Embed Size (px)
Citation preview

ORIGINAL ARTICLE
Cisplatin treatment increases survival and expansion of a highly
tumorigenic side-population fraction by upregulating VEGF/Flt1
autocrine signaling
R Tsuchida1,8, B Das1,2,8, H Yeger3,4, G Koren2, M Shibuya5, PS Thorner3,4, S Baruchel1,2,6
and D Malkin1,2,6,7
1Division of Hematology/Oncology, Department of Paediatrics, The Hospital for Sick Children, Toronto, Ontario, Canada; 2Instituteof Medical Science, University of Toronto, Toronto, Ontario, Canada; 3Division of Pathology, The Hospital for Sick Children,Toronto, Ontario, Canada; 4Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario,Canada; 5Department of Genetics, Institute of Medical Science, University of Tokyo, Division of Genetics, Tokyo, Japan;6Department of Paediatrics, University of Toronto, Toronto, Ontario, Canada and 7Department of Medical Biophysics, Universityof Toronto, Toronto, Ontario, Canada
The cellular and molecular mechanisms of tumor progres-sion following chemotherapy are largely unknown.Here, we demonstrate that cisplatin (CDDP) treatmentupregulates VEGF and Flt1 expression leading to thesurvival and expansion of a highly tumorigenic fraction ofside-population (SP) cells in osteosarcoma (HOS),neuroblastoma (SK-N-BE2) and rhabdomyosarcoma(RH-4) cell lines. In all three lines, we show that CDDPtreatment increases levels of VEGF and Flt1 expression,and induces enhanced clonogenic capacity and increasedexpression of the ‘stemness’-associated genes Nanog,Bmi-1 and Oct-4 in the SP fraction. In HOS, thesechanges are associated with the transformation of anon-tumorigenic osteosarcoma SP fraction to a highlytumorigenic phenotype. Inhibition of Flt1 led to completereduction of tumorigenicity in the HOS SP fraction,and reduction of clonogenic capacity and expressionof stemness genes in the SK-N-BE(2) and RH-4 SPfractions. Treatment with U0126, a specific inhibitor ofMAPK/ERK1,2 completely downregulates CDDP-induced VEGF and Flt1 expression and induction/expansion of SP fraction in all three cell lines, indicatingthat these effects are mediated through MAPK/ERK1,2signaling. In conclusion, we report a novel mechanism ofCDDP-induced tumor progression, whereby the activationof VEGF/Flt1 autocrine signaling leads to the survivaland expansion of a highly tumorigenic SP fraction.Oncogene (2008) 27, 3923–3934; doi:10.1038/onc.2008.38;published online 10 March 2008
Keywords: osteosarcoma; cisplatin; tumorigenic SP;VEGF/Flt1 autocrine
Introduction
Vascular endothelial growth factor (VEGF) is a potentangiogenic agent having tumor-promoting activity.Tumor cells secrete VEGF that increases the prolifera-tion of endothelial cells leading to tumor angiogenesisand subsequent tumor progression (Ferrara et al., 2003).In addition to its effects on endothelial cells, VEGF mayalso act directly on tumor cells in an autocrine mannerto increase survival and proliferation (Fukumura et al.,1998; Dias et al., 2000; Bellamy et al., 2001; Sokeret al., 2001; Strizzi et al., 2001; Bates et al., 2003; Qiet al., 2003; Mercurio et al., 2004; Steiner et al., 2004).We have shown that VEGF binds to its cognate receptorVEGFR-1 (Flt1 also known as fms-like tyrosine kinase-1)in neuroblastoma and rhabdomyosarcoma leading toactivation of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase-1,2 (ERK1,2)and subsequent tumor cell survival (Das et al., 2005;Gee et al., 2005). During hypoxia-induced stress,increased expression of Flt1 rescued the tumor cellsfrom hypoxia-induced cell death (Das et al., 2005). Ourpreliminary data suggested that exposure to cisplatin(cis-diammine-dichloroplatinum (II); CDDP) may in-crease Flt1 expression in osteosarcoma cell lines (Das Bet al.). The role of VEGF autocrine signaling duringoxidative stress. 7th International Symposium onCytokines and Chemokines; 8–9 September 2005; WorldCongress of Gastroenterology, Montreal, Canada).Therefore, we speculated that in the fraction of tumorcells resistant to chemotherapy, treatment-induced Flt1autocrine signaling may provide survival signaling.Recently, a tumor side-population (SP) fraction hasbeen isolated from tumors exhibiting high resistance tochemotherapeutic treatment (Patrawala et al., 2005).Therefore, we studied the potential upregulation of Flt1in the SP fraction and subsequent tumorigenicity.Chemotherapeutic agents have multiple effects on
tumor cells including transient modulation of growthfactor signaling, drug efflux pumps and detoxification
Received 15 August 2007; revised 28 November 2007; accepted 21December 2007; published online 10 March 2008
Correspondence: Dr D Malkin, Division of Hematology/Oncology,Department of Paediatrics, The Hospital for Sick Children, 555University Avenue, Toronto, Ontario, Canada M5G 1X8.E-mail: [email protected] authors contributed equally to this work.
Oncogene (2008) 27, 3923–3934& 2008 Nature Publishing Group All rights reserved 0950-9232/08 $30.00
www.nature.com/onc

systems leading to tumor cell survival (Cara andTannock, 2001; Cole and Tannock, 2004; Kim andTannock, 2005). Recently, Biswas et al. (2007) showedthat chemotherapeutic agents like doxorubicin mayincrease transforming growth factor-b signaling leadingto survival and expansion of highly tumorigenic cells.Chemotherapeutic agents may transform non-tumori-genic into tumorigenic tumor cells. For example, anosteosarcoma cell line HOS, which is non-tumorigenicbecame tumorigenic following acute exposure to heavymetals including platinum (Lin and Costa, 1994; Milleret al., 1998, 2001; Salnikow et al., 1999). Cisplatin, aplatinum drug, has been shown to activate MAPK/ERK1,2 growth signaling pathway leading to eithertumor cell survival or death depending upon the celltypes (Woessmann et al., 2002; Brozovic and Osmak,2007). Our unpublished study suggested that cisplatintreatment may activate the VEGF/Flt1 autocrinesignaling pathway in tumor cell lines including HOS(Das B, Tsuchida R, Yeger H, Malkin D and Baruchel S).The role of VEGF autocrine signaling during oxidativestress. 7th International Symposium on Cytokines andChemokines; 8–9 September 2005; World Congress ofGastroenterology, Montreal, Canada).Thus, chemotherapeutic drugs may induce cellular
signaling pathways involved in tumor growth andprogression leading to tumorigenicity and/or repopulation.Understanding the molecular mechanism involved in theseprocesses may help to develop new combination therapyregimens to reduce the incidence of tumor relapse.Therefore, we investigated the potential role of VEGF/Flt1 autocrine signaling in a CDDP model of drug-induced tumorigenicity. We found that CDDP treatmentactivates VEGF/Flt1 autocrine signaling in the highlytumorigenic SP fraction of cell lines obtained fromosteosarcoma, rhabdomyosarcoma and neuroblastoma.Subsequently, we show that the CDDP-induced SPfraction and its tumorigenic potential can be reduced byinhibiting VEGF/Flt1 autocrine signaling. Furthermore,we found that several stemness genes, including Nanog,are highly expressed in the CDDP-treated SP fractionboth in vitro and in vivo. Our findings highlightthe importance of VEGF/Flt1 autocrine signaling indrug-induced tumorigenicity including the expansion ofan SP fraction expressing self-renewal genes includingNanog and Oct-4.
Results
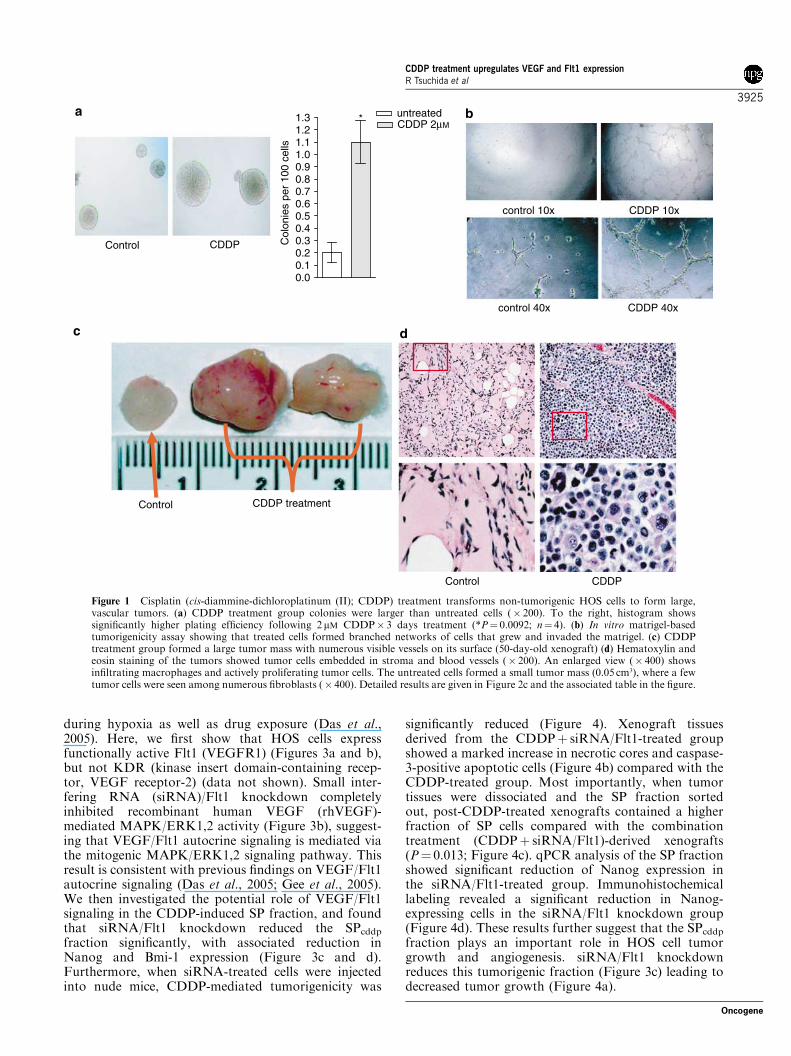
CDDP treatment increases tumorigenic potential ofosteosarcoma HOS cellsWe used a human osteosarcoma cell line, HOS, as amodel to study the role of VEGF/Flt1 autocrinesignaling in CDDP-induced tumorigenicity. This cellline is non-tumorigenic when injected into nude mice(McAllister et al., 1971), and becomes tumorigenicfollowing exposure to heavy metals (Lin and Costa,1994; Miller et al., 2001; Miura et al., 2005). Wefound that exposure of HOS cells to cisplatin(CDDP) (2mM� 3 days) increased in vitro tumorigenicity
(Figures 1a and b) as measured by in vitro matrigel assay(Miller et al., 2001), and induced formation of largetumors when injected into nude mice (2� 106 cells perinjection, n¼ 16; Figures 1c and d).We observed that theuntreated HOS group (10� 106 cells per injection)reached 0.05 cm3 size (four out of eight injection;Figure 1c). Histopathological examination of tumorsat 50 days revealed that the area of inoculation was filledwith fibroblasts and a few scattered tumor cells, whereasthe CDDP-treated tumors showed tumor cells withsurrounding blood vessels (Figure 1d). Consideringthat neovascularization is involved in the progressionof tumor from dormancy to a state of rapid growth(Folkman, 1971), we measured the MVD (microvesseldensity) of the 0.05 cm3 growth using CD34 staining(Gasparini and Harris, 1995) and found that CDDP-treated xenograft showed numerous CD34 stainingmicrovessels, whereas untreated growth did not showevidence of angiogenesis (Supplementary Figure 1).We then investigated the potential role for SP cells in
CDDP-treated HOS cell tumorigenicity. In all the follow-ing assays, 3-day exposure to 2mM CDDP was used as astandard treatment for in vitro as well as in vivo studies.
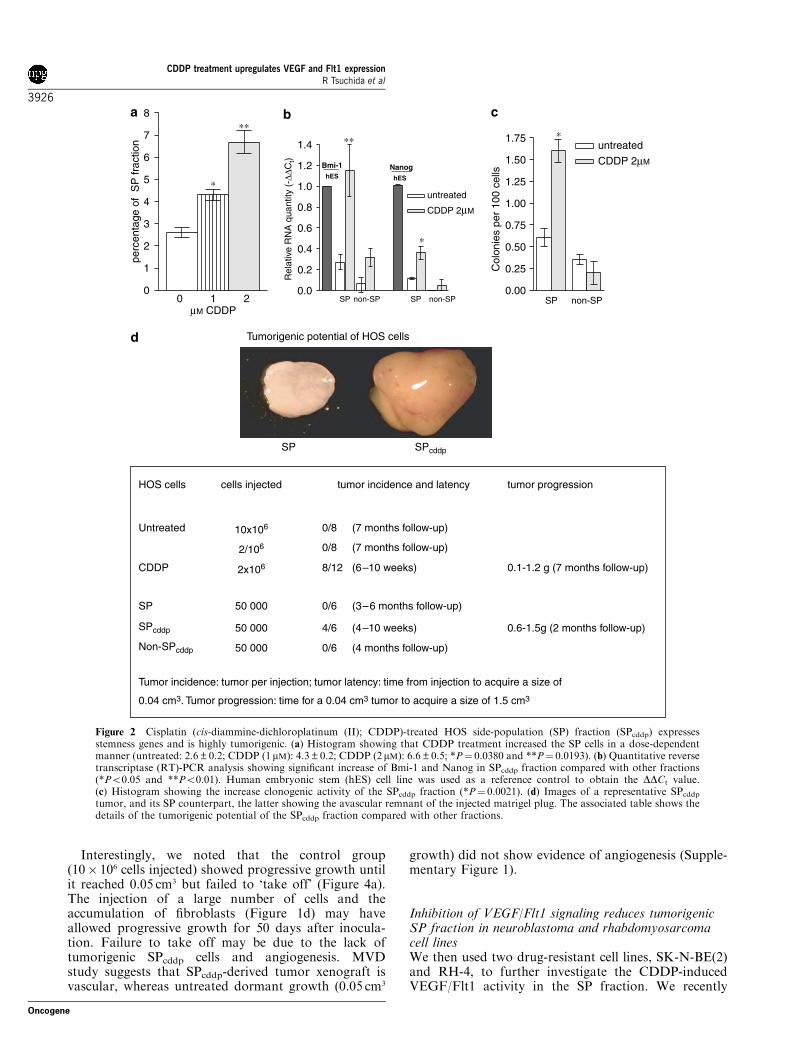
CDDP-treated HOS SP fraction (SPcddp) is highlytumorigenicTumor SP cells characterized by high expression ofdrug efflux pumps such as BCRP1 and MDR1 havebeen found to contain the tumor stem cell fraction(Hirschmann-Jax et al., 2004; Kondo et al., 2004;Patrawala et al., 2005). We show that the HOS cell linecontains a small fraction of SP cells, which can bemaintained in serum-free media containing growthfactors bFGF (basic fibroblast growth factor) andPDGF (platelet-derived growth factor; SupplementaryFigure 2A). Figure 2a shows that CDDP treatmentincreases the SP cell fraction significantly.We then sorted SP, non-SP, SPcddp (SP cells obtained
following 3 days treatment of HOS cells with 2 mMCDDP) and non-SPcddp (non-SP cells obtained follow-ing 3 days treatment of HOS cells with 2 mM CDDP) andinvestigated the expression of the stemness-associatedgenes Nanog and Bmi-1 by quantitative reverse tran-scriptase (RT)-PCR (qPCR). Both these genes arehighly expressed in the SPcddp fraction (Figure 2b).Furthermore, colony assay showed increased clonogenicefficiency of CDDP-treated SP fraction (SPcddp) com-pared with the untreated SP and treated non-SP (non-SPcddp) fractions (Figure 2c). In addition, when SPcddpcells were injected into nude mice, they formed rapidlygrowing tumors (Figure 2d) compared with the SP andnon-SPcddp group. Furthermore, the SPcddp xenograftshowed a 1.5-fold increase in MVD compared withCDDP-treated unsorted HOS cell-derived xenografts(Mann–Whitney test; Supplementary Figure 1).
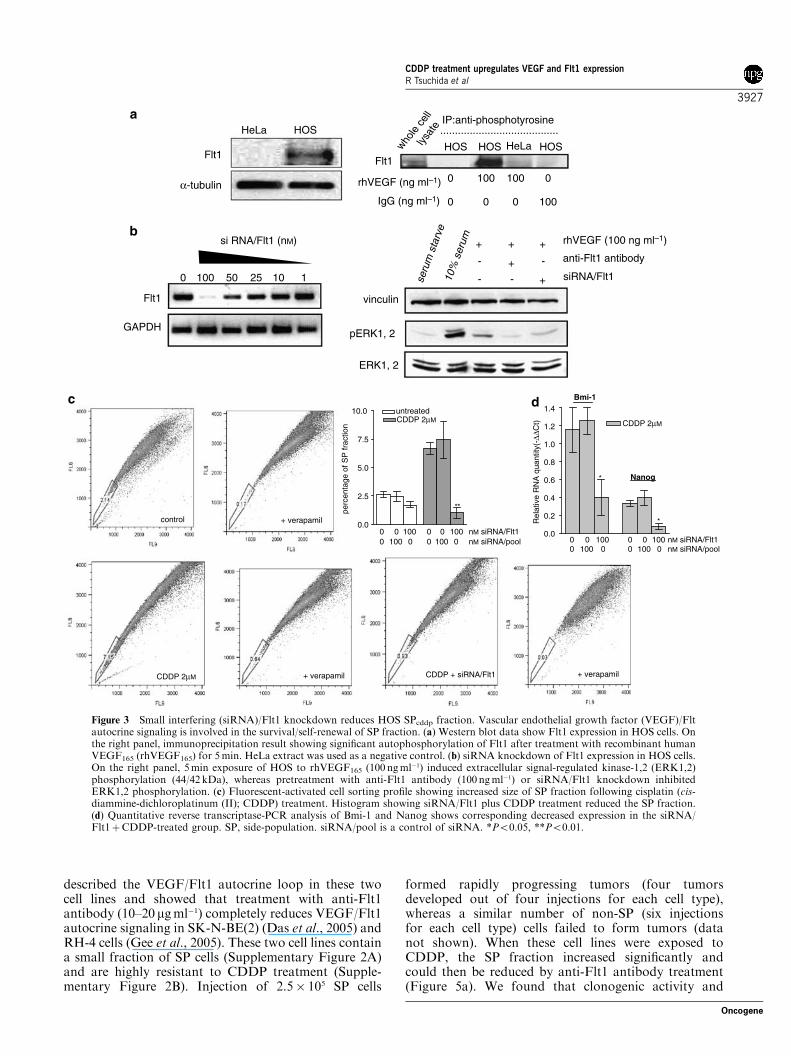
siRNA/Flt1 knockdown reduces HOS SPcddp fraction andsubsequent tumorigenicityPreviously, we reported that VEGF/Flt1 autocrinesignaling is involved in the survival of tumor cells
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3924
Oncogene
during hypoxia as well as drug exposure (Das et al.,2005). Here, we first show that HOS cells expressfunctionally active Flt1 (VEGFR1) (Figures 3a and b),but not KDR (kinase insert domain-containing recep-tor, VEGF receptor-2) (data not shown). Small inter-fering RNA (siRNA)/Flt1 knockdown completelyinhibited recombinant human VEGF (rhVEGF)-mediated MAPK/ERK1,2 activity (Figure 3b), suggest-ing that VEGF/Flt1 autocrine signaling is mediated viathe mitogenic MAPK/ERK1,2 signaling pathway. Thisresult is consistent with previous findings on VEGF/Flt1autocrine signaling (Das et al., 2005; Gee et al., 2005).We then investigated the potential role of VEGF/Flt1signaling in the CDDP-induced SP fraction, and foundthat siRNA/Flt1 knockdown reduced the SPcddpfraction significantly, with associated reduction inNanog and Bmi-1 expression (Figure 3c and d).Furthermore, when siRNA-treated cells were injectedinto nude mice, CDDP-mediated tumorigenicity was
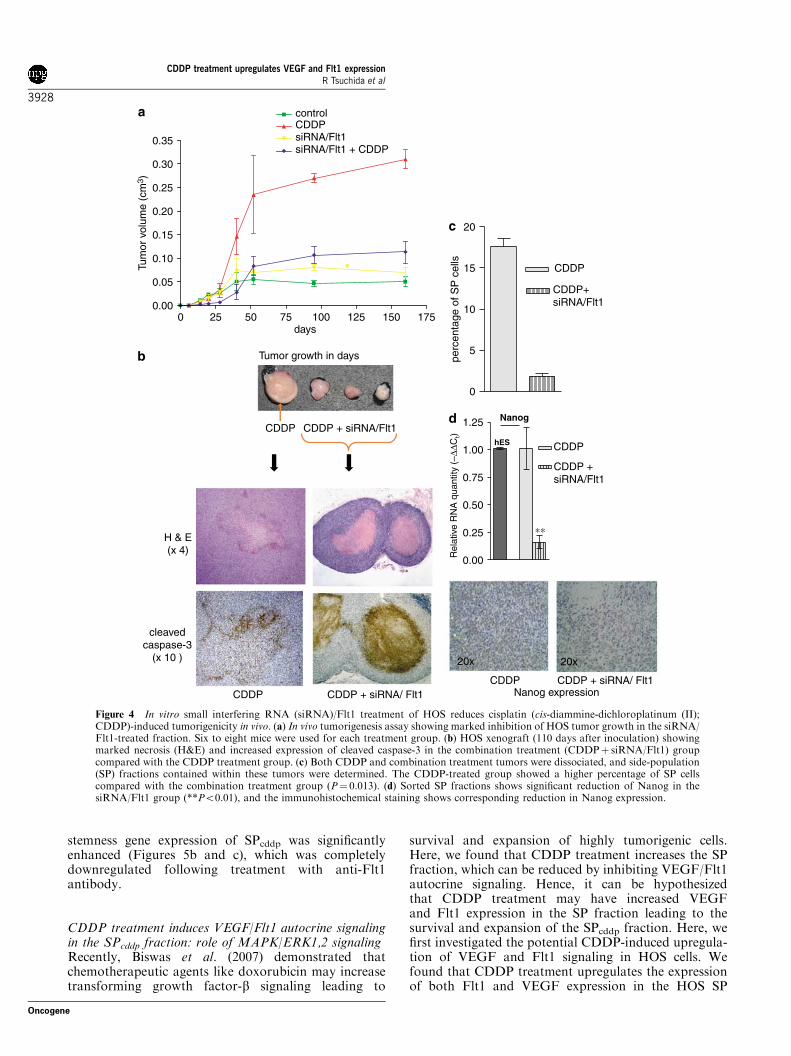
significantly reduced (Figure 4). Xenograft tissuesderived from the CDDPþ siRNA/Flt1-treated groupshowed a marked increase in necrotic cores and caspase-3-positive apoptotic cells (Figure 4b) compared with theCDDP-treated group. Most importantly, when tumortissues were dissociated and the SP fraction sortedout, post-CDDP-treated xenografts contained a higherfraction of SP cells compared with the combinationtreatment (CDDPþ siRNA/Flt1)-derived xenografts(P¼ 0.013; Figure 4c). qPCR analysis of the SP fractionshowed significant reduction of Nanog expression inthe siRNA/Flt1-treated group. Immunohistochemicallabeling revealed a significant reduction in Nanog-expressing cells in the siRNA/Flt1 knockdown group(Figure 4d). These results further suggest that the SPcddpfraction plays an important role in HOS cell tumorgrowth and angiogenesis. siRNA/Flt1 knockdownreduces this tumorigenic fraction (Figure 3c) leading todecreased tumor growth (Figure 4a).
Control
Control
CDDP
CDDP treatment
Control CDDP
0.0
1.31.21.11.00.90.80.70.60.50.40.30.20.1
* untreatedCDDP 2µM
Col
onie
s pe
r 10
0 ce
lls
control 10x CDDP 10x
control 40x CDDP 40x
Figure 1 Cisplatin (cis-diammine-dichloroplatinum (II); CDDP) treatment transforms non-tumorigenic HOS cells to form large,vascular tumors. (a) CDDP treatment group colonies were larger than untreated cells (� 200). To the right, histogram showssignificantly higher plating efficiency following 2 mM CDDP� 3 days treatment (*P¼ 0.0092; n¼ 4). (b) In vitro matrigel-basedtumorigenicity assay showing that treated cells formed branched networks of cells that grew and invaded the matrigel. (c) CDDPtreatment group formed a large tumor mass with numerous visible vessels on its surface (50-day-old xenograft) (d) Hematoxylin andeosin staining of the tumors showed tumor cells embedded in stroma and blood vessels (� 200). An enlarged view (� 400) showsinfiltrating macrophages and actively proliferating tumor cells. The untreated cells formed a small tumor mass (0.05 cm3), where a fewtumor cells were seen among numerous fibroblasts (� 400). Detailed results are given in Figure 2c and the associated table in the figure.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3925
Oncogene
Interestingly, we noted that the control group(10� 106 cells injected) showed progressive growth untilit reached 0.05 cm3 but failed to ‘take off’ (Figure 4a).The injection of a large number of cells and theaccumulation of fibroblasts (Figure 1d) may haveallowed progressive growth for 50 days after inocula-tion. Failure to take off may be due to the lack oftumorigenic SPcddp cells and angiogenesis. MVDstudy suggests that SPcddp-derived tumor xenograft isvascular, whereas untreated dormant growth (0.05 cm3
growth) did not show evidence of angiogenesis (Supple-mentary Figure 1).
Inhibition of VEGF/Flt1 signaling reduces tumorigenicSP fraction in neuroblastoma and rhabdomyosarcomacell linesWe then used two drug-resistant cell lines, SK-N-BE(2)and RH-4, to further investigate the CDDP-inducedVEGF/Flt1 activity in the SP fraction. We recently
SP SPcddp
0.00
0.25
0.50
0.75
1.00
1.25
1.50
1.75untreated
CDDP 2µM
∗
SP non-SP
Col
onie
s pe
r 10
0 ce
llsHOS cells cells injected tumor incidence and latency tumor progression
Untreated 10x106 0/8 (7 months follow-up)
2/106 0/8 (7 months follow-up)
CDDP 2x106 8/12 (6–10 weeks) 0.1-1.2 g (7 months follow-up)
SP 50 000 0/6 (3–6 months follow-up)
SPcddp 50 000 4/6 (4–10 weeks) 0.6-1.5g (2 months follow-up)
Non-SPcddp 50 000 0/6 (4 months follow-up)
Tumor incidence: tumor per injection; tumor latency: time from injection to acquire a size of
0.04 cm3. Tumor progression: time for a 0.04 cm3 tumor to acquire a size of 1.5 cm3
Tumorigenic potential of HOS cells
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
Bmi-1hES
Nanog hES
untreated
CDDP 2µM
SP non-SP SP non-SP
∗∗
∗
Rel
ativ
e R
NA
qua
ntity
(-∆
∆Ct)
0
1
2
3
4
5
6
7
8∗∗
∗
perc
enta
ge o
f S
P fr
actio
n
0 1 2µM CDDP
Figure 2 Cisplatin (cis-diammine-dichloroplatinum (II); CDDP)-treated HOS side-population (SP) fraction (SPcddp) expressesstemness genes and is highly tumorigenic. (a) Histogram showing that CDDP treatment increased the SP cells in a dose-dependentmanner (untreated: 2.6±0.2; CDDP (1 mM): 4.3±0.2; CDDP (2mM): 6.6±0.5; *P¼ 0.0380 and **P¼ 0.0193). (b) Quantitative reversetranscriptase (RT)-PCR analysis showing significant increase of Bmi-1 and Nanog in SPcddp fraction compared with other fractions(*Po0.05 and **Po0.01). Human embryonic stem (hES) cell line was used as a reference control to obtain the DDCt value.(c) Histogram showing the increase clonogenic activity of the SPcddp fraction (*P¼ 0.0021). (d) Images of a representative SPcddptumor, and its SP counterpart, the latter showing the avascular remnant of the injected matrigel plug. The associated table shows thedetails of the tumorigenic potential of the SPcddp fraction compared with other fractions.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3926
Oncogene
described the VEGF/Flt1 autocrine loop in these twocell lines and showed that treatment with anti-Flt1antibody (10–20 mgml�1) completely reduces VEGF/Flt1autocrine signaling in SK-N-BE(2) (Das et al., 2005) andRH-4 cells (Gee et al., 2005). These two cell lines containa small fraction of SP cells (Supplementary Figure 2A)and are highly resistant to CDDP treatment (Supple-mentary Figure 2B). Injection of 2.5� 105 SP cells
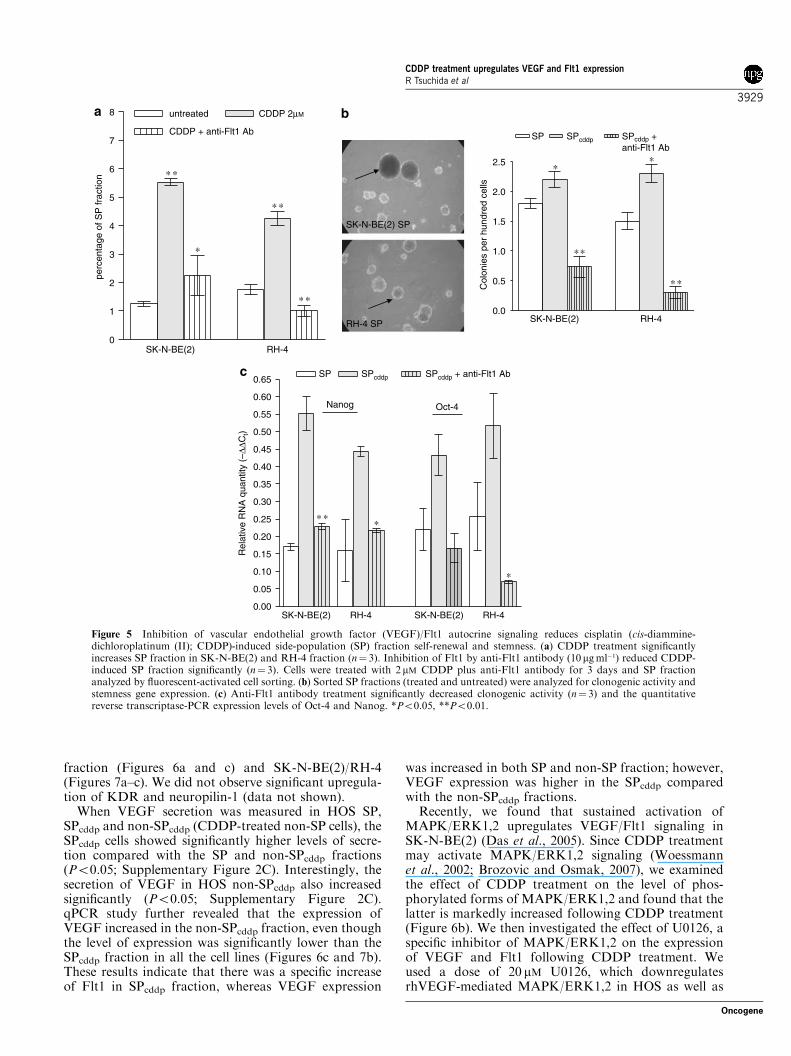
formed rapidly progressing tumors (four tumorsdeveloped out of four injections for each cell type),whereas a similar number of non-SP (six injectionsfor each cell type) cells failed to form tumors (datanot shown). When these cell lines were exposed toCDDP, the SP fraction increased significantly andcould then be reduced by anti-Flt1 antibody treatment(Figure 5a). We found that clonogenic activity and
0 100 100 0
0 0 0 100
Flt1HeLa
........................................
rhVEGF (ng ml–1)
seru
m s
tarv
e
vinculin
pERK1, 2
ERK1, 2
10%
ser
um
+ + +
+- -
+- -
rhVEGF (100 ng ml–1)
anti-Flt1 antibody
siRNA/Flt1
whole
cel
l
l
ysat
e
GAPDH
Flt1
si RNA/Flt1 (nM)
HeLa
Flt1
HOSIP:anti-phosphotyrosine
0.0
2.5
5.0
7.5
10.0
**
untreatedCDDP 2µM CDDP 2µM
perc
enta
ge o
f SP
frac
tion
00
00
100100
00
00
100100
nM siRNA/Flt1nM siRNA/pool 0
00
0100
10000
00
100100
nM siRNA/Flt1nM siRNA/pool
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
Bmi-1
Nanog*
*Rel
ativ
e R
NA
qua
ntity
(-∆∆
Ct)
control + verapamil
+ verapamil + verapamilCDDP + siRNA/Flt1CDDP 2µM
HOS HOSHOS
0 100 50 25 10 1
IgG (ng ml–1)
α-tubulin
Figure 3 Small interfering (siRNA)/Flt1 knockdown reduces HOS SPcddp fraction. Vascular endothelial growth factor (VEGF)/Fltautocrine signaling is involved in the survival/self-renewal of SP fraction. (a) Western blot data show Flt1 expression in HOS cells. Onthe right panel, immunoprecipitation result showing significant autophosphorylation of Flt1 after treatment with recombinant humanVEGF165 (rhVEGF165) for 5min. HeLa extract was used as a negative control. (b) siRNA knockdown of Flt1 expression in HOS cells.On the right panel, 5min exposure of HOS to rhVEGF165 (100 ngml
�1) induced extracellular signal-regulated kinase-1,2 (ERK1,2)phosphorylation (44/42 kDa), whereas pretreatment with anti-Flt1 antibody (100 ngml�1) or siRNA/Flt1 knockdown inhibitedERK1,2 phosphorylation. (c) Fluorescent-activated cell sorting profile showing increased size of SP fraction following cisplatin (cis-diammine-dichloroplatinum (II); CDDP) treatment. Histogram showing siRNA/Flt1 plus CDDP treatment reduced the SP fraction.(d) Quantitative reverse transcriptase-PCR analysis of Bmi-1 and Nanog shows corresponding decreased expression in the siRNA/Flt1þCDDP-treated group. SP, side-population. siRNA/pool is a control of siRNA. *Po0.05, **Po0.01.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3927
Oncogene
stemness gene expression of SPcddp was significantlyenhanced (Figures 5b and c), which was completelydownregulated following treatment with anti-Flt1antibody.
CDDP treatment induces VEGF/Flt1 autocrine signalingin the SPcddp fraction: role of MAPK/ERK1,2 signalingRecently, Biswas et al. (2007) demonstrated thatchemotherapeutic agents like doxorubicin may increasetransforming growth factor-b signaling leading to
survival and expansion of highly tumorigenic cells.Here, we found that CDDP treatment increases the SPfraction, which can be reduced by inhibiting VEGF/Flt1autocrine signaling. Hence, it can be hypothesizedthat CDDP treatment may have increased VEGFand Flt1 expression in the SP fraction leading to thesurvival and expansion of the SPcddp fraction. Here, wefirst investigated the potential CDDP-induced upregula-tion of VEGF and Flt1 signaling in HOS cells. Wefound that CDDP treatment upregulates the expressionof both Flt1 and VEGF expression in the HOS SP
Tumor growth in days
Tum
or v
olum
e (c
m3 )
0 25 50 75 100 125 150 1750.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
controlCDDPsiRNA/Flt1siRNA/Flt1 + CDDP
days
CDDP CDDP + siRNA/ Flt1
cleavedcaspase-3
(x 10 )
H & E(x 4)
0.00
0.25
0.50
0.75
1.00
1.25 Nanog
CDDP
CDDP +siRNA/Flt1
hES
∗∗
Rel
ativ
e R
NA
qua
ntity
(–∆
∆Ct)
CDDP CDDP + siRNA/ Flt1Nanog expression
0
5
10
15
20
CDDP
CDDP+siRNA/Flt1
perc
enta
ge o
f SP
cel
ls
20x 20x
CDDP + siRNA/Flt1CDDP
Figure 4 In vitro small interfering RNA (siRNA)/Flt1 treatment of HOS reduces cisplatin (cis-diammine-dichloroplatinum (II);CDDP)-induced tumorigenicity in vivo. (a) In vivo tumorigenesis assay showing marked inhibition of HOS tumor growth in the siRNA/Flt1-treated fraction. Six to eight mice were used for each treatment group. (b) HOS xenograft (110 days after inoculation) showingmarked necrosis (H&E) and increased expression of cleaved caspase-3 in the combination treatment (CDDPþ siRNA/Flt1) groupcompared with the CDDP treatment group. (c) Both CDDP and combination treatment tumors were dissociated, and side-population(SP) fractions contained within these tumors were determined. The CDDP-treated group showed a higher percentage of SP cellscompared with the combination treatment group (P¼ 0.013). (d) Sorted SP fractions shows significant reduction of Nanog in thesiRNA/Flt1 group (**Po0.01), and the immunohistochemical staining shows corresponding reduction in Nanog expression.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3928
Oncogene
fraction (Figures 6a and c) and SK-N-BE(2)/RH-4(Figures 7a–c). We did not observe significant upregula-tion of KDR and neuropilin-1 (data not shown).When VEGF secretion was measured in HOS SP,
SPcddp and non-SPcddp (CDDP-treated non-SP cells), theSPcddp cells showed significantly higher levels of secre-tion compared with the SP and non-SPcddp fractions(Po0.05; Supplementary Figure 2C). Interestingly, thesecretion of VEGF in HOS non-SPcddp also increasedsignificantly (Po0.05; Supplementary Figure 2C).qPCR study further revealed that the expression ofVEGF increased in the non-SPcddp fraction, even thoughthe level of expression was significantly lower than theSPcddp fraction in all the cell lines (Figures 6c and 7b).These results indicate that there was a specific increaseof Flt1 in SPcddp fraction, whereas VEGF expression
was increased in both SP and non-SP fraction; however,VEGF expression was higher in the SPcddp comparedwith the non-SPcddp fractions.Recently, we found that sustained activation of
MAPK/ERK1,2 upregulates VEGF/Flt1 signaling inSK-N-BE(2) (Das et al., 2005). Since CDDP treatmentmay activate MAPK/ERK1,2 signaling (Woessmannet al., 2002; Brozovic and Osmak, 2007), we examinedthe effect of CDDP treatment on the level of phos-phorylated forms of MAPK/ERK1,2 and found that thelatter is markedly increased following CDDP treatment(Figure 6b). We then investigated the effect of U0126, aspecific inhibitor of MAPK/ERK1,2 on the expressionof VEGF and Flt1 following CDDP treatment. Weused a dose of 20 mM U0126, which downregulatesrhVEGF-mediated MAPK/ERK1,2 in HOS as well as
SK-N-BE(2) SP
RH-4 SP
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
0.65
SK-N-BE(2) RH-4
SP
Nanog
SPcddp
Oct-4
SK-N-BE(2) RH-4
∗
SPcddp + anti-Flt1 Ab
Rel
ativ
e R
NA
qua
ntity
(–∆
∆Ct)
∗
∗∗
0
1
2
3
4
5
6
7
8
∗ ∗
untreated CDDP 2µM
CDDP + anti-Flt1 Ab
SK-N-BE(2) RH-4
∗
∗ ∗
∗ ∗
perc
enta
ge o
f SP
frac
tion
0.0
0.5
1.0
1.5
2.0
2.5
SP SPcddp
SK-N-BE(2) RH-4
∗
SPcddp +anti-Flt1 Ab
Col
onie
s pe
r hu
ndre
d ce
lls
∗∗
∗∗
∗
Figure 5 Inhibition of vascular endothelial growth factor (VEGF)/Flt1 autocrine signaling reduces cisplatin (cis-diammine-dichloroplatinum (II); CDDP)-induced side-population (SP) fraction self-renewal and stemness. (a) CDDP treatment significantlyincreases SP fraction in SK-N-BE(2) and RH-4 fraction (n¼ 3). Inhibition of Flt1 by anti-Flt1 antibody (10 mgml�1) reduced CDDP-induced SP fraction significantly (n¼ 3). Cells were treated with 2 mM CDDP plus anti-Flt1 antibody for 3 days and SP fractionanalyzed by fluorescent-activated cell sorting. (b) Sorted SP fractions (treated and untreated) were analyzed for clonogenic activity andstemness gene expression. (c) Anti-Flt1 antibody treatment significantly decreased clonogenic activity (n¼ 3) and the quantitativereverse transcriptase-PCR expression levels of Oct-4 and Nanog. *Po0.05, **Po0.01.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3929
Oncogene
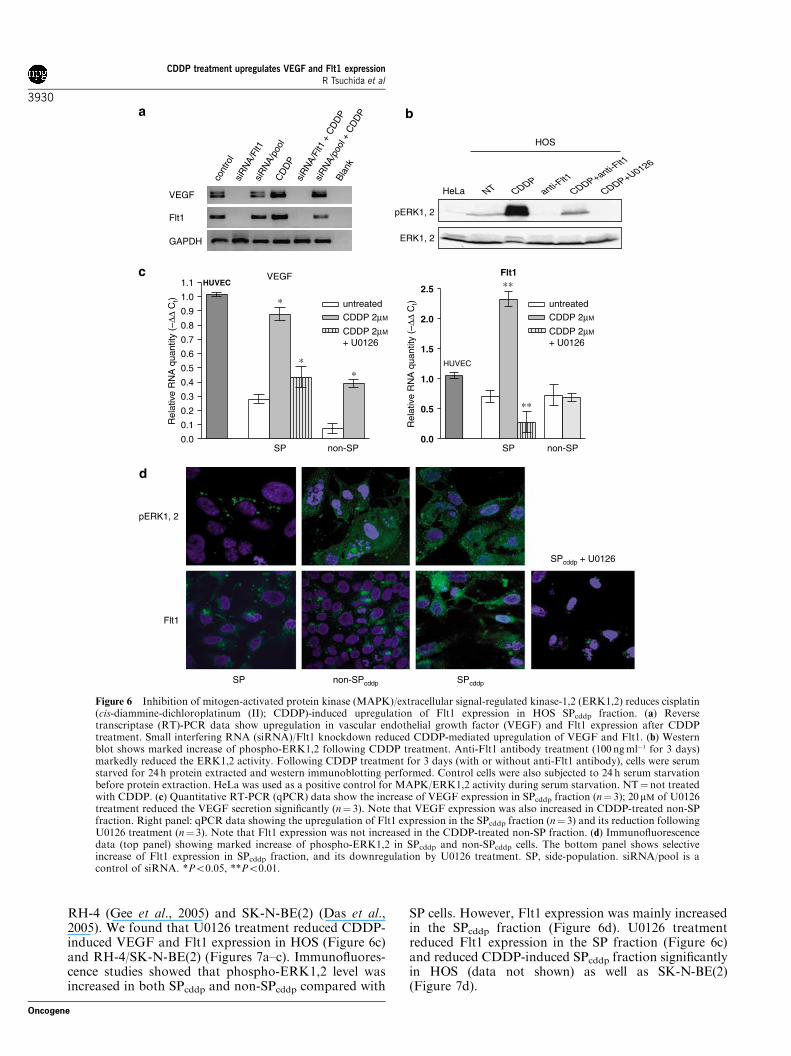
RH-4 (Gee et al., 2005) and SK-N-BE(2) (Das et al.,2005). We found that U0126 treatment reduced CDDP-induced VEGF and Flt1 expression in HOS (Figure 6c)and RH-4/SK-N-BE(2) (Figures 7a–c). Immunofluores-cence studies showed that phospho-ERK1,2 level wasincreased in both SPcddp and non-SPcddp compared with
SP cells. However, Flt1 expression was mainly increasedin the SPcddp fraction (Figure 6d). U0126 treatmentreduced Flt1 expression in the SP fraction (Figure 6c)and reduced CDDP-induced SPcddp fraction significantlyin HOS (data not shown) as well as SK-N-BE(2)(Figure 7d).
pERK1, 2
ERK1, 2
HeLa NT CDDPCDDP+anti-F
lt1
anti-Flt1
CDDP+U0126
SPcddpSP non-SPcddp
pERK1, 2
Flt1
SPcddp + U0126
HOS
0.0
0.5
1.0
1.5
2.0
2.5 ∗∗Flt1
HUVEC
SP non-SP
∗∗R
elat
ive
RN
A q
uant
ity (
–∆∆
Ct)
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
untreated
CDDP 2µM
CDDP 2µM
+ U0126
untreated
CDDP 2µM
CDDP 2µM
+ U0126
VEGF
SP non-SP
∗
∗∗
HUVEC
Rel
ativ
e R
NA
qua
ntity
(–∆
∆ C
t)
cont
rol
siR
NA/
pool
siR
NA/
Flt1
Blan
k
CD
DP
siR
NA/
Flt1
+ C
DD
P
siR
NA/
pool
+ C
DD
P
VEGF
Flt1
GAPDH
Figure 6 Inhibition of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase-1,2 (ERK1,2) reduces cisplatin(cis-diammine-dichloroplatinum (II); CDDP)-induced upregulation of Flt1 expression in HOS SPcddp fraction. (a) Reversetranscriptase (RT)-PCR data show upregulation in vascular endothelial growth factor (VEGF) and Flt1 expression after CDDPtreatment. Small interfering RNA (siRNA)/Flt1 knockdown reduced CDDP-mediated upregulation of VEGF and Flt1. (b) Westernblot shows marked increase of phospho-ERK1,2 following CDDP treatment. Anti-Flt1 antibody treatment (100 ngml�1 for 3 days)markedly reduced the ERK1,2 activity. Following CDDP treatment for 3 days (with or without anti-Flt1 antibody), cells were serumstarved for 24 h protein extracted and western immunoblotting performed. Control cells were also subjected to 24 h serum starvationbefore protein extraction. HeLa was used as a positive control for MAPK/ERK1,2 activity during serum starvation. NT¼not treatedwith CDDP. (c) Quantitative RT-PCR (qPCR) data show the increase of VEGF expression in SPcddp fraction (n¼ 3); 20mM of U0126treatment reduced the VEGF secretion significantly (n¼ 3). Note that VEGF expression was also increased in CDDP-treated non-SPfraction. Right panel: qPCR data showing the upregulation of Flt1 expression in the SPcddp fraction (n¼ 3) and its reduction followingU0126 treatment (n¼ 3). Note that Flt1 expression was not increased in the CDDP-treated non-SP fraction. (d) Immunofluorescencedata (top panel) showing marked increase of phospho-ERK1,2 in SPcddp and non-SPcddp cells. The bottom panel shows selectiveincrease of Flt1 expression in SPcddp fraction, and its downregulation by U0126 treatment. SP, side-population. siRNA/pool is acontrol of siRNA. *Po0.05, **Po0.01.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3930
Oncogene
Discussion
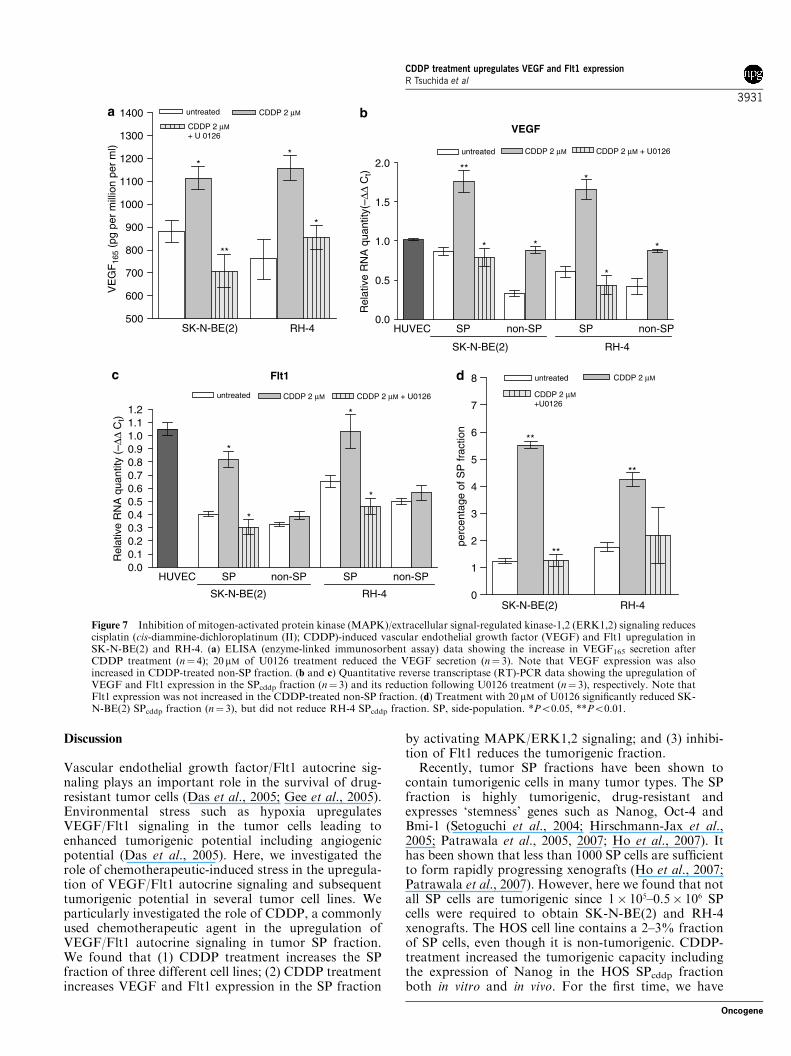
Vascular endothelial growth factor/Flt1 autocrine sig-naling plays an important role in the survival of drug-resistant tumor cells (Das et al., 2005; Gee et al., 2005).Environmental stress such as hypoxia upregulatesVEGF/Flt1 signaling in the tumor cells leading toenhanced tumorigenic potential including angiogenicpotential (Das et al., 2005). Here, we investigated therole of chemotherapeutic-induced stress in the upregula-tion of VEGF/Flt1 autocrine signaling and subsequenttumorigenic potential in several tumor cell lines. Weparticularly investigated the role of CDDP, a commonlyused chemotherapeutic agent in the upregulation ofVEGF/Flt1 autocrine signaling in tumor SP fraction.We found that (1) CDDP treatment increases the SPfraction of three different cell lines; (2) CDDP treatmentincreases VEGF and Flt1 expression in the SP fraction
by activating MAPK/ERK1,2 signaling; and (3) inhibi-tion of Flt1 reduces the tumorigenic fraction.Recently, tumor SP fractions have been shown to
contain tumorigenic cells in many tumor types. The SPfraction is highly tumorigenic, drug-resistant andexpresses ‘stemness’ genes such as Nanog, Oct-4 andBmi-1 (Setoguchi et al., 2004; Hirschmann-Jax et al.,2005; Patrawala et al., 2005, 2007; Ho et al., 2007). Ithas been shown that less than 1000 SP cells are sufficientto form rapidly progressing xenografts (Ho et al., 2007;Patrawala et al., 2007). However, here we found that notall SP cells are tumorigenic since 1� 105–0.5� 106 SPcells were required to obtain SK-N-BE(2) and RH-4xenografts. The HOS cell line contains a 2–3% fractionof SP cells, even though it is non-tumorigenic. CDDP-treatment increased the tumorigenic capacity includingthe expression of Nanog in the HOS SPcddp fractionboth in vitro and in vivo. For the first time, we have
VEGF
0.0
0.5
1.0
1.5
2.0
SK-N-BE(2) RH-4
untreated CDDP 2 µM CDDP 2 µM + U0126
**
*
* *
*
*
HUVEC SP non-SP SP non-SP
Rel
ativ
e R
NA
qua
ntity
(–∆∆
Ct)
0.00.10.20.30.40.50.60.70.80.91.01.11.2
HUVEC SP non-SP SP non-SP
*
*
*
Flt1
CDDP 2 µMuntreated CDDP 2 µM + U0126
*
SK-N-BE(2) RH-4
Rel
ativ
e R
NA
qua
ntity
(–∆
∆ C
t)
0
1
2
3
4
5
6
7
8
**
untreated CDDP 2 µM
SK-N-BE(2) RH-4
**
CDDP 2 µM +U0126
**
perc
enta
ge o
f SP
frac
tion
500
600
700
800
900
1000
1100
1200
1300
1400 untreated CDDP 2 µM
SK-N-BE(2) RH-4
**
**
*
CDDP 2 µM + U 0126
VE
GF
165
(pg
per
mill
ion
per
ml)
Figure 7 Inhibition of mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase-1,2 (ERK1,2) signaling reducescisplatin (cis-diammine-dichloroplatinum (II); CDDP)-induced vascular endothelial growth factor (VEGF) and Flt1 upregulation inSK-N-BE(2) and RH-4. (a) ELISA (enzyme-linked immunosorbent assay) data showing the increase in VEGF165 secretion afterCDDP treatment (n¼ 4); 20mM of U0126 treatment reduced the VEGF secretion (n¼ 3). Note that VEGF expression was alsoincreased in CDDP-treated non-SP fraction. (b and c) Quantitative reverse transcriptase (RT)-PCR data showing the upregulation ofVEGF and Flt1 expression in the SPcddp fraction (n¼ 3) and its reduction following U0126 treatment (n¼ 3), respectively. Note thatFlt1 expression was not increased in the CDDP-treated non-SP fraction. (d) Treatment with 20 mM of U0126 significantly reduced SK-N-BE(2) SPcddp fraction (n¼ 3), but did not reduce RH-4 SPcddp fraction. SP, side-population. *Po0.05, **Po0.01.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3931
Oncogene

demonstrated that drug exposure may actually enhancethe tumorigenic potential of the SP fraction.We found that CDDP treatment led to a significant
increase in the SP fraction including specific upregula-tion of Flt1 and stemness gene expression in the SPfraction. In our experimental model, cells were firsttreated with CDDP, and then SP and non-SP fractionswere sorted and qPCR analysis of Flt1 expression wasperformed. Hence, it can be argued that an apparentincrease of Flt1/stemness gene expression in the SPfraction may be due to either survival (selection) and/orexpansion (induction) of SP cells having Flt1/stemnessgene expression. While selection may explain theincrease of Flt1/stemness gene expressing HOS SPcddpcells, the RH-4 and SK-N-BE(2) are highly resistant toCDDP treatment (Supplementary Figure 1B), andtherefore, apparent selection of Flt1-expressing SPcddpcells is unlikely. Instead, CDDP-mediated stress mayexpand SPcddp fraction. We found that hypoxia-inducedoxidative stress expands highly tumorigenic SP cells(Das et al., 2007) (Das B. The role of VEGF autocrinesignaling in hypoxia and oxidative stress driven ‘stem-ness switch’: implications in solid tumor progression andmetastasis. PhD thesis, Institute of Medical Sciences,University of Toronto, Canada). Earlier, it was reportedthat chemotherapy-induced stress led to the expansionof normal bone marrow stem cells (Richman et al., 1976;Cottler-Fox et al., 2003). Hence, CDDP-induced expan-sion of a highly tumorigenic SPcddp fraction is a likelypossibility. Such a stress-induced expansion of tumori-genic cells would involve autocrine signaling pathwayssuch as VEGF/Flt1 signaling. Our findings that siRNA/Flt1 inhibition significantly decreased the SPcddp frac-tion support this possibility. It is possible that onlytumorigenic SP cells are capable of activating thesignaling pathway leading to expansion, which wouldexplain why the Flt1/stemness gene expression did notchange in the non-tumorigenic non-SPcddp fraction.Overall, our findings in HOS, SK-N-BE(2) and RH-4
cells suggest that CDDP-induced SP fraction propaga-tion is mediated by upregulation of VEGF and itsreceptor Flt1. VEGF/Flt1 autocrine signaling ismediated by MAPK/ERK1,2 signaling, the latter beingfound to be upregulated during stress including follow-ing CDDP treatment (Woessmann et al., 2002; Brozovicand Osmak, 2007). Numerous studies have reported theactivation of the MAPK superfamily including ERK1,2in tumor cells following stress (Brozovic and Osmak,2007). The activation of these signaling pathways maybe involved in cell survival as well as cell deathdepending upon the specific stimulus and cell typeinvolved. We found that ERK1,2 activation followinghypoxia reperfusion injury leads to cell survival inneuroblastoma cell lines (Das et al., 2005). On the otherhand, hypoxia reperfusion-activated MAPK/ERK1,2 incardiac tissue may lead to protection as well as damageto cardiac tissue depending on the degree of stimulationand cell type involved (Behrends et al., 2000). Ranga-nathan et al. (2006) reported that upregulation ofMAPK/ERK1,2 signaling may activate dormant tumorcells leading to the expansion of tumorigenic cells and
subsequent tumor progression. However, no molecularmechanism was identified that accounted for theactivation of the dormant tumors. Here, our findingson MAPK/ERK1,2-mediated activation of VEGF/Flt1signaling in the SP fraction suggest a molecularmechanism by which the mitogenic signaling ofMAPK/ERK1,2 may activate dormant tumor stemcell-like SP cells. In the clinical setting, it may benecessary to first assess the effect of CDDP on thespecific tumor cell type to determine whether the druginduces VEGF and MAPK/ERK1,2 signaling activity.If it does, combination therapy might be consideredbeneficial. Therefore, use of such preclinical data mayhelp to design effective and innovative combinationtherapies.Our findings on the CDDP-induced activation of
VEGF/Flt1 signaling in a subset of tumor SP cellswarrant further investigation including whether otherchemotherapeutic agents may have similar effect. Ourpreliminary investigation suggests that doxorubicinand methotrexate may also increase SP fraction andFlt1 expression in the tumor cells (SupplementaryFigures 3A and B).In summary, we report on a novel mechanism of
CDDP-induced tumorigenicity and demonstrate thateven brief exposure of tumor cells to drugs may lead tothe upregulation of VEGF autocrine signaling. One ofthe important and direct implications of our result isthat primary dormant tumors may become highlytumorigenic following exposure to drugs by activatinga signaling loop between MAPK/ERK1,2 and VEGF/Flt1. Hence, combination therapies that includeeither anti-Flt1 or MAPK/ERK1,2 inhibitors have thepotential benefit to reduce the incidence of drug-inducedtumor cell repopulation and relapse.
Methods
Cell lines and cultureThe human osteosarcoma cell line HOS and neuroblastomacell line SK-N-BE(2) were obtained from the AmericanTissue Culture Collection (ATCC, Rockville, MD, USA).The RH-4 rhabdomyosarcoma cell line was kindly provided byDr Thomas Look (Dana-Farber Cancer Institute, Boston,MA, USA). All cell lines were maintained in the culture mediaas recommended by ATCC.
Reagents and drugsCisplatin (CDDP), doxorubicin and methotrexate were ob-tained from Sigma (St Louis, MO, USA). Recombinanthuman VEGF165 (rhVEGF165), nonspecific goat IgG and aneutralizing antibody against VEGF165 (anti-human mousemonoclonal antibody) were purchased from R&D Systems(Minneapolis, MN, USA). Source of other reagents, anti-bodies and kits are noted with assay descriptions.
Side-population analysisThe protocol was based on that described by Montanaroet al. (2004). Details are given in the Supplementary Methodssection.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3932
Oncogene

Western blot analysis and immunoprecipitationThe overall method has been previously described in detail(Das et al., 2005). Details are given in the SupplementaryMethods section.
Methylcellulose-based clonogenic assayThe assay was performed as previously described (Das et al.,2003). Tumor cells were washed three times with phosphate-buffered saline and then 2� 103 cells were plated in methyl-cellulose media (Stem Cell Inc., Vancouver, BC, USA), andcolonies were counted after 2 weeks using an invertedmicroscope.
Reverse transcriptase-PCRDetails are given in the Supplementary Methods section.
Real-time quantitative RT-PCRReal-time qPCR was performed using TaqMan Gene Expres-sion Assays (Applied Biosystems, Foster City, CA, USA).Details are provided in the Supplementary Methods section.
Small interfering RNASmall interfering RNA knockdown of genes was performedaccording manufacturer’s instructions (details are provided inthe Supplementary Methods section).
ELISA analysisThe measurements of both VEGF were carried out using anELISA kit (R&D Systems) according to the manufacturer’sprotocols. Samples consisted of conditioned media harvestedafter treatment with or without CDDP (2 mM) for 3 days inserum-free media. Fluorescence activity was converted toactual concentration by a standard curve and normalized bycell number.
Immunofluorescence and immunohistochemistryStandard protocol was used as described in the SupplementaryMethods section.
In vitro matrigel tumorigenicity assayTumor cells were incubated for 8 h on top of a matrigel layer.Cells with tumorigenic potential form branched networks ofcells that grow and invade the matrigel structures, whereasnon-tumorigenic cells either form colonies or undergo celldeath (Miller et al., 2001). We modified the assay, where cellswere treated with serum-free medium containing 200 ngml�1
SDF1a (stromal cell-derived factor-1a), and then plated ingrowth factor-free matrigel (BD Bioscience, San Jose, CA,USA).
In vivo tumorigenicity assayTumorigenicity of the HOS cells was measured by tumorincidence (number of tumors/number of injection of 2–10� 106
cells), and latency was measured as time required to formpalpable tumors of more than 0.05 cm3. Details are given in theSupplementary Methods section.
Statistical analysisThe data are presented as mean±s.d. The statistical calcula-tions were performed with GraphPad Prism 4.0 (HearneScientific Software, Chicago, IL, USA) using Student’s t-testfor cell survival assays. A value of Pp0.05 was consideredstatistically significant.
Acknowledgements
This work is supported by the National Cancer Institute ofCanada, the Andrew Mizzoni Cancer Research Fund and theHarry and Hannah Fisher Research Fund. RT and BD aresupported in part by awards of the Hospital for SickChildren’s Research Training Centre and the National CancerInstitute of Canada Fellowship with funds from the Terry FoxFoundation. We thank Dr Meredith Irwin for critical review ofthe manuscript; Sherry Zhao, Shamim Lotif, Micky Tsui, RezaMokhtari, Michael Ho, Suzanne McGovern and Ana Novok-met for technical assistance; and the staff of the animal facilityof the Hospital for Sick Children for their technical support.
References
Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M,Oettgen P et al. (2003). Flt-1-dependent survival characterizes theepithelial–mesenchymal transition of colonic organoids. Curr Biol
13: 1721–1727.Behrends M, Schulz R, Post H, Alexandrov A, Belosjorow S, MichelMC et al. (2000). Inconsistent relation of MAPK activationto infarct size reduction by ischemic preconditioning in pigs. Am
J Physiol Heart Circ Physiol 279: H1111–H1119.Bellamy WT, Richter L, Sirjani D, Roxas C, Glinsmann-Gibson B,Frutiger Y et al. (2001). Vascular endothelial cell growth factor is anautocrine promoter of abnormal localized immature myeloidprecursors and leukemia progenitor formation in myelodysplasticsyndromes. Blood 97: 1427–1434.
Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL et al.(2007). Inhibition of TGF-beta with neutralizing antibodies preventsradiation-induced acceleration of metastatic cancer progression.J Clin Invest 117: 1305–1313.
Brozovic A, Osmak M. (2007). Activation of mitogen-activatedprotein kinases by cisplatin and their role in cisplatin-resistance.Cancer Lett 251: 1–16.
Cara S, Tannock IF. (2001). Retreatment of patients with the samechemotherapy: implications for clinical mechanisms of drugresistance. Ann Oncol 12: 23–27.
Cole S, Tannock IF. (2004). Drug Resistance, 4th edn. McGraw-Hill:Toronto, 390 pp.
Cottler-Fox MH, Lapidot T, Petit I, Kollet O, DiPersio JF, Link Det al. (2003). Stem cell mobilization. Hematology Am Soc Hematol
Educ Program, 419–437.Das B, Tsuchida R, Malkin D, Baruchel S, Yeger H. (2007). Hypoxiaenhances tumor stemness by increasing the invasive and tumorigenicside-population fraction. Stem Cells (under review).
Das B, Yeger H, Baruchel H, Freedman M, Koren G, Baruchel S.(2003). In vitro cytoprotective activity of squalene on a bone marrowversus neuroblastoma model of cisplatin-induced toxicity. Implica-tions in cancer chemotherapy. Eur J Cancer 39: 2556–2565.
Das B, Yeger H, Tsuchida R, Torkin R, Gee MF, Thorner PS et al.(2005). A hypoxia-driven vascular endothelial growth factor/Flt1autocrine loop interacts with hypoxia-inducible factor-1alpha throughmitogen-activated protein kinase/extracellular signal-regulated kinase1/2 pathway in neuroblastoma. Cancer Res 65: 7267–7275.
Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W et al. (2000).Autocrine stimulation of VEGFR-2 activates human leukemic cellgrowth and migration. J Clin Invest 106: 511–521.
Ferrara N, Gerber HP, LeCouter J. (2003). The biology of VEGF andits receptors. Nat Med 9: 669–676.
Folkman J. (1971). Tumor angiogenesis: therapeutic implications. N
Engl J Med 285: 1182–1186.Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N et al.(1998). Tumor induction of VEGF promoter activity in stromalcells. Cell 94: 715–725.
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3933
Oncogene

Gasparini G, Harris AL. (1995). Clinical importance of thedetermination of tumor angiogenesis in breast carcinoma: muchmore than a new prognostic tool. J Clin Oncol 13: 765–782.
Gee MF, Tsuchida R, Eichler-Jonsson C, Das B, Baruchel S, MalkinD. (2005). Vascular endothelial growth factor acts in an autocrinemanner in rhabdomyosarcoma cell lines and can be inhibited withall-trans-retinoic acid. Oncogene 24: 8025–8037.
Hirschmann-Jax C, Foster AE, Wulf GG, Goodell MA, Brenner MK.(2005). A distinct ‘side population’ of cells in human tumorcells: implications for tumor biology and therapy. Cell Cycle 4:203–205.
Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW,Gobel U et al. (2004). A distinct ‘side population’ of cells with highdrug efflux capacity in human tumor cells. Proc Natl Acad Sci USA
101: 14228–14233.Ho MM, Ng AV, Lam S, Hung JY. (2007). Side population in humanlung cancer cell lines and tumors is enriched with stem-like cancercells. Cancer Res 67: 4827–4833.
Kim JJ, Tannock IF. (2005). Repopulation of cancer cells duringtherapy: an important cause of treatment failure. Nat Rev Cancer 5:516–525.
Kondo T, Setoguchi T, Taga T. (2004). Persistence of a smallsubpopulation of cancer stem-like cells in the C6 glioma cell line.Proc Natl Acad Sci USA 101: 781–786.
Lin X, Costa M. (1994). Transformation of human osteoblasts toanchorage-independent growth by insoluble nickel particles. Environ
Health Perspect 102(Suppl 3): 289–292.McAllister RM, Gardner MB, Greene AE, Bradt C, Nichols WW,Landing BH. (1971). Cultivation in vitro of cells derived from ahuman osteosarcoma. Cancer 27: 397–402.
Mercurio AM, Bachelder RE, Bates RC, Chung J. (2004). Autocrinesignaling in carcinoma: VEGF and the alpha6beta4 integrin. Semin
Cancer Biol 14: 115–122.Miller AC, Blakely WF, Livengood D, Whittaker T, Xu J, Ejnik JWet al. (1998). Transformation of human osteoblast cells to thetumorigenic phenotype by depleted uranium-uranyl chloride.Environ Health Perspect 106: 465–471.
Miller AC, Mog S, McKinney L, Luo L, Allen J, Xu J et al. (2001).Neoplastic transformation of human osteoblast cells to thetumorigenic phenotype by heavy metal-tungsten alloy particles:induction of genotoxic effects. Carcinogenesis 22: 115–125.
Miura K, Uniyal S, Leabu M, Oravecz T, Chakrabarti S, Morris VLet al. (2005). Chemokine receptor CXCR4-beta1 integrin axis
mediates tumorigenesis of osteosarcoma HOS cells. Biochem Cell
Biol 83: 36–48.Montanaro F, Liadaki K, Schienda J, Flint A, Gussoni E, KunkelLM. (2004). Demystifying SP cell purification: viability, yield, andphenotype are defined by isolation parameters. Exp Cell Res 298:144–154.
Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K,Tang DG. (2005). Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2� cancer cells aresimilarly tumorigenic. Cancer Res 65: 6207–6219.
Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG.(2007). Hierarchical organization of prostate cancer cells inxenograft tumors: the CD44+{alpha}2{beta}1+ cell population isenriched in tumor-initiating cells. Cancer Res 67: 6796–6805.
Qi L, Robinson WA, Brady BM, Glode LM. (2003). Migration andinvasion of human prostate cancer cells is related to expression ofVEGF and its receptors. Anticancer Res 23: 3917–3922.
Ranganathan AC, Adam AP, Aguirre-Ghiso JA. (2006). Opposingroles of mitogenic and stress signaling pathways in the induction ofcancer dormancy. Cell Cycle 5: 1799–1807.
Richman CM, Weiner RS, Yankee RA. (1976). Increase in circulatingstem cells following chemotherapy in man. Blood 47: 1031–1039.
Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. (1999).Nickel-induced transformation shifts the balance between HIF-1and p53 transcription factors. Carcinogenesis 20: 1819–1823.
Setoguchi T, Taga T, Kondo T. (2004). Cancer stem cells persist inmany cancer cell lines. Cell Cycle 3: 414–415.
Soker S, Kaefer M, Johnson M, Klagsbrun M, Atala A, Freeman MR.(2001). Vascular endothelial growth factor-mediated autocrinestimulation of prostate tumor cells coincides with progression to amalignant phenotype. Am J Pathol 159: 651–659.
Steiner HH, Karcher S, Mueller MM, Nalbantis E, Kunze S, Herold-Mende C. (2004). Autocrine pathways of the vascular endothelialgrowth factor (VEGF) in glioblastoma multiforme: clinical rele-vance of radiation-induced increase of VEGF levels. J Neurooncol
66: 129–138.Strizzi L, Catalano A, Vianale G, Orecchia S, Casalini A, Tassi G et al.(2001). Vascular endothelial growth factor is an autocrine growthfactor in human malignant mesothelioma. J Pathol 193: 468–475.
Woessmann W, Chen X, Borkhardt A. (2002). Ras-mediated activa-tion of ERK by cisplatin induces cell death independently of p53 inosteosarcoma and neuroblastoma cell lines. Cancer Chemother
Pharmacol 50: 397–404.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
CDDP treatment upregulates VEGF and Flt1 expressionR Tsuchida et al
3934
Oncogene





























