Embed Size (px)
Citation preview

144 Biochimica et Biophysica Acta, 997 (1989) 144-153 Elsevier
BBAPRO 33407
H-Nuclear magnetic resonance studies of the neuropeptide head activator *
Rainer Saffrich 1, Hans Robert Kalbitzer 1, Heinz Bodenmfiller 2, Peter Muhn ' , Rfideger Pipkorn '~ and H. Chica Schaller 2
l Max.Planck-lnstitutefor Medical Research, Department of Biophysics, Heidelberg, and 2 Center of Molecular Biology, Heidelberg (ER. (7.)
(Received 5 January 1989)
Key words: NMR, 1H-; Neuropeptide head activator; Nuclear Overhauser effect
The I H-NMR spectrum of the neuropeptide head activator in aqueous solution has been completely assigned by two.dimensional NMR spectroscopy and selective deuteration. The apparent pseudo-first-order exchange rate, kex, of the backbone amide protons and the correspondent activation enthalpies, AH*, were determined. The exchange rates decrease and the activation enthalpies increase from the N-terminal to the C-terminal part of the peptide. The exchange rates vary from 21 to 0.3 s -I at 274 K, the activation enthalpies from 60 to 75 kJ. tool - l . The p£ values of the terminal carboxyl group and of the lyslne amino group have been estimated as 3.3 and 10.3, respectively. The NMR results are in line with a dimeric structure ;n an antisymmetric arrangement of the subunits, forming an antiparallel D-pleated sheet between C-terminal segments. The peptide bonds between pGlu-l, Pro-2 and Pro-3 are predominantly in tram-configuration, in fact no c/s-isomers can be observed spectroscopically. The structure appears to be very stable; in the temperature and pH range studied, i.e., from 274 to 338 K and from pH 0.8 to pH 11.6, there are no spectroscopic indications [or a global structural change.
Introduction
The neuropeptide head activator with the sequence Glp-Pro-Pro-Gly-Gly-Ser-Lys-Val-Ile-Leu-Phe [1] was first isolated from the freshwater coelenterate Hydra attenuata [2], where it initiates head-specific growth and differentiation processes (for a review see ReL 3). Later- on, the head activator was found in tissues from higher organisms, e,g,, in rat intestine, and in bovine or human hypothalamus [4].
The biological activity of synthetic head activator as well as head activator isolated from biological sources appears to be related to its concentration and its purity.
* Presented at the International Conference on Magnetic Resonance in Biological Systems, Madison, 1988.
Abbreviations: DQF-COSY, double quantum filtered correlated spec- troscopy; NMR, nuclear magnetic resonance; NOE, nuclear Over- hauser effect: NOESY, nuclear Overhauser enhancement spec- troscopy; RCT, relayed coherence transfer spectroscopy; ROESY, rotating-frame Overhauser spectroscopy; DSS, 4,4-dimethyl-4-sila- pentane-l-sulfonic acid.
Correspondence: H.R. Kalbitzer, Department of Biophysics, Max- Pluck-Institute for Medical Research, Heidelberg, D-6900, F.R.G.
Highly purified and concentrated head activator shows only minor activity in the biological assay. Circular dichroism and Raman spectroscopy as well as molecular sieve chromatography suggest that the deactivation is caused by a dimerization of the neuropeptide which may be related to a physiological mecha~lism of regu- lation [5]. Physiologically, monomeric (active) neuro- peptide is released from its carrier molecule [6]; mono- meric head activator can also be obtained from the dimer by treatment with concentrated solutions (1 M) of salts such as ammonium sulfate at neutral pH or with moderate concentrations (100 mM) at very low pH [5].
It is commonly accepted that the biological activity of proteins is closely related to their structure in solu- tion. It is an open question whether the same is true for small linear peptides such as head activator, since it is not clear, whether they can be described adequately by one or only a few well-defined conformations or if they occur essentially in random-coil form. Moreover, these conformations may depend on various external factors such as temperature and pH of the solution. Nuclear magnetic resonance (NMR) is certainly one of the best methods available for obtaining such information at atomic resolution. In the present paper we report the results of one- and two-dimensional N M R studies on this topic.
0167-4838/89/$03.50 © 1989 Elsevier Science Publishers B.V. (Biomedical Division)

Materials and Me/hods
Materials Glycine was perdeuterated catalytically from the
non-deuterated amino acid, according to the following procedure. PtO 2 (0.5 g) was prereduced in 30 ml 99.870 D20 with deuterium gas. Glycine (3 g) was dissolved in 50 ml 99.870 D20 and was evaporated to dryness. The product was dissolved again in 30 ml D20 and the reduced platinum catalyst was added. The mixture was reacted at 423 K in a high-pressure vessel for 5 days. The product was separated from the catalyst and re- crystalized from H20. The deuteration of the product (yield approx. 9070) in the a-position was beaer than 9570 as shown by 1H-NMR spectroscopy.
Selectively deuterated head activator was synthesized by Fmoc solid-phase synthesis, non-deuterated head activator was obtained from Bachem (Bubendorf, Switzerland).
NMR-spectroscopy The NMR-samples were prepared by dissolving the
freeze-dried neuropeptide in an appropriate volume of D20 (99.8570) or of normal water with 5 to 1070 D20 added to an end concentration of approximately I mM. The pH was varied by adding an appropriate amount of DCI or KOH to the sample. The pH was measured by a combination glass electrode from Ingold (Frankfurt, F.R.G.) and was not corrected for isotope effects.
The pK-values and chemical shifts, 81 and 82, were obtained by a non-linear three-parameter fit of the experimental data to a modified Henderson-Hassel- balch equation (see e.g., Ref. 7). The errors given corre- spond to a confidence level of 9570.
The ~H-NMR spectra were recorded with a Bruker HX-360 spectrometer operating at 360 MHz, and a Bruker AM-500 spectrometer operating at 500 MHz.
In one-dimensional spectroscopy, the water signal was either suppressed by selective presaturation for typically 0.8 s or it was not excited by using a 1331 pulse [81.
Phase-sensitive DQF-COSY spectra were recorded using the pulse sequences [90~°-ti-90~°-~'-90,-t2]. [9]. Homonuclear RCT spectra were obtained with the pulse sequence [90°-t l-90°-, /2-180°-, /2-90°-t2], [10] using the phase cycling described by Bax and Drobny [11] and a mixing time, ~-, of 40 ms. Phase-sensitive z-filtered ROESY spectra were recorded with the pulse sequence [90x-tt-90x-1--CW-~--90~],, with the ap- propriate phase cycling as described by Rance [12]. The HDO resonance was suppressed by selective irradiation for all times, except acquisition. Peak recognition and pattern search was performed with the program NOPP described earlier [13].
All chemical shift values are referred to 4,4-dimethyl- 4-silapentane-l-sulfol~c acid (DSS) as internal stan-
145
dard. If necessary spectra were simulated using the program PANIC obtained from Bruker (Karlsruhe, F.R.G.).
The exchange rates, kex, of the amide protons were estimated with the relation [14]:
~r/~Pl/2 = 1/72 + kex (1)
which holds for moderately slow exchange (Av: actual linewidth, T2: transverse relaxation time in the absence of exchange). Eqn. 1 applies also in the case of weak coupling for individual transitions, as can be shown by using the density matrix formalism (see e.g., Ref. 15).
The activation enthalpy, a l l * , has been calculated by a non-linear three-parameter fit of the observed relaxation rate 1/T 2" on AH*, on the activation ent- ropy, AS*, and on the phenomenological factor, A, to a modified Eyring equation:
1/T; = kT /h exp( - A H*/ ( RT) + A S * / R ) + A / T e x p ( b / T ) (2)
where k is the Boltzmann constant, h the Planck con- stant, T the absolute temperature, R the gas constant, and b an exponent describing the temperature depen- dence of the viscosity of water (b = 2074.4 K). The first term in Eqn. 2 describes the temperature dependence of kex according to the Eyring relation, the second term approximates the temperature dependence of 1/T a in the absence of exchange. It can be obtained using the following relations:
= a exp (b /T ) (3)
*R = v'~i/( kr) (4)
i /r2 = c,R (5)
Eqn. (3) describes the temperature dependence of the viscosity, 7, of liquids with a and b material constants, and Eqn. 4 describes the dependence of the rotational correlation time, ~'R, on the effective volume V' of the molecule, on the viscosity ,/, and on the temperature T (Stokes-Einstein relation). The general functional de- pendence of Eqns. 3 and 4 is well established. Outside the fast tumbling domain, the proportionality in Eqn. 5 holds only in first approximation, but it is appropriate in our context, i.e., for the use as merely a small correction term.
Results
Assignments to spin systems The 1H-NMR spectrum of the head activator is well
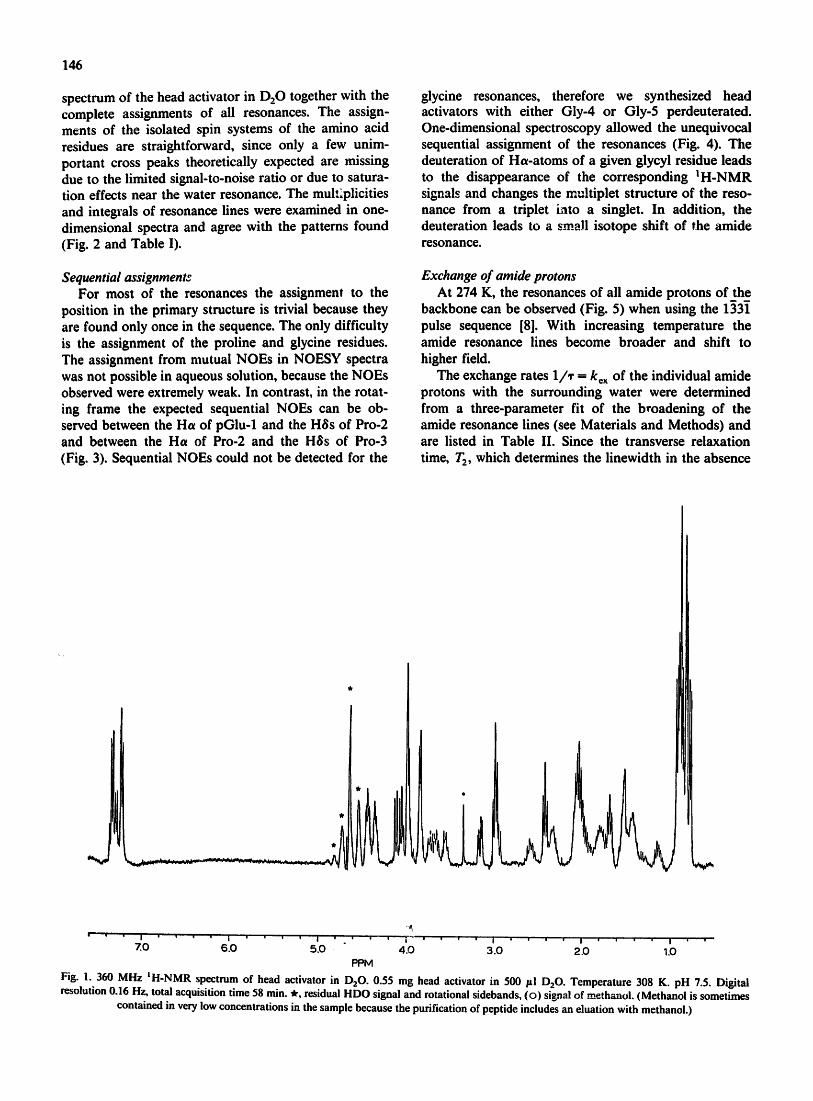
resolved and shows no obvious signs of structural het- erogeneity in the sample (Fig. 1).
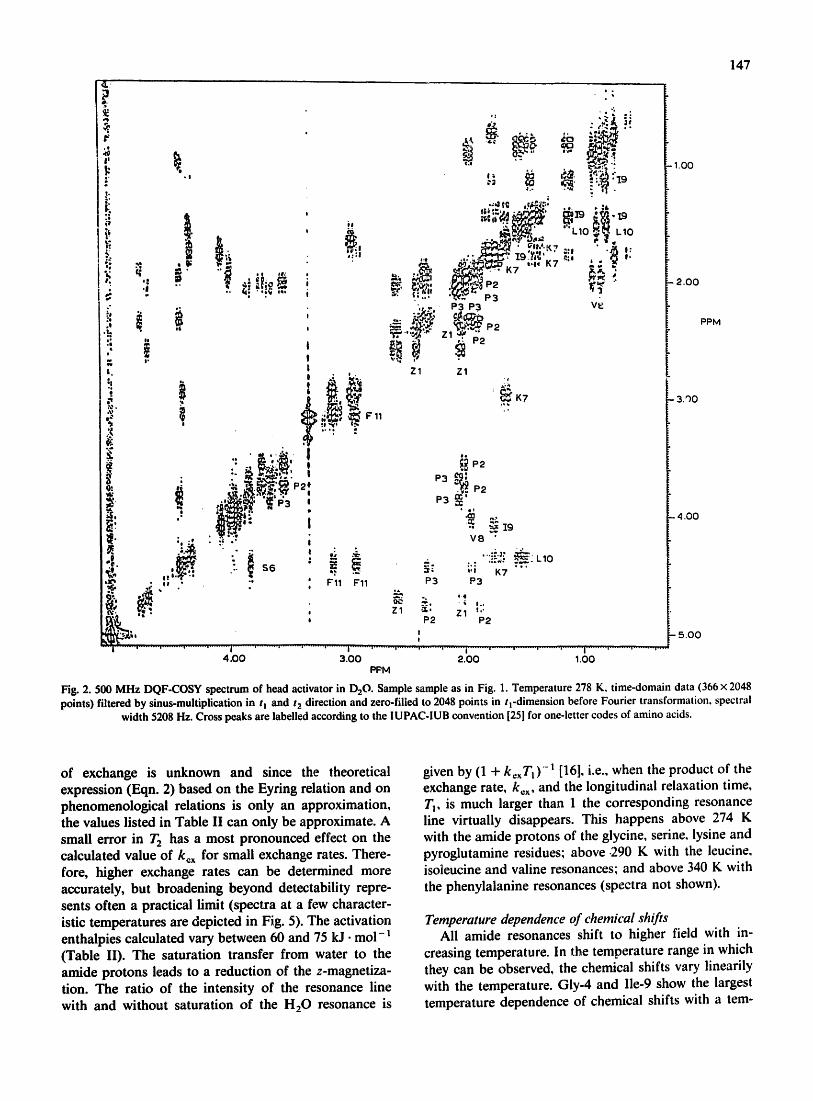
The assignment to spin systems was done from the DQF-COSY- and RCT-spectra in D20 and H20. Fig. 2 shows a phase-sensitive double quantum filtered COSY
146
spectrum of the head activator in D20 together with the complete assignments of all resonances. The assign- ments of the isolated spin systems of the amino acid residues are straightforward, since only a few unim- portant cross peaks theoretically expected are missing due to the limited signal-to-noise ratio or due to satura- tion effects near the water resonance. The mul,~plicities and integrals of resonance fines were examined in one- dimensional spectra and agree with the patterns found (Fig. 2 and Table I).
Sequential assignments For most of the resonances the assignment to the
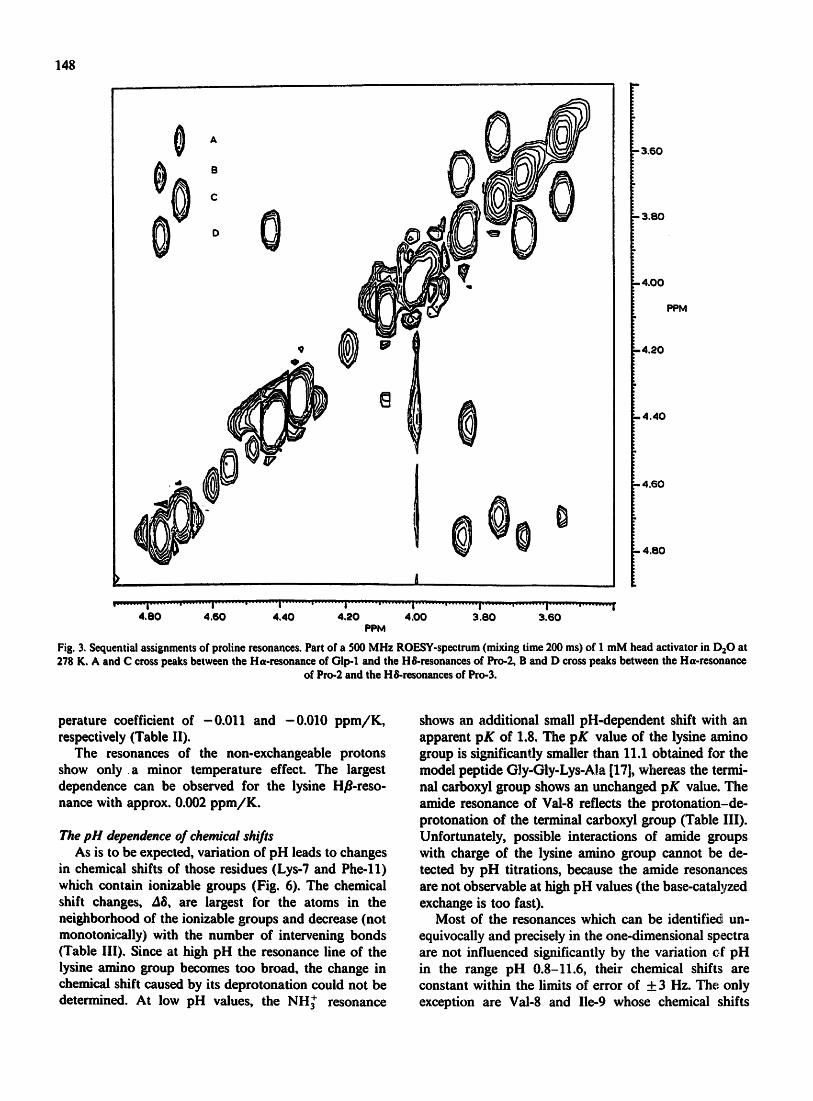
position in the primary structure is trivial because they are found only once in the sequence. The only difficulty is the assignment of the proline and glycine residues. The assignment from mutual NOEs in NOESY spectra was not possible in aqueous solution, because the NOEs observed were extremely weak. In contrast, in the rotat- ing frame the expected sequential NOEs can be ob- served between the Ha of pGlu-1 and the HSs of Pro-2 and between the Ha of Pro-2 and the HSs of Pro-3 (Fig. 3). Sequential NOEs could not be detected for the
glycine resonances, therefore we synthesized head activators with either Gly-4 or Gly-5 perdeuterated. One-dimensional spectroscopy allowed the unequivocal sequential assignment of the resonances (Fig. 4). The deuteration of Ha-atoms of a given glycyl residue leads to the disappearance of the corresponding ~H-NMR signals and changes the multiplet structure of the reso- nance from a triplet iato a singlet. In addition, the deuteration leads to a sm~.ll isotope shift of the amide resonance.
Exchange of amide protons At 274 K, the resonances of all amide protons of the
e
backbone can be observed (Fig. 5) when using the 1331 pulse sequence [8]. With increasing temperature the amide resonance lines become broader and shift to higher field.
The exchange rates 1/1- - kex of the individual amide protons with the surrounding water were determined from a three-parameter fit of the broadening of the amide resonance lines (see Materials and Methods) and are listed in Table II. Since the transverse relaxation time, T 2, which determines the linewidth in the absence
.A
I
" - - " " I . . . . I ' ' ' ' i ' - . ' ' ' | - - " ' ' ' ' I ' ' ' ' I ' ' ' ' I ' '
7.0 6.0 5.0 4.0 3.0 2.0 1.0 PPM
Fig. 1. 360 MHz IH-NMR spectrum of head activator in 1:)20. 0.55 mg head activator in 500 pl D20. Temperature 308 K. pH 7.5. Digital resolution 0.16 Hz, total acquisition time 58 rain. ~, residual HDO signal and rotational sidebands, (o) signal of methanol. (Methanol is sometimes
contained in very low concentrations in the sample because the purification of peptide includes an eluation with methanol.)
147
[ 4."
~,~ , , o_Nb -:h =; .
' ;,~:~1 :. 1.oo ,~ ~ ~:'.-~.~..~
~. ".- ~ 0]-
o ~;' ;= . . . . " °' , :a,
{~. , "" ~ ' " L I O L IO
' ..... :.~a:9~N' ~., , ,~,, *: -" -~ .~ . ., . ~ ,.
,; I t ,_ 2 . 0 0 • " . . i~ i ~.~ , ; . . . .
" '" . P3 !:'" ~. ~. , N~' ~ '¢~.~'~'' ~3 ~3 ,,~ .I o ~ P2 PPM
.t. E~." " " ~ ' " Z l
~' i ' ~ .~ ': t ~ / t" ,o
| Z l Z l ' '
!~ : ~ : ~ ~ - ~,,o ~" ~. ~ . ..
* I ': i~__ ~2 : - -
' . : - 4 .00
I I . " ; v s " ;o :e . : ! . - . " . ,
":'" " ~ ' ":' "~ '":';'J: :~,.~'L10
~ ~ , % .e~ ' "" ' F l l F l l P3 P3
, Z l ~ ' Z l t,. o P2 P2
t 5 . 0 0
. . . . I . . . . . . ' . . . . . . . ' . . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . t . . . . . . . . ' . . . . . . . . ' . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . i . . . . . . . . . ' . . . . . . . . . , ......... , ......... , ......... i . . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . I . . . . . . . . . ' . . . . . . . . . ' . . . . . . . . . ' " "
4 . 0 0 3 .00 2 .00 1.00 PPM
Fig. 2. 500 MHz DQF-COSY spectrum of head activator in D20. Sample sample as in Fig. 1. Temperature 278 K, t ime-domain data (366 x 2048 points) filtered by sinus-multiplication in tn and t~ direction and zero-filled to 2048 points in q-dimension before Fourier transformation, spectral
width 5208 Hz. Cross peaks are labelled according to the IUPAC-IUB convention [25] for one-letter codes of amino acids.
of exchange is unknown and since the theoretical expression (Eqn. 2) based on the Eyring relation and on phenomenological relations is only an approximation, the values listed in Table II can only be approximate. A small error in T 2 has a most pronounced effect on the calculated value of kc~ for small exchange rates. There- fore, higher exchange rates can be determined more accurately, but broadening beyond detectability repre- sents often a practical limit (spectra at a few character- istic temperatures are depicted in Fig. 5). The activation enthalpies calculated vary between 60 and 75 kJ. mol-n (Table II). The saturation transfer from water to the amide protons leads to a reduction of the z-magnetiza- tion. The ratio of the intensity of the resonance line with and without saturation of the H20 resonance is
given by (1 + k~Tt)-1 [16], i.e., when the product of the exchange rate, k~, and the longitudinal relaxation time, 7"1, is much larger than 1 the corresponding resonance line virtually disappears. This happens above 274 K with the amide protons of the glycine, serine, lysine and pyroglutamine residues; above .290 K with the leucine, isoleucine and valine resonances; and above 340 K with the phenylalanine resonances (spectra not shown).
Temperature dependence of chemical shifts All amide resonances shift to higher field with in-
creasing temperature. In the temperature range in which they can be observed, the chemical shifts vary linearily with the temperature. Gly-4 and lie-9 show the largest temperature dependence of chemical shifts with a tem-
148
A
B
0 ° 0
O
¢
0
000 0
" 3 . 6 0
3 . 8 0
- 4 . 0 0
P P M
- 4 . 2 0
• 4 . 4 0
• 4 . 6 0
• 4 . 6 0
" " . . . . . . . . i . . . . . . . . . . . . . . . . . . . i . . . . . . . . . " . . . . . . . ~ ' | . . . . . . . . . . . " . . . . . . . . i . . . . . . . . . " . . . . . . . . . i . . . . . . . . . . . . . . . . . . . i . . . . . . . . . ' . . . . . . . . . I . . . . . . . . . ' . . . . . . . . . I 4 . 0 0 4 . 6 0 4 . 4 0 4 . 2 0 4 . 0 0 3 . 8 0 3 . 6 0
P P M
Fig. 3, Sequential assignments of proline resonances. Part of a 500 MHz ROESY-spectrum (mixing time 200 ms) of 1 mM head activator in DaO at 278 K, A and C cross peaks between the Ha-resonance of Olp-1 and the H&resonances of Pro-2, B and D cross peaks between the Ha-resonance
of Pro-2 and the H&resonances of Pro-3.
perature coefficient of -0.011 and -0.010 ppm/K, respectively (Table If).
The resonances of the non-exchangeable protons show only a minor temperature effect. The largest dependence can be observed for the iysine H~-reso- nance with approx. 0.002 ppm/K.
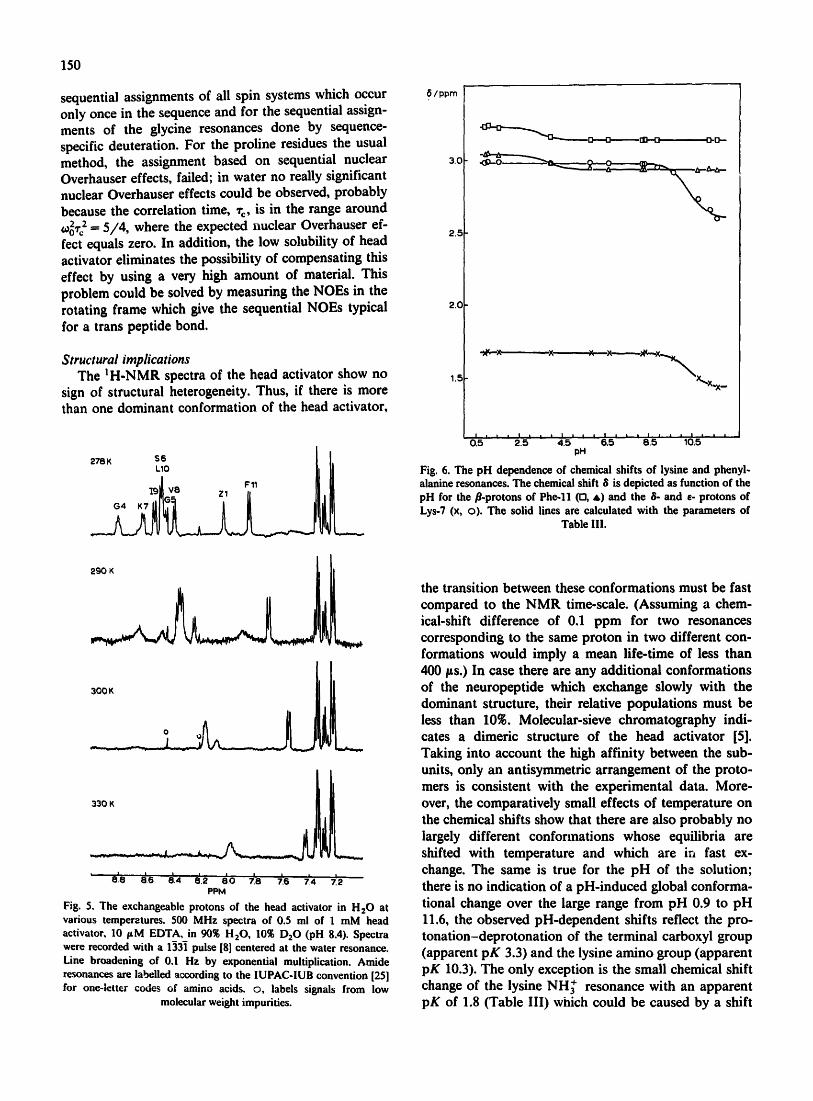
The pH dependence of chemical shifts As is to be expected, variation of pH leads to changes
in chemical shifts of those residues (Lys-7 and Phe-ll) which contain ionizable groups (Fig. 6). The chemical shift changes, A6, are largest for the atoms in the neighborhood of the ionizable groups and decrease (not monotoni~dly) with the number of intervening bonds (Table Ill). Since at high pH the resonance line of the iysine amino group becomes too broad, the change in chemical shift caused by its deprotonation could not be determined. At low pH values, the NH~ resonance
shows an additional small pH-dependent shift with an apparent pK of 1.8. The pK value of the lysine amino group is significantly smaller than 11.1 obtained for the model peptide G]y-Gly-Lys-Ala [17], whereas the termi- nal carboxyl group shows an unchanged pK value. The amide resonance of Val-8 reflects the protonation-de- protonation of the terminal carboxyl group (Table III). Unfortunately, possible interactions of amide groups with charge of the lysine amino group cannot be de- tected by pH titrations, because the amide resonances are not observable at high pH values (the base-catalyzed exchange is too fast).
Most of the resonances which can be identified un- equivocally and precisely in the one-dimensional spectra are not influenced significantly by the variation ef pH in the range pH 0.8-11.6, their chemical shifts are constant within the limits of error of + 3 Hz. The only exception are Val-8 and Ile-9 whose chemical shifts
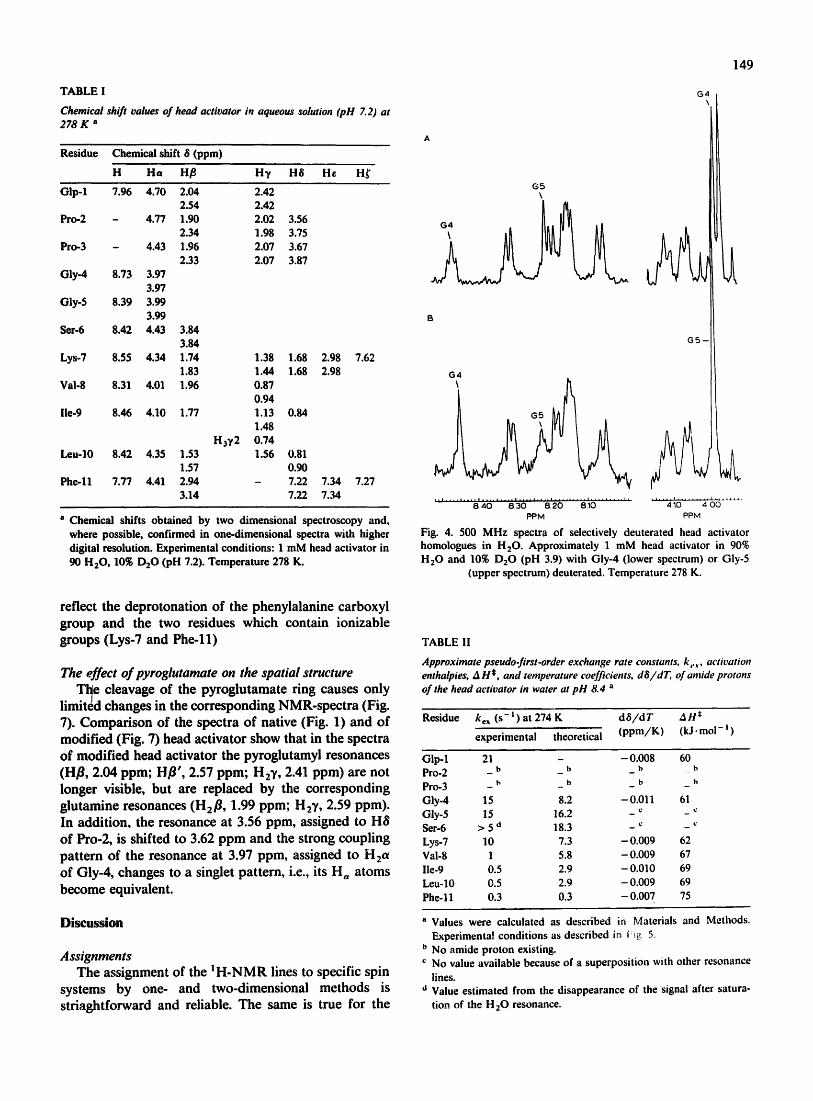
TABLE I
Chemical shift values of head activator in aqueous solution (pH Z2) at 278K a
Residue Chemical shift 6 (ppm)
H Ha Hfl H), H6 He H~
Glp-1 7.96 4.70 2.04 2.54
Pro-2 - 4.77 1.90 2.34
Pro-3 - 4.43 1.96 2.33
Gly-4 8.73 3.97 3.97
Giy-5 8.39 3.99 3.99
Set-6 8.42 4.43 3.84 3.84
Lys-7 8.55 4.34 1.74 1.38 1.68 2.98 7.62 1.83 1.44 1.68 2.98
Val-8 8.31 4.01 1.96 0.87 0.94
lie-9 8.46 4.10 1.77 1.13 0.84 1.48
H3¥2 0.74 Leu-10 8.42 4.35 1.53 1.56 0.81
1.57 0.90 Phe - l l 7.77 4.41 2.94 - 7.22 7.34
3.14 7.22 7.34
2.42 2.42 2.02 1.98 2.07 2.07
3.56 3.75 3.67 3.87
7.27
a Chemical shifts obtained by two dimensional spectroscopy and, where possible, confirmed in one-dimensional spectra with higher digital resolution. Experimental conditions: 1 mM head activator in 90 H 2 0 , 10~o D20 (pH 7.2). Temperature 278 K.
reflect the deprotonation of the phenylalanine carboxyl group and the two residues which contain ionizable groups (Lys-7 and Phe-ll)
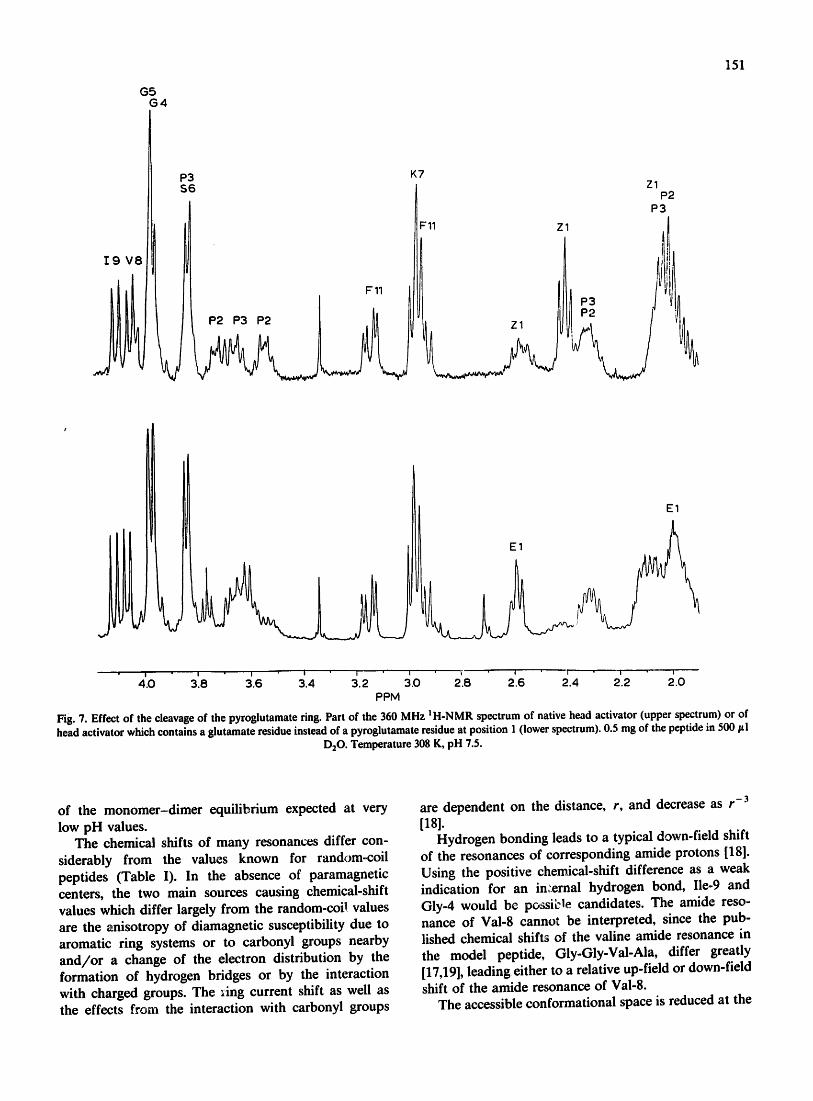
The effect of pyroglutamate on the spatial structure Tl~e cleavage of the pyroglutamate ring causes only
limited changes in the corresponding NMR-spectra (Fig. 7). Comparison of the spectra of native (Fig. 1) and of modified (Fig. 7) head activator show that in the spectra of modified head activator the pyroglutamyl resonances (Hfl, 2.04 ppm; Hfl', 2.57 ppm; H2Y, 2.41 ppm) are not longer visible, but are replaced by the corresponding glutamine resonances (H2fl, 1.99 ppm; H2Y, 2.59 ppm). In addition, the resonance at 3.56 ppm, assigned to H8 of Pro-2, is shifted to 3.62 ppm and the strong coupling pattern of the resonance at 3.97 ppm, assigned to H2ot of Gly-4, changes to a singlet pattern, i.e., its H, atoms become equivalent.
Discussion
Assignments The assignment of the I H-NMR lines to specific spin
systems by one- and two-dimensional methods is striaghtforward and reliable. The same is true for the
G5 \
t 3 4
149
G4
GS--
G4
. . i . . . . . . . . . i . . . . . . . . . | . . . . . . . . . i . . . . . . . . . i . . . . . . . . . i .
8, 40 8.30 8.20 8.10 PPM
I P P M
Fig. 4. 500 M H z spectra of selectively deuterated head activator homologues in H20 . Approximately 1 mM head activator in 90% H 2 0 and 10~ D 2 0 (pH 3.9) with Gly-4 (lower spectrum) or Gly-5
(upper spectrum) deuterated. Temperature 278 K.
TABLE II
Approximate pseudo-first-order exchange rate constants, k,,,. activation enthalpies, A H*. and temperature coefficients, dS/dT, of amide protons of the head activator in water at pH 8.4 a
Residue ke~ ( s - t ) at 274 K d 6 / d T A H *
experimental theoretical ( p p m / K ) (kJ. moi - t )
Glp-1 21 - - 0.008 60 b b b b Pro-2 - - -
Pro-3 - ~' _ b _ b _ t,
Gly-4 15 8.2 - 0.011 61 Gly-5 15 16.2 - ~ - ~ Ser-6 > 5 '~ 18.3 _ c _ Lys-7 10 7.3 - 0.009 62 Vai-8 1 5.8 - 0.009 67 lie-9 0.5 2.9 - 0.010 69 Leu-lO 0.5 2.9 - 0.009 69 Phe-11 0.3 0.3 -0 .007 75
~' Values were calculated as described in Materials and Methods, Experimental conditions as described in ~i~ 5.
b No amide proton existing. c No value available because of a superposition with other resonance
lines. o Value est imated from the disappearance of the signal after satura-
tion of the H 20 resonance.
150
sequential assignments of all spin systems which occur only once in the sequence and for the sequential assign- ments of the glycine resonances done by sequence- specific deuteration. For the proline residues the usual method, the assignment based on sequential nuclear Overhauser effects, failed; in water no really significant nuclear Overhauser effects could be observed, probably because the correlation time, %, is in the range around ,0o2~-) = 5/4, where the expected nuclear Overhauser ef- fect equals zero. In addition, the low solubility of head activator eliminates the possibility of compensating this effect by using a very high amount of material. This problem could be solved by measuring the NOEs in the rotating frame which give the sequential NOEs typical for a trans peptide bond.
Structural implications The 1H-NMR spectra of the head activator show no
sign of structural heterogeneity. Thus, if there is more than one dominant conformation of the head activator,
_ ELLJ L
300K o I
330 K
' 8',8 ~.6 ~,4 ~2 ~,o ' 7,8 ~,e ~4 ' 7,2 PPM
Fig. 5. The exchangeable protons of the head activator in H20 at various temperatures. 500 MHz spectra of 0.5 ml of 1 mM head activator, 10 FM EDTA, in 90~ HzO, 10~ D20 (pH 8.4). Spectra were recorded with a 1331 pulse [8] centered at the water resonance. Line broadening of 0.1 Hz by exponential multiplication. Amide resonances are labelled according to the IUPAC-IUB convention [25] for one-letter codes of amino acids, o, labels signals from low
molecular weight impurities.
6 / ppm
3.e i
2.5
2,0
1,5
D..'..--D " I]g.-n D-D--
' ~ V , - - X ' X 7 ,
:~ ~"x"~x,,.x.x -
i | | | | | | i I J * * I , , , I | | | ! * , ,
0.5 2,,5 - 4.5 6.5 8.5 10.5 pH
Fig. 6. The pH dependence of chemical shifts of lysine and phenyl- alanine resonances. The chemical shift 8 is depicted as function of the pH for the/3-protons of Phe-ll (n, &) and the 8- and e- protons of Lys-7 (x, o). The solid lines are calculated with the parameters of
Table Ill.
the transition between these conformations must be fast compared to the NMR time-scale. (Assuming a chem- ical-shift difference of 0.1 ppm for two resonances corresponding to the same proton in two different con- formations would imply a mean life-time of less than 400 ~s.) In case there are any additional conformations of the neuropeptide which exchange slowly with the dominant structure, their relative populations must be less than 10~. Molecular-sieve chromatography indi- cates a dimeric structure of the head activator [5]. Taking into account the high affinity between the sub- units, only an antisymmetric arrangement of the proto- mers is consistent with the experimental data. More- over, the comparatively small effects of temperature on the chemical shifts show that there are also probably no largely different conformations whose equilibria are shifted with temperature and which are ir~ fast ex- change. The same is true for the pH of the solution; there is no indication of a pH-induced global conforma- tional change over the large range from pH 0.9 to pH 11.6, the observed pH-dependent shifts reflect the pro- tonation-deprotonation of the terminal carboxyl group (apparent pK 3.3) and the lysine amino group (apparent pK 10.3). The only exception is the small chemical shift change of the iysine NH~ resonance with an apparent pK of 1.8 (Table III) which could be caused by a shift
G5 G4
P3 K7 S6
11 Z1
I 9 V 8
P2 P3 P2
Zl P2
P3
151
E1
E1
4.0 3 8 :3.6 .4 3.2 .0 2.8 2.6 2.4 2'.2 2.0 PPM
Fig. 7. Effect of the cleavage of the pyroglutamate ring. Part of the 360 MHz I H-NMR spectrum of native head activator (upper spectrum) or of head activator which contains a glutamate residue instead of a pyroglutamate residue at position 1 (lower spectrum). 0.5 mg of the peptide in 500 #l
D,O. Temperature 308 K, pH 7.5.
of the monomer-dimer equilibrium expected at very low pH values.
The chemical shifts of many resonances differ con- siderably from the values known for random-coil peptides (Table I). In the absence of paramagnetic centers, the two main sources causing chemical-shift values which differ largely from the random-coil values are the anisotropy of diamagnetic susceptibility due to aromatic ring systems or to carbonyi groups nearby and/or a change of the electron distribution by the formation of hydrogen bridges or by the interaction with charged groups. The ,Ang current shift as well as the effects from the interaction with carbonyl groups
-3 are dependent on the distance, r, and decrease as r [181.
Hydrogen bonding leads to a typical down-field shift of the resonances of corresponding arnide protons [18]. Using the positive chemical-shift difference as a weak indication for an in:.ernal hydrogen bond, Ile-9 and Gly-4 would be possiL'le candidates. The arnide reso- nance of Val-8 cannot be interpreted, since the pub- lished chemical shifts of the valine amide resonance in the model peptide, Gly-Gly-Val-Ala, differ greatly [17,19], leading either to a relative up-field or down-field shift of the amide resonance of Val-8.
The accessible conformational space is reduced at the

152
TABLE III
pH dependence of chemical shifts and pK values a
Residue atoms pK b 8 I /ppm b 8 2/ppm b Ag/ppm b
Lys-7 HA8 less than 3 Hz H28 10.3+0.1 ¢ 1.677 1.45 -0.23 H2e 10.3 4.0.2 c 2.980 2.61 -0.37 H3~" 1.8 +0.4 d 7.604 7.621 0.017
Val-8 H 3.2 + 0.1 c 8.296 8.312 0.016 Ha 3.6 +0.3 c 4.007 4.014 0.007 H3Y . H3Y' A8 less than 3 Hz
lie-9 H, Hy1, H38 A8 less than 3 Hz Ha 3.2+0.1 c 4.093 4.103 0.010 H~1,2 3.34.0.1 © 0.721 0.744 0.023 HyI ' 3.34.0.1 c 1.434 1.483 0.049
Leu-10 H38, H38' A8 less than 3 Hz
Phe-ll H 3.34"0.1 © 8.486 7.772 -0.714 Hfi 3.3 + 0.1 c 3.036 2.941 -0.094 Hp 3.3+0.1 c 3.224 3.137 -0.087 |t28 3.2 +0.1 c 7.263 7.222 - 0.041 H~e 3.24.0.1 c 7.354 7.335 -0.019 H~ 3.24.0.1 © 7.295 7.274 -0.021
" Resot~ances not listed cannot be observed with sufficient precision because of overlapping lines. The change of chemical shifts A8 is less ~an 3 Hz for H and H2V of pGlu-1, for H8 and Hg' of Pro-2, for H8' of Pro-3, for H and H2et of Oly-4, for H2a of Gly-5, and for tt2fl of Set-6. Because of exchange broadening the amide resonances of pGlu-I and Gly-4 could only be observed up to pH 7.8.
b Chemical shift values B t, B 2, d~ = 82 - B ! and pK values are the result oY a non-linear fit to ~he modified Henderson-Hasselbalch equation (see Materials and Methods). The sample contained 1 mM head activator in 90~ H20 and 10~ D20. Temperature 278 K. The pH has ~een varied between pH 0.8 and pH 11.6. Because of exchange broadening, the amide resonances of Lys-7 and Val-8 gould oni~ be observed up to pH 8.0, the lie-9 amide resonance up to pH 8.5, ~md the Phe-ll amide resonance up to pH 9.9.
c Only one pX value observable. o Line only observable from pH 0.8 to pH 6.9.
N-terminal segment by the occurrence of the three ring systems of pGlu-pro-pro where the angle, qD, is confined around - 6 0 degrees and the angle, ~, around - 55 and + 145 degrees, respectively [20]. The possible conforma- tions in this part of the peptide chain are influenced by the fact that the peptide bond between proline residues and its N-terminal neighbors often occur as cis-isomers because the difference of free enthalpy, AG °, between both conformations is usually only of the order of a few kJ. mol-I. Spectroscopically, there is no indication that the head activator occurs in more than one conforma- tion in aqueous solution, i.e., the peptide bonds between pGlu-1, Pro-2 and Pro-3 must be stabilized in only one well-defined configuration. Calculations for the di- peptides Xxx-Pro and Pro-Pro show that a trans-peptide bond between amino acid (1) and (2) is characterized by
a strong NOE between Ha( l ) and H28(2) (or H28(1 ) and H28(2)) and missing or very weak NOE's between Ha(l) and Ha(2) as well as H28(1 ) and Ha(2) [21]. In fact, this is exactly the situation observed for pGlu-1, Pro-2 and Pro-3 (Fig. 3), that is the peptide bonds between these residues are in trans-configuration.
Low exchange rates of amide protons and high activation enthalpies, AH*, are typical for amide hy- drogen bonds in proteins [22]. Using these quantities as indicators for possible hydrogen bonds, the expectation that the amide protons are involved in hydrogen bond- ing interactions would increase monotonically from the N-terminal to the C-terminal part of the peptide (Table II). However, one should be aware that additional fac- tors such as solvent accessibility and charge effects can also cause differences in k~x and 8H*. In proteins such as the basic pancreatic inhibitor, AH* varies over a wide range between 55 and 370 kJ. mo1-1 [22]. The pseudo-first-order exchange rates, kex, and activation enthalpies, A H*, in random-coil peptides can be calcu- lated with a phenomenological equation given by Englander et al. [23]. At pH 8.4 and 274 K one obtains an activation enthalpy, A H*, of approx. 60 kJ. mol-1. This value is close to the values obtained experimentally for Glp-1, Gly-4 and Lys-7 (Table II), implying that the peptide amide protons of these residues are probably not involved in hydrogen-bonding interactions. Amide exchange rates are strongly influenced by the nearest neighbors in the sequence. Empirical correction factors are tabulated by Molday et al. [24] and can be used for the calculation of amide exchange rates for 'free' peptide groups. Comparison of these values with the experimen- tal rates for Gly-5 and Lys-7 shows a good agreement between theoretical and experimental values (Table If), indicating that these residues are probably not involved in hydrogen bonding. In contrast, the experimental exchange rates for Val-9, lie-9 and Leu-10 are consider- ably lower than expected, indicating that these residues may be involved in hydrogen bonding. This interpre- tation is supported by an increase of the corresponding activation enthalpies. For Phe-ll , the exchange data cannot be interpreted in such a simple way, because Phe-ll shows a normal kex but an elevated AH*.
A salt bridge between the positively charged amino group of Lys-7 and the terminal carboxyl group o f Phe-11 could stabiliTe the structure. The titration curve of the side-chain amino group of Lys-7 shows no indica- tion of a co-titration with the terminal carboxyl group, suggesting that there is most probably no direct interac- tion between these residues. The ionization state of the carboxyl group of Phe-11 influences the chemical shifts of the backbone protons of lle-9 and Val-8; a rather large change of chemical shift can be found also for the isoleucine 3'2 methyl protons and for one of the 3,1 methylen protons, whereas the side-chain protons of Leu-10 and Val-8 show no pH effect. This may indicate

a close spatial relation between the carboxyl group of Phe-11 and the side-chain of Ile-9. The Ha-protons of Gly-4 are inequivalent, which is probably due to :he steric hindrance by the three neighbored ring systems of pGlu-Pro-Pro. After cleavage of the ring of pGlu-1, the Ha-protons assigned to Gly-4 become equivalent possi- bly because of an increase in motional freedom. The cleavage of the pyroglutamine ring inactivates the head activator. This inactivation could be a local effect or could follow from a global conformational change in- fluencing the proper interaction with the receptor. Since there is no experimental evidence for a global confor- mational change, the local interaction of the head group of the peptide with the receptor is probably important for the proper function of the neuropeptide and is disturbed by the cleavage of the pyroglutamate ring.
Acknowledgements
This project was supported by the Deutsche For- schungsgemeinschaft. We are grateful to H. Zimmer- mann for the perdeuteration of glycine.
References
1 Schaller, H.C. and Bodenmiiller, H. (1981) Proc. Natl. Acad. Sci. USA 78, 7000-7004.
2 Schaller, H.C. (1973) J. Embryol. Exp. Morphol. 29, 27-38. 3 Schailer, H.C. and Bodenmiiiler, H. (1985) Hoppe-Seyler's Biol.
Chem. 366, 1003-1007.
153
4 Bodenmfiller, H. and Schailer, H.C. (1981) Nature 293, 579-580. 5 Bodenmiiller, H., Schilling, E., Zachmann, B. and Schaller, H.C.
(1986) EMBO J. 5, 1825-i829. 6 Schaller, H.C., Roberge, M., Zachmann, B., Hoffmeister, S., Schill-
ing, E. and Bodenmfiller, H. (1986) EMBO J. 5, 1821-1824. 7 Kalbitzer, H.R. and Rfisch, P. (1981) Org. Magn. Reson. 17,
88-91. 8 Hore, P.J. (1983) J. Magn. Reson. 55, 283-300. 9 Rance, M., Sorensen, O.W., Bodenhausen, G., Wagner, G., Ernst,
R.R. and Wiithrich, K. (1983) Biochem. Biophys. Res. Commun. 117, 479-485.
10 Eich, G., Bodenhausen, G. and Ernst, R.R. (1982) J. Am. Chem. Soc. 104, 3731-7332.
11 Bax, A. and Drobny, G. (1985) J. Magn. Reson. 61, 306-320. 12 Rance, M. (1987) J. Magn. Reson. 74, 557-564. 13 Neidig, K.P., Bodenmfiller, H. and Kaibitzer, H.R. (1984) Bio-
chem. Biophys. Res. Commun. 125, 1143-1150. 14 McConneil, H.M. (1958) J. Chem. Phys. 28, 430-431. 15 Kaplan, J l. and Fraenkei, G. (1972) J. Am. Chem. Soc. 94,
2907-2912. 16 Forsen, S. and Hoffman, R.A. (1963) J. Chem. Phys. 39, 2892-2901. 17 Bundi, A. and Wfithrich, K. (1979) Biopolymers 18, 285-297. 18 Wagner, G., Pardi, A. and Wiithrich, K. (1983) J. Am. Chem. Soc.
105, 5948-5949. 19 Bundi, A. and WUthrich, K. (1979) Biopolymers 18, 299-311. 20 Cantor, C.R. and Schimmel, P.R. (1980) Biophysical Chemistry,
Vol. I, W.H. Freeman, San Francisco. 21 Wtithdch, K. (1986) NMR of Proteins and Nucleic Acids, Wiley,
New York. 22 Wagner, G. (1983) Quart. Rev. Biophys. 16, 1-57. 23 Englander, S.W., Downer, N.W. and Teiteibaum, H. (1972) Annu.
Rev. Biochem. 41,903-924. 24 Molday, R.S., Englander, S.W. and Kailen, R.G. (1972) Biochem-
istry. 11, 150-158. 25 IUPAC-IUB (1984) Eur. J. Biochem. 138, 9-37.




























