Embed Size (px)
Citation preview

This article was downloaded by: [Inonu Universitesi]On: 24 August 2014, At: 16:44Publisher: Taylor & FrancisInforma Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House,37-41 Mortimer Street, London W1T 3JH, UK
Polymer-Plastics Technology and EngineeringPublication details, including instructions for authors and subscription information:http://www.tandfonline.com/loi/lpte20
A Model for Molecular Weight Prediction inAcrylonitrile PolymerizationIlknur Atasoy a , Ridvan Berber a & Mehmet Yuceer ba Department of Chemical Engineering, Faculty of Engineering, Ankara University ,Tandogan, Ankara, Turkeyb Department of Chemical Engineering, Faculty of Engineering, Inonu University , Malatya,TurkeyPublished online: 13 Aug 2008.
To cite this article: Ilknur Atasoy , Ridvan Berber & Mehmet Yuceer (2008) A Model for Molecular Weight Prediction inAcrylonitrile Polymerization, Polymer-Plastics Technology and Engineering, 47:9, 874-882, DOI: 10.1080/03602550802189027
To link to this article: http://dx.doi.org/10.1080/03602550802189027
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) containedin the publications on our platform. However, Taylor & Francis, our agents, and our licensors make norepresentations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of theContent. Any opinions and views expressed in this publication are the opinions and views of the authors, andare not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon andshould be independently verified with primary sources of information. Taylor and Francis shall not be liable forany losses, actions, claims, proceedings, demands, costs, expenses, damages, and other liabilities whatsoeveror howsoever caused arising directly or indirectly in connection with, in relation to or arising out of the use ofthe Content.
This article may be used for research, teaching, and private study purposes. Any substantial or systematicreproduction, redistribution, reselling, loan, sub-licensing, systematic supply, or distribution in anyform to anyone is expressly forbidden. Terms & Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

A Model for Molecular Weight Prediction in AcrylonitrilePolymerization
Ilknur Atasoy1, Ridvan Berber1, and Mehmet Yuceer2
1Department of Chemical Engineering, Faculty of Engineering, Ankara University,Tandogan, Ankara, Turkey2Department of Chemical Engineering, Faculty of Engineering, Inonu University, Malatya, Turkey
The number and weight-average molecular weights in acrylonitrilepolymerization have been calculated previously by Peebles[2]. How-ever, the foundations for the two critically important expressionsleading to the calculation of molecular weights were not disclosedin detail, no dynamics were presented, and predictions were not ingood agreement with the experimental data, particularly in termsof polydispersity index. The present work focuses on the same issue,and brings a new rigorous dynamic model, based on the kineticsgiven by Peebles[2]. The new, more detailed model defines the chainlengths in terms of the leading moments of active and dead polymer,provides the prediction of reactor dynamics in compliance with thepractice in industry, and estimates the polydispersity index of thepolymer with better agreement to the experimental data.
Keywords Acrylonitrile polymerization; Average molecularweight prediction; Moment generation
INTRODUCTION
The acrylic fiber industry is growing, particularly indeveloping nations due to the expanding demand for textileproducts at home and for exports. It has been shown thatacrylic fiber production grew at a trend rate of 2.5% overthe last two decades[1]. What makes the acrylic fiberattractive compared to the alternatives are its low specificgravity, soft and wool-like aesthetics, resistance to UVfading, and shape retention after machine washing.
Acrylic fiber is commercially produced by free radicalpolymerization, initiated by a potassium peroxydisulfate-sodium bisulfite-iron redox system. The monomers (acry-lonitrile and a comonomer, vinyl acetate) and watertogether with catalyst (potassium peroxydisulfate), an actu-ator (SO2 in water) and a pH adjusting agent (usually bicar-bonate) are fed to a well-stirred reactor. The terminationmechanism is assumed to be controlled by recombinationof active radicals with no disproportionation taking place.
The kinetics of acrylonitrile polymerization is a lot morecomplicated than that of ordinary polymerization systems,which are well-studied in the literature, due to the redoxinitiation system. Knowledge on the acrylonitrile poly-merization is limited and mainly confined to steady statemodeling.
Probably the most detailed study of the kinetics of thispolymerization was undertaken by Peebles[2], who set upa kinetic model and derived the molecular weight distri-bution as a function of the activator-to-catalyst ratio andcompared the predictions with some experimental datawith particular reference to properties such as dyeabilityand molecular weight. However, no account of the reactordynamics was provided, and the weight-average molecularweight predictions did not seem to be in very good agree-ment with those experimentally obtained from viscositymeasurements. The calculated values were reported to belower than those obtained by the viscosity measurementsby a factor of about 1.5. Computation of the Mw=Mn ratiogave a range of values between 1.5 and 2.0 depending onthe initial catalyst and activator concentrations, whereasthe same ratios obtained from measured specific viscositiesranged from 2.2 to 3.7. This meant that the molecularweight distribution of the actual polymerization wasbroader than that predicted from the assumed mechanismof polymerization. The difference was attributed to the het-erogeneity that was not accounted for in the modeling. Theinfluence of heterogeneity on the mechanism of polymeri-zation is said to be one of the unsolved problems in poly-mer kinetics. Despite the inconsistencies in the molecularweight prediction, the observed and calculated conversionswere in good agreement. Peebles[2], however, derived a cor-relation equation from his experimental data relating theweight-average molecular weight, the conversion, and theactivator concentration.
Literature survey shows that the open knowledge on thepolymerization of acrylonitrile is very limited and parti-cularly confined to the steady state case. In parallel with
Address correspondence to Mehmet Yuceer, Department ofChemical Engineering, Faculty of Engineering, Inonu University,44280 Malatya, Turkey. E-mail: [email protected]
Polymer-Plastics Technology and Engineering, 47: 874–882, 2008
Copyright # Taylor & Francis Group, LLC
ISSN: 0360-2559 print/1525-6111 online
DOI: 10.1080/03602550802189027
874
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

the increase in their share in the worldwide PACN (poly-acrylonitrile) market, China seems to be putting someefforts into the control of PACN reactors[3,4]. However, itis not easy to get access to such information due to lan-guage difficulties. What, therefore, is really missing is areliable dynamic model capable of predicting the averagemolecular weight and its distribution.
This study aims at defining a new dynamic mathemat-ical model for PACN reactors[5]. The motivation was thatthe most detailed study available in the literature was lack-ing in the provision of two critical equations, and its resultsshowed inconsistency with experimental data. Although arather detailed kinetics mechanism was provided and con-servation equations were developed in unsteady state, nodynamic solution was presented, and a thorough under-standing of the reactor dynamics is a must for a better con-trol. By virtue of these facts, our attention was conveyed toa better way of describing the molecular weight distri-bution. Considering the fact that one of the key polymerproperties determining user specifications is the averagemolecular weight and its distribution, this area bears parti-cular importance. In this work, a rigorous dynamic modelwas developed based on the kinetics previously defined byPeebles[2], transients were identified, and the predictionswere compared to the experimental data previouslyreported.
The moment analysis technique employed here waspreviously applied to a number of relatively simple andwell-known polymerization systems, such as polyethyleneand olefins[6–8]. As there are different ways of applying thistechnique in polymerization[9], its use in an industriallyimportant and complex polymerization system aimed atproduction of synthetic fiber is appealing. The results ofthis research concluded that the technique could beemployed in such a setting. Therefore, it not only helps ful-fill an industrial demand but also contributes to academicknowledge. Comparison of the predictions of the newmodel to experimental data taken from literature revealedcloser agreement.
MATHEMATICAL MODEL
Industrial production of polyacrylonitrile is a variantof aqueous dispersion polymerization and takes place incompletely stirred tank reactors with a peroxydisulfate-bisulfite-iron redox system. One of the two feeds consistof the monomer and vinyl acetate, which is a neutralco-monomer helping increase the solubility in the spinningsolution and the diffusion of dye, and affecting the mor-phology of the fiber. The other feed consists of the agentsfor the redox initiation system together with some sul-phuric acid to adjust the pH in the reactor.
Polymerization follows the following steps as in radicaladdition polymerization[2].
Initiation: In the redox initiation system, active radicalsof sulphate and sulfite groups are formed as shown in thefollowing:
S2O�28 þ Feþ2�!k1
SO�24 þ SO��4 þ Feþ3
HSO�3 þ Feþ3�!k2HSO�3 þ Feþ2
ð1Þ
S2O�28 þHSO�3 ! SO�2
4 þ SO��4 þHSO�3 ð2Þ
I �!kd2R ð3Þ
where, I is initiator and R is radical. Reactivity of SO��4
and HSO�3 radicals are the same.Addition: Assuming that the radicals formed in the
previous step react only with a monomer, a monomer isadded to these radicals and produces active radical chainsof a single monomer with end groups of sulphate andsulfite, respectively.
SO��4 þM �!kaR�1 ð4Þ
HSO�3 þM �!kaQ�1 ð5Þ
Propagation: In this step, the active chains keep growing byaddition of monomers.
R�n þM �!kp
R�nþ1 ð6Þ
Q�n þM �!kp
Q�nþ1 ð7Þ
All radicals propagate with the same velocity. Additionand growing rate of monomers to the polymer chain weretaken to be equal.
Termination: Growing chains of active polymers reactwith each other, lose their activity, and turn into inactive,i.e., nongrowing, polymer chains. Termination can resulteither from recombination of the chains (e.g., active poly-mers with n and s monomers coming together to give adead polymer of nþ s monomers) or from the transfer(where the active side of a polymer chain is transferred tothe bisulfite group present in the medium producing a sul-fite radical). Transfer reactions occur only to the activatorand not to the monomer or solvent (water). As stated pre-viously, it was assumed that no branching reactionsoccurred and termination occurred only by coupling. Here,the third mechanism of termination, disproportionation, isneglected by virtue of the findings of previous experimentalstudies[2]. The termination reactions produce polymers withdifferent end groups.
R�n þ R�s �!kr
Tnþs ð8Þ
Q�n þQ�s �!kr
Unþs ð9Þ
Q�n þ R�s �!kr
Wnþs ð10Þ
ACRYLONITRILE POLYMERIZATION MODEL 875
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

The transfer to activator reaction is the following:
R�n þHSO�3 �!ktr
Pn þ SO��3 ð11Þ
Q�n þHSO�3 �!ktr
Sn þ SO��3 ð12Þ
The mathematical model developed is based on the follow-ing simplifying assumptions:
� Polymerization takes place in homogenous phaseunder isothermal conditions with perfect mixing.
� Reactivities of SO��4 ve HSO�3 radicals are thesame.
� All radicals propagate with the same velocity con-stant (kp), undergo termination with the samevelocity constant (kt), and transfer with the samevelocity constant (ktr).
� All species are soluble in the solvent.� Branching reactions do not take place.� Equilibrium is established after 5 – 6� dwell time.� Addition and propagation steps proceed with the
same velocity constant (ka ¼ kp).� All reactions are of second order.
Model-1, Based on the Literature (Peebles’ Model)
The model given by Peebles[2] is probably the mostdetailed model to the present study. It comprises the con-servation equations of each species involved in the reactionsystem with the previous assumptions. Considering acontinuous flow, stirred tank reactor, Peebles wrote theconservation equations as follows. These equations arerewritten for the ease of understanding and unity to thereader (omitting details which can be found in the originalpaper).
½dM�dt¼ �kp½M� ½SO��4 � þ ½HSO�3� þ
X1n¼1
½R�n�
þX1n¼1
½Q�n�!þ ½M�0
h� ½M�
hð13Þ
d½R�1�dt¼ kp½M�½SO��4 � � kp½R�1�½M� � ktr½R�1�½HSO�3 �
� kt½R�1�X1s¼1
½R�s � � kt½R�1�X1s¼1
½Q�s � �½R�1�h¼ 0 ð14Þ
d½R�n�dt¼ kp½M�½R�n�1� � kp½M�½R�n� � ktr½R�n�½HSO�3 �
� kt½R�n�X1s¼1
½R�s � � kt½R�n�X1s¼1
½Q�s � �½R�n�h¼ 0 ð15Þ
d½Q�1�dt¼ kp½M�½HSO�3� � kp½Q�1�½M� � ktr½Q�1�½HSO�3 �
� kt½Q�1�X1s¼1
½R�s ��kt½Q�1�X1s¼1
½Q�s � �½Q�1�h¼ 0
ð16Þ
d½R�n�dt¼ kp½M�½Q�n�1� � kp½M�½Q�n� � ktr½Q�n�½HSO�3 �
� kt½Q�n�X1s¼1
½R�s ��kt½Q�n�X1s¼1
½Q�s � �½Q�n�h¼ 0 ð17Þ
d½Pn�dt¼ ktr½R�n�½HSO�3 � �
½Pn�h¼ 0 ð18Þ
d½Sn�dt¼ ktr½Q�n�½HSO�3 � �
½Sn�h¼ 0 ð19Þ
d½Tn�dt¼ 1
2kt
Xn�1
s¼1
½R�s �½R�n�s� �½Tn�h¼ 0 ð20Þ
(the factor 12 is included so that each species will not be
counted twice)
d½Un�dt¼ 1
2kt
Xn�1
s¼1
½Q�s �½Q�n�s� �½Un�h¼ 0 ð21Þ
d½Wn�dt¼ kt
Xn�1
s¼1
½R�s �½Q�n�s� �½Wn�
h¼ 0 ð22Þ
Probability of propagation is given by,
a¼ kp½M�½R�n�
kp½M�½R�n�þkr½R�n� R1
s¼1½R�s �þkr½R�n� R
1
s¼1½Q�s �þktr½HSO�3 �½R�n� �
½R�n �h
ð23Þ
a ¼ kp½M�
kp½M� þ ktr½HSO�3 � þ kr
P1s¼1
½R�s � þP1s¼1
½Q�s �� �
þ 1h
ð24Þ
Equation (17) is solved to obtain [R�n] and likewise [Q�n]
½Rn�� ¼ an½SO�4
�� ð25Þ
½Qn�� ¼ an½HSO3
�� ð26Þ
Considering the assumption of instantaneous mixing andequilibrium after 6 dwell times, equilibrium concentrationsof some species were found from steady state solutions.
½SO�4�� ¼ k1½Feþ2�½S2O�2
8 �kp½M�
ð27Þ
876 I. ATASOY ET AL.
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

½HSO�3� ¼k1½Feþ2�½S2O�2
8 �kp½M�
:kp½M� þ ktr½HSO�3 �a=ð1� aÞkp½M� � ktr½HSO�3 �a=ð1� aÞ
� �ð28Þ
½S2O�28 � ¼
½S2O�28 �o
1þ k1h½Feþ2� ð29Þ
½HSO�3 � ¼½HSO�3 �o �
k1h Feþ2�½S2O�28
� �o
1þ k1h Feþ2� �� �
1þ h1=2 ktr
k1=2r
!2k1h Feþ2�½S2O�2
8
� �o
1þ k1h Feþ2½ �
!1=2ð30Þ
The equations given to this point form the starting pointfor the model developed in this work.
As for the number-average and weight-average chainlengths, Peebles[2] gives the following:
�rrn ¼2kp½M�
zð1þ 2yÞ ð31Þ
�rrw ¼kp½M�ð3þ 2yÞ
zð1þ yÞ2ð32Þ
where,
z ¼ 2k1kr Feþ2�½S2O�28
� �� �1=2 ð33Þ
z ¼ 2k2kr Feþ3�½HS O�3� �� �1=2 ð34Þ
y ¼ ktr½HS O�3 �
z ð35Þ
Probability of propagation is redefined as follows:
a ¼ kp½M�kp½M� þ zð1þ yÞ ð36Þ
The number-average and weight-average molecular weightsare
Mn ¼ �rrnWm ð37ÞMw ¼ �rrwWm ð38Þ
The equations describing the number-average and weight-average chain lengths (Eqs. 31 and 32) are the most criticalof all the derived model equations because they give theaverage molecular weights through Eqs. (37) and (38).However, no account or explanation of the definitions ofrn and rw were provided. It is to be noted furthermore that,although the equations were written in unsteady state form,no dynamic solution of the model was provided, and tran-sient characteristics of the model were lacking. Therefore, itwas felt that a new model was needed.
New Model
In this work, we have rederived the number and weight-average chain lengths of the polymer, by employing the
moment generating functions[10]. Consequently, we havereached a new dynamic model, which will be described next.
The moments of the molecular weight distribution arederived by means of moment generating functions[10].
Moments of the number chain length distribution aredescribed as follows:,
For living polymers;
Gðr; xÞ ¼X1x¼1
rxPxdG
dt¼X1x¼1
rx dPx
dtð39Þ
For dead polymers;
Fðr; xÞ ¼X1x¼1
rxMxdF
dt¼X1x¼1
rx dMx
dtð40Þ
from which the moments of the living and dead chaindistributions can be obtained by differentiation.
zn ¼@nGð1; xÞ
@rnqn ¼
@nFð1; xÞ@rn
ð41Þ
Variation of monomer concentration with time is asfollows:
d½M�dt¼ �kp½M�
X1x¼1
½R�x� � kp½M�X1x¼1
½Q�x� � kp½M�½SO��4 �
� kp½M�½HSO�3� þ½M�0
h� ½M�0
hð42Þ
where ½R�x� and ½Q�x�, define ½R�n� and ½Q�n�, respectively.Variation in the sum of radical concentration with
time is
dR
dt¼ k1½Feþ2�½S2O�2
8 � � kp½M�½SO��4 � �SO��4
� �h
þ k2 Feþ3�½HSO�3� �
� kp½M� HSO�3� �
þ ktr HSO�3� � X1
n¼1
½R�n� þX1n¼1
½Q�n�" #
�HSO�3� �
hð43Þ
where R defines the sum of HSO�3� �
and SO��4
� �.
Variation in active radical concentration with timehaving one monomer is as follows:
dP1
dt¼ kp½M� SO��4
� �� kp½M�½R�1� � kr½R�1�
X1s¼1
½R�s �
� kr½R�1�X1s¼1
½Q�s � � ktr½R�1�½HSO�3 � �½R�1�h
þ kp½M� HSO�3� �
� kp½M�½Q�1� � kr½Q�1�X1s¼1
½Q�s �
� kr½Q�1�X1s¼1
½R�s � � ktr½Q�1�½HSO�3 � �½Q�1�h
ð44Þ
where P1 stands for the sum of ½R�1� and ½Q�1�.
ACRYLONITRILE POLYMERIZATION MODEL 877
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

Variation in the total active polymer concentration withtime is as follows:
dPx
dt¼ kp½M�½R�x�1� � kp½M�½R�x� � kr½R�x�
X1s¼1
½R�s �
� kr½R�x�X1s¼1
½Q�s � � ktr½R�x�½HSO�3 � �½R�x�h
þ kp½M�½Q�x�1� � kp½M�½Q�x� � kr½Q�x�X1s¼1
½Q�s �
� kr½Q�x�X1s¼1
½R�s � � ktr½Q�x�½HSO�3 � �½Q�x�h
ð45Þ
where ½R�x�1�, ½R�x�, ½Q�x�1�, and ½Q�x� in fact define ½R�n�1�,½R�n�, ½Q�n�1�, and ½Q�n�, respectively. Px stands for the sumof ½R�x� and ½Q�x�.
The change in total dead polymer concentration withtime is
dMx
dt¼ ktr½R�X �½HSO�3 � �
½PX �hþ ktr½Q�X �½HSO�3 �
� ½SX �hþ 1
2kr
XX�1
S¼1
½R�S�½R�X�S� �½TX �
h
þ 1
2kr
XX�1
S¼1
½Q�S�½Q�X�S� �½UX �
h
þ kr
XX�1
S¼1
½R�S�½Q�X�S� �½WX �
hð46Þ
where Mx defines the sum of Px, Sx, Tx, Ux, and Wx. There-fore, the changes in the moments of number chain lengthdistribution functions can be written as follows:
dG
dt¼ kp½M�½SO��4 �rþ kp½M�
X1x¼1
rxþ1 ½R�x� þ ½Q�x�� �
� kp½M�X1x¼1
rx ½R�x� þ ½Q�x�� �
� kr
X1x¼1
rx½R�x�X1s¼1
½R�s � þ ½Q�s �� �
� ktr½HSO�3 �X1x¼1
rx ½R�x� þ ½Q�x�� �
�X1x¼1
rx ½R�x� þ ½Q�x�� �
h
� kr
X1x¼1
rx½Q�x�X1s¼1
½R�s � þ ½Q�s �� �
þ kp½M�½HSO�3�r
ð47Þ
dF
dt¼ ktr½HSO�3 �
X1x¼1
rx½R�x� �X1s¼1
rx ½Px�h
þ ktr½HSO�3 �X1x¼1
rx½Q�x� �X1x¼1
rx ½Sx�h
þ 1
2kr
X1x¼1
rxXx�1
s¼1
½R�s �½R�x�s� �X1x¼1
rx ½Tx�h
þ 1
2kr
X1x¼1
rxXx�1
s¼1
½Q�s �½Q�x�s� �X1x¼1
rx ½Ux�h
þ kr
X1x¼1
rxXx�1
s¼1
½R�s �½Q�x�s� �X1x¼1
rx ½Wx�h
ð48Þ
After some very tedious algebra, we obtain the followingmoment balances for the active and dead polymer chaindistributions.
dZ0
dt¼ kp½M� ½SO��4 � þ ½HSO�3�
� �� krZ
20
� ktr HSO�3� �
Z0 �Z0
hð49Þ
dZ1
dt¼ kp½M� ½SO��4 � þ ½HSO�3�
� �þ kp½M�Z0
� krZ1Z0 � ktr½HSO�3 �Z1 �Z1
hð50Þ
@Z2
@t¼ 2kp½M�Z1 �
Z2
h� krZ0Z2 � ktr½HSO�3 �Z2 ð51Þ
dq0
dt¼ ktr½HSO�3 �Z0 �
q0
hþ 1
2krZ
20 ð52Þ
dq1
dt¼ ktr½HSO�3 �Z1 �
q1
hþ kr
a
ð1� aÞ2½HSO�3�Z0
þ kra
1� a½SO��4 �Z1 ð53Þ
dq2
dt¼ ktr½HSO�3 �Z2 �
q2
hþ kr½SO��4 �
a2
ð1� aÞ3½SO��4 �ð1� aÞ
�
þ2Z0� þ kra
ð1� aÞ ½HSO�3� 3½HSO�3�a
ð1� aÞ3þ Z2
" #
ð54Þ
Equations between Eq. (49) and (54) were obtained withthe aid of the following identities.
X1x¼1
Px
X1n¼1
Pn ¼X1x¼1
Xx�1
i¼1
PiPx�i ð55Þ
X1x¼1
xPx
X1n¼1
Pn ¼1
2
X1x¼1
xXx�1
i¼1
PiPx�i ð56Þ
878 I. ATASOY ET AL.
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

1
2
X1x¼1
ðx2 � xÞXx�1
i¼1
PiPx�i ¼X1x¼1
xPx
!2
þX1n¼1
ðn2 � nÞPn
ð57Þ
Finally, the number and weight-average molecular weightscan be calculated through
Mn ¼q1
q0
�Wm ð58Þ
Mw ¼q2
q1
�Wm ð59Þ
SIMULATION AND RESULTS
The derived model was first solved for the steady stateand then the dynamics were investigated by simulatingthe model in the MATLAB=SIMULINK environment.
Table 1 gives the kinetics constants employed insimulations. Table 2 presents the state variables and initialsteady states for both of the models.
Starting from the initial steady state described, an exten-sive number of dynamic simulations were run in order toobtain the predictions of the model under different operat-ing conditions. In order to compare the model predictionsto experimental values, the operating conditions in simu-lated runs coincided with those presented by Peebles[2],who provided the only experimental data available in theliterature.
Results reveal that the predictions of monomer conver-sion by the suggested model are in very good agreementwith the experimental data extracted from the literature,as reflected in Fig. 1. There appears to be almost no differ-ence in this respect between the predictions of the newmodel and previously reported model.
With respect to the average molecular weight, the theor-etical predictions, from the previous as well as the presentmodel, seem to be not in close agreement with the experi-mental values (as reflected in Fig. 2). Peebles[2] admittedthat his calculated molecular weights were lower than thoseobtained from viscosity measurements by a factor of about1.5. He noted that such discrepancy was indeed consistentwith the observations in one of his earlier works, where theMw=Mn ratio was roughly 2.7. Computation of theMw=Mn ratio by Peebles’s model gave a range of valuesfrom roughly 1.5 to 2.0, depending on the initial catalystand activator concentration. Conversely, the molecularweight ratio obtained from the measured specific viscositiesgave a range of 2.2–3.7. This means that the molecularweight distribution of the actual polymers is broaderthan that predicted from the assumed mechanism of poly-merization. Peebles attributed the difference to the ass-umption of homogenous conditions in the polymerization
TABLE 1Kinetics constants (by Peebles, 1973) and molecular
weights
Constant Value Unit
kp 30000 L=(mol.s)h 4500 sec[Feþ2] 6� 10�7 mol=LK1 50.7 L=(mol.s)ktr 6189 L=(mol.s)kr 8.82� 108 L=(mol.s)Wm 53 g=gmolWater=Monomer Ratio 5 —WSO2 64 g=gmolWK2S2O8 270 g=gmol
TABLE 2State variables and initial steady states
State variables forprevious model(Peebles’ model)
Initial steady statesfor previous model
x0 (mol=L)State variablesfor new model
Initial steady statesfor new model
x0 (mol=L)
[M] 94.18� 10�2 [M] 94.18� 10�2
[SO��4 ] 4.24� 10�12 [R�n] 0[HSO�3] 18.48� 10�12 [Q�n] 0[R�1] 4.23� 10�12 [HSO�3 ] 39.44� 10�4
[Q�1] 18.45� 10�12 [S2O�28 ] 39.36� 10�4
[R�n] 0 [z0] 16� 10�9
[Q�n] 0 [z1] 11.97� 10�6
[HSO�3 ] 3.944� 10�3 [z2] 97.99� 10� 4
[S2O�28 ] 3.936� 10�3 [q0] 23.49� 10�4
[q1] 20.98� 10�1
[q2] 29.58� 102
ACRYLONITRILE POLYMERIZATION MODEL 879
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014
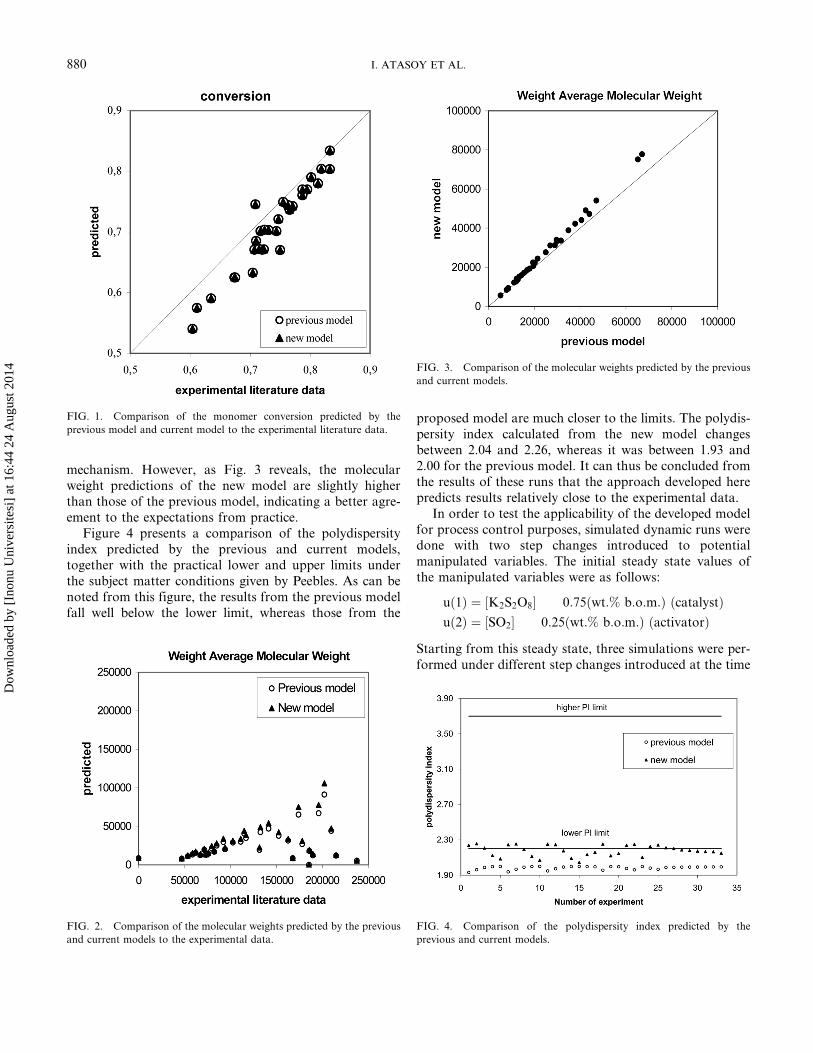
mechanism. However, as Fig. 3 reveals, the molecularweight predictions of the new model are slightly higherthan those of the previous model, indicating a better agre-ement to the expectations from practice.
Figure 4 presents a comparison of the polydispersityindex predicted by the previous and current models,together with the practical lower and upper limits underthe subject matter conditions given by Peebles. As can benoted from this figure, the results from the previous modelfall well below the lower limit, whereas those from the
proposed model are much closer to the limits. The polydis-persity index calculated from the new model changesbetween 2.04 and 2.26, whereas it was between 1.93 and2.00 for the previous model. It can thus be concluded fromthe results of these runs that the approach developed herepredicts results relatively close to the experimental data.
In order to test the applicability of the developed modelfor process control purposes, simulated dynamic runs weredone with two step changes introduced to potentialmanipulated variables. The initial steady state values ofthe manipulated variables were as follows:
uð1Þ ¼ ½K2S2O8� 0:75ðwt:% b:o:m:Þ ðcatalystÞuð2Þ ¼ ½SO2� 0:25ðwt:% b:o:m:Þ ðactivatorÞ
Starting from this steady state, three simulations were per-formed under different step changes introduced at the time
FIG. 2. Comparison of the molecular weights predicted by the previous
and current models to the experimental data.
FIG. 3. Comparison of the molecular weights predicted by the previous
and current models.
FIG. 4. Comparison of the polydispersity index predicted by the
previous and current models.
FIG. 1. Comparison of the monomer conversion predicted by the
previous model and current model to the experimental literature data.
880 I. ATASOY ET AL.
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014
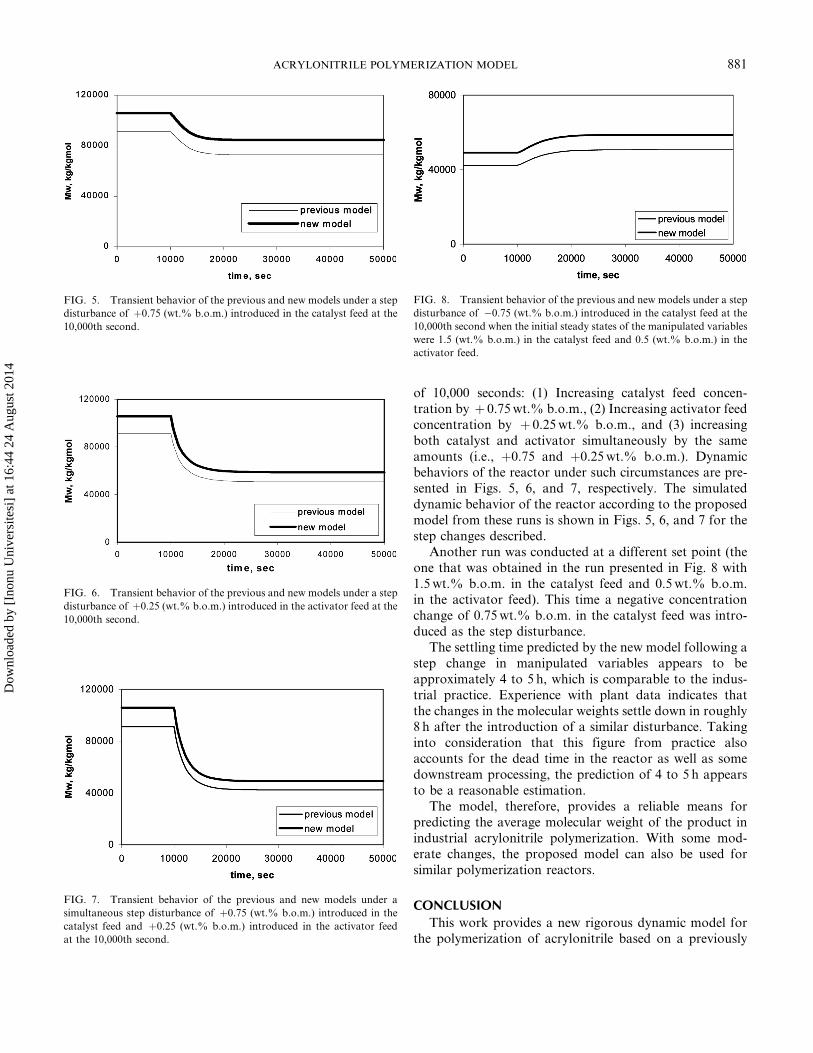
of 10,000 seconds: (1) Increasing catalyst feed concen-tration by þ 0.75 wt.% b.o.m., (2) Increasing activator feedconcentration by þ 0.25 wt.% b.o.m., and (3) increasingboth catalyst and activator simultaneously by the sameamounts (i.e., þ0.75 and þ0.25 wt.% b.o.m.). Dynamicbehaviors of the reactor under such circumstances are pre-sented in Figs. 5, 6, and 7, respectively. The simulateddynamic behavior of the reactor according to the proposedmodel from these runs is shown in Figs. 5, 6, and 7 for thestep changes described.
Another run was conducted at a different set point (theone that was obtained in the run presented in Fig. 8 with1.5 wt.% b.o.m. in the catalyst feed and 0.5 wt.% b.o.m.in the activator feed). This time a negative concentrationchange of 0.75 wt.% b.o.m. in the catalyst feed was intro-duced as the step disturbance.
The settling time predicted by the new model following astep change in manipulated variables appears to beapproximately 4 to 5 h, which is comparable to the indus-trial practice. Experience with plant data indicates thatthe changes in the molecular weights settle down in roughly8 h after the introduction of a similar disturbance. Takinginto consideration that this figure from practice alsoaccounts for the dead time in the reactor as well as somedownstream processing, the prediction of 4 to 5 h appearsto be a reasonable estimation.
The model, therefore, provides a reliable means forpredicting the average molecular weight of the product inindustrial acrylonitrile polymerization. With some mod-erate changes, the proposed model can also be used forsimilar polymerization reactors.
CONCLUSION
This work provides a new rigorous dynamic model forthe polymerization of acrylonitrile based on a previously
FIG. 6. Transient behavior of the previous and new models under a step
disturbance of þ0.25 (wt.% b.o.m.) introduced in the activator feed at the
10,000th second.
FIG. 7. Transient behavior of the previous and new models under a
simultaneous step disturbance of þ0.75 (wt.% b.o.m.) introduced in the
catalyst feed and þ0.25 (wt.% b.o.m.) introduced in the activator feed
at the 10,000th second.
FIG. 8. Transient behavior of the previous and new models under a step
disturbance of �0.75 (wt.% b.o.m.) introduced in the catalyst feed at the
10,000th second when the initial steady states of the manipulated variables
were 1.5 (wt.% b.o.m.) in the catalyst feed and 0.5 (wt.% b.o.m.) in the
activator feed.
FIG. 5. Transient behavior of the previous and new models under a step
disturbance of þ0.75 (wt.% b.o.m.) introduced in the catalyst feed at the
10,000th second.
ACRYLONITRILE POLYMERIZATION MODEL 881
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014

defined kinetics mechanism. The model comprises, inaddition to the regular conservation equations of theinvolved species, the dynamics of the sum of bisulfateand sulfate radicals, the sum of active radicals with singleand n monomers, and the total active and dead polymerconcentrations.
The change in first leading moments of molecular weightdistribution in polymerization is defined by means ofmoment generating functions. The steady state solutionof these moments gave zero, first, and second momentsof active and dead polymers. The weight-average molecularweight of the polymer was then calculated from the deadpolymer moments.
The results from the steady state solution of the rigorousdynamic model were compared to the experimental dataextracted from the literature, and it was seen that the predic-tions were remarkably better than a previous model availablein the literature. The dynamics revealed that the settling timepredicted was comparable to the industrial practice.
The developed model is, therefore, promising in thesense that it provides an effective means to analyze thedynamics of polyacrylonitrile polymerization and to morereliably predict the average molecular weight of the pro-duct. The model can be used for similar polymerizationsystems with minor changes.
NOTATION
a probability of propagationACN acrylonitrileb.o.m based on monomerI initiatork1 rate constant of formation of sulphate radical,
mol=(L.s)k2 rate constant of formation of bisufite radical,
mol=(L.s)ka rate constant of addition of radicals having
monomer, mol=(L.s)kd rate constant of formation of active radicals ,
mol=(L.s)kp rate constant of propagation of radicals having
monomer, mol=(L.s)
kr rate constant of recombination of radicals havingmonomer, mol=(L.s)
ktr rate constant of termination by transfer of radicalshaving monomer, mol=(L.s)
M monomerMx dead polymerMn,w number and weight-average molecular weight,
(kg=kgmol)Px living polymerqn the moment of dead polymerR parameterrn,w number and weight-average chain lengthsR radicalh residence time, sVA vinyl acetateWm molecular weight of monomerzn the moment of active polymer
REFERENCES
1. Masson, J.C.; Matzke, R.R. Jr. In: Acrylic Fiber Technology and
Applications, Marcel Deccer: New York, 1995.
2. Peebles, L.H. A kinetic model of Persulfate-bisulfite-initiated acryloni-
trile polymerization. J. Appl. Polym. Sci. 1973, 17, 113.
3. Pan, L.; Ma, J.; Zhao, G.; Zhang, Z. Advanced process control for
acrylonitrile polymerization. Beijing Huagong Daxue Xuebao, Ziran
Kexubean. 1996, 23, 31.
4. Hu, Q.; Chen, Z.; Shen, L.; Zhu, J. Simulation Training System for the
Whole Process of the Acrylic Plant. Jisuanji Yu Yingyong Huaxue.
1997, 14, 152.
5. Atasoy, I. Dynamic Model of Acrylonitrile Polymerization Reactor,
Masters Thesis, Ankara University, Ankara, Turkey, Dept. of
Chemical Engineering, 2000.
6. Shirodkar, P.P.; Tsien, G.O. A mathematical model for the production
of low density polyethylene in a tubular reactor. Chem. Eng. Sci. 1986,
41, 1031.
7. Zabisky, R.C.M.; Chan, W.M.; Gloor, P.E.; Hamielec, A.E. A kinetic
model for olefin polymerization in high-pressure tubular reactors: A
review and update. Polmy. J. 1992, 33, 2243.
8. Jackson, R.A.; Small, P.A.; Whiteley, K.S. Prediction of weight distri-
butions in branched polymers. J. Polym. Sci. 1973, 11, 1781.
9. Katz, S.; Saidel, G.M. Moments of the size distribution in radical
polymerization. AIChE J. 1967, 13, 319.
10. Sacks, M.E.; Lee, S.I.; Biesenberger, J.A. Effect of temperature varia-
tions on molecular weight distributions: batch chain addition poly-
merization. Chem. Eng. Sci. 1973, 28, 241.
882 I. ATASOY ET AL.
Dow
nloa
ded
by [
Inon
u U
nive
rsite
si]
at 1
6:44
24
Aug
ust 2
014





























