Embed Size (px)
Citation preview

Hum Genet (1991) 88:1-12
Original investigations �9 Springer-Verlag 1991
Chromosome 1 Charcot-Marie-Tooth disease (CMTIB) locus in the Fc7 receptor gene region
Roger V. Lebo l, Phillip F. Chance 2, Peter J. Dyck 3, Ma. Theresa Redila-Flores l, Eric D. Lynch 1, Mitchell S. Golbus l, Thomas D. Bird 4, Mary Claire King 5, Lee A. Anderson 1'5, Jeffrey Hall 5, Joop Wiegant 6, Zharong Jiang l, Paul F. Dazin l, Hope H. Punnett 7, Steven A. Schonberg l, Kevin Moore 8, Marcia M. Shull 9, Sandra Gendler l~ Orest Hurko 11, Robert E. Lovelace 12, Norman Latov 12, James Trofatter 13, and P. Michael Conneally 13
1Departments of Obstetrics, Gynecology, and Pediatrics, U-262, University of California, San Francisco, CA 94143-0720, USA ZDepartment oiPediatrics, University of Utah, Salt Lake City, Utah, USA 3Department of Neurology, Mayo Clinic, Rochester, Minnesota, USA 4Department of Neurology, University of Washington, Seattle, Washington, USA 5 School of Public Health, University of California, Berkeley, California, USA 6Department of Histochemistry and Cytochemistry, Leiden University, Leiden, The Netherlands 7 St. Christopher's Hospital for Children, Philadelphia, Pennsylvania, USA aDNAX Institute of Molecular and Cellular Biology, Palo Alto, California, USA 9 Department of Molecular Genetics, Biochemistry, and Microbiology, University of Cincinnati, Ohio, USA 1~ Cancer Research Fund, London, UK 11 Department of Neurochemistry, Johns Hopkins, Baltimore, Maryland, USA 12Department of Neurology, Columbia University, New York, New York, USA 13Department of Medical Genetics, University of Indiana, Indianapolis, Indiana, USA
Received November 27, 1990 / Revised March 7, 1991
Summary. The Charcot-Marie-Tooth disease (heredit- ary motor and sensory neuropathy) loci have been re- ported to be on at least three chromosomes: 1 (CMT1B, HMSNIB), 17 (CMT1A), and X (CMTX). In this study multipoint linkage analysis of two Duffy-linked families given a combined LOD score of 8.65 to establish that the Duff-y-linked CMT1B gene exists in the 18 centimorgan region between the antithrombin III gene and the Dully/ sodium-potassium ATPase loci. The simultaneous segre- gation of polymorphisms near the CMTIA locus on chro- mosome 17 excludes linkage to this chromosome region in both families. Polymorphic sites that flank the CMT1B gene have been subchromosomally localized to the prox- imal chromosome-1 long arm (lq21.2---,lq25) by spot blot analysis of sorted chromosomes, polymorphic dele- tion analysis, in situ hybridization, and multipoint link- age analysis.
Introduction
Charcot-Marie-Tooth (CMT) disease, also called heredi- tary motor and sensory neuropathy (HMSN) or peroneal muscular atrophy, is a worldwide problem that causes distal muscle weakness and atrophy and peripheral nerve hypertrophy in affected patients (Lovelace and Shapiro 1990). The incidence in western Norway was reported to be 1/2600 for autosomal dominant; 1/26,000 for autoso-
Offprint requests to: R.V. Lebo
mal recessive; and 1/72,000 for X-linked CMT disease (Skre 1974). A similar autosomal dominant frequency has been reported in Spain (Berciano et al. 1986). These prevalence figures are undoubtedly low because undiag- nosed neuropathy frequently can be shown to be inher- ited, mild cases go undetected, and others are probably diagnosed incorrectly.
The CMT subtypes are clinically similar but geneti- cally heterogeneous with different patterns of natural history and clinical electrophysiology. The disease sub- types share abnormal foot structures including pes cavus, distal muscle wasting and atrophy, mild sensory loss, and autonomic disturbances. Abnormalities ar e symmetrical and more severe in the lower limbs. Hereditary motor and sensory neuropathy has been subdivided into two types based on motor nerve conduction velocity. Type I (HMSN1, CMT1) is a demyelinating neuropathy with prolonged motor nerve conduction velocities and hyper- trophic changes on peripheral nerve histology. Type II (HMSN2, CMT2) is a nondemyelinating neuronal disor- der with normal or near normal motor nerve conduction velocities without hypertrophic changes. (Dyck and Lam- bert 1968a, b; Dyck 1984). The CMT1 disorders are manifested in childhood or adolescence and progress slowly but inexorably (Bird and Kraft 1978; Vanasse and Dubowitz 1981).
CMT disease gene loci have been reported to reside on at least three chromosomes. A slow nerve conduction velocity dominant CMT1 (HMSN1; Bird et al. 1980, 1982; Guiloff et al. 1982) gave a combined two-point LOD score with the Dully blood group locus of 3.02 at
2
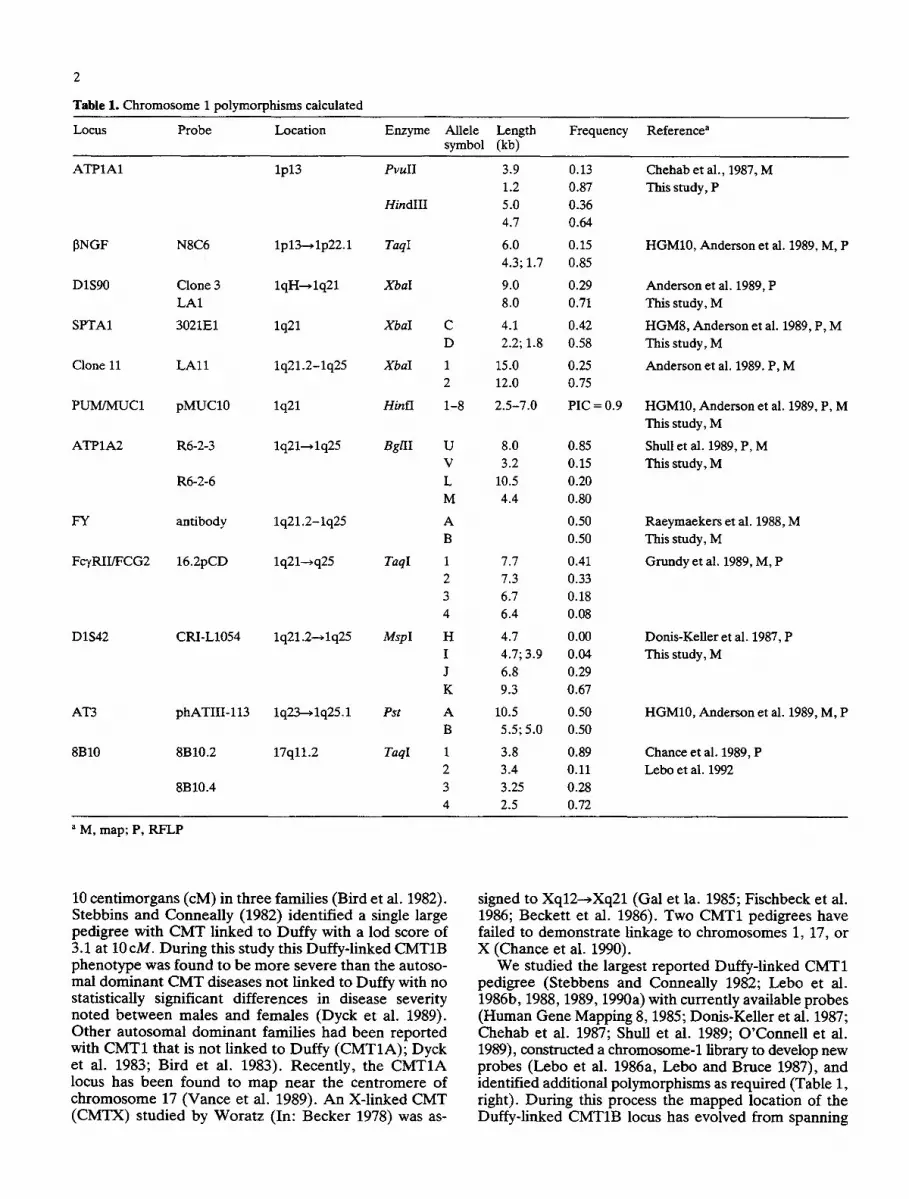
Table 1. Chromosome 1 polymorphisms calculated
Locus Probe Location Enzyme Allele Length Frequency Reference a symbol (kb)
ATP1A1 lp13 PvulI 3.9 0.13 Chehab et al., 1987, M 1.2 0.87 This study, P
HindlII 5.0 0.36 4.7 0.64
TaqI 6.0 0.15 HGM10, Anderson et al. 1989, M, P 4.3; 1.7 0.85
XbaI 9.0 0 . 2 9 Anderson et al. 1989, P 8.0 0.71 This study, M
XbaI C 4.1 0.42 HGM8, Anderson et al. 1989, P, M D 2.2; 1.8 0.58 This study, M
XbaI 1 15.0 0 . 2 5 Anderson et al. 1989. P, M 2 12.0 0.75
Hinfl 1-8 2.5-7.0 PIC = 0.9 HGM10, Anderson et al. 1989, P, M This study, M
BgllI 8.0 Shull et al. 1989, P, M 3.2 This study, M
10.5 4.4
[3NGF N8C6 lp13--~ lp22.1
DIS90 Clone 3 lqH-*1q21 LA1
SPTA1 3021E1 lq21
Clone 11 LAl l 1q21.2-1q25
PUM/MUC1 pMUC10 lq21
ATP1A2 R6-2-3 lq21---~lq25 U 0.85 V 0.15
R6-2-6 L 0.20 M 0.80
FY antibody 1q21.2-1q25 A 0.50 Raeymaekers et al. 1988, M B 0.50 This study, M
Fc'tRII/FCG2 16.2pCD lq21---~q25 TaqI 1 7.7 0.41 Grundy et al. 1989, M, P 2 7.3 0.33 3 6.7 0.18 4 6.4 0.08
D1S42 CRI-L1054 lq21.2---~lq25 MspI H 4.7 0.00 Donis-KeUer et al. 1987, P I 4.7; 3.9 0.04 This study, M J 6.8 0.29 K 9.3 0.67
AT3 phATIII-113 1q23--~1q25.1 Pst A 10.5 0.50 HGM10, Anderson et al. 1989, M, P B 5.5; 5.0 0.50
8B10 8B10.2 17q11.2 TaqI 1 3.8 0.89 Chance et al. 1989, P 2 3.4 0.11 Lebo et al. 1992
8B10.4 3 3.25 0.28 4 2.5 0.72
a M, map; P, RFLP
10 cent imorgans (cM) in three families (Bird et al. 1982). Stebbins and Conneal ly (1982) identified a single large pedigree with C M T linked to Duffy with a lod score of 3.1 at 10 cM. During this study this Duffy-linked CMT1B phenotype was found to be more severe than the autoso- mal dominant C M T diseases not linked to Dul ly with no statistically significant differences in disease severity noted be tween males and females (Dyck et al. 1989). Other autosomal dominant families had been reported with CMT1 that is not linked to Duffy (CMT1A); Dyck et al. 1983; Bird et al. 1983). Recently, the C M T I A locus has been found to map near the centromere of chromosome 17 (Vance et al. 1989). An X-linked CMT (CMTX) studied by Wora tz (In: Becker 1978) was as-
signed to Xq12---~Xq21 (Gal et la. 1985; Fischbeck et al. 1986; Becket t et al. 1986). Two CMT1 pedigrees have failed to demonstrate linkage to chromosomes 1, 17, or X (Chance et al. 1990).
We studied the largest repor ted Dully-l inked CMT1 pedigree (Stebbens and Conneal ly 1982; Lebo et al. 1986b, 1988, 1989, 1990a) with currently available probes (Human Gene Mapping 8, 1985; Donis-KeUer et al. 1987; Chehab et al. 1987; ShuU et al. 1989; O'Connel l et al. 1989), constructed a chromosome-1 library to develop new probes (Lebo et al. 1986a, Lebo and Bruce 1987), and identified additional polymorphisms as required (Table 1, right). During this process the mapped location of the Dully-linked CMT1B locus has evolved f rom spanning

abou t 136cM cover ing 31% of c h r o m o s o m e 1 (lp21---~ lq23; H u m a n G e n e M a p p i n g 8, 1985) to an 18-cM region be tween a n t i t h r o m b i n I I I and the Duffy-sodium-potas - s ium A T P a s e ( A T P 1 A 2 ) loci cover ing 15% of chromo- some 1. A l o n g with ou r results f rom a second CMT1 pedigree , these da ta conf i rm the existence of Duffy- lilaked C M T I B .
Materials and methods
Pedigree analysis
Pedigree 1 is the "Indiana kindred" of Stebbens and ConneaUy (1982) with individuals in five eastern States in the U.S. Pedigree 2 was reported by Bird et al. (1982). We investigated 66 subjects including 27 affected individuals. At-risk individuals were exam- ined by neurologists who determined that propositi had motor nerve conduction velocities of less than 38 m/s, severe distal muscle weakness and atrophy with absent deep tendon reflexes, and foot deformities (Dyck and Lambert 1968a, b; Harding and Thomas 1980). Sural nerve biopsy from a member of each family demon- strated demyelinating neuropathy with substantial peripheral nerve hypertrophy. After informed consent was obtained, periph- eral blood was obtained by venipuncture for DNA extraction and Epstein-Barr (EB) virus transformation of lymphocytes.
Linkage analysis
Pedigree 1 linkage data were handled by the data management program LIPIN (Trofatter et al. 1986). Pedigree 2 data were analyzed by multipoint linkage analysis using version 4.7 of the LINKAGE program package (Lathrop et al. 1984). In contrast to other forms of slow nerve conduction CMT1, all affected members of both pedigrees had a more severe form of CMT with complete concordance between clinical symptoms and nerve conduction vel- ocities. Thus the disease locus was given full penetrance in the multilocus linkage analyses. Loci were recoded to simpler systems to decrease calculation time. The eight-allele mucin locus was analyzed as a four-allele locus with equal allele frequencies (Ott 1985). The four-allele 1054 and ATP1A2 loci were analyzed as three-allele systems since three alleles were segregating in family 1. Linkage analyses were run on a VAX 8800.
Chromosome I library construction
A library was constructed from 12- to 25-kb sorted chromosomal DNA inserts in contrast to the other 2- to 5-kb insert libraries being constructed at the time (Lebo et al. 1986a; Deaven et al. 1986). Sorted chromosomal DNA was partially digested with a fre- quent cutting restriction enzyme to generate overlapping cloned D.NA segments suitable for walking. Five million chromosomes 1 were sorted into Beckman SW27 polypropylene centrifuge tubes in several sorting sessions and pelleted by centrifugation at 25,000 rpm for 4-20 h after each session. The supernatant was decanted to about 3 ml and the tubes stored at 4~ until sorting was resumed into the same tube. After centrifuging the final sorted aliquot, the entire superuatant was gently poured away and the sorted chromo- some pellet digested in 300 ~ proteinase K (10 ~tg/ml). Then the chromosomal DNA was phenol extracted gently on a rotary shaker, ethanol precipitated, and redissolved in 10raM tris-HC1, 1 mM EDTA. More than 90% of control sorted chromosomal DNA was longer than 80kb following extraction. Extracted chromosomal DNA was partially digested with MboI to an average size of 20-kb and dephosphorylated to prevent ligation of two or more frag- ments into the EMBL4 cloning site. The EBM-4 phage were di- gested with BamHI and SalI to excise the insert, ligated to pre- pared chromosome-1 DNA, packaged, and plated on restrictive
host Q359 (Karn et al. 1983). The entire 50,000 plaque library was amplified and stored at -80~ (Lebo et al. 1986a). Of the first 36 plaques mapped, 29 (80%) hybridized to sorted chromosome-1 DNA and 6 hybridized to chromosome-2 DNA. Calculations ac- cording to Maniatis et al. (1982) indicate this chromosome-1 li- brary is 90% complete. In contrast, other characterized sorted chromosome libraries are about 70% pure with 30% contamina- tion from all other chromosomes (see Deaven et al. 1986).
Lymphocyte transformation
Our most recent modifications to the Anderson and Gusella (1984) protocol follow. Concentrated EB virus stock was prepared by centrifuging the supernatant of a 1-week-old B-95 monolayer cul- ture at 800g for 7min, filtering the supernatant through a 0.45 ~tm nitrocellulose filter, centrifuging in ethylene oxide sterilized tubes at 100,000 g for 35 rain, and resuspending the virus pellet in 1% the original volume. Lymphocytes from peripheral blood, collected in acid citrate dextrose vacutainers and transported at ambient tem- perature, were isolated on a ficoll-hypaque step gradient, washed, and resuspended at 6 x 106 to 12 • 106 cells/ml in RPMI-1640 medium with 25 mM hepes-NaOH pH 7.25, 15% fetal calf serum, 0.3 mg/ml glutamine, 1 mM Na pyruvate, 100units/ml penicillin, 100units/ml streptomycin, 50mg/ml gentamicin, and 30mg/ml phytohemagglutinin-P (PHA; Sigma, St. Louis, Mo.) diluted with an equal volume of virus stock. PHA was deleted from the medium after the lymphocytes were transformed.
Karyotyping
Fibroblast cell lines, GM97, GM4618, GM2025, GM201, GM2096A, GM6038, and GM803, for gene mapping were obtained from the NIGHS Human Genetic Mutant Cell Repository, Camden, N.J. (1984). Lymphocyte cell line GM10005 was established from pa- tient GM4618 (Grundy et al. 1989) in this study. Lymphocytes from patient GM97 were karyotyped by early metaphase chromo- some banding to reassign the chromosome breakpoints originally determined in fibroblasts by standard banding. Early metaphase banding of GM201 fibroblasts confirmed the original designation of the chromosome breakpoints. Chromosome breakpoints of GM2096A, GM6038 and GMS03 were published by the Human Genetic Mutant Cell Repository.
Gene mapping and restriction enzyme analysis
Gene mapping by spot blot analysis was according to Lebo and Bruce (1987). Small quantities of chromosome-1 library fragments cloned in EMBL 4 phage were isolated for miniscreening and spot blot mapping (Maniatis et al. 1982) with two modifications. First, the top agarose of a lysed plate was removed and added to 5 ml SM buffer plus CHC13 in 30 ml corex tubes with occasional vortexing to elute the phage. Second, isolated phage were lysed with 200 mg proteinase K in 10mM tris-HCl (pH 7.0), 5 mM EDTA, and 0.5% Sodium dodecyl sulfate (SDS). Fluorescence in situ hybridization and restriction enzyme analysis were according to Lebo et al. (1990b).
Results
Duffy-linked CMT1 pedigree confirmation
W e began s tudying the largest family carrying the on ly form of CMT1 that had b e e n m a p p e d (Fig. 1; S t ebbens and Connea l ly 1982). Neurologica l examina t ion of these pat ients and no rma l individuals at the t ime b lood was drawn for D N A analysis conf i rmed the original diagnosis of heredi ta ry moto r a nd sensory n e u r o p a t h y (CMT) and

.~ ,-,.o ,~"~
.= "~ ~a ~
1 "4--] .'-
,~ = . ~ = . ~ ~
" 0 " " ~ - ~ e . - ~ o
~= = ~ ' = ~ ~
= = =_ ~ - ~ ~ 6 "~ ~ ' ~ ° °
%--;-,- ~ ~'--'', ~
.~_~ ~ o
. . . . . . . ~ ~ o
"" ° [] ..... ".: z ,~ ..
_ ,, ~ ~ ._~'~ ~
o . . . . ~ ~.~ N ~ , ' ,
~mP"
~ , . . .
• ,~ ~ ~ ~ , ~ . - ~ >_ > ~ <

Table 2. Pedigree 1 two-point LOD results a Locus Logt0 likelihood for the recombination values
0.00 0.001 0.01 0.05 0.10 0.20 0.30 0.40
Chromosome 1 (CMT1B)
ATP1A1 -15.13 -8.60 -5.85 -3.31 -2.13 -0.99 -0.42 -0.11 I3-NGF -5.00 -2.39 -1.40 -0.72 -0.44 -0.20 -0.09 -0.04
Clone 3 -5.35 -2.71 -1.71 -0.98 -0.66 -0.36 -0.19 -0.07 SPTA1 -5.69 -3.26 -2.20 -1.31 -0.88 -0.45 -0.21 -0.07 Clone 11 -4.43 -1.44 -0.47 0.14 0.31 0.34 0.23 0.09
PUM -8.77 -0.91 1.07 2.30 2.57 2.32 1.63 0.73 ATP1A2 N.D. -2.96 -1.00 -0.24 0.62 0.73 0.54 0.24
FY -2.33 1.96 2.89 3.28 3.17 2.61 1.83 0.88 Fc3,RII 5.18 5.17 5.09 4.73 4.27 3.27 2.17 0.96 1054 -6.26 -1.92 0.03 1.20 1.50 1.45 1.07 0.54 AT3 -5.71 -1.70 -0.71 -0.07 0.15 0.27 0.25 0.15
Chromosome 17 (CMT1A)
8B10 N.D. -11.69 -7.63 -4.10 -2.59 -1.20 -0.53 -0.17
Assume: penetrance = 0.95; normal allele frequency = 0.9999; CMT allele frequency = 0.0001
9 7 4 6 1 8 2 0 2 5 201 2096A 6 0 3 8 8 0 3
36
3S
34
32
31
22.3 22.2 22.1
13.2 13.1
12 11
qH 21.1 21.2 21.3
22 23 24 25
32
41
42 43 44
1
Fig. 2. Physical and genetic map of the CMT1B gene region. Left Idiogram of the early metaphase chromosome 1 showing the breakpoints of the three reciprocal translocations (97, 4618, and 201) and one interstitial deletion (2025) used to sublocalize cloned DNAs by spot blot analysis of sorted chromosomes. Three addi- tional deletions (2096A, 6038, and 803) excluded genes from the interstitial deletion region when both polymorphic alleles were found in total DNA. The physical location of clone 3 (DIS90) and a-spectrin (SPTA1) were further defined by fluorescence in situ hybridization (Fig. 6). The positions of renin (REN), phosphogly- ceromutase (PGM), and DIS77 are from the HHMI Human Gene
cM Locus - - ]
I I ~- D I S 7 7 �9
- P G M �9
CMT
"NGF
- PUM
- MLAJ1
\ A T 3 �9 HB140
1A2 ; 3
~ cm" / Fo~' Ri141
~-- D I S 7 4
i
Map. Right Linkage map including selected loci across chromo- some 1 and the tested polymorphic markers in the CMT1B gene region are drawn to scale. The CMT1B and Fc7 receptor loci are located in the 6-cM region between the Duffy locus (FY) and clone 1054. Linkage analysis by O'Connell et al. (1989) established a continuous chromosome-1 sex-averaged linkage map of 464cM. The positions of L1054 and antithrombin III (AT3) were added by Donis-Keller et al. (1987). The relative position of clone 3 was added by Anderson et al. (1989) and segregation in family 1, and the relative position of clone 11 by linkage in family 1

Fig. 3. Giemsa-banded rearranged chromosomes. The reciprocal translocation in cell line 97 (left) was found to have a breakpoint in lq21.2 instead of lq12 (NIGMS 1984) by analysis of longer peripheral blood lymphocyte chromosomes. Similar analysis confirmed the interstitial deletion breakpoints in cell line 2025 (right)
Fig. 4 A-C. Flow histograms of normal and derivative chromo- somes. Top view of a 64 • 128 channel electronic histogram of dual laser-excited chromosomes from cell lines (A) 4618, (B) 97,and (C) 2025. The chromomycin fluorescence signals increase linearly on the abscissa and Hoechst fluorescence on the ordinate. The hght intensity of each dot in the three-dimensional histogram is linearly proportional to the number of chromosomes recorded in
each channel. Derivative chromosomes 1 and X (DER1 and DERX in cell lines 4618 and 97) and chromosome 1 carrying a de- letion (DELl in cell line 2025) are resolved uniquely and sorted from their normal homologous chromosomes. These histograms each illustrate data accumulated from about 250,000 chromosomes analyzed in 2 rain
further classified the disease as slow nerve conduction type 1 (Dyck et al. 1989). This project Duffy typed 41 subjects including 19 affected individuals in family 1 with complete agreement in the 18 originally repor ted indi- viduals who were tested at two locations. Two-point link- age analysis further supported CMT1 linkage to the Duffy locus in this large family (lod = 3.28; Fig. 1, Table 1).
Finding a DNA fragment in the C M T I B gene region
Because the possible Duffy blood group location spanned a large region of chromosome 1, several polymorphic probes had to be tested to identify one linked to CMT1B. Two-point lod scores of the first two available probes, an- tithrombin III (AT3) and G-nerve growth factor (NGFB), failed to reveal linkage to CMT1B (Tables 1, 2). When a sodium-potassium ATPase gene (ATP 1A1) was mapped to lp13 (Fig. 2A, Table 1), our ATP1A1 polymorphic studies found the informative polymorphisms and ex- cluded the ATP1A1 gene from the immediate CMT1B vicinity (Tables 1, 2).
Having exhausted the available chromosome 1 probes, we constructed an 80% pure chromosome-1 library from flow-sorted chromosomes to obtain more chromosome sequences (see Materials and methods). Four cell lines with chromosome-1 rearrangements were used to sub-
localize the chromosome-1 library clones. To obtain a more accurate map, the chromosome breakpoints in two of these cell lines had to be redefined by higher resolu- tion prometaphase karyotypes of peripheral blood lym- phocyte chromosomes (GM97 in Fig. 3; GM4618 in Grun- dy et al. 1989). Derivative chromosomes carrying chro- mosome-1 regions were sorted from each other and the normal chromosome 1 onto nitrocellulose filter paper and the denatured D N A hybridized to labeled D N A probe (Figs. 4, 5). Of the 20 clones sublocalized by spot blot analysis (Fig. 2A, left), polymorphisms were found for clone 3 (D1S90; HGM10) and clone 11, which both mapped to lq21.2---~lq25. Clone 3 did not cosegregate with CMT1 while clone 11 suggested linkage at some dis- tance (Table 2). In situ hybridization further sublocalized the constant clone 3 polymorphic locus to band lq21 with zero to four homologous sequences in the variable lqh heterochromatic region (Fig. 6, top; Fig. 2B, left). Together these data suggested the CMT1 locus was dis- tal to clone 3 on chromosome lq.
Sublocalizing CMTIB on chromosome lq
A second sodium-potassium ATPase gene (ATP1A2; Table 1) became a candidate gene for slow nerve con- duction CMT1 because it was mapped to chromosome lg

Fig.5. Duplicate spot blot filters of sorted translocated chromo- somes. Unique DNA probes hybridize to homologous sequences in denatured, sorted chromosomal DNA. Derivative chromosome 1 DNA from 4618 hybridized specifically to antithrombin III probe while the control reciprocal tanslocation derivative X did not. Since we have reassigned the 4618 translocation breakpoint, this result and other mapping data place antithrombin III in the lq23--, lq25.1 chromosome region. Results were obtained in dupli- cate to improve reliability
(Yang-Feng et al. 1988), enzyme deficiency could cause decreased nerve conduction, and A T P 1 A 2 message was found in rat brain (Orlowski and Lingrel 1988). The Fcy receptor gene family became candidates for CMT1B when these genes were mapped to lq21.2---~lq25 (Table 1), and one of these genes was found to be expressed on the surface of the myelin sheath in Schwann cells (Vedeler 1987). We sublocalized the addit ional probes mapped to this chromosome region with our sorted chro- mosome mapping panel and by polymorphic restriction enzyme analysis of total D N A f rom three cell lines carry- ing interstitial deletions of ch romosome 1. I f total D N A f rom a cell with an interstitial deletion of ch romosome 1 carries two polymorphic f ragments of a D N A clone, that clone must lie outside the deleted ch romosome region. This composite map is illustrated in Fig. 2 (left).
Collected, sublocalized polymorphic probes were tested for segregation with the C M T I B pheno type in family 1 (Fig. 1). The candidate FcTRII polymorphic site cosegre- gated with the C M T I B phenotype in each informative meiosis (lod = 5 . 1 8 at 0 =0 .000; Table 2). Multilocus linkage analysis was applied only to the loci closest to the CMT1B gene (Tables 1, 3; Figs. 1, 7) because available computer t ime on the V A X 8800 could not analyze the data with additional markers . Assuming that FeyRI I is 3 cM distal to CMT1B, the mult ipoint locat ion score is 6.6. This logarithmic score indicates that the likelihood of linkage of CMT1B to FcTRII is about 106.6 to 1 (or 4,000,000 to 1). Because no recombination is observed be- tween FcTRII and CMT1B, the multipoint location score is slightly higher when these loci are placed together (6.9 at 0 = 0.00). Because the C M T I A locus has since been mapped to chromosome 17, we tested the segregation of seven polymorphisms with five probes: 17cen - LEW301 (D17S58; TaqI/BgllI) - 1.5 cM -8BIO.2(TaqI) /8BIO.4 (TaqI) - 0 . 5 cM -pA1041(D17S71; MspI/PvuII) - 5 cM - C M T 1 A - 3 c M - I O E 4 ( B a m H I ) (Lebo et al. 1992). The multipoint linkage score using all but the 8B10 poly- morphisms were uninformative. In contrast the 8B10 polymorphisms alone excluded linkage of CMT1B to about 15 cM (Tables 2). Because 8B10 is about 5 c M from CMT1A on chromosome 17, these data exclude the CMT1B locus f rom chromosome 17.
Note in individual III-9 that a phase known recom- binant occurs between the Duf fy /ATP1A2 chromosome
Fig. 6. Fluorescence in situ hybridization. Top Chromosomal DNA is stained red with propidium iodide and biotinylated clone 3 pro- be is detected simultaneously by yellow-green fluoreseein signal conjugated to avidin D. The entire complex is DNA-[biotin-DNA]- [avidin.D-FITC]- [biotinylated-goat-antiavidin.D]-[avidin.D-FITC]. Clone 3 probe hybridizes only to band lq21 in both chromosomes of some individuals. Clone 3 hybridizes to the same number of extra homologous segments (1 to 4) in the lqH chromosome re- gion of the same homologous chromosome in other individuals (c.f. the single smaller spot on each chromosome arm). Note three of the four chromatids in the two homologous chromosomes are labeled. The lighter signal on the upper chromatid of the left chro- mosome was out of the focal plane and lost during reproduction. As expected, clone 3 restriction fragment length polymorphisms segregate with the more intense distal signal, the only constant loca- tion in each chromosome studied. Bottom left Blue DAPI-banded metaphase chromosomes make it possible to identify the recta- phase chromosomes in the karyotype. DAPI stains the variable lqH heterochromatic region intensely to clearly delineate the non- variable region. Bottom right Yellow-green FITC-labeled a-spectrin and clone 3 probe hybridized to region lq21 with the paler yellow color of the chromosomes observed from the DAPI-stained DNA
region and the FcTRII /CMT1B chromosome region indi- cating that CMT1B is distal to Duf fy /ATPIA2 . As ex- pected, this conclusion is supported by the most likely orders and location scores (Table 3, bo t tom) showing that this order is 128.8-fold more likely than placing CMT1B proximal to Duffy /ATP1A2. Analysis of the distal loci are less helpful. Note that a recombinat ion oc- curs between CMT1B and probe 1054 f rom individual I II-6 to IV-3 where the FcTRII allele is uninformative.

Fig. 7. Most informative chromosome 1 polymorphic restriction fragments. The polymorphic restriction fragments with their length and our alpha-numeric designations used to test cosegregation with the CMT1B phenotype
CMT1B is proximal to the antithrombin III (AT3) locus but is only 12-fold more likely to be proximal to the 1054 locus than distal (Table 3, top). Taken together these data indicate that the CMTIB locus is in the 18-cM re- gion between AT3 and Duffy/ATP1A2 spanning about 4% of the 464-cM chromosome-1 genetic map. These two flanking markers lie in proximal chromosome lq (lq21.2---~lq25) representing about 15% of the chromo- some-1 physical map (Fig. 2).
CMTIB family 2 from the same geographic region
Family 2 was reported to have a two-point lod score of 2.1 at 0 = 0.00 between the Duffy blood group locus and CMT1B (Bird et al. 1982). Our analysis of nine DNA polymorphisms in the CMT1B gene region confirmed the Duffy result (Fig. 8) to give a multipoint location score of 2.1. While the closer polymorphic loci segregate faithfully with the CMT1 locus, the more distal a-spec- trin (SPTA1) and antithrombin III (AT3) loci each re- combine in different meioses (Fig. 8). Linkage analysis with chromosome-17 polymorphisms clearly excluded linkage to the CMT1A locus on chromosome 17 with a
lod score < -3.0 (Chance et al. 1990). Assuming Fc3,RII and 1054 are at the same locus, multipoint linkage analy- sis of both families gave a location score of 8.4 (Table 4, top). When the Fcy RII locus was assumed to be proxi- mal to 1054, the more likely order, multipoint analysis of both families gave a location score of 8.7 (Table 4, bot- tom). In addition, family 2 had the most severe form of CMT observed by Chance et al. (1990) just as family 1 had a more severe form of CMT than other HMSN type- I patients reported by Dyck et al. (1989). We then traced the origin of these families to Wales and Northern Ire- land, which both have shorelines on the Irish Sea. In contrast, the CMT1A families with lod scores > 3.0 trace their origins throughout Europe to England, Germany, Austria (Chance et al. 1990), Belgium (Raeymaekers et al. 1988), and France (Patel et al. 1990).
Discussion
Our linkage studies prove one form of CMT1 is linked to the Duffy blood group locus. Previously this association has been attributed to incorrect Duffy typing or chance

Table 3. Multipoint linkage analysis with CMT1B pedigree 1
Condition: Most --> Least likely Recombinat ion fractions - 2 In Location score be tween adjacent loci
0 = 0 . 5
1 AT3-1054-CMT1B-FY-ATP1A2 0.12 0.03 0.03 0.00
2 AT3-1054-FY-ATP1A2-CMT1B 0.12 0.06 0.00 0.09
3 AT3-CMT1B- 1054-FY-ATP1A2 0.10 0.02 0.06 0.00
4 CMT1B-AT3-1054-FY-ATP1 A2 0.19 0.00 0.12 0.06
Relative order ratios 10 (xy) where x = more likely location score and y = least likely location score
Order versus 4 Order versus 3 Order versus 2
1 588.8 1 12.0 1 2.5
2 239.9 2 4.9
3 49.0
315.858
296.225 4.27
298.006 3.88
301.194 3.19
308.798 1.50
0 = 0.5
1 CMTIB-FY-A T PI A 2- SPT A 1- PU M 0.06 0.00 0.06 0.03
2 FY-ATP 1A2-CMT1B-SPTA1-PUM 0.00 0.02 0.04 0.03
3 FY-ATP 1A2-SPTA1 -PUM-CMT1B 0.00 0.06 0.03 0.18
4 FY-ATPIA2-SPTA1-CMT1 B-PUM 0.00 0.06 0.01 0.02
Relative order ratios 10 (xy) where x = more likely location score and y = least likely location score
Order versus 4 Order versus 3 Order versus 2
1 46,773.0 1 1480.2 1 128.8
2 363.1 2 11.5
3 31.6
319.986
294.075 5.63
303.781 3.52
308.674 2.46
315.582 0.96
I I I
IV
2112 - a - ~ ' U U s ~ - 3
2 2 " ~ ' - R I 1 s T I q l - J - s r'~' ~, Bgl I I
'E5 '6
:11:
(-) I §
' ()
2 1
,i'." 2 3
~
2
§
'1 1 1
(-) I §
3[]
ii "
(-) 1§ (-) l§
Fig. 8. CMT1B family 2 pedigree. No recombi- nat ion is observed with the polymorphisms in the CMT1B gene region but the tt-spectrin (ATPA1) locus and ant i thrombin III (AT3) each recombine once. Individual 1-2 traces her ancestor with CMT to Northern Ireland
10
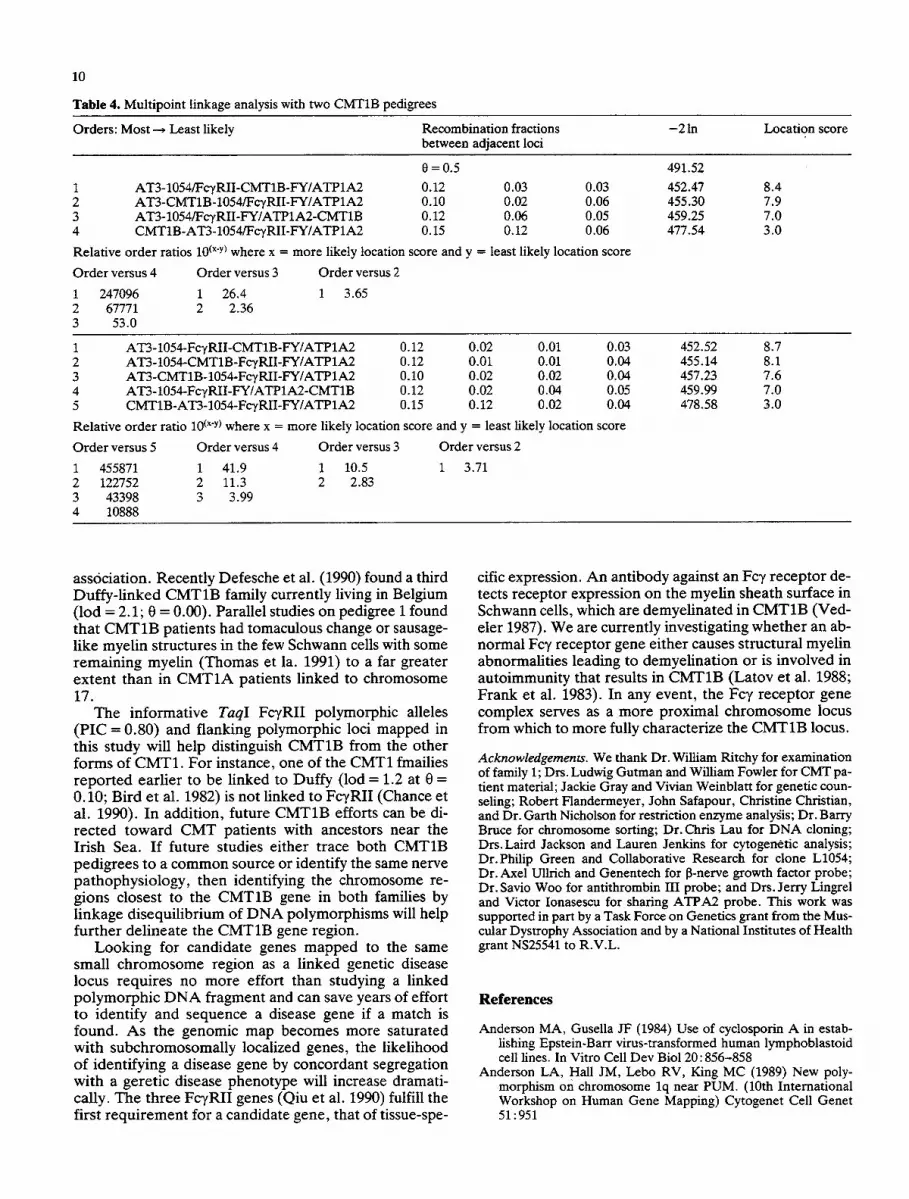
Table 4. Multipoint linkage analysis with two CMT1B pedigrees
Orders: Most ~ Least likely Recombination fractions - 2 In Location score between adjacent loci
0 = 0.5 1 AT3-1054/FcyRII-CMT1B-FY/ATP1A2 0.12 0.03 0.03 2 AT3-CMT1B- 1054/FcTRII-FY/ATP1A2 0.10 0.02 0.06 3 AT3-1054/FcyRII-FY/ATP1A2-CMT1B 0.12 0.06 0.05 4 CMT1B-AT3-1054/FeyRII-FY/ATP1A2 0.15 0.12 0.06
Relative order ratios 10 (x'y) where x = more likely location score and y = least likely location score
Order versus 4 Order versus 3 Order versus 2
1 247096 1 26.4 1 3.65 2 67771 2 2.36 3 53.0
491.52 452.47 8.4 455.30 7.9 459.25 7.0 477.54 3.0
1 AT3-1054-FcyRII-CMTIB-FY/ATPIA2 0.12 0.02 0.01 0.03 2 AT3-1054-CMT1B-FcTRII-FY/ATP1A2 0.12 0.01 0.01 0.04 3 AT3-CMTIB- 1054-FcTRII-FY/ATP1A2 0.10 0.02 0.02 0.04 4 AT3-1054-FcyRII-FY/ATP1A2-CMT1B 0.12 0.02 0.04 0.05 5 CMT1B-AT3-1054-FcyRII-FY/ATP1A2 0.15 0.12 0.02 0.04
Relative order ratio 10 (x'y) where x = more likely location score and y = least likely location score
Order versus 5 Order versus 4 Order versus 3 Order versus 2
1 455871 1 41.9 1 10.5 1 3.71 2 122752 2 11.3 2 2.83 3 43398 3 3.99 4 10888
452.52 8.7 455.14 8.1 457.23 7.6 459.99 7.0 478.58 3.0
associat ion. Recent ly Defesche et al. (1990) found a third Duf fy - l inked C M T 1 B family current ly living in Belgium ( lod = 2.1; 0 = 0.00). Parallel studies on pedigree 1 found tha t C M T 1 B patients had tomaculous change or sausage- like myel in structures in the few Schwann cells with some remain ing myel in (T hom a s et la. 1991) to a far greater ex ten t t han in C M T I A patients l inked to ch romosome 17.
The informat ive TaqI F c y R I I po lymorphic alleles ( P I C = 0.80) and f lanking po lymorph ic loci mapped in this s tudy will help distinguish C M T 1 B f rom the o ther fo rms o f C M T 1 . For instance, one o f the C M T 1 fmailies r e p o r t e d earl ier to be l inked to D u l l y (lod = 1.2 at 0 = 0.10; Bird et al. 1982) is not l inked to F c y R I I (Chance et al. 1990). I n addit ion, future C M T 1 B efforts can be di- rec ted t oward C M T patients with ancestors near the I r ish Sea. I f future studies ei ther t race bo th C M T 1 B ped ig rees to a c o m m o n source or identify the same nerve pa thophys io logy , then identifying the ch romosome re- gions closest to the C M T 1 B gene in bo th families by l inkage disequi l ibr ium of D N A po lymorph i sms will help fu r the r del ineate the C M T I B gene region.
L o o k i n g for candidate genes m a p p e d to the same small c h r o m o s o m e region as a l inked genetic disease locus requires no m o r e effort than s tudying a linked p o l y m o r p h i c D N A f ragment and can save years of effort to ident i fy and sequence a disease gene if a match is found . A s the genomic map becomes m o r e saturated with subch romosoma l ly localized genes, the l ikelihood o f ident i fying a disease gene by concordan t segregat ion with a geret ic disease pheno type will increase dramati- cally. The three F e v R I I genes (Qiu et al. 1990) fulfill the first r equ i r emen t for a candidate gene, that o f tissue-spe-
cific expression. A n ant ibody against an Fcy r ecep to r de- tects receptor expression on the myel in sheath surface in Schwann cells, which are demyel ina ted in C M T I B (Ved- eler 1987). We are current ly investigating whe th e r an ab- normal Fcy receptor gene ei ther causes s tructural myel in abnormali t ies leading to demyel ina t ion o r is involved in au to immuni ty that results in C M T 1 B (La tov et al. 1988; F rank et al. 1983). In any event , the Fcv recep to r gene complex serves as a more proximal c h r o m o s o m e locus f rom which to more fully character ize the C M T 1 B locus.
Acknowledgements. We thank Dr. William Ritchy for examination of family 1; Drs. Ludwig Gutman and William Fowler for CMT pa- tient material; Jackie Gray and Vivian Weinblatt for genetic coun- seling; Robert Flandermeyer, John Safapour, Christine Christian, and Dr. Garth Nicholson for restriction enzyme analyr Dr. Barry Bruce for chromosome sorting; Dr. Chris Lau for DNA cloning; Drs.Laird Jackson and Lauren Jenkins for cytogerretic analysis; Dr.Philip Green and Collaborative Research for clone L1054; Dr. Axel Ullrich and Genentech for 13-nerve growth factor probe; Dr. Savio Woo for antithrombin III probe; and Drs. Jerry Lingrel and Victor Ionasescu for sharing ATPA2 probe. This work was supported in part by a Task Force on Genetics grant from the Mus- cular Dystrophy Association and by a National Institutes of Health grant NS25541 to R.V.L.
References
Anderson MA, GuseUa JF (1984) Use of cyclosporin A in estab- lishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro Cell Dev Biol 20 : 856-858
Anderson LA, Hall JM, Lebo RV, King MC (1989) New poly- morphism on chromosome lq near PUM. (10th International Workshop on Human Gene Mapping) Cytogenet Cell Genet 51:951

11
Becker PE (1978) Humangenetik: ein kurzes handbuch. Thieme, Stuttgart, pp 425-431
Becket J, Holden JJA, Simpson NE, White BN, MacLeod PM (1986) Localization of X-linked dominant Charcot-Marie-Tooth disease (CMT2) to Xq13. J Neurogenet 3:225-231
Berciano J, Combarros O, Figols J, Calleja J, Cabello A, Silos I, Coria F (1986) Hereditary motor and sensory neuropathy type II: clinicopathological study of a family. Brain 109:897- 914
Bird TD, Kraft GH (1978) Charcot-Marie-Tooth disease: data for genetic counseling relating age to risk. Clin Genet 14: 43-49
Bird TD, Ott J, Giblett ER (1980) Linkage of Charcot-Marie- Tooth neuropathy to the Duffy locus on chromosome 1. Am J Hum Genet 32: 99A
Bird TD, Ott J, Giblett ER (1982) Evidence for linkage oI Char- cot-Marie-Tooth neuropathy to the Dully locus on chromo- some 1. Am J Hum Genet 34: 388-394
Bird TD, Ott J, Giblett ER, Chance PF, Sumi SM, Kraft GH (1983) Genetic linkage evidence for heterogeneity in Charcot- Marie-Tooth Neuropathy (HMSN type I). Ann Neurol 14: 679-684
Chance PF, Bird TD, O'Connell P, Lipe H, Lalouel J-M, Leppert M (1990) Genetic linkage and heterogeneity in Type I Charcot- Marie-Tooth disease (Hereditary Motor and Sensory Neurop- athy Type I). Am J Hum Genet 47: 915-925
Chehab FF, Kan YW, Law ML, Hartz J, Kao F-T, Blostein R (1987) Human placental Na+-K§ alpha subunit: eDNA cloning, tissue expression, DNA polyrnorphism, and chromo- somal localization. Proc Nail Acad Sci USA 84: 7901-7905
Deaven LL, Dilla MA van, Bartholdi MF, Carrano AV, Cram LS, Fuscoe JC, Gray JW, Hildebrand CE, Moyzis RK, Perlman J (1986) Construction of human chromosome-specific DNA lib- raries from flow-sorted chromosomes. Cold Spring Harbor Syrup Quant Biol 51 : 159-167
Defesche JC, Hoogendijk JE, Visser M de, Visser O de, Bolhuis PA (1990) Genetic linkage of hereditary motor and sensory neuropathy type I (Charcot-Marie-Tooth disease) to markers of chromosomes 1 and 17. Neurology 40:1450-1453
Donis-Keller H, Green P, Helms C, Cartinhour S, Weiffenbach B, Stephens K, Keith TP, Bowden DW, Smith DR, Lander ES, Botstein D, Akots G, Rediker KS, Gravius T, Brown VA, Ris- ing M.B, Parker C, Powers JA, Watt DE, Kauffman ER, Bricker A, Phipps P, Muller-Kahle H, Braman JC, Knowlton RG, Barker DF, Crooks SM, Lincoln SE, Daly MJ, Abraham- son J (1987) A genetic linkage map of the human genome. Cell 51 : 319-337
Dyck PJ (1984) Inherited neuronal degeneration and atrophy. In: Diseases of the peripheral nervous system. Saunders, Philadel- phia, pp 1609-1630
Dyck PJ, Lambert EH (1968a) Lower motor primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneu- ropathies. Arch Neurol 18: 603-618
Dyck P J, Lambert EH (1968b) Lower motor and primary sensory neuron disease with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal de- generations. Arch Neurol 18 : 619-625
Dyck P J, Ott J, Moore SB, Swanson CJ, Lambert EH (1983) Link- age evidence for genetic heterogeneity among kinships with hereditary motor and sensory neuropathy Type I. Mayo Clin Proc 58: 430-435
Dyck PJ, Karnes JL, Lambert EH (1989) Longitudinal study of neuropathic deficits and nerve conduction abnormalities in hereditary motor and sensory neuropathy type 1. Neurology 39:1302-1308
Fischbeck KH, ar-Rushdi N, Perieak-Vance M, Rozear M, Roses AD, Fryns JP (1986) X-linked neuropathy: gene localization with DNA probes. Ann Neurol 20: 527-532
Frank MM, Lawley TJ, Hamburger MI, Brown EJ (1983) Immu- noglobulin G Fc receptor-mediated clearance in autoimmune diseases. Ann Intern Med 98: 206-218
Gal A, Mueke J, Theile H, Wieacker PF, Ropers HH, Wienker "IT (1985) X-linked dominant Charcot-Marie-Tooth disease: suggestion of linkage with a cloned DNA sequence from the proximal Xq. Hum Genet 70: 38-42
Grundy HO, Peltz GA, Moore KW, Golbus MS, Jackson LG, Lebo RV (1989) The polymorphie Fe7 receptor II gene maps to human chromosome lq. Immunogenetics 29: 331-339
Guiloff RJ, Thomas PK, Contreras M, Armitage S, Schwartz G, Sedgwick EM (1982) Linkage of autosomal dominant type I hereditary motor and sensory neuropathy to the Duff-y locus on chromosome 1. Ann Hum Genet 46: 25-27
Harding AE, Thomas PK (1980) Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J Med Genet 17: 329-336
Human Gene Mapping 8 (1985) 8th International Workshop on Human Gene Mapping. Cytogenet Cell Genet 40: 67-106
Human Gene Mapping 10 (1989) 10th International Workshop on Human Gene Mapping. Cytogenet Cell Genet 51 : 67-90
Karn J, Brenner S, Barnett L (1983) New bacteriophage lambda vectors with positive selection for cloned inserts. Methods En- zymol 101:3-19
Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81 : 3443-3446
Latov N, Hays AP, Sherman WH (1988) Peripheral neuropathy and anti-MAG antibodies. CRC Crit Rev Neurobiol 3:301- 332
Lebo RV, Bruce BD (1987) Gene mapping with sorted chromo- somes. Methods Enzymol 151 : 292-313
Lebo RV, Anderson LA, Lau Y-F, Flandermeyer R, Kan YW (1986) Flow sorting analysis of normal and abnormal human genomes. Cold Spring Harbor Symp Quant Biol 51:169- 176
Lebo R, Anderson L, Lau YF, Carver V, Conneally PM (1986b) Linkage analysis of Charcot-Marie-Tooth syndrome. Muscle Nerve 9 (5S) : 235
Lebo RV, Dyck PJ, Christian CC, Flandermeyer RR, Golbus MS, Hurko O, Ploeg M van der, Anderson LA, King MC, Chance PF, Bird RD, Jackson LG, Kan YW, Bruce BD, Lovelace RE, Dickoff D, Blostein R, Sadler JE, Green P, Schonberg SA, Ionasescu V, Olney RK, Gutman L, Kadasi L, Ferak V, Fow- ler WM, Conneally PM (1988) The multilocus Charcot-Marie- Tooth syndrome. Am J Hum Genet 43 : A149
Lebo RV, Dyck PJ, Chance PF, King MC, Lynch E, Redila-Flores M, Golbus MS, Anderson LA, Christian CC, Flandermeyer RR, van der Ploeg M, Weigant J, Hall JM, Hurko O, Corn- blath DR, Johns DR, Bird TD, Kan YW, Bruce BD, Jackson LG, Epstein E, Peltz G, Moore K, Shull MM, Lingrel JB, Green P, Gendler S, Latov N, Lovelace RE, Diekoff D, Blo- stein R, Schonberg SA, Jenkins LS, Ionasescu V, Gutman L, Nicholson G, Kadasi L, Ferak V, Fowler WM, Trofatter J, Conneally PM (1989) Chromosome 1 Charcot-Marie-Tooth locus in FcyRII gene region. Am J Hum Genet 45 :A148
Lebo RV, Conneally PM, Flandermeyer RR, Christian C, Golbus MS, Lovelace RE, Anderson LA, Chance PF, Bird TD, Bruce BD, Slomick RN, Dickoff D, Sadler JE, Carver V, Sehonberg SA, Fowler WM, Ionasescu V, Kadasi L, Dyck PJ (1990a) The multilocus Charcot-Marie-Tooth syndrome. In: Lovelace RE, Shapiro HK (eds) Charcot-Marie-Tooth disorders: pathophysi- ology, molecular genetics, and therapy. Liss, New York, pp 307-334
Lebo RV, Anderson LA, DiMauro S, Lynch E, Hwang P, Flet- terick R (1990b) Rare McArdle disease locus polymorphic site on 11q13 contains CpG sequence. Hum Genet 86:17-24
Lebo RV, Lynch ED, O'Connell P, Bird TD, Golbus MS, Barker DF, Chance PF (1992) Multicolor in situ hybridization and linkage analysis cytogenetically order CMTIA gene region loci. Am J Hum Genet 47:A188 (in press)
Lovelace RE, Shapiro HK (1990) Charcot-Marie-Tooth disorder: pathophysiology, molecular genetics, and therapy. Liss, New York, pp 100-101

12
Maniatis T, Fritsch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY
National Institute of General Medical Sciences (1984) Human Ge- netic Mutant Cell Repository Catalog of cell lines. Public Health Service, National Institutes of Health, NIH publ no. 84-2011
O'Connell P, Lathrop GM, Nakamura Y, Leppert ML, Ardinger RH, Murray JL, Lalouel J-M, White R (1989) Twenty-eight loci form a continuous linkage map of markers for human chro- mosome 1. Genomics 4:12-20
Orlowski J, Lingrel JB (1988) Tissue-specific & developmental regulation of rat Na,K-ATPase catalytic a isoform and 13 sub- unit mRNA. J Biol Chem 263 : 10436-10442
Ott J (1985) Analysis of human genetic linkage. Johns Hopkins University Press, Baltimore, pp 200-203
Patel PI, Franc<) B, Garcia C, Slaugenhaupt SA, Nakamura Y, Ledbetter DH, Chakravarti A, Lupski JR (1990) Genetic map- ping of autosomal dominant Charcot-Marie-Tooth Disease in a large French-Acadian kindred: identification of new linked markers on chromosome 17. Am J Hum Genet 46: 801-809
Qiu WQ, Bruin D de, Brownstein BH, Pearse R, Ravetch JV (1990) Organization of the human and mouse low-affinity Fe /R genes: duplication and recombination. Science 248:732-735
Raeymaekers P, Van Broeckhoven C, Backhovens H, Wehnert A, Muylle L, De Jonghe P, Gheuens J, Vandenberghe A (1988) The Duffy blood group is linked to the alpha-spectrin locus in a large pedigree with autosomal dominant inheritance of Char- cot-Marie-Tooth disease type 1. Hum Genet 78: 76-78
Shull MM, Pugh DG, Lingrel JI3 (1989) Characterization of hu- man Na,K-ATPase a2 gene and identification of intragenic restriction fragment length polymorphisms. J Biol Chem 264: 17532-17543
Skre H (1974) Genetic and clinical aspects of Charcot-Marie- Tooth disease. Clin Genet 6: 98-118
Stebbens NB, Conneally PM (1982) Linkage of dominantly inher- ited Charcot-Marie-Tooth neuropathy to the Duffy locus in an Indiana family. Am J Hum Genet 34:195A
Thomas FP, Lebo RV, Ding X-S, Lee SS, Latov N, Hayes AP (1991) Tomaculous neuropathy in chromosome 1 Charcot- Marie-Tooth syndrome (HMSNIB). Neurology 41:339
Trofatter JA, Haines JL, Conneally PM (1986) LIPIN: an interac- tive data entry and management program for LIPED. Am J Hum Genet 39:147-148
Vanasse M, Dubowitz (1981) Dominantly inherited peroneal mus- cular atrophy (hereditary motor and sensory neuropathy type I) in infancy and childhood. Muscle Nerve 4: 26-30
Vance JM, Nicholson GA, Yamaoka LH, Stajich J, Stewart CS, Speer MC, Hung WY, Roses AD, Barker D, Pericak-Vance MA (1989) Linkage of Charcot-Marie-Tooth neuropathy type l a to chromosome 17. Exp Neurol 104:186-189
Vedeler CA (1987) Demonstration of Fcy receptors on human peripheral nerve fibres. J Neuroimmunol 15:207-216
Yang-Feng TL, Schneider JW, Lindgren Z, Schull MM, Benz E J, Lingrel JB, Francke U (1988) Chromosomal localization of human Na,K-ATPase tt and 13 subunit genes. Genomics 2:128- 138





























