Embed Size (px)
Citation preview

Seediscussions,stats,andauthorprofilesforthispublicationat:https://www.researchgate.net/publication/51553224
AndréL,GouziF,ThireauJ,etal.Carbonmonoxideexposureenhancesarrhythmiaaftercardiacstress:involvementofoxidativestress
ARTICLEinARCHIVFÜRKREISLAUFFORSCHUNG·AUGUST2011
ImpactFactor:5.41·DOI:10.1007/s00395-011-0211-y·Source:PubMed
CITATIONS
16
READS
50
17AUTHORS,INCLUDING:
JeromeThireau
FrenchInstituteofHealthandMedicalRese…
53PUBLICATIONS908CITATIONS
SEEPROFILE
AlainLacampagne
FrenchInstituteofHealthandMedicalRese…
109PUBLICATIONS2,979CITATIONS
SEEPROFILE
SylvainRichard
FrenchInstituteofHealthandMedicalRese…
161PUBLICATIONS3,509CITATIONS
SEEPROFILE
OlivierCazorla
FrenchInstituteofHealthandMedicalRese…
84PUBLICATIONS2,266CITATIONS
SEEPROFILE
Availablefrom:FaresGouzi
Retrievedon:03February2016

ORIGINAL CONTRIBUTION
Carbon monoxide exposure enhances arrhythmia after cardiacstress: involvement of oxidative stress
Lucas Andre • Fares Gouzi • Jerome Thireau • Gregory Meyer • Julien Boissiere •
Martine Delage • Aldja Abdellaoui • Christine Feillet-Coudray • Gilles Fouret •
Jean-Paul Cristol • Alain Lacampagne • Philippe Obert • Cyril Reboul •
Jeremy Fauconnier • Maurice Hayot • Sylvain Richard • Olivier Cazorla
Received: 3 February 2011 / Revised: 22 July 2011 / Accepted: 24 July 2011
� Springer-Verlag 2011
Abstract Arrhythmias following cardiac stress are a key
predictor of death in healthy population. Carbon monoxide
(CO) is a ubiquitous pollutant promoting oxidative stress
and associated with hospitalization for cardiovascular dis-
ease and cardiac mortality. We investigated the effect of
chronic CO exposure on the occurrence of arrhythmic
events after a cardiac stress test and the possible involve-
ment of related oxidative stress. Wistar rats exposed
chronically (4 weeks) to sustained urban CO pollution
presented more arrhythmic events than controls during
recovery after cardiac challenge with isoprenaline in vivo.
Sudden death occurred in 22% of CO-exposed rats versus
0% for controls. Malondialdehyde (MDA), an end-product
of lipid peroxidation, was increased in left ventricular tis-
sue of CO-exposed rats. Cardiomyocytes isolated from
CO-exposed rats showed higher reactive oxygen species
(ROS) production (measured with MitoSox Red dye),
higher diastolic Ca2? resulting from SR calcium leak and
an higher occurrence of irregular Ca2? transients (mea-
sured with Indo-1) in comparison to control cells after a
high pacing sequence. Acute treatment with a ROS scav-
enger (N-acetylcysteine, 20 mmol/L, 1 h) prevented this
sequence of alterations and decreased the number of
arrhythmic cells following high pacing. Chronic CO
exposure promotes oxidative stress that alters Ca2?
homeostasis (through RYR2 and SERCA defects) and
thereby mediates the triggering of ventricular arrhythmia
after cardiac stress that can lead to sudden death.
Keywords Calcium � Electrocardiography � Signal
transduction � Tachyarrhythmias
Introduction
The clinical importance of an arrhythmic phenotype fol-
lowing cardiac stress is well recognized. Ventricular
ectopic beats (VEB) during recovery represent a better
predictor of increased risk of death in healthy populations
than ventricular ectopy triggered only during exercise [18].
However, the origin and factors that influence the occur-
rence of post-cardiac stress ectopic beats are still unknown.
Mitochondria could play a key role since mitochondrial
dysfunction produces reactive oxygen species (ROS) that
F. Gouzi and J. Thireau equally contributed to this work.
Electronic supplementary material The online version of thisarticle (doi:10.1007/s00395-011-0211-y) contains supplementarymaterial, which is available to authorized users.
L. Andre � F. Gouzi � J. Thireau � J. Boissiere � A. Abdellaoui �A. Lacampagne � J. Fauconnier � M. Hayot � S. Richard �O. Cazorla (&)
INSERM U1046 Physiologie & Medecine Experimentale Coeur
Muscles, Universite Montpellier1, CHU Arnaud de Villeneuve,
34295 Montpellier, France
e-mail: [email protected]
L. Andre � F. Gouzi � J. Thireau � J. Boissiere � A. Abdellaoui �A. Lacampagne � J. Fauconnier � M. Hayot � S. Richard �O. Cazorla
INSERM U1046, Universite Montpellier2,
34295 Montpellier, France
G. Meyer � J. Boissiere � P. Obert � C. Reboul
EA-4278, Universite Avignon et des Pays de Vaucluse,
84000 Avignon, France
M. Delage � J.-P. Cristol
Department of Biochemistry, Lapeyronie Hospital,
34295 Montpellier, France
C. Feillet-Coudray � G. Fouret
INRA UMR 866, 34060 Montpellier, France
123
Basic Res Cardiol
DOI 10.1007/s00395-011-0211-y

can promote calcium (Ca2?) leak through Ryanodine
receptors (RyR), leading to irregular Ca2? transients.[3, 6,
10, 24, 42].
Air pollution promotes oxidative stress [5, 9] and is
involved in the development of arrhythmic events that
trigger heart attack and increase the risk of cardiovascular
death [26, 44]. Epidemiological studies have associated air
pollution with decreased heart rate variability, increased
propensity toward arrhythmic events [43] and sudden car-
diac death [16, 32, 46]. Carbon monoxide (CO), an ubiq-
uitous air pollutant that is found in many common sources
(secondhand smoke, vehicular exhaust, industrial emis-
sions…), is closely associated with hospital admissions for
cardiovascular disease [23] and cardiac mortality [40]. In
urban areas, ambient CO usually varies between 2 and
40 ppm, but under certain circumstances, such as during
heavy traffic, its concentration may reach levels as high as
150–200 ppm [51]. We have recently shown in an exper-
imental animal model that chronic exposure to CO levels,
which mimic pollution in urban areas, induces a heart
failure, like remodeling of Ca2? homeostasis in ventricular
myocytes in basal conditions accounting for a high
occurrence of in vivo premature ventricular contractions in
healthy rats [4]. The pro-arrhythmogenic phenotype in CO-
exposed rats was further enhanced under b-adrenergic
stimulation. These defects were suspected to be associated
with increased tissular oxidative stress. However, the link
between CO exposure and the related oxidative stress on
the occurrence of arrhythmic events after a cardiac stress
test is unknown.
We investigated in a rat model the signaling path-
way(s) that associates chronic CO exposure and post-car-
diac stress arrhythmic phenotype. In vivo chronic CO
exposure increased the number of ventricular ectopic beats
and the risk of sudden cardiac death during recovery from a
pharmacological cardiac challenge. Moreover, we found
that ROS production was significantly higher in cardio-
myocytes isolated from chronically exposed rats than in
controls after a protocol of rapid pacing change that altered
Ca2? homeostasis resulting in an arrhythmic phenotype.
Methods
Detailed information on the methodology is available as
online-only data supplement.
Animal model of chronic CO exposure
Experiments complied with the guide for Care and Use of
Laboratory Animals published by the US National Insti-
tutes of Health (NIH Publications No. 85-23, revised 1996)
and the European Union Council Directives for care of
laboratory animals, and were approved by the French
Ministry of Agriculture.
Male Wistar rats (13 week/old, n = 36) were exposed to
CO in airtight container for 4 weeks as previously descri-
bed [4]. Briefly, rats were exposed to 30 ppm CO for a
12-h period that also included 5 peaks at 100 ppm (1 h
each) to reproduce environmentally relevant air quality
variations [51]. For the remaining 12 h, rats were exposed
to filtered air (\1 ppm CO). CO content was continuously
monitored with an infrared aspirative CO analyzer
(CHEMGARD, NEMA 4 Version, MSA). Rats exposed to
CO pollution were compared with rats exposed only to
filtered ambient air (CO \ 1 ppm; control, n = 27).
Experiments were performed 24 h after the last CO expo-
sure to avoid CO acute effects.
ECGs were recorded by Holter telemetry during the
recovery from a b-adrenergic challenge following a bolus
of 1 mg kg-1 isoprenaline (iso) injected intraperitoneally
(IP). Ventricular ectopic beats including isolated, sequen-
tial or repetitive extra-systoles, and ventricular tachycardia
([20 successive VEB) were counted over a 20-min period
starting when the heart rate was 15% below the maximal
iso-induced rate to avoid rhythmic storm.
Cell isolation
Single intact rat ventricular cardiomyocytes were isolated
by enzymatic digestion from the inner sub-endocardial
layer of the left ventricle (LV) as previously described
[33].
Mitochondrial ROS production
Mitochondrial ROS production was measured using
MitoSOX Red [12]. Single rat cardiomyocytes were
loaded with 5 lmol/L MitoSOX Red at room temperature
for 15 min. MitoSOX Red fluorescence was measured at
583 nm following excitation at 488 nm using a Zeiss
LSM 510 inverted confocal microscope with a 409 lens.
In order to reproduce an exercise test protocol, cardio-
myocytes were subjected to workload changes. Cardio-
myocytes were paced at 0.5 Hz for 5 min, followed by
5 min at high pacing (4 Hz, HP), and then back to 0.5 Hz
for 5 min. MitoSOX Red fluorescence was measured at
the end of each phase. For each cell, the value in quies-
cent cells was set at 0% and background noise was
subtracted.
Anti-oxidant and mitochondrial complex activities
Catalase, total superoxide dismutase (SOD), Complex I and
V activities were measured in rat cardiomyocytes as
described elsewhere [14].
Basic Res Cardiol
123

Ca2? imaging in intact cardiomyocytes
Cardiomyocytes were loaded with Indo-1 AM (10 lM
molecular probes, France) at room temperature for
30 min to monitor intracellular Ca2?. Cells were illumi-
nated at 305 nm using a xenon arc lamp. Sarcomere
length (SL) and Indo-1 fluorescence emitted at 405 and
480 nm were simultaneously recorded (IonOptix system,
Hilton, USA). In some experiments, cells were pre-trea-
ted with the ROS scavenger N-acetylcysteine (20 mmol/L
for 1 h).
For Ca2? sparks, myocytes were loaded for 30 min
with the fluorescent Ca2? indicator Fluo-4 AM (5 lM,
molecular probes). Spontaneous Ca2? sparks (in quies-
cent cells) were recorded in line-scan mode (1.54 ms/
line, 512 pixels 9 3,000 lines) with a Zeiss LSM 510
confocal microscope (639 lens) [13]. Data were ana-
lyzed using ImageJ 1.41 coupled with SparkMaster.
Ca2?-spark frequency was calculated for each cardio-
myocytes, based on scans performed on 10 successive
images collected at 3–4 different line locations and
represented as the number of sparks per lm per second
(events lm-1 s-1).
Western blotting and protein kinase A activity
Proteins were separated using 2–20% SDS–PAGE and
blotted onto PVDF membranes (Protran, Schleichen and
Schuele, Dassel, Germany). Membranes were incubated
with the primary anti-RyR-2 (Covalab, France), -Phospho
Ser2809-RyR-2 (A010-30, Badrilla, UK), -SERCA-2a
(A010-20, Badrilla, UK) and -PLB antibodies (Badrilla,
UK) overnight at 4�C. Immunodetection was carried out
using the ECL Plus System (Amersham Pharmacia,
England).
Malondialdehyde (MDA), an end-product of lipid per-
oxidation, was measured in myocardial tissue isolated
either from the sub-endocardium of hearts that have been
unpaced for 5 min and washed out in Ca2? free solution or
from hearts mounted in Langendorff apparatus and paced
to 7 Hz for 15 min prior rapid dissection. Total protein
samples (5 lg) from each rat heart were dotted onto
nitrocellulose membranes, and dot blot analyses were
performed. The membranes were incubated with the pri-
mary anti-MDA antibody (Academy Biomedical, Houston,
TX, USA) for 1 h at room temperature. Immunodetection
was revealed with ECL. Optical density of each dot was
quantified after scanning and MDA content was expressed
relative to Ponceau S staining.
Protein kinase A (PKA) activity was measured using
a non-radioactive PKA activity assay kit according to
the manufacturer’s recommendations (Assay Designs,
France).
Statistical analysis
Data were analyzed using one-way or 2-way ANOVA
between groups. When significant interactions were found,
a Bonferroni post-hoc test was applied with P \ 0.05
(Statview 5.0). For comparing the number of arrhythmic
cells between control and CO-exposed rats a Chi-square
test was used. Data are presented as mean ± SEM.
Results
Impact of CO on in vivo ventricular arrhythmic events
after pharmacological cardiac challenge in a rat model
To investigate the impact of chronic CO exposure in the
occurrence of arrhythmic events during recovery after a
cardiac challenge, we used a previously characterized
animal model of chronic CO exposure [4]. Rats exposed to
CO for 4 weeks (n = 9) and controls (exposed to filtered
air) (n = 9) were challenged in vivo with a IP bolus of
1 mg kg-1 isoprenaline, a dose that induced a homoge-
neous and rapid increase of the heart rate in all animals. We
then measured the number of VEB occurring during the
recovery phase when the heart rate started to recuperate
(Fig. 1a). All animals from control and CO-exposed groups
exhibited VEB. Although the mean heart rate was com-
parable in control and CO-exposed rats (367 ± 1 and
365 ± 1 bpm, respectively), CO-exposed animals pre-
sented about 2.5 times more VEB than controls (Fig. 1a).
Qualitative analysis of the arrhythmic events showed that
control rats developed exclusively isolated ventricular
extra-systoles, whereas CO-exposed rats presented more
complex arrhythmic events, such as salvos of extra-systoles
(Fig. 1b). Moreover, in CO-exposed animals, we observed
one animal with non-sustained ventricular tachycardia and
two animals with sustained ventricular tachycardia that
degenerated into ventricular fibrillation and led to death
during the recovery period (Fig. 1c). These findings con-
firm that chronic CO exposure is associated with higher
occurrence of VEB and cardiac death during recovery after
cardiac challenge.
Impact of CO on arrhythmic events in rat ventricular
cardiomyocytes after high-frequency pacing
We then investigated the cellular mechanisms of the CO-
associated ventricular arrhythmia observed in vivo using
LV cardiomyocytes isolated from CO-exposed (n = 4) and
control rats (n = 5). Cardiomyocytes, which had been
loaded with Indo-1 to measure cytosolic Ca2?, were elec-
trically stimulated at 0.5 Hz (pre-HP) and submitted to an
abrupt increase in stimulation frequency to 4 Hz (high
Basic Res Cardiol
123

pacing, HP) followed by a return to 0.5 Hz (post-HP)
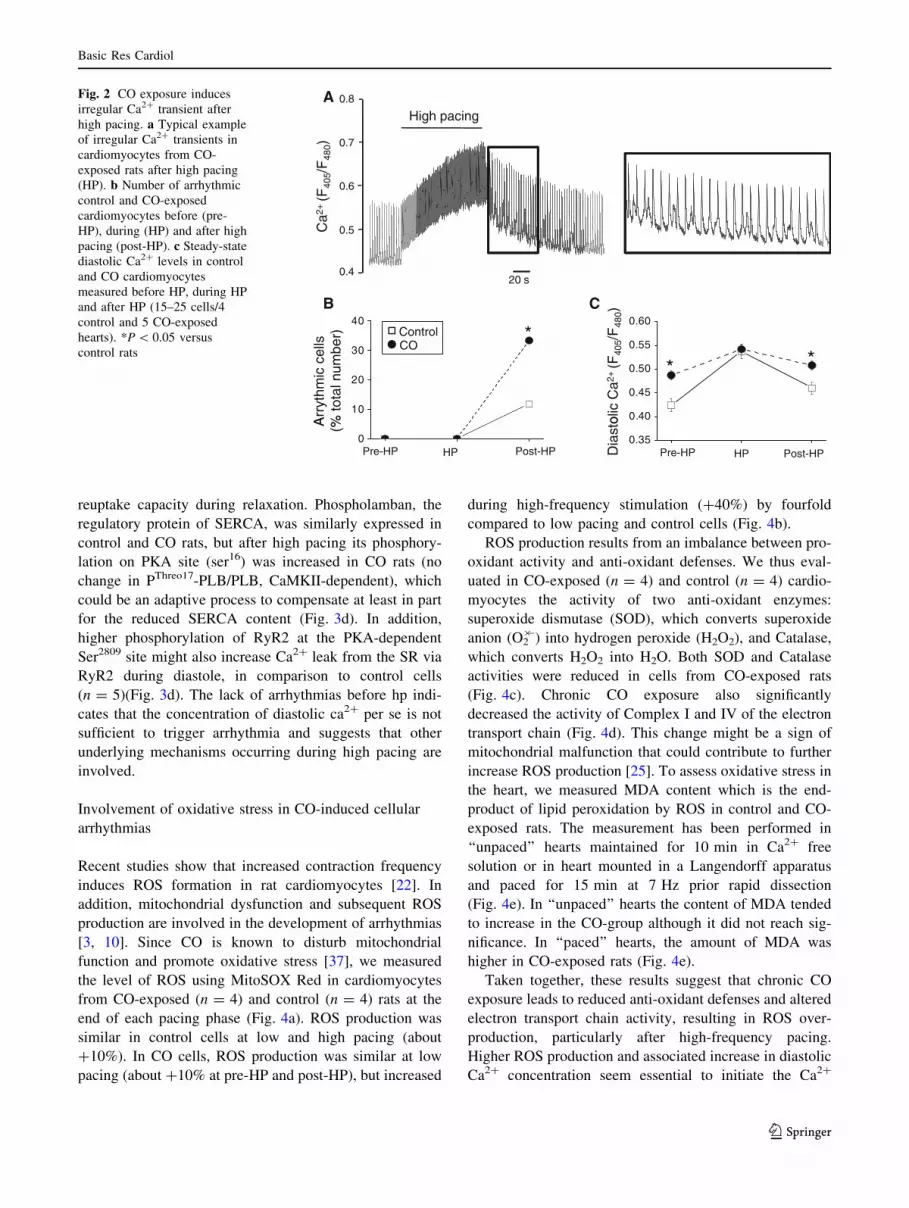
(Fig. 2a). Occurrence of cellular arrhythmic events was
quantified by measuring the number of irregular Ca2?
transients. During the pre-HP and HP phases, no irregular
Ca2? transient was observed in cardiomyocytes from both
control and CO-exposed rats. Conversely, during the post-
HP period, irregular Ca2? transients (Fig. 2a) occurred
spontaneously in about 10% of control cardiomyocytes and
30% of cardiomyocytes isolated from CO-exposed rats
(Fig. 2b).
Since diastolic intracellular Ca2? overload can trigger
spontaneous Ca2? waves and irregular Ca2? transients
[47], we then measured the diastolic Ca2? concentration in
these cells. During pre-HP, diastolic Ca2? levels were
significantly higher in cardiomyocytes from CO-exposed
rats than in control animals (Fig. 2c). Rapid pacing
increased the diastolic Ca2? concentration in both groups,
but more in controls than in CO-exposed rats and thus the
maximal diastolic Ca2? level in the two groups was com-
parable at HP. After HP, the diastolic Ca2? concentration
decreased to almost pre-HP levels in both groups, but
remained significantly higher in cardiomyocytes from CO-
exposed rats than in control cells (Fig. 2c). Calcium
homeostasis was further studied by measuring spontaneous
Ca2?-sparks, as an index of diastolic sarcoplasmic reticu-
lum (SR) Ca2? leak via RyR2. Ca2?-sparks frequency was
higher in CO-exposed rats than in control animals
(Fig. 3a). SR Ca2? leak in CO-exposed rats was also
addressed by measuring tetracaine-sensitive changes in
diastolic Ca2? as previously described by Shannon et al.
[41] (Fig. 3b). After high pacing, stimulation was stopped,
Na?/Ca2? exchanger (NCX) was blocked and diastolic
Ca2? was measured after 1 min. Diastolic Ca2? was higher
in CO-exposed rats. The protocol was repeated in presence
of tetracaine during the quiescent phase to block RyR.
Diastolic Ca2? decreased only in CO-exposed rats. The
difference of [Ca2?] between the two protocols, which
reflects the SR Ca2? leak, was higher in CO-exposed rats.
SR Ca2? load measured after caffeine pulse was decreased
in CO-exposed rats (Fig. 3c). Furthermore, cardiomyocytes
from CO-exposed rats (n = 5) showed also reduced Serca-
2a expression, which might result in a weaker Ca2?
Fig. 1 Arrhythmic phenotype
following cardiac challenge in
CO-exposed rats. a Isoprenaline
was injected in awaken rats
equipped for surface ECG
recordings. Heart beat
frequency increased rapidly,
remained stable (60 min on this
example) and then decreased to
basal values. Spontaneous
rhythmic disorders were
investigated over a period of
20 min starting at a heart rate
15% below the maximal
isoprenaline-induced heart rate.
b Ventricular ectopic beats
(VEB) were counted. An
example of isolated extra-
systoles is shown (left panel). In
CO-exposed rats we observed
more complex arrhythmic
events such as salvos of extra-
systoles (right panel).C Ventricular tachycardia and
fibrillation were observed in
three CO-exposed rats that led
to death in two of them (n = 9
rats per group). *P \ 0.05
versus control rats
Basic Res Cardiol
123
reuptake capacity during relaxation. Phospholamban, the
regulatory protein of SERCA, was similarly expressed in
control and CO rats, but after high pacing its phosphory-
lation on PKA site (ser16) was increased in CO rats (no
change in PThreo17-PLB/PLB, CaMKII-dependent), which
could be an adaptive process to compensate at least in part
for the reduced SERCA content (Fig. 3d). In addition,
higher phosphorylation of RyR2 at the PKA-dependent
Ser2809 site might also increase Ca2? leak from the SR via
RyR2 during diastole, in comparison to control cells
(n = 5)(Fig. 3d). The lack of arrhythmias before hp indi-
cates that the concentration of diastolic ca2? per se is not
sufficient to trigger arrhythmia and suggests that other
underlying mechanisms occurring during high pacing are
involved.
Involvement of oxidative stress in CO-induced cellular
arrhythmias
Recent studies show that increased contraction frequency
induces ROS formation in rat cardiomyocytes [22]. In
addition, mitochondrial dysfunction and subsequent ROS
production are involved in the development of arrhythmias
[3, 10]. Since CO is known to disturb mitochondrial
function and promote oxidative stress [37], we measured
the level of ROS using MitoSOX Red in cardiomyocytes
from CO-exposed (n = 4) and control (n = 4) rats at the
end of each pacing phase (Fig. 4a). ROS production was
similar in control cells at low and high pacing (about
?10%). In CO cells, ROS production was similar at low
pacing (about ?10% at pre-HP and post-HP), but increased
during high-frequency stimulation (?40%) by fourfold
compared to low pacing and control cells (Fig. 4b).
ROS production results from an imbalance between pro-
oxidant activity and anti-oxidant defenses. We thus eval-
uated in CO-exposed (n = 4) and control (n = 4) cardio-
myocytes the activity of two anti-oxidant enzymes:
superoxide dismutase (SOD), which converts superoxide
anion (O2�-) into hydrogen peroxide (H2O2), and Catalase,
which converts H2O2 into H2O. Both SOD and Catalase
activities were reduced in cells from CO-exposed rats
(Fig. 4c). Chronic CO exposure also significantly
decreased the activity of Complex I and IV of the electron
transport chain (Fig. 4d). This change might be a sign of
mitochondrial malfunction that could contribute to further
increase ROS production [25]. To assess oxidative stress in
the heart, we measured MDA content which is the end-
product of lipid peroxidation by ROS in control and CO-
exposed rats. The measurement has been performed in
‘‘unpaced’’ hearts maintained for 10 min in Ca2? free
solution or in heart mounted in a Langendorff apparatus
and paced for 15 min at 7 Hz prior rapid dissection
(Fig. 4e). In ‘‘unpaced’’ hearts the content of MDA tended
to increase in the CO-group although it did not reach sig-
nificance. In ‘‘paced’’ hearts, the amount of MDA was
higher in CO-exposed rats (Fig. 4e).
Taken together, these results suggest that chronic CO
exposure leads to reduced anti-oxidant defenses and altered
electron transport chain activity, resulting in ROS over-
production, particularly after high-frequency pacing.
Higher ROS production and associated increase in diastolic
Ca2? concentration seem essential to initiate the Ca2?
A
/F48
0)
High pacing0.8
0.7
0.6
CC
a2+
(F40
5
20 s
0.5
0.4
BA
rryt
hmic
cel
ls%
tota
l num
ber) Control
CO
0 40
0.45
0.50
0.55
0.60
* *
olic
Ca2
+(F
405/
F48
0)
10
20
30
40
*
HP
A (%
Post-HPPre-HP0.35
.
Dia
sto
HP Post-HPPre-HP0
Fig. 2 CO exposure induces
irregular Ca2? transient after
high pacing. a Typical example
of irregular Ca2? transients in
cardiomyocytes from CO-
exposed rats after high pacing
(HP). b Number of arrhythmic
control and CO-exposed
cardiomyocytes before (pre-
HP), during (HP) and after high
pacing (post-HP). c Steady-state
diastolic Ca2? levels in control
and CO cardiomyocytes
measured before HP, during HP
and after HP (15–25 cells/4
control and 5 CO-exposed
hearts). *P \ 0.05 versus
control rats
Basic Res Cardiol
123

waves responsible for cellular arrhythmias in CO-exposed
cardiomyocytes.
Finally, in order to further test the relative importance of
oxidative stress in the arrhythmic process, we incubated
CO-exposed cardiomyocytes with 20 mmol/L N-acetyl-
cysteine (NAC) (a ROS scavenger) for 1 h before testing
the effect of high-frequency pacing. NAC treatment (1)
reduced the number of arrhythmic cells after HP to a level
similar to that of control cardiomyocytes (Fig. 5a), and (2)
normalized both diastolic Ca2? concentration (Fig. 5b) and
PKA-dependent phosphorylation of RyR2 (Fig. 5c) in CO-
exposed cardiomyocytes. Consistently, PKA activity,
which was increased in co-exposed cardiomyocytes, was
normalized by NAC treatment (Fig. 5d).
A3
m/s
ec
*Control
0
1
2
Spa
rks/
100µ
m
Control CO
CO
10 µ
m
CB
4
6
8
10
leak
(ra
tio*1
00)
*
0.2
0.3
0.4
Ca
2+ c
onte
nt
*0.5
0.6
0.7
0.8
#*
control CO*
tolic
Ca2
+ra
tiohi
gh p
acin
g
500 ms
0
2S
R C
a2+
0.0
0.1
SR
Control COControl CO
3 40 4
0.3
0.4
Dia
staf
ter
0Ca, 0Na 0Ca, 0Na+ tetracaine
0
1
2
*
0
1
2
3
4
*
0.0
0.2
.
Control CO
Ser
ca c
onte
nt
RyR
con
tent
P-R
yR /
RyR
D
Control CO Control CO
Control
RyR
Serca
P-RyRCO
2
4
6
8
PLB
con
tent
PLB
PThreo17-PLB
PSer16-PLB
2
4
6
*
er16
-PLB
/ P
LB
0.5
1.0eo
17-P
LB /
PLB
0Control CO
CSQ 0Control CO
PS
e
0.0Control CO
PT
hre
Fig. 3 CO-associated alterations of Ca2? homeostasis. a Typical
examples of spontaneous Ca2? sparks in cardiomyocytes from control
and CO-exposed rats. The frequency of sparks appearance increased
by three times in CO-exposed myocytes (30–38 cells/3 control and
CO-exposed hearts). b SR Ca2? leak in CO-exposed rats was also
addressed by measuring tetracaine-sensitive changes in diastolic
Ca2?. After high pacing for 3 min, stimulation was stopped, Na?/
Ca2? exchanger was blocked by a solution of 0Ca2?/0Na? and
diastolic Ca2? was measured after 1 min. The protocol was repeated
in presence of tetracaine (1 mM) during the quiescent phase to block
RyR (left panel). The difference of [Ca2?] between the two protocols
reflects the SR Ca2? leak (right panel) (12 cells/3 hearts). *P \ 0.05
versus control rats, #P \ 0.05 versus without tetracaine. c SR Ca2?
load measured after caffeine pulse (10 mM) was decreased in CO-
exposed rats (12 cells/3 hearts). d Expression/phosphorylation of
RyR-2, Serca-2a and PLB in control and CO-exposed cardiomyocytes
were analyzed by western blotting. RyR-2, Serca-2a and PLB
contents were normalized to Calsequestrin (CSQ). After high pacing
the level of PKA-dependent Ser2809 RyR-2 phosphorylation (P-RyR)
was normalized to total RyR-2 and the PKA-dependent Ser16-
phosphorylated PLB and CamKII-dependent threo17-phosphorylated
PLB were normalized to PLB (5 hearts each). *P \ 0.05 versus
control rats
Basic Res Cardiol
123

Discussion
Air pollution is suspected to be involved in sudden death
by increasing ventricular arrhythmic events. VEB preva-
lence during recovery from cardiac stress is a valuable
predictor of mortality. In this study, we find higher VEB
occurrence during recovery after cardiac challenge in an
experimental rat model of chronic exposure to urban
atmospheric CO pollution that leads in some cases to
sudden death. Moreover, higher ROS production (and thus
oxidative stress) seems to be essential together with
increased diastolic Ca2? concentration to initiate the Ca2?
waves responsible for the arrhythmic phenotype in single
cardiomyocytes isolated from CO-exposed rats. The
Control
Post-HPPre-HPA
CO
B CControl 12060
O2-
prod
uctio
n(%
res
ting
leve
l) CO
20
40
60
80
100
120
**
0
10
20
30
40
50 *
HP Post-HPPre-HP
Act
ivity
(U
/mg
prot
ein)
SOD Catalase00
3 3
*
E
MDA
Ponceau
Unpaced
MDA
Ponceau
Paced
0
1
2
0
1
2
Control CO
MD
A c
onte
nt
Control CO
MD
A c
onte
nt
D
Control
CO
3
4
5
6
20
30
400
600
800
Act
ivity
(U
/g p
rote
in)
Complex IVComplex I
Act
ivity
(U
/g p
rote
in)
Act
ivity
(U
/g p
rote
in)
Citrate synthase0
1
2
3
0
10 *
0
200
400
*
Fig. 4 Effects of CO pollution
on mitochondrial ROS
production. a Typical images
showing Mitosox Red
fluorescence (indicator of
superoxide production) in
cardiomyocytes from control
and CO-exposed rats at rest and
after high pacing (HP).
b Production of O2�2 in control
and CO-exposed
cardiomyocytes measured at the
end of each pacing phase
before, during and after HP
relative to the value at rest
(n = 22–30 cells/4 control and
4 CO-exposed hearts).
c Myocardial anti-oxidant
defences were evaluated by
measuring SOD and Catalase
activity in cardiomyocytes from
control and CO-exposed rats (4
hearts per group). d Myocardial
citrate synthase (CS), Complex I
and Complex IV activities in
cardiomyocytes from control
and CO-exposed rats (4 hearts
per group). e Malondialdehyde
(MDA) content in myocardial
tissue isolated from the sub-
endocardium of hearts that have
been either unpaced for about
5 min in Ca2? free solution or
paced to 7 Hz for 15 min using
Langendorff apparatus.
*P \ 0.05 versus control rats
Basic Res Cardiol
123

variations in diastolic Ca2? handling were normalized to
control levels, when CO-exposed cardiomyocytes were
treated with a ROS scavenger before high pacing.
CO exposure increases ROS production after high
pacing
Air pollution is known to promote oxidative stress [9, 43].
Traffic-related air pollutants, such as particles, CO and
nitrogen species, have been associated with increased
systemic inflammation and oxidative stress [9, 43]. The
relative contribution of each pollutant was not investigated.
In order to control the level and origin of CO, we used an
experimental model in which rats were exposed to CO
levels that mimic those recorded in urban areas. This ani-
mal model allowed us to study precisely the effects of
chronic CO exposure on ROS production and its conse-
quences on heart rhythm independently of the other
pollutants.
One key finding of this study is that chronic CO expo-
sure potentiates at high-frequency pacing severe mito-
chondrial alterations leading to exacerbated ROS
production in cardiomyocytes and related oxidative stress
at the tissue level. The deleterious effects of CO were not
observed at low pacing in cells and in unpaced hearts. This
suggests that despite reduced antioxidant defences, the cell
is able to control the level of ROS production under basal
conditions. Moreover, the activity of other cytosolic anti-
oxidant enzymes such as peroxiredoxin or glutathione
peroxidase-1 have not been tested and could compensate
for the decrease in SOD and catalase activities [54]. The
effect of CO directly at the cardiomyocyte level might be
due to the fact that, under condition of chronic CO expo-
sure, CO competes with oxygen for binding to hemoglobin
(Hb), resulting in decreased cardiomyocyte oxygenation.
Tissue hypoxia could be worsened by CO binding also to
cardiac myoglobin [37], leading to disturbed cardiac
mitochondrial oxidation [20]. This is, however. unlikely in
our experimental conditions due to the modest reduction of
oxygen in arterial blood resulting from CO binding to Hb
(data not shown). Moreover, since we did not observe
electrocardiographic signs of ischemia in CO-exposed rats,
the involvement of the vascular system in vivo due to CO-
induced vasoconstriction linked to ROS production [30]
can also be excluded. Alternatively, CO binding to Cyto-
chrome p-450 might inhibit directly the Complex IV of the
electron transport chain, leading to ROS production [18,
37]. This hypothesis is supported by the reduced Complex
IV activity we observed in cardiomyocytes from CO-
exposed rats (Fig. 4d).
The present results contrast with the described cyto-
protective effect of CO at low dose and acute treatment in
Fig. 5 Effect of acute NAC
treatment on CO-associated
Ca2? handling alterations.
a Percentage of arrhythmic
cardiomyocytes isolated from
control (5 hearts), CO-exposed
(5 hearts) and CO-exposed rats
treated with NAC for 1 h (CO-
NAC) (5 hearts) during
recovery after high pacing
(15–25 cells). b Diastolic Ca2?
levels in control, CO-exposed
and CO-NAC cardiomyocytes
after high pacing (35–50 cells).
c Level of PKA-dependent
Ser2809 RyR-2 phosphorylation
normalized to total RyR-2
(n = 5 hearts). d PKA activity
was measured in cardiac tissue
from control and CO-exposed
and CO-NAC rats. *P \ 0.05
versus control rats
Basic Res Cardiol
123

cardiovascular diseases [17] with anti-oxidant, anti-
inflammatory, and anti-apoptotic properties [36]. However,
our model of chronic moderate CO exposure (30 ppm/day
plus five 1-h peaks at 100 ppm, 4 weeks) highlights the
potential harmful effects of low-dose CO gas inhalation or
synthetic CO-releasing molecules on cardiac function if
used as chronic treatments.
CO-mediated ROS production triggers irregular Ca2?
transients
Excessive ROS production plays a pivotal role in triggering
cellular and ventricular arrhythmic events through changes
in RyR2 activity [13, 48]. In CO-exposed cardiomyocytes,
arrhythmias were observed in association with increased
diastolic Ca2? level and ROS overproduction induced by
high pacing. This supports the concept of a crosstalk
between Ca2? and ROS as a central element in CO-medi-
ated Ca2? handling disorders in which Ca2?/ROS poten-
tiate each other [15]. Our results are in line with a previous
study in isolated guinea-pig cardiomyocytes showing that
perfusion of 1 mM H2O2 increased after 5 min diastolic
Ca2? and few minutes later, cells became automatic [21].
Acute treatment with the broad range antioxidant NAC
prevented diastolic Ca2? overload and irregular Ca2?
transients in CO-exposed cardiomyocytes (Fig. 5a). This is
consistent with the beneficial effects of NAC treatment on
RyR2-mediated diastolic SR Ca2? leak and arrhythmias
reported in Duchenne muscular dystrophy cardiomyocytes
[13]. Antioxidant strategies are also beneficial in various
rhythmic pathologies. In a sheep model, an anti-oxidant
cocktail (vitamin C and deferoxamine) reduced the number
of ventricular tachycardia/fibrillation events induced by
ischemia–reperfusion [28].
Oxidation of ion transport systems that participate in
cardiac excitation–contraction coupling can potentially
lead to Ca2? overload [11, 52]. However, other post-
translational modifications of RyR2 may be involved since
ROS can affect also protein phosphorylation through
inhibition of protein phosphatases [38] and activation of
PKA [34, 8]. Indeed, CO-exposed cardiomyocytes show
PKA-dependent hyper-phosphorylation of RyR2 that is
reverted following NAC treatment (Fig. 3c). Although this
is controversial, PKA-dependent RyR2 hyper-phosphory-
lation may lead to SR Ca2? leak that contributes to
increasing diastolic Ca2? levels [50, 31], which may be
involved in the contribution of sustained sympathetic
activation on arrhythmogenesis [35]. Additional pathway
may involve the Ca2?/calmodulin-dependent serine/threo-
nine kinase-d (CaMKIId), which is activated by Ca2?-
calmodulin at high Ca2? concentration, particularly when
heart rate increases. The impact of RyR2 phosphorylation
by CaMKII is controversial ranging from stabilizing RyR2
activity [53] to RyR2 disturbance resulting in Ca2? leak
[2]. A recent study showed that RyR2 phosphorylation by
CaMKIId is required for normal force–frequency response
in mice [29]. Its possible involvement in arrhythmia in CO-
exposed rats should be carefully investigated.
The reduced expression of SERCA-2a (responsible for
Ca2? reuptake in the SR during diastole) in CO-exposed
cardiomyocytes may also contribute to increasing dia-
stolic Ca2? concentration and the reduction in SR Ca2?
load. However, in our study acute NAC treatment nor-
malized diastolic Ca2? level indicating that a reversible
post-translational modification is involved. We show here
that the phosphorylation level of PLB on PKA site is
increased in CO-exposed rats, which should at least in
part compensate for the decrease in SERCA content.
Further phosphorylation on PLB after NAC treatment is
unlikely, considering the decrease of PKA activity in
presence of NAC. Other direct or indirect pathways
induced by ROS over-production may impact on SERCA-
2a function after chronic CO exposure, since previous
reports have shown that ROS inhibit SERCA-2a activity
and expression [1]. This pathway will require further
studies.
It has been reported that ‘‘millimolar concentrations of
ROS can cause increase in the Ca2? and Na? permeabil-
ity’’ but there is no consensus regarding potential effects on
Ca2? channels and on the Na?/Ca2? exchanger [19]. We
can just point out that decreased activities of IcaL and NCX
would not be in favor of proarrhythmogenicity. Although
ROS were reported to increase the late sodium current [45,
49] favoring Na? and Ca2? overloads responsible for
DADs/EADs, we have no evidence for prolonged AP
repolarization and no change in L-type Ca2? current in
subendocardial cells [4]. Finally, most of studies report
acute effects of moderate to high concentrations of H2O2,
whereas the present study describes effects resulting from
remodeling after long-term exposure to CO enhanced
during/after high pacing. Considering that we did not focus
on these mechanisms in the present study, we cannot
completely exclude their contribution to the increase dia-
stolic Ca2?.
CO level in human and incidence on cardiovascular
disease
Diastolic Ca2? overload induces oscillations of the cell
membrane potential that trigger delayed after-depolariza-
tions [47]. We provide direct evidence that sustained CO
exposure promotes irregular Ca2? transients in vitro and
arrhythmic events in vivo during recovery after cardiac
challenge in an experimental animal model. Ventricular
arrhythmias occurring during recovery after a cardiac stress
is common even in healthy subjects as indicated by the
Basic Res Cardiol
123

presence of VEB in all control rats (&12 VEB/h). How-
ever, these events are exclusively isolated VEB. The fact
that CO-exposed rats had 2.5 times more VEB (&36 VEB/
h) and, more importantly, more complex events such as
repetitive extra-systoles and ventricular tachycardia (3/9
CO animals and none in control rats) indicate that CO
exposure is pro-arrhythmogenic. Nevertheless, the link
between human CO levels and arrhythmias remains to be
determined. In an exploratory investigation, we measured
various functional parameters in non-smoker healthy vol-
unteers who performed an incremental maximal exercise
test on ergocycle (Supplemental data Figure S1A). Age,
gender, VO2max, and maximal heart rate were not corre-
lated with the number of arrhythmic events during recovery
after exercise (Supplemental data, Table S1). Although the
air was the same in the hospital, volunteers have been
exposed prior to their arrival at the hospital to various
levels of environmental CO (secondhand smoke, vehicular
exhaust, industrial emissions…) linked to different lifestyle
(living/working in the urban and/or non-urban locations of
the Montpellier region). In order to assess CO exposure, we
measured carboxyhemoglobin level (HbCO), which is
recognized as a biological marker of recent CO exposure
[39]. Conversely, we found a positive correlation
(R2 = 0.76, P \ 0.05) between the blood level of HbCO
and the number of VEB (Figure S1B). Similarly, the blood
level of HbCO was linearly correlated (R2 = 0.82,
P \ 0.01) with that of isoprostane, an index of oxidative
stress, consistent with a pro-oxidative effect of CO expo-
sure (Figure S1C). Since oxidative stress plays a critical
role in arrhythmogenesis, we then evaluated whether the
number of VEB after exercise was also associated with the
concentration of isoprostane (Figure S1D). Although a
positive linear correlation between isoprostane concentra-
tion and VEB number did not reach statistical significance
(P = 0.07), most probably due to the small number of
subjects available for this analysis (n = 17), the results
suggest, however, a link between (CO-induced) oxidative
stress and the occurrence of VEB in healthy human sub-
jects. Those results have to be confirmed in more subjects,
but they are in line with a recent study performed in a large
cohort of patients from the Framingham Heart study [7]. In
this 4 years follow-up study, a close relation between
exhaled CO, considered as an indicator of total blood CO
concentration, and risk of developing future cardiovascular
diseases and metabolic syndrome was established. This
work was based on the hypothesis that the measured CO
reflected the endogenous CO production without consid-
ering exogenous environmental CO. The effect of CO
pollution on the post-exercise arrhythmic phenotype could
be even more deleterious in elderly populations [27] or in
populations with pathologies, such as metabolic syndrome
or heart failure, which are known to generate oxidative
stress. This remains to be determined in epidemiological
studies.
Conclusions
The present study provides evidence that sustained expo-
sure to urban CO pollution directly influences the occur-
rence of arrhythmic events during recovery after cardiac
challenge. We show a link between CO exposure, oxidative
stress, diastolic Ca2? and arrhythmic events during cardiac
stress recovery. Since post-cardiac stress irregular beats are
a key factor for predicting sudden cardiac death [18], our
findings suggest that chronic CO exposure represents a
potential risk factor for triggering cardiac events and thus
should be taken into account for the prevention of cardio-
vascular risk in clinical practice.
Acknowledgments This work was supported by a French National
Research Agency grant (COMYOCARD). SR, JF, AL, and OC are
scientists from the Centre National de la Recherche Scientifique. We
thank Sandrine Gayrard for technical assistance.
Conflict of interest None declared.
References
1. Adachi T, Matsui R, Xu S, Kirber M, Lazar HL, Sharov VS,
Schoneich C, Cohen RA (2002) Antioxidant improves smooth
muscle sarco/endoplasmic reticulum Ca(2 ?)-ATPase function
and lowers tyrosine nitration in hypercholesterolemia and
improves nitric oxide-induced relaxation. Circ Res 90:1114–
1121. doi:10.1161/01.RES.0000019757.57344.D5
2. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM (2005)
Ca2?/calmodulin-dependent protein kinase modulates cardiac
ryanodine receptor phosphorylation and sarcoplasmic reticulum
Ca2? leak in heart failure. Circ Res 97:1314–1322. doi:
10.1161/01.RES.0000194329.41863.89
3. Akar FG, Aon MA, Tomaselli GF, O’Rourke B (2005) The
mitochondrial origin of postischemic arrhythmias. J Clin Invest
115:3527–3535. doi:10.1172/JCI25371
4. Andre L, Boissiere J, Reboul C, Perrier R, Zalvidea S, Meyer G,
Thireau J, Tanguy S, Bideaux P, Hayot M, Boucher F, Obert P,
Cazorla O, Richard S (2010) Carbon Monoxide Pollution Pro-
motes Cardiac Remodeling and Ventricular Arrhythmia in
Healthy Rats. Am J Respir Crit Care Med 181:587–595. doi:
10.1164/rccm.200905-0794OC
5. Araujo JA (2010) Particulate air pollution, systemic oxidative
stress, inflammation, and atherosclerosis. Air Qual Atmos Health
4:79–93. doi:10.1007/s11869-010-0101-8
6. Brown DA, O’Rourke B (2010) Cardiac mitochondria
and arrhythmias. Cardiovasc Res 88:241–249. doi:10.1093/cvr/
cvq231
7. Cheng S, Lyass A, Massaro JM, O’Connor GT, Keaney JF Jr,
Vasan RS (2010) Exhaled carbon monoxide and risk of metabolic
syndrome and cardiovascular disease in the community. Circu-
lation 122:1470–1477. doi:10.1093/cvr/cvq231
Basic Res Cardiol
123

8. de Lamirande E, O’Flaherty C (2008) Sperm activation: role of
reactive oxygen species and kinases. Biochim Biophys Acta
1784:106–115. doi:10.1016/j.bbapap.2007.08.024
9. Delfino RJ, Staimer N, Tjoa T, Gillen DL, Polidori A, Arhami M,
Kleinman MT, Vaziri ND, Longhurst J, Sioutas C (2009) Air
pollution exposures and circulating biomarkers of effect in a
susceptible population: clues to potential causal component
mixtures and mechanisms. Environ Health Perspect 117:1232–
1238. doi:10.1289/journla.ehp.0800194
10. Di Lisa F, Kaludercic N, Carpi A, Menabo R, Giorgio M (2009)
Mitochondrial pathways for ROS formation and myocardial
injury: the relevance of p66(Shc) and monoamine oxidase. Basic
Res Cardiol 104:131–139. doi:10.1007/s00395-009-0008-4
11. Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera
SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR,
Shirokova N, Protasi F, Dirksen R, Hamilton SL (2008) RyR1
S-nitrosylation underlies environmental heat stroke and sudden
death in Y522S RyR1 knockin mice. Cell 133:53–65. doi:
10.1016/j.cell.2008.02.042
12. Fauconnier J, Andersson DC, Zhang SJ, Lanner JT, Wibom R,
Katz A, Bruton JD, Westerblad H (2007) Effects of palmitate on
Ca(2?) handling in adult control and ob/ob cardiomyocytes:
impact of mitochondrial reactive oxygen species. Diabetes
56:1136–1142. doi:10.2337/db06-0739
13. Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki
S, Marks AR, Lacampagne A (2010) Leaky RyR2 trigger ven-
tricular arrhythmias in Duchenne muscular dystrophy. Proc Natl
Acad Sci USA 107:1559–1564. doi:10.1073/pnas.0908540107
14. Feillet-Coudray C, Sutra T, Fouret G, Ramos J, Wrutniak-Cabello
C, Cabello G, Cristol JP, Coudray C (2009) Oxidative stress in
rats fed a high-fat high-sucrose diet and preventive effect of
polyphenols: Involvement of mitochondrial and NAD(P)H oxi-
dase systems. Free Radic Biol Med 46:624–632. doi:10.1016/
j.freeradbiomed.2008.11.020
15. Feissner RF, Skalska J, Gaum WE, Sheu SS (2009) Crosstalk
signaling between mitochondrial Ca2? and ROS. Front Biosci
14:1197–1218
16. Finkelstein MM (2002) Pollution-related mortality and educa-
tional level. Jama 288:830. doi:10.1001/jama.288.7.830
17. Foresti R, Bani-Hani MG, Motterlini R (2008) Use of carbon
monoxide as a therapeutic agent: promises and challenges.
Intensive Care Med 34:649–658. doi:10.1007/s00134-008-1011-1
18. Frolkis JP, Pothier CE, Blackstone EH, Lauer MS (2003) Fre-
quent ventricular ectopy after exercise as a predictor of death.
N Engl J Med 348:781–790. doi:10.1056/NEJMoa022353
19. Gen W, Tani M, Takeshita J, Ebihara Y, Tamaki K (2001)
Mechanisms of Ca2? overload induced by extracellular H2O2 in
quiescent isolated rat cardiomyocytes. Basic Res Cardiol 96:623–
629. doi:10.1007/s003950170014
20. Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart
failure. J Clin Invest 115:500–508. doi:10.1172/JCI200524408
21. Goldhaber JI, Liu E (1994) Excitation-contraction coupling in
single guinea-pig ventricular myocytes exposed to hydrogen
peroxide. J Physiol 477(Pt 1):135–147
22. Heinzel FR, Luo Y, Dodoni G, Boengler K, Petrat F, Di Lisa F,
de Groot H, Schulz R, Heusch G (2006) Formation of reactive
oxygen species at increased contraction frequency in rat cardio-
myocytes. Cardiovasc Res 71:374–382. doi:10.1016/j.cardiores.
2006.05.014
23. Henry CR, Satran D, Lindgren B, Adkinson C, Nicholson CI,
Henry TD (2006) Myocardial injury and long-term mortality
following moderate to severe carbon monoxide poisoning. Jama
295:398–402. doi:10.1001/jama.295.4.398
24. Heusch G, Boengler K, Schulz R (2010) Inhibition of mito-
chondrial permeability transition pore opening: the Holy Grail of
cardioprotection. Basic Res Cardiol 105:151–154. doi:10.1007/
s00395-009-0080-9
25. Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N,
Uchida K, Arimura K, Egashira K, Takeshita A (1999) Mito-
chondrial electron transport complex I is a potential source of
oxygen free radicals in the failing myocardium. Circ Res
85:357–363
26. Kaiser J (2005) Epidemiology. How dirty air hurts the heart.
Science 307:1858–1859. doi:10.1126/science.307.5717.1858b
27. Kakarla SK, Fannin JC, Keshavarzian S, Katta A, Paturi S,
Nalabotu SK, Wu M, Rice KM, Manzoor K, Walker EM Jr.,
Blough ER (2010) Chronic acetaminophen attenuates age-asso-
ciated increases in cardiac ROS and apoptosis in the Fischer
Brown Norway rat. Basic Res Cardiol 105:535–544. doi:
10.1007/s00395-010-0094-3
28. Karahaliou A, Katsouras C, Koulouras V, Nikas D, Niokou D,
Papadopoulos G, Nakos G, Sideris D, Michalis L (2008) Ven-
tricular arrhythmias and antioxidative medication: experimental
study. Hellenic J Cardiol 49:320–328
29. Kushnir A, Shan J, Betzenhauser MJ, Reiken S, Marks AR (2010)
Role of CaMKIIdelta phosphorylation of the cardiac ryanodine
receptor in the force frequency relationship and heart failure. Proc
Natl Acad Sci USA 107:10274–10279. doi:10.1073/pnas.1005
843107
30. Lamon BD, Zhang FF, Puri N, Brodsky SV, Goligorsky MS,
Nasjletti A (2009) Dual pathways of carbon monoxide-mediated
vasoregulation: modulation by redox mechanisms. Circ Res
105:775–783. doi:10.1161/CIRCRESAHA.109.197434
31. Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX,
Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer
WJ, Kass RS, Morley G, Marks AR (2008) Leaky Ca2? release
channel/ryanodine receptor 2 causes seizures and sudden cardiac
death in mice. J Clin Invest 118:2230–2245. doi:10.1172/
JCI35346
32. Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH,
Anderson GL, Kaufman JD (2007) Long-term exposure to air
pollution and incidence of cardiovascular events in women.
N Engl J Med 356:447–458
33. Mou YA, Reboul C, Andre L, Lacampagne A, Cazorla O (2009)
Late exercise training improves non-uniformity of transmural
myocardial function in rats with ischaemic heart failure. Car-
diovasc Res 81:555–564. doi:10.1093/cvr/cvn229
34. O’Flaherty C, de Lamirande E, Gagnon C (2006) Reactive oxy-
gen species modulate independent protein phosphorylation
pathways during human sperm capacitation. Free Radic Biol Med
40:1045–1055. doi:10.1016/j.freeradbiomed.2005.10.055
35. Oikonomidis DL, Tsalikakis DG, Baltogiannis GG, Tzallas AT,
Xourgia X, Agelaki MG, Megalou AJ, Fotopoulos A, Papalois A,
Kyriakides ZS, Kolettis TM (2010) Endothelin-B receptors and
ventricular arrhythmogenesis in the rat model of acute myocar-
dial infarction. Basic Res Cardiol 105:235–245
36. Piantadosi CA (2002) Biological chemistry of carbon monoxide.
Antioxid Redox Signal 4:259–270. doi:10.1089/1523086027536
66316
37. Piantadosi CA (2008) Carbon monoxide, reactive oxygen sig-
naling, and oxidative stress. Free Radic Biol Med 45:562–569.
doi:10.1016/j.freeradbiomed.2008.05.013
38. Rhee SG, Bae YS, Lee SR, Kwon J (2000) Hydrogen peroxide: a
key messenger that modulates protein phosphorylation through
cysteine oxidation. Sci STKE:PE1. doi:10.1126/stke.2000.53.pe1
39. Rudra CB, Williams MA, Sheppard L, Koenig JQ, Schiff MA,
Frederick IO, Dills R (2010) Relation of whole blood carboxy-
hemoglobin concentration to ambient carbon monoxide exposure
estimated using regression. Am J Epidemiol 171:942–951. doi:
10.1093/aje/kwq022
Basic Res Cardiol
123

40. Samoli E, Touloumi G, Schwartz J, Anderson HR, Schindler C,
Forsberg B, Vigotti MA, Vonk J, Kosnik M, Skorkovsky J,
Katsouyanni K (2007) Short-term effects of carbon monoxide on
mortality: an analysis within the APHEA project. Environ Health
Perspect 115:1578–1583. doi:10.1289/ehp.10375
41. Shannon TR, Ginsburg KS, Bers DM (2002) Quantitative
assessment of the SR Ca2 ? leak-load relationship. Circ Res
91:594–600. doi:10.1161/01.RES.0000036914.12686.28
42. Shiva S, Gladwin MT (2009) Nitrite mediates cytoprotection
after ischemia/reperfusion by modulating mitochondrial function.
Basic Res Cardiol 104:113–119. doi:10.1007/s00395-009-0009-3
43. Simkhovich BZ, Kleinman MT, Kloner RA (2008) Air pollution and
cardiovascular injury epidemiology, toxicology, and mechanisms.
J Am Coll Cardiol 52:719–726. doi:10.1016/j.jacc.2008.05.029
44. Simkhovich BZ, Marjoram P, Kleinman MT, Kloner RA (2007)
Direct and acute cardiotoxicity of ultrafine particles in young
adult and old rat hearts. Basic Res Cardiol 102:467–475. doi:
10.1007/s00395-007-0681-0
45. Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L (2006)
Blocking late sodium current reduces hydrogen peroxide-induced
arrhythmogenic activity and contractile dysfunction. J Pharmacol
Exp Ther 318:214–222. doi:10.1124/jpet.106.101832
46. Stieb DM, Szyszkowicz M, Rowe BH, Leech JA (2009) Air
pollution and emergency department visits for cardiac and
respiratory conditions: a multi-city time-series analysis. Environ
Health 8:25. doi:10.1186/1476-069X-8-25
47. Ter Keurs HE, Boyden PA (2007) Calcium and arrhythmogene-
sis. Physiol Rev 87:457–506. doi:10.1152/physrev.00011.2006
48. Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A,
Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ,
Carnes CA, Gyorke S (2008) Redox modification of ryanodine
receptors contributes to sarcoplasmic reticulum Ca2? leak in
chronic heart failure. Circ Res 103:1466–1472. doi:10.1161/
CIRCRESAHA.108.184457
49. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T,
Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L,
Maier LS (2011) Reactive oxygen species-activated Ca/calmod-
ulin kinase IIdelta is required for late I(Na) augmentation leading
to cellular Na and Ca overload. Circ Res 108:555–565. doi:
10.1161/CIRCRESAHA.110.221911
50. Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cer-
vantes D, Coromilas J, Landry DW, Marks AR (2004) Protection
from cardiac arrhythmia through ryanodine receptor-stabilizing
protein calstabin2. Science 304:292–296. doi:10.1126/science.
1094301
51. Wright GR, Jewczyk S, Onrot J, Tomlinson P, Shephard RJ
(1975) Carbon monoxide in the urban atmosphere: hazards to
the pedestrian and the street-worker. Arch Environ Health
30:123–129
52. Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H (2008)
Bidirectional regulation of Ca2? sparks by mitochondria-derived
reactive oxygen species in cardiac myocytes. Cardiovasc Res
77:432–441. doi:10.1093/cvr/cvm047
53. Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG,
Xiao RP, Cheng H (2007) Ca2?/calmodulin kinase II-dependent
phosphorylation of ryanodine receptors suppresses Ca2? sparks
and Ca2? waves in cardiac myocytes. Circ Res 100:399–407.
doi:10.1161/01.RES.0000258022.13090.55
54. Zhao W, Fan GC, Zhang ZG, Bandyopadhyay A, Zhou X, Kra-
nias EG (2009) Protection of peroxiredoxin II on oxidative stress-
induced cardiomyocyte death and apoptosis. Basic Res Cardiol
104:377–389. doi:10.1007/s00395-008-0764-6
Basic Res Cardiol
123





























