Embed Size (px)
Citation preview

,1 ,1
*University Medical Center Hamburg-Eppendorf, Children’s Hospital – Biochemistry, Hamburg, Germany
�Department of Medical Biochemistry and Developmental Biology, Biomedicum, Institute of Biomedicine, University of Helsinki,
Finland
�Research Center Borstel, Borstel, Germany
§Institute of Neuropathology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
¶Department of Neuroscience, Albert Einstein College of Medicine, Bronx, New York, USA
**Institute of Biochemistry, University of Kiel, Kiel, Germany
��Faculte de Medecine RTH Laennec, Institut de la Sante et de la Recherche Medicale, Lyon, France
Neuronal ceroid lipofuscinoses (NCL), also collectivelycalled Batten disease, are a group of autosomal recessivelyinherited neurodegenerative diseases that affect both childrenand adults. They are characterized by the accumulation ofautofluorescent lipopigment as well as aggregated proteinssuch as subunit c of mitochondrial ATP synthase orsphingolipid activator proteins (Tyynela 2005). In NCLpatients, visual failure leading to blindness, seizures andprogressive mental and physical deterioration are common(Santavuori et al. 2001).
Received April 14, 2008; revised manuscript received May 8, 2008;accepted May 13, 2008.Address correspondence and reprint requests to Thomas Braulke,
University Medical Center Hamburg-Eppendorf, Children’s Hospital –Biochemistry, Martinistrasse 52, Hamburg, 20246, Germany.E-mail: [email protected] authors contributed equally to this work.Abbreviations used: BMP, bis(monoacylglycero)phosphate; ctsd,
cathepsin D; galcer, galactosylceramide; GFAP, glial fibrillary acidicprotein; HPTLC, high performance thin layer chromatography;MPR300, 300 kDa mannose 6-phosphate receptor; MS, mass spec-trometry; NCL, neuronal ceroid lipofuscinoses; proSAP, prosaposin;TLC, thin layer chromatography.
Abstract
The neuronal ceroid lipofuscinoses comprise a group of
inherited severe neurodegenerative lysosomal disorders
characterized by lysosomal dysfunction and massive accu-
mulation of fluorescent lipopigments and aggregated proteins.
To examine the role of lipids in neurodegenerative processes
of these diseases, we analysed phospho- and glycolipids in
the brains of ctsd)/) and nclf mice, disease models of
cathepsin D and CLN6 deficiency, respectively. Both ctsd)/)and nclf mice exhibited increased levels of GM2 and GM3
gangliosides. Immunohistochemically GM2 and GM3 staining
was found preferentially in neurons and glial cells, respec-
tively, of ctsd)/) mice. Of particular note, a 20-fold elevation
of the unusual lysophospholipid bis(monoacylglycero)phos-
phate was specifically detected in the brain of ctsd)/) mice
accompanied with sporadic accumulation of unesterified
cholesterol in distinct cells. The impaired processing of the
sphingolipid activator protein precursor, an in vitro cathepsin D
substrate, in the brain of ctsd)/) mice may provide the
mechanistic link to the storage of lipids. These studies show
for the first time that cathepsin D regulates the lysosomal
phospho- and glycosphingolipid metabolism suggesting that
defects in the composition, trafficking and/or recycling of
membrane components along the late endocytic pathway may
be critical for the pathogenesis of early onset neuronal ceroid
lipofuscinoses.
Keywords: bis(monoacylglycero)phosphate, cathepsin D,
GM2 ganglioside, lysosomes, mouse brain, neuronal ceroid
lipofuscinosis.
J. Neurochem. (2008) 106, 1415–1425.
JOURNAL OF NEUROCHEMISTRY | 2008 | 106 | 1415–1425 doi: 10.1111/j.1471-4159.2008.05497.x
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425 1415

Currently, nine human NCL genes have been identified(Siintola et al. 2006a). CTSD, CLN1 and CLN2 encode thesoluble lysosomal enzymes cathepsin D (ctsd), palmitoyl-protein thioesterase 1 and tripeptidyl-peptidase I, respec-tively, whereas the function of the fourth soluble lysosomalCLN5 protein is unknown. CLN3, CLN6, CLN7, CLN8and the chloride transport protein, ClC-6 are polytopicmembrane proteins localized in the late endosomal/lyso-somal compartment, in the endoplasmic reticulum andendoplasmic reticulum-Golgi-intermediate compartment(Kyttala et al. 2006; Poet et al. 2006; Siintola et al.2007). CLN3 appears to be a multifunctional proteininvolved in the regulation of lysosomal acidification,lysosomal arginine import, apoptosis and vesicular mem-brane traffic (Kyttala et al. 2006). The functions of CLN6,CLN7 and CLN8 are unknown.
The pathomechanisms causing neurodegeneration indifferent NCLs are poorly understood. Whereas the intra-lysosomal accumulation of non-metabolized substrates isthe primary manifestation of the disease, there is, however,a lack of understanding of the association between storagematerial and neurodegeneration (Walkley 1998; Cooper2003). Thus, determining the downstream alterations inbiochemical and cellular pathways brought on by theprimary gene defect is essential for a complete understand-ing of pathogenesis. Among these, secondary changes inthe lipid composition of brain material for CLN1, CLN2,CLN3 and CLN8 patients and in the ovine CLN6 modelhave been reported (Bourre et al. 1979; Palmer et al. 1985;Svennerholm et al. 1987; Kakela et al. 2003; Hermanssonet al. 2005a). In this study, lipid profiles have beenexamined in different areas of brains from mouse modelsfor CLN6 [nclf; (Bronson et al. 1998)] and ctsd)/) (Saftiget al. 1995), differing in the severity of the disease.Ctsd)/) mice are normal at birth, manifest seizures andbecome blind near the terminal stage at postnatal day26 ± 1. The storage of autophagosome/autolysosome-likebodies was found in neuronal perikarya, (Koike et al.2005). Nclf mice, in contrast, are clinically normal until8 months of age, at which time they develop progressiveretinal atrophy followed by paresis, spastic paralysis andearly death by 12 months of age (Bronson et al. 1998). Inthis paper, we report the presence of increasing amounts ofGM2 and GM3 ganglioside in brains of primarily ctsd)/)and nclf mice. The most striking finding was that of highlyelevated levels of bis(monoacylglycero)phosphate (BMP),also known as lysobisphosphatidic acid, in brain andcultured neuronal and glial cells of ctsd)/) mice. The datademonstrate that the deficiency of cathpsin D leads tocharacteristic alterations in the lipid composition ofendosomal/lysosomal membranes in brain cells and suggestthat neuronal BMP storage is a significant factor in theearly onset of neurodegeneration in the ctsd)/) modelmice.
Material and methods
AnimalsCln6-defective mice (B6.Cg-Cln6nclf) were obtained from The
Jackson Laboratory (Bar Habor, ME, USA). Nclf and age-matched
control mice were used at an age of 5, 12, 19, 25 and 52 weeks,
respectively. Mice deficient for cathepsin D (ctsd)/)) in a C57BL/
6-129SV mixed background (Saftig et al. 1995) and their wild-type
littermates were used at postnatal day 24. The animals were
maintained and killed according to the institutional guidelines in
animal facilities of the University Medical Center Hamburg-
Eppendorf. Tissues were either immediately frozen and kept at
)25�C or lower prior to analysis or were fixed in 4% paraformal-
dehyde and stored in phosphate buffer.
AntibodiesPolyclonal antibodies against galactosylceramide (galcer) and
human 300 kDa mannose 6-phosphate receptor (MPR300) were
raised in rabbits (Korner et al. 1995; Brade et al. 2000). The
polyclonal goat antiserum against human saposin D was kindly
provided by Dr. K. Sandhoff (University of Bonn, Bonn, Germany).
The monoclonal antibody 6C4 against BMP was described
previously (Kobayashi et al. 1998) and was a generous gift by
Dr. J. Gruenberg (University of Geneva, Geneva, Switzerland). A
mouse monoclonal IgM antibody (MoAb 10–11) against GM2 was
kindly provided by Dr. P. Livingston (Memorial Sloan Kettering,
NY, USA) and a mouse IgG antibody (DH2) to GM3 ganglioside
was kindly provided by Dr. S. Hakomori (University of Seattle,
WA, USA). The anti-mouse lysosomal associated membrane
protein-1 (1D4B) antibody was obtained from the Developmental
Studies Hybridoma Bank under the auspices of NICHD and
maintained by the Department of Biological Sciences, University of
Iowa (Iowa, IA, USA). The monoclonal antibodies against glial
fibrillary acidic protein (GFAP) and NeuN were obtained from
Dako (Glostrup, Denmark) and Chemicon (Temecula, CA, USA),
respectively. Anti-mouse IgG-Cy3 and anti-rabbit IgG-Alexa488
conjugates were obtained from Sigma (St. Louis, MO, USA) and
Invitrogen (Carlsbad, CA, USA), respectively, and the peroxidase-
conjugated goat-anti-rabbit IgG was from Jackson Immuno-
Research Laboratories (West Grove, PA, USA) and Dianova
(Hamburg, Germany).
Lipid extraction and analysisTotal lipids were extracted from water homogenates obtained from
cerebrum, cerebellum and brain stem by chloroform : methanol
(v/v, 1 : 2) as described previously (Fujita et al. 1996). An aliquot
of the total lipid extract was simultaneously desalted and separated
into a neutral and an acidic lipid fractions using reverse-phase Bond
Elut C18 columns (Varian, Palo Alto, CA, USA) as described
(Kyrklund 1987). Each fraction was analysed by thin layer
chromatography (TLC) using high performance thin layer chroma-
tography plates (HPTLC 60; Merck, Darmstadt, Germany) devel-
oped in chloroform : methanol : water (v/v/v; 65 : 25 : 4). Lipids
were visualized by anisaldehyde spray and heating. The total lipid-
sialic acid content was measured directly on the anionic fraction
(Fujita et al. 1996). For ganglioside analysis, aliquots of the anioniclipid fraction corresponding to 3 mg fresh tissue weight were
spotted on HPTLC plates using a Camag Linomat 4 system. The
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425� 2008 The Authors
1416 | S. Jabs et al.

plates were developed in chloroform : methanol : 0.2% CaCl2(v/v/v; 45 : 55 : 10). Gangliosides were visualized using the sialic
acid specific resorcinol-hydrochlorhydric acid reagent. Quantitative
evaluation of the ganglioside patterns was performed by scanning
the plates at 580 nm with a Camag TLC Scanner II/CATS software
system (Fujita et al. 1996). The concentration of individual
gangliosides was then calculated from their proportion in relation
to the total lipidic sialic acid concentration, followed by correction
for the number of sialic acids in each compound.
Mass spectrometry (MS) was used to determine molecular
species of lipids in the dissected brain areas. Total lipid extracts
(Folch et al. 1957) and the collected fractions of anionic lipids of
homogenates of whole cerebrum were analysed by liquid chroma-
tography–mass spectrometry as described previously (Kakela et al.2003; Hermansson et al. 2005b) and by direct infusion tandem MS
(MS/MS) to detect sulfatides and gangliosides as precursors of
the lipid-class specific m/z 97 and 290 fragments, respectively
(Hsu et al. 1998; Tsui et al. 2005). The very high mass di- and
trisialoganglioside GD and GT, were detected as doubly charged
ions.
TLC-immunostaining of galactosylceramideImmunostaining of galcer was carried out after TLC as described
previously (Brade et al. 2000). Briefly, glycolipids were separated
on silica gel 60 TLC plates with aluminium support (Merck), in a
solvent system of chloroform : methanol : 25% aqueous NH4OH
(v/v/v, 65 : 35 : 5). For TLC-immunostaining, the plates were
incubated with blocking buffer (1% polyvinylpyrrolidone and
0.075% non-fat dry milk in 50 mM Tris–HCl, pH 7.4, 200 mM
NaCl) for 1 h at 20�C and then incubated with rabbit anti-galcer
(diluted 1 : 400) overnight 20�C. After five washings (5 min each)
in washing buffer (50 mM Tris–HCl, pH 7.4, 200 mM NaCl), the
plates were incubated with peroxidase-conjugated goat anti-rabbit
IgG diluted 1 : 1,000 in blocking buffer for 2 h at 20�C, washedfour times as before and fifth time in substrate buffer (0.1 M sodium
citrate buffer, pH 4.5). Bound antibody was then detected by
incubation with substrate buffer containing 4-chloro-1-naphthol
(3 mg/mL in MeOH) and hydrogen peroxide.
Preparation and cultivation of neurons and glial cellsHippocampal neurons were prepared from mice brains at embryonic
day 17 (Quitsch et al. 2005) and plated at a density of approxi-
mately 500 cells/mm2 on glass coverslips coated with poly-L-lysine.
Cells were grown in neurobasal medium (Invitrogen) supplemented
with 2% B27 (Invitrogen), 0.5 mM glutamine and 12.5 lMglutamate for 14 days. Glial cells were isolated at birth as described
previously (Adcock et al. 2004).
ImmunohistochemistryCryosections from brain were cut to a thickness of 7 lm and
immunostained with monoclonal antibodies to GFAP, BMP and
NeuN, according to published protocols (Glatzel et al. 2001).
Selected sections were double-stained for BMP and GFAP or BMP
and NeuN using peroxidase-coupled (Stept ABC Complex; Dako)
or alkaline phosphatase-coupled antibodies (Histofine; Nichirei
Biosciences, Tokyo, Japan) and diaminobenzidine or fast red as
chromogens, respectively. Fast red staining was visualized by
fluorescence and bright field microscopy using a Zeiss Axiostar
microscope (Oberkochen, Germany). For GM2 and GM3 immuno-
histochemistry, mouse brain was first fixed in 4% paraformaldehyde
in 0.1 M phosphate buffer for 6 h followed by storage in 0.1 M
phosphate buffer at 4�C. Filipin histochemistry and immunocyto-
chemical staining of GM2 and GM3 was performed as described
previously (McGlynn et al. 2004).
Total lipids
Neutral lipids
Anionic lipids
St (b) (a)
–/– –/– +/+
–/– +/+
+/+ –/– +/+ ctsd
chol
galcer
sulfatides
gangliosides
PC PS SM
PE
Fig. 1 Lipid profiles in the brain of wild-type and ctsd)/) mice. (a)
Total lipid extracts from cerebrum of 24-day-old wild-type (+/+) and
ctsd)/) mice were loaded on a C18 hydrophobic chromatography
column, and anionic and neutral lipids were eluted successively with
methanol : water (12 : 1) followed by chloroform : methanol (1 : 2),
respectively. The equivalent of 1 mg brain tissue of total and neutral
lipids and the equivalent of 2 mg brain tissue of anionic lipids were
separated on high performance thin layer chromatographic plates and
detected with the anisaldehyde spray reagent. The Rfs of lipid markers
are indicated (chol, cholesterol; galcer, galactosylceramide; PE, eth-
anolamine phosphoglycerides; PC, choline phosphoglycerides; PS,
serine phosphoglycerides; SM, sphingomyelin). The upper arrow
indicates the band of the initially unknown anionic lipid accumulating in
ctsd)/) mice brain, the lower arrow indicates the band corresponding
to gangliosides GM2 and GM3, not separated in this system. (b)
Galcer thin layer chromatography (TLC) immunostaining. Aliquots of
standard (ST) galcer and neutral lipid fractions of wild-type (+/+) and
ctsd)/) mice were separated by TLC and visualized by immuno-
staining with mouse anti galcer antibody. The identity of the immu-
noreactive lipid species at the front of the plates is unknown. ctsd)/),
cathepsin D deficiency.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425
BMP accumulation in cathepsin D deficiency | 1417

Other methodsDouble immuno-fluorescence microscopy and western blot analysis
were described previously (Heine et al. 2007).
Results
General lipid patterns in nclf, ctsd)/) and control miceTo compare the general lipid profile of brain tissue in the twomouse models differing in severity and course of the disease,aliquots of the total lipid extract and of the two fractions fromthe reverse-phase C18-columns containing either neutral oracidic lipids were studied by HPTLC, using anisaldehyde, a
general lipid spray reagent, for visualization. Obviousalterations were only observed in the cerebrum of ctsd)/)mice (Fig. 1a). In the neutral lipid fraction, the mostconspicuous finding was a reduction in the content of galcer,the major typical myelin lipid. This observation wasconfirmed by TLC immunostaning (Fig. 1b). No evidentalteration in the content of the other main lipids – unesterifiedcholesterol, phosphatidylethanolamine, phosphatidylcholineor sphingomyelin – could be detected. Among the anioniclipids, sulfatides also appeared reduced, in accordance withthe concomitant reduced level of galcer. Two other abnor-malities were evident. First, there was a clear indication for asignificant increase in the minor monosialogangliosides(lower arrow on Fig. 1a). Second, a compound not discer-nible in extracts from nclf or wild-type mice was observed inctsd)/) mice. It appeared as a smear (indicated by the upperarrow on Fig. 1a), migrating faster than sulfatides (andslower than phosphatidylethanolamine), and showed with theanisaldehyde spray a colour suggestive of a phosphoglyce-ride. The mobility and acidic properties of this compoundwere similar to those of BMP. Very similar lipid profiles werealso observed in the brain stem and cerebellum of 24-day-oldctsd)/) mice (not shown).
Accumulation of minor monosialogangliosides in the brainof nclf and ctsd)/)miceAs expected from initial testing of the acidic lipid fraction,more striking changes were observed in the monosialogan-glioside pattern of ctsd)/) mice (Fig. 2a). In wild-type mice,the minor monosialogangliosides, GM3 and GM2, werebarely detectable, constituting less than 1% of the totalgangliosides. Already at the age of 24 days, ctsd)/) miceshowed a clear increase in the concentration of GM2 andGM3 and also in GD3. Quantification by HPTLC combinedwith total sialic acid measurements showed that the contents
(c)
(b)
(a)
Fig. 2 Ganglioside profiles in the cerebral hemispheres of wild-type
and ctsd)/) mice. Total lipids from cerebrum of wild-type (+/+) and
ctsd)/) mice were extracted and separated by C18 hydrophobic
chromatography. Eluted fractions containing gangliosides (corre-
sponding to 3 mg of wet tissue) were analysed by high performance
thin layer chromatography and resorcinol-hydrochlorhydric acid
staining. (a) Ganglioside patterns obtained in cerebrum of two ctsd)/)and two wild-type mice (24 days of age). (b) The chromatogram was
quantified by densitometric scanning at 580 nm. (c) The ganglioside-
containing fraction isolated from cerebrum of wild-type (WT) and
ctsd)/) mice were analysed by mass spectroscopy. The gangliosides
were detected as precursors of m/z 290. As an insert, enlarged partial
spectra aquired by using higher collision energy are also shown. All
major ganglioside species had a 18:0 acyl and sphingosine (d18:1)
backbone. The gangliosides were detected either as singly charged
(1)) or doubly charged (2)) ions. Multiple minor cationized species are
present especially for GT1. Intensities of the spectra were normalized
to the main GD1 species. ctsd)/), cathepsin D deficiency.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425� 2008 The Authors
1418 | S. Jabs et al.

of GM2 and GM3 in ctsd)/) mice were approximately 20-and 6-fold higher, respectively, than those in wild-type mice(Fig. 2b), which was also confirmed by MS (Table 1;Fig. 2c). According to the MS/MS, the quantitatively impor-tant ganglioside species had 18:0 acyl and a sphingosine(d18:1) backbone. In addition, HPTLC showed also a 5-foldincrease in the minor ganglioside GD3 in ctsd)/) mice.
In brain tissue of 5 to 52-week-old nclf mice, a small andprogressive rise in the concentration of GM2 and GM3gangliosides occurred (Fig. 3). In 25-week-old animals, theconcentration of GM3 was about 4- to 5-fold to that observedin wild-type mice (90 nmol/g wet tissue vs. 20 nmol/g wet
tissue, respectively) and that of GM2 was doubled. At theage of 52 weeks, nclf mice showed 9- and 4-fold higherlevels of GM3 and GM2, respectively, compared with wild-type mice (measured by MS/MS; Fig. 3b).
Accumulation of BMP in the brain of ctsd)/) miceTo identify the unknown lipid species accumulating in brainof ctsd)/) mice (Fig. 1a) and to further document diffe-rences in ganglioside patterns, MS was performed on theanionic lipid fractions (Table 1). This allowed final identi-fication of the unknown compound as BMP. Furthermore,a striking increase in this lipid in ctsd)/) mouse brain –20-fold compared with the minute normal level – wasdocumented. The most abundant BMP species were dipoly-unsaturated, 44:12 (22:6n-3/22:6n-3), constituting half of thislipid (Fig. 4). According to MS, the level of sulfatides wasalso confirmed to be slightly lower in the ctsd)/) brain(Table 1).
Neuronal and glial lipid storage in ctsd)/) miceLight microscopic studies of GM2 and GM3 ganglioside-immunoreactive sections showed staining in neurons andglial cells, widely scattered throughout the cerebral cortex,subcortical areas, brain stem and to a lesser degree in thecerebellum of 24-day-old ctsd)/) mice. Higher magnifica-tions of the immunostained sections showed the presence ofGM2-labelled vesicles in the cytoplasm, and many of thecortical neurons exhibiting this staining were clearly pyra-midal in morphology (Fig. 5b and c). GM3 staining, incontrast, appeared to be mostly in glial cells (Fig. 5e and f).Immunohistochemical staining of brain tissue from controlwild-type mice revealed no immunoreactivity for either ofthese gangliosides (Fig. 5a and d).
A uniform strong BMP labelling was also found immu-nohistochemically across the brain of ctsd)/) mice. BMP-positive structures were found both in the cortex andhippocampus in cells compatible with reactive astrocytesand neurons (Fig. 6). The neuronal and astroglial identity ofthese cells was confirmed by double labelling experimentsusing antibodies directed against the astroglial and neuronalmarkers GFAP and NeuN, respectively (Fig. 7).
BMP accumulates in cultured ctsd-deficient neuronal andglial cellsTo analyse whether cultured brain cells accumulate BMP andto determine its subcellular localization, hippocampalneurons and glial cells were isolated from ctsd)/+ andctsd)/) mice and analysed by double immunofluorescencemicroscopy. After culturing for 12 days, both glial cells(Fig. 8a) and neuronal cells (Fig. 8b) showed strong BMPstaining in large vesicular structures which were partiallyimmunoreactive for MPR300.
In fibroblasts and extraneural organs of patients withNiemann-Pick disease, increased levels of BMP were found
Table 1 Relative intensities of main lipid species in the anionic brain
lipid fractions of ctsd)/) and wild-type control mice, and the ratio
ctsd)/) and control for each lipid
Lipid species Chains
Relative intensities
Control ctsd)/) ctsd)/) : control
PA 34:1 16:0/18:1 1.76 1.47 0.8
PA 36:1 18:0/18:1 1.68 0.97 0.6
PA 38:4 18:0/20:4 1.29 1.31 1.0
PA 40:6 18:0/22:6 3.60 2.44 0.7
BMP 38:4 18:0/20:4 0.07 0.86 11.7
BMP 40:8 20:4/20:4 0.05 1.07 22.0
BMP 42:10 20:4/22:6 0.06 1.18 19.8
BMP 44:12 22:6/22:6 0.14 2.69 18.7
PG 36:4 16:0/20:4 1.28 0.79 0.6
Sulf 18:0 18:0 2.50 3.00 1.2
Sulf 22:0 22:0 1.77 0.54 0.3
Sulf 24:1 24:1 2.19 0.97 0.4
Sulf 24:0 24:0 1.27 0.95 0.7
Sulf-OH 18:0 18:0 2.03 1.07 0.5
Sulf-OH 24:0 24:0 1.96 1.10 0.6
PS 34:1 16:0/18:1 2.22 2.92 1.3
PS 36:4 16:0/20:4 0.84 0.93 1.1
PS 36:2 18:1/18:1 2.64 1.89 0.7
PS 36:1 18:0/18:1 4.83 2.99 0.6
PS 38:4 18:0/20:4 3.41 3.02 0.9
PS 38:1 18:0/20:1 1.46 0.54 0.4
PS 40:7 18:1/22:6 1.87 1.62 0.9
PS 40:6 18:0/22:6 23.31 25.43 1.1
PS 40:4 18:0/22:4 3.17 3.45 1.1
PI 36:4 16:0/20:4 3.44 4.39 1.3
PI 38:5 18:1/20:4 2.68 3.39 1.3
PI 38:4 18:0/20:4 16.04 18.68 1.2
PI 40:6 18:0/22:6 0.98 1.36 1.4
GM3 18:0 18:0/d18:1 0.17 0.93 5.3
GM2 18:0 18:0/d18:1 0.07 0.90 12.4
GM1 18:0 18:0/d18:1 1.22 1.02 0.8
GD1 18:0 18:0/d18:1 8.61 5.03 0.6
GT1 18:0 18:0/d18:1 1.39 1.10 0.8
PA, phosphatidic acid, BMP, bis(monoacylglycero)phosphate; PG,
phosphatidylglycerol, Sulf, sulfatide, Sulf-OH, alpha-hydroxysulfatide,
PS, phosphatidylserine, PI, phosphatidylinositol; ctsd, cathepsin D.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425
BMP accumulation in cathepsin D deficiency | 1419

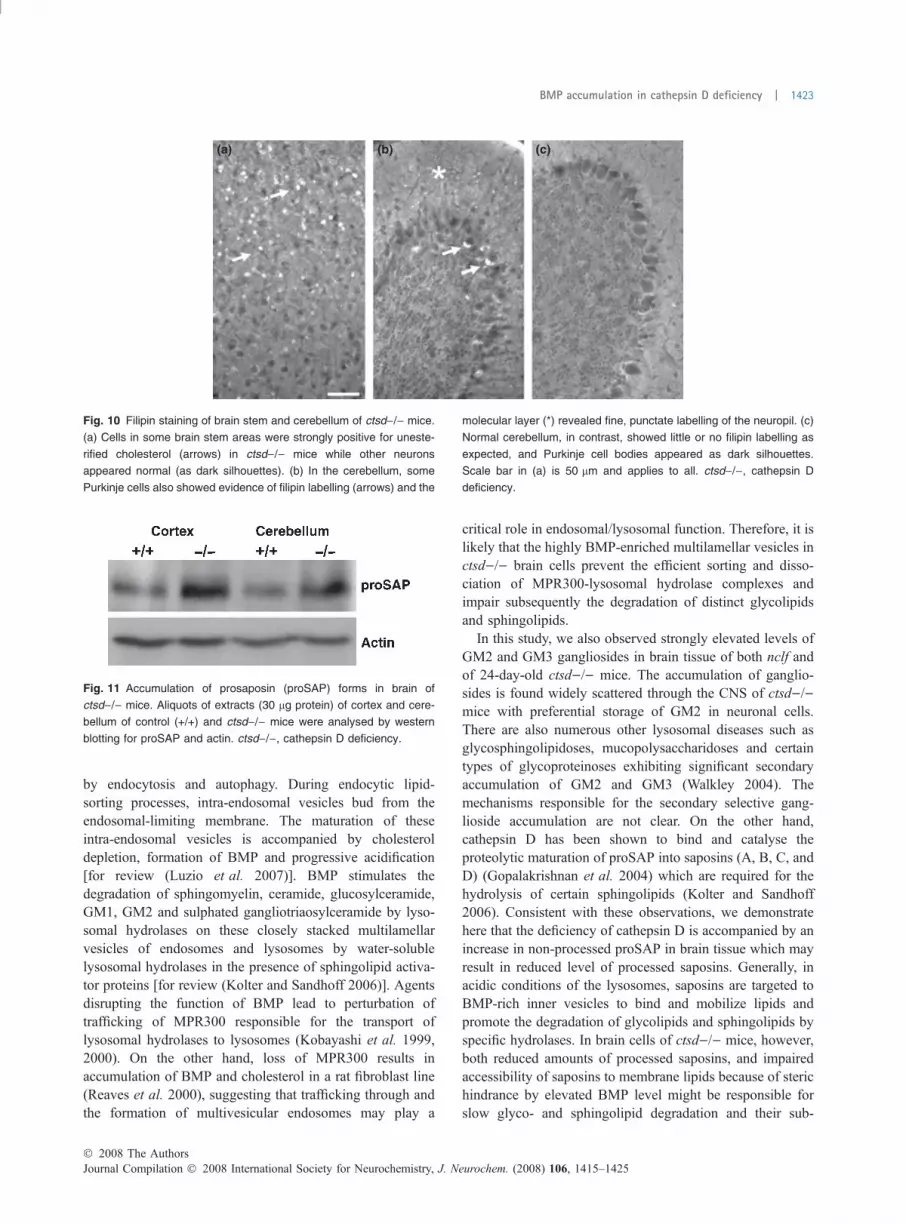
to be accompanied with storage of cholesterol (Vanier 1983),(Kobayashi et al. 1999). Filipin staining for cholesterol incultured neurons of ctsd)/) mice were not altered comparedwith wild-type cells (Fig. 9). Filipin staining of cerebralcortex, subcortical areas, cerebellum and brain stem ofctsd)/) mice, however, provided evidence of substantialaccumulation of unesterified cholesterol in scattered neuronsand glia (Fig. 10). At times, small filipin-positive punctawere observed in the neuropil and not clearly associated withindividual cells. This was a particularly prominent feature of
the molecular layer of the cerebellar cortex. Many neurons,for example in brain stem and elsewhere, were clearly notstained for filipin, indicating cell-selective differences to theresponse to cathepsin D deficiency.
Accumulation of prosaposin in the brain of ctsd)/) miceProsaposin (proSAP) is a precursor of saposins A to D whichare essential cofactors in the degradation of sphingolipids(Kolter and Sandhoff 2006). Because cathepsin D has beenshown to cleave the 60–73 kDa proSAP precursor into the
(a) (b)
Fig. 3 Ganglioside profiles in the cerebral hemispheres of wild-type
and nclf mice. (a) Ganglioside profiles of 5-, 12-, 19- and 25-week-old
mice. Gangliosides were prepared and analysed as described in Fig-
ure 2. As a control, cerebrum of 25-week-old wild-type (WT) mice was
used. For the ages of 5 weeks and 25 weeks, brains of two different
WT and nclf mice were analised, showing similar patterns of gan-
gliosides. The positions of ganglioside markers are indicated. (b) The
ganglioside-containing fractions from the cerebrum of 52-week-old WT
and nclf mice were analysed by mass spectroscopy as described in the
legend of Fig. 2.
(a)
(b) Fig. 4 MS analysis of anionic brain lipid
fractions. Averaged mass spectra covering
the elution range of phosphatidic acid
(PA) and bis(monoacylglycero)phosphate
(BMP) of negative-mode LC–MS runs of
anionic lipid fractions from (a) wild-type and
(b) ctsd)/) mice brain. The intensity scale
of the spectra normalized to BMP 44:12
of the ctsd)/) brain is shown. LC/MS,
liquid chromatography/mass spectrometry.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425� 2008 The Authors
1420 | S. Jabs et al.

8–11 kDa saposins in vitro (Gopalakrishnan et al. 2004),western blot analyses for proSAP were carried out. Figure 11showed increased amounts of proSAP in extracts of cortexand cerebellum of ctsd)/) mice.
Discussion
The major finding of the present study is that the anioniclipid BMP accumulates in brain tissue of mice deficient in thelysosomal aspartyl protease cathepsin D. The ctsd)/) mouse
represents a severe form of NCL with early onset seizures,blindness and death at postnatal day 26 ± 1 (Saftig et al.1995). Along with storage of granular osmiophilic depositsand the mitochondrial ATP synthase subunit c, accumulationof autophagic structures in neurons in the brain has beendescribed (Koike et al. 2005). The pathological changes andthe course of the disease in ctsd)/) mice resemble therecently reported congenital form of NCL in humansassociated with enzymatically inactive mutant cathepsin D(Siintola et al. 2006b). BMP is enriched in internal mem-branes of multivesicular endosomes and lysosomes account-ing for 15% of total organelle phospholipids (Kobayashiet al. 1998). In acidic lipid fractions of brain tissues of24-day-old ctsd)/) mice, different species of BMP withvarious fatty acid compositions were 11- to 22-fold increasedcompared with wild-type controls. The accumulation ofBMP was found throughout the brain of ctsd)/) mice both inneuronal and glial cells. Furthermore, double immunofluo-rescence microscopy of cultured primary neuronal and glialcells of ctsd)/) mice provide evidence that BMP is localizedboth in MPR300-positive endosomes and in the cholesterol-negative lysosomal compartment. BMP is not synthesizedde novo but is rather formed during degradation ofphosphatidylglycerol and cardiolipin in the endosomal/lysosomal compartment (Somerharju and Renkonen 1980;Amidon et al. 1996). Because of its unusual sn1, sn1¢configuration, BMP appears to be resistant to most lysosomalphospholipases (Matsuzawa and Hostetler 1979). It is likelythat the deficiency of cathepsin D impairs the proteolyticactivation of lysosomal enzymes functioning in the synthesisor degradation of BMP with its subsequent accumulation.Furthermore, in fibroblasts of patients with various forms ofNCL, alterations in lysosomal pH have been reported (Hol-opainen et al. 2001) which may also occur in ctsd)/) brain
(a) (b) (c)
(d) (e) (f)
Fig. 5 Accumulation of GM2 and GM3 gangliosides in neurons and
glial cells of ctsd)/) mice. Brain sections of control mice (a and d) and
ctsd)/) mice (b, c and e, f) were stained for GM2 (a–c) and GM3
gangliosides (d–f). GM2 ganglioside was found primarily in neurons
(b,c; arrows) whereas GM3 was most prominently found in glial-type
cells. Calibration bar in (f) equals 10 lm and applies to all panels.
(a) (b)
(c) (d)
Fig. 6 Immunostaining for bis(monoacyl-
glycero)phosphate (BMP) in ctsd)/) mouse
brain. Cryosections of 24-day-old ctsd)/)mice show strong BMP immunoreactivity in
the cortex and hippocampus (c, d) when
compared with ctsd)/+ (a, b). Scale bars
are 50 lm. ctsd)/), cathepsin D deficiency.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425
BMP accumulation in cathepsin D deficiency | 1421

cells and impair the activities of lysosomal hydrolasesinvolved in the metabolism of BMP. This may subsequentlylead to the accumulation of BMP. Neither in brain tissue ofnclf mice, the murine model of the variant late infantile NCLdisease CLN6, or in Cln3exon1)6del mice (S. Jabs andM.Vanier, unpublished data) nor in the cerebral cortex ofpatients with juvenile CLN3, the accumulation of BMP was
observed (Kahma et al. 1976; Kakela et al. 2003). There arealso no reports on BMP storage in other NCL mouse modelsso far. In brain tissue, however, from patients with infantileNCL (mutant CLN1) with an onset age of 8–18 month,moderately elevated levels of BMP were detected (Kahmaet al. 1976; Kakela et al. 2003). In addition, increased level ofBMP in association with marked elevations of cholesterolwere found in the liver but not in the brain of patients withNiemann-Pick diseases or in spm, Niemann-Pick Type C1 orC2 defective mice and murine models of the lysosomal lipidstorage disease of Niemann-Pick type C (Rouser et al. 1968;Vanier 1983; Nakashima et al. 1984; Sleat et al. 2004). Thesedata suggest that alterations in brain BMP level might bespecific and associated with severe, early onset forms of NCL.It remains to be determined whether BMPmight be suitable asbiomarker for these NCL forms.
For degradation, glycolipids and sphingolipids of plasmamembranes are transported to acidic compartments of the cell
(a) (b) (c)
(d) (e) (f)
Fig. 7 Accumulation of bis(monoacylglyc-
ero)phosphate (BMP) in neurons and glial
cells of ctsd)/) mice. Cryosections of
24-day-old ctsd)/) hippocampi were dou-
ble-labelled for BMP (a, d) and either the
astroglial marker glial fibrillary acidic protein
(GFAP) (b) or the neuronal marker NeuN
(e). Analysis of the merged immunostains
(c, f) show BMP immunoreactivity in hip-
pocampal neurons (N) as well in white mat-
ter (W). Scale bar in (a) is 50 lm. ctsd)/),
cathepsin D deficiency.
Fig. 8 Localization of bis(monoacylglycero)phosphate (BMP) in cul-
tured brain cells. Glial cells (a) and neurons (b) were isolated from
control (ctsd)/+) and ctsd)/) mice and cultured for 12 days. The cells
were double-immunostained for BMP (red) and the 300 kDa mannose
6-phosphate receptor (MPR300; green), a Golgi/endosomal marker.
Scale bars: 10 lm. ctsd)/), cathepsin D deficiency.
Fig. 9 Different subcellular localization of bis(monoacylglycero)
phosphate (BMP) and cholesterol in hippocampal neurons. Hippo-
campal primary neurons were isolated from control (ctsd)/+) and
ctsd)/) mice and cultured for 12 days. Neurons were double-labelled
for BMP (red) and filipin (blue) staining unesterified cholesterol. Note
the accumulation of bright BMP vesicles in the perikarya; filipin
staining is detrimental to the labelling of BMP. Scale bars: 10 lm.
ctsd)/), cathepsin D deficiency.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425� 2008 The Authors
1422 | S. Jabs et al.
by endocytosis and autophagy. During endocytic lipid-sorting processes, intra-endosomal vesicles bud from theendosomal-limiting membrane. The maturation of theseintra-endosomal vesicles is accompanied by cholesteroldepletion, formation of BMP and progressive acidification[for review (Luzio et al. 2007)]. BMP stimulates thedegradation of sphingomyelin, ceramide, glucosylceramide,GM1, GM2 and sulphated gangliotriaosylceramide by lyso-somal hydrolases on these closely stacked multilamellarvesicles of endosomes and lysosomes by water-solublelysosomal hydrolases in the presence of sphingolipid activa-tor proteins [for review (Kolter and Sandhoff 2006)]. Agentsdisrupting the function of BMP lead to perturbation oftrafficking of MPR300 responsible for the transport oflysosomal hydrolases to lysosomes (Kobayashi et al. 1999,2000). On the other hand, loss of MPR300 results inaccumulation of BMP and cholesterol in a rat fibroblast line(Reaves et al. 2000), suggesting that trafficking through andthe formation of multivesicular endosomes may play a
critical role in endosomal/lysosomal function. Therefore, it islikely that the highly BMP-enriched multilamellar vesicles inctsd)/) brain cells prevent the efficient sorting and disso-ciation of MPR300-lysosomal hydrolase complexes andimpair subsequently the degradation of distinct glycolipidsand sphingolipids.
In this study, we also observed strongly elevated levels ofGM2 and GM3 gangliosides in brain tissue of both nclf andof 24-day-old ctsd)/) mice. The accumulation of ganglio-sides is found widely scattered through the CNS of ctsd)/)mice with preferential storage of GM2 in neuronal cells.There are also numerous other lysosomal diseases such asglycosphingolipidoses, mucopolysaccharidoses and certaintypes of glycoproteinoses exhibiting significant secondaryaccumulation of GM2 and GM3 (Walkley 2004). Themechanisms responsible for the secondary selective gang-lioside accumulation are not clear. On the other hand,cathepsin D has been shown to bind and catalyse theproteolytic maturation of proSAP into saposins (A, B, C, andD) (Gopalakrishnan et al. 2004) which are required for thehydrolysis of certain sphingolipids (Kolter and Sandhoff2006). Consistent with these observations, we demonstratehere that the deficiency of cathepsin D is accompanied by anincrease in non-processed proSAP in brain tissue which mayresult in reduced level of processed saposins. Generally, inacidic conditions of the lysosomes, saposins are targeted toBMP-rich inner vesicles to bind and mobilize lipids andpromote the degradation of glycolipids and sphingolipids byspecific hydrolases. In brain cells of ctsd)/) mice, however,both reduced amounts of processed saposins, and impairedaccessibility of saposins to membrane lipids because of sterichindrance by elevated BMP level might be responsible forslow glyco- and sphingolipid degradation and their sub-
(a) (b) (c)
Fig. 10 Filipin staining of brain stem and cerebellum of ctsd)/) mice.
(a) Cells in some brain stem areas were strongly positive for uneste-
rified cholesterol (arrows) in ctsd)/) mice while other neurons
appeared normal (as dark silhouettes). (b) In the cerebellum, some
Purkinje cells also showed evidence of filipin labelling (arrows) and the
molecular layer (*) revealed fine, punctate labelling of the neuropil. (c)
Normal cerebellum, in contrast, showed little or no filipin labelling as
expected, and Purkinje cell bodies appeared as dark silhouettes.
Scale bar in (a) is 50 lm and applies to all. ctsd)/), cathepsin D
deficiency.
Fig. 11 Accumulation of prosaposin (proSAP) forms in brain of
ctsd)/) mice. Aliquots of extracts (30 lg protein) of cortex and cere-
bellum of control (+/+) and ctsd)/) mice were analysed by western
blotting for proSAP and actin. ctsd)/), cathepsin D deficiency.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425
BMP accumulation in cathepsin D deficiency | 1423

sequent accumulation. Thus, the impaired processing ofproSAP provides a possible mechanistic link between storageof lipids and the deficiency of the lysosomal proteasecathepsin D.
In conclusion, these studies show for the first time thatcathepsin D regulates the lysosomal phospho- and glyco-sphingolipid metabolism. Whatever the precise mechanisminvolved in the accumulation of BMP and GM2/GM3 in thebrain of ctsd)/) mice, our results suggest that defects in thecomposition, trafficking and/or recycling of membranecomponents along the late endocytic pathway may be criticalfor the pathogenesis of early onset NCL.
Acknowledgements
This work was supported by the Commission of the European
Communities (Grant No. 503051) to SJ, AQ, BK, JT and TB and
Deutsche Forschungsgemeinschaft (Research Unit 885) to MG and
TB. RK and JT were supported by the Academy of Finland
(Fellowship No. 111 261 and 214343).
References
Adcock K. H., Brown D. J., Shearer M. C., Shewan D., Schachner M.,Smith G. M., Geller H. M. and Fawcett J. W. (2004) Axonbehaviour at Schwann cell – astrocyte boundaries: manipulation ofaxon signalling pathways and the neural adhesion molecule L1 canenable axons to cross. Eur. J. Neurosci. 20, 1425–1435.
Amidon B., Brown A. and Waite M. (1996) Transacylase and phos-pholipases in the synthesis of bis(monoacylglycero)phosphate.Biochemistry 35, 13995–14002.
Bourre J., Haltia M., Daudu O., Monge M. and Baumann N. (1979)Infantile form of so-called neuronal ceroid lipofuscinosis: lipidbiochemical studies, fatty acid analysis of cerebroside sulfatidesand sphingomyelin, myelin density profile and lipid composition.Eur. Neurol. 18, 312–321.
Brade L., Vielhaber G., Heinz E. and Brade H. (2000) In vitro charac-terization of anti-glucosylceramide rabbit antisera. Glycobiology10, 629–636.
Bronson B. T., Donahue L. R., Johnson K. R., Tanner A., Lane P. W. andFaust J. R. (1998) Neuronal ceroid lipofuscinosis (nclf), a newdisorder of the mouse linked to chromosome 9. Am. J. Med. Genet.77, 289–297.
Cooper J. D. (2003) Progress towards understanding the neurobiology ofBatten disease or neuronal ceroid lipofuscinosis. Curr. Opin.Neurol. 16, 121–128.
Folch J., Lees M. and Sloane-Stanley G. (1957) A simple method for theisolation and purification of total lipides from animal tissue. J. Biol.Chem. 226, 497–509.
Fujita N., Suzuki K., Vanier M. T., Popko B., Maeda N., Klein A.,Henseler M., Sandhoff K., Nakayasu H. and Suzuki K. (1996)Targeted disruption of the mouse sphingolipid activator proteingene: a complex phenotype, including severe leukodystrophy andwide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 5,711–725.
Glatzel M., Heppner F. L., Albers K. M. and Aguzzi A. (2001) Sym-pathetic innervation of lymphoreticular organs is rate limiting forprion neuroinvasion. Neuron 31, 25–34.
Gopalakrishnan M. M., Grosch H.-W., Locatelli-Hoops S., Werth N.,Smolenova E., Nettersheim M., Sandhoff K. and Hasilik A. (2004)
Purified recombinant human prosaposin forms oligomers that bindprocathepsin D and affect its autoactivation. Biochem. J. 383,507–515.
Heine C., Quitsch A., Storch S., Martin Y., Lonka L., Lehesjoki A.-E.,Mole S. E. and Braulke T. (2007) Topology and endoplasmicreticulum retention signals of the lysosomal storage disease-relatedmembrane protein CLN6. Mol. Membr. Biol. 24, 74–87.
Hermansson M., Kakela R., Berghall M., Lehesjoki A.-E., Somerharju P.and Lahtinen U. (2005a) Mass spectrometric analysis revealschanges in phospholipid, neutral sphingolipid and sulfatidemolecular species in progressive epilepsy with mental retardation,EPMR, brain: a case study. J. Neurochem. 95, 609–617.
Hermansson M., Uphoff A., Kakela R. and Somerharju P. (2005b)Quantitative analysis of complex lipidomes by liquid chromatog-raphy/mass spectrometry. Anal. Chem. 77, 2166–2175.
Holopainen J. M., Saarikoski J., Kinnunen P. K. J. and Jarvela I. (2001)Elevated lysosomal pH in neuronal ceroid lipofuscinoses (NCLs).Eur. J. Biochem. 268, 5851–5856.
Hsu F.-F., Bohrer A. and Turk J. (1998) Electrospray ionization tan-dem mass spectrometric analysis of sulfatide. Determination offragmentation patterns and characterization of molecular speciesexpressed in brain and in pancreatic islets. Biochim. Biophys. Acta1392, 202–216.
Kahma K., Brotherus J., Haltia M. and Renkonen O. (1976) Low andmoderate concentrations of lysobisphosphatidic acid in brain andliver of patients affected by some storage diseases. Lipids 11,539–544.
Kakela R., Somerharju P. and Tyynela J. (2003) Analysis of phospho-lipid molecular species in brains from patients with infantile andjuvenile neuronal-ceroid lipofuscinosis using liquid chromatogra-phy–electrospray ionization mass spectrometry. J. Neurochem. 84,1051–1065.
Kobayashi T., Stang E., Fang K., de Moerloose P., Parton R. and Gru-enberg J. (1998) A lipid associated with the antiphospholipidsyndrome regulates endosome structure and function. Nature 392,193–197.
Kobayashi T., Beuchat M., Lindsay M., Frias S., Palmiter R., SakurabaH., Parton R. and Gruenberg J. (1999) Late endosomal membranesrich in lysobisphosphatidic acid regulate cholesterol transport. Nat.Cell Biol. 1, 113–118.
Kobayashi T., Vischer U., Rosnoblet C., Lebrand C., Lindsay M., PartonR., Kruithof E. and Gruenberg J (2000) The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments inhuman endothelial cells. Mol. Biol. Cell 11, 1829–1843.
Koike M., Shibata M., Waguri S. et al. (2005) Participation of autophagyin storage of lysosomes in neurons from mouse models of neuronalceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 167, 1713–1728.
Kolter T. and Sandhoff K. (2006) Sphingolipid metabolism diseases.Biochim. Biophys. Acta 1758, 2057–2079.
Korner C., Nurnberg B., Uhde M. and Braulke T. (1995) Mannose6-phosphate/insulin-like growth factor II receptor fails to interactwith G-proteins. J. Biol. Chem. 270, 287–295.
Kyrklund T. (1987) Two procedures to remove polar contaminants froma crude brain lipid extract by using prepacked reversed-phasecolumns. Lipids 22, 274–277.
Kyttala A., Lahtinen U., Braulke T. and Hofmann S. L. (2006) Func-tional biology of the neuronal ceroid lipofuscinoses (NCL) pro-teins. Biochim. Biophys. Acta 1762, 920–933.
Luzio J., Pryor P. and Bright N. (2007) Lysosomes: fusion and function.Nat. Rev. Mol. Cell Biol. 8, 622–632.
Matsuzawa Y. and Hostetler K. (1979) Degradation of bis(monoacyl-glycero)phosphate by an acid phosphodiesterase in rat liver lyso-somes. J. Biol. Chem. 254, 5997–6001.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425� 2008 The Authors
1424 | S. Jabs et al.

McGlynn R., Dobrenis K. and Walkley S. U. (2004) Differentialsubcellular localization of cholesterol, gangliosides, and glycos-aminoglycans in murine models of mucopolysaccharide storagedisorders. J. Comp. Neurol. 480, 415–425.
Nakashima S., Nagata K., Banno Y., Sakiyama T., Kitagawa T.,Miyawaki S. and Nozawa Y. (1984) A mouse model for Niemann-Pick disease: phospholipid class and fatty acid composition ofvarious tissues. J. Lipid Res. 25, 219–227.
Palmer D. N., Husbands D. and Jolly R. (1985) Phospholipid fatty acidsin brains of normal sheep and sheep with ceroid-lipofuscinosis.Biochim. Biophys. Acta 834, 159–163.
Poet M., Kornak U., Schweizer M. et al. (2006) Lysosomal storagedisease upon disruption of the neuronal chloride transport proteinClC-6. Proc. Natl Acad. Sci. USA 103, 13854–13859.
Quitsch A., Berhorster K., Liew C. W., Richter D. and Kreienkamp H.-J.(2005) Postsynaptic Shank antagonizes dendrite branching inducedby the leucine-rich repeat protein densin-180. J. Neurosci. 25,479–487.
Reaves B., Row P., Bright N., Luzio J. and Davidson H. (2000) Loss ofcation-independent mannose 6-phosphate receptor expressionpromotes the accumulation of lysobisphosphatidic acid in multil-amellar bodies. J. Cell Sci. 113, 4099–4108.
Rouser G., Kritchevsky G., Knudson A. G. J. and Simon G. (1968)Accumulation of a glycerolphospholipid in classical Niemann-Pickdisease. Lipids 3, 287–290.
Saftig P., Hetman M., Schmahl W. et al. (1995) Mice deficient for thelysosomal proteinase cathepsin D exhibit progressive atrophy ofthe intestinal mucosa and profound destruction of lymphoid cells.EMBO J. 14, 3599–3608.
Santavuori P., Vanhanen S. and Autti T. (2001) Clinical and neurora-diological diagnostic aspects of neuronal ceroid lipofuscinosesdisorders. Eur. J. Paediatr. Neurol. 5 (Suppl A), 157–161.
Siintola E., Lehesjokia A.-E. and Mole S. E. (2006a) Molecular geneticsof the NCLs – status and perspectives. Biochim. Biophys. Acta1762, 857–864.
Siintola E., Partanen S., Stromme P., Haapanen A., Haltia M., MaehlenJ., Lehesjoki A. and Tyynela J. (2006b) Cathepsin D deficiencyunderlies congenital human neuronal ceroid-lipofuscinosis. Brain129, 1353–1356.
Siintola E., Topcu M., Aula N. et al. (2007) The novel neuronal ceroidlipofuscinosis gene MFSD8 encodes a putative lysosomal trans-porter. Am. J. Hum. Genet. 81, 136–146.
Sleat D., Wiseman J., El-Banna M., Price S., Verot L., Shen M., TintG., Vanier M., Walkley S. and Lobel P. (2004) Genetic evidencefor nonredundant functional cooperativity between NPC1 andNPC2 in lipid transport. Proc. Natl Acad. Sci. USA 101,5886–5891.
Somerharju P. and Renkonen O. (1980) Conversion of phosphatidyl-glycerol lipids to bis(monoacylglycero)phosphate in vivo. Biochim.Biophys. Acta 618, 407–419.
Svennerholm L., Fredman P., Jungbjer B., Mansson J., Rynmark B.,Bostrom K., Hagberg B., Noren L. and Santavuori P. (1987)Large alterations in ganglioside and neutral glycosphingolipidpatterns in brains from cases with infantile neuronal ceroidlipofuscinosis/polyunsaturated fatty acid lipidosis. J. Neurochem.49, 1772–1783.
Tsui Z.-C., Chen Q.-R., Thomas M., Samuel M. and Cui Z. (2005) Amethod for profiling gangliosides in animal tissues using electro-spray ionization-tandem mass spectrometry. Anal. Biochem. 341,251–258.
Tyynela J.(2005) Neuronal ceroid lipofuscinoses, in Lysosomes (Saftig P.(ed.), pp. 82–99. Landes Bioscience/Eurekah.com, Georgetown,TX, USA.
Vanier M. T. (1983) Biochemical studies in Niemann-Pick disease. I.Major sphingolipids of liver and spleen. Biochim. Biophys. Acta750, 178–184.
Walkley S. U. (1998) Cellular pathology of lysosomal storage disorders.Brain Pathol. 8, 175–193.
Walkley S. U. (2004) Secondary accumulation of gangliosides in lyso-somal storage disorders. Sem. Cell Dev. Biol. 15, 433–444.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 106, 1415–1425
BMP accumulation in cathepsin D deficiency | 1425





























