Embed Size (px)
Citation preview

Quelles sont les probléma0ques en bio-‐informa0que du séquençage haut débit
pour le diagnos0c des maladies géné0ques
Pr. Stéphane BEZIEAU, service de Géné0que, CHU Nantes et Université de Nantes

La solution technologique : le séquençage nouvelle génération = NGS
Approche « shotgun », on séquence la cible en une seule étape, par petits fragments, puis on assemble par bioinformatique (alignement) puis on compare par rapport à la séquence de référence, encore par bioinformatique.
ADN génomique
Fragmentation puis enrichissement
Séquençage de l’ensemble des fragments en un seul temps (séquençage massif parallèle)
Analyse bioinformatique Assemblage par alignement sur la
séquence de référence, puis analyse

• La technologie existe (exomes & genomes) • Des logiciels sont développés activement pour annoter, trier, classifier les variants
UN GENOME : 3 Milliards 200 millions de paires de bases
EXOME : 1,5 % DU GENOME (23 000 Gènes)
48 MILLIONS DE PAIRES DE BASES
VARIATIONS INTER-INDIVIDUELLES APPELEES POLYMORPHISMES
3 millions par Génome
QU’EST CE QUE L’ON ATTEND POUR L’EXOME EN ROUTINE ?
panel exome génome
…AGCTTTAGGCTAGGGCCCCTTAGTATATATATTATATATGGGTGTGTGTAAGTCCCCAAA….
…AGCTTTAGGCTAGGGCCCCTTAGTATATATA/GTTATATATGGGTGTGTGTAAGTCCCCAAA….
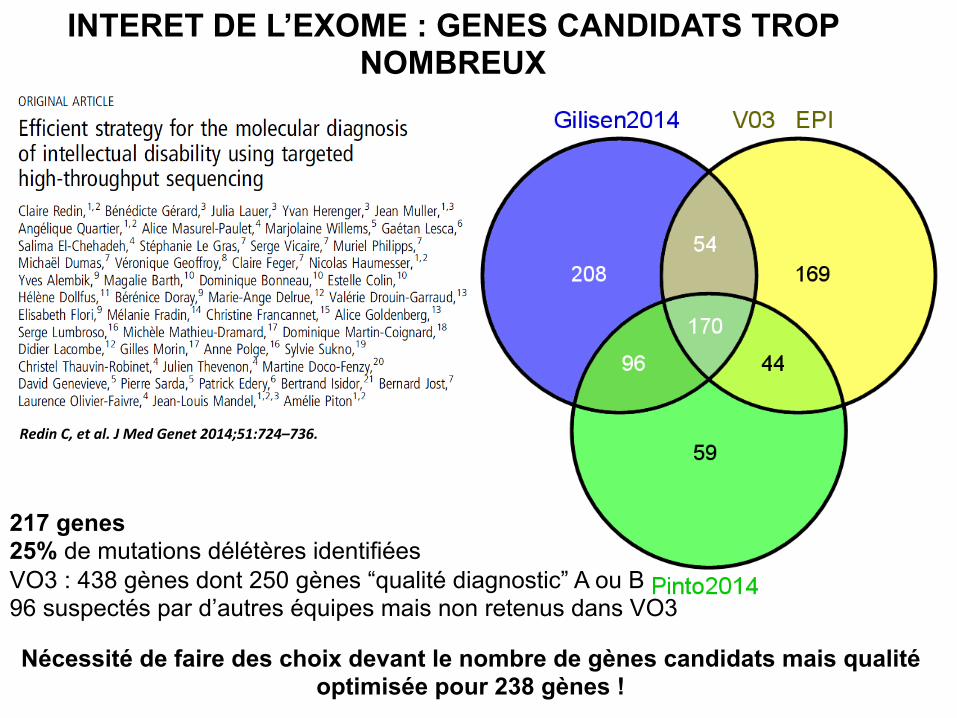
INTERET DE L’EXOME : GENES CANDIDATS TROP NOMBREUX
217 genes 25% de mutations délétères identifiées VO3 : 438 gènes dont 250 gènes “qualité diagnostic” A ou B 96 suspectés par d’autres équipes mais non retenus dans VO3
Redin C, et al. J Med Genet 2014;51:724–736.
Nécessité de faire des choix devant le nombre de gènes candidats mais qualité optimisée pour 238 gènes !
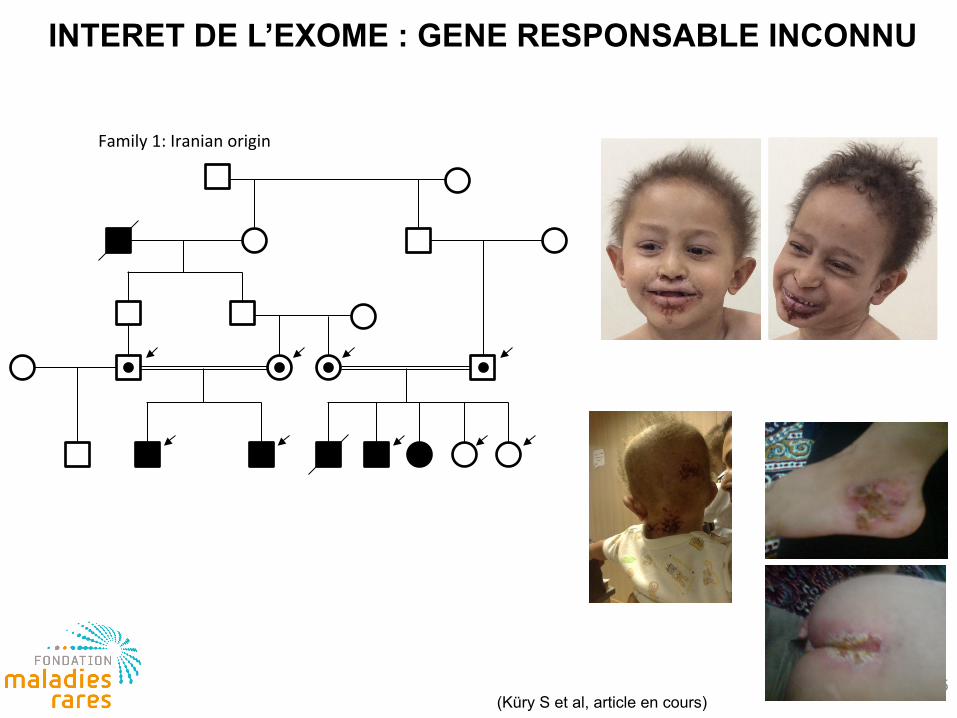
5
Family 1: Iranian origin
(Küry S et al, article en cours)
INTERET DE L’EXOME : GENE RESPONSABLE INCONNU

Séquençage Haut-Débit: 2 technologies (aujourd’hui)
Illumina MiSeq, NextSeq and HiSeq series: séquençage par synthèse, lectures 36- 250 pb 2 à 11 jours de run env.; plusieurs déclinaisons de débit: 1Gb > 3 Tb et plus
Life Ion Torrent technology: PGM and Proton Séquençage par semi-conducteur, lectures 100-400pb, ->1 Gb/run, 3h de run (PGM) ; exome (Proton) Le taux d’erreur élevé de cette technologie peut être traité en constitutionnel (on attend des hétérozygotes donc un taux de mutation de 50%) mais plus délicat en somatique.
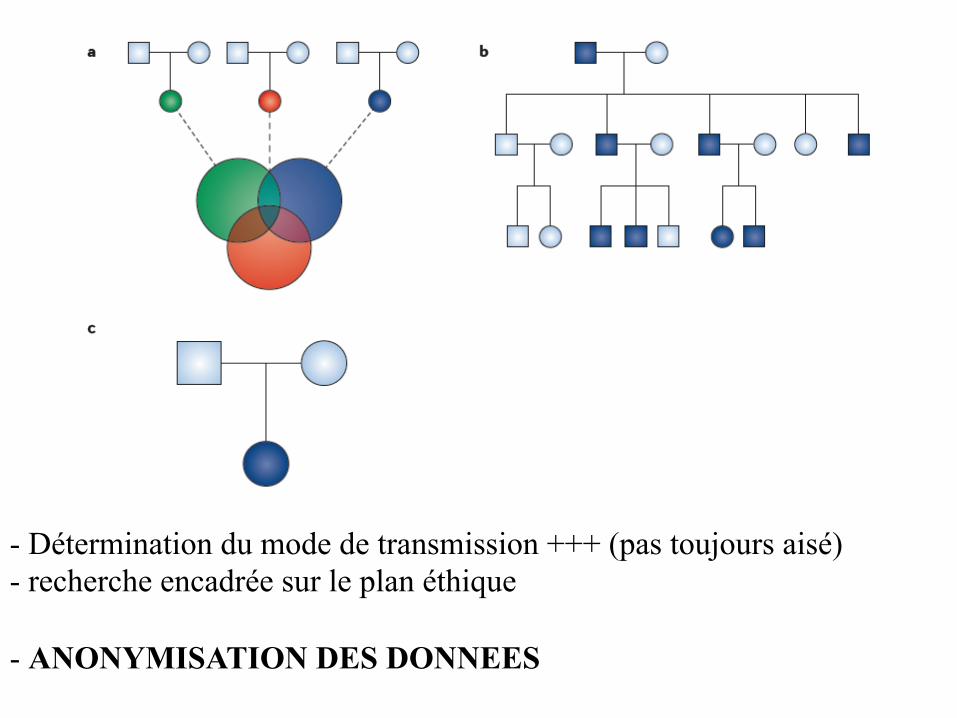
- Détermination du mode de transmission +++ (pas toujours aisé) - recherche encadrée sur le plan éthique - ANONYMISATION DES DONNEES
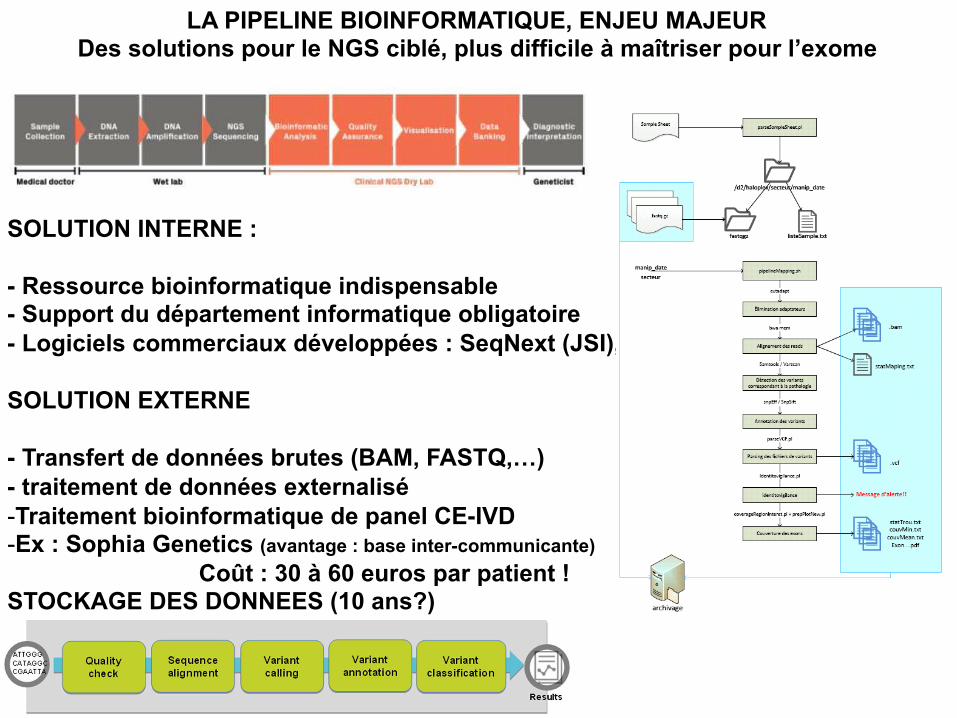
LA PIPELINE BIOINFORMATIQUE, ENJEU MAJEUR Des solutions pour le NGS ciblé, plus difficile à maîtriser pour l’exome
SOLUTION INTERNE : - Ressource bioinformatique indispensable - Support du département informatique obligatoire - Logiciels commerciaux développées : SeqNext (JSI), Sure Call SOLUTION EXTERNE - Transfert de données brutes (BAM, FASTQ,…) - traitement de données externalisé - Traitement bioinformatique de panel CE-IVD - Ex : Sophia Genetics (avantage : base inter-communicante)
Coût : 30 à 60 euros par patient ! STOCKAGE DES DONNEES (10 ans?)
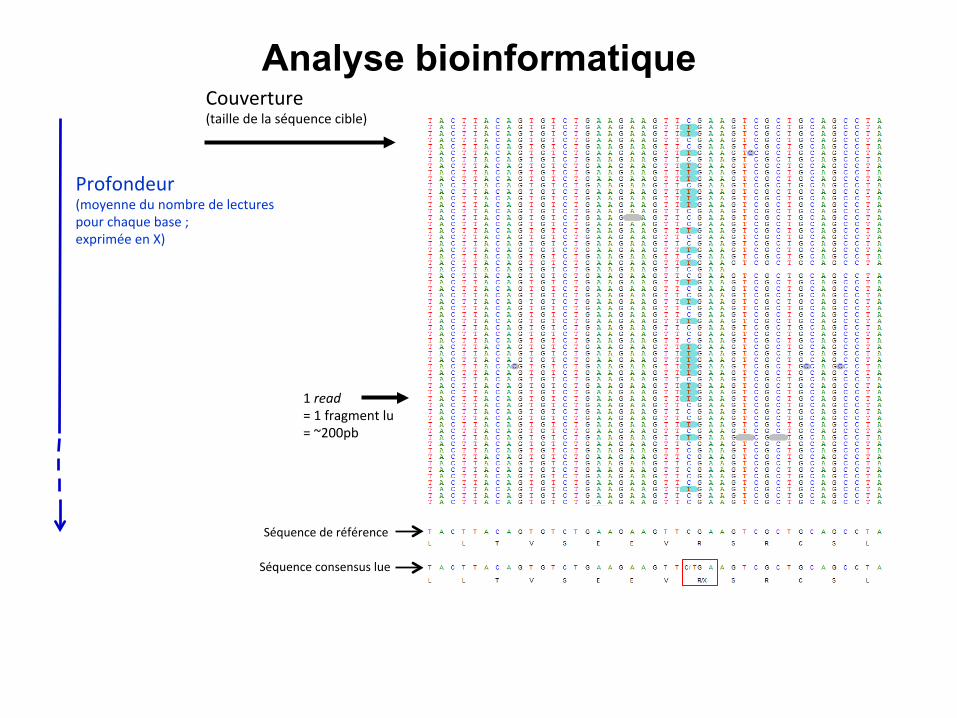
Séquence de référence
1 read = 1 fragment lu = ~200pb
Séquence consensus lue
Couverture (taille de la séquence cible)
Profondeur (moyenne du nombre de lectures pour chaque base ; exprimée en X)
Analyse bioinformatique

10
NGS ciblé
Mutation SLC39A4 c.192+19G>A
Validation Sanger indispensable
en exome ou en ciblé
Notion indispensable en NGS : couverture et profondeur de couverture (ex : 50 X)
Seuil diagnostic minimum 30 X Exome 50X, 100 X de moyenne

Exemple de défaut de couverture : MLH1 exon 16
Séquençage Sanger obligatoire car région non couverte
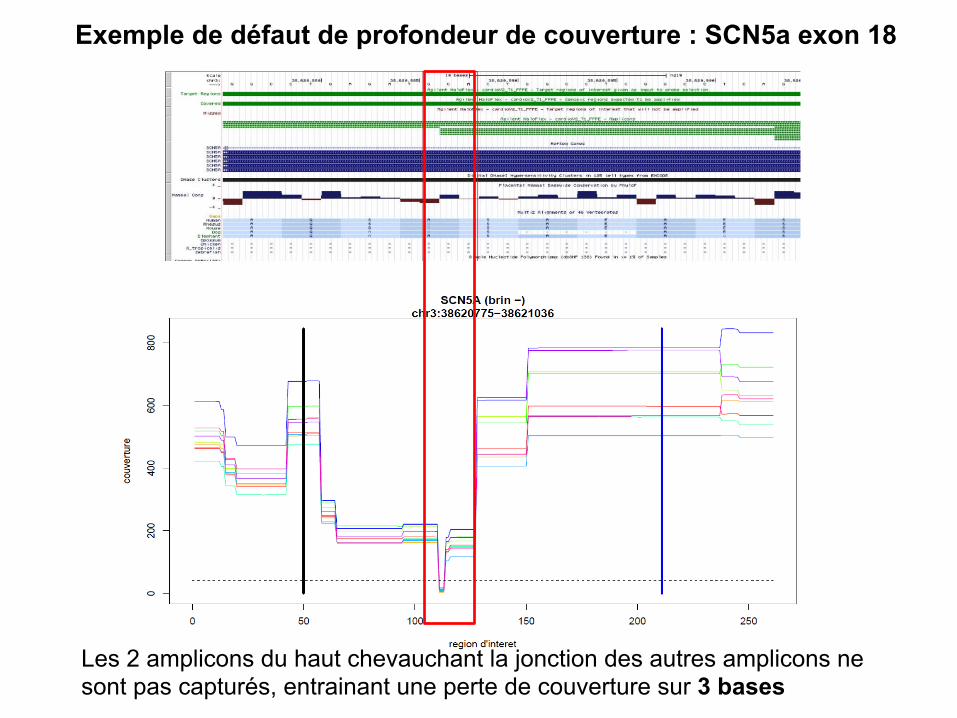
Les 2 amplicons du haut chevauchant la jonction des autres amplicons ne sont pas capturés, entrainant une perte de couverture sur 3 bases
Exemple de défaut de profondeur de couverture : SCN5a exon 18
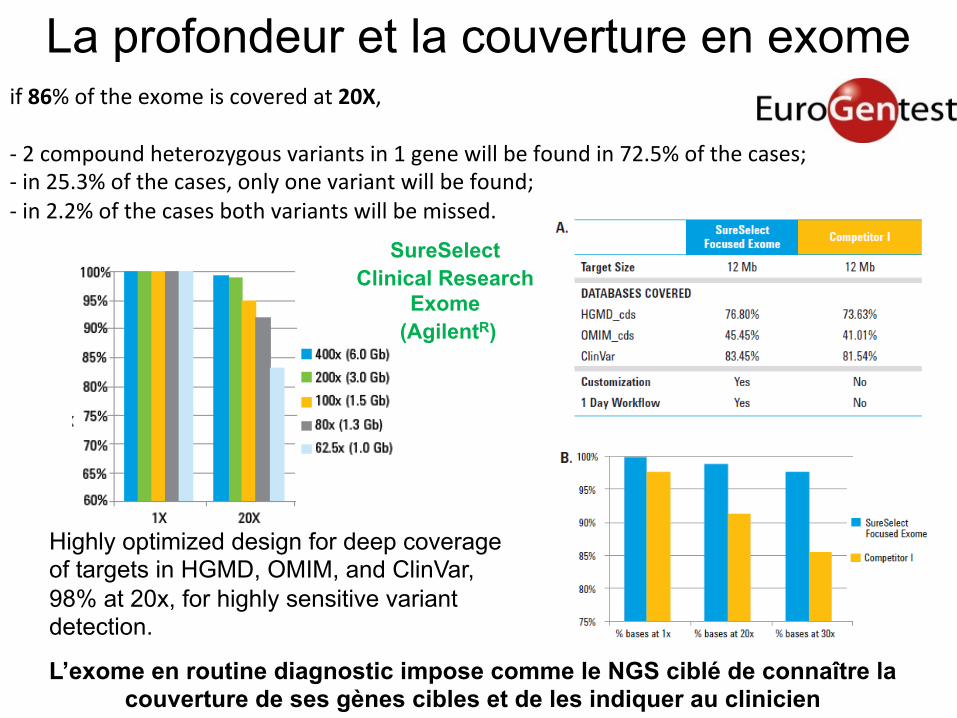
La profondeur et la couverture en exome if 86% of the exome is covered at 20X, -‐ 2 compound heterozygous variants in 1 gene will be found in 72.5% of the cases; -‐ in 25.3% of the cases, only one variant will be found; -‐ in 2.2% of the cases both variants will be missed.
Highly optimized design for deep coverage of targets in HGMD, OMIM, and ClinVar, 98% at 20x, for highly sensitive variant detection. L’exome en routine diagnostic impose comme le NGS ciblé de connaître la
couverture de ses gènes cibles et de les indiquer au clinicien
SureSelect
Clinical Research Exome
(AgilentR)

DDDCCC?<CCCCDE>DACD;DBDCDC>;C>D?@C9C7@CCDACCADDDAB,=C@ @IL31_4368:1:1:1001:20031/2 AGCAATTAATAGAGTGAAGAGAAAACCTACAAAATGGGAGAAAATATTTGTAAA + ?@FBEFECFCE8EC<>C@AE>CDDEEFCEEE@@+ED=CDDE4EE<@CDD+C?CC @IL31_4368:1:1:1001:7078/2 TGGGTGTGTGTGTGTTTGCGTGCGCGCTTCTGGGTGTAGTTATGTGTGTGTCTG + ??>D7DD?AD?EDECEAB@<@,7@A9'>7><BC;=A31?A.;<>>B)A708>>4 @IL31_4368:1:1:1001:11670/2 AGGATGATGACAGCAACTTTCATATGGATTTCATCGTGGCTGCATCCAACCTCC + ;EEE,FEAEFEFFFECEFCEFA>FCEEC@@ECEBCDFFAFD9E;9E@E?BD@?C @IL31_4368:1:1:1001:15110/2 GGAGTACAAGAAGGAAGGCATTGAGTGGACGTTCATTGACTTTGGGATGGACCT + FCEFCGGEGFECEEGCGCAAFEC=EAFGFEE?8BAA<C=DA=4:EE>9@6;4B> @IL31_4368:1:1:1002:6807/2 AGTCTTTGGACTTGCAAGAGAAGCCCAGCATTAAGAAAGAGACCCTCCTCAAAA +
FASTQ LES FORMATS DE FICHIER EN NGS
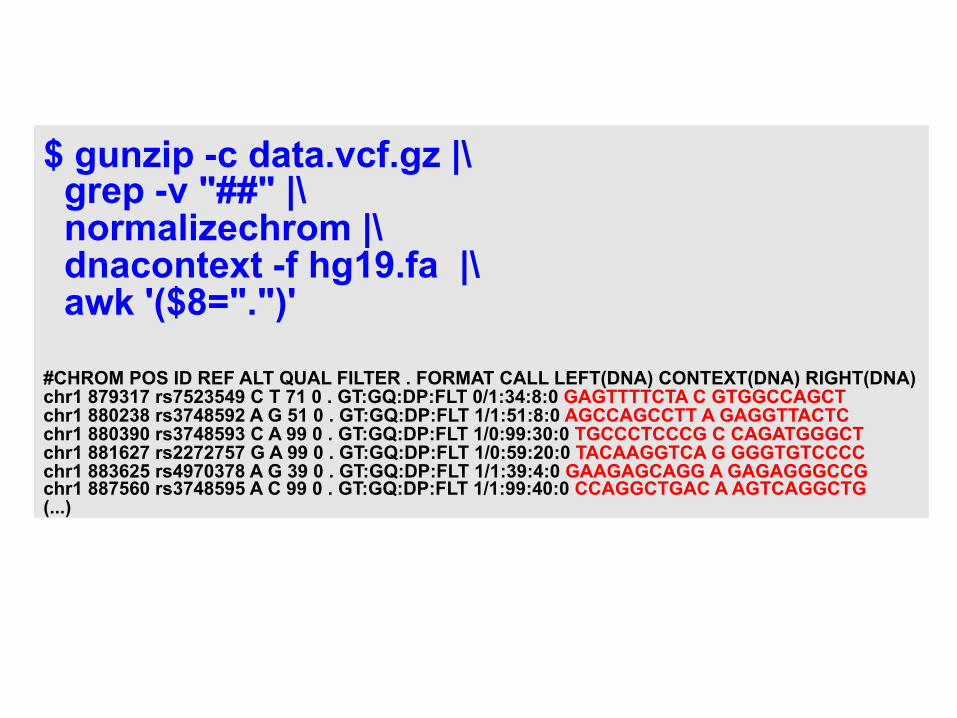
$ gunzip -c data.vcf.gz |\ grep -v "##" |\ normalizechrom |\ dnacontext -f hg19.fa |\ awk '($8=".")' #CHROM POS ID REF ALT QUAL FILTER . FORMAT CALL LEFT(DNA) CONTEXT(DNA) RIGHT(DNA) chr1 879317 rs7523549 C T 71 0 . GT:GQ:DP:FLT 0/1:34:8:0 GAGTTTTCTA C GTGGCCAGCT chr1 880238 rs3748592 A G 51 0 . GT:GQ:DP:FLT 1/1:51:8:0 AGCCAGCCTT A GAGGTTACTC chr1 880390 rs3748593 C A 99 0 . GT:GQ:DP:FLT 1/0:99:30:0 TGCCCTCCCG C CAGATGGGCT chr1 881627 rs2272757 G A 99 0 . GT:GQ:DP:FLT 1/0:59:20:0 TACAAGGTCA G GGGTGTCCCC chr1 883625 rs4970378 A G 39 0 . GT:GQ:DP:FLT 1/1:39:4:0 GAAGAGCAGG A GAGAGGGCCG chr1 887560 rs3748595 A C 99 0 . GT:GQ:DP:FLT 1/1:99:40:0 CCAGGCTGAC A AGTCAGGCTG (...)
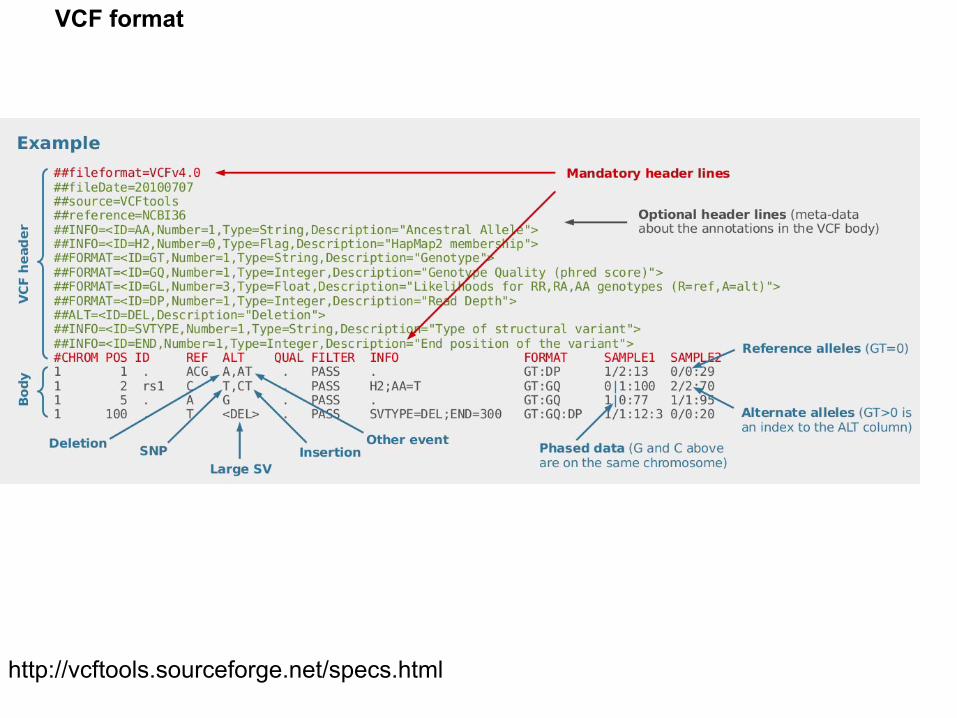
http://vcftools.sourceforge.net/specs.html
VCF format

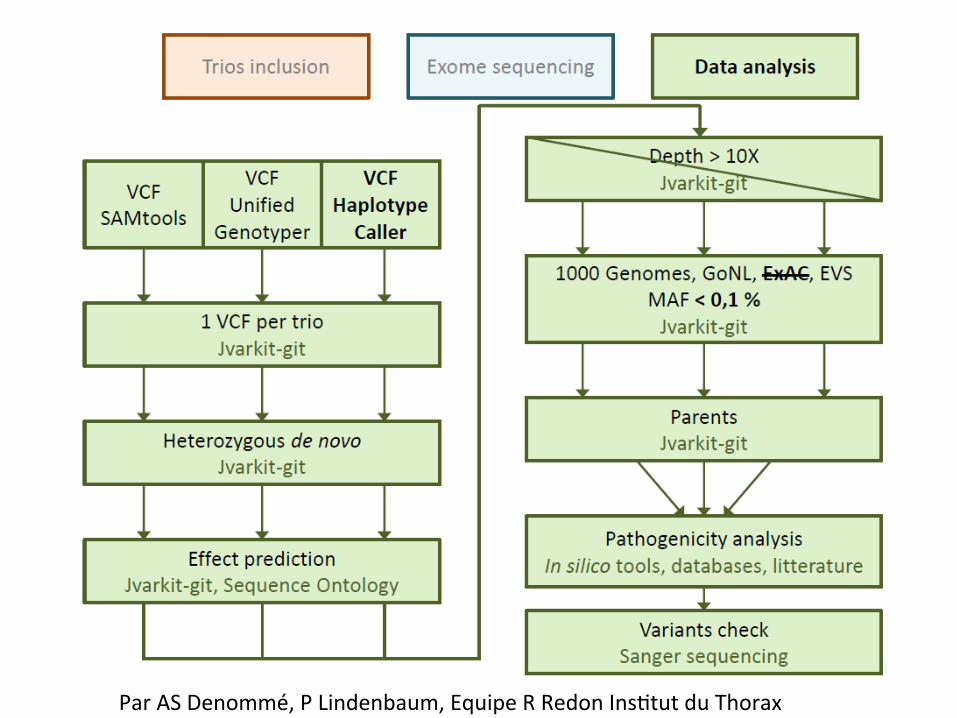
Par AS Denommé, P Lindenbaum, Equipe R Redon Ins0tut du Thorax
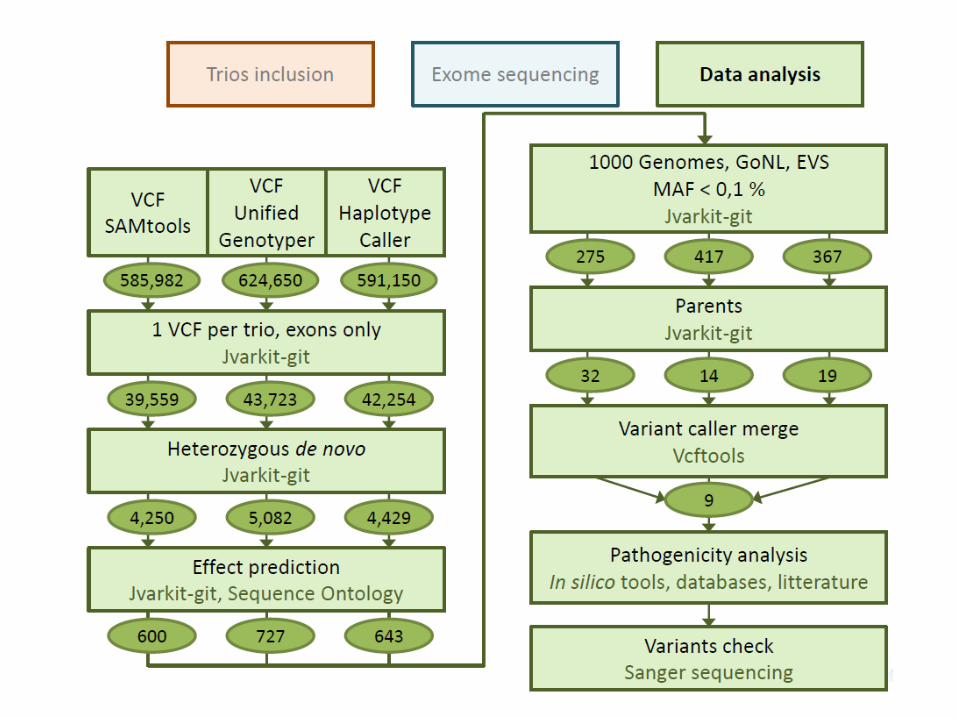


Contrôle qualité : 24 laboratoires européens participants
Test du pipeline bioinformatique seul : 60 % de concordance parfaite Dr Simon Patton, EMQN, 7 novembre 2014
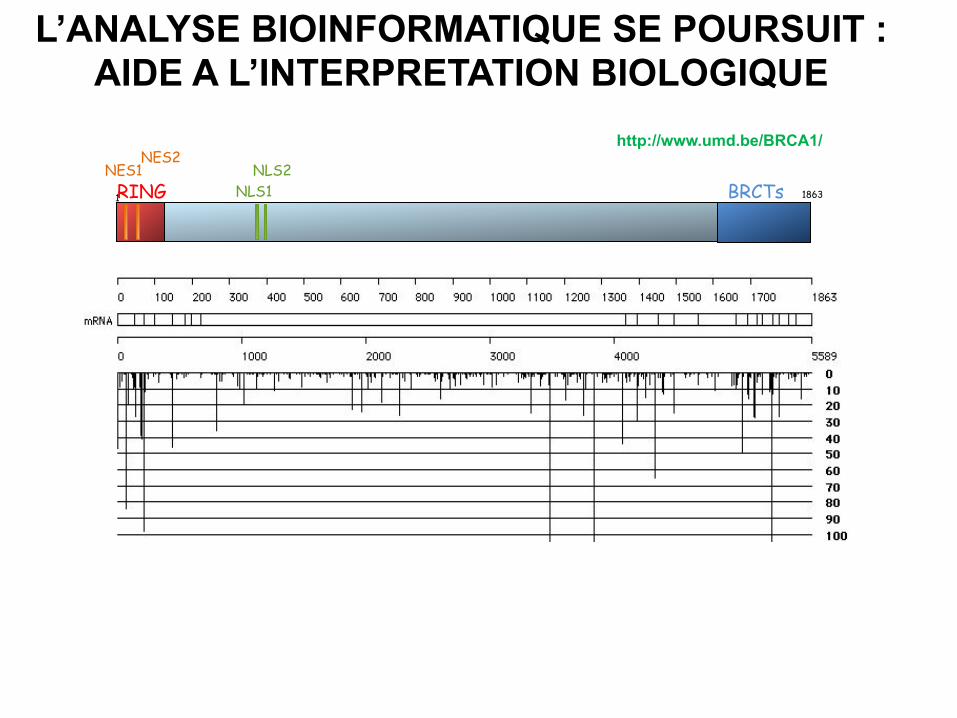
L’ANALYSE BIOINFORMATIQUE SE POURSUIT : AIDE A L’INTERPRETATION BIOLOGIQUE
RING BRCTs NES1
NES2
NLS1 NLS2
1 1863
http://www.umd.be/BRCA1/
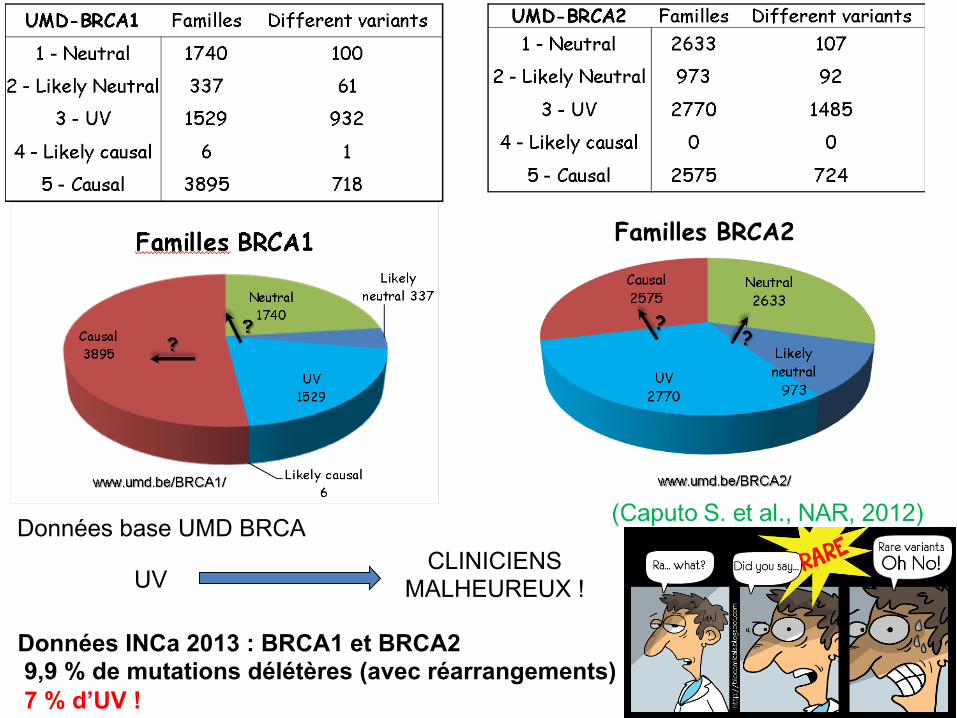
CLINICIENS MALHEUREUX ! UV
Données base UMD BRCA
Données INCa 2013 : BRCA1 et BRCA2 9,9 % de mutations délétères (avec réarrangements) 7 % d’UV !

A l’échelle de l’exome, more…..
Exome
40 639 variants
80 000 avec les artefacts
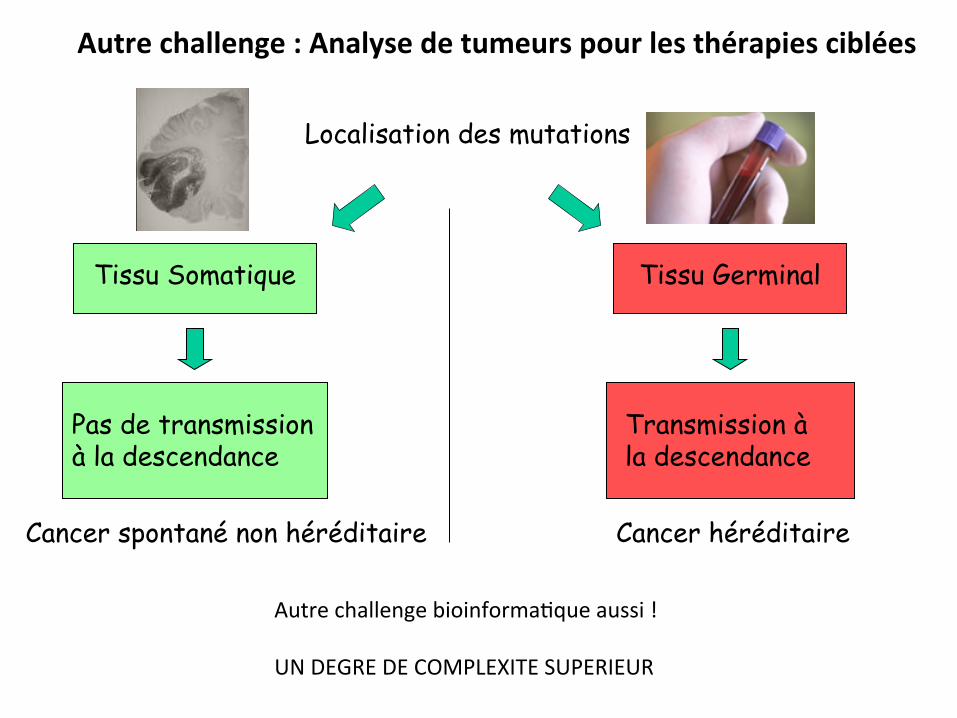
Localisation des mutations
Tissu Somatique Tissu Germinal
Pas de transmission à la descendance
Transmission à la descendance
Cancer héréditaire Cancer spontané non héréditaire
Autre challenge : Analyse de tumeurs pour les thérapies ciblées
Autre challenge bioinforma0que aussi ! UN DEGRE DE COMPLEXITE SUPERIEUR
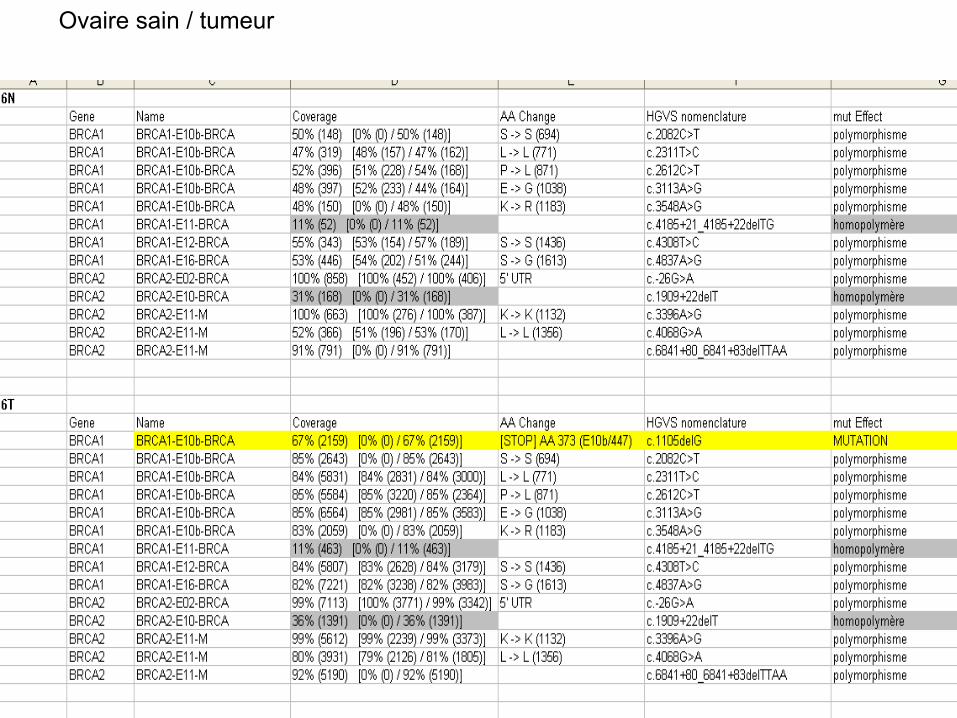
Ovaire sain / tumeur
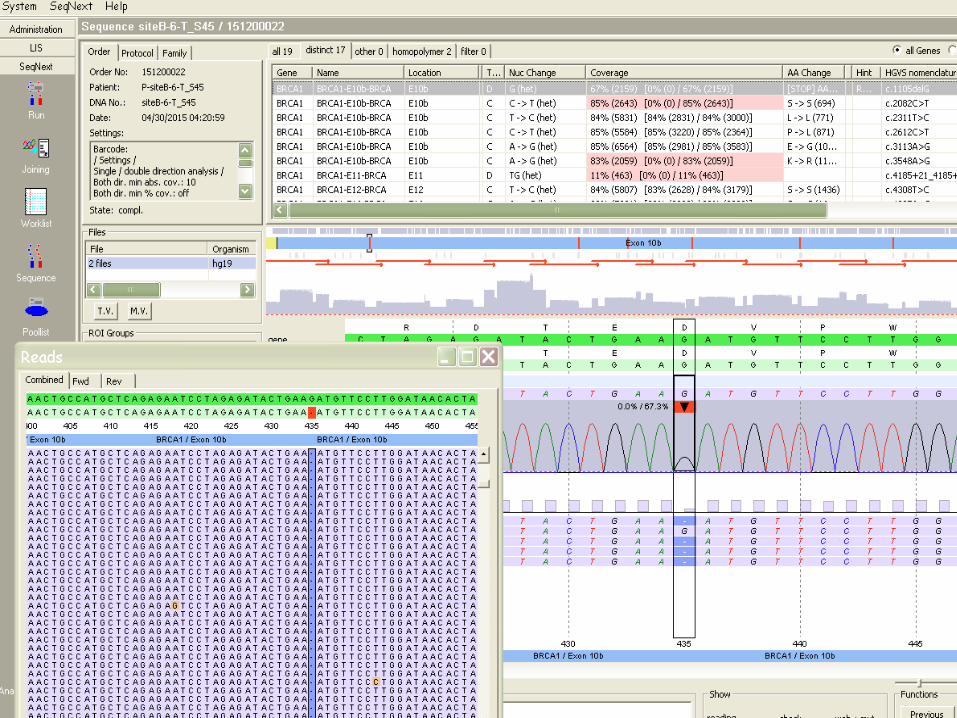
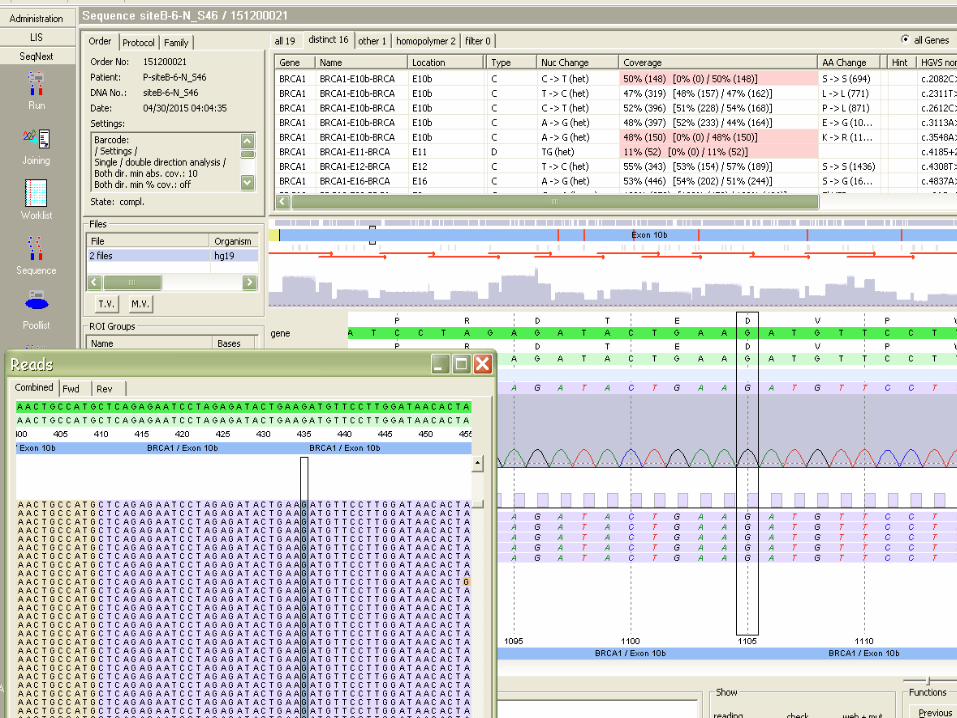

APRES L’EXOME, LE GENOME ! -‐ INTERPRETATION PLUS COMPLEXE
-‐ MAIS AVANTAGE TECHNOLOGIQUE : PAS DE CAPTURE DES EXONS
-‐ AUTRE VARIATIONS VISIBLES : COPY NUMBER VARIATION (CNV)
-‐ INTERPRETATION DES VARIATIONS HORS DES SEQUENECS CODANTES !
Extrait de « comprendre la génétique et ses enjeux » par G. Matthijs et J. Vermeesch, avec l’aimable autorisation des auteurs





























