Embed Size (px)
Citation preview

© The Author 2013. Published by Oxford University Press. All rights reserved. For Permissions, please email: [email protected]
Carcinogenesis vol.35 no.4 pp.923–934, 2014doi:10.1093/carcin/bgt407Advance Access publication December 9, 2013
FAT10, an ubiquitin-like protein, confers malignant properties in non-tumorigenic and tumorigenic cells
Yun Gao1,2,†, Steven Setiawan Theng3,4,†, Jingli Zhuo1,†, Wei Bing Teo3, Jianwei Ren1 and Caroline G.L.Lee1,3,4,5,* 1Division of Medical Sciences, Humphrey Oei Institute of Cancer Research, National Cancer Centre Singapore, Singapore 169610, Singapore, 2Department of Endocrinology and Metabolism, Institute of Endocrinology, Liaoning Provincial Key Laboratory of Endocrine Diseases, The First Affiliated Hospital of China Medical University, Shenyang, 110001, People’s Republic of China, 3Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore 119077, Singapore and 4NUS Graduate School for Integrative Sciences and Engineering, 28 Medical Drive #05-01, Singapore 117597, Singapore and 5Cancer and Stem Cell Biology, Duke-NUS Graduate Medical School Singapore, Singapore 169547, Singapore
*To whom correspondence should be addressed. Tel: +65 6436 8353; Email: [email protected]
FAT10 (HLA-F-adjacent transcript 10) is an ubiquitin-like modi-fier, which has been implicated in immune response and cancer development. In particular, the hypothesis of FAT10 as a mediator of tumorigenesis stems from its ability to associate with a spindle checkpoint protein Mad2 during mitosis and cause aneuploidy, a hallmark of cancer cells. Furthermore, FAT10 is overexpressed in several carcinomas types, including that of liver and colon. Nevertheless, direct evidence linking FAT10 to cell malignant trans-formation and progression is lacking. Here, we demonstrate that high FAT10 expression enhanced the proliferative, invasive, migra-tory and adhesive functions of the transformed cell line, HCT116. These observations were consistently demonstrated in an immor-talized, non-tumorigenic liver cell line NeHepLxHT. Importantly, FAT10 can induce malignant transformation as evidenced from the anchorage-independent growth as well as in vivo tumor-form-ing abilities of FAT10-overexpressing NeHepLxHT cells, whereas in rapidly proliferating HCT116, increased FAT10 further aug-mented tumor growth. FAT10 was found to activate nuclear factor-κB (NFκB), which in turn upregulated the chemokine receptors CXCR4 and CXCR7. Importantly, small interfering RNA deple-tion of CXCR7 and CXCR4 attenuated cell invasion of FAT10-overexpressing cells, indicating that the CXCR4/7 is crucial for the FAT10-dependent malignant phenotypes. Taken together, our data reveal novel functions of FAT10 in malignant transformation and progression, via the NFκB-CXCR4/7 pathway.
Introduction
FAT10 (HLA-F-adjacent transcript 10) is an 18 kDa ubiquitin-like modifier, which comprises two tandem head-to-tail ubiquitin-like domains. Similar to other ubiquitin-like modifier proteins, FAT10 contains a C-terminus diglycine motif, which is essential for covalent conjugation to itself and other substrates (1,2). FAT10 can also form non-covalent interactions with other proteins (3). Unlike ubiquitin, which is well characterized in cellular functions (4), the physiological functions of FAT10 are unclear, as the substrates and interaction part-ners of FAT10 remain largely unknown, with few exceptions includ-ing p53 (5), Mad2 (3,6), the E1 ubiquitin-activating enzyme (UBA6)
(7), p62 (1), histone deacetylase 6 (HDAC6) (8), huntingtin (9) and UBA6-specific E2 ubiquitin-conjugating enzymes (USE-1) (10).
Although the physiological functions of FAT10 remain to be elu-cidated, emerging evidence suggests that FAT10 may be implicated in tumorigenesis and malignancy. Upregulation of FAT10 was found in various carcinomas, including hepatocellular, gastrointestinal and gynecological cancers (11–16) as well as glioma (17). Among these studies, an association between increased FAT10 expression and pro-gression of disease was found in hepatocellular carcinoma (HCC) (16), gastric cancer (13), colon cancer (14) and glioma (17) patients. Interestingly, FAT10 expression can be induced by carcinogens in murine models (18,19), as well as by proinflammatory cytokines in cancer cell lines and immune cells (6,15), raising a possibility that FAT10 may function as a mediator of inflammation-associated tumo-rigenesis. An indirect link of FAT10’s involvement in tumorigenesis comes from studies which showed that FAT10 is able to bind and deregulate the function of the spindle assembly checkpoint protein Mad2 during mitosis (3) to induce abbreviated mitotic phase and ane-uploidy (6,20), a hallmark of many solid cancers (21). Nevertheless, it remains unclear whether an aberrant increase in FAT10 levels can pro-mote malignant transformation, forming the basis for this study. We demonstrate that FAT10 overexpression in an immortalized, non-tum-origenic liver cell line, NeHepLxHT, conferred malignant properties, including increased cell proliferation, anchorage-independent growth, resistance to apoptosis as well as invasion in vitro. Importantly, FAT10-overexpressing NeHepLxHT cells induced tumor formation in nude mice, whereas the wild-type (WT) and empty vector-transfected cells remained non-tumorigenic. Furthermore, these promalignant proper-ties of FAT10 were consistently demonstrated in a tumorigenic colo-rectal cell line, HCT116 as well. Genome-wide expression analyses of FAT10-overexpressing NeHepLxHT and HCT116 cells revealed that nuclear factor-κB (NFκB)-associated networks were dysregulated in common. FAT10 overexpression was further found to activate NFκB, which in turn upregulated CXCR4/CXCR7, facilitating the proinva-sive and migratory functions of FAT10 in NeHepLxHT.
Taken together, our data argue for an important role for FAT10 in promoting malignancy in non-cancerous as well as tumor cells. This study would provide a constructive framework with which to further explore the mechanisms of FAT10 in cancer progression.
Materials and methods
Cell culture and antibodieshTERT-immortalized hepatocyte cell line NeHepLxHT and the human colo-rectal cancer cell line HCT116 were purchased from Creative Bioarray (Long Island, NY) and American Type Culture Collection (Rockville, MD), respec-tively. NeHepLxHT were cultured in Dulbecco’s modified Eagle’s medium/F12 medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS), dexamethasone, insulin (Lonza, Walkersville, MD) and 50 μg/ml G418 (Promega, Fitchburg, WI), whereas HCT116 was cultured in McCoy’s 5A medium (Sigma–Aldrich, St Louis, MO) supplemented with 10% FBS and 800 μg/ml hygromycin. All cell lines were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Rabbit anti-FAT10 polyclonal antibodies were generated as described previ-ously (11). Anti-PCNA, anti-β-actin, anti-Ikβ-α as well as secondary antibod-ies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), whereas anti-p65 and anti-IKKβ were from BD Biosciences (San Diego, CA) and Cell Signaling Technology, respectively. Immunoreactive proteins were visualized using the Enhance Chemiluminescent Detection System kit (GE Health sys-tem, Piscataway, NJ). The FITC-PCNA kit for flow cytometry was from BD Biosciences.
Human HCC samplesThirteen human HCC tissue samples from anonymized patients were obtained from the National Cancer Centre of Singapore Tissue Repository, which had obtained written informed consent from study participants. This study has
Abbreviations: FAT10, HLA-F-adjacent transcript 10; FBS, fetal bovine serum; HCC, hepatocellular carcinoma; IPA, Ingenuity Pathway Analyses; NFκB, nuclear factor-κB; PBS, phosphate-buffered saline; siRNA, small inter-fering RNA; WT, wild-type.
†These authors contributed equally to this work.
923
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

Y.Gao et al.
prior approval from the National Cancer Centre of Singapore Institutional Review Board (NCC_IRB_No_2003/415/B).
RNA was extracted from these samples and real-time PCR analysis for FAT10 expression performed as described under ‘Real-time quantitative RT-PCR’.
Establishment of FAT10-overexpressing cell linesHuman FAT10 complementary DNA was cloned into the p4.1 vector (Invitrogen). To generate stable NeHepLxHT cell lines, FAT10-p4.1 or empty p4.1 vector were transfected into NeHepLxHT cells using Lipofectamine2000 (Invitrogen) and selected using 200 μg/ml hygromycin (Sigma–Aldrich). Stable FAT10-overexpressing as well as FAT10-knockdown HCT116 cell lines were generated previously as described (6,20).
siRNA transfectionA total of 250 nM of small interfering RNA (siRNA) against CXCR4, CXCR7 or p65 (Santa Cruz Biotechnology) were electroporated into 2 × 106 FAT10-overexpressing NeHepLxHT cells at 180 V for 100 ms. The Universal Negative Control 1 siRNA (Ambion), and no siRNA treatment were included as controls. Cells were harvested at 48 h and used for invasion assay as described. A total of 100 nM of FAT10 siRNA (Ambion) were transfected into FAT10-overexpressing HCT116 cells using siPORT-Amine reagent (Ambion). For transient overexpression of pCMV4-3 HA/Iκβ-α plasmid (AddGene, Cambridge, MA), Lipofectamine2000 (Invitrogen) was used.
Cell proliferationCells were seeded on six wells and cell proliferation was determined by trypan blue exclusion cell counting method at various timepoints (24–192 h). Cell doubling time was computed using the software at http://www.doubling-time.com/compute.php.
Anchorage-independence growth was investigated using the soft agar col-ony formation assay. Briefly, 10 000 NeHepLxHT cells or 5000 HCT116 cells were mixed with 0.4% agarose (Invitrogen) in complete media and seeded on top of a 0.7% agarose layer. Complete media was replaced every 3–4 days for 3 weeks after which colonies were stained with 0.01% methylene blue stain dissolved in 40% methanol and quantified.
Ectopic xenograft tumor model1 × 107 NeHepLxHT cells or 5 × 106 HCT116-derived stable cells were injected subcutaneously into 6-week-old male BALB/c nude mice (Biological Resource Centre, Singapore). FAT10-overexpressing and vector control cells were injected into the right and left flank, respectively, of the mice. At least six mice were used per treatment group. The tumors were monitored by measur-ing two perpendicular diameters using a digital caliper twice weekly and the tumor volume was determined using the formula 0.5 × a × b2, where a and b are the largest and smallest diameters of the tumor, respectively. At the end of the study (8 weeks for NeHepLxHT3 study; 3 weeks for HCT116 study), all mice were euthanized. This experiment was approved by the SingHealth Institutional Animal Care and Use Committee (Institutional Animal Care and Use Committee Protocol no.: 2009/SHS/504) and conducted in accordance to guidelines.
Cell death analysisTo determine cell death, 1 × 106 NeHepLxHT and 0.4 × 106 HCT116 cells were cultured on 6-well plates for 18 h followed by incubation with 20 μM camptothecin (Sigma–Aldrich) for 24 and 18 h, respectively. Subsequently, the cells were harvested, stained with annexin-V-PE and 7-aminoactinomycin D and analyzed using FACSCalibur flow cytometer (BD Biosciences).
Matrigel invasion assayThe invasion assays were performed using the Matrigel-coated Invasion Chambers (BD Biosciences). 5 × 104 NeHepLxHT or HCT116 cells were seeded on the matrigel layer in medium containing 0.1% FBS. Ten percent-age of FBS was used as chemoattractant at the bottom transwell chamber. After incubation for 36 h at 37°C, cells were fixed with methanol. Cells on the upper surface of each membrane were removed with cotton swabs, whereas the invaded cells on the underside were stained with Diff Quick (Siemens Healthcare Diagnostics, Deerfield, IL) and manually quantified based on five distinct fields (×100 magnification).
Scratch-wound migration assayCell migration was assessed using the scratch-wound assay. Confluent mon-olayers of NeHepLxHT and HCT116 cells were scratched with a pipette tip and monitored for 24 and 15 h, respectively. The migration distance of NeHepLxHT cells from the edge of the wound to the furthest migration point was measured in five random fields (×200 magnification). For the HCT116 migration study, the distance across the gap was measured, based on five fields (×100 magnification). Alternatively, cells were scratched by WoundMaker™,
imaged by Incucyte Zoom (Essen Bioscience), and analyzed using Incucyte Zoom software that tracks the cell confluency of the wounded area. The per-centage increase in the confluency of wounded area from start to endpoint (12 or 24 h) was computed.
Cell adhesion assay5 × 105 NeHepLxHT cells or 2 × 105 HCT116 cells were seeded on collagen IV-coated (Sigma–Aldrich) 6-well plates and incubated for 30 min at 37°C. Four wash steps using phosphate-buffered saline (PBS) were performed to remove non-adherent cells. The adhered cells were harvested by trypsiniza-tion, and quantified using a Beckman cell coulter counter (Beckman Coulter, Indianapolis, IN).
Phalloidin immunofluorescenceNeHepLxHT or HCT116 cells were seeded on collagen-coated or poly-l-ly-sine-coated coverslips, respectively, for 24–48 h. Cells were then fixed with 4% paraformaldehyde in PBS for 15 min, washed with PBS, permeabilized in 0.2% PBS-Triton X-100 for 10 min and blocked with 1% bovine serum albumin for 1 h at room temperature. To visualize filamentous actin, the cells were stained with rhodamine-conjugated phalloidin antibody (Invitrogen) for 30 min at 37°C followed by washing with PBS-Triton X-100. The cover-slips were then mounted onto the glass slides using mounting medium (Olink Bioscience, Uppsala, Sweden or Sigma–Aldrich) and images were obtained using Olympus IX51 microscope.
Microarray analyses
Microarray analyses were performed on WT NeHepLxHT and HCT116 cells as well as their respective stable FAT10-overexpressing cell lines (NeHepLxHT FAT10-A/B/C, HCT116 FAT-A/B), using HumanHT-12v4 Expression Beadchips (Illumina) according to the manufacturer’s instructions and analyzed using the Partek software (Partek, St Louis, MO). Genes show-ing an average of at least 2-fold change in all FAT10-overexpressing clones compared with WT and with P < 0.05 were considered to be differentially expressed.
Ingenuity Pathway Analyses (IPA) (www.ingenuity.com) was used to iden-tify the most significant biological functions, diseases and canonical pathways. The significant overrepresented functions, diseases and pathways were identi-fied by the right-tailed Fisher’s exact test using a threshold P < 0.05. Network analyses were performed by IPA using all the genes with ≥2-fold change in gene expression and P < 0.05. Briefly, IPA overlays test genes onto a global molecular network developed from information contained in the Ingenuity knowledge base. Networks of uploaded genes are then algorithmically gener-ated based on their connectivity. The score assigned to each network is the negative log of the P value and is calculated by the right-tailed Fisher’s exact test using the hypergeometric distribution.
Real-time quantitative reverse transcription–polymerase chain reactionTotal RNA was extracted from NeHepLxHT cells using TRIzol rea-gent (Invitrogen). Next, reverse transcription–polymerase chain reac-tion was performed using oligo(dT)17 primer and SuperScript II kit (Invitrogen). A total of 10–100 ng of complementary DNA were then used for real-time PCR using QuantiTect SYBR Green PCR kit (Qiagen, Hilden, Germany) and carried out on the 7500 Fast Real-Time PCR sys-tem (Applied Biosystems, Foster City, CA). The primers which were designed using VectorNTI software (Invitrogen) are specific for genes encoding CXCR4 (forward primer: 5′ GCCTTATCCTGCCTGGTATTGTC 3′; reverse primer: 5′ GCGAAGAAAGCCAGGATGAGGAT 3′), CXCR7 (forward primer: 5′ CACAGCACAGCCAGGAAGG 3′; reverse primer: 5′ GTTCCCTGGCTCTGAGTAGTCGA 3′) and FAT10 (for-ward primer: 5′ CAATGCTTCCTGCCTCTGTG 3′; reverse primer: 5′ TGCCTCTTTGCCTCATCACC 3′). Glyceraldehyde 3-phosphate dehydroge-nase (forward primer: 5′ AGCACCCCTGGCCAAGGTCA 3′; reverse primer: 5′ GCAGTGGGGACACGGAAGGC 3′) was used as an internal control.
NFκB activity
The Great EscAPe-secreted alkaline phosphatase NFκB reporter assay (Clontech, Mountain View, CA) was used to assess NFκB transcriptional activity. NeHepLxHT cells were cotransfected with pNFκB-secreted alka-line phosphatase/green fluorescent protein construct using lipofectamine2000 (Invitrogen). Thirty-six hours posttransfection, cell conditioned media were col-lected and incubated with the substrate provided according to the manufacturer’s instructions. Absorbance readings of NFκB activity were taken with Fluoroskan Ascent FL Microplate Fluorometer (Thermo Scientific, St Leon-Rot, Germany) and normalized to green fluorescent protein activity. Additionally, immunofluo-rescence was employed to track NFκB localization. Cells were incubated with anti-p65 antibody overnight, followed by AlexaFluor488 secondary antibody for 1 h. Cell nuclei were stained by 4′,6-diamidino-2-phenylindole contained in
924
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

FAT10’s role in malignant transformation
the mounting media (Olink Bioscience). Cytoplasmic nuclear fractionation was also performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL).
Statistical analysisStatistical analyses were performed using unpaired Student’s t-test. Differences between sample means were considered significant if P < 0.05.
Results
FAT10 promotes cell proliferation and survival as well as induces cel-lular transformationThe malignant transformative potential of FAT10 was investigated by stably overexpressing FAT10 in immortalized, non-tumorigenic NeHepLxHT liver cells (FAT10-A, FAT10-B, FAT10-C) as well as HCT116 colorectal cancer cells (FAT-A, FAT-B). WT or empty vec-tor-transfected cells (V-Ctrl) were included as controls. As shown in Figure 1A (upper panel), the basal expression of FAT10 in WT and vector control cells is low or undetectable by western blot, in con-trast to the FAT10-overexpressing clones, which showed relatively abundant FAT10. In NeHepLxHT cells, a differential expression of FAT10 can be observed in the stable clones, with FAT10-A and -B expressing much higher FAT10 levels than FAT10-C. The FAT10 lev-els in the FAT10-overexpressing stable cells are clinically relevant, as they fall within the range of FAT10 messenger RNA levels observed from 13 human HCC samples (Figure 1A, lower left panel). Increased FAT10 levels in both NeHepLxHT and HCT116 significantly aug-mented cell proliferation in vitro (P < 0.05), as determined by the trypan blue exclusion cell counting method over a period of up to 8 days expressed as cell numbers (Figure 1B, top left and middle pan-els) or doubling time (Figure 1B, bottom left and middle panels). In addition, a dose-dependent effect of FAT10 on proliferation could be observed in NeHepLxHT FAT10-overexpressing clones (FAT10-A, -B, -C) (Figure 1B, left), whereby FAT10 levels correlated with cell proliferation. Of note, FAT10 knockdown (‘FATi’) in HCT116 cells, which overexpress FAT10, attenuated proliferation to similar levels observed in WT HCT116 (Figure 1B, top-middle panel), indicating the important role of FAT10 in modulating cell growth. This notion is also supported by the increased expression of the proliferation marker, proliferating cell nuclear antigen in FAT-A/B cells as detected by flow cytometry (Figure 1B, top panel, far right) and western blot (Figure 1B, bottom panel, far right).
Since FAT10 confers proliferative properties, we investigate its effect on cell survival in response to cellular stresses, such as lack of a solid substrate for adherence as well as genotoxic agents. The ability to survive under anchorage-independent conditions is one of the hall-marks of transformed malignant cells (22). As evident in Figure 1C (left panel), negligible number of colonies on semisolid soft agar were observed in the untransfected (WT) (between 0 and 2) and empty vec-tor-transfected (V-Ctr1 and V-Ctr2) non-tumorigenic NeHepLxHT (between 0 and 3). In contrast, significantly more colonies were observed in three different clones of NeHepLxHT cells stably express-ing different levels of FAT10 (72.7 ± 11.2 cells in FAT10-A, 59.0 ± 9.5 cells in FAT10-B and 28.8 ± 4.5 cells in FAT10-C), suggesting that FAT10 can induce anchorage-independent growth and malignant transformation in non-tumorigenic cells in a FAT10 dose-dependent manner. In the already transformed HCT116 cells, FAT10 overex-pression significantly increased the number of colonies (52 ± 16.7 cells in WT, 273 ± 63.2 cells in FAT-A, 258 ± 43.5 cells in FAT10-B) growing on soft agar and this increase can be attenuated by siRNA against FAT10 (63 ± 8.9 cells in FAT10i) (Figure 1C, right panel). To evaluate the effect of FAT10 on cell death resistance, NeHepLxHT were treated for 24 h with 20 μM camptothecin, a cytotoxic chemo-therapeutic agent, which inhibits DNA replication (23). Annexin-V staining showed that FAT10 overexpression in NeHepLxHT con-ferred a protective effect against camptothecin-induced cell death (6.1 ± 1.9% in FAT10-A and 6.3 ± 2.4% in FAT10-B) compared with WT (17.4 ± 3.1%) and vector control (18.9 ± 2.9%) cells. A protective effect of FAT10 on cell death resistance was similarly observed in
HCT116 (2.4 ± 0.3% in FAT-A, 3.5 ± 0.3% in FAT-B versus 6.9 ± 1.4% in WT and 7.48 ± 0.3 in FATi) (Figure 1D). These observations indi-cate that FAT10 overexpression not only promotes cell proliferation and cell survival in both non-tumorigenic as well as tumorigenic cells, it can also induce transformation in non-tumorigenic cells.
FAT10 promotes cell invasion, migration and adhesionBesides cell proliferation and cell survival, malignant transforma-tion of cells encompasses other important attributes, such as the gain of invasive and migratory capabilities. FAT10 overexpression significantly augmented the invasion of NeHepLxHT cells across matrigel by 3.6- to 8.4-fold compared with V-Ctr1, with a dose-dependent effect observed (Figure 2A, left panel). In HCT116 FAT10-overexpressing cells, the proinvasive function of FAT10 was similarly observed (at least 5-fold compared with WT) (Figure 2A, right panel). Cell migration in both cell lines was also significantly increased by FAT10 overexpression, as evident from the scratch-wound-healing assay (Figure 2B). In addition, we investigated the effect of FAT10 on cell adhesion, since cell adhesion to extracellular matrix is involved in the invasion and migration processes. Cells were seeded on col-lagen IV and allowed to adhere for 30 min. A greater number of adherent cells were observed in FAT10-overexpressing NeHepLxHT and HCT116 stable clones compared with their respective controls (Figure 2C), indicating that FAT10 promotes cell adhesion. To rule out non-specific adhesion, cells were also seeded on bovine serum albumin-coated wells. No adherent NeHepLxHT and HCT116 cells were observed (data not shown).
As cell invasion and motility requires dynamic actin cytoskeletal remodeling and the formation of protrusive structures, we exam-ined the actin cytoskeleton of the cells using rhodamine–phalloidin staining. Overall, our immunofluorescence data indicate that FAT10-overexpressing cells exhibited more intense filamentous actin staining than WT and vector control cells (Figure 2D). As shown in Figure 2D, FAT10-overexpressing NeHepLxHT and HCT116 cells exhibited prominent lamellipodia migratory structures, which are broad exten-sion of the plasma membrane (24), consistent with the heightened motilities of these cells. Moreover, actin filaments were also observed to be enriched at the lamellipodial structures, especially in FAT10-overexpressing NeHepLxHT cells, consistent with the requirement of active cytoskeletal turnover (including polymerization and depolym-erization of actin filaments) at the edge of migrating cells (24).
Our findings indicate that besides its role in cell proliferation and survival, FAT10 confers invasive and migratory malignant properties to non-tumorigenic as well as tumorigenic cells.
FAT10 promotes tumor formation in nude miceHaving demonstrated the malignant properties of FAT10 in vitro, an important question to address is whether the tumorigenic properties can be recapitulated in vivo. Control NeHepLxHT cells (WT or V-Ctr) or FAT10-overexpressing cells (FAT10-A or FAT10-C) were injected subcutaneously into the left and right flanks of nude mice, respec-tively. No tumor formation was observed at the sites where control NeHepLxHT cells were injected (Figure 3, left panel). In stark con-trast, both FAT10-overexpressing clones induced tumor formation at all injected sites, demonstrating the ability of FAT10 to initiate tumo-rigenesis. Furthermore, FAT10 overexpression in HCT116 greatly promoted the growth of tumors in nude mice compared with WT HCT116 (Figure 3, right panel), indicating that the growth-promot-ing promalignant functions of FAT10 extends to tumorigenic cells as well. Of note, a consistent dose-dependent effect of FAT10 on tumor growth was observed in NeHepLxHT cells.
FAT10 overexpression leads to dysregulation of NFκB-associated signaling network and promotes invasion and migration via CXCR4/CXCR7In order to elucidate the molecular mechanisms by which FAT10 exerts its malignant effects, we performed expression microar-ray analyses on FAT10-overexpressing and control NeHepLxHT
925
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

Y.Gao et al.
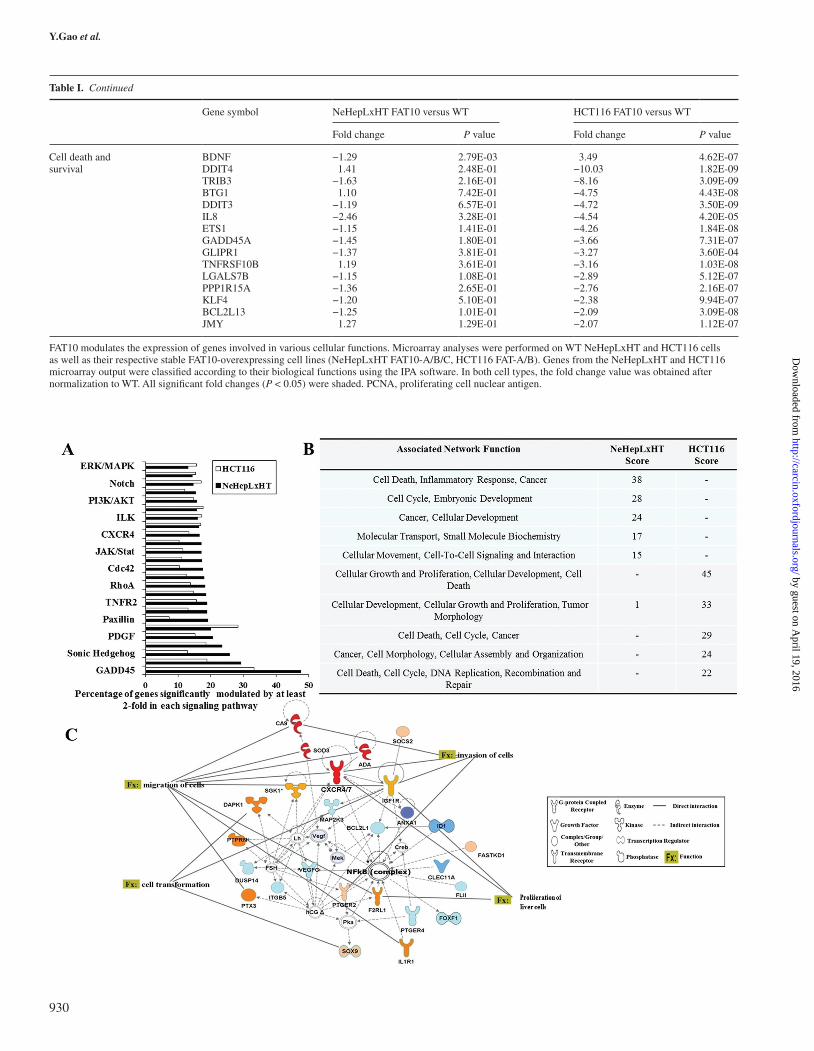
and HCT116 cells. A total of 724 genes (334 upregulated and 390 downregulated genes) were significantly altered (P < 0.05) by at least 2-fold in FAT10-overexpressing NeHepLxHT cells (FAT10-A, -B, -C) compared with V-Ctr (Supplementary Figure S1A, available at Carcinogenesis Online). In HCT116, 720 upregulated and 935 downregulated genes were detected to be altered (Supplementary Figure S1B, available at Carcinogenesis Online). Using the IPA software, genes from the microarray output were classified accord-ing to their cellular functions, including proliferation as well as migration and invasion. The genes under each biological process are shown in Table I. Despite the inherent molecular differences between the two cell lines NeHepLxHT and HCT116 cells, FAT10 overex-pression induced gene expression changes in common canonical
signaling pathways, which are often activated in pathological condi-tions (Figure 4A). IPA Molecular Network Analyses revealed that different top-associated biological networks were dysregulated by FAT10 in NeHepLxHT compared with HCT116 (Figure 4B). In the non-transformed NeHepLxHT cells, the top-associated network was ‘Cell Death, Inflammatory Response and Cancer’, consistent with the role of FAT10 in the early events of inflammation-associated tumori-genesis (6). The top-associated network in the transformed HCT116 cells was found to be ‘Cellular Growth and Proliferation, Cellular Development, Cell Death’ suggesting that FAT10 role extends beyond the early events of inflammation-associated tumorigenesis to influence cell growth, proliferation and death in transformed can-cer cells. Though different networks were associated with FAT10
Fig. 1. FAT10 promotes cell proliferation and cell survival. (A) (Left) FAT10 protein (top) and messenger RNA (mRNA) (bottom) expression in WT and various stably transfected NeHepLxHT cells (V-Ctrl1/2: vector control 1/2; FAT10-A, B and C: different clones stably expressing FAT10). FAT10 mRNA expression is normalized against β-actin and FAT10 mRNA expression in various human HCC patient samples were included to show the range of FAT10 expression in the tumors of HCC patients. (Right) FAT10 protein (top) and mRNA (bottom) expression in WT and various stably transfected HCT116 cells (FAT-A, B: different clones stably expressing FAT10; FAT-i: FAT10-overexpressing HCT116 cells stably transfected with siRNA against FAT10). FAT10 mRNA expression is normalized against β-actin. (B) Proliferation of WT NeHepLxHT (left), WT HCT116 (middle) and the various stably transfected cell lines were determined by trypan blue exclusion cell counting method. Cells were seeded on six wells and the number of cells was quantified at various timepoints, with up to 8 days for NeHepLxHT cells and 79 h for HCT116 (top panel). The cell doubling time (d) in hours was calculated as d = log2/k, where k is the growth constant determined by the slope of the linearly regressed line (bottom panel). FAT10-overexpressing cells have significantly shorter cell doubling time compared with WT. HCT116 WT and FAT-A/B cells were also stained with proliferative cell nuclear antigen (PCNA) and the expression of PCNA was determined through flow cytometry (far right, top panel) and western blot (far right, bottom panel). (C) Anchorage-independent survival and growth was accessed by suspending NeHepLxHT (left) and HCT116 (right) cells in soft agar and monitoring these cells for 3 weeks before staining and quantification. The colonies are photographed at ×20 magnification. Representative pictures are shown. A significantly greater proportion of FAT10-overexpressing cells exhibit the ability to survive and grow under anchorage-independent condition compared with WT. (D) Cell death profiles of NehepLxHT (left) or HCT116 (right) cells which were treated with 20 μM camptothecin for 24 h. Annexin V fluorescein staining was used to identify dead cells by fluorescence-activated cell sorting analysis. All bar charts show mean and standard deviation from triplicate experiments. *P < 0.05, **P < 0.01.
926
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

FAT10’s role in malignant transformation
in NeHepLxHT and HCT116 cells, closer examination revealed NFκB as a central key nodal molecule in the top pathways in both NeHepLxHT (Figure 4C) and HCT116 (Supplementary Figure S2, available at Carcinogenesis Online) cells. Hence, the NFκB-associated pathway(s) may be an important mechanism by which FAT10 exerts its promalignant effects.
To further elucidate the role of FAT10 in the events leading to malignant transformation, the top network in non-transformed NeHepLxHT cells was further examined. As shown in Figure 4C, the NFκB complex represents a key nodal hub in a network of 26 mol-ecules, of which the chemokine receptors CXCR4 and CXCR7 are among the genes which exhibited the greatest differential gene expres-sion (4.13x and 25.31x, respectively) between FAT10-expressing and vector control cells.
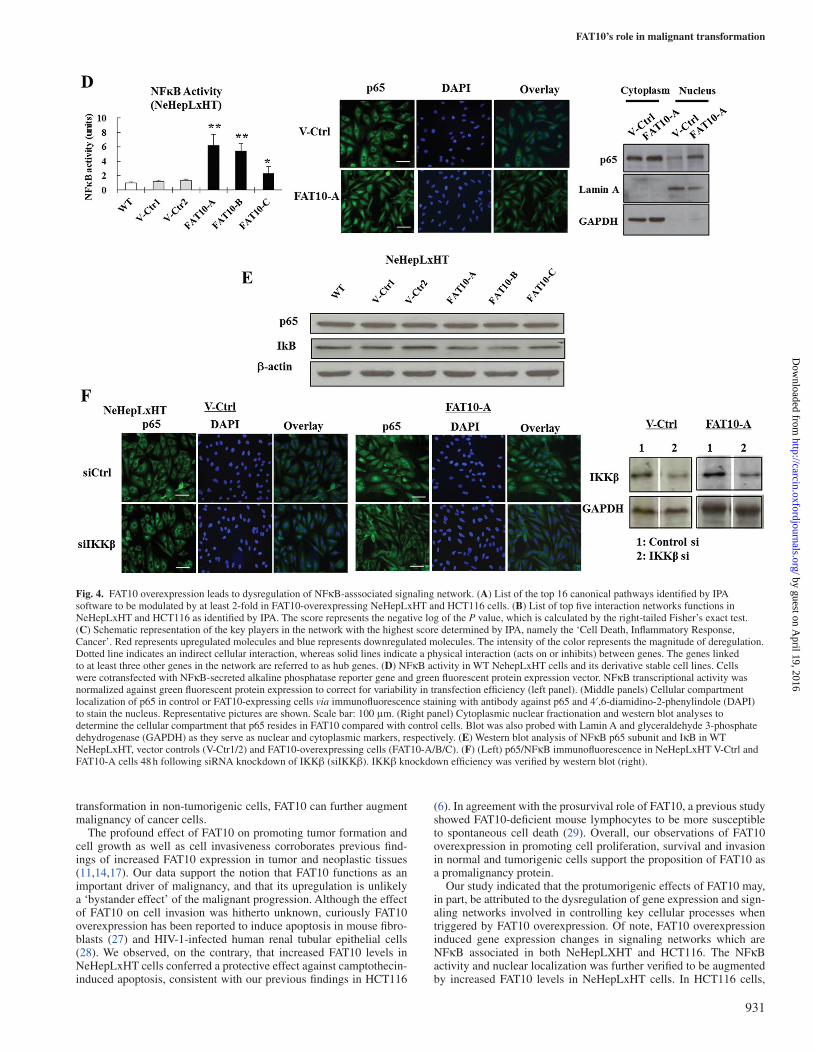
Figure 4D provides evidence that the NFκB transcriptional activ-ity is indeed significantly enhanced in the presence of upregulated FAT10 in NeHepLxHT cells, according to the NFκB reporter assay data (left panel). This observation was confirmed via immunofluores-cence and cellular fractionation, which showed higher nuclear locali-zation of p65/NFκB (Figure 4D, middle and right panels). Western blot analyses showed that the expression of p65 remained unchanged in FAT10-expressing NeHepLxHT cells. On the other hand, the levels of IκB, an inhibitor of NFκB, were reduced in FAT10-overexpressing NeHepLxHT clones (Figure 4E). Silencing of IKKβ, using IKKβ siRNA, an inhibitor of IκB, inhibited p65 nuclear translocation in FAT10-overexpressing cells (Figure 4F, middle panel). IKKβ knock-down efficiency was verified by western blot (Figure 4F, right panel). Although not to the same extent as was observed in NeHepLxHT,
Fig. 2. FAT10 promotes cell invasion, migration and adhesion. (A) Invasion profiles of WT NehepLxHT (left), WT HCT116 (right) and their respective stable derivative cell lines. Cells were assayed for invasion through matrigel for 36 h for NeHepLxHT cells and for 15 h for HCT116, after which the invaded cells were stained and quantified. FAT10 overexpression in NeHepLxHT and HCT116 significantly augmented cell invasion compared with WT cells. Representative pictures of the invaded cells are shown below the graphs. The data shown is representative of three experiments. (B) Cell migration profiles of NehepLxHT (left) or HCT116 (right) as assessed by scratch-wound-healing assay. Confluent NeHepLxHT and HCT116 cell monolayers were scratched and monitored for 24 and 15 h, respectively. The direction of cell migration is arrowed. The furthest distance which the NeHepLxHT cells migrated from the edge of scratch was measured in five random fields (×200). For HCT116, the gap of the artificial wound was measured in five fields (×100), and an inverse correlation of the distance of gap and migration was performed. Both graphs show the migration index relative to WT, based on three independent experiments. Representative pictures of the invaded cells are shown below the graphs. All data shown are representative of three experiments. (C) Profiles of the adhesive properties of WT NehepLxHT cells, WT HCT116 and their respective derivative cell lines. Cells were allowed to adhere to collagen IV-coated wells for 2 h, after which adhered cells were quantified in five random fields (×200). Graph shows the mean and standard deviation of the number of adhered cells. FAT10-overexpressing NeHepLxHT and HCT116 promoted significant adhesion to collagen compared with WT. The data shown are representative of three experiments. (D) Actin cytoskeleton profiles in NeHepLxHT and HCT116 cells. The cells were seeded on poly-l-lysine-coated plates, allowed to adhere for 24 h and stained with rhodamine-conjugated phalloidin to visualize filamentous actin. Lamellipodia structures are arrowed. Representative pictures (×400) are shown. Scale bar represents 50 μm (NeHepLxHT) and 20 μm (HCT116). *P < 0.05, **P < 0.01, ***P < 0.001.
927
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

Y.Gao et al.
similar increase in nuclear localization of p65 was observed in HCT116 cells when FAT10 was overexpressed (Supplementary Figure S3, right, available at Carcinogenesis Online). This increase in nuclear-localized p65 can be reversed when siRNA against FAT10 (FAT10 si) was introduced (Supplementary Figure S3, right bottom, available at Carcinogenesis Online). However, unlike in NeHepLxHT cells where the p65 protein levels remained unchanged upon FAT10 expression, in HCT116 cells, p65 expression is significantly induced in the presence of FAT10 and this increase in p65 protein expression can be attenuated by the introduction of siRNA. Taken together, our data indicate that increased FAT10 indirectly augmented NFκB activ-ity by alleviating the inhibition on NFκB.
The chemokine receptors, CXCR4 and CXCR7, which were found to be very highly upregulated in FAT10 cells and were pre-viously reported to activate NFκB (25,26), were further examined to evaluate if these chemokine receptors mediate FAT10’s invasive and migration properties. Consistent with the microarray analyses, real-time reverse transcription–polymerase chain reaction revealed that both CXCR4 and CXCR7 were significantly upregulated in the non-transformed NeHepLxHT that were stably expressing the FAT10 gene (Figure 5A). To evaluate our hypothesis that increased FAT10 heightens NFκB activity, which, in turn, promotes the expres-sion of CXCR4 and CXCR7 and mediates malignant transformation of cells, the NFκB pathway was silenced using siRNA against p65. Upon silencing p65, the transcript expression of both CXCR4 and CXCR7 in FAT10-expressing NeHepLxHT cells were significantly reduced (Figure 5B). Similar reduction of CXCR7 expression in FAT10-expressing NeHepLxHT cells was observed when IκB was introduced (Figure 5C). Notably, inhibition of the NFκB pathway via siRNA depletion of p65 (Figure 5D) as well as overexpression of IκB (Figure 5E) significantly attenuated cell invasion and migration
properties in NeHepLxHT cells overexpressing FAT10. Furthermore, siRNA silencing of CXCR4 and CXCR7 reduced cell invasion in FAT10-overexpressing NeHepLxHT clones (Figure 5F, left). However, only the inhibition of CXCR7 but not CXCR4 significantly inhibited cell migration (Figure 5F, right). Knockdown of neither CXCR4 nor CXCR7 has any effect on cell proliferation (Supplementary Figure S4, available at Carcinogenesis Online). Overall, our data suggest that NFκB and CXCR4/7 are key mediators by which FAT10 confers invasive and migratory properties in non-tumorigenic cells, thereby contributing to its malignant progression. A proposed model of the molecular mechanisms by which FAT10 confers cell malignancy is illustrated in Figure 6.
Discussion
Thus far, the role of FAT10 in tumorigenesis has been largely cir-cumstantial mainly based on correlative, expression studies of FAT10 in tumor samples (11,13,17). FAT10 was previously implicated in the induction of aneuploidy—a common characteristic of solid tumors (21)—by binding and deregulating the spindle checkpoint protein Mad2 and preventing it from localizing to the kinetochores during metaphase. Although FAT10 has also recently been reported to promote tumor progression in the transformed HepG2 cells (16), it remains unknown whether FAT10 can initiate tumorigenesis. In this study, we provide definitive evidence of FAT10’s promalig-nant roles, particularly in tumor initiation and cell growth by using an immortalized, non-tumorigenic liver cell line (NeHepLxHT). Similar malignant properties conferred by FAT10 were consistently observed in tumorigenic colorectal (HCT116) (Figures 1 and 2) as well as liver (HepG2) (16) cells, indicating that the effects of FAT10 are not cell type-specific, and that in addition to inducing malignant
Fig. 3. FAT10 promotes tumor formation in nude mice. Stably transfected NeHepLxHT cells (V-Ctr1, FAT10-A/C) as well as HCT116 (WT, FAT-A/B) were injected subcutaneously into the left and right flanks of at least six nude mice, respectively. Eight weeks after injection of NeHepLxHT or 3 weeks after injection of HCT116, the mice were killed and the weight of their subcutaneous primary tumors were measured. FAT10 overexpression in NeHepLxHT and HCT116 significantly promoted tumor growth compared with WT. No tumor can be observed in WT NehepLxHT and V-Ctr1 cells. *P < 0.05, **P < 0.01, ***P < 0.001.
928
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

FAT10’s role in malignant transformation
Table I. FAT10 modulates the expression of genes involved in various cellular functions
Gene symbol NeHepLxHT FAT10 versus WT HCT116 FAT10 versus WT
Fold change P value Fold change P value
Proliferation CA9 9.43 2.35E-03 1.01797 8.57E-01IL1R1 4.66 1.31E-03 −1.0194 8.86E-01LIN28B 3.45 3.02E-04 1.02364 8.06E-01E2F5 3.32 9.88E-03 −3.09738 6.80E-05IGFL3 3.17 1.39E-03 1.16623 2.41E-01CDC16 2.39 6.19E-03 1.04227 5.77E-01CDC23 2.24 7.79E-03 1.61041 1.12E-04EP300 2.04 1.32E-03 −1.03673 7.25E-01E2F7 −2.91 3.00E-03 1.27941 1.12E-02PDE4B −1.01 9.59E-01 17.39 1.82E-08NRIP1 1.27 4.21E-01 6.54 1.65E-06S100A4 −1.19 4.22E-01 6.14 2.06E-04SERPINB5 −1.05 8.24E-01 4.21 2.82E-06EREG −1.12 6.10E-02 3.91 9.44E-04FGFBP1 1.09 7.67E-02 3.74 2.17E-05SLC2A1 1.18 6.77E-01 3.59 2.91E-07ID1 −8.24 8.81E-03 3.03 8.94E-09CDC25A −1.63 4.33E-02 2.79 1.15E-07SGK1 3.07 7.34E-03 2.78 1.99E-08PCNA −1.92 7.98E-02 2.71 1.88E-07SKP2 −1.24 1.06E-01 2.7 5.41E-07MYD88 1.11 3.46E-01 2.43 3.12E-08E2F2 −1.72 2.85E-01 2.11 1.31E-06
Malignant transformation
SMAD3 2.45 4.04E-03 −2.11 5.74E-05FOS 3.68 2.84E-04 −2.75 9.52E-06CDCA7 3.87 1.88E-04 −1.05 5.90E-01JUN 2.48 1.99E-03 −2.57 5.60E-08RUNX2 3.21 1.67E-03 1.05 6.82E-01MYCBP2 2.16 2.62E-03 −1.20 1.13E-01WNT5A 2.13 1.56E-03 −1.01 9.35E-01TGFB3 2.06 1.90E-03 1.21 1.67E-02
Migration and invasion
CXCR7 25.31 1.01E-03 1.04 6.96E-01PDPN 4.8 4.97E-04 1.09 3.96E-01SPANXC 4.61 3.39E-03 −1.04 5.54E-01CXCR4 4.13 1.39E-03 1.42 5.03E-05IGFBP3 3.73 8.68E-03 4.40 9.96E-07ITGAV 2.58 1.67E-03 1.24 1.12E-01HMGB3 2.45 9.15E-03 1.73 1.07E-03VSNL1 −1.12 1.34E-01 13.01 2.59E-08S100A4 −1.19 4.22E-01 6.14 2.06E-04AXIN2 −1.11 4.74E-01 5.02 5.44E-08IGFBP3 4.40 9.96E-07 4.4 6.81E-08SERPINB5 −1.05 8.24E-01 4.21 2.82E-06CAV1 −1.76 1.08E-01 3.05 7.12E-06RRM2 −1.54 3.88E-01 2.43 3.12E-08MSX2 −1.01 9.84E-01 2.1 4.62E-04TCF7L2 1.03 7.79E-01 2 5.98E-05SOX9 2.43 8.24E-03 2 1.47E-04NDRG1 −2.17 2.07E-02 −3.99 3.01E-08ANXA10 −1.30 6.01E-02 −6.37 6.12E-05AKAP12 1.54 3.52E-01 -7.59 2.80E-08
Cytoskeleton rearrangement and adhesion
TUBB2B 7.98 9.20E-03 −1.02 8.75E-01FAM65B 7.13 8.08E-03 1.07 6.07E-01TNIK 6.04 4.17E-05 1.17 2.18E-01IQGAP2 4 5.51E-03 1.13 2.82E-01TUBA4A 3.1 8.01E-03 1.30 4.39E-01TUBB4 2.73 1.97E-03 2.30 5.20E-04ICAM2 2.36 4.99E-03 −1.08 3.45E-01LPXN −2.9 1.81E-03 −1.85 1.19E-04FEZ1 −1.02 8.28E-01 3.22 2.59E-08TUBB2C −2.32 2.34E-02 3.17 2.06E-04ITGA6 1.04 5.76E-01 2.97 5.44E-08TUBB4Q −1.55 6.56E-02 2.22 6.81E-08ARPM1 −1.10 1.62E-01 2.19 2.82E-06MAPK13 1.12 1.96E-01 2.06 7.12E-06TUBA1A −1.93 5.23E-03 2 3.12E-08KRT7 −1.51 2.10E-02 −2.28 1.27E-04KRT86 −1.89 3.03E-01 −5.81 5.98E-05KRT81 1.05 8.57E+01 −7.18 1.47E-04
929
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from
Y.Gao et al.
Gene symbol NeHepLxHT FAT10 versus WT HCT116 FAT10 versus WT
Fold change P value Fold change P value
Cell death and survival
BDNF −1.29 2.79E-03 3.49 4.62E-07DDIT4 1.41 2.48E-01 −10.03 1.82E-09TRIB3 −1.63 2.16E-01 −8.16 3.09E-09BTG1 1.10 7.42E-01 −4.75 4.43E-08DDIT3 −1.19 6.57E-01 −4.72 3.50E-09IL8 −2.46 3.28E-01 −4.54 4.20E-05ETS1 −1.15 1.41E-01 −4.26 1.84E-08GADD45A −1.45 1.80E-01 −3.66 7.31E-07GLIPR1 −1.37 3.81E-01 −3.27 3.60E-04TNFRSF10B 1.19 3.61E-01 −3.16 1.03E-08LGALS7B −1.15 1.08E-01 −2.89 5.12E-07PPP1R15A −1.36 2.65E-01 −2.76 2.16E-07KLF4 −1.20 5.10E-01 −2.38 9.94E-07BCL2L13 −1.25 1.01E-01 −2.09 3.09E-08JMY 1.27 1.29E-01 −2.07 1.12E-07
FAT10 modulates the expression of genes involved in various cellular functions. Microarray analyses were performed on WT NeHepLxHT and HCT116 cells as well as their respective stable FAT10-overexpressing cell lines (NeHepLxHT FAT10-A/B/C, HCT116 FAT-A/B). Genes from the NeHepLxHT and HCT116 microarray output were classified according to their biological functions using the IPA software. In both cell types, the fold change value was obtained after normalization to WT. All significant fold changes (P < 0.05) were shaded. PCNA, proliferating cell nuclear antigen.
Table I. Continued
930
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from
FAT10’s role in malignant transformation
transformation in non-tumorigenic cells, FAT10 can further augment malignancy of cancer cells.
The profound effect of FAT10 on promoting tumor formation and cell growth as well as cell invasiveness corroborates previous find-ings of increased FAT10 expression in tumor and neoplastic tissues (11,14,17). Our data support the notion that FAT10 functions as an important driver of malignancy, and that its upregulation is unlikely a ‘bystander effect’ of the malignant progression. Although the effect of FAT10 on cell invasion was hitherto unknown, curiously FAT10 overexpression has been reported to induce apoptosis in mouse fibro-blasts (27) and HIV-1-infected human renal tubular epithelial cells (28). We observed, on the contrary, that increased FAT10 levels in NeHepLxHT cells conferred a protective effect against camptothecin-induced apoptosis, consistent with our previous findings in HCT116
(6). In agreement with the prosurvival role of FAT10, a previous study showed FAT10-deficient mouse lymphocytes to be more susceptible to spontaneous cell death (29). Overall, our observations of FAT10 overexpression in promoting cell proliferation, survival and invasion in normal and tumorigenic cells support the proposition of FAT10 as a promalignancy protein.
Our study indicated that the protumorigenic effects of FAT10 may, in part, be attributed to the dysregulation of gene expression and sign-aling networks involved in controlling key cellular processes when triggered by FAT10 overexpression. Of note, FAT10 overexpression induced gene expression changes in signaling networks which are NFκB associated in both NeHepLXHT and HCT116. The NFκB activity and nuclear localization was further verified to be augmented by increased FAT10 levels in NeHepLxHT cells. In HCT116 cells,
Fig. 4. FAT10 overexpression leads to dysregulation of NFκB-asssociated signaling network. (A) List of the top 16 canonical pathways identified by IPA software to be modulated by at least 2-fold in FAT10-overexpressing NeHepLxHT and HCT116 cells. (B) List of top five interaction networks functions in NeHepLxHT and HCT116 as identified by IPA. The score represents the negative log of the P value, which is calculated by the right-tailed Fisher’s exact test. (C) Schematic representation of the key players in the network with the highest score determined by IPA, namely the ‘Cell Death, Inflammatory Response, Cancer’. Red represents upregulated molecules and blue represents downregulated molecules. The intensity of the color represents the magnitude of deregulation. Dotted line indicates an indirect cellular interaction, whereas solid lines indicate a physical interaction (acts on or inhibits) between genes. The genes linked to at least three other genes in the network are referred to as hub genes. (D) NFκB activity in WT NehepLxHT cells and its derivative stable cell lines. Cells were cotransfected with NFκB-secreted alkaline phosphatase reporter gene and green fluorescent protein expression vector. NFκB transcriptional activity was normalized against green fluorescent protein expression to correct for variability in transfection efficiency (left panel). (Middle panels) Cellular compartment localization of p65 in control or FAT10-expressing cells via immunofluorescence staining with antibody against p65 and 4′,6-diamidino-2-phenylindole (DAPI) to stain the nucleus. Representative pictures are shown. Scale bar: 100 μm. (Right panel) Cytoplasmic nuclear fractionation and western blot analyses to determine the cellular compartment that p65 resides in FAT10 compared with control cells. Blot was also probed with Lamin A and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as they serve as nuclear and cytoplasmic markers, respectively. (E) Western blot analysis of NFκB p65 subunit and IκB in WT NeHepLxHT, vector controls (V-Ctr1/2) and FAT10-overexpressing cells (FAT10-A/B/C). (F) (Left) p65/NFκB immunofluorescence in NeHepLxHT V-Ctrl and FAT10-A cells 48 h following siRNA knockdown of IKKβ (siIKKβ). IKKβ knockdown efficiency was verified by western blot (right).
931
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

Y.Gao et al.
Fig. 5. Role of p65-CXCR4/7 in modulating the malignant properties of FAT10. (A) Messenger RNA levels of CXCR4 and CXCR7 in WT NeHepLxHT and its derivative stable cell lines as assessed by real-time reverse transcription–polymerase chain reaction. FAT10 cells expressed significantly higher levels of CXCR4 and CXCR7 compared with WT. (B) Role of NFκB in modulating CXCR4/7 expression in WT NehepLxHT cells or its derivative stable cell lines. Cells were electroporated with siRNAs against p65 (si-p65) control siRNA (si-Ctr) and cultured for 36 h before harvesting for RNA extraction and real-time reverse transcription–polymerase chain reaction. NFκB p65 depletion significantly reduced the messenger RNA levels of CXCR4 and CXCR7. (C) The expression of CXCR4 and CXCR7 in NeHepLxHT vector control (V-Ctr1) and FAT10-overexpressing cells (FAT10-A) 48 h after IκB was introduced. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control. (D) p65 knockdown significantly reduced invasion and migration in FAT10 cells. FAT10-A NeHepLxHT cells were treated with siRNA against p65/NFκB (left panel) and invasion (middle panel) and migration (right panel) assays were performed. (E) IκB upregulation significantly reduced invasion and migration in FAT10 cells. IκB was overexpressed in FAT10-A NeHepLxHT cells and invasion (middle panel) and migration (right panel) assays were performed. Cont: Vehicle control-treated cells were transfected with empty plasmid. Representative pictures of cell invasion and migration are shown alongside the respective graphs. (F) Inhibition of CXCR7 significantly attenuated invasion as well as migration properties, whereas attenuation of CXCR4 inhibited invasive but not migration properties of FAT10-A cells. FAT10-A NehepLxHT cells were treated with siRNAs against CXCR4 and/or CXCR7 and invasion and migration assays were performed. No siRNA (No si) and universal negative control siRNA (Cont si) were included as controls. Graphs represent mean ± SD of three replicates. *P < 0.05, **P < 0.01, ***P < 0.001.
932
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

FAT10’s role in malignant transformation
FAT10 overexpression enhanced both the protein expression and nuclear localization of NFκB p65 subunit. This is in agreement with the requirement of FAT10 for tumor necrosis factor-α-induced NFκB activation in renal cells (30) and is consistent with the association of FAT10 with inflammation where FAT10 expression is inducible by cytokines tumor necrosis factor-α and interferon-γ in immune cells as well as cancer cells (3,6,15). FAT10 is also highly expressed in organs of the immune system and inflammation-associated cancers (11). Interestingly, putative NFκB binding sites have been found at the FAT10 promoter region (31), and indeed in colorectal cells, the tumor necrosis factor-α-dependent induction of FAT10 expression requires NFκB activation (6). These observations suggest a potential positive feedback system in cells surrounded by an inflammation-associated microenvironment in which cytokine-induced FAT10 induces NFκB, which in turn stimulates FAT10 production. A closer examination of the NFκB network activated in FAT10-overexpressing NeHepLxHT cells confirmed the upregulation of chemokine receptors CXCR4 and CXCR7, in part through FAT10-induced NFκB, consistent with NFκB as a known transcription factor for both receptors (32,33). On the other hand, CXCR4 and CXCR7 have also been reported to activate NFκB (25,26), pointing to a potential positive feedback loop in sus-taining NFκB activities. Importantly, we show that CXCR4/7 medi-ates the invasive and migratory phenotypes of FAT10-overexpressing NeHepLxHT cells, consistent with the reported functions of CXCR4 and CXCR7 in various cancers (34–37). The gene expression changes induced by FAT10 overexpression, including CXCR4 and CXCR7, are novel mechanisms by which FAT10 induces malignant progres-sion. Further elucidation of the FAT10-induced NFκB and CXCR4/7 as well as other signaling pathways identified in the microarray analy-ses is warranted to further understand the complex process of cancer development.
Taken together, this study demonstrates novel promalignant func-tions of FAT10, as evident from the ability of FAT10 to initiate tumorigenesis in non-transformed cells and promote the malignant progression of tumor cells. Further dissection of the mechanisms involved could contribute to new insights in cancer progression, as well as the development of novel anticancer strategies.
Supplementary material
Supplementary Figures S1–S4 can be found at http://carcin.oxford-journals.org/
Funding
National Medical Research Council (NMRC/1306/2011) as well as block funding from National Cancer Centre, Singapore.
Conflict of Interest Statement: None declared.
References
1. Aichem,A. et al. (2012) The proteomic analysis of endogenous FAT10 sub-strates identifies p62/SQSTM1 as a substrate of FAT10ylation. J. Cell Sci., 125, 4576–4585.
2. Hipp,M.S. et al. (2005) FAT10, a ubiquitin-independent signal for protea-somal degradation. Mol. Cell. Biol., 25, 3483–3491.
3. Liu,Y.C. et al. (1999) A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc. Natl Acad. Sci. USA, 96, 4313–4318.
4. Herrmann,J. et al. (2007) Ubiquitin and ubiquitin-like proteins in protein regulation. Circ. Res., 100, 1276–1291.
5. Li,T. et al. (2011) FAT10 modifies p53 and upregulates its transcriptional activity. Arch. Biochem. Biophys., 509, 164–169.
6. Ren,J. et al. (2011) FAT10 mediates the effect of TNF-α in inducing chro-mosomal instability. J. Cell Sci., 124(Pt 21), 3665–3675.
7. Pelzer,C. et al. (2010) FAT10: activated by UBA6 and functioning in pro-tein degradation. Subcell. Biochem., 54, 238–246.
8. Kalveram,B. et al. (2008) The ubiquitin-like modifier FAT10 interacts with HDAC6 and localizes to aggresomes under proteasome inhibition. J. Cell Sci., 121(Pt 24), 4079–4088.
9. Nagashima,Y. et al. (2011) FAT10 protein binds to polyglutamine proteins and modulates their solubility. J. Biol. Chem., 286, 29594–29600.
10. Aichem,A. et al. (2010) USE1 is a bispecific conjugating enzyme for ubiq-uitin and FAT10, which FAT10ylates itself in cis. Nat. Commun., 1, 13.
11. Lee,C.G. et al. (2003) Expression of the FAT10 gene is highly upregulated in hepatocellular carcinoma and other gastrointestinal and gynecological cancers. Oncogene, 22, 2592–2603.
Fig. 6. Proposed model of the molecular mechanisms by which FAT10 promotes cell malignancy. Hypothetical model of the molecular mechanism of malignant transformation of non-tumorigenic NehepLxHT liver cells by FAT10 integrating previous reported knowledge with current data.
933
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from

Y.Gao et al.
12. Qing,X. et al. (2011) Increased expression of FAT10 in colon benign, premalignant and malignant epithelial neoplasms. Exp. Mol. Pathol., 90, 51–54.
13. Ji,F. et al. (2009) FAT10 level in human gastric cancer and its relation with mutant p53 level, lymph node metastasis and TNM staging. World J. Gastroenterol., 15, 2228–2233.
14. Yan,D.W. et al. (2010) Ubiquitin D is correlated with colon cancer progres-sion and predicts recurrence for stage II-III disease after curative surgery. Br. J. Cancer, 103, 961–969.
15. Lukasiak,S. et al. (2008) Proinflammatory cytokines cause FAT10 upregu-lation in cancers of liver and colon. Oncogene, 27, 6068–6074.
16. Liu,L. et al. (2013) As an independent prognostic factor, FAT10 promotes hepatitis B virus-related hepatocellular carcinoma progression via Akt/GSK3beta pathway. Oncogene.
17. Yuan,J. et al. (2012) Increased expression of FAT10 is correlated with pro-gression and prognosis of human glioma. Pathol. Oncol. Res., 18, 833–839.
18. Yamashita,S. et al. (2002) Profiling and selection of genes differentially expressed in the pylorus of rat strains with different proliferative responses and stomach cancer susceptibility. Carcinogenesis, 23, 923–928.
19. Oliva,J. et al. (2008) Fat10 is an epigenetic marker for liver preneoplasia in a drug-primed mouse model of tumorigenesis. Exp. Mol. Pathol., 84, 102–112.
20. Ren,J. et al. (2006) FAT10 plays a role in the regulation of chromosomal stability. J. Biol. Chem., 281, 11413–11421.
21. Holland,A.J. et al. (2009) Boveri revisited: chromosomal instability, ane-uploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol., 10, 478–487.
22. Thiery,J.P. (2002) Epithelial-mesenchymal transitions in tumour progres-sion. Nat. Rev. Cancer, 2, 442–454.
23. Liu,L.F. et al. (2000) Mechanism of action of camptothecin. Ann. N. Y. Acad. Sci., 922, 1–10.
24. Lauffenburger,D.A. et al. (1996) Cell migration: a physically integrated molecular process. Cell, 84, 359–369.
25. Huang,C.Y. et al. (2009) Stromal cell-derived factor-1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF-kappaB-dependent pathways. J. Cell. Physiol., 221, 204–212.
26. Mosley,C.A. et al. (2009) Recent patents regarding the discovery of small molecule CXCR4 antagonists. Expert Opin. Ther. Pat., 19, 23–38.
27. Raasi,S. et al. (2001) The ubiquitin-like protein FAT10 forms covalent con-jugates and induces apoptosis. J. Biol. Chem., 276, 35334–35343.
28. Ross,M.J. et al. (2006) Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J. Am. Soc. Nephrol., 17, 996–1004.
29. Canaan,A. et al. (2006) FAT10/diubiquitin-like protein-deficient mice exhibit minimal phenotypic differences. Mol. Cell. Biol., 26, 5180–5189.
30. Gong,P. et al. (2010) The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J. Am. Soc. Nephrol., 21, 316–326.
31. Zhang,D.W. et al. (2006) p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene, 25, 2318–2327.
32. Tarnowski,M. et al. (2010) Regulation of expression of stromal-derived factor-1 receptors: CXCR4 and CXCR7 in human rhabdomyosarcomas. Mol. Cancer Res., 8, 1–14.
33. Katoh,M. et al. (2010) Integrative genomic analyses of CXCR4: transcrip-tional regulation of CXCR4 based on TGFbeta, Nodal, Activin signaling and POU5F1, FOXA2, FOXC2, FOXH1, SOX17, and GFI1 transcription factors. Int. J. Oncol., 36, 415–420.
34. Zheng,K. et al. (2010) Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res., 29, 31.
35. Heinrich,E.L. et al. (2012) Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med., 10, 68.
36. Gao,Z. et al. (2010) Pancreatic stellate cells increase the invasion of human pancreatic cancer cells through the stromal cell-derived factor-1/CXCR4 axis. Pancreatology, 10, 186–193.
37. Liang,Z. et al. (2005) Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res., 65, 967–971.
Received February, 18, 2013; revised November 12, 2013; accepted November 29, 2013
934
by guest on April 19, 2016
http://carcin.oxfordjournals.org/D
ownloaded from





























