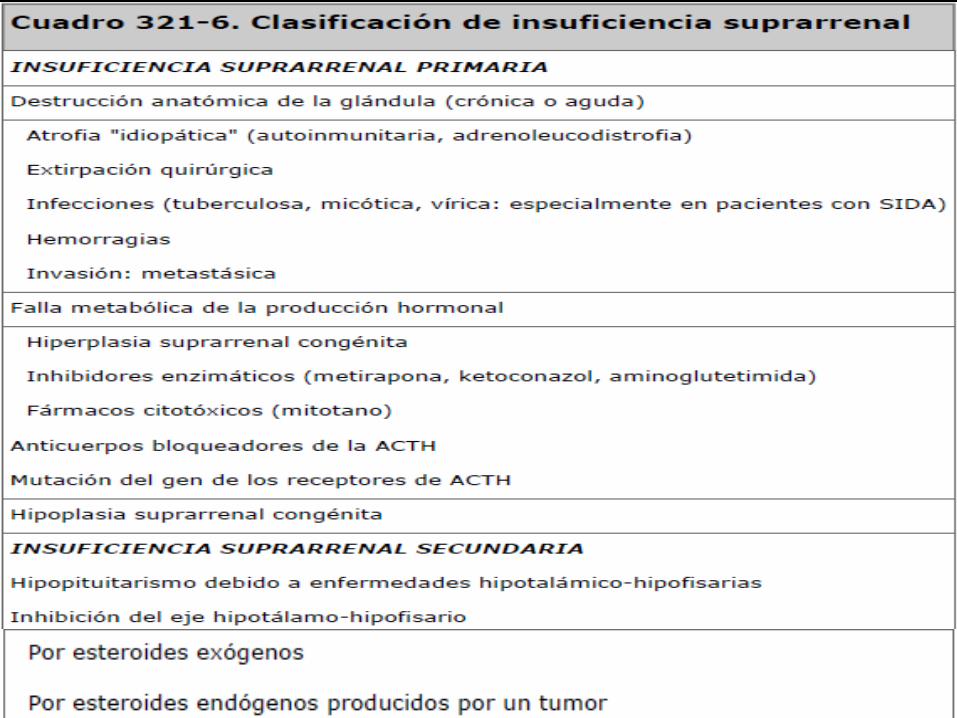
INSUFICIENCIA SUPRARRENAL
Síndrome causado por un defecto en la secreción esteroidea de la corteza adrenal
17
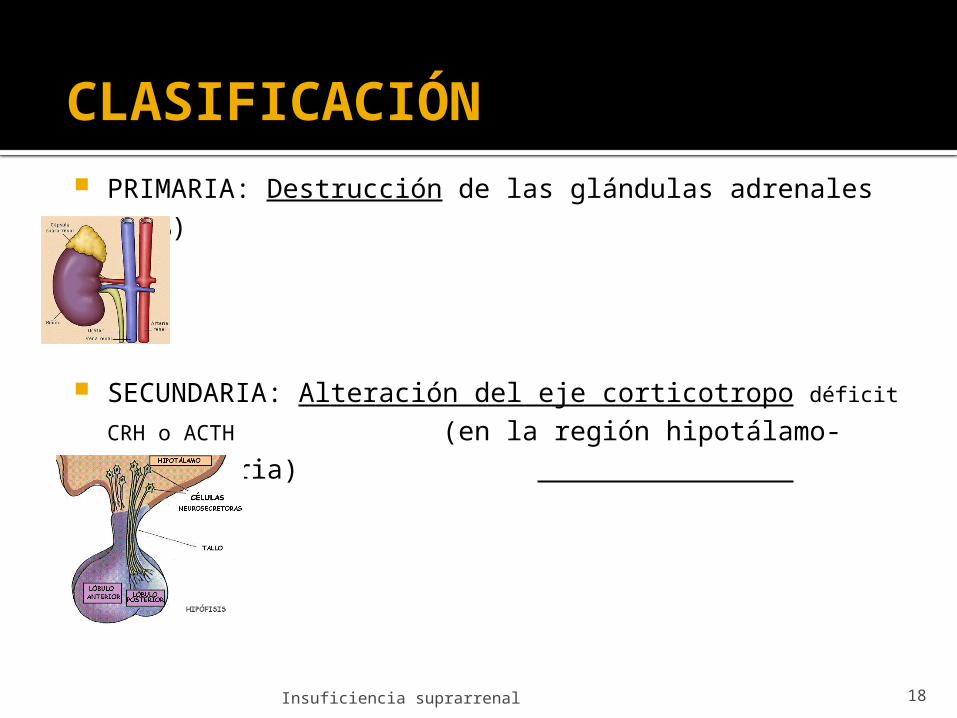
CLASIFICACIÓN PRIMARIA: Destrucción de las glándulas adrenales (90%)
SECUNDARIA: Alteración del eje corticotropo déficit CRH o ACTH (en la región hipotálamo-
hipofisaria)
Insuficiencia suprarrenal 18

INSUFICIENCIA CORTICO SUPRARRENAL SECUNDARIA
Glucocorticoides en exceso Déficit de ACTHPanhipopituitarismo

Síntomas y signos muy parecidos a la ISR primaria, pero carece de hiperpigmentación.
Concentraciones séricas de ACTH :1ria : elevadas2ria : disminuidas o ausentes
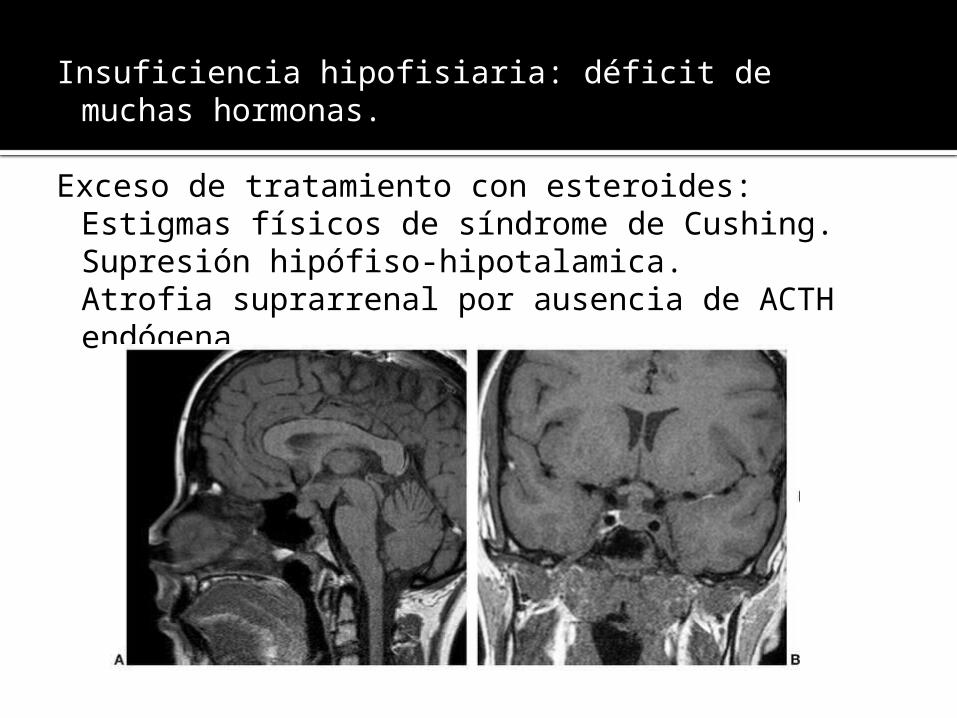
Insuficiencia hipofisiaria: déficit de muchas hormonas.
Exceso de tratamiento con esteroides: Estigmas físicos de síndrome de Cushing.Supresión hipófiso-hipotalamica.Atrofia suprarrenal por ausencia de ACTHendógena

INSUFICIENCIA CORTICO SUPRARRENAL AGUDA
Procesos que la conllevan:1. Sepsis.2. Estrés quirúrgico.3. Destrucción hemorrágica aguda de ambas glándulas.4. En niños por septicemia por pseudomonas.5. En adultos con tx anticoagulante o con trastornos de la
coagulación.

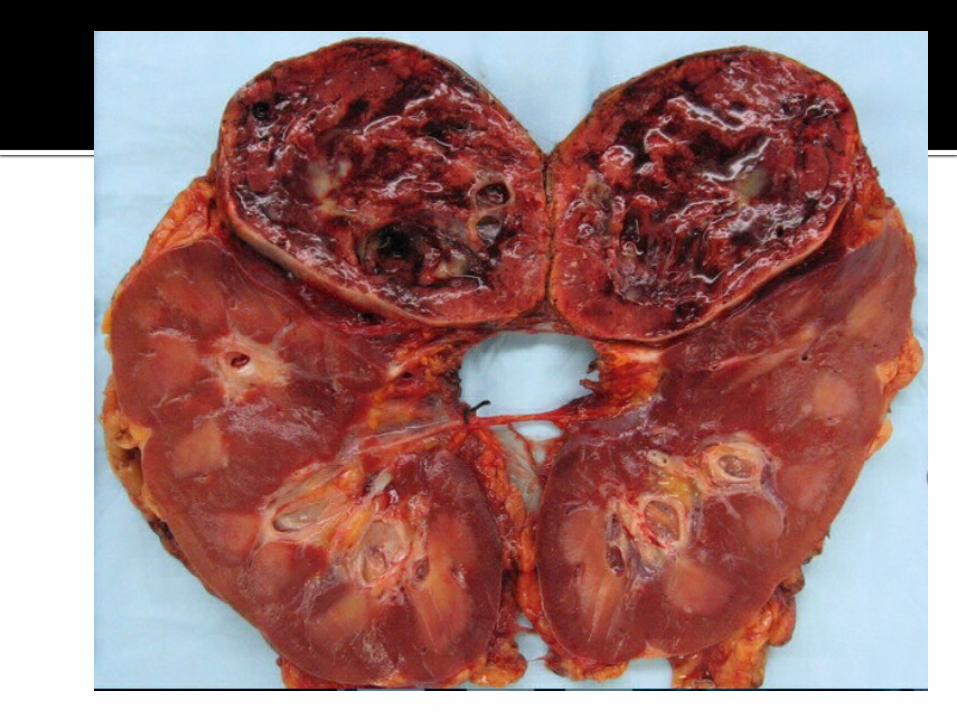
SINDROME DE WATERHOUSE-F
Es una insuficiencia de las glándulas suprarrenales, debido a sangrado dentro de dichas glándulas.
Es causado por una infección Meningocóccica severa.
Es de muy rara frecuencia, si se presenta el cuadro clínico es muy mal pronóstico.
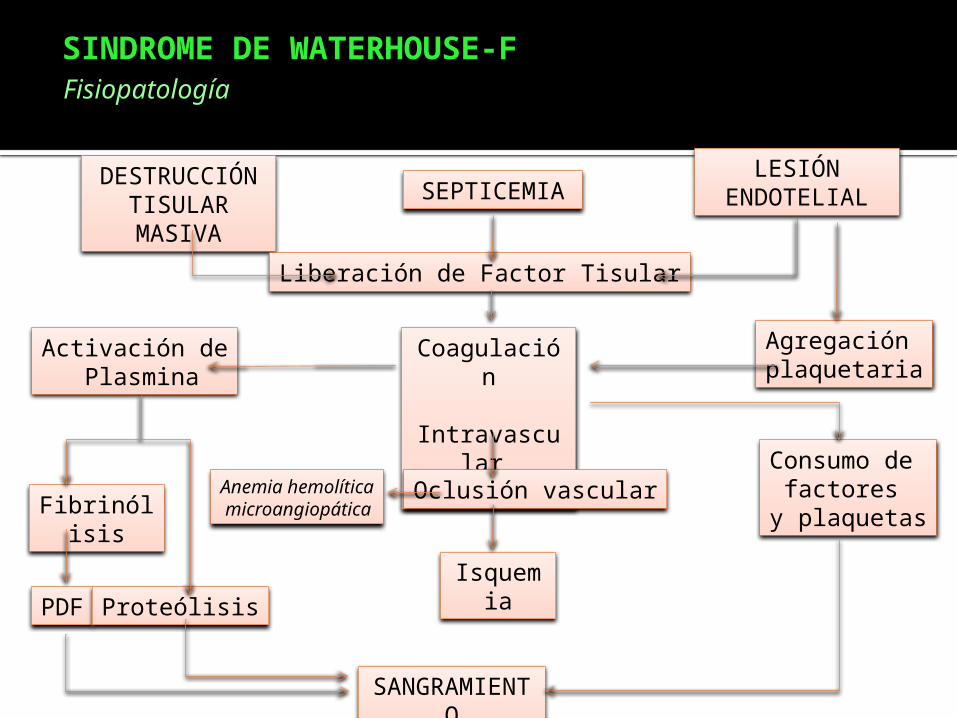
SINDROME DE WATERHOUSE-F
DESTRUCCIÓN TISULAR MASIVA
SEPTICEMIALESIÓN
ENDOTELIAL
Liberación de Factor Tisular
Coagulación
Intravascular
Diseminada
Activación de Plasmina
Fibrinólisis
PDF Proteólisis
SANGRAMIENTO
Oclusión vascular
Isquemia
Agregación plaquetaria
Consumo de factores
y plaquetasAnemia hemolítica microangiopática
Fisiopatología

SINDROME DE WATERHOUSE-F
CLÍNICA DEL PACIENTE:Dolor abdominal o torácicoFiebreNáuseas y vómitosSíntomas psiquiátricosLesiones hemorrágicas en pielDeshidrataciónCianosis (Trombosis) Shock
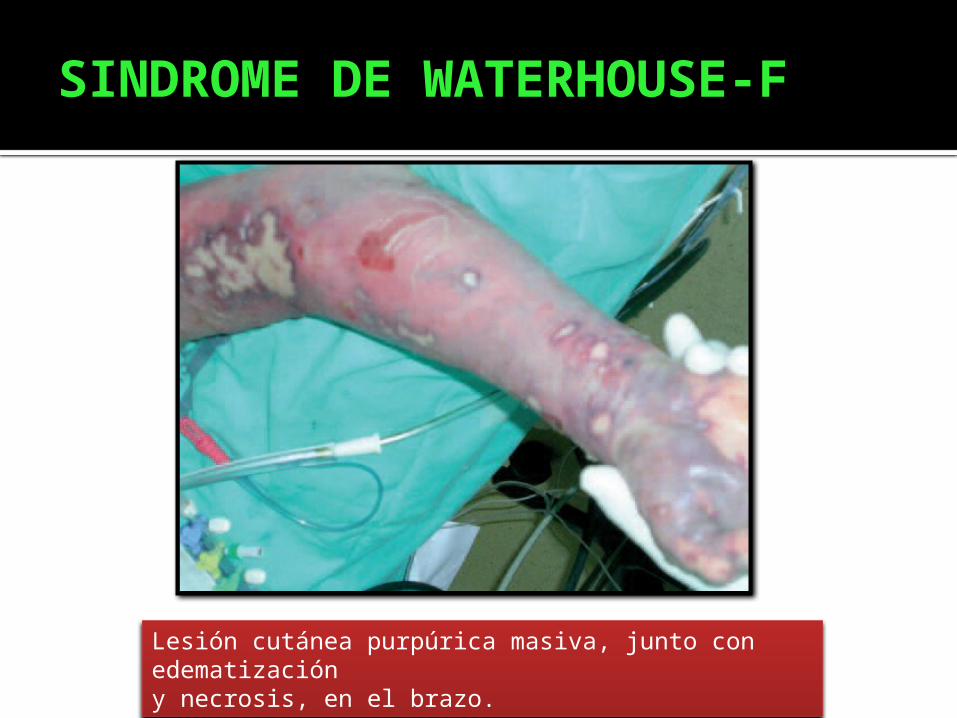
SINDROME DE WATERHOUSE-F
Lesión cutánea purpúrica masiva, junto con edematizacióny necrosis, en el brazo.
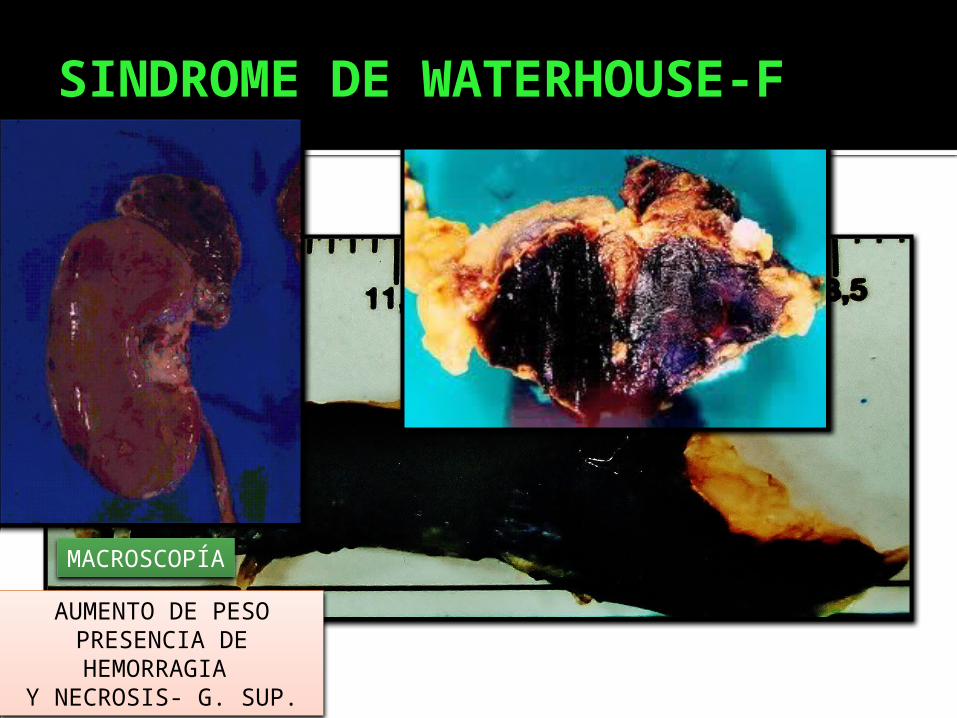
SINDROME DE WATERHOUSE-F
MACROSCOPÍA
AUMENTO DE PESO PRESENCIA DE HEMORRAGIA
Y NECROSIS- G. SUP.
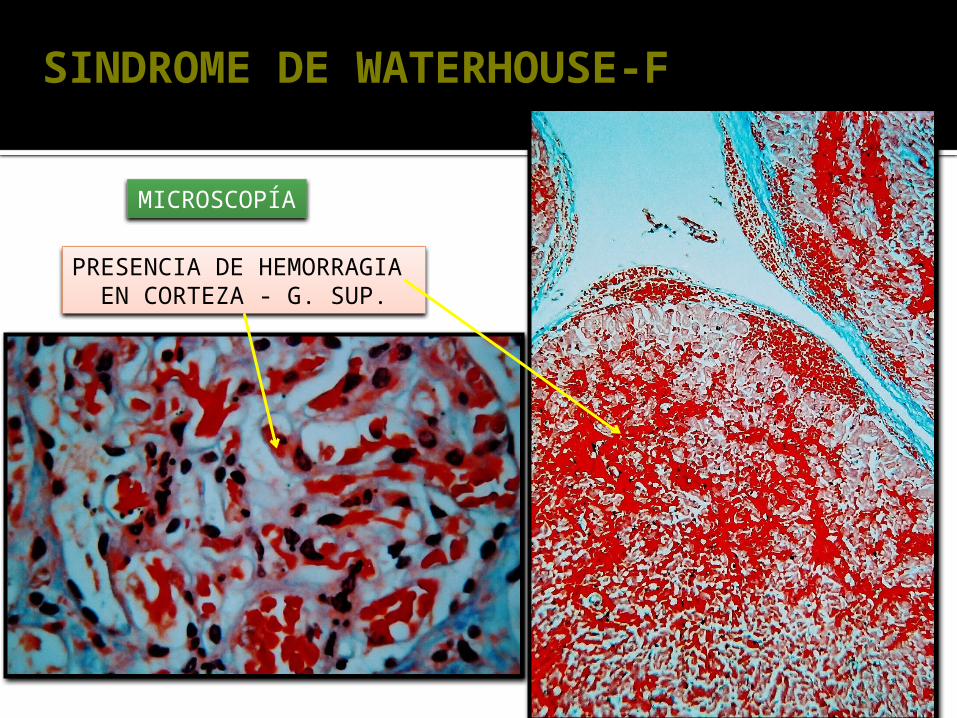
SINDROME DE WATERHOUSE-F
MICROSCOPÍA
PRESENCIA DE HEMORRAGIA EN CORTEZA - G. SUP.
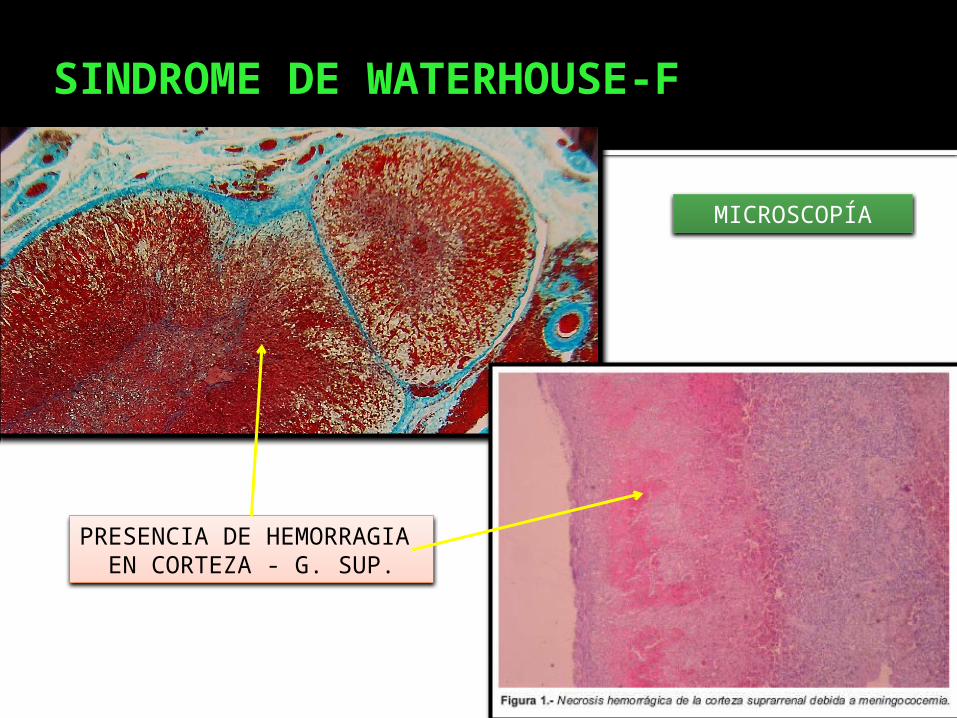
SINDROME DE WATERHOUSE-F
MICROSCOPÍA
PRESENCIA DE HEMORRAGIA EN CORTEZA - G. SUP.
6. Interrupción brusca de los esteroides en pacientes con atrofia suprarrenal por tx prolongado con esteroides.
7. Fármacos que inhiben la síntesis de los esteroides (fenitoína, rifampicina).
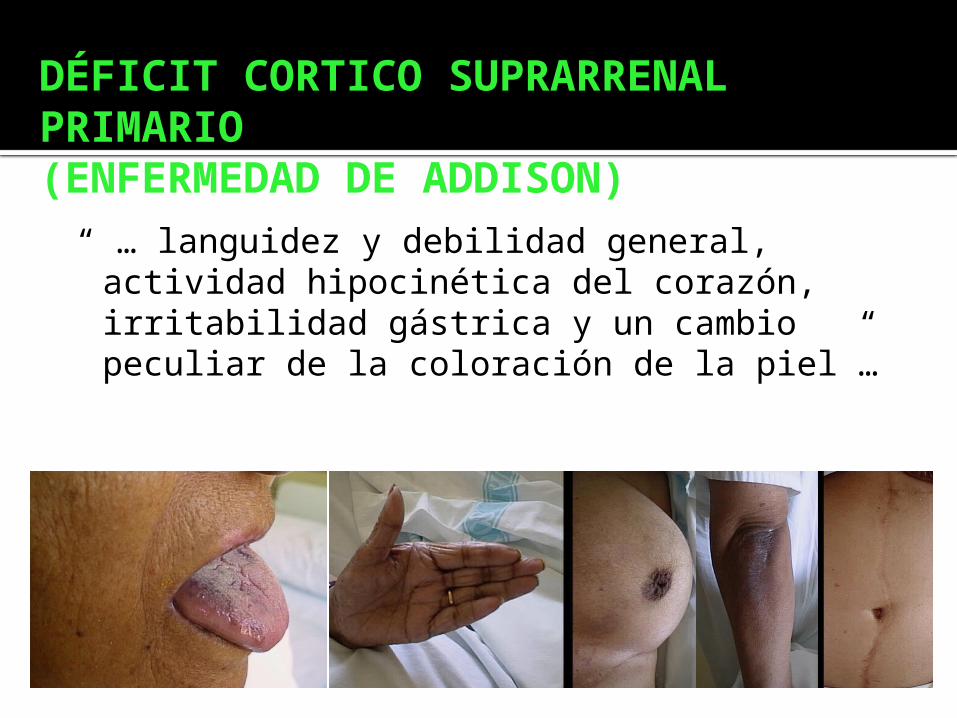
DÉFICIT CORTICO SUPRARRENAL PRIMARIO(ENFERMEDAD DE ADDISON)
“ … languidez y debilidad general, actividad hipocinética del corazón, irritabilidad gástrica y un cambio peculiar de la coloración de la piel”…

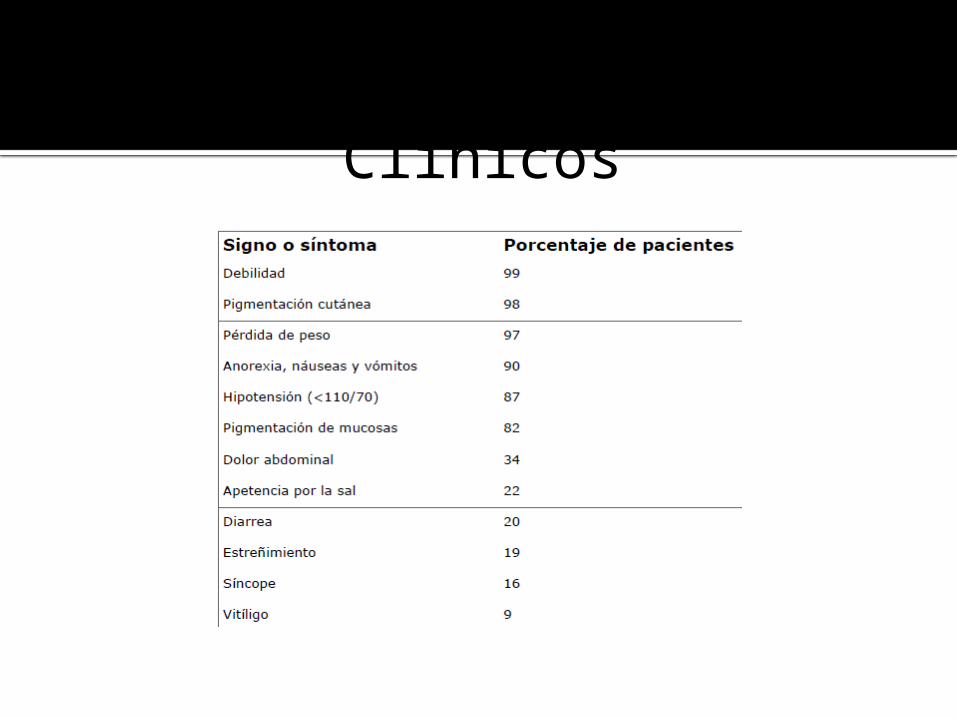
Signos y Síntomas Clínicos
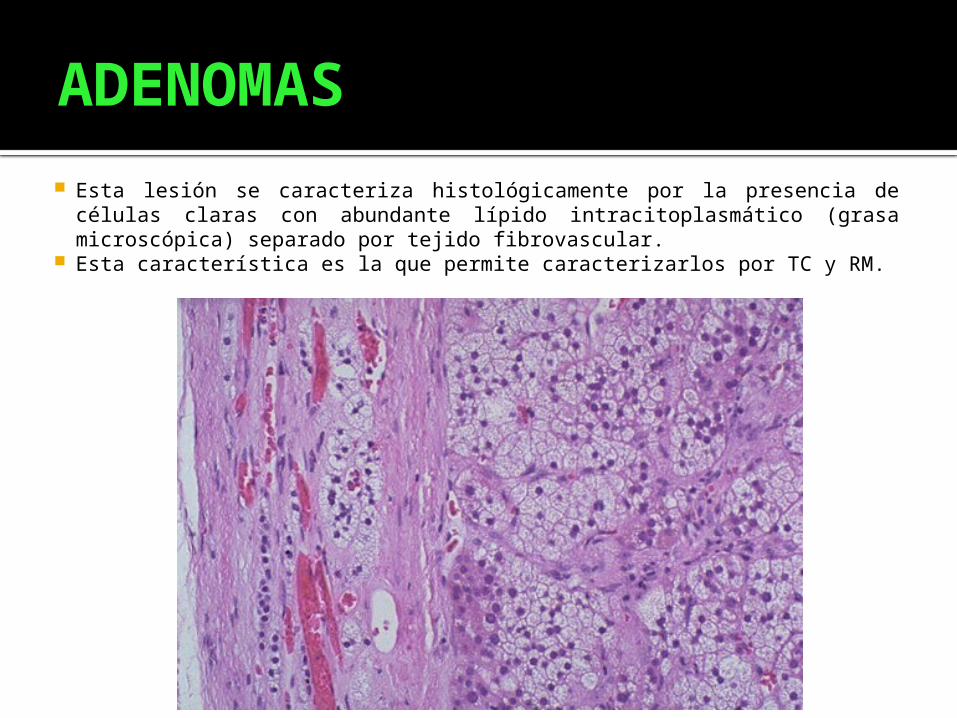
ADENOMAS
Esta lesión se caracteriza histológicamente por la presencia de células claras con abundante lípido intracitoplasmático (grasa microscópica) separado por tejido fibrovascular.
Esta característica es la que permite caracterizarlos por TC y RM.
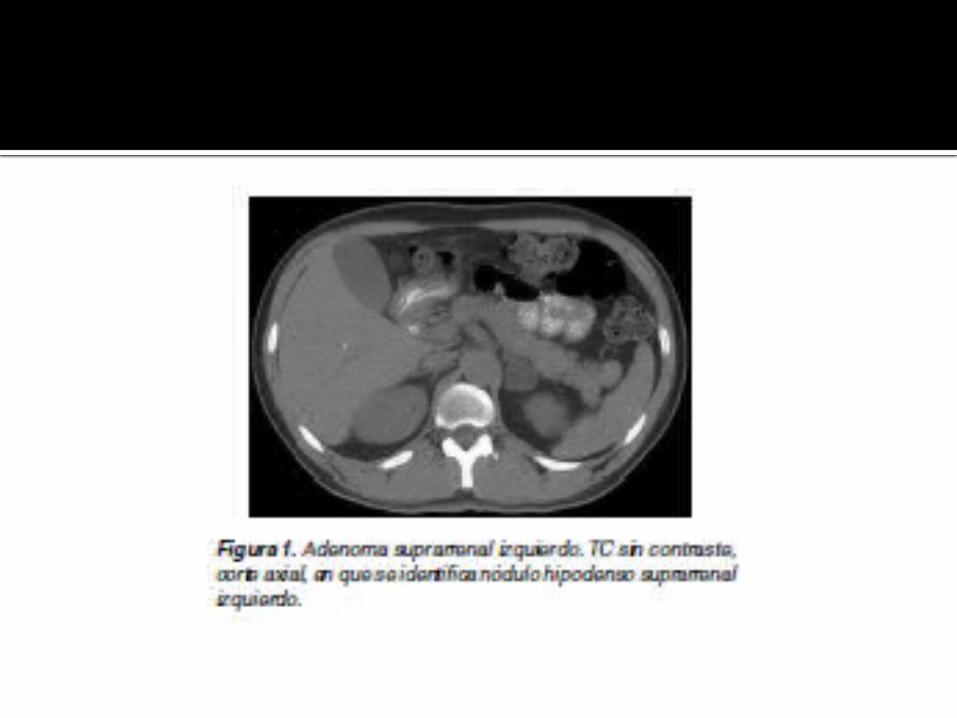

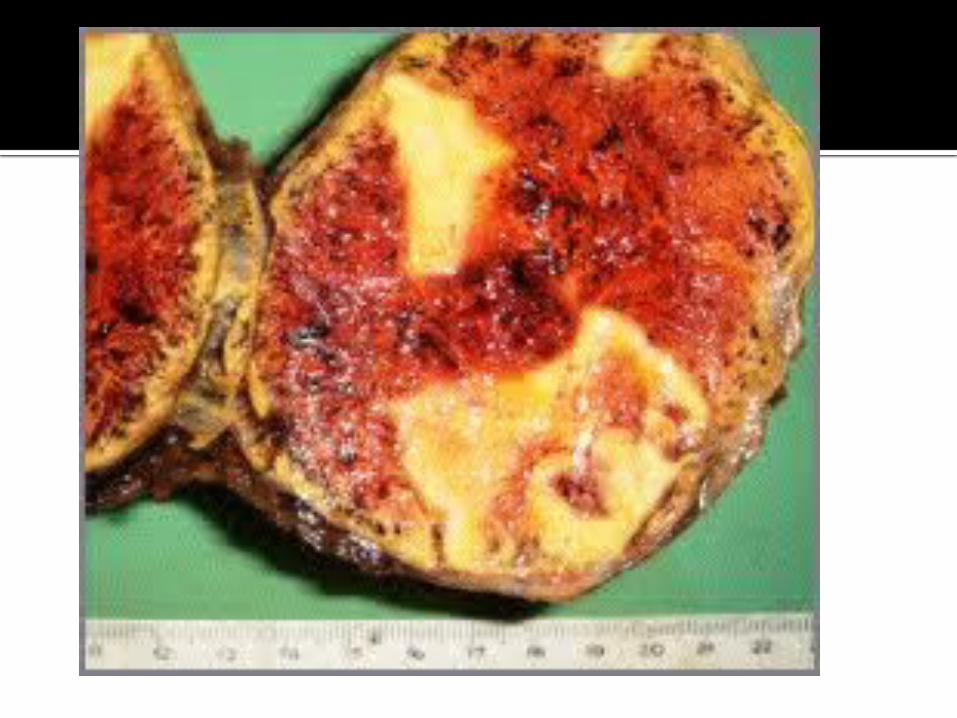
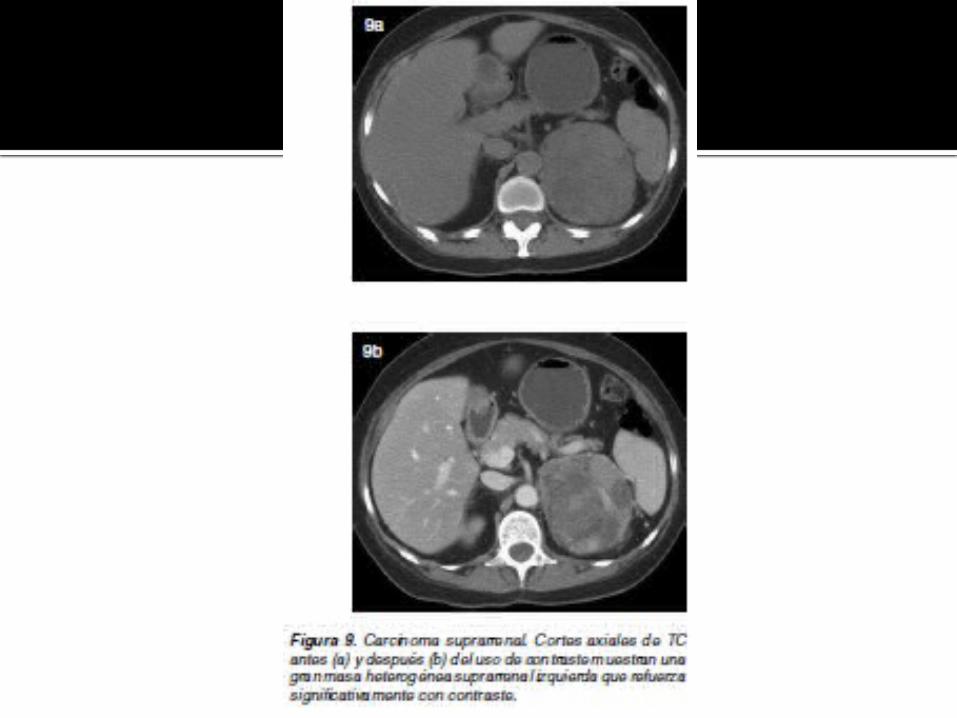
CARCINOMA SUPRARRENAL
Tumor raro (1 en 1.000.000), originado de la corteza suprarrenal, en general entre la cuarta y séptima décadas de la vida.
Es hiperfuncionante en un 40% de los casos, manifestándose más comúnmente como síndrome de Cushing.
Puede presentarse con dolor abdominal y masa palpable. Al diagnóstico, el tumor es grande, en general mayor a 6 cm,
alcanzando en algunos casos hasta 20 cm. Calcifican en un 30%; puede observarse invasión directa de
órganos vecinos, de la vena renal y vena cava inferior. Además, pueden observarse adenopatías y metástasis a
distancia, como por ejemplo en pulmón.
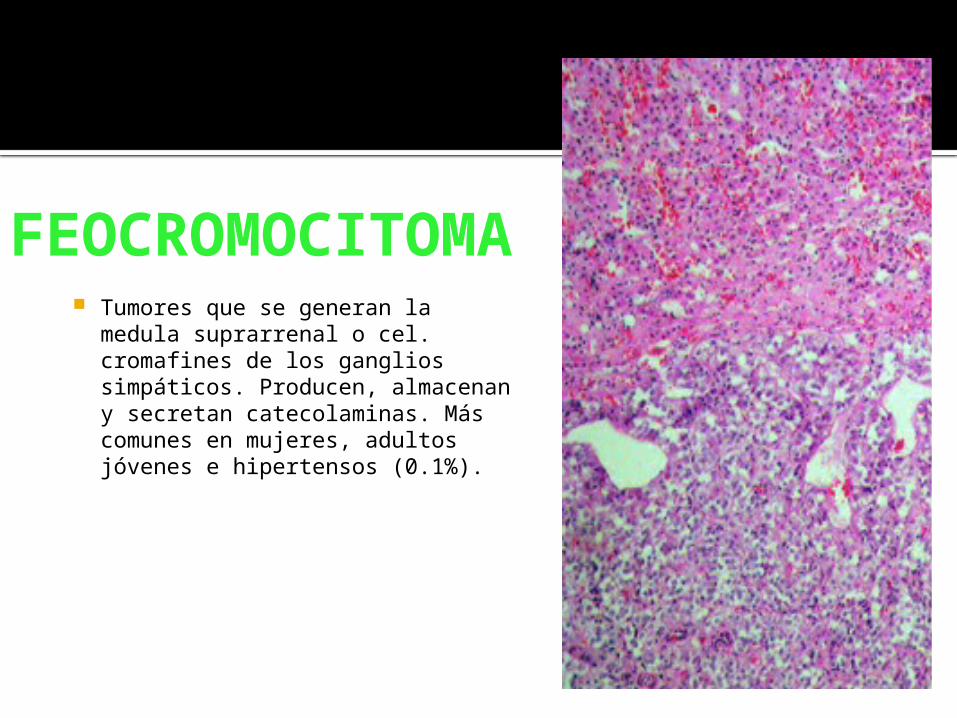
FEOCROMOCITOMA
Tumores que se generan la medula suprarrenal o cel. cromafines de los ganglios simpáticos. Producen, almacenan y secretan catecolaminas. Más comunes en mujeres, adultos jóvenes e hipertensos (0.1%).
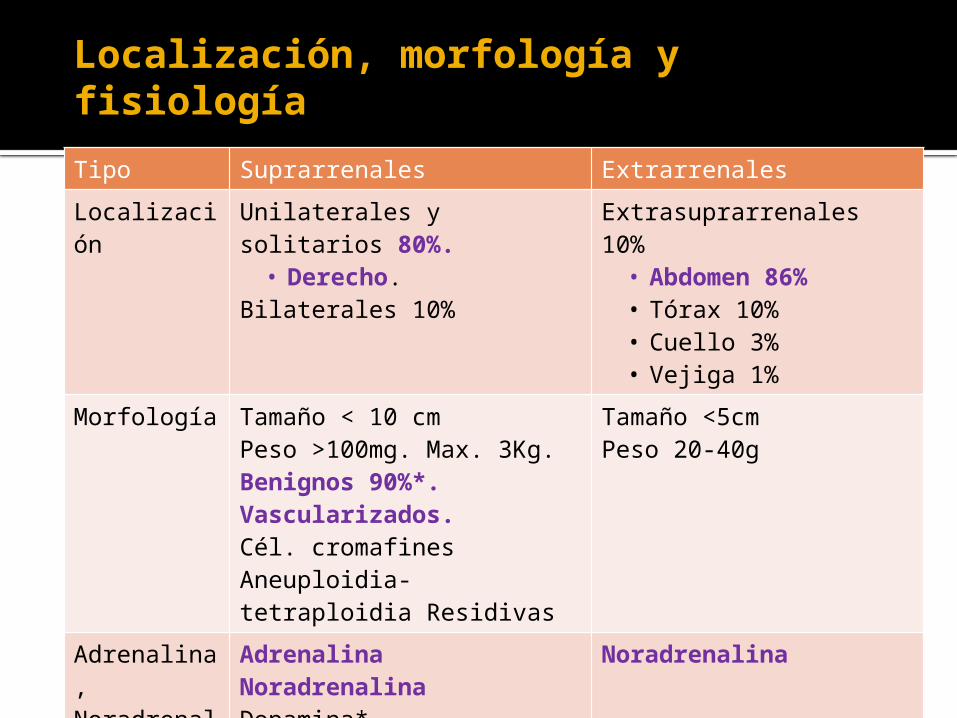
Localización, morfología y fisiología
HARRISON. Medicina Interna. Enfermedades de la corteza suprarrenal. 16a edición. Vol. II. México. 2006. 2340-2363.
Tipo Suprarrenales Extrarrenales
Localización Unilaterales y solitarios 80%.
• Derecho. Bilaterales 10%
Extrasuprarrenales 10%• Abdomen 86%• Tórax 10%• Cuello 3% • Vejiga 1%
Morfología Tamaño < 10 cmPeso >100mg. Max. 3Kg. Benignos 90%*. Vascularizados.Cél. cromafinesAneuploidia-tetraploidia Residivas
Tamaño <5cmPeso 20-40g
Adrenalina, Noradrenalina y Dopamina
AdrenalinaNoradrenalinaDopamina*HVA*
Noradrenalina
MEN se deben a alteraciones genéticas de un gen supresor de tumor y un protooncoogen.
Estos síndromes se caracterizan por predisposición en la transformación neoplásica en múltiples tejidos endocrinos y por alteraciones patológicas de tejidos no endocrinos .
Dentro de cada uno de los tejidos endocrinos implicados se desarrolla una hiperplasia pre neoplásica difusa que precede de la visión microscópica o aparición de un carcinoma multifocal.
Neoplasia endocrina múltiple
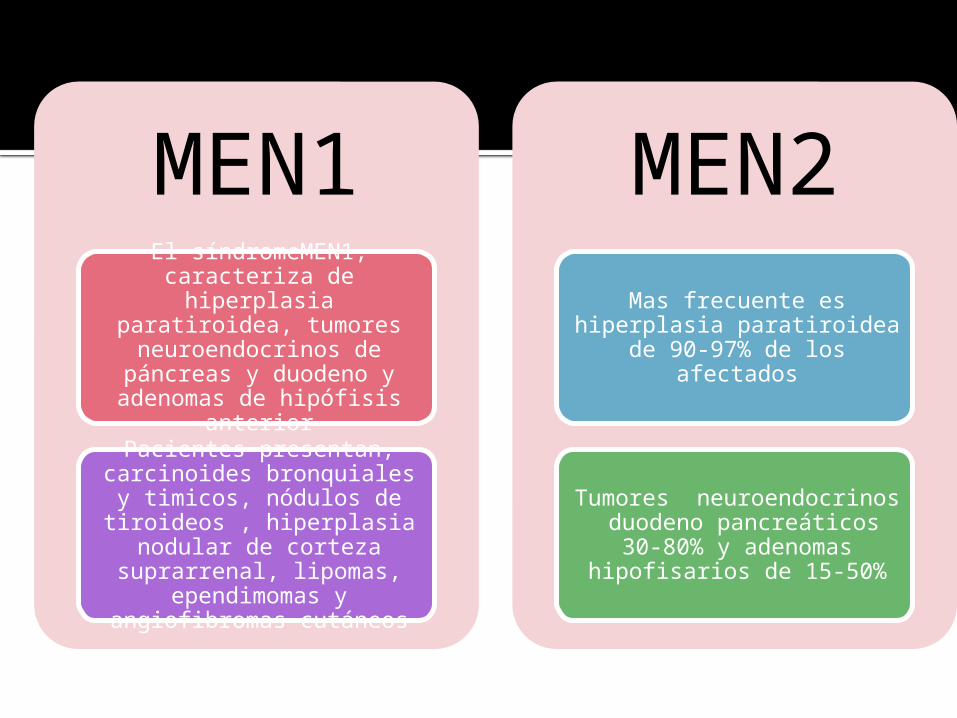
MEN1El síndromeMEN1,
caracteriza de hiperplasia paratiroidea, tumores
neuroendocrinos de páncreas y duodeno y adenomas de
hipófisis anterior
Pacientes presentan, carcinoides bronquiales y
timicos, nódulos de tiroideos , hiperplasia
nodular de corteza suprarrenal, lipomas,
ependimomas y angiofibromas cutáneos
MEN2
Mas frecuente es hiperplasia paratiroidea de 90-97% de
los afectados
Tumores neuroendocrinos duodeno pancreáticos 30-
80% y adenomas hipofisarios de 15-50%

Etiologia
Síndromes de neoplasia endocrina múltiple tipo 2MEN2A Y B, CMTF presentan penetrancia completa todos los individuos con el alelo desarrollan CMT El 25% de los casos de CMT son
familiares, el 42% de los afectados desarrollan feocromocitomas multifocales o bilaterales y se asocian a hiperplasia de la medula suprarrenal .
El 10-35% desarrollan hiperparatiroidismo secundario a hiperplasia asimétrica.
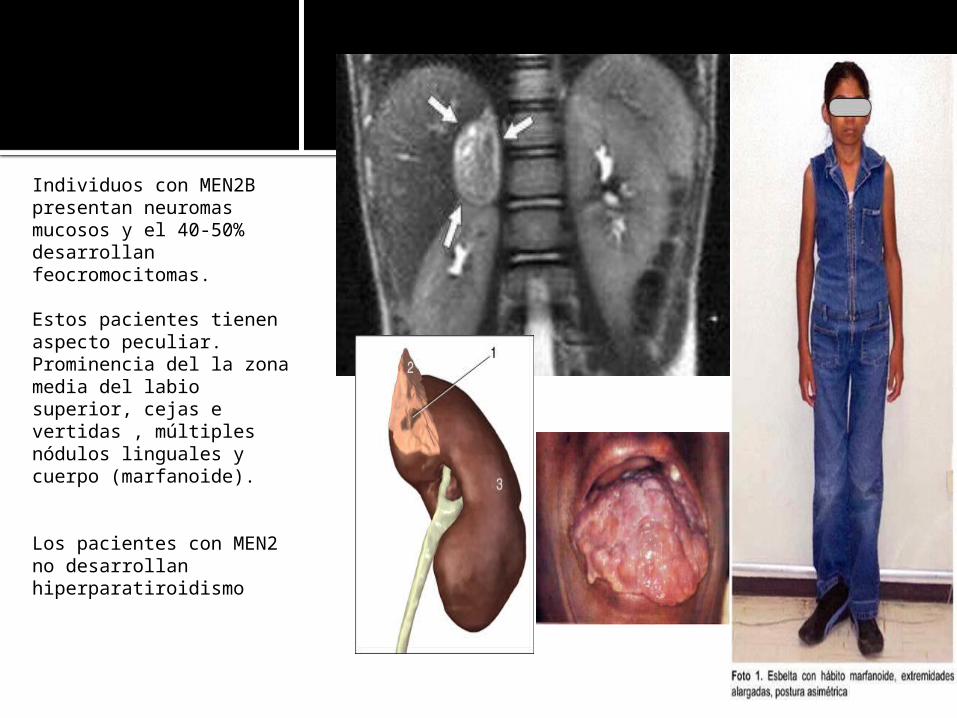
Individuos con MEN2B presentan neuromas mucosos y el 40-50% desarrollan feocromocitomas.
Estos pacientes tienen aspecto peculiar.Prominencia del la zona media del labio superior, cejas e vertidas , múltiples nódulos linguales y cuerpo (marfanoide).
Los pacientes con MEN2 no desarrollan hiperparatiroidismo























