Embed Size (px)
Citation preview

Cell Damage Following Carbon Monoxide Releasing MoleculeExposure: Implications for Therapeutic Applications
Ian C. Winburn1,2,3, Kishan Gunatunga1, Robert D. McKernan1, Robert J. Walker3, Ivan A. Sammut1 and Joanne C. Harrison1
1Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand, 2Pfizer UK, Tadworth, UK and 3Department ofMedicine, University of Otago, Dunedin, New Zealand
(Received 1 June 2011; Accepted 3 January 2012)
Abstract: The cytoprotective properties of carbon monoxide (CO) gas and CO-releasing molecules (CORMs) are well established.Despite promising pre-clinical results, little attention has been paid to the toxicological profile of CORMs. The effects of CORM-2and its CO-depleted molecule (iCORM-2) (20–400 lM) were compared in primary rat cardiomyocytes and two cell lines [humanembryonic kidney (HeK) and Madine-Darby canine kidney Cells (MDCK)]. Cells were assessed for cell viability, apoptosis, necro-sis, cytology, mitochondrial energetics, oxidative stress and cell cycle arrest markers. In separate experiments, the anti-apoptoticeffects of CORM-2 and i-CORM-2 treatment were compared against CO gas treatment in HeK and MDCK lines. H2O2-inducedcellular damage, measured by lactate dehydrogenase (LDH) release from primary cardiomyocytes, was reduced by 20 lM CORM-2; LDH activity, however, was directly inhibited by 400 lM CORM-2. Both CORM-2/iCORM-2 and CO gas decreased cisplatin-induced caspase-3 activity in MDCK and HeK cells suggesting an anti-apoptotic effect. Conversely, both CORM-2 and iCORM-2induced significant cellular toxicity in the form of decreased cell viability, abnormal cell cytology, increased apoptosis and necrosis,cell cycle arrest and reduced mitochondrial enzyme activity. Comparison of these markers after CO gas administration to MDCKcells found significantly less cellular toxicity than in 100 lM CORM-2/iCORM-2-treated cells. CO gas did not have an adverseeffect on mitochondrial energetics and integrity. Release of CO by low concentrations of intact CORM-2 molecules provides cyto-protective effects. These results show, however, that the ruthenium-based CORM by-product, iCORM-2, is cytotoxic and suggestthat the accumulation of iCORM-2 would seriously limit any clinical application of the ruthenium-based CORMs.
Carbon monoxide (CO), iron and biliverdin are products ofthe catabolism of haem by the enzyme haem oxygenase (HO)[1]. Both CO administration and HO-1 induction have beendemonstrated to afford cytological and organ protection in anumber of models [2,3]. CO provides cytoprotection via anumber of different mechanisms that include anti-inflammatory[4], vasodilatory [5,6], anti-coagulative [7], anti-apoptotic [7,8]and anti-proliferative/fibrotic pathways [3].The anti-apoptotic properties of CO have been demonstrated
in both in vivo and in vitro models to involve modulation ofintrinsic pathways, extrinsic pathways and also up-regulationof protective anti-apoptotic Bcl-XL/Bax interaction [6–9]. COexerts a variety of effects on mitochondrial function dependenton its concentration, duration of exposure and cell type stud-ied. Both endogenous and exogenous CO have been demon-strated to diminish cellular respiration via inhibition ofcomplex IV [10,11]. However, inhibition of complex IV byCO may account in part for some of its cytoprotective effects,by creating a ‘metabolic hypoxia’ [12] and activating anti-inflammatory p38 MAPK pathways via ROS release [13].Exposure of rats to CO (50 ppm; 7 days) transiently increaseshepatic mitochondrial oxidative stress and activates calcium-dependent mitochondrial pore transition that may predisposecells to activation of intrinsic apoptotic pathways. CO-medi-ated increases in superoxide dismutase-2 expression, however,
may counterbalance the increased ROS generation renderingcells more tolerant to on-going elevated oxidative stress [14].Furthermore, CO promotes cardiac mitochondrial biogenesisindependent of NO with increased copy numbers of mitochon-drial DNA (coding for complexes I–V) observed 24 hr afterCO exposure [15].Three distinct methods of delivering CO have been utilised in
studies to date: The simplest method involves gaseous delivery atlow concentrations, with COHb levels of 10–20% being achievedin human beings and large animal models after 250 ppm COexposure, without deleterious effects [16,17]. A second methodof delivery is the use of CO gas saturated physiological solutions[18], which whilst applicable in a transplant setting would havelimited application as a therapeutic agent. Alternatively, CO maybe delivered to organ systems by chemical agents that releaseCO. These include methylene chloride [19] and more recently,the CO-releasing molecules (CORMs) employed both in in vitroand in vivo studies [20].Although various CORMs have been demonstrated to offer
cytoprotection in a wide range of models [21–23], minimaldata exist that evaluate the potential toxicity of the spent resid-ual parent donor molecule in these systems. Both CORM-2(tricarbonyldichlororuthenium II) and CORM-3 are ruthenium-based CO donor molecules that will inevitably expose organsystems to ruthenium. A number of toxic chemotherapiesafford their mechanism of action to ruthenium, and it could besuggested that CO-depleted CORM-2 or CORM-3 may alsohave a similar detrimental impact on cellular function [24,25].Despite an abundance of evidence that supports a role for
CO as a cytoprotective agent, it is essential that any proposed
Author for correspondence: Joanne C. Harrison, Department ofPharmacology and Toxicology, University of Otago, PO Box 913,Dunedin, Otago 9054, New Zealand (fax +64 3 479 9140, [email protected]).
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
Basic & Clinical Pharmacology & Toxicology Doi: 10.1111/j.1742-7843.2012.00856.x

therapeutic strategy does not induce toxicity either as a directconsequence of CO exposure or secondary to the mode of COadministration. Previous in vitro studies have shown CO gasto have anti-apoptotic effects at 50–400 ppm [8,26,27] andCORM-3 at 1–500 lM [28,29]. In vivo 500 ppm CO gasexposure protects against murine lung ischaemia-reperfusioninjury [30] whereas low doses of 20 ppm have been shown toincrease allograft survival [31]. Based upon the cytotoxicproperties of ruthenium, we hypothesise that CORM-2 depletedof its cytoprotective CO would display cellular toxicity andthat active CORM-2 may consequently be less cytoprotectivethan CO gas at 20 ppm. Here, we investigate this hypothesisby comparing the active CORM-2 molecule, the CO-depletedform iCORM-2 and 20 ppm CO gas exposure on primary ratcardiomyocytes, canine renal epithelial (MDCKs) and HeKcell lines using the lower concentrations of CORM-2 and COthat had shown protective effects. We report the effects of theCORM-2 on cellular morphology, enzyme activity, cell viabil-ity, mitochondrial energetics, cell cycle arrest and ability ofthis compound to protect against or induce apoptosis.
Materials and Methods
Materials. All inorganic and organic chemicals used in the followingprotocols were purchased from Sigma (Auckland, New Zealand) orBDH Laboratory Supplies (Palmerston North, New Zealand), unlessotherwise specified. Medical grade gases and 20 ppm CO gas (in 5%CO2 in air) were obtained from BOC (Auckland/Dunedin, NewZealand). All cell culture plasticware was purchased from GreinerBio-One GmbH (CellstarTM; Frickenhausen, Germany) or CorningIncorporated Life Sciences (CostarTM; Lowell, MA, USA). HeK cells(HeK; CCL-1573TM) and Madine-Darby Canine Kidney cells repre-sentative of renal distal tubule cells (MDCK; CCL-34TM) wereobtained from ATCC (Rockville, MD, USA).
Isolation of primary rat cardiomyocytes. Male Lewis rats (250–350 g) were housed at 25°C under controlled light: dark cycles andfed ad libitum on a standard rat diet prior to experimentation. All pro-cedures were conducted in accordance with the regulations of theOtago University Committee on Ethics in the Care and Use of Labora-tory Animals. Rats were anaesthetised with diethyl-ether, the abdomi-nal cavity exposed, 0.2 ml heparin (5000 IU/ml) administered via thevena cava and a midline thoracotomy performed. The heart wasexcised, placed into ice-cold Ca2+-free filtered (0.2 lm) Tyrode buffer(137 mM NaCl, 2 mM KCl, 1 mM MgCl2·6H2O, 0.3 mM NaH2-
PO4·H2O, 12 mM NaHCO3 and 5.5 mM glucose saturated with 95%O2 and 5% CO2, pH 7.4) and perfused using a constant flow perfusionsystem at 37°C with Tyrode buffer until the buffer ran clear. The ven-tricular myocardium was then digested using a type I collagenase aspreviously described [32]. The cardiac ventricular tissue was then agi-tated in 1 mM Ca2+-containing, oxygenated (95% O2/5% CO2) Tyrodebuffer and the cardiomyocytes pelleted by centrifugation at 8 9 g(1 min. 9 2) to produce a typical isolation yield of 70–90% of rod-shaped cells. Cells were maintained at 37°C in sterile phenol red freeDulbecco's Modification of Eagle's Medium (DMEM) supplementedwith 5 mM of sodium pyruvate, 5% New Born Calf serum (NBCS)and 1% penicillin/amphotericin and used within 24 hr.
Cell treatment protocols. Primary cardiomyocytes were aliquoted insix-well plates (1 9 105 cells/3 ml) at 37°C and 5% CO2 (humid-
ity > 95%). HeK cells were cultured in DMEM supplemented with5% NBCS, 18 mM sodium bicarbonate, 13.4 mM glucose and 1%penicillin/amphotericin at 37°C and 5% CO2 in air (humidity > 95%).The same medium supplemented with 5 mM of sodium pyruvate with-out additional glucose was used for MDCK cell culture. MDCK andHeK were seeded at 1 9 105 cells/ml and cultured in 24-well platesto 80% confluence.Carbon monoxide releasing molecule-2 is insoluble in water but sol-
uble in DMSO. MDCK and HeK cells were exposed to 20 lMCORM-2 in 0.2% DMSO and 100 lM CORM-2 in 1% DMSO orinactivated CORM-2 dissolved in equivalent concentrations of DMSO.Concentrations of DMSO up to 0.4% had no deleterious effect on cellviability or cytology (data not shown); however, 1% DMSO wasrequired to fully dissolve the 100 lM CORM-2. Hence, wells contain-ing control cells were exposed to 0.2% or 1% DMSO as appropriate.Inactive (CO depleted) CORM-2 (iCORM-2) was produced by prepar-ing CORM-2 (20 and 100 lM) in DMSO as described above andleaving it in a sealed, sterile container exposed to light for 48 hr.Nitrogen gas was persufflated through the solution to displace anyresidual dissolved CO.The effects of CO gas on MDCK and HEK cells were examined by
culturing these lines in a dedicated incubator with a constant environ-ment of 20 ppm CO in 5% CO2 in air. All cells were incubated witheach treatment regime for a further 48 hr.
Lactate dehydrogenase activity assay. Lactate dehydrogenase (LDH)activity in primary cardiomyocyte cell supernatants was measured by acommercial spectrophotometric assay according to the manufacturer'sinstructions (DG1340-K; Sigma Diagnostics, Auckland, New Zealand).The primary cardiomyocytes were pre-treated with CORM-2 (20–400 lM) as above, 20 min. prior to H2O2 (0.1–1.0 mM) exposure.The effects of CORM-2 and iCORM-2 were compared at 20 lM.LDH activity was calculated from the mean kinetic rate of conversionof NADH to NAD+ based on the millimolar absorptivity of 6.22 at340 nm of NADH at 25°C. The extent of CORM-2‘s effect on LDHactivity was separately determined in 1.0% (w/v) sodium dodecylsulphate (SDS) lysed cell extracts collected by centrifugation(12,000 x g, 10 min.) from harvested cardiomyocytes. Cell lysate wasincubated with CORM-2 (20 and 400 lM) in a final volume of 0.5 mlat 37°C and assayed for LDH activity on three separate occasions asabove. Results were normalised against DMSO vehicle control.
Crystal violet cell adhesion assay. The loss of cell adhesion foranchorage-dependent cell lines, such as MDCK and HeK cells, repre-sents a surrogate marker of imminent cellular demise which can bemeasured by the crystal violet cell adhesion (CVCA) assay [33]. AfterCORM-2/iCORM-2/CO incubation, growth media were decanted andcells fixed in 96% ethanol and stained in 0.05% crystal violet solutionin 20% ethanol. Excess stain was washed off with distilled water, andcells solubilised in a final volume of 2 ml 0.1% acetic acid in 50%ethanol. Sample solute (100 ll) was collected after 24 hr from eachwell and absorbance measured at 585 nm.
Cytological evaluation. MDCK cells were seeded as described above,onto 7 mm sterile, glass coverslips (Zeiss, Oberkochen, Germany)in individual wells of a 24-well plate. After 24 hr, CORM-2 ori-CORM-2 (20 or 100 lM) or control (0.2 or 1% DMSO) in freshmedia was added to each well and cells cultured for a further 24 hr.Cover slips were fixed in chilled (4°C) acetone for 20 min. and storedat �20°C. Cells were stained with Harris haematoxylin, matured andcounterstained in 1% eosin-Y (in 70% ethanol). Qualitative cytologicalevaluations were performed in a double-blind manner on five individ-ual coverslip samples per group, using an Axioplan-2 microscope and
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
2 IAN C. WINBURN ET AL.

recorded with the Axiovision 3.1 image analysis system (Carl ZeissLtd., Göttingen, Germany).
Mitochondrial complex activity assays. Cells were cultured in T25flasks until 95% confluent and exposed to CO, CORM-2, iCORM-2 orvehicle as above. Cells were stripped using 0.1% trypsin, pelleted at18 x g (5 min. at 4°C) and resuspended in 1 ml of mitochondrial isola-tion buffer (225 mM mannitol, 75 mM sucrose, 10 mM Tris base,2 mM EGTA and 0.1 mM PMSF; pH 7.2 at 4°C). This cell suspensionwas stored at �80°C for further analysis. Cell suspensions were freeze-thawed three times in liquid nitrogen to ensure complete cellular andmitochondrial lysis. Cell lysate protein concentrations were determinedusing a modified Bradford assay and diluted in isolation buffer to 1 mg/ml. Assays for mitochondrial enzyme kinetics, complex I (NADH-ubi-quinone oxidoreductase, EC 1.6.99.3), complex V (ATPase, EC 3.6.1.3)as well as citrate synthase (EC 4.1.3.7) a mitochondrial matrix enzyme,were performed essentially as described earlier [34–36]. Mitochondrialenzyme activities were assessed using a SpectraMax-Plus 96-wellspectrophotometer (Molecular Devices, Crawley, UK) at 30°C. Opticalpath-lengths were corrected for microplate use and enzyme activitiesexpressed as nmol NADH/min./mg protein for complex I and V and asnmol aconitate/min./mg protein for citrate synthase.
Assessment of caspase 3/7 activity. Preliminary studies confirmed thatexposure to 20 lM cisplatin reproducibly produced apoptosis in bothcell lines as previously shown [37]. To compare CO gas and CORM-2-mediated protection against cisplatin-induced apoptosis, both MDCKand HeK cells were cultured as above in individual black-walled 96-well plates. Once the cells had reached 80% confluence, they weretreated with 20 lM cisplatin dissolved in sterile 0.9% saline for 1 hrprior to exposure to 20 ppm CO gas or the addition of CORM-2/iCORM-2 (20 or 100 lM). Control (no additions) and vehicle groups(1% DMSO) were also evaluated. Cells were then incubated for a totalof 16 hr after cisplatin administration and then assayed for caspase-3/7activity using the EnzoLyteTM AFC Caspase 3/7 Assay kit (AnaSpec,San Jose, CA, USA) according to the manufacturer's instructions.Upon cleavage by caspase-3 and or -7, Ac-DEVD-AFC liberates theAFC fluorophore which after 45 min. incubation was detected at exci-tation/emission wavelengths of 380/500 nm, respectively.
Assessment of apoptotic versus necrotic cell death. The relative con-tribution of apoptotic versus necrotic cell death, as a result of exposureto CORM-2, was assessed by annexin V FITC and propidium iodidefluorescence-activated cell sorting (FACS) analysis. MDCK cells werecultured for 24 hr in six-well plates as above and then incubated in thepresence of CORM-2/iCORM-2 (20 or 100 lM) for 24 hr. Cells weredetached using 2 ml of 0.1% trypsin, washed (92) in cold PBS andthen resuspended at 1 9 106 cells/ml in binding buffer (0.1 M Hepes,1.4 M NaCl, 25 mM CaCl2, pH 7.4). 100 ll of the cell suspensionwas incubated with 5 ll of FITC annexin V and 10 ll of PI (Invitro-gen, Eugene, OR, USA) in the dark for 15 min. at room temperature.Samples were diluted in binding buffer and analysed by flow cytometryusing a FACSCaliburTM flow cytometer (BD, La Jolla, CA, USA) andFlowJo software, v9.3.3 (Treestar Inc., San Carlos, CA, USA).
Cell cycle profiles. FACS analysis was used to determine the effect ofCORM-2 on MDCK cell cycle profiles. Cells were treated and har-vested as described in the section above, then washed (92) in coldPBS with 0.1% BSA (FACS buffer). Cells (1 9 106 cells/ml) werere-suspended in 200 ll FACS buffer and then fixed and permeabilisedusing 600 ll of ice-cold ethanol (2 hr). After two further washes inFACS buffer, cells were re-suspended in FACS buffer containing100 ng/ml PI (Invitrogen) and RNase (Invitrogen) and incubated in
the dark for 2 hr. Cells were rewashed and re-suspended in 300 llFACS buffer and analysed by flow cytometry as described using Win-MDI software, v2.8 (Scripps Research Institute, La Jolla, CA, USA)to quantify the percentage of cells existing in the G1, S and G2/Mphase of the cell cycle [38].
Statistics. Data are expressed mean ± S.E.M. Statistical analysis wasperformed using SigmaStat v2.03 (SPSS, Chicago, IL, USA). Statisti-cal significance was determined across groups for each of the variablesusing one-way ANOVA with post hoc analysis conducted using a Bon-ferroni test to compare the effect of compounds on cell viability at asingle time-point. A p-value of <0.05 was considered significant.
Results
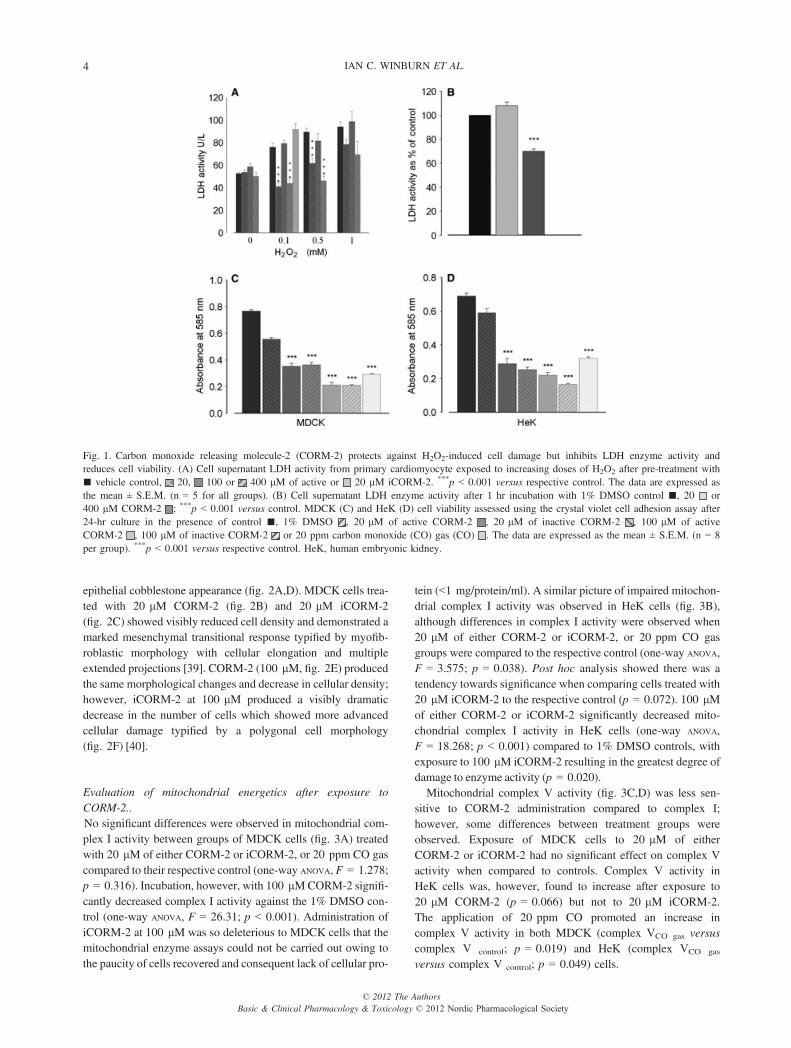
Primary cardiomyocyte cell viability by LDH assay.Exposure of primary cardiomyocytes to 20, 100 and 400 lMCORM-2 prior to treatment with increasing concentrations ofH2O2 did not produce an obvious concentration-dependentresponse (fig. 1A). Twenty and 400 lM CORM-2 appeared tosignificantly reduce LDH activity in supernatants from cellsexposed to 0.1 and 0.5 mM H2O2 compared to respective con-trols. However, no such protective effect was observed afterpre-incubation with 100 lM CORM-2. CORM-2 treatmentfailed to reduce LDH release observed after exposure of cellsto the highest concentration of H2O2 (1 mM). Examination ofthe effects of CORM-2 on cellular LDH activity in SDS-derived cardiomyocyte lysate preparations showed (fig. 1B)that at 20 lM, CORM-2 did not significantly affect LDHactivity compared to control. However, 400 lM CORM-2 wasfound to significantly inhibit LDH enzyme activity by 29%compared control cell lysate.
MDCK and HeK cell viability after exposure to CORM-2.Cell viability was assessed using the CVCA assay in MDCK(fig. 1C) and HeK (fig. 1D) cells exposed to active CORM-2,iCORM-2 or 20 ppm of CO. Treatment with either the COdonor molecule or CO gas reduced cell viability in both celllines (p < 0.001 against the respective control). MDCK cellsexposed to either CORM-2 or iCORM-2 at 100 lM weremore adversely (p < 0.001) affected than cells treated with20 lM CORM-2 or iCORM-2. CO gas at 20 ppm reducedMDCK cell viability to a lesser extent than 100 lM CORM-2/iCORM-2 (p < 0.001). Treatment with iCORM-2 was notassociated with any reduction in MDCK cell viability whencompared to the active preparation at the same concentration.Similarly, HeK cell viability was also significantly reducedafter exposure to 20 ppm CO or CORM-2/i-CORM-2 againstvehicle controls. Exposure of HeK cells to 100 lM iCORM-2reduced cell number by the greatest degree, more so than theactive CORM-2 (100 lM p < 0.001). There was no differencein the HeK cell viability between cells treated with 20 or100 lM active CORM-2 (p = 0.226).
Cytological evaluation after exposure to CORM-2.Vehicle control treated MDCK cells (0.2% and 1% DMSO) wereobserved to form a uniform monolayer with a characteristic
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
CARBON MONOXIDE DONOR MOLECULE TOXICITY 3
epithelial cobblestone appearance (fig. 2A,D). MDCK cells trea-ted with 20 lM CORM-2 (fig. 2B) and 20 lM iCORM-2(fig. 2C) showed visibly reduced cell density and demonstrated amarked mesenchymal transitional response typified by myofib-roblastic morphology with cellular elongation and multipleextended projections [39]. CORM-2 (100 lM, fig. 2E) producedthe same morphological changes and decrease in cellular density;however, iCORM-2 at 100 lM produced a visibly dramaticdecrease in the number of cells which showed more advancedcellular damage typified by a polygonal cell morphology(fig. 2F) [40].
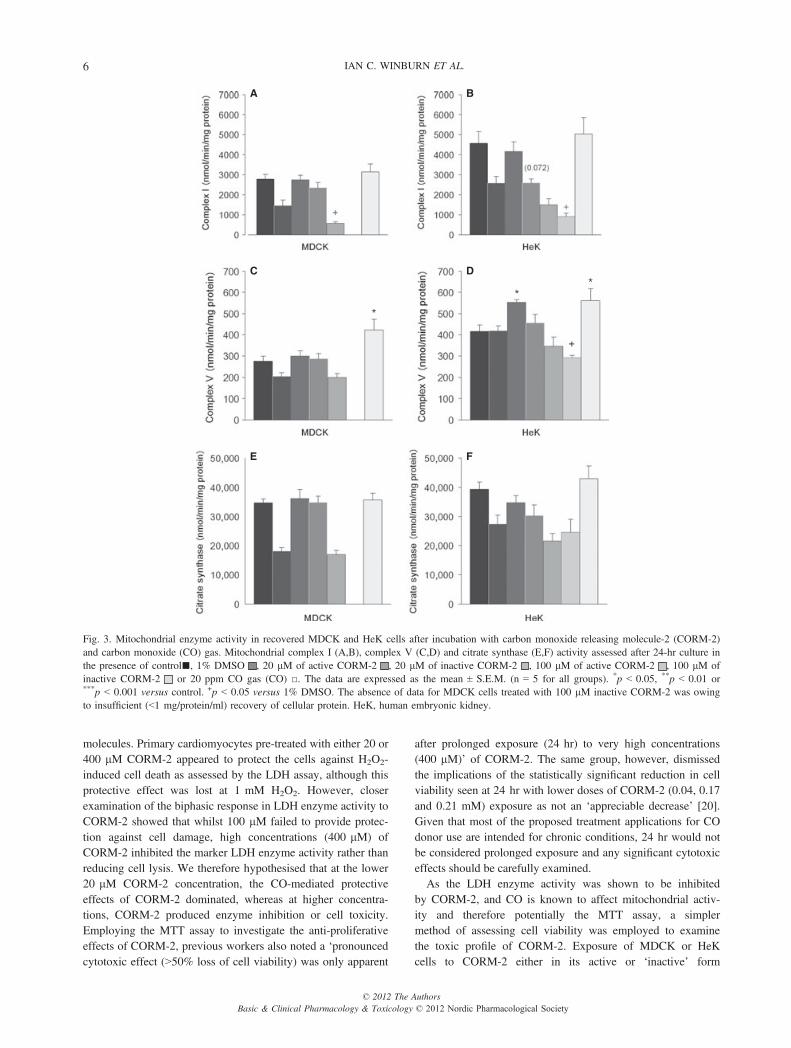
Evaluation of mitochondrial energetics after exposure toCORM-2..No significant differences were observed in mitochondrial com-plex I activity between groups of MDCK cells (fig. 3A) treatedwith 20 lM of either CORM-2 or iCORM-2, or 20 ppm CO gascompared to their respective control (one-way ANOVA, F = 1.278;p = 0.316). Incubation, however, with 100 lMCORM-2 signifi-cantly decreased complex I activity against the 1% DMSO con-trol (one-way ANOVA, F = 26.31; p < 0.001). Administration ofiCORM-2 at 100 lM was so deleterious to MDCK cells that themitochondrial enzyme assays could not be carried out owing tothe paucity of cells recovered and consequent lack of cellular pro-
tein (<1 mg/protein/ml). A similar picture of impaired mitochon-drial complex I activity was observed in HeK cells (fig. 3B),although differences in complex I activity were observed when20 lM of either CORM-2 or iCORM-2, or 20 ppm CO gasgroups were compared to the respective control (one-way ANOVA,F = 3.575; p = 0.038). Post hoc analysis showed there was atendency towards significance when comparing cells treated with20 lM iCORM-2 to the respective control (p = 0.072). 100 lMof either CORM-2 or iCORM-2 significantly decreased mito-chondrial complex I activity in HeK cells (one-way ANOVA,F = 18.268; p < 0.001) compared to 1% DMSO controls, withexposure to 100 lM iCORM-2 resulting in the greatest degree ofdamage to enzyme activity (p = 0.020).Mitochondrial complex V activity (fig. 3C,D) was less sen-
sitive to CORM-2 administration compared to complex I;however, some differences between treatment groups wereobserved. Exposure of MDCK cells to 20 lM of eitherCORM-2 or iCORM-2 had no significant effect on complex Vactivity when compared to controls. Complex V activity inHeK cells was, however, found to increase after exposure to20 lM CORM-2 (p = 0.066) but not to 20 lM iCORM-2.The application of 20 ppm CO promoted an increase incomplex V activity in both MDCK (complex VCO gas versuscomplex V control; p = 0.019) and HeK (complex VCO gas
versus complex V control; p = 0.049) cells.
Fig. 1. Carbon monoxide releasing molecule-2 (CORM-2) protects against H2O2-induced cell damage but inhibits LDH enzyme activity andreduces cell viability. (A) Cell supernatant LDH activity from primary cardiomyocyte exposed to increasing doses of H2O2 after pre-treatment with■ vehicle control, 20, 100 or 400 lM of active or 20 lM iCORM-2. ***p < 0.001 versus respective control. The data are expressed asthe mean ± S.E.M. (n = 5 for all groups). (B) Cell supernatant LDH enzyme activity after 1 hr incubation with 1% DMSO control ■, 20 or400 lM CORM-2 ; ***p < 0.001 versus control. MDCK (C) and HeK (D) cell viability assessed using the crystal violet cell adhesion assay after24-hr culture in the presence of control ■, 1% DMSO , 20 lM of active CORM-2 , 20 lM of inactive CORM-2 , 100 lM of activeCORM-2 , 100 lM of inactive CORM-2 or 20 ppm carbon monoxide (CO) gas (CO) . The data are expressed as the mean ± S.E.M. (n = 8per group). ***p < 0.001 versus respective control. HeK, human embryonic kidney.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
4 IAN C. WINBURN ET AL.

Mitochondrial citrate synthase activity (fig. 3E) was notadversely affected after the administration of 20 lM of eitherCORM-2 or the iCORM-2 or 20 ppm CO gas (MDCK:one-way ANOVA, F = 0.100; p = 0.960, HeK: one-way ANOVA,F = 2.771; p = 0.075 versus control). There was no significantdifference in the severity of citrate synthase impairment whentreatment with 100 lM of CORM-2 or the iCORM-2 was com-pared with the respective 1% DMSO control in either cell type.
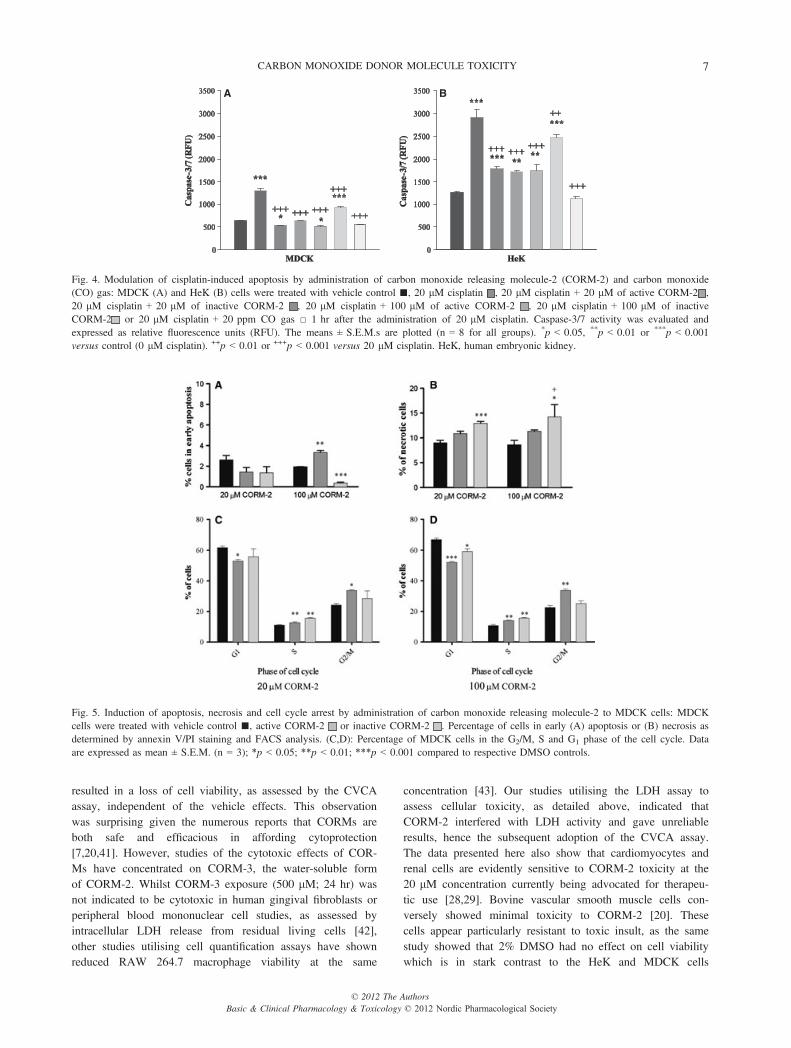
The effect of CORM-2 and CO gas on cisplatin-inducedapoptosis.As expected, the addition of 20 lM cisplatin to cultured MDCKand HeK cells increased (p < 0.001) activities of the apoptoticmarker enzymes, caspase-3/7 (fig. 4). Cisplatin-stimulated cas-pase activity in MDCK cells (fig. 4A) was conversely decreased(one-way ANOVA, F = 106.400; p < 0.001) in the presence ofeither CORM-2 (20 and 100 lM p < 0.001), i-CORM-2 (20and 100 lM p < 0.001) or 20 ppm CO gas (p < 0.001). Similarresults were obtained in HeK cells (fig. 4B) after eitherCORM-2 (20 and 100 lM p < 0.001), i-CORM-2 (20 lMp < 0.001, 100 lM p = 0.017) or 20 ppm CO gas (p < 0.001)administration (one-way ANOVA, F = 51.747; p < 0.001). Onlytreatment of MDCK cells with 100 lM of iCORM-2 failed todecrease cisplatin-induced caspase-3/7 activity to levelsobtained in the non-cisplatin-treated control group (p < 0.001).This was in contrast to HeK cells where the cisplatin-inducedincrease in caspase-3/7 activity remained elevated abovecontrols even in the presence of 20 and 100 lM CORM-2 oriCORM-2 (20 lM CORM-2 p < 0.001; 100 lM CORM-2p = 0.004; 20 lM iCORM-2 p = 0.016; 100 lM iCORM-2p < 0.001). Treatment with 20 ppm CO gas inhibited the cis-platin-induced apoptotic response and resulted in a statistically
equivalent caspase-3/7 activity in both cell types when com-pared to controls (p = 1.000).
The effect of CORM-2 on apoptotic and necrotic cell death.Treatment of MDCK cells with 20 lM CORM-2 or iCORM-2resulted in no change in apoptotic markers compared tocontrol (0.2% DMSO) (fig. 5A). Whereas 100 lM CORM-2increased apoptosis, application of 100 lM iCORM-2appeared to decrease apoptotic cell numbers compared to 1%DMSO. Additionally, both 20 and 100 lM iCORM-2 causeda much larger, significant increase in PI staining, reported aspercentage of total cells undergoing necrosis compared to con-trol (fig. 5B). No significant change in late apoptosis wasobserved in any treatment group (results not shown).
Cell cycle arrest.FACS analysis of cell cycle profiles showed that treatment ofMDCK cells with 20 or 100 lM CORM-2 resulted in a signif-icant decrease in the percentage of cells in the G1 phase,whilst significantly increasing the percentage of cells in boththe S and G2/M phases (fig. 5C,D). Whilst cells treated with100 lM of iCORM-2 also showed a small significant reduc-tion in the percentage of cells in the G1 phase compared tocontrol, 20 or 100 lM iCORM-2 significantly increased thenumber of cells in the S phase (fig. 5C,D).
Discussion
There are a number of studies employing metal carbonyl COdonor molecules in cellular and organ protection applications.However, few have intensively examined the toxicity of theresidual as well as the active form of these CO donor
A B C
D E F
Fig. 2. Photomicrographs of MDCK cells after exposure to carbon monoxide releasing molecule-2 (CORM-2). MDCK cells were cultured on glasscover slips for 24 hr and then exposed and maintained in medium containing respective vehicle control (A,D), or 20 lM CORM-2 (B), 20 lMiCORM-2 (C), 100 lM CORM-2 (E) or 100 lM iCORM-2 (F) for a further 24 hr and stained with H&E. Scale bar shown represents 20 lm.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
CARBON MONOXIDE DONOR MOLECULE TOXICITY 5
molecules. Primary cardiomyocytes pre-treated with either 20 or400 lM CORM-2 appeared to protect the cells against H2O2-induced cell death as assessed by the LDH assay, although thisprotective effect was lost at 1 mM H2O2. However, closerexamination of the biphasic response in LDH enzyme activity toCORM-2 showed that whilst 100 lM failed to provide protec-tion against cell damage, high concentrations (400 lM) ofCORM-2 inhibited the marker LDH enzyme activity rather thanreducing cell lysis. We therefore hypothesised that at the lower20 lM CORM-2 concentration, the CO-mediated protectiveeffects of CORM-2 dominated, whereas at higher concentra-tions, CORM-2 produced enzyme inhibition or cell toxicity.Employing the MTT assay to investigate the anti-proliferativeeffects of CORM-2, previous workers also noted a ‘pronouncedcytotoxic effect (>50% loss of cell viability) was only apparent
after prolonged exposure (24 hr) to very high concentrations(400 lM)’ of CORM-2. The same group, however, dismissedthe implications of the statistically significant reduction in cellviability seen at 24 hr with lower doses of CORM-2 (0.04, 0.17and 0.21 mM) exposure as not an ‘appreciable decrease’ [20].Given that most of the proposed treatment applications for COdonor use are intended for chronic conditions, 24 hr would notbe considered prolonged exposure and any significant cytotoxiceffects should be carefully examined.As the LDH enzyme activity was shown to be inhibited
by CORM-2, and CO is known to affect mitochondrial activ-ity and therefore potentially the MTT assay, a simplermethod of assessing cell viability was employed to examinethe toxic profile of CORM-2. Exposure of MDCK or HeKcells to CORM-2 either in its active or ‘inactive’ form
*
**
A B
C D
E F
Fig. 3. Mitochondrial enzyme activity in recovered MDCK and HeK cells after incubation with carbon monoxide releasing molecule-2 (CORM-2)and carbon monoxide (CO) gas. Mitochondrial complex I (A,B), complex V (C,D) and citrate synthase (E,F) activity assessed after 24-hr culture inthe presence of control■, 1% DMSO , 20 lM of active CORM-2 , 20 lM of inactive CORM-2 , 100 lM of active CORM-2 , 100 lM ofinactive CORM-2 or 20 ppm CO gas (CO) □. The data are expressed as the mean ± S.E.M. (n = 5 for all groups). *p < 0.05, **p < 0.01 or***p < 0.001 versus control. +p < 0.05 versus 1% DMSO. The absence of data for MDCK cells treated with 100 lM inactive CORM-2 was owingto insufficient (<1 mg/protein/ml) recovery of cellular protein. HeK, human embryonic kidney.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
6 IAN C. WINBURN ET AL.
resulted in a loss of cell viability, as assessed by the CVCAassay, independent of the vehicle effects. This observationwas surprising given the numerous reports that CORMs areboth safe and efficacious in affording cytoprotection[7,20,41]. However, studies of the cytotoxic effects of COR-Ms have concentrated on CORM-3, the water-soluble formof CORM-2. Whilst CORM-3 exposure (500 lM; 24 hr) wasnot indicated to be cytotoxic in human gingival fibroblasts orperipheral blood mononuclear cell studies, as assessed byintracellular LDH release from residual living cells [42],other studies utilising cell quantification assays have shownreduced RAW 264.7 macrophage viability at the same
concentration [43]. Our studies utilising the LDH assay toassess cellular toxicity, as detailed above, indicated thatCORM-2 interfered with LDH activity and gave unreliableresults, hence the subsequent adoption of the CVCA assay.The data presented here also show that cardiomyocytes andrenal cells are evidently sensitive to CORM-2 toxicity at the20 lM concentration currently being advocated for therapeu-tic use [28,29]. Bovine vascular smooth muscle cells con-versely showed minimal toxicity to CORM-2 [20]. Thesecells appear particularly resistant to toxic insult, as the samestudy showed that 2% DMSO had no effect on cell viabilitywhich is in stark contrast to the HeK and MDCK cells
A B
Fig. 4. Modulation of cisplatin-induced apoptosis by administration of carbon monoxide releasing molecule-2 (CORM-2) and carbon monoxide(CO) gas: MDCK (A) and HeK (B) cells were treated with vehicle control ■, 20 lM cisplatin , 20 lM cisplatin + 20 lM of active CORM-2 ,20 lM cisplatin + 20 lM of inactive CORM-2 , 20 lM cisplatin + 100 lM of active CORM-2 , 20 lM cisplatin + 100 lM of inactiveCORM-2 or 20 lM cisplatin + 20 ppm CO gas □ 1 hr after the administration of 20 lM cisplatin. Caspase-3/7 activity was evaluated andexpressed as relative fluorescence units (RFU). The means ± S.E.M.s are plotted (n = 8 for all groups). *p < 0.05, **p < 0.01 or ***p < 0.001versus control (0 lM cisplatin). ++p < 0.01 or +++p < 0.001 versus 20 lM cisplatin. HeK, human embryonic kidney.
Fig. 5. Induction of apoptosis, necrosis and cell cycle arrest by administration of carbon monoxide releasing molecule-2 to MDCK cells: MDCKcells were treated with vehicle control ■, active CORM-2 or inactive CORM-2 . Percentage of cells in early (A) apoptosis or (B) necrosis asdetermined by annexin V/PI staining and FACS analysis. (C,D): Percentage of MDCK cells in the G2/M, S and G1 phase of the cell cycle. Dataare expressed as mean ± S.E.M. (n = 3); *p < 0.05; **p < 0.01; ***p < 0.001 compared to respective DMSO controls.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
CARBON MONOXIDE DONOR MOLECULE TOXICITY 7

where concentrations above 0.4% reduced cell viability (datanot shown). Similarly, 10 nM CORM-2 has also been previ-ously shown to induce cell death in HL-1 heart cells [44].As iCORM-2 was not used with the HL-1 heart cells, it isimpossible to conclude whether the observed toxicity wasinduced by CO release or the result of the ruthenium-basedCORM-2. The hypothesis that toxicity may be mediated bythe core molecular structure of the CORM is supported bythe current study where no differences in cell viability, asassessed by the CVCA assay, were observed in MDCK cellstreated with either iCORM-2 or the active CORM-2.Several studies have included the use of iCORMs as nega-
tive controls, although there is evidence that challenges theassertion that these molecules are truly ‘inactive’. The additionof iCORM-3 (10 mg/kg intra-peritoneal) to cisplatin-treatedrats resulted in more severe histological scoring of renal tissuewhen compared to rats exposed to cisplatin alone [28]. AsCORM-3 is water-soluble, this study supports the hypothesisthat CORM-related toxicity is independent of the solvent usedor the minor structural modifications between CORM-2 andCORM-3. Further histological evidence of iCORM-3 toxicitywas seen as increases in sinusoidal and laminar injuries com-pared to controls in rat livers subjected to cold storage prior totransplantation [45]. Evidence of iCORM toxicity is providedby studies showing that stress response proteins, like HO-1,are induced after iCORM-3 exposure in diabetic rat aorta [46]and in ischaemic rat renal tissue [47], further indicating thatthese ‘inactive’ CORMs, although lacking CO release, are notwithout biological effect. Other studies have utilised rutheniumchloride as a negative control when evaluating the propertiesof CORM-2 or CORM-3, possibly to mitigate against theconfounding effects that ‘inactive’ CORM compoundsexert [48]. However, when microglia cells were exposed toIFN-c-induced neuro-inflammation, 75 lM ruthenium chlorideadministration resulted in significantly greater cell toxicitycompared to control cells [49]. These findings suggest thatruthenium may be partly responsible for toxicity associatedwith CORM-2/-3 administration.The CVCA data suggest that exposure to 20 ppm CO gas
reduced cell number. The lack of adverse effect of CO gas onmitochondrial energetics at this low dose, however, suggeststhat the reduced cell number may not stem from a toxic effectbut from an anti-proliferative effect. Indeed, CO has beendemonstrated to reduce smooth muscle cell and T-lymphocyteproliferation via a cGMP-dependent down-regulation of endo-thelial-derived mitogens [50,51].MDCK cells exposed to 20 lM CORM-2 or iCORM-2
underwent morphological changes typical of endothelial tomesenchymal transition [39] with 100 lM iCORM-2 resultingin more advanced damage typified by the polygonal appear-ance of the cells [40]. Administration of iCORM-2 at 100 lMimpaired MDCK cells to such an extent that insufficient cellswere retrieved to run the mitochondrial complex activityassays. The profound cell loss witnessed after iCORM-2administration suggests that the cytological effects observedare independent of the anti-proliferative properties of CO.Other authors have also described disruptions in histological
architecture after iCORM-3 exposure, particularly in the kid-ney and liver [45].Previous published data have indicated that CORM-3 (20
and 100 lM) does not significantly impair complex I activityin isolated rat cardiac mitochondria [52]. The work suggested,however, that as iCORM-3 had no effect, CO release fromlow (20 and 100 lM) levels of CORM-3 must be interact-ing with mitochondrial uncoupling proteins and adeninedinucleotide transporters to disrupt membrane potentials. Thereis previous work by Alonso et al. [11] to show that CO(� 500 ppm) does not induce complex I impairment and theapplication of CO gas to MDCK and HeK cells in our study,produced a similar absence of effect supporting the conclusionthat the reduction in complex I activity was not a direct effectof CO. In the current study, mitochondrial complex I activitywas, however, impaired after the administration of CORM-2(100 lM) or iCORM-2 (20 lM) to renal epithelial cells. Thisnovel finding suggests that this impairment may be a conse-quence of the ruthenium-based molecule inhibiting themitochondrial Ca2+ uniporter in a similar fashion to the non-competitive inhibitor, ruthenium red (ammoniated rutheniumoxychloride). Ruthenium red, at nano to micromolar concen-trations, induces mitochondrial toxicity by disrupting the Ca2+
uniporter, inactivating Ca2+-dependent mitochondrial enzymes[53,54] and impairing complex I activity [55]. Rutheniumcompounds have demonstrated significant mitochondrial toxic-ity [56–59] adding support to the hypothesis that the ruthe-nium base of CORM-2 may be responsible for the impairmentof complex I activity observed in this study.The finding that iCORM-2 inhibited complex V activity at
a concentration of 100 lM in HeK cells is especially signifi-cant given the fact that exposure of both cell lines to 20 ppmCO gas resulted in heightened complex V activity. CO con-taining CORM-2 (20 lM) also raised complex V activity inHeK cells. A possible explanation is that, in mice and humanbeings, 24-hr exposure to low concentrations of CO increasesactivity and levels of complex V protein and may also pro-mote mitochondrial biogenesis, secondary to an increase inmitochondrial ROS, cGMP expression and Akt signalling[15,60]. However, the failure of CO or CORM-2 to alter com-plex I or citrate synthase activity in MDCK and HeK cellschallenges the assertion that CO promotes mitochondrial bio-genesis. Citrate synthase, a mitochondrial matrix enzyme, hasbeen used to provide a quantitative estimation of mitochon-drial integrity. Previous studies examining CO-mediated bio-genesis have only quantified mitochondrial DNA copies andgene products [15,60,61]. The data presented here, however,provide evidence that citrate synthase activity related to cellu-lar protein was unaffected in CO or CORM-2-treated cells.This finding suggests that the metal carbonyl in CORM-2 andiCORM-2 can directly inhibit complex I whilst CO gas canstimulate complex V, rather than mitochondrial biogenesis.Both CO gas (20 ppm) and CORM-2 (at 20 and 100 lM)
protected against cisplatin-induced apoptosis in both cell linesas assessed by the caspase activity assay. These findings aresimilar to previously published data examining the effects of1–50 lM CORM-3 treatment in the LLC-PK1 proximal
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
8 IAN C. WINBURN ET AL.

tubule cell line [6]. Surprisingly, we also found that 20 lMiCORM-2 also reduced caspase activity, although this effectwas diminished at the higher concentration. This finding fur-ther casts doubt over the ‘inactivity’ of iCORM-2. Althoughprotection against cisplatin-induced apoptosis by iCORM-3was not reported in the LLC-PK1 proximal tubule cells, 300–500 lM iCORM-3 has been demonstrated to reduce caspase-3 and -7 activities in a xenogeneic pig to primate model[28,29]. iCORM-3 has, however, also been shown to induceHO-1 expression which is documented to protect against cis-platin and ischaemia-reperfusion injury-induced apoptosis[28,47,62,63]. Evidence that CO scavenging oxyhaemoglobindoes not completely inhibit the anti-apoptotic effect of theCORMs supports the suggestion that CORM-2 protection isnot entirely consequent to CO release [64]. These findingssuggest that both the active and ‘inactive’ CORM moleculesprovide some cytoprotection independently of CO.Annexin V binding was decreased by iCORM-2 at 100 lM
whilst the active form increased apoptosis in the absence of anapoptotic inducing agent. PI staining indicative of necrosis,however, confirmed that the reduction in apoptotic values areattributable to the increase in necrosis consequent to iCORM-2administration (20 and 100 lM). Both CORM-2 as well asiCORM-2 (20 and 100 lM) also produced significant increasesin the percentage of cells arrested in the S phase of the cell cyclesuggesting that this effect is not attributable to CO. Studies haveshown that oxidative stress can increase DNA replication[65,66]. Therefore, we hypothesise that oxidative stress causedby exposure of MDCK cells to the ruthenium (II) metal centreof CORM-2/iCORM-2 may be causing this cellular effect. Asentry into the mitosis is blocked at the G2 checkpoint whenchromosomal DNA is damaged, the increased cell number pres-ent in the G2/M phase after CORM-2 (20 and 100 lM) expo-sure could be an indicator of DNA damage by the molecule.Interestingly, iCORM-2 had no effect on G2/M population num-bers suggesting a CO-mediated effect.As our results clearly show cell responses to both active
and inactive CORM-2, we suggest that these effects may bemediated by the transition metal base of the donor molecule.Although mechanisms of ruthenium handling by the kidneyare uncertain, it is likely that ruthenium is handled in a similarway to other transition metals, such as iron [67]. Transferrin-bound iron enters proximal tubule cells via either basolateralmembrane transport or endocytic uptake from the luminal sur-face [68]. Subsequent accumulation and/or release of ironfrom lysosomes may promote iron-catalysed ROS productionand subsequent cellular damage [69]. Whilst other CO donormolecules have been investigated, such as the boranocarbon-ate-related CORMs, these may release borane ‘BH3’ to pro-duce a BH3(OH) anion thereby forming borate or boric acid.Whilst borate may be considered safe in terms of acute toxic-ity, its chronic effects on a number of organ systems make itless than ideal as a therapeutic agent [70].The primary findings from this study provide strong evi-
dence that exposure of primary cardiomyocytes, MDCK andHeK cells to ruthenium metal carbonyls but not CO gasresults in cytotoxic effects. A reduction in cell viability was
observed with CO gas, which we suggest may result as a con-sequence of the anti-proliferative effects afforded by CO. Thisstudy shows that a much narrower margin exists between thecytoprotective and cytotoxic concentration of CORM-2 in cellsthan previously reported with the ruthenium-based CORM-2in bovine vascular smooth muscle cells or CORM-3 inRAW264.7 monocyte macrophages [20,71]. Thus, accumula-tion of the ruthenium containing carbonyls in vivo, after theliberation of CO, could limit any clinical application of thesemolecules.
AcknowledgementsThis work was supported by funding for a Clinical
Research Training Fellowship from the New Zealand HealthResearch Council (ICW) and utilised equipment funded by theNew Zealand Lottery Health (IAS & JCH).
References
1 Tenhunen R, Marver HS, Schmid R. The enzymatic conversion ofheme to bilirubin by microsomal heme oxygenase. Proc Natl AcadSci U S A 1968;61:748–55.
2 Nakao A, Choi AM, Murase N. Protective effect of carbon monox-ide in transplantation. J Cell Mol Med 2006;10:650–71.
3 Neto JS, Nakao A, Toyokawa H, Nalesnik MA, Romanosky AJ,Kimizuka K et al.Low-dose carbon monoxide inhalation preventsdevelopment of chronic allograft nephropathy. Am J Physiol RenalPhysiol 2006;290:F324–34.
4 Chora AA, Fontoura P, Cunha A, Pais TF, Cardoso S, Ho PPet al.Heme oxygenase-1 and carbon monoxide suppress autoim-mune neuroinflammation. J Clin Invest 2007;117:438–47.
5 Sammut IA, Foresti R, Clark JE, Exon DJ, Vesely MJ, Sarathchan-dra P et al.Carbon monoxide is a major contributor to the regula-tion of vascular tone in aortas expressing high levels of haemeoxygenase-1. Br J Pharmacol 1998;125:1437–44.
6 Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiy-ama T et al.Protection of transplant-induced renal ischemia-reper-fusion injury with carbon monoxide. Am J Physiol Renal Physiol2004;287:F979–89.
7 Chlopicki S, Olszanecki R, Marcinkiewicz E, Lomnicka M, Mot-terlini R. Carbon monoxide released by CORM-3 inhibits humanplatelets by a mechanism independent of soluble guanylate cyclase.Cardiovasc Res 2006;71:393–401.
8 Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM.Carbon monoxide protects against hyperoxia-induced endothelialcell apoptosis by inhibiting reactive oxygen species formation.J Biol Chem 2007;282:1718–26.
9 Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide dif-ferentially modulates STAT1 and STAT3 and inhibits apoptosisvia a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependentSTAT3 pathway during anoxia-reoxygenation injury. J Biol Chem2005;280:8714–21.
10 D'Amico G, Lam F, Hagen T, Moncada S. Inhibition of cellularrespiration by endogenously produced carbon monoxide. J Cell Sci2006;119:2291–8.
11 Alonso JR, Cardellach F, Lopez S, Casademont J, Miro O. Carbonmonoxide specifically inhibits cytochrome C oxidase of human mito-chondrial respiratory chain. Pharmacol Toxicol 2003;93:142–6.
12 Nystul TG, Roth MB. Carbon monoxide-induced suspendedanimation protects against hypoxic damage in Caenorhabditiselegans. Proc Natl Acad Sci U S A 2004;101:9133–6.
13 Zuckerbraun BS, Chin BY, Bilban M, d'Avila JC, Rao J, BilliarTR et al.Carbon monoxide signals via inhibition of cytochrome c
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
CARBON MONOXIDE DONOR MOLECULE TOXICITY 9

oxidase and generation of mitochondrial reactive oxygen species.FASEB J 2007;21:1099–106.
14 Piantadosi CA, Carraway MS, Suliman HB. Carbon monoxide,oxidative stress, and mitochondrial permeability pore transition.Free Radic Biol Med 2006;40:1332–9.
15 Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new acti-vating role for CO in cardiac mitochondrial biogenesis. J Cell Sci2007;120:299–308.
16 Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich Ret al.Effects of carbon monoxide inhalation during experimentalendotoxemia in humans. Am J Respir Crit Care Med 2005;171:354–60.
17 Moore BA, Overhaus M, Whitcomb J, Ifedigbo E, Choi AM,Otterbein LE et al.Brief inhalation of low-dose carbon monoxideprotects rodents and swine from postoperative ileus. Crit Care Med2005;33:1317–26.
18 Nakao A, Schmidt J, Harada T, Tsung A, Stoffels B, Cruz RJet al.A single intraperitoneal dose of carbon monoxide-saturatedringer's lactate solution ameliorates postoperative ileus in mice.J Pharmacol Exp Ther 2006;319:1265–75.
19 Martins PN, Reuzel-Selke A, Jurisch A, Atrott K, Pascher A, Prat-schke J et al.Induction of carbon monoxide in the donor reducesgraft immunogenicity and chronic graft deterioration. TransplantProc 2005;37:379–81.
20 Motterlini R, Clark JE, Foresti R, Sarathchandra P, Mann BE, GreenCJ. Carbon monoxide-releasing molecules: characterization ofbiochemical and vascular activities. Circ Res 2002;90:E17–24.
21 Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, MannBE et al.Cardioprotective actions by a water-soluble carbon mon-oxide-releasing molecule. Circ Res 2003;93:e2–8.
22 Sandouka A, Balogun E, Foresti R, Mann BE, Johnson TR, TayemY et al.Carbon monoxide-releasing molecules (CO-RMs) modulaterespiration in isolated mitochondria. Cell Mol Biol (Noisy-le-grand) 2005;51:425–32.
23 Sandouka A, Fuller BJ, Mann BE, Green CJ, Foresti R, MotterliniR. Treatment with CO-RMs during cold storage improves renalfunction at reperfusion. Kidney Int 2006;69:239–47.
24 Menezes CS, de Paula Costa LC, de Melo RA, V , Ferreira MJ,Vieira CU et al.Analysis in vivo of antitumor activity, cytotoxicityand Interaction between plasmid DNA and the cis-dichloro-tetra-amine-ruthenium(III) chloride. Chem Biol Interact 2007;167:116–24.
25 Kostova I. Ruthenium complexes as anticancer agents. Curr MedChem 2006;13:1085–107.
26 Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W.Carbon monoxide inhibits apoptosis in vascular smooth musclecells. Cardiovasc Res 2002;55:396–405.
27 Queiroga CS, Almeida AS, Martel C, Brenner C, Alves PM,Vieira HL. Glutathionylation of adenine nucleotide translocaseinduced by carbon monoxide prevents mitochondrial membranepermeabilization and apoptosis. J Biol Chem 2010;285:17077–88.
28 Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R. Protec-tion against cisplatin-induced nephrotoxicity by a carbon monox-ide-releasing molecule. Am J Physiol Renal Physiol 2006;290:F789–94.
29 Vadori M, Seveso M, Besenzon F, Bosio E, Tognato E, Fante Fet al.In vitro and in vivo effects of the carbon monoxide-releasingmolecule, CORM-3, in the xenogeneic pig-to-primate context. Xe-notransplantation 2009;16:99–114.
30 Zhang X, Shan P, Otterbein LE, Alam J, Flavell RA, Davis RJet al.Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activatedprotein kinase pathway and involves caspase 3. J Biol Chem2003;278:1248–58.
31 Nakao A, Faleo G, Nalesnik MA, Seda-Neto J, Kohmoto J,Murase N. Low-dose carbon monoxide inhibits progressive chronicallograft nephropathy and restores renal allograft function. Am JPhysiol Renal Physiol 2009;297:F19–26.
32 Lawrence CL, Billups B, Rodrigo GC, Standen NB. The KATPchannel opener diazoxide protects cardiac myocytes during meta-bolic inhibition without causing mitochondrial depolarization orflavoprotein oxidation. Br J Pharmacol 2001;134:535–42.
33 Mickuviene I, Kirveliene V, Juodka B. Experimental survey ofnon-clonogenic viability assays for adherent cells in vitro. ToxicolIn Vitro 2004;18:639–48.
34 Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA,Murphy MP et al.Targeting an antioxidant to mitochondriadecreases cardiac ischemia-reperfusion injury. FASEB J 2005;19:1088–95.
35 Sammut IA, Jayakumar J, Latif N, Rothery S, Severs NJ, Smolen-ski RT et al.Heat stress contributes to the enhancement of cardiacmitochondrial complex activity. Am J Pathol 2001;158:1821–31.
36 Vranyac-Tramoundanas A, Harrison JC, Clarkson AN, Kapoor M,Winburn IC, Kerr DS et al.Domoic acid impairment of cardiacenergetics. Toxicol Sci 2008;105:395–407.
37 Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and reno-protective strategies. Kidney Int 2008;73:994–1007.
38 Darzynkiewicz Z, Juan G, Bedner E. Determining cell cycle stagesby flow cytometry. Curr Protoc Cell Biol 2001;Chapter 8:Unit.
39 Mariasegaram M, Tesch GH, Verhardt S, Hurst L, Lan HY, Nikol-ic-Paterson DJ. Lefty antagonises TGF-beta1 induced epithelial-mesenchymal transition in tubular epithelial cells. Biochem Bio-phys Res Commun 2010;393:855–9.
40 Prunotto M, Compagnone A, Bruschi M, Candiano G, ColombattoS, Bandino A et al.Endocellular polyamine availability modulatesepithelial-to-mesenchymal transition and unfolded protein responsein MDCK cells. Lab Invest 2010;90:929–39.
41 Foresti R, Hammad J, Clark JE, Johnson TR, Mann BE, Friebe Aet al.Vasoactive properties of CORM-3, a novel water-soluble car-bon monoxide-releasing molecule. Br J Pharmacol 2004;142:453–60.
42 Song H, Zhao H, Qu Y, Sun Q, Zhang F, Du Z et al.Carbon mon-oxide releasing molecule-3 inhibits concurrent tumor necrosis fac-tor-alpha- and interleukin-1beta-induced expression of adhesionmolecules on human gingival fibroblasts. J Periodontal Res2011;46:48–57.
43 Desmard M, Davidge KS, Bouvet O, Morin D, Roux D, Foresti Ret al.A carbon monoxide-releasing molecule (CORM-3) exerts bac-tericidal activity against Pseudomonas aeruginosa and improvessurvival in an animal model of bacteraemia. FASEB J 2009;23:1023–31.
44 Czibik G, Sagave J, Martinov V, Ishaq B, Sohl M, Sefland I et al.Cardioprotection by hypoxia-inducible factor 1 alpha transfectionin skeletal muscle is dependent on haem oxygenase activity inmice. Cardiovasc Res 2009;82:107–14.
45 Pizarro MD, Rodriguez JV, Mamprin ME, Fuller BJ, Mann BE,Motterlini R et al.Protective effects of a carbon monoxide-releas-ing molecule (CORM-3) during hepatic cold preservation. Cryobi-ology 2009;58:248–55.
46 Rodella L, Lamon BD, Rezzani R, Sangras B, Goodman AI, FalckJR et al.Carbon monoxide and biliverdin prevent endothelial cellsloughing in rats with type I diabetes. Free Radic Biol Med 2006;40:2198–205.
47 Vera T, Henegar JR, Drummond HA, Rimoldi JM, Stec DE. Pro-tective effect of carbon monoxide-releasing compounds in ische-mia-induced acute renal failure. J Am Soc Nephrol 2005;16:950–8.
48 Megias J, Busserolles J, Alcaraz MJ. The carbon monoxide-releas-ing molecule CORM-2 inhibits the inflammatory response inducedby cytokines in Caco-2 cells. Br J Pharmacol 2007;150:977–86.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
10 IAN C. WINBURN ET AL.

49 Bani-Hani MG, Greenstein D, Mann BE, Green CJ, Motterlini R.A carbon monoxide-releasing molecule (CORM-3) attenuates lipo-polysaccharide- and interferon-gamma-induced inflammation inmicroglia. Pharmacol Rep 2006;58(Suppl):132–44.
50 Morita T, Perrella MA, Lee ME, Kourembanas S. Smooth musclecell-derived carbon monoxide is a regulator of vascular cGMP.Proc Natl Acad Sci U S A 1995;92:1475–9.
51 Otterbein LE, Zuckerbraun BS, Haga M, Liu F, Song R, UshevaA et al.Carbon monoxide suppresses arteriosclerotic lesions associ-ated with chronic graft rejection and with balloon injury. Nat Med2003;9:183–90.
52 Lo IL, Boczkowski J, Zini R, Salouage I, Berdeaux A, Motterlini Ret al.A carbon monoxide-releasing molecule (CORM-3) uncouplesmitochondrial respiration and modulates the production of reactiveoxygen species. Free Radic Biol Med 2011;50:1556–64.
53 McCormack JG, Denton RM. Influence of calcium ions on mamma-lian intramitochondrial dehydrogenases. Methods Enzymol 1989;174:95–118.
54 Johnston JD, Brand MD. Stimulation of the respiration rate of ratliver mitochondria by sub-micromolar concentrations of extrami-tochondrial Ca2 + . Biochem J 1987;245:217–22.
55 Martin M, Macias M, Escames G, Reiter RJ, Agapito MT, OrtizGG et al.Melatonin-induced increased activity of the respiratorychain complexes I and IV can prevent mitochondrial damageinduced by ruthenium red in vivo. J Pineal Res 2000;28:242–8.
56 Meinicke AR, Bechara EJ, Vercesi AE. Ruthenium red-catalyzeddegradation of peroxides can prevent mitochondrial oxidativedamage induced by either tert-butyl hydroperoxide or inorganicphosphate. Arch Biochem Biophys 1998;349:275–80.
57 Landgraf G, Gellerich FN, Wussling MH. Inhibitors of SERCAand mitochondrial Ca-uniporter decrease velocity of calcium wavesin rat cardiomyocytes. Mol Cell Biochem 2004;257:379–86.
58 Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di LFet al.Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiaccells. EMBO J 2001;20:4998–5007.
59 Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation inadult rat cardiomyocytes: time-resolved decoding of rapid mito-chondrial calcium spiking imaged with targeted photoproteins.J Biol Chem 2006;281:28058–67.
60 Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, CherryAD, Wester TE et al.Carbon monoxide, skeletal muscle oxidative
stress, and mitochondrial biogenesis in humans. Am J PhysiolHeart Circ Physiol 2009;297:H392–9.
61 Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R,Neviere R. Carbon monoxide rescues mice from lethal sepsis bysupporting mitochondrial energetic metabolism and activatingmitochondrial biogenesis. J Pharmacol Exp Ther 2009;329:641–8.
62 Chok MK, Ferlicot S, Conti M, Almolki A, Durrbach A, Loric Set al.Renoprotective potency of heme oxygenase-1 induction in ratrenal ischemia-reperfusion. Inflamm Allergy Drug Targets 2009;8:252–9.
63 Wagner M, Cadetg P, Ruf R, Mazzucchelli L, Ferrari P, RedaelliCA. Heme oxygenase-1 attenuates ischemia/reperfusion-inducedapoptosis and improves survival in rat renal allografts. Kidney Int2003;63:1564–73.
64 Heo JM, Kim HJ, Ha YM, Park MK, Kang YJ, Lee YS et al.YS 51, 1-(beta-naphtylmethyl)-6,7-dihydroxy-1,2,3,4,-tetrahy-droisoquinoline, protects endothelial cells against hydrogenperoxide-induced injury via carbon monoxide derived from hemeoxygenase-1. Biochem Pharmacol 2007;74:1361–70.
65 Janssen YM, Van HB, Borm PJ, Mossman BT. Cell and tissueresponses to oxidative damage. Lab Invest 1993;69:261–74.
66 Lee HC, Yin PH, Lu CY, Chi CW, Wei YH. Increase of mito-chondria and mitochondrial DNA in response to oxidative stress inhuman cells. Biochem J 2000;348(Pt 2):425–32.
67 Kratz F, Hartmann M, Keppler B, Messori L. The binding proper-ties of two antitumor ruthenium(III) complexes to apotransferrin.J Biol Chem 1994;269:2581–8.
68 Alfrey AC, Hammond WS. Renal iron handling in the nephroticsyndrome. Kidney Int 1990;37:1409–13.
69 Harris DC, Tay YC, Chen J, Chen L, Nankivell BJ. Mechanismsof iron-induced proximal tubule injury in rat remnant kidney. AmJ Physiol 1995;269:F218–24.
70 United States Environmental Protection Agency. Guidance for theregistration of pesticides products containing boric acid and boroncontaining salts as the active ingredient. R E D Facts 1993;EPA-738-F-93-006.
71 Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R.Carbon monoxide-releasing molecules (CO-RMs) attenuate theinflammatory response elicited by lipopolysaccharide in RAW264.7murine macrophages. Br J Pharmacol 2005;145:800–10.
© 2012 The AuthorsBasic & Clinical Pharmacology & Toxicology © 2012 Nordic Pharmacological Society
CARBON MONOXIDE DONOR MOLECULE TOXICITY 11





























